
Submitted:
10 July 2023
Posted:
10 July 2023
You are already at the latest version
Abstract
Whole-genome sequencing (WGS) was used for genomic characterization of 110 strains of Listeria innocua isolated from 23 cattle farms, 8 beef abattoirs, and 48 retail outlets in Gauteng province, South Africa. In silico multilocus sequence typing (MLST) was used to identify the isolates' sequence types (STs). BLAST-based analyses were used to identify antimicrobial and virulence genes. The study also linked the detection of the genes to the origin (sources and types of samples) of the L. innocua isolates. The study detected 12 STs, 13 resistance genes, and 23 virulence genes. Of the 12 STs de-tected, ST637 (26.4%), ST448 (20%), 537 (13.6%), and 1085 (12.7%) were predominant, and the frequency varied significantly (P<0.05). All 110 isolates of L. innocua were car-riers of one or more antimicrobial resistance genes, with resistance genes lin (100%), fosX (100%), and tet(M) (30%) being the most frequently detected (P<0.05). Of the 23 virulence genes recognized, 13 (clpC, clpE, clpP, hbp1.svpA, hbp2, iap/cwhA, lap, lpeA, lplA1, lspA, oatA, pdgA, and prsA2) were found in all 110 isolates of L. innocua. Overall, diversity and significant differences were detected in the frequencies of STs, resistance, and virulence genes according to the origins (source and sample type) of the L. innocua isolates. This, being the first genomic characterization of L. innocua recovered from the three levels (farm, abattoir, and retail outlets) of the beef production system in South Africa, provides data on the organism's distribution and potential food safety implications..
Keywords:
beef production chain
; Listeria innocua
; whole-genome sequencing
; sequence type
; an-timicrobial resistance genes
; virulence gene
; South Africa
1. Introduction
Listeria species consist of a group of non-spore-forming Gram-positive facultative anaerobic coccobacilli [1]. There are 21 species of Listeria documented since 2020, but few are known to be pathogenic to animals and/or humans [2]. Listeria monocytogenes is the only recognized human pathogen and is also pathogenic to animals [3,4]. After con- sumption, several contaminated food types, such as milk and milk products, vegetables, meat, and meat products, have been implicated in listeriosis sporadic cases and/or out- breaks [5,6,7]. Some clinical manifestations of listeriosis in humans include fever, muscle aches, nausea, vomiting, stomach cramps, diarrhoea, abortion, preterm birth, stillbirth in pregnant women, meningitis or encephalitis, and death [8,9].
Currently, Listeria ivanovii is known to cause listeriosis in animals [10]. Although rare cases of human and animal infections were reported [11,12,13], L. innocua is not recognized as a human pathogen. Listeria innocua co-exists in the same food and environmental niches as L. monocytogenes, thus serving as indicator organisms for L. monocytogenes [14,15].
L. innocua has been documented to share some virulence factors with L. monocyte- genes [16], including some Listeria pathogenic islands (Listeria pathogenic island (LIPI)-1, LIPI-3, and LIPI-4) [17,18]. Virulence genes contribute to the virulence and pathogenicity of L. monocytogenes. Several virulence genes have been reported in strains of L. innocua, but the prevalence and roles of these virulence genes have not been elucidated [19].
Classical multi-locus sequence typing (MLST) targeting six to eight housekeeping/virulence genes and core genome-based (cg) MLST genotyping have been essential in epidemiological investigation and surveillance of L. monocytogenes isolates [20]. Numerous STs have been documented, mainly for L. monocytogenes. Since these housekeeping genes are also present in other Listeria spp., STs have also been assigned to L. innocua and other Listeria spp. [21,22]. Furthermore, some STs of L. monocytogenes have been associated with human listeriosis, and their occurrence and distribution are affected by geographical locations and food types [23,24].
Strain typing techniques such as traditional serotyping, multi-virulence-locus sequence typing (MVLST), MLST, multilocus variable number tandem repeat analysis (MLVA), and pulse-field gel electrophoresis (PFGE), among others, have been used to detect and characterize Listeria spp. [25]. However, whole-genome sequencing (WGS) and various in silico analyses based on WGS are currently the methods of choice for molecular sub-typing for Listeria spp. as they provide a higher resolution of strains over the other methods [26,27,28].
The use and abuse of antimicrobial agents in humans and animals have resulted in the expansion of bacterial antimicrobial resistance, and some bacteria are resistant to multiple antimicrobials [29]. The genes that encode antimicrobial resistance (AMR) in Listeria spp. are well documented [22,30]. Still, it is also known that the resistance genes carried by bacteria may not be expressed, limiting their importance in assessing their clinical or therapeutic significance [31]. Variable frequencies of AMR genes have been reported in strains of L. monocytogenes, L. innocua, and other Listeria spp. [32,33]. Hosain et al. [34] have highlighted the potential negative impact of AMR on therapy in feed ani- mals and humans. To date, there has not been any information on the genomic carriage of resistance genes by L. innocua in South Africa.
Between 2017 and 2018, South Africa experienced the world’s largest outbreak of human listeriosis, caused by ‘polony,’ a ready-to-eat (RTE) beef product [35], and L. mon- ocytogenes ST6 was determined to be the aetiological agent [24]. A few studies using poly- merase chain reaction (PCR) has characterized L. monocytogenes strains recovered from meat and meat products across the country [36] and MLVA genotypes of Listeria monoc- ytogenes and L. innocua isolated from farms, abattoirs, and retail in Gauteng province [37], Mpumalanga and North West provinces [38] were reported.
To the best of our knowledge, there is a dearth of information on the genomic char- acteristics of L. innocua strains in the country. Mafuna et al. [39] used WGS to character- ize 38 isolates of L. innocua recovered from the country’s meat and food processing facili- ties, while ElZowalaty et al. [40] reported the genomic sequence of one strain of L. innocua isolated from a healthy goat. The genomic characteristic of L. innocua at the levels of the beef production chain is currently unknown.
Therefore, the specific objectives of this study were to use WGS to characterize strains of L. innocua isolates from samples collected from cattle farms, beef abattoirs, and retail outlets in Gauteng, South Africa, to elucidate the diversity in the profiles of their sequence types, resistance genes, and virulence genes. The study also investigated the relationships of the profiles with the sources and sample types from which the isolates originated, and the profiles.
2. Materials and Methods
2.1. Study Design and Sources of Samples
Three cross-sectional studies were conducted at three levels of the beef production system in Gauteng province by collecting samples from 23 cattle farms (March-October, 2021), eight beef abattoirs (May-September, 2022), and 48 retail outlets (October 2019 – April 2021). The sample size was determined as recommended by Thrusfield [41]. De- tails on the types and number of samples collected from the sources mentioned earlier and types of samples in the current study have been documented [42].
2.2. Isolation, Identification of L. innocua, and Determination of AMR
All 110 isolates were previously identified (bacteriological and multiplex PCR) as L. innocua as described [43,44,45]. The confirmed isolates of L. innocua were inoculated in 50% brain heart infusion (BHI)/50% glycerol and stored at -20oC until subjected to whole genome sequencing (WGS) analyses. The number of isolates of L. innocua used in this study was 110 (11.1%) from 990 samples. The prevalence of L. innocua at the three levels of beef production was 10.4% (34/328), 5.7% (15/262), and 15.3% (61/400) for cattle farms, beef abattoirs, and retail outlets, respectively. The current study assessed all the isolates recovered from these sources and the types of samples.
In an earlier study [42] on the 110 isolates of L. innocua used in the current study, their phenotypic AMR to 16 antimicrobial agents was determined using the disc diffusion method. The antimicrobial agents used comprised penicillin (10 units), amoxicillin clavu- lanic acid (30 μg), ampicillin (10 μg), cephalothin (30 μl), cefotaxime (30 μg), streptomycin (25 μg), gentamicin (10 μg), kanamycin (30 μg), tetracycline (30 μg), doxycycline (30 μg), nalidixic acid (30 μg), ciprofloxacin (5 μg), enrofloxacin (5 μg), azithromycin (15 μg), clindamycin (10 μg), and sulfamethoxazole-trimethoprim (23.75/1.25 μg).
2.3. Whole-Genome Sequencing, Genomic Analysis, Assembly, and Annotation
DNA extraction was done using the Qiagen DNAEasy Blood & Tissue kit, manual, Gram-positive protocol as per the manufacturer’s instructions. All isolates were sequenced either on an Illumina NextSeq platform (150-bp paired-end reads; Illumina or on an Illumina MiSeq platform (250-bp paired-end reads; Illumina, Inc., San Diego, CA) using the Nextera XT library preparation kit per the manufacturer’s instructions.
Quality control, including adapter removal of the raw data, was done using BBDuk (v.38.91; https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbduk- uide/; sourceforge.net/projects/bbmap/). SPAdes v.3.15.3 [46] created a de novo assembly of each isolate. Only contigs longer than 500 bp were retained for further analysis. Completeness and contamination of the de novo assemblies were assessed with CheckM v.1.1.3 [47], and taxonomic classification was done using GTDB-Tk v.1.7.0 [48].
2.4. In Silico MLST
MLST STs were determined using the MLST tool (http://github.com/tseemann/mlst) [49] which makes use of the PubMLST website (https://pubmlst.org/) developed by Keith Jolley (Jolley & Maiden 2010, BMC Bioinformatics, 11:595) and sited at the University of Oxford.
2.5. Resistance, and Virulence Profiles
ABRicate (https://github.com/tseemann/abricate) [50] was used to detect antimicrobial resistance genes, and virulence factors in species of interest. Abricate was run with default parameters, and the NCBI database was selected for AMR detection. This database was locally updated on 2 November 2022 and, at the time of usage, including 6,334 sequences (doi: 10.1128/AAC.00483-19). For virulence factors, the “vfdb” database was used, updated on 2 November 2022, and containing 4,332 sequences (doi:10.1093/nar/gkv1239).
2.6. Construction of the Phylogenetic Tree for L. innocua Isolates and Correlation with Source and Type of Samples
2.7. Data Analysis
All data analyses were done using R v.4.1.2 [52] implemented In RStudio v.2022.2.3.492 [53]. Distance matrices were calculated using the “daisy” function with the “gower” parameter specified to determine Gower distances with the R package “cluster” [54]. Minimum spanning trees were calculated using the “ape” package [55], with the “mst” function, and visualized using “igraph” [56] and “ggnetwork” [57]. R packages ggstatsplot [58], ggsci [59], and ggpubr [60] were further used for data analyses and visualization. Balloon plots were constructed using the ggballoonplot function available from ggpubr. The ggstatsplot function, ggscatterstats, was implemented to perform correlation analyses based on Pearson's correlation coefficient. Pearson's Chi-squared Test for Count Data, implemented by the chisq.test, was used to test for associations. Pearson's correlation coefficients were calculated using the cor function. Bar charts were produced using the ggstatplot function ggbarstats and Pearson's chi-squared test used to test for significant differences.
3. Results
3.1. Frequency of STs of L. innocua
For samples collected from the three levels of beef production, 12 STs were detected in the 110 isolates of L. innocua, and no ST could be assigned in two isolates (1.8%) (1 each from the farm and retail outlet sources). The frequency of STs found was as follows: ST637 (29 isolates, 26.4%), ST448 (22, 20%), ST537 (15, 13.6%), ST1085 (14, 12.7%), ST1489 (8, 7.3%), ST1482 (7, 6.4%), ST1008 (5, 4.5%), ST1610 (4, 3.6%), and ST1619, ST602, ST1087 and ST43 (1 isolate each, 0.9%). The differences in the frequencies of STs observed across all the samples were statistically significant (P<2.2e-16).
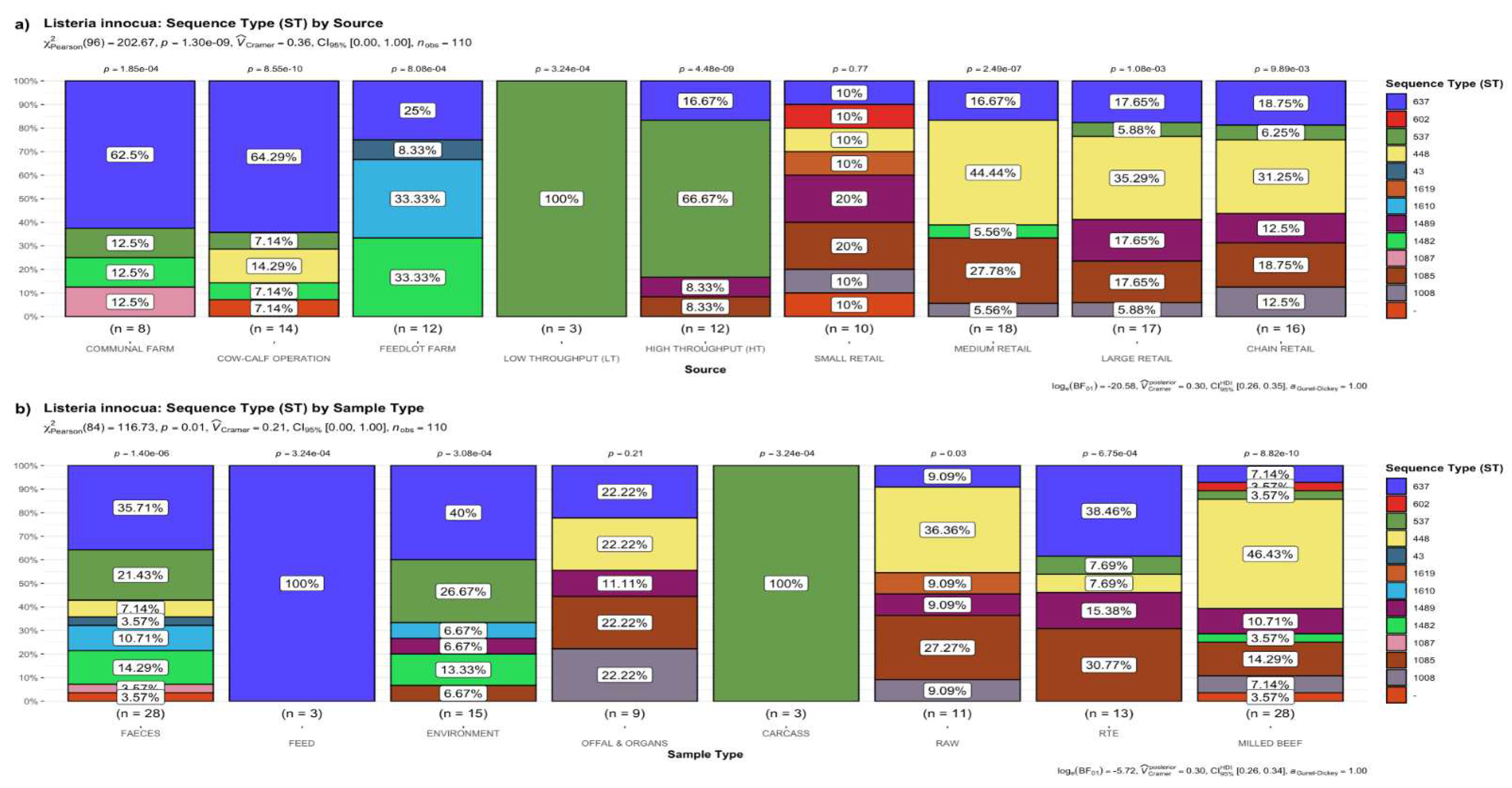
3.1.1. Distribution of STs by the Source of Samples
The proportion of STs in L. innocua isolates within each of the nine sources varied significantly (P<0.05), except for those recovered from the small retail outlets (P=0.77), as shown in Figure 1a.
The associations of STs with the nine sources were statistically significantly higher for ST1087 (communal farm), ST1482 (feedlot), ST1610 (feedlot), ST43 (feedlot), ST537 (low throughput abattoir), ST537 (high throughput abattoir), ST1619 (small retail outlet), ST602 (small retail outlet), ST448 (medium retail outlet), ST537 and ST637 (cow-calf farm).
3.1.2. Distribution of STs by the Type of Sample
The distribution of STs among L. innocua isolates according to the types of samples from which they originated is shown in Figure 1b. Within each of the 8 sample types, the proportion of STs of L. innocua was highly statistically significantly different (P<0.05) except for Offal and Organs.
Across the eight food types, the association of STs with certain sample types was statistically significant (P<0.05) for ST637 (feed), ST537 (carcass), ST448 (milled beef), ST1619 (raw beef), and ST1008 (offal and organs) (Supplementary data: Table S1).
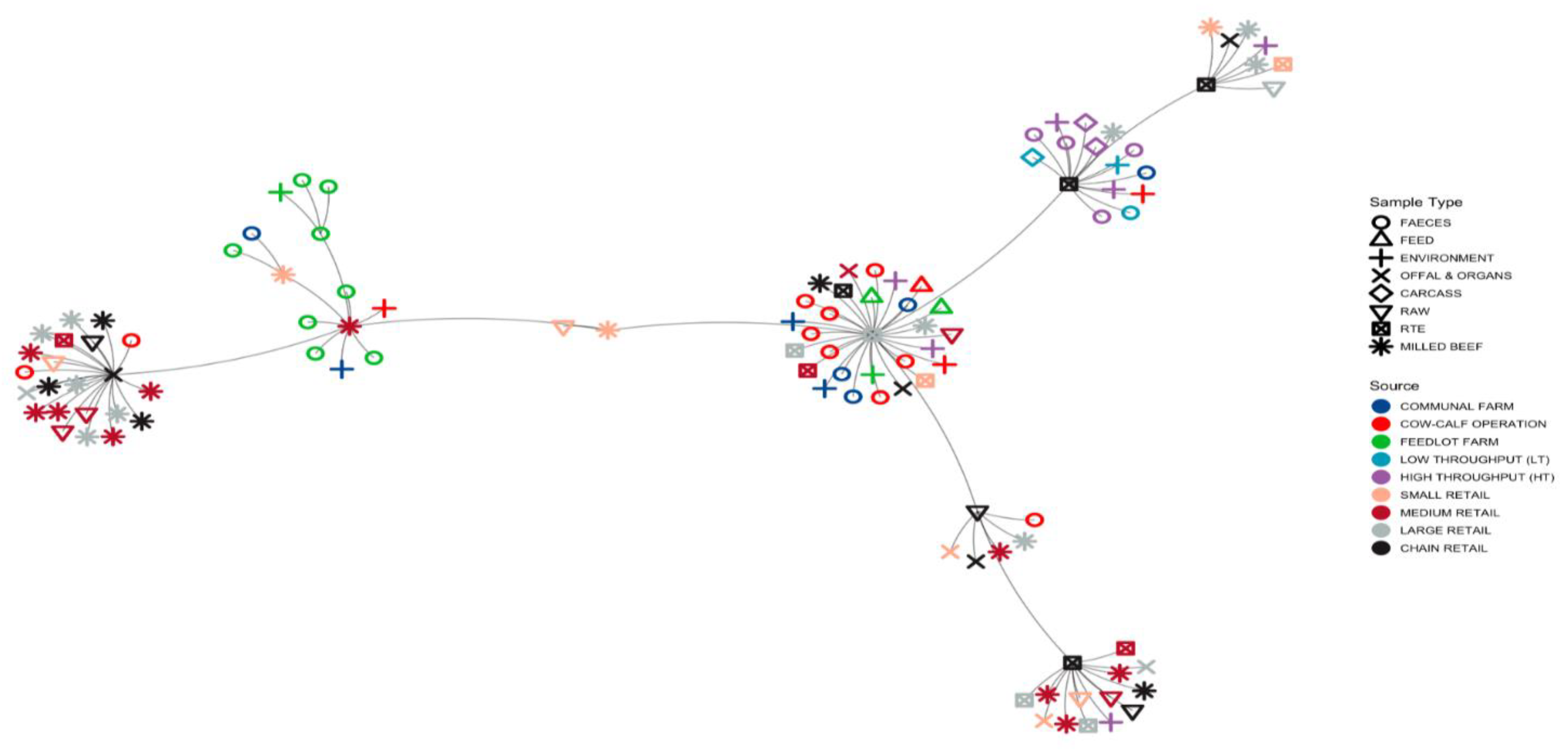
3.1.3. Minimum Spanning Tree (MST) Based on ST Profiles
The MST of the STs of L. innocua isolates is displayed in Figure 2. Clear clustering of the samples based on the ST profiles was evident. The origin of the samples based on “Sample Type” and “Source” further highlighted the composition of the clusters. Overall, seven distinct clusters with two larger clusters appear to contain isolates from different sample types and sources, while the smaller clusters were homogenous. Three of the clusters were predominantly filled with samples from retail outlets. This indicates a propensity of certain ST profiles in this environment. This is the case with clusters biased toward farming or production environments.
Figure 1.
Frequency of L. innocua sequence types by source and sample type. L. innocua STs grouped by source: The STs displayed a significant deviation from the ex- pected distribution across Sources. All Sources, except for Small Retail, were found to be significantly associated with specific STs (Figure 1a). L. innocua STs grouped by sample type: A significant deviation from the expected ST distribution was also found for the ST by Sample type distribution. All sample types, except Offal & Organs, were found to be significantly associated with certain STs (Figure 1b).
Figure 1.
Frequency of L. innocua sequence types by source and sample type. L. innocua STs grouped by source: The STs displayed a significant deviation from the ex- pected distribution across Sources. All Sources, except for Small Retail, were found to be significantly associated with specific STs (Figure 1a). L. innocua STs grouped by sample type: A significant deviation from the expected ST distribution was also found for the ST by Sample type distribution. All sample types, except Offal & Organs, were found to be significantly associated with certain STs (Figure 1b).
The clustering of samples based on ST is evident. The shape and colour of each sample indicate the Sample Type and Source, respectively. The smaller clusters are relat- ively homogenous regarding Sample Type and Source. In particular, three smaller clusters are associated with the sample from Retail environments.
3.2. Detection of Antimicrobial Resistance Genes in L. innocua
Thirteen resistance genes were detected in the 110 L. innocua isolates from the sources tested. They were as follows: fosX, 110 (100%), lin, 110 (100%), tet(M), 33 (30%), dfrG, 9 (8.2%), ImuD, 6 (5.5%), mphB, 5 (4.5%), mefA, 4 (3.6%), msrD, 4 (3.6%), tet(S), 4 (3.6%), ant.6.1a, 1 (0.9%), InuG, 1 (0.9%), vatB, 1 (0.9%), and vga, 1 (0.9%). The differences were statistically significant (p<0.05). The details of the sources, sample types, sequence types, and resistance genes identified in the 110 L. innocua isolates are shown in Supple- mentary data: Table S1.
3.2.1. Distribution Frequency of Antimicrobial Resistance Genes by Source
Balloon plots of the 13 resistance genes distribution by source are in Figure 3. Within each of the nine sources investigated, high diversity of the resistance genes was detected in feedlots, 61.5% (8/13), and small retail outlets, 53.8% (7/13), compared with the low diversity of 33.3% (3/9) found in communal farms and LT abattoirs. The differences were statistically significant (P=0.0302). Across the nine sources, the frequency of resistance genes (fosX and lin) was high, 100% (9/9) but low, 11.1% (1/9) for six genes (vatB, vgaB, msrD, InuD, mefA, and ant6.1a). The difference was statistically significant (P=0.0004).
3.2.2. Distribution Frequency of Antimicrobial Resistance Genes by Type of Sample
Figure 4 shows the frequency distribution of resistance genes by the type of samples. Faeces, environmental samples, and offal and organ samples yielded L. innocua with high carriage of resistance genes at 69.2% (9/13), 69.2% (9/13), and 61.5% (8/13), respectively, compared with that for feeds and carcasses, 23.1% (3/13). The difference was statistically significant (P=0.018). Across the 8 sample types, resistance genes fosX, lin, and tet(M) were each detected in 8 (100%), indicating co-occurrence, while only 1 (12.5%) sample type was positive for genes vatB, vgaB, InuG, and ant.6.1a. The difference was statistically significant (P=0.0014).
3.2.3. Patterns of Multiple Antimicrobial Resistance Genes
For the 110 isolates of L. innocua recovered from the farms, abattoirs, and retail outlets, nine antimicrobial resistance gene patterns with a range from 2-7 resistance genes per pattern were as follows, foxX-lin, 65 (59.1%), fosX-lin-tetM, 27 (24.5%), dfrG- fosX-lin, 8 (7.3%), fosX-lin-Inu.G-tet.M, 2 (1.8%), dfrG-fosX-lin-tetM, 1 (0.9%), fosX-lin-InuD-tet.M, 1 (0.9%), fosX-lin-tetM-vat.B-vga.B, 1 (0.9%), ant.6.Ia-fosX-lin-tet.M-mph.B, 1 (0.9%), and fosX-lin-InuD-mefA-mph.B-msrD-tet.S, 4 (3.6%) (p<0.05).
3.2.4. MST for AMR
The MST for the AMR profiles of L. innocua isolates according to the sources and sample types is shown in Figure 5. This analysis grouped the isolates into two major clusters with more than 25 isolates each and four minor clusters. The largest cluster contained isolates from all sample types except carcass and all sample sources, while the second largest cluster contained isolates from all sample types and sources. The two smaller clusters are predominantly associated with farming and retail environments. The clustering observed indicates the sharing of AMR profiles across all sectors. Each minor cluster comprised 8, 4, 1, and 1 isolates from limited sample types and sources. The smaller clusters contained samples from similar environments, highlighting some unique AMR profiles in isolates associated with these environments.
3.2.5. Putative Resistance Phenotypes
The frequency of putative resistance phenotypes detected in the 110 isolates of L. innocua consisted of trimethoprim, 8.2% (9/110), tetracycline, 33.6% (37/110), streptomycin, 0.9% (1/110), streptogramin, 0.9% (1/110), macrolide, 4.5% (5/110), lincosamide, 100% (110/110), fosfomycin, 100% (110/110), and erythromycin, 3.6% (4/110). The differences in the frequencies of putative resistance phenotypes were statistically significant (p<0.05).
Putativce phenotypes by the source of isolates
The frequency of putative resistance phenotypes detected by the source of the isolates is shown in Figure 6a. For each of the nine sources studied, the frequencies of the putative resistance phenotypes were significantly associated with each particular source (p<0.05).
Putative resistance phenotypes by the sample type
The frequency of putative resistance phenotypes detected is shown in Figure 6b. All eight sample types showed significant under- or over-representation of resistance pheno- type (p<0.05).
3.3. Detection of Putative Virulence Genes
Twenty-three putative virulence factors (clpC, clpE, clpP, fbpA, gtcA, hbp1.svpA, hbp2, iap. cwhA, lap, llsA, llsB, llsD, llsG llsH, llsP, llsX, llsY, lpeA, lplA1, lspA, oatA, pdgA, and prsA2) previously identified in L. monocytogenes were detected across the 110 isolates of L. innocua from farms, abattoirs, and retail outlets.
Among the 34 isolates from cattle farms, 19 (82.6%) of 23 genes were detected, except for llsB, llsD, llsP, and llsY. The frequency of detection of virulence factors was from 0% (0/34) to 100% (34/34) in 14 genes.
For the 15 abattoir isolates, all 23 (100%) virulence genes were detected, with a range from 6.7% (1/15) for llsB, llsD, llsP, and llsY to 100% (15/15) for 15 genes.
For the 61 isolates of L. innocua from retail outlets assessed for virulence genes, all 23 (100%) genes were detected, and the frequency range was from 8.2% (1/61) for llsP to 100% (61/61) in 14 genes (p<0.05).
Overall, the frequency of virulence genes detected across the three levels of beef production ranged from 82.6% - 100%, but the differences were not statistically signifi- cant (P=0.109); however, the frequency of the types of virulence genes varied significantly- in 14 genes (P<0.001).
Supplementary data: Table S2 shows the details of the sources, sample types, sequence types, and virulence factor genes identified in the 110 L. innocua isolates.
3.3.1 Occurrence of virulence genes by source
Within each of the nine sources, all (100%) the isolates of L. innocua that originated from HT abattoirs, small retail outlets, and large supermarkets were positive for the 23 virulence genes detected, while 19 (82.6%) of the 23 virulence genes were detected in five sources (communal farms, cow-calf farms, feedlots, LT abattoirs, and medium retail outlets) (P=0.1085) (Figure 7).
3.3.2. Virulence Genes by Sample Type
Within the food types, all the samples that originated from sample types, 5 (55.6%) samples (environment, offal and organs, raw beef, RTE, and milled beef) were each positive for all (100%) 23 virulence genes. (Figure 8). However, only 15 (65.2%) of the 23 virulence genes were found in feed samples (P=0.038).
3.3.3. MST Based on Virulence Gene Profiles
The MST based on the virulence gene profile of each isolate is shown in Figure 9. As with the previous trees, certain profiles are unique to similar environments. A similar clustering pattern was observed for the virulence factor profile here; two large clusters are evident. Four distinct clusters, with two larger clusters each, made up of isolates from all the different sample sources and types, while the two smaller clusters, each con- tained five (from 4 different sample types and three sources) and seven (from 3 sample types and 3 sources) isolates, respectively. The large clusters indicate the sharing of viru- lence gene profiles across various environments, with smaller clusters alluding to unique profiles for certain environments.
3.4. Distribution of Virulence Genes by Sequence Type
For the 108 isolates for which the STs were determined, five patterns of carriage of virulence genes by ST were detected (Figure 10).
Pattern 1 comprised 44 (40.7%) isolates recovered from the farms, abattoirs, and outlets distributed across three STs. The four STs were 637 (n=29, 26.9%), 1085 (n=14, 13%) and 602 (n=1, 0.9%). Fifteen (65.2%) of the 23 virulence genes were detected: clpC, clpE, clpP, fbpA, gtcA, hbp1.svp, hbp2, iap.cwhA, lap, IpeA, IpIA1, IspA, oatA, pdgA, and prsA2.
Pattern 2: consists of 51 (47.2%) isolates recovered from farms, abattoirs, and retail outl- ets, spread across six STs. These are STs 448 (n=22, 20%), 537 (n=15, 13.6%), 1482 (n=7, 6.4%), 1008 (n=5, 4.5%), 1619 (n=1, 0.9%), and 43 (n=1, 0.9%). A total of 19 (82.6%) of the 23 virulence genes were detected in this pattern: clpC, clpE, clpP, fbpA, gtcA, hbp1.svp, hbp2, iap.cwhA, lap, llsA, llsG, llsH, llsX, IpeA, IpIA1, IspA, oatA, pdgA, and prsA2
Pattern 3:has only 8 (7.4%) isolates recovered from abattoirs and retail outlets. All (100%) of the 23 STs were detected.
Pattern 4: consists of 4 (3.7%) isolates from farms and detected ST 1610 only. Eighteen (78.3%) virulence genes were found: clpC, clpE, clpP, fbpA, hbp1.svpA, hbp2, iap.cwhA, lap, llsA, llsG, llsH, llsX, IpeA, IpIA1, IspA, oatA, pdgA, and prsA2.
Pattern 5: composed of 1 (0.9%) isolate from 18 (78.3%) virulence genes detected were clpC, clpE, clpP, fbpA, gtcA, hbp1.svp, hbp2, iap.cwhA, lap, llsA, llsG, llsH, IpeA, IpIA1, IspA, oatA, pdgA, and prsA2.
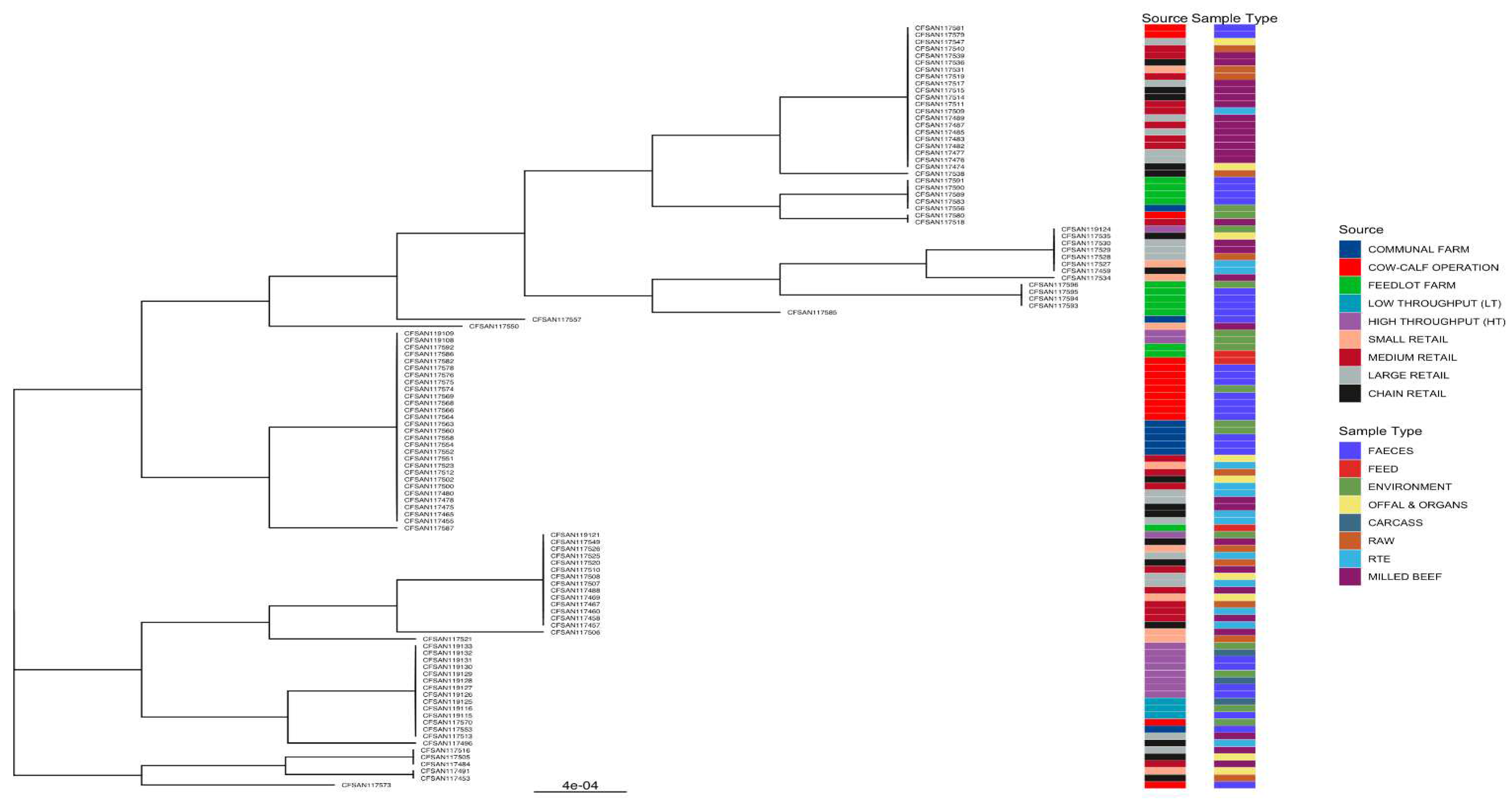
3.5. Phylogenies of L. innocua Isolates According to the STs
The genetic relationships of the L. innocua isolates recovered from three beef production levels and several types of samples are shown in Figure 11. The tree indicated grouping based on phylogenetic relatedness with distributions across various environments for the groups. The isolates formed five distinct clusters, which appear to be a mix of the different sample types and sources. Some smaller clusters indicated a propensity for specific environments, with the larger clusters containing a diverse spread of sample origins. This could indicate an inclination toward certain L. innocua iso- lates to environments with others distributed across disjoint origins. Overall, the phylo- genetic tree suggests that the positioning of the isolates of L. innocua isolates in the phylo- genetic tree did not depend on the sample type or source.
Similar to the MST trees, there seems to be a subset of isolates unique to particular origins, with the larger isolate groupings, found distributed across many sample origins.
Figure 3.
Balloon plots showing the distribution of resistance genes by source.
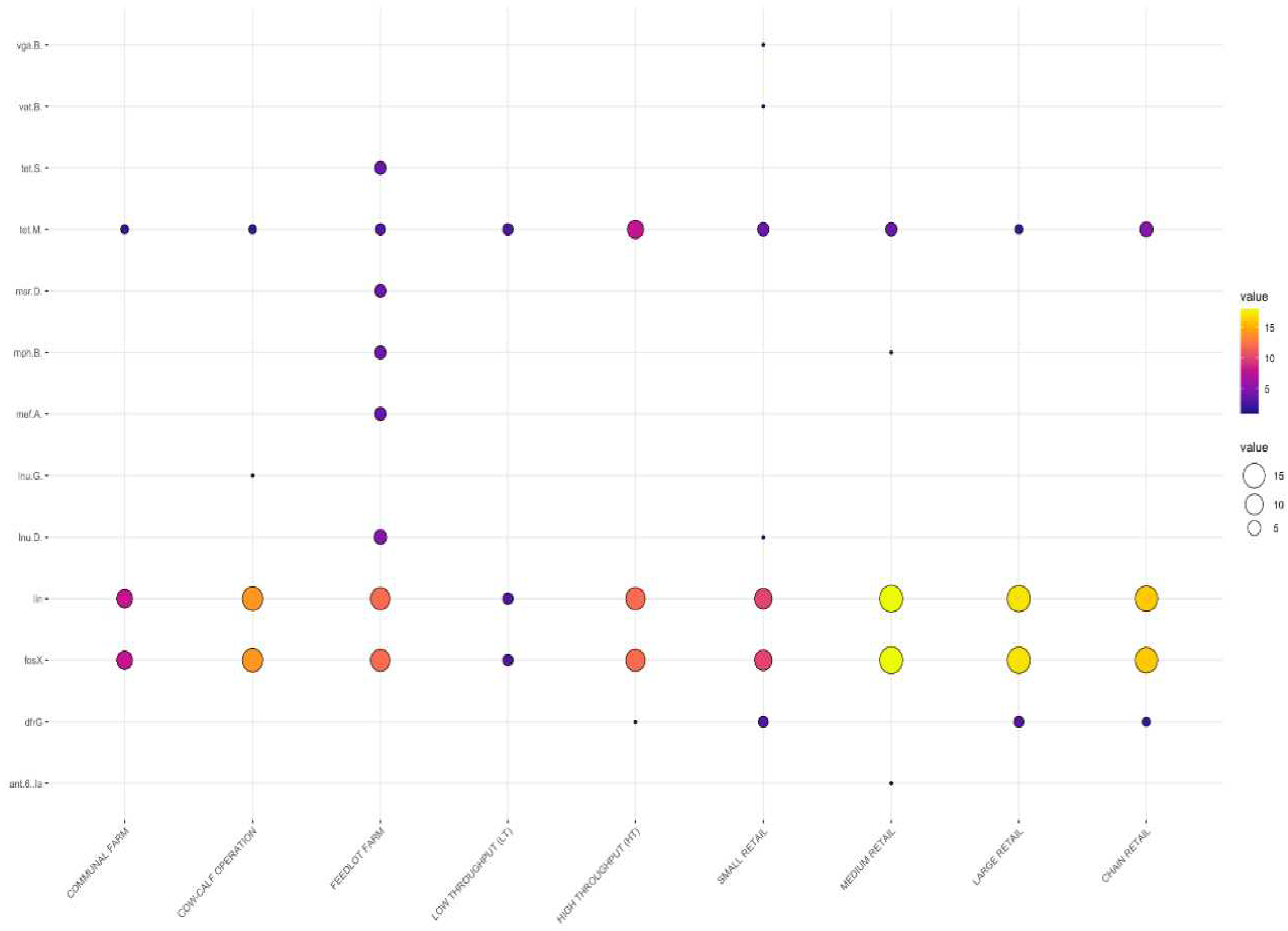
Figure 4.
Balloon plots showing the distribution of resistance genes by sample type.
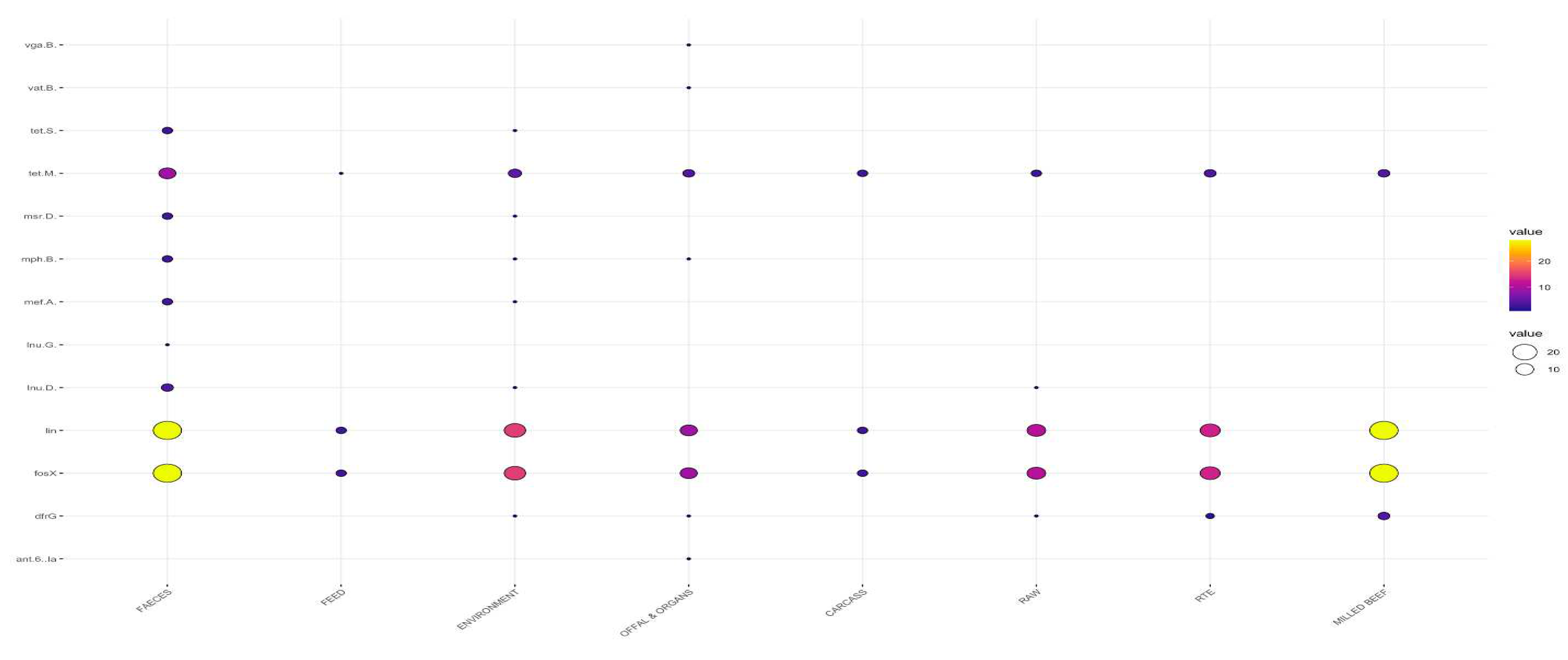
Figure 5.
Minimum spanning tree for AMR of L. innocua isolates.
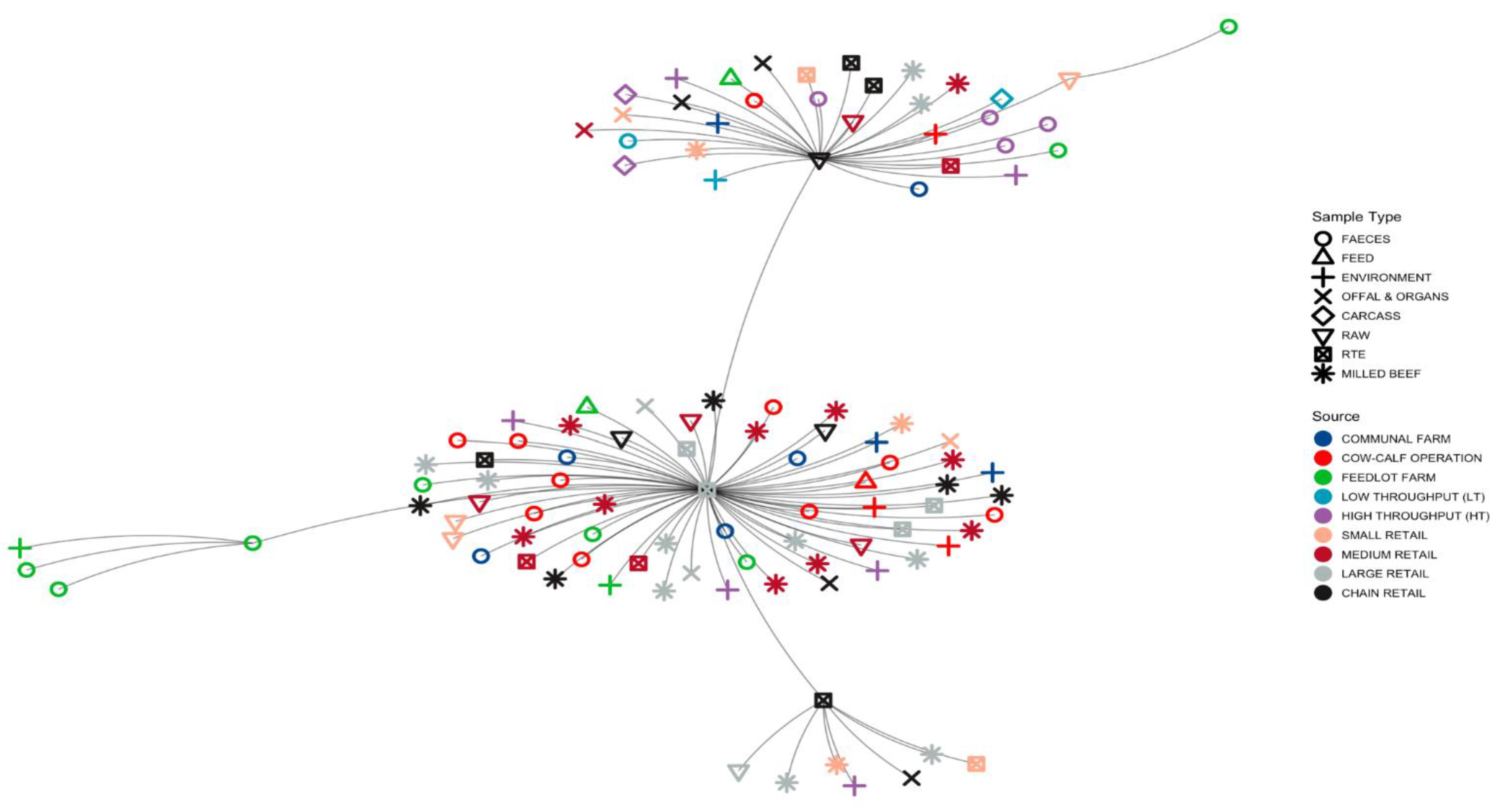
Figure 6.
Putative resistance of L. innocua by source and type of samples.
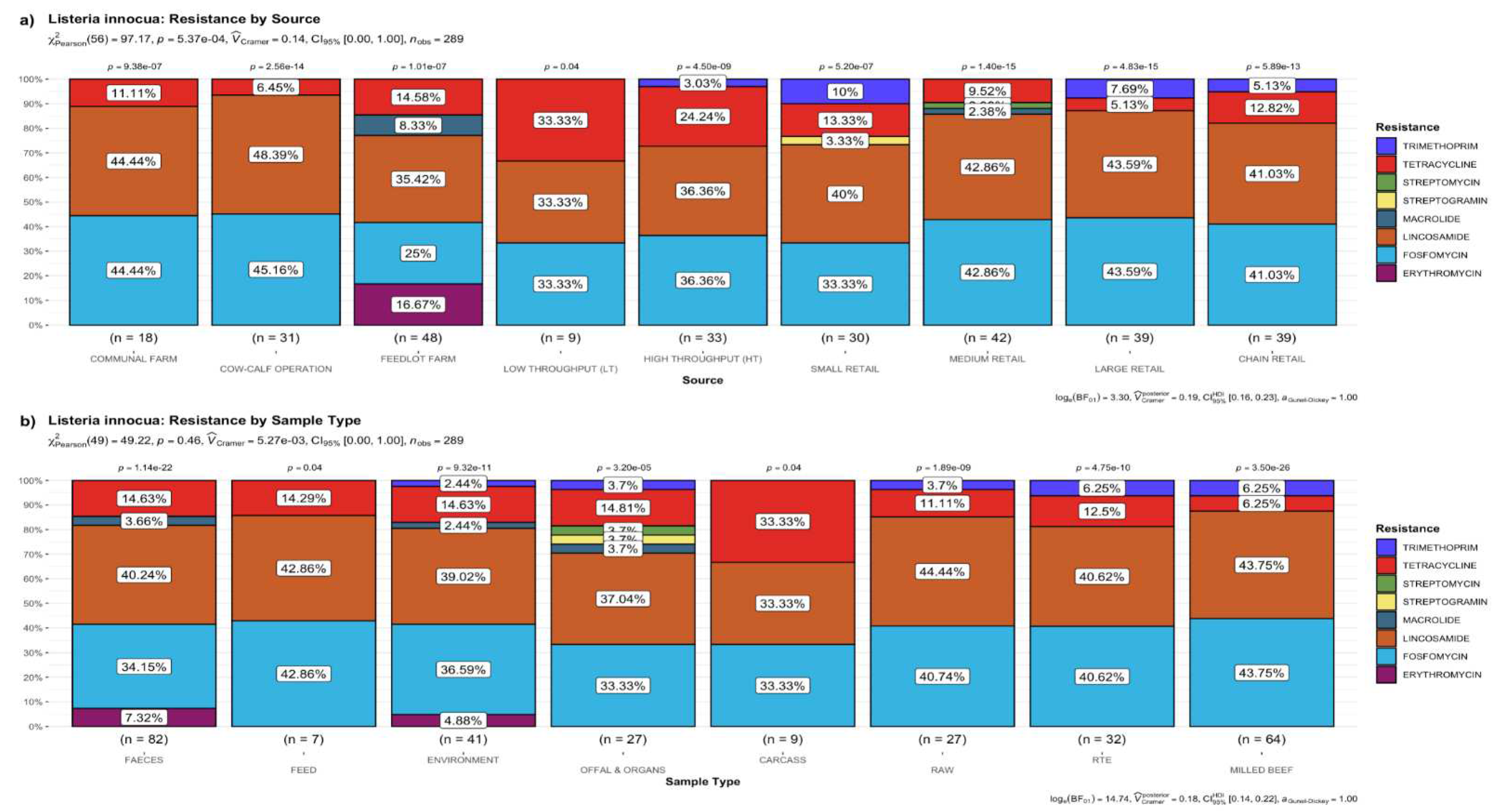
Figure 7.
Balloon plot of virulence factors by source.
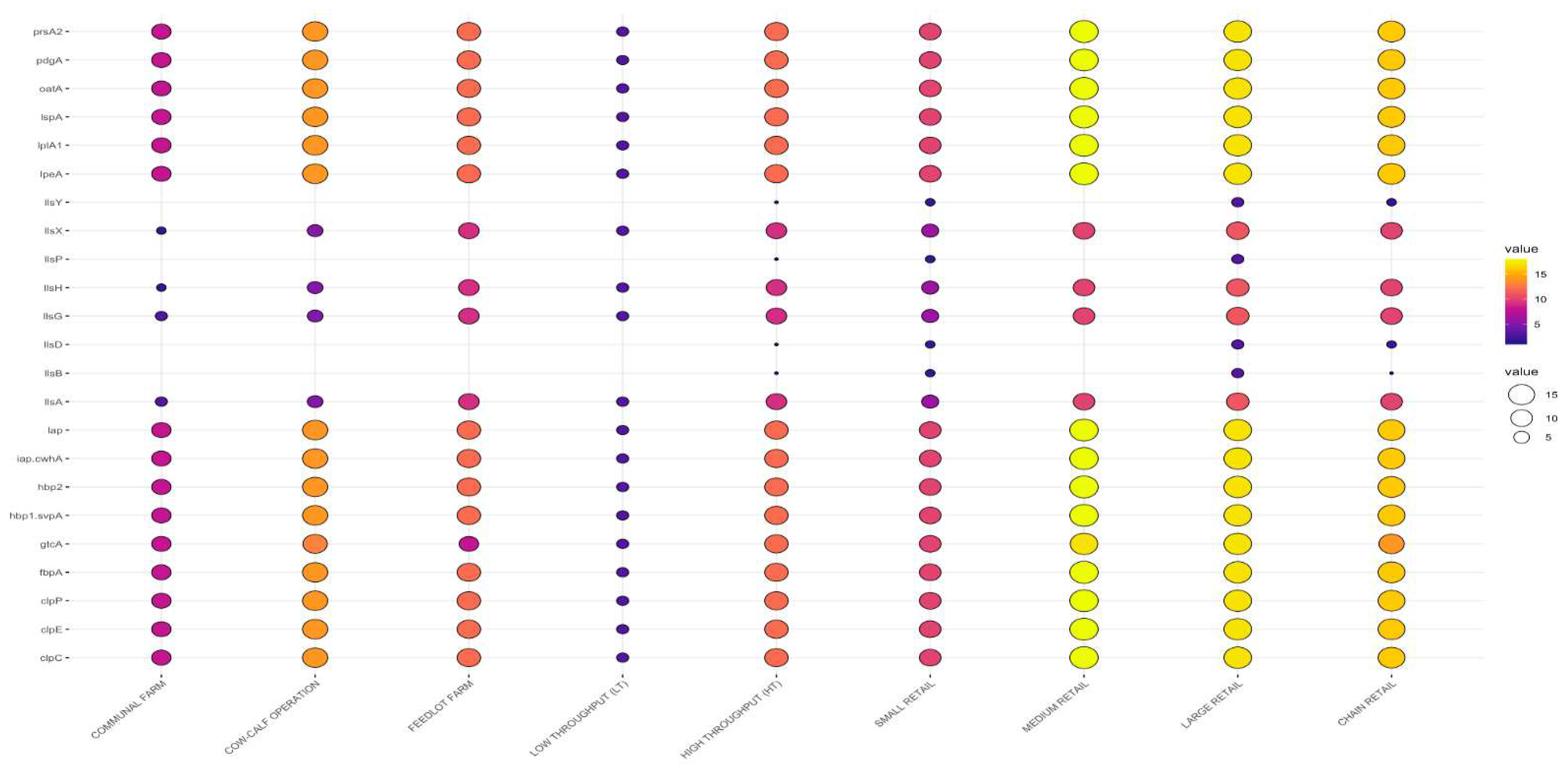
Figure 8.
Balloon plot of virulence factors by sample type.

Figure 9.
Minimum spanning tree of the virulence gene profiles of L. innocua isolates.
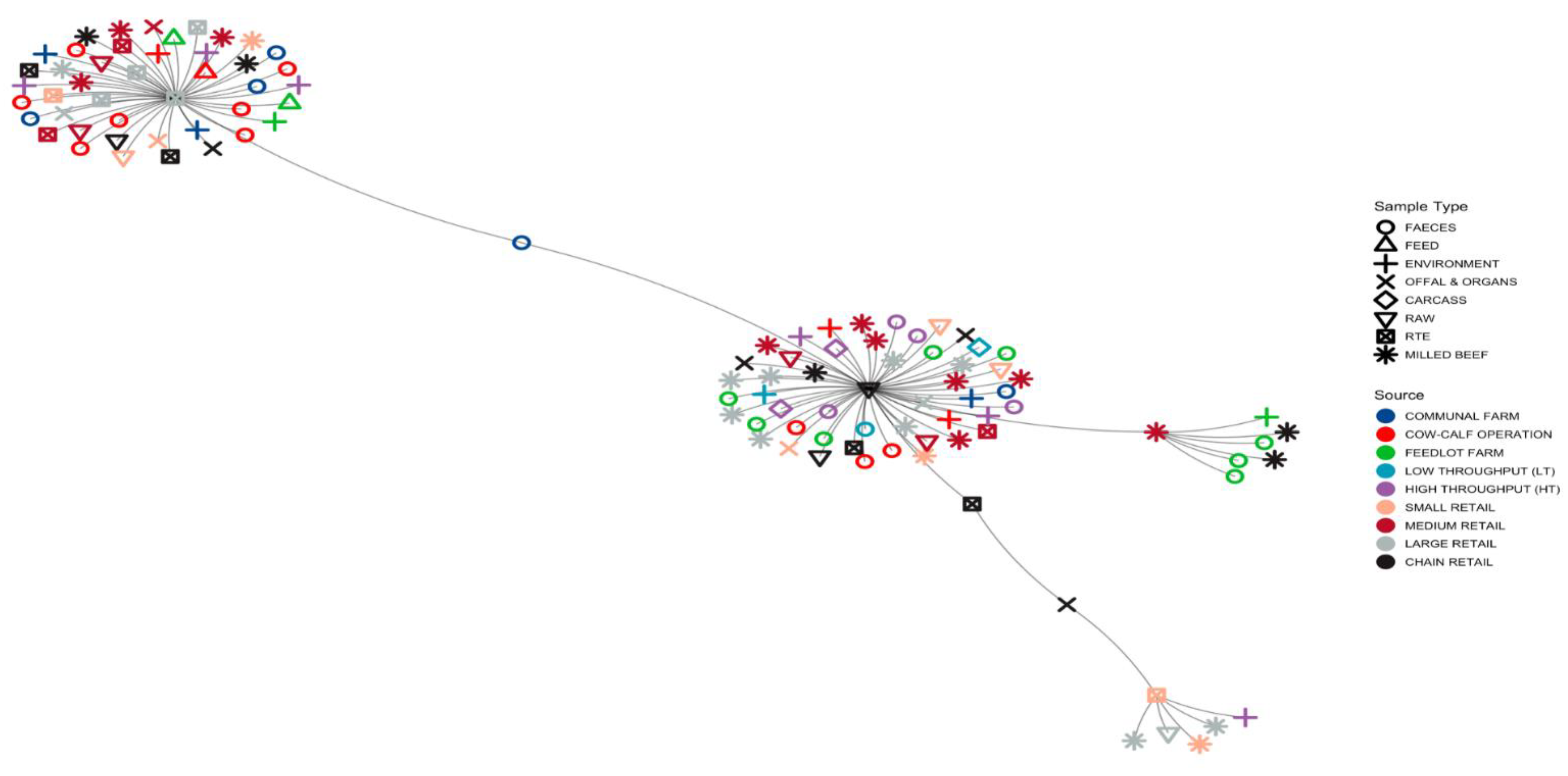
Figure 10.
Balloon plots of virulence genes and ST of L. innocua isolates.
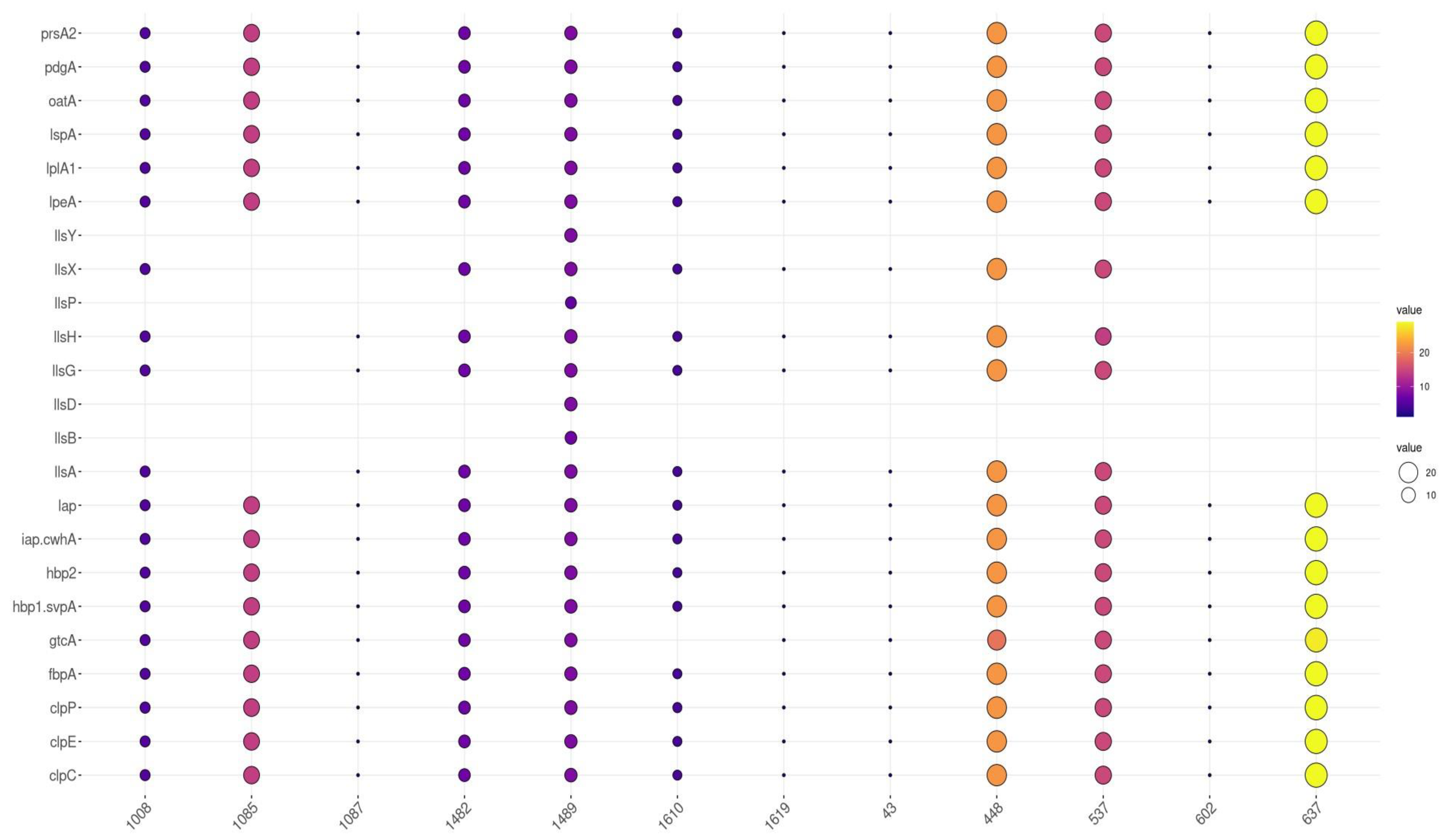
Figure 11.
Phylogenic tree of 110 isolates of L. innocua by source and type of samples.
4. Discussion
The current study is the first comprehensive study undertaken in South Africa on L. innocua recovered from three levels of cattle production (cattle farms), beef abattoirs (cattle slaughter), and beef/beef products retailing (retail outlets), concerning the genomic characterization of sequence types, resistance genes, and virulence genes. Both L. monocytogenes and L. innocua occupy the same niche in foods [14,15]; the detection of L. innocua, indicates the possible presence of L. monocytogenes in foods. Unlike the present study, the few published genomic characterization of Listeria species were studies cond- ucted on L. monocytogenes strains recovered from the 2017-2018 large outbreak of human listeriosis [35,61], the report by Mafuna et al. [39] on the strains of L. innocua and L. welsh- imeri isolated from meat and food processing facilities in the country and the sequencing of one isolate of L. innocua from a healthy goat [40]. The current study provides important data on L. innocua in the country's farm-abattoir-retail association. In other countries, L. innocua isolates recovered from meat are characterized using molecular methods [22,62,63]. It is interesting that in our study, the predominant STs of L. innocua detected differed significantly as to the source of isolates, ST637 (cattle farms), ST537 (abattoirs), and
ST448 (retail outlets). This is in comparison to the L. innocua isolates obtained from retail outlets in Gauteng province, where nine STs were identified, of which ST448 (33.3%), ST1085 (23.3%), and ST637 (15%) were prevalent. Mafuna et al. [39] also identified nine STs, of which the most common were ST537 (56%) and ST1085. Also, only four STs (ST537, ST637, 448, and 1085) were common in both studies. The differences in the STs detected between both studies may be explained partly by the types of samples collected (beef versus meats), the source (retail outlets versus food processing facilities), and the number of locations (one province versus nine provinces). Reports by others have documented diversity in the STs, and their frequencies are affected by the geographical location, source, and types of samples from where the isolates originate, among other factors [23,36]. In our study, resistance genes fosX (100%), lin (100%), and tet(M) (30%) were predominantly detected. Similarly, Hanes and Huang [64] reported that in the USA, from 2010 through 2021, data analysis identified fosX, lin, abc-f, and tet(M) as the four most AMR genes found in L. monocytogenes. Compared with published reports on resistance genes in L. innocua, the distribution of the resistance genes varied considerably [32,33].
In our study, it is important that there was a high diversity of resistance genes detected in the isolates of L. innocua obtained from feedlots (61.5%) compared to the low diversity of resistance genes found in the isolates from communal farms. This is no surprise because animals at intensively managed feedlots receive cattle from diverse sources (farms and auctions) mostly experience antibiotic pressure to control infections and disease. On the other hand, the communal farms in South Africa rear fewer cattle (<10 per herd) in extensive or semi-intensive management systems with minimal antimicrobial agents use, often dictated by financial limitations posed to farmers by the cost of treatment.
The significantly higher diversity of AMR genes detected in L. innocua recovered from faecal and environmental samples may be explained partly by the fact that some of the faecal samples were pooled from around the feeding areas and environmental water and effluent samples; thus, a sample may have originated from several animals. Reports by others support our findings, where the frequency and distribution of resistance genes of L. innocua varied considerably by the types of samples from where the isolates origin- ated [34,65].
Our study also revealed that the frequency of resistance genes was significantly affected by the STs of the L. innocua isolates in five STs 637, 1482, 537, 1008, and 1489. It is also interesting to have detected ST-specific AMR genes as demonstrated by the presence of gene dfrG only in L. innocua ST1489 and the four isolates that belonged to ST1610 were each carrier of multi-drug resistance (MDR) genes (fosX, lin, inuD, metA, mph, msrD, and tetS) in all four ST 1610 isolates. The association of resistance genes with STs has been documented by others [66]. Regardless of the STs, it is of potential therapeutic significance that nine MDR genes were detected in our study ranging from two to seven genes per isolate. Palaiodimou et al. [66] have also reported the occurrence of MDR genes bcrABC, emrC, and qacH and emphasized the risk of the AMR and MDR transfer to other bacteria, including L. monocytogenes [62,67].
Of the three predominant resistance genes (fosX, lin, and tet(M), putative resistance to fosfomycin and tetracycline appears to be pertinent to South Africa because these antimicrobial agents are inexpensive, readily available, and used by farmers on livestock in the country [68,69]. However, tetracycline is the country's most frequently used antimicrobial gene on livestock. Therefore, the detection of 30% of the L. innocua isolates recovered from the three levels of sampling (farm, abattoirs, and retail outlets), and the putative resistance encoded by tet(M) and tet(S) genes based on WGS was 33.6%. Therefore, there is a potential for tetracycline-resistant L. innocua strains to enter the human food chain. It is relevant to mention that the prevalence of phenotypic tetracycline resistance exhibited by the same isolates of L. innocua using the disc diffusion method was 36.8% [42]. Interestingly, this phenotypic resistance correlates well with the putative resistance to tetracycline due to both tet(M) and tet(S) genes, suggesting that the genes may have been responsible partly for the resistance detected. These findings suggest that tetracycline resistance may have been acquired with the potential for these antimicrobial genes to be transferred to commensal and pathogenic bacteria through the food chain, in addition to the fact that antimicrobial resistance in L. monocytogenes may have an adverse effect on the effective treatment of listeriosis in humans, as mentioned by Escolar et al. [62]. Studies have been reported on the resistance of bacterial pathogens, such as E. coli, Salmonella, and Listeria species, to tetracyclines in the livestock industry in South Africa [36,69,70]. The resistance of bacteria to tetracycline in South Africa has been attributed to the unregulated use of veterinary drugs, including tetracycline, in the country. This is attributed to the existing Fertilizers, Farm, and Agricultural and Stock Remedy Act (Act 36, 1947), which legalizes the use of certain antimicrobial agents, such as sulphonamides and trimethoprim, to be purchased over-the-counter, and they are used for treatment and as growth promoters [71]. Interestingly, the phenotypic resistance exhibited to tetracycline (36.8%) determined by Gana [42] correlates well with the putative resistance to tetracycline due to both tet(M) and tet(S) genes based on WGS on the same isolates, suggesting that the genes may have been responsible partly for the resistance detected. Other studies have similarly reported the correlation between phenotypic resistance and the carriage of corresponding encoding resistance genes [67,72,73]. However, a lack of correlation between these variables has also been reported by others [62]. It has been documented that bacteria may possess resistance genes but not express them, or they may be lost, thus limiting their application to their therapeutic implications and significance [74,75].
L. innocua is considered non-pathogenic. Previous analyses have suggested that L. monocytogenes and L. innocua evolved from a common virulent ancestor. During the evolution, consecutive loss of virulence genes critical to host adaptation were associated with the emergence of L. innocua [18]. Rare, atypical L. innocua strains that harbor LIPI-1 and inlA and are hemolytic and weakly virulent may represent an intermediary evolutionary stage. In addition, rare, atypical L. monocytogenes strains resulted from the spontaneous loss of virulence genes and were nonhemolytic [76]. In the present study, 23 virulence factors were detected in the 110 isolates using WGS, thus providing a spectrum of the virulence factors carried by the isolates, unlike PCR which provided information specific only to the primers targeted [77,78]. Unlike L. innocua, the ability of L. monocytogenes to cause listeriosis is known to be multifaceted and has been attributed to six virulence genes prfA, plcA, hly, mpl, actA, and plcB, which are located in the PrfA-dependent virulent gene cluster known as LIPI-1 [78], other Listeria pathogenicity islands, namely LIPI-3 and LIPI-4, Internalins (inl) genes, and other virulence genes as reported by Glimour et al. [79]. None of our L. innocua isolates contained virulence genes and are therefore classified as non-pathogenic [80]. However, it has emerged that some strains of L. innocua have been demonstrated to contain virulence genes that have contributed to their weak virulence [18]. Some of the factors documented by others in strains of L. innocua include the carriage of virulence factors, such as LGI2, LGI3, LIPI-3, and LIPI-4 [17,39,81]. It was noteworthy to have detected that the grouping of virulence genes by the STs revealed five distinct patterns. For example, pattern 1 (44 isolates) comprised three STs (637, 1085, and 602) consisting of 15 virulence factors, and pattern 2 (51 isolates) consisted of 6 STs (448, 537, 1482, 1008, 1619, and 43), with 15 virulence factors. According to the STs, this distribution of virulence genes has been documented in L. monocytogenes, where some are more associated with listeriosis depending on the virulence genes they carry, as reported in a recent outbreak of human listeriosis was caused by L. monocytogenes, ST6 [24,35]. Notwithstanding the high frequency of virulence genes in L. innocua isolates recovered from the three levels of beef production in South Africa, it is important to note that the presence/absence of virulence genes in L. monocytogenes was not a predictor of the virulence potential of L. monocytogenes [82]. Similarly, we should interpret the presence of virulence genes in L. innocua with caution. Further assessments, including hemolytic and virulence assays, on our L. innocua strains are needed.
5. Conclusions
For the first time in South Africa, this study provided a comprehensive genomic characterization of resistance and virulence genes in L. innocua isolated from three levels (production, processing, and retailing) of the beef industry using WGS. The MSTs using the profiles of the STs, AMRs, and virulence genes revealed a diversity in their spread and clustering regardless of the sources and sample types from where the L. innocua isolates originated. Furthermore, the phylogenies confirmed the genetic relatedness of the L. innocua isolates from the three sampling levels. The high frequency of resistance genes tet(M) and fosX observed in this study suggests that the use of tetracycline and fosfomycin in the livestock industry in the country and its role in the development of bacterial antimicrobial resistance should be reviewed. Finally, caution is needed in extrapolating the data based on the presence and absence of genes to the potential phenotype (i.e., resistance and virulence potential). The study has provided invaluable data on the status of L. innocua in the cattle industry food chain in the country.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1. Sources, types of samples, sequence types, and resistance genes detected in 110 L. innocua isolates. Table S2. Sources, types of samples, sequence types, and virulence genes detected in L. innocua isolates.
Author Contributions
Conceptualization, A.A.A., and N.G.; methodology, J.G., Y.C., R.E.P., A.A.A., and NG; software, R.E.P., Y.C., and NG; validation, R.E.P., and Y.C.; formal analysis, R.E.P., Y.C., A.A.A., and JG; resources, A.A.A., N.G., and R.M.; data curation, A.A.A., JG, and R.E.P.; writing—original draft preparation, A.A.A., J.G., N.G., and R.E.P.; writing—review and editing, A.A.A., J.G., N.G., R.E.M., Y.C. and R.M.; visualization, R.E.P., Y. C., N.G., J.G., and A.A.A.; supervision, A.A.A., N.G., and R.M.; project administration, A.A.A., N.G., and J.G.; funding acquisition, A.A.A., N.G., and R.M.. All authors have read and agreed to the published version of the manuscript.
Funding
The Red Meat Research and Development, South Africa (RMRD-SA) provided the funding for the project (Research grant REC138-19, awarded on January 01, 2019).
Institutional Review Board Statement
The study was approved and conducted under terms approved by the University Pretoria, South Africa: Animal Ethic Committee (#REC 138-19; 03/05/2020), and the Research Ethic Committee (04/11/2019); Section 20 (#12/11/1/1/8 (1131); 22/10/2019 ).
Data Availability Statement
All the data are contained within the article and the supplementary materials.
Acknowledgments
We thank ARC-OVR technicians (Kuda Jwamba, Makhado Lavhesani, Carol Matau, and Lesego Mashiane) for their assistance. We gratefully appreciate the sequencing team at the Public Health Laboratory in the U.S. South Carolina Department of Health & Environmental Control for generating the whole genome sequencing data for the Listeria isolates. We thank Maria Balkey for data management and submissions to NCBI.
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the official policy of the Department of Health and Human Services, the U.S. Food and Drug Administration, or the U.S. Government.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the study’s design; in the collection, analysis, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Gebretsadik, S., Kassa, T., Alemayehu, H., Huruy, K., and Kebede, N. . Isolation and characterization of Listeria monocytogenes and other Listeria species in foods of animal origin in Addis Ababa, Ethiopia. J Infect Publ Health, 2011 4, 22-29. [CrossRef]
- Carlin, C.R., Liao, J., Weller, D., Guo, X., Orsi, R. and Wiedmann, M. . Listeria cossartiae sp. nov., Listeria immobilis sp. nov., Listeria portnoyi sp. nov. and Listeria rustica sp. nov., isolated from agricultural water and natural environments. Inter J Syst Evolu Microbiol, 2021, 71, 004795.
- Castro, H., Jaakkonen, A., Hakkinen, M., Korkeala, H. and Lindström, M. Occurrence, persistence, and contamination routes of Listeria monocytogenes genotypes on three Finnish dairy cattle farms: a longitudinal study. Appl Environ Microbiol 2018, 84, e02000-17. [CrossRef]
- Murray, E.G.D., Webb, R.A. and Swann, M.B.R. A disease of rabbits characterised by a large mononuclear leucocytosis, caused by a hitherto undescribed bacillus Bacterium monocytogenes (n. sp.). J Patho Bacteriol 1926, 29, 407-439. [CrossRef]
- Molla, B., Yilma, R. and Alemayehu, D. Listeria monocytogenes and other Listeria species in retail meat and milk products in Addis Ababa, Ethiopia. Ethiopia J Hlth Dev 2004, 18, 208-212. [CrossRef]
- Zoellner, C., Wiedmann, M. and Ivanek, R., An Assessment of Listeriosis Risk Associated with a Contaminated Production Lot of Frozen Vegetables Consumed under Alternative Consumer Handling Scenarios. J Food Prot 2019, 82, 2174-2193. [CrossRef]
- Di Pinto, A., Novello, L., Montemurro, F., Bonerba, E. and Tantillo, G. Occurrence of Listeria monocytogenes in ready-to-eat foods from supermarkets in Southern Italy. New Microbiol 2010, 33, 249-252.
- Acciari, V., Iannetti, L., Schirone, M., Neri, D., Visciano, P., Acciari, V.A., Centorotola, G., Mangieri, M.S., Torresi, M., Santarelli, G.A. and Di Marzio, V. Listeria monocytogenes in poultry: Detection and strain characterization along an integrated production chain in Italy. 2020, pubag.nal.usda.gov.
- Kureljušić, J., Rokvić, N., Jezdimirović, N., Kureljušić, B., Pisinov, B. and Karabasil, N. Isolation and detection of Listeria monocytogenes in poultry meat by standard culture methods and PCR. In IOP Conference Series: Earth Environ Sci 2017, 85, 012069. IOP Publishing. iopscience.iop.org.
- Rocha, C.E., Mol, J.P., Garcia, L.N., Costa, L.F., Santos, R.L. and Paixão, T.A. Comparative experimental infection of Listeria monocytogenes and Listeria ivanovii in bovine trophoblasts. PLoS One, 2017, 12, p.e0176911. [CrossRef]
- Perrin, M., Bemer, M. and Delamare, C. Fatal case of Listeria innocua bacteremia. J Clin Microbiol 2003, 41, 5308-5309. [CrossRef]
- Favaro, M., Sarmati, L., Sancesario, G. and Fontana, C. First case of Listeria innocua meningitis in a patient on steroids and eternecept. JMM Case Rep 2014, 1. e003103. [CrossRef]
- Gradovska, S., Šteingolde, Ž., Ķibilds, J., Meistere, I., Avsejenko, J., Streikiša, M., Alksne, L., Terentjeva, M. and Bērziņš, A. Genetic diversity and known virulence genes in Listeria innocua strains isolated from cattle abortions and farm environment. Veter Anim Sci, 2023, 19, .100276.
- Seçil, A.B.A.Y., Aydin, F. and Sumerkan, A.B. Molecular typing of Listeria spp. isolated from different sources. Ankara Üniversitesi Veteriner Fakültesi Dergisi, 2012, 59, 183-190.
- Chen, J., Chen, Q., Jiang, L., Cheng, C., Bai, F., Wang, J., Mo, F. and Fang, W. Internalin profiling and multilocus sequence typing suggest four Listeria innocua subgroups with different evolutionary distances from Listeria monocytogenes. BMC Microbiol, 2010, 10, 1-16. [CrossRef]
- Disson, O., Moura, A. and Lecuit, M.. Making sense of the biodiversity and virulence of Listeria monocytogenes. Trends Microbiol, 2021 29, 811-822. [CrossRef]
- Clayton, E.M., Daly, K.M., Guinane, C.M., Hill, C., Cotter, P.D. and Ross, P.R. Atypical Listeria innocua strains possess an intact LIPI-3. BMC Microbiol, 2014, 14.1-9. [CrossRef]
- Moura, A., Disson, O., Lavina, M., Thouvenot, P., Huang, L., Leclercq, A., Fredriksson-Ahomaa, M., Eshwar, A.K., Stephan, R. and Lecuit, M. Atypical hemolytic Listeria innocua isolates are virulent, albeit less than Listeria monocytogenes. Infection and immunity, 2019, 87, e00758-18. [CrossRef]
- Johnson, J., Jinneman, K., Stelma, G., Smith, B.G., Lye, D., Messer, J., Ulaszek, J., Evsen, L., Gendel, S., Bennett, R.W. and Swaminathan, B. Natural atypical Listeria innocua strains with Listeria monocytogenes pathogenicity island 1 Genes. Appl Envirol Microbial.. 2004, 70, 4256-66. [CrossRef]
- Chen, Y., Gonzalez-Escalona, N., Hammack, T.S., Allard, M.W., Strain, E.A. and Brown, E.W. Core genome multilocus sequence typing for identification of globally distributed clonal groups and differentiation of outbreak strains of Listeria monocytogenes. Appl Environ Microbiol, 2016, 82, 6258-6272. [CrossRef]
- Kaszoni-Rückerl, I., Mustedanagic, A., Muri-Klinger, S., Brugger, K., Wagner, K.H., Wagner, M. and Stessl, B. Predominance of distinct Listeria innocua and Listeria monocytogenes in recurrent contamination events at dairy processing facilities. Microorganisms, 2020 10; 234. [CrossRef]
- Oswaldi, V., Lüth, S., Dzierzon, J., Meemken, D., Schwarz, S., Feßler, A.T., Félix, B. and Langforth, S. Distribution and characteristics of Listeria spp. in pigs and pork production chains in Germany. Microorganisms. 2022, 26,:512.
- Tomáštíková, Z., Gelbíčová, T. and Karpíšková, R. Population structure of Listeria monocytogenes isolated from human listeriosis cases and from ready-to-eat foods in the Czech Republic. J. Food Nutri Res. 2019 1, 58.
- Olanya, O.M., Hoshide, A.K., Ijabadeniyi, O.A., Ukuku, D.O., Mukhopadhyay, S., Niemira, B.A. and Ayeni O. Cost estimation of listeriosis (Listeria monocytogenes) occurrence in South Africa in 2017 and its food safety implications. Food Contr. 2019 1, 231-9. [CrossRef]
- Hyden, P., Pietzka, A., Lennkh, A., Murer, A., Springer, B., Blaschitz, M., Indra, A., Huhulescu, S., Allerberger, F., Ruppitsch, W. and Sensen, C.W. Whole genome sequence-based serogrouping of Listeria monocytogenes isolates. J. Biotech ,2016, 235, 181-186. [CrossRef]
- Schmid, D., Allerberger, F., Huhulescu, S., Pietzka, A., Amar, C., Kleta, S., Prager, R., Preussel, K., Aichinger, E. and Mellmann, A. Whole genome sequencing as a tool to investigate a cluster of seven cases of listeriosis in Austria and Germany, 2011–2013. Clin Microbiol Infect, 2014, 20, 431-436. 20. [CrossRef]
- Kwong, J.C., McCallum, N., Sintchenko, V. and Howden, B.P., 2015. Whole genome sequencing in clinical and public health microbiology. Path, 2015, 47, 199-210. [CrossRef]
- Segerman, B. The most frequently used sequencing technologies and assembly methods in different time segments of the bacterial surveillance and RefSeq genome databases. Front Cellu Iinfect microbiol, 2020, 10, .527102. [CrossRef]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482. [CrossRef]
- Chowdhury, A.S., Call, D.R., Broschat S.L., PARGT: a software tool for predicting antimicrobial resistance in bacteria. Scientific Reports. 2020 1,11033. [CrossRef]
- Pfeffer, K., Matsuyama, T., Kündig, T.M., Wakeham, A., Kishihara, K., Shahinian, A., Wiegmann, K., Ohashi, P.S., Krönke, M. and Mak, T.W. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to Listeria. monocytogenes infection. Cell. 1993, 7, 457-67. [CrossRef]
- Shamloo, E., Hosseini, H., Moghadam, Z.A., Larsen, M.H., Haslberger, A and Alebouyeh,M. Importance of Listeria monocytogenes in food safety: a review of its prevalence, detection, and antibiotic resistance. Iran J. Vet Res. 2019;20, 241.
- Wu, L., Bao, H., Yang, Z., He, T., Tian, Y., Zhou, Y., Pang, M., Wang, R. and Zhang, H. Antimicrobial susceptibility, multilocus sequence typing, and virulence of Listeria isolated from a slaughterhouse in Jiangsu, China. BMC Microbiol. 2021 21, 1-3. [CrossRef]
- Hosain, M.Z., Kabir, S.L. and Kamal, M.M. Antimicrobial uses for livestock production in developing countries. Vet World. 2021,;14, 210. [CrossRef]
- Allam, M., Tau, N., Smouse, S.L., Mtshali, P.S., Mnyameni, F., Khumalo, Z.T., Ismail, A., Govender, N., Thomas, J., and Smith, A.M. Whole-genome sequences of Listeria monocytogenes sequence type 6 isolates associated with a large foodborne outbreak in South Africa, 2017 to 2018. Geno Annon, 2018, 6, e00538-18. [CrossRef]
- Matle, I., Mbatha, K.R., Lentsoane, O., Magwedere, K., Morey, L. and Madoroba, E. Occurrence, serotypes, and characteristics of Listeria monocytogenes in meat and meat products in South Africa between 2014 and 2016. J F00d Safe, 2019, 39, e12629. [CrossRef]
- Gana, J., Gcebe, N., Pierneef, R., Moerane, R. and Adesiyun, A.A. Multiple-Locus Variable-Number Tandem Repeat Analysis Genotypes of Listeria monocytogenes Isolated from Farms, Abattoirs, and Retail in Gauteng Province, South Africa. J. Food Prot. 2022,, 85,1249-57. [CrossRef]
- Manqele, A., Gcebe, N., Pierneef, R.E., Moerane, R. and Adesiyun, A.A. Identification of Listeria species and Multilocus Variable-Number Tandem Repeat Analysis (MLVA) Typing of Listeria innocua and Listeria monocytogenes Isolates from Cattle Farms and Beef and Beef-Based Products from Retail Outlets in Mpumalanga and North West Provinces, South Africa. Pathogens 2023, 12, 147. [CrossRef]
- Mafuna, T., Matle, I., Magwedere, K., Pierneef, R.E. and Reva, O.N. Comparative Genomics of Listeria Species Recovered from Meat and Food Processing Facilities. Microbiol Spect, 2022, 10, e01189-22. [CrossRef]
- ElZowalaty, M.E., Hickman, R.A., Moura, A., Lecuit, M., Zishiri, O.T., Noyes, N. and Järhult, J.D. Genome sequence of Listeria innocua strain MEZLIS26, isolated from a goat in South Africa. Microbiol Res Anno. 2019, 8, e00991-19. [CrossRef]
- Thrusfield, M. Sample size determination. Veterinary Epidemiology 2007, 3, 185-189.
- Gana, J. Prevalence, risk factors and molecular characterization of Listeria species from cattle farms, beef abattoir and retail outlets in Gauteng, South Africa. Ph.D. Thesis, University of Pretoria, 2022 .
- Doumith, M., Buchrieser, C., Glaser, P., Jacquet, C. and Martin, P. Differentiation of the major Listeria monocytogenes serovars by multiplex PCR. J Clin Microbiol 2004, 42, 3819-3822. [CrossRef]
- Soumet, C., Ermel, G., Fach, P. and Colin, P. Evaluation of different DNA extraction procedures for the detection of Salmonella from chicken products by polymerase chain reaction. Letters Appl Microbiol 1994, 19, 294-298. [CrossRef]
- Jamali, H., Chai, L.C. and Thong, K.L. Detection and isolation of Listeria spp. and Listeria monocytogenes in ready-to-eat foods with various selective culture media. Food Contr 2013, 32, 19-24. [CrossRef]
- Bankevich, A., Nurk, S., Antipov, D., Gurevich, A.A., Dvorkin, M., Kulikov, A.S., Lesin, V.M., Nikolenko, S.I., Pham, S., Prjibelski, A.D. and Pyshkin, A.V. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comp Bio, 2012, 19, 455-477.
- Parks, D.H., Imelfort, M., Skennerton, C.T., Hugenholtz, P. and Tyson, G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome research, 2015, 25, 1043-1055. [CrossRef]
- Chaumeil, P.A., Mussig, A.J., Hugenholtz, P. and Parks, D.H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. 2020, 1925-1927. [CrossRef]
- Seemann T, mlst Github https://github.com/tseemann/mlst.
- Seemann T, Abricate, Github https://github.com/tseemann/abricate.
- Yu, G. Using ggtree to visualize data on tree-like structures. Current protocols in bioinformatics,2020, 69 .e96. [CrossRef]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.,2021. https://www.R-project.org/.
- RStudio Team. RStudio: Integrated Development Environment for R. RStudio, PBC, Boston, MA.,2022 http://www.rstudio.
- Maechler, M. Finding groups in data: Cluster analysis extended Rousseeuw et al. R package version, 2019, 2, .242-248.
- Paradis, E. and Schliep, K.. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics, 2019, 35, 526-528. [CrossRef]
- Csardi, G. and Nepusz, T. The igraph software package for complex network research. InterJournal, complex systems, 2006, 1695, 1-9.
- Briatte, F., Bojanowski, M., Canouil, M., Charlop-Powers, Z., Fisher, J., Johnson, K. and Rinker, T. 2020. ggnetwork.
- Patil, I. Visualizations with statistical details: The'ggstatsplot'approach. J Open Sou Soft, 2021, 61, 3167. [CrossRef]
- Xiao, N. ggsci: scientific journal and Sci-Fi themed color palettes for'ggplot2'. R package version, 2018, 2, 9.
- Kassambara, A. and Kassambara, M.A.. Package ‘ggpubr’. R package version 0.1, 2020, 6, 0.
- Kaptchouang Tchatchouang, C.D., Fri, J., De Santi, M., Brandi, G., Schiavano, G.F., Amagliani, G. and Ateba, C.N. Listeriosis outbreak in South Africa: a comparative analysis with previously reported cases worldwide. Microorganisms. 2020, 8, 135. [CrossRef]
- Escolar, C., Gómez, D., del Carmen Rota García, M., Conchello, P. and Herrera, A. Antimicrobial resistance profiles of Listeria monocytogenes and Listeria innocua isolated from ready-to-eat products of animal origin in Spain. Foodborn Path Dis, 2017, 14, 357-363.
- Nayak, D.N., Savalia, C.V., Kalyani, I.H., Kumar, R. and Kshirsagar, D.P. Isolation, identification, and characterization of Listeria spp. from various animal origin foods. Veter World, 2015, 8, 695. [CrossRef]
- Hanes, R.M. and Huang, Z. Investigation of Antimicrobial Resistance Genes in Listeria monocytogenes from 2010 through to 2021. Inter J Environ Res Pub Health, 2022, 19, 5506. [CrossRef]
- Jorgensen, J., Bland, R., Waite-Cusic, J. and Kovacevic, J.. Diversity and antimicrobial resistance of Listeria spp. and Listeria monocytogenes clones from produce handling and processing facilities in the Pacific Northwest. Food Contr, 2021, 123, 107665.
- Palaiodimou, L., Fanning, S. and Fox, E.M. Genomic insights into persistence of Listeria species in the food processing environment. J Appl Microbiol, 2021, 131, 082-2094. [CrossRef]
- Mpondo, L., Ebomah, K.E. and Okoh, A.I., 2021. Multidrug-resistant Listeria species shows abundance in environmental waters of a key district municipality in South Africa. Inter J Envirol Res Publ Health, 2021, 18, 481. [CrossRef]
- Henton, M.M., Eagar, H.A., Swan, G.E. and Van Vuuren, M.. Part VI. Antibiotic management and resistance in livestock production. SAMJ: South Afri Med J, 2011, 101, 583-586.
- Mupfunya, C.R., Qekwana, D.N. and Naidoo, V. Antimicrobial use practices and resistance in indicator bacteria in communal cattle in the Mnisi community, Mpumalanga, South Africa. Vetr Med Sci, 2021, 7, 112-121. [CrossRef]
- Adesiyun, A.A., Nkuna, C., Mokgoatlheng-Mamogobo, M., Malepe, K. and Simanda, L. Food safety risk posed to consumers of table eggs from layer farms in Gauteng Province, South Africa: Prevalence of Salmonella species and Escherichia coli, antimicrobial residues, and antimicrobial resistant bacteria. J Food Safe, 2020, 40, e12783. [CrossRef]
- Van, T.T.H., Yidana, Z., Smooker, P.M. and Coloe, P.J. Antibiotic use in food animals worldwide, with a focus on Africa: Pluses and minuses. J Glob Antimicrob Resist 2020, 20, 170–177. [CrossRef]
- Assisi, C., Forauer, E., Oliver, H.F. and Etter, A.J. Genomic and transcriptomic analysis of biofilm formation in persistent and transient Listeria monocytogenes isolates from the retail deli environment does not yield insight into persistence mechanisms. Foodborn Path Dis, 2021,18, 179-188. [CrossRef]
- Wilson, A., Gray, J., Chandry, P.S. and Fox, E.M. Phenotypic and genotypic analysis of antimicrobial resistance among Listeria monocytogenes isolated from Australian food production chains. Genes, 2018,9, 80. [CrossRef]
- Salyers, A.A. and Amabile-Cuevas, C.F. Why are antibiotic resistance genes so resistant to elimination?. Antimicrob age chemo,1997, 41, 2321-2325. [CrossRef]
- Hummel, A.S., Hertel, C., Holzapfel, W.H. and Franz, C.M.. Antibiotic resistances of starter and probiotic strains of lactic acid bacteria. Appl Environ Microbiol, 2007, 73.730-739. [CrossRef]
- Maury, M.M., Chenal-Francisque, V., Bracq-Dieye H., Han L., Leclercq, A., Vales, G., Moura, A., Gouin, E., Scortti, M., Disson, O., Vázquez-Boland, J.A., Lecuit, M. Spontaneous loss of virulence in natural populations of Listeria monocytogenes. Infect Immun, 2017, 85(11): e00541-17. /. [CrossRef]
- Liu, D., Lawrence, M.L., Austin, F.W. and Ainsworth, A.J. A multiplex PCR for species-and virulence-specific determination of Listeria monocytogenes. J Microbiol Meth, 2007, 71, 133-140. [CrossRef]
- Rabinovich, L., Sigal, N., Borovok, I., Nir-Paz, R. and Herskovits, A.A. Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell, 2012, 150, .792-802. [CrossRef]
- Gilmour, M.W., Graham, M., Van Domselaar, G., Tyler, S., Kent, H., Trout-Yakel, K.M., Larios, O., Allen, V., Lee, B. and Nadon, C.. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genomics, 2010, 11, 1-15. [CrossRef]
- Wurtzel, O., Sesto, N., Mellin, J.R., Karunker, I., Edelheit, S., Bécavin, C., Archambaud, C., Cossart, P. and Sorek, R.. Comparative transcriptomics of pathogenic and non-pathogenic Listeria species. Mol sys bio, 2012, 8.583.
- Gradovska, S., Šteingolde, Ž., Ķibilds, J., Meistere, I., Avsejenko, J., Streikiša, M., Alksne, L., Terentjeva, M., Bērziņš, A. Genetic diversity and known virulence genes in Listeria innocua strains isolated from cattle abortions and farm environment. Vet Anim Sci, 2022, 19:100276. [CrossRef]
- Nielsen, E.M., Björkman, J.T., Kiil, K., Grant, K., Dallman, T., Painset, A., Amar, C., Roussel, S., Guillier, L., Félix, B., Rotariu, O., Perez-Reche, F., Forbes, K., Strachan, N.. Closing gaps for performing a risk assessment on Listeria monocytogenes in ready-to-eat (RTE) foods: activity 3, the comparison of isolates from different compartments along the food chain and from humans using whole genome sequencing (WGS) analysis. EFSA Supporting Publications 2017, 14, 1151E https://efsa.onlinelibrary.wiley.com/doi/abs/10.2903/sp.efsa.2017.EN-1151).
Figure 2.
Minimum spanning tree for the sequence types of L. innocua isolates according to the source and type of sample.
Figure 2.
Minimum spanning tree for the sequence types of L. innocua isolates according to the source and type of sample.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



