
Submitted:
04 July 2023
Posted:
05 July 2023
You are already at the latest version
Abstract
Prostate-specific membrane antigen (PSMA) has been identified as a target for the development of theranostic agents. In our current work, we describe the design and synthesis of novel N-[N-[(S)-1,3-dicarboxypropyl]carbamoyl]-(S)-L-lysine (DCL) urea-based PSMA inhibitors with a chlorine-substituted aromatic fragment at the lysine ε-nitrogen atom, a dipeptide as peptide fragment of the linker, including two phenylalanine residues in the L-configuration, and 3- or 4-(tributylstannyl)benzoic acid as prosthetic group in their structures for radiolabeling. The standard compounds ([127I]PSMA-m-IB and [127I]PSMA-p-IB) for comparative and characterization studies were firstly synthesized using two alternative synthetic approaches. An important advantage of the alternative synthetic approach in which the prosthetic group (NHS-activated esters of compounds) is first conjugated with the polypeptide sequence, followed by replacement of the Sn(Bu)3 group with radioiodine, is that the radionuclide is introduced in the final step of synthesis, thereby minimizing operating time with iodine-123 during the radiolabeling process. The obtained DCL urea-based PSMA inhibitors were radiolabeled with iodine-123. The radiolabeling optimization results showed that the radiochemical yield of [123I]PSMA-p-IB was higher than those of [123I]PSMA-m-IB, which were 74.9± 1.0% and 49.4 ± 1.2%, respectively. The radiochemical purity of [123I]PSMA-p-IB after purification was greater than 99.50%. The initial preclinical evaluation of [123I]PSMA-p-IB demonstrated a considerable affinity and specific binding to PC-3 PIP (PSMA-expressing cells) in vitro. The in vivo biodistribution of this new radioligand [123I]PSMA-p-IB showed less accumulation than [177Lu]Lu-PSMA-617 in several normal organs (liver, kidney and bone). These results warrants further preclinical development including toxicology evaluation and experiments in tumor-bearing mice.
Keywords:
DCL ligand
; iodine radioisotopes
; radiolabeled pharmaceuticals
; targeted delivery
; prostatic specific membrane antigen
; prostate cancer
1. Introduction
Prostate cancer is currently one of the most commonly reported oncological diseases in men population [1]. Although the developments and practical uses of radiopharmaceuticals which are mediated by prostate-specific membrane antigen (PSMA) for the diagnosis and endoradiotherapy of prostate cancer have recently shown good results, further debelopment continues in a search of new, more efficient targeting agents capable of enhancing of effect of treatment.
PSMA functions as a human neuropeptidase glutamate-preferring carboxypepti-dase II (GCP II). PSMA has been identified as a target for the development of theranostic agents. PSMA is overexpressed in prostate cancer cells compared to healthy prostate cells. PSMA expression also exists in numerous tissue types, including the testis, ovary, brain, salivary gland, small intestine, colon, liver, spleen, breast, kidney tissue, and normal prostate tissues have the greatest expression [1,2]. The level of PSMA expression corresponds with tumor aggressiveness. PSMA is a target for delivery, prostate cancer diagnostics, and intraoperative guidance due to its high expression in prostate cancer [3,4].
Small molecule ligands are one of three classes of PSMA targeting molecules. Small molecules have a number of advantages over antibodies and aptamers, including: ease synthesis and modification, absence of immunogenicity, improved pharmacokinetic properties, and rapid clearance from normal tissues [5–9]. In recent years, there has been an increase in the number of publications devoted to optimizing the structure of the linker incorporating the vector fragment with a diagnostic agent, in addition to optimizing the structure of the ligand itself [10]. As a result, a variety of highly promising therapeutic and diagnostic conjugates have been developed [11–16].
Urea-based ligands are the most widely developed at present [10,17,18]. The advantages of urea-based ligands include significant potential for further modification, adequate bioavailability in comparison with ligands based on phosphinic and phosphonic acids [19], and this type of ligand is more stable than thiol-based ligands [20,21]. N-[N-[(S)-1,3-dicarboxypropyl]carbamoyl]-(S)-L-lysine (DCL) is one of the most developed urea-based ligands due to its promising potential [17,18]. A number of publications confirmed that modification of the DCL ligand structure affects its affinity for PSMA. We assume that the linker modification in DCL with a dipeptide fragment containing two phenylalanine residues improves the affinity properties of the PSMA ligand by taking advantage of the hydrophobic interaction with the S1 hydrophobic pocket of PSMA [22,23].
123I, as a gamma-ray emitter with an energy of 159 keV, is an ideal radionuclide for use in single photon emission computerized tomography (SPECT) diagnostics. The gamma emission of 123I enables for excellent imaging with low background activity (80% efficiency for a 1-inch-thick crystal). It delivers substantially lower absorbed doses to the patients while maintaining comparable activity to 131I [24].
The commonly used method for radioiodination of peptides was direct labeling. Radioiodine was oxidized in situ using an oxidizing agent to form I+ ions, which then attack the activated phenolic ring of the amino acid tyrosine in proteins and produced a stable covalent bond. This is not a problem when using prosthetic groups in the labeling of targeting protein or peptide. Radioiodination of a conjugate, in which the peptide is conjugated with a prosthetic group before iodination, is considered an appropriate idea [25].
Thus, this work aimed to synthesize novel DCL urea-based with chlorine-substituted aromatic fragment at the lysine ε-nitrogen atom, a dipeptide as peptide fragment of the linker including two phenylalanine residues in the L-configuration, and 4- or 3-(tributylstannyl)benzoic acid as prosthetic group in its structures. The general structure of the obtained ligands is shown in Figure 1. These ligands were studied as novel PSMA ligands by conducting radiolabeling optimization with iodine-123. The [123I]PSMA-p-IB ligand was tested in the initial preclinical evaluation. As a comparison of the biodistribution of [123I]PSMA-p-IB in normal mice, we used [177Lu]Lu-PSMA-617, which is known to have demonstrated promising results in clinical studies.
2. Results
2.1. Synthesis
For the synthesis of target compounds 19-22 (PSMA-m-TBSB, PSMA-p-TBSB, [127I]PSMA-m-IB, and [127I]PSMA-p-IB, a scheme including the sequential solution of five synthetic objectives was chosen (Schemes 1–3):
- synthesis of PSMA vector 6, which is a derivative of DCL urea modified with ε-aminocaproic acid (Ahx) and succinic acid (Suc) (Scheme 1);
- formation of a peptide linker (compound 8) using Solid Phase Peptide Synthesis (SPPS) (Scheme 2) and further connection of the vector fragment (compound 6) with a dipeptide linker (Scheme 2);
- modification of C-terminal fragment of the polypeptide sequence (compound 11) to connect it with the prosthetic groups of N-succinimidyl 3-(tributylstannyl)benzoate (m-STBSB)/compound 13 and N-succinimidyl 4-(tributylstannyl)benzoate m-STBSB/compound 16 (Scheme 2);
- obtaining of NHS-activated esters of m-STBSB (compound 13) and m-STBSB (compound 16) prosthetic groups (Scheme 3);
- combining a PSMA ligand with m-STBSB or p-STBSB prosthetic group, as well as the synthesis of [127I]PSMA-m-IB (compound 21) or [127I]PSMA-p-IB (compound 22 conjugates in two alternative ways (Scheme 4).
Figure 2 demonstrated the denotations of structural fragments of synthesized compounds used in the text (on the example of the compound 21, [127I]PSMA-m-IB). This denotation of fragment structure is employed in the characterization results of the resulting compounds, which are presented in the materials and methods section.
The initial stages of the vector fragment 6 synthesis (Scheme 1) were realized by previously described methods [26]. Compound 6 was prepared by coupling of succinic anhydrides with the tert-butylated compound 5 (Scheme 1); the resulting products contained a free carboxylic group was suitable for further addition of the peptide fragment.
The second stage of the synthesis consisted in assembling the peptide sequence Phe(L)-Phe(L) to obtain highly specific PSMA vectors (compounds 11), using SPPS on cross-linked styrene-divinylbenzene (1%) copolymer matrix (2-CTC resin) (Scheme 2). The selected reaction sequence was a classical scheme of peptide synthesis:
1) immobilization of an N-substituted amino acid onto a solid-phase substrate;
2) removal of the protective group;
3) modification of NH2- group of amino acid (stages 2 and 3 are repeated the required number of times to assemble the necessary peptide sequence);
4) removal of the modified amino acid sequence from the 2-CTC resin [27].
The using of 2-CTC resin allows to keep acid-labile functional groups intact, since the removal of amino acid sequence from the resin proceeds under mild conditions (in this case DCM/TFA – 99,25%/0,75.% V/V; the reaction does not affect СOOBut groups labile to acids) [28].
Then the vector fragment was attached to the immobilized on a 2-CTC resin dipeptide 8 using HOBt/HBTU/DIPEA as activating agents. After that, the modified peptide was removed from the polymer matrix by DCM/TFA treatment (99.25%-0.75%, V/V). As a result, compound 9 was isolated as an individual stereoisomer, which was confirmed by 1H and 13C NMR spectral data, LCMS and HRMS (Figure S4–S7 in Supplementary Information)
At the third stage, it was supposed to introduce a fragment of NH2(CH2)3NHFmoc into compound 9 by a peptide synthesis reaction, according to an optimized technique [26], and then to obtain compound 11 by Fmoc deprotection. During the synthesis of compound 10, it was found that the presence of a Phe-Phe-(СH2)3-Fmoc fragment in the molecule leads to the appearance of strong gelation properties of the target compound, which greatly complicates its isolation and purification [29]. Nevertheless, product 10 was isolated as an individual stereoisomer with an 88% yield (Figures S8-S11 in Supplementary Information). Next, the Fmoc protection was removed to obtain product 11 (Scheme 2).
The fourth stage was the preparation of NHS-activated esters of prosthetic groups p-STBSB and p-STBSB (Scheme 3). The synthesis was carried out similarly to the method described in the article [30], with some modifications (see Material and methods).
At the final stage, the protective tert-butyl groups of compound 11 were removed by TFA action (Scheme 4.). During the synthesis, the effectiveness of target conjugates 21, 22 obtaining was evaluated with comparing two alternative synthetic approaches (Scheme 4, Table 1). The first approach consisted in conjugation of the polypeptide sequence 18 with NHS-activated esters of m-S[127I]IB 14) and p-S[127I]IB (17) prosthetic groups (Way A). The second approach consisted in the reaction of the polypeptide sequence 18 with the obtained NHS-activated esters of compounds m-STBSB (13) and p-STBSB (16), followed by the replacing the Sn(Bu)3 group with 127I (Way B). The pros and cons of each of the approaches, as well as the total yield relative to compound 18 are shown in Table 1. From the data presented, it can be seen that the total yield relative to the prosthetic group does not differ significantly depending on the chosen pathway. It should also be emphasized the important advantage of Way B, relative to Way A - the radionuclide is introduced at the last stage of synthesis, thus minimizing the time of operation with 123I.
2.2. Radiolabeling Optimization of PSMA-p-TBSB with 123I: Physico-chemical Study
PSMA-p-TBSB (compound 20) was used as a model for studying radiolabeling optimization with 123I and initial preclinical evaluation of these novel PSMA-targeting ligand. Radiolabeling of the novel PSMA-targeting ligand by 123I was conducted via electrophilic radioiodination reaction by incubation with [123I]NaI in the presence of chloramine-T as an oxidizing agent. This radiolabeling reaction produced [123I]PSMA-p-IB (compound 23). The scheme of the radiolabeling reaction is shown as Figure 3. Radiolabeling optimization of this novel PSMA-targeting ligand with 123I was performed by investigating the effect of the PSMA ligand amount, the reaction time, and the oxidizing agent amount.
To study the effect of the PSMA ligand amount to the radiochemical yield, a fixed reaction time of 5 min and oxidizing agent amount of 40 µg was used. The radiochemical yields (RCY), determined by radio-iTLC as a function of PSMA ligand amount are presented in Figure 4. The reaction was quite efficient at a low mass of PSMA ligand (0.73-5 nmol or 1-7 µg). Generally, it appeared that the use of a larger molar amount of PSMA ligand in the radiolabeling reaction improved the labeling yield. However, no increase was found, instead a significant decrease (p = 0.0072) in RCY occurred when the amount of PSMA ligand was increased from 10 nmol to 50 nmol. The radiochemical yield on the use of 10 nmol amount of PSMA ligand was 75.9 ± 1.0 %.
Reaction time of the radiolabeling was studied using fixed amounts of 7 µg (5 nmol) PSMA ligand and 40 μg (176 nmol) of oxidizing agent. It appears that the 30 seconds reaction process has produced a radiochemical yield of 61.9 ± 0.4%. The increase in radiochemical yield occurred during the increase in reaction time until the use of a 10-minute reaction time produced RCY 71.7 ± 0.3%. Radiochemical yield started to slightly decrease when the reaction time was extended to 30 min. The radiochemical yield as a function of reaction time is presented in Figure 5.
The effect of the amount of oxidizing agent on the yield of [123I]PSMA-p-IB radiolabeling was studied as shown in Figure 6. The reactions were carried out using a fixed PSMA ligand 5 nmol and for a fixed time of 5 minutes. The data clearly show that increasing the amount of chloramine-T as an oxidizing agent from 10 µg to 40 µg can significantly improve the yield of 61.3 ± 0.5% to 69.2 ± 0.2%. Increasing the amount of chloramine-T in the reaction to 80 µg did not increase the radiolabeling results; even further additions up to 150 µg resulted in significantly decreased radiolabeling results. The yields for each radiolabeling condition of PSMA-p-TBSB ligands by 123I are presented in Table 2.
Based on the results of the radiolabeling optimization study, the following labeling conditions were considered to be optimal: amount of PSMA-p-TBSB ligand 10 nmol, amount of oxidizing agent 40 µg, and reaction time 5 minutes. The [123I]PSMA-p-IB radiolabeling results obtained under these conditions were 75.9 ± 1.0%. We also performed radio-HPLC analysis of radiolachemical yield to compare with the optimum result of radiochemical yield analysis using radio-iTLC-SG glass fiber sheet. The radio-HPLC chromatograms of [123I]PSMA-p-IB without purification were displayed in Figure S45 (in Supplementary Information). The results of the two methods (Figure 7) showed no significant difference with 99% confidence interval (by two-tailed t-test, p < 0.01). The radio-iTLC of [123I]PSMA-p-IB ligand based on the results of the radiolabeling optimization is shown in Figure S46 (in Supplementary Information). As a comparison, we conducted a radiolabeling study of PSMA-m-STB with 123I to produce [123I]PSMA-m-IB (compound 24) under the optimum radiolabeling conditions of [123I]PSMA-p-IB. The obtained radiochemical yield of the [123I]PSMA-m-IB was 49.41 %. We recognized that the [123I]PSMA-p-IB radiochemical yield was higher than [123I]PSMA-m-IB. Subsequently, the [123I]PSMA-p-IB was used as a model to perform initial preclinical evaluations of this new PSMA-targeting ligand.
2.3. Radiochemical Purity and Shelf-Life Stability
After radiolabeling process, the [123I]PSMA-p-IB was separated from the impurities in the reaction mixture. The radiolabeled PSMA was purified using using Sep-Pak® C18 cartridge. The quality control of [123I]PSMA-p-IB radiochemical purity was performed by radio-iTLC and radio-HPLC methods. The radio-iTLC and radio-HPLC chromatograms of pure [123I]PSMA-p-IB were shown in Figure S47 (in Supplementary Information). The radiochemical purity of the [123I]PSMA-p-IB obtained using radio-iTLC method was 99.50 ± 0.7%. While using radio-HPLC showed a [123I]PSMA-p-IB radiochemical purity of 100 ± 0.0%, which was not significantly different (99% confidence level) from the radiochemical purity obtained by radio-iTLC. Radiochemical purities of [123I]PSMA-p-IB analyzed by different methods were shown in Figure 8.
After three days of storage at temperature -20 ºC, the shelf-life stability of [123I]PSMA-p-IB in ethanol was investigated. Shelf-life stability needs to be evaluated because during storage, radiolysis may cause radiopharmaceuticals to decompose, resulting in radiochemical impurities. The results of radio-HPLC analysis of [123I]PSMA-p-IB showed that the purity obtained at 100% from the purification process did not change at all after 3 days. The radio-HPLC chromatogram of [123I]PSMA-p-IB after 3 days storage period was presented in Figure S48 (in Supplementary Information).
2.4. Lipophilicity
The lipophilicity of [123I]PSMA-p-IB was determined by its equilibrium distribution after thorough shaking in a two-phase system consisting of n-octanol and water. Small aliquots from both phases were collected and analyzed in an automated gamma-counter in order to calculate distribution coefficients Log(D). The distribution coefficient Log(D) of the [123I]PSMA-p-IB was 0.99. The results obtained indicate that this ligand was lipophilic. The hydrophobic characteristics of this ligand was influenced by the linker structure which was a dipeptide fragment containing isotope 123I, two phenylalanine residues in the L-configuration, and the presence of the chlorine substituents on the aromatic group of the ε-amino group of lysine [10].
2.5. In Vitro Cell Binding Assay
In vitro cell binding test of [123I]PSMA-p-IB was performed using PC-3 PIP (PSMA-positive) and PC-3 (PSMA-negative) cell lines (Figure 9). The level of binding of [123I]PSMA-p-IB to PC-3 PIP was significantly (p < 0.0001, unpaired two-tailed t-test) higher than to PC-3 cells. To investigate the specificity of [123I]PSMA-p-IB toward the receptor, the blocking of the receptors by adding 500x molar excess of unlabeled PSMA ligand was conducted. When the active site of PSMA was partially saturated with unlabeled PSMA ligand, positive PSMA-expressing PC-3 PIP cells showed a significant (p = 0.0002) decrease in [123I]PSMA-p-IB accumulation. On the other hand, there was no significant (p < 0.05) different between non-blocked and blocked negative PSMA-expressing PC-3 cells, that means [123I]PSMA-p-IB demonstrated a slight non-specific accumulation by the negative PSMA-expressing PC-3 cells.
2.6. Binding Affinity
The binding affinity of [123I]PSMA-p-IB was evaluated using PC-3 PIP cells. The saturation experiment of [123I]PSMA-p-IB on PC-3 PIP cells is presented in Figure 10. The equilibrium dissociation constant (KD) for [123I]PSMA-p-IB was 103 ± 30 nM.
2.7. In Vivo Biodistribution
A comparative biodistribution study was conducted between [123I]PSMA-p-IB and [177Lu]Lu-PSMA-617 in normal mice. The used [177Lu]Lu-PSMA-617 was obtained in high radiochemical purity (>95%). The radio-iTLC and radio-HPLC of [177Lu]Lu-PSMA-617 yield were presented in Figure S49 (in Supplementary Information).
The mice were sacrificed at 4 h post-injection (p.i.) by cervical dislocation. A side-by-side comparison of the biodistribution of [123I]PSMA-p-IB and [177Lu]Lu-PSMA-617 in normal mice is shown in Figure 11. Biodistribution of [123I]PSMA-p-IB 4 h p.i. in CD1 mice reflects low accumulation in almost all normal organs, except in salivary gland, gastrointestinal tract, and rest of body. There were significant differences between the uptake of [123I]PSMA-p-IB and [177Lu]Lu-PSMA-617 in several normal organs. Low accumulations of both ligands were detected in muscle and bone. However, it was determined that the uptake of [177Lu]Lu-PSMA-617 in muscle (0.1 ± 0.1 %ID/g) was noticeably lower than that of [123I]PSMA-p-IB (0.2 ± 0.0 %ID/g). Meanwhile, [123I]PSMA-p-IB accumulation in bone was significantly lower than [177Lu]Lu-PSMA-617 accumulation, respectively 0.3 ± 0.1 and 0.6 ± 0.1 %ID/g. The activity of [123I]PSMA-p-IB in blood was 1.3 ± 0.5 %ID/g. This uptake was significantly higher than [177Lu]Lu-PSMA-617 uptake, which was 0.1 ± 0.0 %ID/g. The activity in the blood for [123I]PSMA-p-IB experiment was associated with PSMA-p-TBSB labeled with 123I which is non-residualizing and is thought to result in rapid excretion of radiometabolites. Additionally, this ligand lipophilicity might lead it to bind to proteins in the blood. The [123I]PSMA-p-IB accumulation of 5.2 ± 1.7 %ID/g was observed in the salivary glands. This value was clearly higher than [177Lu]Lu-PSMA-617 accumulation in the same organ 0.1 ± 0.0 %ID/g. This [123I]PSMA-p-IB accumulation could be due to radioiodine catabolites. The activity in the whole gastrointestinal tract (with contents) and small intestine for [123I]PSMA-p-IB, was 12.4 ± 0.8 and 1.1 ± 0.2 %ID/g, respectively. Meanwhile, the activity of [177Lu]Lu-PSMA-617 in the whole gastrointestinal tract (with contents) and small intestine was 1.2 ± 0.8 and 0.2 ± 0.1 %ID/g, respectively.
Remarkable results were observed whereby the accumulation of [123I]PSMA-p-IB (0.9 ± 0.1 %ID/g) in the liver was substantially lower than that of [177Lu]Lu-PSMA-617 (2.3 ± 0.2 %ID/g). In addition, also there was difference in the accumulation of radioligands in the kidney organ, where the uptake of [123I]PSMA-p-IB (1.0 ± 0.1 %ID/g) was significantly less than that of [177Lu]Lu-PSMA-617 (6.1 ± 3.7 %ID/g).
3. Discussion
A number of publications confirmed that modification of the DCL ligand structure affects its affinity for PSMA. We assume that the linker modification in DCL with a dipeptide fragment containing two phenylalanine residues improves the affinity properties of the PSMA ligand by taking advantage of the hydrophobic interaction with the S1 hydrophobic pocket of PSMA [22,23]. Other evidence showing that modification of the aromatic moiety at the ε-amino group of lysine with halogen has an effect on the affinity [10]. Thus for the synthesis we choose novel DCL urea-based PSMA-ligand with chlorine-substituted aromatic fragment at the lysine ε-nitrogen atom, a dipeptide as peptide fragment of the linker including two phenylalanine residues in the L-configuration.
The assembly of Phe(L)-Phe(L) peptide sequence to obtain highly specific PSMA vectors was carried out using SPPS on cross-linked styrene-divinylbenzene copolymer matrix (2-CTC resin) (Scheme 2). 2-CTC resin was chosen for solid-phase synthesis, as it allows applying the concept of Fmoc\But SPPS and minimizing possible side reactions. In addition, it allows to keep acid-labile functional groups intact, since the removal of amino acid sequence from the resin proceeds under mild conditions (in this case DCM/TFA – 99,25%/0,75.% V/V; the reaction does not affect СOOBut groups labile to acids) [28]
The preparation of NHS-activated esters of prosthetic groups p-STBSB and m-STBSB (Scheme 3) was carried out similarly to the method described in the article [30]. The most significant of these modifications were: 1) the method of the compound 13 synthesiss, which allowed significantly increased the yield; 2) the methods of compounds 13-17 isolation.
At the final stage of the synthesis (Scheme 4), the protective tert-butyl groups of compound 11 were removed. According to literature data [31], But group can be removed by TFA (or HCl in AcOH or in dioxane) with acidolysis of carboxylate esters -COOBut. When TFA acts on tert-Bu-containing compounds, tert-Bu+ cations are formed, which are capable of being captured by TFA to form a strong alkylating agent CF3COOBut, which can cause side alkylation reactions. A possible way to avoid alkylation side reactions is to add scavenger molecules to the reaction mixture. The best absorbers of tert-butyl cations are trialkylsilanes (R3SiH), such as triethyl- and triisopropylsilane (TIPS). Water is also an effective scavenging agent for tert-butyl cations [32].
It should be noted that the possible side processes can be not only alkylation and acylation reactions of the peptide sequence, but also reactions of intramolecular condensation of the DCL ligand, leading to the formation of three isomeric five-membered heterocycles [33].
Compounds containing non-radioactive iodine (compound 21 and 22) were obtained in the study as standard compounds for comparison and characterization, and are therefore applicable regardless of the other technique. Meanwhile, in studying these novel ligands as candidates for radiopharmaceuticals, the radioiodination process was carried out through the electrophilic radioiodination method in the presence of chloramine-T as an oxidizing agent.
As a model for studying radiolabeling optimization and initial preclinical evaluation of these new ligands as radiopharmaceutical candidates, a ligand in its prosthetic group containing 4-tributylstannyl was used. Radiolabeling is accomplished through an electrophilic radioiodination reaction in the presence of [123I]NaI and chloramine-T. The electrophilic species (HO*I, H2O*I), generated from radioiodide and the oxidant, react directly with the aromatic moiety of prosthetic group to be labelled [34]. The iododestannylation reactions was used because usually give high product yields [38]. Chloramine-T was used as an oxidizing agent in this experiment because it allows radiolabeling to occur under mild conditions so that the peptides will not be damaged due to the influence of the reaction temperature.
The study on the effect of the amount of PSMA ligand on radiolabeling yields in general indicate that the use of a larger molar amount of PSMA-p-TBSB in a radiolabeling reaction increases the labelling yield until it reached its optimum point when using 10 nmol of PSMA-p-TBSB ligand. Findings from time effect studies demonstrate that when using oxidizing agents in the iododestannylation process of peptide, the correct reaction time is critical. Longer reaction times can diminish RCY due to undesired overoxidation, which results in chlorination and oxidative denaturation [35]. In addition, the optimal yield of [123I]PSMA-p-IB ligand is dependent upon an appropriate amount of chloramine-T as an oxidizing agent in this radiolabeling procedure. Elemental iodine was formed by the oxidation of sodium iodide with oxidizing agents to produce H2OI+ and HOI from sodium iodide. Excess amount of chloramine-T in the reaction caused a decrease in yield which may be due to the formation of undesirable oxidative side polymerization [35]. 10 nmol PSMA ligand, 40 µg chloramine-T, and carried out in a reaction time of 5 minutes was considered as the optimum condition in the PSMA-p-TBSB radiolabeling process with 123I. The labeling yield obtained under these method was 75.9 ± 1.0%.
Following the purification procedure [123I]PSMA-p-IB with a high radiochemical purity of > 99.50% was achieved. This value was validated utilizing radio-iTLC and radio-HPLC techniques. During storage, radiolysis may cause radiopharmaceuticals to decompose, resulting in radiochemical impurities. Radiolysis generates free radicals, which is one of the primary causes of radiolabeled preparation degradation. Radiolysis can result in the breakdown of chemical bonds between the radionuclide and the molecule, leading to the formation of radiochemical impurities [35]. Shelf-life stability needs to be evaluated because an impurity can become the main tracer circulating in the bloodstream, resulting in excessive body background, obscuring the diagnosis of disease, and increasing the patient's radiation exposure. The investigation revealed that the purity of [123I]PSMA-p-IB did not change at all during its shelf-life after three days following the purification procedure.
The major purpose of the in vitro investigation was the evaluation of the ability of [123I]PSMA-p-IB to bind to prostate cancer cells dependent on the PSMA surface representation. The binding of [123I]PSMA-p-IB to PSMA-expressing PC-3 PIP cells was receptor mediated. The equilibrium dissociation constant was still at the level of nanomole. This affinity measurement revealed that [123I]PSMA-p-IB binds to PSMA-expressing cells with a considerable affinity.
There were numerous variables that could impact the acquisition of this KD value. The introduction of hydrophobic functional groups in the linkers in these new ligands is thought to increase the binding affinity of PSMA in addition to the binding characteristics of glutamate or glutamate-like residues to the S1' pocket and the simultaneous inhibition of the zinc binuclear active site, which is an indispensable and minimum requirement for PSMA inhibitors. We suspect that the presence of chlorine substituents on the aromatic group of the ε-amino lysine group increased the lipophilic properties of the ligand, which could be advantageous when interacting with the S1 hydrophobic pocket of PSMA. Chlorine has a higher π value relative to hydrogen [37]. In addition, the chemical structure size of this ligand that interact with the S1 hydrophobic pocket is crucial. [123I]PSMA-p-IB contains quite large substituents at the linker, namely two phenylalanine residues in the L-configuration and chlorine on the aromatic group of the ε-amino group of lysine. As reported by Lundmark et al., the size of the substituent in the linker in a PSMA ligand has a significant effect on the value of KD [38].
To examine the biodistribution characteristics of this new ligand, the biodistribution of [123I]PSMA-p-IB in normal mice was compared to that of [177Lu]Lu-PSMA-617 which is known to have demonstrated promising results in clinical studies. The uptakes of [123I]PSMA-p-IB in normal organs, except in the salivary gland, gastrointestinal tract, and body, were low. At 4 hours after injection, the activity of [123I]PSMA-p-IB in blood and salivary glands was significantly higher than [177Lu]Lu-PSMA-617 uptake. This value was correlated with PSMA-p-IB that had been 123I-labeled, which is non-residualizing and is thought to cause radiometabolites to be excreted quickly. This advantage has the potential to be exploited to increase the ratio between activity concentrations in tumors and normal organs. The lipophilic nature of this ligand may also predispose it to bind to proteins in the blood. Meanwhlie the high [123I]PSMA-p-IB accumulation in salivary glands could be due to radioiodine catabolites. Radioactive accumulation was observed in several organs capable of concentrating iodine by Na/I symporter. This experiment was carried out without giving potassium iodide to the drinking water of mice in the days before the experiment. It is useful for evaluating the true biodistribution. The network that expresses the Na/I symporter which plays a role in the processing of radioiodinated peptide metabolites was not blocked, so it might affect the uptake value of radioiodinated peptide in this organ [39]. The accumulation of [123I]PSMA-p-IB in bone tissue was lower than the accumulation of [177Lu]Lu-PSMA-617 in bone. Its phenomenon can be explained by the fact that Lu3+, as a lanthanoid, exhibits some chemical similarities with Ca2+, which has a high uptake in bone tissue. Despite the presence of endogenous PSMA expression in the kidney, the [123I]PSMA-p-IB biodistribution pattern suggested a low uptake in this organ. This value was significantly lower than that of [177Lu]Lu-PSMA-617. It was also discovered that the accumulation of [123I]PSMA-p-IB in the liver was significantly less than that of [177Lu]Lu-PSMA-617. According to other previous studies, high uptake in these organs is a known concern. Hillier et al. [40] have also attempted to develop radiolabeled ligands with prosthetic groups as molecular imaging pharmaceuticals for prostate cancer, namely [123I]MIP-1072 and [123I]MIP-1095. Those ligands have high affinity to PSMA-expressing cells. However, the results of preclinical evaluation of the two ligands showed high accumulation in the liver and kidney of NCr nude mice bearing LNCaP xenografts. In liver, the accumulation of [123I]MIP-1072 and [123I]MIP-1095 after 4 h after injection was 2.17 ± 0.67 and 7.82 ± 1.01 %ID/g, respectively. Meanwhile in kidney, the accumulation of [123I]MIP-1072 and [123I]MIP-1095 after 4 h was 35.7 ± 18.7 and 77.5 ± 16.5 %ID/g, respectively [40]. The low [123I]PSMA-p-IB accumulation in the kidney and liver is an encouraging finding. The accumulation of [123I]PSMA-p-IB in liver and kidney 4 h p.i. was 0.9 ± 0.1 and 1.0 ± 0.1 %ID/g, respectively. This will undoubtedly benefit [123I]PSMA-p-IB ligand when used as a diagnostic for prostate cancer, as the location of the kidney is close to the prostate gland, thereby optimizing the prostate image during imaging due to the low accumulation of [123I]PSMA-p-IB in the kidney. It was therefore hypothesized that [123I]PSMA-p-IB provides low retention of activity in excretory organs.
4. Materials and Methods
All starting compounds are commercially available reagents. The initial stages of the synthesis of the vector fragment 1–5 (Scheme 1) was made by methods previously developed by our scientific group [26]. For compounds 12, 15, the reaction and purification conditions are given in [30]. 1H NMR was measured at Bruker Avance spectrometer operating at 400 MHz for 1H using CDCl3, DMSO-d6 as solvents. Chemical shifts are reported in δ units to 0.01 ppm precision with coupling constants reported to 0.1 Hz precision using residual solvent as an internal reference.13C NMR was measured at Bruker Avance spectrometer operating at 100 MHz using DMSO-d6 as solvents. Chemical shifts are reported in δ units to 0.1 ppm precision using residual solvent as an internal reference. NMR spectra were processed and analyzed using Mnova software (Mestrelab Research, Spain). High resolution mass spectra were recorded on the Orbitrap Elite high resolution mass spectrometer. Solutions of samples in acetonitrile with 1% formic acid were introduced into the ionization source by electrospray. For HPLC analysis system with Shimadzu Prominence LC-20 column and a convection fraction collector connected with a single quadrupole mass spectrometer Shimadzu LCMS-2020 with dual ionization source DUIS-ESI-APCI were used. The analytical and preparative column was Phenomenex Luna 3 µm C18 100 Å. Preparative chromatographic separation of substances was carried out using the INTERCHIM puriFlash 430 chromatograph.
The radionuclidic purtity of 123I was measured by Canberra γ - spectrometer equipped with a germanium detector (HPGe) and Genie2000 software. The detector was previously calibrated using standard point source. Solution with activity value more than 25 MBq were measured by an ionization chamber using dose calibrator RIS-1A, Amplituda, Russia. Radio-iTLC was performed using an iTLC-scanner miniGITA Single (Elysia Raytest, Straubenhardt, Germany). The samples in vitro and vivo test were measured by thallium-activated sodium iodide detector (NaI(Tl) using automated gamma-counter Wizard 1480 (Pelkin Elmer, Waltham, MA, USA).
Purifications of the radiolabeling yield were performed by Sep-Pak® C18 cartridge, catalogue number WAT036815, (Waters, Milford, Massachusetts, USA). Thin-layer chromatography (TLC) analysis was performed using iTLC glass microfiber chromatography sheet impregnated with a silica gel (iTLC SG fiber sheet) (Agilent Technologies, Inc., Folsom, CA, USA) and iTLC silica gel 60 F254 aluminium plates (iTLC SG 60 F254) (Merck KGaA, Darmstadt, Germany)
Data on radiolabeling, binding specificity, and biodistribution were analyzed by ANOVA test with Turkey’s post-hoc analysis and two-tailed t-test to determine any significant difference using GraphPad Prism (version 9.5.0 for Windows; GraphPad Software, La Jolla, CA, USA).
4.1. Chemistry
Compound 6. DIPEA (1.4 eq; 244 μL; 1.4 mmol) and succinic anhydride (1.02 eq; 102 mg; 1.02 mmol) were added to a solution of compound 5 (1 eq; 725 mg; 1.0 mmol) in 20 mL of DCM. The mixture was stirred for 12 h, then MeOH (2 eq.) was added and the resulting mixture was stirred for 1 h. Then the solvent was removed under reduced pressure, the residue was dissolved in DCM and extracted with (1) 0.1 M HCl (2 * 30 mL), (2) brine (2 * 30 mL). Then the organic fraction was dried over Na2SO4, and concentrated under reduced pressure to obtain final compound 6 as yellow oil (801 mg, yield 97%).
1HNMR (400 MHz, DMSO-d6, δ): 12.06 (br.s., 1Н, X4C(O)OH), 7.81 (t, J = 5.2Hz, m) &7.77 (t, J = 5.2Hz, n) (1H, X3NHk, m + n, m/n = 3/2), 7.40 (t, J = 7.7Hz, X8He, n), 7.37–7.27 (m, X8Hd + X8He(m)), 7.26-7.21 (m, 1H, X8Ht, m + n), 7.19–7.10 (m, 1H, X8Hg, m + n), 6.34–6.20 (m, 2Н, K2NH + E1NH, m + n), 4.56 (s, n) & 4.48 (s, m) (2H, X8Ha, m + n, m/n = 3/2), 4.07–4.00 (m, 1H, E1Ha, m + n), 4.00–3.90 (m, 1H, K2Ha, m + n), 3.22 (t, J = 7.3 Hz, n) & 3.19 (t, J = 7.3Hz, m) (2H, K2He, m + n, m/n = 3/2), 3.01 (q, J = 6.4, 12.7 Hz, m) & 2.96 (q, J = 6.4, 12.7 Hz, n) (2H, X3He, m + n, m/n = 3/2), 2.44–2.38 (m, 2H, X4Hb, m + n), 2.36 (t, J = 7.4Hz, X3Ha, m), 2.31–2.25 (m, 2H, X4Ha, m + n), 2.25–2.15 (m, E1Hg + X3Ha(n)), 1.91–1.80 (m, 1Н, E1Hb(a)), 1.72–1.63 (m, 1Н, E1Hb(b)), 1.63–1.56 (m, 1Н, K2Hb(a)), 1.40–1.35 (m, 27H, tBu), 1.56–1.15 (m, 11Н, K2Hb(b) + X3Hb + X3Hd+ K2Hd + K2Hg + X3Hg, m + n).
13C NMR (100 MHz, DMSO-d6, δ): 173.93 (X4Cg), 172.26 (K2C(n)), 172.23 (K2C(m)), 172.22 (X3C(n)), 172.19 (X3C(m)), 171.95 (E1C), 171.47 (E1Cg), 170.76 (X4C(m)), 170.73 (X4C(n)), 157.18 (U(m)), 157.16 (U(n)), 141.20 (X9Cb(m)), 140.80 (X9Cb(n)), 133.45 (X9Ce(n)), 133.10 (X9Ce(m)), 130.63 (X9Cd(n)), 130.26 (X9Cd(m)), 127.24 (X9Ct(m)), 127.17 (X9Ck(n)), 126.88 (X9Ck(m)), 126.34 (X9Ct(n)), 126.08 (X9Cg(m)), 124.99 (X9Cg(n)), 80.59 (E1tBu), 80.42 (K2tBu(m)), 80.33 (K2tBu(n)), 79.77 (E1dtBu), 53.01 (K2Ca(n)), 52.88 (K2Ca(m)), 52.20 (E1Ca(m)), 52.18 (E1Ca(n)) 49.63 (X9Ca(n)), 47.11 (X9Ca(m)), 46.83 (K2Ce(m)), 45.20 (K2Ce(n)),38.49 (X3Ce(m)), 38.43 (X3Ce(n)), 32.34 (X3Ca(n)), 31.95 (X3Ca(m)), 31.83 (K2Cb), 30.93 (E1Cg), 30.06 (X4Ca), 29.25 (X4Cb), 29.13 (X3Cd(m)), 29.04 (X3Cd(n)), 27.75 (tBuE1), 27.69 (K2Cd(m)), 27.66 (tBuK2), 27.64 (tBuE1g+E1Cb), 26.72 (K2Cd(n)), 26.23 (X3Cg(m)), 26.15 (X3Cg(n)), 24.76 (X3Cb(m)), 24.63 (X3Cb(n)), 22.45 (K2Cg(n)), 22.27 (K2Cg(m)).
ESI-MS C41H65ClN4O11: m/z calcd. for [M+H+]+: 825.44, found: 825.45
Compound 8. Activation of 2-CTC. The suspension of 2-СTC (1 eq.; 1g; 1.2–1.4 mmol/g; 100–200 mesh) in DCM (10 mL) was stirred for 10 min, after that the mixture was purged with Ar, then SOCl2 (3 eq.; 305 µL; 4.2 mmol) was added dropwise, and then DMF (16 µL; 5% V/V to SOCl2) was added and stirred at 40 °C for 4 h. After that the resin was filtered off and transferred to a polypropylene reactor, washed with DMF (3 * 10 mL, 1 min) and DCM (3 * 10 mL, 1 min).
The addition of FmocPhe(L)-OH. To the mixture of CTC-2 (1 eq.; 1g; 1.2–1.4 mmol/g; 100–200 mesh) in DMF (10 mL) FmocPhe(L)-OH (2 eq.; 1.085 g; 2.8 mmol) and DIPEA (10 eq.; 2.44 mL; 14 mmol) were added, and the mixture was stirred for 2 h. Then the resin was filtered off, washed with MeOH (3 * 10 mL, 5 min), DCM (3*10 mL, 1 min), DMF (3 * 10 mL, 1 min), and DCM (3 * 10 mL, 1 min).
Fmoc-deprotection. FmocPhe(L) on a 2-CTC resin (1 eq.) was washed with DMF (2*15 mL, 1min), then 4-methylpiperidine in DMF (20%/80% V/V, 15 mL) was added and the mixture was stirred for 15 min, then the resin was filtered off, washed with DMF (3 * 15 mL, 1 min), 4-methylpiperidine in DMF (20%/80% V/V, 15 mL) was added and the mixture stirred for 15 min. After the resin was filtered off, washed with DMF (3*15 mL, 1 min), and DCM (3 * 15 mL, 1 min).
The addition of FmocPhe(L)-OH. To the mixture of NH2-Phe(L) on a CTC-2 resin (1 eq.) in DMF (15 mL) FmocPhe(L)-OH (2 eq.; 1.085 g; 2.8 mmol), HOBt (0.5 eq.; 95 mg; 0.7 mmol), HBTU (2 eq.; 1.062 g; 2.8 mmol) and DIPEA (3 eq.; 0.73 mL; 4.2 mmol) were added and the mixture wasstirred for 2 h. Then the resin was filtered off, washed with DMF (3 * 15 mL, 1 min) and DCM (3 * 15 mL, 1 min).
Fmoc-deprotection. Fmoc-Phe(L)Phe(L) on a CTC-2 resin (1 eq.) was washed with DMF (2 * 15 mL, 1 min), then 4-methylpiperidineine in DMF (20%/80% V/V, 15 mL) was added and the mixture was stirred for 15 min, then the resin was filtered off, washed with DMF (3 * 15 mL, 1 min), 4-methylpiperidine in DMF (20%/80% V/V, 15 mL) was added and the mixture was stirred for 15 min. After the resin was filtered off, washed with DMF (3 * 15 mL, 1 min), DCM (3 * 15 mL, 1 min). Thus, the NH2-Phe(L)Phe(L) dipeptide was obtained on 2-CTC resin (~1.4 mmol).
Compound 9. To the NH2-Phe(L)Phe(L) dipeptide on a 2-CTC (1 eq; 0.54 mmol) in 7 ml DMF compound 6 (1.2 eq; 535 mg; 0.648 mmol), HOBt (0.5 eq; 37 mg; 0. 27 mmol), HBTU (2 eq; 410 mg; 1.08 mmol) and DIPEA (3 eq; 282 µl; 1.62 mmol) were added. The mixture was stirred for 4 hours. The solvent was removed by filtration and the resin was washed three times with DMF (7 ml), three times with DCM (7 ml), then dried from traces of solvents. DCM/TFA mixture (99.25%/0.75%, 11 ml) was added to the resin and left under stirring for 15 minutes, after which the resin was filtered off from the solution. The solvent was removed under reduced pressure and the residue was reevaporated with DCM and then purified by column chromatography (Puriflash on a PF-15C18AQ-F0025 column (15μ 40g): H2O(80%)/MeCN(20%) => H2O(0%)/MeCN(100%) for 15 minutes, after MeCN(100%) for 5 minutes Compound 9 was obtained as a white amorphous substance (460 mg, 76% yield).
1HNMR (400 MHz, DMSO-d6, δ): 12.71 (br.s., 1Н, F6COOH), 8.30-8.22 (br.d., 1Н, F5NHmn), 8.09-8.03 (br.d., 1Н, F6NHmn), 7.79 (t, J=5.4 Hz, m) & 7.75 (t, J=5.4 Hz, n) (1H, X3NHk), 7.40 (t, J=7.7Hz, X8Hdn), 7.36-7.29 (m, X8Hen+X8Hdm+X8Hem), 7.29-7.08 (m, 12H, F6He+F6Hd+X8Htmn+F5He+F6Hk+F5Hk+F5Hd +X8Hgmn), 6.37-6.18 (m, 2Н, K2NH+E1NH, m+n), 4.55 (s, X8Han), 4.53-4.44 (m, F6Ha+X8Ham), 4.44-4.36 (m, 1H, F5Ha), 4.08-3.99 (m, 1H, E1Ha), 3.99-3.90 (m, 1H, K2Ha), 3.22 (t, J=7.3 Hz, n) & 3.17 (t, J=7.3 Hz, m) (2H, K2He), 3.11-3.02 (m, 1Н, F6Hb(a)), 3.02-2.90 (m, 4Н, F6Hb(b)+X3Hemn+F5Hb(a)), 2.71-2.60 (m, 1Н, F5Hb(b)), 2.35 (t, J=7.4 Hz, X3Ham), 2.30-2.10 (m, X4Hbmn+E1Hg+X4Hamn+X3Han), 1.92-1.80 (m, 1Н, E1Hb(a)), 1.71-1.62 (m, 1Н, E1Hb(b)), 1.62-1.54 (m, 1Н, K2Hb(a)), 1.54-1.10 (m, 11Н, X3Hb+K2Hb(b)+X3Hd+K2Hd+K2Hg+X3Hg, m+n), 1.40-1.34 (m, 27H, tBu).
13C NMR (100 MHz, DMSO-d6, δ): 172.77 (F6C), 172.25 (K2C(n)), 172.21 (K2C(m)), 172.14 (X3C(n)), 172.12 (X3C(m)), 171.93 (E1C), 171.45 (E1Cd), 171.42 (X4Cg(mn)), 171.37 (F5C(mn)), 171.16 (X4C(m)), 171.15 (X4C(n)) 157.15 (U(m)), 157.14 (U(n)), 141.19 (X8Cb(m)), 140.78 (X8Cb(n)), 138.13 (F5Cg), 137.57 (F6Cg), 133.44 (X8Ck(n)), 133.09 (X8Ck(m)), 130.61 (X8Cd(n)), 130.25 (X8Cd(m)), 129.18 (F6Cd+F5Cd), 128.23 (F6Ce), 127.99 (F5Ce), 127.22 (X8Ct(m)), 127.16 (X8Ce(n)), 126.87 (X8Ce(m)), 126.45 (F6Ck), 126.32 (X8Ct(n)), 126.18 (F5Ck), 126.07 (X8Cg(m)), 124.96 (X8Cg(n)), 80.58 (E1tBu), 80.41 (K2tBu(m)), 80.32 (K2tBu(n)), 79.76 (E1dtBu), 53.67 (F5Ca), 53.63 (F6Ca), 53.00 (K2Ca(n)), 52.87 (K2Ca(m)), 52.19 (E1Ca), 49.61 (X8Ca(n)), 47.10 (X8Ca(m)), 46.80 (K2Ce(m)), 45.20 (K2Ce(n)), 38.53 (X3Ce(m)), 38.47 (X3Ce(n)), 37.34 (F5Cb), 36.61 (F6Cb), 32.33 (X3Ca(n)), 31.95 (X3Ca(m)), 31.83 (K2Cb), 30.93 (E1Cg), 30.82 (X4Ca), 30.75 (X4Cb), 29.12 (X3Cd(m)), 29.02 (X3Cd(n)), 27.75 (tBuE1), 27.66 (tBuK2+K2Cd(m)), 27.63 (tBuE1d+E1Cb), 26.72 (K2Cd(n)), 26.29 (X3Cg(m)), 26.19 (X3Cg(n)), 24.76 (X3Cb(m)), 24.62 (X3Cb(n)), 22.44 (K2Cg(n)) 22.27 (K2Cg(m)).
ESI-MS C59H83ClN6O13: m/z calcd. for [M+H+]+: 1119.58, found: 1119.45.
HRMS (m/z, ESI): calcd. for C59H83ClN6O13 - [M+H+]+: 1119.5779, found: 1119.5746.
Compound 10. To a solution of compound 9 (1 eq.; 300 mg; 0.268 mmol) in 10 ml of DMF TFA*NH2(CH2)3NHFmoc (1.1 eq.; 121 mg; 0.294 mmol), DIPEA (2.5 eq.; 117 μl; 0.67 mmol) in 10 ml DMF, followed by HOBt (1 eq.; 36 mg; 0.268 mmol) and HBTU (1.5 eq.; 152 mg; 0.402 mmol) were added. The mixture was stirred for 12 hours under an inert atmosphere. The solvent was then removed under reduced pressure and the residue was reevaporated twice with DCM, dissolved in 30 ml of DCM and the extraction was carried out: 1) H2O (2*30 ml), 2) saturated NaCl solution (2*30 ml). Then the organic fraction was dried over Na2SO4, the solvent was removed, and the residue was purified using a column chromatography method (Puriflash on a column (15μ 40g)); DCM(100%)/MeOH(0%) => DCM(90%)/MeOH(10%) for 30 minutes, after MeOH (100%) for 5 minutes. Compound 10 was obtained as a pale-yellow amorphous substance (330 mg, 88% yield).
1HNMR (400 MHz, DMSO-d6, δ): 8.35-8.25 (br.d., 1Н, F5NHmn), 8.22-8.13 (m, 1Н, F6NHmn), 7.93 (t, J=5.4 Hz, X3NHkm), 7.91-7.84 (m, X3NHkn+FmocHt), 7.67 (d, J = 7.5 Hz, 2H, FmocHd), 7.60-7.52 (m, 1H, X7NH), 7.45-7.35 (m, FmocHk +X8Hdn), 7.35-7.08 (m, X8Hen+FmocHe+X8Hdm +X8Hem+X7NHd+F6He+F6Hd+X8Htmn+F5He+F6Hk +F5Hk+F5Hd +X8Hgmn), 6.36-6.21 (m, 2Н, K2NH+E1NH, m+n), 4.54 (s, n) & 4.47 (s, m) (2H, X8Ha, m+n), 4.44-4.36 (m, 1H, F6Ha), 4.36-4.25 (m, 3H, F5Ha+FmocHa), 4.20 (t, J = 6.9 Hz, 1H, FmocHb), 4.08-3.99 (m, 1H, E1Ha), 3.99-3.90 (m, 1H, K2Hamn), 3.21 (t, J=7.3 Hz, n) & 3.16 (t, J=7.3 Hz, m) (2H, K2He, m+n), 3.11-2.84 (m, 9Н, F6Hb(a)+X7Hg+X7Ha+X3He(mn)+F6Hb(b)+F5Hb(a)), 2.70-2.60 (m, 1Н, F5Hb(b)), 2.40-2.10 (m, 8H, X3Ham+X4Hbmn+E1Hg+X4Hamn+X3Han), 1.92-1.80 (m, 1Н, E1Hb(a)), 1.72-1.62 (m, 1Н, E1Hb(b)), 1.62-1.55 (m, 1Н, K2Hb(a)), 1.54-1.10 (m, 13Н, X7Hb+X3Hb+K2Hb(b)+X3Hd +K2Hd+K2Hg +X3Hg, m+n), 1.40-1.34 (m, 27H, tBu).
13C NMR (100 MHz, DMSO-d6, δ): 172.79 (X4Cg(n)), 172.76 (X4Cg(m)), 172.24 (K2C(n)), 172.20 (K2C(m)), 172.15 (X3C(n)), 172.13 (X3C(m)), 171.92 (E1C), 171.57 (X4C(mn)), 171.45 (E1Cd), 171.11 (F5C), 170.67 (F6C) 157.15 (U(mn)), 156.12 (C(O)Fmoc), 143.94 (FmocCg), 141.16 (X8Cb(m)), 140.77 (X8Cb(n)+FmocCte), 138.12 (F6Cg), 138.02 (F5Cg), 133.44 (X8Ck(n)), 133.09 (X8Ck(m)), 130.59 (X8Cd(n)), 130.23 (X8Cd(m)), 129.04 (F6Cd+F5Cd), 128.16 (F6Ce), 128.06 (F5Ce), 127.63 (FmocCk), 127.21 (X8Ct(m)), 127.15 (X8Ce(n)), 127.09 (FmocCt), 126.86 (X8Ce(m)), 126.29 (F6Ck+X8Ct(n)), 126.24 (F5Ck), 126.05 (X8Cg(m)), 125.17 (FmocCe), 124.95 (X8Cg(n)), 120.13 (FmocCd), 80.59 (E1tBu), 80.42 (K2tBu(m)), 80.33 (K2tBu(n)), 79.77 (E1dtBu), 65.32 (FmocCa), 54.94 (F5Ca), 54.40 (F6Ca), 53.01 (K2Ca(n)), 52.87 (K2Ca(m)), 52.20 (E1Ca), 49.62 (X8Ca(n)), 47.11 (X8Ca(m)), 46.80 (FmocCb+K2Ce(m)), 45.19 (K2Ce(n)), 38.65 (X3Ce(m)), 38.60 (X3Ce(n)), 37.90 (X7Cg), 37.11 (F5Cb), 36.82 (F6Cb), 36.39 (X7Ca), 32.32 (X3Ca(n)), 31.95 (X3Ca(m)), 31.82 (K2Cb), 30.93 (E1Cg), 30.69 (X4Ca), 30.58 (X4Cb), 29.20 (X7Cb), 29.05 (X3Cd(m)), 28.96 (X3Cd(n)), 27.74 (tBuE1), 27.65 (tBuK2+K2Cd(m)), 27.62 (tBuE1d+E1Cb), 26.69 (K2Cd(n)), 26.31 (X3Cg(m)), 26.22 (X3Cg(n)), 24.73 (X3Cb(m)), 24.60 (X3Cb(n)), 22.43 (K2Cg(n)) 22.25 (K2Cg(m)).
ESI-MS C77H101ClN8O14: m/z calcd. for [M+H+]+: 1397.72, found: 1397.65
HRMS (m/z, ESI): calcd. for C77H101ClN8O14 - [M+H]+ 1397.7199, found: 1397.7210
Compound 11. Compound 10 (1 eq; 200 mg; 0.143 mmol) was dissolved in Et2NH/DMF (20 eq Et2NH, 10 ml DMF) mixture and stirred for 20 minutes, then the solvent was removed under reduced pressure and the residue was reevaporated with DCM three times. The product was precipitated with Et2O and washed twice with Et2O (2 ml). The residue was purified by reverse phase column chromatography (Puriflash PF-15C18AQ-F0025 (15μ 35g): H2O*TFA(0.1%) (80%)/MeCN(20%) => H2O*TFA(0.1%) (0 %)/MeCN(100%) for 30 minutes after MeCN (100%) for 5 minutes. Compound 11 was obtained as a *TFA salt as a white amorphous solid (160 mg, 89% yield).
1HNMR (400 MHz, DMSO-d6, δ): 8.37 (d, J=7.3 Hz, 1Н, F5NHmn), 8.23-8.15 (br.d, 1Н, F6NHmn), 8.00 (t, J=5.4 Hz, m) & 7.97 (t, J=5.4 Hz, n) ) (1H, X3NHk, m+n), 7.82 (br.s, 3H, X7NHd), 7.77-7.69 (m, 1H, X7NH), 7.42-7.09 (m, 14H, X8Hdn+X8Hen +X8Hdm+X8Hem+F6He+F6Hd +X8Htmn+F5He+F6Hk+F5Hk+F5Hd+X8Hgmn), 6.39-6.23 (m, 2Н, K2NHm+ K2NHn+E1NHm +E1NHn), 4.55 (s, n) & 4.47 (s, m) (2H, X8Ha, m+n), 4.43-4.33 (m, 1H, F6Ha), 4.33-4.23 (m, 1H, F5Ha), 4.08-3.99 (m, 1H, E1Ha), 3.99-3.90 (m, 1H, K2Ham+K2Han), 3.26-3.11 (m, 3H, K2Hemn+X7Hg(a)), 3.11-2.85 (m, 6Н, F6Hb(a)+X7Hg(b)+X3He(mn)+F6Hb(b)+F5Hb(a)), 2.78-2.61 (m, 3Н, X7Ha+F5Hb(b)), 2.40-2.10 (m, 8H, X3Ham+X4Hbmn+E1Hg +X4Hamn+X3Han), 1.92-1.80 (m, 1Н, E1Hb(a)), 1.73-1.61 (m, 3Н, X7Hb+E1Hb(b)), 1.62-1.55 (m, 1Н, K2Hb(a)), 1.54-1.10 (m, 11Н, X3Hb+K2Hb(b)+X3Hd+K2Hd +K2Hg+X3Hg, m+n), 1.40-1.34 (m, 27H, tBu).
13C NMR (100 MHz, DMSO-d6, δ): 172.91 (X4Cg(n)), 172.88 (X4Cg(m)), 172.24 (K2C(n)), 172.20 (K2C(m)), 172.15 (X3C(n)), 172.13 (X3C(m)), 171.92 (E1C), 171.61 (X4C(mn)), 171.46 (E1Cd), 171.23 (F5C), 171.09 (F6C), 157.18 (U(m)), 157.16 (U(n)), 141.18 (X8Cb(m)), 140.78 (X8Cb(n)), 138.08 (F6Cg), 138.02 (F5Cg), 133.41 (X8Ck(n)), 133.07 (X8Ck(m)), 130.61 (X8Cd(n)), 130.25 (X8Cd(m)), 129.05 (F6Cd+F5Cd), 128.20 (F6Ce), 128.07 (F5Ce), 127.20 (X8Ct(m)), 127.15 (X8Ce(n)), 126.86 (X8Ce(m)), 126.34 (F6Ck), 126.31 (X8Ct(n)), 126.25 (F5Ck), 126.06 (X8Cg(m)), 124.97 (X8Cg(n)), 80.55 (E1tBu), 80.38 (K2tBu(m)), 80.30 (K2tBu(n)), 79.77 (E1dtBu), 55.10 (F5Ca), 54.53 (F6Ca), 53.01 (K2Ca(n)), 52.88 (K2Ca(m)), 52.19 (E1Ca), 49.62 (X8Ca(n)), 47.10 (X8Ca(m)), 46.81 (K2Ce(m)), 45.22 (K2Ce(n)), 38.64 (X3Ce(m)), 38.58 (X3Ce(n)), 36.92 (F5Cb), 36.79 (F6Cb), 36.58 (X7Cg), 35.81 (X7Ca), 32.33 (X3Ca(n)), 31.95 (X3Ca(m)), 31.79 (K2Cb), 30.92 (E1Cg), 30.66 (X4Ca), 30.57 (X4Cb), 29.05 (X3Cd(m)), 28.97 (X3Cd(n)), 27.75 (tBuE1), 27.66 (tBuK2+K2Cd(m)), 27.63 (tBuE1d), 27.57 (E1Cb), 27.09 (X7Cb), 26.70 (K2Cd(n)), 26.32 (X3Cg(m)), 26.23 (X3Cg(n)), 24.74 (X3Cb(m)), 24.60 (X3Cb(n)), 22.45 (K2Cg(n)) 22.26 (K2Cg(m)).
ESI-MS C62H91ClN8O12: m/z calcd. for [M+H+]+: 1175.65, found: 1175.6
HRMS (m/z, ESI): calcd. for C62H91ClN8O12 - [M+H]+ 1175,6518, found: 1175.6520
Compound 13. Compound 12 (1 eq; 1800 mg; 4.376 mmol, calculated assuming that 12 is only a mono-stanylated derivative) was dissolved in 40 ml dry DCM. Then NHS (1.2 eq; 604 mg; 5.25 mmol), DMAP (0.1 eq; 53 mg; 0.438 mmol) and EDC*HCl (1.1 eq; 924 mg; 4.82 mmol) in DMF (4 ml) were added dropwise. The mixture was stirred overnight. After the reaction proceeded, the solvent was removed on a rotary evaporator, the residue was dissolved in DCM (100 ml) and transferred to a separating funnel, washed twice with H2O and then with saturated NaCl solution. The organic layer was dried over Na2SO4. The solvent was removed on a rotary evaporator and the residue was purified using a column chromatography (Puriflash on a column (40μ-60 μ 120g)); P.E. (97%) / E.A. (3%) for 7 minutes, then P.E. (97 %)/E.A.(3%) => P.E.(60%)/E.A.(40%) for 40 minutes, then P.E.(60%)/E.A.( 40%) => P.E.(0%)/E.A.(100%) for 5 minutes, then E.A.(100%) for 10 minutes. As a result, a fraction was isolated, which is a pale-yellow transparent oily substance (m = 1729 mg, 78%).
1HNMR (400 MHz, DMSO-d6, δ): 8.18-8.06 (m., 1Н, 2), 8.05-7.97 (m, 1H, 4), 7.96-7.82 (m, 1H, 6), 7.65-7.55 (m, 1H, 5), 2.89 (s, 4H, 9), 1.63-1.39 (m, 6H, 11), 1.35-1.21 (m, 6H, 12), 1.20-1.00 (m, 6H, 10), 0.84 (t, J = 7.3 Hz, 9H, 13)
13C NMR (100 MHz, DMSO-d6, δ): 170.33 (8), 162.14 (7), 143.46 (6), 143.24 (1), 137.05 (2), 129.64 (3), 128.84 (4), 124.07 (5), 28.52 (11), 26.67 (12), 25.57 (9), 13.48 (10), 9.36 (13).
ESI-MS C23H35NO4118Sn: m/z calcd. for [M+Na+]+: 530,15,found: 530.10
HRMS (m/z, ESI): calcd for C23H35NO4120Sn - [M+Na+]+ 532,1480, found: 532,1487
Compound 14. I2 (1 eq; 97.5 mg; 0.384 mmol) was dissolved in 0.1N NaOH (2300 µl = V1), AcOH (3%) in CHCl3 (2300 µl = V2) was added (V1 = V2), then tert-butyl hydroperoxide (TBHP) in CHCl3 (the solution is prepared in advance by adding 1800 µl of 70% TBHP in water to 11209 µl of CHCl3, after which Na2SO4 is added to the prepared mixture to bind water) (11513 µl), then 13 (1 eq; 195 mg; 0.383 mmol) in CHCl3 (3900 µl) were added. The mixture was stirred for 30 minutes, the solvent was then removed under reduced pressure. The product was precipitated with Н2O and washed twice with Н2O (2 ml) and twice with P.E. (2 ml). The residue was purified by reverse phase column chromatography (Puriflash PF-15C18AQ-F0025 (15μ 40g): H2O(90%)/MeCN(10%) => H2O(0 %)/MeCN(100%) for 30 minutes after MeCN (100%) for 15 minutes. As a result, a fraction was isolated, which is the target substance in the form of a white powder (m = 95 mg, 72%).
1H NMR (400 MHz, DMSO-d6, δ): 8.34 (t, J = 1.7 Hz, 1H, 2), 8.21 (ddd, J = 7.9, 1.7, 1.0 Hz, 1H, 6), 8.10 (ddd, J = 7.9, 1.7, 1.0 Hz, 1H, 4), 7.45 (t, J = 7.9 Hz, 1H, 5), 2.90 (s, 4H, 9).
13C NMR (100 MHz, DMSO-d6, δ): 170.22 (8), 160.60 (7), 144.11 (6), 137.87 (2), 131.62 (3), 129.30 (5), 126.41 (4), 95.53 (1), 25.57 (9).
Compound 16. Compound 15 (1 eq; 1380 mg; 3.35 mmol, calculated assuming that 15 is only a mono-stanylated derivative) was dissolved in 15 ml dry THF. Then NHS (1.2 eq; 463 mg; 4.02 mmol), and DCC (1 eq; 691 mg; 3.35 mmol) in THF (15 mL) were added dropwise. The mixture was stirred overnight. The precipitated dicyclohexylurea was removed by filtration through a fritted funnel. The precipitate was washed with 2×6 ml of THF, then the solvent was removed on a rotary evaporator. The residue was purified using a column chromatography (Puriflash on a column (40μ-60 μ 120g)); P.E. (97%) / E.A. (3%) for 7 minutes, then P.E. (97 %)/E.A.(3%) => P.E.(60%)/E.A.(40%) for 40 minutes, then P.E.(60%)/E.A.( 40%) => P.E.(0%)/E.A.(100%) for 5 minutes, then E.A.(100%) for 10 minutes. As a result, a fraction was isolated, which is a pale-yellow transparent oily substance (m = 772 mg, 45%).
1HNMR (400 MHz, DMSO-d6, δ): 8.05-7.93 (m., 1Н, 3), 7.82-7.66 (m, 1H, 4), 2.89 (s, 4H, 7), 1.65-1.39 (m, 6H, 9), 1.36-1.21 (m, 6H, 10), 1.20-1.00 (m, 6H, 8), 0.84 (t, J = 7.3 Hz, 9H, 11)
13C NMR (100 MHz, DMSO-d6, δ): 170.33 (6), 162.10 (5), 153.06 (1), 137.16 (2), 128.53 (3), 124.01 (4), 28.52 (9), 26.67 (10), 25.56 (7), 13.51(8), 9.37 (11).
HRMS (m/z, ESI): calcd for C23H35NO4120Sn - [M+Na+]+ 532,1480, found: 532,1485
Compound 17. I2 (1 eq; 75 mg; 0.295 mmol) was dissolved in 0.1N NaOH (1770 µl = V1), AcOH (3%) in CHCl3 (1770 µl = V2) was added (V1 = V2), then tert-butyl hydroperoxide (TBHP) in CHCl3 (the solution is prepared in advance by adding 1264 µl of 70% TBHP in water to 7872 µl of CHCl3, after which Na2SO4 is added to the prepared mixture to bind water) (8856 µl), then 16 (1 eq; 150 mg; 0.295 mmol) in CHCl3 (3000 µl) were added. The mixture was stirred for 30 minutes, the solvent was removed under reduced pressure. The product was precipitated with Н2O and washed twice with Н2O (2 ml) and twice with P.E./E.A. (80/20) (2 ml). As a result, a fraction was isolated, which is the target substance in the form of a white powder (m = 98.5 mg, 97%).
1HNMR (400 MHz, DMSO-d6, δ): 8.06 (d, J = 8.5 Hz, 2H, 3), 7.83 (d, J = 8.5 Hz, 1H, 2), 2.89 (s, 4H, 7).
13C NMR (100 MHz, DMSO-d6, δ): 170.28 (6), 161.69 (5), 138.65 (2), 131.39 (3), 123.88 (4), 105.01 (1), 25.58 (7).
Compound 18. Compound 11 (1 eq.; 228.5 mg; 177.1 mmol) was dissolved in the mixture of DCM/TFA/TIPS/H2O (46.25%/46.25%/2.5%/5%; V/V respectively, 8 mL). The mixture was stirred for 3 h, then the solvent was removed under reduced pressure and the residue was re-evaporated with DCM three times. The product was precipitated with Et2O and washed twice with Et2O (1 mL). After the compound was purified by column chromatography (Puriflash on the column PF-15C18HP-F0012(15μ 20g), eluent: H2O(90%)/MeCN (10%) => H2O(0%)/MeCN (100%) for 30 min, after MeCN (100%) for 5 min. The individual compound 18 was obtained as white amorphous solid (159 mg, yield 80%).
1HNMR (400 MHz, DMSO-d6, δ): 8.36 (d, J=7.3 Hz, 1Н, F5NHmn), 8.24-8.16 (m, 1Н, F6NHm+F6NHn), 7.96 (t, J=5.4 Hz, m) & 7.93 (t, J=5.4 Hz, n) (1H, X3NHk, m+n), 7.75-7.58 (m, 4H, X7NH+X7NH3+d), ), 7.42-7.09 (m, 14H, X8Hdn+X8Hen+X8Hdm+ X8Hem+F6He+F6Hd+X8Htmn+F5He+F6Hk+F5Hk+F5Hd+X8Hgmn), 6.39-6.25 (m, 2Н, K2NHm+K2NHn +E1NHm+E1NHn), 4.55 (s, n) & 4.47 (s, m) (2H, X8Ha, m+n), 4.42-4.33 (m, 1H, F6Ha), 4.33-4.23 (m, 1H, F5Ha), 4.14-3.98 (m, 2H, E1Ha+K2Ham+K2Han), 3.25-3.10 (m, 3H, K2Hemn+X7Hg(a)), 3.10-2.85 (m, 6Н, F6Hb(a) +X7Hg(b)+X3He(mn)+F6Hb(b)+F5Hb(a)), 2.78-2.58 (m, 3Н, X7Ha+F5Hb(b)), 2.40-2.10 (m, 8H, X3Ham+X4Hbmn +E1Hg +X4Hamn+X3Han), 1.97-1.85 (m, 1Н, E1Hb(a)), 1.76-1.56 (m, 4Н, E1Hb(b)+X7Hb +K2Hb(a)), 1.56-1.11 (m, 11Н, X3Hb+K2Hb(b)+X3Hd+ K2Hd+K2Hg+X3Hg, m+n).
13C NMR (100 MHz, DMSO-d6, δ): 174.61 (K2C(n)), 174.57 (K2C(m)), 174.27 (E1C(mn)), 173.84 (E1Cd), 173.06 (X4Cg(n)), 173.03 (X4Cg(m)), 172.23 (X3C), 171.70 (X4C(mn)), 171.33 (F5C), 171.24 (F6C), 159.03 (C(O)TFA), 158.66 (C(O)TFA), 158.30 (C(O)TFA), 157.92 (C(O)TFA), 157.36 (U), 141.27 (X8Cb(m)), 140.87 (X8Cb(n)), 138.09 (F6Cg), 138.03 (F5Cg), 133.46 (X8Ck(n)), 133.11 (X8Ck(m)), 130.66 (X8Cd(n)), 130.30 (X8Cd(m)), 129.07 (F6Cd+F5Cd), 128.28 (F6Ce), 128.16 (F5Ce), 127.24 (X8Ct(m)), 127.19 (X8Ce(n)), 126.90 (X8Ce(m)), 126.44 (F6Ck), 126.34 (X8Ct(n))+F5Ck), 126.14 (X8Cg(m)), 125.02 (X8Cg(n)), 117.00 (CF3), 114.17 (CF3), 55.15 (F5Ca), 54.58 (F6Ca), 52.33 (K2Ca(n)), 52.20 (K2Ca(m)), 51.72 (E1Ca), 49.67 (X8Ca(n)), 47.21 (X8Ca(m)), 46.93 (K2Ce(m)), 45.39 (K2Ce(n)), 38.72 (X3Ce(m)), 38.65 (X3Ce(n)), 36.94 (F5Cb), 36.82 (F6Cb), 36.72 (X7Cg), 35.79 (X7Ca), 32.34 (X3Ca(n)), 31.93 (X3Ca(m)), 31.85 (K2Cb), 30.72 (X4Ca), 30.59 (X4Cb), 29.97 (E1Cg), 29.11 (X3Cd(m)), 29.01 (X3Cd(n)), 27.85 (K2Cd(m)), 27.59 (E1Cb), 27.23 (X7Cb), 26.80 (K2Cd(n)), 26.34 (X3Cg(m)), 26.26 (X3Cg(n)), 24.77 (X3Cb(m)), 24.63 (X3Cb(n)), 22.56 (K2Cg(n)) 22.40 (K2Cg(m)).
ESI-MS C50H67СlN8O12: m/z calcd. for [M+H+]+: 1007.46,found: 1007.55
HRMS (m/z, ESI): calcd. for C50H67СlN8O12 - [M+H]+ 1007.4640, found: 1007.4622
Compound 19. Compound 18 (1 eq.; 30 mg; 26.75 μmol) and DIPEA (6 eq.; 28 μl; 160.5 μmol) were dissolved in DMF (2 mL). Compound 13 (1 eq.; 14 mg; 26.75 μmol) was added to the obtained mixture and the system was purged with argon. The mixture was stirred for 6 h and the solvent was evaporated under reduced pressure. The residue was then purified by column chromatography (Puriflash on a column of PF-15C18HP-F0012(15μ 20g), eluent: H2O(90%)/MeCN(10%) => H2O(0%)/MeCN(100%) for 20 min after MeCN (100%) for 5 min. Compound 19 was obtained as white powder (28 mg, yield 75%).
1HNMR (400 MHz, DMSO-d6, δ): 13.30-11.30 (br.s, 3H, COOH), 8.45-8.36 (m, 1H, X7NHδ), 8.33-8.25 (m, 1Н, F5NHmn), 8.24-8.14 (m, 1Н, F6NHm+F6NHn), 7.97-7.83 (m, 2H, X3NHζmn+2), 7.78-7.70 (m, 1H, 6), 7.69-7.59 (m, 1H, X7NH), 7.59-7.50 (m, 1H, 4), 7.45-7.08 (m, 15H, 5+X8Hδn+X8Hεn+X8Hδm+X8Hεm+F6Hε+F6Hδ+X8Hηmn+F5Hε+F6Hζ+F5Hζ+F5Hδ +X8Hγmn), 6.38-6.24 (m, 2Н, K2NHm+K2NHn +E1NHm+E1NHn), 4.54 (s, n) & 4.47 (s, m) (2H, X8Hα, m+n), 4.44-4.37 (m, 1H, F6Hα), 4.37-4.27 (m, 1H, F5Hα), 4.14-3.98 (m, 2H, E1Hα+K2Hαm+K2Hαn), 3.26-2.84 (m, 11H, K2Hεmn+X7Hγ(a))+F6Hβ(a) +X7Hγ(b) +X3Hε(mn) +F6Hβ(b)+X7Hα+F5Hβ(a)), 2.70-2.60 (m, 1Н, F5Hβ(b)), 2.40-2.10 (m, 8H, X3Hαm+X4Hβmn+E1Hγ +X4Hαmn+X3Hαn), 1.97-1.85 (m, 1Н, E1Hβ(a)), 1.76-1.67 (m, 1Н, E1Hβ(b)), 1.67-1.13 (m, 26Н, X7Hβ+K2Hβ(a)+8+X3Hβ +K2Hβ(b)+X3Hδ+9+ K2Hδ+K2Hγ+X3Hγ, m+n), 1.13-0.95 (m, 6H, 10), 0.83 (t, J = 7.1 Hz, 9H, 11).
13C NMR (100 MHz, DMSO-d6, δ): 174.54 (K2C(n)), 174.51 (K2C(m)), 174.22 (E1C(mn)), 173.80 (E1Cδ), 172.74 (X4Cγ(n)), 172.68 (X4Cγ(m)), 172.12 (X3C), 171.50 (X4C(mn)), 171.12 (F5C), 170.78 (F6C), 166.60 (7), 157.26 (U), 141.50 (6), 141.22 (X8Cβ(m)), 140.80 (X8Cβ(n)), 138.90 (1), 138.10 (F6Cγ), 138.00 (F5Cγ), 135.53 (2), 134.73 (3), 133.92 (5), 133.40 (X8Cζ(n)), 133.04 (X8Cζ(m)), 130.59 (X8Cδ(n)), 130.23 (X8Cδ(m)), 129.04 (F6Cδ+F5Cδ), 128.16 (F6Cε), 128.04 (F5Cε), 127.75 (4), 127.20 (X8Cη(m)), 127.12 (X8Cε(n)), 126.83 (X8Cε(m)), 126.29 (F6Cζ+X8Cη(n)), 126.22 (F5Cζ), 126.08 (X8Cγ(m)), 124.95 (X8Cγ(n)), 54.86 (F5Cα), 54.41 (F6Cα), 52.25 (K2Cα(n)), 52.14 (K2Cα(m)), 51.68 (E1Cα), 49.61 (X8Cα(n)), 47.13 (X8Cα(m)), 46.85 (K2Cε(m)), 45.31 (K2Cε(n)), 38.64 (X3Cε(m)), 38.57 (X3Cε(n)), 37.09 (F5Cβ), 36.86 (F6Cβ), 36.69 (X7Cγ), 35.48 (X7Cα), 32.28 (X3Cα(n)), 31.86 (X3Cα(m)), 31.81 (K2Cβ), 30.67 (X4Cα), 30.56 (X4Cβ), 29.97 (E1Cγ), 29.07 (X3Cδ(m)+X7Cβ), 28.95 (X3Cδ(n)), 28.60 (9), 27.80 (K2Cδ(m)), 27.60 (E1Cβ), 26.70 (K2Cδ(n)+10), 26.28 (X3Cγ(m)), 26.21 (X3Cγ(n)), 24.72 (X3Cβ(m)), 24.57 (X3Cβ(n)), 22.53 (K2Cγ(n)) 22.34 (K2Cγ(m)), 13.58 (8), 9.20 (11).
HRMS (m/z, ESI): calcd. for C69H97ClN8O13Sn - [M+H]+ 1401,5958, found: 1401,5990.
Compound 20. Compound 18 (1 eq.; 38 mg; 34 μmol) and DIPEA (6 eq.; 30 μl; 174.36 μmol) were dissolved in DMF (2 mL). Compound 16 (1 eq.; 17.3 mg; 34 μmol) was added to the obtained mixture, the system was purged with argon and the mixture was stirred for 6 h. The solvent was evaporated under reduced pressure and the residue was then purified by column chromatography (Puriflash on a column of PF-15C18HP-F0012(15μ 20g), eluent: H2O(90%)/MeCN(10%) => H2O(0%)/MeCN(100%) for 20 min after MeCN (100%) for 5 min. Compound 19 was obtained as white powder (30.5 mg, yield 64%).
1HNMR (400 MHz, DMSO-d6, δ): 12.95-11.50 (br.s, 3H, COOH), 8.44-8.34 (m, 1H, X7NHδ), 8.34-8.26 (m, 1Н, F5NHmn), 8.25-8.15 (m, 1Н, F6NHm+F6NHn), 7.92 (t, J=5.4 Hz, m) & 7.89 (t, J=5.4 Hz, n) (1H, X3NHζ, m+n), 7.75 (d, J=7.5 Hz, 2H, 3), 7.70-7.60 (m, 1H, X7NH), 7.51 (d, J=7.5 Hz, 2H, 2), 7.41-7.07 (m, 14H, X8Hδn+X8Hεn+X8Hδm+X8Hεm+F6Hε+F6Hδ+X8Hηmn +F5Hε+F6Hζ+F5Hζ +F5Hδ+X8Hγmn), 6.39-6.22 (m, 2Н, K2NHm+K2NHn +E1NHm+E1NHn), 4.54 (s, n) & 4.46 (s, m) (2H, X8Hα, m+n), 4.44-4.37 (m, 1H, F6Hα), 4.37-4.27 (m, 1H, F5Hα), 4.14-3.98 (m, 2H, E1Hα +K2Hαm+K2Hαn), 3.25-2.84 (m, 11H, K2Hεmn+X7Hγ(a))+F6Hβ(a) +X7Hγ(b) +X3Hε(mn) +F6Hβ(b)+X7Hα+ F5Hβ(a)), 2.70-2.60 (m, 1Н, F5Hβ(b)), 2.40-2.10 (m, 8H, X3Hαm+X4Hβmn+E1Hγ +X4Hαmn+X3Hαn), 1.96-1.84 (m, 1Н, E1Hβ(a)), 1.77-1.67 (m, 1Н, E1Hβ(b)), 1.67-1.13 (m, 26Н, X7Hβ+K2Hβ(a)+6+X3Hβ +K2Hβ(b)+X3Hδ+7+ K2Hδ+K2Hγ+X3Hγ, m+n), 1.13-0.95 (m, 6H, 8), 0.83 (t, J = 7.1 Hz, 9H, 9).
13C ЯМР (100 MHz, DMSO-d6, δ): 174.56 (K2C(mn)), 174.26 (E1C(mn)), 173.85 (E1Cδ), 172.77 (X4Cγ(n)), 172.72 (X4Cγ(m)), 172.16 (X3C), 171.53 (X4C(mn)), 171.18 (F5C), 170.81 (F6C), 166.50 (5), 157.29 (U), 145.70 (4), 141.23 (X8Cβ(m)), 140.83 (X8Cβ(n)), 138.12 (F6Cγ), 138.02 (F5Cγ), 136.10 (3), 134.29 (1), 133.42 (X8Cζ(n)), 133.06 (X8Cζ(m)), 130.60 (X8Cδ(n)), 130.25 (X8Cδ(m)), 129.06 (F6Cδ+F5Cδ), 128.19 (F6Cε), 128.06 (F5Cε), 127.21 (X8Cη(m)), 127.13 (X8Cε(n)), 126.84 (X8Cε(m)), 126.36 (2), 126.31 (F6Cζ+X8Cη(n)), 126.23 (F5Cζ), 126.10 (X8Cγ(m)), 124.97 (X8Cγ(n)), 54.89 (F5Cα), 54.47 (F6Cα), 52.29 (K2Cα(n)), 52.20 (K2Cα(m)), 51.76 (E1Cα), 49.65 (X8Cα(n)), 47.16 (X8Cα(m)), 46.88 (K2Cε(m)), 45.33 (K2Cε(n)), 38.65 (X3Cε(m)), 38.59 (X3Cε(n)), 37.09 (F5Cβ), 36.87 (F6Cβ), 36.71 (X7Cγ), 35.48 (X7Cα), 32.30 (X3Cα(n)), 31.89 (X3Cα(m)), 31.83 (K2Cβ), 30.69 (X4Cα), 30.58 (X4Cβ), 30.08 (E1Cγ), 29.07 (X3Cδ(m)), 29.01 (X7Cβ), 28.97 (X3Cδ(n)), 28.62 (7), 27.82 (K2Cδ(m)), 27.73 (E1Cβ), 26.71 (K2Cδ(n)+8), 26.29 (X3Cγ(m)), 26.22 (X3Cγ(n)), 24.74 (X3Cβ(m)), 24.59 (X3Cβ(n)), 22.55 (K2Cγ(n)) 22.36 (K2Cγ(m)), 13.59 (6), 9.23 (9).
HRMS (m/z, ESI): calcd. for C69H97ClN8O13Sn - [M+H]+ 1401,5958, found: 1401,5978.
Compound 21. Way A. Compound 18 (1 eq.; 32.5 mg; 29 μmol) and DIPEA (6 eq.; 30 μl; 174.36 μmol) were dissolved in DMF (4 mL). Compound 14 (1 eq.; 10 mg; 29 μmol) was added to the odtained mixture and the system was purged with argon. The mixture was stirred for 6 h. The solvent was evaporated under reduced pressure and the residue was then purified by column chromatography (Puriflash on a column of PF-15C18HP-F0012(15μ 20g), eluent: H2O(90%)/MeCN(10%) => H2O(0%)/MeCN(100%) for 20 min after MeCN (100%) for 5 min. Compound 19 was obtained as white powder (28.6 mg, yield 80%).
Way B. I2 (1 eq; 3.4 mg; 13.136 μmol) was dissolved in 0.1N NaOH (79 µl = V1), AcOH (3%) in CHCl3 (79 µl = V2) was added (V1 = V2), then tert-butyl hydroperoxide (TBHP) in CHCl3 (the solution is prepared in advance by adding 1800 µl of 70% TBHP in water to 11209 µl of CHCl3, after which Na2SO4 is added to the prepared mixture to bind water) (394 µl) and then 19 (1 eq; 18.4 mg; 13.136 μmol l) in DMF (131 µl) were added. The mixture was stirred for 30 minutes. Next, the solvent was removed under reduced pressure and the residue was purified by reverse phase column chromatography (Puriflash PF-15C18AQ-F0012 (15μ 20g): H2O(90%)/MeCN(10%) => H2O(0 %)/MeCN(100%) for 30 minutes after MeCN (100%) for 5 minutes. Compound 21 was obtained as white powder (10.5 mg, yield 65%).
1HNMR (400 MHz, DMSO-d6, δ): 12.74-11.94 (br.s, 3H, COOH), 8.55-8.47 (m, 1H, X7NHδ), 8.29 (d, J = 7.4 Hz, 1Н, F5NH), 8.22-8.13 (m, 1Н, F6NHmn+2), 7.93-7.80 (m, 3H, X3NHζmn+4+6), 7.67-7.58 (m, 1H, X7NH), 7.42-7.08 (m, 15H, X8Hδn+5+X8Hεn+ X8Hδm+X8Hεm+F6Hε+F6Hδ +X8Hηmn+F5Hε+F6Hζ+F5Hζ+F5Hδ+X8Hγmn), 6.37-6.23 (m, 2Н, K2NHm +K2NHn +E1NHm+E1NHn), 4.55 (s, n) & 4.47 (s, m) (2H, X8Hα, m+n), 4.44-4.37 (m, 1H, F6Hα), 4.37-4.28 (m, 1H, F5Hα), 4.14-3.98 (m, 2H, E1Hα+K2Hαm+K2Hαn), 3.26-2.84 (m, 11H, K2Hεmn+X7Hγ(a)) +F6Hβ(a) +X7Hγ(b)+X3Hε(mn)+F6Hβ(b)+X7Hα+F5Hβ(a)), 2.70-2.60 (m, 1Н, F5Hβ(b)), 2.40-2.10 (m, 8H, X3Hαm+X4Hβmn+E1Hγ +X4Hαmn+X3Hαn), 1.97-1.85 (m, 1Н, E1Hβ(a)), 1.76-1.67 (m, 1Н, E1Hβ(b)), 1.67-1.11 (m, 14Н, X7Hβ+K2Hβ(a)+X3Hβ+K2Hβ(b)+X3Hδ+ K2Hδ+K2Hγ+X3Hγ, m+n).
13C NMR (100 MHz, DMSO-d6, δ): 174.56 (K2C(n)), 174.53 (K2C(m)), 174.25 (E1C(mn)), 173.81 (E1Cδ), 172.82 (X4Cγ(n)), 172.77 (X4Cγ(m)), 172.16 (X3C), 171.55 (X4C(mn)), 171.16 (F5C), 170.75 (F6C), 164.71 (7), 157.30 (U), 141.24 (X8Cβ(m)), 140.82 (X8Cβ(n)), 139.68 (6), 138.13 (F6Cγ), 138.02 (F5Cγ), 136.59 (3), 135.63 (2), 133.42 (X8Cζ(n)), 133.07 (X8Cζ(m)), 130.61 (X8Cδ(n)), 130.54 (5), 130.25 (X8Cδ(m)), 129.06 (F6Cδ+F5Cδ), 128.19 (F6Cε), 128.08 (F5Cε), 127.21 (X8Cη(m)), 127.14 (X8Cε(n)), 126.85 (X8Cε(m)), 126.64 (4), 126.31 (F6Cζ+X8Cη(n)), 126.25 (F5Cζ), 126.09 (X8Cγ(m)), 124.97 (X8Cγ(n)), 94.74 (1), 54.96 (F5Cα), 54.45 (F6Cα), 52.29 (K2Cα(n)), 52.16 (K2Cα(m)), 51.68 (E1Cα), 49.65 (X8Cα(n)), 47.17 (X8Cα(m)), 46.89 (K2Cε(m)), 45.33 (K2Cε(n)), 38.67 (X3Cε(m)), 38.60 (X3Cε(n)), 37.09 (F5Cβ), 36.97 (X7Cγ), 36.86 (F6Cβ), 36.55 (X7Cα), 32.28 (X3Cα(n)), 31.86 (X3Cα(m)), 31.81 (K2Cβ), 30.67 (X4Cα), 30.56 (X4Cβ), 29.97 (E1Cγ), 29.07 (X3Cδ(m)), 28.95 (X3Cδ(n)), 28.87 (X7Cβ), 27.80 (K2Cδ(m)), 27.60 (E1Cβ), 26.70 (K2Cδ(n), 26.28 (X3Cγ(m)), 26.21 (X3Cγ(n)), 24.72 (X3Cβ(m)), 24.57 (X3Cβ(n)), 22.53 (K2Cγ(n)) 22.34 (K2Cγ(m)).
ESI-MS C57H70СlIN8O13: m/z calcd. for [M+H+]+: 1237.38, found: 1237.55
HRMS (m/z, ESI): calcd. for C57H70СlIN8O13 - [M+H]+ 1237.3868, found: 1237.3873
Compound 22. Way A. Compound 18 (1 eq.; 20 mg; 18 μmol) and DIPEA (6 eq.; 19 μl; 107 μmol) were dissolved in DMF (4 mL). Compound 17 (1 eq.; 6.1 mg; 18 μmol) was added to the obtained mixture and the system was purged with argon. The mixture was stirred for 6 h, the solvent was then evaporated under reduced pressure. The residue was purified by column chromatography (Puriflash on a column of PF-15C18HP-F0012(15μ 20g), eluent: H2O(90%)/MeCN(10%) => H2O(0%)/MeCN(100%) for 20 min after MeCN (100%) for 5 min. Compound 19 was obtained as white powder (13 mg, yield 59%).
Way B. I2 (1 eq; 5.4 mg; 21.42 μmol) was dissolved in 0.1N NaOH (129 µl = V1), AcOH (3%) in CHCl3 (129 µl = V2) was added (V1 = V2), then tert-butyl hydroperoxide (TBHP) in CHCl3 (the solution is prepared in advance by adding 1800 µl of 70% TBHP in water to 11209 µl of CHCl3, after which Na2SO4 is added to the prepared mixture to bind water) (643 µl), then 20 (1 eq; 30 mg; 21.42 μmol l) in DMF (214 µl) were added. The mixture was stirred for 30 minutes and the solvent was then removed under reduced pressure. The residue was purified by reverse phase column chromatography (Puriflash PF-15C18AQ-F0012 (15μ 20g): H2O(90%)/MeCN(10%) => H2O(0 %)/MeCN(100%) for 30 minutes after MeCN (100%) for 5 minutes. Compound 21 was obtained as white powder (11.7 mg, yield 44%).
1HNMR (400 MHz, DMSO-d6, δ): 12.74-11.94 (br.s, 3H, COOH), 8.54-8.45 (m, 1H, X7NHδ), 8.38-8.28 (m, 1Н, F5NH), 8.26-8.15 (m, 1Н, F6NHmn+2), 7.93 (t, J=5.4 Hz, m) & 7.90 (t, J=5.4 Hz, n) (1H, X3NHζ, m+n), 7.86-7.81 (m, 2H, 3), 7.68-7.58 (m, 3H, X7NH+2), 7.41-7.09 (m, 14H, X8Hδn+X8Hεn+X8Hδm+X8Hεm+F6Hε+F6Hδ+X8Hηmn+F5Hε+F6Hζ+F5Hζ +F5Hδ+X8Hγmn), 6.37-6.23 (m, 2Н, K2NHm+K2NHn +E1NHm+E1NHn), 4.54 (s, n) & 4.47 (s, m) (2H, X8Hα, m+n), 4.44-4.35 (m, 1H, F6Hα), 4.35-4.27 (m, 1H, F5Hα), 4.14-3.98 (m, 2H, E1Hα+K2Hαm+K2Hαn), 3.26-2.84 (m, 11H, K2Hεmn+X7Hγ(a)) +F6Hβ(a) +X7Hγ(b) +X3Hε(mn)+F6Hβ(b)+X7Hα+F5Hβ(a)), 2.70-2.60 (m, 1Н, F5Hβ(b)), 2.40-2.10 (m, 8H, X3Hαm+X4Hβmn+E1Hγ +X4Hαmn+X3Hαn), 1.97-1.85 (m, 1Н, E1Hβ(a)), 1.76-1.11 (m, 15Н, E1Hβ(b)+X7Hβ+K2Hβ(a)+X3Hβ+K2Hβ(b) +X3Hδ+K2Hδ+K2Hγ+X3Hγ, m+n).
13C NMR (100 MHz, DMSO-d6, δ): 174.57 (K2C(n)), 174.54 (K2C(m)), 174.25 (E1C(mn)), 173.82 (E1Cδ), 172.80 (X4Cγ(n)), 172.75 (X4Cγ(m)), 172.16 (X3C), 171.53 (X4C(mn)), 171.17 (F5C), 170.76 (F6C), 165.51 (5), 157.28 (U), 141.23 (X8Cβ(m)), 140.83 (X8Cβ(n)), 139.68 (6), 138.11 (F6Cγ), 138.01 (F5Cγ), 137.19 (3), 133.97 (2), 133.42 (X8Cζ(n)), 133.07 (X8Cζ(m)), 130.61 (X8Cδ(n)), 130.25 (X8Cδ(m)), 129.06 (F6Cδ+F5Cδ), 128.19 (F6Cε), 128.08 (F5Cε), 127.21 (X8Cη(m)), 127.14 (X8Cε(n)), 126.85 (X8Cε(m)), 126.31 (F6Cζ+X8Cη(n)), 126.25 (F5Cζ), 126.09 (X8Cγ(m)), 124.97 (X8Cγ(n)), 98.73 (1), 54.96 (F5Cα), 54.45 (F6Cα), 52.29 (K2Cα(n)), 52.16 (K2Cα(m)), 51.68 (E1Cα), 49.65 (X8Cα(n)), 47.17 (X8Cα(m)), 46.89 (K2Cε(m)), 45.33 (K2Cε(n)), 38.67 (X3Cε(m)), 38.60 (X3Cε(n)), 37.09 (F5Cβ), 36.88 (X7Cγ+F6Cβ), 36.55 (X7Cα), 32.28 (X3Cα(n)), 31.86 (X3Cα(m)), 31.81 (K2Cβ), 30.67 (X4Cα), 30.56 (X4Cβ), 30.01 (E1Cγ), 29.07 (X3Cδ(m)), 28.95 (X3Cδ(n)), 28.87 (X7Cβ), 27.80 (K2Cδ(m)), 27.60 (E1Cβ), 26.70 (K2Cδ(n), 26.28 (X3Cγ(m)), 26.21 (X3Cγ(n)), 24.72 (X3Cβ(m)), 24.57 (X3Cβ(n)), 22.53 (K2Cγ(n)) 22.34 (K2Cγ(m)).
ESI-MS C57H70СlIN8O13: m/z calcd. for [M+H+]+: 1237.38, found: 1237.55
HRMS (m/z, ESI): calcd. for C57H70СlIN8O13 - [M+H]+ 1237.3868, found: 1237.3890
4.2. Iodine-123 Production
The 123I was obtained from Tosmk Polytechnic University R-7M cyclotron facility. 123I was produced by irradiating [122Te]TeO2 target by deuteron 13.6 MeV with current of 20 µA. As a target, 99.6% enriched-[122Te]TeO2 and 1% of Al2O3 were deposited on platinum backing plate. The irradiation was carried out for 3 hours. After irradiation, the 123I was isolated from the target by dry distillation methods. The target was heated, then 123I was distilled out of the matrix and carried by the sweep gas to a receiving vessel filled with alkaline and iodine solutions where it was trapped as [123I]NaI.
4.3. Radiolabeling Optimization of PSMA-TBSB with 123I
The 123I was available in 0.01 M NaOH with an average batch activity of approx. 732 MBq/mL. This An average 123I activity used for every experiment was 25 MBq. An amount of PSMA solution 1 mg/mL previously dissolved in CH3OH/CH3COOH, 95/5 (v/v) was added to the 123I solution. 10 µL chloramine-T in Milli-Q water was used as an oxidizing agent. The reaction was performed at room temperature. The reaction time was calculated from the time chloramine-T was added to the mixed solution and vortexed carefully. To quench the reaction, 10 μl of sodium metabisulfite solution was added to the reaction mixture. The amount of sodium metabisulfite used was 2 (two) times that of the oxidizing agent used. After that, 5 μl of NaI solution 10 mg/mL was added to the reaction mixture. To test the radiolabeling yield, a radio-iTLC was performed by spotting 2 μl onto iTLC glass microfiber chromatography sheet impregnated with a silica gel (iTLC-SG fiber sheet) and eluted with a developing solution of CH3CN/H2O, 95/5 (v/v). Under these conditions, the [123I]PSMA-p-IB had a Rf = 0.1-0.3, while free radioiodine moved with the front of the developing solution (Rf ≥ 0.75). The percent radiolabeling efficiency of the radioligand was calculated. All data are expressed as mean ± SD.
In order to investigate the optimal radiolabeling condition, the study was performed by varying the peptide amount, the reaction time, and the oxidizing agent amount. The influence of the PSMA ligand (PSMA-p-TBSB) amount (at 0.73 nmol, 3 nmol, 5 nmol, 10 nmol and 50 nmol) was investigated at a fixed reaction time of 5 min and oxidizing agent amount of 40 µg. The effect of the reaction time was studied by applying time variation of 0.5 min, 5 min, 10 min and 30 min to the process of reactions with the peptide and the oxidizing agent amount used are fixed, respectively 5 nmol and 40 µg, respectively. Whereas the study of oxidizing agent amount on the radiolabeling optimization was conducted by using 10 µg, 40 µg, 80 µg and 150 µg in variation with a fixed reaction time of 5 min and peptide amount of 5 nmol. All reactions were carried out at room temperature and the pH was adjusted to a value of 5-6, with the addition of each 1% CH3COOH solution as much as one tenth of the volume of [123I]NaI used.
4.4. Radiolabeling of [177Lu]Lu-PSMA-617
Ultrapure and metal-free buffers for radiosynthesis were prepared using high-quality Milli-Q water and pretreated with Chelex 100 resin sodium form (Sigma-Aldrich, USA). The compound of PSMA-617 used herein was synthesized by the method as published by Benesova et al. [41]. Ammonium acetate buffer (0.2 M, pH 5.5, Merck, Germany) in an amount of 80 µL was added in a LoBind Eppendorf tube containing 5 nmol of the PSMA-617 in Milli-Q water (1 nmol/µl). After the addition of 177Lu (5 µL, 25 MBq, IRT-T Nuclear Research Reactor of Tomsk Polytechnic University), the reaction mixture was vortexed and incubated for 30 min at 80 °C. Radiochemical yield and purity were determined by using radio-ITLC and radio-HPLC. The used iTLC method was glass fiber sheets (Agilent Technologies, Inc., Folsom, CA, USA) eluted in 0.2 M citric acid with pH of 2.0. Performing radio-iTLC analysis in this system provides retention of the 177Lu-labeled PSMA ligand molecules at the point of application, while free 177Lu3+ ion migrate with the solvent front. While the HPLC technique employed for [177Lu]Lu-PSMA-617 was identical to that used to analyse [123I]PSMA-p-IB.
4.5. Radiochemical Purity and Shelf Life Stability
After the optimal radiolabeling condition was achieved, the radiolabeled PSMA was purified using Sep-Pak® C18 cartridge. The cartridge was previously pre-equilibrated with 10 mL ethanol, then ethanol/water 9/10 (v/v), and followed by passing 10 mL Milli-Q water. Then, the radiolabeled mixture was loaded into the C18 cartridge. The cartridge was rinsed by passing 10 mL of Milli-Q water 3 times. The [123I]PSMA-p-IB was purified by passing 1 mL of ethanol and the drops coming from the cartridge was collected.
The purity of the [123I]PSMA-p-IB was determined by performing radio-iTLC and radio-HPLC. Radio-iTLC was performed using TLC silica gel 60 F254 aluminium plates (iTLC-SG 60 F254) with a developing solution of CH3CN/H2O, 95/5 (v/v). Radio-HPLC was performed using Agilent 1200 Series HPLC Systems (Agilent Technologies, USA) with Luna С18(2) column 5 µm, 100 Å, 250×4.6 mm. The radio detector was raytest Gabi Star with Serial Nr.: #30685 raytest GINA star 20.04.09 Firmw. The concentration gradient: 0 min 95% A (5% B), 5 min 80% A (20% B), 10 min 65% A (35% B), 15 min 50% A (50% B), 25 min 20% A (80% B), 30 min 0 % A (100% B), 32 min 95% A (5% B), where system A - 0.1% TFA in water, and system B - 0.1% TFA in acetonitrile, flow rate 1 mL/min). The [177Lu]Lu-PSMA-617 purity was also determined by radio-iTLC and radio-HPLC. The radio-iTLC and radio-HPLC methods used are described in sub-section of 5.5.
The shelf life stability of [123I]PSMA-p-IB with ethanol solvent was examined by storing it in a fridge for three days. After three days of storage, the sample’s stability was assessed using radio-HPLC.
4.6. Lipophilicity Assay: Log(D)
Lipophilicity of the 123I-PSMA was determined as the logarithm of partition coefficient, log(D), of the 123I-PSMA compound between n-octanol and water [42]. 500 µL of n-octanol was added to a eppendorf tube containing the same volume of Milli-Q water. 10 pmol of 123I-PSMA was added to the eppendorf tube containing n-octanol and Milli-Q water. The mixture was vigorously vortexed for 3 min and then centrifuged for 5 min. The activity concentration of 100 µL of both phase was then measured by gamma counter. Each measurement was repeated in triplicate.
4.7. In Vitro Cell Binding Assay
Binding specificity test of [123I]PSMA-p-IB was performed against human prostate cancer cell lines, PSMA-positive PC-3 PIP and PSMA-negative PC-3. The PSMA-expressing isogenic human prostate carcinoma PC-3-PIP cell line was obtained from Dr Warren Heston, Cleaveland Clinic. While PC-3 cell line was purchased from the American Type Culture Collection (ATCC; LGC Promochem, Borås, Sweden). The cell lines were cultured as published by Lundmark et al. [38].
Two sets (six wells) of dishes were used for each cell line. One day prior to the experiment, three dishes containing approximately 0,7×106 cells per dish were seeded. One set of dishes (three dishes) for each cell line added with 500-fold molar excess of unlabeled PSMA ligand to saturate PSMA receptors 30 min before adding the [123I]PSMA-p-IB. An equal volume of complete media was added to another set of dishes for each cell line. Then, all cells were incubated with 1 nM concentration of [123I]PSMA-p-IB for 1 hour at 37 oC. After incubation, the medium and 1 mL PBS solution used to wash the dish was collected. The cells were detached by treatment with 500 µL trypsine and incubated 10 min at 37 oC, then were collected in tubes. Cell-associated radioactivity was measured using a gamma counter and displayed as a percentage of cell-associated activity.
4.8. Affinity Measurements Using Saturation Binding Experiments
The binding kinetics of [123I]PSMA-p-IB to living PC-3 PIP cells was measured by the equilibrium dissociation constant (KD) using saturation binding experiments. Several concentrations in the range of ~0.1 × KD to ~10 × KD were used in this experiment. The radioactive ligand was added from the same stock solution. To achieve a lower concentration, less of the radioactive solution was added to the dishes and media was added to compensate for the rest of the volume. Four dishes (3 non-blocked and 1 blocked) were used for each concentration. The blocked dish was used to account for non-specific binding. Twenty (20) µM of unlabeled ligand was added to each of blocked dish. Incubations were conducted at 4 °C for 4 hours. The solution was removed from the cell after incubation, followed by rinsing. Then, 500 µl of trypsin was added to the cell dish, and the discharge of the cells was awaited. Following the discharge of all cells, 1 mL of medium was added to each dish. Afterwards, one-third of the sample volume was taken to the cell counter, and the remaining two-thirds of the sample volume was measured using the gamma counter. The KD was determined using a nonlinear regression analysis implemented in GraphPad Prism.
4.9. In Vivo Biodistribution
All applicable international and national guidelines of Russian Federation for the care and the use of the animals were followed during planning and execution of animal experiments. The animal study protocol was approved by the Ethics Committee of Siberian State Medical University, Tomsk, Russia (protocol code 2, 20220927).
The biodistribution of the [123I]PSMA-p-IB and [177Lu]Lu-PSMA-617 in normal mice were evaluated in 8 female CD1 mice (2 groups) of 6 weeks old with an average weight of 31.8 ± 3.6 g. The mice were housed and cared for under standard conditions prior to use. Four (4) mice were injected intravenously through the tail vein with 40 kBq (80 pmol) of [123I]PSMA-p-IB in 100 µL PBS with 10% ethanol. While the remaining 4 mice were intravenously (i.v.) injected with 130 kBq (80 pmol) of [177Lu]Lu-PSMA-617 in 100 µL PBS with 1% BSA per mouse. The mice were sacrificed 4 h after injection. Cervical dislocation has been employed to sacrifice anesthetized mice. The blood, tissues and organs (salivary gland, lung, liver, spleen, small intestine, kidney, muscle, and bone) were excised, collected and weighed. The 123I and 177Lu activities were simultaneously measured using gamma-counter. The activity uptake was expressed as the percentage of injected activity per gram of organ (%ID/g).
5. Conclusion
Thus, two novel DCL urea-based PSMA inhibitors with chlorine-substituted aromatic fragment at the lysine ε-nitrogen atom, L-Phe-L-Phe dipeptide linker, and 3- or 4-(tributylstannyl)benzoic acid as prosthetic groups for radioiodination were firstly synthesized using two alternative synthetic schemes. These inhibitors were studied as novel PSMA ligands by conducting radiolabeling optimization with Iodine-123. The [123I]PSMA-p-IB ligand was tested in the initial preclinical evaluation. The novel PSMA-targeting radioligand [123I]PSMA-p-IB demonstrated a considerable affinity and specific binding to PSMA-expressing cells in vitro. Low accumulation in normal organs during an in vivo test indicates that this novel PSMA inhibitor has the potential to be a promising novel PSMA-targeting radioligand, which warrants further study.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, experimental data.
Author Contributions
Coceptualization, M.S.L. M.S.Y.; methodology, M.S.L. L.A.H.; formal analysis, L.A.H., M.S.L., E.P., V.B., F.Y., M.S.Y., E.S.; investigation, L.A.H., M.S.L., E.P., V.B., F.Y., E.S., S.A.P., N.Y.Z., A.E.M., N.I.V., E.K.B., V.G.N., V.T., A.O., M.S.Y.; resources, M.S.L. M.S.Y., E.S.; data curation, L.A.H., M.S.L.; writing-original draft preparation, L.A.H., M.S.L., E.P., V.B., F.Y., M.S.Y., E.S.; writing-review and editing, L.A.H., M.S.L.; supervision, M.S.L., M.S.Y.; project administration, M.S.L.; funding acquition, M.S.L., M.S.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the TPU development program Priority 2030 (Priority-2030-NIP/IS-042-375-2023); Russian Science Foundation for Grant No: 22-15-00098, https://rscf.ru/project/22-15-00098/ (Synthesis of PSMA ligands) and Grant No: 21-13-00023; Grant of the President of the Russian Federation (MK-3748.2022.1.3); and in the part of NMR study by the M.V. Lomonosov Moscow State University Program of Development.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Machulkin, A.E.; Uspenskaya, A.A.; Zyk, N.U.; Nimenko, E.A.; Ber, A.P.; Petrov, S.A.; Polshakov, V.I.; Shafikov, R.R.; Skvortsov, D.A.; Plotnikova, E.A.; et al. Synthesis, Characterization, and Preclinical Evaluation of a Small-Molecule Prostate-Specific Membrane Antigen-Targeted Monomethyl Auristatin e Conjugate. J. Med. Chem. 2021, 64, 17123–17145. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Kuratsukuri, K.; Landas, S.; Imaida, K.; Rovito, P.M.; Wang, C.Y.; Haas, G.P. Expression of Prostate-Specific Membrane Antigen in Normal and Malignant Human Tissues. World J. Surg. 2006, 30, 628–636. [Google Scholar] [CrossRef]
- Wang, X.; Yin, L.; Rao, P.; Stein, R.; Harsch, K.M.; Lee, Z.; Heston, W.D.W. Targeted Treatment of Prostate Cancer. J. Cell. Biochem. 2007, 102, 571–579. [Google Scholar] [CrossRef]
- Machulkin, A.E.; Shafikov, R.R.; Uspenskaya, A.A.; Petrov, S.A.; Ber, A.P.; Skvortsov, D.A.; Nimenko, E.A.; Zyk, N.U.; Smirnova, G.B.; Pokrovsky, V.S.; et al. Synthesis and Biological Evaluation of PSMA Ligands with Aromatic Residues and Fluorescent Conjugates Based on Them. J. Med. Chem. 2021, 64, 4532–4552. [Google Scholar] [CrossRef]
- Machulkin, A.E.; Skvortsov, D.A.; Ivanenkov, Y.A.; Ber, A.P.; Kavalchuk, M. V.; Aladinskaya, A. V.; Uspenskaya, A.A.; Shafikov, R.R.; Plotnikova, E.A.; Yakubovskaya, R.I.; et al. Synthesis and Biological Evaluation of PSMA-Targeting Paclitaxel Conjugates. Bioorganic Med. Chem. Lett. 2019, 29, 2229–2235. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Kantoff, P.; Vogelzang, N.J.; Mega, A.; Fleming, M.T.; Stephenson, J.J.; Frank, R.; Shore, N.D.; Dreicer, R.; McClay, E.F.; et al. Phase 1 Study of PSMA ADC, an Antibody-Drug Conjugate Targeting Prostate-Specific Membrane Antigen, in Chemotherapy-Refractory Prostate Cancer. Prostate 2019, 79, 604–613. [Google Scholar] [CrossRef]
- Teo, M.Y.; Morris, M.J. Prostate-specific membrane antigen-directed therapy for metastatic castration-resistant prostate cancer. Cancer J (United States). 2016, 22(5), 47–352. [CrossRef]
- Fan, X.; Guo, Y.; Wang, L.; Xiong, X.; Zhu, L.; Fang, K. Diagnosis of Prostate Cancer Using Anti-PSMA Aptamer A10-3.2-Oriented Lipid Nanobubbles. Int. J. Nanomedicine 2016, 11, 3939–3950. [Google Scholar] [CrossRef]
- Cho, S.; Zammarchi, F.; Williams, D.G.; Havenith, C.E.G.; Monks, N.R.; Tyrer, P.; D’Hooge, F.; Fleming, R.; Vashisht, K.; Dimasi, N.; et al. Antitumor Activity of MEDI3726 (ADCT-401), a Pyrrolobenzodiazepine Antibody-Drug Conjugate Targeting PSMA, in Preclinical Models of Prostate Cancer. Mol. Cancer Ther. 2018, 17, 2176–2186. [Google Scholar] [CrossRef]
- Zyk, N.Y.; Ber, A.P.; Nimenko, E.A.; Shafikov, R.R.; Evteev, S.A.; Petrov, S.A.; Uspenskaya, A.A.; Dashkova, N.S.; Ivanenkov, Y.A.; Skvortsov, D.A.; et al. Synthesis and Initial in Vitro Evaluation of PSMA-Targeting Ligands with a Modified Aromatic Moiety at the Lysine ε-Nitrogen Atom. Bioorganic Med. Chem. Lett. 2022, 71, 128840. [Google Scholar] [CrossRef]
- Schäfer, M.; Bauder-Wüst, U.; Leotta, K.; Zoller, F.; Mier, W.; Haberkorn, U.; Eisenhut, M.; Eder, M. A Dimerized Urea-Based Inhibitor of the Prostatespecific Membrane Antigen for 68Ga-PET Imaging of Prostate Cancer. EJNMMI Res. 2012, 2, 1–11. [Google Scholar] [CrossRef]
- Schottelius, M.; Wirtz, M.; Eiber, M.; Maurer, T.; Wester, H.-J. [111In]PSMA-I&T: Expanding the Spectrum of PSMA-I&T Applications towards SPECT and Radioguided Surgery. EJNMMI Res. 2015, 5, 68. [Google Scholar] [CrossRef]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Baum, R.P.; Yildiz, A.; Beykan, S.; Kulkarni, H.R.; Lassmann, M.; Klette, I.; Eiber, M.; et al. 68Ga-and 177Lu-Labeled PSMA i and T: Optimization of a PSMA-Targeted Theranostic Concept and First Proof-of-Concept Human Studies. J. Nucl. Med. 2015, 56, 1169–1176. [Google Scholar] [CrossRef]
- Leamon, C.P.; Reddy, J.A.; Bloomfield, A.; Dorton, R.; Nelson, M.; Vetzel, M.; Kleindl, P.; Hahn, S.; Wang, K.; Vlahov, I.R. Prostate-Specific Membrane Antigen-Specific Antitumor Activity of a Self-Immolative Tubulysin Conjugate. Bioconjug. Chem. 2019, 30, 1805–1813. [Google Scholar] [CrossRef]
- Kularatne, S.A.; Zhou, Z.; Yang, J.; Post, C.B.; Low, P.S. Design, Synthesis, and Preclinical Evaluation of Prostate-Specific Membrane Antigen Targeted 99mTc-Radioimaging Agents. Mol. Pharm. 2009, 6, 790–800. [Google Scholar] [CrossRef]
- Kularatne, S.A.; Venkatesh, C.; Santhapuram, H.K.R.; Wang, K.; Vaitilingam, B.; Henne, W.A.; Low, P.S. Synthesis and Biological Analysis of Prostate-Specific Membrane Antigen-Targeted Anticancer Prodrugs. J. Med. Chem. 2010, 53, 7767–7777. [Google Scholar] [CrossRef]
- Glutamate, U.I.; Kozikowski, A.P.; Nan, F.; Conti, P.; Zhang, J.; Ramadan, E.; Bzdega, T.; Wroblewska, B.; Neale, J.H. Design of Remarkably Simple, Yet Potent Urea-Based Inhibitors of Glutamate Carboxypeptidase II (NAALADase). J. Med. Chem. 2001, 44, 298–301. [Google Scholar]
- Maresca, K.P.; Hillier, S.M.; Femia, F.J.; Keith, D.; Barone, C.; Joyal, J.L.; Zimmerman, C.N.; Kozikowski, A.P.; Barrett, J.A.; Eckelman, W.C.; et al. A Series of Halogenated Heterodimeric Inhibitors of Prostate Specific Membrane Antigen (PSMA) as Radiolabeled Probes for Targeting Prostate Cancer. J. Med. Chem. 2009, 52, 347–357. [Google Scholar] [CrossRef]
- 19 Jackson, P.F.; Cole, D.C.; Slusher, B.S.; Stetz, S.L.; Ross, L.E.; Donzanti, B.A.; Trainor, D.A. Design, Synthesis, and Biological Activity of a Potent Inhibitor of the Neuropeptidase N-Acetylated α-Linked Acidic Dipeptidase. J. Med. Chem. 1996, 39, 619–622. [Google Scholar] [CrossRef]
- Majer, P.; Jackson, P.F.; Delahanty, G.; Grella, B.S.; Ko, Y. Sen; Li, W.; Liu, Q.; Maclin, K.M.; Poláková, J.; Shaffer, K.A.; et al. Synthesis and Biological Evaluation of Thiol-Based Inhibitors of Glutamate Carboxypeptidase II: Discovery of an Orally Active GCP II Inhibitor. J. Med. Chem. 2003, 46, 1989–1996. [Google Scholar] [CrossRef]
- Machulkin, A.E.; Ivanenkov, Y.A.; Aladinskaya, A. V.; Veselov, M.S.; Aladinskiy, V.A.; Beloglazkina, E.K.; Koteliansky, V.E.; Shakhbazyan, A.G.; Sandulenko, Y.B.; Majouga, A.G. Small-Molecule PSMA Ligands. Current State, SAR and Perspectives. J. Drug Target. 2016, 24, 679–693. [Google Scholar] [CrossRef]
- Kuo, H.T.; Pan, J.; Zhang, Z.; Lau, J.; Merkens, H.; Zhang, C.; Colpo, N.; Lin, K.S.; Bénard, F. Effects of Linker Modification on Tumor-to-Kidney Contrast of 68 Ga-Labeled PSMA-Targeted Imaging Probes. Mol. Pharm. 2018, 15, 3502–3511. [Google Scholar] [CrossRef] [PubMed]
- Uspenskaya, A.A.; Machulkin, A.E.; Nimenko, E.A.; Shafikov, R.R.; Petrov, S.A.; Skvortsov, D.A.; Beloglazkina, E.K.; Majouga, A.G. Influence of the Dipeptide Linker Configuration on the Activity of PSMA Ligands. Mendeleev Commun. 2020, 30, 756–759. [Google Scholar] [CrossRef]
- Mettler, F.A.; Guiberteau, M.J. Thyroid, Parathyroid, and Salivary Glands. Essentials Nucl. Med. Mol. Imaging 2019, 85–115. [Google Scholar] [CrossRef]
- Wilbur, D.S.; Chyan, M.K.; Hamlin, D.K.; Vessella, R.L.; Wedge, T.J.; Hawthorne, M.F. Reagents for Astatination of Biomolecules. 2. Conjugation of Anionic Boron Cage Pendant Groups to a Protein Provides a Method for Direct Labeling That Is Stable to in Vivo Deastatination. Bioconjug. Chem. 2007, 18, 1226–1240. [Google Scholar] [CrossRef]
- Petrov, S.A.; Machulkin, A.E.; Uspenskaya, A.A.; Zyk, N.Y.; Nimenko, E.A.; Garanina, A.S.; Petrov, R.A.; Polshakov, V.I.; Grishin, Y.K.; Roznyatovsky, V.A.; et al. Polypeptide-Based Molecular Platform and Its Docetaxel/Sulfo-Cy5-Containing Conjugate for Targeted Delivery to Prostate Specific Membrane Antigen. Molecules 2020, 25, 5784. [Google Scholar] [CrossRef]
- Bollhagen, R.; Schmiedberger, M.; Barlos, K.; Grell, E. A New Reagent for the Cleavage of Fully Protected Peptides Synthesised on 2-Chlorotrityl Chloride Resin. J. Chem. Soc., Chem. Commun. 1994, 2559. [CrossRef]
- Behrendt, R.; White, P.; Offer, J. Advances in Fmoc Solid-Phase Peptide Synthesis. J. Pept. Sci. 2016, 22, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Petrov, S.A.; Machulkin, A.E.; Petrov, R.A.; Tavtorkin, A.N.; Bondarenko, G.N.; Legkov, S.A.; Nifant’ev, I.E.; Dolzhikova, V.D.; Zyk, N. V.; Majouga, A.G.; et al. Synthesis and Organogelating Behaviour of Urea- and Fmoc-Containing Diphenylalanine Based Hexaamide. J. Mol. Struct. 2021, 1234. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Zalutsky, M.R. Preparation of N-Succinimidyl 3-[*I]Iodobenzoate: An Agent for the Indirect Radioiodination of Proteins. Nat. Protoc. 2006, 1, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Isidro-Llobet, A.; Alvarez, M.; Albericio, F. Amino Acid-Protecting Groups. Chem. Rev 2009, 109, 2455–2504. [Google Scholar] [CrossRef]
- Yang, Y. Peptide Global Deprotection/Scavenger-Induced Side Reactions. In Side Reactions in Peptide Synthesis; Elsevier Inc.; ISBN: 978-0-12-801009-9, 2016; pp. 43–75 ISBN 9780128010099.
- Martin, S.; Tönnesmann, R.; Hierlmeier, I.; Maus, S.; Rosar, F.; Ruf, J.; Holland, J.P.; Ezziddin, S.; Bartholomä, M.D. Identification, Characterization, and Suppression of Side Products Formed during the Synthesis of [177Lu]Lu-PSMA-617. J. Med. Chem. 2021, 64, 4960–4971. [Google Scholar] [CrossRef]
- Eersels, J.L.H.; Travis, M.J.; Herscheid, J.D.M. Manufacturing I-123-Labelled Radiopharmaceuticals. Pitfalls and Solutions. J. Label. Compd. Radiopharm. 2005, 48, 241–257. [Google Scholar] [CrossRef]
- 35 Petrov, S.A.; Yusubov, M.S.; Beloglazkina, E.K.; Nenajdenko, V.G. Synthesis of Radioiodinated Compounds. Classical Approaches and Achievements of Recent Years. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.D.A.; Silva, J.L.; Ramos, M.P.S.; Oliveira, I.M. De; Felgueiras, C.F.; Herrerias, R.; Júnior, C.L.Z.; Mengatti, J.; Fukumori, N.T.O.; Matsuda, M.M.N. Radiochemical stability of radiopharmaceutical preparations. 2011 International Nuclear Atlantic Conference - INAC 2011, Belo Horizonte,MG, Brazil, October 24-28, 2011, ASSOCIAÇÃO BRASILEIRA DE ENERGIA NUCLEAR – ABEN.
- Fujita, T.; Iwasa, J.; Hansch, C. A New Substituent Constant, Ir, Derived from Partition Coefficients. J. Am. Chem. Soc. 1964, 86, 5175–5180. [Google Scholar] [CrossRef]
- Lundmark, F.; Olanders, G.; Rinne, S.S.; Abouzayed, A.; Orlova, A.; Rosenström, U. Design, Synthesis, and Evaluation of Linker-Optimised PSMA-Targeting Radioligands. Pharmaceutics 2022, 14, 1098. [Google Scholar] [CrossRef]
- Vorobyeva, A.; Sсhulga, A.; Konovalova, E.; Güler, R.; Mitran, B.; Garousi, J.; Rinne, S.; Löfblom, J.; Orlova, A.; Deyev, S.; Tolmachev, V. Comparison of tumor-targeting properties of directly and indirectly radioiodinated designed ankyrin repeat protein (DARPin) G3 variants for molecular imaging of HER2. Int J Oncol. 2019, 54, 1209–1220. [Google Scholar] [CrossRef]
- Hillier, S. M.; Maresca, K. P.; Femia, F. J.; Marquis, J. C.; Foss, C. A.; Nguyen, N.; Zimmerman, C. N.; Barrett, J. A.; Eckelman, W. C.; Pomper, M. G.; Joyal, J. L.; Babich, J. W. Preclinical evaluation of novel glutamate-urea-lysine analogues that target prostate-specific membrane antigen as molecular imaging pharmaceuticals for prostate cancer. Cancer Res. 2009, 69(17), 6932–6940. [Google Scholar] [CrossRef]
- Benešová, M.; Bauder-Wüst, U.; Schäfer, M.; Klika, K. D.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Linker Modification Strategies to Control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic Properties of DOTA-Conjugated PSMA Inhibitors. J. Med. Chem. 2016, 59(5), 1761–1775. [CrossRef]
- Oroujeni, M.; Abouzayed, A.; Lundmark, F.; Mitran, B.; Orlova, A.; Tolmachev, V.; Rosenström, U. Evaluation of Tumor-Targeting Properties of an Antagonistic Bombesin Analogue RM26 Conjugated with a Non-Residualizing Radioiodine Label Comparison with a Radiometal-Labelled Counterpart. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef]
Figure 1.
The general structure of the obtained ligands
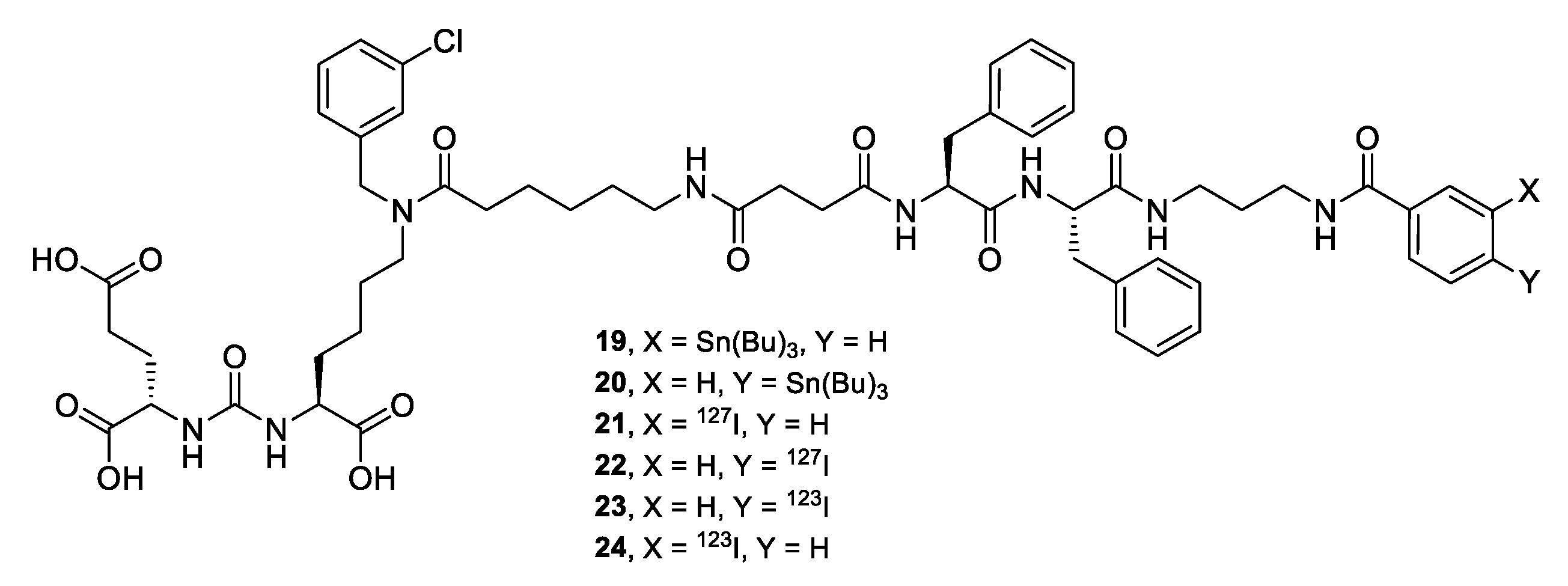
Figure 2.
Denotations of structural fragments of synthesized compounds (on the example of [127I]PSMA-m-IB (21)).
Figure 2.
Denotations of structural fragments of synthesized compounds (on the example of [127I]PSMA-m-IB (21)).
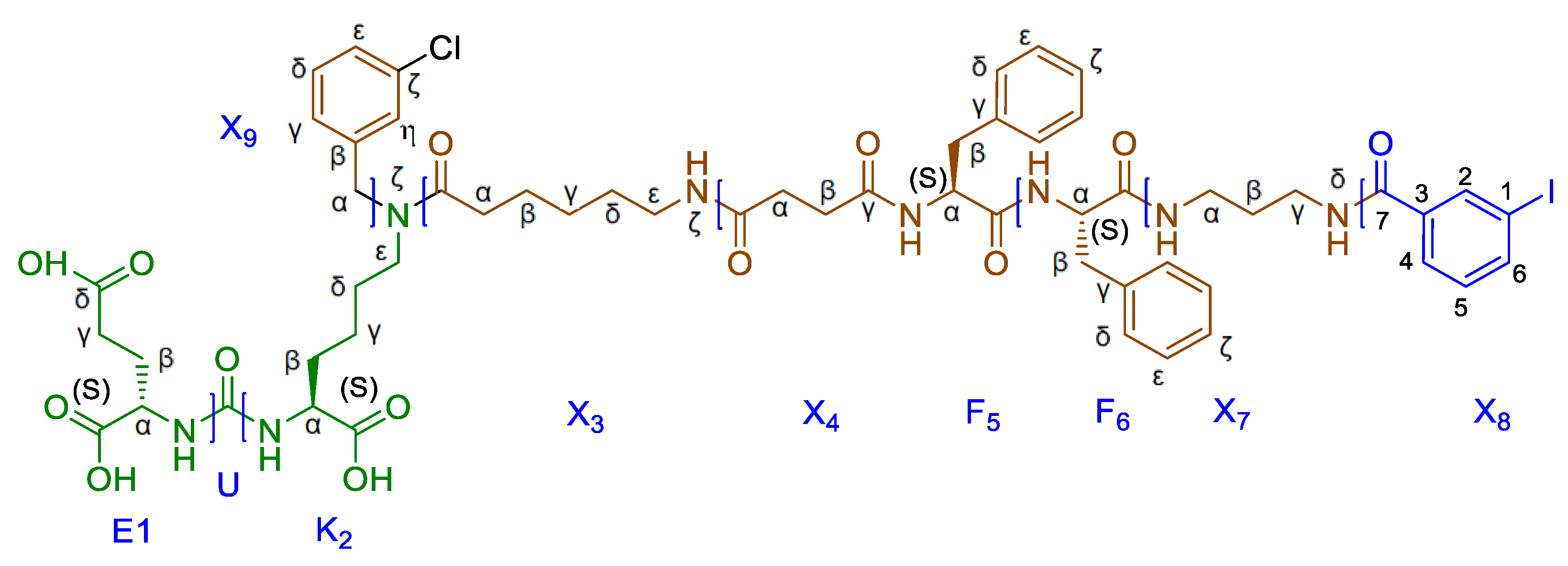
Scheme 1.
Synthesis of PSMA-vector fragment 6. Reagents and conditions: (a) (1) thriphosgene, DCM, −78 °C; (2) H-Lyz(Cbz)-O-tBu·HCl, Et3N, 20 °C; (b) H2, Pd/C (10%), MeOH; (c) (1) 3-Cl-C6H4-CHO, DCM; (2) NaBH4; (d) PyBOP, DIPEA, DMF, N3(CH2)5COOH; (e) THF/H2O, Ph3P, 50 °C; (f) (1) Succinic anhydride, DCM, DIPEA; (2) MeOH; (3) HCl (0.1M). All amino acids have L-configuration.
Scheme 1.
Synthesis of PSMA-vector fragment 6. Reagents and conditions: (a) (1) thriphosgene, DCM, −78 °C; (2) H-Lyz(Cbz)-O-tBu·HCl, Et3N, 20 °C; (b) H2, Pd/C (10%), MeOH; (c) (1) 3-Cl-C6H4-CHO, DCM; (2) NaBH4; (d) PyBOP, DIPEA, DMF, N3(CH2)5COOH; (e) THF/H2O, Ph3P, 50 °C; (f) (1) Succinic anhydride, DCM, DIPEA; (2) MeOH; (3) HCl (0.1M). All amino acids have L-configuration.
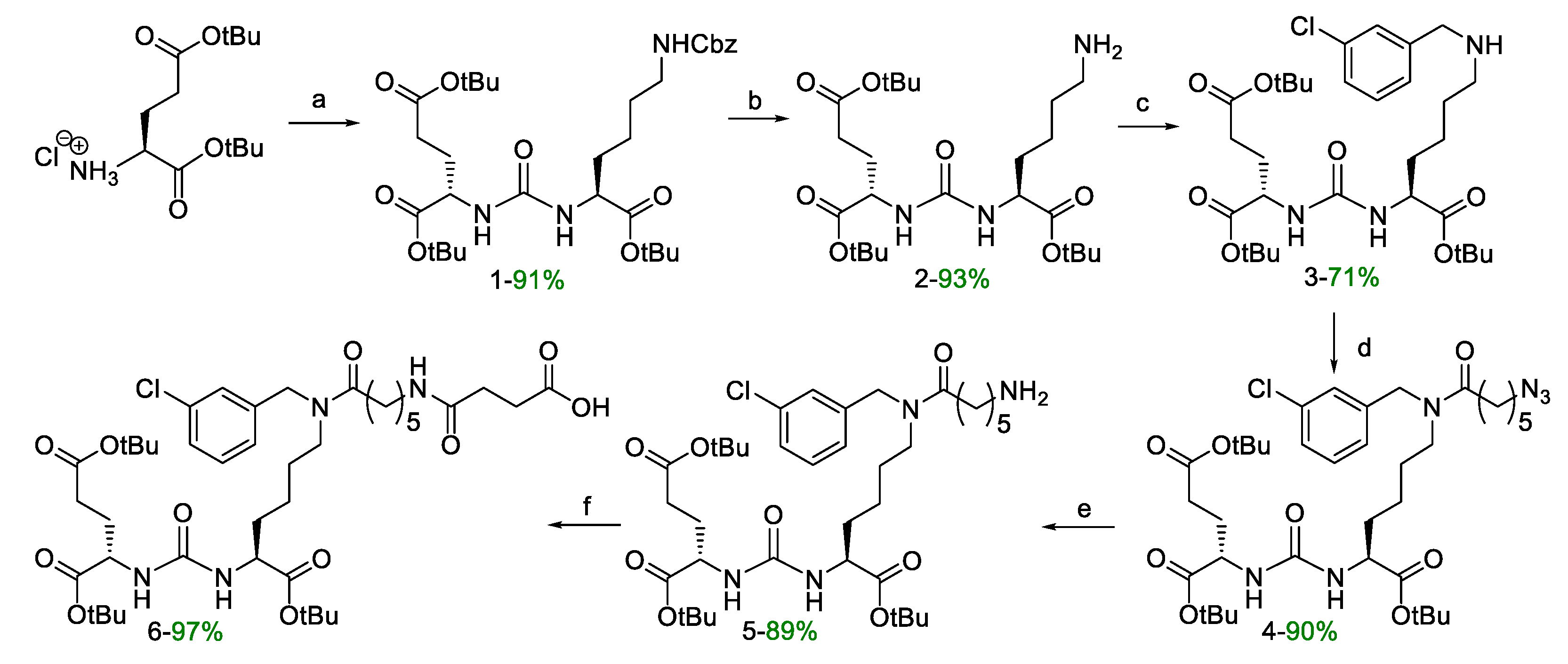
Scheme 2.
Synthesis of the polypeptide sequence 11. (a) (1) FmocPhe-OH(L), DIPEA, DMF; (2) 4-methylpiperidine/DMF; (b) (1) FmocPhe-OH(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF. (c) (1) compound 6, HBTU, HOBt, DIPEA, DMF; (2) DCM/TFA (99.25%/0.75%; V/V); (d) FmocNH(CH2)3NH3+TFA-, HBTU, HOBt, DIPEA, DMF; (e) Et2NH/DMF.
Scheme 2.
Synthesis of the polypeptide sequence 11. (a) (1) FmocPhe-OH(L), DIPEA, DMF; (2) 4-methylpiperidine/DMF; (b) (1) FmocPhe-OH(L), HBTU, HOBt, DIPEA; (2) 4-methylpiperidine/DMF. (c) (1) compound 6, HBTU, HOBt, DIPEA, DMF; (2) DCM/TFA (99.25%/0.75%; V/V); (d) FmocNH(CH2)3NH3+TFA-, HBTU, HOBt, DIPEA, DMF; (e) Et2NH/DMF.
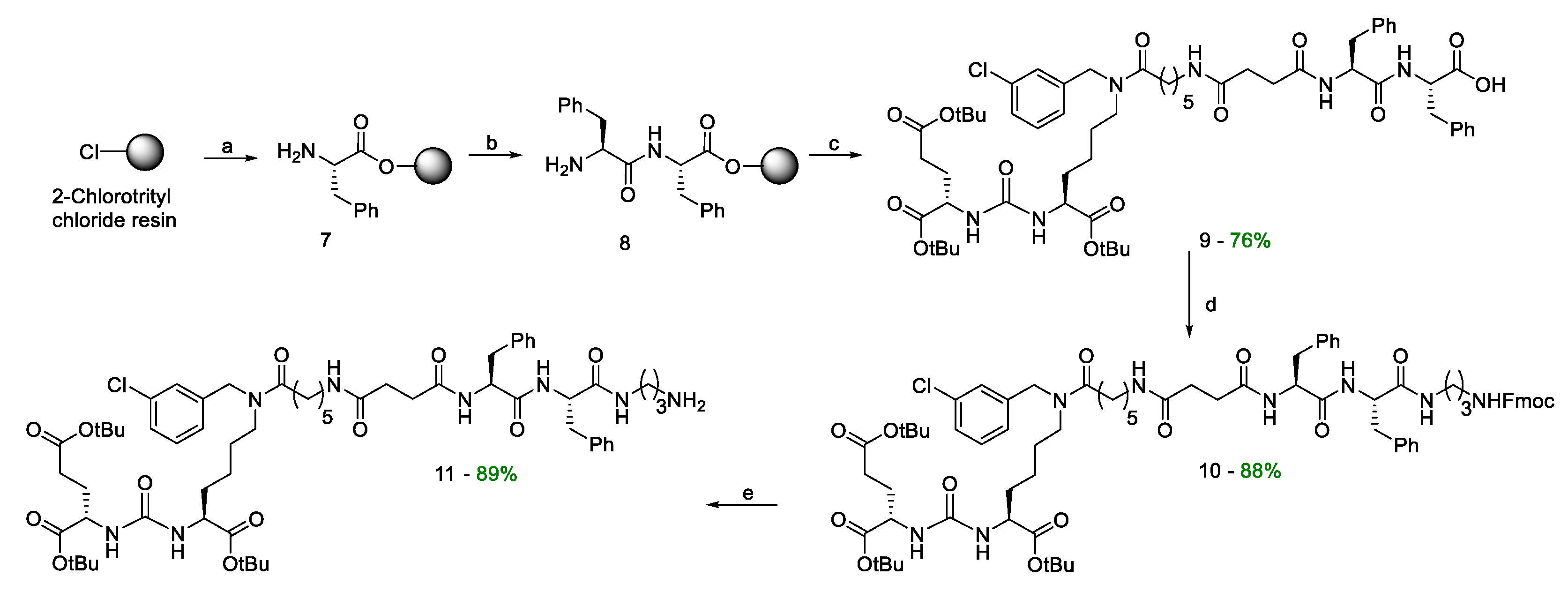
Scheme 3.
Synthesis of prosthetic groups (compounds 13, 14, 16 and 17). (a) (1) BuLi, THF, -78 oC, Ar; (2) (Bu)3SnCl, THF, -78 oC, Ar; (b) NHS, EDC*HCl, DMAP, DCM, Ar, r.t.; (c) NHS, DCC, THF, Ar, r.t.; (d) (1) I2 in 0.1 NaOH V1; (2) AcOH (3%) in CHCl3 V2; V1=V2; (3) TBHP/CHCl3 (10% w/v).
Scheme 3.
Synthesis of prosthetic groups (compounds 13, 14, 16 and 17). (a) (1) BuLi, THF, -78 oC, Ar; (2) (Bu)3SnCl, THF, -78 oC, Ar; (b) NHS, EDC*HCl, DMAP, DCM, Ar, r.t.; (c) NHS, DCC, THF, Ar, r.t.; (d) (1) I2 in 0.1 NaOH V1; (2) AcOH (3%) in CHCl3 V2; V1=V2; (3) TBHP/CHCl3 (10% w/v).
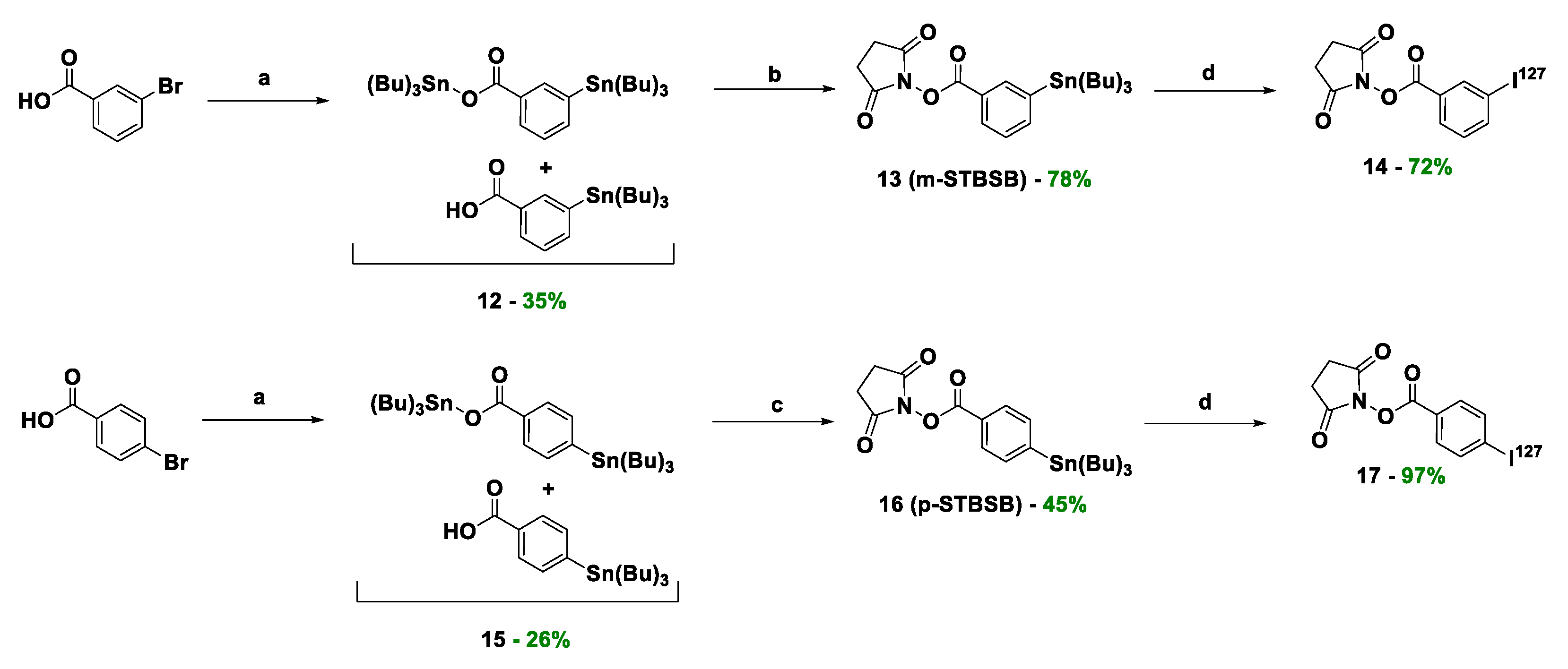
Scheme 4.
Synthesis of PSMA-m-TBSB (19), PSMA-p-TBSB (20); [127I]PSMA-m-IB (21) and [127I]PSMA-p-IB (22). (a) DCM/TFA/TIPS/H2O (46.25%/46.25%/5%/2.5% V/V); (b) 14/17, DIPEA, DMF (c) 13/16, DIPEA, DMF (d) (1) I2 in 0.1 NaOH V1, (2) AcOH (3%) in CHCl3 V2; V1=V2, (3) TBHP/CHCl3 (10% w/v).
Scheme 4.
Synthesis of PSMA-m-TBSB (19), PSMA-p-TBSB (20); [127I]PSMA-m-IB (21) and [127I]PSMA-p-IB (22). (a) DCM/TFA/TIPS/H2O (46.25%/46.25%/5%/2.5% V/V); (b) 14/17, DIPEA, DMF (c) 13/16, DIPEA, DMF (d) (1) I2 in 0.1 NaOH V1, (2) AcOH (3%) in CHCl3 V2; V1=V2, (3) TBHP/CHCl3 (10% w/v).
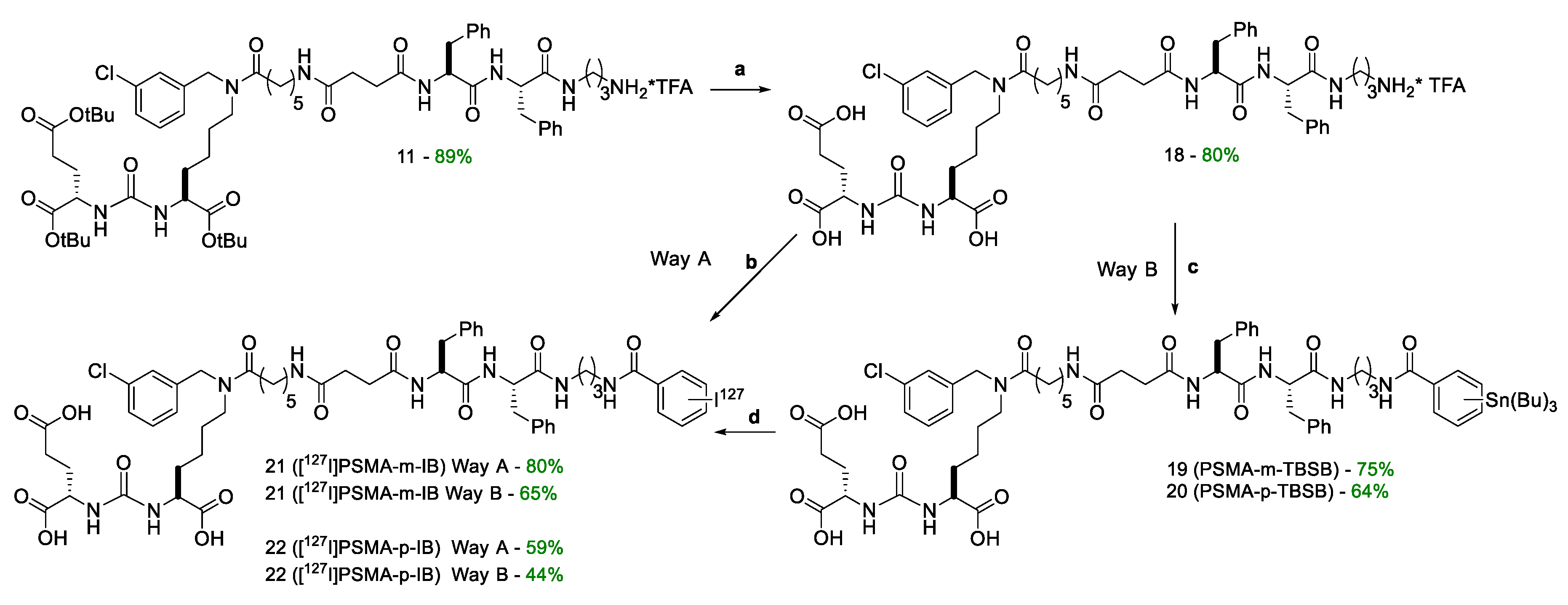
Figure 3.
Radiolabeling scheme of PSMA-p-TBSB (compound 20) ligand by 123I to obtain [123I]PSMA-p-IB (compound 23)
Figure 3.
Radiolabeling scheme of PSMA-p-TBSB (compound 20) ligand by 123I to obtain [123I]PSMA-p-IB (compound 23)
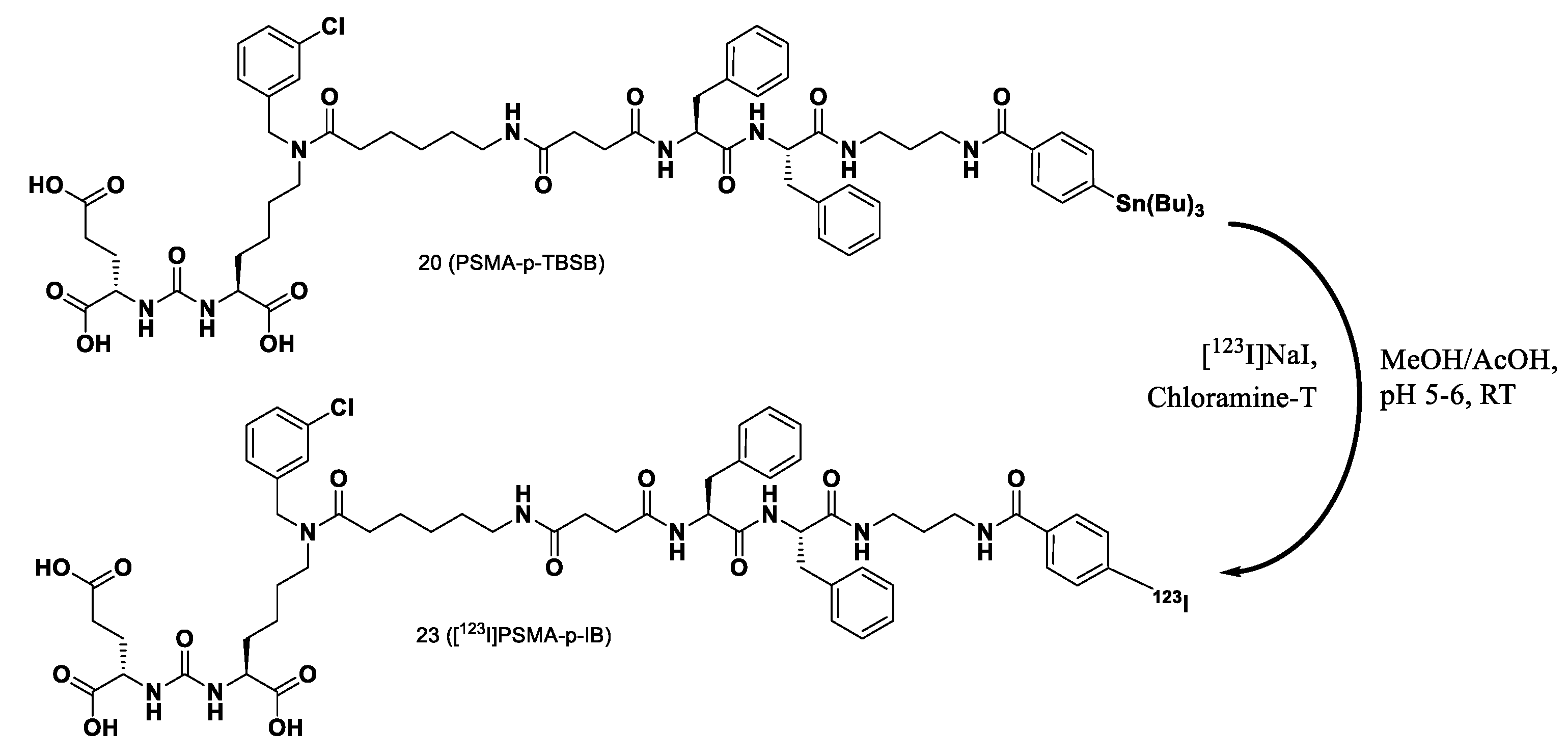
Figure 4.
Radiochemical yields as a function of the amount of PSMA ligand. The comparison study was performed by ANOVA test with Turkey’s post-hoc analysis (95% confidence interval)
Figure 4.
Radiochemical yields as a function of the amount of PSMA ligand. The comparison study was performed by ANOVA test with Turkey’s post-hoc analysis (95% confidence interval)
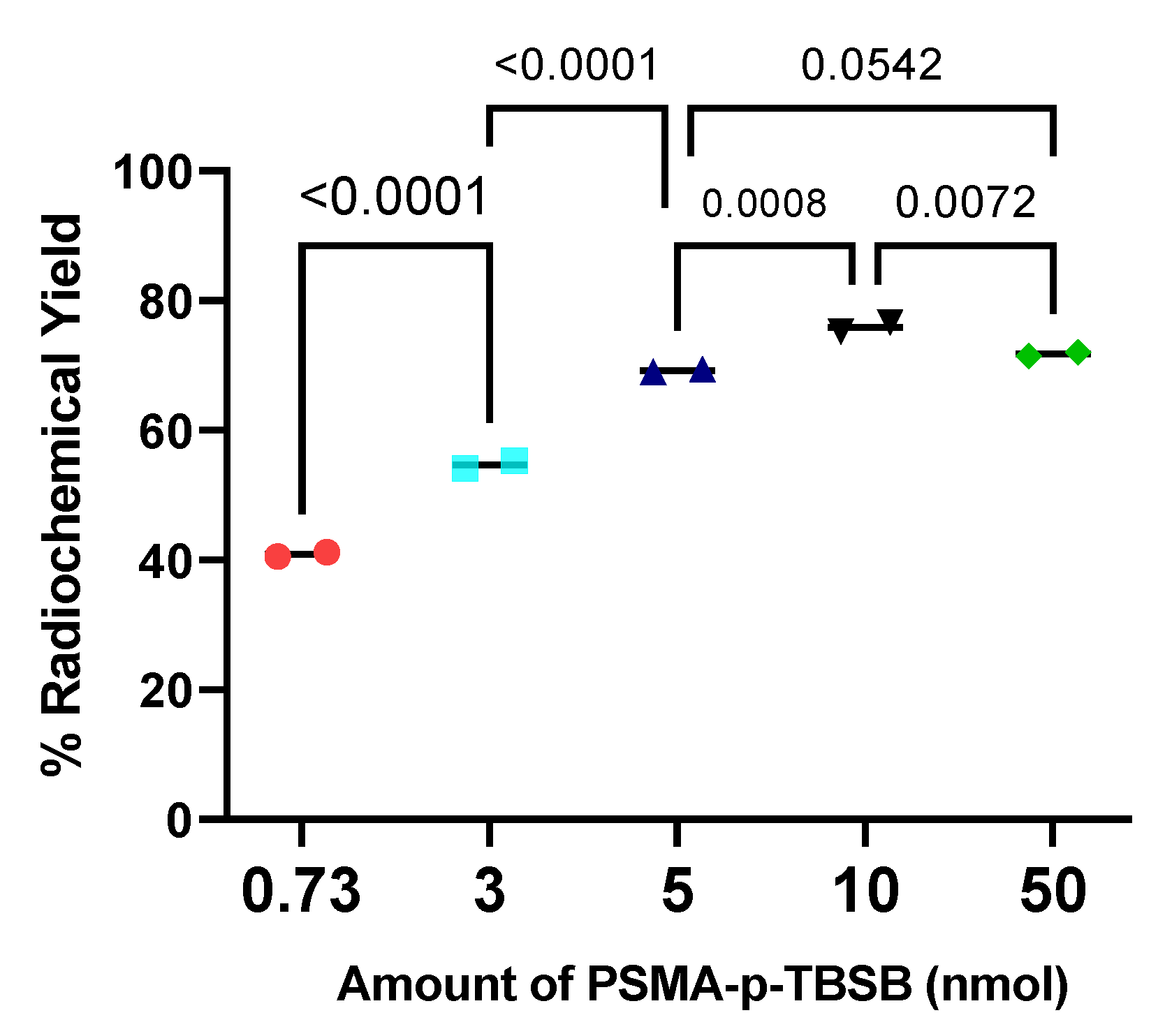
Figure 5.
Radiochemical yields as a function of reaction time. The comparison study was performed by ANOVA test with Turkey’s post-hoc analysis (95% confidence interval)
Figure 5.
Radiochemical yields as a function of reaction time. The comparison study was performed by ANOVA test with Turkey’s post-hoc analysis (95% confidence interval)
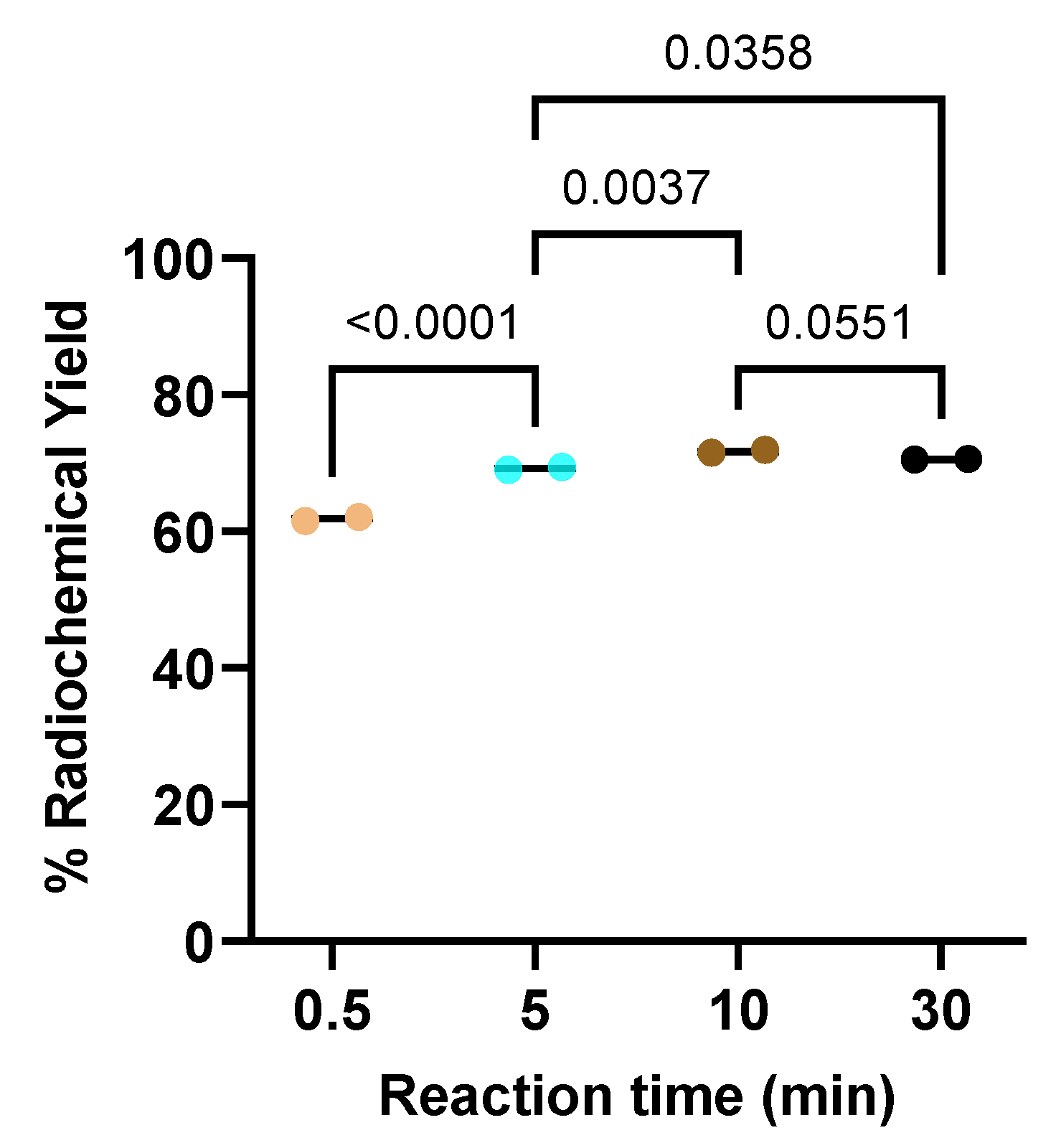
Figure 6.
Radiochemical yields as a function of oxidizing agent amount. The comparison study was performed by ANOVA test with Turkey’s post-hoc analysis (95% confidence interval)
Figure 6.
Radiochemical yields as a function of oxidizing agent amount. The comparison study was performed by ANOVA test with Turkey’s post-hoc analysis (95% confidence interval)
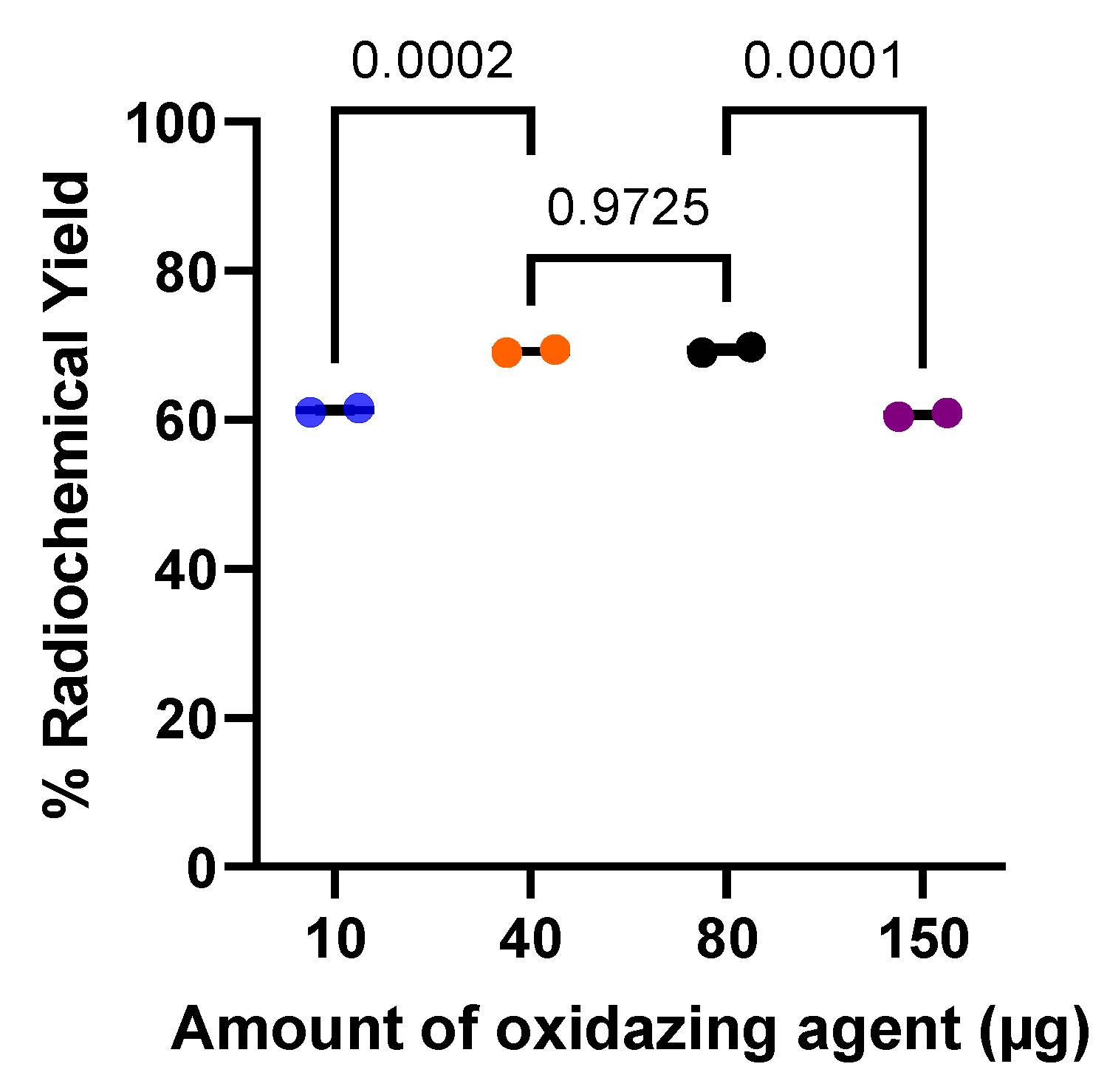
Figure 7.
The radiochemical yields as denoted above are radiochemical yield of [123I]PSMA-p-IB analyzed by different methods. ns marks a not significant difference by two-tailed t-test (99% confidence interval)
Figure 7.
The radiochemical yields as denoted above are radiochemical yield of [123I]PSMA-p-IB analyzed by different methods. ns marks a not significant difference by two-tailed t-test (99% confidence interval)
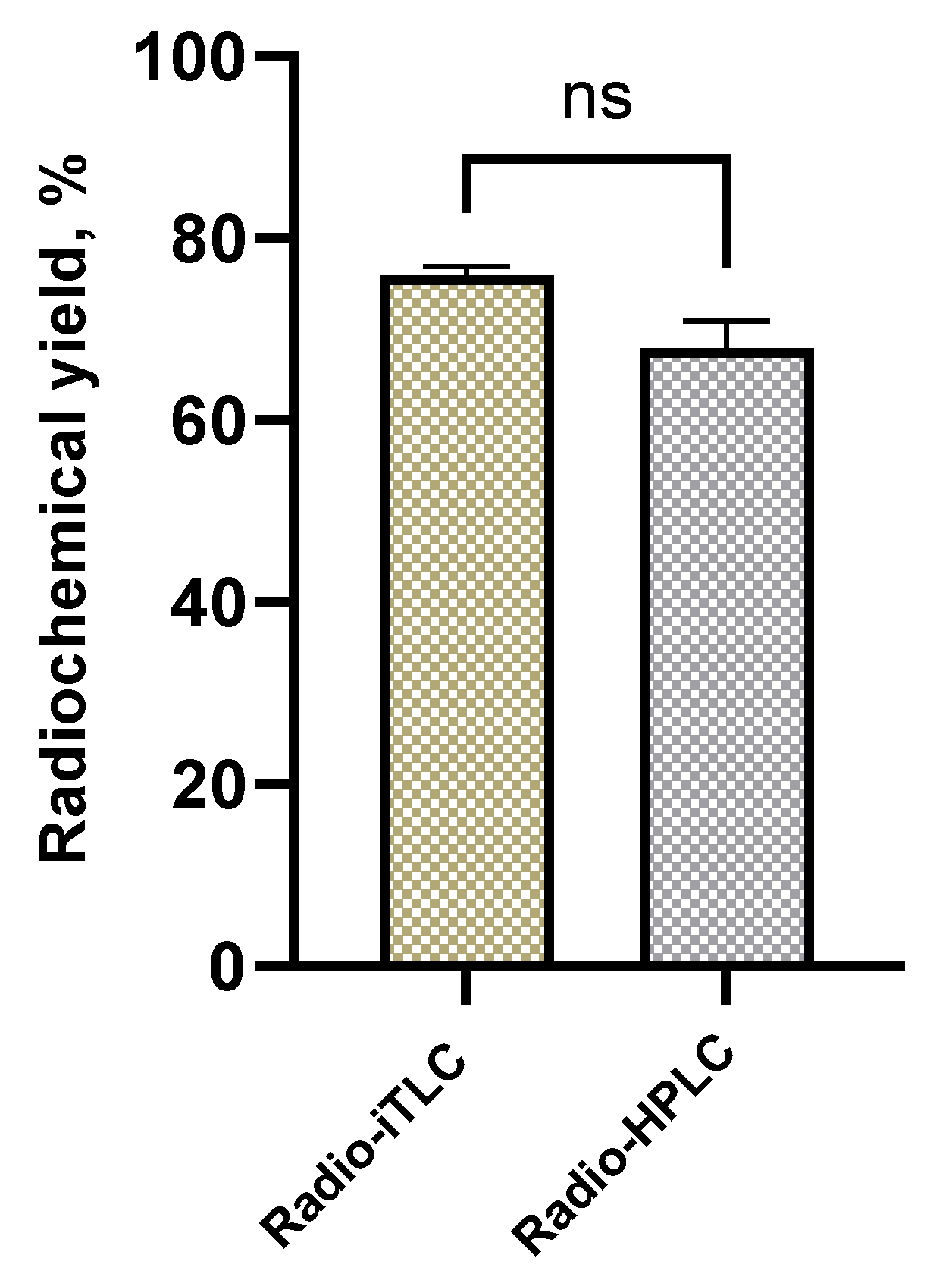
Figure 8.
Radiochemical purity of [123I]PSMA-p-IB analyzed by different methods. ns marks a not significant difference different by two-tailed t-test (99% confidence interval)
Figure 8.
Radiochemical purity of [123I]PSMA-p-IB analyzed by different methods. ns marks a not significant difference different by two-tailed t-test (99% confidence interval)
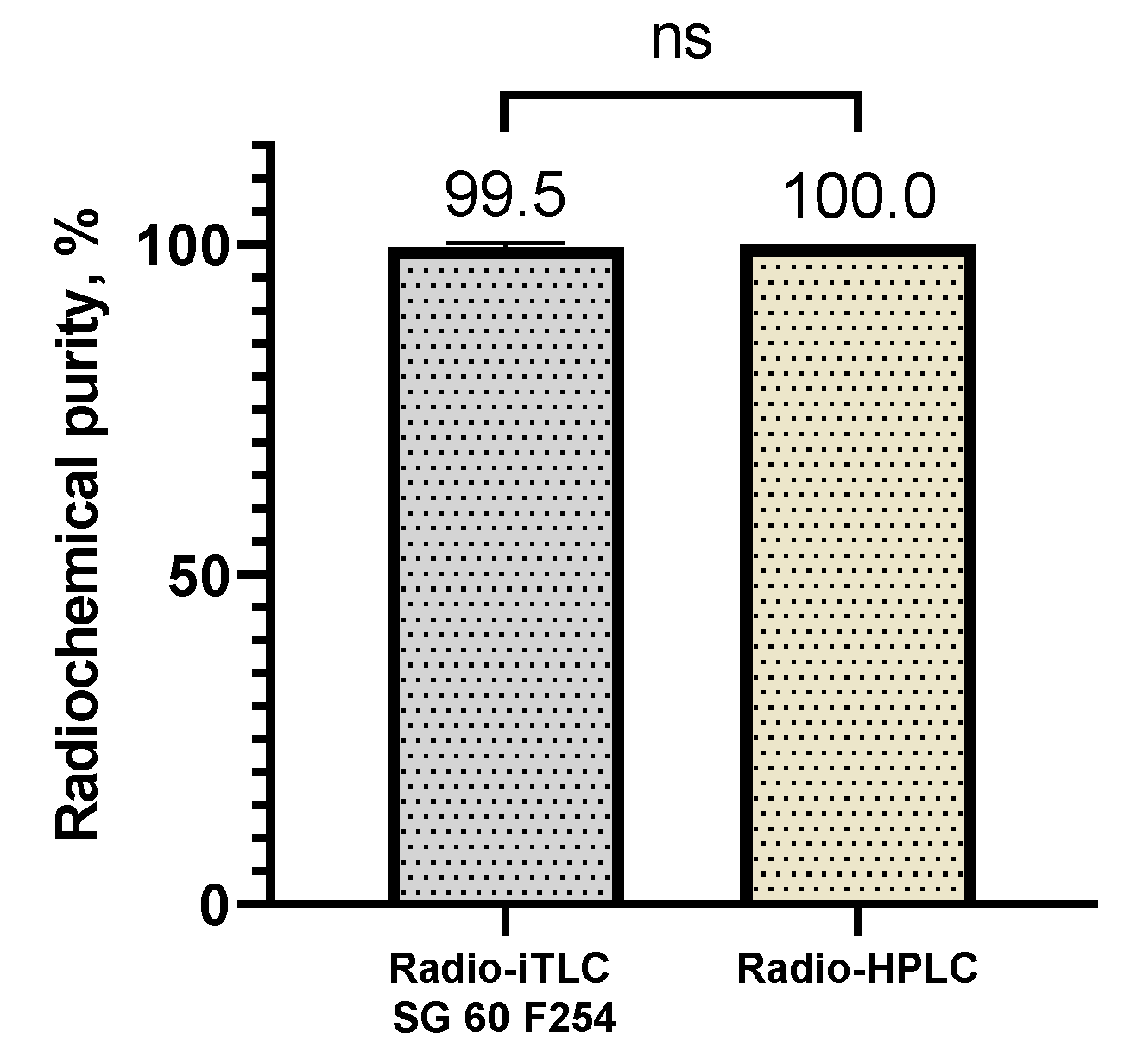
Figure 9.
Binding specificity of [123I]PSMA-p-IB. For blocking, a 500-fold molar excess of non-labeled PSMA ligand was added to the blocked groups. The final concentration of radiolabeled compound was 1 nM.
Figure 9.
Binding specificity of [123I]PSMA-p-IB. For blocking, a 500-fold molar excess of non-labeled PSMA ligand was added to the blocked groups. The final concentration of radiolabeled compound was 1 nM.
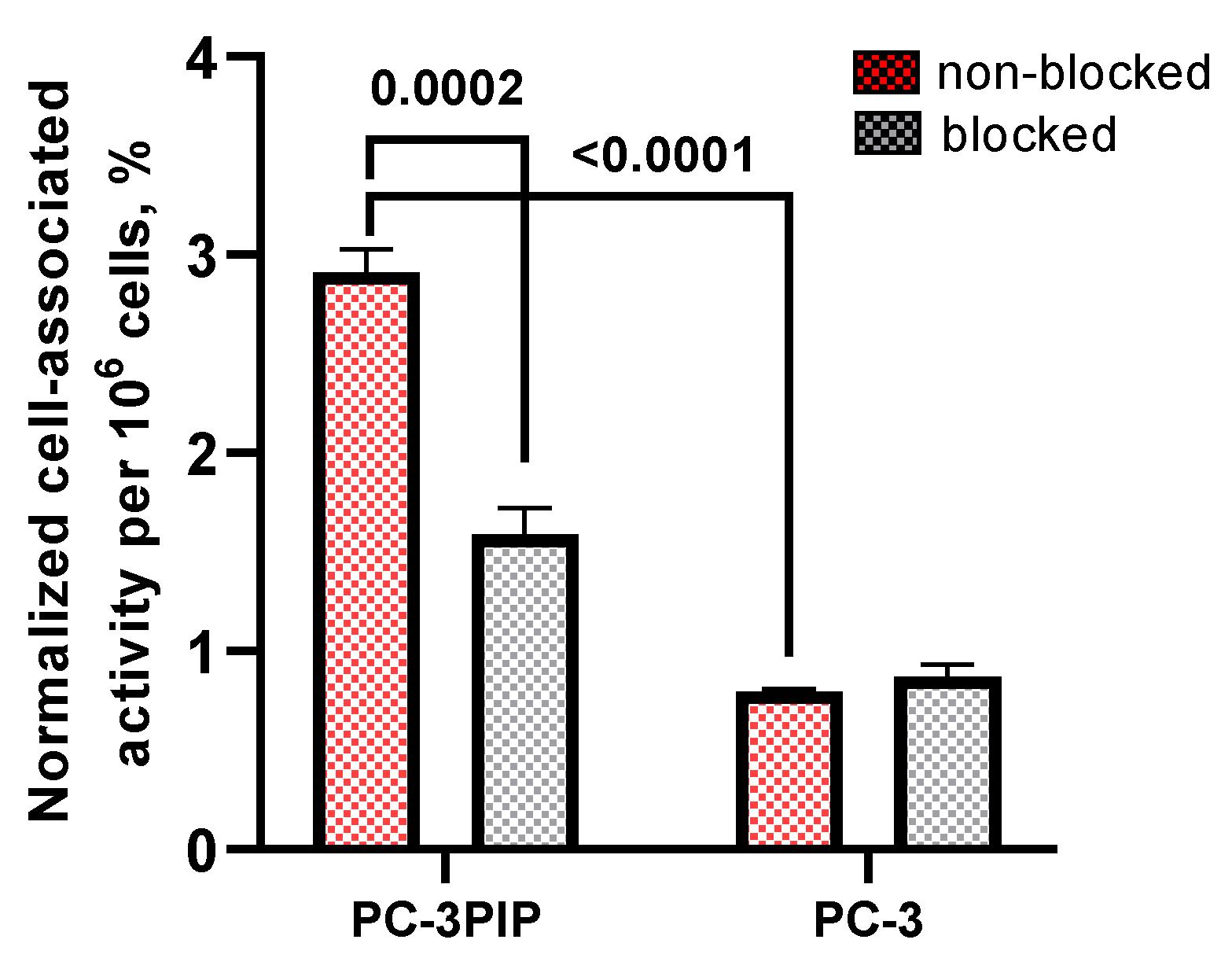
Figure 10.
The saturation experiment of [123I]PSMA-p-IB on PC-3 PIP cells.
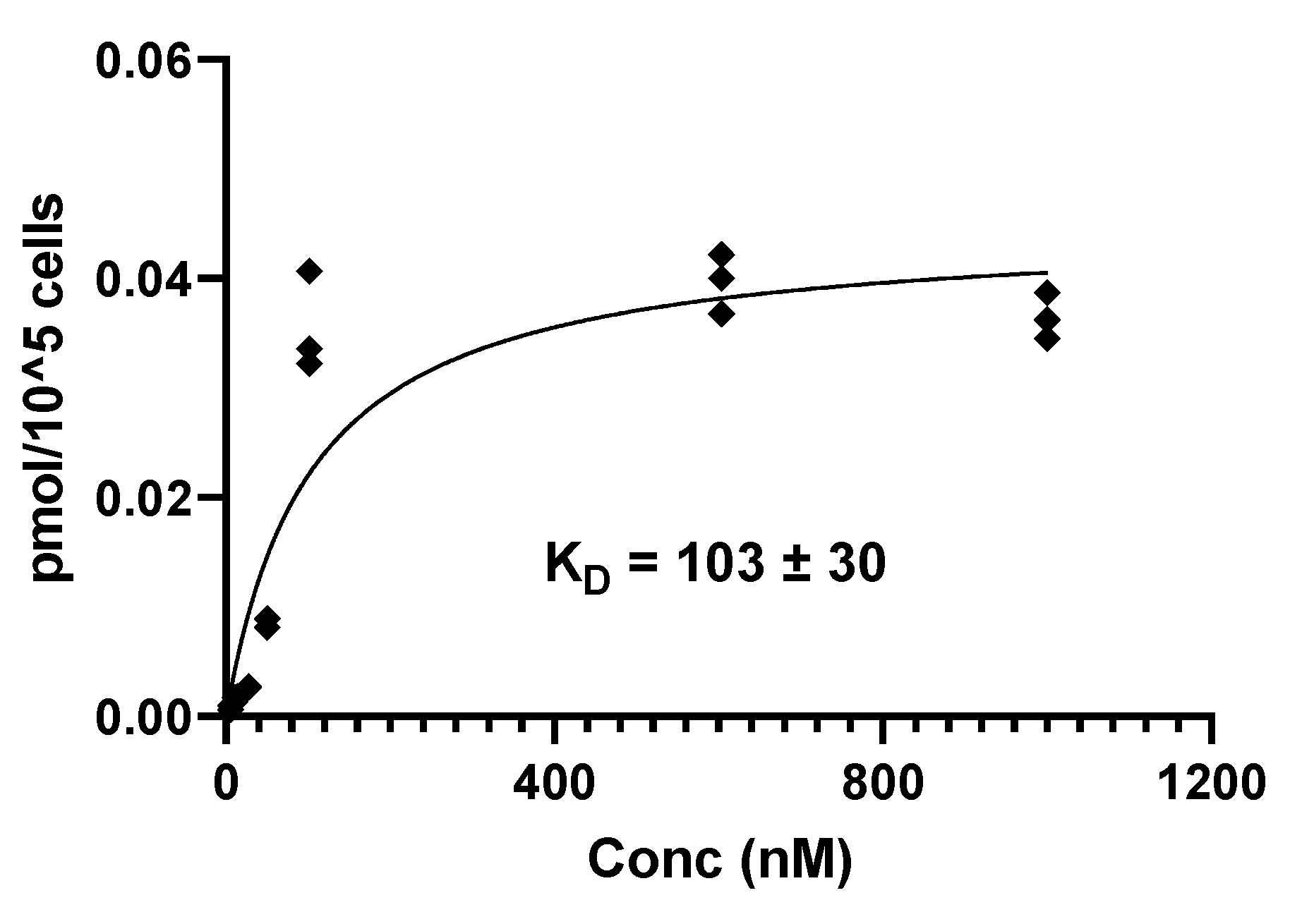
Figure 11.
Biodistribution of [123I]PSMA-p-IB and [127Lu]Lu-PSMA-617 4 h post-injection (p.i.) in CD1 mice. *Data for the gastrointestinal tract (including the contents) and the body are presented as %ID per whole sample. The comparison study was performed by unpaired 2-tailed t-tests (95% confidence interval).
Figure 11.
Biodistribution of [123I]PSMA-p-IB and [127Lu]Lu-PSMA-617 4 h post-injection (p.i.) in CD1 mice. *Data for the gastrointestinal tract (including the contents) and the body are presented as %ID per whole sample. The comparison study was performed by unpaired 2-tailed t-tests (95% confidence interval).
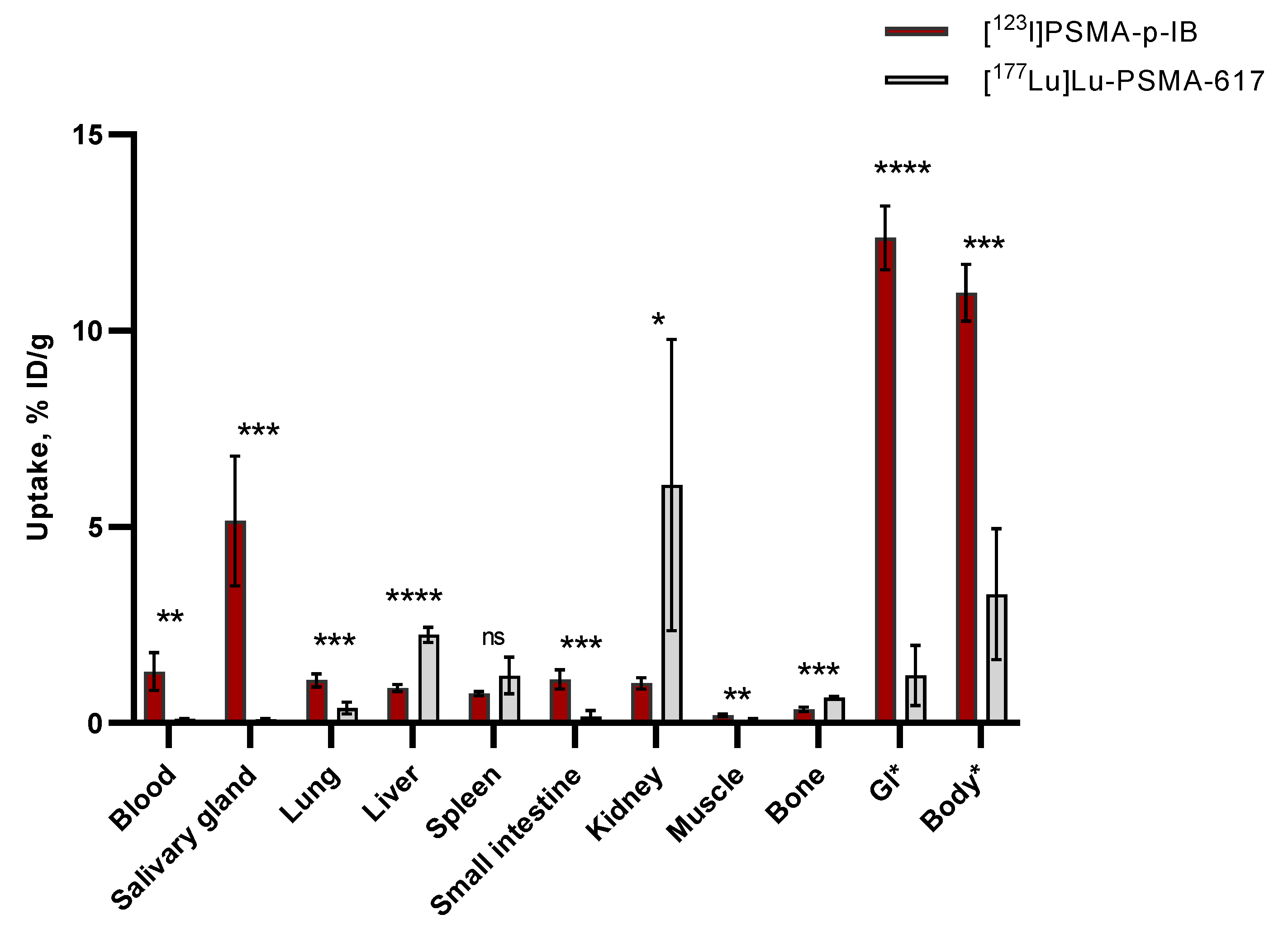

Table 1.
Comparison of alternative methods for obtaining the conjugate 21 (Scheme 4).
Table 1.
Comparison of alternative methods for obtaining the conjugate 21 (Scheme 4).
| Parameters | Way A | Way B |
|---|---|---|
| Yield of iodized product by prosthetic group (based on m-STBSB) | 57.6% | 48.8% |
| Number of stages with peptidomimetic / number of stages with 123I | 1 / 2 | 2 / 1 |
Table 2.
Radiochemical yield from radiolabeling optimization studies of [123I]PSMA-p-IB ligand. Radiochemical yields were based on iTLC SG fiber sheet analysis.
Table 2.
Radiochemical yield from radiolabeling optimization studies of [123I]PSMA-p-IB ligand. Radiochemical yields were based on iTLC SG fiber sheet analysis.
|
Variation of PSMA-p-TBSB amounts* |
Variation of reaction time** |
Variation of oxidizing agent amount*** |
|||||||
| Amount of peptide (nmol) |
RCY (%) | Time (min) |
RCY (%) |
Amount of chloramine-T (µg) |
RCY (%) |
||||
| 0.73 | 40.9 ± 0.5 | 0.5 | 61.9 ± 0.4 | 10 | 61.3 ± 0.5 | ||||
| 3 | 54.7 ± 0.8 | 5 | 69.2 ± 0.2 | 40 | 69.2 ± 0.2 | ||||
| 5 | 69.2 ± 0.2 | 10 | 71.7 ± 0.3 | 80 | 69.4 ± 0.5 | ||||
| 10 | 75.9 ± 1.0 | 30 | 70.6 ± 0.1 | 150 | 60.7 ± 0.4 | ||||
| 50 | 72.8 ± 0.4 | ||||||||
* Reactions were carried out using a fixed chloramine-T 40 µg and for a fixed time of 5 minutes; ** Reactions were carried out using a fixed PSMA-p-TBSB 5 nmol and chloramine-T 40 µg; *** Reactions were carried out using a fixed PSMA-p-TBSB 5 nmol and for a fixed time of 5 minutes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



