
Submitted:
06 July 2023
Posted:
07 July 2023
You are already at the latest version
Abstract
The optimal therapy for patients with non-metastatic biochemically relapsed prostate cancer (BRPC-M0) after local therapy is elusive. Thus, the evaluation of new non-toxic compounds in BRPC-M0 patients is warranted. PectaSol® modified citrus pectin (P-MCP) is a food supplement generally recognized as safe (GRAS) by the Food and Drug Administration (FDA) and is a competitive inhibitor of galectin-3 protein, involved in cancer pathogenesis. P-MCP treatment of BRPC-M0 patients for 6 months led to a 75% prostate-specific antigen doubling time (PSADT) improvement. To determine the longer-term effects of P-MCP we investigated an additional 12 months of treatment (phase II) with oral P-MCP at 4.8 grams X 3/day in patients with no disease progression in the previous 6 months. Of the 46 patients that entered the second phase, 7 patients withdrew consent and continued therapy independently, and 39 received another 12 months of therapy. After a total of 18 months P-MCP treatment, 62% (n=24) had a decreased/stable PSA, 90% (n=35) had improved PSADT, and 85% (n=33) had a durable long-term response, with a decreased/stable PSA, and/or improvement of PSADT, and negative scans. No patient had grade 3/4 toxicity. In conclusion, P-MCP may have long term durable efficacy, and is safe, in BRPC-M0.

Keywords:
modified citrus pectin
; non-metastatic biochemically relapsed prostate cancer
1. Introduction
Globally, prostate cancer is the second most common solid tumor [1] and cause of cancer-related death in men from Westernized countries [2]. Though its etiology needs further elucidation [3], with an age-standardized incidence rate of 31 per 100,000 [1] and approximately 1.5 million new cases reported in 2020 [1,4], prostate cancer incidence is increasing worldwide [4,5] and presents a significant economic burden [6]. With the rapid evolution of treatment options [7], however, and with changes in screening protocols [8], prostate cancer-related mortality patterns have stabilized [5]. While primary treatments for localized prostate cancer, including radical prostatectomy and radiation therapy, and androgen suppression therapy (AST) for advanced cases [9], are often initially effective, subsequent cancers typically develop [2]. Within 10 years of treatment, approximately 20–50% of patients will experience biochemical relapse prostate cancer (BRPC) [9,10,11], diagnosed through rising prostate-specific antigen (PSA) [12], which is a glycoprotein found exclusively in healthy and neoplastic prostate cells [13]. While patients with BRPC often remain asymptomatic and free of clinical evidence of disease for years [14], BRPC and corresponding PSA doubling time (PSADT) [15] following localized treatment [10] can be predictive of future metastases and mortality [10,13,16]. The typically prolonged disease natural history of BRPC [17], however, means that elevating PSA or shortening PSADT do not indicate an imminent mortality risk [17]. With multiple definitions reported in the literature [18], including patients experiencing variable risk factors and treatment efficacies [19], definitive BRPC treatment remain poorly established [10,20], and is often reliant on intuitive care [21].
The variation within the BRPC patient group, inconsistency in definitions within the literature and the protracted manifestation of the disease, present challenges to establishing effective patient management and salvage treatments [17]. While there are various localized therapy-specific protocols in place for BRPC [10,11], such as defined by The Phoenix Criteria [22], some treatment options are controversial [17,21]. Premature treatment with androgen deprivation treatment (ADT) in response to elevating PSA, for example, has uncertain benefits to survival potential though may negatively impact quality of life and increase the chances of comorbidities developing [10]. For example, ADT is associated with increased risk of cardiovascular (CV) adverse events [23,24], insulin resistance, dyslipidemia, obesity, and bone health disorders [14,25]. Characteristics of the BRPC patient population confound the identification of potential treatments [21]. BRPC’s prolonged natural history [17] has led to a lack of validated endpoints that can be measured within conventional clinical trial time frames. Dynamic PSA measurements [26], including PSADT [27], have been proposed and used as surrogate endpoints, both as predictive and stratification factors for clinical disease progression [17,28] and an increasing PSADT is sought to determine the efficacy of active compounds in clinical studies [24,29,30].
There is an urgent need to discover and evaluate new treatments for effective BRPC management [2,10,11,31]. Natural compounds have been extensively explored for potential anti-prostate cancer activities e.g., [32,33,34,35] through selective targeting of key molecules or signaling pathways, such as androgen signaling [36] that might help reduce cancer as dysregulated cell proliferation [37]. Many such compounds modulate key signaling pathways affecting cell proliferation [33,38] and immune responses [39], and are plant-derived. The polyphenols curcumin, tannic acid and flavonoids [33,40], for example, have antioxidant and anti-inflammatory effects [41], and complex polysaccharides, like modified citrus pectin (MCP) [42], have anti-inflammatory, antitumor, and antimetastatic activities [42,43,44].
Pectins comprise carbohydrate-soluble fiber found in plant cell walls and are indigestible to humans in their unmodified form [43]. Modified citrus pectin (MCP), with a shorter polysaccharide units, are water soluble [43] and have demonstrated significant anticancer activity [42,43,45,46], potentially by disrupting tumor-promoting signaling by binding galactose-containing side chains to the carbohydrate recognition domains of galectins [42,47,48]. Galectins comprise an evolutionarily conserved family of endogenous glycan-binding proteins and they play multifunctional roles in tumor progression [42] through modulating and recalibrating inter and intracellular signaling [49]. Galectins affect cellular responses, including cell aggregation, growth, differentiation, apoptosis and proliferation [42,49,50] and may promote immune evasion of cancer cells [51]. Galectins, including Galectin1 (GAL1) and GAL3, have therefore been explored as potentially effective therapeutic targets for cancer patients [42,52,53].
PectaSol® Modified Citrus Pectin (P-MCP; EcoNugenics Inc, Santa Rosa, CA, USA) is a commercially available polysaccharide and is a Galectin-3 (Gal-3) inhibitor, binding to the Galectin-3 carbohydrate recognition domain [42,47]. Derived from the pith of citrus fruit peels and classified by the US-FDA as generally regarded as safe (GRAS), P-MCP is a food supplement with demonstrated cytotoxicity towards, and inhibition of, cancer cells [43,54], including prostate cancer cells [34,45,46] suggesting its viability in delaying disease progression [42].
In clinical trials, P-MCP delivered as oral therapy to BRPC patients has had a demonstrable effect on prostate-specific antigen (PSA) levels, leading to a lengthening of PSADT in the majority of participants and suggesting a reduction in tumor progression [34,45]. An increase in PSADT with P-MCP oral therapy at both six [34] and 12 [45] months, suggests the benefits may be perpetuated with extended treatment. Using the participant cohort of Keizman et al. 2021, which had demonstrated benefits from 6 months of P-MCP therapy, we herein report the influence of extending treatment to a total of 18 months treatment on BRPC indicators. We aim to establish whether the benefits of oral P-MCP therapy observed at 6 months [34] are sustained over a longer time period.
2. Methods and Materials
Study design: The eligibility criteria are described in our previous publication [34].
Briefly, patients with BRPC-M0, rising PSA post-primary therapy (surgery and/or radiation) and negative scans were included. All patients had a normal level of serum testosterone > 150 ng/ml, and Eastern Cooperative Oncology Group (ECOG) performance status ≤ 2 at study entry. All participating patients signed an Institutional Review Board (IRB)-approved consent form. Study participants were recruited between 2013 to 2019 from 5 medical centers in Israel (Meir, Rabin, Rambam, Soroka, and Tel-Aviv Sourasky). Patients were given 4.8 grams of P-MCP to take orally, times 3 per day for the duration of the study. The P-MCP was provided by PectaSol-C ®, EcoNugenics, Santa Rosa, CA, USA, in packs of 270 capsules. Patients without evidence of disease progression or dose-limiting toxicity after 6 months of therapy (n=46), entered the second long term phase of the study, and were given an additional 12 months of treatment. A total of 39 patients completed the full 18 months of treatment. The study design is described in Figure 1.
Evaluation of disease status: Patients underwent monthly visits for toxicities, physical examinations, and serum PSA, with baseline measurements taken prior to starting the P-MCP treatment protocol. A positron emission tomography (PET) - prostate-specific membrane antigen (PSMA) scan was conducted after 6 and 18 months in patients without clinical or PSA progression, or earlier upon clinical or PSA progression. The primary efficacy endpoint was the rate of patients without PSA progression (defined as an increase of ≥ 25% from baseline) and/or patients with improvement (lengthening) of PSADT versus baseline. The post-baseline PSADT was calculated using baseline PSA measurements obtained at the start of the study and every month during treatment. Secondary endpoints included the rate of patients without radiologic progression and toxicity, and with treatment benefits according to the PSADT risk grouping (e.g., poor < 3 months, intermediate 3-8.99 months, and good ≥9 months) [34].
Duration of treatment: Treatment as per the protocol continued for 12 additional months or until biochemical or clinical disease progression or dose-limiting toxicity. Disease progression was defined as biochemical progression, and/or new radiological evidence of metastases.
Safety evaluation of toxicity: Toxicity was defined according to the National Cancer Institute (NCI) Common Toxicity Criteria with treatments terminated at grades 3/4 and patients monitored weekly until ≤ grade 1 before restarting therapy. Treatment would be discontinued upon the recurrence of a same grade 3/4 event, and for any toxicity requiring longer than 4 weeks to recover to ≤ grade 1.
Statistical analysis: Comparisons between pre- and post-treatment endpoint parameters, and within groups were analyzed using the Wilcoxon Signed Rank test for abnormally distributed data, or the two-tailed Student t-test for normally distributed data, with results reported as number, percentage, mean or median, and standard deviation (SD). A p-value ≤ 0.05 was considered statistically significant.
Regulatory Considerations: The research was conducted in accordance with the approval by the IRB committee of our institution. The study was registered at clinicaltrials.gov (NCT01681823).
3. Results
Patients: Of the 46 patients that displayed a benefit from the initial 6 months of therapy (in terms of stabilization/decrease of PSA, and/or improvement of PSADT, and with negative scans), and that were eligible the second phase of additional 12 months therapy (to a total of 18 months of treatment), 7 patients withdrew consent during the first month of the additional year of therapy, and chose to continue the effective therapy independently due to the travel distance to monthly medical visits. Thus, 39 patients were treated as per the protocol for a total of 18 months of treatment. Patient pre-treatment characteristics are summarized in Table 1.
Long-term outcome as determined by PSA level, PSADT, and disease progression: After the 39 patients had received 18 months of treatment, 85% (n=33) demonstrated a decreased or stable PSA (Figure 2) and/or improvement of PSADT (54%, n = 21) and negative scans (90%, n = 35). Median PSADT improved significantly compared to baseline (p = 0.003), from a median pre-treatment PSADT of 10.3 (median range = 1.4 – 54.6) months to a median post-treatment PSADT of 43.5 (median range = 3.5 – 981.0) months (Table 1 and Table 2).
The benefits of 18 months of therapy in terms of PSA stabilization or decrease, and/or PSADT lengthening were seen in all PSADT risk groups (Table 2). There was also an improvement in PSADT risk grouping after 18 months of treatment (Table 2, Figure 3). After 18 months of P-MCP therapy, all patients with a pre-treatment PSADT risk grouping of poor (<3 months), improved their PSADT to intermediate-good risk, and most patients (77%) with a pre-treatment risk PSADT of intermediate improved their PSADT risk to good (Table 2, Figure 3). In patients with a pre-treatment PSADT risk of good, 91% retained their risk grouping and 87% of which had an improved PSADT. After 18 months of therapy, no patients remained in the poor PSADT risk group, and the proportion of patients with a PSADT risk of good (≥ 9.00) increased from 59% (n=23) at baseline to 87% (n=34) after 18 months of therapy (Figure 3). The pre-treatment (baseline) distribution of PSADT risk groupings improved after 18 months of P-MCP therapy, with the proportion of patients in the PSADT poor risk group reducing from 8% to 0. Similarly, the proportion of patients in the intermediate PSADT risk group reduced from 33% to 13% and the good risk group saw an increase from 59% to 87% (Table 1 and Table 2). There was a significant change in the median PSADT after 18 months of P-MCP therapy (Table 2) as compared to baseline (Table 2), for patients with a pre-treatment PSADT good risk (45.9 versus 14.7 months, p=0.027) and intermediate risk (22.75 versus 5.1 months, p=0.0015). Disease progression during the 18 months of therapy was observed in 18% (n=7) of patients, 8% (n=3) of which had PSA progression only (without radiological progression) and 10% (n=4) presented with both PSA and radiologic progression.
Toxicity and compliance: None of the patients had grade 3/4 toxicity during the 18 months of therapy. Grade 1 toxicity was observed as transient and reversible bloating that did not require treatment discontinuation. This grade 1 toxicity was noted in 20% (n=12) of patients during the first six months of therapy, and in 23% (n=9) during the additional 12 months of treatment.
4. Discussion
Prostate cancer is a leading cause of death in men worldwide [1], with tumor progression after primary treatment presenting a persistent therapeutic challenge [10,20]. As such, determining effective treatment for the slow, and often cryptic, progression of biochemical relapse prostate cancer (BRPC) [14] has proven problematic [2,10,11,20]. The anticancer properties of natural products [32,33,34,35] offer important investigative potential for cancer therapies, including in relation to long-term BRPC treatment [34,45]. Building on the promising results of Keizman et al.’s (2021) 6-month clinical study into the efficacy of PectaSol® Modified Citrus Pectin (P-MCP; EcoNugenics Inc, Santa Rosa, CA, USA) [34], we document a lengthening of PSADT within the same participant cohort after sustained P-MCP treatment for 18 months.
Consistent with investigations of other natural products [30,35], treatment with P-MCP for a total of 18 months led to an increase in PSADT for 90% of the participants, as compared to pre-treatment baseline. In the preceding 6-months of P-MCP therapy, three quarters of the participant cohort demonstrated a lengthening of PSADT, suggesting that the additional 12-months of treatment increased the likelihood of efficacy. PSADT is an important measure of BRCP-M0 progression and predictor of future metastases [10,13,15,16,25] and, while a shortening of PSADT is not necessarily indicative of imminent risk [17], a lengthening of PSADT can be used to indicate effective therapeutic management [27]. Findings therefore suggest that oral therapy with P-MCP three times daily is potentially effective in managing BRPC-M0 patients over a sustained period.
BRPC-M0 patients within the study were categorized into one of the three PSADT risk groupings [16,20], with risk staying stable or improving with P-MCP treatment for all but 5% of participants. In the absence of placebo control group, the proportion of participants that may have been stable, improved or reduced their PSADT risk grouping of over an 18-month period without P-MCP therapy is unknown. The improvement of all participants in the poor risk category at baseline, however, suggests that P-MCP is beneficial for BRPC-M0 patients at highest risk of metastasis. Such findings should be further validated with a higher sample size for the poor risk category. Additionally, as with the previous study, no long-term or significant toxic effects were recorded, suggesting P-MCP can be safely taken over a prolonged period.
A major limitation of the study is the lack of placebo (control) arm, which would have have provided confirmation that the recorded PSADT lengthening was a consequence of P-MCP therapy. Using only the patient cohort from Keizman et al. (2021) that demonstrated beneficial responses P-MCP by 6 months of therapy may have provided a skewed sample population by eliminating participants who were unresponsive to treatment. It is therefore possible that the 90% benefit rate observed in this study will be higher than that for a randomly selected cohort of BRCP-M0 patients. Additionally, the low sample size within the PSADT poor risk grouping (n = 3) means that benefits of P-MCP for this cohort need further validation. Furthermore, although retrospective studies have shown that PSADT is a strong predictor of metastasis-free survival, overall survival [11,13] or both [28,30], further validation is required to establish whether change in PSADT is an acceptable endpoint for clinical trials in this patient population. Investigating the mechanisms of PSADT lengthening in BRPC patients, such as by galectin inhibition [42], was beyond the scope of the study but is a key avenue for future research.
Overall, we demonstrated a sustained benefit of P-MCP therapy for the majority of our study cohort, without toxicity, over an 18-month period. This indicates that P-MCP treatment should be continued for the long-term to ensure maximum benefit and BRPC-M0 management. Further double-blind placebo controlled clinical studies are planned to optimize the therapeutic use of P-MCP in this patient population.
Author Contributions
DK: MF, AP, IK, ER, DS, IL, RM, OY, DM, KR, AS, HD and IE contributed equally to this work. DK and IE devised the concept and designed the study. DK, MF, AP, IK, ER, DS, IL, RM, OY, DM, KR and HD conducted the most experiments. DK and IE contributed to the manuscript writing. All authors reviewed the manuscript and signed-off on its accuracy.
Funding
This study received funding from EcoNugenics Inc., Santa Rosa, CA, USA. EcoNugenics was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of Meir Medical Center (Protocol 019-2-12 MMC. Current ongoing approval date 23/08/2019). The study was registered at clinicaltrials.gov (NCT01681823).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in the study are available with the investigator Dr. Daniel Keizman.
Conflicts of Interest
IE discloses being the developer of the sponsoring dietary supplement company but holds no ownership in the company. The other authors declare no potential conflicts of interest.
References
- Gandaglia, G.; Leni, R.; Bray, F.; Fleshner, N.; Freedland, S.J.; Kibel, A.; Stattin, P.; Van Poppel, H.; La Vecchia, C. Epidemiology and Prevention of Prostate Cancer. European Urology Oncology 2021, 4, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, A.N.; Usman, A.; Morgans, A.; VanderWeele, D.J.; Sosman, J.; Wu, J.D. Past, Current, and Future of Immunotherapies for Prostate Cancer. Front. Oncol. 2019, 9, 884. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J Oncol 2019, 10, 63–89. [Google Scholar] [CrossRef]
- Wang, L.; Lu, B.; He, M.; Wang, Y.; Wang, Z.; Du, L. Prostate Cancer Incidence and Mortality: Global Status and Temporal Trends in 89 Countries From 2000 to 2019. Front. Public Health 2022, 10, 811044. [Google Scholar] [CrossRef]
- Zhai, Z.; Zheng, Y.; Li, N.; Deng, Y.; Zhou, L.; Tian, T.; Yang, S.; Hao, Q.; Song, D.; Wu, Y.; et al. Incidence and Disease Burden of Prostate Cancer from 1990 to 2017: Results from the Global Burden of Disease Study 2017. Cancer 2020, 126, 1969–1978. [Google Scholar] [CrossRef]
- Roehrborn, C.G.; Black, L.K. The Economic Burden of Prostate Cancer: ECONOMIC BURDEN OF PROSTATE CANCER. BJU International 2011, 108, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Swami, U.; McFarland, T.R.; Nussenzveig, R.; Agarwal, N. Advanced Prostate Cancer: Treatment Advances and Future Directions. Trends in Cancer 2020, 6, 702–715. [Google Scholar] [CrossRef]
- Catalona, W.J. Prostate Cancer Screening. Medical Clinics of North America 2018, 102, 199–214. [Google Scholar] [CrossRef]
- Heidenreich, A.; Bastian, P.J.; Bellmunt, J.; Bolla, M.; Joniau, S.; van der Kwast, T.; Mason, M.; Matveev, V.; Wiegel, T.; Zattoni, F.; et al. EAU Guidelines on Prostate Cancer. Part II: Treatment of Advanced, Relapsing, and Castration-Resistant Prostate Cancer. European Urology 2014, 65, 467–479. [Google Scholar] [CrossRef]
- Paller, C.J.; Antonarakis, E.S. Management of Biochemically Recurrent Prostate Cancer after Local Therapy: Evolving Standards of Care and New Directions. Clin Adv Hematol Oncol 2013, 11, 14–23. [Google Scholar]
- Simon, N.I.; Parker, C.; Hope, T.A.; Paller, C.J. Best Approaches and Updates for Prostate Cancer Biochemical Recurrence. American Society of Clinical Oncology Educational Book 2022, 352–359. [Google Scholar] [CrossRef]
- Heijnsdijk, E.A.M.; Bangma, C.H.; Borràs, J.M.; de Carvalho, T.M.; Castells, X.; Eklund, M.; Espinàs, J.A.; Graefen, M.; Grönberg, H.; Lansdorp-Vogelaar, I.; et al. Summary Statement on Screening for Prostate Cancer in Europe: Prostate Cancer Screening in Europe. Int. J. Cancer 2018, 142, 741–746. [Google Scholar] [CrossRef]
- Arlen, P.M.; Bianco, F.; Dahut, W.L.; D’Amico, A.; Figg, W.D.; Freedland, S.J.; Gulley, J.L.; Kantoff, P.W.; Kattan, M.W.; Lee, A.; et al. Prostate Specific Antigen Working Group Guidelines on Prostate Specific Antigen Doubling Time. Journal of Urology 2008, 179, 2181–2186. [Google Scholar] [CrossRef]
- Pound, C.R. Natural History of Progression After PSA Elevation Following Radical Prostatectomy. JAMA 1999, 281, 1591. [Google Scholar] [CrossRef]
- Aggarwal, R.; Heller, G.; Hillman, D.; Xiao, H.; Picus, J.; Wang, J.; Taplin, M.E.; Dorff, T.; Appleman, L.J.; Weckstein, D.; et al. LBA63 PRESTO: A Phase III, Open-Label Study of Androgen Annihilation in Patients (Pts) with High-Risk Biochemically Relapsed Prostate Cancer (AFT-19). Annals of Oncology 2022, 33, S1428. [Google Scholar] [CrossRef]
- Freedland, S.J.; Humphreys, E.B.; Mangold, L.A.; Eisenberger, M.; Dorey, F.J.; Walsh, P.C.; Partin, A.W. Risk of Prostate Cancer–Specific Mortality Following Biochemical Recurrence After Radical Prostatectomy. JAMA 2005, 294, 433. [Google Scholar] [CrossRef]
- Zhou, P.; Chen, M.-H.; McLeod, D.; Carroll, P.R.; Moul, J.W.; D’Amico, A.V. Predictors of Prostate Cancer–Specific Mortality After Radical Prostatectomy or Radiation Therapy. JCO 2005, 23, 6992–6998. [Google Scholar] [CrossRef]
- Cookson, M.S.; Aus, G.; Burnett, A.L.; Canby-Hagino, E.D.; D’Amico, A.V.; Dmochowski, R.R.; Eton, D.T.; Forman, J.D.; Goldenberg, S.L.; Hernandez, J.; et al. Variation in the Definition of Biochemical Recurrence in Patients Treated for Localized Prostate Cancer: The American Urological Association Prostate Guidelines for Localized Prostate Cancer Update Panel Report and Recommendations for a Standard in the Reporting of Surgical Outcomes. Journal of Urology 2007, 177, 540–545. [Google Scholar] [CrossRef]
- Van den Broeck, T.; van den Bergh, R.C.N.; Arfi, N.; Gross, T.; Moris, L.; Briers, E.; Cumberbatch, M.; De Santis, M.; Tilki, D.; Fanti, S.; et al. Prognostic Value of Biochemical Recurrence Following Treatment with Curative Intent for Prostate Cancer: A Systematic Review. European Urology 2019, 75, 967–987. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Feng, Z.; Trock, B.J.; Humphreys, E.B.; Carducci, M.A.; Partin, A.W.; Walsh, P.C.; Eisenberger, M.A. The Natural History of Metastatic Progression in Men with Prostate-Specific Antigen Recurrence after Radical Prostatectomy: Long-Term Follow-up: METASTATIC PROGRESSION IN PSA-RECURRENT PROSTATE CANCER. BJU International 2012, 109, 32–39. [Google Scholar] [CrossRef]
- Paller, C.J.; Antonarakis, E.S.; Eisenberger, M.A.; Carducci, M.A. Management of Patients with Biochemical Recurrence After Local Therapy for Prostate Cancer. Hematology/Oncology Clinics of North America 2013, 27, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Roach, M.; Hanks, G.; Thames, H.; Schellhammer, P.; Shipley, W.U.; Sokol, G.H.; Sandler, H. Defining Biochemical Failure Following Radiotherapy with or without Hormonal Therapy in Men with Clinically Localized Prostate Cancer: Recommendations of the RTOG-ASTRO Phoenix Consensus Conference. International Journal of Radiation Oncology*Biology*Physics 2006, 65, 965–974. [Google Scholar] [CrossRef]
- Keating, N.L.; O’Malley, A.J.; Smith, M.R. Diabetes and Cardiovascular Disease During Androgen Deprivation Therapy for Prostate Cancer. JCO 2006, 24, 4448–4456. [Google Scholar] [CrossRef]
- Keizman, D.; Zahurak, M.; Sinibaldi, V.; Carducci, M.; Denmeade, S.; Drake, C.; Pili, R.; Antonarakis, E.S.; Hudock, S.; Eisenberger, M. Lenalidomide in Nonmetastatic Biochemically Relapsed Prostate Cancer: Results of a Phase I/II Double-Blinded, Randomized Study. Clinical Cancer Research 2010, 16, 5269–5276. [Google Scholar] [CrossRef] [PubMed]
- Zelefsky, M.J.; Ben-Porat, L.; Scher, H.I.; Chan, H.M.; Fearn, P.A.; Fuks, Z.Y.; Leibel, S.A.; Venkatraman, E.S. Outcome Predictors for the Increasing PSA State After Definitive External-Beam Radiotherapy for Prostate Cancer. JCO 2005, 23, 826–831. [Google Scholar] [CrossRef]
- Suzman, D.L.; Zhou, X.C.; Zahurak, M.L.; Lin, J.; Antonarakis, E.S. Change in PSA Velocity Is a Predictor of Overall Survival in Men with Biochemically-Recurrent Prostate Cancer Treated with Nonhormonal Agents: Combined Analysis of Four Phase-2 Trials. Prostate Cancer Prostatic Dis 2015, 18, 49–55. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.V.; Moul, J.W.; Carroll, P.R.; Sun, L.; Lubeck, D.; Chen, M.-H. Surrogate End Point for Prostate Cancer-Specific Mortality After Radical Prostatectomy or Radiation Therapy. JNCI Journal of the National Cancer Institute 2003, 95, 1376–1383. [Google Scholar] [CrossRef]
- Gleason, D.F.; Mellinger, G.T. The Veterans Administration Cooperative Urological Research Group Members of this group areS0022534717598894-57c401330b4195dbd1713587ed540477 Lino J. Arduino Veterans Administration Center (VAC), Des Moines, Iowa S0022534717598894-61e5a9195ff74e58ab72b9f6a7b48125 John C. Bailar Veterans Administration Central Office (VACO), Washington, D. C. S0022534717598894-1936883d21db441268a90e6e79675523 Leslie E. Becker VAC, Leavenworth, Kansas S0022534717598894-3619ae2ed4ad7e7734f8222e5502f779 Henry I. Berman Veteran Prediction of Prognosis for Prostatic Adenocarcinoma by Combined Histological Grading and Clinical Staging. Journal of Urology 1974, 111, 58–64. [Google Scholar] [CrossRef]
- Rosenbaum, E.; Zahurak, M.; Sinibaldi, V.; Carducci, M.A.; Pili, R.; Laufer, M.; DeWeese, T.L.; Eisenberger, M.A. Marimastat in the Treatment of Patients with Biochemically Relapsed Prostate Cancer: A Prospective Randomized, Double-Blind, Phase I/II Trial. Clinical Cancer Research 2005, 11, 4437–4443. [Google Scholar] [CrossRef]
- Paller, C.J.; Ye, X.; Wozniak, P.J.; Gillespie, B.K.; Sieber, P.R.; Greengold, R.H.; Stockton, B.R.; Hertzman, B.L.; Efros, M.D.; Roper, R.P.; et al. A Randomized Phase II Study of Pomegranate Extract for Men with Rising PSA Following Initial Therapy for Localized Prostate Cancer. Prostate Cancer Prostatic Dis 2013, 16, 50–55. [Google Scholar] [CrossRef]
- Gillessen, S.; Bossi, A.; Davis, I.D.; de Bono, J.; Fizazi, K.; James, N.D.; Mottet, N.; Shore, N.; Small, E.; Smith, M.; et al. Management of Patients with Advanced Prostate Cancer. Part I: Intermediate-/High-Risk and Locally Advanced Disease, Biochemical Relapse, and Side Effects of Hormonal Treatment: Report of the Advanced Prostate Cancer Consensus Conference 2022. European Urology 2023, 83, 267–293. [Google Scholar] [CrossRef]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. Natural Compounds in Prostate Cancer Prevention and Treatment: Mechanisms of Action and Molecular Targets. Cells 2020, 9, 460. [Google Scholar] [CrossRef]
- Bilgin, S.; Erden Tayhan, S.; Yıldırım, A.; Koç, E. Investigation of the Effects of Isoeugenol-Based Phenolic Compounds on Migration and Proliferation of HT29 Colon Cancer Cells at Cellular and Molecular Level. Bioorganic Chemistry 2023, 130, 106230. [Google Scholar] [CrossRef]
- Keizman, D.; Frenkel, M.; Peer, A.; Kushnir, I.; Rosenbaum, E.; Sarid, D.; Leibovitch, I.; Mano, R.; Yossepowitch, O.; Margel, D.; et al. Modified Citrus Pectin Treatment in Non-Metastatic Biochemically Relapsed Prostate Cancer: Results of a Prospective Phase II Study. Nutrients 2021, 13, 4295. [Google Scholar] [CrossRef]
- Pantuck, A.J.; Leppert, J.T.; Zomorodian, N.; Aronson, W.; Hong, J.; Barnard, R.J.; Seeram, N.; Liker, H.; Wang, H.; Elashoff, R.; et al. Phase II Study of Pomegranate Juice for Men with Rising Prostate-Specific Antigen Following Surgery or Radiation for Prostate Cancer. Clinical Cancer Research 2006, 12, 4018–4026. [Google Scholar] [CrossRef]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen Signaling in Prostate Cancer. Cold Spring Harb Perspect Med 2017, 7, a030452. [Google Scholar] [CrossRef]
- Salehi; Fokou; Yamthe; Tali; Adetunji; Rahavian; Mudau; Martorell; Setzer; Rodrigues; et al. Phytochemicals in Prostate Cancer: From Bioactive Molecules to Upcoming Therapeutic Agents. Nutrients 2019, 11, 1483. [CrossRef] [PubMed]
- Li, F.; Li, S.; Li, H.-B.; Deng, G.-F.; Ling, W.-H.; Wu, S.; Xu, X.-R.; Chen, F. Antiproliferative Activity of Peels, Pulps and Seeds of 61 Fruits. Journal of Functional Foods 2013, 5, 1298–1309. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Tang, L.; Chen, H.; Wu, C.; Zhao, M.; Yang, Y.; Chen, X.; Liu, G. Resveratrol Inhibits TGF-Β1-Induced Epithelial-to-Mesenchymal Transition and Suppresses Lung Cancer Invasion and Metastasis. Toxicology 2013, 303, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Li, A.-N.; Li, S.; Zhang, Y.-J.; Xu, X.-R.; Chen, Y.-M.; Li, H.-B. Resources and Biological Activities of Natural Polyphenols. Nutrients 2014, 6, 6020–6047. [Google Scholar] [CrossRef] [PubMed]
- Kausar, H.; Jeyabalan, J.; Aqil, F.; Chabba, D.; Sidana, J.; Singh, I.P.; Gupta, R.C. Berry Anthocyanidins Synergistically Suppress Growth and Invasive Potential of Human Non-Small-Cell Lung Cancer Cells. Cancer Letters 2012, 325, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Mariño, K.V.; Cagnoni, A.J.; Croci, D.O.; Rabinovich, G.A. Targeting Galectin-Driven Regulatory Circuits in Cancer and Fibrosis. Nat Rev Drug Discov 2023. [Google Scholar] [CrossRef] [PubMed]
- Jun Yan; Katz, A. PectaSol-C Modified Citrus Pectin Induces Apoptosis and Inhibition of Proliferation in Human and Mouse Androgen-Dependent and- Independent Prostate Cancer Cells. Integr Cancer Ther 2010, 9, 197–203. [CrossRef]
- Demotte, N.; Wieërs, G.; Van Der Smissen, P.; Moser, M.; Schmidt, C.; Thielemans, K.; Squifflet, J.-L.; Weynand, B.; Carrasco, J.; Lurquin, C.; et al. A Galectin-3 Ligand Corrects the Impaired Function of Human CD4 and CD8 Tumor-Infiltrating Lymphocytes and Favors Tumor Rejection in Mice. Cancer Research 2010, 70, 7476–7488. [Google Scholar] [CrossRef] [PubMed]
- Guess, B.W.; Scholz, M.C.; Strum, S.B.; Lam, R.Y.; Johnson, H.J.; Jennrich, R.I. Modified Citrus Pectin (MCP) Increases the Prostate-Specific Antigen Doubling Time in Men with Prostate Cancer: A Phase II Pilot Study. Prostate Cancer Prostatic Dis 2003, 6, 301–304. [Google Scholar] [CrossRef]
- Pienta, K.J.; Nailk, H.; Akhtar, A.; Yamazaki, K.; Replogle, T.S.; Lehr, J.; Donat, T.L.; Tait, L.; Hogan, V.; Raz, A. Inhibition of Spontaneous Metastasis in a Rat Prostate Cancer Model by Oral Administration of Modified Citrus Pectin. JNCI Journal of the National Cancer Institute 1995, 87, 348–353. [Google Scholar] [CrossRef]
- Glinskii, O.V.; Sud, S.; Mossine, V.V.; Mawhinney, T.P.; Anthony, D.C.; Glinsky, G.V.; Pienta, K.J.; Glinsky, V.V. Inhibition of Prostate Cancer Bone Metastasis by Synthetic TF Antigen Mimic/Galectin-3 Inhibitor Lactulose-l-Leucine. Neoplasia 2012, 14, 65–73. [Google Scholar] [CrossRef]
- Stegmayr, J.; Lepur, A.; Kahl-Knutson, B.; Aguilar-Moncayo, M.; Klyosov, A.A.; Field, R.A.; Oredsson, S.; Nilsson, U.J.; Leffler, H. Low or No Inhibitory Potency of the Canonical Galectin Carbohydrate-Binding Site by Pectins and Galactomannans. Journal of Biological Chemistry 2016, 291, 13318–13334. [Google Scholar] [CrossRef]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the Hallmarks of Cancer: Galectins as Multifunctional Mediators of Tumor Progression. Journal of Experimental Medicine 2020, 217, e20182041. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.-Y.; Rabinovich, G.A.; Liu, F.-T. Galectins: Structure, Function and Therapeutic Potential. Expert Rev. Mol. Med. 2008, 10, e17. [Google Scholar] [CrossRef]
- Méndez-Huergo, S.P.; Blidner, A.G.; Rabinovich, G.A. Galectins: Emerging Regulatory Checkpoints Linking Tumor Immunity and Angiogenesis. Current Opinion in Immunology 2017, 45, 8–15. [Google Scholar] [CrossRef]
- Dong, R.; Zhang, M.; Hu, Q.; Zheng, S.; Soh, A.; Zheng, Y.; Yuan, H. Galectin-3 as a Novel Biomarker for Disease Diagnosis and a Target for Therapy (Review). Int J Mol Med 2017. [Google Scholar] [CrossRef] [PubMed]
- Nangia-Makker, P.; Hogan, V.; Honjo, Y.; Baccarini, S.; Tait, L.; Bresalier, R.; Raz, A. Inhibition of Human Cancer Cell Growth and Metastasis in Nude Mice by Oral Intake of Modified Citrus Pectin. JNCI Journal of the National Cancer Institute 2002, 94, 1854–1862. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Wu, J.M. Changes in Cell Growth, Cyclin/Kinase, Endogenous Phosphoproteins and Nm23 Gene Expression in Human Prostatic JCA-1 Cells Treated with Modified Citrus Pectin. Biochem Mol Biol Int 1995, 37, 833–841. [Google Scholar] [PubMed]
Figure 1.
Study design.
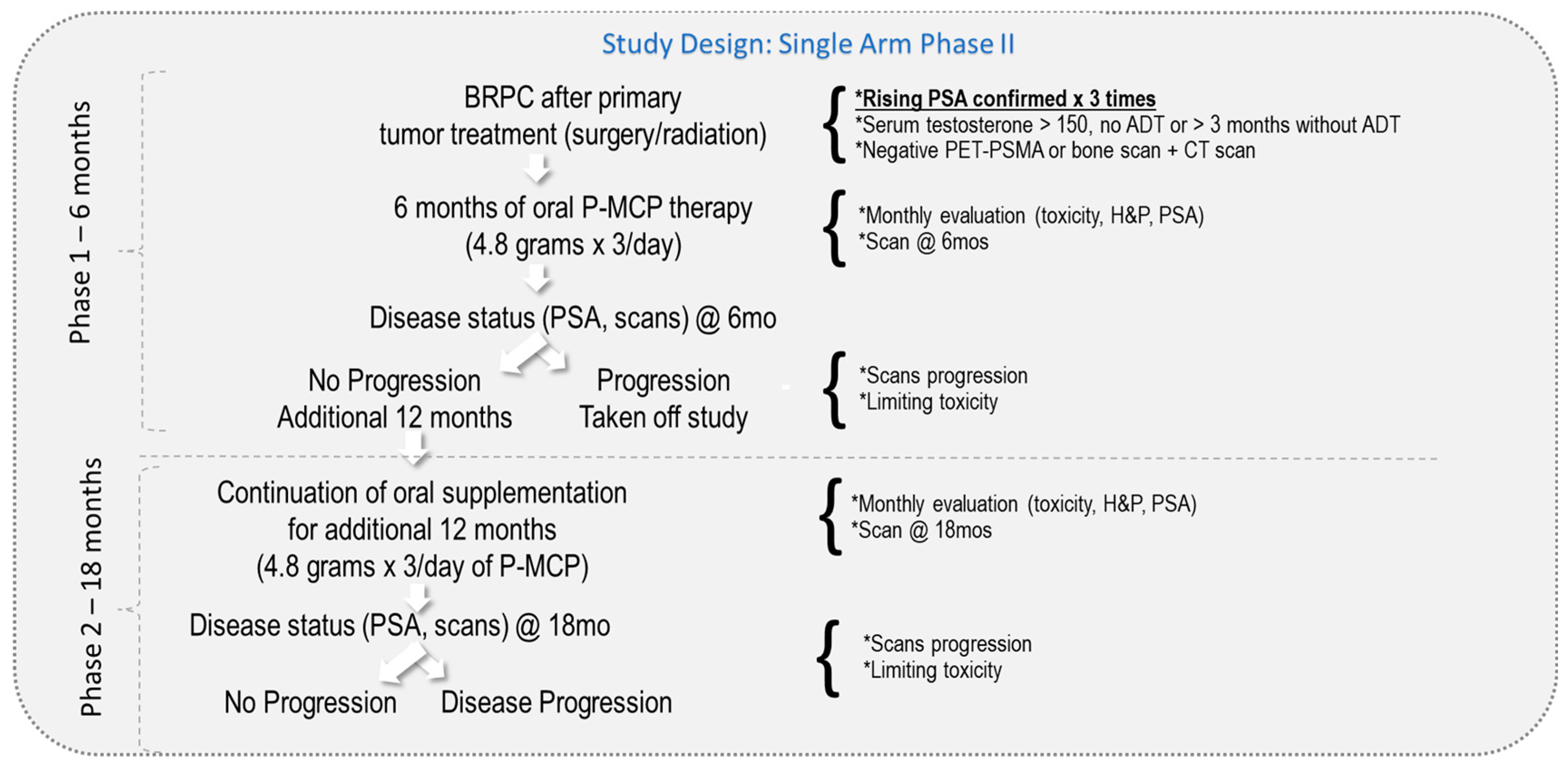
Figure 2.
Disease progression status after 18 months of PectaSol® Modified Citrus Pectin (P-MCP) therapy.
Figure 2.
Disease progression status after 18 months of PectaSol® Modified Citrus Pectin (P-MCP) therapy.
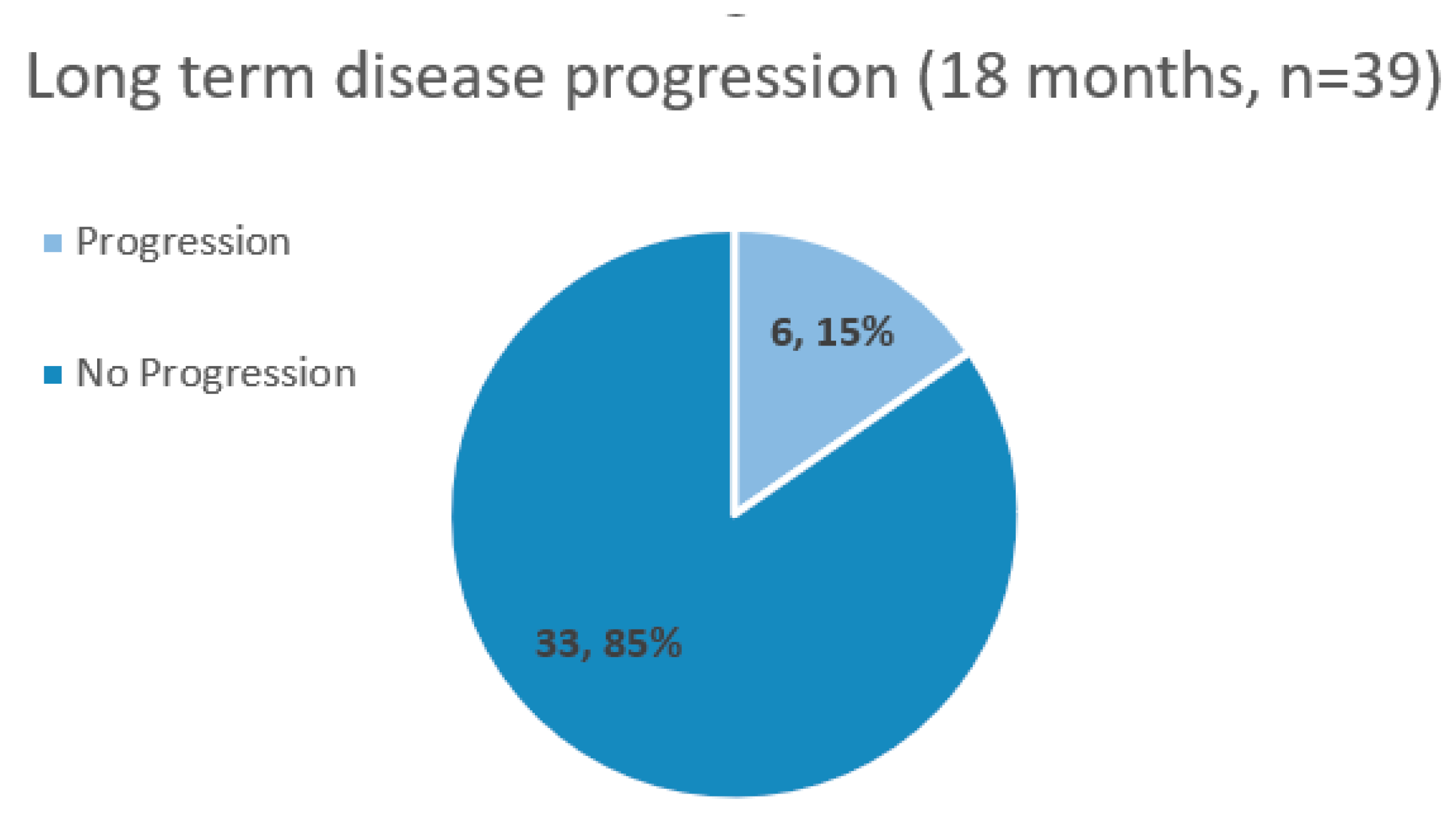
Figure 3.
The number of patients in each PSADT risk group at baseline and after 18 months of PectaSol® Modified Citrus Pectin (P-MCP) therapy.
Figure 3.
The number of patients in each PSADT risk group at baseline and after 18 months of PectaSol® Modified Citrus Pectin (P-MCP) therapy.
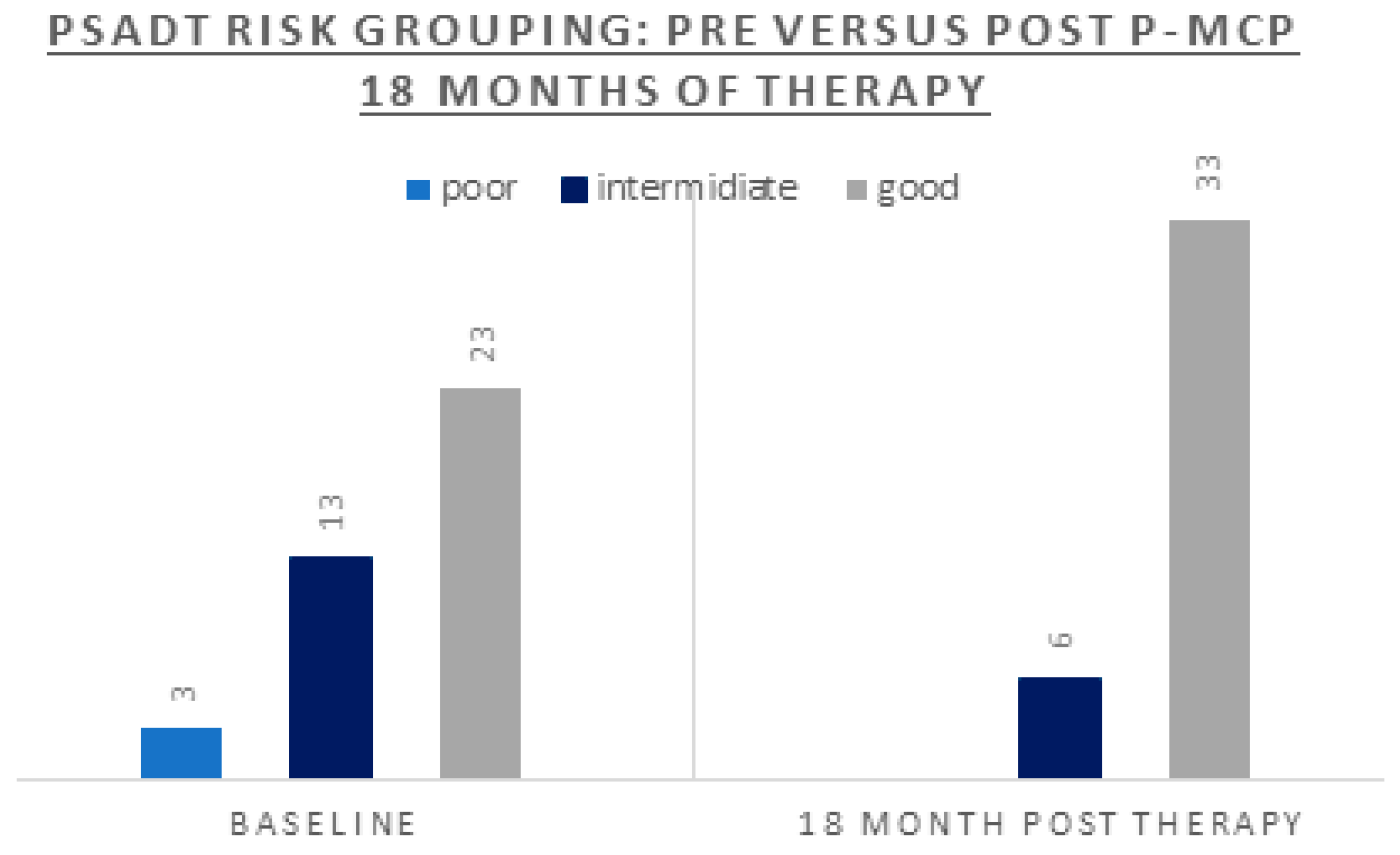

Table 1.
Pre-treatment (baseline) patient characteristics.
| Parameter | n = 39 |
|---|---|
| Age (years): Median (range) | 75 (52 - 88) |
| Gleason: % (n) 6 7 8-10 |
41% (n=16) 38% (n=15) 21% (n=8) |
| Local therapy: % (n) Radical prostatectomy Radiation therapy Surgery+RT |
18% (n=7) 54% (n=21) 28% (n=11) |
| Prior ADT | 38% (n=15) |
| PSA (ng/ml): Median (range) | 4.1 (0.28-30) |
| PSADT (months) risk grouping: % (n) Poor <3 Intermediate 3-8.99 Good ≥9 |
8% (n=3) 33% (n=13) 59% (n=23) |
| PSADT (months): Median (range) Whole cohort Poor PSADT risk Intermediate risk Good risk |
10.3 (1.4 – 55) 1.6 (1.4 – 1.8) 5.12 (3.5 – 8.2) 14.74 (9.10 – 54.6) |
Table 2.
Treatment characteristics and responses after 18 months of PectaSol® Modified Citrus Pectin (P-MCP) therapy.
Table 2.
Treatment characteristics and responses after 18 months of PectaSol® Modified Citrus Pectin (P-MCP) therapy.
| Parameter | Whole cohort (n=39) |
According to Pre-study PSADT (months) risk grouping |
||
|---|---|---|---|---|
| Poor < 3.00 (n=3) |
Intermediate 3.00-8.99 (n=13) |
Good ≥ 9.00 (n=23) |
||
| Overall response to therapy (decrease or stabilization of PSA, and/or lengthening of PSADT, with negative scans) |
85% (n=33) |
66% (n=2) |
77% (n=10) |
91% (n=21) |
| PSA response Stable/decreased Progression |
54% (n=21) 46% (n=18) |
67% (n=2) 33% (n=1) |
23% (n=3) 77% (n=10) |
70% (n=16) 30% (n=7) |
| PSADT (months): Median (range) | 43.5 (3.5-981) | 9.8 (6-200) | 18.3 (6.7-500) | 47.7 (3.5-981) |
| PSADT (months) risk grouping: % (n) | 0% (n=0) | 13% (n=5) | 87% (n=34 | |
| PSADT lengthening | 90% (n=35) | 100% (n=3) | 92% (n=12) | 87% (n=20) |
| Change to a better PSADT risk grouping | 36% (n=14) | 100% (n=3) | 85% (n=11) | not applicable |
| Radiologic response Negative scans Disease progression |
90% (n=35) 10% (n = 4) |
67% (n=2) 33% (n=1) |
75% (n=11) 15% (n=2) |
96% (n=22) 4% (n=1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



