Submitted:
04 July 2023
Posted:
05 July 2023
You are already at the latest version
Abstract
Lipofuscin accumulates with age as intracellular fluorescent granules originating from incomplete lysosomal digestion of phagocytosed and autophagocytosed material. The purpose of this review is to provide an update on the current understanding of the role of oxidative stress and/or lysosomal dysfunction in lipofuscin accumulation and its consequences, particularly for retinal pigment epithelium (RPE). Next, the fluorescence of lipofuscin, spectral changes induced by oxidation, and its contribution to retinal fluorescence are discussed. This is followed by reviewing recent developments in fluorescence imaging of the retina, and the current evidence on the prognostic value of retinal fluorescence for the progression of age-related macular degeneration (AMD), the major blinding disease affecting elderly people in developed countries. The evidence of lipofuscin oxidation in vivo, and the evidence of increased oxidative damage in AMD retina ex vivo lead to the conclusion that imaging of spectral characteristics of lipofuscin fluorescence may serve as a useful biomarker of oxidative damage which can be helpful in assessing the efficacy of potential antioxidant therapies in retinal degenerations associated with accumulation of lipofuscin and increased oxidative stress. Finally, amendments to currently used fluorescence imaging instruments are suggested, to be more sensitive and specific for imaging spectral char-acteristics of lipofuscin fluorescence.
Keywords:
lipofuscin
; retina
; fundus autofluorescence
; fluorescence imaging
; oxidative stress
; photooxida-tion
; oxidation
; biomarker
; age-related macular degeneration
1. Introduction
Lipofuscin accumulates in postmitotic cells as autofluorescent deposits due to incomplete lysosomal digestion of autophagocytosed and phagocytosed material. The purpose of this review is to provide an update on the current understanding of the role of oxidative stress and lysosomal dysfunction in lipofuscin formation and its effects on cells as well as their fluorescence properties. Next, the retinal pigment epithelium (RPE) is considered as the main site of lipofuscin accumulation in the retina, and the roles of oxidative stress, lysosomal dysfunction, and vitamin A derivatives in the formation of RPE lipofuscin are discussed, as well as its structure and composition, and effects on function and viability of RPE cells and neighbouring structures. The fluorescence of RPE lipofuscin and its changes upon oxidation are discussed in detail.
Next, sources of fluorescence in the retina other than lipofuscin are discussed, recent developments in fluorescence imaging of the retina, as well as the current evidence on the prognostic value of retinal fluorescence for the progression of age-related macular degeneration (AMD), the major blinding disease affecting elderly people in developed countries.
In the retina, RPE lipofuscin provides the major contribution to fluorescence emission excited by UV, blue, and green light. RPE lipofuscin has the potential to increase oxidative stress and is susceptible to oxidative degradation, which results in spectral changes in its fluorescence characteristics: a decrease in its yellow-red fluorescence and a concomitant increase in its blue-green fluorescence. The evidence of lipofuscin oxidation in vivo, and the evidence of increased oxidative stress and oxidative damage in AMD retina are reviewed leading to the conclusion that imaging of spectral characteristics of lipofuscin fluorescence may serve as a useful biomarker of oxidative damage in vivo which can be useful in assessing the efficacy of potential antioxidant therapies in AMD and other retinal degenerations associated with accumulation of lipofuscin and increased oxidative stress. Finally, amendments to currently used fluorescence imaging instruments based on two-photon excitation are suggested, which could make them more sensitive and specific for imaging spectral characteristics of lipofuscin fluorescence emission while minimizing the excitation of other retinal fluorophores and photosensitizers.
2. Lipofuscin: formation, composition, and potential deleterious effects
Lipofuscin is an intracellular deposit composed of lipids, proteins, and carbohydrates, which are cross-linked and highly modified by end-products of glycation and/or lipid peroxidation [1,2,3,4,5,6]. It accumulates with age in the form of small yellow-orange granules surrounded by a lipid membrane as a result of incomplete lysosomal digestion of autophagocytosed and phagocytosed material. It is present in most organs of the body and in most types of cells, especially if they are highly metabolically active and/or are postmitotic [7,8,9,10,11].
The lipofuscin content is split during cell division into daughter cells, thereby diluting it. This option is not available in the postmitotic cells, so the lipofuscin level per cell can considerably increase with age [12]. It has been reported that the lipofuscin increase in cardiomyocytes is 0.6% of cell volume per decade, whereas, in motor neurons of centenarians, up to 75% of cell volume can be occupied by lipofuscin [1]. Because of its age-related accumulation and colour, lipofuscin is also known as the age pigment.
The theory of the lysosomal origin of lipofuscin is supported by an increased accumulation of lipofuscin in diseases caused by dysfunction of autophagy and/or lysosomal degradation [13,14,15,16,17,18,19,20,21,22,23,24,25,26]. Among them, there is a group of neurodegenerative genetic disorders called neuronal ceroid lipofuscinosis (NCL), which is characterized by the formation of intracellular deposits in neurons and other cell types. The deposits are often referred to as ceroid to distinguish their abnormally early accumulation from the age-related accumulation of lipofuscin. Usually, NCL affects the brain and retina, often leading to vision loss, motor and cognitive regression, seizures, and premature death. It is a rare disease with an incidence of 1-3 per 100,000 and a prevalence of about 2-4 per million.
Inhibition of lysosomal enzymes in experimental animals and in cells in vitro also results in the accumulation of lipofuscin-like deposits [27,28]. Accumulation of such deposits can be also induced in an earlier step than lysosomal degradation by exposure of cultured cells to inhibitors of autophagy [29,30,31]. If both - autophagy and lysosomal activities are inhibited, lipofuscin-like material can rapidly accumulate in the cytosol [32].
Consistently, a decrease in lipofuscin accumulation can be achieved by upregulation of autophagy by inhibition of the mammalian target of rapamycin complex 1 (MTORC1) by rapamycin or by pharmacological activation of the transcriptional control of transcription factor EB (TFEB), which stimulates the expression of autophagy proteins, lysosomal membrane proteins and lysosomal hydrolases [33,34,35,36,37,38,39,40,41].
An increased lipofuscin accumulation is also a characteristic feature of many, but not all, diseases associated with increased oxidative stress, and it has been demonstrated in numerous experiments in vitro and in vivo that accumulation of lipofuscin can be accelerated by increased oxidative stress [9,26,42,43,44]. For example, it has been shown that lipofuscin accumulates more rapidly in cultured cells or in animals exposed to increased oxygen tension, redox-active iron ions, or depleted of antioxidants [31,45,46,47].
Based on experimental findings on cultured rat cardiomyocytes, Brunk and colleagues proposed that autophagy and lysosomal degradation of organelles producing reactive species and containing redox-active iron ions, such as mitochondria, can increase oxidative stress by facilitating lipid peroxidation [48]. Subsequently, the formation of oxidized and crosslinked biomolecules makes them no longer susceptible to lysosomal degradation. Iron chelators and antioxidants, such as vitamin E can effectively inhibit lipofuscin accumulation by preventing lipid peroxidation.
While there is some evidence that cells can survive and apparently still perform their function despite having a high lipofuscin content [49], there is also a growing body of evidence showing that lipofuscin can compromise cell function or even lead to cell death, suggesting that accumulation of lipofuscin cannot be viewed as a benign feature of ageing of postmitotic cells, particularly if they exhibit high metabolism and/or are under oxidative stress conditions [17,50]. For example, it has been shown on confluent fibroblasts and astrocytes that inhibition of autophagy by 3-methyladenine or inhibition of lysosomal enzymes by leupeptin leads to a rapid intracellular accumulation of lipofuscin-like material and eventually results in apoptotic cell death [51]. In cultured human fibroblasts, accumulation of lipofuscin is progressing rapidly but once it reaches a certain level, it stops and cells start to die [52]. Reaching that threshold can be accelerated by supplementing the culture medium with mitochondria oxidized by exposure to ultraviolet light. Oxidized mitochondria are phagocytosed and form lipofuscin even more rapidly, and the lipofuscin exerts a toxic effect upon reaching a similar level as that which causes toxicity in cells not exposed to exogenously oxidized mitochondria.
One of the potentially deleterious effects of lipofuscin is a decreased efficiency of lysosomal degradation of newly (auto)phagocytosed material. This effect can be due to the competition between lipofuscin and newly formed (auto)phagosomes for the primary lysosomes delivering the hydrolytic enzymes [45,50]. Autophagy is essential for the maintenance of cellular homeostasis [4,40,53]. This is particularly important in the case of mitochondria, for which autophagy dysfunction means that damaged mitochondria, with impaired ATP production and excessive production of superoxide, can accumulate. It has been proposed that, in the case of cardiac myocytes, such overloading with damaged mitochondria may increase oxidative stress and lead to cell death, eventually contributing to heart failure [53].
Another potential deleterious effect of lipofuscin is increased oxidation of biomolecules within lipofuscin and formation of toxic products, which can cause lysosomal membrane permeabilization, act as inhibitors of lysosomal proton pumps or hydrolases, or diffuse outside the lipofuscin granule, and damage cellular organelles and cytoplasmic molecules [54,55,56].
Such a scenario may occur after the autophagy of mitochondrion (mitophagy) and its fusion with the lysosome. The low lysosomal pH can facilitate lipid oxidation mediated by the superoxide radical anion produced by mitochondria because the superoxide radical anion becomes protonated, and, in the protonated form it can dismutate much more rapidly to the hydrogen peroxide than in the non-protonated form. Lysosomal degradation of autophagocytosed mitochondria can release iron ions from the Fe-S centres and metalloproteins, which are abundant in mitochondria [57,58,59]. Indeed, it has been reported for lipofuscin from many different tissues that it contains redox-active iron [60]. The interaction of ferrous iron with hydrogen peroxide leads to the decomposition of hydrogen peroxide releasing the hydroxyl radical. The hydroxyl radical is the most reactive free radical formed in the human body: it can react with different types of biomolecules and damage them with bimolecular rate constants close to diffusion-controlled limits. Importantly, the hydroxyl radical can initiate lipid peroxidation, which is a chain reaction: it can be initiated by a single free radical, such as the hydroxyl radical or the protonated form of superoxide, leading to the oxidation of numerous lipid molecules via propagation steps. The initial products of lipid oxidation, lipid hydroperoxides, can undergo further reactions forming a wide range of products, including small end-products of lipid oxidation with reactive carbonyl groups, which can form adducts with the lysosomal proteins and with lipids with amine groups, such as phosphatidylethanolamine, thereby contributing to the formation of material not susceptible to the degradation by lysosomal enzymes.
Extensive lipid peroxidation can lead to the permeability of the lysosomal membrane and leakage of proteolytic enzymes, such as cathepsin D, into the cytosol, where they can damage cellular components [56]. As a result of oxidative modifications of biomolecules, there is a formation of cross-links between proteins, nucleic acids, carbohydrates, and lipids, which renders them no longer susceptible to lysosomal degradation. Some of the end-products of lipid oxidation are hydrophilic enough to diffuse outside the lipofuscin granules damaging biomolecules in the cytoplasm and cellular organelles.
It is plausible that the oxidation and lysosomal dysfunction can potentiate effects of each other: inhibition of lysosomal degradative enzymes provides time and environment for oxidation of (auto)phagocytosed material to proceed, resulting in the formation of material no longer susceptible to lysosomal degradation; certain oxidative modifications can inhibit directly the lysosomal enzymes and/or lysosomal proton pumps. The latter can lead to an increase in lysosomal pH, thereby decreasing the activity of several lysosomal enzymes. It can be suggested that once the accumulation of lipofuscin starts affecting the lysosomal degradation of newly (auto)phagocytosed material, the process of further lipofuscin accumulation, and accumulation of damaged organelles, such as mitochondria, can spiral out of control, leading to the massive cell dysfunction, loss of cell viability, and, eventually, can contribute to numerous pathologies.
3. Lipofuscin fluorescence
The characteristic feature of all lipofuscins is their golden-yellow fluorescence when excited with ultraviolet A or visible light [61,62,63,64]. There are numerous reports of fluorescence emission spectra of lipofuscin from different tissues. Not all of them include spectra corrected for the changes in sensitivity of detection for different emission wavelengths, or specify whether or not such a correction was done, which can lead to confusion [65,66].
In cases where care was taken to consider if the spectral correction was needed, it has been shown that the spectral range of fluorescence emission varies depending on the tissue from which the lipofuscin is derived. For example, it has been reported that the emission from lipofuscin in rat brain neurons exhibits maxima at 570 nm or 600 nm, from the adrenal glands – at 660 nm [42,61], whereas from the kidneys – at 620 nm [64,67]. Fluorescence emissions with maxima at about 600 nm were observed in the resected neocortex of 18-28 years old patients with drug-resistant epilepsy and in the neocortex from the autopsy of two human cadavers [42].
In attempts to identify the components of lipofuscin responsible for its fluorescence, several model systems have been employed attempting to create fluorophores with characteristics similar to those of lipofuscin. For example, different products with fluorescent properties have been synthesized by incubation of end-products of lipid peroxidation, such as 4HNE, MDA, or epoxy-heptenal with proteins or amino acids [68,69,70]. These small lipid-derived aldehydes modify the protein structure and form chromophores with the ability to fluoresce. Consistently, oxidation of polyunsaturated lipids, such as arachidonic acid, triggered by iron ions in the presence of glycine, leads to the formation of products with fluorescent properties [71]. Also, proteins modified by nonenzymatic glycation exhibit fluorescence. It has been demonstrated that there are some similarities in fluorescence features between those synthetic products and the fluorescence of oxidized subcellular components [72]. However, in most cases, the obtained fluorophores exhibit the emission maxima corresponding to blue light from the range of 400-490 nm, which shift towards longer wavelengths only upon a considerable increase of their concentrations [73]. In contrast, autoxidation of docosahexaenoic acid (DHA) results in the formation of products with an emission maximum at about 610 nm when excited with 488 nm light and broad-band fluorescence with a maximum in blue when excited with UV light [74].
4. Lipofuscin in the retina
4.1. Retinal pigment epithelium (RPE) is the major site of lipofuscin accumulation in the retina
The retina is a thin highly organized tissue sandwiched between the vitreous and the choroid (Figure 1). Its neural part is separated from the highly vascularised choroid by the monolayer of RPE cells. RPE cells provide the blood-retina barrier and are responsible for the uptake of vitamin A from the blood and photoreceptive neurons, its storage, and its enzymatic transformation to the chromophore of visual pigments, 11-cis-retinal. RPE cells are also responsible for the trans-epithelial transport of nutrients and waste products between the photoreceptive part of the retina and the fenestrated choriocapillaris of the choroidal blood supply. Moreover, RPE cells are responsible for the molecular renewal of photoreceptive neurons, which includes phagocytosis and lysosomal degradation of distal tips of photoreceptor outer segments (POS). Photoreceptive neurons – rods and cones, are considered the most metabolically active cells in the body. Despite that, the major site of lipofuscin accumulation in the retina is the RPE, where, based on quantification of areas of cellular cross-sections, lipofuscin can occupy up to 19% of the cytoplasmic area in RPE from cadavers of 81-90 years of age [75,76].
The massive accumulation of lipofuscin in the RPE is mainly due to the high daily phagocytic load, with some of the phagocytosed material accumulating over a lifetime as a result of incomplete lysosomal digestion [75,79,80,81,82,83]. Every day about 7-10% of POS is shed and phagocytosed by the RPE. POS in rod photoreceptors contain stacks of flattened discs made from a lipid bilayer densely populated by visual pigments. The dense stack of 600-1,000 discs is surrounded by the plasma membrane, forming a cylindrical rod-like shape, 2 µm in diameter and 24-35 µm long [84,85]. The outer segments of cone photoreceptors are tapered towards the distal end and are also made of stacks of discs, but the discs are continuous with the plasma membrane and are formed by its deep infoldings, allowing for the diffusion of membrane components from the basal side to the distal tip and vice versa. These structural differences in POS of rods and cones may have important consequences for their phagocytosis and lysosomal degradation: in the case of rods, RPE phagocytoses the oldest discs, whereas, in the case of cones, the phagocytosed material is a mixture of old and recently added lipids and proteins. Upon phagocytosis, an individual phagosome content is approximately 10-fold greater in diameter than the diameter of a typical lysosome [86]. A single RPE cell, depending on its location in the retina, phagocytoses daily tips of 12 to 43 photoreceptors, suggesting a massive phagocytic load of up to 324 µm3 POS per cell per day [87].
4.2. Oxidative stress, lysosomal dysfunction, and vitamin A derivatives as contributors to the accumulation of RPE lipofuscin
Similarly to lipofuscin in other cell types, it has been demonstrated that oxidative stress induced by excessive iron or depletion of antioxidants and/or autophagy/lysosomal dysfunction can increase the accumulation of RPE lipofuscin (reviewed in [88]). For example, a rapid lipofuscin accumulation in the RPE of rats and dogs can be induced by intravitreal injection of an inhibitor of lysosomal protease, leupeptin [27,89,90], or by intravitreal injection of ferrous sulphate or ferric ammonium citrate [91,92,93].
In addition, the lysosomal degradation in the RPE can be hindered by oxidative damage to the phagocytosed material due to the photosensitizing properties of retinaldehydes. Following the photoexcitation of visual pigment, which is the first step leading to visual perception, the Schiff base linkage between the protein and the isomerized chromophore is hydrolyzed, resulting in a release of all-trans-retinaldehyde from the protein, opsin [88,94,95,96,97,98]. The lysosomal degradation of the visual pigments can lead to the release of their intact chromophore, 11-cis-retinaldehyde. Overall, both retinaldehydes exhibit similar photosensitizing properties but the absorption spectrum of all-trans-retinaldehyde extends more into the visible range than that of 11-cis-retinaldehyde. Upon absorption of ultraviolet or blue light, retinaldehyde can form an excited triplet state. In the presence of oxygen, the photoexcited triplet state can interact with oxygen leading to the generation of reactive oxygen species, such as singlet oxygen and superoxide. The ability of superoxide to initiate lipid peroxidation in the lysosomal environment was discussed in Section 1. Singlet oxygen is an electronically excited form of oxygen molecule O2, which can oxidize biomolecules, including unsaturated fatty acids leading to the formation of lipid hydroperoxides. Interaction of the lipid hydroperoxide with iron ion leads to its decomposition and formation of lipid radicals, which can initiate a chain of lipid oxidation. POS contain high concentrations of polyunsaturated fatty acids, such as DHA and arachidonic acid, with 6 and 4 unsaturated double bonds, respectively, and therefore are very susceptible to lipid peroxidation [79,99].
It has been also shown that photoexcited all-trans-retinaldehyde can inactivate the ATP-binding cassette transporter rim protein (ABCR, also known as ABCA4), which is present in the rims of POS discs [100]. This protein facilitates the enzymatic reduction of all-trans-retinaldehyde by transporting it (usually as a Schiff base adduct with phosphatidylethanolamine (PE), N-retinylidene-phosphatidylethanolamine (NRPE)) from the intradiscal leaflet of the lipid bilayer to the cytoplasmic site, where it can serve as a substrate for retinol dehydrogenase 8 (RDH8) and other oxidoreductases [101]. ABCA4 can also transport in the same direction the 11-cis-retinaldehyde adduct with PE. In addition to its expression in photoreceptors, ABCA4 is also expressed in the RPE, where it co-localizes with endolysosomal proteins and is thought to aid in the recycling of retinaldehydes from phagocytosed tips of POS [102]. Therefore, inactivation of ABCA4 may lead to a decreased clearance of both retinaldehydes.
PE is an abundant phospholipid in POS membranes, accounting for about 35% of all lipids [99]. PE reacts with all-trans-retinaldehyde forming NRPE with a greater rate than the rate of all-trans-retinaldehyde hydrolysis from opsin, therefore it can reduce the risk of this reactive aldehyde reacting with proteins [79,88,97,99,103]. NRPE can interact with another molecule of retinaldehyde forming a pyridinium bisretinoid, abbreviated as A2PE, as well as other bisretinoids [104,105,106]. These bisretinoids are not degradable by the lysosomal enzymes except for the hydrolysis of the phosphate or fatty acid chains from PE. In the case of A2PE, this leads to the formation of A2E or lysoA2PE. The presence of bisretinoids makes RPE lipofuscin rather unique among lipofuscins from other cells. Several different bisretinoids have been detected in isolated lipofuscin and in the human retina [107,108,109,110,111,112].
Considering the release of photoreactive retinaldehydes upon exposure of the retina to light, it is not surprising to see an increased accumulation of RPE lipofuscin in response to rearing in light/dark cycle as opposed to rearing in dark or in response to short-term exposures to light causing damage to photoreceptors (reviewed in [88]). For example, an 18-hour exposure to 3000-3200 lx cool-white fluorescent light can increase lipofuscin accumulation in the RPE of Japanese quail (Coturnix Japonica) [113]. Consistently, a delayed clearance of all-trans-retinaldehyde in POS and/or 11-cis-retinaldehyde in phagolysosomes in the RPE results in an accelerated accumulation of RPE lipofuscin, which has been observed in the genetically-modified mice with deleted genes responsible for synthesis of enzymes involved in clearance of retinaldehydes, such as single gene knockouts: abca4-/-, rdh8-/-, and rdh12-/-; double knockouts: abca4(-/-)rdh8(-/-), abca4(-/-)rdh12(-/-) and rdh8(-/-) rdh12(-/-); and triple knockouts abca4(-/-)rdh8(-/-)rdh12(-/-) [102,114,115,116,117,118,119,120,121,122].
The presence of vitamin A and its derivatives plays a critical role in the accumulation of lipofuscin in the RPE [123]. Rats fed with a vitamin A-depleted diet accumulate considerably less RPE lipofuscin than rats with normal vitamin A intake [124,125,126], even when they are subjected to increased oxidative stress by depletion of vitamin E, or injection of iron, or their lysosomal degradation is inhibited by leupeptin [91,92,126]. It has been shown that deficiency in vitamin A leads not only to a decrease of lipofuscin-like fluorescence but also to a decrease in phagosomal volume in the RPE, which can be ascribed to fewer phagosomes as well as their smaller size [126,127]. Even in the case of abca4-/- knockout mice or abca4(-/-)rdh12(-/-) double knockout mice, the systemic depletion of vitamin A or pharmacological inhibition of synthesis of 11-cis-retinaldehyde results in a decreased accumulation of lipofuscin [128,129,130,131,132,133,134], whereas supplementation with vitamin A increases it [135].
As discussed in Section 1, oxidative stress and impaired lysosomal degradation can enhance each other’s effects and accelerate the formation of lipofuscin. In addition to the end-products of lipid oxidation, such as malondialdehyde (MDA) or4-hydroxy nonenal (4HNE), which can affect the susceptibility of POS to lysosomal degradation, RPE lipofuscin contains A2E, which can also inhibit lysosomal enzymes either directly or via inhibition of lysosomal ATP-dependent proton pumps [136,137,138]. Moreover, in case the RPE function is compromised, ameliorating the causes of RPE dysfunction can reduce lipofuscin accumulation. For example, the increased accumulation of lipofuscin in albino abca4-/- knockout mice can be partly inhibited by overexpression of the complement regulatory protein, complement receptor 1-like protein y (CRRY) in the RPE [102]. CRRY prevents the cleavage of complement components C3 and C5, thereby preventing the formation of the membrane attack complex (MAC) and its deleterious effects on RPE cell function and viability.
4.3. Structure and composition of RPE lipofuscin
As a result of incomplete lysosomal digestion of POS and intracellular organelles, lipofuscin accumulates in the RPE in the form of compact granules exhibiting Gaussian distribution of sizes, with the mean diameters reported as 0.69 ± 0.63 µm and 0.74 ± 0.24 [6,66]. RPE lipofuscin is an amorphous material of heterogeneous composition with the main components being lipids and extensively modified proteins [6,66,99,139,140,141]. Oxidative modifications of proteins include carboxyethylpyrrole (CEP) and iso[4]levuglandin E2, as well as adducts with other end-products of lipid peroxidation: MDA and 4HNE. Moreover, lipofuscin proteins include oxidized methionine to sulfoxide and sulfone, adducts with advanced glycation end-products (AGEs), and are modified by peroxynitrite forming nitrotyrosine.
It has been determined that there is 0.77 mmol of lipids per mg of protein in lipofuscin granules isolated from human cadavers below 40 years of age [99]. This ratio decreases with age so lipofuscin from cadavers above the age of 45 years contains only 0.41 mmol lipids per mg protein. The lipid fraction of lipofuscin is composed mainly of free fatty acids accounting for about 40%, followed by phosphatidylcholines accounting for ~30%, phosphatidylethanolamines (~13%), phosphatidylinositol (~7%), phosphatidylserine (~4%) and diacylglycerols (~3%). Free fatty acids and fatty acids esterified in lipids contain a high proportion of polyunsaturated fatty acids, with arachidonic acid accounting for 16-22% of free fatty acids and 10-13% of total phospholipids. DHA accounts for about 11-15% of free fatty acids, and 7-9% of fatty acids in phospholipids. As mentioned before, the lipophilic fraction of lipofuscin contains various bisretinoids and their oxidation products [104,105,106]. Interestingly, it has been demonstrated that solubilizing the superficial proteins by treatment with sodium dodecyl sulphate (SDS) or proteinase K removes about 80% of proteins, whereas the content of A2E and isoA2E decreases only by about 14%, and the content of all-trans-retinaldehyde dimer-PE decreases by about 7% [6].
The bisretinoid content in lipofuscin varies across the human retina, with some bisretinoids, including A2E and its oxidation product monofuran-A2E, being more abundant in the retinal periphery than in the macula [110,111,112].
The near-field scanning optical microscopy (NSOM) images of lipofuscin topography show that the lipofuscin surface appears to be composed of small particles 10-30 nm in diameter [142,143]. Due to the high content of polyunsaturated fatty acids, lipofuscin can be easily distinguished from melanosomes by comparing histological sections stained with and without osmium tetroxide and examined with transmission electron microscopy (TEM) [144]. Due to melanin content, melanosome granules appear electron dense without osmication, whereas lipofuscin granules require osmium tetroxide staining to appear electron dense. Atomic force microscopy (AFM) investigation of the lipofuscin granules have shown that the surface of the granules is smooth when examined in height images (within 4 nm resolution); however, the phase images show particle-like structures appearing on the surface, about 50 nm in diameter, separated from each other by thin layers [145]. A mixture of chloroform with methanol solubilizes partly the surface of the granules creating 20-40 nm deep indentations of the same diameter as the observed particles. The mass spectrum of the solubilized material revealed phosphatidylcholines and phosphatidylethanolamines as the main constituents of the extract. A more recent study of lipofuscin granules by AFM confirmed the existence of sub-structures with sizes of 30-40 and 100-120 nm [146].
4.4. Distribution of lipofuscin in the human RPE
Lipofuscin granules have been detected in the RPE as young as that of 16- and 17-month-old children and their concentration increases rapidly with age in people of White ethnic origin [147,148]. It was suggested that the rapid increase in lipofuscin accumulation in the first two decades of life is due to increased exposure to ultraviolet light transmitted through the lens, which can increase oxidative stress in the retina. By the age of 20 years, there is usually sufficient build-up of lenticular chromophores to absorb most of the ultraviolet light so it can no longer reach the retina. However, there is great inter-individual variability in the transmission properties of the lens [149]. In some individuals, the transmission window around 320 nm is still present above the age of 60 years.
Initially, lipofuscin granules appear in the basal area of the cells, whereas the melanosomes are present in the apical portion [75,82,147,148]. Later in life, both - melanosomes and lipofuscin become distributed more evenly throughout the cell, and this effect becomes apparent first in the posterior part of the fundus and spreads centrifugally towards the periphery.
The faster accumulation of lipofuscin in people of White ethnic origin than in Blacks has been attributed to the greater optical density of melanin in the iris and choroid of Blacks than of Whites [77,148]. As a result of the greater density of melanin in people with dark pigmentation, less light is transmitted through the iris and the anterior sclera-choroid complex, and less light is transmitted to the posterior sclera. A part of the light that reaches the sclera is reflected back to the choroid where it can be absorbed by melanin preventing it from reaching RPE and photoreceptors.
Weiter et al. have shown that there is a negative correlation between the lipofuscin and melanin contents in the RPE [148]. The lipofuscin and melanin contents have been evaluated in the fovea, perifovea, paramacula, and equator. In the centre of the fovea, where the RPE melanin density is the greatest, the lipofuscin density is about 30% smaller than its maximal density in the perifovea located at half the distance from the fovea to optic disc. From the area of its maximal density at the perifovea, lipofuscin density decreases towards the equator. In contrast, melanin density decreases from the fovea to the parafovea, remains at a similar level until the paramacula, and then increases towards the equator.
Wing et al. have demonstrated that the greatest accumulation of lipofuscin appears in the proximity of the optic disc on the nasal side and at a similar distance from the centre of the fovea on the temporal side [147]. These areas of greatest lipofuscin accumulation are close to the area with the greatest density of rods, which is 3-6 mm from the centre of the fovea [150].
RPE lipofuscin accumulates at increased rates in several retinal diseases, including Stargardt’s disease [151], Best’s vitelliform macular dystrophy [152,153], and some types of retinitis pigmentosa [154,155]. It is not clear whether there is an increased accumulation of lipofuscin in age-related macular degeneration (AMD), with some studies providing evidence of decreased fluorescence and concentration of lipofuscin granules in this disease, and an increase in complex granules called melanolipofuscin in comparison with age-matched healthy retinas [156,157].
4.5. Effects of RPE lipofuscin on the function and viability of RPE cells and photoreceptors
As in other tissues, the presence of lipofuscin in the RPE raises the question whether lipofuscin can affect the function and viability of the RPE and neighbouring cells, which rely on the RPE for support. It has been demonstrated that there is a correlation between lipofuscin density in the RPE and loss of underlying photoreceptors in people of Caucasian origin but there is no such correlation for people of Black ethnic origin [158]. It has been suggested that there may be a threshold density of lipofuscin, above which the deleterious effects of lipofuscin affect RPE cell function and subsequently, the function and viability of photoreceptors are compromised. Due to the slow accumulation of lipofuscin in Blacks, the threshold may not be achieved, and therefore there is no correlation between the lipofuscin concentration and photoreceptor loss in this population. To date, there are no subsequent studies confirming the findings from this report but there are studies contradicting these findings (reviewed by Curcio [159]). The main argument of Curcio against RPE lipofuscin affecting the viability of photoreceptors is that the age-related loss of rods is not co-localized with the area where the accumulation of lipofuscin is the greatest. The most pronounced loss of rods occurs in a ring surrounding the fovea from 0.5 to 2 mm from its centre and is no longer detectable 8 mm from the fovea centre, whereas the greatest accumulation of lipofuscin, measured by its fluorescence, occurs 2-4 mm from the fovea centre.
Like in other cell types, it can be expected that RPE lipofuscin can affect autophagy and contribute to the accumulation of damaged mitochondria. This effect can be exacerbated in the RPE because of the composition of RPE lipofuscin: it has been shown that a component of lipofuscin, A2E exhibits detergent-like properties and disrupts the structure of the lipid membrane causing its permeability [160,161]. Moreover, while A2E does not directly affect the activity of lysosomal hydrolases, A2E can inhibit the vacuolar H+-ATPase (v-ATPase) [138,162]. V-ATPase acts as a proton pump which provides low lysosomal pH needed for activation of lysosomal hydrolases. As a result of A2E effects, the lysosomal degradation of phagocytosed material can be compromised resulting in increased accumulation of phagolysosomes with non-digested contents and/or permeable membranes [17,138]. It can be expected that this can affect the phagosomal and autophagy pathways, and there is a growing body of evidence suggesting that the impairment of autophagy and, resulting from that, accumulation of damaged mitochondria can contribute to the development of AMD [38,163].
Testing the potential deleterious effects of RPE lipofuscin on mitochondria, phagocytosis, autophagy, and other cellular processes is facilitated by cell culture models. Several reports demonstrated that lipofuscin accumulation can be induced in cultured RPE cells by supplementing cells with lipofuscin isolated from human RPE cells post mortem or with POS isolated from retinas, usually bovine or rodent [80,164,165,166,167,168,169,170]. Consistent with other studies on lipofuscin accumulation as a result of phagocytosis of oxidized organelles, feeding cultured RPE cells with POS oxidized by exposure to ultraviolet light leads to 50-fold faster lipofuscin accumulation than in cells fed non-oxidized POS [166]. This effect is further enhanced by culturing cells in 40% oxygen in comparison to cells cultured under 8% oxygen whereas supplementation of RPE cells with antioxidants: α-tocopheol, lycopene, or lutein and zeaxanthin inhibits lipofuscin accumulation (reviewed in [171]). Also, enrichment of POS with retinaldehyde, 4HNE, MDA or oxidizable phospholipids, such as PE with polyunsaturated fatty acyl chains or phosphatidylcholine (PC) with monounsaturated fatty acyl chains, leads to an increased accumulation of lipofuscin [172,173,174].
The cell culture model has also enabled numerous studies investigating the effects of various factors, such as lysosomal enzymes, on lipofuscin accumulation. For example, it has been shown that cathepsin D plays a particularly important role in lysosomal degradation, with its monomeric form being more effective in preventing the formation of lipofuscin than its multimeric form [175,176]. The cell culture model of lipofuscin accumulation also allowed for testing various pharmacological approaches to inhibit it. For example, it has been shown that flunarizine and centrophenoxine can decrease lipofuscin formation in cells fed with bovine POS, presumably by decreasing Ca2+ overload, which can occur under physiological conditions as a result of exposure to all-trans-retinaldehyde [177,178]. Upregulation of autophagy by rapamycin has been shown to prevent the accumulation of lipofuscin in cultured RPE cells fed with POS, whereas inhibitors of autophagy increase lipofuscin formation [35].
Importantly, the RPE cell culture model enables to study of the effects of exposure of lipofuscin-laden cells to light. It has been demonstrated in several studies that exposure to visible light of cultured RPE cells with phagocytosed RPE lipofuscin leads to various deleterious effects [6,107,179,180,181]. For example, an exposure for 48 h of such cells in the photosensitizer-free medium to 2.8 mW/cm2 blue light (400-500 nm) providing fluence of 484 J/cm2 leads to about 50% loss of cell viability [6]. Sublethal exposures of lipofuscin-laden cells to light lead to degradation of lipofuscin, exocytosis of lipofuscin, cytoskeletal changes, decreased phagocytosis of POS, inhibition of antioxidant enzymes (superoxide dismutase and catalase) and lysosomal enzymes (cathepsin D, N-acetyl-β-glucosamidase, acid phosphatase), loss of lysosomal integrity, DNA damage, and enhanced accumulation of lipid peroxidation-derived aldehydes, such as MDA and 4HNE, and damage to mitochondrial and nuclear DNA [74,107,179,180,182,183].
It has been shown that isolated lipofuscin is susceptible to light-induced autooxidation: the exposure of a suspension of lipofuscin granules to narrow-band light results in oxygen consumption, the rates of which, when normalized to equal fluxes of incidents photons, monotonically increase with decreasing wavelength within the range of 600 to 280 nm [184]. Some of the consumed oxygen is reduced to superoxide radical, which then dismutates to hydrogen peroxide [184,185]. It has been determined that hydrogen peroxide accounts for only about 1% of oxygen consumed during the exposure of lipofuscin to blue light, indicating that the majority of oxygen is used for the oxidation of intragranular components [184]. As mentioned before, lipofuscin contains high concentrations of polyunsaturated fatty acids, and therefore they are likely to be the primary target of oxidation. Indeed, it has been shown that exposure of lipofuscin to light leads to the generation of lipid hydroperoxides and end-products of lipid peroxidation, such as MDA. It has been also shown that light exposure of lipofuscin in the presence of proteins or lipid vesicles with incorporated unsaturated fatty acids or cholesterol results in increased oxygen consumption, indicating that oxidation is not confined to the lipofuscin granule but reactive oxygen species can diffuse outside the granule and damage extra-granular molecules [184,185,186]. In the case of cholesterol, different hydroperoxides are formed, including 5α-cholesterol hydroperoxide, which is a specific product of cholesterol interaction with singlet oxygen [184].
Chloroform-methanol extraction of lipofuscin allows for the separation of the chloroform-soluble lipophilic fraction and chloroform-insoluble material [187]. Both fractions exhibit substantial photoreactivity.
The lipophilic extract of lipofuscin exhibits a broad absorption spectrum monotonically increasing with decreasing wavelength from 640 to 280 nm [188]. This mixture of numerous components includes potent photosensitizer(s), which, upon photoexcitation, form an excited triplet state with the ability to efficiently transfer the excitation energy to oxygen leading to the formation of singlet oxygen [189,190]. The quantum yields of singlet oxygen generation by photoexcited lipophilic extract of lipofuscin depend on the excitation wavelength: excitation with ultraviolet light of 355 nm wavelength or blue light from the range of 420 to 440 nm results in about 8 and 5% of absorbed photons utilized for singlet oxygen production, respectively. Superoxide is a minor product generated by the photoexcited lipophilic extract of lipofuscin. The quantum yield of blue light-induced generation of superoxide is only ~0.1%, which is about 50 times smaller than that of singlet oxygen [191].
One of the lipophilic components of lipofuscin is A2E [105,192]. Based on the quantification of A2E in lipofuscin granules and the absorption spectra of A2E and lipophilic extract of lipofuscin, it has been estimated that A2E provides only 0.8% contribution to the absorption of visible light by the lipophilic fraction of lipofuscin [97,107]. Considering A2E contribution to the absorption spectrum of lipofuscin extract and its very weak photosensitizing properties, it has been evaluated that i) A2E contributes at most one singlet oxygen molecule per 300 singlet oxygen molecules generated by lipofuscin; and ii) A2E contributes at most one superoxide molecule per 384 superoxide molecules generated by lipofuscin [193].
A2E has attracted a great deal of attention due to the simple way it can be synthesized so its effects on RPE cells can be studied in vitro [192]. Most of the studies on cultured cells used A2E solubilized in dimethylsulphoxide, and added to the culture medium, so A2E was free to diffuse to various organelles (reviewed in [88,97,193]). Under physiological conditions, A2E is present in the lipofuscin granule, and it appears to be strongly anchored within that granule, so even treatment with SDS, which leads to the removal of most identifiable proteins from the granule, does not affect the concentration of A2E remaining in the granule [6]. Therefore, it is questionable whether some of the deleterious effects of A2E on mitochondria, DNA, or transport proteins, observed when A2E is delivered in solution, are relevant to the situation in vivo where A2E is encapsulated within the lipofuscin granule.
A2E is susceptible to oxidation and, as a result, it forms various oxidation products, which include epoxides, furanoid oxides, cyclic peroxides, and carbonyls [108,109,194,195,196,197,198,199,200,201]. Several of these products have been identified in the RPE from human cadavers. Studies on cultured RPE cells, where solubilized oxidation products of A2E were delivered to cells in solution, or where A2E-laden RPE cells were exposed to light to cause its oxidation in situ, have demonstrated several detrimental effects of A2E oxidation products including DNA damage, induction of pro-angiogenic factors, activation of the complement cascade and other pro-inflammatory pathways (reviewed in [105,202,203]). It remains to be shown whether these A2E oxidation products can stimulate these deleterious effects in vivo or whether they are safely trapped in the lipofuscin granule. It has been reported that oxidized A2E can react with other oxidized A2E molecules or A2E itself forming high-molecular weight products, which are more hydrophobic than A2E, and therefore more likely to remain in the granule [204,205].
Interestingly, oxidation products of DHA, an abundant component of POS membranes and lipofuscin, exhibit potent photosensitizing properties upon exposure to ultraviolet or blue light [188]. The absorption spectrum of a mixture of products of DHA oxidation exhibits an increasing absorption with decreasing wavelength in a range of 280-600 nm. Photoexcitation of oxidized DHA with 355 nm or blue light leads to the formation of a triplet state similar to the triplet state of lipophilic extract of lipofuscin. The triplet state is quenched by oxygen, which leads to the photosensitized generation of singlet oxygen. Exposure of oxidized DHA to visible light leads to the photosensitized generation of superoxide. The quantum yields of generation of singlet oxygen and superoxide are 2.4- and 3.6-fold greater, respectively than those of the lipophilic extract of lipofuscin. This is consistent with lipofuscin containing a mixture of chromophores contributing to the absorption of light, some of which, such as A2E, have very weak photosensitizing properties, whereas others, such as oxidized DHA are very potent photosensitizers.
Chloroform-insoluble components of lipofuscin also exhibit the ability to photosensitize the generation of singlet oxygen, superoxide, and oxidation of exogenous lipids and proteins [187]. Interestingly, when studied at the same concentration of dry mass, both soluble and insoluble fractions of lipofuscin demonstrate no age-related changes in photoreactivity, even though lipofuscin granules become more photoreactive with age. What changes with age is the ratio of insoluble to soluble components per lipofuscin granule, which increases with age. Therefore, the age-related increase in the photoreactivity of lipofuscin granules can be attributed to the increase in the insoluble part. However, to date, no successful attempts have been reported to identify the visible light-absorbing chromophores of the insoluble part, which could be responsible for the observed photoreactivity.
As discussed above, numerous studies in vitro point to the potential phototoxic effects of lipofuscin in the RPE in vivo, which is exposed daily to light in the presence of high oxygen tensions of about 70 mm Hg [206], thus providing ideal conditions for the photoexcitation of photosensitizers of lipofuscin, and subsequent formation of reactive oxygen species, which can cause oxidation of the lipofuscin components and damage cellular proteins, lipids and nucleic acids. While it is clear that lipofuscin exhibits photosensitizing properties and can affect cell function and viability in vitro upon exposure to light, the evidence for the contribution of lipofuscin to light-induced retinal injury in vivo is rather limited. The retina is protected from exposure to excessive fluxes of light by various mechanisms (reviewed in [88]). They include limiting fluxes of light reaching the retina by the eyelid and eyebrows, and the action of the iris regulating the pupil size. Moreover, the high concentrations of enzymatic and low-molecular-weight antioxidants, detoxification enzymes, and efflux transporters can prevent oxidative damage to biomolecules and/or remove damaged products. Thus, it may be argued that the retina is equipped well to defend itself from the noxious species generated by lipofuscin, and the deleterious effects occur only when the capacity of these cellular defences is exceeded.
One of the circumstantial pieces of evidence pointing to lipofuscin contribution in light-induced injury is the age-related increase in susceptibility to retinal photodamage in rats reared under dim cyclic-light as opposed to rats reared in dark [207]. It can be suggested that rats reared under dim cyclic light may have an increased lipofuscin content in the RPE but this was not evaluated in the study.
It has been shown in numerous studies that mice with abnormal trafficking of retinaldehydes and accelerated lipofuscin accumulation, such as Abca4(-/-)Rdh8(-/-) double knockout, are very susceptible to light-induced retinal injury [117,118,129,130,178,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227]. In most of these studies, 30-60 minute exposure of Abca4(-/-)Rdh8(-/-) double knockout mice with dilated pupils to light from a desk lamp equipped in a fluorescent bulb led to a substantial photoreceptors loss [117,118,129,130,178,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,226]. This loss of photoreceptors was effectively prevented by inhibitors of the synthesis of 11-cis-retinaldehyde and/or scavenging of retinaldehydes by amine compounds such as retinylamine. Therefore, these studies provided solid evidence for retinaldehydes being responsible for light-induced retinal injury but have not provided evidence for the lipofuscin contribution to that injury.
It has been recognized very early in the studies on light-induced injury to the retina that RPE is the primary target for the threshold injury induced by light of 441 nm and longer wavelengths applied to the macaque retina [228]. More recent studies on macaques demonstrated that exposure of the retina to 460, 488, 544, 568, and 594 nm laser light can lead to bleaching of RPE fluorescence and, at higher retinal radiant exposures, varying from about 30 to 300 J/cm2 for the shortest and longest wavelengths, respectively, to the disruption of the RPE mosaic, suggesting that photoreactivity of lipofuscin could contribute to the RPE injury [229,230,231,232]. Interestingly, these studies showed that light-induced retinal injury occurs at much lower radiant exposure thresholds for the cyan, green, yellow, and orange lasers than expected based on previous reports by Ham and colleagues [233]. A possible explanation for the discrepancy in the observed radiant exposure thresholds for injury could be the different concentrations of RPE lipofuscin, making macaques with more lipofuscin more susceptible to light injury; however, lipofuscin was not quantified in any of these studies.
To date, two reports suggest that RPE cells can be the primary target of light-induced injury in abca4-/- knockout mice with an increased accumulation of lipofuscin [225,227]. One of them demonstrated that a 15-minute exposure to blue light (430 nm wavelength, 50 mW/cm2 irradiance) of pigmented wild-type and abca4-/- knockout mice results in a loss of some photoreceptors and no significant loss of RPE cells in the wild-type mice, whereas the knockout mice exhibit a massive loss of RPE cells in the central retina with the loss of photoreceptors not greater than that in the wild-type mice [225]. It was interpreted that the damage to the RPE in the knockout mice was caused by lipofuscin. While this can be true, it cannot be excluded that the abnormal trafficking of all-trans-retinaldehyde due to the absence of ABCR protein in the POS and RPE, affects RPE cells more than photoreceptors. Such a possibility is supported by experiments of Wu et al. on susceptibility to light damage of albino wild-type and abca4-/- knockout mice of different ages [234]. They have shown that 8-month-old abca4+/+ mice have similar concentrations of A2E isomers as 2-month-old abca4-/- mice but are much less susceptible to RPE cell loss induced by exposure of the retina to 430 nm light than the young knockout mice.
A follow-up study by Fang et al. provided another circumstantial piece of evidence by demonstrating that upon pharmacological removal of RPE lipofuscin from 12-month-old abca4-/- mice, the exposure to blue light is less damaging [227]. The mice were pre-treated with soraprazan, a drug developed to treat gastroesophageal reflux, which, due to its ability to decrease the lipofuscin content, was renamed Remofuscin. Remofuscin was injected intravitreally and, after 28 days, led to decreased levels of LF and melanosomes in the RPE when quantified by fluorescence and transmission electron microscopy. Exposure to blue light was also performed 28 days after Remofuscin injection. Mice pre-treated with Remofuscin retained about twice more RPE and photoreceptor nuclei than those treated with DMSO used as a vehicle or without pre-treatment. Still, some doubts remain regarding the role of lipofuscin in mediating light-induced injury because Remofuscin has been shown to generate reactive oxygen species, such as superoxide, upon exposure to blue light. Therefore, it can be suggested that mice pre-treated with Remofuscin may have upregulated antioxidant and detoxification defences, making them more resistant to light-induced injury than mice pre-treated with the vehicle.
Teussing et al. have demonstrated in a small clinical study on five Stargardt’s disease patients with identified disease-related mutations in ABCA4 gene that protecting one eye from light by wearing for at least 11 months a black contact lens during waking hours results in a smaller percentage of hypofluorescent pixels in the fundus fluorescence image, corresponding to focal losses of the RPE, in comparison with the unprotected fellow eye [235]. Like in studies on rodents described above, it is not clear whether the protective effect was due to preventing the deleterious effects of retinaldehydes and/or lipofuscin.
Another study on the potentially deleterious effects of lipofuscin accumulation has been done on Abca4(-/-)Rdh8(-/-) double knockout mice reared in the dark [17]. Rearing in the dark slows down the light-induced retinal degeneration in these double knockout mice while still enabling the massive accumulation of lipofuscin in the RPE and preventing its photodegradation, as opposed to rearing under low-intensity cyclic light (below 10 lx), which can cause a total loss of photoreceptors and RPE degeneration by the age of 6 months as shown by Maeda et al. [17,118]. Interestingly, Abca4(-/-)Rdh8(-/-) double knockout mice used by Pan et al. have demonstrated similar loss of photoreceptors and RPE nuclei at 12 months of age independently whether the mice were housed in dark or in cyclic light of about 10 lx illuminance [17]. At the old age of 26 months, the thickness of the layer with photoreceptor nuclei in these double knockout mice decreases by about 40% in comparison with the wild-type mice, whereas the number of RPE nuclei decreases by about 30%. The remaining RPE cells in the 24-month-old Abca4(-/-)Rdh8(-/-) mice are significantly larger and with more nuclei per cell than in wild-type mice and, unlike healthy-looking wild-type RPE cells, exhibit stress fibres.
The fluorescence of lipofuscin in 3 months old Abca4(-/-)Rdh8(-/-) mice is significantly greater than in 33-month-old wild-type mice [17]. At 12 months, the double-knockout mice reared in the dark have about 5-fold greater fluorescence than their counterparts raised in cyclic light. At 20 months, the dark-reared double knockout mice exhibit massive accumulation of RPE lipofuscin, and, in some RPE cells, anti-galectin-3 positive staining, which, in some small areas, colocalizes with staining for lysosomal marker Lamp1, suggesting loss of lysosomal membrane integrity [17,236].
Interestingly, the flat-mounts of the Abca4(-/-)Rdh8(-/-) eyecups with removed neural retinas exhibit anti–phospho-Ser358 MLKL staining in RPE already at 2 months, and that staining density strongly increases with age, whereas, in the wild type RPE, it is virtually undetectable even at 27 months [17]. MLKL stands for mixed lineage kinase domain-like protein, which is a protein essential for triggering necroptosis and is also involved in facilitating endosomal trafficking and generation of extracellular vesicles [237]. In 12- and 26-month-old Abca4(-/-)Rdh8(-/-) mice, the anti–phospho-Ser358 MLKL staining is present not only in the cytoplasmic areas but also in the plasma membrane [17]. Retinal cross-sections from 20-month-old double knockouts exhibit the anti–phospho-Ser358 MLKL staining not only in the RPE layer but also in retinal layers occupied by photoreceptors: the outer and inner segment layers and outer nuclear layer, where the staining appears punctate. Interestingly, the anti–phospho-Ser358 MLKL staining in 26-month-old RPE can be completely prevented by a single intravitreal injection of necroptosis inhibitor, necrostatin 7 (Nec7) a week prior to the dissection. However, injecting another necroptosis inhibitor, namely necrostatin-1 (Nec1; an inhibitor of receptor-interacting serine/threonine-protein kinase 1 (RIPK1)), or a vehicle has no effect. Nec7 is not an inhibitor of RIPK1 and its mechanism of action as a necroptosis inhibitor is unclear. Nevertheless, it also protects from RPE cell loss in these double knockout mice when injected once a month for 6 months starting from the age of 7 months.
The double knockout Abca4(-/-)Rdh8(-/-) neural retinas include large clusters of lipofuscin and melanosomes enclosed in structures of about 5-10 µm in diameter [17]. The photoreceptor layer in proximity to such structures appears more disorganized with fewer nuclei than elsewhere, and the remaining nuclei stain positive for terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL), suggesting that these photoreceptors are undergoing an apoptotic type of cell death. Lipofuscin-containing debris outside RPE detected in these double knockout mice vary in diameter from about 1-20 µm to much larger structures forming sub-retinal deposits which usually are surrounded by anti–phospho-Ser358 MLKL staining, suggesting that phosphorylated MLKL may promote shedding parts of photoreceptors and RPE as a way to minimize necroptotic cell death [17].
The anti–phospho-Ser358 MLKL staining has been also detected in the human retinas from 74-, 80- and 86-year-old donors affected by dry AMD, but not in the normal retina from a 42-year-old donor [17]. The staining in AMD retinas spread from the RPE to the outer nuclear layer and appears either punctate or covers larger areas, some of which co-stain for Iba1, a marker of microglia. It is stated by the authors that there is a correlation between the accumulated lipofuscin and staining of anti–phospho-Ser358 MLKL and Iba1 but no data supporting that statement are provided. The authors suggest that lipofuscin induces the permeability of the lysosomal membrane and subsequent leakage of lysosomal enzymes, which triggers the formation of atypical necrosome, not dependent on RIPK1 nor RIPK3, which can phosphorylate MLKL. Phosphorylation of MLKL enables its oligomerization and insertion into the lysosomal or plasma membranes forming pores, which lead to membrane permeability, with subsequent shedding of cell fragments or cell death by atypical necroptosis.
Another study on abca4(-/-)rdh8(-/-) double knockout mice, which provides another piece of evidence of the deleterious effect of lipofuscin, used supplementation with a form of vitamin A, where a hydrogen atom at carbon 20 is substituted by deuterium to prevent the formation of bisretinoids [238]. This strategy leads to a substantial decrease in lipofuscin fluorescence, which in 3- month-old mice is about 75% less intense than in the retinas of mice fed with normal vitamin A. The effect is similar for A2E, isoA2E, and their oxidation products, which accumulation was quantified between 1 and 18 months and has been shown to be decreased by about 75% in comparison with mice fed normal vitamin A. Interestingly, in both treatment groups, the bisretinoids increased up to the age of 8 months and then reached a plateau. There were no differences at the ages of 7 and 18 months between the normal and deuterated vitamin A-supplemented mice in electroretinogram amplitudes of a- and b-waves, which reflect functions of photoreceptors and bipolar cells, respectively. The age-related decrease in a- and b-waves was similar in both groups. Supplementation with deuterated vitamin A was associated with improved dark-adaptation in 12-month-old mice in comparison with mice supplemented with normal vitamin A, where 30 minutes after photobleaching mice recovered 71% and 53% of the fully dark-adapted b-wave amplitude for deuterated and normal vitamin A, respectively. It has been stated by the authors that the deuterated vitamin A partly prevented age-related loss of retinal thickness measured at 12 and 18 months when compared with retinal thickness at 3 months. The retinal thickness in mice supplemented with deuterated vitamin A decreased only by 5% and 3% at 12 and 18 months, respectively. The retinal thickness in mice on normal vitamin A decreased by 17% and 15% at 12 and 18 months, respectively. The thickness was measured 1 mm from the optic disc, but it is not specified whether and how the eyes were oriented before enucleation, so it is not clear whether these measurements were taken from the same location in each retina. It is important because there is a different susceptibility to photoreceptor loss in the superior and inferior retina of rodents. Interestingly, there were no statistically significant differences in the thickness of the outer nuclear layer between the treatment groups. The outer nuclear layer is made of cell bodies of photoreceptors and its thickness can reflect their density. Importantly, deuterated vitamin A completely prevented focal degenerative changes starting to appear at 12 months in mice supplemented with normal vitamin A, where RPE cell loss or hypertrophy, the appearance of pigmented cells in the photoreceptor layers was detectable in every eye examined from twelve 18-month-old mice, suggesting that lipofuscin bisretinoids can lead to deleterious effects in the retina in vivo.
The beneficial effects of deuterated vitamin A were also shown in experiments on abca4(-/-) single knockout mice [239]. In comparison with 9-month-old animals supplemented with normal vitamin A, age-matched animals supplemented with deuterated vitamin A had about 50% decreased cytoplasmic volumes occupied by lipofuscin granules in the RPE and intensities of fundus fluorescence excited by 488 nm laser, as well as a 5-fold decreased levels of A2E. The eyecups from knockout mice supplemented with normal vitamin A had 2.2-fold increased mRNA levels of complement component C3 and 2.0- and 2.3-fold decreased mRNA levels of complement factor B and complement factor properdin in comparison with wild-type mice on the same supplement. Importantly, supplementation of abca4-/- with deuterated vitamin A resulted in the same levels of mRNA for all three complement factors as those in the eyecups from wild-type supplemented with normal vitamin A. These findings are significant considering the associations of complement proteins with AMD [240,241].
Altogether, while there is a large body of evidence on the deleterious effects of RPE lipofuscin in cultured cells exposed to light levels exceeding typical light levels reaching the retina in vivo, the evidence suggesting that lipofuscin can exert deleterious effects in the retina in vivo is still rather limited and requires further exploration.
5. Fluorescence of RPE lipofuscin
When excited with light, isolated RPE lipofuscin emits a characteristic golden-yellow fluorescence with a broad maximum at about 580-630 nm for lipofuscin in suspension and at about 610-630 nm for lipofuscin dried on a microscope slide [63]. The emission peaks are at similar wavelengths irrespectively whether lipofuscin granules are excited with 364 nm or 476 nm light. For lipofuscin isolated from 5-29 years old donors, the emission spectra exhibit shoulders at 470, 550, and 680 nm. The contributions of 470 and 550 nm shoulders to the emission spectrum are less pronounced for lipofuscin from 30-49-year-old donors and are no longer noticeable for lipofuscin from the group of donors above 50 years of age. In contrast, the contribution from the 680 nm shoulder seems to increase with age.
Spectrally resolved confocal microscopy with a greater sensitivity of fluorescence detection than in experiments of Boulton et al. [63] enabled to show that there is a large variation in the emission maxima between individual lipofuscin granules with the wavelengths corresponding to the maxima varying from 513 to 645 nm [142,145,242]. It has been demonstrated that varying the excitation wavelength from 532 to 400 nm does not affect the long-wavelength downward slope of the emission, and the emission maximum remains the same for the excitations with 400, 457, or 488 nm, and shifts slightly towards longer wavelengths for excitations with 514 or 532 nm. With the increasing excitation wavelength, the emission spectrum becomes narrower, suggesting a decreasing number of excitable fluorophores as the excitation wavelength increases. Studies of fluorescence emission and polarization spectra upon excitation with 400 nm light have shown that the emission is significantly polarized for 430 nm wavelength and that polarisation decreases rapidly with increasing wavelength until reaching a relatively steady low value at 575 nm, which remains at that level till at least 725 nm (the end wavelength of the detection range used in the study). These changes in polarization for different emission wavelengths suggest that there are different fluorophores contributing to the emission in the blue and green spectral ranges than the fluorophores contributing to the emission in the yellow-red spectral range. It appears that at least some fluorophores emitting in the blue-green spectral range are also the chromophores absorbing the excitation light, whereas at least some fluorophores emitting in the yellow-red spectral range are excited via energy transfer from blue-light-absorbing chromophores. The simultaneous collection of NSOM images of fluorescence and topography of lipofuscin granules shows that orange-emitting fluorophore(s), are not uniformly distributed within the granule [142,143,146].
A2E and other bisretinoids are considered the major emitters of yellow and red fluorescence from photoexcited RPE lipofuscin [105,243,244]. Quantum yields of A2E fluorescence have been studied in Triton X-100 micelles in aqueous solution as well as in various organic solvents and shown to be the greatest in Triton X-100 where 1.1% of absorbed photons are used for emission of fluorescence [245,246]. A direct comparison of fluorescence quantum yields of A2E with the lipophilic extract of lipofuscin shows that fluorescence emission from the lipophilic extract of lipofuscin is 70-fold greater than for A2E [190,247]. Thus, the small contribution to the absorption of lipofuscin and the findings of Haralampus-Grynaviski et al. suggest that to be the major emitter of fluorescence in the lipofuscin granule, A2E must act as an energy acceptor from other blue-light-absorbing chromophores [145]. While the blue-light-absorbing chromophores, which can act as energy donors for A2E have not been identified, it can be suggested that some products of oxidation of DHA can be the potential candidates [74,188]. Lipofuscin contain proteins modified by products of lipid oxidation, such as 4HNE, MDA, and therefore it can be expected that they contribute to lipofuscin fluorescence excited by UV light [68,69,70,140,248].
It has been demonstrated that exposure of lipofuscin isolated from human RPE to visible light results in substantial changes in the fluorescence spectra [74,249]. For experiments shown in Figure 2, lipofuscin granules were suspended in PBS, exposed to 9.76 mW/cm2 visible light, and their fluorescence emission spectra were measured at selected times for up to 10.5 h [74]. As a result of exposure to visible light, there is a dose-dependent decrease in the intensity of fluorescence above 560 nm. The emission in the range of 530-550 nm exhibits changes dependent on the excitation wavelength: an excitation with 360 nm light results in a small, dose-dependent increase in fluorescence followed by a plateau, whereas an excitation with 488 nm causes an initial increase in the fluoresce emission in that range followed by a decrease. The decrease in the long-wavelength fluorescence during the photodegradation of lipofuscin is accompanied by a concomitant increase in the emission in the range of 380–520 nm observed upon excitation with 360 nm light.
Because bisretinoids are the major emitter of lipofuscin fluorescence [105,106,145], it is plausible that some of the observed changes in fluorescence during lipofuscin photodegradation can be ascribed to light-induced oxidation of bisretinoids, which are prone to oxidative degradation.
To compare the changes of fluorescence upon photodegradation of lipofuscin and A2E, a suspension of lipofuscin granules and a suspension of proteoliposomes made of cardiolipin, bovine serum albumin and A2E were exposed to visible light under the same conditions [249]. Photodegradation of A2E proteoliposomes led to a decrease in fluorescence above 540 nm, which appears to be an isosbestic point below which an increased fluorescence occurs. Some of the A2E photodegradation products are hydrophilic enough to diffuse out of the liposomes and exhibit that short-wavelength fluorescence. The emission maximum of the A2E photodegradation products appears at a shorter wavelength than the emission maximum of the lipofuscin photodegradation products. Another source of short-wavelength fluorescence appearing during the photodegradation of lipofuscin can be oxidized DHA [74].
An addition of A2E to the suspension of photodegraded lipofuscin granules results in an expected increase in the long-wavelength emission and in a decrease in the short-wavelength emission, providing additional evidence for energy transfer to A2E from the chromophores absorbing 365 nm light used for photoexcitation [249].
Spectral changes of fluorescence emission similar to those observed during photodegradation of lipofuscin suspension have been observed upon exposure to visible light of lipofuscin-laden RPE cells, ARPE-19 (Figure 3) [74]. Cells were exposed to the same irradiance as the suspension of lipofuscin but, unlike the exposure of lipofuscin in suspension, which was continuous, the radiant exposure of cells was fractionated into 45-minute daily exposures delivering the same total dose after 14 days as that delivered after 10.5 hours of irradiation of lipofuscin in suspension. This approach was needed to ensure the maintenance of cell viability and function. Interestingly, upon exposure to light, the cells assembled vesicles containing multiple lipofuscin granules, which appeared floating above the cell monolayer. As a result of the removal of lipofuscin from cells, both - the density of lipofuscin granules inside cells and the fluorescence intensity of cells solubilized in Triton X-100 considerably decreased. There was a marked increase in the ratio of blue-green emission to yellow-red emission.
6. Fluorescence of the retina:
6.1. Sources of fluorescence in the retina
In addition to lipofuscin, the retina contains a number of other fluorophores. Like all other cells from different tissues, retinal cells contain NAD(P)H, flavins and porphyrins, which emit fluorescence upon absorption of light. NAD(P)H requires excitation with ultraviolet light and emits broad-band fluorescence ranging from 400 up to 650 nm with maxima at ~ 440 and 460 nm for the protein-bound and free state, respectively [250]. Flavins can be excited by ultraviolet light and blue light and emit in the range of 470-650 nm with maxima at about 445 and 540 nm, respectively [251,252]. The fluorescence of flavins and NAD(P)H has been investigated as a way of monitoring the metabolic status of cells. Porphyrins appear transiently in the multistep biosynthetic pathways of hemes, which serves as a prosthetic group of many proteins, including cytochromes, cytochrome c oxidase, catalase, peroxidases, and normally porphyrins do not accumulate in substantial concentrations [253].
Photoreceptors, Müller cells and RPE contain considerable concentrations of retinoids, which also exhibit fluorescence properties [254,255]. In particular, retinyl palmitate, which serves as a storage form for vitamin A from which the chromophores for visual pigments are synthesized, can accumulate in particularly high concentrations in the RPE and emits broad range fluorescence between 400-650 nm with a maximum at ~490 nm when excited with ultraviolet light [254,255,256].
In the RPE, in addition to lipofuscin, there are other pigment granules, which can emit fluorescence: melanosomes and melanolipofuscin [63,257]. Melanosome number in human RPE cells decreases with age whereas melanolipofuscin increases [75,258].
Studies of pigment granules isolated by differential centrifugation from human RPE revealed that melanosomes, when excited with 364 nm light, emit broad-band fluorescence with emission peaks at 440 and 560 nm and a tail extending beyond 700 nm [63,257]. A comparison of the intensity of fluorescence at their corresponding maxima showed that the fluorescence intensity of RPE melanosomes is 3.7 to 6.7-fold smaller than for lipofuscin. The excitation maxima of the isolated melanosomes appear at 350 and 450 nm. Isolated melanolipofuscin granules also emit broad-band fluorescence, extending beyond 700 nm, with the main emission peak at about 560 nm and shoulders at 460 and 680 nm. The excitation spectrum of melanolipofuscin is similar to that of lipofuscin with a broad maximum at about 400 nm and a sharp peak at 460 nm.
In contrast to the findings on isolated pigment granules described by Docchio et al. and Boulton et al. [63,257], the evaluation of pigment granules in RPE cells by high-resolution structured illumination microscopy (SIM) and detection of fluorescence excitable by 488 nm laser, allowing for the lateral resolution improvement to 110 nm, demonstrated that human RPE cells do contain melanosomes granules, both spherical or spindle-shaped, which do not fluorescence when excited with blue light [259]. These findings are based on an analysis of an impressive number of 193,096 individual pigment granules in 450 RPE cells. Bermond and colleagues also identified different types of melanolipofuscin granules, some of which exhibit non-fluorescent cores similar to melanosomes surrounded by a fluorescent coat, others appear as fluorescent inclusions inside the non-fluorescent granules, while some other granules appear highly fluorescent throughout. It is possible that some of these granules classified as melanolipofuscin by Bermond and colleagues have a high enough density to pass through 2 M sucrose together with melanosomes but do not appear as different to melanosomes by transmission electron microscopy, the techniques used by Boulton and colleagues for isolation and testing the purity of pigment granules, respectively.
Unlike lipofuscin, for which photooxidation results in an increase of the short-wavelength fluorescence and a decrease in the long-wavelength fluorescence, photooxidation of melanosomes leads to an increase in their fluorescence throughout the visible and near-infrared [74,260,261].
Due to the high concentration of polyunsaturated fatty acids, especially in photoreceptor outer segments, lipofuscin, drusen, and Bruch’s membrane it can be expected that products of lipid oxidation and their adducts with proteins can contribute to retinal fluorescence [68,69,70,74,140,248].
In addition to the fluorophores present in the normal retina, certain pathologies result in the accumulation of additional fluorophores. In the case of impaired phagocytosis of POS by RPE, such as that occurring in the Royal College of Surgeons (RCS) rats, there is an accumulation of material with lipofuscin-like fluorescence at the interface of POS and RPE [125]. RCS rats carry a mutation in MERTK gene coding a receptor Mer tyrosine kinase (MerTK) [262]. MerTK is a widely expressed receptor involved in phagocytosis of shed POS by the RPE as well as apoptotic bodies by macrophages and microglia [262,263,264,265]. Mutations in human MERTK can cause early-onset retinal degenerations including rod-cone dystrophy (also known as retinitis pigmentosa) and cone-rod dystrophy [266]. There are a number of other conditions affecting the human retina where the accumulation of non-phagocytosed POS can occur and has been documented in a number of case reports, such as detachment of the neural retina from the RPE [267], central serous chorioretinopathy [268], vitelliform macular dystrophy [269] or optic nerve pit maculopathy with serous macular detachment [270].
In the cases of intra-retinal or sub-retinal haemorrhages, the metabolic degradation of haemoglobin can result in the accumulation of biliverdin and bilirubin which, when excited by UVA or blue light, exhibit broad-band fluorescence emission with maxima at about 470-480 and 520-540 nm, respectively, and the long-wavelength tail extending well above 600 nm, especially for bilirubin bound to serum albumin or erythrocyte ghosts [271,272,273,274].
Sub-retinal and sub-RPE deposits, which accumulate with age and some of them are a hallmark of AMD, such as reticular pseudodrusen, basal laminar deposits, basal linear deposits and drusen, can fluorescence [275,276,277]. The fluorescent properties of drusen appear highly variable, ranging from homogenous fluorescence to highly irregular with hypo- and hyper-fluorescent areas, and some with no fluorescence.
6.2. Imaging of fluorescence in the retina
Fluorescence of the retina can be detected in the human eye non-invasively in vivo [254,278]. Because it does not involve exogenous fluorophores, it is often referred to as autofluorescence. In an elegant series of experiments, Delori et al. have demonstrated that fundus autofluorescence is mainly due to the fluorescence of retinal structures behind photoreceptors and exhibits characteristics of RPE lipofuscin [278]. These experiments involved 30 White volunteers ranging in age from 21 to 67 years, with a distribution of 6 individuals per decade. The fluorescence excitation was obtained from a xenon arc lamp and one of the fluorescence filters transmitting light centred at 430, 450, 470, 490, 510, 530 or 550 nm. The area of the retina exposed to the excitation was a 3° in diameter centred at the fovea or at 7° temporal to the fovea, while the emission was collected from an area of 2° (corresponding to 585 µm) in diameter. The emission intensity was corrected for the spectral characteristics of the detection system. The emission spectra from the human fundus exhibited maxima at 620-630 nm for all excitation wavelengths except for 550 nm, which caused a small shift of the maximum of about 10 nm towards longer wavelengths. The excitation spectrum recorded for the emission at 620 nm exhibited a maximum at 510 nm. For the excitation with 430 or 470 nm, some emission spectra showed additional fluorescence with the broad emission between 520-580 nm, indicating that more fluorophores contribute to the emission induced by blue light than by green light.
Delori and colleagues compared the spectral characteristics of the fluorescence emission from human fundus 7° temporal from the fovea in vivo with the spectral characteristics of RPE explants taken at 36-33° from the fovea of post-mortem donor eyes fixed in 4% paraformaldehyde and demonstrated that they are similar [278].
They also demonstrated that the topographical distribution of fundus fluorescence and an age-related increase in fluorescence intensity is consistent with the quantification of lipofuscin granules in the RPE sections examined by TEM [75,278]. Fundus fluorescence in vivo excited by light of 510 or 550 nm wavelength and monitored at 620 nm shows an age-related statistically significant increase both in the fovea and at 7° temporal to the fovea. For excitation with 470 nm light, only the fluorescence at 7°exhibits a statistically significant age-related increase whereas fluorescence at the fovea does not. For the emission obtained by excitation with 430 nm light, there is no correlation with age in that age range of 21-67 years.
For all excitation wavelengths, the fluorescence at the fovea is 41 to 75% less intense than at 7° away from the fovea [278]. This effect is more pronounced for excitations with blue light than for green light of 510 or 550 nm wavelengths, indicating that, apart from lower lipofuscin content in the fovea, additional factor(s) are involved. Macular pigment present in the inner retina can efficiently absorb light of 430 and 470 nm wavelengths, but much less green light of 510 nm wavelength, and its absorption of 550 nm light is negligible. The difference spectrum obtained by subtracting the fluorescence excitation spectrum from the fovea from the spectrum for the 7° allowed the authors to calculate the absorption spectrum of the chromophores absorbing blue light and demonstrate that it matches the absorption spectrum of the macular pigment, thus providing an additional explanation for the lower fluorescence emission from the fovea than outside this area for the excitation with blue light.
On the temporal side from the fovea, the fluorescence intensity remains high up to 15° and then gradually declines towards the equator [278].
As an additional test of the site of origin of retinal fluorescence, Delori and colleagues compared fundus fluorescence at 10-15° temporal to the fovea from dark-adapted and light-adapted volunteers and demonstrated that the difference in emission spectra can be explained by the visual pigment of rods absorbing partly the 450 nm light used for excitation and emitted fluorescence [278]. This provided evidence that the fluorescence originates behind POS.
To examine the origin of fluorescence in retinal layers behind POS, Marmorstein et al. investigated fluorescence properties of non-fixed cryo-sections from human retina-choroid-sclera complex isolated post-mortem from normal eyes of 81, 83, and 87 years of age. They used 364, 488, 568, and 633 nm as the excitation wavelengths of the laser scanning confocal microscope [275]. The emission spectra were not corrected for the changes in detection sensitivity at different wavelengths, but as they were collected with a Hamamatsu R6357 detector, which exhibits similar radiant sensitivity and quantum efficiency for different wavelengths from the range of 400-800nm, it can be expected that the reported wavelengths of the emission maxima are close to the true values. They have demonstrated that RPE emits strong fluorescence when excited with any of the four wavelengths, and the emission spectra maxima shift towards the longer wavelength as the excitation wavelength increases to yellow (568 nm) or red (633 nm) light [275]. For the excitations with 364 or 488 nm light, the emission maxima for the RPE are at the same wavelength of 555 nm. For the excitations with 568 or 633 nm, the emission maxima are at 615 and 655 nm, respectively. The Bruch’s membrane and sub-RPE deposits: basal laminar, basal linear deposits, and drusen, also emit fluorescence but with the emission maxima at shorter wavelengths than for the RPE. The most pronounced difference has been seen for the excitation with 364 nm, where the emission maximum for Bruch’s membrane/sub-RPE deposits is at 485 nm, which is 70 nm shorter than the wavelength for the emission maximum from the RPE. For other excitation wavelengths, the differences are only 5-15 nm.
The intensities of the emission maxima of the Bruch’s membrane/sub-RPE deposits are about 10-fold smaller than for the RPE for excitations with 633 and 568 nm [275]. For the excitation with 488 nm, the emission maximum of the RPE is still 40 and 60% greater than for Bruch’s membrane and sub-RPE deposits, respectively. However, the emission intensity maximum of Bruch’s membrane is similar to that of the RPE for the excitation with 364 nm. Because the emission spectrum of Bruch’s membrane is narrower than that of the RPE, it can be expected that, when measuring total fluorescence intensity integrated over a wide spectral range, the contribution of Bruch’s membrane can be smaller than that judged by the intensity of its spectral maximum. The emission spectrum of sub-RPE deposits excited by 364 nm light is similar to that of Bruch’s membrane but its maximum is about 15% smaller.
All in all, Marmorstein and colleagues have demonstrated that RPE fluorescence strongly dominates for excitations with 568 or 633 nm light; excitation with 488 nm brings about strong emission of not only RPE but also considerable contributions from Bruch’s membrane and sub-RPE deposits, which become comparable to the RPE emission upon photoexcitation with 364 nm light [275]. These studies were done on 8 µm thin tissue cross-sections, therefore there was no issue with the RPE pigments preventing the excitation light from reaching the Bruch’s membrane and sub-RPE deposits, as well as filtering the emitted light. This issue is encountered when imaging the fluorescence en face mainly due to the filtering effect of RPE pigment granules [75,258,279]. Moreover, this filtering effect is likely to change with ageing because i) there is an overall loss of melanin; ii) there is an accumulation of lipofuscin and complex granules containing both melanin and lipofuscin; and iii) melanosomes redistribute from their initial location in the apical portion of the RPE and lipofuscin redistributes from its initial location at the basal site to become distributed more evenly throughout the cell.
Another study of fluorescence of RPE and sub-RPE deposits has been done on paraformaldehyde-fixed retinal cross-sections from Caucasian eyes and employed multiphoton laser scanning microscopy using 960 nm wavelength from 140 femtosecond laser as the excitation source [277]. Following such multiphoton excitation, sub-RPE deposits exhibit a broad emission maximum at about 560 nm whereas the emission maximum from the RPE is at about 610 nm.
In a study on twenty 20 µm thick flat-mounts from human RPE-Bruch’s membrane complexes without retinal pathologies, Smith et al. used narrow-band excitation from mercury arc lamp and dichroic filters with transmission centred at 436 and 480 nm wavelengths, and measured fluorescence emission spectra en face at the fovea, at perifovea (2 mm superior to the fovea), and at the periphery (10 to 12 mm superior to the fovea) using a hyperspectral camera, which recorded a fluorescence emission spectrum every 10 nm in the range of 420-720 nm from each pixel in the imaged area [280]. The emission spectra were corrected for the spectral changes in the quantum efficiency of the detector. The areas devoid of RPE allowed for the collection of emission spectra from Bruch’s membrane, which exhibited a broad flat maximum at about 550 nm when excited with either 436 or 480 nm. The position of that maximum was similar to the emission maximum from the Bruch’s membrane/sub-RPE deposits at about 540-545 nm obtained by photoexcitation with 488 nm laser reported by Marmorstein et al. on non-fixed cryo-cross-sections from human retinas-choroid-sclera complex described earlier [275]. The authors did not provide information on the age of donors but did provide some examples where the donor’s age was stated [280]. This included an emission spectrum from the perifovea of a 47-year-old donor, which exhibited a global maximum at about 560-570 nm and the whole spectrum could be fitted by a combination of four Gaussian curves, with the maxima at about 525, 555, 600 and 640 nm. The fitting procedure was done for the emission spectra from each pixel. As stated by the authors, the most consistent emission maxima obtained by excitation with 436 nm were between 525-540 nm and 550-575nm. The emission maxima obtained by excitation with 480 nm were 20-30 nm shifted towards longer wavelengths. Local maxima or shoulders could be seen at 600 and 640 nm in the perifovea and periphery for both excitation wavelengths. A small maximum appeared also at 700 nm. Importantly, the authors developed a mathematical procedure for finding the best-fitting combination of four Gaussian curves and an emission spectrum of Bruch’s membrane to the emission spectrum from each pixel across a considerable retinal area. Absorption by RPE melanin of the excitation light and emitted light was considered when evaluating the contribution of Bruch’s membrane emission to the total emission of the RPE-Bruch’s membrane complex. This then allowed for mapping the contributions of individual fluorophores, or rather groups of chromophores, across the imaged area.
Using structured illumination microscopy (SIM) on RPE-Bruch’s membrane flat-mounts from 15 Caucasian donors, Bermond et al. demonstrated that, in the fovea, melanolipofuscin is more abundant than lipofuscin, lipofuscin predominates in the perifovea, whereas in the near periphery, the abundance of both types of pigment granules is similar [259,281]. Overall, the granule densities and intensities of RPE fluorescence appear rather uniform, suggesting that RPE cells can control granule numbers [281]. RPE cells with the lowest granule numbers and low fluorescence intensity excited by 488 nm light contain more melanolipofuscin than lipofuscin, whereas lipofuscin predominates over melanolipofuscin in cells with intense fluorescence and high granule load.
The spectrofluorometer developed by Delori at al. mentioned earlier collected emission spectra from the human eye in vivo from a retinal area of about 2° in diameter [278]. In parallel, Delori et al. and Ruckmann et al. have developed a confocal scanning laser ophthalmoscope (SLO), where the fluorescence was excited by laser light and allowed for detailed mapping of fluorescence topography across relatively large square retinal areas of 30° in length [282,283,284,285].
Since the pioneering work of Delori and von Rückmann’s groups, fundus autofluorescence imaging has been further improved, commercialized, and has become a part of routine clinical evaluation of the retina [286,287,288,289]. Typical current imaging systems employ either scanning lasers or lamps equipped with fundus cameras and vary with excitation wavelengths from 435 to 610 nm for imaging of lipofuscin fluorescence, and 780 - 795 nm excitation for imaging of melanosomes fluorescence. Both lipofuscin and melanosomes can emit fluorescence upon excitation with 633 nm light.
While the collection of the whole emission spectra was done in the initial instrument developed by Delori et al. [278], this option has not been made available in commercial instruments. Later studies demonstrated that using photoexcitation with 450 nm-light emitting diode (LED) and recording emission in two spectral channels: green (510-560) and yellow-red (560-700) allows seeing that retinal areas vary in the ratios of short-wavelength to long-wavelength emission [290,291,292,293]. Therefore, the development of a hyperspectral camera with the ability to collect the emission spectra has been of particular importance [294]. The recent applications of the hyperspectral camera for ex vivo imaging of the RPE-Bruch’s membrane explants and human eyes in vivo have demonstrated its great potential for differentiating emission from different retinal fluorophores [254,295,296].
Combining scanning laser ophthalmoscopy with adaptive optics (AOSLO) to minimize the aberrations of the anterior segment of the eye allows for a selective collection of fluorescence emission from RPE cells excited with blue or near-infrared light [297]. Further information can be extracted by adding confocal laser scanning reflectance imaging and optical coherence tomography (OCT) to visualize individual RPE cells and their morphometry, identify areas with drusen or subretinal drusenoid deposits that can help in the explanation of the apparent disruption of the RPE cell mosaic seen in these areas in fluorescence emission imaged with AOSLO.
Another fluorescent technique, which can add complementary information on fluorophores is imaging of fluorescence lifetimes [254,295,298]. Upon a short excitation pulse, in the range of pico- or even femtoseconds, and absorption of photons by fluorophores of the same type, the intensity of emission from fluorophores in a dilute solution decays in an exponential manner, with a rate of decay being a reciprocal of the fluorescence lifetime. Each type of fluorophore has its own fluorescence lifetime, which can change due to the interaction of the fluorophores with the solvent or other solutes. Oxygen is a well-recognized quencher of fluorescence because it can act as an energy acceptor from the excited electronic states. As a result of energy transfer from a fluorophore to an energy acceptor, the fluorescence lifetime is decreased, and consequently the total integrated intensity of fluorescence is decreased.
As described earlier, energy transfer can occur from UV- and blue-light absorbing chromophores of lipofuscin to A2E [145]. If these chromophores exhibit fluorescence, the loss of A2E in lipofuscin due to photooxidation can increase their fluorescence lifetime. This is consistent with the experimental results of Semenov et al. who have shown that exposure of lipofuscin-laden ARPE-19 cells to sub-lethal doses of visible light results in an increase of lipofuscin fluorescence lifetime and this effect can be prevented by supplementation of cells with zeaxanthin [299]. The effect of zeaxanthin can be due to its ability to quench excited triplet states of photosensitizers and singlet oxygen thereby preventing photooxidation of A2E.
Moreover, melanin is a well-recognized acceptor of energy from excited electronic states (reviewed in [77]). Therefore, it can be expected that fluorophores in proximity to melanin within the melanolipofuscin granule will exhibit shorter fluorescence lifetimes. The quenching effect of melanin on A2E fluorescence was demonstrated by Guan et al. who isolated pigment granules from RPE cells, quantified A2E in melanosomes, melanolipofuscin and lipofuscin per mg of dry granule mass and showed that, despite having a substantial content of A2E of 3.5±0.1 µg/mg, which was similar to A2E content in lipofuscin (3.7-6.7 µg/mg), melanosomes did not emit detectable fluorescence upon photoexcitation of melanosomes with 488 nm laser [300].
Due to the numerous fluorophores present in the retina, and their different environments, it can be expected that the fluorescence decay is a superposition of tens, if not more, of different exponential decays. For the lipofuscin granules, there is a need to fit with at least four exponential decays to get a reasonably good fit [190,257]. The same is true for melanolipofuscin and melanosomes.
In currently used clinical fluorescence lifetime imaging systems, the fluorescence is excited by pulsed laser emitting visible or near-infrared light, and the decay is measured in two or more different spectral channels [295,301,302].
When measuring the fluorescence from the fundus, the age-related changes in absorption properties and fluorescence of the lens need to be taken into consideration [303,304,305,306]. With age, there is a progressive build-up of lenticular chromophores absorbing initially ultraviolet, then violet, blue light, and eventually even longer-wavelength light [149]. Some of these chromophores have fluorescence properties, which can interfere with measurements of the fluorescence from the fundus by absorbing some excitation/emission light and by emitting their own fluorescence, thereby shifting the acquired emission spectrum and altering the fluorescence decay kinetics [303,304,305,306]. Moreover, with age there is an increased risk of aggregation of lens crystallins, causing light scatter that eventually becomes visible as opacities known as cataracts, all of which affect both the intensity and spectrum of detected fluorescence [303]. While in their initial instrument, Delori et al. measured the actual spectral transmission of the anterior segment and accounted for it to extract the fundus fluorescence [278], the later studies, where the effects of the anterior segment were considered, used the normative data of van de Kraats and van Norren to account for age-related decrease in spectral transmission of the lens, and therefore did not account for the great variability in the lens chromophores/fluorophores between individuals [149,307]. A recent clinical study clearly demonstrated that lens opacities can affect the measurements of quantitative fundus autofluorescence [305].
By increasing the excitation wavelength to near infrared, the effects of absorption and scatter by the lens can be minimized. To enable the excitation of retinal fluorophores, the photon density needs to be high enough to allow for two-photon excitation. This can be achieved by employing short pulses of high-intensity near-infrared laser. The most recent development in clinical imaging of fundus fluorescence is spectral imaging of fluorescence emitted in the range of 400-700 nm upon intense pulse excitation with two photons of near-infrared wavelengths and the use of adaptive optics [217,254,255,295,308,309,310,311,312,313,314,315,316,317,318,319,320,321,322]. The emitted light can be spectrally analyzed, which enables the detection of specific fluorophores. For example, abca4(-/-)rdh8(-/-) double knockout mice with accumulated lipofuscin show the greatest intensity of fundus fluorescence collected in 594–646 nm range upon two-photon excitation with 780 nm pulsed laser. Rpe65(-/-) knockout mice, which accumulate relatively low amounts of lipofuscin but a lot of retinyl esters in the RPE, exhibit less intense emission in 594–646 nm range than abca4(-/-)rdh8(-/-) but more intense emission in the 400–550 nm range [295]. This technique has been tested for safety in imaging of the retinas of human volunteers with healthy eyes and it can be expected to be used soon to image diseased retinas (Figure 4). In addition to retinyl esters and lipofuscin, there are a number of fluorophores and photosensitizers in the RPE which can be excited by absorption of two photons of 780 nm wavelength, including retinoids, flavins, and porphyrins.
Summing up, fundus autofluorescence techniques allow visualizing fluorescence distribution, its spectral characteristics and lifetimes, and are widely used for mapping the areas of geographic atrophy in various retinal diseases including Stargardt’s disease and age-related macular degeneration, where the loss of RPE cells is followed by their clearance resulting in distinct hypofluorescent areas visible with excitation with blue, green, as well as near-infrared light [323,324,325,326,327,328,329,330,331,332,333].
6.3. Age-related changes in retinal fluorescence
Lipofuscin-like autofluorescence of the fundus appears very early during development. Fundus autofluorescence excited with 532 nm laser from confocal SLO has been imaged in 13 premature infants who were screened for retinopathy of prematurity and showed that fluorescence is detectable at that early age, appears first in the posterior pole and develops centrifugally towards the periphery over the course of the following 30 weeks [334]. Importantly, using 532 nm as the excitation wavelength avoids the confounding effect of the macular pigment.
In addition to the previously discussed studies by Feeney-Burns, Delori and colleagues [75,278], a number of other studies in vivo and ex vivo demonstrated an age-related increase in the fluorescence of the fundus and RPE-Bruch’s membrane complex [147,148,284,335,336] (Table 1). Wing et al. used 44 human eyes from 35 donors, ranging in age from a 6-week premature newborn to 88 years, and measured fluorescence from retinal cross-sections through the entire retina including the fovea and optic disc [147]. The excitation was provided by a xenon arc lamp and a filter with transmission centred at 380 nm. The emission was collected using a band-pass filter transmitting blue light from the range of 460-480 nm. The results have shown that the RPE fluorescence increase occurs particularly fast in the first and second decades of life, then it appears to slow down followed by an increase in people above the age of 60 years. They also showed that there is an age-related increase in emission intensity across the whole retina when results were analyzed based on four age groups: premature-12-month-old, 17 months-30 years; 31-60 years old, and 61-88 years old. The fluorescence emission from tissues of the youngest age group appeared green suggesting that it was mostly due to retinoids and not lipofuscin. There was about a 40% increase in fluorescence emission intensity in the oldest age group in comparison with 31-60 years group.
Weiter and colleagues evaluated lipofuscin fluorescence at 470 nm excited by 365 nm light in the RPE across the retina post mortem from 19 White donors, ranging in age from 2 weeks old to 88 years old, and from 19 Black donors, from 6.5 years old to 90 years old [148]. A linear fit of the data showed a statistically significant increase in emission intensity with age only for Whites, with r2 ranging from 0.45 to 0.65 depending on the location in the retina, and p=0.0001. The actual data were not shown.
Measurements of fundus fluorescence in vivo excited by 488 nm laser and collected from the fovea and an area 7-15° temporal to the fovea in healthy retinas of 33 White subjects 6-78 yeays of age demonstrated a linear increase with age up to about 60 years, above which the emission appeared to plateau [284]. The emission was collected using a long-pass filter transmitting light above 521 nm to beyond 650 nm.
Okubo et al. measured fluorescence in 5 µm thick sections from the formalin-fixed 8 mm in diameter circles centred on the fovea of 88 donors ranging in age from 1-98 years [337]. Excitation was provided by a mercury lamp equipped with a filter transmitting 450-490 nm light, and emission was collected after passing through 520 nm long-pass filter. In this study, the emission also showed a linear increase up to the age of 60 years, followed by a plateau. A similar increase followed by a plateau was obtained from morphometric quantification of the cytoplasmic volume occupied by lipofuscin on other sections from the same retinas imaged with TEM.
Consistent findings were reported by Delori and colleagues from a study on 145 participants ranging in age from 15 to 80 years where the fundus fluorescence was excited by 470 nm and the emission integrated above 520 nm [335]. The emission intensities reached a maximum for the age group in their 7th decade and remained at the same level in the 8th decade. In contrast, when the excitation of the fundus of the same subjects was performed with a xenon arc lamp equipped with a broad-band transmission filter centred at 550 nm, and the emission was collected in the range of 650-750 nm, a linear increase in fluorescence occurred up to the age of 70 years, followed by steep decreases in all 5 locations examined: the fovea, and at 7° temporal, nasal, superior and inferior to the fovea. The rate of age-related decrease in fluorescence at the temporal site of elderly fundi was about 2.5-fold greater than the rate of the fluorescence increase in younger fundi.
The largest study of fundus fluorescence as a function of age was done by Greenberg and colleagues on 277 subjects of different ethnicities aged 5-60 years and involved the examination of 374 healthy eyes [338]. The excitation was with a 488 nm laser and emission was collected from 500-680 nm. A fluorescence standard was incorporated into the imaging instrument to allow for image analysis to compensate for changes in the laser power and detector gain. To account for the age-related changes in the transmission of the anterior segment of the eye, the normative data of van de Kraats and van Norren were used [307]. The age-related increase in fundus fluorescence was the greatest for Whites, followed by Indians, Hispanics, Blacks, and Asians.
Ach et al. analyzed fluorescence from RPE-Bruch’s membrane flat-mounts from 20 donors divided into two age groups: one with 10 donors from 16-51 years of age (average age of 40 years), and another with 10 donors from 82-90 years of age (average age of 85 years), [336]. The fluorescence was excited by light from a mercury arc lamp and filter transmitting 460-490 nm wavelengths. The emission was collected above 505 nm. The emission intensity significantly increased in the elderly group in comparison with the younger group by 51% in the central fovea (a circle centred in the fovea with a 0.6 mm radius), by 45% in the areas of parafovea, perifovea, and perimacula (0.601-3 mm from the foveal centre), and by and 56% in the periphery (more than 3 mm from the centre of the fovea), respectively. This difference between the two groups is similar to that in measurements in vivo by Delori et al. using similar excitation and emission wavelengths in age groups of about 55 and 80 years of age where the intensity of fluorescence in the eldest group is about 60% greater than in the 55-year-old group [335].
In a follow-up study to the studies of Ach and Smith et al. [280,336] described above and in section 5.2 on 20 flat-mounts from human RPE-Bruch’s membrane complexes, the data from 6 eyes were excluded due to the loss of RPE cells and/or the inability of the mathematical algorithm to recover sufficient spectra for the fitting procedure, and the analysis was performed on tissues from 14 donors ranging in age from 16 to 90 years [339]. The acquisition of hyperspectral data and processing was the same as described in their previous report with the excitation wavelengths of either 436 or 480 nm and emission collected using a hyperspectral camera collecting emission from the range of 410-720 nm passing through the long-pass emission filters transmitting light above 460 or 510 nm, respectively [280]. The data were analyzed based on dividing the flat-mounts into two age groups: below 51 years of age and above 80 years of age, with 7 donors in each of them [339]. Interestingly, there were no significant differences between the wavelengths at which maxima of fitted individual curves appeared for the two age groups. The comparison of the relative contributions of the fitted curves to the overall fluorescence was not the subject of the analysis; nevertheless, the two examples, where the graphs were provided, show that spectra with the emission maximum at about 570 nm dominate in the emission in a flat-mount from 16-year old, whereas in the 36-year-old, the dominant spectrum exhibits a maximum at about 520-530 nm [339]. It would be of great interest to see if there is an age-dependent change in the contributions of spectra with the short- and long-wavelength maxima.
The evaluation of the emission maxima as a function of age in vivo was done by Schultz et al. who compared the fluorescence emission maxima in 44 persons below 40 years of age (average age of 24 years) and 18 persons above 40 years of age (average age of 67.5 years) [340]. The fluorescence was excited by a picosecond laser diode emitting 473 nm wavelength with a repetition rate of 80 MHz and it was coupled with a commercial laser scanning ophthalmoscope. Fluorescence emission was collected from each pixel by the time-correlated single photon counting in two spectral channels: green (498-560 nm) and yellow-red (560-720 nm). The ratio of photons collected in each channel was calculated and the emission maximum was evaluated based on the calibration curve [341]. The calibration was prepared by measuring the fluorescence emission ratio from both channels of healthy human fundus and lens in a cohort of healthy volunteers and plotting it against the actual emission maxima from the human fundus and lens published by Delori [278] and Zuclich [342], respectively [341]. The emission peaks were evaluated for the fovea, and the inner and outer rings surrounding the fovea in a standard grid [340]. For the young group, the emission maxima were at 602±16, 614±12, and 621±11 nm for the fovea, inner and outer rings, respectively. For the elderly group, the emission maxima were at 599±17, 611±11, and 614±11, respectively. While it appears that there is a trend of slight decreases with age in the wavelength corresponding to the fluorescence maxima in all three locations, the differences are not statistically significant. Based on spectral changes in lipofuscin fluorescence upon photodegradation, it can be expected that a shorter excitation wavelength and monitoring short-wavelength emission in about 470-490 nm spectral range is more likely to show statistically significant age-related decrease in the calculated emission maxima.
Altogether, there is an age-related increase in fundus fluorescence intensity at least to the age of 60 years and is most pronounced in people of White ethnicity [284,335,337,338] (Table 1). Emission in the blue range 460-480 nm shows an age-related increase even in the age range of 60 to 88 years [147]. At least three studies demonstrated that the emission collected >520 nm reaches a plateau above the age of 60 years [284,335,337]. Fluorescence emission excited by 550 nm light and collected in the red range of 650-750 nm exhibits an-age related increase up to the age of 70 years followed by a steep decrease [335]. Therefore, it appears that there is an age-related decrease in fluorophores emitting long-wavelength fluorescence and an increase in fluorescence from fluorophores emitting short-wavelength light, [339,340].

Table 1.
Studies of the relationship of fundus fluorescence with age.
| Study on normal human eyes in vivo and ex vivo | Excitation (nm) | Emission (nm) | Age-related changes in fluorescence intensity |
| Cross-sections from 19 White donors, 2 weeks-88 years of age, and 19 Black donors, 6.5-90 years old [148] | 365 | 470 | Age-related increase for Whites No correlation with age for Blacks |
| Cross-sections from 44 human eyes from 35 donors, 6-week premature newborn - 88 years [147] | 380 | 460-480 | A fast increase in the 1st and 2nd decades of life, then slowing down followed by an increase in people above the age of 60 years; about a 40% increase in fluorescence emission intensity in the oldest age group 61-88 in comparison with 31-60 years group |
| 30 participants, 21-67 years of age [278] | 430 | 620 | No significant correlation with age |
| RPE-Bruch’s membrane flat-mounts from 20 donors divided into two age groups: 16-51 years of age (10 donors, average age of 40 years), and 82-90 years of age (10 donors, average age of 85 years) [336] | 460-490 | >505 | Increased in the 82-90 years-old group in comparison with the 16-51 years-old group |
| 33 white participants 6-78 years of age [284] | 488 | >521 | linear increase with age from 6 to about 60 years, above 60 the emission appeared to plateau |
| Formalin-fixed 8 mm in diameter circles centred on the fovea of 88 donors ranging in age from 1-98 years [337] | 450-490 | >520 | a linear increase up to the age of 60 years, followed by a plateau; supported by TEM quantification of LF |
| 277 participants of different ethnicities from 5-60 years of age [338] | 488 | 500-680 | The age-related increase in fundus fluorescence was the greatest for Whites, followed by Indians, Hispanics, Blacks, and Asians |
| 145 participants, 15-80 years of age [335] | 470 | >520 | Intensities reached a maximum for the age group in their 7th decade and remained at the same level in the 8th decade |
| 30 participants, 21-67 years of age [278] | 470, 510 or 550 | 620 | Positive correlation with age |
| 145 participants, 15-80 years of age [335] | 550 | 650-750 | A linear increase in fluorescence occurred up to the age of 70 years, followed by a steep decrease |
| 44 participants below 40 years of age (average age of 24 years) and 18 participants above 40 years of age (average age of 67.5 years); calculated emission maxima based on emission of fluorescence in two spectral channels [340] | 473 | 498-560 and 560-720 | For the young group, the emission maxima were at 602±16, 614±12, and 621±11 nm for the fovea, inner and outer ring, respectively. For the elderly group the emission maxima were at 599±17, 611±11, and 614±11, respectively |
6.3. Fundus autofluorescence in age-related macular degeneration (AMD)
AMD is the leading cause of vision loss in people above the age of 50 in developed countries with an estimated prevalence of 288 million worldwide by 2040 [343,344,345,346,347]. Multiple factors are involved in its development and progression including the genetic component, cardiovascular risk factors, and lifestyle. AMD can be classified into several stages with an early stage considered when drusen appear of medium size, between 63 and 125 µm, but there are no pigmentary abnormalities in the retina [347]. Intermediate AMD is diagnosed when there are drusen larger than 125 µm and/or pigmentary abnormalities. These stages of AMD are predictors for its progress into late forms, which affect vision: i) dry (atrophic) form, characterized by loss of RPE and photoreceptors; ii) wet (neovascular) form, characterized by the growth of abnormal blood vessels from the choroid into the retina, which can leak fluid and other blood components (exudates); or iii) a combination of both. More recently, reticular pseudodrusen has been identified as another risk factor for progression to late AMD, particularly the atrophic form [348]. The atrophy initially appears in a small retinal area and is known as geographic atrophy, which expands as the disease progresses. There are several studies investigating the effect of AMD on fundus autofluorescence ex vivo and in vivo and using the findings to monitor the progression of the disease in vivo.
6.4.1. Sources of fluorescence in the AMD retina examined ex vivo
Marmorstein et al. compared fluorescence properties of non-fixed cryosections from human retinas-choroid-sclera explants isolated postmortem from three normal eyes (age 81, 83 and 87 years) and three eyes with AMD (age 74, 84, and 92 years) [275]. All AMD eyes had extensive drusen and one eye had choroidal neovascularization. The cryosections were cut from 2 mm wide strips centred on the fovea. Like in cryosection from normal eyes, excitation with 568 and 633 nm laser of cryosections from AMD eyes induced fluorescence in the RPE, Bruch’s membrane, and sub-RPE deposits (drusen, basal linear, and basal laminar deposits); however, the ratio of fluorescence from Bruch’s membrane and sub-RPE deposits to the RPE fluorescence was about twice greater than in normal eyes. For the excitation with 488 nm, the intensity maximum of Bruch’s membrane fluorescence was similar to that of the RPE and about twice greater than for sub-RPE deposits.
Upon excitation with 364 nm light of AMD cryosections, Bruch’s membrane/sub-RPE deposits emitted strong fluorescence with intensity maxima at similar wavelengths of 485-490 nm as for normal retinas [275]. However, in contrast to normal eyes, where the intensity maxima were similar for Bruch’s membrane and RPE, in AMD eyes the intensity maximum was about 1.8-fold greater for Bruch’s membrane than for RPE. The emission spectrum for sub-RPE deposits was similar in shape but the emission maximum was about twice smaller than for Bruch’s membrane. The emission maximum of the RPE fluorescence induced by excitation with 364 nm was at 540 nm, which is 15 nm shifted towards shorter wavelengths in comparison with the normal retina.
The number of tissue donors was small, yet the images of fluorescence excited by 488 nm light in the cryosections showed strikingly greater fluorescence emission at 545nm, 555 nm, and 605 in the RPE and sub-RPE deposits in AMD eyes than in normal eyes [275]. This difference between AMD and normal eyes was even more apparent when fluorescence was excited by 364 nm light and emission was monitored at 485, 555, 625, and 695 nm.
Rudolf et al. examined sections of the AMD retinas with geographic atrophy from 10 donor eyes, ranging in age from 73-93 years (average age of 87 years), and demonstrated that the areas adjacent to the geographic atrophy exhibit non-uniform fluorescence in the RPE monolayer with some cells exhibiting substantially decreased fluorescence excited by 488 nm laser with emission collected after passing via filter transmitting light above 500 nm [349]. They also showed some fluorescent cells present in the neural retina and RPE cells stacked one on top of another in the areas adjacent to geographic atrophy areas. This stacking of cells can explain the increased fluorescence seen at the edges of geographic atrophy when imaged en face [350]. Moreover, Zanzottera et al. showed that edges of the geographic atrophy contain RPE cells or their parts in the neural retina, thick basal laminar deposits, and some flattened RPE cells adjacent to Bruch’s membrane, which are separated from the photoreceptors by basal laminar deposits [350].
Bonilha et al. examined fundus fluorescence of 32 eyes from 17 donors, 65-97 years of age, with atrophic AMD and 10 eyes from 10 donors with no eye disease, 57-95 years of age, which was followed by analyses of histologic sections in the areas of geographic atrophy [351]. The fundus fluorescence was imaged upon excitation with 488 nm and 787 nm lasers from a commercial laser scanning ophthalmoscope. The fluorescence of histological sections was acquired with filters with excitation and transmission properties close to those of typical clinical instruments: the emission of fluorescence excited with 480 nm light was collected in the range of 500-680 nm; whereas the emission of fluorescence excited by 750 nm light was collected above 800 nm. The RPE present away from the border of geographic atrophy had non-fluorescent melanosomes present in the apical portions of the cells, a high concentration of fluorescent granules excited by blue and near-infrared light, and appeared similar to the RPE in the age-matched control eyes. Close to the atrophy border, the RPE cells were hypertrophic and filled with granules emitting in the green-yellow-red spectral range as well as in the near infrared. In those cells, the infrequent non-fluorescent melanosomes were dispersed throughout the cell. The morphology and pigmentation of the RPE close to the border were frequently nonuniform. Next to the intact RPE appeared cells sloughed into the subretinal area or aggregates of granules shed into the basal laminar deposit. Some RPE cells appeared stacked into a bilayer or appeared as individual cells with no contact with other RPE cells or even translocated into the neural retina.
In a series of studies of human RPE flat-mounts from AMD and normal eyes examined by SIM and confocal fluorescence microscopy, Bermond et al. demonstrated that AMD-affected RPE cells show an increased variability in size and a significantly increased granule load, and this can be seen in all retinal locations examined: the fovea, perifovea, and near periphery [157,259,281]. In comparison with normal retinas, the relative contribution of lipofuscin to all pigment granules decreases in the AMD retina, whereas melanolipofuscin contribution increases, particularly in the fovea. AMD-affected RPE cells exhibit decreased fluorescence collected within the range of 510-750 nm when excited by 488 nm laser in comparison with normal RPE [157]. These findings can explain the results of quantitative measurements of fundus autofluorescence intensity in AMD patients and healthy controls, which revealed that there is lower overall fluorescence in AMD retina induced by photoexcitation with 488 nm laser [156]. This effect was pronounced particularly in patients with reticular pseudodrusen, which are lipid-rich deposits between POS and RPE.
In another study where the imaging of fluorescence was done en face on RPE-Bruch’s membrane-choroid flat-mounts, it has been observed that RPE cells from aged or AMD retinas sequester fluorescent granules – lipofuscin and melanolipofuscin into packets, 2.5 to 20.9 µm in diameter leaving some cytoplasmic areas, or even entire RPE cells, almost devoid of fluorescent granules [20]. Cross-sections of these flat mounts revealed that some cells form mushroom cap-like structures filled with lipofuscin, which protrude into the area of POS. Another study showed lipofuscin/melanolipofuscin granules shed into basal laminar deposit [350]. It has been proposed that the removal of the lipofuscin-laden packets from cells is a mechanism of control of intracellular levels of lipofuscin but it may contribute to the sub-RPE deposits [20,349,350,352,353]. Interestingly, similar packets with lipofuscin were observed in vitro above lipofuscin-laden confluent ARPE-19 cell monolayer after exposure to visible light, which caused spectral changes in lipofuscin fluorescence [74].
In another study where the fluorescence imaging of RPE-Bruch’s membrane was performed en face on flat mounts, five AMD eyes were used: one with early AMD, one with geographic atrophy, and three with exudative AMD [354]. Twelve areas with drusen were imaged in total. Out of the total of 25 drusen analyzed, 12 were small (≤63 μm in diameter), 8 were intermediate (64–124 μm), and 5 were large (≥125 μm). As before [336], the explants were excited with 436 or 480 nm and emission filters were transmitting light above 460 nm and 510 nm, respectively. The hyperspectral camera was used for image acquisition every 10 nm from 420-720 nm. The fluorescence of the RPE from AMD eyes was similar to that from normal eyes. Using the same fitting procedure as the one used previously on explants from normal eyes, a new spectrum specific for drusen has been identified. With the excitation of 436 nm, the emission maxima of that spectrum were at 510 nm for 10 drusen, one druse exhibited a maximum at 500 nm, and one druse at 490 nm. Excitation with 480 nm led to a less intense emission with the maximum at 520 nm for the majority of drusen, followed by some drusen emitting with the maximum at 540 nm and one druse at 530 nm. Other sub-RPE deposits (basal laminar and basal linear deposits) emitted similar fluorescence as drusen.
Subsequent studies by Mohammed, Tong, and colleagues imaged hyperspectral autofluorescence of RPE-Bruch’s membrane-choroid explants in pre-selected retinal locations (fovea, perifovea, mid-periphery) as well as in all identified drusen [296,355]. The explants were obtained post mortem from AMD patients 81-88 years of age. For the excitation, they used four dichroic filters transmitting narrow-band light from a mercury arc lamp centred at 436, 450, 480, or 505 nm. The emission spectra were collected from each pixel by the hyperspectral camera as in their previous study described above [354]. Using their mathematical fitting procedure, they showed that contribution from drusen fluorescence dominated the emission in AMD RPE-Bruch’s membrane flat-mounts excited by 436 nm, and its contribution decreased as the excitation wavelength increased [296,355]. This approach allowed for even better differentiation of drusen and sub-RPE deposits than the one based on the use of only two excitation wavelengths. Importantly, the authors showed that excitation with 450 nm light provides similar spectral characteristics of the drusen spectrum as that obtained with 436 nm excitation. This is an important finding because using a longer excitation wavelength is safer for the retina in vivo considering the light-induced reactivity of retinaldehydes, flavins, porphyrins and lipofuscin, [6,77,88,96,98,188,356,357,358].
Clearly, there are distinct changes in the AMD retina in comparison with the age-matched control retina, which affect the fluorescence. The contribution from Bruch’s membrane and sub-RPE deposits to retinal fluorescence increases in AMD; there are changes in the distribution of pigment granules in RPE cells, with an overall increase in fluorescent granule concentration, but with decreased concentrations of lipofuscin and increased melanolipofuscin, which results in a decreased fluorescence excited by 488 nm light in the affected RPE cells. In the AMD retina, some RPE cells are hypertrophic, cells/cell fragments filled with fluorescent granules are present in the neural retina, while some sub-RPE deposits include fluorescent granules. Some RPE cells are stacked on other RPE cells, while in the areas of atrophy, there is a complete absence of RPE cells.
6.4.2. Fluorescence characteristics of AMD retina in vivo
It has been observed already in the initial studies of fundus autofluorescence, that the emission spectrum in areas with drusen is shifted toward shorter wavelengths [340,359,360,361]. Hammer et al. analyzed 43 eyes of 39 White patients with AMD characterized by drusen and absence of choroidal neovascularization or atrophy [361]. As mentioned earlier, pigmentary changes and drusen, which are visible in a typical fundus examination and recorded with colour fundus camera, are considered as early or intermediary stages of AMD [347]. Consistently with the findings on RPE-Bruch’s membrane explants and histological sections of the retina described above, they demonstrated that drusen and hyperpigmentation were associated with shorter emission wavelengths for fundus autofluorescence. Retinal areas with subretinal drusenoid deposits (SDD; also known as reticular pseudodrusen) as determined by OCT also exhibit shorter peak emission wavelengths than the surrounding areas [362].
Borrelli et al.’s study on 25 eyes of 18 patients diagnosed with an atrophic form of AMD has shown that some areas devoid of RPE (as seen by OCT) exhibit substantial green fluorescence when excited with 450 nm light [291,363]. From the OCT images included in their paper, it appears that these green-fluorescent areas correspond to areas of severely damaged photoreceptors and absent RPE: there is no characteristic hypo-reflective layer corresponding to the outer nuclear layer and no characteristic hyper-reflective layers corresponding to the outer limiting membrane and the ellipsoid area of the inner segment. Some of the areas of increased green fluorescence appear in OCT as hyper-reflective in locations corresponding to the inner retinal layers: the inner nuclear layer or even the inner plexiform layer; some other areas are hyper-reflective at the area corresponding to the non-existing outer nuclear layer through RPE. It has been concluded that the residual debris and drusen-like deposits are the origins of the green-emitting fluorophores.
Several studies compared the emission spectra in normal and AMD eyes in vivo including one which demonstrated a statistically significant shift in the emission maximum towards shorter wavelength [276,340,359]. Schultz et al. compared the wavelength of the fluorescence emission maxima in 18 persons above 40 years of age (average age of 67.5 years) and 63 patients with early AMD (average age of 74 years) [340]. As described before for the comparison of emission maxima in two age groups, the fluorescence was excited by a picosecond laser diode emitting 473 nm wavelength, the fluorescence emission was collected in two spectral channels: green (498-560 nm) and yellow-red (560-720 nm), and the calibration was used to determine the emission maxima [341]. AMD retinas exhibited maxima at 571±26, 596±17, and 602±16 nm for the fovea, inner and outer ring, respectively [340]. These values are 28, 15, and 12 nm shifted towards shorter wavelengths in comparison with the emission maxima of the healthy retinas at the corresponding locations, and the differences were statistically significant.
In another study by the same group, fundus fluorescence lifetimes and emission intensities in the two spectral channels were examined on three patients with an atrophic form of AMD [298]. It has been determined that the area of complete RPE and outer retinal atrophy (cRORA) had a stronger emission in the green (498-560 nm) channel than in yellow-red (560-720 nm) channel, with a particularly strong emission from drusen. Some drusen were surrounded by hypofluorescent areas. Areas with drusen without overlying RPE exhibited longer fluorescence lifetimes than other areas of cRORA. This is consistent with the previous studies by Hammer, Schweitzer et al. demonstrating that fluorescence lifetimes in AMD patients are longer than in controls [364,365]. In these studies, the fluorescence was excited with a 448 nm laser, and emission was monitored in two spectral ranges: 490-560 nm and 560-700 nm.
6.4.3. Current evidence for the prognostic value of fundus fluorescence characteristics for AMD progression
The characteristics of fundus fluorescence as a potential predictive biomarker for AMD progression have been investigated in several studies. A longitudinal study of 121 eyes of 71 patients with intermediate AMD followed over a median period of 15 months (interquartile range of 9-21 months) has demonstrated that a decrease in autofluorescence in a standardized ring surrounding the fovea precedes the progression to the atrophic, but not to the neovascular, form of AMD [366]. The quantification of fundus fluorescence was done with excitation with a 488 nm laser and the emission was collected between 500 and 680 nm using a commercial instrument combining SLO and OCT. Another study from the same group and using the same instrument demonstrated on 43 eyes of 26 patients with atrophic AMD that there is a positive correlation between the progression of the geographic atrophy area over a period of 12 months and the intensity of fluorescence at the junctional zone in the inferior retina but not elsewhere [367]. The junctional zone was defined by the area of the retina adjacent to the area of geographic atrophy, which had still a distinguishable ellipsoid layer in OCT images. Interestingly, the expansion of geographic atrophy occurred towards the outside areas with decreased fluorescence and this effect was statistically significant also only for the inferior retina.
The prognostic value of fluorescence lifetimes in the area of large soft hyperfluorescent drusen with overlying RPE has been demonstrated by Schwanengel et al. in a study of 40 eyes from 38 patients with intermediate AMD examined by colour fundus camera, OCT, and fluorescence excited by 473 nm laser and emission collected in two spectral channels: green (498-560 nm) and yellow-red (560-720 nm) [341,368] (Figure 5). Over the follow-up of up to 6 years, the retinas which progressed to choroidal neovascularisation or cRORA, exhibited longer fluorescence lifetimes of the drusen areas at baseline than the retinas with no disease progression. These differences were statistically significant when monitored in either, green or yellow-red channels.
The peak emission wavelength of the drusen at baseline was shorter in eyes that exhibited AMD progression than in eyes with no progression but this difference was not statistically significant [368]. In another study by the same group, 39 AMD patients were followed for up to 6 years to determine the progression of the disease. During the follow-up visits, a shortening of the peak emission wavelength was observed and reached statistical significance in the fovea and inner ring in months 37-72 of the follow-up [341]. Interestingly, 10 AMD patients, who progressed to the outer retina atrophy or to cRORA in the follow-up, had the fluorescence emission maxima at significantly shorter wavelengths in all three retinal locations: centre, inner and outer rings, than patients who did not progress in their disease.
It could be argued that the spectral shift of the emission maximum to shorter wavelengths is due to SDD, drusen, and other deposits, such as basal laminar deposit, which accumulate in the Bruch’ membrane and are already known as prognostic factors for AMD progression [369,370]. While the contribution from such sub-RPE deposits can indeed contribute to the observed decrease in a wavelength corresponding to the fluorescence emission maximum, there is some evidence that this decrease is due to spectral changes in fluorescence of the RPE [371,372,373,374]. Feldman et al. reported fluorescence spectra of suspensions of RPE cells from two normal eyes and two AMD eyes [371]. All four samples showed broad emission upon excitation with 430 nm, overall similar in shape, but with a hypsochromic shift in the emission maxima from AMD-affected RPE. Normal RPE exhibited maxima at 563 and 564 nm, whereas the maxima for the RPE from AMD eyes were at 528 and 540 nm [371]. Follow up study using excitation with 488 nm laser of RPE cell suspensions from 19 normal eyes (age 27-74 years) and 12 AMD (age 59-88 years) demonstrated substantial spectral differences between the emission spectra of RPE from normal and AMD-affected eyes [372,373,374]. There was a clear emission maximum at 592 nm for normal RPE. RPE from eyes with AMD showed a flat maximum extending from about 550 to 590 nm. The ratios of integrated green-yellow fluorescence (530-580 nm) to orange-red fluorescence (600-650 nm) were 1.04±0.11 and 1.73±0.16 for RPE from normal and AMD eyes, respectively.
7. Retinal spectral fluorescence characteristics as a potential in vivo biomarker of oxidative damage and efficacy of potential antioxidant therapies
7.1. Current evidence for photooxidation of lipofuscin in vivo
As discussed in the previous sections, lipofuscin is the major contributor to fundus fluorescence excited by UVA and visible light, but photooxidation of lipofuscin in vitro leads to spectral changes in its emission spectrum, namely a decrease in its long-wavelength yellow-orange fluorescence and an increase in its blue-cyan fluorescence.
While exposure to light can accelerate lipofuscin accumulation in the retina and its fluorescence, there is a growing body of evidence suggesting that photooxidation can affect fluorescent components of lipofuscin in animal and human retinas also in vivo and, as a consequence, can lead to a decrease in lipofuscin fluorescence. For example, raising pigmented wild-type and abca4-/- mice under cyclic light initially, up to 18 weeks, accelerates the accumulation of A2E in comparison to mice raised in the dark, with the knockout mice having 12-fold greater levels of A2E than their wild-type counterparts [197,375]. However, this difference becomes smaller over longer rearing times and depends on the intensity of light the animals are exposed to [197,375]. This is particularly apparent in albino mice: 7-month-old albino abca4-/- mice raised under 30 lx cyclic light exhibit similar levels of A2E as pigmented counterparts but have about 30% greater levels of oxidized A2E to oxiranes [197]. The level of A2E-derived oxiranes in pigmented 7-month-old abca4-/- mice is almost two-fold greater in mice raised under 120 lx than under 30 lx, while the levels of intact A2E are similar.
In another study, it has been shown that the fundus fluorescence excited with 488 nm laser of 9-month-old albino BALB/cJ mice decreases by about 20% after seven daily two-hour exposures to 430 ± 30 nm light from a 1 mW halogen source [376]. The decrease in fluorescence emission is accompanied by about an 18% decrease in A2E extracted from the eyecups. A similar decrease in A2E of about 24% has been also observed in 8-month-old albino C57BL/CJc2j mice subjected to the same light exposure protocol in comparison with not-exposed control mice. Comparison of A2E levels in pigmented C57BL/CJ and albino C57BL/CJc2j mice raised in dark or under cyclic light (40-60 lx from white LEDs) has shown that the six-month-old albino mice raised under cyclic light have almost twice lower levels of A2E than their counterparts raised in the dark whereas the difference between pigmented mice raised under cyclic light or in dark is smaller than 10%. It has been also shown that the levels of A2E and other bisretinoids are several-fold smaller in albino abca4-/- knockout mice raised for 8 months under cyclic light than in mice raised in the dark but this light-dependent decrease, together with a decrease in fundus fluorescence, can be partly prevented by supplementing the animals with vitamin E. It has been shown in a follow-up study that daily administration of an iron chelator deferiprone from the age of 2 to 6 months, can partly inhibit loss of bisretinoids and fluorescence decrease in albino abca4-/- knockout mice and bisretinoids loss in agouti 129/SvJ mice [377].
A rearing light-dependent fluorescence decrease has been also observed by Pan and colleagues in pigmented mice, both wild type and mice with deleted genes coding for two proteins involved in clearance of all-trans-retinaldehyde, abca4(-/-)rdh8(-/-) [17]. The double knockout mice raised under a cyclic 10 lx light for 12 months exhibit a 2.8-fold lower intensity of RPE fluorescence emission in comparison to mice of the same genetic background raised in the dark, but it is not clear what the spectral range of the emission was measured because the excitation and emission details have not been provided.
Moreover, it has been shown that RPE fluorescence in the macaque retina in vivo decreases by about 20% as a result of a single radiant exposure of 210 J/cm2 to the light of 568 nm wavelength with no indication of any damage to the RPE monolayer [229,230,231]. It has been also demonstrated that exposure of fixed human RPE ex vivo to a much smaller dose of only 30.6 J/cm2 of 568 nm light leads to fluorescence photobleaching. The decrease in fluorescence in the macaque retina in vivo without damaging the RPE has been also observed after exposure to 460, 488, 544, and 594 nm lasers [232].
There are reports on changes in retinal fluorescence dependent on light exposure in human retinas as well. It has been shown that translocation of the re-attached neural retina after detachment reveals areas of increased fluorescence matching the shape of blood vessels, which, after the translocation, no longer prevent the incident light from reaching the RPE behind them suggesting that the fluorescence in the other areas has been partially bleached by exposure to light [378,379].
As mentioned previously, Teussing et al. studied the effects of wearing a black contact lens in one eye of Stargardt’s disease patients during waking hours and compared it with the unprotected eye [235]. They imaged fundus fluorescence excited by a 488 nm laser and have shown that the average increase in the percentage of hypofluorescent pixels in the covered eye was 2.5%/year whereas the average increase in the unprotected eye was 8.1%/year, which is 3.2-fold greater.
As the retina ages, the cumulative exposure to light increases, and therefore it can be expected that at a certain age, the effects on fluorescence of oxidative degradation of lipofuscin can overshadow the effects of accumulation of fresh lipofuscin. Indeed, changes in fundus fluorescence consistent with oxidative degradation of lipofuscin have been observed in studies investigating the effect of age on the fluorescence of the human fundus (Table 1). For fluorescence excited with UV light (365 or 380 nm) and emission detected in the blue range of the visible spectrum (at about 470 nm), there is an age-related increase in the emission intensity even in the eldest eyes studied, at the age of 88 or 90 years [147,148]. In studies where fluorescence was excited by long-wavelength blue light (470-490 nm) and the emission was collected at above 505 or 520 nm, an age-related increase in fluorescence was observed until the 6th or 7th decade when it appeared to reach a plateau [284,335,337]. The long-wavelength fluorescence emission collected in the range of 650-750 nm exhibits a linear increase up to the age of 70 years followed by a steep decrease after that age [335]. These studies on age-dependence of fluorescence intensity have not assessed the spectral characteristics of the emission so it is not clear whether the decreased emission was due to removal of lipofuscin, spectral changes in the emission spectra, or both. However, the study by Delori showing that there is an age-dependent decrease in fluorescence emission in the range of 650-750 nm between the age of 70 and 80 years, while the same group of individuals exhibited a plateau of emission collected above 520 nm suggest that spectral changes in the fluorescence emission do occur [335]. Schultz and colleagues also demonstrated a small spectral shift of the emission maximum towards shorter wavelengths in a group of 18 subjects above the age of 40 years in comparison with a group of 44 subjects below the age of 40 but the differences did not reach statistical significance [340]. This study was based on collecting emission in two spectral channels 498-560 nm and 560-720 nm and the fluorescence was excited by 470 nm light.
Based on the spectral changes observed during photodegradation of RPE lipofuscin, it can be suggested that by using a shorter excitation wavelength, such as 460 nm, and collecting emissions in the short wavelength channel where the fluorescence of degraded lipofuscin is increased, such as 470-490 nm (Figure 2) [74], the age-related differences could be more pronounced.
7.2. Current evidence for increased oxidative stress and oxidative damage in AMD retina
It has been shown that the AMD retina is under increased oxidative stress in comparison with the age-matched retina and there is a growing body of evidence suggesting that the oxidative stress and products of lipid and bisretinoid oxidation can contribute to the development and progression of AMD by activation of the complement cascade, and other proinflammatory and proangiogenic signalling (reviewed in [193,380,381,382,383,384,385]). Firstly, RPE affected by early, atrophic, and exudative AMD exhibit about five-fold increased content of total iron and iron chelateable by desferrioxamine in comparison with RPE in age-matched normal retinas [386]. It has been shown in experiments on mice that the retinal iron overload, caused by intravitreal injection of ferric ammonium citrate into the eye, results in i) the modification of proteins by a characteristic product of DHA oxidation forming CEP; ii) the upregulation of interleukin-1β (IL-1β) and cluster of differentiation 68 (CD68) expression; and iii) the photoreceptor and RPE atrophy [93,387]. Inhibiting lipid peroxidation by supplementing animals with a form of DHA where the hydrogen atoms are replaced by deuteriums, thereby making it substantially less susceptible to the propagation of lipid peroxidation than normal DHA, can effectively prevent the CEP formation, upregulation of both inflammatory markers and cell loss [387,388,389,390,391]. Notably, upregulation of IL-1β can create a vicious cycle leading to excessive accumulation of intracellular iron because it has been shown to upregulate RPE iron importers and downregulate iron exporters [392]. Sterling et al. have shown that high-fat-diet-induced obesity, a risk factor for AMD, drives systemic and local inflammatory circuits upregulating IL-1beta [392]. Moreover, with regard to the pro-inflammatory signalling, it has been shown that an 8-hour exposure of ARPE-19 cells to IL-1β or A2E results in the upregulation of a potent pro-inflammatory enzyme, cyclooxygenase-2 [393,394].
Secondly, in comparison with age-matched normal human retinas, AMD-affected retinas exhibit a greater level of protein modifications by oxidized lipids, such as CEP which has been detected in photoreceptor outer segments, RPE, Bruch’s membrane and drusen [395,396]. It has been shown that CEP can stimulate angiogenesis in the chick embryo and rat cornea and in a mouse model of laser-induced chorioretinal neovascularization [397,398]. Interestingly, administration of anti-VEGF antibodies only partly blocks CEP–protein adduct-induced angiogenesis in vivo, while it completely blocks angiogenesis induced by VEGF, suggesting an involvement of an additional, VEGF-independent, angiogenesis pathway. While CEP does not upregulate VEGF in ARPE-19 cells in vitro, another product of lipid oxidation, 4HNE does so at a concentration of 1 µM [399]. Also, it has been shown that exposure to blue light of A2E-laden ARPE-19 leads to an increased expression of VEGF mRNA and protein [400,401]. VEGF is a potent angiogenic factor that promotes choroidal neovascularization and anti-VEGF treatments have been established as effective in preventing neovascularisation and its deleterious consequences in the neovascular form of AMD [402]. In addition to triggering pro-angiogenic signalling, oxidized lipids can also trigger pro-inflammatory signalling. For example, the phagocytosis of oxidized POS by cultured ARPE-19 causes 2.8- and 3.2-fold increased expression of proinflammatory cytokines: interleukin 8 (IL-8; also known as the neutrophil chemotactic factor) and monocyte chemoattractant protein-1 (MCP-1, also known as CCL2), respectively, in comparison to cells fed naïve POS [403]. IL-8 and MCP-1 are potent chemoattractants for phagocytes and are elevated in the vitreous of patients with retinal neovascularization [404]. Intravitreal injection of 4HNE into the mouse eye results in an increased formation of C3a in the RPE in comparison with the vehicle-treated mice suggesting that 4HNE can activate the complement cascade [405]. Notably, C3a accumulation is present in Bruch’s membrane in AMD-affected eyes [406].
Products of A2E oxidation, such as peroxy-A2E and furano-A2E, and A2E-laden cells exposed to blue light can activate the complement cascade in the human serum, as observed by monitoring the cleavage products of complement component C3, which are known to trigger inflammatory responses and formation of the membrane attack complex (MAC) which can cause cell death [104,407,408].
Thirdly, several reports demonstrate that in RPE from AMD retinas, the damage to mitochondrial DNA is greater than in age-matched normal RPE and the mitochondrial DNA damage increases with the disease progression [409,410,411,412,413,414,415]. Interestingly, the mitochondrial DNA damage is significantly higher in RPE from AMD retinas with the homozygous complement factor H (FH) variant Y402 (rs1061170), which is associated with an increased risk for AMD, than in retinas with H402 variant of CFH [412].
Moreover, RPE from AMD retinas exhibits upregulation of two important antioxidant enzymes, glutathione peroxidases (GPXs): extracellular GPX3 and mitochondrial isoform of GPX4, which catalyze the reduction of hydrogen peroxide and lipid hydroperoxides by glutathione [416].
Finally, the role of oxidative stress in the progression of AMD is underscored by the results of two large clinical trials Age-Related Eye Disease Study (AREDS) and AREDS2 showing that patients with moderate AMD receiving a mixture of antioxidants including vitamin C, vitamin E, zinc and carotenoids were at a smaller risk of progression to advanced AMD, especially if their dietary intake of these antioxidants was low [417,418,419,420].
The increased accumulation of iron and oxidative damage to biomolecules in AMD retina provide evidence of an increased oxidative environment in the AMD retina. Therefore, it can be expected that LF can be oxidized in such an environment, and this can be detected by measurement of fluorescence emission spectra or a ratio of cyan (about 470 nm) to orange-red (620-650 nm) fluorescence.
Considering the above-mentioned products of oxidation which can activate the complement cascade and upregulate VEGF, an effective antioxidant therapy can decrease the risk of the formation of such oxidation products and therefore act upstream of these deleterious effects thereby preventing complement activation and excessive secretion of VEGF which are held responsible for the development of AMD. There is room for improvement in AREDS2 supplementation with Vitamin E, vitamin C, lutein, zeaxanthin, zinc and copper. Other approaches to inhibit oxidative damage to the retina include inhibitors of the retinoid cycle, such as Emixustat (known also as ACU-449), which is tested for slowing down the progression of AMD and Stargardt’s disease in several clinical trials (ClinicalTrials.gov Identifiers: NCT03033108, NCT03772665, NCT02130531, and NCT01002950), while improved derivatives of Emixustat are being developed [421]. Another approach to decrease the oxidative damage induced by all-trans-retinaldehyde upon release from visual pigments is by inhibiting delivery of vitamin A to the retina by compounds such as Tinlarebant tested in clinical trials NCT05266014 and NCT05244304, STG-001 (NCT04489511), and Fenretinide (NCT00429936).
Previously mentioned Remofuscin which has been shown in animal studies to stimulate lipofuscin removal from the RPE is also tested in a clinical trial (EudraCT registration: 2018-001496-20 STARTT) and is likely to affect the fluorescence of the retina.
Also, the deuterated form of vitamin A (C20-D3-Vitamin A also known as ALK-001) discussed earlier has been shown in animal studies to slow down the formation of bisretinoids, which otherwise can facilitate lipofuscin formation by inhibition of lysosomal degradation and is currently tested in three clinical trials: NCT03845582, NCT02402660, NCT04239625), is likely to affect the fluorescence of the retina.
Another approach to decrease the oxidative damage in the retina is the use of deuterated polyunsaturated fatty acids such as DHA, where hydrogen atoms at bis-allylic sites are replaced by deuteriums [387,388,389,390,391]. Supplementation with deuterated DHA is expected to inhibit lipofuscin accumulation by inhibiting lipid peroxidation, which can be initiated not only by retinaldehyde and its derivatives, but also by reactive oxygen species generated during phagocytosis, and by other compounds, such as iron, which accumulates in the AMD retina.
7.3. RPE lipofuscin fluorescence: intensity and spectral characteristics as a potential biomarker of oxidative damage to the retina in vivo.
There is a growing body of evidence demonstrating that: i) RPE lipofuscin is the major fluorophore in the retina; ii) oxidation of RPE lipofuscin results in characteristic changes in its emission spectrum; iii) oxidation of RPE lipofuscin can occur in vivo; iv) there is increased oxidative damage in the AMD retina; v) there is a spectral shift of fluorescence emission maximum towards shorter wavelengths in AMD retina; vi) a shorter wavelength of the emission maximum is a risk factor for the AMD progression. Therefore, it can be expected that spectral changes in fluorescence emission due to lipofuscin oxidation in the AMD retina can be a contributing factor to the observed changes in fluorescence in the AMD retina. It is tempting to suggest that the characteristics of lipofuscin fluorescence can be used in clinics to monitor the oxidative damage in AMD and other retinal degenerations associated with lipofuscin accumulation and to evaluate the efficacy of potential antioxidant therapies.
It appears that the recent developments in imaging techniques, which include combined OCT and fluorescence imaging of the fundus with subcellular resolution make such monitoring in vivo of the spectral changes in fluorescence emission of lipofuscin feasible [254,295,322]. The combined use of OCT and SLO allows for the identification of areas with SDD or drusen, which can also be a source of fluorescence. The subcellular resolution of fluorescence imaging is enabled by TPEF-SLO, which limits the area of the excitation, and imaging of both - fluorescence and reflectance [254,295,322]. The latter is used to identify the retinal layer where the laser light is focused so it is the origin of the fluorescence. Moreover, due to excitation with near-infrared lasers, the artifacts introduced by the anterior segment of the eye are minimized. Importantly, it has been demonstrated that such imaging can be done safely for the human eye in vivo [254,295,322].
However, current protocols for excitation and collection of emitted fluorescence from the human fundus in vivo are not optimal to detect the spectral changes occurring in lipofuscin fluorescence due to oxidation. Lipofuscin oxidation results in a decrease in its yellow-red fluorescence with a concomitant increase in the emission of its blue fluorescence (Figure 2) [74]. Optimizing the excitation wavelength and the spectral ranges of collected emission can improve the specificity for monitoring these changes and thereby improve the sensitivity with which the progression of oxidation occurs. The safety aspects need to be considered as well. All-trans-retinal, which transiently accumulates in the photoreceptor outer segments after photoexcitation of visual pigments, is a potent photosensitizer absorbing visible light up to 460 nm. Absorption of flavins extends even more towards longer wavelengths.
Boguslawski and colleagues have demonstrated that safe imaging of the human fundus in vivo by TPEF-SLO provides a sufficient signal-to-noise ratio for informative imaging of the reflectance and fluorescence emission when collected in distinct spectral windows obtained by bandpass filters: 400–600 nm, 594–646 nm, 500–540 nm, and 400–550 nm [254,295,322]. However, the excitation source used in this instrument providing 76 fs pulses of 780 nm wavelength excites multiple retinal fluorophores and photosensitizers, including not only lipofuscin but also retinoids, flavins, and porphyrins. While this has its own advantages, including the ability to monitor the retinoid cycle in vivo, it can be argued that for imaging of oxidation-induced changes in lipofuscin fluorescence, TPEF-SLO using excitation with femtosecond pulses of 920 nm near-infrared laser [314] could provide a better option by being more specific for excitation of lipofuscin.
Such an excitation with 55 fs pulses of 920 nm light fired at 80 MHz repetition rate combined with AO-SLO with imaging of both reflectance and fluorescence of various retinal layers was used in a study by Sharma and colleagues on anaesthesized macaques in vivo [312,314]. This elegant study demonstrated the great potential of such a technique for visualizing fluorophores in the individual layers of the retina but, despite the emitted fluorescence was collected over a broad range of 400-680 nm, to achieve the sufficiently high signal-to-noise ratio required radiant exposure 3.5-fold greater than the maximal permissible exposure for that wavelength given by the American National Standard for Safe Use of Lasers ANSI Z136.1-2014. 2014.
To make their TPEF radiant exposures safe, Boguslawki and colleagues employed a dispersion precompensation unit, which enabled the two-photon excitation to be more efficient, and reduced the pulse repetition rate from the typically used 80 MHz to 6 MHz, which enabled to increase the peak power and therefore increase the yield of fluorescence [295,322]. Therefore, it appears that TPEF with 920 nm light can also be amended to be safe for the retina. The spectral channels for the acquisition of emitted fluorescence would also benefit from collecting short-wavelength fluorescence from the 470-500 nm range and long-wavelength fluorescence from 560-700 nm range. The intensity of long-wavelength fluorescence would provide information on the presence of lipofuscin, whereas the ratio of the emissions in these two spectral channels would provide information on the oxidation state of lipofuscin.
8. Conclusions
In conclusion, despite substantial research efforts, lipofuscin remains rather enigmatic. Numerous components derived from retinoids have been identified in RPE lipofuscin, but not many other components of interest, such as those which exhibit strong photosensitizing properties and may be derived from the oxidation of polyunsaturated fatty acids or are not soluble in organic solvents. The contribution of such components to lipofuscin fluorescence is also not known. Oxidized DHA, which is a likely component of lipofuscin, fluoresces upon excitation with UV or 488 nm blue light. When excited with UV light, oxidized DHA exhibits its fluorescence emission maximum in blue and extends its tail into the yellow-red spectral range. It can be expected that at least some of these not-identified components derived from DHA oxidation can be common between lipofuscin from RPE and other tissues rich in DHA, such as the brain.
It is well-established that oxidative stress and lysosomal dysfunction play a major role in lipofuscin accumulation. Less is known about the effects lipofuscin exerts on cells under physiological conditions. It appears cells can accumulate substantial amounts of lipofuscin before reaching a certain threshold above which the cells can no longer cope, and the deleterious properties of lipofuscin manifest themselves. Alternatively, when lipofuscin starts exhibiting toxic properties, which could be induced by exposure to light, the cells remove it but it is unclear what mechanisms are involved in the removal of packets of lipofuscin from RPE cells and how it contributes to SDD, drusen, or other sub-RPE deposits in Bruch’s membrane.
Lipofuscin is the major contributor to the fluorescence of the retina. Imaging of RPE lipofuscin fluorescence is widely employed in clinics to monitor the progression of atrophic AMD, Stargardt’s disease, bestrophinopathies, and some forms of retinitis pigmentosa associated with lipofuscin accumulation and RPE cell loss. Oxidation of RPE lipofuscin results in spectral changes in its fluorescence emission: a decrease in its yellow-red fluorescence with a concomitant increase in the emission of its blue fluorescence. Lipofuscin oxidation can occur in vivo. However, the excitation wavelength in currently used fluorescence imaging instruments in clinical settings is too long so the fluorescence emission cannot be collected in the blue spectral range where the most pronounced fluorescence increase occurs upon oxidation. Recent developments in TPEF-SLO for safe imaging of the human retina suggest that this technique has the potential to be useful for imaging of oxidation state of lipofuscin in vivo and therefore for monitoring the oxidative damage in AMD and other retinal degenerations associated with lipofuscin accumulation, and for evaluation of the efficacy of potential antioxidant therapies.
Finally, it would be of interest to compare the effects of oxidation on fluorescence properties of RPE lipofuscin with lipofuscin from other tissues. While bisretinoids may be a unique feature of RPE lipofuscin, polyunsaturated lipids can be common components for many types of lipofuscin, especially those from the brain, which is rich in DHA and other polyunsaturated fatty acids. It well may be that oxidation-induced spectral changes in lipofuscin from other tissues may be a biomarker of oxidative damage and the formation of highly harmful end-products of lipid oxidation.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
4HNE, 4-hydroxynonenal; ABCR, ATP-binding cassette transporter rim protein; AGEs, advanced glycation end-products; AFM, atomic force microscopy; AMD, age-related macular degeneration; AOSLO, adaptive optics scanning laser ophthalmoscopy; AREDS, Age-Related Eye Disease Study; CEP, carboxyethylpyrrole; CRRY, complement receptor 1-like protein y; cRORA, complete RPE and outer retinal atrophy; DHA, docosahexaenoic acid; GPX, glutathione peroxidase; IL-1β, interleukin-1β; IL-8, interleukin 8; MTORC1, mammalian target of rapamycin complex 1; MAC, membrane attack complex; MCP-1, monocyte chemoattractant protein-1; MDA, malondialdehyde; MerTK, Mer tyrosine kinase; MLKL, mixed lineage kinase domain-like protein; NCL, neuronal ceroid lipofuscinosis; Nec7, necrostatin 7; NRPE, N-retinylidene-phosphatidylethanolamine; OCT, optical coherence tomography; NSOM, near-field scanning optical microscopy; POS, photoreceptor outer segments; PE, phosphatidylethanolamine; RCS, Royal College of Surgeons; RDH8, retinol dehydrogenase 8; RIPK1, receptor-interacting serine/threonine-protein kinase 1; RPE, retinal pigment epithelium; SDD, subretinal drusenoid deposits; SDS, sodium dodecyl sulphate; SIM, structured illumination microscopy; SLO, scanning laser ophthalmoscope; TEM, transmission electron microscopy; TFEB, transcriptional control of transcription factor EB; TPEF, two photon excited fluorescence; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labelling; VEGF, vascular endothelial growth factor
References
- Yin, D. Biochemical basis of lipofuscin, ceroid, and age pigment-like fluorophores. Free Radic Biol Med 1996, 21, 871–888. [Google Scholar] [CrossRef]
- Terman, A.; Gustafsson, B.; Brunk, U.T. Autophagy, organelles and ageing. J Pathol 2007, 211, 134–143. [Google Scholar] [CrossRef]
- Jung, T.; Bader, N.; Grune, T. Lipofuscin: formation, distribution, and metabolic consequences. Ann N Y Acad Sci 2007, 1119, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.A.; Brunk, U.T. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal 2010, 12, 503–535. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Wang, Q.; Hu, H.; Shen, Y.; Fan, C.; Chen, P.; Ma, Y.; Wu, H.; Xiang, M. Restoring autophagic flux attenuates cochlear spiral ganglion neuron degeneration by promoting TFEB nuclear translocation via inhibiting MTOR. Autophagy 2019, 15, 998–1016. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Gugiu, B.; Renganathan, K.; Davies, M.W.; Gu, X.; Crabb, J.S.; Kim, S.R.; Rozanowska, M.B.; Bonilha, V.L.; Rayborn, M.E.; et al. Retinal pigment epithelium lipofuscin proteomics. Mol Cell Proteomics 2008, 7, 1397–1405. [Google Scholar] [CrossRef]
- Krog, S.; Ludvigsen, T.P.; Nielsen, O.L.; Kirk, R.K.; Lykkegaard, K.; Wulff, E.M.; Moller, J.E.; Pedersen, H.D.; Olsen, L.H. Myocardial Changes in Diabetic and Nondiabetic Nonhuman Primates. Vet Pathol 2020, 57, 332–343. [Google Scholar] [CrossRef]
- Kakimoto, Y.; Okada, C.; Kawabe, N.; Sasaki, A.; Tsukamoto, H.; Nagao, R.; Osawa, M. Myocardial lipofuscin accumulation in ageing and sudden cardiac death. Sci Rep 2019, 9, 3304. [Google Scholar] [CrossRef]
- Moreno-Garcia, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front Neurosci 2018, 12, 464. [Google Scholar] [CrossRef]
- Couve, E.; Schmachtenberg, O. Autophagic activity and aging in human odontoblasts. J Dent Res 2011, 90, 523–528. [Google Scholar] [CrossRef]
- Sulzer, D.; Mosharov, E.; Talloczy, Z.; Zucca, F.A.; Simon, J.D.; Zecca, L. Neuronal pigmented autophagic vacuoles: lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J Neurochem 2008, 106, 24–36. [Google Scholar] [CrossRef]
- Jung, T.; Hohn, A.; Grune, T. Lipofuscin: detection and quantification by microscopic techniques. Methods Mol Biol 2010, 594, 173–193. [Google Scholar] [CrossRef] [PubMed]
- Simonati, A.; Williams, R.E. Neuronal Ceroid Lipofuscinosis: The Multifaceted Approach to the Clinical Issues, an Overview. Front Neurol 2022, 13, 811686. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nelvagal, H.R.; Lange, J.; Cooper, J.D. Glial Dysfunction and Its Contribution to the Pathogenesis of the Neuronal Ceroid Lipofuscinoses. Front Neurol 2022, 13, 886567. [Google Scholar] [CrossRef]
- Nelvagal, H.R.; Lange, J.; Takahashi, K.; Tarczyluk-Wells, M.A.; Cooper, J.D. Pathomechanisms in the neuronal ceroid lipofuscinoses. Biochim Biophys Acta Mol Basis Dis 2020, 1866, 165570. [Google Scholar] [CrossRef]
- Valdez, C.; Wong, Y.C.; Schwake, M.; Bu, G.; Wszolek, Z.K.; Krainc, D. Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet 2017, 26, 4861–4872. [Google Scholar] [CrossRef]
- Pan, C.; Banerjee, K.; Lehmann, G.L.; Almeida, D.; Hajjar, K.A.; Benedicto, I.; Jiang, Z.; Radu, R.A.; Thompson, D.H.; Rodriguez-Boulan, E.; et al. Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Klein, Z.A.; Takahashi, H.; Ma, M.; Stagi, M.; Zhou, M.; Lam, T.T.; Strittmatter, S.M. Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron 2017, 95, 281–296 e286. [Google Scholar] [CrossRef]
- Kohlschutter, A.; Schulz, A. CLN2 Disease (Classic Late Infantile Neuronal Ceroid Lipofuscinosis). Pediatr Endocrinol Rev 2016, 13 Suppl 1, 682–688. [Google Scholar]
- Ach, T.; Tolstik, E.; Messinger, J.D.; Zarubina, A.V.; Heintzmann, R.; Curcio, C.A. Lipofuscin redistribution and loss accompanied by cytoskeletal stress in retinal pigment epithelium of eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci 2015, 56, 3242–3252. [Google Scholar] [CrossRef]
- Nilsson, M.I.; MacNeil, L.G.; Kitaoka, Y.; Suri, R.; Young, S.P.; Kaczor, J.J.; Nates, N.J.; Ansari, M.U.; Wong, T.; Ahktar, M.; et al. Combined aerobic exercise and enzyme replacement therapy rejuvenates the mitochondrial-lysosomal axis and alleviates autophagic blockage in Pompe disease. Free Radic Biol Med 2015, 87, 98–112. [Google Scholar] [CrossRef]
- Simonati, A.; Pezzini, F.; Moro, F.; Santorelli, F.M. Neuronal Ceroid Lipofuscinosis: The Increasing Spectrum of an Old Disease. Curr Mol Med 2014, 14, 1043–1051. [Google Scholar] [CrossRef]
- Vidal-Donet, J.M.; Carcel-Trullols, J.; Casanova, B.; Aguado, C.; Knecht, E. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS One 2013, 8, e55526. [Google Scholar] [CrossRef] [PubMed]
- Piyanova, A.; Albayram, O.; Rossi, C.A.; Farwanah, H.; Michel, K.; Nicotera, P.; Sandhoff, K.; Bilkei-Gorzo, A. Loss of CB1 receptors leads to decreased cathepsin D levels and accelerated lipofuscin accumulation in the hippocampus. Mech Ageing Dev 2013, 134, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Baltazar, G.C.; Coffey, E.E.; Tu, L.A.; Lim, J.C.; Beckel, J.M.; Patel, S.; Eysteinsson, T.; Lu, W.; O’Brien-Jenkins, A.; et al. Lysosomal alkalinization, lipid oxidation, and reduced phagosome clearance triggered by activation of the P2X7 receptor. FASEB J 2013, 27, 4500–4509. [Google Scholar] [CrossRef] [PubMed]
- Kohan, R.; Cismondi, I.A.; Oller-Ramirez, A.M.; Guelbert, N.; Anzolini, T.V.; Alonso, G.; Mole, S.E.; de Kremer, D.R.; de Halac, N.I. Therapeutic approaches to the challenge of neuronal ceroid lipofuscinoses. Curr Pharm Biotechnol 2011, 12, 867–883. [Google Scholar] [CrossRef] [PubMed]
- 10.2174/138920111795542633.
- Katz, M.L.; Shanker, M.J. Development of lipofuscin-like fluorescence in the retinal pigment epithelium in response to protease inhibitor treatment. Mech Ageing Dev 1989, 49, 23–40. [Google Scholar] [CrossRef]
- Ivy, G.O.; Schottler, F.; Wenzel, J.; Baudry, M.; Lynch, G. Inhibitors of lysosomal enzymes: accumulation of lipofuscin-like dense bodies in the brain. Science 1984, 226, 985–987. [Google Scholar] [CrossRef]
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One 2011, 6, e23367. [Google Scholar] [CrossRef]
- Lee, S.J.; Park, M.H.; Kim, H.J.; Koh, J.Y. Metallothionein-3 regulates lysosomal function in cultured astrocytes under both normal and oxidative conditions. Glia 2010, 58, 1186–1196. [Google Scholar] [CrossRef]
- Stroikin, Y.; Dalen, H.; Loof, S.; Terman, A. Inhibition of autophagy with 3-methyladenine results in impaired turnover of lysosomes and accumulation of lipofuscin-like material. Eur J Cell Biol 2004, 83, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Hohn, A.; Sittig, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin is formed independently of macroautophagy and lysosomal activity in stress-induced prematurely senescent human fibroblasts. Free Radic Biol Med 2012, 53, 1760–1769. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Nagy, M.; Fenton, W.A.; Horwich, A.L. Absence of lipofuscin in motor neurons of SOD1-linked ALS mice. Proc Natl Acad Sci U S A 2014, 111, 11055–11060. [Google Scholar] [CrossRef]
- Martinez-Cisuelo, V.; Gomez, J.; Garcia-Junceda, I.; Naudi, A.; Cabre, R.; Mota-Martorell, N.; Lopez-Torres, M.; Gonzalez-Sanchez, M.; Pamplona, R.; Barja, G. Rapamycin reverses age-related increases in mitochondrial ROS production at complex I, oxidative stress, accumulation of mtDNA fragments inside nuclear DNA, and lipofuscin level, and increases autophagy, in the liver of middle-aged mice. Exp Gerontol 2016, 83, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Tzekov, R.; Li, H.; McDowell, J.H.; Gao, G.; Smith, W.C.; Tang, S.; Kaushal, S. Inhibition or Stimulation of Autophagy Affects Early Formation of Lipofuscin-Like Autofluorescence in the Retinal Pigment Epithelium Cell. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra Rao, S.; Fliesler, S.J. Monitoring basal autophagy in the retina utilizing CAG-mRFP-EGFP-MAP1LC3B reporter mouse: technical and biological considerations. Autophagy 2022, 18, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Yu, M.; Liu, Y.; Weh, E.; Pawar, M.; Li, L.; Besirli, C.G.; Schwendeman, A.A. Synthetic high-density lipoprotein nanoparticles delivering rapamycin for the treatment of age-related macular degeneration. Nanomedicine 2022, 44, 102571. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Blasiak, J.; Liton, P.; Boulton, M.; Klionsky, D.J.; Sinha, D. Autophagy in age-related macular degeneration. Autophagy 2022, 1–13. [Google Scholar] [CrossRef]
- Li, W.W.; Wang, H.J.; Tan, Y.Z.; Wang, Y.L.; Yu, S.N.; Li, Z.H. Reducing lipofuscin accumulation and cardiomyocytic senescence of aging heart by enhancing autophagy. Exp Cell Res 2021, 403, 112585. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Bajaj, L.; Lotfi, P.; Pal, R.; Ronza, A.D.; Sharma, J.; Sardiello, M. Lysosome biogenesis in health and disease. J Neurochem 2019, 148, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Hakvoort, K.; Otto, L.; Haeren, R.; Hoogland, G.; Schijns, O.; Vink, H.; Klein, D.; van Zandvoort, M.; Rijkers, K. Shedding light on human cerebral lipofuscin: An explorative study on identification and quantification. J Comp Neurol 2021, 529, 605–615. [Google Scholar] [CrossRef]
- McElnea, E.M.; Hughes, E.; McGoldrick, A.; McCann, A.; Quill, B.; Docherty, N.; Irnaten, M.; Farrell, M.; Clark, A.F.; O’Brien, C.J.; et al. Lipofuscin accumulation and autophagy in glaucomatous human lamina cribrosa cells. BMC Ophthalmol 2014, 14, 153. [Google Scholar] [CrossRef]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes and oxidative stress in aging and apoptosis. Biochim Biophys Acta 2008, 1780, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Liton, P.B.; Lin, Y.; Luna, C.; Li, G.; Gonzalez, P.; Epstein, D.L. Cultured porcine trabecular meshwork cells display altered lysosomal function when subjected to chronic oxidative stress. Invest Ophthalmol Vis Sci 2008, 49, 3961–3969. [Google Scholar] [CrossRef] [PubMed]
- Marzabadi, M.R.; Sohal, R.S.; Brunk, U.T. Effect of alpha-tocopherol and some metal chelators on lipofuscin accumulation in cultured neonatal rat cardiac myocytes. Anal Cell Pathol 1990, 2, 333–346. [Google Scholar]
- Marzabadi, M.R.; Sohal, R.S.; Brunk, U.T. Effect of ferric iron and desferrioxamine on lipofuscin accumulation in cultured rat heart myocytes. Mech Ageing Dev 1988, 46, 145–157. [Google Scholar] [CrossRef]
- Brunk, U.T.; Jones, C.B.; Sohal, R.S. A novel hypothesis of lipofuscinogenesis and cellular aging based on interactions between oxidative stress and autophagocytosis. Mutat Res 1992, 275, 395–403. [Google Scholar] [CrossRef]
- Shevtsova, Z.; Garrido, M.; Weishaupt, J.; Saftig, P.; Bahr, M.; Luhder, F.; Kugler, S. CNS-expressed cathepsin D prevents lymphopenia in a murine model of congenital neuronal ceroid lipofuscinosis. Am J Pathol 2010, 177, 271–279. [Google Scholar] [CrossRef]
- Reeg, S.; Grune, T. Protein Oxidation in Aging: Does It Play a Role in Aging Progression? Antioxid Redox Signal 2015, 23, 239–255. [Google Scholar] [CrossRef]
- Stroikin, Y.; Dalen, H.; Brunk, U.T.; Terman, A. Testing the “garbage” accumulation theory of ageing: mitotic activity protects cells from death induced by inhibition of autophagy. Biogerontology 2005, 6, 39–47. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T.; Nilsson, E.; Docke, W.D.; Brunk, U.T. Lipofuscin accumulation and ageing of fibroblasts. Gerontology 1995, 41 Suppl 2, 95–108. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Gustafsson, B.; Brunk, U.T. The involvement of lysosomes in myocardial aging and disease. Curr Cardiol Rev 2008, 4, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Epstein, D.L.; Liton, P.B. Intralysosomal iron induces lysosomal membrane permeabilization and cathepsin D-mediated cell death in trabecular meshwork cells exposed to oxidative stress. Invest Ophthalmol Vis Sci 2010, 51, 6483–6495. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Gomez, N.M.; Lim, J.C.; Guha, S.; O’Brien-Jenkins, A.; Coffey, E.E.; Campagno, K.E.; McCaughey, S.A.; Laties, A.M.; Carlsson, L.G.; et al. The P2Y12 Receptor Antagonist Ticagrelor Reduces Lysosomal pH and Autofluorescence in Retinal Pigmented Epithelial Cells From the ABCA4(-/-) Mouse Model of Retinal Degeneration. Front Pharmacol 2018, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, C.; Kravic, B.; Meyer, H. Repair or Lysophagy: Dealing with Damaged Lysosomes. J Mol Biol 2020, 432, 231–239. [Google Scholar] [CrossRef]
- Kurz, T.; Eaton, J.W.; Brunk, U.T. Redox activity within the lysosomal compartment: implications for aging and apoptosis. Antioxid Redox Signal 2010, 13, 511–523. [Google Scholar] [CrossRef]
- Hohn, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin-bound iron is a major intracellular source of oxidants: role in senescent cells. Free Radic Biol Med 2010, 48, 1100–1108. [Google Scholar] [CrossRef]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol 2008, 129, 389–406. [Google Scholar] [CrossRef]
- Grubman, A.; Pollari, E.; Duncan, C.; Caragounis, A.; Blom, T.; Volitakis, I.; Wong, A.; Cooper, J.; Crouch, P.J.; Koistinaho, J.; et al. Deregulation of biometal homeostasis: the missing link for neuronal ceroid lipofuscinoses? Metallomics 2014, 6, 932–943. [Google Scholar] [CrossRef]
- Mochizuki, Y.; Park, M.K.; Mori, T.; Kawashima, S. The difference in autofluorescence features of lipofuscin between brain and adrenal. Zoolog Sci 1995, 12, 283–288. [Google Scholar] [CrossRef]
- Katz, M.L.; Robison, W.G., Jr.; Herrmann, R.K.; Groome, A.B.; Bieri, J.G. Lipofuscin accumulation resulting from senescence and vitamin E deficiency: spectral properties and tissue distribution. Mech Ageing Dev 1984, 25, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Docchio, F.; Dayhaw-Barker, P.; Ramponi, R.; Cubeddu, R. Age-related changes in the morphology, absorption and fluorescence of melanosomes and lipofuscin granules of the retinal pigment epithelium. Vision Res 1990, 30, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Kikugawa, K.; Beppu, M.; Sato, A.; Kasai, H. Separation of multiple yellow fluorescent lipofuscin components in rat kidney and their characterization. Mech Ageing Dev 1997, 97, 93–107. [Google Scholar] [CrossRef]
- Eldred, G.E.; Miller, G.V.; Stark, W.S.; Feeney-Burns, L. Lipofuscin: resolution of discrepant fluorescence data. Science 1982, 216, 757–759. [Google Scholar] [CrossRef]
- Warburton, S.; Southwick, K.; Hardman, R.M.; Secrest, A.M.; Grow, R.K.; Xin, H.; Woolley, A.T.; Burton, G.F.; Thulin, C.D. Examining the proteins of functional retinal lipofuscin using proteomic analysis as a guide for understanding its origin. Mol Vis 2005, 11, 1122–1134. [Google Scholar] [PubMed]
- Kikugawa, K.; Beppu, M.; Kato, T.; Yamaki, S.; Kasai, H. Accumulation of autofluorescent yellow lipofuscin in rat tissues estimated by sodium dodecylsulfate extraction. Mech Ageing Dev 1994, 74, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Kikugawa, K.; Beppu, M. Involvement of lipid oxidation products in the formation of fluorescent and cross-linked proteins. Chem Phys Lipids 1987, 44, 277–296. [Google Scholar] [CrossRef]
- Kikugawa, K.; Kato, T.; Beppu, M.; Hayasaka, A. Fluorescent and cross-linked proteins formed by free radical and aldehyde species generated during lipid oxidation. Adv Exp Med Biol 1989, 266, 345–356; discussion 357. [Google Scholar] [CrossRef]
- Hidalgo, F.J.; Zamora, R. Modification of bovine serum albumin structure following reaction with 4,5(E)-epoxy-2(E)-heptenal. Chem Res Toxicol 2000, 13, 501–508. [Google Scholar] [CrossRef]
- d’Ischia, M.; Costantini, C.; Prota, G. Lipofuscin-like pigments by autoxidation of polyunsaturated fatty acids in the presence of amine neurotransmitters: the role of malondialdehyde. Biochim Biophys Acta 1996, 1290, 319–326. [Google Scholar] [CrossRef]
- Eldred, G.E.; Katz, M.L. The autofluorescent products of lipid peroxidation may not be lipofuscin-like. Free Radic Biol Med 1989, 7, 157–163. [Google Scholar] [CrossRef]
- Yin, D.Z.; Brunk, U.T. Microfluorometric and fluorometric lipofuscin spectral discrepancies: a concentration-dependent metachromatic effect? Mech Ageing Dev 1991, 59, 95–109. [Google Scholar] [CrossRef]
- Rozanowska, M.B.; Rozanowski, B. Photodegradation of Lipofuscin in Suspension and in ARPE-19 Cells and the Similarity of Fluorescence of the Photodegradation Product with Oxidized Docosahexaenoate. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Feeney-Burns, L.; Hilderbrand, E.S.; Eldridge, S. Aging human RPE: morphometric analysis of macular, equatorial, and peripheral cells. Invest Ophthalmol Vis Sci 1984, 25, 195–200. [Google Scholar]
- Hayasaka, S. Aging changes in lipofuscin, lysosomes and melanin in the macular area of human retina and choroid. Jpn J Ophthalmol 1989, 33, 36–42. [Google Scholar] [PubMed]
- Rozanowska, M. Properties and Functions of Ocular Melanins and Melanosomes. In Melanins and Melanosomes; Biosynthesis, Biogenesis, Physiological and Pathological Functions, Borovansky, J., Riley, P.A., Eds.; Wiley-Blackwell: Singapore, 2011; pp. 187-224.
- Yanoff, M.; Duker, J.S. Ophthalmology: Expert Consult: Online and Print; Elsevier Health Sciences: 2013.
- Lewandowski, D.; Sander, C.L.; Tworak, A.; Gao, F.; Xu, Q.; Skowronska-Krawczyk, D. Dynamic lipid turnover in photoreceptors and retinal pigment epithelium throughout life. Prog Retin Eye Res 2022, 89, 101037. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; McKechnie, N.M.; Breda, J.; Bayly, M.; Marshall, J. The formation of autofluorescent granules in cultured human RPE. Invest Ophthalmol Vis Sci 1989, 30, 82–89. [Google Scholar]
- Katz, M.L.; Drea, C.M.; Eldred, G.E.; Hess, H.H.; Robison, W.G., Jr. Influence of early photoreceptor degeneration on lipofuscin in the retinal pigment epithelium. Exp Eye Res 1986, 43, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Feeney-Burns, L.; Eldred, G.E. The fate of the phagosome: conversion to ‘age pigment’ and impact in human retinal pigment epithelium. Trans Ophthalmol Soc U K (1962) 1983, 103 ( Pt 4), 416–421. [Google Scholar]
- Feeney, L. Lipofuscin and melanin of human retinal pigment epithelium. Fluorescence, enzyme cytochemical, and ultrastructural studies. Invest Ophthalmol Vis Sci 1978, 17, 583–600. [Google Scholar]
- Pugh, E.N., Jr.; Lamb, T.D. Amplification and kinetics of the activation steps in phototransduction. Biochim Biophys Acta 1993, 1141, 111–149. [Google Scholar] [CrossRef]
- Rodieck, R.W. The first steps in seeing; Sinauer Associates Inc.: Sunderland, Massachusetts, 1998. [Google Scholar]
- de Araujo, M.E.G.; Liebscher, G.; Hess, M.W.; Huber, L.A. Lysosomal size matters. Traffic 2020, 21, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Snodderly, D.M.; Sandstrom, M.M.; Leung, I.Y.; Zucker, C.L.; Neuringer, M. Retinal pigment epithelial cell distribution in central retina of rhesus monkeys. Invest Ophthalmol Vis Sci 2002, 43, 2815–2818. [Google Scholar]
- Rozanowska, M.; Rozanowski, B.; Boulton, M. Photobiology of the retina: Light damage to the retina. (accessed on 23 March 2023). In Photobiological Sciences Online: http://www.photobiology.info, Smith, K.C., Ed.; American Society for Photobiology: Herndon, VA, USA, 2009.
- Katz, M.L.; Rice, L.M.; Gao, C.L. Reversible accumulation of lipofuscin-like inclusions in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 1999, 40, 175–181. [Google Scholar]
- Ivy, G.O.; Ihara, Y.; Kitani, K. The protease inhibitor leupeptin induces several signs of aging in brain, retina and internal organs of young rats. Arch Gerontol Geriatr 1991, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Stientjes, H.J.; Gao, C.L.; Christianson, J.S. Iron-induced accumulation of lipofuscin-like fluorescent pigment in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 1993, 34, 3161–3171. [Google Scholar] [PubMed]
- Katz, M.L.; Christianson, J.S.; Gao, C.L.; Handelman, G.J. Iron-induced fluorescence in the retina: dependence on vitamin A. Invest Ophthalmol Vis Sci 1994, 35, 3613–3624. [Google Scholar] [PubMed]
- Liu, Y.; Bell, B.A.; Song, Y.; Kim, H.J.; Sterling, J.K.; Kim, B.J.; Poli, M.; Guo, M.; Zhang, K.; Rao, A.; et al. Intraocular iron injection induces oxidative stress followed by elements of geographic atrophy and sympathetic ophthalmia. Aging Cell 2021, 20, e13490. [Google Scholar] [CrossRef]
- Rozanowska, M.B.; Czuba-Pelech, B.; Rozanowski, B. Is There an Optimal Combination of AREDS2 Antioxidants Zeaxanthin, Vitamin E and Vitamin C on Light-Induced Toxicity of Vitamin A Aldehyde to the Retina? Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Rozanowska, M.B.; Czuba-Pelech, B.; Landrum, J.T.; Rozanowski, B. Comparison of Antioxidant Properties of Dehydrolutein with Lutein and Zeaxanthin, and their Effects on Cultured Retinal Pigment Epithelial Cells. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Rozanowska, M.; Handzel, K.; Boulton, M.E.; Rozanowski, B. Cytotoxicity of all-trans-retinal increases upon photodegradation. Photochem Photobiol 2012, 88, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Rozanowska, M.; Sarna, T. Light-induced damage to the retina: role of rhodopsin chromophore revisited. Photochem Photobiol 2005, 81, 1305–1330. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Rozanowska, M.; Rozanowski, B. Retinal photodamage. J Photochem Photobiol B 2001, 64, 144–161. [Google Scholar] [CrossRef] [PubMed]
- Bazan, H.E.; Bazan, N.G.; Feeney-Burns, L.; Berman, E.R. Lipids in human lipofuscin-enriched subcellular fractions of two age populations. Comparison with rod outer segments and neural retina. Invest Ophthalmol Vis Sci 1990, 31, 1433–1443. [Google Scholar]
- Sun, H.; Nathans, J. ABCR, the ATP-binding cassette transporter responsible for Stargardt macular dystrophy, is an efficient target of all-trans-retinal-mediated photooxidative damage in vitro. Implications for retinal disease. J Biol Chem 2001, 276, 11766–11774. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Garces, F.A.; Scortecci, J.F.; Molday, L.L. Structure and function of ABCA4 and its role in the visual cycle and Stargardt macular degeneration. Prog. Retin. Eye Res. 2022, 89. [Google Scholar] [CrossRef]
- Lenis, T.L.; Hu, J.; Ng, S.Y.; Jiang, Z.; Sarfare, S.; Lloyd, M.B.; Esposito, N.J.; Samuel, W.; Jaworski, C.; Bok, D.; et al. Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc Natl Acad Sci U S A 2018, 115, E11120–E11127. [Google Scholar] [CrossRef]
- Hong, J.D.; Salom, D.; Kochman, M.A.; Kubas, A.; Kiser, P.D.; Palczewski, K. Chromophore hydrolysis and release from photoactivated rhodopsin in native membranes. Proc Natl Acad Sci U S A 2022, 119, e2213911119. [Google Scholar] [CrossRef]
- Zhao, J.; Kim, H.J.; Ueda, K.; Zhang, K.; Montenegro, D.; Dunaief, J.L.; Sparrow, J.R. A vicious cycle of bisretinoid formation and oxidation relevant to recessive Stargardt disease. J Biol Chem 2021, 296, 100259. [Google Scholar] [CrossRef]
- Kim, H.J.; Sparrow, J.R. Bisretinoid phospholipid and vitamin A aldehyde: shining a light. J Lipid Res 2021, 62, 100042. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Montenegro, D.; Zhao, J.; Sparrow, J.R. Bisretinoids of the Retina: Photo-Oxidation, Iron-Catalyzed Oxidation, and Disease Consequences. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.; Elliott, M.H.; Floor, E.; Truscott, T.G.; Zareba, M.; Sarna, T.; Shamsi, F.A.; Boulton, M.E. Photocytotoxicity of lipofuscin in human retinal pigment epithelial cells. Free Radic Biol Med 2001, 31, 256–265. [Google Scholar] [CrossRef]
- Avalle, L.B.; Wang, Z.; Dillon, J.P.; Gaillard, E.R. Observation of A2E oxidation products in human retinal lipofuscin. Exp Eye Res 2004, 78, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.P.; Matsuda, H.; Itagaki, Y.; Nakanishi, K.; Sparrow, J.R. Characterization of peroxy-A2E and furan-A2E photooxidation products and detection in human and mouse retinal pigment epithelial cell lipofuscin. J Biol Chem 2005, 280, 39732–39739. [Google Scholar] [CrossRef]
- Ablonczy, Z.; Higbee, D.; Anderson, D.M.; Dahrouj, M.; Grey, A.C.; Gutierrez, D.; Koutalos, Y.; Schey, K.L.; Hanneken, A.; Crouch, R.K. Lack of correlation between the spatial distribution of A2E and lipofuscin fluorescence in the human retinal pigment epithelium. Invest Ophthalmol Vis Sci 2013, 54, 5535–5542. [Google Scholar] [CrossRef] [PubMed]
- Adler, L.t.; Boyer, N.P.; Anderson, D.M.; Spraggins, J.M.; Schey, K.L.; Hanneken, A.; Ablonczy, Z.; Crouch, R.K.; Koutalos, Y. Determination of N-retinylidene-N-retinylethanolamine (A2E) levels in central and peripheral areas of human retinal pigment epithelium. Photochem Photobiol Sci 2015, 14, 1983–1990. [Google Scholar] [CrossRef]
- Kotnala, A.; Senthilkumari, S.; Wu, G.; Stewart, T.G.; Curcio, C.A.; Halder, N.; Singh, S.B.; Kumar, A.; Velpandian, T. Retinal Pigment Epithelium in Human Donor Eyes Contains Higher Levels of Bisretinoids Including A2E in Periphery than Macula. Invest Ophthalmol Vis Sci 2022, 63, 6. [Google Scholar] [CrossRef]
- Fite, K.V.; Bengston, L.; Donaghey, B. Experimental light damage increases lipofuscin in the retinal pigment epithelium of Japanese quail (Coturnix coturnix japonica). Exp Eye Res 1993, 57, 449–460. [Google Scholar] [CrossRef]
- Boyer, N.P.; Thompson, D.A.; Koutalos, Y. Relative Contributions of All-Trans and 11-Cis Retinal to Formation of Lipofuscin and A2E Accumulating in Mouse Retinal Pigment Epithelium. Invest Ophthalmol Vis Sci 2021, 62, 1. [Google Scholar] [CrossRef]
- Adler, L.t.; Boyer, N.P.; Chen, C.; Ablonczy, Z.; Crouch, R.K.; Koutalos, Y. The 11-cis Retinal Origins of Lipofuscin in the Retina. Prog Mol Biol Transl Sci 2015, 134, e1–e12. [Google Scholar] [CrossRef]
- Boyer, N.P.; Higbee, D.; Currin, M.B.; Blakeley, L.R.; Chen, C.; Ablonczy, Z.; Crouch, R.K.; Koutalos, Y. Lipofuscin and N-retinylidene-N-retinylethanolamine (A2E) accumulate in retinal pigment epithelium in absence of light exposure: their origin is 11-cis-retinal. J Biol Chem 2012, 287, 22276–22286. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Golczak, M.; Maeda, T.; Palczewski, K. Limited roles of Rdh8, Rdh12, and Abca4 in all-trans-retinal clearance in mouse retina. Invest Ophthalmol Vis Sci 2009, 50, 5435–5443. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Maeda, T.; Golczak, M.; Palczewski, K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J Biol Chem 2008, 283, 26684–26693. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Sun, W.; Zhang, H.; Baehr, W.; Palczewski, K. Redundant and unique roles of retinol dehydrogenases in the mouse retina. Proc Natl Acad Sci U S A 2007, 104, 19565–19570. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Maeda, T.; Imanishi, Y.; Kuksa, V.; Alekseev, A.; Bronson, J.D.; Zhang, H.; Zhu, L.; Sun, W.; Saperstein, D.A.; et al. Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J Biol Chem 2005, 280, 18822–18832. [Google Scholar] [CrossRef] [PubMed]
- Mata, N.L.; Tzekov, R.T.; Liu, X.; Weng, J.; Birch, D.G.; Travis, G.H. Delayed dark-adaptation and lipofuscin accumulation in abcr+/- mice: implications for involvement of ABCR in age-related macular degeneration. Invest Ophthalmol Vis Sci 2001, 42, 1685–1690. [Google Scholar]
- Weng, J.; Mata, N.L.; Azarian, S.M.; Tzekov, R.T.; Birch, D.G.; Travis, G.H. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 1999, 98, 13–23. [Google Scholar] [CrossRef]
- Eldred, G.E. Vitamins A and E in RPE lipofuscin formation and implications for age-related macular degeneration. Prog Clin Biol Res 1989, 314, 113–129. [Google Scholar]
- Katz, M.L.; Drea, C.M.; Robison, W.G., Jr. Relationship between dietary retinol and lipofuscin in the retinal pigment epithelium. Mech Ageing Dev 1986, 35, 291–305. [Google Scholar] [CrossRef]
- Katz, M.L.; Eldred, G.E.; Robison, W.G., Jr. Lipofuscin autofluorescence: evidence for vitamin A involvement in the retina. Mech Ageing Dev 1987, 39, 81–90. [Google Scholar] [CrossRef]
- Katz, M.L.; Norberg, M. Influence of dietary vitamin A on autofluorescence of leupeptin-induced inclusions in the retinal pigment epithelium. Exp Eye Res 1992, 54, 239–246. [Google Scholar] [CrossRef]
- Katz, M.L.; Norberg, M.; Stientjes, H.J. Reduced phagosomal content of the retinal pigment epithelium in response to retinoid deprivation. Invest Ophthalmol Vis Sci 1992, 33, 2612–2618. [Google Scholar]
- Dobri, N.; Qin, Q.; Kong, J.; Yamamoto, K.; Liu, Z.; Moiseyev, G.; Ma, J.X.; Allikmets, R.; Sparrow, J.R.; Petrukhin, K. A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. Invest Ophthalmol Vis Sci 2013, 54, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Golczak, M.; Chen, Y.; Okano, K.; Kohno, H.; Shiose, S.; Ishikawa, K.; Harte, W.; Palczewska, G.; Maeda, T.; et al. Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat Chem Biol 2011, 8, 170–178. [Google Scholar] [CrossRef]
- Maeda, T.; Maeda, A.; Matosky, M.; Okano, K.; Roos, S.; Tang, J.; Palczewski, K. Evaluation of potential therapies for a mouse model of human age-related macular degeneration caused by delayed all-trans-retinal clearance. Invest Ophthalmol Vis Sci 2009, 50, 4917–4925. [Google Scholar] [CrossRef] [PubMed]
- Golczak, M.; Maeda, A.; Bereta, G.; Maeda, T.; Kiser, P.D.; Hunzelmann, S.; von Lintig, J.; Blaner, W.S.; Palczewski, K. Metabolic basis of visual cycle inhibition by retinoid and nonretinoid compounds in the vertebrate retina. J Biol Chem 2008, 283, 9543–9554. [Google Scholar] [CrossRef] [PubMed]
- Radu, R.A.; Han, Y.; Bui, T.V.; Nusinowitz, S.; Bok, D.; Lichter, J.; Widder, K.; Travis, G.H.; Mata, N.L. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci 2005, 46, 4393–4401. [Google Scholar] [CrossRef] [PubMed]
- Golczak, M.; Kuksa, V.; Maeda, T.; Moise, A.R.; Palczewski, K. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci U S A 2005, 102, 8162–8167. [Google Scholar] [CrossRef]
- Radu, R.A.; Mata, N.L.; Nusinowitz, S.; Liu, X.; Sieving, P.A.; Travis, G.H. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc Natl Acad Sci U S A 2003, 100, 4742–4747. [Google Scholar] [CrossRef]
- Radu, R.A.; Yuan, Q.; Hu, J.; Peng, J.H.; Lloyd, M.; Nusinowitz, S.; Bok, D.; Travis, G.H. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Invest Ophthalmol Vis Sci 2008, 49, 3821–3829. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; O’Neil, J.; Miyagi, M.; West, K.; Hoff, H.F. Hydroxynonenal inactivates cathepsin B by forming Michael adducts with active site residues. Protein Sci 2002, 11, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Bermann, M.; Schutt, F.; Holz, F.G.; Kopitz, J. Does A2E, a retinoid component of lipofuscin and inhibitor of lysosomal degradative functions, directly affect the activity of lysosomal hydrolases? Exp Eye Res 2001, 72, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Holz, F.G.; Schutt, F.; Kopitz, J.; Eldred, G.E.; Kruse, F.E.; Volcker, H.E.; Cantz, M. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci 1999, 40, 737–743. [Google Scholar]
- Warburton, S.; Davis, W.E.; Southwick, K.; Xin, H.; Woolley, A.T.; Burton, G.F.; Thulin, C.D. Proteomic and phototoxic characterization of melanolipofuscin: correlation to disease and model for its origin. Mol Vis 2007, 13, 318–329. [Google Scholar]
- Schutt, F.; Bergmann, M.; Holz, F.G.; Kopitz, J. Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Invest Ophthalmol Vis Sci 2003, 44, 3663–3668. [Google Scholar] [CrossRef]
- Schutt, F.; Ueberle, B.; Schnolzer, M.; Holz, F.G.; Kopitz, J. Proteome analysis of lipofuscin in human retinal pigment epithelial cells. FEBS Lett 2002, 528, 217–221. [Google Scholar] [CrossRef]
- Clancy, C.M.R.; Krogmeier, J.R.; Pawlak, A.; Rozanowska, M.; Sarna, T.; Dunn, R.C.; Simon, J.D. Atomic force microscopy and near-field scanning optical microscopy measurements of single human retinal lipofuscin granules. J. Phys. Chem. B 2000, 104, 12098–12101. [Google Scholar] [CrossRef]
- Krogmeier, J.R.; Clancy, C.M.; Pawlak, A.; Rozanowska, M.; Sarna, T.; Simon, J.D.; Dunn, R.C. Mapping the distribution of emissive molecules in human ocular lipofuscin granules with near-field scanning optical microscopy. J Microsc 2001, 202, 386–390. [Google Scholar] [CrossRef]
- Gouras, P.; Brown, K.R.; Mattison, J.A.; Neuringer, M.; Nagasaki, T.; Ivert, L. The Ultrastructure, Spatial Distribution, and Osmium Tetroxide Binding of Lipofuscin and Melanosomes in Aging Monkey Retinal Epithelium. Curr Eye Res 2018, 43, 1019–1023. [Google Scholar] [CrossRef]
- Haralampus-Grynaviski, N.M.; Lamb, L.E.; Clancy, C.M.; Skumatz, C.; Burke, J.M.; Sarna, T.; Simon, J.D. Spectroscopic and morphological studies of human retinal lipofuscin granules. Proc Natl Acad Sci U S A 2003, 100, 3179–3184. [Google Scholar] [CrossRef] [PubMed]
- Petrukhin, A.N.; Astaf’ev, A.A.; Zolotavin, P.N.; Fel’dman, T.B.; Dontsov, A.E.; Sarkisov, O.M.; Ostrovsky, M.A. Heterogeneity of structure and fluorescence of single lipofuscin granule from retinal pigment epithelium of human donor eyes: study with the use of atomic force microscopy and near-field microscopy. Dokl Biochem Biophys 2005, 405, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Wing, G.L.; Blanchard, G.C.; Weiter, J.J. The topography and age relationship of lipofuscin concentration in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 1978, 17, 601–607. [Google Scholar] [PubMed]
- Weiter, J.J.; Delori, F.C.; Wing, G.L.; Fitch, K.A. Retinal pigment epithelial lipofuscin and melanin and choroidal melanin in human eyes. Invest Ophthalmol Vis Sci 1986, 27, 145–152. [Google Scholar]
- Artigas, J.M.; Felipe, A.; Navea, A.; Fandino, A.; Artigas, C. Spectral transmission of the human crystalline lens in adult and elderly persons: color and total transmission of visible light. Invest Ophthalmol Vis Sci 2012, 53, 4076–4084. [Google Scholar] [CrossRef]
- Curcio, C.A.; Sloan, K.R.; Kalina, R.E.; Hendrickson, A.E. Human photoreceptor topography. J Comp Neurol 1990, 292, 497–523. [Google Scholar] [CrossRef]
- Gliem, M.; Muller, P.L.; Birtel, J.; Herrmann, P.; McGuinness, M.B.; Holz, F.G.; Charbel Issa, P. Quantitative Fundus Autofluorescence and Genetic Associations in Macular, Cone, and Cone-Rod Dystrophies. Ophthalmol Retina 2020, 4, 737–749. [Google Scholar] [CrossRef]
- Bakall, B.; Radu, R.A.; Stanton, J.B.; Burke, J.M.; McKay, B.S.; Wadelius, C.; Mullins, R.F.; Stone, E.M.; Travis, G.H.; Marmorstein, A.D. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2). Exp Eye Res 2007, 85, 34–43. [Google Scholar] [CrossRef]
- Zhang, Q.; Small, K.W.; Grossniklaus, H.E. Clinicopathologic findings in Best vitelliform macular dystrophy. Graefes Arch Clin Exp Ophthalmol 2011, 249, 745–751. [Google Scholar] [CrossRef]
- Jauregui, R.; Chan, L.; Oh, J.K.; Cho, A.; Sparrow, J.R.; Tsang, S.H. Disease asymmetry and hyperautofluorescent ring shape in retinitis pigmentosa patients. Sci Rep 2020, 10, 3364. [Google Scholar] [CrossRef]
- Jauregui, R.; Park, K.S.; Duong, J.K.; Sparrow, J.R.; Tsang, S.H. Quantitative Comparison of Near-infrared Versus Short-wave Autofluorescence Imaging in Monitoring Progression of Retinitis Pigmentosa. Am J Ophthalmol 2018, 194, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Gliem, M.; Muller, P.L.; Finger, R.P.; McGuinness, M.B.; Holz, F.G.; Charbel Issa, P. Quantitative Fundus Autofluorescence in Early and Intermediate Age-Related Macular Degeneration. JAMA Ophthalmol 2016, 134, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Bermond, K.; von der Emde, L.; Tarau, I.S.; Bourauel, L.; Heintzmann, R.; Holz, F.G.; Curcio, C.A.; Sloan, K.R.; Ach, T. Autofluorescent Organelles Within the Retinal Pigment Epithelium in Human Donor Eyes With and Without Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 2022, 63, 23. [Google Scholar] [CrossRef] [PubMed]
- Dorey, C.K.; Wu, G.; Ebenstein, D.; Garsd, A.; Weiter, J.J. Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci 1989, 30, 1691–1699. [Google Scholar]
- Curcio, C.A. Photoreceptor topography in ageing and age-related maculopathy. Eye (Lond) 2001, 15, 376–383. [Google Scholar] [CrossRef]
- De, S.; Sakmar, T.P. Interaction of A2E with model membranes. Implications to the pathogenesis of age-related macular degeneration. J Gen Physiol 2002, 120, 147–157. [Google Scholar] [CrossRef]
- Schutt, F.; Bergmann, M.; Holz, F.G.; Kopitz, J. Isolation of intact lysosomes from human RPE cells and effects of A2-E on the integrity of the lysosomal and other cellular membranes. Graefes Arch Clin Exp Ophthalmol 2002, 240, 983–988. [Google Scholar] [CrossRef]
- Bergmann, M.; Schutt, F.; Holz, F.G.; Kopitz, J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J 2004, 18, 562–564. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef]
- Boulton, M.; Marshall, J. Repigmentation of human retinal pigment epithelial cells in vitro. Exp Eye Res 1985, 41, 209–218. [Google Scholar] [CrossRef]
- Rakoczy, P.; Kennedy, C.; Thompson-Wallis, D.; Mann, K.; Constable, I. Changes in retinal pigment epithelial cell autofluorescence and protein expression associated with phagocytosis of rod outer segments in vitro. Biol Cell 1992, 76, 49–54. [Google Scholar] [CrossRef]
- Brunk, U.T.; Wihlmark, U.; Wrigstad, A.; Roberg, K.; Nilsson, S.E. Accumulation of lipofuscin within retinal pigment epithelial cells results in enhanced sensitivity to photo-oxidation. Gerontology 1995, 41 Suppl 2, 201–212. [Google Scholar] [CrossRef]
- Wassell, J.; Ellis, S.; Burke, J.; Boulton, M. Fluorescence properties of autofluorescent granules generated by cultured human RPE cells. Invest Ophthalmol Vis Sci 1998, 39, 1487–1492. [Google Scholar]
- Boulton, M.E. Studying melanin and lipofuscin in RPE cell culture models. Exp Eye Res 2014, 126, 61–67. [Google Scholar] [CrossRef]
- Zhang, Q.; Presswalla, F.; Calton, M.; Charniga, C.; Stern, J.; Temple, S.; Vollrath, D.; Zacks, D.N.; Ali, R.R.; Thompson, D.A.; et al. Highly Differentiated Human Fetal RPE Cultures Are Resistant to the Accumulation and Toxicity of Lipofuscin-Like Material. Invest Ophthalmol Vis Sci 2019, 60, 3468–3479. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.O. Phagocytosis of light- and dark-adapted rod outer segments by cultured pigment epithelium. Science 1978, 202, 526–528. [Google Scholar] [CrossRef]
- Nilsson, S.E.; Sundelin, S.P.; Wihlmark, U.; Brunk, U.T. Aging of cultured retinal pigment epithelial cells: oxidative reactions, lipofuscin formation and blue light damage. Doc Ophthalmol 2003, 106, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Tzekov, R.; McDowell, J.H.; Smith, W.C.; Tang, S.; Kaushal, S. Formation of lipofuscin-like material in the RPE Cell by different components of rod outer segments. Exp Eye Res 2013, 112, 57–67. [Google Scholar] [CrossRef]
- Kaemmerer, E.; Schutt, F.; Krohne, T.U.; Holz, F.G.; Kopitz, J. Effects of lipid peroxidation-related protein modifications on RPE lysosomal functions and POS phagocytosis. Invest Ophthalmol Vis Sci 2007, 48, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Krohne, T.U.; Stratmann, N.K.; Kopitz, J.; Holz, F.G. Effects of lipid peroxidation products on lipofuscinogenesis and autophagy in human retinal pigment epithelial cells. Exp Eye Res 2010, 90, 465–471. [Google Scholar] [CrossRef]
- Rakoczy, P.E.; Baines, M.; Kennedy, C.J.; Constable, I.J. Correlation between autofluorescent debris accumulation and the presence of partially processed forms of cathepsin D in cultured retinal pigment epithelial cells challenged with rod outer segments. Exp Eye Res 1996, 63, 159–167. [Google Scholar] [CrossRef]
- Escrevente, C.; Falcao, A.S.; Hall, M.J.; Lopes-da-Silva, M.; Antas, P.; Mesquita, M.M.; Ferreira, I.S.; Cardoso, M.H.; Oliveira, D.; Fradinho, A.C.; et al. Formation of Lipofuscin-Like Autofluorescent Granules in the Retinal Pigment Epithelium Requires Lysosome Dysfunction. Invest Ophthalmol Vis Sci 2021, 62, 39. [Google Scholar] [CrossRef]
- Zhang, L.; Hui, Y.N.; Wang, Y.S.; Ma, J.X.; Wang, J.B.; Ma, L.N. Calcium overload is associated with lipofuscin formation in human retinal pigment epithelial cells fed with photoreceptor outer segments. Eye (Lond) 2011, 25, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Maeda, T.; Golczak, M.; Chou, S.; Desai, A.; Hoppel, C.L.; Matsuyama, S.; Palczewski, K. Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J Biol Chem 2009, 284, 15173–15183. [Google Scholar] [CrossRef] [PubMed]
- Wiktor, A.; Sarna, M.; Wnuk, D.; Sarna, T. Lipofuscin-mediated photodynamic stress induces adverse changes in nanomechanical properties of retinal pigment epithelium cells. Sci Rep 2018, 8, 17929. [Google Scholar] [CrossRef] [PubMed]
- Olchawa, M.M.; Furso, J.A.; Szewczyk, G.M.; Sarna, T.J. Lipofuscin-mediated photic stress inhibits phagocytic activity of ARPE-19 cells; effect of donors’ age and antioxidants. Free Radic Res 2017, 51, 799–811. [Google Scholar] [CrossRef]
- Zareba, M.; Skumatz, C.M.; Sarna, T.J.; Burke, J.M. Photic injury to cultured RPE varies among individual cells in proportion to their endogenous lipofuscin content as modulated by their melanosome content. Invest Ophthalmol Vis Sci 2014, 55, 4982–4990. [Google Scholar] [CrossRef]
- Godley, B.F.; Shamsi, F.A.; Liang, F.Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J Biol Chem 2005, 280, 21061–21066. [Google Scholar] [CrossRef]
- Shamsi, F.A.; Boulton, M. Inhibition of RPE lysosomal and antioxidant activity by the age pigment lipofuscin. Invest Ophthalmol Vis Sci 2001, 42, 3041–3046. [Google Scholar]
- Rozanowska, M.; Jarvis-Evans, J.; Korytowski, W.; Boulton, M.E.; Burke, J.M.; Sarna, T. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J Biol Chem 1995, 270, 18825–18830. [Google Scholar] [CrossRef]
- Boulton, M.; Dontsov, A.; Jarvis-Evans, J.; Ostrovsky, M.; Svistunenko, D. Lipofuscin is a photoinducible free radical generator. J Photochem Photobiol B 1993, 19, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Rozanowska, M.; Korytowski, W.; Rozanowski, B.; Skumatz, C.; Boulton, M.E.; Burke, J.M.; Sarna, T. Photoreactivity of aged human RPE melanosomes: a comparison with lipofuscin. Invest Ophthalmol Vis Sci 2002, 43, 2088–2096. [Google Scholar] [PubMed]
- Rozanowska, M.; Pawlak, A.; Rozanowski, B.; Skumatz, C.; Zareba, M.; Boulton, M.E.; Burke, J.M.; Sarna, T.; Simon, J.D. Age-related changes in the photoreactivity of retinal lipofuscin granules: role of chloroform-insoluble components. Invest Ophthalmol Vis Sci 2004, 45, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Rózanowska, M.B.; Pawlak, A.; Rózanowski, B. Products of ˙docosahexaenoate oxidation as contributors to photosensitising properties of retinal lipofuscin. Int. J. Mol. Sci. 2021, 22, 3525. [Google Scholar] [CrossRef]
- Rozanowska, M.; Wessels, J.; Boulton, M.; Burke, J.M.; Rodgers, M.A.J.; Truscott, T.G.; Sarna, T. Blue light-induced singlet oxygen generation by retinal lipofuscin in non-polar media. Free Radic. Biol. Med. 1998, 24, 1107–1112. [Google Scholar] [CrossRef]
- Gaillard, E.R.; Atherton, S.J.; Eldred, G.; Dillon, J. Photophysical studies on human retinal lipofuscin. Photochem Photobiol 1995, 61, 448–453. [Google Scholar] [CrossRef]
- Pawlak, A.; Wrona, M.; Rozanowska, M.; Zareba, M.; Lamb, L.E.; Roberts, J.E.; Simon, J.D.; Sarna, T. Comparison of the aerobic photoreactivity of A2E with its precursor retinal. Photochem Photobiol 2003, 77, 253–258. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res 2005, 80, 595–606. [Google Scholar] [CrossRef]
- Różanowska, M.; Różanowski, B. Visual transduction and age-related changes in lipofuscin. In Ophthalmology Research: The Visual Transduction Cascade, Tombran-Tink, J., Barnstable, C.J., Eds.; The Humana Press Inc.: Totowa, NJ, 2008; pp. 405–446.
- Zhou, J.; Jang, Y.P.; Kim, S.R.; Sparrow, J.R. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A 2006, 103, 16182–16187. [Google Scholar] [CrossRef]
- Ben-Shabat, S.; Itagaki, Y.; Jockusch, S.; Sparrow, J.R.; Turro, N.J.; Nakanishi, K. Formation of a nonaoxirane from A2E, a lipofuscin fluorophore related to macular degeneration, and evidence of singlet oxygen involvement. Angew Chem Int Ed Engl 2002, 41, 814–817. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Vollmer-Snarr, H.R.; Zhou, J.; Jang, Y.P.; Jockusch, S.; Itagaki, Y.; Nakanishi, K. A2E-epoxides damage DNA in retinal pigment epithelial cells. Vitamin E and other antioxidants inhibit A2E-epoxide formation. J Biol Chem 2003, 278, 18207–18213. [Google Scholar] [CrossRef] [PubMed]
- Radu, R.A.; Mata, N.L.; Bagla, A.; Travis, G.H. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt’s macular degeneration. Proc Natl Acad Sci U S A 2004, 101, 5928–5933. [Google Scholar] [CrossRef] [PubMed]
- Yakovleva, M.A.; Sakina, N.L.; Kononikhin, A.S.; Feldman, T.B.; Nikolaev, E.N.; Dontsov, A.E.; Ostrovsky, M.A. Detection and study of the products of photooxidation of N-retinylidene-N-retinylethanolamine (A2E), the fluorophore of lipofuscin granules from retinal pigment epithelium of human donor eyes. Dokl Biochem Biophys 2006, 409, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Keller, L.M.; Dillon, J.; Gaillard, E.R. Oxidation of A2E results in the formation of highly reactive aldehydes and ketones. Photochem Photobiol 2006, 82, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, E.R.; Avalle, L.B.; Keller, L.M.; Wang, Z.; Reszka, K.J.; Dillon, J.P. A mechanistic study of the photooxidation of A2E, a component of human retinal lipofuscin. Exp Eye Res 2004, 79, 313–319. [Google Scholar] [CrossRef]
- Dillon, J.; Wang, Z.; Avalle, L.B.; Gaillard, E.R. The photochemical oxidation of A2E results in the formation of a 5,8,5’,8’-bis-furanoid oxide. Exp Eye Res 2004, 79, 537–542. [Google Scholar] [CrossRef]
- Kim, H.J.; Sparrow, J.R. Bisretinoids: More than Meets the Eye. Adv Exp Med Biol 2019, 1185, 341–346. [Google Scholar] [CrossRef]
- Ueda, K.; Kim, H.J.; Zhao, J.; Sparrow, J.R. Bisretinoid Photodegradation Is Likely Not a Good Thing. Adv Exp Med Biol 2018, 1074, 395–401. [Google Scholar] [CrossRef]
- Murdaugh, L.S.; Avalle, L.B.; Mandal, S.; Dill, A.E.; Dillon, J.; Simon, J.D.; Gaillard, E.R. Compositional studies of human RPE lipofuscin. J Mass Spectrom 2010, 45, 1139–1147. [Google Scholar] [CrossRef]
- Murdaugh, L.S.; Mandal, S.; Dill, A.E.; Dillon, J.; Simon, J.D.; Gaillard, E.R. Compositional studies of human RPE lipofuscin: mechanisms of molecular modifications. J Mass Spectrom 2011, 46, 90–95. [Google Scholar] [CrossRef]
- Ahmed, J.; Braun, R.D.; Dunn, R., Jr.; Linsenmeier, R.A. Oxygen distribution in the macaque retina. Invest Ophthalmol Vis Sci 1993, 34, 516–521. [Google Scholar]
- Organisciak, D.T.; Darrow, R.M.; Barsalou, L.; Darrow, R.A.; Kutty, R.K.; Kutty, G.; Wiggert, B. Light history and age-related changes in retinal light damage. Invest Ophthalmol Vis Sci 1998, 39, 1107–1116. [Google Scholar] [PubMed]
- Maeda, A.; Maeda, T.; Golczak, M.; Imanishi, Y.; Leahy, P.; Kubota, R.; Palczewski, K. Effects of potent inhibitors of the retinoid cycle on visual function and photoreceptor protection from light damage in mice. Mol Pharmacol 2006, 70, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Maeda, T.; Imanishi, Y.; Sun, W.; Jastrzebska, B.; Hatala, D.A.; Winkens, H.J.; Hofmann, K.P.; Janssen, J.J.; Baehr, W.; et al. Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. J Biol Chem 2006, 281, 37697–37704. [Google Scholar] [CrossRef] [PubMed]
- Shiose, S.; Chen, Y.; Okano, K.; Roy, S.; Kohno, H.; Tang, J.; Pearlman, E.; Maeda, T.; Palczewski, K.; Maeda, A. Toll-like receptor 3 is required for development of retinopathy caused by impaired all-trans-retinal clearance in mice. J Biol Chem 2011, 286, 15543–15555. [Google Scholar] [CrossRef]
- Chen, Y.; Okano, K.; Maeda, T.; Chauhan, V.; Golczak, M.; Maeda, A.; Palczewski, K. Mechanism of all-trans-retinal toxicity with implications for stargardt disease and age-related macular degeneration. J Biol Chem 2012, 287, 5059–5069. [Google Scholar] [CrossRef]
- Okano, K.; Maeda, A.; Chen, Y.; Chauhan, V.; Tang, J.; Palczewska, G.; Sakai, T.; Tsuneoka, H.; Palczewski, K.; Maeda, T. Retinal cone and rod photoreceptor cells exhibit differential susceptibility to light-induced damage. J Neurochem 2012, 121, 146–156. [Google Scholar] [CrossRef]
- Maeda, T.; Golczak, M.; Maeda, A. Retinal photodamage mediated by all-trans-retinal. Photochem Photobiol 2012, 88, 1309–1319. [Google Scholar] [CrossRef]
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy protects the retina from light-induced degeneration. J Biol Chem 2013, 288, 7506–7518. [Google Scholar] [CrossRef]
- Chen, Y.; Palczewska, G.; Mustafi, D.; Golczak, M.; Dong, Z.; Sawada, O.; Maeda, T.; Maeda, A.; Palczewski, K. Systems pharmacology identifies drug targets for Stargardt disease-associated retinal degeneration. J Clin Invest 2013, 123, 5119–5134. [Google Scholar] [CrossRef]
- Kohno, H.; Chen, Y.; Kevany, B.M.; Pearlman, E.; Miyagi, M.; Maeda, T.; Palczewski, K.; Maeda, A. Photoreceptor proteins initiate microglial activation via Toll-like receptor 4 in retinal degeneration mediated by all-trans-retinal. J Biol Chem 2013, 288, 15326–15341. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Palczewska, G.; Golczak, M.; Kohno, H.; Dong, Z.; Maeda, T.; Palczewski, K. Two-photon microscopy reveals early rod photoreceptor cell damage in light-exposed mutant mice. Proc Natl Acad Sci U S A 2014, 111, E1428–E1437. [Google Scholar] [CrossRef] [PubMed]
- Sawada, O.; Perusek, L.; Kohno, H.; Howell, S.J.; Maeda, A.; Matsuyama, S.; Maeda, T. All-trans-retinal induces Bax activation via DNA damage to mediate retinal cell apoptosis. Exp Eye Res 2014, 123, 27–36. [Google Scholar] [CrossRef]
- Wu, X.; Yu, G.; Luo, C.; Maeda, A.; Zhang, N.; Sun, D.; Zhou, Z.; Puntel, A.; Palczewski, K.; Lu, Z.R. Synthesis and evaluation of a nanoglobular dendrimer 5-aminosalicylic Acid conjugate with a hydrolyzable schiff base spacer for treating retinal degeneration. ACS Nano 2014, 8, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wu, X.; Ayat, N.; Maeda, A.; Gao, S.Q.; Golczak, M.; Palczewski, K.; Lu, Z.R. Multifunctional PEG retinylamine conjugate provides prolonged protection against retinal degeneration in mice. Biomacromolecules 2014, 15, 4570–4578. [Google Scholar] [CrossRef]
- Puntel, A.; Maeda, A.; Golczak, M.; Gao, S.Q.; Yu, G.; Palczewski, K.; Lu, Z.R. Prolonged prevention of retinal degeneration with retinylamine loaded nanoparticles. Biomaterials 2015, 44, 103–110. [Google Scholar] [CrossRef]
- Parmar, T.; Parmar, V.M.; Arai, E.; Sahu, B.; Perusek, L.; Maeda, A. Acute Stress Responses Are Early Molecular Events of Retinal Degeneration in Abca4-/-Rdh8-/- Mice After Light Exposure. Invest Ophthalmol Vis Sci 2016, 57, 3257–3267. [Google Scholar] [CrossRef]
- Schur, R.M.; Gao, S.; Yu, G.; Chen, Y.; Maeda, A.; Palczewski, K.; Lu, Z.R. New GABA modulators protect photoreceptor cells from light-induced degeneration in mouse models. FASEB J 2018, 32, 3289–3300. [Google Scholar] [CrossRef]
- Parmar, T.; Parmar, V.M.; Perusek, L.; Georges, A.; Takahashi, M.; Crabb, J.W.; Maeda, A. Lipocalin 2 Plays an Important Role in Regulating Inflammation in Retinal Degeneration. J Immunol 2018, 200, 3128–3141. [Google Scholar] [CrossRef]
- Fang, Y.; Tschulakow, A.; Taubitz, T.; Illing, B.; Biesemeier, A.; Julien-Schraermeyer, S.; Radu, R.A.; Jiang, Z.; Schraermeyer, U. Fundus autofluorescence, spectral-domain optical coherence tomography, and histology correlations in a Stargardt disease mouse model. FASEB J 2020, 34, 3693–3714. [Google Scholar] [CrossRef]
- Yu, G.; Gao, S.Q.; Dong, Z.; Sheng, L.; Sun, D.; Zhang, N.; Zhang, J.; Margeivicus, S.; Fu, P.; Golczak, M.; et al. Peptide Derivatives of Retinylamine Prevent Retinal Degeneration with Minimal Side Effects on Vision in Mice. Bioconjug Chem 2021, 32, 572–583. [Google Scholar] [CrossRef]
- Fang, Y.; Taubitz, T.; Tschulakow, A.V.; Heiduschka, P.; Szewczyk, G.; Burnet, M.; Peters, T.; Biesemeier, A.; Sarna, T.; Schraermeyer, U.; et al. Removal of RPE lipofuscin results in rescue from retinal degeneration in a mouse model of advanced Stargardt disease: Role of reactive oxygen species. Free Radical Biology and Medicine 2022, 182, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Ham, W.T., Jr.; Ruffolo, J.J., Jr.; Mueller, H.A.; Clarke, A.M.; Moon, M.E. Histologic analysis of photochemical lesions produced in rhesus retina by short-wave-length light. Invest Ophthalmol Vis Sci 1978, 17, 1029–1035. [Google Scholar]
- Morgan, J.I.; Hunter, J.J.; Masella, B.; Wolfe, R.; Gray, D.C.; Merigan, W.H.; Delori, F.C.; Williams, D.R. Light-induced retinal changes observed with high-resolution autofluorescence imaging of the retinal pigment epithelium. Invest Ophthalmol Vis Sci 2008, 49, 3715–3729. [Google Scholar] [CrossRef]
- Morgan, J.I.; Hunter, J.J.; Merigan, W.H.; Williams, D.R. The reduction of retinal autofluorescence caused by light exposure. Invest Ophthalmol Vis Sci 2009, 50, 6015–6022. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.J.; Morgan, J.I.; Merigan, W.H.; Sliney, D.H.; Sparrow, J.R.; Williams, D.R. The susceptibility of the retina to photochemical damage from visible light. Prog Retin Eye Res 2012, 31, 28–42. [Google Scholar] [CrossRef]
- Zhang, J.; Sabarinathan, R.; Bubel, T.; Williams, D.R.; Hunter, J.J. Action spectrum for photochemical retinal pigment epithelium (RPE) disruption in an in vivo monkey model. Optical Interactions with Tissue and Cells Xxvii 2016, 9706. [Google Scholar] [CrossRef]
- Ham, W.T., Jr.; Mueller, H.A.; Ruffolo, J.J., Jr.; Guerry, D., 3rd; Guerry, R.K. Action spectrum for retinal injury from near-ultraviolet radiation in the aphakic monkey. Am J Ophthalmol 1982, 93, 299–306. [Google Scholar] [CrossRef]
- Wu, L.; Ueda, K.; Nagasaki, T.; Sparrow, J.R. Light damage in Abca4 and Rpe65rd12 mice. Invest Ophthalmol Vis Sci 2014, 55, 1910–1918. [Google Scholar] [CrossRef]
- Teussink, M.M.; Lee, M.D.; Smith, R.T.; van Huet, R.A.; Klaver, C.C.; Klevering, B.J.; Theelen, T.; Hoyng, C.B. The effect of light deprivation in patients with Stargardt disease. Am J Ophthalmol 2015, 159, 964–972 e962. [Google Scholar] [CrossRef]
- Aits, S.; Kricker, J.; Liu, B.; Ellegaard, A.M.; Hamalisto, S.; Tvingsholm, S.; Corcelle-Termeau, E.; Hogh, S.; Farkas, T.; Holm Jonassen, A.; et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015, 11, 1408–1424. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017, 47, 51–65 e57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Robinson, K.; Washington, I. C20D3-Vitamin A Prevents Retinal Pigment Epithelium Atrophic Changes in a Mouse Model. Transl Vis Sci Technol 2021, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Charbel Issa, P.; Barnard, A.R.; Herrmann, P.; Washington, I.; MacLaren, R.E. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc Natl Acad Sci U S A 2015, 112, 8415–8420. [Google Scholar] [CrossRef]
- Allingham, M.J.; Loksztejn, A.; Cousins, S.W.; Mettu, P.S. Immunological Aspects of Age-Related Macular Degeneration. In Age-related Macular Degeneration,, Chew, E.Y., Swaroop, A., Eds.; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2021; Volume 1256, pp. 143–190. [Google Scholar]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res 2010, 29, 95–112. [Google Scholar] [CrossRef]
- Haralampus-Grynaviski, N.M.; Lamb, L.E.; Simon, J.D.; Krogmeier, J.R.; Dunn, R.C.; Pawlak, A.; Rozanowska, M.; Sarna, T.; Burke, J.M. Probing the spatial dependence of the emission spectrum of single human retinal lipofuscin granules using near-field scanning optical microscopy. Photochem Photobiol 2001, 74, 364–368. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Gregory-Roberts, E.; Yamamoto, K.; Blonska, A.; Ghosh, S.K.; Ueda, K.; Zhou, J. The bisretinoids of retinal pigment epithelium. Prog Retin Eye Res 2012, 31, 121–135. [Google Scholar] [CrossRef]
- Eldred, G.E.; Lasky, M.R. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature 1993, 361, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Ragauskaite, L.; Heckathorn, R.C.; Gaillard, E.R. Environmental effects on the photochemistry of A2-E, a component of human retinal lipofuscin. Photochem Photobiol 2001, 74, 483–488. [Google Scholar] [CrossRef]
- Lamb, L.E.; Ye, T.; Haralampus-Grynaviski, N.M.; Williams, T.R.; Pawlak, A.; Sarna, T.; Simon, J.D. Primary photophysical properties of A2E in solution. J. Phys. Chem. B 2001, 105, 11507–11512. [Google Scholar] [CrossRef]
- Reszka, K.; Eldred, G.E.; Wang, R.H.; Chignell, C.; Dillon, J. The photochemistry of human retinal lipofuscin as studied by EPR. Photochem Photobiol 1995, 62, 1005–1008. [Google Scholar] [CrossRef]
- Kopitz, J.; Holz, F.G.; Kaemmerer, E.; Schutt, F. Lipids and lipid peroxidation products in the pathogenesis of age-related macular degeneration. Biochimie 2004, 86, 825–831. [Google Scholar] [CrossRef]
- Dontsov, A.E.; Sakina, N.L.; Golubkov, A.M.; Ostrovsky, M.A. Light-induced release of A2E photooxidation toxic products from lipofuscin granules of human retinal pigment epithelium. Dokl Biochem Biophys 2009, 425, 98–101. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Szmacinski, H.; Nowaczyk, K.; Johnson, M.L. Fluorescence lifetime imaging of free and protein-bound NADH. Proc Natl Acad Sci U S A 1992, 89, 1271–1275. [Google Scholar] [CrossRef]
- Croce, A.C.; Bottiroli, G. Autofluorescence spectroscopy and imaging: a tool for biomedical research and diagnosis. Eur. J. Histochem. 2014, 58, 320–337. [Google Scholar] [CrossRef]
- Croce, A.C.; Ferrigno, A.; Bottiroli, G.; Vairetti, M. Autofluorescence-based optical biopsy: An effective diagnostic tool in hepatology. Liver Int 2018, 38, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Layer, G.; Reichelt, J.; Jahn, D.; Heinz, D.W. Structure and function of enzymes in heme biosynthesis. Protein Sci 2010, 19, 1137–1161. [Google Scholar] [CrossRef]
- Palczewska, G.; Wojtkowski, M.; Palczewski, K. From mouse to human: Accessing the biochemistry of vision in vivo by two-photon excitation. Prog Retin Eye Res 2023, 93, 101170. [Google Scholar] [CrossRef] [PubMed]
- Palczewska, G.; Maeda, T.; Imanishi, Y.; Sun, W.; Chen, Y.; Williams, D.R.; Piston, D.W.; Maeda, A.; Palczewski, K. Noninvasive multiphoton fluorescence microscopy resolves retinol and retinal condensation products in mouse eyes. Nat Med 2010, 16, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Szweda, L.I. Age-related increase in liver retinyl palmitate. Relationship to lipofuscin. J Biol Chem 1994, 269, 8712–8715. [Google Scholar] [CrossRef]
- Docchio, F.; Boulton, M.; Cubeddu, R.; Ramponi, R.; Barker, P.D. Age-related changes in the fluorescence of melanin and lipofuscin granules of the retinal pigment epithelium: a time-resolved fluorescence spectroscopy study. Photochem Photobiol 1991, 54, 247–253. [Google Scholar] [CrossRef]
- Pollreisz, A.; Messinger, J.D.; Sloan, K.R.; Mittermueller, T.J.; Weinhandl, A.S.; Benson, E.K.; Kidd, G.J.; Schmidt-Erfurth, U.; Curcio, C.A. Visualizing melanosomes, lipofuscin, and melanolipofuscin in human retinal pigment epithelium using serial block face scanning electron microscopy. Exp Eye Res 2018, 166, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Bermond, K.; Wobbe, C.; Tarau, I.S.; Heintzmann, R.; Hillenkamp, J.; Curcio, C.A.; Sloan, K.R.; Ach, T. Autofluorescent Granules of the Human Retinal Pigment Epithelium: Phenotypes, Intracellular Distribution, and Age-Related Topography. Invest Ophthalmol Vis Sci 2020, 61, 35. [Google Scholar] [CrossRef]
- Taubitz, T.; Fang, Y.; Biesemeier, A.; Julien-Schraermeyer, S.; Schraermeyer, U. Age, lipofuscin and melanin oxidation affect fundus near-infrared autofluorescence. EBioMedicine 2019, 48, 592–604. [Google Scholar] [CrossRef]
- Kayatz, P.; Thumann, G.; Luther, T.T.; Jordan, J.F.; Bartz-Schmidt, K.U.; Esser, P.J.; Schraermeyer, U. Oxidation causes melanin fluorescence. Invest Ophthalmol Vis Sci 2001, 42, 241–246. [Google Scholar]
- D’Cruz, P.M.; Yasumura, D.; Weir, J.; Matthes, M.T.; Abderrahim, H.; LaVail, M.M.; Vollrath, D. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet 2000, 9, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Hamm, G.; Maglennon, G.; Williamson, B.; Macdonald, R.; Doherty, A.; Jones, S.; Harris, J.; Blades, J.; Harmer, A.R.; Barton, P.; et al. Pharmacological inhibition of MERTK induces in vivo retinal degeneration: a multimodal imaging ocular safety assessment. Arch Toxicol 2022, 96, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Zihni, C.; Georgiadis, A.; Ramsden, C.M.; Sanchez-Heras, E.; Haas, A.J.; Nommiste, B.; Semenyuk, O.; Bainbridge, J.W.B.; Coffey, P.J.; Smith, A.J.; et al. Spatiotemporal control of actomyosin contractility by MRCKbeta signaling drives phagocytosis. J Cell Biol 2022, 221. [Google Scholar] [CrossRef]
- Zheng, L.; Jia, J.; Chen, Y.; Liu, R.; Cao, R.; Duan, M.; Zhang, M.; Xu, Y. Pentoxifylline alleviates ischemic white matter injury through up-regulating Mertk-mediated myelin clearance. J Neuroinflammation 2022, 19, 128. [Google Scholar] [CrossRef]
- Audo, I.; Mohand-Said, S.; Boulanger-Scemama, E.; Zanlonghi, X.; Condroyer, C.; Demontant, V.; Boyard, F.; Antonio, A.; Mejecase, C.; El Shamieh, S.; et al. MERTK mutation update in inherited retinal diseases. Hum Mutat 2018, 39, 887–913. [Google Scholar] [CrossRef]
- Kaga, T.; Fonseca, R.A.; Dantas, M.A.; Yannuzzi, L.A.; Spaide, R.F. Optical coherence tomography of bleb-like subretinal lesions after retinal reattachment surgery. Am J Ophthalmol 2001, 132, 120–121. [Google Scholar] [CrossRef]
- Spaide, R.F.; Klancnik, J.M., Jr. Fundus autofluorescence and central serous chorioretinopathy. Ophthalmology 2005, 112, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Spaide, R.F.; Noble, K.; Morgan, A.; Freund, K.B. Vitelliform macular dystrophy. Ophthalmology 2006, 113, 1392–1400. [Google Scholar] [CrossRef]
- Laud, K.; Visaetsilpanonta, S.; Yannuzzi, L.A.; Spaide, R.F. Autofluorescence imaging of optic pit maculopathy. Retina 2007, 27, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Fathi, P.; Roslend, A.; Mehta, K.; Moitra, P.; Zhang, K.; Pan, D. UV-trained and metal-enhanced fluorescence of biliverdin and biliverdin nanoparticles. Nanoscale 2021, 13, 4785–4798. [Google Scholar] [CrossRef] [PubMed]
- Sawa, M.; Ober, M.D.; Spaide, R.F. Autofluorescence and retinal pigment epithelial atrophy after subretinal hemorrhage. Retina 2006, 26, 119–120. [Google Scholar] [CrossRef]
- Piccolino, F.C.; Borgia, L.; Zinicola, E.; Iester, M.; Torrielli, S. Pre-injection fluorescence in indocyanine green angiography. Ophthalmology 1996, 103, 1837–1845. [Google Scholar] [CrossRef]
- Glushko, V.; Thaler, M.; Ros, M. The fluorescence of bilirubin upon interaction with human erythrocyte ghosts. Biochim Biophys Acta 1982, 719, 65–73. [Google Scholar] [CrossRef]
- Marmorstein, A.D.; Marmorstein, L.Y.; Sakaguchi, H.; Hollyfield, J.G. Spectral profiling of autofluorescence associated with lipofuscin, Bruch’s Membrane, and sub-RPE deposits in normal and AMD eyes. Invest Ophthalmol Vis Sci 2002, 43, 2435–2441. [Google Scholar]
- Hammer, M.; Konigsdorffer, E.; Liebermann, C.; Framme, C.; Schuch, G.; Schweitzer, D.; Strobel, J. Ocular fundus auto-fluorescence observations at different wavelengths in patients with age-related macular degeneration and diabetic retinopathy. Graefes Arch Clin Exp Ophthalmol 2008, 246, 105–114. [Google Scholar] [CrossRef]
- Schultz, R.; Gamage, K.; Messinger, J.D.; Curcio, C.A.; Hammer, M. Fluorescence Lifetimes and Spectra of RPE and Sub-RPE Deposits in Histology of Control and AMD Eyes. Invest Ophthalmol Vis Sci 2020, 61, 9. [Google Scholar] [CrossRef] [PubMed]
- Delori, F.C.; Dorey, C.K.; Staurenghi, G.; Arend, O.; Goger, D.G.; Weiter, J.J. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci 1995, 36, 718–729. [Google Scholar] [PubMed]
- Sarna, T.; Burke, J.M.; Korytowski, W.; Rozanowska, M.; Skumatz, C.M.; Zareba, A.; Zareba, M. Loss of melanin from human RPE with aging: possible role of melanin photooxidation. Exp Eye Res 2003, 76, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.T.; Post, R.; Johri, A.; Lee, M.D.; Ablonczy, Z.; Curcio, C.A.; Ach, T.; Sajda, P. Simultaneous decomposition of multiple hyperspectral data sets: signal recovery of unknown fluorophores in the retinal pigment epithelium. Biomed Opt Express 2014, 5, 4171–4185. [Google Scholar] [CrossRef]
- Bermond, K.; Berlin, A.; Tarau, I.S.; Wobbe, C.; Heintzmann, R.; Curcio, C.A.; Sloan, K.R.; Ach, T. Characteristics of normal human retinal pigment epithelium cells with extremes of autofluorescence or intracellular granule count. Ann Eye Sci 2021, 6. [Google Scholar] [CrossRef]
- Webb, R.H.; Hughes, G.W.; Delori, F.C. Confocal scanning laser ophthalmoscope. Appl Opt 1987, 26, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- von Ruckmann, A.; Fitzke, F.W.; Bird, A.C. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol 1995, 79, 407–412. [Google Scholar] [CrossRef]
- von Ruckmann, A.; Fitzke, F.W.; Bird, A.C. Fundus autofluorescence in age-related macular disease imaged with a laser scanning ophthalmoscope. Invest Ophthalmol Vis Sci 1997, 38, 478–486. [Google Scholar]
- von Ruckmann, A.; Fitzke, F.W.; Bird, A.C. Distribution of pigment epithelium autofluorescence in retinal disease state recorded in vivo and its change over time. Graefes Arch Clin Exp Ophthalmol 1999, 237, 1–9. [Google Scholar] [CrossRef]
- Reiter, G.S.; Told, R.; Baratsits, M.; Hecht, A.; Schlanitz, F.G.; Sacu, S.; Schmidt-Erfurth, U. Repeatability and reliability of quantitative fundus autofluorescence imaging in patients with early and intermediate age-related macular degeneration. Acta Ophthalmol. 2019, 97, E526–E532. [Google Scholar] [CrossRef]
- Fischer, J.; Otto, T.; Delori, F.; Pace, L.; Staurenghi, G. Scanning Laser Ophthalmoscopy (SLO). In High Resolution Imaging in Microscopy and Ophthalmology: New Frontiers in Biomedical Optics, Bille, J.F., Ed.; Cham (CH), 2019; pp. 35–57.
- Orellana-Rios, J.; Yokoyama, S.; Bhuiyan, A.; Gao, L.; Otero-Marquez, O.; Smith, R.T. Translational Retinal Imaging. Asia Pac J Ophthalmol (Phila) 2020, 9, 269–277. [Google Scholar] [CrossRef]
- Schmitz-Valckenberg, S.; Pfau, M.; Fleckenstein, M.; Staurenghi, G.; Sparrow, J.R.; Bindewald-Wittich, A.; Spaide, R.F.; Wolf, S.; Sadda, S.R.; Holz, F.G. Fundus autofluorescence imaging. Prog Retin Eye Res 2021, 81, 100893. [Google Scholar] [CrossRef]
- Borrelli, E.; Battista, M.; Zuccaro, B.; Sacconi, R.; Brambati, M.; Querques, L.; Prascina, F.; Sadda, S.R.; Bandello, F.; Querques, G. Spectrally Resolved Fundus Autofluorescence in Healthy Eyes: Repeatability and Topographical Analysis of the Green-Emitting Fluorophores. J Clin Med 2020, 9. [Google Scholar] [CrossRef]
- Borrelli, E.; Lei, J.; Balasubramanian, S.; Uji, A.; Cozzi, M.; Sarao, V.; Lanzetta, P.; Staurenghi, G.; Sadda, S.R. Green emission fluorophores in eyes with atrophic age-related macular degeneration: a colour fundus autofluorescence pilot study. Br J Ophthalmol 2018, 102, 827–832. [Google Scholar] [CrossRef]
- Dysli, C.; Muller, P.L.; Birtel, J.; Holz, F.G.; Herrmann, P. Spectrally Resolved Fundus Autofluorescence in ABCA4-Related Retinopathy. Invest Ophthalmol Vis Sci 2019, 60, 274–281. [Google Scholar] [CrossRef]
- Muller, P.L.; Dysli, C.; Hess, K.; Holz, F.G.; Herrmann, P. Spectral Fundus Autofluorescence Excitation and Emission in Abca4-Related Retinopathy. Retina 2020, 40, 2332–2342. [Google Scholar] [CrossRef]
- Gao, L.; Smith, R.T.; Tkaczyk, T.S. Snapshot hyperspectral retinal camera with the Image Mapping Spectrometer (IMS). Biomed Opt Express 2012, 3, 48–54. [Google Scholar] [CrossRef]
- Boguslawski, J.; Palczewska, G.; Tomczewski, S.; Milkiewicz, J.; Kasprzycki, P.; Stachowiak, D.; Komar, K.; Marzejon, M.J.; Sikorski, B.L.; Hudzikowski, A.; et al. In vivo imaging of the human eye using a 2-photon-excited fluorescence scanning laser ophthalmoscope. J Clin Invest 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Ach, T.; Curcio, C.A.; Smith, R.T. Hyperspectral autofluorescence characterization of drusen and sub-RPE deposits in age-related macular degeneration. Ann Eye Sci 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Granger, C.E.; Yang, Q.; Song, H.; Saito, K.; Nozato, K.; Latchney, L.R.; Leonard, B.T.; Chung, M.M.; Williams, D.R.; Rossi, E.A. Human Retinal Pigment Epithelium: In Vivo Cell Morphometry, Multispectral Autofluorescence, and Relationship to Cone Mosaic. Invest Ophthalmol Vis Sci 2018, 59, 5705–5716. [Google Scholar] [CrossRef] [PubMed]
- Schultz, R.; Hasan, S.; Curcio, C.A.; Smith, R.T.; Meller, D.; Hammer, M. Spectral and lifetime resolution of fundus autofluorescence in advanced age-related macular degeneration revealing different signal sources. Acta Ophthalmol 2022, 100, e841–e846. [Google Scholar] [CrossRef]
- Semenov, A.N.; Maksimov, E.G.; Moysenovich, A.M.; Yakovleva, M.A.; Tsoraev, G.V.; Ramonova, A.A.; Shirshin, E.A.; Sluchanko, N.N.; Feldman, T.B.; Rubin, A.B.; et al. Protein-Mediated Carotenoid Delivery Suppresses the Photoinducible Oxidation of Lipofuscin in Retinal Pigment Epithelial Cells. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Guan, Z.; Li, Y.; Jiao, S.; Yeasmin, N.; Rosenfeld, P.J.; Dubovy, S.R.; Lam, B.L.; Wen, R. A2E Distribution in RPE Granules in Human Eyes. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Dysli, C.; Wolf, S.; Berezin, M.Y.; Sauer, L.; Hammer, M.; Zinkernagel, M.S. Fluorescence lifetime imaging ophthalmoscopy. Prog Retin Eye Res 2017, 60, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Palczewska, G.; Boguslawski, J.; Stremplewski, P.; Kornaszewski, L.; Zhang, J.; Dong, Z.; Liang, X.X.; Gratton, E.; Vogel, A.; Wojtkowski, M.; et al. Noninvasive two-photon optical biopsy of retinal fluorophores. Proc Natl Acad Sci U S A 2020, 117, 22532–22543. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Brauer, J.L.; Meller, D.; Hammer, M. Impact of cataract on the spectral measurement of fundus autofluorescence. Graefes Arch Clin Exp Ophthalmol 2022, 260, 2057–2059. [Google Scholar] [CrossRef]
- Berlin, A.; Clark, M.E.; Swain, T.A.; Fischer, N.A.; McGwin, G., Jr.; Sloan, K.R.; Owsley, C.; Curcio, C.A. Impact of the Aging Lens and Posterior Capsular Opacification on Quantitative Autofluorescence Imaging in Age-Related Macular Degeneration. Transl Vis Sci Technol 2022, 11, 23. [Google Scholar] [CrossRef]
- Reiter, G.S.; Schwarzenbacher, L.; Schartmuller, D.; Roggla, V.; Leydolt, C.; Menapace, R.; Schmidt-Erfurth, U.; Sacu, S. Influence of lens opacities and cataract severity on quantitative fundus autofluorescence as a secondary outcome of a randomized clinical trial. Scientific Reports 2021, 11. [Google Scholar] [CrossRef]
- Brauer, J.L.; Schultz, R.; Klemm, M.; Hammer, M. Influence of Lens Fluorescence on Fluorescence Lifetime Imaging Ophthalmoscopy (FLIO) Fundus Imaging and Strategies for Its Compensation. Transl Vis Sci Technol 2020, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- van de Kraats, J.; van Norren, D. Optical density of the aging human ocular media in the visible and the UV. J Opt Soc Am A Opt Image Sci Vis 2007, 24, 1842–1857. [Google Scholar] [CrossRef]
- Hunter, J.J.; Masella, B.; Dubra, A.; Sharma, R.; Yin, L.; Merigan, W.H.; Palczewska, G.; Palczewski, K.; Williams, D.R. Images of photoreceptors in living primate eyes using adaptive optics two-photon ophthalmoscopy. Biomed Opt Express 2010, 2, 139–148. [Google Scholar] [CrossRef]
- Palczewska, G.; Golczak, M.; Williams, D.R.; Hunter, J.J.; Palczewski, K. Endogenous fluorophores enable two-photon imaging of the primate eye. Invest Ophthalmol Vis Sci 2014, 55, 4438–4447. [Google Scholar] [CrossRef]
- Palczewska, G.; Dong, Z.; Golczak, M.; Hunter, J.J.; Williams, D.R.; Alexander, N.S.; Palczewski, K. Noninvasive two-photon microscopy imaging of mouse retina and retinal pigment epithelium through the pupil of the eye. Nat Med 2014, 20, 785–789. [Google Scholar] [CrossRef]
- Sharma, R.; Yin, L.; Geng, Y.; Merigan, W.H.; Palczewska, G.; Palczewski, K.; Williams, D.R.; Hunter, J.J. In vivo two-photon imaging of the mouse retina. Biomed Opt Express 2013, 4, 1285–1293. [Google Scholar] [CrossRef]
- Sharma, R.; Schwarz, C.; Williams, D.R.; Palczewska, G.; Palczewski, K.; Hunter, J.J. In Vivo Two-Photon Fluorescence Kinetics of Primate Rods and Cones. Invest Ophthalmol Vis Sci 2016, 57, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C.; Sharma, R.; Fischer, W.S.; Chung, M.; Palczewska, G.; Palczewski, K.; Williams, D.R.; Hunter, J.J. Safety assessment in macaques of light exposures for functional two-photon ophthalmoscopy in humans. Biomed Opt Express 2016, 7, 5148–5169. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Williams, D.R.; Palczewska, G.; Palczewski, K.; Hunter, J.J. Two-Photon Autofluorescence Imaging Reveals Cellular Structures Throughout the Retina of the Living Primate Eye. Invest Ophthalmol Vis Sci 2016, 57, 632–646. [Google Scholar] [CrossRef] [PubMed]
- Marcos, S.; Werner, J.S.; Burns, S.A.; Merigan, W.H.; Artal, P.; Atchison, D.A.; Hampson, K.M.; Legras, R.; Lundstrom, L.; Yoon, G.; et al. Vision science and adaptive optics, the state of the field. Vision Res 2017, 132, 3–33. [Google Scholar] [CrossRef]
- Sharma, R.; Schwarz, C.; Hunter, J.J.; Palczewska, G.; Palczewski, K.; Williams, D.R. Formation and Clearance of All-Trans-Retinol in Rods Investigated in the Living Primate Eye With Two-Photon Ophthalmoscopy. Invest Ophthalmol Vis Sci 2017, 58, 604–613. [Google Scholar] [CrossRef]
- Murashova, G.A.; Mancuso, C.A.; Sakami, S.; Palczewski, K.; Palczewska, G.; Dantus, M. Epi-direction detected multimodal imaging of an unstained mouse retina with a Yb-fiber laser. Proc SPIE Int Soc Opt Eng 2017, 10069. [Google Scholar] [CrossRef]
- Murashova, G.A.; Mancuso, C.A.; Canfield, J.L.; Sakami, S.; Palczewski, K.; Palczewska, G.; Dantus, M. Multimodal nonlinear optical imaging of unstained retinas in the epi-direction with a sub-40 fs Yb-fiber laser. Biomed Opt Express 2017, 8, 5228–5242. [Google Scholar] [CrossRef] [PubMed]
- Palczewska, G.; Stremplewski, P.; Suh, S.; Alexander, N.; Salom, D.; Dong, Z.; Ruminski, D.; Choi, E.H.; Sears, A.E.; Kern, T.S.; et al. Two-photon imaging of the mammalian retina with ultrafast pulsing laser. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Palczewska, G.; Kern, T.S.; Palczewski, K. Noninvasive Two-Photon Microscopy Imaging of Mouse Retina and Retinal Pigment Epithelium. Methods Mol Biol 2019, 1834, 333–343. [Google Scholar] [CrossRef]
- Tang, J.A.H.; Granger, C.E.; Kunala, K.; Parkins, K.; Huynh, K.T.; Bowles-Johnson, K.; Yang, Q.; Hunter, J.J. Adaptive optics fluorescence lifetime imaging ophthalmoscopy of in vivo human retinal pigment epithelium. Biomed Opt Express 2022, 13, 1737–1754. [Google Scholar] [CrossRef]
- Boguslawski, J.; Tomczewski, S.; Dabrowski, M.; Komar, K.; Milkiewicz, J.; Palczewska, G.; Palczewski, K.; Wojtkowski, M. In vivo imaging of the human retina using a two-photon excited fluorescence ophthalmoscope. STAR Protoc 2023, 4, 102225. [Google Scholar] [CrossRef]
- Wolf-Schnurrbusch, U.E.; Wittwer, V.V.; Ghanem, R.; Niederhaeuser, M.; Enzmann, V.; Framme, C.; Wolf, S. Blue-light versus green-light autofluorescence: lesion size of areas of geographic atrophy. Invest Ophthalmol Vis Sci 2011, 52, 9497–9502. [Google Scholar] [CrossRef]
- Burke, T.R.; Duncker, T.; Woods, R.L.; Greenberg, J.P.; Zernant, J.; Tsang, S.H.; Smith, R.T.; Allikmets, R.; Sparrow, J.R.; Delori, F.C. Quantitative fundus autofluorescence in recessive Stargardt disease. Invest Ophthalmol Vis Sci 2014, 55, 2841–2852. [Google Scholar] [CrossRef]
- Duncker, T.; Greenberg, J.P.; Ramachandran, R.; Hood, D.C.; Smith, R.T.; Hirose, T.; Woods, R.L.; Tsang, S.H.; Delori, F.C.; Sparrow, J.R. Quantitative fundus autofluorescence and optical coherence tomography in best vitelliform macular dystrophy. Invest Ophthalmol Vis Sci 2014, 55, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Duncker, T.; Stein, G.E.; Lee, W.; Tsang, S.H.; Zernant, J.; Bearelly, S.; Hood, D.C.; Greenstein, V.C.; Delori, F.C.; Allikmets, R.; et al. Quantitative Fundus Autofluorescence and Optical Coherence Tomography in ABCA4 Carriers. Invest Ophthalmol Vis Sci 2015, 56, 7274–7285. [Google Scholar] [CrossRef]
- Duncker, T.; Tsang, S.H.; Lee, W.; Zernant, J.; Allikmets, R.; Delori, F.C.; Sparrow, J.R. Quantitative fundus autofluorescence distinguishes ABCA4-associated and non-ABCA4-associated bull’s-eye maculopathy. Ophthalmology 2015, 122, 345–355. [Google Scholar] [CrossRef]
- Duncker, T.; Tsang, S.H.; Woods, R.L.; Lee, W.; Zernant, J.; Allikmets, R.; Delori, F.C.; Sparrow, J.R. Quantitative Fundus Autofluorescence and Optical Coherence Tomography in PRPH2/RDS- and ABCA4-Associated Disease Exhibiting Phenotypic Overlap. Invest Ophthalmol Vis Sci 2015, 56, 3159–3170. [Google Scholar] [CrossRef]
- Marsiglia, M.; Lee, W.; Mahajan, V.B.; Zernant, J.; Delori, F.C.; Tsang, S.H.; Sparrow, J.R. Quantitative autofluorescence as a clinical tool for expedited differential diagnosis of retinal degeneration. JAMA Ophthalmol 2015, 133, 219–220. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Duncker, T.; Woods, R.; Delori, F.C. Quantitative Fundus Autofluorescence in Best Vitelliform Macular Dystrophy: RPE Lipofuscin is not Increased in Non-Lesion Areas of Retina. Adv Exp Med Biol 2016, 854, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Pfau, M.; Goerdt, L.; Schmitz-Valckenberg, S.; Mauschitz, M.M.; Mishra, D.K.; Holz, F.G.; Lindner, M.; Fleckenstein, M. Green-Light Autofluorescence Versus Combined Blue-Light Autofluorescence and Near-Infrared Reflectance Imaging in Geographic Atrophy Secondary to Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 2017, 58, BIO121–BIO130. [Google Scholar] [CrossRef] [PubMed]
- Schuerch, K.; Woods, R.L.; Lee, W.; Duncker, T.; Delori, F.C.; Allikmets, R.; Tsang, S.H.; Sparrow, J.R. Quantifying Fundus Autofluorescence in Patients With Retinitis Pigmentosa. Invest Ophthalmol Vis Sci 2017, 58, 1843–1855. [Google Scholar] [CrossRef]
- Chen, L.; Messinger, J.D.; Ferrara, D.; Freund, K.B.; Curcio, C.A. Fundus Autofluorescence in Neovascular Age-Related Macular Degeneration: A Clinicopathologic Correlation Relevant to Macular Atrophy. Ophthalmol Retina 2021, 5, 1085–1096. [Google Scholar] [CrossRef]
- Salcedo-Villanueva, G.; Lopez-Contreras, Y.; Gonzalez, H.L.A.; Romo-Aguas, J.C.; Garcia-Aguirre, G.; Cernichiaro-Espinosa, L.A.; Martinez-Castellanos, M.A.; Quiroz-Mercado, H. Fundus autofluorescence in premature infants. Sci Rep 2021, 11, 8823. [Google Scholar] [CrossRef]
- Delori, F.C.; Goger, D.G.; Dorey, C.K. Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci 2001, 42, 1855–1866. [Google Scholar] [PubMed]
- Ach, T.; Huisingh, C.; McGwin, G., Jr.; Messinger, J.D.; Zhang, T.; Bentley, M.J.; Gutierrez, D.B.; Ablonczy, Z.; Smith, R.T.; Sloan, K.R.; et al. Quantitative autofluorescence and cell density maps of the human retinal pigment epithelium. Invest Ophthalmol Vis Sci 2014, 55, 4832–4841. [Google Scholar] [CrossRef]
- Okubo, A.; Rosa, R.H., Jr.; Bunce, C.V.; Alexander, R.A.; Fan, J.T.; Bird, A.C.; Luthert, P.J. The relationships of age changes in retinal pigment epithelium and Bruch’s membrane. Invest Ophthalmol Vis Sci 1999, 40, 443–449. [Google Scholar] [PubMed]
- Greenberg, J.P.; Duncker, T.; Woods, R.L.; Smith, R.T.; Sparrow, J.R.; Delori, F.C. Quantitative fundus autofluorescence in healthy eyes. Invest Ophthalmol Vis Sci 2013, 54, 5684–5693. [Google Scholar] [CrossRef] [PubMed]
- Ben Ami, T.; Tong, Y.; Bhuiyan, A.; Huisingh, C.; Ablonczy, Z.; Ach, T.; Curcio, C.A.; Smith, R.T. Spatial and Spectral Characterization of Human Retinal Pigment Epithelium Fluorophore Families by Ex Vivo Hyperspectral Autofluorescence Imaging. Transl Vis Sci Technol 2016, 5, 5. [Google Scholar] [CrossRef]
- Schultz, R.; Schwanengel, L.; Klemm, M.; Meller, D.; Hammer, M. Spectral fundus autofluorescence peak emission wavelength in ageing and AMD. Acta Ophthalmol 2022, 100, e1223–e1231. [Google Scholar] [CrossRef] [PubMed]
- Schultz, R.; Klemm, M.; Meller, D.; Hammer, M. Spectral calibration of fluorescence lifetime imaging ophthalmoscopy. Acta Ophthalmol 2022, 100, e612–e613. [Google Scholar] [CrossRef]
- Zuclich, J.A.; Previc, F.H.; Novar, B.J.; Edsall, P.R. Near-UV/blue light-induced fluorescence in the human lens: potential interference with visual function. J Biomed Opt 2005, 10, 44021. [Google Scholar] [CrossRef]
- Rein, D.B.; Wittenborn, J.S.; Burke-Conte, Z.; Gulia, R.; Robalik, T.; Ehrlich, J.R.; Lundeen, E.A.; Flaxman, A.D. Prevalence of Age-Related Macular Degeneration in the US in 2019. JAMA Ophthalmol 2022. [CrossRef]
- Creuzot-Garcher, C.P.; Srour, M.; Baudin, F.; Daien, V.; Dot, C.; Nghiem-Buffet, S.; Girmens, J.F.; Coulombel, N.; Ponthieux, A.; Delcourt, C. Incidence and Prevalence of Neovascular Age-Related Macular Degeneration in France between 2008 and 2018: The LANDSCAPE Study. Ophthalmol Sci 2022, 2, 100114. [Google Scholar] [CrossRef]
- Keenan, T.D.L.; Cukras, C.A.; Chew, E.Y. Age-Related Macular Degeneration: Epidemiology and Clinical Aspects. In Age-related Macular Degeneration, Advances in Experimental Medicine and Biology, Chew, E.Y., Swaroop, A., Eds.; Advances in Experimental Medicine and Biology; Springer Nature Switzerland AG: Cham, Switzerland, 2021; Volume 1256, pp. 1–31.
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (AMD): associations with cardiovascular disease phenotypes and lipid factors. Eye Vis (Lond) 2016, 3, 34. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [PubMed]
- Agron, E.; Domalpally, A.; Cukras, C.A.; Clemons, T.E.; Chen, Q.; Lu, Z.; Chew, E.Y.; Keenan, T.D.L.; Areds; Groups, A.R. Reticular Pseudodrusen: The Third Macular Risk Feature for Progression to Late Age-Related Macular Degeneration: Age-Related Eye Disease Study 2 Report 30. Ophthalmology 2022, 129, 1107–1119. [CrossRef]
- Rudolf, M.; Vogt, S.D.; Curcio, C.A.; Huisingh, C.; McGwin, G., Jr.; Wagner, A.; Grisanti, S.; Read, R.W. Histologic basis of variations in retinal pigment epithelium autofluorescence in eyes with geographic atrophy. Ophthalmology 2013, 120, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Zanzottera, E.C.; Ach, T.; Huisingh, C.; Messinger, J.D.; Freund, K.B.; Curcio, C.A. Visualizing Retinal Pigment Epithelium Phenotypes in the Transition to Atrophy in Neovascular Age-Related Macular Degeneration. Retina 2016, 36 Suppl 1, S26–S39. [Google Scholar] [CrossRef]
- Bonilha, V.L.; Bell, B.A.; Hu, J.; Milliner, C.; Pauer, G.J.; Hagstrom, S.A.; Radu, R.A.; Hollyfield, J.G. Geographic Atrophy: Confocal Scanning Laser Ophthalmoscopy, Histology, and Inflammation in the Region of Expanding Lesions. Invest Ophthalmol Vis Sci 2020, 61, 15. [Google Scholar] [CrossRef] [PubMed]
- Zanzottera, E.C.; Messinger, J.D.; Ach, T.; Smith, R.T.; Freund, K.B.; Curcio, C.A. The Project MACULA Retinal Pigment Epithelium Grading System for Histology and Optical Coherence Tomography in Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 2015, 56, 3253–3268. [Google Scholar] [CrossRef]
- Gambril, J.A.; Sloan, K.R.; Swain, T.A.; Huisingh, C.; Zarubina, A.V.; Messinger, J.D.; Ach, T.; Curcio, C.A. Quantifying Retinal Pigment Epithelium Dysmorphia and Loss of Histologic Autofluorescence in Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 2019, 60, 2481–2493. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Ben Ami, T.; Hong, S.; Heintzmann, R.; Gerig, G.; Ablonczy, Z.; Curcio, C.A.; Ach, T.; Smith, R.T. Hyperspectral Autofluorescence Imaging of Drusen and Retinal Pigment Epithelium in Donor Eyes with Age-Related Macular Degeneration. Retina 2016, 36 Suppl 1, S127–S136. [Google Scholar] [CrossRef]
- Mohammed, T.; Tong, Y.; Agee, J.; Challa, N.; Heintzmann, R.; Hammer, M.; Curcio, C.A.; Ach, T.; Ablonczy, Z.; Smith, R.T. Ex Vivo Hyperspectral Autofluorescence Imaging and Localization of Fluorophores in Human Eyes with Age-Related Macular Degeneration. Vision (Basel) 2018, 2. [Google Scholar] [CrossRef]
- Rozanowski, B.; Cuenco, J.; Davies, S.; Shamsi, F.A.; Zadlo, A.; Dayhaw-Barker, P.; Rozanowska, M.; Sarna, T.; Boulton, M.E. The phototoxicity of aged human retinal melanosomes. Photochemistry and photobiology 2008, 84, 650–657. [Google Scholar] [CrossRef]
- Rozanowski, B.; Burke, J.M.; Boulton, M.E.; Sarna, T.; Rozanowska, M. Human RPE melanosomes protect from photosensitized and iron-mediated oxidation but become pro-oxidant in the presence of iron upon photodegradation. Invest Ophthalmol Vis Sci 2008, 49, 2838–2847. [Google Scholar] [CrossRef]
- Teussink, M.M.; Lambertus, S.; de Mul, F.F.; Rozanowska, M.B.; Hoyng, C.B.; Klevering, B.J.; Theelen, T. Lipofuscin-associated photo-oxidative stress during fundus autofluorescence imaging. PLoS One 2017, 12, e0172635. [Google Scholar] [CrossRef] [PubMed]
- Arend, O.; Weiter, J.J.; Goger, D.G.; Delori, F.C. [In vivo fundus fluorescence measurements in patients with age related macular degeneration]. Ophthalmologe 1995, 92, 647–653. [Google Scholar]
- Delori, F.C.; Fleckner, M.R.; Goger, D.G.; Weiter, J.J.; Dorey, C.K. Autofluorescence distribution associated with drusen in age-related macular degeneration. Invest Ophthalmol Vis Sci 2000, 41, 496–504. [Google Scholar]
- Hammer, M.; Schultz, R.; Hasan, S.; Sauer, L.; Klemm, M.; Kreilkamp, L.; Zweifel, L.; Augsten, R.; Meller, D. Fundus Autofluorescence Lifetimes and Spectral Features of Soft Drusen and Hyperpigmentation in Age-Related Macular Degeneration. Transl Vis Sci Technol 2020, 9, 20. [Google Scholar] [CrossRef]
- Weber, S.; Simon, R.; Schwanengel, L.S.; Curcio, C.A.; Augsten, R.; Meller, D.; Hammer, M. Fluorescence Lifetime and Spectral Characteristics of Subretinal Drusenoid Deposits and Their Predictive Value for Progression of Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 2022, 63, 23. [Google Scholar] [CrossRef]
- Borrelli, E.; Nittala, M.G.; Abdelfattah, N.S.; Lei, J.; Hariri, A.H.; Shi, Y.; Fan, W.; Cozzi, M.; Sarao, V.; Lanzetta, P.; et al. Comparison of short-wavelength blue-light autofluorescence and conventional blue-light autofluorescence in geographic atrophy. Br J Ophthalmol 2018, 103, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Hammer, M.; Quick, S.; Klemm, M.; Schenke, S.; Mata, N.; Eitner, A.; Schweitzer, D. In-vivo and in-vitro investigations of retinal fluorophores in age - related macular degeneration by fluorescence lifetime imaging. Multiphoton Microscopy in the Biomedical Sciences Ix 2009, 7183. [Google Scholar] [CrossRef]
- Schweitzer, D.; Quick, S.; Schenke, S.; Klemm, M.; Gehlert, S.; Hammer, M.; Jentsch, S.; Fischer, J. [Comparison of parameters of time-resolved autofluorescence between healthy subjects and patients suffering from early AMD]. Ophthalmologe 2009, 106, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Reiter, G.S.; Hacker, V.; Told, R.; Schranz, M.; Krotka, P.; Schlanitz, F.G.; Sacu, S.; Pollreisz, A.; Schmidt-Erfurth, U. Longitudinal Changes in Quantitative Autofluorescence during Progression from Intermediate to Late Age-Related Macular Degeneration. Retin.-J. Retin. Vitr. Dis. 2021, 41, 1236–1241. [Google Scholar] [CrossRef]
- Reiter, G.S.; Told, R.; Baumann, L.; Sacu, S.; Schmidt-Erfurth, U.; Pollreisz, A. Investigating a Growth Prediction Model in Advanced Age-Related Macular Degeneration with Solitary Geographic Atrophy Using Quantitative Autofluorescence. Retin.-J. Retin. Vitr. Dis. 2020, 40, 1657–1664. [Google Scholar] [CrossRef]
- Schwanengel, L.S.; Weber, S.; Simon, R.; Lehmann, T.; Augsten, R.; Meller, D.; Hammer, M. Changes in drusen-associated autofluorescence over time observed by fluorescence lifetime imaging ophthalmoscopy in age-related macular degeneration. Acta Ophthalmol 2022. [CrossRef]
- Chen, L.; Yang, P.; Curcio, C.A. Visualizing lipid behind the retina in aging and age-related macular degeneration, via indocyanine green angiography (ASHS-LIA). Eye (Lond) 2022, 36, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Myers, C.E.; Lee, K.E.; Gangnon, R.E.; Sivakumaran, T.A.; Iyengar, S.K.; Klein, B.E. Small Drusen and Age-Related Macular Degeneration: The Beaver Dam Eye Study. J Clin Med 2015, 4, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Feldman, T.B.; Yakovleva, M.A.; Arbukhanova, P.M.; Borzenok, S.A.; Kononikhin, A.S.; Popov, I.A.; Nikolaev, E.N.; Ostrovsky, M.A. Changes in spectral properties and composition of lipofuscin fluorophores from human-retinal-pigment epithelium with age and pathology. Anal Bioanal Chem 2015, 407, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Yakovleva, M.A.; Feldman, T.B.; Arbukhanova, P.M.; Borzenok, S.A.; Kuzmin, V.A.; Ostrovsky, M.A. The fluorescence lifetime of lipofuscin granule fluorophores contained in the retinal pigment epithelium cells from human cadaver eyes in normal state and in the case of visualized pathology. Dokl Biochem Biophys 2017, 474, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Feldman, T.B.; Yakovleva, M.A.; Larichev, A.V.; Arbukhanova, P.M.; Radchenko, A.S.; Borzenok, S.A.; Kuzmin, V.A.; Ostrovsky, M.A. Spectral analysis of fundus autofluorescence pattern as a tool to detect early stages of degeneration in the retina and retinal pigment epithelium. Eye (Lond) 2018, 32, 1440–1448. [Google Scholar] [CrossRef]
- Yakovleva, M.A.; Radchenko, A.S.; Feldman, T.B.; Kostyukov, A.A.; Arbukhanova, P.M.; Borzenok, S.A.; Kuzmin, V.A.; Ostrovsky, M.A. Fluorescence characteristics of lipofuscin fluorophores from human retinal pigment epithelium. Photochem Photobiol Sci 2020, 19, 920–930. [Google Scholar] [CrossRef]
- Mata, N.L.; Weng, J.; Travis, G.H. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A 2000, 97, 7154–7159. [Google Scholar] [CrossRef]
- Ueda, K.; Zhao, J.; Kim, H.J.; Sparrow, J.R. Photodegradation of retinal bisretinoids in mouse models and implications for macular degeneration. Proc Natl Acad Sci U S A 2016, 113, 6904–6909. [Google Scholar] [CrossRef]
- Ueda, K.; Kim, H.J.; Zhao, J.; Song, Y.; Dunaief, J.L.; Sparrow, J.R. Iron promotes oxidative cell death caused by bisretinoids of retina. Proc Natl Acad Sci U S A 2018, 115, 4963–4968. [Google Scholar] [CrossRef]
- Pandya, V.B.; Ho, I.V.; Hunyor, A.P. Does unintentional macular translocation after retinal detachment repair influence visual outcome? Clin Exp Ophthalmol 2012, 40, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Shiragami, C.; Shiraga, F.; Yamaji, H.; Fukuda, K.; Takagishi, M.; Morita, M.; Kishikami, T. Unintentional displacement of the retina after standard vitrectomy for rhegmatogenous retinal detachment. Ophthalmology 2010, 117, 86–92 e81. [Google Scholar] [CrossRef] [PubMed]
- Hollyfield, J.G. Age-related macular degeneration: the molecular link between oxidative damage, tissue-specific inflammation and outer retinal disease: the Proctor lecture. Invest Ophthalmol Vis Sci 2010, 51, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.X.; Stiles, T.; Douglas, C.; Ho, D.; Fan, W.; Du, H.; Xiao, X. Oxidative stress, innate immunity, and age-related macular degeneration. AIMS Mol Sci 2016, 3, 196–221. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog Retin Eye Res 2017, 60, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Handa, J.T.; Cano, M.; Wang, L.; Datta, S.; Liu, T. Lipids, oxidized lipids, oxidation-specific epitopes, and Age-related Macular Degeneration. Biochim Biophys Acta Mol Cell Biol Lipids 2017, 1862, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.; Datta, S.; Wang, L.; Pegany, R.; Cano, M.; Handa, J.T. The impact of lipids, lipid oxidation, and inflammation on AMD, and the potential role of miRNAs on lipid metabolism in the RPE. Exp Eye Res 2019, 181, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Karunadharma, P.P.; Kapphahn, R.J.; Stahl, M.R.; Olsen, T.W.; Ferrington, D.A. Dissecting Regulators of Aging and Age-Related Macular Degeneration in the Retinal Pigment Epithelium. Oxid Med Cell Longev 2022, 2022, 6009787. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.; Milam, A.H.; Dunaief, J.L. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch’s membrane. Arch Ophthalmol 2003, 121, 1099–1105. [Google Scholar] [CrossRef]
- Liu, Y.; Bell, B.A.; Song, Y.; Zhang, K.; Anderson, B.; Axelsen, P.H.; Bohannan, W.; Agbaga, M.P.; Park, H.G.; James, G.; et al. Deuterated docosahexaenoic acid protects against oxidative stress and geographic atrophy-like retinal degeneration in a mouse model with iron overload. Aging Cell 2022, 21, e13579. [Google Scholar] [CrossRef] [PubMed]
- von Krusenstiern, A.N.; Robson, R.N.; Qian, N.; Qiu, B.; Hu, F.; Reznik, E.; Smith, N.; Zandkarimi, F.; Estes, V.M.; Dupont, M.; et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat Chem Biol 2023. [CrossRef]
- Shchepinov, M.S. Polyunsaturated Fatty Acid Deuteration against Neurodegeneration. Trends Pharmacol Sci 2020, 41, 236–248. [Google Scholar] [CrossRef]
- Firsov, A.M.; Fomich, M.A.; Bekish, A.V.; Sharko, O.L.; Kotova, E.A.; Saal, H.J.; Vidovic, D.; Shmanai, V.V.; Pratt, D.A.; Antonenko, Y.N.; et al. Threshold protective effect of deuterated polyunsaturated fatty acids on peroxidation of lipid bilayers. FEBS J 2019, 286, 2099–2117. [Google Scholar] [CrossRef]
- Rosell, M.; Giera, M.; Brabet, P.; Shchepinov, M.S.; Guichardant, M.; Durand, T.; Vercauteren, J.; Galano, J.M.; Crauste, C. Bis-allylic Deuterated DHA Alleviates Oxidative Stress in Retinal Epithelial Cells. Antioxidants (Basel) 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Sterling, J.K.; Baumann, B.; Foshe, S.; Voigt, A.; Guttha, S.; Alnemri, A.; McCright, S.J.; Li, M.; Zauhar, R.J.; Montezuma, S.R.; et al. Inflammatory adipose activates a nutritional immunity pathway leading to retinal dysfunction. Cell Rep 2022, 39, 110942. [Google Scholar] [CrossRef]
- Lukiw, W.J.; Mukherjee, P.K.; Cui, J.G.; Bazan, N.G. A2E selectively induces cox-2 in ARPE-19 and human neural cells. Curr Eye Res 2006, 31, 259–263. [Google Scholar] [CrossRef]
- Chahal, S.; Rani, P.; Kiran; Sindhu, J.; Joshi, G.; Ganesan, A.; Kalyaanamoorthy, S.; Mayank; Kumar, P.; Singh, R.; et al. Design and Development of COX-II Inhibitors: Current Scenario and Future Perspective. ACS Omega 2023, 8, 17446–17498. [CrossRef]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A 2002, 99, 14682–14687. [Google Scholar] [CrossRef]
- Gu, X.; Meer, S.G.; Miyagi, M.; Rayborn, M.E.; Hollyfield, J.G.; Crabb, J.W.; Salomon, R.G. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J Biol Chem 2003, 278, 42027–42035. [Google Scholar] [CrossRef] [PubMed]
- Renganathan, K.; Ebrahem, Q.; Vasanji, A.; Gu, X.; Lu, L.; Sears, J.; Salomon, R.G.; Anand-Apte, B.; Crabb, J.W. Carboxyethylpyrrole adducts, age-related macular degeneration and neovascularization. Adv Exp Med Biol 2008, 613, 261–267. [Google Scholar] [CrossRef]
- Ebrahem, Q.; Renganathan, K.; Sears, J.; Vasanji, A.; Gu, X.; Lu, L.; Salomon, R.G.; Crabb, J.W.; Anand-Apte, B. Carboxyethylpyrrole oxidative protein modifications stimulate neovascularization: Implications for age-related macular degeneration. Proc Natl Acad Sci U S A 2006, 103, 13480–13484. [Google Scholar] [CrossRef] [PubMed]
- Ayalasomayajula, S.P.; Kompella, U.B. Induction of vascular endothelial growth factor by 4-hydroxynonenal and its prevention by glutathione precursors in retinal pigment epithelial cells. Eur J Pharmacol 2002, 449, 213–220. [Google Scholar] [CrossRef]
- Zhou, J.; Cai, B.; Jang, Y.P.; Pachydaki, S.; Schmidt, A.M.; Sparrow, J.R. Mechanisms for the induction of HNE- MDA- and AGE-adducts, RAGE and VEGF in retinal pigment epithelial cells. Exp Eye Res 2005, 80, 567–580. [Google Scholar] [CrossRef]
- Yanagi, Y.; Inoue, Y.; Iriyama, A.; Jang, W.D. Effects of yellow intraocular lenses on light-induced upregulation of vascular endothelial growth factor. J Cataract Refract Surg 2006, 32, 1540–1544. [Google Scholar] [CrossRef] [PubMed]
- Ung, C.; Lains, I.; Miller, J.W.; Kim, I.K. Current Management of Age-Related Macular Degeneration. In Age-related Macular Degeneration From Clinic to Genes and Back to Patient Management. Advances in Experimental Medicine and Biology, Chew, E.Y., Swaroop, A., Eds.; Springer Nature Switzerland AG: Cham, Switzerland, 2019; Volume 1256, pp. 295–314.
- Higgins, G.T.; Wang, J.H.; Dockery, P.; Cleary, P.E.; Redmond, H.P. Induction of angiogenic cytokine expression in cultured RPE by ingestion of oxidized photoreceptor outer segments. Invest Ophthalmol Vis Sci 2003, 44, 1775–1782. [Google Scholar] [CrossRef]
- Hernandez, C.; Segura, R.M.; Fonollosa, A.; Carrasco, E.; Francisco, G.; Simo, R. Interleukin-8, monocyte chemoattractant protein-1 and IL-10 in the vitreous fluid of patients with proliferative diabetic retinopathy. Diabet Med 2005, 22, 719–722. [Google Scholar] [CrossRef]
- Wang, L.; Cano, M.; Datta, S.; Wei, H.; Ebrahimi, K.B.; Gorashi, Y.; Garlanda, C.; Handa, J.T. Pentraxin 3 recruits complement factor H to protect against oxidative stress-induced complement and inflammasome overactivation. J Pathol 2016, 240, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A 2006, 103, 2328–2333. [Google Scholar] [CrossRef]
- Ng, E.S.Y.; Kady, N.; Hu, J.; Dave, A.; Jiang, Z.; Pei, J.; Gorin, M.B.; Matynia, A.; Radu, R.A. Membrane Attack Complex Mediates Retinal Pigment Epithelium Cell Death in Stargardt Macular Degeneration. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Pauer, G.J.; Hagstrom, S.A.; Bok, D.; DeBenedictis, M.J.; Bonilha, V.L.; Hollyfield, J.G.; Radu, R.A. Evidence of complement dysregulation in outer retina of Stargardt disease donor eyes. Redox Biol 2020, 37, 101787. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Fisher, C.R.; Kowluru, R.A. Mitochondrial Defects Drive Degenerative Retinal Diseases. Trends Mol Med 2020, 26, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Ebeling, M.C.; Polanco, J.R.; Qu, J.; Tu, C.; Montezuma, S.R.; Ferrington, D.A. Improving retinal mitochondrial function as a treatment for age-related macular degeneration. Redox Biol 2020, 34, 101552. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog Retin Eye Res 2020, 79, 100858. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Kapphahn, R.J.; Leary, M.M.; Atilano, S.R.; Terluk, M.R.; Karunadharma, P.; Chen, G.K.; Ratnapriya, R.; Swaroop, A.; Montezuma, S.R.; et al. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp Eye Res 2016, 145, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J Neurosci 2015, 35, 7304–7311. [Google Scholar] [CrossRef]
- Lin, H.; Xu, H.; Liang, F.Q.; Liang, H.; Gupta, P.; Havey, A.N.; Boulton, M.E.; Godley, B.F. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Invest Ophthalmol Vis Sci 2011, 52, 3521–3529. [Google Scholar] [CrossRef] [PubMed]
- Karunadharma, P.P.; Nordgaard, C.L.; Olsen, T.W.; Ferrington, D.A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest Ophthalmol Vis Sci 2010, 51, 5470–5479. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Kapphahn, R.J.; Zhang, M.; Qian, S.; Montezuma, S.R.; Shang, P.; Ferrington, D.A.; Qu, J. Quantitative Proteomics of Human Retinal Pigment Epithelium Reveals Key Regulators for the Pathogenesis of Age-Related Macular Degeneration. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Chew, E.Y.; Clemons, T.E.; Agron, E.; Domalpally, A.; Keenan, T.D.L.; Vitale, S.; Weber, C.; Smith, D.C.; Christen, W.; Group, A.R. Long-term Outcomes of Adding Lutein/Zeaxanthin and omega-3 Fatty Acids to the AREDS Supplements on Age-Related Macular Degeneration Progression: AREDS2 Report 28. JAMA Ophthalmol 2022, 140, 692–698. [Google Scholar] [CrossRef]
- Evans, J.R.; Lawrenson, J.G. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst Rev 2017, 7, CD000254. [Google Scholar] [CrossRef] [PubMed]
- Age-Related Eye Disease Study 2 Research, G.; Chew, E.Y.; Clemons, T.E.; Sangiovanni, J.P.; Danis, R.P.; Ferris, F.L., 3rd; Elman, M.J.; Antoszyk, A.N.; Ruby, A.J.; Orth, D.; et al. Secondary analyses of the effects of lutein/zeaxanthin on age-related macular degeneration progression: AREDS2 report No. 3. JAMA Ophthalmol 2014, 132, 142–149. [Google Scholar] [CrossRef]
- Group, A.R.; Chew, E.Y.; Clemons, T.; SanGiovanni, J.P.; Danis, R.; Domalpally, A.; McBee, W.; Sperduto, R.; Ferris, F.L. The Age-Related Eye Disease Study 2 (AREDS2): study design and baseline characteristics (AREDS2 report number 1). Ophthalmology 2012, 119, 2282–2289. [Google Scholar] [CrossRef]
- Bassetto, M.; Zaluski, J.; Li, B.; Zhang, J.; Badiee, M.; Kiser, P.D.; Tochtrop, G.P. Tuning the Metabolic Stability of Visual Cycle Modulators through Modification of an RPE65 Recognition Motif. J Med Chem 2023, 66, 8140–8158. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A diagram showing a cross-section of the eye and a photograph of the fundus with depicted areas of the fovea and macula. Modified from [77]. The diameter of the fovea is 1.5 mm. A 0.5 mm thick ring surrounding the fovea is known as parafovea, and a 1.5 mm thick ring surrounding the parafovea is known as perifovea [78].
Figure 1.
A diagram showing a cross-section of the eye and a photograph of the fundus with depicted areas of the fovea and macula. Modified from [77]. The diameter of the fovea is 1.5 mm. A 0.5 mm thick ring surrounding the fovea is known as parafovea, and a 1.5 mm thick ring surrounding the parafovea is known as perifovea [78].
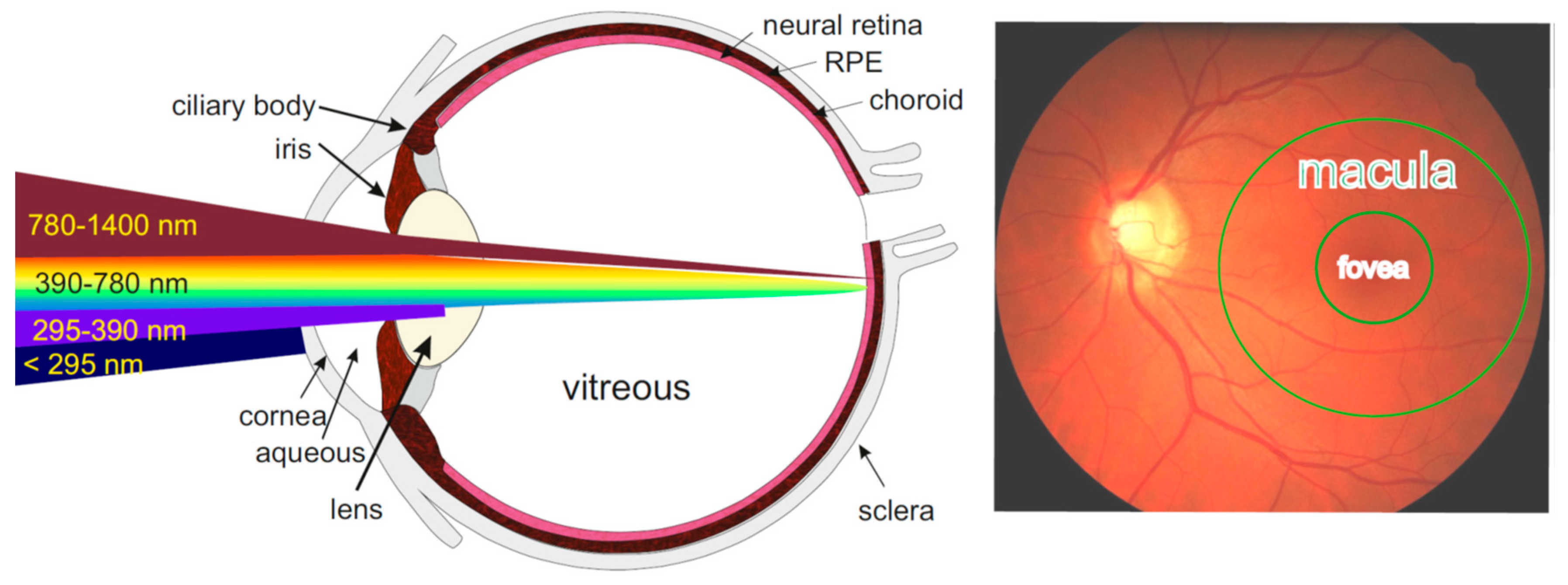
Figure 2.
A and B: Representative fluorescence emission spectra of lipofuscin granules isolated from human RPE and suspended in PBS before and after the indicated times of irradiation with visible light (9.76 mW/cm2). Fluorescence was excited with 360 nm (A, D, F) or 488 nm (B, E, G) light. C: Fluorescence emission spectra from B corrected for the spectral changes in the detection sensitivity. D and E: Kinetics of changes of fluorescence emission intensity monitored at the indicated wavelengths, normalized to the value before irradiation and expressed as a percentage. F and G: fluorescence intensity ratios at the indicated wavelengths. The ratios of fluorescence intensities were calculated based on uncorrected spectra in the case of excitation with 360 nm and on corrected spectra in the case of excitation with 488 nm light. The Fluorescence of the solvent (PBS) was recorded to show its Raman emission peaks. From [74].
Figure 2.
A and B: Representative fluorescence emission spectra of lipofuscin granules isolated from human RPE and suspended in PBS before and after the indicated times of irradiation with visible light (9.76 mW/cm2). Fluorescence was excited with 360 nm (A, D, F) or 488 nm (B, E, G) light. C: Fluorescence emission spectra from B corrected for the spectral changes in the detection sensitivity. D and E: Kinetics of changes of fluorescence emission intensity monitored at the indicated wavelengths, normalized to the value before irradiation and expressed as a percentage. F and G: fluorescence intensity ratios at the indicated wavelengths. The ratios of fluorescence intensities were calculated based on uncorrected spectra in the case of excitation with 360 nm and on corrected spectra in the case of excitation with 488 nm light. The Fluorescence of the solvent (PBS) was recorded to show its Raman emission peaks. From [74].
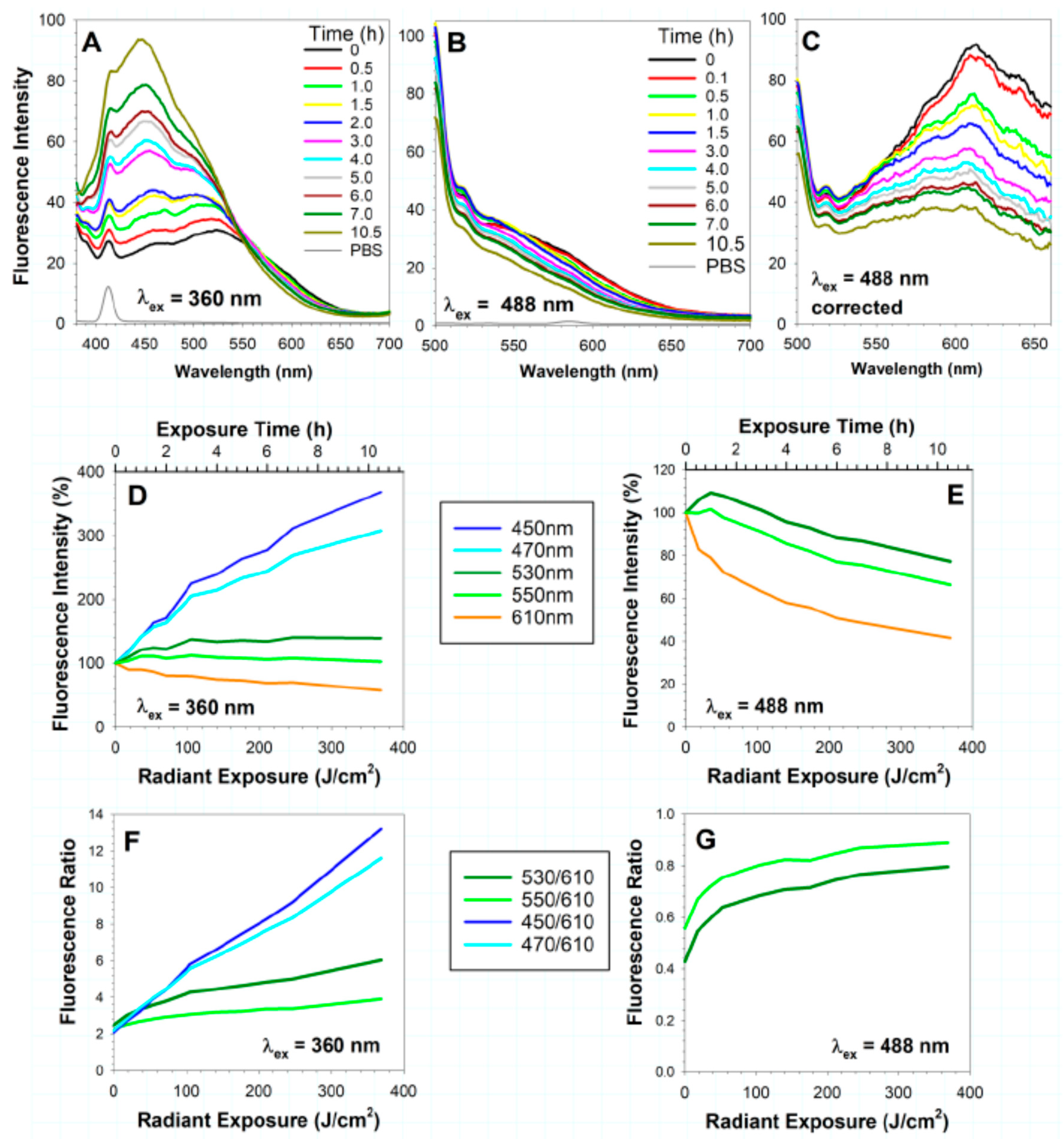
Figure 3.
Top panel: fluorescence of lipofuscin-laden ARPE-19 cell monolayer kept in the dark )left panel) or exposed daily for 14 days to 45 minutes of 9.76 mW/cm2 visible light providing a total dose of 369 J/cm2 (right panel). Fluorescence images were acquired using a fluorescence microscope with blue light excitation and a colour camera capturing the colour of lipofuscin fluorescence as it appears to the human eye. The gain used for collecting fluorescence images was two-fold smaller for lipofuscin-laden cells not exposed to light than for cells exposed to light. Bottom panel A and B: Representative fluorescence emission spectra from ARPE-19 cells with and without lipofuscin (LF) kept in dark or exposed to light as above and then solubilized in Triton X-100 (Tx). Fluorescence was induced by photoexcitation with 360 nm (A) or 488 nm (B) light. C: Fluorescence emission spectra from (B), corrected for the spectral changes in the detector sensitivity. Modified from [74].
Figure 3.
Top panel: fluorescence of lipofuscin-laden ARPE-19 cell monolayer kept in the dark )left panel) or exposed daily for 14 days to 45 minutes of 9.76 mW/cm2 visible light providing a total dose of 369 J/cm2 (right panel). Fluorescence images were acquired using a fluorescence microscope with blue light excitation and a colour camera capturing the colour of lipofuscin fluorescence as it appears to the human eye. The gain used for collecting fluorescence images was two-fold smaller for lipofuscin-laden cells not exposed to light than for cells exposed to light. Bottom panel A and B: Representative fluorescence emission spectra from ARPE-19 cells with and without lipofuscin (LF) kept in dark or exposed to light as above and then solubilized in Triton X-100 (Tx). Fluorescence was induced by photoexcitation with 360 nm (A) or 488 nm (B) light. C: Fluorescence emission spectra from (B), corrected for the spectral changes in the detector sensitivity. Modified from [74].
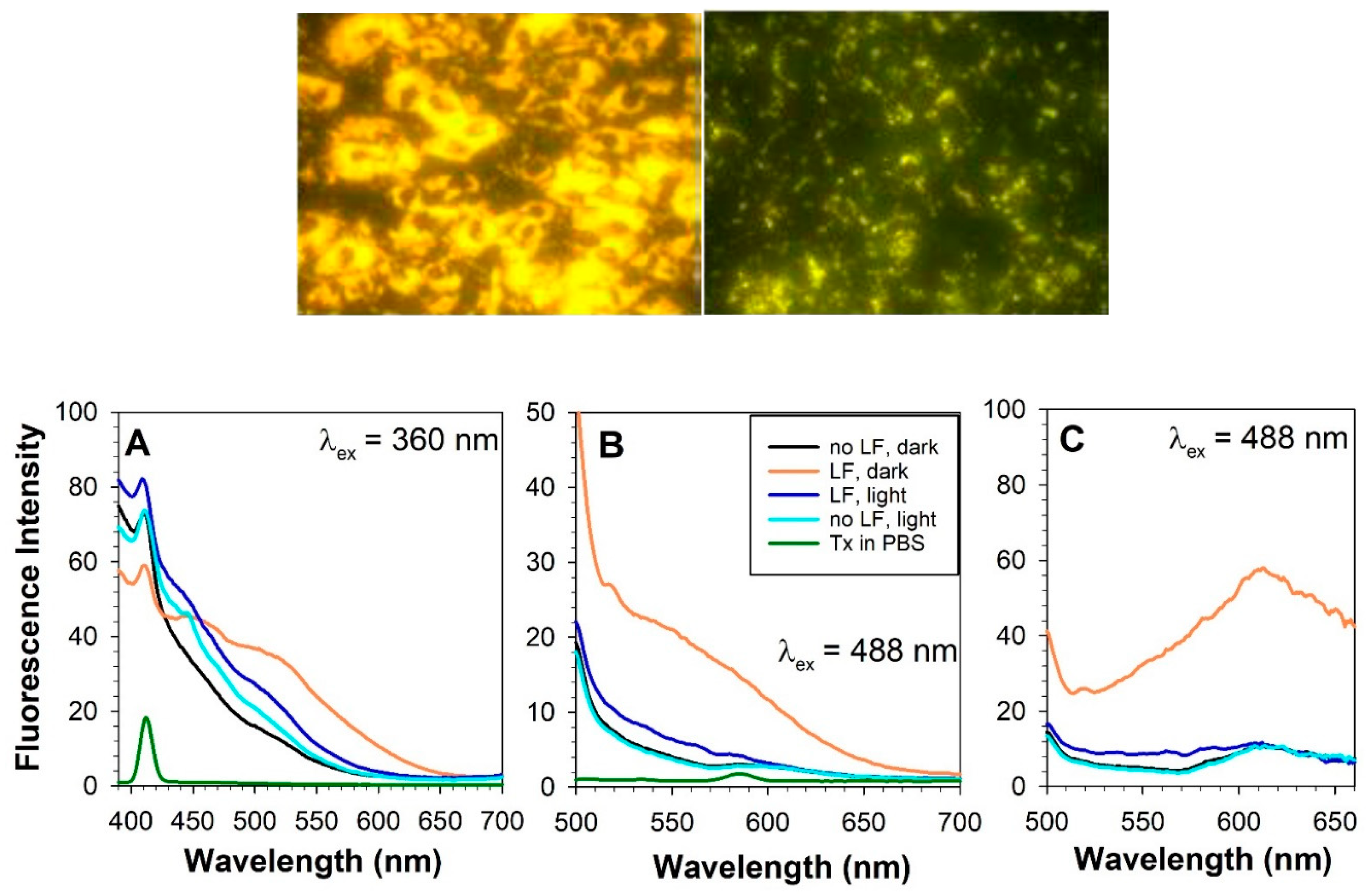
Figure 4.
Top panel: imaging of two-photon-excited fluorescence (TPEF) by scanning laser ophthalmoscope (SLO) in the fundus of a healthy eye of a human volunteer. The laser system provided 76 fs pulses of 780 nm wavelength. The images of the scanned areas were acquired simultaneously in fluorescence channels at the indicated spectral ranges and in a reflectance channel. Bottom panel: the integrated fluorescence intensity in the four spectral ranges normalized to the integrated fluorescence intensity in the range of 400–700 nm. Adapted from Boguslawski et al. In vivo imaging of the human eye using a 2-photon-excited fluorescence scanning laser ophthalmoscope. J Clin Invest 2022, 132, doi:10.1172/JCI154218 licensed under a Creative Commons Attribution 4.0 International License. Copyright Boguslawski et al. 2022 [295].
Figure 4.
Top panel: imaging of two-photon-excited fluorescence (TPEF) by scanning laser ophthalmoscope (SLO) in the fundus of a healthy eye of a human volunteer. The laser system provided 76 fs pulses of 780 nm wavelength. The images of the scanned areas were acquired simultaneously in fluorescence channels at the indicated spectral ranges and in a reflectance channel. Bottom panel: the integrated fluorescence intensity in the four spectral ranges normalized to the integrated fluorescence intensity in the range of 400–700 nm. Adapted from Boguslawski et al. In vivo imaging of the human eye using a 2-photon-excited fluorescence scanning laser ophthalmoscope. J Clin Invest 2022, 132, doi:10.1172/JCI154218 licensed under a Creative Commons Attribution 4.0 International License. Copyright Boguslawski et al. 2022 [295].
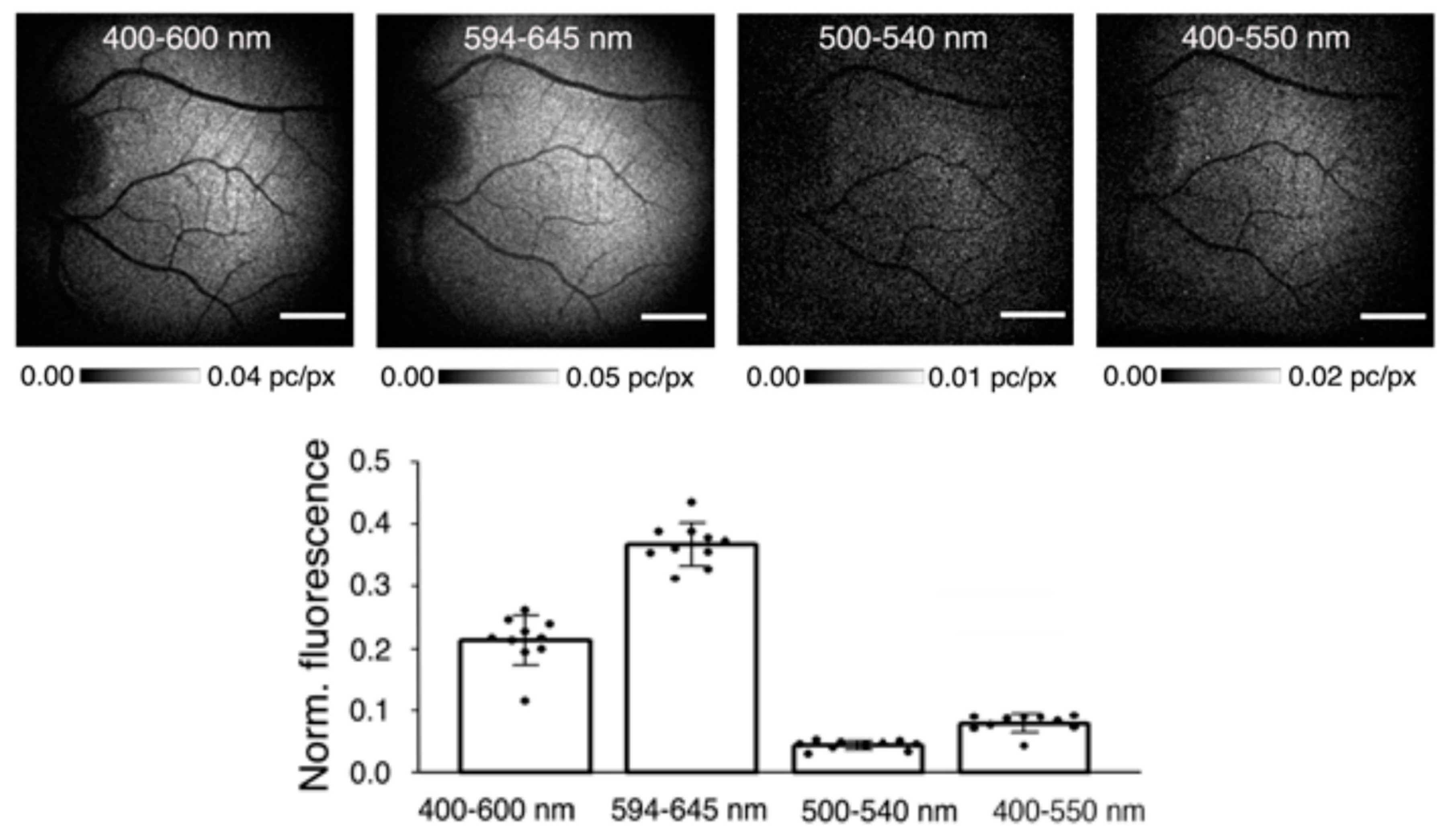
Figure 5.
Multimodal imaging of a 72-year-old patient at baseline (panels on the left) and 52 months later (panels on the right); a, b: colour fundus photography (CFP); c–f: OCT at the green lines in a and b; g, h: fundus autofluorescence (FAF) intensity; i, j: imaging of mean FAF lifetimes in the short-wavelength spectral channel (498-560 nm; SSC); k, l: imaging of mean FAF lifetimes in the long-wavelength spectral channel (560-720 nm; LSC); m, n: peak emission wavelength (PEW; calculated based on calibration in the range of 540–670 nm). Arrows point to drusen with different fates. Light purple arrowhead: druse with short mean fluorescence lifetime and indifferent PEW at baseline, which collapsed over follow-up leaving ectopic RPE hyperreflective foci in OCT. The lifetime changed from short to long and the PEW decreased. White arrowhead: hyperautofluorescent druse with a short lifetime and PEW, which disappeared over the follow-up not leaving RPE distortion. Black arrowhead: a druse growing over the follow-up with increasing mean fluorecence lifetime and decreasing PEW. From Schwanengel, L.S.; Weber, S.; Simon, R.; Lehmann, T.; Augsten, R.; Meller, D.; Hammer, M.: Changes in drusen-associated autofluorescence over time observed by fluorescence lifetime imaging ophthalmoscopy in age-related macular degeneration. Acta Ophthalmol 2022, doi:10.1111/aos.15238 under the Creative Commons Attribution License © 2022 Schwanengel et al. [368].
Figure 5.
Multimodal imaging of a 72-year-old patient at baseline (panels on the left) and 52 months later (panels on the right); a, b: colour fundus photography (CFP); c–f: OCT at the green lines in a and b; g, h: fundus autofluorescence (FAF) intensity; i, j: imaging of mean FAF lifetimes in the short-wavelength spectral channel (498-560 nm; SSC); k, l: imaging of mean FAF lifetimes in the long-wavelength spectral channel (560-720 nm; LSC); m, n: peak emission wavelength (PEW; calculated based on calibration in the range of 540–670 nm). Arrows point to drusen with different fates. Light purple arrowhead: druse with short mean fluorescence lifetime and indifferent PEW at baseline, which collapsed over follow-up leaving ectopic RPE hyperreflective foci in OCT. The lifetime changed from short to long and the PEW decreased. White arrowhead: hyperautofluorescent druse with a short lifetime and PEW, which disappeared over the follow-up not leaving RPE distortion. Black arrowhead: a druse growing over the follow-up with increasing mean fluorecence lifetime and decreasing PEW. From Schwanengel, L.S.; Weber, S.; Simon, R.; Lehmann, T.; Augsten, R.; Meller, D.; Hammer, M.: Changes in drusen-associated autofluorescence over time observed by fluorescence lifetime imaging ophthalmoscopy in age-related macular degeneration. Acta Ophthalmol 2022, doi:10.1111/aos.15238 under the Creative Commons Attribution License © 2022 Schwanengel et al. [368].
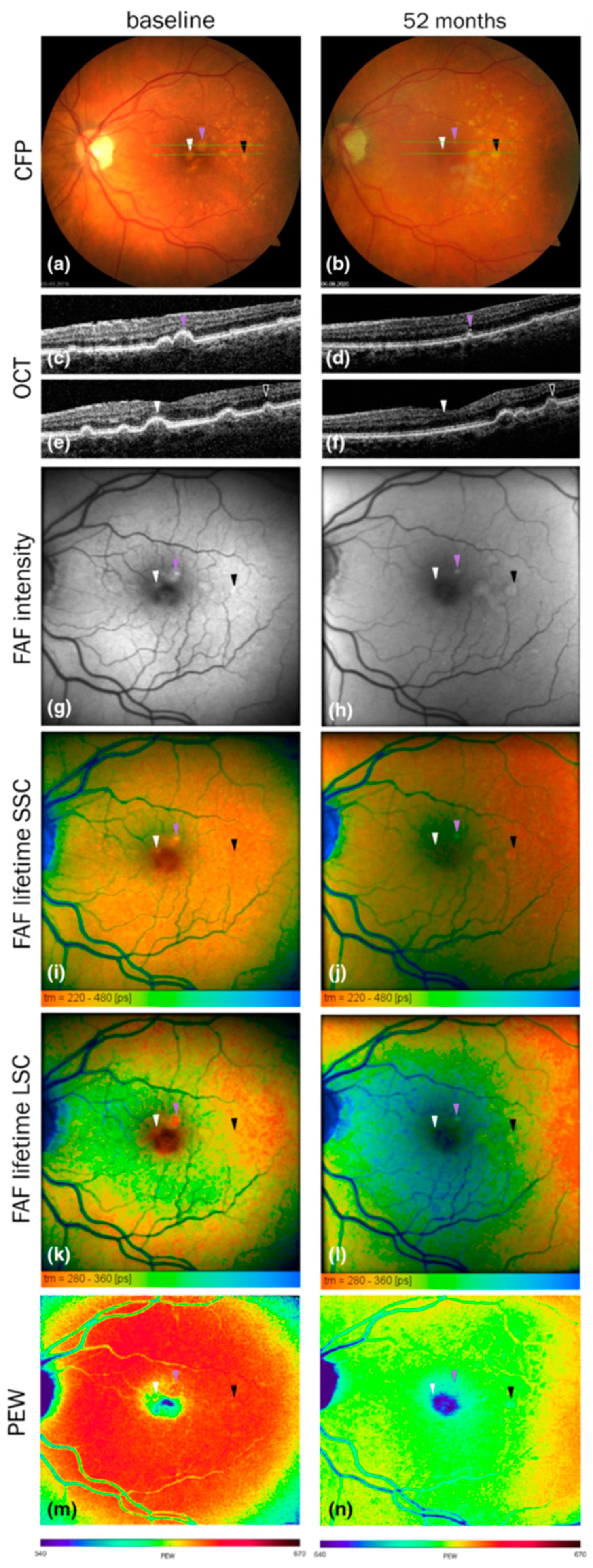
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



