
Submitted:
03 July 2023
Posted:
05 July 2023
You are already at the latest version
Abstract
The effects of hypothermia on neonatal encephalopathy may vary regionally, spatially, and cytopathologically in the gyrencephalic neocortex with manifestations potentially influenced by seizures that alter the severity and distribution of neuropathology. We developed a neonatal piglet survival model of hypoxic-ischemic (HI) encephalopathy and hypothermia with continuous encephalographic (cEEG) monitoring for seizures to study injury in neocortex. Neonatal piglets were randomized to naïve, HI-normothermia (NT), overnight hypothermia (HT) initiated 2 hours after HI, sham-NT, or sham-HT treatments. Some piglets within sham and HI groups received cEEG monitoring during recovery. Survival was 2-7 days (piglets were unmedicated and those with poor recovery and unresolving seizures were euthanized early); there was no differences in survival among groups (p>0.078). Neuropathology was assessed by hematoxylin and eosin staining and immunohistochemistry for RNA Binding FOX-1 Homolog 3 (Rbfox3/NeuN). Normal and ischemic-necrotic neurons were counted (layers II-VI collectively) in somatosensory, motor, and prefrontal cortices, identified by cytoarchitecture and connectomics, and in inferior parietal cortex by layer. Seizure burden was determined. HI-NT piglets had reduced normal/total neuron ratio and increased ischemic-necrotic/total neuron ratio relative to naïve, sham-NT, and sham-HT piglets in anterior and posterior motor and somatosensory cortices. Frontal cortex was vulnerable in the prefrontal lateral bank after HI-NT with ischemic-necrosis. HI-HT piglets had higher normal/total neuron ratios and lower ischemic-necrotic/total neuron ratios than HI-NT piglets in anterior/posterior motor and somatosensory cortices. Total normal neuron density in layer III of inferior parietal cortex was reduced in HI-NT piglets compared to sham piglets and was protected by HT. Laminar analysis of Rbfox3 in somatosensory cortex revealed three types of neurons: Rbfox3-positive/normal, Rbfox3-positive/ischemic-necrotic, and Rbfox3-depleted. HI piglets had increased Rbfox3-depleted/total neuron ratio in layers II and III compared to sham-NT. Cortical neuron Rbfox3-depletion was partly rescued by HT. Seizure burden was more severe in HI piglets compared to sham piglets, but HI-NT and HI-HT piglets were similar. We conclude that 1) the neonatal piglet neocortex has a suprasylvian spatial vulnerability to HI and seizures; 2) HT protects against neuropathology in functionally different regions of the neonatal gyrencephalic neocortex; 3) neurons in neonatal neocortex have a limited cytopathology repertoire seen by H&E staining; 4) higher seizure burden correlates with more ischemic-necrotic neurons in somatosensory cortex; 5) seizure presence associates with damage spread to inferior parietal cortex; 6) seizure presence is insensitive to HT, and 7) Rbfox3 immunophenotyping identifies a novel neuronal RNA splicing protein nuclear-depletion pathology that appears reversible by HT. This work demonstrates that HT protection of neocortex in neonatal HI is topographic and laminar, seizure unmitigating, and restores depleted neuronal RNA splicing factor.
Keywords:
cell death
; corticostriatal projection
; motor cortex
; neocortical pyramidal neuron
; neonatal encephalopathy
; RNA binding protein
1. INTRODUCTION
Moderate to severe hypoxic-ischemic (HI) encephalopathy (HIE) is caused by reduced brain oxygen and blood supply from birth asphyxia and occurs in about 1 to 3 infants in every 1000 live term births in America and Western Europe with greater prevalence in eastern countries [1]. Nearly one million infants die worldwide each year from HIE after perinatal asphyxia [2]. Hypothermia (HT) is the only approved treatment for neonatal HIE in western countries [3]. However, approximately one-third of survivors who receive whole body or head cooling still have, compared to peers, moderate-to-severe impairments in executive, visuospatial, and motor functions, language, and emotional maturity years later [4,5,6]. Because these functions are forged neocortically [7,8,9], these childhood outcomes suggest that effects of HT on the gyrencephalic cerebral cortex are less therapeutic than in subcortical brain regions or that different neocortical areas or cell layers are affected variably by the injury and the cooling. The neocortex indeed segregates into highly sensitive and less vulnerable regions after neonatal HI with the peri-Rolandic and watershed patterns [10,11,12], and cortical laminar necrosis can be seen associating with increased risk for spasticity [13], though with severe insults total neocortical damage can prevail [14]. The idea of differential regional protection with HT is supported by studies of infants cooled for neonatal HIE with follow-up MRI [15]. MRI of HIE cooled infants also suggests that the pattern of neocortical insensitivity to HT appears predictive of long-term epilepsy originating in watershed territories of neocortex [16]. Experimentally, gyrencephalic animals are propitious for fully examining the organization of neocortex [17] and its pathobiology, including cortical neuron cell death [18]. Neonatal piglets, like infant humans, present with a neocortical selective vulnerability, including laminar necrosis, after asphyxic cardiac arrest [19,20]. Piglets (< 2 weeks of age) with hypovolemic ischemia and normothermia (NT) or mild HT (1 hour) treatments show reduced histologic damage in deep layers of temporal and occipital cortex with HT but not in frontal or parietal cortex and not in the superficial cell layers of any cortical region [21].
HIE commonly causes neonatal seizures, even in cooled newborns [22,23]. Seizure severity or burden in infants is associated with amount of brain injury and adverse neurodevelopmental outcome [24,25]. These seizures can be overt or subtle and undetectable by clinical observation [26,27]. Amplitude-integrated EEG helps with the problem of seizure detection and is used in neonatal intensive care units [28]. Seizure monitoring is critical because rewarming from therapeutic HT places infants at risk for seizures [29].
There is debate about whether seizures have additional direct damaging effects on the immature brain separate from the primary underlying HIE that instigates seizures. Some clinical studies report that neonatal seizures independently associate with poor outcome by causing additional brain damage [25,30]. However, others find that neonatal seizures do not exert an deleterious impact independent of the underlying HIE [31]. Neonatal rhesus monkeys with term intrauterine partial asphyxia develop seizures about 24 hours after the insult and then severe neuropathology involving telencephalon, brainstem, and cerebellum [32,33]. In neonatal piglets (24-49 hours old) with severe hypoxia-induced seizures, seizure activity did not exacerbate cortical gray matter lactate/pyruvate ratios at 6 hours after the insult, but higher cerebral cortical damage grade was seen in piglets that developed electroconvulsive activity, burst suppression, or persistently low EEG amplitudes [34]. In newborn (14-72 hours old) posthypoxic piglets recovered under NT or mild HT for 3 hours, neuropathology grade in neocortex did not differ among NT and HT piglets, but after excluding piglets with seizures, HT animals had less damage than NT piglets suggesting that seizures cause more neuropathology [35]. In piglets (7-10 days old) with HI brain damage from asphyxic cardiac arrest, we described neocortical injury as topographic, selectively restricted, and laminar or distributed and panlaminar; the latter pattern was present in piglets with clinical seizures and responsiveness to diazepam [19,20]. Subsequently, greater cellular neuropathology was found in piglets with clinical seizures compared to piglets without clinical seizures [36]. Discordant outcomes in clinical and experimental settings could be related to seizures not being uniform due to differences in species (and strain), genetic background, semiology, detection methods, anatomical locations, gyrencephaly and network connectivity, tissue oxygenation, and metabolic depletion and recovery [37,38]. Resolving the question of whether seizures in gyrencephalic neonatal animals with HIE independently add or contribute to brain damage or reflect the underlying severity of encephalopathy is important because answers can instruct on clinical management needs [39]. Commonly used anticonvulsant therapies for neonates are not strongly proven in efficacy and may be harmful [40] with neurotoxic potential in neonatal animals, including non-human primates [41,42,43].
We developed a neonatal piglet model that combines global HI and HT, the standard of clinical care, with continuous EEG (cEEG) monitoring during survival for neuropathological assessments. We addressed several questions with our gyrencephalic large animal model: 1) does HT protect neocortical areas differentially after neonatal HI; 2) do cEEG-confirmed seizures associate with worse neuropathology after neonatal HI; 3) does HT after neonatal HI protect against seizures and neocortical neuropathology; 4) is there clear secondary seizure-related encephalopathy distinguishable from HIE evidenced by topographical and laminar damage in neocortex; and 5) is there a seizure-related cytopathology phenotype, identified by hematoxylin and eosin (H&E) or immunostaining, in neonatal neocortex divisible from the typically predominant HIE ischemic-necrosis? We examined the localization of RNA Binding FOX-1 Homolog 3 (Rbfox3/NeuN) because the gene for this RNA splicing protein is seizure-associated with loss of function mutations causing neonatal and childhood epilepsy [38,44,45,46]. This work can advance the understanding of the effects of HT on the gyrencephalic cerebral cortex after HI and the relationships among neonatal seizures, neocortical neuronal injury, and molecular pathology.
2. MATERIALS AND METHODS
2.1. Neocortical Cytoarchitecture and Connectomics in Normal Piglets
The animal protocols were reviewed and approved on 06/06/2023 by the Institutional Animal Use and Care Committee of the Johns Hopkins University (protocol number SW23M119). Neonatal Yorkshire piglets (2 to 5-days old, 1-2 kg, males) were used for descriptive normative neuroanatomy and tract-tracing experiments to identify, using cytology and connectivity, the locations of the motor cortex and somatosensory cortex. We wanted to verify these regions for placement of cEEG electrodes and neuropathological assessments. Naïve piglets (n=3) were deeply anesthetized with pentobarbital 50 mg/kg and phenytoin 6.4 mg/kg (SomnaSol) and, after thoracotomy and left myocardial puncture and aortic catheterization, ice-cold 100 mM phosphate-buffered saline (pH 7.4) was perfused (~2 liters) for body exsanguination followed by freshly prepared 4% paraformaldehyde (PF) in 100 mM phosphate buffer (pH 7.4) for brain fixation (~4 liters). Appropriate tissue fixation was judged by the stiffness of the body and immovability of the jaw and limbs. The piglets were decapitated, and the head was placed in 4% PF for overnight. The following day the calvarium was removed carefully by rongeur, and the brain was extracted from the skull base, placed in PF for overnight, and then immersed in 20% glycerol for cryoprotection.
The brains were bisected mid-sagittally, and individual cerebral hemispheres were frozen and serially cut on a sliding microtome into 40 µm thick floating sections in sagittal, horizontal, and coronal planes. Every 10th section was mounted on a glass microscope slide for Nissl (cresyl violet, CV) staining to study neocortical cytology. Every 11th section was used for visualization of cytochrome C oxidase enzyme activity by histochemistry, as described [19,20,47,48], for visualization of neocortical mitochondrial metabolic activity in situ.
Other neonatal piglets (n=7) were used for brain tract-tracing experiments to confirm the locations of somatosensory and motor cortices for cEEG electrode placement. Anesthesia was induced by a nose cone with isoflurane 5% and 50% nitrous oxide in 50% oxygen. The piglets were intubated, and the anesthetic was changed to isoflurane 1.5-2% and 70% nitrous oxide in 30% oxygen. Using sterile technique, catheters were placed in the external jugular vein and femoral artery. A fentanyl 20 µg/kg intravenous (iv) bolus was given followed by 20 µg/kg/h. Additional fentanyl boluses of 10-20 µg/kg were given as needed to mitigate discomfort.
Each piglet was mounted securely in a stereotaxic frame (Kopf Instruments, Tujunga, CA) in a flat skull position. The head surface was shaved and washed with chlorhexidine antiseptic scrub and 70% alcohol, draped, and then painted with povidone-iodine solution. With a scalpel (10 blade), a midline incision was made in the scalp carefully avoiding damage to the vulnerable superior sagittal sinus below. The scalp was reflected with clamps. Fascia was cleared with a bone spatula to visualize surface landmarks on the skull for craniotomies. Craniotomies were made using a high-speed dental drill with a 0.5 mm fine bit and a gentle sweeping hand motion (to carefully remove skull bone layers) rather than a vertical compression to avoid cortical injury. Stereotaxic coordinates were selected from the atlas of Salinas-Zeballos et al [49]. Because this atlas uses a 3-day-old (1.15 kg) piglet as a reference, targeting adjustments were made based on the neonatal piglet size and brain neuroanatomical histology. Brain regional stereotaxic coordinates were: motor cortex (2 mm anterior to bregma, 1 mm lateral, 2 mm ventral to cortical surface), anterior somatosensory cortex (1 mm anterior to bregma, 10 mm lateral, 2 mm ventral to cortical surface), middle somatosensory cortex (at bregma, 17 mm lateral, 2 mm ventral to cortical surface), posterior somatosensory cortex (5 mm posterior to bregma, 15 mm lateral, 2 mm ventral to cortical surface), and striatum (10 mm anterior to bregma, 10 mm lateral, 15 mm ventral to cortical surface). Motor cortex was verified by evoked muscle contractions upon cortical stimulation, while somatosensory cortex was verified by somatosensory-evoked potentials as described [50]. We used a combination of track-tracers including recombinant neurotropic viruses genetically encoding different fluorescent proteins and non-genetic connectivity reporters such as wheat germ agglutinin-horseradish peroxidase (WGA-HRP) and FluoroGold (FG) [47,51,52]. The viruses used were adeno-associated virus-enhanced green fluorescent protein (AAV-eGFP, Vector BioLabs), adenovirus-cyan fluorescent protein (AV-CFP, GenTarget Inc) and lentivirus-red fluorescent protein (LV-RFP, Amsbio). All recombinant viruses drove the expression of their fluorescent protein under the control of the cytomegalovirus promoter for high neural cell expression. Commercially prepared WGA-HRP (Vector Laboratories) and FG (Fluorochrome LLC, Denver, CO) were used. A Hamilton microsyringe (blunt tip) was used for injections. Tracer injections (10-20 µl) were delivered slowly by discontinuous pressure pulses; after the final pulse the needle was allowed to sit in place for 10 minutes before withdrawal. The tracers were used at the following concentrations: AV-CFP (1x109 IFU/ml), AAV-eGFP (1x1012 genome copies/ml), LV-RFP (1x108 IFU/ml), FG (5%), and WGA-HRP (20%). The craniotomies were sealed with bone wax, the scalp was sutured, and the piglets emerged from anesthesia and were extubated. Piglet survival times were 3 or 7 days at which time they were perfusion-fixed with 4% PF, and their brains were prepared for frozen sectioning.
Tracer visualization in piglet brain sections was done by direct fluorescence, immunohistochemistry, and enzyme histochemistry [47,51,52]. eGFP, RFP, and FG were observed by their fluorescence. Highly specific antibodies were also used for the detection of RFP (BioVision), FG (Millipore) and CFP (BioVision). WGA-HRP was detected by enzyme histochemistry [53] with neutral red counterstaining.
2.2. Neonatal Piglet Model of Encephalopathy
Neonatal Yorkshire piglets (2 to 3-days old, 1-2 kg, males) were randomized to one of four experimental groups: sham normothermia (NT), sham HT, HI-NT, or HI-HT. An unanesthetized unoperated group of piglets was a naive control. The group sizes were sham-NT (n=6), sham-HT (n=10), HI-NT (n=8), HI-HT (n=10), and naïve (n=6).
Anesthesia was induced with isoflurane 5% and 50% nitrous oxide in 50% oxygen delivered by a nose cone. After intubation, the anesthetic was changed to isoflurane 1.5-2% and 70% nitrous oxide in 30% oxygen. Sterile catheters were placed in the external jugular vein and femoral artery. A fentanyl bolus (20 µg/kg iv) was given followed by 20 µg/kg/h. Additional fentanyl (10-20 µg/kg boluses) was given as needed for discomfort management.
Some piglets received craniotomies for placement of sterile, epidural 4-lead electrode telemetry arrays for cEEG (Stellar Telemetry, TSE Systems, Inc., Chesterfield MO). The placement (LJM, NRD) of the Stellar Telemetry real-time continuous recording electrode arrays (Figure 1A–C) was determined from previous neuroanatomical mapping of the topographic distribution of neocortical pathology in newborn piglets that survived for 1-4 days after HI [19,36] and key confirmatory neuroanatomical experiments shown here. The EEG recording arrays had 4 bipolar channels with each electrode attached securely to a miniature cranial screw inserted epidurally through a craniotomy made at stereotaxically determined coordinates. Each electrode-epidural screw assembly was secured firmly to the surrounding bone by low-heat, quickset acrylic cement (Figure 1A–C). The immobility of each electrode was confirmed with forceps. The placement was bilaterally symmetrical in the right and left hemispheres. The anterior most channel (designated right or left hemisphere anterior) had electrode placement 15 mm anterior to bregma and 3 mm lateral to the midline and was paired with an electrode placed 10 mm anterior to bregma and 6 mm lateral to the midline. The anterior channel recorded EEG from anterior primary motor cortex and anterior primary somatosensory cortex. The posterior most channel (designated right or left hemisphere posterior) had electrode placement 8 mm anterior to bregma and 3 mm lateral to the midline and was paired with an electrode 4 mm anterior to bregma and 6 mm lateral to the midline. The posterior channel recorded EEG from the posterior motor cortex and posterior primary somatosensory cortex. The electrode array transmitter (Figure 1C) was inserted subdermally in a nape pocket. The left- and right-side electrode wire leads were loosely braided and flanked laterally under each side of the scalp. The array antenna was secured to the posterior most point of the scalp incision with suture and emerged externally. The surgical incision was closed with 3.0 suture. Upon completion of surgery, the isoflurane was discontinued, and the fentanyl was decreased to 10 µg/kg/h. All piglets received vecuronium 0.2 mg/kg/h to prevent ventilatory efforts during hypoxia-asphyxia and shivering during HT. The same anesthetic regimen was given to all HI and sham piglets. The cortical electrode placement for each piglet was confirmed visually (LJM) after brain perfusion-fixation (Figure 1D,E).
2.3. Global HI Injury and Mild HT
The inhaled oxygen was deceased to 10% for 45 minutes. Then, 5 minutes of room air was supplied to reoxygenate the heart to improve cardiac resuscitation in our model. The endotracheal tube was then clamped to induce 8 minutes of asphyxia [54,55,56]. With this protocol, piglets develop severe bradycardia and hypotension with heart rates < 60 beats per minute or mean arterial blood pressure (MAP) less than 45 mmHg. The piglets were resuscitated with an inhaled oxygen concentration of 50%, manual chest compressions, and epinephrine (100 µg/kg iv). Piglets that were not resuscitated within 3 minutes of chest compressions were excluded. Sham procedure piglets received 50% inhaled oxygen for 3 minutes. After return of spontaneous circulation (ROSC) or time equivalent in shams, the 70% nitrous oxide in 30% oxygen was restarted.
NT in neonatal piglets is rectal temperature in the range of 37.5-39.0°C [57,58,59]. In piglets randomized to receive HT, whole body HT was started 2 hours after ROSC using ice packs and a cooling blanket to a goal rectal temperature of 34°C [54]. This change in temperature is like the 4°C decrease in clinical HT [60]. HT was initiated 2 hours after resuscitation to mimic clinical delays in cooling [61].
Three hours after ROSC, ketamine (10 mg/kg/h iv) was started, and the nitrous oxide was decreased to 33% in 33% oxygen and 33% air. For the remainder of the anesthetic, fentanyl and ketamine were increased in 10 µg/kg/h and 10 mg/kg/h increments, respectively, with additional as needed boluses of fentanyl 10 µg/kg and ketamine 10 mg/kg for comfort. Dopamine was given when needed to maintain the MAP above 40 mmHg during the overnight HT or NT protocols.
Rewarming to NT began in HT piglets at 20 hours from onset by increasing the temperature of the water circulating through the blanket. The rate of rewarming was 0.5°C/h, which is the clinical rewarming rate at Johns Hopkins Hospital NICU [60]. Piglets reached their NT target temperature of 38.5°C/h at ~ 29 hours from onset. Vecuronium infusion was stopped 15 hours after ROSC to allow time for the neuromuscular blockade to wear off before extubating. Piglets then emerged from anesthesia and were extubated. After regaining postural and ambulatory control, the piglets were returned to their cages with free access to milk and continued overnight supervision and intermittent video monitoring (LJM, Supplementary Material Video 1).
2.4. Piglet Survival and Perfusion-Fixation
The piglets survived for 2-7 days. They did not receive anticonvulsant medications. The humane treatment and survivability of piglets required their independent ambulation and feeding (drinking milk) ad libitum. Piglets that became non-ambulatory and unable to drink, had sustained general tonic-clonic seizures > 20 mins (Supplementary Material Videos 3 and 4), or deemed to be in status epilepticus [62] were euthanized before the 7-day endpoint. All piglets in this cohort had their brains prepared optimally for neuropathology. There were no postmortem delays before animal perfusion-fixation. Piglets were euthanized with SomnaSol iv. Sham procedure piglets were euthanized as time matches for HI piglets throughout the 2 to 7-day period. After deep anesthesia, but prior to fatal cardiorespiratory arrest, the cEEG electrode arrays were quickly removed from the skull before perfusion fixation with 4% PF (LJM). All piglets achieved uniform fixation. Afterwards, they were decapitated, and the head was immersed in 4% PF for overnight. The following day the calvarium was removed (LJM) and the brain with the intact dura mater was examined and photographed (Figure 1D); afterwards, the dura was removed carefully, and the brain was again examined for cortical damage and photographed (Figure 1E). This protocol was followed strictly to determine that the cEEG electrode cranial screws were only epidural without underlying neocortical parenchymal damage (Figure 1D,E). The brain was then removed from the skull base and returned to PF for overnight. The following day the brain was immersed in 20% glycerol. Each brain, left and right cerebral hemispheres, was blocked in the coronal plane (Figure 2A–C) from the frontal lobes to the hindbrain, including the telencephalon, diencephalon, midbrain, pons, and medulla with cerebellum, and paraffin processed in tissue cassettes. The paraffinized brain blocks were cut on a rotary microtome into 10-µm sections and mounted on glass slides for hematoxylin and eosin (H&E) staining (Figure 2) and immunohistochemistry (Figure 3).
2.5. H&E Neuropathology and Cell Counting
H&E-stained brain sections, the standard for clinical HIE postmortem neuropathological diagnosis [63], were used for neuropathological assessments. The assessments were done blinded to piglet treatment. An investigator (JKL) used for neuronal counting one H&E-stained section from each of the blocks containing: 1) the frontal lobe with the dorsomedial anterior motor cortex and gyrus rectus and the dorsolateral and inferior prefrontal cortex (Figure 2A,D); 2) the anterior striatum and the overlying central motor cortex and the anterior primary somatosensory cortex (Figure 2B,E); and 3) the mid-thalamus with the overlying posterior motor cortex and somatosensory cortex (Figure 2C,F). In separate piglets, the identities of these neocortical regions were confirmed by cytology (Figure 4) and connectivity (Figure 5). The primary somatosensory and motor regions were specifically assessed for neuropathology because they have reproducible vulnerability to HI in many variations of this swine model of asphyxic cardiac arrest [19,20,36,56,64], and these brain regions in piglet have high endogenous mitochondrial metabolism and sodium/potassium ATPase activity [19,20] and blood flow [65]. Another investigator (LJM) analyzed the inferior parietal neocortex for neuropathology (Figure 2B,E). This area was assessed because previous topographic mapping suggests that this region in piglet is typically not vulnerable to the primary HI insult, but in the presence of clinical seizures, damage spreads into this area of neocortex [19,20].
A profile counting based approach was used for quantification of the amount of neuronal damage in each piglet brain. Individual neuron profiles were counted microscopically in the medial and lateral banks of the anterior and mid-parietal somatosensory cortex and middle and posterior motor cortex. The anterior motor cortex was counted medial to the dorsolateral prefrontal cortex (Figure 2D). The identification of these regions was a consensus between two investigators (JKL, LJM). At 400x magnification, five vertical rows of microscope fields spanning cortical layers II through VI in each gyral bank were counted independent of laminar specification (Figure 2D–F). In the inferior parietal cortex, the number of morphologically normal neuron profiles was counted (LJM) in 10 horizontally arranged 1000x microscopic specifically in layers II, III, and V of the superior bank. These counts were done at a high magnification for improved resolution and careful assessment of neuronal cell death phenotype [66]. Additionally, 10 microscope fields at 400x were counted (JKL) in putamen gray matter (Figure 2B) to assess the efficacy of HT protection as described before [54,64,67,68].
Counted neuronal profiles in H&E-stained sections were classified by their microscopic appearances [54,56,66,69]. Normal neurons (Figure 2G) had a size of 8-15 μm in diameter and non-vacuolated cytoplasm, interpreted as intact membranous organelles without swelling, and an open nucleus (not condensed, darkly basophilic, or pyknotic) with at least one nucleolus and gossamer chromatin strands dispersed in a finely particulate nucleoplasmic matrix. Other neurons (Figure 2H) were unlike normal neurons with a stark basophilic cytoplasm and few vacuoles or dilated cisterns, notably at a perinuclear location, seemingly intact cytoplasmic and nuclear membranes, and a prominent darky basophilic (blue-dark purple) nucleoplasmic matrix but non-pyknotic nucleus with a nucleolus [54,56]. Because neurons typified by enhanced basophilia might survive [70,71] and may correspond to neurons with enhanced chromatin-DNA template activity and RNA synthesis [72], they were combined into a total normal neuron category for analysis. The ischemic-necrotic neuron had a hematoxylin (blue-purple)-stained, angular, and pyknotic nucleus, angular soma, vacuolated and eosinophilic (red-pink) cytoplasm, and absence of perinuclear pallor (Figure 2I). These cells undergo dissolution of the plasma and nuclear membranes and nucleoplasmic matrix speckling [59,69]. Cells undergoing the apoptosis-necrosis continuum had ≥ 5 nuclear fragments of irregularly shaped chromatin clumps, eosinophilic cytoplasm, some cytoplasmic vacuolation, but seemingly intact cell membrane (Figure 2J) [59,69]. Apoptotic cells could be identified as neurons because of their size and residual cytoplasm (Figure 2K) or they were cell type non-identifiable and were round and small profiles with eosinophilic, condensed cytoplasm, chromatin clumps (≤4 crescent-shaped or round clumps), and cell surface that often was withdrawn from the surrounding neuropil [59,69].
Rbfox3 Immunohistochemistry (IHC)
Immunoperoxidase IHC, with diaminobenzidine (DAB) as chromogen, was done on piglet brain paraffin sections as described [48,73,74,75,76] to localize Rbfox3 (previously known by its antibody called NeuN) using a mouse monoclonal antibody (Millipore, Clone A60). This antibody was characterized for specificity in pig brain homogenates using western blotting (Figure 3A, inset) and the results were similar as described for human and mouse brain [77,78]. Nissl counterstaining with CV was done for cellular and laminar identifications in piglet neocortex and for profile counting of total neurons.
The Rbfox3-stained brain sections were used for cell counting in somatosensory cortex (Figure 3). Counting was done specifically in layers II, III, and V. The CV counterstaining defined layer identification by cell morphology, distribution, and packing densities as well as other landmarks such as distances from deep layer I (a neuron poor layer in piglet) and the very distinctive polymorphic layer VI just above the subcortical white matter (Figure 3A–C). After layer identification from positive cell morphology and packing densities (Figure 3A), neuronal counts were made in 1000x non-overlapping microscopic fields distributed horizontally intralaminarly (Figure 2E). Total neurons were counted based on the CV counterstaining in each microscopic field, and then neurons were classified as normal Rbfox3-positive with nuclear and cytoplasmic staining (Figure 3B inset), ischemic-necrotic Rbfox3-positive with pyknotic and clumped nuclear immunoreactivity (Figure 3C inset), and neurons with nuclear depletion of Rbfox3 immunoreactivity but otherwise appeared morphologically normal from the CV staining (Figure 3E).
2.6. EEG and Video Analysis
cEEG data were acquired for up to 7 days after HI (Stellar Telemetry, TSE Systems, Inc.). The EEG data were analyzed using Notocord-hem software (NOTOCORD Systems, Version 4.4.0.3, 2020, Philadelphia, PA). An investigator (CTP, senior neurology resident) with experience in reading human EEG analyzed the piglet EEG with the guidance of two American Board of Psychiatry and Neurology epileptologists (EKR, CWH) who are experienced in analyzing human neonatal and experimental EEG. These investigators were blinded to the treatment group and clinical video recordings. Electrographic seizure activity was defined as waveform activity with features of epileptiform pattern (spike wave, polyspikes, sharp wave, rhythmicity) of at least 10 seconds with confidence in evolution of waveform in frequency, morphology, or location. Seizure activity that clustered in time was counted as a single seizure if clustering occurred within 60 seconds of an event. Seizure start times, stop times and waveform characteristics, including morphology, frequency, and focality, were documented within each recording. The seizure burden was defined as the percentage of the total EEG recording with seizure activity. Another investigator (LJM) who video recorded piglet behavior during recovery used the recordings to corroborate electrographic seizures and rule out motion artifact. Seizures were allowed to self-resolve without anti-seizure medications, to identify the natural biology of brain injury with seizures, but piglets with continuous clinical seizure activity (Supplementary Videos 3, 4) were euthanized.
2.7. Sample Size and Statistical Analysis of Data
In a prior report [64] of neonatal piglets with HI injury that received 29 hours of anesthesia like the current study, the mean difference in ischemic necrotic neurons within the motor cortex between HI-NT and sham-NT piglets was 100 with a within-group standard deviation of 5. A sample size of 4 would generate power > 0.9. We increased the sample size to allow for some variability in our estimates.
The differences between mean ratios of normal-to-total and ischemic necrotic-to-total neurons and the numbers of apoptotic and apoptosis-necrosis continuum cells within the frontal, motor, and somatosensory cortices determined from H&E-stained sections were estimated using linear mixed models with random intercepts to account for measurements within the same piglet (33 comparisons for each cell category). Sham-NT was the reference group, and these analyses were adjusted for survival duration.
The counts representing the number of normal neurons (cells/mm2) in the inferior parietal cortex from the H&E-stained sections and the total neurons and NeuN (Rbfox3)-nuclear depleted neurons in somatosensory cortex were analyzed separately outside of the linear mixed model. Individual comparisons of group means were made between independent groups (e.g., Sham-NT vs Sham-NT, Sham-NT vs HI-NT) using a two-sample t-test and comparisons among means of all four groups were done with ANOVA.
Arterial blood pH, partial pressure of carbon dioxide (PaCO2), partial pressure of oxygen (PaO2), MAP, hemoglobin, and glucose levels at baseline and 1, 3, 20, 24, and 28 hours after ROSC were analyzed by 2-way repeated measures analysis of variance with post-hoc Tukey tests. The blood gas and MAP data for 42 minutes hypoxia and 7-8 minutes of asphyxia between HI piglets destined to receive NT or HT were compared by t-tests. Blood gas data were analyzed from the 7-minute and the MAP data were analyzed from the 8-minute time points of asphyxia. These data are graphed as means with 95% confidence intervals.
Non-parametric data, including survival duration and the ratio of normal-to-total neurons among naïve control and anesthetized sham groups, were analyzed by Kruskal Wallis analysis of variance on ranks. The difference in seizure burden between all sham and all HI piglets were analyzed by Mann Whitney tests. Spearman correlations evaluated the relationship between seizure burden and the ischemic necrotic-to-total neuron ratio. These data are graphed as box or scatter plots.
3. RESULTS
3.1. Neonatal Piglet Gyrencephaly, Cytoarchitecture, Chemoarchitecture, and Connectivity
Like human brain [79,80,81], the piglet brain has distinct cortical gyral and sulcul patterns that segregate into functional localizations including motor, somatosensory, and visual cortices (Figures 1E, 2A–F and 4A). These cortical functional localizations in pig have been identified by electrophysiology [50,82,83] and fMRI-BOLD (P Liu, D Jiang, D Liu, LJ Martin, JK Lee, unpublished observations). Histologically, by Nissl and Rbfox3 staining, the motor cortex is discernable by the prominent layers V and VI and attenuated layer IV typical of the agranular neocortex (Figures 3B and 4B). In contrast, the somatosensory cortex is distinguished by its prominent layer IV and attenuated layer V (Figure 3A and Figure 4C,D). In piglet, as in human [84], the cytological transition between motor cortex and somatosensory cortex can be identified (Figure 4D).
To confirm our Nissl- and H&E-based histological frame of reference for the parcellation of the neonatal piglet neocortex, we also did in situ (tissue section) metabolic mapping for complex IV cytochrome C oxidase enzyme activity in the axial plane (Figure 4E–G) as we have shown before in the coronal plane, along with, as a negative control, its complete inhibition by potassium cyanide [20]. This assay shows that mitochondrial activity varies throughout different regions of piglet neocortex. The highest activity levels in neocortex appear to be in visual (Figure 4E), somatosensory (Figure 4F), and inferior frontal (Figure 4G) cortices.
The connectivity experiments further supported our identifications of motor and somatosensory cortex in neonatal piglets (Figure 5). Various genetic tract-tracers were injected unilaterally into different neocortical regions (Figure 5A), while FG was injected unilaterally subcortically into the striatum (Figure 5B). FG retrogradely labeled corticostriatal projection neurons in layer V of motor cortex were observed (Figure 5B inset). Injections of AV-CFP robustly infected pyramidal neurons in layer V in motor cortex (lateral bank) as visualized with antibody to CFP (Figure 5C), including the human-like Betz neurons (Figure 5C inset), thus confirming the primary motor cortex identity of the cortical area that was studied here neuropathologically. Pyramidal neuron dendrites originating from neurons in layer V could be visualized spanning the entire width of cortex to layer I (Figure 5D). In central putamen, single axons from motor corticostriatal projection neurons could be identified (Figure 5E). LV-RFP injections into somatosensory cortex (Figure 5A,G,H) transduced neurons in the lateral bank and revealed that somatosensory corticostriatal projection axons travel in the external capsule to target lateral putamen in a dorsal-ventral ladder-like pattern (Figure 5F). Antibody detection of RFP was more sensitive than direct fluorescence and revealed the fine terminal fields of corticothalamic projections to ventral posterolateral thalamic nucleus (Figure 5H,I) which, in human and monkey, corresponds to somatosensory afferents from body. AAV-eGFP injections into somatosensory cortex showed, by retrograde labeling, extensive cortical-cortical connections with primary somatosensory cortex, including layer V of posterior cingulate cortex (Figure 5J) and layer III of primary olfactory cortex (data not shown). Injections of WGA-HRP into posterior somatosensory cortex confirmed the lateral external capsule trajectory of corticostriatal preterminal axons and their entry into lateral putamen and extensive deposition of synaptic terminals in central putamen (Figure 5L). Retrogradely filled neurons in the ventral posterolateral nucleus of thalamus were also seen with WGA-HRP (Figure 5M), thus confirming the primary somatosensory identity of the cortical area that was studied here neuropathologically.
3.2. Neonatal Piglet Brain Injury Model
Forty piglets were used for the HI experiments. There was an 85% protocol completion rate with survival (see Supplemental Material on animal attrition and Video 1). Six sham-NT, 10 sham-HT, 8 HI-NT, and 10 HI-HT piglets completed their protocols and were analyzed for histologic brain injury.
3.2.1. Pathophysiology
Both HI groups had significantly decreased PaO2 during the hypoxia and asphyxia (Figure 6A) and corresponding significantly increased PaCO2 during the hypoxia and asphyxia (Figure 6B). Both HI groups had severe hypotension (< 40 mm Hg) and arterial blood acidosis (pH < 6.9) during asphyxia (Figure 6C,D).
Target hypothermic core temperature was achieved in 3 hours (Figure 6E). Rewarming from this target temperature began at 20 hours of cooling and continued through 29 hours for emergence from anesthesia and extubating. MAP and arterial blood pH did not differ significantly among the treatment groups at any time point during HT. Time and treatment interactively affected the PaCO2 (p=0.006). Post-hoc tests showed that sham-HT piglets had higher PaCO2 than did HI-NT piglets at 1 hour from cooling onset (p=0.038; Figure 6B). The PaO2 also varied across time (p<0.001) with higher 1-hour PaO2 in HI piglets than in sham piglets that would later be HT (p=0.049; Figure 6A). Hemoglobin levels varied by time (p<0.001). HI-HT piglets had higher hemoglobin levels than did HI-NT piglets at 3 hours (p=0.029) and 20 hours (p=0.002) of HT (Figure 6F). Hemoglobin levels did not differ between groups during NT or rewarming. Glucose was interactively affected by time and treatment (p<0.001). At 1 hour from onset of cooling (Figure 6G), the HI group had higher glucose levels than sham piglets that would receive HT (HI-NT: p=0.013; HI-HT: p=0.042). Sham piglets at 20 hours of HT had higher glucose than did sham-NT piglets (p=0.048) and HI-NT pigs (p=0.034). Glucose was also higher in HI-HT piglets than in HI-NT pigs at 20 hours (p=0.047). During rewarming at 24 hours, the HI-HT piglets had higher glucose than HI-NT piglets (p=0.018). Glucose levels did not differ among groups at 28 hours and prior to emergence from anesthesia.
3.2.2. HT Protects Primary Somatosensory Cortex after HI
H&E-stained paraffin sections (10 µm thick) were used for neuron profile counting (Figure 2). The survival times of piglets for brain neuropathological assessments did not differ among treatment groups (p=0.078; Supplementary Material Figure 1).
HI-NT piglets had a lower normal-to-total neuron ratio in the somatosensory cortex within the anterior parietal cortex (Figure 2B,E) in the medial (p<0.001) and lateral (p<0.001) banks relative to sham-NT piglets after adjusting for survival duration (Figure 7A). Loss of normal neurons (Figure 2G) also occurred in the mid-parietal somatosensory cortex (Figure 2C,F) in both gyral banks (medial: p<0.001; lateral: p<0.001). The ratio of normal-to-total neurons for HI-HT piglets was not statistically different from that of sham-NT piglets (Figure 7A).
We next examined the phenotypes of neocortical neurodegeneration in HI piglets seen by H&E staining. Classic ischemic-necrosis (Figure 2I) was the predominant cell death phenotype seen in neocortex compared to apoptosis and hybrid continuum death (Figures 2J,K and 7). Neuronal ischemic necrosis was present in layer II-VI if the neocortical injury pattern was panlaminar (Figure 7E) or precisely in layer III and upper layer IV if the injury pattern was laminar (Figure 7F), as we have described before [19]. The dominant presentation of neuronal ischemic necrosis in neocortex (Figure 2I) is also seen in putamen [48,56,73]. HI-NT increased ischemic necrosis in the somatosensory cortex relative to sham-NT (Figure 7B). In the anterior parietal somatosensory cortex (Figure 2B,C), HI-NT increased the ratio of ischemic-necrotic neurons in the medial (p<0.001) and lateral (p<0.001) banks. HI-NT piglets also had more ischemic necrosis in the medial (p<0.001) and lateral (p=0.001) banks of the somatosensory gyrus in the mid-parietal cortex (Figure 2C,F).
Though apoptosis (Figure 2K) was less common than ischemic-necrotic neurons, HI-NT increased somatosensory cortical apoptosis to exceed that of sham-NT piglets (lateral bank of anterior parietal cortex: p=0.003; medial bank of mid-parietal cortex: p=0.013) (Figure 7C). Apoptosis was seen in all neocortical layers.
Sham-HT piglets had less apoptosis than did sham-NT piglets (p=0.036). Cell death by the apoptosis-necrosis continuum (Figure 2J) was most common in the medial bank of the mid-parietal somatosensory cortex in HI-NT piglets (p=0.026; Figure 7D). HI-HT piglets had no statistical differences in ischemic-necrotic, apoptotic, or apoptosis-necrosis continuum cells relative to sham-NT.
3.2.3. HT Protects Primary Motor Cortex after HI
HI-NT piglets had reduced normal-to-total neuron ratios in the motor cortex than sham-NT piglets (Figure 8). This effect varied by anteroposterior level (Figure 2A–C). Normal neurons were significantly lost in HI-NT piglets at the anterior parietal (medial bank: p=0.010; lateral p=0.001) and mid-parietal (medial, p=0.003; lateral, p=0.001) levels of motor cortex (Figure 8A). HI-HT piglets had no statistical difference in the normal neuron ratio compared to sham-NT. The frontal anterior-most motor cortex and the gyrus rectus and inferior prefrontal cortex (Figure 2A) showed no difference in normal neurons after adjusting for survival duration (p>0.05).
HI-NT piglets had a higher ratio of ischemic necrotic-to-total neurons in the anterior and mid-parietal motor cortex than did sham-NT piglets (Figure 8B). In anterior parietal motor cortex (Figure 2B) this effect was seen in the medial gyral bank (p<0.009) and lateral bank (p=0.001) after adjusting for survival duration (Figure 8B). The mid-parietal motor cortex (Figure 2B) showed a similar trend with more ischemic necrosis in the medial (p=0.003) and lateral (p<0.001) banks. By contrast, the frontal cortex (Figure 2A) showed no differences in ischemic necrotic cell counts among groups (p>0.05) and less apoptosis in sham-HT piglets relative to sham-NT piglets (Figure 8C, p=0.020). The cell death continuum manifested more often in HI-NT piglets than sham-NT piglets (Figure 8D, p=0.001). HI-HT piglets showed no differences in ischemic-necrotic, apoptotic, or apoptosis-necrosis continuum cell death when compared to sham-NT piglets (Figure 8B–D).
To examine the potential effects of anesthesia with and without HT, we compared the ratio of normal-to-total neurons among naïve unanesthetized (n=6), sham-NT (n=6), and sham-HT (n=10) piglets. The ratio of normal neurons did not differ between anterior and mid-parietal somatosensory and motor cortices among the unanesthetized and sham procedure piglets (p>0.05 for all comparisons, data not shown).
3.3. Neuropathology in Neocortex of HI Piglets Involves Laminar Vulnerability and Depletion of Nuclear Rbfox3
Because our other neuropathological assessments in HI piglets (Figure 7 and Figure 8) were done independent of layer, we examined the inferior parietal cortex and the somatosensory cortex more closely for specific laminar pathology. The piglet neocortex has very discernible layers (Figures 3A,B, 4B–D, 5C,J and 7E). The inferior parietal gyrus is easily identifiable for characterization in piglets (Figure 2B,C). This region was interrogated for neuropathology by H&E staining (Figure 9) because it appears to be damaged in seizing piglets but not as primary HI injury [19,20]. Layer III neurons in a sham-NT piglet are illustrated in Figure 9A. All neuron profiles present are normal, and the neuropil is intact without perineuronal swelling or spongiform encephalopathy (Figure 9A). In HI-NT piglets, layer III of inferior parietal cortex was severely depleted of normal neurons (Figure 9B,C) with only ischemic-necrotic neuronal tombstones remaining and extensive encephalopathic spongiform changes in the neuropil (Figure 9B) similar to the pattern shown for somatosensory cortex (Figure 2I). Layer III was protected in HI-HT piglets compared to HI-NT piglets (Figure 9C).
We used IHC for Rbfox3 as a neuronal marker to further verify the identities of somatosensory and motor cortices in piglets (Figure 3A,B) and to reveal potential aberrancies in its neocortical neuron localization after HI. The NeuN antibody [85] used to detect Rbfox3 in pig brain was specific as shown by western blotting after SDS-PAGE where expected proteins with mobilities at 46 and 48 kDa were seen (Figure 3A inset). Total neuron counting was done using the Nissl counterstain. In sham piglet neocortex, Rbfox3 defined all six layers with particular discernment depending on known functional localizations (Figure 3A,B). In the somatosensory cortex, layer IV was prominently thick (Figure 3A). In contrast, the motor cortex had an attenuated layer IV and prominent layers V and VI (Figure 3B). Rbfox3 had nuclear and cytoplasmic localizations (Figure 3B, inset, E). Within the nucleus, Rbfox3 was present in numerous subnuclear particles (Figure 3B,E).
Rbfox3 was then used as a marker to count neurons (Figure 10A-C). In HI-NT piglets compared to sham-NT piglets (Figure 10), significant loss of total neurons was detected in layer II (p<0.0001) and layer III (p<0.0001), while layer V had significant loss (p< 0.0005) but less vulnerability than layers II and III. In HI-HT piglets compared to HI-NT piglets, neuronal densities were partially rescued in all layers (Figure 10A–C).
In HI piglets, Rbfox3 positivity in the somatosensory cortex discriminated additional patterns of injury unseen by the H&E staining. Layer II neurons in somatosensory cortex were depleted of Rbfox3 while, in the same tissue section, layer II neurons in adjacent motor cortex were positive (Figure 3C,D). In the somatosensory cortex, layer II, III, and V neurons were characterized by their Rbfox3 staining patterns. Rbfox3 immunoreactivity was affected by HI and HT treatments (Figure 10D–F). Sham-HT piglets compared to sham-NT piglets had significantly higher Rbfox3-depleted-to-total neuron ratios in layers II (p=0.002), III (p=0.01), and V (p=0.001). HI-NT piglets compared to sham-NT (Figure 10D–F) had significantly higher Rbfox3-depleted-to-total neuron ratios in layers II (p<0.0001), III (p<0.0001), and V (p<0.0001). The increased Rbfox3-depleted-to-total neuron ratio seen in HI-NT piglets was mitigated in HI-HT piglets in layer V (Figure 10D–F).
3.4. HT Reduces the Burden of Neuronal Injury in the Putamen after HI
Neuronal damage was assessed in putamen in the target areas of motor cortex and somatosensory cortex (Figure 5F,L). HT had incomplete protective effects in the putamen after HI. Significant loss of normal neurons occurred in piglets that received HI with either HT (p=0.008) or NT (p<0.001) compared to sham-NT piglets after adjusting for survival duration (Supplementary Material Figure 2A). In concert, increases in ischemic necrosis was detected in HI-HT piglets (p=0.014) and HI-NT piglets (p<0.001) (Supplementary Material Figure 2B). The number of cells with apoptosis or the apoptosis-necrosis continuum did not differ (Supplementary Material Figure 2C,D).
3.5. Electrographic and Clinical Seizures
Eighteen piglets had cEEG and video monitoring. The cEEG determined seizure burden was not affected by temperature in this small cohort of piglets. Seizure burden was higher in all HI piglets compared to all sham piglets (p=0.019; Figure 11A). Normal piglet EEG patterns showed activity of various frequencies, primarily high (>10 Hz), and low voltage (0.1-0.2 mV), with variations in frequency and amplitude throughout all leads but no clear epileptiform activity or evolutionary pattern suggestive of seizure activity (Figure 11B). Clinical seizures usually emerged within 24 hours after extubating during the second day after HI. They often appeared during sleep, consisting of orofacial twitching, tongue movements (Supplemental Video 2), rooting movement of snout and head, repeated jerks of the head and legs, sudden arousal, and then clonic movements (see Supplementary Material Videos 3,4). The clonic movements spread from the head region to the shoulder and forelegs to form generalized seizures (see Supplementary Material Video 4). Some piglets developed fictive running movements when the seizure was believed to progress to status epilepticus (see Supplementary Material Videos 3,4). EEG confirmed the presence of seizures with rhythmic spike-wave complexes of higher voltage (>0.4 mV) in specific neocortical areas that generalized to other areas of neocortex (Figure 11C, Supplemental Video 4). Some EEG detected seizures appeared subclinical or equivocally clinical (Supplemental Video 5).
Greater seizure burden correlated with more ischemic-necrosis in the somatosensory cortex at the anterior parietal (medial bank: r=0.57; p=0.015; n=18) and mid-parietal (medial: r=0.69; p=0.002; lateral: r=0.62; p=0.006) levels. (Figure 12) Ischemic necrosis in the lateral bank of the frontal motor cortex also correlated with seizure burden (r=0.48; p=0.045). Additionally, more ischemic necrosis in putamen correlated with higher seizure burden (r=0.60, p=0.009; Figure 12E). The seizure burden did not correlate with the amount of ischemic-necrosis in motor cortex at the anterior parietal or mid-parietal levels, medial bank of the frontal motor cortex, or in the lateral bank of anterior parietal somatosensory cortex (p>0.05 for all comparisons).
4. DISCUSSION
We developed, using neonatal piglets, a new survival model of global HI with remote cEEG monitoring during recovery from overnight HT to study neurological phenotypes and neocortical neuropathology. The salient findings of this work are: 1) piglets are a translationally relevant neonatal large non-primate animal model to study the cytoarchitecture and connectomics of the developing gyrencephalic cerebral cortex; 2) the neonatal piglet neocortex has a suprasylvian spatial vulnerability to HI and seizures that damages somatosensory, motor, inferior parietal, and prefrontal cortex; 3) HT initiated 2 hours after HI protects variably against neuropathology in functionally different regions of the neonatal gyrencephalic neocortex but does not protect against electroencephalographic seizures; 4) by H&E staining, neurons in neonatal neocortex of piglets have a limited repertoire in their morphologic degeneration (ischemic-necrosis is the predominant form) in HIE with and without seizures; 5) higher seizure burden correlates with more ischemic-necrotic neurodegeneration and neuron loss; and 6) nuclear depletion of cortical neuron RNA splicing protein Rbfox3 is a novel immunophenotype of neonatal HIE with seizures, and it could be a molecular mechanism involved in seizure origin.
4.1. Animal Models to Study Neocortical Organization, HIE, and Seizures in the Neonatal Brain
An important aspect of experimental work is its translational applicability to human disease and injury. In human neonatal HIE, the peri-Rolandic area of neocortex is often damaged [10,11,12,14,86], meaning the frontal lobe-located primary motor cortex in the anterior bank of the central sulcus of Rolando and the parietal lobe-located primary somatosensory cortex in the posterior bank and bottom of this sulcus. These regions are contiguous as Brodmann area 4 merges with Brodmann area 3a. The ansate sulcus in piglet, not present in rodents because of their lissencephaly [87], is homologous to the human central sulcus [88]. In neonatal piglet, we identified cytologically the agranular motor cortex by its attenuated layer IV and prominent layer V and large Betz pyramidal neurons, and its projections to central putamen. Similarly, we identified the somatosensory cortex by cytology and its connectivity with putamen, thalamus, and cortical-cortical connections. The cytology is like what has been described for human [77,84,89,90]. We did limited evoked motor responses and somatosensory-evoked potential recordings to assist in identifying these regions, and they corresponded to the electrophysiological mapping of others in pig [82,91].
Human infants with HIE plus seizures are likely to have an abnormal brain MRI and persistent neurodevelopmental impairments despite having received therapeutic HT [94]; therefore, a better understanding of the mechanisms of neonatal seizures and long-lasting functional deficits ensuing from neonatal HI brain injury in translationally relevant animal models can help guide development of therapeutics adjuvant to HT. Animal models will drive the understanding of relationships between neonatal HIE, seizure co-morbidity, neuropathology, and impairments in cognitive, motor, and affective functioning. In mice and rats, seizures early in life can lead to cognitive, social, and behavioral disorders [92] with changes in hippocampal synaptic plasticity and memory [93]; however, rodents are lissencephalic [87] and have very different brain developmental chronicity and primary sensory-driven, including binocularity of vision, social structure compared to humans. To best model human neonates, a time-equivalent gyrencephalic species with a structurally similar six-layered neocortex is desirable [95]. Term fetal rhesus monkeys subjected to partial asphyxia have seizures that aggravate the neuropathology of HIE [33]. Near-term fetal sheep have been used as HIE models to show therapeutic efficacy of HT [96] and that status epilepticus following severe HI associates with greater brain injury [97]. However, generally sheep models have been short survivals without extended monitoring for neurological outcomes. Ideally, it is best for injured animals to survive with an independent upright and mobile status for feeding and observer assessments of posture, muscle tone, balance, gait, and cognition. At a cellular level, neocortical pyramidal neurons and interneurons in animals should have archetypal biology like human neurons. For example, though gyrencephalic, ferrets have layer V pyramidal neurons with very different biophysical properties compared to human neurons that can affect excitability and excitotoxicity [98]. We have shown that pig cortical neurons derived from directed differentiation of neural stem cells have, compared to human neurons, more similar responses to excitotoxins and oxidative stress than mouse cortical neurons [18].
Therefore, we sought to develop a model that comprehensively integrates clinically relevant HIE in a large gyrencephalic animal, a therapeutic standard of clinical care (HT), survival, cEEG, neurologic phenotypes, and robust neocortical cellular neuropathology. The size of the neonatal piglet cranium and the brain gyrencephaly allows precise transcranial electrode placement to monitor EEG in specific gyri in the immediate post-HI and HT periods for many days. This contrasts to the cEEG monitoring in rodents that begins long after HI [99] because newborn pups cannot be separated from a dam for extended periods for EEG monitoring [100,101]. Because of longstanding ethical and moral concerns about using nonhuman primates in research [102], particularly infant rhesus monkeys [103,104], our work here and other work [105] demonstrates that the piglet is a realistic, clinicopathophysiologically relevant, and legitimate neuroanatomical and neuropathological alternative for survival modeling of neonatal HIE with the standard of care, co-morbid seizures, and long-term neurological outcomes.
The ability to monitor continuously post-HI EEG in neonatal piglets will provide a key method to study the molecular and cellular interactions between HI, seizures, and secondary brain injury with a physiologic read-out for testing adjuvant treatments to HT. This effort has importance because the clinical management of neonatal seizures is debated [23]. Nearly half of neonatal seizures from HIE are subclinical [106]. A randomized clinical trial of neonates cooled for HIE showed no difference in death or 2-year disability, including blindness, deafness, deficits in cognition and language, as well as motor, social-emotional, and adaptive skills between children who did or did not receive anti-seizure medication (phenobarbital or phenytoin) for electrographic or clinical seizures [106]. However, site discrepancies in seizure management caused study under-powering and premature ending; thus, uncertainty remains in the predictive relationship between treating neonatal seizures and persistent childhood disability [106].
We found that the timing for general tonic-clonic clinical seizures in piglets corresponded with seizures in neonatal human and monkey, which typically begin within 1-2 days after birth injury [32,107,108]. Video recordings confirmed seizure activity, including general tonic-clonic seizures in some piglets. Among a subset of piglets monitored by EEG, HI increased the electrographic seizure burden. Though some piglets needed to be euthanized prior of the endpoint survival of 7 days because of unresolving seizures, animals surviving to 7 days and beyond could be subjected to future neurological examination and cognitive testing. Cognitive status assessment using a spatial T-maze is feasible to test for deficits that may be related to HIE and seizures. We found previously that cognitive deficits in the T-maze are associated with hippocampal CA1 interneuron attrition and CA3 neuron loss in piglets with HI, though clinical seizures were not seen in this non-cooled cohort [105].
4.2. Our Piglet Pathophysiology Mimics Clinical Neonatal HIE
Our hypoxia-asphyxia, CPR, and therapeutic HT protocol is clinically relevant. It caused severe hypotension with respiratory and metabolic acidosis. PaCO2 levels after resuscitation were like clinical observations [61,109]. The target temperatures during HT and rewarming were strictly monitored and achievable. Hemoglobin levels were higher during HT in HI piglets and then showed no differences once rewarming began. This is consistent with expected increases in measured hematocrit during HT [110,111]. Stress induced hyperglycemia occurred immediately after resuscitation from HI as expected with severe injury [112]. Subsequent glucose levels tended to be higher in HT groups. Early hyperglycemia is associated with greater brain injury on MRI and poorer neurodevelopmental outcomes [112]. This early hyperglycemia is likely related to the sympathoadrenal response engaged during the asphyxia and was observed in both HI groups. Hyperglycemia was also seen at the end of cooling in HI and sham piglets suggesting sustained sympathetic activity or decreased tissue glucose utilization. However, we did not administer insulin in our protocols for safety reasons because hypoglycemia is also associated with poor neurologic outcomes, cardiovascular instability, and seizures [112,113]. Instead, the glucose content in the IV fluids was adjusted to reduce the incidence of hyperglycemia.
4.3. The Neonatal Piglet Neocortex has a Suprasylvian and Prefrontal Vulnerability to HIE
Many children surviving neonatal HIE and HT have, compared to peers without history of injury, IQ deficits and moderate-to-severe impairments in attention-executive, visuospatial, and emotional-social functions, and language skills [4,6]. The neocortex drives these functions [7], and the prefrontal cortex reigns supreme [8,9]; thus, these persisting childhood deficits suggest that: 1) the neocortex has regional topographic vulnerability in neonatal HIE; 2) vulnerable neocortical areas are insensitive or less responsive to HT compared to deep gray matter; and 3) damage to the neocortex can develop after the injury-cooling period perhaps with delayed and remote and trans-synaptic degenerative features. These possibilities are consistent with clinical observations and with elegant observations in HIE rodents [114,115]. Term HIE infants show selective neocortical highlighting on T1-MRI involving superior peri-Rolandic, posterior cingulate, and insular cortex [14]. These abnormal signals can take several days to manifest and peak during the second week after the insult [14]. The peri-Rolandic watershed damage in HIE correlates with verbal IQ at 4 years of age [116]. Though gyrencephalic, the pig brain is not furnished with a Rolandic central sulcus or an extensive Sylvian lateral fissure, but there is a Sylvian sulcus and superior to this is the primary somatosensory cortex and posterior motor cortex homologous to the human peri-Rolandic cortex [88]. Their vulnerability to neonatal HI is shown here and before [19,20]. These regions were protected after HI by overnight HT. We also examined for the first time in our piglet model the prefrontal anterior motor cortex and the inferior orbitofrontal cortex. Interestingly, in contrast to the vulnerability of the posterior regions of motor cortex, the anterior motor cortex was not significantly damaged by HI. The cortical area assessed here also included the gyrus rectus; it corresponds to Brodmann area 11 and functions in decision-making in context to reward [117]. The lateral inferior prefrontal was damaged by HI and was protected by HT. This area roughly corresponds to Brodmann areas 10 and 47 in human [81]; these cortical regions have executive and cognitive flexibility functions [118,119]. Close examination of these areas (Figure 2A) with MRI in clinical neonatal HIE could yield new information relevant to the functional deficits seen in school age children with early life HIE and cooling.
We also examined quantitatively for the first time in our piglet model the inferior parietal cortex. Piglets with seizures had extensive damage in this area consistent with previous observation based on clinical seizures in non-cooled HI piglets [19,20,36]. This neocortical region may correspond to the vestibular/auditory cortex identified in monkey, cat, and human [82,120], allowing for the possibility that damage to this region relates to emergence of generalized tonic clonic seizures and decerebrate posturing through release of the lateral vestibular nucleus and activation of the lateral vestibulospinal pathway that strongly excites antigravity extensors [121,122,123]. Lateral vestibular nucleus neurons (Deiter’s neurons) forming the vestibulospinal tract are tonically active [123]. Like the descending influences of suprasegmental pathways on spinal cord through inhibitory interneurons [121,124], the cerebral cortex projects to the lateral vestibular nucleus mediating inhibitory modulation [125]. This pathway, alone or in combination with damage to cerebellum, another potent inhibitory modulator of Deiter’s neurons [122,123], could be inactivated in HI piglets with neocortical damage. “Released” central pattern generators in brainstem [126,127] could also be involved in the fictive locomotor activity we observed and documented by video recording in some piglets with seizures (see Supplementary Material Videos 1-3).
We specifically did a laminar analysis of layers II, III, and V because layer-specific cortical-cortical connectivity can mediate seizure propagation [128], and layers II and III have emergent synaptic mechanisms controlling pyramidal neurons in the gyrencephalic brain that became human-specific [95,129]. Moreover, layer III frontal cortical neurons show dendritic spine depletion in human epilepsy [130]. Layers II, III, and V showed very significant loss of neurons in HI-NT piglets, consistent with our previous work [19]. Layers II and III appeared more vulnerable than layer V. Pervious work in a rat model of epilepsy has shown that layer III is preferentially vulnerable [131]. A recent RNAseq study of human epilepsy identified layer II-III Cux1-positive principal neurons and layer V Fexf2-positive neurons as possible drivers of excitability perturbations in neocortex [132].
Seizures appeared unmitigated by HT in piglets after HI in this study with small group sizes. HT was initiated 2 hours after HI to approximate delays encountered in hospital practice. Other work in piglets suggested that HT could prevent clinical seizures, but a lesser insult and slightly older animals were used [133]. Clinically, 40-70% of neonates cooled for HIE have seizures [22,94]. Clinical seizures and abnormal EEG in human neonates associate with injury in the neocortex and basal ganglia [94,108]. We also found that greater seizure burden correlated with more ischemic necrosis in the anterior and mid-parietal somatosensory cortex, frontal motor cortex, interior parietal cortex, and putamen; moreover, HT reduced ischemic necrotic neuron number. Thus, a dichotomy emerges with HT affecting the burden of neuronal cell body damage but not seizure burden. Perhaps seizures are independent of the primary HI brain damage that is responsive to HT. However, these HT-rescued neuronal cell bodies may be functional, non-functional, or aberrantly functional. Preservation of a particular domain of a neuron does not necessarily mean preservation of appropriate function. The cellular and molecular pathobiology for the inability of HT to protect against seizures needs identification. Perhaps there is a relationship to Rbfox3.
4.4. Neocortical Neuronal Degeneration in Piglet after HI and HT has a Limited Structural Phenotype Repertoire
This study is our first detailed multiregional examination of neurodegeneration in piglet neocortex after HI and overnight HT and co-morbid seizures. In addition to determining whether there is a differential regional and laminar vulnerability of neocortex in HI piglets with and without cooling and whether cooling protected neocortex, we examined neuronal cell death phenotypes. Classic ischemic-necrotic neurodegeneration was the predominant phenotype in neocortex independent of temperature and seizure presence. More ischemic necrosis in the anterior and mid-parietal somatosensory cortex, frontal motor cortex, and putamen correlated with higher seizure burden. Importantly, seizure presence did not alter neurodegeneration morphology seen by H&E staining, and, though we did detect apoptotic and continuum forms of cell degeneration in piglet neocortex, the predominance of these phenotypes also did not appear to be influenced by seizures, survival, or treatment.
An ischemic-necrotic neuron majority in cerebral cortex is consistent with our observations in piglet striatum [56,73,133]. One expectation was that the cooling would slow down the kinetics of neuronal injury and shift the ischemic necrosis to hybrid continuum or apoptotic forms of cell death [134] (see Figure 2). We observed neuronal cell death phenotype shifting in other forms of degeneration in mouse systems [135,136,137]. This was not the case in piglet neocortex within our 2 to 7-day survival period. An immutable form of neuronal cell death also appears to be the case in putamen where we saw partial protection with HT. Absence of cell death phenotype switching was also seen in our previous studies of HT protection of putamen with piglet survivals of 3 and 6 hours [68] and 1 and 10 days [67], though in these cohorts, with a less severe insult, clinical seizures were not observed. These observations are meaningful because they would be relevant to adjuvant therapy to HT. If this holds true for human neonatal HIE and cooling, though it might not, because of human and non-primate neuron species differences in cell death mechanisms [18], then regardless of targeted temperature management and presence or absence of seizures after HI, subacute therapies targeting cell death will likely need to be modulators of cellular necrosis.
4.5. Rbfox3 Immunophenotyping Identifies a New Form of Neocortical Neuropathology in Neonatal HIE
The neuron-specific nuclear protein NeuN is Rbfox3 and is a member of the RNA-binding protein Rbfox-1 gene family [138]. Broadly, RNA-binding proteins have been critical for the evolution of neocortical expansion and transition from lissencephaly to gyrencephaly [139]. Rbfox3 is found in subnuclear structures called speckles (interchromatin clusters); it shuttles between the nucleoplasm and nuclear matrices [140]. Rbfox3 functions in mRNA splicing and in cryptic exon suppression [141]. Rbfox family members target several epilepsy-candidate genes encoding for glutamic acid decarboxylase, GABA receptors, glutamate transporters, potassium channels, and sodium channels [38,142]. Mutations in the Rbfox3 gene have been identified in patients with neonatal epilepsy [38,44,46], developmental delay, and language/speech disorders [143,144]. Knockout mice show that Rbfox3 is required for maintenance of the balance of excitatory-inhibitory circuitry [145] by regulating vesicle-associated membrane protein 1 expression [146]. Another downstream function of Rbfox3 protein is assembly of the axon initial segment which is a key axonal microdomain involved in neuronal excitability [147].
We used a monoclonal antibody that is well characterized by others [85,148] and us [77,78] (Figure 3A inset) to detect Rbfox3 in piglet brain sections. In normal piglets, virtually all neocortical neurons had nuclear positivity for Rbfox3 in somatosensory, motor, and inferior parietal cortices. As in other species [148], piglet neocortical neurons had nuclear and cytoplasmic Rbfox3 immunoreactivity (Figure 3B inset, F). Rbfox proteins in the cytoplasm regulate gene expression by binding to 3’-untranslated regions in target mRNA [149]. Rbfox proteins in the nucleus bind to intronic consensus UGCAUG sequences that flank alternate exons to regulate pre-mRNA splicing [150]. We identified three types of neurons in sham and HI piglet neocortex: Rbfox3-positive/normal, Rbfox3-positive/ischemic-necrotic, and Rbfox3-nuclear depleted neurons. Rbfox3-depleted neurons appeared in layers II, III, and V in the HI pigs. The reason for the loss of immunopositivity for Rbfox3 is unclear. The NeuN antibody epitope maps to an N-terminal 16 amino acid sequence with a tyrosine-proline residue pair being critical for binding [148]. With tyrosine being part of the epitope, antibody binding could be phosphorylation dependent. Depletion of Rbfox3 staining in subsets of neurons that appear normal (not ischemic-necrotic, apoptotic, or continuum) by Nissl staining in piglet somatosensory cortex could be biochemically and functionally tantamount to somatic de novo loss of function mutations in the Rbfox3 gene in neocortical neuron ensembles with altered excitability and function caused by exonic “poisons” as in Dravet syndrome [151]. If this turns out to be the case, and because Rbfox3 mutations are a known cause of epilepsy in infants [44,46], then this Rbfox3 nuclear depletion phenotype could be a molecular mechanism for seizures in HIE piglets. Our Rbfox3 observations in primary somatosensory cortex are also pertinent because of the propinquity to the perisylvian epileptic network proposed in human [152]. In future experiments we will do RNAseq to examine RNA splicing regulated nonsense mediated decay of epilepsy-related gene targets and axon initial segment integrity of neocortical neurons subtypes in our piglets.
4.6. Study Caveats and Future Directions
This work has noteworthy experimental design and interpretational limitations. The treatment group sizes are small, and only male piglets were studied. The HT was not done for 72 hours like the clinical protocol [60,61]. The H&E neuropathology assessment was limited, being based only on the neuronal cell body and entirely exclusive of damage to dendrites, axons, and other components in the neuropil compartment. The piglet seizures were allowed to self-resolve without pharmacologic treatment, and piglets with continuous clinical seizures were euthanized. We did not confirm the clinical and encephalographic seizures by blocking them with known anti-epileptics. We also could not determine unequivocally whether seizures induced secondary brain injury and propagated additional seizures or if the seizures strictly resulted from the primary HI injury. Lastly, we did not identify seizure mechanisms at cellular, connectome, or molecular levels, though we gleaned evidence that cellular necrosis, layers II-IV, and Rbfox3 might be involved. The interface between lower layer III and upper layer IV interests us particularly. In the future, the neurologic and neuropathologic (cellular and connectome-wide) outcomes of treating post-HI seizures with different anti-seizure medications and layer-specific bulk RNAseq will be done in piglets with and without HT.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Information on Animal Attrition in study. Supplementary Figure 1. Piglet survival among treatment groups. Supplementary Figure 2. HT reduces the burden of neuropathology in piglet putamen after HI. Supplementary Video 1. This video shows our piglet survival model and cEEG with a piglet upright and ambulatory, feeding independently, and engaging with cage toys. Supplementary Video 2. This video shows an HI piglet sleeping and manifesting orofacial seizures with tongue, lip and snout movements. Supplementary Video 3. This video shows an HI piglet with orofacial movements and fictive running. This piglet was euthanized immdeiately after this video. Supplementary Video 4. This video shows and HI piglet with clinical generalized tonic-clonic seizures manifesting as fictive running. This piglet was euthanized immediately after this video. Supplementary Video 5. This video shows one sham piglet (dark green wrap, ear tag 330) and one HI piglet (cow-pattern wrap, ear tag 331). The wraps cover and protect the jugular line that is tunneled to the back for access. The video pans from the piglets to the running cEEG monitor. Each piglet has a 4-channel EEG. The left EEG is the sham piglet. The right EEG is the HI piglet. The motion artifact of the sham pig is observed but otherwise the EEG pattern is normal. The HI piglet appears sleeping and shows intermittent polyspiking in all 4 EEG channels against a normal background. Clinically, though the HI pig shows some minor tremulousness, there are no clinical seizures evident. Further inquiries can be directed to the corresponding author (LJM).
Author Contributions
LJM and JKL: study concept and design, data acquisition and analysis, manuscript writing, and funding. CTP: seizure burden analysis. EKR and CWH: EEG consultation. CEO and LJM: EEG acquisition. MWC, CEO, and EK: model generation, supervision, and intensive care. JP: data analysis and biostatistician. PT, BL, MVN, LJM: technical support for tract-tracing. ZJY and LJM: generation of WGA-HRP tract-tracing pigs. SA, BL, NRD, and LJM: piglet model generation and care. VO and MVN: paraffin histology; XL and LJM: in vivo cortical physiology. RCK: manuscript editing.
Funding
This work was funded by NIH NINDS grants R01 NS113921 and R01 NS107417.
Institutional Review Board Statement
The study was conducted in accordance with the guidelines of the Helsinki Declaration and the United States Public Health Service Policy on the Humane Care and Use of Laboratory Animals and the Guide for the Care and Use of Laboratory Animals. The animal protocol was reviewed and approved on 06/06/2023 by the Institutional Animal Use and Care Committee of the Johns Hopkins University (protocol number SW23M119).
Acknowledgments
The authors are grateful for the clinical consultations with the veterinarian staff of the JHU Department of Molecular and Comparative Pathobiology. JKL discloses that she is a paid consultant for the United States Food and Drug Administration and for Edwards LifeSciences.
References
- Liu, L.; Johnson, H.L.; Cousens, S.; et al. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet (London, England). 2012, 379, 2151-2161.
- Lawn, J., Shibuya, K., Stein, C. No cry at birth: global estimates of intrapartum stillbirths and intrapartum-related neonatal deaths. Bull. World Health Organ. 2005, 83, 409-417.
- Shankaran, S.; Laptook, A.R.; Ehrenkranz, R.A.; Tyson, J.E.; McDonald, S.A.; Donovan, E.F.; Fanaroff, A.A.; Poole, W.K.; Wright, L.L.; Higgins, R.D. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N. Engl. J. Med. 2005, 353, 1574–84. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N. Engl. J. Med. 2012, 366, 2085–92. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, D. , Strohm, B., Marlow, N., et al. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N. Engl. J. Med. 2014, 371, 371,140–149. [Google Scholar]
- Lee-Kelland, R., Jary, S., Tonks, J., Cowan, F.M., Thoresen, M., Chakkarapani, E. School-age outcomes of children without cerebral palsy cooled for neonatal hypoxic-ischaemic encephalopathy in 2008-2010. Arch. Dis. Child. Fetal Neonatal Ed. 2020, 105, 8-13. [CrossRef]
- Geschwind, N. Specializations of the human brain. Sci. Am. 1979, 241, 180–99. [Google Scholar] [CrossRef] [PubMed]
- Kolb, B.; Mychasiuk, R.; Muhammad, A.; Li, Y.; Frost, D.O.; Gibb, R. Experience and the developing prefrontal cortex. Proc. Natl. Acad. Sci. USA. 2012, 2 (Suppl 2), 17186–93. [Google Scholar] [CrossRef]
- Teffer, K.; Semendeferi, K. Human prefrontal cortex: evolution, development, and pathology. Prog. Brain Res. 2012, 195, 191–218. [Google Scholar] [CrossRef]
- Barkovich, A.J. MR and CT evaluation of profound neonatal and infantile asphyxia. Am. J. Neuroradiol. 1992, 13, 959–72. [Google Scholar]
- Roland, E.H.; Poskitt, K.; Rodriguez, E.; Lupton, B.A.; Hill, A. Perinatal hypoxic-ischemic thalamic injury: clinical features and neuroimaging. Ann. Neurol. 1998, 44, 161–6. [Google Scholar] [CrossRef]
- Miller, S.P.; Ramaswamy, V.; Michelson, D.; Barkovich, A.J.; Holshouser, B.; Wycliffe, N.; Glidden, D.V.; Deming, D.; Partridge, J.C.; Wu, Y.W.; Ashwal, S.; Ferriero, D.M. Patterns of brain injury in term neonatal encephalopathy. J. Pediatr. 2005, 146, 453–60. [Google Scholar] [CrossRef]
- van der Knaap, M.S.; Smit, L.S.; Nauta, J.J.; Lafeber, H.N.; Valk, J. Cortical laminar abnormalities--occurrence and clinical significance. Neuropediatrics. 1993, 24, 143–8. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, M.A. The asphyxiated term infant. In, MRI of the neonatal brain. Rutherford MA, editor. eBook 2002-2023.
- Weeke, L.C.; Groenendaal, F.; Mudigonda, K.; Blennow, M.; Lequin, M.H.; Meiners, L.C.; van Haastert, I.C.; Benders, M.J.; Hallberg, B.; de Vries, L.S. A novel magnetic resonance imaging score predicts neurodevelopmental outcome after perinatal asphyxia and therapeutic hypothermia. J. Pediatr. 2018, 192, 33-40.e2. [CrossRef]
- Xu, Q.; Chau, V.; Sanguansermsri, C.; Muir, K.E.; Tam, E.W.Y.; Miller, S.P.; Wong, D.S.T.; Chen, H.; Wong, P.K.H.; Zwicker, J.G.; Poskitt, K.J.; Hill, A.; Roland, E.H. Pattern of brain injury predicts long-term epilepsy following neonatal encephalopathy. J. Child Neurol. 2019, 34, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Kaas, J.H. Neocortex in early mammals and its subsequent variations. Ann. NY. Acad. Sci. 2011, 1225, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Chang, Q. DNA damage response and repair, DNA methylation, and cell death in human neurons and experimental animal neurons are different. J. Neuropathol. Exp. Neurol. 2018, 77, 636–655. [Google Scholar]
- Martin, L.J.; Brambrink, A.; Koehler, R.C.; Traystman, R.J. Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. J. Comp. Neurol. 1997, 377, 262–285. [Google Scholar] [CrossRef]
- Martin, L.J.; Brambrink, A.; Koehler, R.C.; Traystman, R.J. Neonatal asphyxic brain injury is neural system preferential and targets sensory-motor networks. In: Fetal and neonatal brain injury: mechanisms, management, and the risks of practice. Stevenson DK, Sunshine P, editors, Oxford University Press, 1997, pp 374-399.
- Laptook, A.R.; Corbett, R.J.; Sterett, R.; Burns, D.K.; Garcia, D.; Tollefsbol, G. Modest hypothermia provides partial neuroprotection when used for immediate resuscitation after brain ischemia. Pediatr. Res. 1997, 42, 17–23. [Google Scholar] [CrossRef]
- Glass, H.C.; Wusthoff, C.J.; Shellhaas, R.A.; et al. Risk factors for EEG seizures in neonates treated with hypothermia: a multicenter cohort study. Neurology. 2014, 82, 1239–1244. [Google Scholar] [CrossRef]
- Shetty, J. Neonatal seizures in hypoxic-ischaemic encephalopathy--risks and benefits of anticonvulsant therapy. Dev. Med. Child Neurol. 2015, 3, 0–3. [Google Scholar] [CrossRef]
- Schmid, R.; Tandon, P.; Stafstrom, C.E.; Holmes, G.L. Effects of neonatal seizures on subsequent seizure-induced brain injury. Neurology. 1999, 53, 1754–61. [Google Scholar] [CrossRef]
- Miller, S.P.; Weiss, J.; Barnwell, A.; Ferriero, D.M.; Latal-Hajnal, B.; Ferrer-Rogers, A.; Newton, N.; Partridge, J.C.; Glidden, D.V.; Vigneron, D.B.; Barkovich, A.J. Seizure-associated brain injury in term newborns with perinatal asphyxia. Neurology. 2002, 58, 542–8. [Google Scholar] [CrossRef]
- Panayiotopoulos, C.P. Idiopathic generalized epilepsies: a review and modern approach. Epilepsia. 2005, 9, 1–6. [Google Scholar] [CrossRef]
- Samanta, D. Recent advances in the diagnosis and treatment of neonatal seizures. Neuropediatrics. 2021, 2021 52, 73–83. [Google Scholar] [CrossRef]
- Tao, J.D.; Mathur, A.M. Using amplitude-integrated EEG in neonatal intensive care. J. Perinatol. 2010, 30 Suppl, S73–81. [Google Scholar] [CrossRef]
- Chalak, L.F.; Pappas, A.; Tan, S.; Das, A.; Sánchez, P.J.; Laptook, A.R.; Van Meurs, K.P.; Shankaran, S.; Bell, E.F.; Davis, A.S.; et al. Association of increased seizures during rewarming with abnormal neurodevelopmental outcomes at 2-year follow-up: A nested multisite cohort study. JAMA Neurol. 2021, 78, 1–10. [Google Scholar] [CrossRef]
- Glass, H.C.; Glidden, D.; Jeremy, R.J.; Barkovich, A.J.; Ferriero, D.M.; Miller, S.P. Clinical neonatal seizures are independently associated with outcome in infants at risk for hypoxic-ischemic brain injury. J. Pediatr. 2009, 155, 318–23. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.M. , Guillet, R., Shankaran, S., Laptook, A.R., McDonald, S.A., Ehrenkranz, R.A., Tyson, J.E., O'Shea, T.M., Goldberg, R.N., Donovan, E.F.; et al. Clinical seizures in neonatal hypoxic-ischemic encephalopathy have no independent impact on neurodevelopmental outcome: secondary analyses of data from the neonatal research network hypothermia trial. J. Child Neurol. 2011, 26, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Ranck, J.B. Jr.; Windle, W.F. Brain damage in the monkey, Macaca mulatta, by asphyxia neonatorum. Exp. Neurol. 1959, 1, 130–54. [Google Scholar] [CrossRef] [PubMed]
- Brann, A.W. Jr.; Myers, R.E. Central nervous system findings in the newborn monkey following severe in utero partial asphyxia. Neurology. 1975, 25, 327–38. [Google Scholar] [CrossRef]
- Thoresen, M.; Hallström, A.; Whitelaw, A.; Puka-Sundvall, M.; Løberg, E.M.; Satas, S.; Ungerstedt, U.; Steen, P.A.; Hagberg, H. Lactate and pyruvate changes in the cerebral gray and white matter during posthypoxic seizures in newborn pigs. Pediatr. Res. 1998, 44, 746–54. [Google Scholar] [CrossRef]
- Haaland, K.; Løberg, E.M.; Steen, P.A.; Thoresen, M. Posthypoxic hypothermia in newborn piglets. Pediatr. Res. 1997, 41(4 Pt 1) Pt 1, 505–12. [Google Scholar] [CrossRef]
- Brambrink, A.M.; Ichord, R.N.; Martin, L.J.; Koehler, R.C.; Traystman, R.J. Poor outcome after hypoxia-ischemia in newborns is associated with physiological abnormalities during early recovery. Possible relevance to secondary brain injury after head trauma in infants. Exp. Toxicol. Pathol. 1999, 51, 151–62. [Google Scholar] [CrossRef]
- Löscher, W. Animal models of seizures and epilepsy: past, present, and future role for the discovery of antiseizure drugs. Neurochem. Res. 2017, 42, 1873–1888. [Google Scholar] [CrossRef]
- Gracie, L.; Rostami-Hochaghan, D.; Taweel, B.; Mirza, N. ; SAGAS Scientists' Collaborative. The seizure-associated genes across species (SAGAS) database offers insights into epilepsy genes, pathways and treatments. Epilepsia. 2022, 63, 2403–2412. [Google Scholar] [CrossRef]
- Kossoff, E. Neonatal seizures due to hypoxic-ischemic encephalopathy: should we care? Epilepsy Curr. 2011, 11, 147–8. [Google Scholar] [CrossRef]
- Zhou, K.Q.; McDouall, A.; Drury, P.P.; Lear, C.A.; Cho, K.H.T.; Bennet, L.; Gunn, A.J.; Davidson, J.O. Treating seizures after hypoxic-ischemic encephalopathy -current controversies and future directions. Int. J. Mol. Sci. 2021, 22, 7121. [Google Scholar] [CrossRef]
- Bittigau, P.; Sifringer, M.; Genz, K.; Reith, E.; Pospischil, D.; Govindarajalu, S.; Dzietko, M.; Pesditschek, S.; Mai, I.; Dikranian, K.; Olney, J.W.; Ikonomidou, C. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc. Natl. Acad. Sci. USA. 2002, 99, 15089–94. [Google Scholar] [CrossRef] [PubMed]
- Forcelli, P.A.; Janssen, M.J.; Vicini, S.; Gale, K. Neonatal exposure to antiepileptic drugs disrupts striatal synaptic development. Ann. Neurol. 2012, 72, 363–72. [Google Scholar] [CrossRef]
- Noguchi, K.K.; Fuhler, N.A.; Wang, S.H.; Capuano, S. 3rd; Brunner, K.R.; Larson, S.; Crosno, K.; Simmons, H.A.; Mejia, A.F.; Martin, L.D.; Dissen, G.A.; Brambrink, A.; Ikonomidou, C. 3rd; Brunner, K.R.; Larson, S.; Crosno, K.; Simmons, H.A.; Mejia, A.F.; Martin, L.D.; Dissen, G.A.; Brambrink, A.; Ikonomidou, C. Brain pathology caused in the neonatal macaque by short and prolonged exposures to anticonvulsant drugs. Neurobiol. Dis. 2021, 149, 105245. [Google Scholar] [CrossRef]
- Lal, D.; Reinthaler, E.M.; Altmüller, J.; Toliat, M.R.; Thiele, H.; Nürnberg, P.; Lerche, H.; Hahn, A.; Møller, R.S.; Muhle, H.; et al. RBFOX1 and RBFOX3 mutations in rolandic epilepsy. PLoS One. 2013, 8, e73323. [Google Scholar] [CrossRef]
- Bobbili, D.R.; Lal, D.; May, P.; Reinthaler, E.M.; Jabbari, K.; Thiele, H.; Nothnagel, M.; Jurkowski, W.; Feucht, M.; Nürnberg, P.; et al. Exome-wide analysis of mutational burden in patients with typical and atypical Rolandic epilepsy. Eur. J. Hum. Genet. 2018, 26, 258–264. [Google Scholar] [CrossRef]
- Numis, A.L.; da Gente, G.; Sherr, E.H.; Glass, H.C. Whole-exome sequencing with targeted analysis and epilepsy after acute symptomatic neonatal seizures. Pediatr. Res. 2022, 91, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Al-Abdulla, N.A.; Martin, L.J. Apoptosis of retrogradely degenerating neurons occurs in association with the accumulation of perikaryal mitochondria and oxidative damage to the nucleus. Am. J. Pathol. 1998, 153, 447–456. [Google Scholar] [PubMed]
- Martin, L.J.; Brambrink, A.M.; Price, A.C.; Kaiser, A.; Agnew, D.M.; Ichord, R.N.; Traystman, R.J. Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol. Dis. 2000, 7, 169–191. [Google Scholar]
- Salinas-Zeballos, M.E.; Zeballos, G.A.; Gootman, P.M. A stereotaxic atlas of the developing swine (Sus scrofa) forebrain. In, Swine in biomedical research, Tumbleson ME, editor, Plenum Press, New York, USA, 1986, pp. 887–906.
- Rose, V.C.; Shaffner, D.H.; Gleason, C.A.; Koehler, R.C.; Traystman, R.J. Somatosensory evoked potential and brain water content in post-asphyxic immature piglets. Pediatr. Res. 1995, 37, 661–6. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Kaiser, A.; Price, A.C. Motor neuron degeneration after sciatic nerve avulsion in adult rat evolves with oxidative stress and is apoptosis. J. Neurobiol. 1999, 40, 185–201. [Google Scholar] [CrossRef]
- Natale, J.E.; Cheng, Y.; Martin, L.J. Thalamic neuron apoptosis emerges rapidly after cortical damage in immature mice. Neuroscience. 2002, 112, 665–676. [Google Scholar] [CrossRef]
- Mesulam, M.M. Tetramethyl benzidine for horseradish peroxidase neurohistochemistry: a non-carcinogenic blue reaction product with superior sensitivity for visualizing neural afferents and efferents. J. Histochem. Cytochem. 1978, 26, 106–17. [Google Scholar] [CrossRef]
- Lee, J.K.; Santos, P.T.; Chen, M.W.; et al. Combining hypothermia and oleuropein subacutely protects subcortical white matter in a swine model of neonatal hypoxic-ischemic encephalopathy. J. Neuropathol. Exp. Neurol. 2021, 80, 182–198. [Google Scholar] [CrossRef]
- El Demerdash, N.; Chen, M.W.; O'Brien, C.E.; et al. Oleuropein activates neonatal neocortical proteasomes, but proteasome gene targeting by AAV9 is variable in a clinically relevant piglet model of brain hypoxia-ischemia and hypothermia. Cells. 2021, 10. [Google Scholar] [CrossRef]
- Chen, M.W.; Santos, P.; Kulikowicz, E.; Koehler, R.C.; Lee, J.K.; Martin, L.J. Targeting the mitochondrial permeability transition pore for neuroprotection in a piglet model of neonatal hypoxic-ischemic encephalopathy. J. Neurosci. Res. 2021, 99, 1550–1564. [Google Scholar] [CrossRef]
- Dudgeon, D.L.; Randall, P.A.; Hill, R.B.; McAfee, J.G. Mild hypothermia: its effect on cardiac output and regional perfusion in the neonatal piglet. J. Pediatr. Surg. 1980, 15, 805–10. [Google Scholar] [CrossRef] [PubMed]
- Busija, D.W.; Leffler, C.W. Hypothermia reduces cerebral metabolic rate and cerebral blood flow in newborn pigs. Am. J. Physiol. 1987, 253(4 Pt 2) Pt 2, H869–73. [Google Scholar] [CrossRef]
- Laptook, A.R.; Corbett, R.J.; Sterett, R.; Burns, D.K.; Tollefsbol, G.; Garcia, D. Modest hypothermia provides partial neuroprotection for ischemic neonatal brain. Pediatr. Res. 1994, 35(4 Pt 1) Pt 1, 436–42. [Google Scholar] [CrossRef]
- Carrasco, M.; Perin, J.; Jennings, J.M.; et al. Cerebral autoregulation and conventional and diffusion tensor imaging magnetic resonance imaging in neonatal hypoxic-ischemic encephalopathy. Pediatr. Neurol. 2018, 82, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, M.M.; Tekes, A.; Perin, J.; et al. Later cooling within 6 h and temperatures outside 33-34 degrees C are not associated with dysfunctional autoregulation during hypothermia for neonatal encephalopathy. Pediatr. Res. 2021, 89, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Wylie, T.; Sandhu, D.S.; Murr, N. Status epilepticus. In, StatPearls [Internet]. Treasure Island, Florida, StatPearls Publishing, 2022.
- Endisch, C.; Westhall, E.; Kenda, M.; Streitberger, K.J.; Kirkegaard, H.; Stenzel, W.; Storm, C.; Ploner, C.J.; Cronberg, T.; Friberg, H.; Englund, E.; Leithner, C. Hypoxic-ischemic encephalopathy evaluated by brain autopsy and neuroprognostication after cardiac arrest. JAMA Neurol. 2020, 77, 1430–1439. [Google Scholar] [CrossRef]
- O'Brien, C.E.; Santos, P.T.; Kulikowicz, E.; et al. Hypoxia-ischemia and hypothermia independently and interactively affect neuronal pathology in neonatal piglets with short-term recovery. Dev. Neurosci. 2019, 41, 17–33. [Google Scholar] [CrossRef]
- Liu, D.; Jiang, D.; Tekes, A.; Kulikowicz, E.; Martin, L.J.; Lee, J.K.; Liu, P.; Qin, Q. Multi-parametric evaluation of cerebral hemodynamics in neonatal piglets using non-contrast-enhanced magnetic resonance imaging methods. J. Magn. Reason. Imaging. 2021, 54, 1053–1065. [Google Scholar] [CrossRef]
- Martin, L.J. Neuronal cell death in nervous system development, disease, and injury. Int J. Mol. Med. 2001, 7, 455–478. [Google Scholar] [CrossRef]
- Agnew, D.M.; Koehler, R.C.; Guerguerian, A.M.; Shaffner, D.H.; Traystman, R.J.; Martin, L.J.; Ichord, R.N. Hypothermia for 24 hours after asphyxic cardiac arrest in piglets provides striatal neuroprotection that is sustained 10 days after rewarming. Pediatr. Res. 2003, 54, 1–10. [Google Scholar] [CrossRef]
- Mueller-Burke, D.; Koehler, R.C.; Martin, L.J. Rapid NMDA receptor phosphorylation and oxidative stress precede striatal neurodegeneration after hypoxic ischemia in newborn piglets and are attenuated with hypothermia. Int. J. Devl. Neurosci. 2008, 26, 67–76. [Google Scholar] [CrossRef]
- Martin, L.J.; Al-Abdulla, N.A.; Brambrink, A.M.; Kirsch, J.R.; Sieber, F.E.; Portera-Cailliau, C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res. Bull. 1998, 46, 281–309. [Google Scholar] [CrossRef]
- Ingvar, M.; Morgan, P.F.; Auer, R.N. The nature and timing of excitotoxic neuronal necrosis in the cerebral cortex, hippocampus and thalamus due to flurothyl-induced status epilepticus. Acta Neuropathol. 1988, 75, 362–9. [Google Scholar] [CrossRef] [PubMed]
- Rubleva, E.Y.; Savulev, Y.I.; Pylaev, A.S. Comparative electron microscopic and autoradiographic studies of "dark" and "light" neurons of the cerebral cortex. Neurosci. Behav. Physiol. 1980, 10, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Doebler, J.A.; Anthony, A. Cytophotometric analysis of neuronal chromatin and RNA changes in oxotremorine-treated rats. Proc. Soc. Exp. Biol. Med. 1986, 181, 41–48. [Google Scholar] [CrossRef]
- Martin, L.J.; Brambrink, A.M.; Lehmann, C.; Portera-Cailliau, C.; Koehler, R.; Rothstein, J.; Traystman, R.J. Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann. Neurol. 1997, 42, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Katzenelson, A.; Koehler, R.C.; Chang, Q. The olfactory bulb in newborn piglet is a reservoir of neural stem and progenitor cells. PLoS One. 2013, 8, e81105. [Google Scholar] [CrossRef]
- Lee, J.K.; Wang, B.; Reyes, M.; et al. Hypothermia and rewarming activate a macroglial unfolded protein response independent of hypoxic-ischemic brain injury in neonatal piglets. Dev. Neurosci. 2016, 38, 277–294. [Google Scholar] [CrossRef]
- Lee, J.K.; Liu, D.; Jiang, D.; et al. Fractional anisotropy from diffusion tensor imaging correlates with acute astrocyte and myelin swelling in neonatal swine models of excitotoxic and hypoxic-ischemic brain injury. J. Comp. Neurol. 2021, 529, 2750–2770. [Google Scholar] [CrossRef]
- Martin, L.J. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 2000, 7, 613–622. [Google Scholar] [CrossRef]
- Martin, L.J.; Price, A.C.; McClendon, K.B.; Al-Abdulla, N.A.; Subramaniam, J.R.; Wong, P.C.; Liu, Z. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J. Neurochem. 2003, 85, 234–247. [Google Scholar] [CrossRef] [PubMed]
- von Economo, C.; Koskinas, G.N. Atlas of cytoarchitectonics of the adult human cerebral cortex. Triarhou LC, editor and translator, 1926, 182 pp.
- Zilles, K.; Amunts, K. Centenary of Brodmann's map--conception and fate. Nat. Rev. Neurosci. 2010, 11, 139–45. [Google Scholar] [CrossRef] [PubMed]
- Elston, G.N.; Garey, L.J. The cytoarchitectonic map of Korbinian Brodmann: arealisation and circuit specialization. In: Geyer S, Turner R, editors. Microstructural parcellation of the human cerebral cortex. Springer, Berlin, Heidelberg. 2013. [Google Scholar] [CrossRef]
- Adrian, E.D. Afferent areas in the brain of ungulates. Brain. 1943, 66, 89–103. [Google Scholar] [CrossRef]
- Sauleau, P.; Lapouble, E.; Val-Laillet, D.; Malbert, C.H. The pig model in brain imaging and neurosurgery. Animal. 2009, 3, 1138–51. [Google Scholar] [CrossRef]
- Martin, L.J. DNA methylation in motor neuron biology and epigeneticopathy in ALS. In, Handbook of Clinical Neurology, Szyf M, editor, 2023, Elsevier.
- Mullen, R.J.; Buck, C.R.; Smith, A.M. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992, 116, 201–11. [Google Scholar] [CrossRef]
- Rutherford, M.A.; Azzopardi, D.; Whitelaw, A.; Cowan, F.; Renowden, S.; Edwards, A.D.; Thoresen, M. Mild hypothermia and the distribution of cerebral lesions in neonates with hypoxic-ischemic encephalopathy. Pediatrics. 2005, 116, 1001–6. [Google Scholar] [CrossRef]
- Krieg, W.J. Connections of the cerebral cortex; the albino rat; topography of cortical areas. J. Comp. Neurol. 1946, 84, 221–275. [Google Scholar] [CrossRef]
- Minervini, S.; Accogli, G.; Pirone, A.; Graïc, J.M.; Cozzi, B.; Desantis, S. Brain mass and encephalization quotients in the domestic industrial pig (Sus scrofa). PLoS One. 2016, 11, e0157378. [Google Scholar] [CrossRef]
- White, L.E.; Andrews, T.J.; Hulette, C.; Richards, A.; Groelle, M.; Paydarfar, J.; Purves, D. Structure of the human sensorimotor system. I: Morphology and cytoarchitecture of the central sulcus. Cereb. Cortex. [CrossRef]
- Martin, L.J.; Adams, D.A.; Niedzwiecki, M.V.; Wong, M. Aberrant DNA and RNA methylation occur in spinal cord and skeletal muscle of human SOD1 mouse models of ALS and in human ALS: targeting DNA methylation is therapeutic. Cells. 2022, 11, 3448. [Google Scholar] [CrossRef]
- Craner, S.L.; Ray, R.H. Somatosensory cortex of the neonatal pig: I. Topographic organization of the primary somatosensory cortex (SI). J. Comp. Neurol. 1991; 306, 24–38. [Google Scholar] [CrossRef]
- Bernard, P.B.; Benke, T.A. Early life seizures: evidence for chronic deficits linked to autism and intellectual disability across species and models. Exp. Neurol. 2015, 263, 72–78. [Google Scholar]
- Cornejo, B.J.; Mesches, M.H.; Coultrap, S.; Browning, M.D.; Benke, T.A. A single episode of neonatal seizures permanently alters glutamatergic synapses. Ann. Neurol. 2007, 61, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.K.; Hwang-Bo, S.; Seo, Y.M.; Youn, Y.A. Clinical seizures and unfavorable brain MRI patterns in neonates with hypoxic ischemic encephalopathy. Medicine (Baltimore). 2021, 100, e25118. [Google Scholar] [CrossRef]
- Defelipe, J. The evolution of the brain, the human nature of cortical circuits, and intellectual creativity. Front. Neuroanat. 2011, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Gunn, A.J.; Gunn, T.R.; de Haan, H.H.; Williams, C.E.; Gluckman, P.D. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J. Clin. Invest. 1997, 99, 248–56. [Google Scholar] [CrossRef] [PubMed]
- Drury, P.P.; Davidson, J.O.; van den Heuij, L.G.; Wassink, G.; Gunn, E.R.; Booth, L.C.; Bennet, L.; Gunn, A.J. Status epilepticus after prolonged umbilical cord occlusion is associated with greater neural injury in fetal sheep at term-equivalent. PLoS One. 2014, 9, e96530. [Google Scholar] [CrossRef]
- Beaulieu-Laroche, L.; Brown, N.J.; Hansen, M.; Toloza, E.H.S.; Sharma, J.; Williams, Z.M.; Frosch, M.P.; Cosgrove, G.R.; Cash, S.S.; Harnett, M.T. Allometric rules for mammalian cortical layer 5 neuron biophysics. Nature. 2021, 600. 274-278. [CrossRef]
- Gailus, B.; Naundorf, H.; Welzel, L.; et al. Long-term outcome in a noninvasive rat model of birth asphyxia with neonatal seizures: Cognitive impairment, anxiety, epilepsy, and structural brain alterations. Epilepsia. 2021, 62, 2826–2844. [Google Scholar] [CrossRef]
- Wagley, P.K.; Williamson, J.; Skwarzynska, D.; Kapur, J.; Burnsed, J. Continuous Video Electroencephalogram during hypoxia-ischemia in neonatal mice. J. Vis. Exp. 2020, 160. [Google Scholar]
- Rensing, N.; Moy, B.; Friedman, J.L.; Galindo, R.; Wong, M. Longitudinal analysis of developmental changes in electroencephalography patterns and sleep-wake states of the neonatal mouse. PLoS One. 2018, 13, e0207031. [Google Scholar] [CrossRef]
- Pirzada, N. The ethical dilemma of non-human primate use in biomedical research. Voices in Bioethics. 2022, 8. [Google Scholar] [CrossRef]
- Martin, L.J.; Spicer, D.M.; Lewis, M.H.; Gluck, J.P.; Cork, L.C. Social deprivation of infant rhesus monkeys alters the chemoarchitecture of the brain: I. Subcortical regions. J. Neurosci. 1991, 11, 3344–58. [Google Scholar] [CrossRef]
- Martin, L.J. , Cork, L.C. The non-human primate striatum undergoes marked prolonged remodeling during postnatal development. Front. Cell. Neurosci. 2014, 8, 294. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kulikowicz, E.; Santos, P.T.; Koehler, R.C.; Martin, L.J.; Lee, J.K. Spatial T-maze identifies cognitive deficits in piglets 1 month after hypoxia-ischemia in a model of hippocampal pyramidal neuron loss and interneuron attrition. Behav. Brain Res. 2019, 369, 111921. [Google Scholar] [CrossRef] [PubMed]
- Hunt, R.W.; Liley, H.G.; Wagh, D.; Schembri, R.; Lee, K.J.; Shearman, A.D.; Francis-Pester, S.; deWaal, K.; Cheong, J.Y.L. ; Olischar. M.; et al. Newborn electrographic seizure trial investigators. Effect of treatment of clinical seizures vs electrographic seizures in full-term and near-term neonates: A randomized clinical trial. JAMA Netw. Open. 2021, 4, e2139604. [Google Scholar] [CrossRef]
- Sun, H.; Juul, H.M.; Jensen, F.E. Models of hypoxia and ischemia-induced seizures. J. Neurosci. Methods. 2016, 260, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Glass, H.C.; Nash, K.B.; Bonifacio, S.L.; et al. Seizures and magnetic resonance imaging-detected brain injury in newborns cooled for hypoxic-ischemic encephalopathy. J. Pediatr. 2011, 159, 731-735.e731.
- Lee, J.K.; Perin, J.; Parkinson, C.; O'Connor, M.; Gilmore, M.M.; Reyes, M.; Armstrong, J.; Jennings, J.M.; Northington, F.J.; Chavez-Valdez, R. Relationships between cerebral autoregulation and markers of kidney and liver injury in neonatal encephalopathy and therapeutic hypothermia. J. Perinatol. 2017, 37, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Mallet, M.L. Pathophysiology of accidental hypothermia. QJM. 2002, 95, 775–785. [Google Scholar] [CrossRef]
- Escalda, A.; Marques, M.; Silva-Carvalho, L.; et al. Hypothermia-induced haemostatic and biochemical phenomena. An experimental model. Platelets. 1993, 4, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Yang, J.J.; Liou, Y.M. Early blood glucose level post-admission correlates with the outcomes and oxidative stress in neonatal hypoxic-ischemic encephalopathy. Antioxidants (Basel). 2021, 11. [Google Scholar] [CrossRef]
- Thornton, P.S.; Stanley, C.A.; De Leon, D.D.; et al. Recommendations from the pediatric endocrine society for evaluation and management of persistent hypoglycemia in neonates, infants, and children. J. Pediatr. 2015, 167, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, W.; Ishida, A.; Lange, M.S.; Gabrielson, K.L.; Wilson, M.A.; Martin, L.J.; Blue, M.E.; Johnston, M.V. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J. Neurosci. 2000, 20, 7994–8004. [Google Scholar] [CrossRef] [PubMed]
- Northington, F.J.; Ferriero, D.M.; Graham, E.M.; Traystman, R.J.; Martin, L.J. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol. Dis. 2001, 8, 207–219. [Google Scholar] [CrossRef]
- Steinman, K.J.; Gorno-Tempini, M.L.; Glidden, D.V.; Kramer, J.H.; Miller, S.P.; Barkovich, A.J.; Ferriero, D.M. Neonatal watershed brain injury on magnetic resonance imaging correlates with verbal IQ at 4 years. Pediatrics. 2009, 123, 1025–30. [Google Scholar] [CrossRef] [PubMed]
- Kringelbach, M.L.; Rolls, E.T. The functional neuroanatomy of the human orbitofrontal cortex: evidence from neuroimaging and neuropsychology. Prog. Neurobiol. 2004, 72, 341–72. [Google Scholar] [CrossRef] [PubMed]
- Wikenheiser, A.M.; Schoenbaum, G. Over the river, through the woods: cognitive maps in the hippocampus and orbitofrontal cortex. Nat. Rev. Neurosci. 2016, 17, 513–23. [Google Scholar] [CrossRef] [PubMed]
- Fettes, P.W.; Moayedi, M.; Dunlop, K.; Mansouri, F.; Vila-Rodriguez, F.; Giacobbe, P.; Davis, K.D.; Lam, R.W.; Kennedy, S.H.; Daskalakis, Z.J.; Blumberger, D.M.; Downar, J. Abnormal functional connectivity of frontopolar subregions in treatment-nonresponsive major depressive disorder. Biol. Psychiatry Cogn. Neurosci. Neuroimaging. 2018, 3, 337–347. [Google Scholar] [CrossRef]
- Akbarian, S.; Grüsser, O.J.; Guldin, W.O. Corticofugal connections between the cerebral cortex and brainstem vestibular nuclei in the macaque monkey. J. Comp. Neurol. 1994, 339, 421–37. [Google Scholar] [CrossRef]
- Sherrington, C.S. The integrative actions of the nervous system. Yale University Press, 1906, New Haven, CT.
- Sukoff, M.H.; Ragatz, R.E. Cerebellar stimulation for chronic extensor-flexor rigidity and opisthotonus secondary to hypoxia. Report of two cases. J. Neurosurg. 1980, 53, 391–6. [Google Scholar] [CrossRef]
- Stitt, I.M.; Wellings, T.P.; Drury, H.R.; Jobling, P.; Callister, R.J.; Brichta, A.M.; Lim, R. Properties of Deiters' neurons and inhibitory synaptic transmission in the mouse lateral vestibular nucleus. J. Neurophysiol. 2022, 128, 131–147. [Google Scholar] [CrossRef]
- Armstrong, D.M. The supraspinal control of mammalian locomotion. J. Physiol. 1988, 405, 1–37. [Google Scholar] [CrossRef]
- Wilson, V.J.; Zarzecki, P.; Schor. R.H.; Isu, N.; Rose, P.K.; Sato, H.; Thomson, D.B.; Umezaki, T. Cortical influences on the vestibular nuclei of the cat. Exp. Brain Res. 1999, 125, 1–13. [Google Scholar] [CrossRef]
- Pavlidis, E.; Cantalupo, G.; Cattani, L.; Tassinari, C.A.; Pisani, F. Neonatal seizure automatism and human inborn pattern of quadrupedal locomotion. Gait Posture. 2016, 49, 232–234. [Google Scholar] [CrossRef]
- Silvestri, R.; Walters, A.S. Rhythmic movements in sleep disorders and in epileptic seizures during sleep. Sleep Science Practice. 2020, 4, 5. [Google Scholar] [CrossRef]
- Connors, B.W.; Pinto, D.J.; Telfeian, A.E. Local pathways of seizure propagation in neocortex. Int. Rev. Neurobiol. 2001, 45, 527–46. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.R.E.; Zhao, H.T.; Park, J.M.; Dipoppa, M.; Monsalve-Mercado, M.M.; Dahan, J.B.; Rodgers, C.C.; Lejeune, A.; Hillman, E.M.C.; Miller, K.D.; Bruno, R.M.; Polleux, F. A human-specific modifier of cortical connectivity and circuit function. Nature. 2021, 599, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Multani, P.; Myers, R.H.; Blume, H.W.; Schomer, D.L.; Sotrel, A. Neocortical dendritic pathology in human partial epilepsy: a quantitative Golgi study. Epilepsia. 1994, 35, 728–36. [Google Scholar] [CrossRef]
- Du, F.; Eid, T.; Lothman, E.W.; Köhler, C.; Schwarcz, R. Preferential neuronal loss in layer III of the medial entorhinal cortex in rat models of temporal lobe epilepsy. J. Neurosci. 1995, 15, 6301–13. [Google Scholar] [CrossRef]
- Pfisterer, U.; Petukhov, V.; Demharter, S.; Meichsner, J.; Thompson, J.J.; Batiuk, M.Y.; Asenjo-Martinez, A.; Vasistha, N.A.; Thakur, A.; Mikkelsen, J. , et al. Identification of epilepsy-associated neuronal subtypes and gene expression underlying epileptogenesis. Nat. Commun. 2020, 11, 5038. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, B.; Lee, J.H.; Armstrong, J.S.; Kulikowicz, E.; Bhalala, U.S.; Martin, L.J.; Koehler, R.C.; Yang, Z.J. Additive neuroprotection of a 20-HETE inhibitor with delayed therapeutic hypothermia after hypoxia-ischemia in neonatal piglets. Dev. Neurosci. 2015, 37, 376–389. [Google Scholar] [CrossRef]
- Martin, L.J. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals (Basel). 2010, 3, 839–915. [Google Scholar] [CrossRef]
- Martin, L.J; Chen, K.; Liu, Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J. Neurosci. 2005, 25, 6449–59. [Google Scholar] [CrossRef]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–46. [Google Scholar] [CrossRef]
- Martin, L.J.; Fancelli, D.; Wong, M.; Niedzwiecki, M.; Ballarini, M.; Plyte, S.; Chang, Q. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 433. [Google Scholar] [CrossRef]
- Kim, K.K.; Adelstein, R.S.; Kawamoto, S. Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J. Biol. Chem. 2009, 284, 31052–61. [Google Scholar] [CrossRef] [PubMed]
- Salamon, I.; Rasin, M.R. Evolution of the neocortex through RNA-binding proteins and post-transcriptional regulation. Front. Neurosci. 2022; 10, 803107. [Google Scholar] [CrossRef]
- Dent, M.A.; Segura-Anaya, E.; Alva-Medina, J.; Aranda-Anzaldo, A. NeuN/Fox-3 is an intrinsic component of the neuronal nuclear matrix. FEBS Lett. 2010, 584, 2767–71. [Google Scholar] [CrossRef]
- Duan, W.; Zhang, Y.P.; Hou, Z.; Huang, C.; Zhu, H.; Zhang, C.Q.; Yin, Q. Novel Insights into NeuN: from Neuronal Marker to Splicing Regulator. Mol. Neurobiol. 2016, 53, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L.; Wexler, E.; Wahnich, A.; Friedrich, T.; Vijayendran, C.; Gao, F.; Parikshak, N.; Konopka, G.; Geschwind, D.H. RBFOX1 regulates both splicing and transcriptional networks in human neuronal development. Hum. Mol. Genet. 2012, 21, 4171–86. [Google Scholar] [CrossRef] [PubMed]
- Utami, K.H.; Hillmer, A.M.; Aksoy, I.; Chew, E.G.; Teo, A.S.; Zhang, Z.; Lee, C.W.; Chen, P.J.; Seng, C.C.; Ariyaratne, P.N.; et al. Detection of chromosomal breakpoints in patients with developmental delay and speech disorders. PLoS One. 2014, 9, e90852. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.J.; Morgan, A.T.; Perez, E.R.; Scheffer, I.E. New genes for focal epilepsies with speech and language disorders. Curr. Neurol. Neurosci. Rep. 2015, 15, 35. [Google Scholar] [CrossRef]
- Wang, H.Y.; Hsieh, P.F.; Huang, D.F.; Chin, P.S.; Chou, C.H.; Tung, C.C.; Chen, S.Y.; Lee, L.J.; Gau, S.S.; Huang, H.S. RBFOX3/NeuN is required for hippocampal circuit balance and function. Sci. Rep. 2015, 5, 17383. [Google Scholar] [CrossRef]
- Huang, D.F.; Lee, C.Y.; Chou, M.Y.; Yang, T.Y.; Cao, X.; Hsiao, Y.H.; Wu, R.N.; Lien, C.C.; Huang, Y.S.; Huang, H.P.; Gau, S.S.; Huang, H.S. Neuronal splicing regulator RBFOX3 mediates seizures via regulating Vamp1 expression preferentially in NPY-expressing GABAergic neurons. Proc. Natl. Acad. Sci. USA. 2022, 119, e2203632119. [Google Scholar] [CrossRef]
- Jacko, M.; Weyn-Vanhentenryck, S.M.; Smerdon, J.W.; Yan, R.; Feng, H.; Williams, D.J.; Pai, J.; Xu, K.; Wichterle, H.; Zhang, C. Rbfox splicing factors promote neuronal maturation and axon initial segment assembly. Neuron. 2018, 97, 853-868.e6. [CrossRef]
- Maxeiner, S.; Glassmann, A.; Kao, H.T.; Schilling, K. The molecular basis of the specificity and cross-reactivity of the NeuN epitope of the neuron-specific splicing regulator, Rbfox3. Histochem. Cell Biol. 2014, 141, 43–55. [Google Scholar] [CrossRef]
- Dredge, B.K.; Jensen, K.B. NeuN/Rbfox3 nuclear and cytoplasmic isoforms differentially regulate alternative splicing and nonsense-mediated decay of Rbfox2. PLoS One. 2011, 6, e21585. [Google Scholar] [CrossRef] [PubMed]
- Damianov, A.; Ying, Y.; Lin, C.H.; Lee, J.A.; Tran, D.; Vashisht, A.A.; Bahrami-Samani, E.; Xing, Y.; Martin, K.C.; Wohlschlegel, J.A.; Black, D.L. Rbfox proteins regulate splicing as part of a large multiprotein complex LASR. Cell. 2016, 165, 606–19. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; Engel, K.L.; Ramamurthy, A.; Cochran, J.N.; Roovers, J.; Stamberger, H.; Lim, N.; Schneider, A.L.; Hollingsworth, G. ; Holder. D.H.; et al. Aberrant inclusion of a poison exon causes Dravet syndrome and related SCN1A-associated genetic epilepsies. Am. J. Hum. Genet. 2018, 103, 1022–1029. [Google Scholar] [CrossRef]
- Halász, P.; Kelemen, A.; Rosdy, B.; Rásonyi, G.; Clemens, B.; Szűcs, A. Perisylvian epileptic network revisited. Seizure. 2019, 65, 31–41. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Surgical placement of electrodes for cEEG in neonatal piglets. A. Cranial screw placement. B. EEG electrode array attachment. C. Isolated EEG electrode array showing the round transmitter (on left) and the naked electrodes and electrodes with attached cranial screws and acrylic cement placodes. D. Exposed brain of a 4%PF perfusion fixed piglet with calvarium removed and intact dura mater showing dural placement sites of cranial screws seen as focal hemorrhagic damage. E. The same brain shown in D with dura mater removed and no damage to the underlying neocortical parenchyma. The optimal quality of the brain perfusion fixation is also demonstrated by the absence of residual blood in cortical vessels and discrete contouring of the cortical surface.
Figure 1.
Surgical placement of electrodes for cEEG in neonatal piglets. A. Cranial screw placement. B. EEG electrode array attachment. C. Isolated EEG electrode array showing the round transmitter (on left) and the naked electrodes and electrodes with attached cranial screws and acrylic cement placodes. D. Exposed brain of a 4%PF perfusion fixed piglet with calvarium removed and intact dura mater showing dural placement sites of cranial screws seen as focal hemorrhagic damage. E. The same brain shown in D with dura mater removed and no damage to the underlying neocortical parenchyma. The optimal quality of the brain perfusion fixation is also demonstrated by the absence of residual blood in cortical vessels and discrete contouring of the cortical surface.
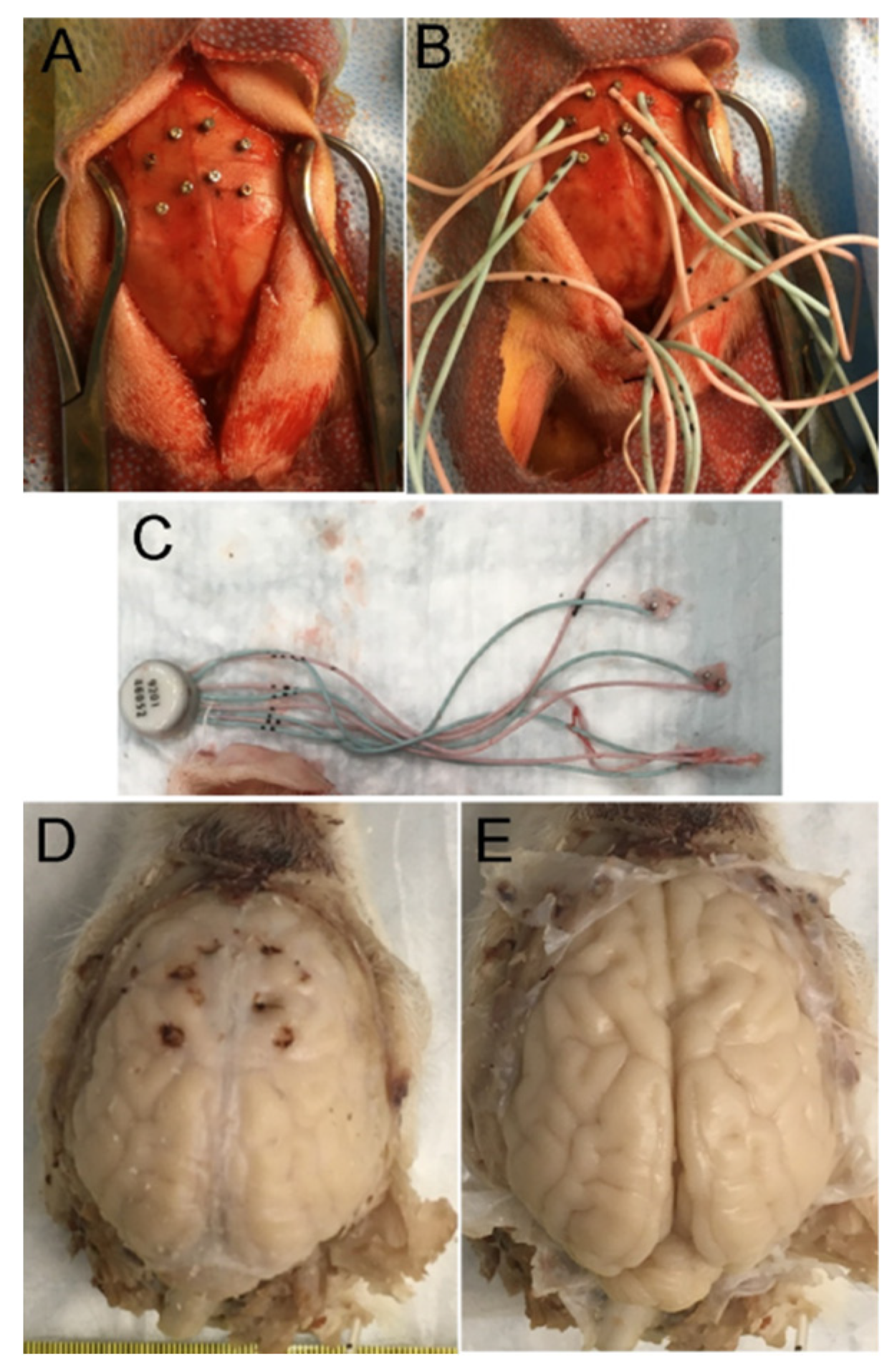
Figure 2.
Piglet brain levels used for neuron counting in three neuroanatomic levels, H&E counting strategies, and neuronal injury classification. A-C. H&E-stained hemibrain sections at the levels of frontal cortex (A), posterior frontal-anterior parietal cortex and striatum (B), mid-parietal cortex, posterior striatum, and thalamus (C). Brain regions pertinent to the study are identified. Medial is at left. Superior is at top. D-F. Neurons were counted in rows of adjacent microscope fields in cortical layers II-VI or in specific layers in inferior parietal cortex. In frontal cortex (A,D) counts were done in the medial-most gyrus comprised of the anterior motor cortex dorsally and the ventrally located gyrus rectus and laterally in the inferior frontal cortex. At a striatal level (B,E) counts were done in the medial and lateral banks of the motor cortex and somatosensory cortex and in layers II, III, and V of inferior parietal cortex. At a thalamic level (C,F) counts were done in the medial and lateral banks of the posterior motor cortex and somatosensory cortex. G-K. Neuron morphology classification by H&E staining (see methods for descriptions): normal neuron (G, arrow), injured neuron (H, arrow), ischemic-necrotic degenerating neurons (I, arrows), apoptosis-necrosis continuum degenerating neuron (J, arrow), and apoptotic neuron (K, arrow). Scale bars (in µm): G, 10; H, 10; I, 8.75; J, 10; K, 25.
Figure 2.
Piglet brain levels used for neuron counting in three neuroanatomic levels, H&E counting strategies, and neuronal injury classification. A-C. H&E-stained hemibrain sections at the levels of frontal cortex (A), posterior frontal-anterior parietal cortex and striatum (B), mid-parietal cortex, posterior striatum, and thalamus (C). Brain regions pertinent to the study are identified. Medial is at left. Superior is at top. D-F. Neurons were counted in rows of adjacent microscope fields in cortical layers II-VI or in specific layers in inferior parietal cortex. In frontal cortex (A,D) counts were done in the medial-most gyrus comprised of the anterior motor cortex dorsally and the ventrally located gyrus rectus and laterally in the inferior frontal cortex. At a striatal level (B,E) counts were done in the medial and lateral banks of the motor cortex and somatosensory cortex and in layers II, III, and V of inferior parietal cortex. At a thalamic level (C,F) counts were done in the medial and lateral banks of the posterior motor cortex and somatosensory cortex. G-K. Neuron morphology classification by H&E staining (see methods for descriptions): normal neuron (G, arrow), injured neuron (H, arrow), ischemic-necrotic degenerating neurons (I, arrows), apoptosis-necrosis continuum degenerating neuron (J, arrow), and apoptotic neuron (K, arrow). Scale bars (in µm): G, 10; H, 10; I, 8.75; J, 10; K, 25.
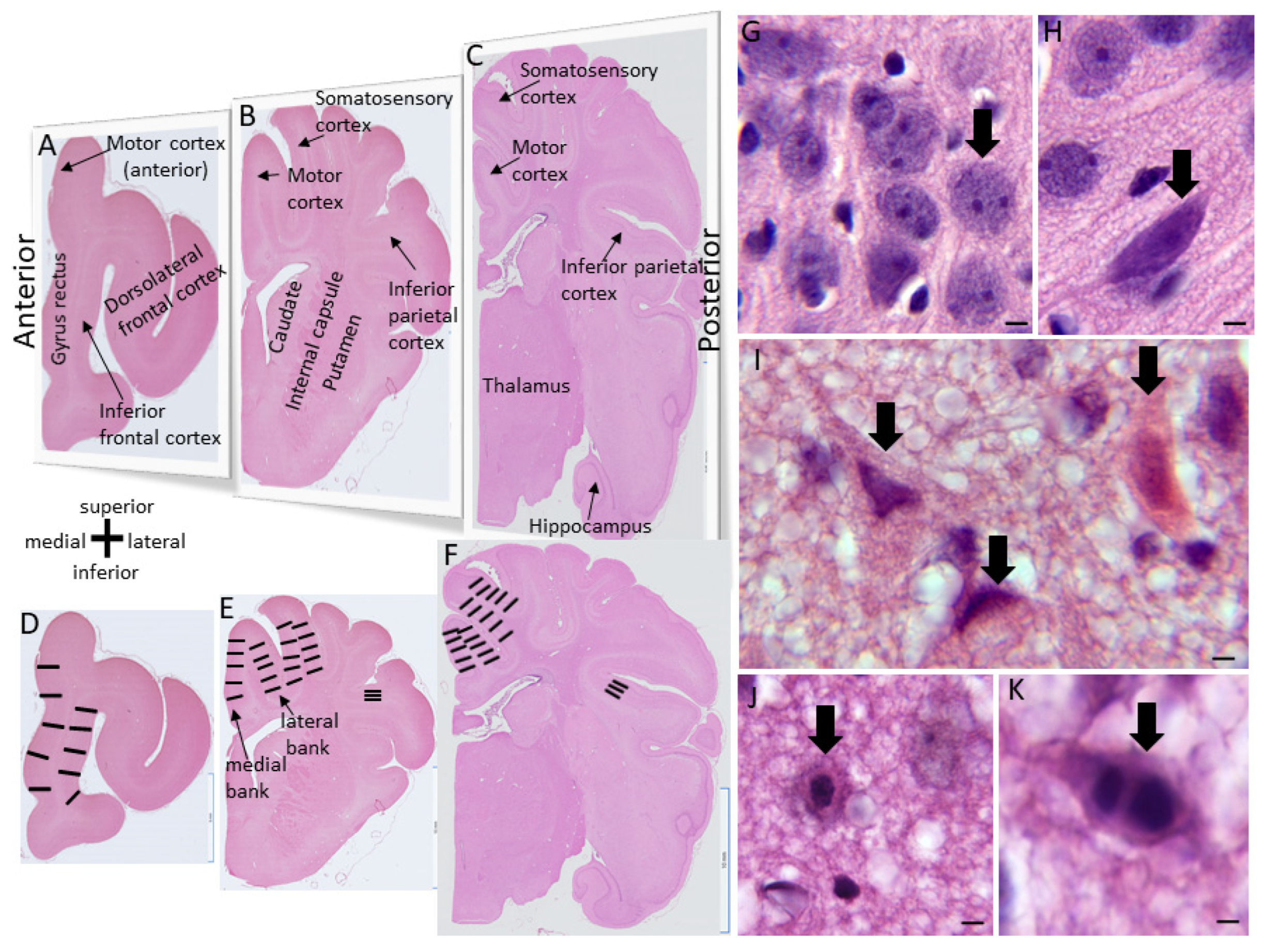
Figure 3.
Rbfox3 detection in piglet neocortex with NeuN antibody and immunoperoxidase immunohistochemistry. A. In sham piglet somatosensory cortex, RbFox3-positive cells have a laminar distribution (brown immunoreactivity with blue Nissl counterstaining). Numbers identify the cortical layers. The thick layer IV is discernable. Inset shows antibody specificity in western blot of crude homogenates of piglet somatosensory cortex with prominent band detection at 46-48 kDa. B. With a prominent layer V and an attenuated layer IV, RbFox3 immunostaining in sham piglet motor cortex is distinct from that seen in the somatosensory cortex. Inset shows the nuclear and cytoplasmic staining for Rbfox3 in piglet neocortical neurons. C. In HI-NT piglets, Rbfox3 can be found selectively depleted in layer II neurons of somatosensory cortex, yet, across the sulcus (asterisk), the motor cortex layer II neurons are positive. Other pyramidal neurons (inset) in layers III-V have nuclear clumping of Rbfox3 when undergoing ischemic-necrosis. Hatched box is shown at higher magnification in D. D. HI-NT piglet with selective depletion of Rbfox3 in layer II, yet the neurons appear morphologically normal. E. Somatosensory cortex layer II of an HI-HT piglet with depletion of Rbfox3 in some neurons (solid arrow) and partial rescue of Rbfox3 positivity in other neurons (solid thin arrows). Scale bars (in µm): A (same for B,C), 160; B inset, 9; C inset, 10; D, 18; E, 7.
Figure 3.
Rbfox3 detection in piglet neocortex with NeuN antibody and immunoperoxidase immunohistochemistry. A. In sham piglet somatosensory cortex, RbFox3-positive cells have a laminar distribution (brown immunoreactivity with blue Nissl counterstaining). Numbers identify the cortical layers. The thick layer IV is discernable. Inset shows antibody specificity in western blot of crude homogenates of piglet somatosensory cortex with prominent band detection at 46-48 kDa. B. With a prominent layer V and an attenuated layer IV, RbFox3 immunostaining in sham piglet motor cortex is distinct from that seen in the somatosensory cortex. Inset shows the nuclear and cytoplasmic staining for Rbfox3 in piglet neocortical neurons. C. In HI-NT piglets, Rbfox3 can be found selectively depleted in layer II neurons of somatosensory cortex, yet, across the sulcus (asterisk), the motor cortex layer II neurons are positive. Other pyramidal neurons (inset) in layers III-V have nuclear clumping of Rbfox3 when undergoing ischemic-necrosis. Hatched box is shown at higher magnification in D. D. HI-NT piglet with selective depletion of Rbfox3 in layer II, yet the neurons appear morphologically normal. E. Somatosensory cortex layer II of an HI-HT piglet with depletion of Rbfox3 in some neurons (solid arrow) and partial rescue of Rbfox3 positivity in other neurons (solid thin arrows). Scale bars (in µm): A (same for B,C), 160; B inset, 9; C inset, 10; D, 18; E, 7.
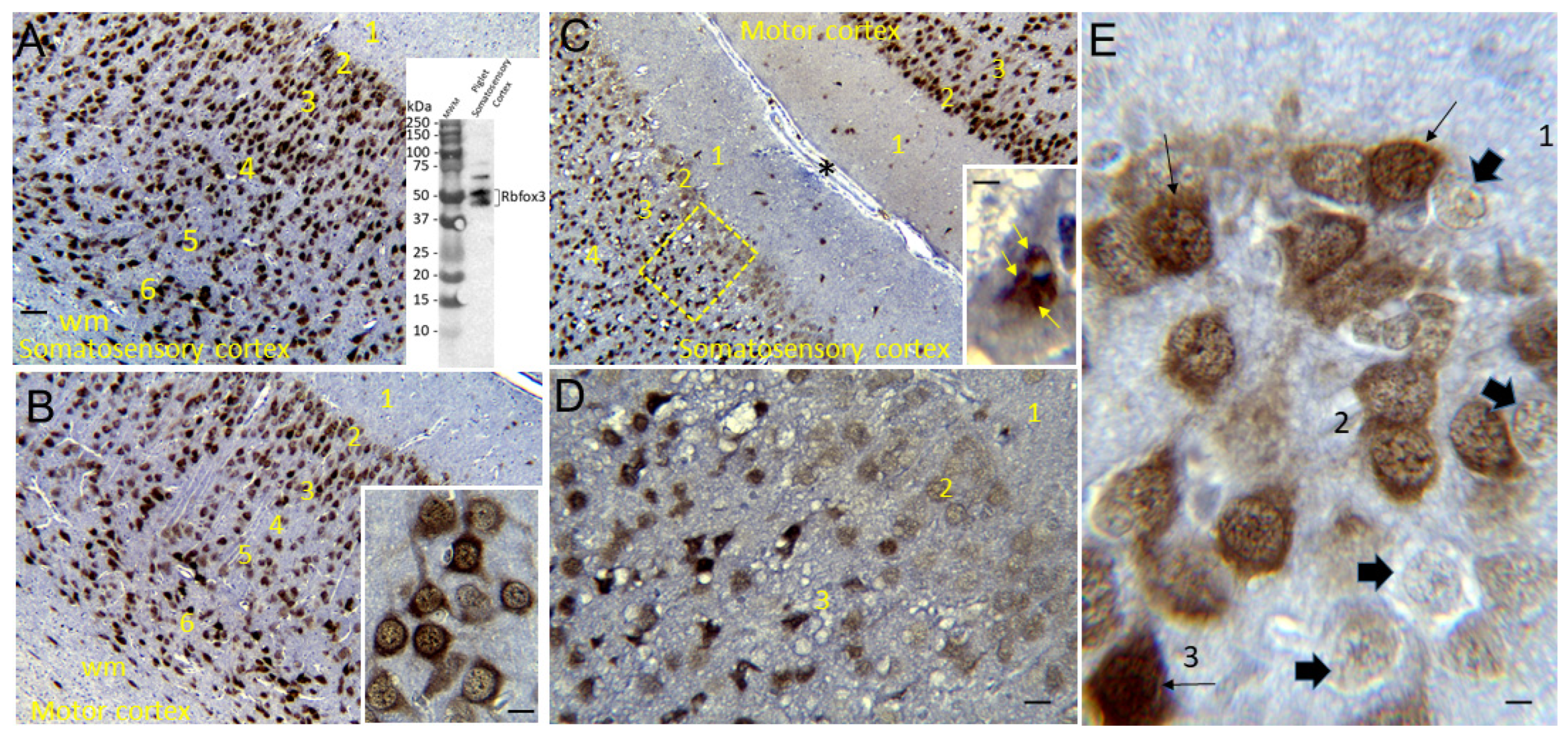
Figure 4.
Neonatal piglet neocortex gyrencephaly, cytoarchitectonics, and chemoarchitectonics. A. Perfusion fixed (4% PF) piglet brain illustrating the gyrencephalic cerebral cortex from a dorsal view. Anterior is at top. Gyri corresponding to the motor cortex, somatosensory cortex, and visual cortex are identified. B. Nissl staining of a piglet brain sagittal section (40 µm) showing the prominent layer V of motor cortex. C. Nissl staining of a piglet brain sagittal section (40 µm) showing the prominent layer IV of somatosensory cortex. D. Nissl staining of a piglet brain sagittal section (40 µm) showing the transition from motor cortex (left) to somatosensory cortex (right). The motor cortex has a conspicuous layer V and an inconspicuous layer IV (agranular), while the somatosensory cortex has a prominent layer IV and an attenuated layer V. E-F. Piglet brain axial sections (anterior is at top) showing that the enzyme activity of cytochrome C oxidase (complex IV) is differentially enriched in different regions of cerebral cortex. The most inferior-most section with the centrum semiovale is G. Scale bars: B and C, 105 µm; D, 54 µm; E (same for F, G), 2 mm.
Figure 4.
Neonatal piglet neocortex gyrencephaly, cytoarchitectonics, and chemoarchitectonics. A. Perfusion fixed (4% PF) piglet brain illustrating the gyrencephalic cerebral cortex from a dorsal view. Anterior is at top. Gyri corresponding to the motor cortex, somatosensory cortex, and visual cortex are identified. B. Nissl staining of a piglet brain sagittal section (40 µm) showing the prominent layer V of motor cortex. C. Nissl staining of a piglet brain sagittal section (40 µm) showing the prominent layer IV of somatosensory cortex. D. Nissl staining of a piglet brain sagittal section (40 µm) showing the transition from motor cortex (left) to somatosensory cortex (right). The motor cortex has a conspicuous layer V and an inconspicuous layer IV (agranular), while the somatosensory cortex has a prominent layer IV and an attenuated layer V. E-F. Piglet brain axial sections (anterior is at top) showing that the enzyme activity of cytochrome C oxidase (complex IV) is differentially enriched in different regions of cerebral cortex. The most inferior-most section with the centrum semiovale is G. Scale bars: B and C, 105 µm; D, 54 µm; E (same for F, G), 2 mm.
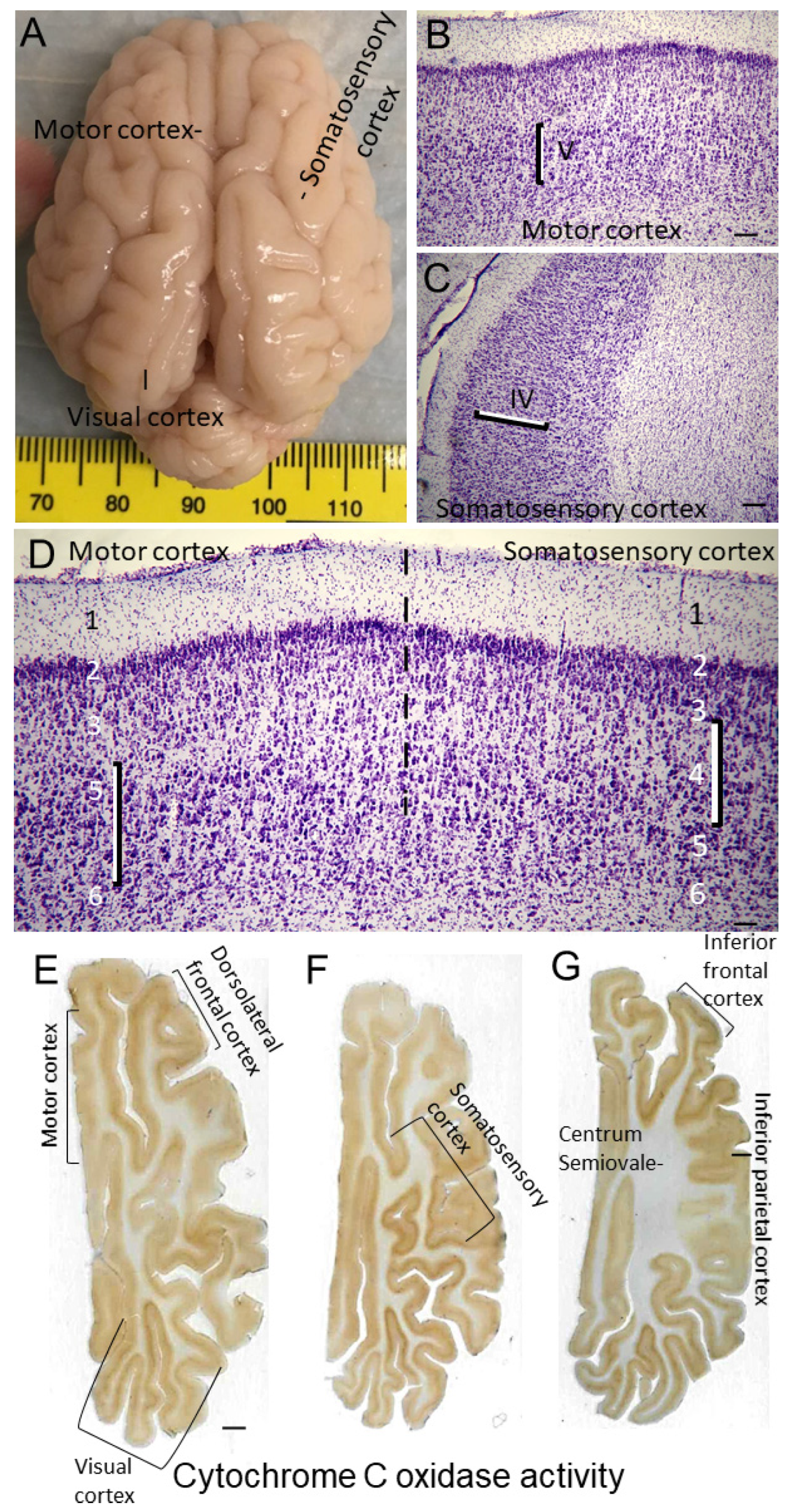
Figure 5.
Neonatal piglet neocortical connectome. A. Perfusion fixed (4% PF) piglet brain (gross) with color-coded buttons identifying locations of injection sites for AV-CFP (blue) in right motor cortex, and AAV-eGFP (green), LV-RFP (red) and WGA-HRP (black) in right somatosensory cortex. Black line in left cerebrum identifies approximate level of brain slab shown in B. B. Piglet gross brain coronal slab identifying subcortical location of injection site for FluoroGold (FG, yellow) in left striatum. Inset shows FG retrogradely labeled (white arrows) motor cortex corticostriatal projection neurons in layer V (direct fluorescence of FG). C. CFP labeling of motor cortex (brown, antibody detection of CFP by immunohistochemistry with diaminobenzidine chromogen and blue Nissl counterstaining). Layer V neurons are positive (arrows) with inset showing a gigantocellular Betz cell (arrow) strongly positive for CFP. Area in hatched box is shown at higher magnification in D. D. Superficial layers of the lateral bank of the motor cortex gyrus showing numerous CFP-labeled apical dendrites of layer V neurons extending through layers IV, III, II, and I to the cortical surface. E. Single-axon resolution labeling (arrows) of CFP motor corticostriatal projections in putamen. F,G. RFP labeling of somatosensory corticostriatal projections to putamen seen in the external capsule (open arrow) and in white matter stria (solid arrow) in the lateral putamen (direct fluorescence of RFP). RFP labeling near the somatosensory cortex (G injection site for LV-RFP (open arrow, direct fluorescence of RFP). H. Somatosensory cortex injection site for LV-RFP (brown, arrow) seen by antibody detection of RFP. Box in ventral thalamus is shown in I. I. RFP anterograde labeling of fine somatosensory corticothalamic terminal field plexus (arrows) in the ventroposterolateral thalamic nucleus. J. Corticocortical projections identified by eGFP-positive (direct fluorescence) retrogradely labeled neurons concentrated in layer V of cingulate cortex after AAV-eGFP injection in somatosensory cortex. K. HRP labeled single axon (arrows) in lateral putamen (near external capsule) resulting from injection of WGA-HRP in ipsilateral somatosensory cortex (neutral red counterstaining). L. Within central putamen, the HRP-labeled somatosensory corticostriatal terminal field is robust and extensive (arrow, black dots represent individual presynaptic terminals). Putamen contralateral to injections of WGA-HRP in somatosensory cortex was essentially negative, indicating negligible crossed corticostriatal projections (data not shown). M. WGA-HRP retrogradely labeled neuron (arrow) in thalamic ventroposterolateral nucleus that projects to somatosensory cortex. Scale bars: B, 1.75 mm; B inset 30 µm; C, 293 µm; C inset, 35 µm; D, 25 µm; E, 8.5 µm; F, 10 µm; G, 10 µm; H, 2.6 mm; I, 7 µm; J, 44 µm; K, 12 µm; L, 21 µm; M, 10.5 µm.
Figure 5.
Neonatal piglet neocortical connectome. A. Perfusion fixed (4% PF) piglet brain (gross) with color-coded buttons identifying locations of injection sites for AV-CFP (blue) in right motor cortex, and AAV-eGFP (green), LV-RFP (red) and WGA-HRP (black) in right somatosensory cortex. Black line in left cerebrum identifies approximate level of brain slab shown in B. B. Piglet gross brain coronal slab identifying subcortical location of injection site for FluoroGold (FG, yellow) in left striatum. Inset shows FG retrogradely labeled (white arrows) motor cortex corticostriatal projection neurons in layer V (direct fluorescence of FG). C. CFP labeling of motor cortex (brown, antibody detection of CFP by immunohistochemistry with diaminobenzidine chromogen and blue Nissl counterstaining). Layer V neurons are positive (arrows) with inset showing a gigantocellular Betz cell (arrow) strongly positive for CFP. Area in hatched box is shown at higher magnification in D. D. Superficial layers of the lateral bank of the motor cortex gyrus showing numerous CFP-labeled apical dendrites of layer V neurons extending through layers IV, III, II, and I to the cortical surface. E. Single-axon resolution labeling (arrows) of CFP motor corticostriatal projections in putamen. F,G. RFP labeling of somatosensory corticostriatal projections to putamen seen in the external capsule (open arrow) and in white matter stria (solid arrow) in the lateral putamen (direct fluorescence of RFP). RFP labeling near the somatosensory cortex (G injection site for LV-RFP (open arrow, direct fluorescence of RFP). H. Somatosensory cortex injection site for LV-RFP (brown, arrow) seen by antibody detection of RFP. Box in ventral thalamus is shown in I. I. RFP anterograde labeling of fine somatosensory corticothalamic terminal field plexus (arrows) in the ventroposterolateral thalamic nucleus. J. Corticocortical projections identified by eGFP-positive (direct fluorescence) retrogradely labeled neurons concentrated in layer V of cingulate cortex after AAV-eGFP injection in somatosensory cortex. K. HRP labeled single axon (arrows) in lateral putamen (near external capsule) resulting from injection of WGA-HRP in ipsilateral somatosensory cortex (neutral red counterstaining). L. Within central putamen, the HRP-labeled somatosensory corticostriatal terminal field is robust and extensive (arrow, black dots represent individual presynaptic terminals). Putamen contralateral to injections of WGA-HRP in somatosensory cortex was essentially negative, indicating negligible crossed corticostriatal projections (data not shown). M. WGA-HRP retrogradely labeled neuron (arrow) in thalamic ventroposterolateral nucleus that projects to somatosensory cortex. Scale bars: B, 1.75 mm; B inset 30 µm; C, 293 µm; C inset, 35 µm; D, 25 µm; E, 8.5 µm; F, 10 µm; G, 10 µm; H, 2.6 mm; I, 7 µm; J, 44 µm; K, 12 µm; L, 21 µm; M, 10.5 µm.
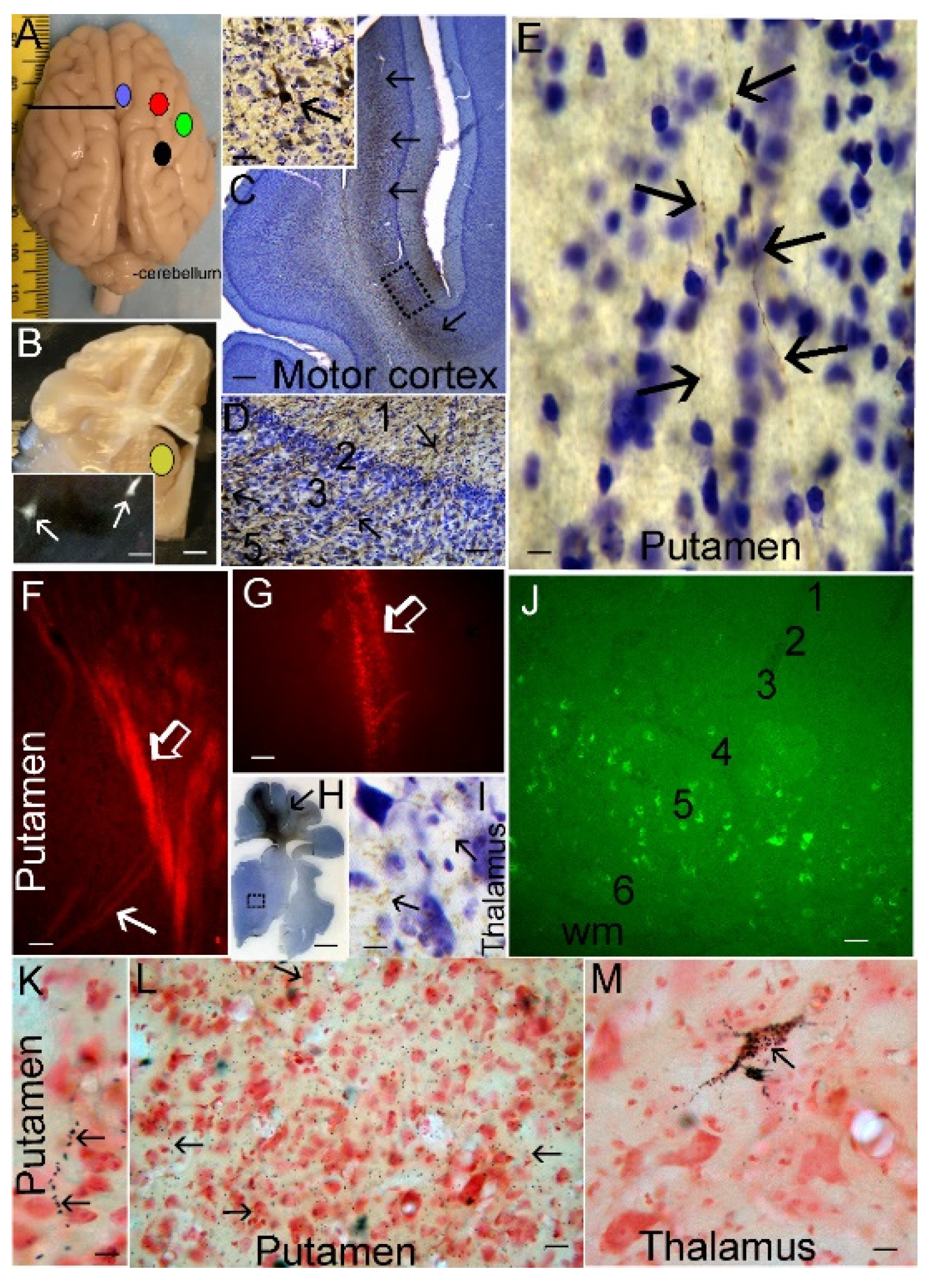
Figure 6.
Physiology of HI and sham piglets. Blood gas, hemoglobin, and glucose data are at 7-min asphyxia. Temperature and blood pressure data are at 8-min asphyxia. The blue arrows along the x-axis show the beginning and maintenance of HT. The purple arrows show rewarming. A. Arterial partial pressure of carbon dioxide (PaCO2) was interactively affected by time and treatment (p=0.006). B. Arterial partial pressure of oxygen (PaO2) varied across time (p<0.001). Mean arterial blood pressure (C), arterial pH (D), and core body temperature (E) were affected by treatment. F. Hemoglobin levels also varied across time (p<0.001). G. Blood glucose was interactively affected by time and treatment (p<0.001). For all parameters group sizes were: sham normothermia (n=6), sham hypothermia (n=10), HI normothermia (n=8), and HI hypothermia (n=10). *p<0.05 in post-hoc tests. Data are shown as means with 95% confidence intervals.
Figure 6.
Physiology of HI and sham piglets. Blood gas, hemoglobin, and glucose data are at 7-min asphyxia. Temperature and blood pressure data are at 8-min asphyxia. The blue arrows along the x-axis show the beginning and maintenance of HT. The purple arrows show rewarming. A. Arterial partial pressure of carbon dioxide (PaCO2) was interactively affected by time and treatment (p=0.006). B. Arterial partial pressure of oxygen (PaO2) varied across time (p<0.001). Mean arterial blood pressure (C), arterial pH (D), and core body temperature (E) were affected by treatment. F. Hemoglobin levels also varied across time (p<0.001). G. Blood glucose was interactively affected by time and treatment (p<0.001). For all parameters group sizes were: sham normothermia (n=6), sham hypothermia (n=10), HI normothermia (n=8), and HI hypothermia (n=10). *p<0.05 in post-hoc tests. Data are shown as means with 95% confidence intervals.
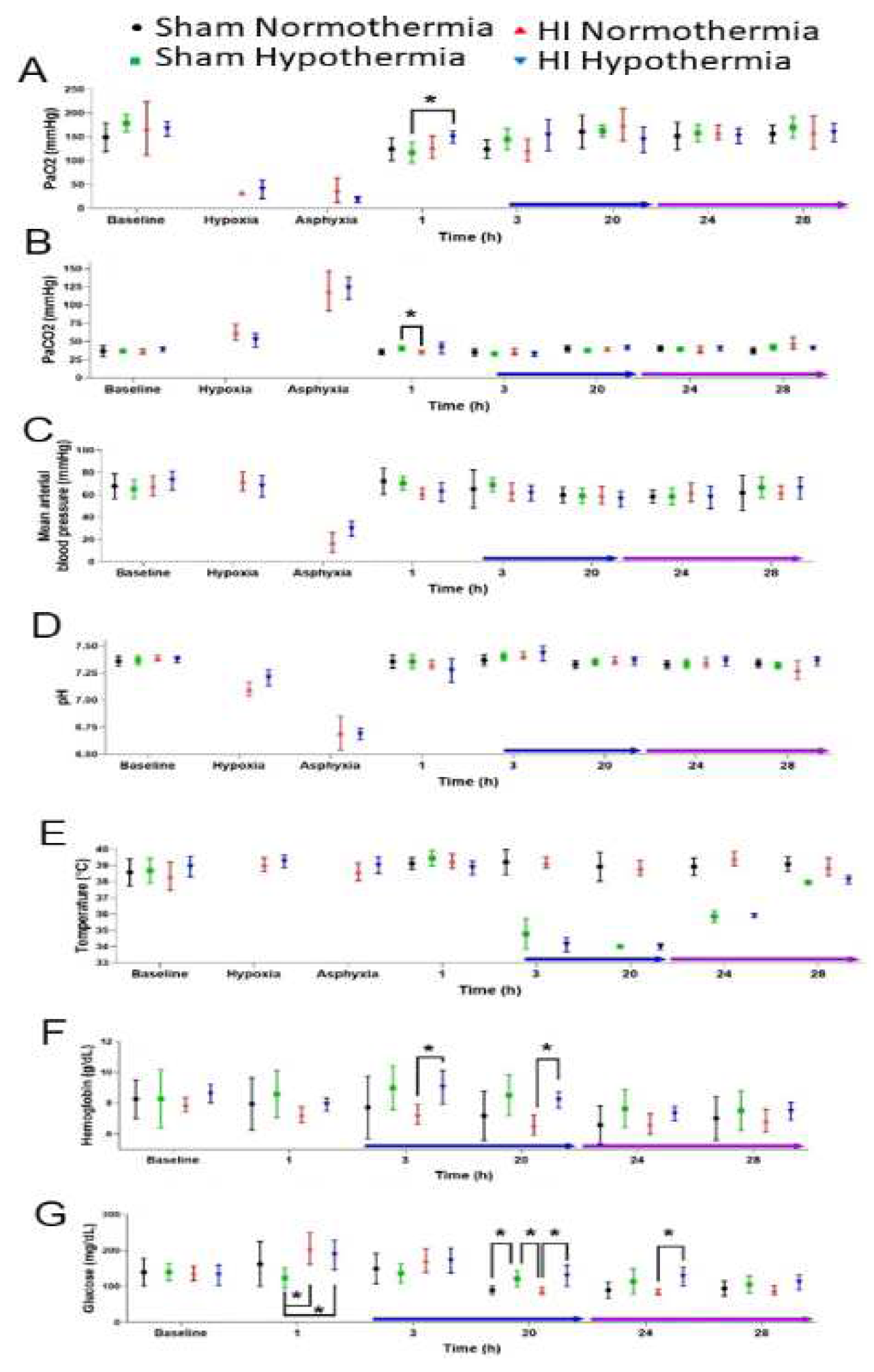
Figure 7.
Primary somatosensory neocortex was protected in hypoxia-ischemia (HI)-HT piglets. A. HI-NT piglets had decreased normal-to-total neuron ratios in the anterior parietal cortex level (medial bank of the somatosensory gyrus: p<0.001; lateral: p<0.001) and in the mid-parietal level (medial: p<0.001; lateral: p<0.001). B. Ischemic necrosis was greatest in HI-NT piglets in the anterior parietal (medial: p<0.001; lateral: p<0.001) and mid-parietal (medial: p<0.001; lateral: p=0.001) somatosensory cortex. C. HI-NT piglets had increased apoptosis in somatosensory cortex (medial mid-parietal: p=0.013; lateral anterior parietal: p=0.003). Sham-HT piglets had less apoptosis than did sham-NT piglets in mid-parietal cortex (p=0.036). D. Apoptosis-necrosis continuum cell degeneration was most common in the medial bank of the mid-parietal somatosensory cortex in HI-NT piglets (p=0.026). Data are shown with means and 95% CIs. *p<0.05. E. H&E staining of the somatosensory cortex of an HI piglet showing clear selective laminar pathology in lower layer III and upper layer IV and more subtle damage in layer II as delineated by the black lines. F. H&E staining of the somatosensory cortex of an HI piglet showing clear panlaminar necrosis. Scale bar in E (same for F), 63 µm.
Figure 7.
Primary somatosensory neocortex was protected in hypoxia-ischemia (HI)-HT piglets. A. HI-NT piglets had decreased normal-to-total neuron ratios in the anterior parietal cortex level (medial bank of the somatosensory gyrus: p<0.001; lateral: p<0.001) and in the mid-parietal level (medial: p<0.001; lateral: p<0.001). B. Ischemic necrosis was greatest in HI-NT piglets in the anterior parietal (medial: p<0.001; lateral: p<0.001) and mid-parietal (medial: p<0.001; lateral: p=0.001) somatosensory cortex. C. HI-NT piglets had increased apoptosis in somatosensory cortex (medial mid-parietal: p=0.013; lateral anterior parietal: p=0.003). Sham-HT piglets had less apoptosis than did sham-NT piglets in mid-parietal cortex (p=0.036). D. Apoptosis-necrosis continuum cell degeneration was most common in the medial bank of the mid-parietal somatosensory cortex in HI-NT piglets (p=0.026). Data are shown with means and 95% CIs. *p<0.05. E. H&E staining of the somatosensory cortex of an HI piglet showing clear selective laminar pathology in lower layer III and upper layer IV and more subtle damage in layer II as delineated by the black lines. F. H&E staining of the somatosensory cortex of an HI piglet showing clear panlaminar necrosis. Scale bar in E (same for F), 63 µm.
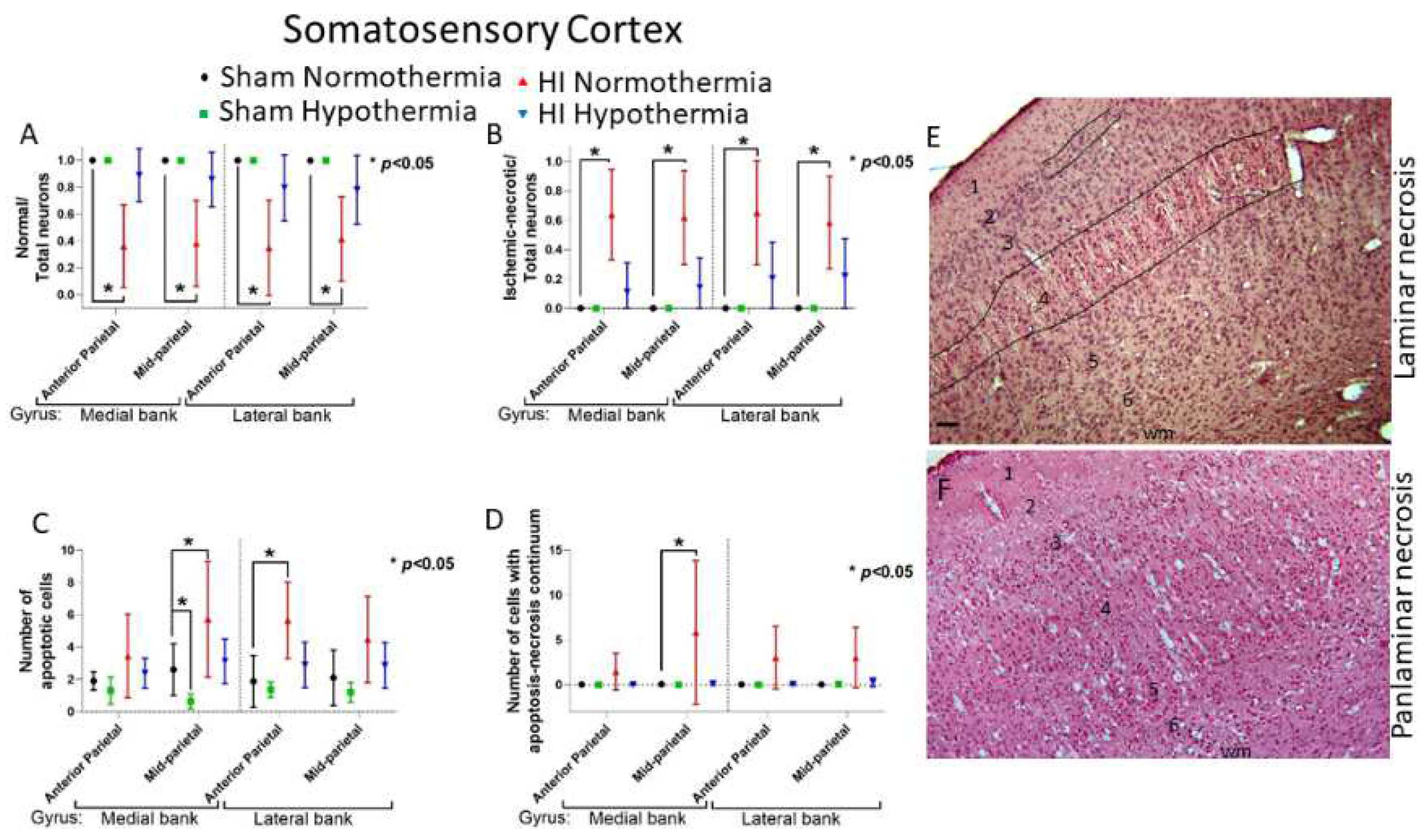
Figure 8.
Motor cortex damage after HI varied regionally. A. HI-NT piglets had a decreased normal-to-total neuron ratio relative to sham-NT piglets at the anterior parietal level in the medial (p=0.010) and lateral (p=0.001) cortical banks. The loss of normal neurons after HI also occurred in mid-parietal motor cortex (medial bank, p=0.003; lateral bank, p=0.001). Neuron loss was not detected in frontal cortex. B. Compared to sham-NT piglets, cortical ischemic necrosis increased in HI-NT piglets in anterior and mid-parietal but not frontal cortex. In anterior parietal motor cortex, ischemic necrosis was greater in HI-NT piglets compared to sham-NT piglets (medial, p<0.009; lateral, p=0.001). In mid-mid-parietal motor cortex, more ischemic necrosis occurred in HI-NT piglets compared to sham pigs (medial, p=0.003; lateral, p<0.001). C. Sham-HT pigs had less apoptosis in frontal cortex than sham-NT pigs (p=0.020). D. HI-NT piglets had more apoptosis-necrosis continuum cell degeneration than shams (p=0.001). Data are shown with means and 95% CIs. *p<0.05. Hypoxia-ischemia, HI.
Figure 8.
Motor cortex damage after HI varied regionally. A. HI-NT piglets had a decreased normal-to-total neuron ratio relative to sham-NT piglets at the anterior parietal level in the medial (p=0.010) and lateral (p=0.001) cortical banks. The loss of normal neurons after HI also occurred in mid-parietal motor cortex (medial bank, p=0.003; lateral bank, p=0.001). Neuron loss was not detected in frontal cortex. B. Compared to sham-NT piglets, cortical ischemic necrosis increased in HI-NT piglets in anterior and mid-parietal but not frontal cortex. In anterior parietal motor cortex, ischemic necrosis was greater in HI-NT piglets compared to sham-NT piglets (medial, p<0.009; lateral, p=0.001). In mid-mid-parietal motor cortex, more ischemic necrosis occurred in HI-NT piglets compared to sham pigs (medial, p=0.003; lateral, p<0.001). C. Sham-HT pigs had less apoptosis in frontal cortex than sham-NT pigs (p=0.020). D. HI-NT piglets had more apoptosis-necrosis continuum cell degeneration than shams (p=0.001). Data are shown with means and 95% CIs. *p<0.05. Hypoxia-ischemia, HI.
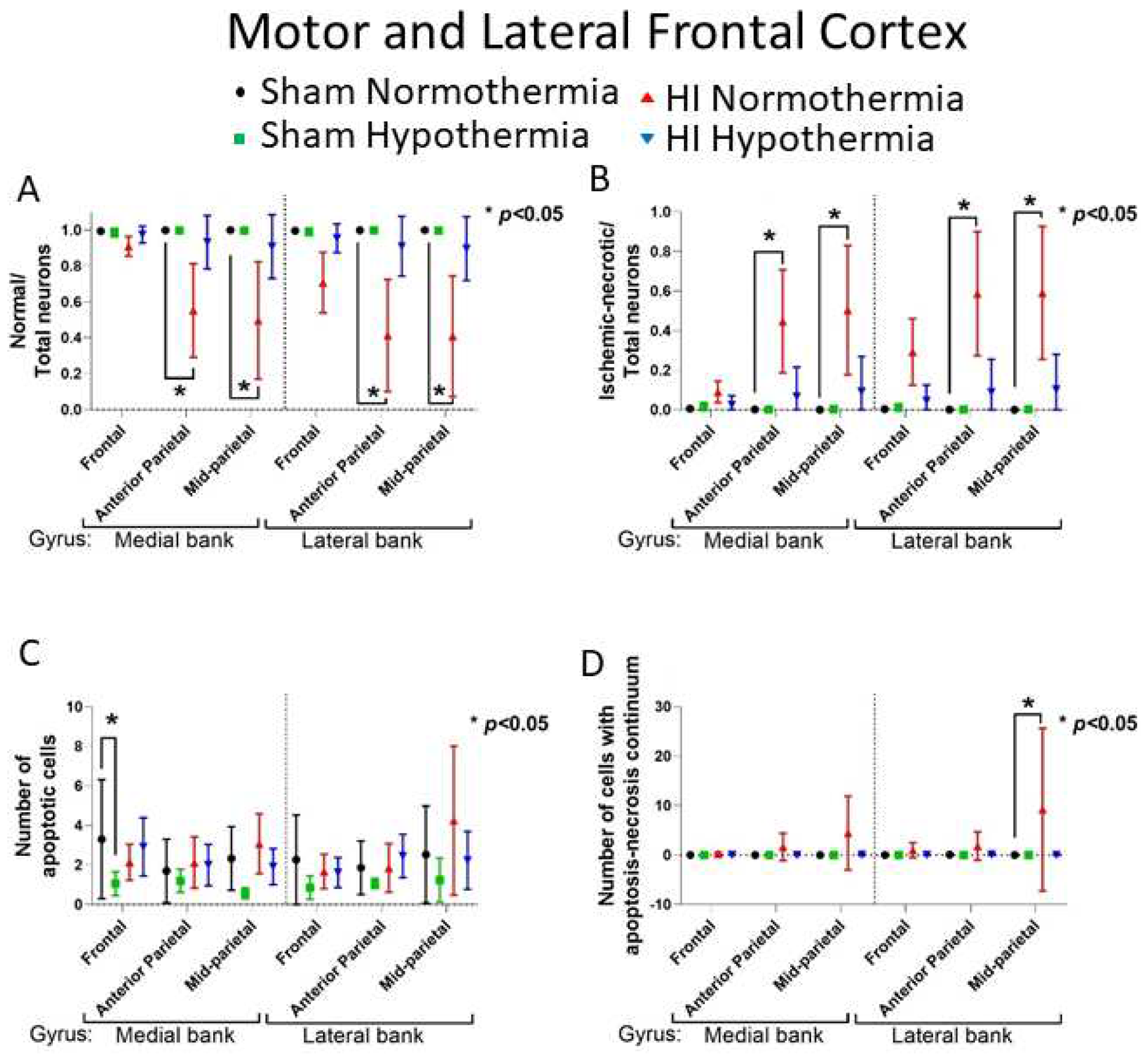
Figure 9.
Layer III has prominent neuronal loss in the inferior parietal neocortex of HI-NT piglets. A. In sham-NT piglets, the H&E staining shows pristine undamaged neuronal cell body (arrows) and neuropil composition. Scale bar (same for B), 13 µm. B. In HI-NT piglets, layer III is devastated with only residual ischemic-necrotic neurons (arrows) remaining in a field of spongiform neuropil. C. HI-NT piglets had a severe loss of normal neurons in layer III of inferior parietal cortex compared to sham-NT piglets (p<0.001). Normal neuron number was partly rescued in HI-HT compared to HI-NT piglets (p<0.001). Box plots show median values with IQR and 5-95th percentile whiskers.
Figure 9.
Layer III has prominent neuronal loss in the inferior parietal neocortex of HI-NT piglets. A. In sham-NT piglets, the H&E staining shows pristine undamaged neuronal cell body (arrows) and neuropil composition. Scale bar (same for B), 13 µm. B. In HI-NT piglets, layer III is devastated with only residual ischemic-necrotic neurons (arrows) remaining in a field of spongiform neuropil. C. HI-NT piglets had a severe loss of normal neurons in layer III of inferior parietal cortex compared to sham-NT piglets (p<0.001). Normal neuron number was partly rescued in HI-HT compared to HI-NT piglets (p<0.001). Box plots show median values with IQR and 5-95th percentile whiskers.
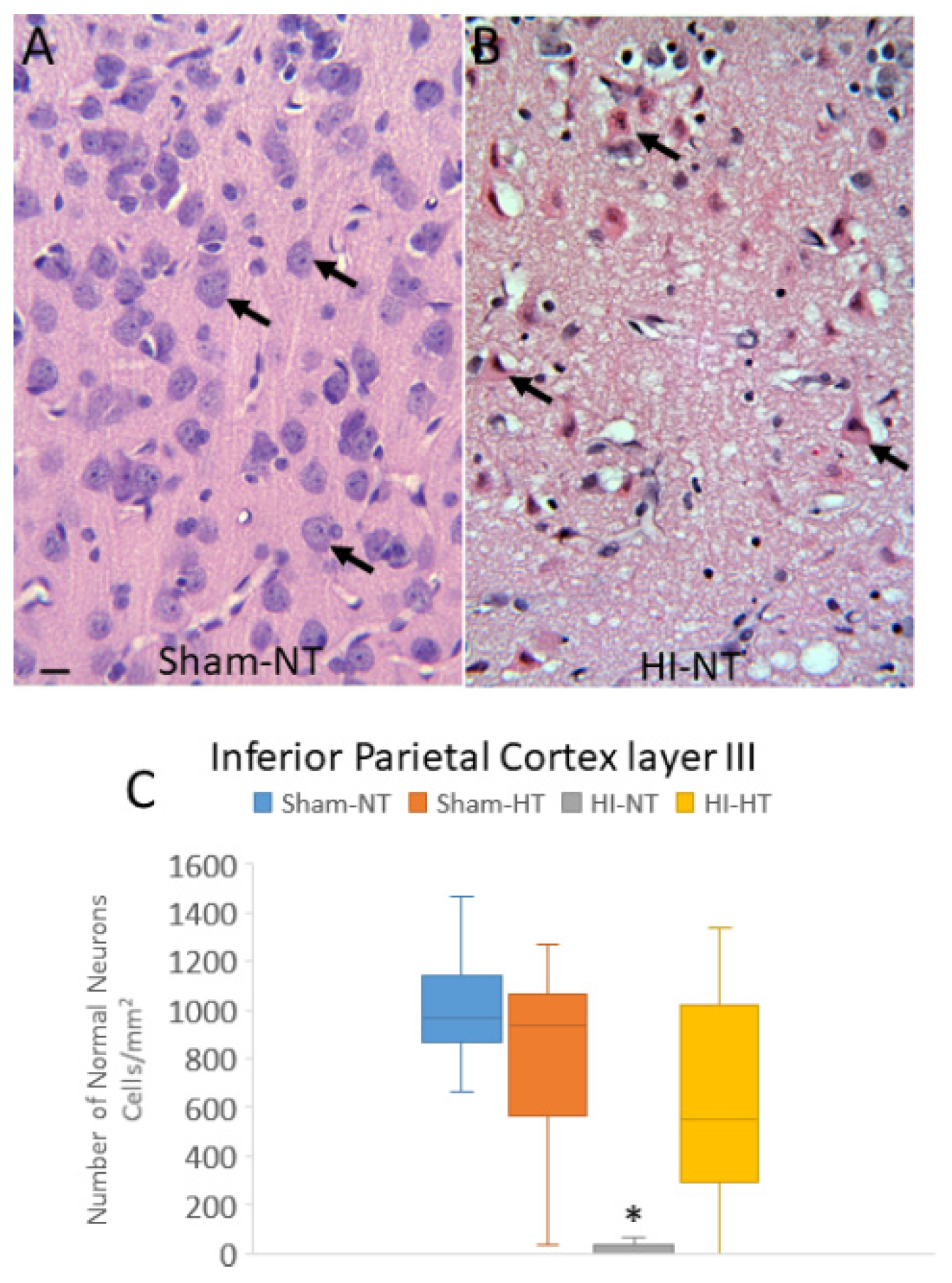
Figure 10.
Somatosensory cortex in HI-NT piglets has differential laminar vulnerability and nuclear depletion of Rbfox3/NeuN immunoreactivity. Brain sections immunostained for Rbfox3 and CV counterstained were used to count total neurons (A-C) and neurons depleted of Rbfox3 nuclear positivity (D-F) in layers II, III, and V of somatosensory cortex. Box-and-whisker plots show the median and interquartile ranges. A-C. In sham piglets, layer II has the most neurons. In HI-NT piglets, total neuron counts were reduced (*p<0.001) compared to sham-NT in layers II (A), III (B), and V (C). In HI-HT piglets, total neuron counts were reduced (**p<0.01) compared to sham-HT in layers II (A) and III (B). There was significant (+p=0.009) rescue of total neurons in HI-HT piglets compared to HI-NT piglets. D-F. In HI-NT piglets, the ratio of Rbfox3-depleted to total neurons increased in all layers compared to sham-NT (p<0.001). In HI-HT piglets, the ratio of Rbfox3-depleted to total neurons increased in layer II compared to sham-NT (p<0.001). There was significant (+p=0.001) rescue of total neurons in HI-HT piglets compared to HI-NT piglets in layer V.
Figure 10.
Somatosensory cortex in HI-NT piglets has differential laminar vulnerability and nuclear depletion of Rbfox3/NeuN immunoreactivity. Brain sections immunostained for Rbfox3 and CV counterstained were used to count total neurons (A-C) and neurons depleted of Rbfox3 nuclear positivity (D-F) in layers II, III, and V of somatosensory cortex. Box-and-whisker plots show the median and interquartile ranges. A-C. In sham piglets, layer II has the most neurons. In HI-NT piglets, total neuron counts were reduced (*p<0.001) compared to sham-NT in layers II (A), III (B), and V (C). In HI-HT piglets, total neuron counts were reduced (**p<0.01) compared to sham-HT in layers II (A) and III (B). There was significant (+p=0.009) rescue of total neurons in HI-HT piglets compared to HI-NT piglets. D-F. In HI-NT piglets, the ratio of Rbfox3-depleted to total neurons increased in all layers compared to sham-NT (p<0.001). In HI-HT piglets, the ratio of Rbfox3-depleted to total neurons increased in layer II compared to sham-NT (p<0.001). There was significant (+p=0.001) rescue of total neurons in HI-HT piglets compared to HI-NT piglets in layer V.
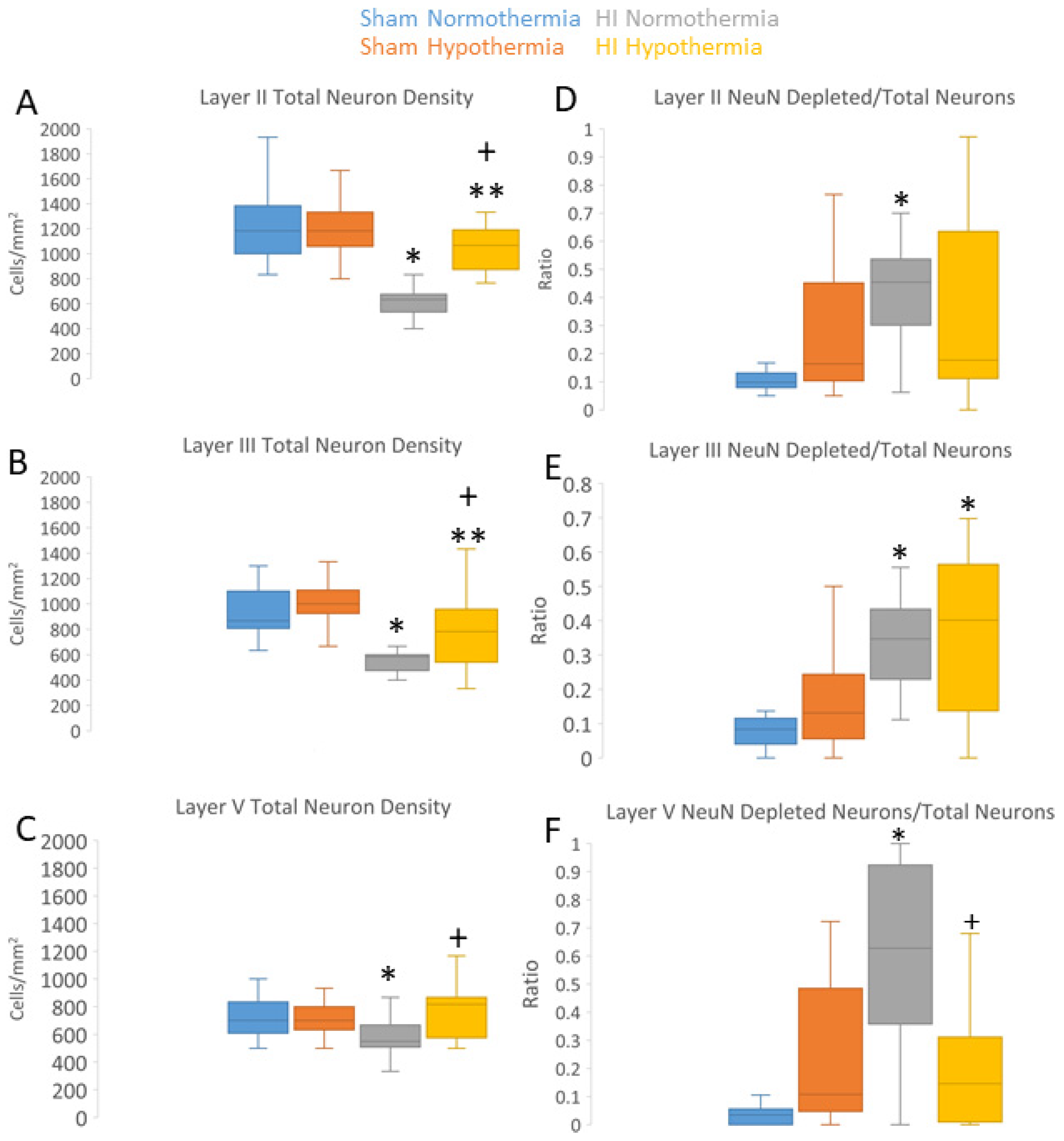
Figure 11.
Seizure assessment in piglets. A. Seizure burden was greater in hypoxia-ischemia (HI) piglets compared to sham piglets (p=0.019). Sham-NT (black), sham-HT (green), HI-NT (red), and HI-HT (blue). *p<0.05. The whiskers show the 5-95th percentiles. B. Representative 10-second electrograms of neocortical activity in right anterior, right posterior, left anterior and left posterior leads in a sham-NT piglet. EEG shows background activity of a variety of frequencies and amplitudes throughout all leads. No rhythmic waveforms are present. C. EEG of HI piglet with seizures showing presence of epileptiform appearing rhythmic spike wave complexes (2-3 Hz) generalized in all four leads over 10 seconds. .
Figure 11.
Seizure assessment in piglets. A. Seizure burden was greater in hypoxia-ischemia (HI) piglets compared to sham piglets (p=0.019). Sham-NT (black), sham-HT (green), HI-NT (red), and HI-HT (blue). *p<0.05. The whiskers show the 5-95th percentiles. B. Representative 10-second electrograms of neocortical activity in right anterior, right posterior, left anterior and left posterior leads in a sham-NT piglet. EEG shows background activity of a variety of frequencies and amplitudes throughout all leads. No rhythmic waveforms are present. C. EEG of HI piglet with seizures showing presence of epileptiform appearing rhythmic spike wave complexes (2-3 Hz) generalized in all four leads over 10 seconds. .
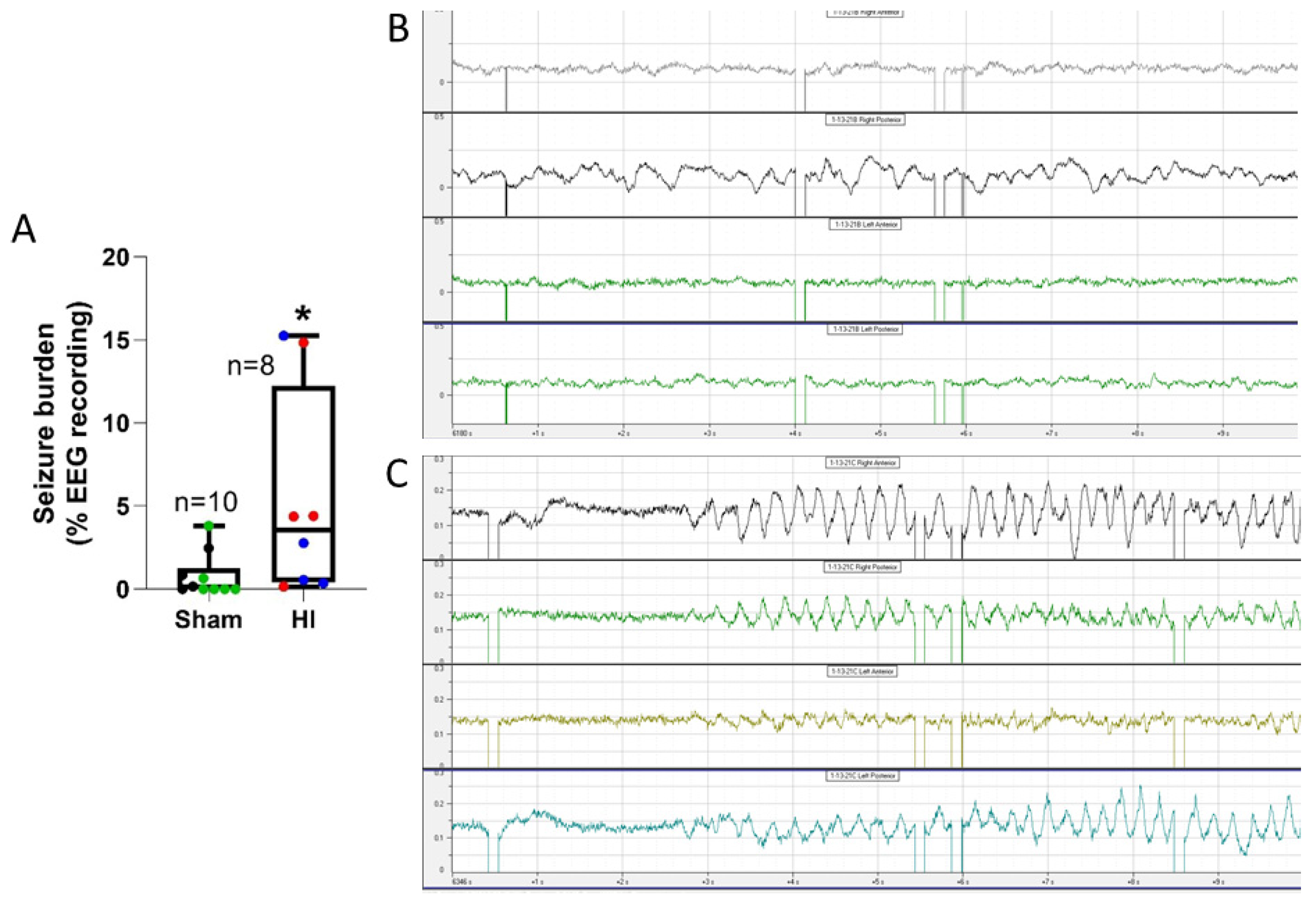
Figure 12.
Piglets with greater seizure burden had more ischemic necrosis in the somatosensory cortex of (A, B) mid-parietal cortex (medial bank: r=0.69, p=0.002, n=18; lateral bank: r=0.62, p=0.006), (C) medial bank of anterior parietal cortex (r=0.57, p=0.015, n=18), (D) lateral bank of frontal motor cortex (r=0.48, p=0.045), and (E) putamen (r=0.60, p=0.009). Sham normothermia, black circles. Sham hypothermia, green squares, hypoxia-ischemia (HI) normothermia, red triangles. HI hypothermia, blue inverted triangle.
Figure 12.
Piglets with greater seizure burden had more ischemic necrosis in the somatosensory cortex of (A, B) mid-parietal cortex (medial bank: r=0.69, p=0.002, n=18; lateral bank: r=0.62, p=0.006), (C) medial bank of anterior parietal cortex (r=0.57, p=0.015, n=18), (D) lateral bank of frontal motor cortex (r=0.48, p=0.045), and (E) putamen (r=0.60, p=0.009). Sham normothermia, black circles. Sham hypothermia, green squares, hypoxia-ischemia (HI) normothermia, red triangles. HI hypothermia, blue inverted triangle.
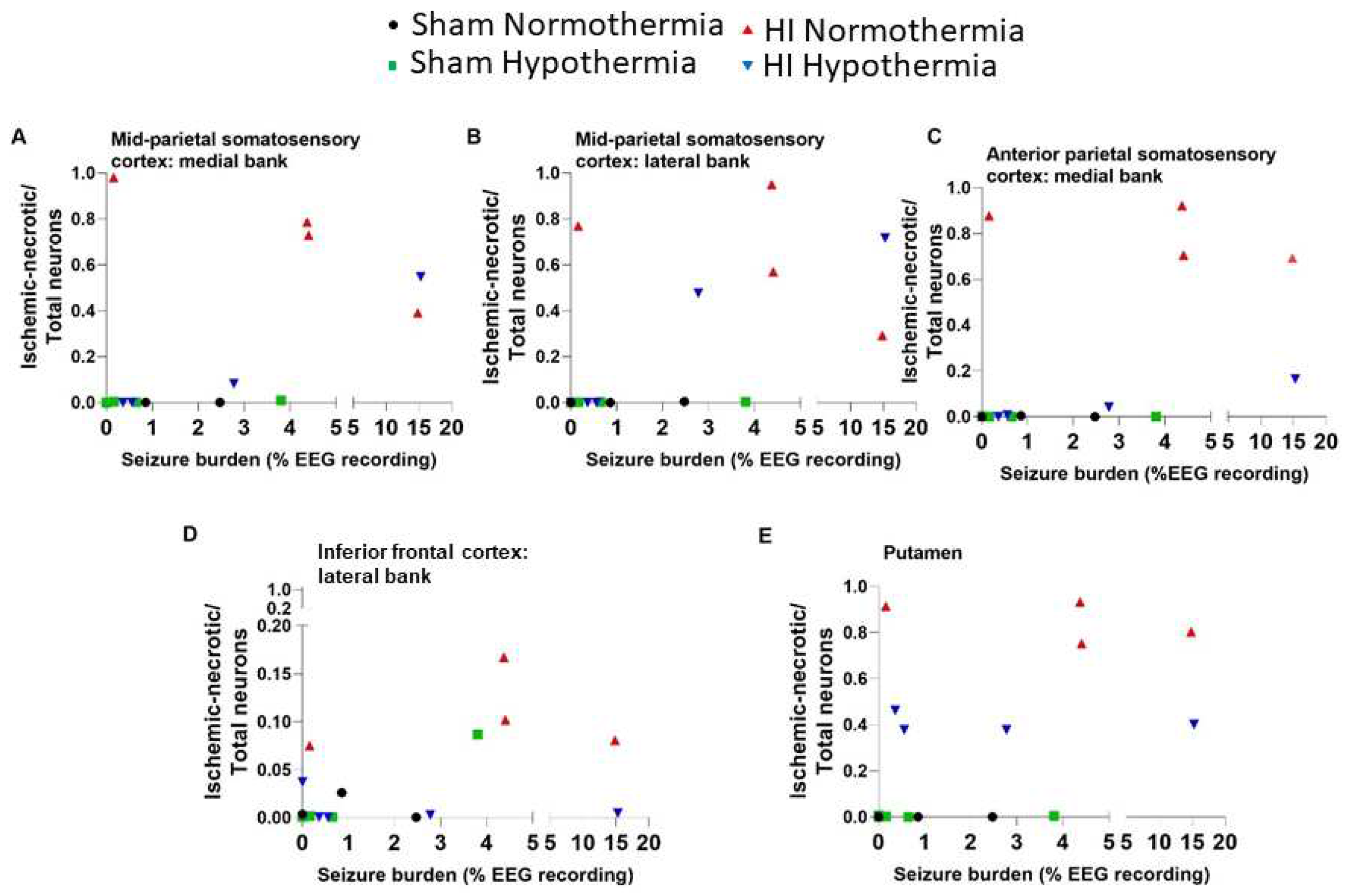
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



