
Submitted:
30 June 2023
Posted:
06 July 2023
You are already at the latest version
Abstract
Conductive hearing losses are typically present in disorders of the external/middle ear. However, there is a rare group of inner ear conditions called third windows that can also generate a conductive hearing loss. This is due to an abnormal connection between the middle and the inner ear or between the inner ear and the cranial cavity. X-linked gusher disorder is an extremely rare congenital inner ear dysplastic syndrome with such an abnormal connection due to a characteristic incomplete cochlear partition type 3 and an incomplete internal auditory meatus fundus. The disorder is inherited in an X-linked fashion due to the mutation of the POU3F4 gene. We present 2 siblings diagnosed with the condition and their long term follow ups. They both presented with audiovestibular symptoms and showed progressive mixed losses and bilateral vestibular weakness. They were treated with cochlear implant, digital amplification and with vestibular rehabilitation. Significant others around them were involved in their journey with the medical team and in both, a very favourable outcome was achieved. This is the first time that we are reporting evolving audiovestibular function with vestibular quantification in X-linked gusher disorder and emphasize on the multidisciplinary holistic approach to manage these children effectively.
Keywords:
Third window
; X-linked gusher
; hearing loss
; audiovestibular
; POU3F4
1. Introduction
X-linked gusher disease is a rare genetic disorder that presents with a conductive element in a hearing loss measured by pure tone audiometry. First identified by Nance in 1971, this entity was considered as a congenital stapedial fixation disorder, where on surgery, a perilymphatic gusher was identified [1]. The disorder is characterized by classical radiological appearances based on high resolution CT scan (HRCT). Phelps et al in 1991 studied and observed the CT phenotype of an X-linked gusher disorder with 3 features – bulbous internal auditory meatus (IAM), incomplete separation of the coils of the cochlea from the IAM and wide first and second parts of the intratemporal facial nerve [2]. Subsequently an absent bony modiolus was identified [3].
The clue that the disorder is genetically inherited was suggested by observing the disorder in males. The gene responsible for the disorder was identified in 1995 [4] as the POU3F4 gene. The POU family of genes are a family of eukaryotic transcription factors regulating neuroendocrine function of which the POU3F4 gene is a key factor for mesenchymal integrity, spiral ganglion functioning, spiral ligament structure and stria vascularis activities [5]. Mutations in this gene lead to an otological phenotype. The gene was mapped to chromosome Xq13-q21.1 in 1988 [6]. The mutation was designated DFNX2 by gene mapping and is the second in a group of five X-linked non syndromic genetic deafness group [7].
X-linked gusher disorder in its most severe form with severe to profound hearing loss can be progressive always occurring in males given its mode of inheritance. However, females may be obligatory carriers presenting with a milder phenotype [3,8]. There had been one case report where typical X-linked gusher associated radiological findings were detected in a female suggesting a separate mode of inheritance yet to be identified [9]. X-linked gusher DFNX2 is the commonest of the 5 X-linked non syndromic hearing loss group comprising about 50% [10]. The X-linked group cumulatively constitutes about 1-5% of non syndromic hearing loss [10]. DFNX2 is very rare [11] and true prevalence is unknown. Given that 1 in 1100 live births will show a congenital bilateral permanent hearing loss of which 80% are non-syndromic genetic and X-linked recessive hearing losses account for only 5% of this non syndromic group of which 50% is DFNX2, the rough prevalence will be 0.0045 per 1100 live births. The condition is defined by the typical cochlear dysplasia [12].
The hearing loss encountered is heterogenous ranging from conductive to sensory cochlear hearing loss, but the mixed variety is the commonest [4,13]. The cochlear hearing loss can be intuitively inferred by the cochlear dysplasia. The conductive component has been postulated to be due to a third window effect that essentially arises due to a direct communication between the subarachnoid space in the cranial cavity and the perilymphatic space in the inner ear due to the incomplete cochlear partition and absent modiolus at the fundus of the IAM [14,15]. In a series of rare third window abnormalities in children, DFNX2 accounted for about a fifth of all rare third window disorders [11]. Vestibular function quantified with objective vestibulometry had been reported only once [11].
In this study we present two siblings who were diagnosed with the condition and underwent full behavioural/objective audiological and objective vestibular quantification. These siblings were reported earlier as part of a cohort [11] with summarized phenotypes. This paper describes the clinical features, clinical/objective audiovestibular quantification and treatment outcomes of these siblings over a period of time. The paper highlights the importance of follow ups and a vestibular phenotype in a rare disorder of the inner ear generating a conductive hearing loss. To our knowledge, a vestibular phenotype with quantification of vestibular function that includes the suppression head impulse test and long-term management outcomes have not been studied in X-linked gusher disorder in children that we are reporting in this paper.
2. Methods
2 children out of a total of 53 third window disorders in a tertiary paediatric vestibular centre in the UK over a period of 3.5 years were diagnosed with X-linked gusher disorder comprising of 3.7% of all third window disorders. Both children were diagnosed with detailed anamnesis, exclusion of other conditions and imaging studies (HRCT/ T2 drive MRI). Both would show mutations in the POU3F4 gene.
Pure tone audiometry, tympanometry, acoustic reflexes, transient otoacoustic emissions were performed to quantify hearing. Videonystagmography (VNG) with and without optic fixation (nystagmus, smooth pursuits and saccades, head shake, head heave, ocular counter roll and the mastoid vibration test), vestibulospinal test battery (VST) with and without foam cushion (Romberg, Unterberger and tandem gait), video head impulse test (vHIT), suppression head impulse test (SHIMP), cervical vestibular evoked myogenic potential test (cVEMP) and measurement of the subjective visual vertical (SVV) were utilized to quantify the vestibular system. Our own laboratory norms were used when analyzing vHIT, SHIMP and cVEMP results (our vHIT VOR gains range from 0.9 to 1 in horizontal canals; 0.6-0.8 in vertical canals and our VEMP laboratory norms include asymmetry up to 26% and thresholds of 80 ≤ dBnHL). The first author performed all vestibular tests for consistency. We did not perform ocular vestibular evoked myogenic potentials (oVEMP) as paediatric norms are hardly available and we are gathering our own norms. These tests were repeated several times as part of audiovestibular surveillance. All children also underwent neurological, paediatric, cardiological and development assessment as mandated in investigating children with suspected vestibular disorders. The families, the sensory services, the school and significant others were all involved in the journey of the children effectively.
Informed signed consent was obtained from both children and their legal guardian for reporting their conditions to scientific journals.
3. Results
Sibling A (SA):
This child did not undergo the universal newborn hearing screening as it was in its infancy in the country of study. He was referred due to delayed speech and motor development at the age of 27 months and a severe mixed hearing loss was observed in both ears. His tympanometry was bilaterally normal as was stapedial reflex on the right only and his transient otoacoustic emissions were absent bilaterally. He was fitted immediately with digital amplification and monitored twice every year. The hearing loss progressed over the next years especially on the left ear becoming profound (Table 1, Figure 1) that necessitated a cochlear implant on that side after 7 years. The right side also showed deterioration, but he refused an implant as he was deriving good benefits from the hearing aid on that side. His speech and communication abilities further improved with speech and language therapy. He went to a mainstream school and did well with his academia and a developed a good positive insight for the future and looking forward to university life.
HRCT and T2 drive MRI demonstrated bulbous end of IAM with dilatation, absent cochlear modiolus with intact septa, incomplete cochlear partition type III and incomplete IAM fundus gene (Figure 2) that confirmed the diagnosis of an X-linked gusher disorder. He would then show the POU3F4 mutation by genetic typing.
HRCT (A) and T2 Drive MRI (B) in the siblings (SA and SB) showing bilateral bulbous dilatation involving the fundus of the internal auditory canal (arrow) and bilateral incomplete separation of the basal turn of the cochlea (arrow head) from the fundus of the internal acoustic canal (dotted arrow) classical of X-linked gusher disorder
He perceived balance problems and was apprehensive in playground activities when he was a toddler. He was fond of dancing but was unable to indulge in twists/turns that dancing demanded. He was diagnosed with bilateral vestibular dysfunction 12 years after diagnosis showing an initial six semicircular canal involvement in the vHIT, absent cVEMP on the left with low thresholds on the right, abnormal VST but normal SHIMP, SVV and VNG (Table 2 and Table 3 and Figure 3 and Figure 4). Vestibular rehabilitation was provided. His vestibular function improved over the years subjectively and objectively in the vHIT to the point of resuming his dancing and overcoming his apprehension with balance.
Sibling B (SB)
Sibling B failed his newborn hearing screening and showed a moderate mixed hearing loss on both sides. He was fitted at the age of 6 weeks and was regularly monitored. There was a mild speech delay and he received speech and language therapy. His hearing loss also showed progression and fluctuation and became severe over the years (Table 4 and Figure 5). His tympanometry was normal as were his stapedial reflexes but transient otoacoustic emissions were absent on both sides. His amplification was continuously revised, and his speech became age appropriate as were his communication skills. He too went to mainstream school and doing quite well with his academia. He has a good positive insight for the future and wants to pursue a career in audiology as he believes that our input from such an early period has changed his life.
HRCT and T2 drive MRI was identical to his brother (Figure 6) confirming the diagnosis of an X-linked gusher disorder. He would also show the POU3F4 mutation by genetic typing.
His motor skills were delayed and like his brother, he was also apprehensive about playground activities. He was diagnosed with bilateral vestibular weakness 10 years after diagnosis (Table 5 and Table 6 and Figure 7 and Figure 8) involving the lateral semicircular canals on the vHIT, reduced amplitudes and lowered thresholds in the cVEMP, abnormal VST but normal SHIMP, SVV and VNG. He received vestibular rehabilitation. He did not complain of any balance problems since then and has good balance function confirmed by latest/recent objective tests.
4. Discussion
X-linked gusher disorder is diagnosed by a characteristic phenotype of congenital mixed hearing loss with normal middle ear studies and by a pathognomonic HRCT demonstratable cochlear dysplasia. The hearing loss is present at birth and may progress over a period of time. The disorder is due to a mutation in the POU3F4 gene with 51 different loci identified [16]. Our siblings fulfilled the criteria to satisfy the diagnosis of an X-linked gusher disorder.
The typical feature of an X-linked gusher disorder in HRCT is an incomplete cochlear partition type 3. The Brn 4 gene in the animal model later called the POU3F4 participates in the remodelling of the otic capsule [17]. A breach in this remodelling leads to the incomplete cochlear partition. There are 3 types of incomplete cochlear partition [18]. Although the 3 different varieties resemble each other in terms of an incomplete partition and that the cochlea is separate from the vestibule, the essential difference between the 3 is in the way the modiolus and the IAM fundus are formed. In type I, the modiolus is completely absent with its interscalar septa, in type II, only the apical modiolus is absent with its septa whilst in type III, the modiolus is absent but the septa are present. Type II is also associated with dilated vestibular aqueduct18. In X-linked gusher disorder, the type III is uniquely observed in addition to a dilated IAM and an incomplete IAM fundus/cochlea partition, i.e. bony deformity of the fundus [13] that makes it pathognomonic of the condition. The siblings in this series exhibit these classical features.
Other cochlear structural abnormalities defined in X-linked gusher disorder are bulbous fundal end of the IAM, thin otic capsule, enlarged superior vestibular nerve canal, enlarged labyrinthine facial nerve canal, enlarged singular nerve canal, vestibule with cystic appearance, semicircular canal with cystic appearance, dilated vestibular aqueduct, dysplasia of oval window, dysplasia of round window and abnormal stapes [12]. The most consistent features are dilated internal auditory meatus, incomplete separation of the basal turn of the cochleae from the fundi of the IAC that includes defects in the lamina cribrosa, bulbous fundal end of the IAM and a thin otic capsule. It should be noted that these consistent abnormalities are absent in cochlear partition I and II (except a dilated vestibular aqueduct in type II) and that explains the third window effect in X-linked gusher disorder discussed below. Unlike HRCT, MRI observations in the condition have hardly been reported. T2 MRI in X-linked gusher disorder shows a bulbous internal auditory canal, an incomplete separation of the basal turn of the cochlea with the fundus and a fluid filled cochlear cavity without the modiolus or the spiral lamina [19]. An MRI also may show a hypothalamic lesion that has been reported in the condition [20]. Our siblings showed these consistent features in their HRCT and MRI to establish a radiological diagnosis and did not show any brain abnormality.
An intriguing and typical phenotypical feature observed in X-linked gusher disorder is a conductive element to the hearing loss that it presents with which was universally observed in all case reports [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,18,19,20,21,22]. Conductive hearing loss not originating from the external or the middle ear can be due to the third window phenomenon that is generated by an inner ear group of disorders called third window disorders. Third window disorders are structural abnormalities in the bony otic capsule that establish a connection between the middle/inner ear or the inner ear/cranial cavity [11]. The Minor group in 1998 identified the first third window prototype superior semicircular canal dehiscence [23] and this has since then seen intense research, criteria elaborated and several inner ear disorders defined [11]. However, Snik et al in 1995 [21] whilst investigating an air bone gap in X-linked gusher disorder believed that a physiologically normal third window exists that is comprised of the cochleovestibular aqueducts, vascular and neural channels and the otic capsule that contribute to bone conduction and stimulation of the cochlear travelling wave. They correctly postulated that a pathological third window effect may be created if there are abnormalities in these structures that in X-linked gusher include a dilated internal auditory meatus, the incomplete partition type III and an incomplete bony fundus of the IAM. These three structural deficits lead to an abnormal communication between the subarachnoid space (cranial cavity) and the perilymphatic space (inner ear) that defines a third window.
The third window effect was later detailed by Merchant et al in 2008 [24]. When sound waves travel through the middle ear, part of the energy is shunted by the pathological third window leading to a drop in air conduction thresholds. The cochlear travelling wave set up from the stapes footplate/oval window interface instead of travelling from the oval to the round window, now travels from an oval window to the third window that offers less resistance. The net result is a lowering of bone conduction thresholds and the appearance of an air bone gap. Anatomical third windows sparing the cochlea for example the bony canal dehiscence group do not affect cochlear function, however, third windows with cochlear dysplasia affect cochlear function [11]. Therefore, in such an instance, there will be an added cochlear element to the third window conductive element generating the typical mixed pattern as in DFNX2. As a result of the third window, the cochlear element also shows fluctuation when measured with bone conduction as observed in our siblings. In these subjects, there may be absent otoacoustic emissions reflecting the sensory hearing loss but usually at least one sided stapedial reflex is preserved unless profound sensory hearing loss with intact tympanometry [25]. Both the siblings in this series demonstrated mixed hearing loss, absent otoacoustic emissions and stapedial reflexes were bilaterally preserved except in one sibling where stapedial reflex was present only in one ear.
Other typical third window symptoms are conductive dysacusis, autophony, Hennebert’s and Tullio’s phenomenon, disequilibrium, dizziness/vertigo, tinnitus/pulsatile tinnitus, misophonia and gaze induced tinnitus [11,14]. It is important to note that these reported symptoms may be difficult to glean from children and may be different in children. This is likely due to the effect of the third window on a developing vestibular system and a developing skull that contains the bony otic capsule where the dysplasia is located [26]. In addition, for a child, its cognitive reaction, emotional reaction, effect on schoolwork and social life and on overall development should be factored in. The children in this series did not complain of any sound induced audiovestibular symptoms even when older except autophony. They both had delayed speech and motor development and struggled in their playground activities as toddlers. Once they were made aware of the pathology, they reconciled to their conditions very well to do well in life and develop a positive insight for the future. The observation that characteristic third window symptoms may be absent in children suggest that the possibility of a third window in a child is not just based on anamnesis but rather on a more holistic and an emerging clinical scenario. Indeed, this holistic approach is quite essential to investigate and then confirm a third window for effective management and avoids missing the diagnosis.
The mixed hearing loss encountered in X-linked gusher disorder is progressive [13,22]. This is likely due to the continuous third window effect on the growing cochlear deformity that may lead to a heightened cochlear deficit. The siblings in this series both presented with progressive hearing loss. They also presented with abnormal audiological behaviour from a very early age, i.e. deficiencies in hearing as observed by their carers and a subjective hearing loss when they were older to report this. The hearing loss does affect speech perception and acquisition unless treated very early to maximize speech development that we observed in our series. Both the siblings acquired excellent speech because of this early intervention.
The entire publication spectrum regarding X-linked gusher disorder has investigated the hearing loss and the radiological phenotype in some detail but all but one made no mention of a vestibular phenotype and neither had these studies performed any objective vestibular quantification in this condition. The first author reported vestibular findings in their study on rare third window disorders in children with the two cases reported in this study but did not elaborate on evolution of vestibular function or a SHIMP test [11]. The siblings reported in the present study were followed up for over 10 years and vestibular function was objectively quantified along with detailed anamnesis to assess compensated vestibular function. This makes this study quite unique and suggests that the vestibular phenotype should not be overlooked. The authors believe that a vestibular deficit will be due to the chronic third window effect that is transmitted across the membranous labyrinth due to the abnormal inner ear-intracranial cavity connection in the absence of a structural anatomical defect in vestibular anatomy that has also been proposed elsewhere [27]. VNG examination that entailed eye movements which were spontaneous or provoked with removal of optic fixation were normal in both children; however, the VST battery eliminating visual fixation and proprioception were abnormal in both children.
The hallmark of diagnosing a third window is a characteristic vestibular evoked myogenic test (VEMP) that when cervical (cVEMP) assesses the saccule and when ocular assesses the utricle (oVEMP). Both these tests show elevated amplitudes and lowered thresholds in mobile third windows due to hyperstimulation of the otolith sensors as a result of the third window effect on the acoustic energy transmission [26]. Studies are limited to investigate VEMPs in third window disorders in children [28,29,30,31]. In children VEMPs can be quite heterogenous and depend on the level of involvement of the utricle and the saccule. A damaged or a weak otolith sensor will still be stimulated at the VEMP stimulus but might not return the same amplitude as a normal sensor will. As suggested earlier, due to the chronic third window effect in X-linked gusher disorder on the vestibular system because of the dysplastic cochlea, the vestibular organs might be inherently weak which is a distinct probability. Consequently, it is probable that VEMPS in children might not show the typical hyperstimulation parameters that is well established in diagnosis of adult third windows and may be absent altogether especially in this condition [11]. In the current series, cVEMP findings were varied with one child showing normal amplitude on one side and absent amplitude on the other whilst the other showed significantly reduced amplitudes. The thresholds when measured in the ears that did return an amplitude were lower than normal as per our laboratory norms. This reduction of amplitudes can be considered as indicative of reduced saccular function [32]. However, reduced thresholds may indicate a third window in spite of reduced amplitudes. It is suggested that thresholds are probably better indicators of diagnosing third windows across all age groups [26]. Since studies regarding VEMP norms in children are rather limited, we recommend that individual laboratory-based norms are established to make informed inferences that were considered in the present study noting that they might be quite different from published adult norms [11].
The video head impulse test (vHIT) had revolutionized vestibular diagnostics in recent times [33]. Normative data may be different from those obtained in adults and like VEMPs individual laboratory norms must be established [34]. Low vestibulo-ocular reflex (VOR) gain along with overt and covert saccades indicate vestibular semicircular canal high frequency weakness in all six semicircular canals [33], however, saccades alone are also important to consider [35] and saccades with normal VOR gain have also been reported in vestibular weakness [36,37]. vHIT studies are limited in third windows and especially in X-linked gusher disorder and it has been shown that they can be deranged [27,38]. One sibling in this study had saccades on all canals with low VOR gain in the vertical canals to start with and the other sibling showed normal gains but with saccades on the lateral canals only in the initial testing. Recovery of VOR gain and reduction and clustering/dispersion of saccades are features of vestibular compensation indicating that vestibular function may recover over a given period of time [39]. Both showed recovery when tested three years later after active intervention following the recovery pattern. This recovery of function was also indicated by a subjective corroboration of adequate balance by the child.
The suppression head impulse test (SHIMP) is a new test in the vestibular test battery that is useful for assessing vestibular compensation and is used as an adjunct to the vHIT [40]. This has been hardly studied in children [41]. This paper is reporting SHIMP results in X-linked disorder for the first time. Our children in the series demonstrated normal SHIMPs with accepted laboratory-based norms in the second assessment with normal peak saccadic velocities and asymmetry suggesting good compensation.
We followed these children up for a period of seventeen years and the older child has been transitioned to the adult services formally, the younger one is still under follow up. They were fitted early with hearing aids. They underwent regular monitoring of hearing loss and digital amplification adjusted accordingly given the progressive nature of their hearing losses. The older sibling was implanted later on one ear and did not want a second implant whilst the younger one is doing very well with bilateral hearing aids. The outcome was development of normal speech and communication skills that is reported after early intervention for hearing loss in a child [42]. However, it is equally important to engage parents and significant others in the management algorithm that is not standardized as yet [43]. This family centred engagement is crucial to maximize outcome from intervention. The families of the siblings in the current series were engaged in every step of their entire journey time as were their school and facilities for their extracurricular activities. In addition, sensory services were involved for real world and classroom support. As they grew older, they were counselled cognitively by the medical team to reconcile, accept and anticipate their futures with their condition. Both children achieved their creative capabilities and are doing well in their academia. The authors therefore stress and emphasize that diagnosing and managing a complex disorder like X-linked gusher disorder is way beyond fitting an implant or a hearing aid and is significantly multidisciplinary and holistic for maximizing a positive outcome.
Similarly, a vestibular weakness is required to be addressed early. Both the siblings underwent a course of vestibular rehabilitation and recovered well with their balance. They received situational counselling as to how to avoid provocation that could lead to vestibular decompensation. They could indulge in playground activities and one child resumed his hobby of dancing.
The limitation of this study is its small number that of course leaves questions for generalization. However, we believe that these two cases provide a rather important insight to this very complex disorder. It might be difficult to launch dedicated controlled and blind studies given the rarity of the condition and thus, snapshots like this yield valuable information. We recommend scrupulous follow ups and a holistic and cognitive approach to manage children with X-linked gusher disorder.
5. Conclusions
X-linked gusher disorder is a rare genetic disorder in children that manifests with an audiovestibular phenotype. The hearing loss is usually a mixed one due to a third window effect as a result of characteristic bony deformities. Vestibular function hitherto unreported may be weak and can be quantified by objective vestibular tests. The condition is progressive and evolves over a period of time where hearing loss progresses, and vestibular weakness undergoes compensation rendering children asymptomatic. Early audiovestibular intervention is pivotal with multidisciplinary engagement in a holistic way involving significant others in the care. This achieves maximal favourable outcome underpinning the importance of regular follow ups of these children.
Author Contributions
Conceptualization, SDG.; methodology, SDG, RM, JH, JS.; software, SDG, AL, JI.; validation, SDG., AL, JS and JI.; formal analysis, SDG, RM, JI, AL, JH, JS.; investigation, SDG, RM, JH, AL, JI, JS.; resources, SDG.; data curation, SDG, AL, JI, RM, JH, JS.; writing—original draft preparation, SDG.; writing—review and editing, SDG, AL, JI, RM, JI, JS.; visualization, SDG.; supervision, SDG.; project administration, SDG.; funding acquisition, SDG. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Verbal/Written informed consent was obtained from all participants and their mother who participated in this study.
Data Availability Statement
All data presented in article.
Acknowledgments
This study was supported by Division of Surgery, Alder Hey Children’s NHS Foundation Trust, UK.
Conflicts of Interest
None declared.
References
- Nance WE, SetleffR, McLedd A, SweeneyA, Cooper C, McDonnell F. X-linked deafness with congenital fixation of the stapedial footplate and perilymphatic gusher. Birth Defects. 1971; 7:64-69.
- Phleps PD, Reardon W, Pembrey M, et al. X-linked deafness, stapes gushers, and a distinctive defect of the inner ear. Neuroradiology. 1991;33:326–330.
- Talbot JM, Wilson DF. Computed tomographic diagnosis of X-linked congenital mixed deafness, fixation of the stapedial footplate, and perilymphatic gusher. Am J Otol .1994;15:177–182.
- de Kok YJ, van der Maarel SM, Bitner-Glindzicz. et al. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science. 1995 Feb 3;267(5198):685-8. [CrossRef]
- Tekin AM, Matulic M, Wuyts W. et al. A New Pathogenic Variant in POU3F4 Causing Deafness Due to an Incomplete Partition of the Cochlea Paved the Way for Innovative Surgery. Genes (Basel). 2021 Apr 21;12(5):613. [CrossRef]
- Wallis C, Ballo R, Wallis G. et al. X-linked mixed deafness with stapes fixation in a Mauritian kindred: linkage to Xq probe pDP34. Genomics. 1988 Nov;3(4):299-301. [CrossRef]
- Van Camp G, Smith RJH. Hereditary Hearing Loss Homepage. https://hereditaryhearingloss.org. June 2023. 20 June.
- Saylisoy S, Incesulu A, Gurbuz MK et al. Computed tomographic findings of X-linked deafness: a spectrum from child to mother, from young to old, from boy to girl, from mixed to sudden hearing loss. J Comput Assist Tomogr. 2014; Jan-Feb;38(1):20-4. [CrossRef]
- Papadaki E, Prassopoulos P, Bizakis J. et al. X-linked deafness with stapes gusher in females. Eur J Radiol. 1998 Nov;29(1):71-5. [CrossRef]
- Petersen MB, Wang Q, Willems PJ. Sex-linked deafness. Clin Genet. 2008; 73:14–23. [CrossRef]
- Dasgupta S, Ratnayake S, Crunkhorn R. et al. Audiovestibular Quantification in Rare Third Window Disorders in Children. Front Neurol. 2020 Sep 16;11:954. [CrossRef]
- Hong R, Du Q, Pan Y. New Imaging Findings of Incomplete Partition Type III Inner Ear Malformation and Literature Review. AJNR Am J Neuroradiol. 2020 Jun;41(6):1076-1080. [CrossRef]
- Kumar S, Mawby T, Sivapathasingam V, Humphries J, Ramsden J. X-linked deafness: A review of clinical and radiological findings and current management strategies. World J Otorhinolaryngol 2016; 6(1): 19-22. [CrossRef]
- Ho ML, Moonis G, Halpin CF. et al. Spectrum of Third Window Abnormalities: Semicircular Canal Dehiscence and Beyond. AJNR Am J Neuroradiol. 2017 Jan;38(1):2-9. [CrossRef]
- Scarpa A, Ralli M, Cassandro C. et al. Inner-Ear Disorders Presenting with Air-Bone Gaps: A Review. J Int Adv Otol. 2020 Apr;16(1):111-116. [CrossRef]
- Du W, Han MK, Wang DY. et al. A POU3F4 Mutation Causes Nonsyndromic Hearing Loss in a Chinese X-linked Recessive Family. Chin Med J (Engl). 2017 Jan 5;130(1):88-92. [CrossRef]
- Phippard D, Heydemann A, Lechner M. et al). Changes in the subcellular localization of the Brn4 gene product precede mesenchymal remodeling of the otic capsule. Hear Res. 1998;120:77-85. [CrossRef]
- Sennaroğlu L, Bajin MD. Classification and Current Management of Inner Ear Malformations. Balkan Med J. 2017 Sep 29;34(5):397-411. [CrossRef]
- W.X. Gong, R.Z. Gong, B. Zhao HRCT and MRI findings in X-linked non-syndromic deafness patients with a POU3F4 mutation. Int. J. Pediatr. Otorhinolaryngol. 2014; 78 (10): pp. 1756-1762.
- Prat Matifoll JA, Wilson M, Goetti R. et al. A Case Series of X-Linked Deafness-2 with Sensorineural Hearing Loss, Stapes Fixation, and Perilymphatic Gusher: MR Imaging and Clinical Features of Hypothalamic Malformations. AJNR Am J Neuroradiol. 2020 Jun;41(6):1087-1093. [CrossRef]
- Snik AF, Hombergen GC, Mylanus EA. et al.. Air-bone gap in patients with X-linked stapes gusher syndrome. Am J Otol. 1995 Mar;16(2):241-6.
- Tang A, Parnes LS. X-linked progressive mixed hearing loss: computed tomography findings. Ann Otol Rhinol Laryngol. 1994;103:655–657.
- Minor L B, Solomon D, Zinreich J S. et al. Sound- and/or pressure-induced vertigo due to bone dehiscence of the superior semicircular canal. Arch Otolaryngol Head Neck Surg. 1998;124(03):249–258.
- Merchant SN, Rosowski JJ. Conductive hearing loss caused by third-window lesions of the inner ear. Otol Neurotol. 2008 Apr;29(3):282-9. [CrossRef]
- Hong RS, Metz CM, Bojrab DI. et al. Acoustic Reflex Screening of Conductive Hearing Loss for Third Window Disorders. Otolaryngol Head Neck Surg. 2016 Feb;154(2):343-8. [CrossRef]
- Wackym PA, Agrawal Y, Ikezono T. et al. Editorial: Third Window Syndrome. Front Neurol. 2021 Jun 18;12:704095. [CrossRef]
- Tikka T, Slim MAM, Gaggini M. et al. Video Head Impulse Test (vHIT) Findings in Patients With Superior Semicircular Canal Dehiscence: A Case-Control Study. J Int Adv Otol. 2021 Mar;17(2):103-108. [CrossRef]
- Wackym PA, Balaban C, Zhang P. et al. Third window syndrome: surgical management of cochlea-facial nerve dehiscence. Front Neurol. 2019; 10:1281. [CrossRef]
- Bulck PV, Leupe PJ, Forton GEJ. Children with posterior semicircular canal dehiscence: a case series. Int J Pediatric Otorhinolaryngol. 2019; 123:51–6. [CrossRef]
- Zhou G, Gopen Q. Characteristics of vestibular evoked myogenic potentials in children with enlarged vestibular aqueduct. Laryngoscope. 2011; 121:220–5. [CrossRef]
- Zhou G, Gopen Q, Poe DS. Clinical and diagnostic characterisation of canal dehiscence syndrome: a great otologic mimicker. Otol Neurotol. 2007; 28:920–26. [CrossRef]
- Rosengren SM, Colebatch JG, Young AS. et al. Vestibular evoked myogenic potentials in practice: Methods, pitfalls and clinical applications. Clinical Neurophysiology Practice. 2019;Volume 4, Pages 47-6. [CrossRef]
- Halmyagi GM, Chen L, MacDougall HG. et al. The video head impulse test. Front Neurol. 2017; 8:258. [CrossRef]
- Bachmann K, Sipos K, Lavender K. et al. Video head impulse testing in a pediatric population: normative findings. J Am Acad Audiol. 2018; 29:417–26. [CrossRef]
- Curthoys IS Manzari L Clinical application of the head impulse test of semicircular canal function. Hear Balance Commun. 2017; 15:113–26. [CrossRef]
- Perez-Fernandez N, Eza-Nunez P. Normal gain of VOR with refixation saccades in patients with unilateral vestibulopathy. J Int Adv Otol. 2015; 11:133–7. [CrossRef]
- Korsager LE, Faber CE, Schmidt JH. et al. Refixation saccades with normal gain values: a diagnostic problemin the video head impulse test: a case report. Front Neurol. 2017; 8:81. [CrossRef]
- Dasgupta S, Ratnayake SAB. Functional and objective audiovestibular evaluation of children with apparent semicircular canal dehiscence–a case series in a pediatric vestibular center. Front Neurol. 2019; 10:306. [CrossRef]
- Fu W, He F, Wei D. et al. Recovery Pattern of High-Frequency Acceleration Vestibulo-Ocular Reflex in Unilateral Vestibular Neuritis: A Preliminary Study. Front Neurol. 2019 Mar 7;10:85. [CrossRef]
- MacDougall H.G., McGarvie L.A., Halmagyi G.M. et al. Weber K.P. A new saccadic indicator of peripheral vestibular function based on the video head impulse test. Neurology. 2016;87:410–418. [CrossRef]
- Nguyen J, Berger J, Curthoys I. et al. Vestibular testing in children - The suppression head impulse (SHIMP) test. Int J Pediatr Otorhinolaryngol. 2021; Dec;151:110921. [CrossRef]
- Christine Yoshinaga-Itano, Principles and Guidelines for Early Intervention After Confirmation That a Child Is Deaf or Hard of Hearing, The Journal of Deaf Studies and Deaf Education. 2014;Volume 19, Issue 2 : Pages 143–175. [CrossRef]
- Wright, B., Hargate, R., Garside, M. et al. A systematic scoping review of early interventions for parents of deaf infants. BMC Pediatr. 2021; 467 (2021). [CrossRef]
Figure 1.
SA - Serial pure tone audiometry.
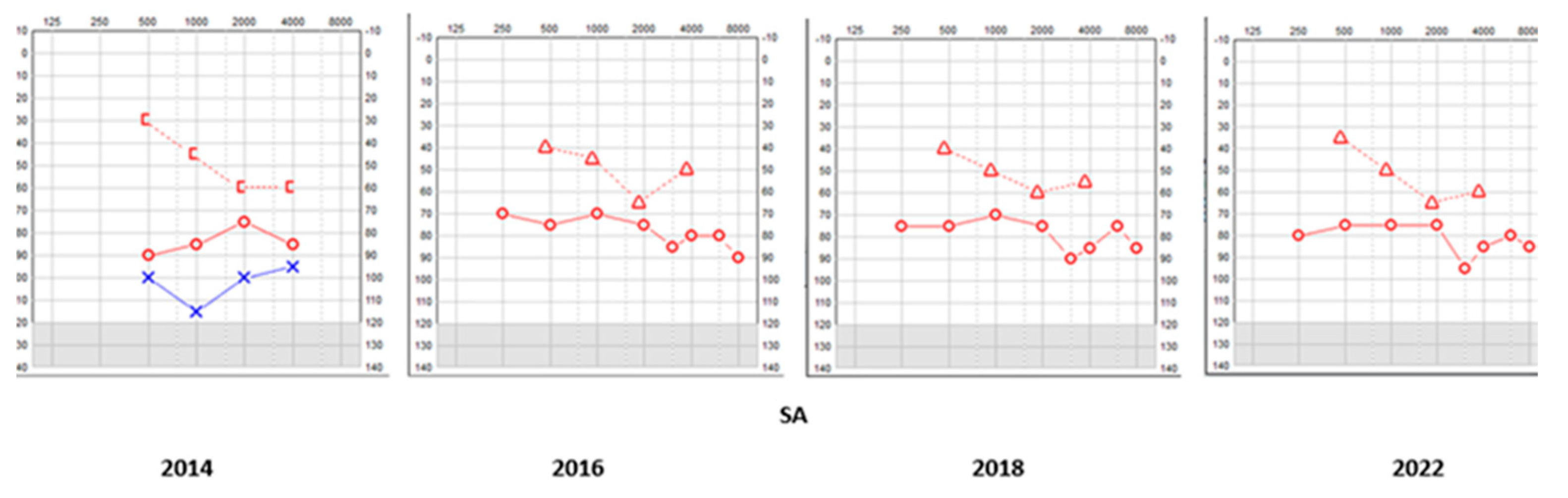
Figure 2.
SA HRCT and MRI
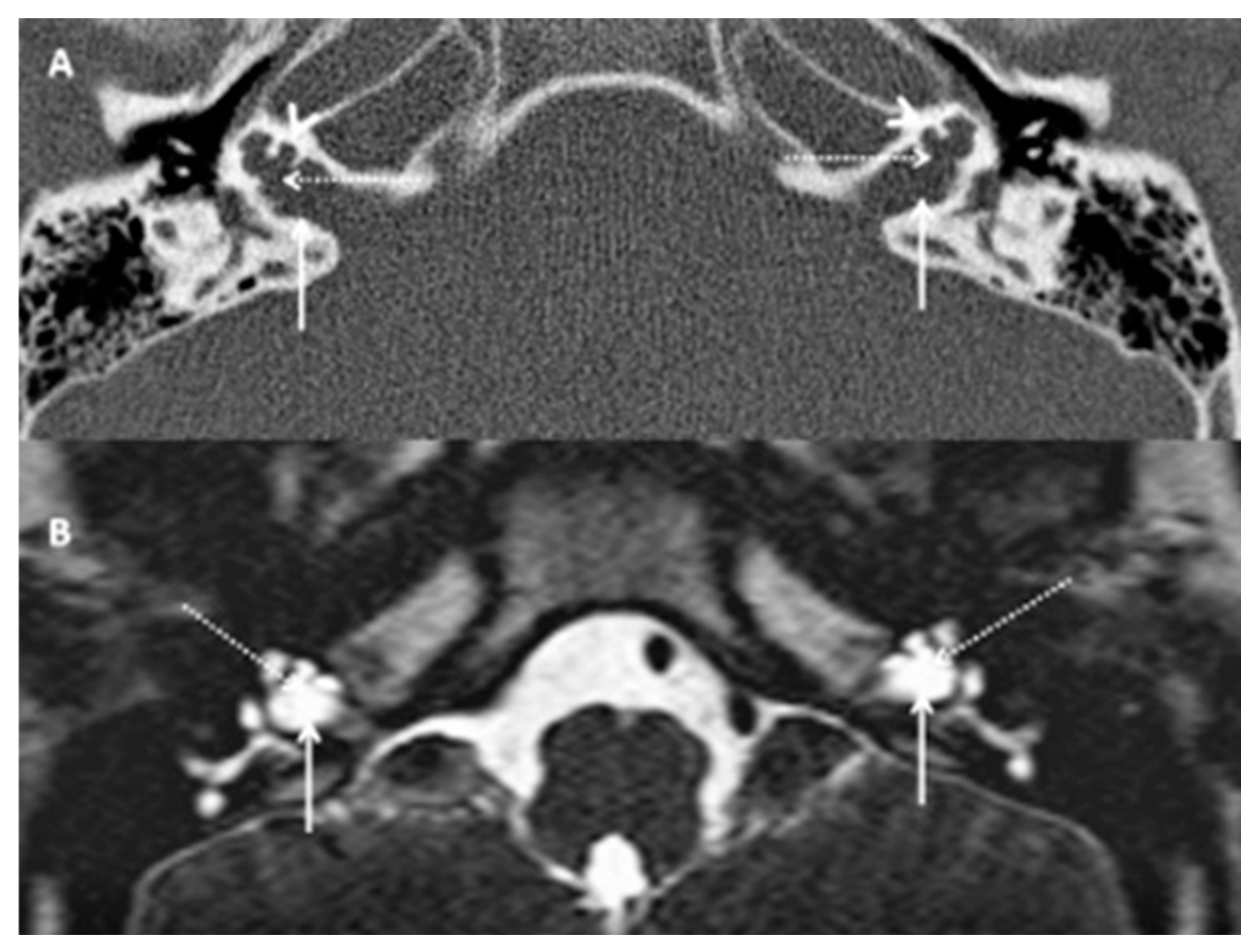
Figure 3.
SA - cVEMP cVEMP 2019, absent response left and low threshold on right
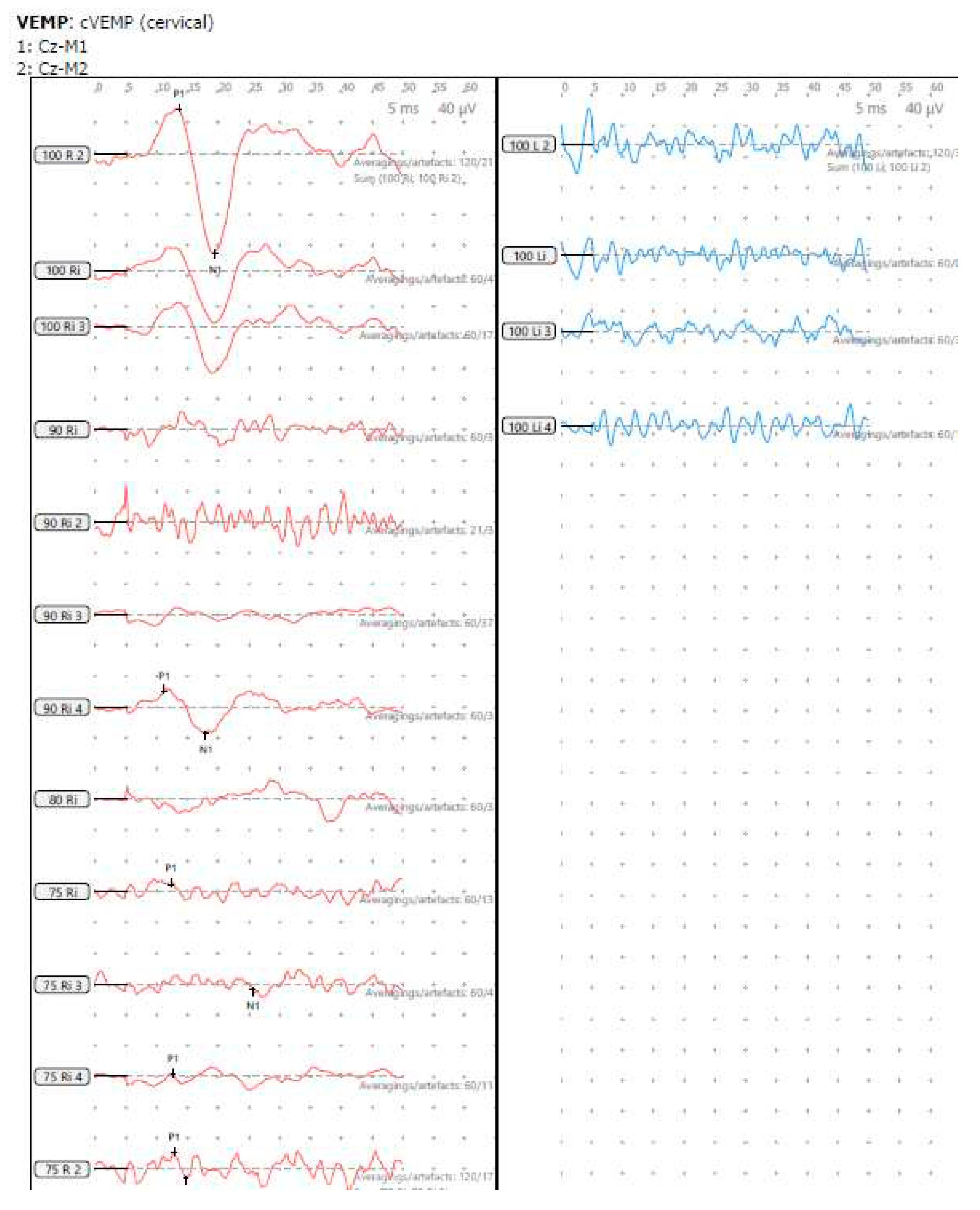
Figure 4.
SA - vHIT and SHIMP. A – vHIT 2016, note low VOR gain and catch-up saccades (arrow); B – vHIT 2019, note recovery of VOR gain and absence and reduction of saccades (arrow); C – SHIMP 2019, normal asymmetry and peak saccadic velocities
Figure 4.
SA - vHIT and SHIMP. A – vHIT 2016, note low VOR gain and catch-up saccades (arrow); B – vHIT 2019, note recovery of VOR gain and absence and reduction of saccades (arrow); C – SHIMP 2019, normal asymmetry and peak saccadic velocities
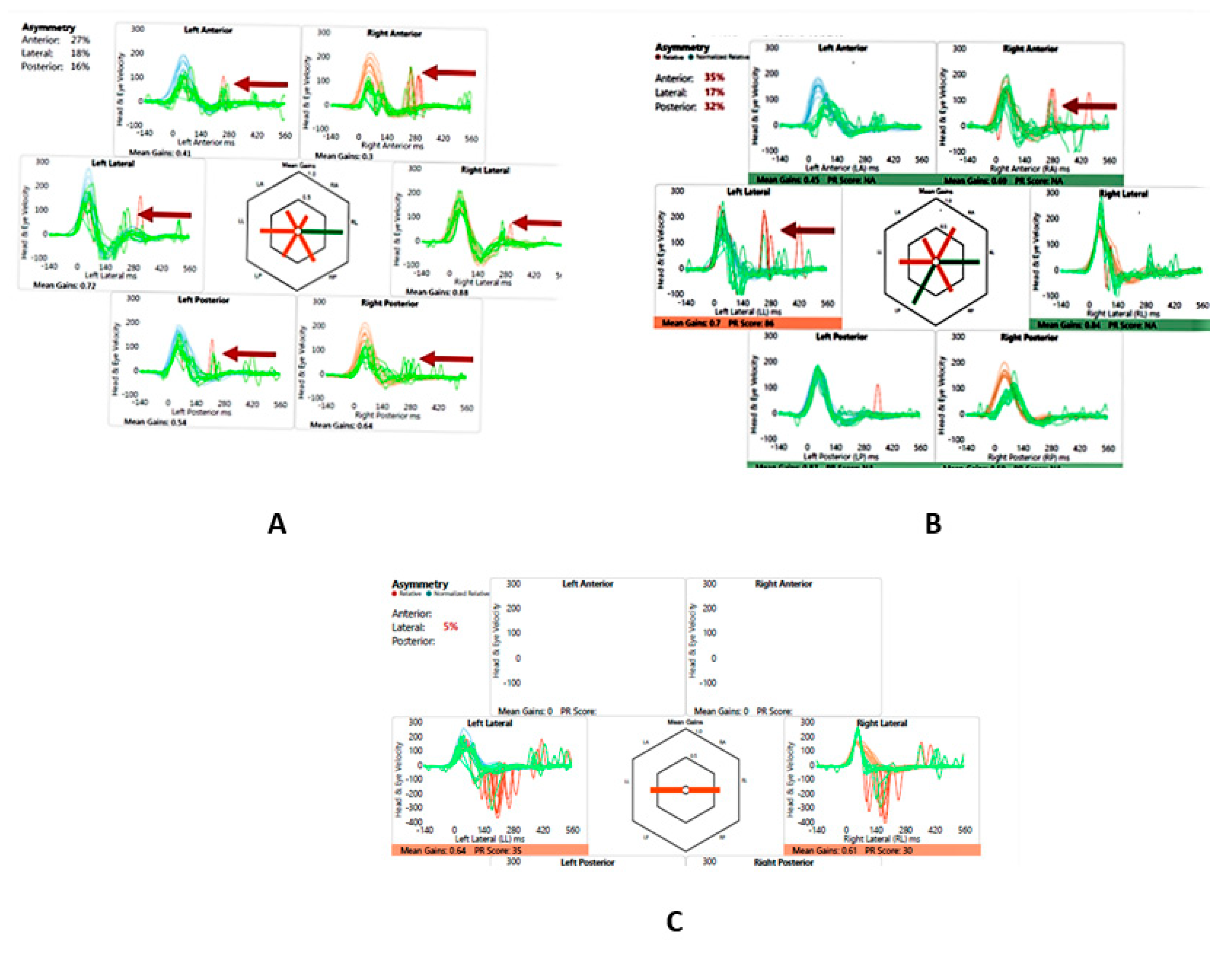
Figure 5.
SB - serial pure tone audiometry
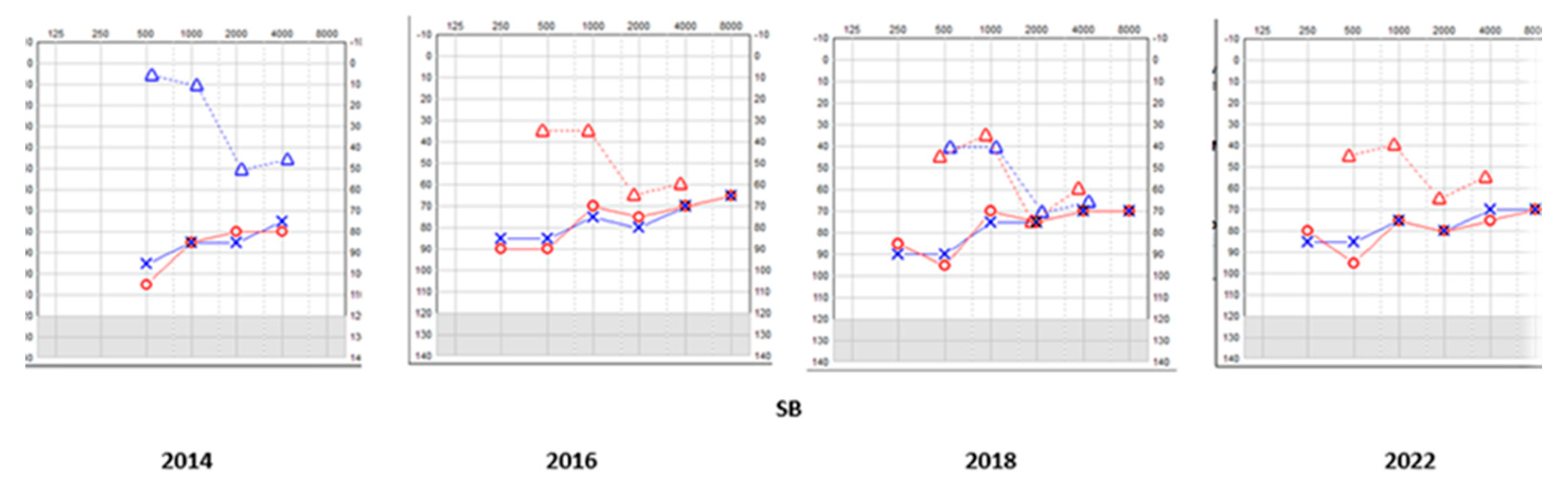
Figure 6.
SB HTCT and MRI. HRCT (A) and T2 Drive MRI (B) showing bilateral bulbous dilatation involving the fundus of the internal auditory canal (arrow) and bilateral incomplete separation of the basal turn of the cochlea (arrow head) from the fundus of the internal acoustic canal (dotted arrow) classical of X-linked gusher disorder and similar to SA in Figure 2
Figure 6.
SB HTCT and MRI. HRCT (A) and T2 Drive MRI (B) showing bilateral bulbous dilatation involving the fundus of the internal auditory canal (arrow) and bilateral incomplete separation of the basal turn of the cochlea (arrow head) from the fundus of the internal acoustic canal (dotted arrow) classical of X-linked gusher disorder and similar to SA in Figure 2
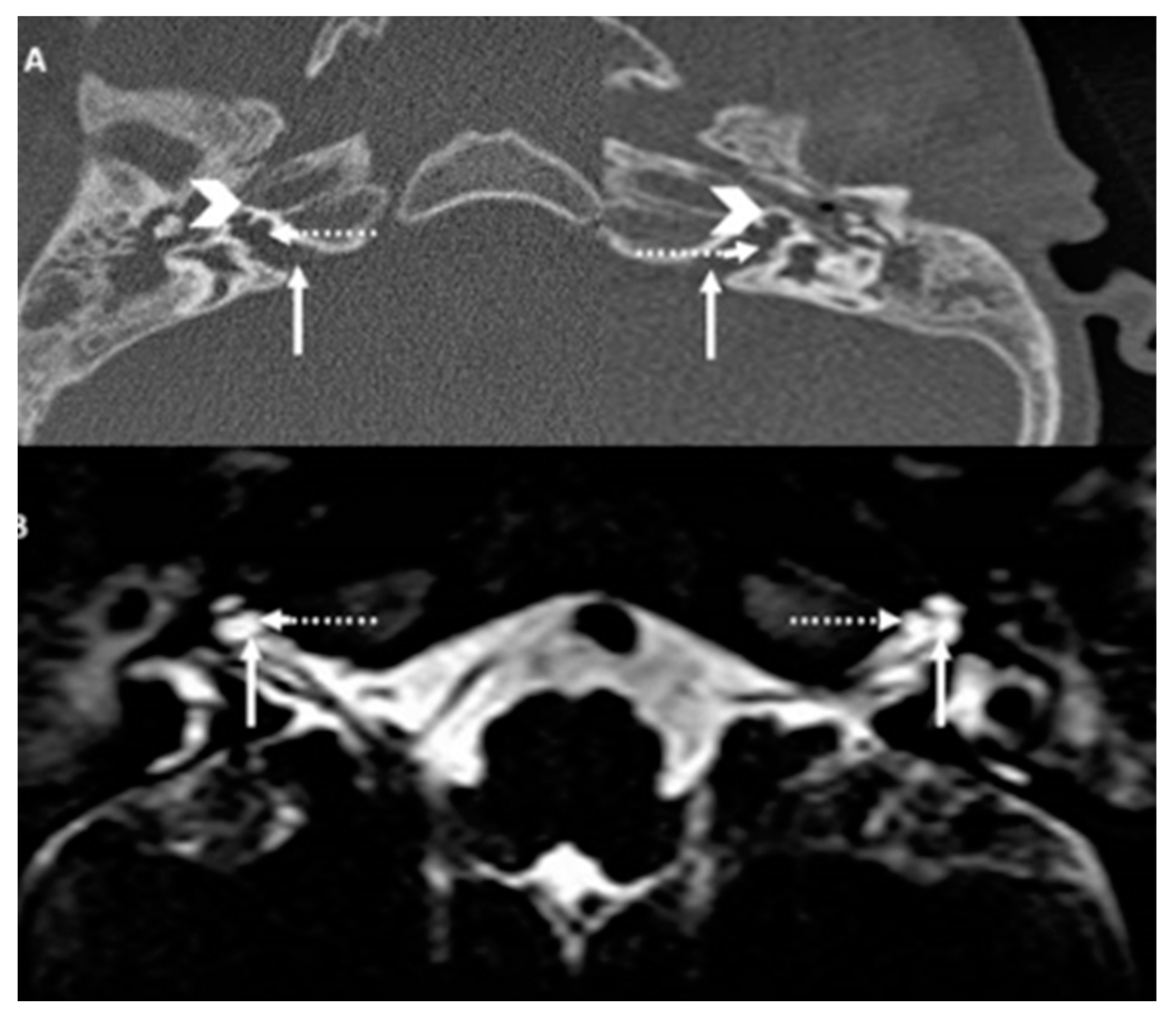
Figure 7.
SB – cVEMP. cVEMP 2019, reduced amplitudes and low threshold both sides
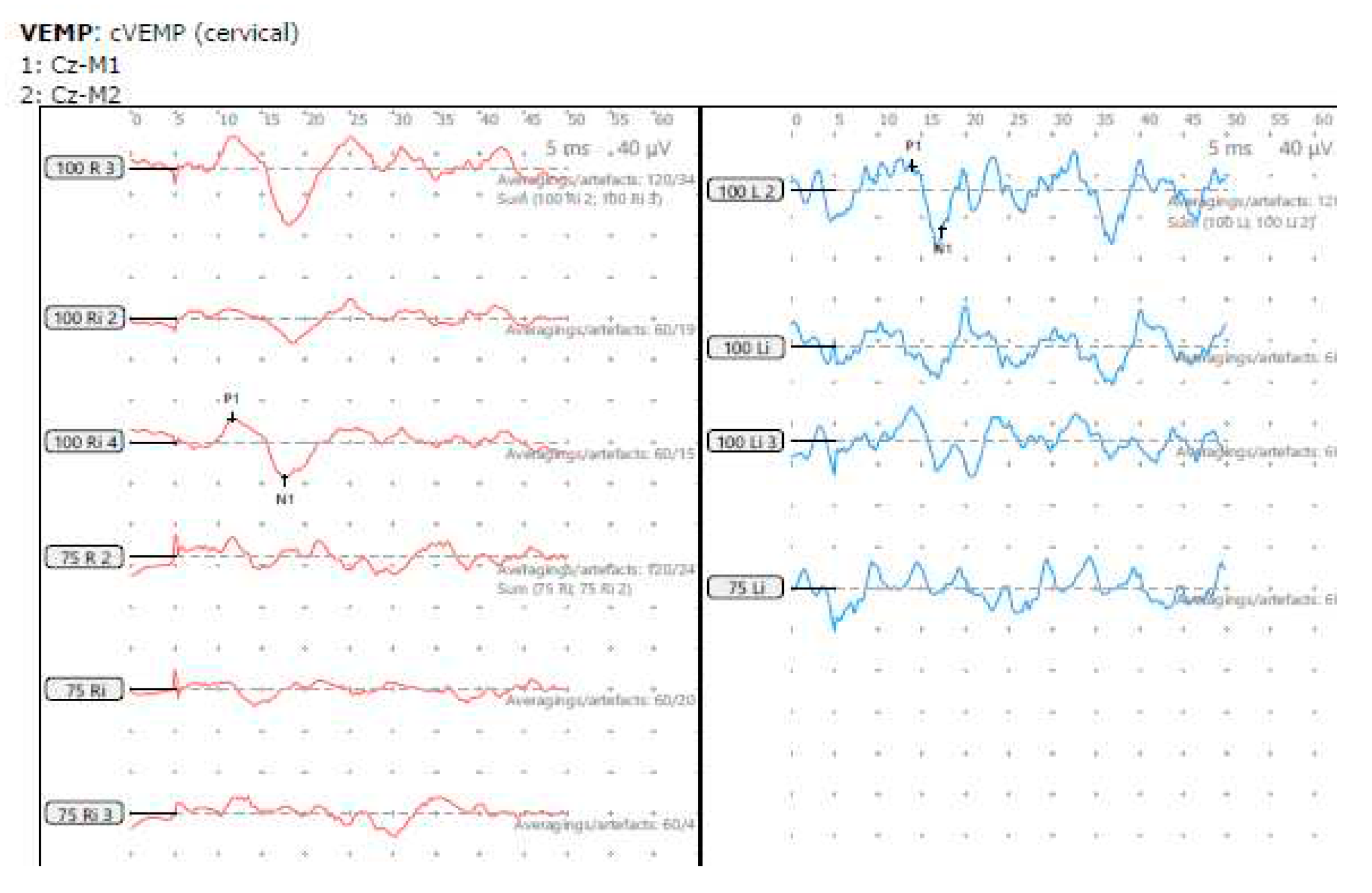
Figure 8.
SB - vHIT, cVEMP and SHIMP. A – vHIT 2016, note catch-up saccades (arrow); B - vHIT 2019, note reduction of saccades (arrow); C – SHIMP 2019, normal asymmetry and peak saccadic velocities
Figure 8.
SB - vHIT, cVEMP and SHIMP. A – vHIT 2016, note catch-up saccades (arrow); B - vHIT 2019, note reduction of saccades (arrow); C – SHIMP 2019, normal asymmetry and peak saccadic velocities
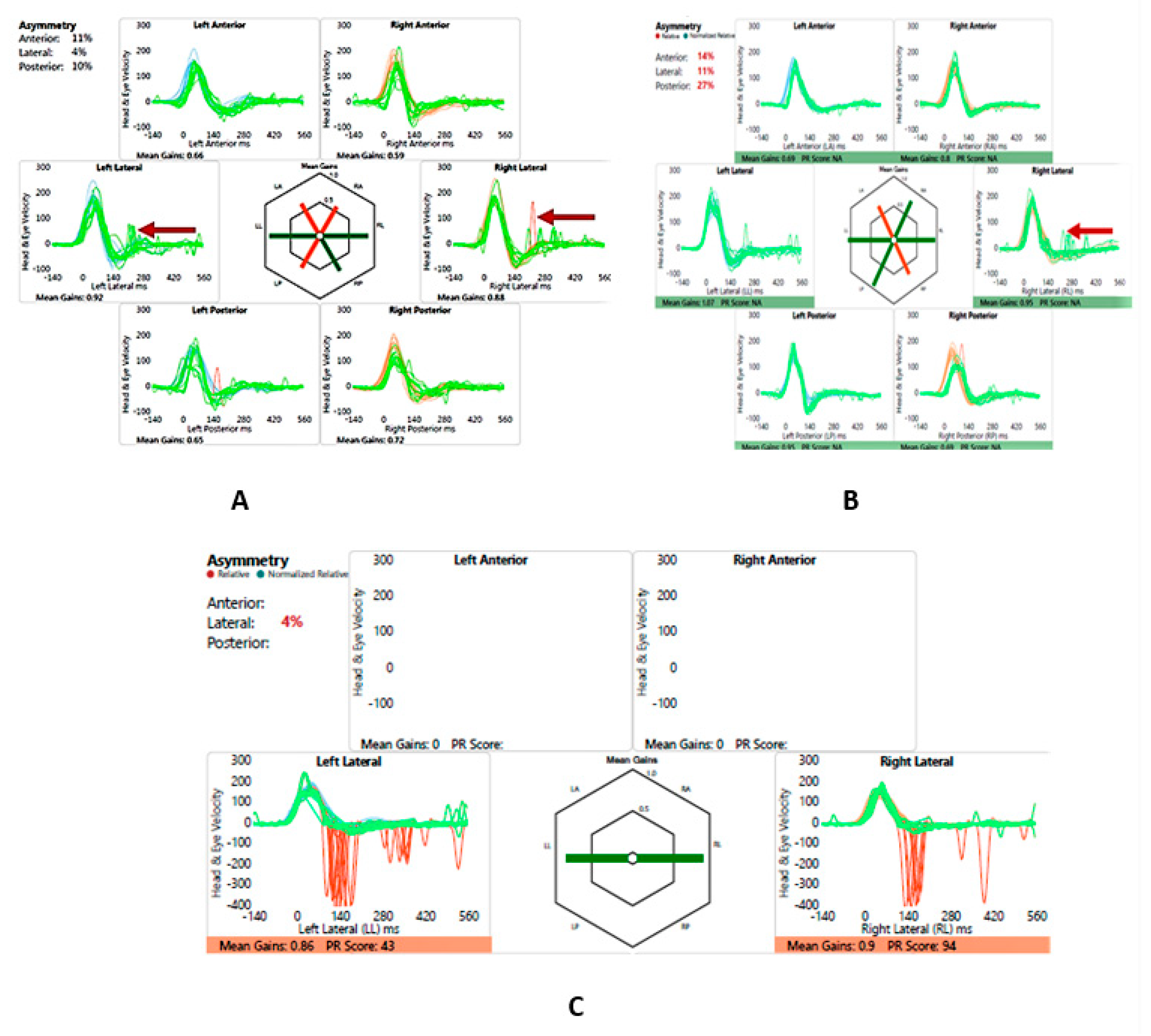

Table 1.
SA - Hearing symptoms and audiological test results (air and bone conduction thresholds masked whenever necessary and averaged between 500 Hz, 1- kHz in dBHL).
Table 1.
SA - Hearing symptoms and audiological test results (air and bone conduction thresholds masked whenever necessary and averaged between 500 Hz, 1- kHz in dBHL).
| Symptoms | Tymp/AR/OAE | Year | AC R/L | BC | AB |
|---|---|---|---|---|---|
| Hearing difficulties; speech delay. | Normal tymp ART absent L OAE absent bil |
2022 | 75/NT | 52.5 | 27.5 |
| 2018 | 76.25/NT | 51.25 | 25 | ||
| 2014 | 75/NT | 50 | 25 | ||
| 2010 | 81.25/ 100 | 48.75 | 35 |
Year – Year of test; AC R/L – air conduction thresholds right and left; BC R/L – bone conduction thresholds right and left; AB – air bone gap; Tymp – tympanometry; AR – acoustic reflex test; bil – bilateral ; NT – no threshold detected; note the fluctuation in BC and AB gap thereof typical of a third window disorder
Table 2.
SA - Vestibular/balance symptoms and vestibular function tests (VNG and VEMP ).
| Symptoms | VNG/VST/SVV | cVEMP AA/RA R μV | cVEMP AA/RA L μV | Asymmetry | cVEMP threshold dBnHL |
|---|---|---|---|---|---|
| Delayed motor development; unsteadiness; postural instability; difficult ambulation in darkness; difficulty in reading; difficulty in playground activities | Normal; abnormal VST | 92.5/2.9 | Nil | 100% | 75 (R) |
VNG – Videonystagmography without optic fixation assessing headshake, head heave, ocular counter rolling and presence absence of any spontaneous or provocation nystagmus and smooth pursuits/saccades; VST – static posturography in different conditions; SVV – subjective visual vertical; AA – absolute amplitude; RA – rectified amplitude; R – right; L – Left.
Table 3.
SA - Video head impulse test and suppression head impulse test.
| Year | VOR L/R ASCC | VOR L/R LSCC | VOR L/R PSCC | Saccades | SHIMP |
|---|---|---|---|---|---|
| 2016 | 0.41/0.3 | 0.72/0.88 | 0.54/0.64 | All canals dispersed | |
| 2019 | 0.5/0.69 | 0.7/0.84 | 0.87/0.59 | Only in left lateral and right anterior some clustering | 241-2660/sec PSV L and 243-3060/sec PSV R; 5% asymmetry |
VOR L/R – VOR gain left and right; ASCC – anterior or superior semicircular canal; LSCC – lateral semicircular canal; PSCC – posterior semicircular canal; PSV – peak saccadic velocity of SHIMP saccades; note the improvement of VOR gain with time; R – right; L – Left
Table 4.
SB - Hearing symptoms and audiological test results (air and bone conduction thresholds masked whenever necessary and averaged between 500 Hz, 1- kHz in dBHL).
Table 4.
SB - Hearing symptoms and audiological test results (air and bone conduction thresholds masked whenever necessary and averaged between 500 Hz, 1- kHz in dBHL).
| Symptoms | Tymp/AR/OAE | Year | AC R/L | BC | AB |
|---|---|---|---|---|---|
| Hearing difficulties; speech delay |
Normal tymp ART present bil OAE absent bil |
2022 | 81.25/83.3 | 51.25 | 27.5 |
| 2018 | 78.75/78.75 | 53.75 | 22.5 | ||
| 2014 | 76.25/75 | 48.75 | 27.5 | ||
| 2010 | 87.5/85 | 27.5 | 56.25 |
Year – Year of test; AC R/L – air conduction thresholds right and left; BC R/L – bone conduction thresholds right and left; AB – air bone gap; Tymp – tympanometry; AR – acoustic reflex test; bil – bilateral ; NT – no threshold detected; note the fluctuation in BC and AB gap thereof typical of a third window disorder
Table 5.
SB - Vestibular/balance symptoms and vestibular function tests (VNG and VEMP ).
| Symptoms | VNG/VST/SVV | cVEMP AA/RA R μV |
cVEMP AA/RA L μV | Asymmetry | cVEMP threshold dBnHL |
|---|---|---|---|---|---|
| Delayed motor development; unsteadiness; postural instability; autophony | Normal; abnormal VST | 57.3/1.1 | 58.8/0.8 | 15% | 75 (R+L) |
VNG – Videonystagmography without optic fixation assessing headshake, head heave, ocular counter rolling and presence absence of any spontaneous or provocation nystagmus and smooth pursuits/saccades; VST – static posturography in different conditions; SVV – subjective visual vertical; AA – absolute amplitude; RA – rectified amplitude; R – right; L – Left
Table 6.
SB - Video head impulse test and suppression head impulse test.
| Year | VOR L/R ASCC | VOR L/R LSCC | VOR L/R PSCC | Saccades | SHIMP |
|---|---|---|---|---|---|
| 2016 | 0.66/0.59 | 0.88/0.92 | 0.65/0.72 | Only laterals dispersed | |
| 2019 | 0.68/0.8 | 1.07/0.95 | 0.95/0.69 | Only lateral R clustered | 287-3580/sec PSV L and 342-4140/sec PSV R; 4% asymmetry |
VOR L/R – VOR gain left and right; ASCC – anterior or superior semicircular canal; LSCC – lateral semicircular canal; PSCC – posterior semicircular canal; PSV – peak saccadic velocity of SHIMP saccades; note the improvement of VOR gain with time; R – right; L – Left
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



