
Submitted:
10 July 2023
Posted:
11 July 2023
You are already at the latest version
Abstract
The affordable production of valuable antioxidant extracts from plant resources depends on the availability of enabling technologies, such as hydrodynamic cavitation (HC), characterized by unrivalled effectiveness, efficiency, and scalability in the extraction of natural products. As sources of phytochemicals showing biological effects on human health, including anti-cancer properties, pomegranate (Punica granatum L.) fruits were processed using HC. Fractions collected during the process (M1-M5) were lyophilized (L), filtered (A), or used as such (C), for identifying their best conservation practice. Biochemical profile and in vitro antioxidant power were investi-gated by spectrophotometric and chromatographic assays. The antiproliferative effect of L-M3, resulting from just 15 minutes of extraction, was tested on human breast cancer line (AU565-PAR) and peripheral blood mononuclear (PBMCs) cells from healthy donors. M3-M4 fractions from L and C series revealed the highest antiradical activity and phenolic content. Cell growth, death, and redox state were assessed, showing that the pomegranate extract M3 significantly reduced tumor cell proliferation and intracellular oxygen reactive species. No effect on PBMCs was de-tected. Thus, the use of antioxidants isolated using HC as adjuvants in anticancer therapies might be advantageous and affordable; moreover, since oxidative stress contributes to cancer devel-opment, its reduction might be the leading mechanism.

Keywords:
anti-cancer
; antiradicals
; bioactive metabolites
; green extraction
; hydrodynamic cavitation
; phytochemicals
; pomegranate
1. Introduction
Nowadays, cancer is a growing disease worldwide. Infographics on the World Cancer Day website estimate that 10 million people died of cancer in 2020. The World Health Organization (WHO) on its website has confirmed that cancer is one of the leading causes of death worldwide. The report 'The Numbers of Cancer in Italy - 2020' by the Italian Medical Oncologists Association (AIOM) indicates that there were an estimated 18 million cases of cancer in 2018. In terms of prevention, the focus is on interventions aimed at reducing the incidence of the disease through reduced exposure to risk factors, and through attempts to increase health resistance at individual level. Early detection of cancer and appropriate treatments increase the chances of survival and cure. Type and stage of cancer suggest the most suitable therapy to be applied; to date, the most commonly used treatments are chemotherapy, radiotherapy, hormone therapy, immunotherapy, surgery, etc., either as single strategy and possibly in combination [1]. Standard protocols, however, have several downsides, including not being able to distinguish between healthy and cancerous cells. In fact, the main expected feature from any antitumor treatments should be that they have few toxic effects and the ability to kill tumor cells without damaging healthy tissues. In this regard, in recent years, there has been an increased demand for research and screening of new anti-cancer agents, including plant bioactive substances, that have few side effects during tumor management. About 35,000 phytoconstituents have been obtained from terrestrial and marine sources, to be potentially employed in standard cancer therapies [2]. It is known in the literature that various plant metabolites, such as flavonoids, phenolics, terpenoids, saponins, and alkaloids, show potent chemoprotective activity against different types of cancer cells, triggering apoptosis and cell cycle arrest, modulation of tumor-suppressive microRNAs, inhibition of oncogenes, and anti-apoptotic factors [3]. The mechanisms of action of these bioactive compounds are different and may include: i) reduction of oxidative stress-mediated DNA damage; ii) induction of cell cycle checkpoint arrest in G1, G1/S, S and G2/M phase to promote apoptosis; iii) reduction of anti-apoptotic protein levels; iv) induction of the expression of several cell cycle inhibitors, such as p53, p21 and p27, and apoptotic markers, such as the cell death agonist BCL2-associated X protein (BAX), Caspase3, Caspase7, Caspase 8, Caspase 9, etc [4,5].
Punica granatum L. (pomegranate) belongs to the family Lythraceae (Punicoideae subfamily) and it is considered as an ornamental tree indigenous to Mediterranean regions and Iran. Since ancient times, this plant has found application in traditional medicine, such as in treatment of gastrointestinal, cardiovascular, and endocrine diseases. Indeed, seed, flower, bark, peel, juice, and leaves of pomegranate are rich in potentially bioactive compounds, making it a functional food [6,7,8]. Numerous biological studies, focused on the phytocomplexes extracted from pomegranate fruit, have demonstrated the nutraceutical and nutritional properties of this plant extract. Acids, sugars, vitamins, minerals, and secondary metabolites, like simple phenols, flavonoids, anthocyanins and tannins, are considered the main phytochemicals of pomegranate derivatives that present anti-inflammatory, antibacterial, anticarcinogenic, antihypertensive, and antiviral activities [9,10]. In particular, several studies showed that pomegranate, containing high amount of polyphenols, exhibits anti-inflammatory effects and absence of toxicity, representing an excellent adjuvant to be used in combination with standard anti-cancer therapies [11], As well as the remarkable antioxidant capacity of P. granatum fruit peel was shown to reduce the breast cancer cell proliferation, inducing apoptosis [12].
In this study, phytocomplexes were isolated from pomegranate fruits using an extraction technique based on hydrodynamic cavitation (HC) processes, consisting in the creation of a periodic depression in a liquid mixture, for example by means the circulation of the liquid through a nozzle of suitable geometric shape. Vapor filled nano- and micro-bubbles, created when the pressure of the circulating liquid or liquid-solid mixture falls below the vapor pressure, subsequently grow and eventually implode under the external force produced in the recovered pressure area. The implosion events release extremely intense energy pulses in the form of pressure shock-waves, hydraulic jets, extreme transient heating, and chemical dissociation reactions [13]. The remarkable effectiveness, efficiency, yield and scalability of HC in the field of the extraction of natural products have been widely proven [14], as well as HC was recently suggested for the first time for the application to the extraction of whole pomegranate fruits [15]. Extracts collected at different times during the HC-based process were biochemically characterized. Then, the possible antiproliferative capacity of the most antioxidant extract was evaluated against the human breast cancer line (AU565-PAR), as well as possible cytotoxic effects were evaluated on peripheral blood mononuclear cells (PBMCs) from healthy donors.
2. Materials and Methods
2.1. Raw Materials and Extraction Process
Whole pomegranate fruits from the Wonderful variety were purchased from a local producer in Apulia region, south-eastern Italy, in mid-November, i.e. in the second half of the harvest period, and processed the day after.
According to a recent comprehensive review, research has not yet identified a sufficiently effective, efficient, affordable and scalable technique for the extraction of whole pomegranate fruits, including the byproducts left after squeezing the juice, although industry has progressed independently, offering important pomegranate-based ingredients widely used in food supplements [15]. In the same review, for the first time HC was suggested as a promising extraction technique, based on the previous experience with plenty of natural products.
In this study, the extraction of whole pomegranate fruits in water was performed using a semi-industrial scale (200 liters) HC pilot device, optimized for food applications. The details of the HC-based extractor, comprising a closed hydraulic circuit with a centrifugal pump and a circular Venturi-shaped reactor as the key components, along with the meaning of the cavitation number as a measure of cavitation intensity and regime, were those described in a previous study [16]. No active heat dissipation method was applied. Power and energy consumption were measured by means of a three-phase digital power meter (IME, Milan, Italy, model D4-Pd). An amount of 48 kg of whole fresh pomegranate fruit, including the thick peel, was ground in ice to coarse pieces, with maximum linear size around 10 mm, then pitched in the HC device, where water was added until a volume including ice equal to 100 liters. The initial temperature was 21.5°C. After filtering with a 200 µm sieve (stainless steel mesh), five samples (M1-M5 fractions) were collected at different extraction times, temperatures, and specific energy consumption, as shown in Table 1, and stored in sterile bottles at −20 °C until analysis. The HC process was smooth, with a fairly constant cavitation number of 0.11 to 0.12, ensuring optimal cavitation yield [14].
After thawing the extracts on ice, their volume was measured and characteristics, such as viscosity, color, and presence of particles/filaments, were assessed. Based on the obtained volume, aliquots of samples were prepared and subjected to the following analyses; in detail, pure extracts were kept as they were (CRUDE; C), freeze-dried and resuspended in water (LYOPHILIZED; L), and filtered with nitex (0.45μm) to produce the aqueous extract (AQUEOUS; A).
2.2. Total Phenolic Content Analysis
Folin-Ciocalteu spectrophotometric assay was performed to determine the amount of simple phenolic compounds present in the extracts, following the method of Impei and colleagues [17]. After incubation with the reagents, an ELISA microplate reader (Sunrise, Tecan, Austria) was used to read the absorbance of the samples at 760 nm. The concentration of the phenolic component was estimated by a calibration curve (0–3 mg/L; R2=0.9975), adequately prepared using gallic acid (GA) as standard. Results were expressed as micrograms (µg) of gallic acid equivalent per milligram (mg) of sample fresh weight (µg GAE/mg SFW).
2.3. Total Flavonoids Content Analysis
Aluminum chloride colorimetric method was applied to quantify flavonoids, as described in Di Marco and colleagues [18]. For this assay, the absorbance of the samples was measured at 415 nm (using the same ELISA microplate reader), and the concentration of the total flavonoid content was calculated based on a standard curve of quercetin (Q; 0–5 mg/L; R2=0.9975). Results were reported as µg of quercetin equivalent per mg of sample fresh weight (µg QE/mg SFW).
2.4. Determination of Condensed Tannin Content
The concentration of tannins in the samples was determined spectrophotometrically, using the method described by Weidner et al. [19] and following the modifications reported in Impei et al. [17]. The absorbance of the sample was measured at 510 nm. Pure catechin was used for the preparation of standard calibration curve (C; 0-300 mg/L; R2=0.9891). The amount of total condensed tannins was expressed as µg of catechin equivalent per mg of sample fresh weight (µg CE/mg SFW).
2.5. Determination of Specific Anthocyanins by Spectrophotometric Analysis
According to Giusti and Wrolstad [20] method, two buffers (0.025 M Potassium Chloride Buffer, pH 1.0; 0.4 M Sodium Acetate Buffer, pH 4.5) were prepared. Fifty µL of each of them were separately mixed with 50 µL of the extract. Then, the samples were incubated, in the dark, for 15 minutes. The absorbance of both solutions was measured at 510 nm (for Cyanidin), 523 nm (for Delphinidine), 557 nm (for Malvidine), 505 nm (for Pelargonidin), 496 nm (for Pelargonidin 3-glucoside), 511 nm (for Peonidin), 520 nm (for Petunidin 3-glucoside), and 700 nm (as background), by a UV/Vis spectrophotometer (Varian Cary 50 Bio UV-Vis, The Netherlands). Equation (1) was used for measuring the absorbance of the sample:
where x is the wavelength at which each single anthocyanin absorbs. Subsequently, the concentration of the single anthocyanin was estimated according to Equation (2):
where A is the absorbance measured for the sample at the wavelength of the single anthocyanin, MW is the molecular weight of the single anthocyanin (i.e., Cyanidin, 287.24; Delphinidine, 303.24; Malvidine, 331.29; Pelargonidin, 271.24; Pelargonidin 3-glucoside, 433.4; Peonidin, 301.27; Petunidin 3-glucoside, 479.41), ε is the molar absorptivity of the anthocyanin (i.e., Cyanidin, 24600; Delphinidine, 34700; Malvidine, 36200; Pelargonidin, 17800; Pelargonidin 3-glucoside, 15600; Peonidin, 37200; Petunidin 3-glucoside, 18900), and l is the length of the cuvette employed for the realization of the test. Results were reported as µg of each anthocyanin per mg of sample fresh weight.
A = [(Ax − A700) pH1.0 − (Ax − A700) pH4.5],
C = (A × MW × dilution factor × 1000)/(ε × l),
2.6. In Vitro Antiradical Assay
FRAP (2,4,6-tris 2-pyridyl-s-triazine; Sigma Aldrich), and ABTS (2,2′-azino-bis-3-ethylbenzothiazoline-6-sulfonic acid; Sigma-Aldrich) assays were carried out according to Benzie and Strain [21], and Re et al. [22] guidelines, after the application of the modifications reported in Gismondi et al. [23], for evaluating the antioxidant properties of the samples. Free radical scavenging activity was expressed as µg of ascorbic acid equivalents per mg of sample fresh weight (µg AAE/mg) for the FRAP test and as micromoles of ascorbic acid equivalents per mg of sample fresh weight (µmol AAE/mg) for ABTS, according to adequate calibration curves produced with pure ascorbic acid.
2.7. HPLC-DAD Analysis
To delineate the biochemical profile of each extract, High Pressure Liquid Chromatographic system associated with a Diode Array Detector (HPLC-DAD) was used for the analysis. The system was equipped with a CBM-20A controller, an LC-20 AD pump, a SIL-20a HT autosampler, and an SPD-M20A diode array detector (DAD) (Shimadzu, Kyoto, Japan). A Luna 3u C18(2) column (150 mm × 4.60 mm × 3 μm) (Phenomenex, Torrance, CA, USA) and mobile phases consisting of 1% formic acid (v/v) (phase A) and MeOH (phase B), at a flow rate of 0.95 mL per minute, were used for the chromatographic separation. Each sample (20 μL) was injected in the instrument and the column temperature was set at 40 °C. The elution started at 15% B solvent; this condition was maintained for 20 min; then, it was linearly increased up to 35% B solvent in 20 min and up to 90% in 55 min; at 70 min, the pump B value was reported at the initial condition (15%). Data acquisition was obtained using LAB-SOLUTION software (Shimadzu). Flavonoid compounds (i.e., Resveratrol; Quercetine-3-glucoside; Myricetin; Quercetin; Genistein; Kaempferol; Chrysin; Epicatechin) and phenolics (i.e., Gallic acid; 3-Hydroxytyrosol; Vanillic acid; Rosmarinic acid; 4-Hydroxybenzoic acid; Chlorogenic acid; Caffeic acid; Syringic acid; ρ-Coumaric acid; Salicylic acid; 1,1-Dimethylallyl caffeate; Caffeic acid phenethyl ester; 5,7-Dimethoxycumarin) were quantified in the samples at 280 nm. Qualitative and quantitative determination of each metabolite was carried out comparing their retention times (minutes), absorption spectra, and peak areas with those of pure standard molecules (Sigma-Aldrich, Milan, Italy). The results were expressed as ng of standard equivalent per mg of sample fresh weight.
2.8. Cell Culture
In this study the human breast cancer cell line AU565 was used (kindly provided by professor Ira-Ida Skvortsova, Medical University of Innsbruck Tyrolean Cancer Research Institute, Innsbruck, Austria). The cell line was cultured in RPMI-1640 medium supplemented with 10% (v/v) heat inactivated fetal bovine serum (FBS), L-glutamine (2 mM), Penicillin-Streptomycin (100 mg/mL) (all from Sigma, MO, USA). AU565 were maintained at 37 °C in a humidified 5% CO2 atmosphere and passaged three times a week after detachment with trypsin (0.05%) and EDTA solution (0.02%) in PBS (Sigma). In addition, cytotoxic effects on peripheral blood mononuclear cells (PBMCs) from healthy donors (HD) were evaluated. Three HD were obtained from individuals attending the local blood Transfusion Unit of Policlinic of “Tor Vergata” in Rome, who were referred to the Virology Unit of PTV for screening and provided written informed consent. The study was performed in accordance with the ethical principles of the Declaration of Helsinki and the Guidelines for Good Clinical Practice. The ethical committee of Tor Vergata University/Hospital approved this study (protocol number: COVID_SEET prot.7562/2020).
PBMCs from heparinized blood samples were isolated by density gradient centrifugation (Pancoll human, PAN-Biotech, Aidenbach, Bavaria) and cultured at a density of 0.1 x 106/500 µL in RPMI 1640 (PAN Biotech, Aidenbach, Bavaria) enriched with 2 mM L-glutamine, 100 U/mL penicillin, 0.1 mg/mL streptomycin, 12% fetal bovine serum in the presence of recombinant human interleukin-2 (IL-2) 20 U/mL (all from Sigma, MO, USA).
2.9. M3 Treatments
To verify the effect on PBMCs and AU565 of M3 extract from L series, which was chosen due to its high content in secondary metabolites among all samples, cells were cultured at a concentration of 0.03x106/500µL for 18 h at 37 °C in a humidified atmosphere containing 5% CO2. Then, they were treated, or not, with the M3-L sample at different concentrations, starting from 0.06 mg/mL to 50 mg/mL for 48 h. In addition to the control conditions, a treatment was also carried out with ddH2O, as a control vehicle for M3, and with etoposide, an inhibitor of DNA synthesis forming a complex with topoisomerase II. After incubation, cells were detached with a trypsin solution analyzed. Similarly, PBMCs were treated, or not, with M3 at the same concentrations reported above for the same timing.
2.10. Cell Viability and Apoptosis Assay
Alive and dead cells were counted by optic microscopy at 48 h of treatment, after staining with 10% Trypan Blue (EuroClone S.p.A., Italy). The percentage of apoptotic cells was estimated by flow cytometry (CytoFLEX; Beckman Coulter, Inc.), measuring the number of hypodiploid nuclei on 150.000 events. In detail, cells were harvested, washed in PBS, and incubated for 45 min in 70% v/v ethanol on ice. Then, they were washed with PBS and stained with propidium iodide (1.25 µg/mL). In addition, early apoptosis by annexin V (ANX-V) staining was evaluated. In detail, cells were harvested, washed, incubated for 15 min with ANX-V and with 7-aminoactinomycin D (7AAD) vitality marker on ice, and analysed. Data acquisition and analysis were performed by CytExpert 2.4 (Beckman Coulter, Inc.).
2.11. Reactive Oxygen Species (ROS) Production Assay
ROS production was evaluated by DCFDA/H2DCFDA-Cellular ROS Assay Kit (ab113851, abcam). The DCFDA assay protocol is based on the fluorescence of this marker after interaction with ROS, considering an excitation/emission spectrum fixed at 485 nm/535 nm. After detachment, AU565 cells were incubated for 30 minutes at 37 °C with 20 µM DCFDA and then washed with 1X Buffer according to the manufacturer’s instructions. The analysis was carried out by flow cytometry (CytoFLEX; Beckman Coulter, Inc). Alive cells were selected using a morphological gate and the mean fluorescence intensity (MFI) of DCFDA was assessed in relation to the different treatments, as shown in Figure S1.
2.12. Statistical Analysis
The results were reported as means ± standard deviation (SD) of 3 independent measurements. Data were subjected to a one-way analysis of variance (ANOVA) and a post-hoc lowest standard deviations (LSD) test (Excel software); p values were indicated as follows: *p<0.05; **p<0.01; ***p<0.001. The relationship existing between classes of phytochemicals and antioxidant power was estimated by Pearson’s correlation coefficients, using PAST software (v4.03).
3. Results
3.1. Biochemical Characterization of Pomegranate Extracts
The content of total phenols, flavonoids, and condensed tannins of M1-M5 fractions from crude (C), lyophilized (L) and aqueous (A) pomegranate extracts was a nalyzed by spectrophotometric analysis (Figure 1). Considering all the studied samples, the extreme values for the total phenols were 0.079 µg GAE/mg in M1 from A and 0.387 µg GAE/mg in M3 from L (Figure 1, panel A), while those of flavonoids were 1.756 µg QE/mg in M1 from A and 2.889 µg QE/mg in M5 from L (Figure 1, panel B). Condensed tannins ranged from 0.366 µg CE/mg in M5 from L to 0.722 µg CE/mg in M5 from A (Figure 1, panel C). In general, for total phenols, we observed lower values in all fractions of A, compared to C and L, albeit not significantly. C and L, instead, always showed more similar values, in all M fractions. For flavonoids, we did not observe a specific trend, but the samples from A presented the lowest values, like total phenols; on the other hand, for the condensed tannins we could see that all fractions from A had significantly higher values compared to C and L.
Through spectrophotometric assays, the content of 7 anthocyanins in the various fractions (M1-M5) from C, L, and A extracts was also evaluated (Figure 2). Cyanidin showed similar values in the three extracts at all extraction times, while the amount of delphinidin, peonidin, pelargonidin, pelagordinin-3-glucoside, and petunidin-3-glucoside tended to be overlapping and comparable for L and C in all fractions, but lower than the respective A fractions. An exception was represented by pelagordinin-3-glucoside, whose measurements appeared more variable in C and L procedures at different time points. It was also possible to observe an opposite trend for malvidin, whose C values were higher than those of the respective L and A fractions.
HPLC-DAD was used to evaluate and quantify 21 specific plant secondary metabolites, as phenolics and flavonoids (Table S1). Gallic acid, genistein, 1,1-dimethylallyl caffeate, and chrysin were not detected in any pomegranate sample. Aqueous fractions, if compared at all time points from C and L extracts, lacked four molecules: 3-hydroxytyrosol, quercetin, 5,7-dimethoxycoumarin, and kaempferol. Other compounds (e.g., caffeic acid phenethyl ester, kaempferol, quercetin, and 5,7-dimethoxycoumarin) were detected in traces in C and L fractions (that is always <1.5 ng/mg of plant fresh weight) and only at late extraction times (i.e., M3-M5). Rosmarinic acid and myricetin showed low values in all samples (with a maximum of 6.82 ng/mg in M2 from L), but both molecules were not detectable in M1 from A samples. Generally, the highest values, among all samples, were achieved by epicatechin (maximum values: 606.14 ng/mg in M3 from C, 535.67 ng/mg in M5 from L, and 642.98 ng/mg in M3 from A), 3-hydroxytyrosol (maximum values: 556.45 ng/mg in M4 from C, and 475.93 ng/mg in M3 from L), chlorogenic acid (maximum values: 263.73 ng/mg in M2 from C, 228.93 ng/mg in M3 from L, and 358.02 ng/mg in M4 from A), quercetin-3-O-glucoside (maximum values: 223.11 ng/mg in M3 from C, 189.47 ng/mg in M3 from L, and 275.08 ng/mg in M4 from A), 4-hydroxybenzoic acid (maximum values: 129.56 ng/mg in M4 from C, 160.67 ng/mg in M5 from L, and 217.31 ng/mg in M1 from A), and salicylic acid (maximum values: 188.72 ng/mg in M3 from C, 135.93 ng/mg in M5 from L, and 172.69 ng/mg in M3 from A). Salicylic acid tended to be less concentrated in all lyophilized fractions, ranging from 4.93 ng/mg in M5 from L to 188.72 ng/mg in M2 from C. Resveratrol reached its highest concentration in M3 from L. Among the phenolics, two molecules were found, albeit in traces, only in some fractions: 5,7-dimethoxycoumarin in M4 and M5 from C (at 0.26 ng/mg and 0.47 ng/mg, respectively) and M4 and M5 from L (at 0.29 ng/mg and 0.42 ng/mg, respectively), and caffeic acid phenethyl ester in M3, M4, and M5 from C (at 0.13 ng/mg, 0.29 ng/mg, and 0.27 ng/mg, respectively), M5 from L (at 0.15 ng/mg), and M1 from A (at 1.46 ng/mg). In the aqueous fractions, p-Coumaric acid was detected only in M4 and M5. Overall, the samples presenting the highest values of the investigated molecules were the M3 fractions from C, L, and A extracts, followed by M5 from L, and M4 from A.
Antioxidant activity of pomegranate samples was measured by ABTS and FRAP assays and correlated in Figure 3 (panel A). C and L samples showed all a high antiradical power during both the tests. The mean value of the FRAP assay for all fractions of these two extracts was overlapping (i.e., 274.8 µg AAE/mg for C and 275.2 µg AAE/mg for L); similarly, this occurred also for ABTS (462.77 µmol AAE/mg for C and 462.65 µmol AAE/mg for L). Indeed, the samples tended to be grouped in the upper right quadrant of the graph shown in Figure 3. Differently, the fractions of the aqueous extract had high ABTS values but low FRAP values; anyway, they could be clearly distinguished in the graph by their respective ABTS values. Among all, M4 from A extract showed the best antiradical power (i.e., 94.55 µg AAE/mg for FRAP and 432.92 µmol AAE/mg for ABTS). In order to understand which group of secondary metabolites from pomegranate mainly influenced the antioxidant potential of the extracts, Pearson's correlation analysis was carried out. The results indicated a positive correlation between antioxidant effect (measured by ABTS or FRAP) and phenolics (i.e., simple phenols and flavonoids), while a negative link existed between antiradical power and condensed tannins (Figure 3, panel B).
3.2. Effects of M3 Treatment on Growth Inhibition in Cancer Cells and PBMCs
Based on the higher concentration of phytochemicals in M3 from L series, the biological effect of this pomegranate sample on AU565 and PBMCs growth and proliferation was assessed. After 48 h of exposure, increasing doses of M3, starting from 3.75 mg/mL, very significantly inhibited AU565 growth compared to CTR, and the same result was found with the etoposide treatment (ETO). Compared to AU565, PBMCs appear to be susceptible to M3 but a drastic reduction in cell growth was neither evident and nor statistically significant, as well as comparable with etoposide treatment (Figure 4). The cytotoxicity of the treatments was also evaluated in term of effective concentration 30% (EC30) and lethal dose 30% (LD30) (Table 2).
3.3. Effects of M3 Treatment on the Induction of Apoptosis in Cancer Cells and PBMCs
To test whether M3 treatments could affect cell death in the investigated cancer cell line, an analysis of apoptosis was performed by assessing the content of hypodiploid DNA using Propidium Iodide staining and analysis by flow cytometry. Histograms in Figure 5 represent the fold change of dead cells per each concentration of treatment compared to their control. Increasing doses of M3 induce apoptosis in AU565, in particular M3 at the concentration of 25 mg/mL (p<0.050) and 50 mg/mL (p<0.01) induced apoptosis in a statistically significant way compared to the control. Interestingly, there was no difference in apoptosis rate for PBMCs.
To confirm this observation, the effect of M3 on cell death was investigated through staining of intact cells with the ANX-V/ 7AAD, after 4h to treatment. This method enables distinguishing early apoptotic cells (ANX-V+/7AAD−) and late apoptotic/necrotic ones (ANX-V+/7AAD+). M3 at 25 mg/mL induce early apoptosis and late apoptosis/necrosis compared to their control, as shown in Figure 6 and Table 3.
3.4. Effects of M3 Treatment on the Modulation of ROS Production in Cancer Cells
Analysis of ROS production assessed by ab113851 DCFDA/H2DCFDA in AU565 cells showed a high median fluorescence intensity in the control conditions. Treatment with increasing doses of M3 induced a significant (p<0.01) reduction in median fluorescence intensity at 3.75 mg/mL and highly significant (p<0.001) at 5 mg/mL and 10 mg/mL.
4. Discussion
In P. granatum, the availability of appreciable levels of nutraceutics, such as polyphenols, tannins and anthocyanins, fostered the interest toward the consumption of this functional food with significant health promoting properties [24]. In our study, the content of phenolics and flavonoids, measured by spectrophotometric assays, was constant across C and L extracts, while the same classes of secondary metabolites tended to decrease in the A fractions, albeit not significantly, perhaps due to the filtering procedure applied on these samples. In general, the fractions that retained greater amounts of phytochemicals were those from C and L series and, in particular, M2, M3, and M4 for total phenols and M3, M4 and M5 for flavonoids. By contrast, all aqueous extracts were significantly rich in condensed tannins. The latter, among all tannins, are the ones that bear the major medicinal properties. Condensed tannins form complexes with proteins, enzymes, and polymers, such as cellulose and hemicelluloses. The stability of the tannin-protein complex depends on several factors and are dynamic and time-dependent [25]. It is possible that the filtering performed on the A samples eliminated the protein component of the pomegranate, allowing a release of the tannins present in the extracts. A portion of these tannins could be correlated with epicatechin, whose detection by HPLC-DAD revealed the highest values precisely in the aqueous fractions. Indeed, catechins are considered as precursors of condensed tannins [25].
Anthocyanins, components typical in pomegranate and known for their antioxidant activity as scavengers for free radicals [26], together with the condensed tannins, were particularly abundant in the aqueous fractions, compared to C and L ones, except for malvidin, whose concentration tended to decrease in L and A extracts, perhaps due to its molecular instability [27]. As anthocyanins are water-soluble phenolic pigments and represent a subclass of flavonoids, the decrease of the total flavonoids detected in the aqueous extracts might not be related to the loss of other compounds from the same molecular class. The samples richest in anthocyanins were M3 and M4 for all the extracts, as well as for total phenol, flavonoid, and condensed tannin content. To have a fingerprint of the extracts, specific phenolics and flavonoids were monitored by HPLC-DAD. Interesting was the absence of gallic acid in all the samples, although it represents a peculiar unit of the tannins [28]. Thus, this simple phenol could be degraded during the cavitation process, or absent in its free form. In the aqueous extracts, vanillic acid, epicatechin, and caffeic acid showed the highest values of the dataset in M3, as well as chlorogenic acid and quercetine-3-O-glucoside in the M4 fraction. The phenolic and flavonoid compounds, which characterized the biochemical profiles of the C and L fractions, reflected the high antioxidant activity of these samples, detected with FRAP and ABTS tests, as also confirmed by Pearson’s correlation analysis. It is noteworthy that just 5 minutes of process time (sample M1) were enough to produce relatively high contents of bioactive compounds, likely due to the immediate extraction of pomegranate juice. The apparent, slight decline in the content of bioactive compounds for samples collected after M3 or M4 could suggest a degradation of certain heat sensitive compounds, unbalanced by further extraction from peel and seeds. Finally, M3 was chosen instead of M4 because the respective contents of bioactive compounds, as well as the levels of antioxidant activity, were indistinguishable, while M3 was produced with only 15 minutes of process time (against 25 minutes for M4), at the temperature of about 30 °C (36 °C for M4), and with much lower energy consumption (38 Wh per kg of fresh raw material) than M4 (62 Wh/kg), thus allowing higher affordability and productivity to the HC-based extraction method.
Numerous research papers have provided extensive evidence on the anti-cancer properties of pomegranate products. A number of studies have suggested that the whole pomegranate fruit, as well as its juice and oil, are promising chemopreventive/chemotherapeutic agents by exerting anti-inflammatory, antiproliferative, and anticancer effects through modulation of multiple signaling pathways [29]. Pomegranate constituents have been shown to: inhibit UVB-induced DNA damage in skin cancer [30], reduce serum PSA levels [31], inhibit STAT3 phosphorylation and NFκB activation in prostate cancer [31], inhibit DNA adduct formation in lung cancer [32], and inhibit COX-2 expression, AKT phosphorylation and NFκB DNA binding activity in colon cancer [33].
In this study, the potential anticancer capacity of pomegranate fruit HC extract was evaluated. As described in other studies [11,12] pomegranate contains high amount of polyphenols with anti-inflammatory and therapeutic effects on different breast cancer, suggesting its use in combination with standard therapies. We observed that M3-L sample reduced cell proliferation and induced apoptosis on AU565 breast cancer line. In particular, the in vitro experiments showed that starting from 3.75 mg/mL of M3 a significative decrease of viability of tumor cells was detectable, together with the enhancement of cell death. Interestingly, the same cellular phenomena in PBMCs appeared to be comparable to those of the untreated control at all concentrations tested, confirming that only cancer cells were susceptible to pomegranate exposure.
To confirm that the treatments induced apoptosis, annexin V/PI assay was carried out on AU565 and PBMCs, distinguishing early apoptotic cells from late apoptotic/necrotic ones [34]. The treatment with M3 determined a significant increase in the percentage of annexin V-positive cells compared to the control at 25 mg/mL. Moreover, at this concentration, the levels of late apoptotic/necrotic cells was higher respect to the untreated sample. Clearly, these experiments asserted that plant treatment was able to specifically induce cell death by apoptosis. Finally, also the intracellular ROS concentration was measured, demonstrating a significant decrease of the oxidative status after treatment with pomegranate sample.
Our evidence finds support in the literature as pomegranate juice has the highest antioxidant capacity compared to other polyphenol-rich beverages, such as red wine, grape juice, blueberry juice, and green tea [35]. It is known, indeed, that pomegranate extract contains a broad spectrum of nutraceutics with strong antioxidant capacity. These compounds would exert their antiradical activity in several ways, including removing or neutralizing free radicals, chelating metals, influencing cell signaling pathways, and modulating gene expression [36,37]. Studies on Caco-2 colon cancer cells treated with punicalagin and ellagic acid, obtained from pomegranate, have shown pro-apoptotic and oxidative stress-reducing effects, providing information about the possible molecular mechanism of action underlying the bioactivity of P. granatum [38].
5. Conclusions
According to all the previous data and taking into consideration the potential use of plant derivatives as food supplements with healthy effects, the lyophilized form of the pomegranate extract obtained using HC would represent the best solution to preserve intact antioxidants and related antiradical properties, considering its stability due to the lack of water responsible for oxidative reactions degrading phytochemicals. Likely acting through the reduction of oxidative stress, which in turn contributes to cancer development, pomegranate-derived antioxidant phytochemicals obtained using HC during just 15 minutes of process time and with very low energy consumption, could be effective and affordable adjuvants in anticancer therapies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Gating strategy by flow cytometry to assess the reactive oxygen species in AU565 treated with M3 extracts. Representation of the distribution of AU565 alive cells by scatter plot (FSC vs SSC): (a) CTR; (c) M3 extract 3.5 mg/mL; (e) M3 extract 5 mg/mL; (g) M3 extract 10 mg/mL. Representative histogram of mean fluorescence intensity (MFI) of DCFDA (FITC-A) to assess reactive oxygen species (ROS) in AU565 alive cells: (b) CTR; (d) M3 extract 3.5 mg/mL; (f) M3 extract 5 mg/mL; (h) M3 extract 10 mg/ml. The representations shown are of a representative experiment of three performed; Table S1: Phenolic and flavonoid compounds detected in pomegranate extracts by HPLC-DAD. Concentrations of secondary metabolites present in the fractions (M1-M5) of the crude, lyophilized and aqueous extracts were reported. Results were expressed as µg per mg of plant fresh weight.
Author Contributions
Conceptualization, Antonella Minutolo, Angelo Gismondi, Mauro Centritto and Francesco Meneguzzo; Data curation, Antonella Minutolo, Angelo Gismondi, Rossella Chirico, Gabriele Di Marco, Marialaura Fanelli and Francesco Meneguzzo; Investigation, Alessia D'Agostino and Lorenzo Albanese; Methodology, Lorenzo Albanese, Federica Zabini and Francesco Meneguzzo; Resources, Mauro Centritto and Francesco Meneguzzo; Supervision, Antonella Minutolo, Angelo Gismondi, Claudia Matteucci and Francesco Meneguzzo; Validation, Rossella Chirico, Gabriele Di Marco, Vita Petrone and Marialaura Fanelli; Writing – original draft, Antonella Minutolo, Angelo Gismondi, Rossella Chirico, Marialaura Fanelli and Francesco Meneguzzo; Writing – review & editing, Antonella Canini, Sandro Grelli, Federica Zabini and Claudia Matteucci.
Funding
The researchers belonging to the National Research Council of Italy were funded by the PNRR (National Recovery and Resilience Plan) - Mission 4, Component 2, Investment 1.3, Italian Ministry of University and Research, funded by the European Union NextGenerationEU - Project PE0000003 (PE10 ON Foods: Research and innovation network on food and nutrition Sustainability, Safety and Security - Working ON Foods; Spoke 2 “Smart and circular food system and distribution”), Concession Decree No. 1550 of 11 October 2022.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Colli, L.M.; Machiela, M.J.; Zhang, H.; Myers, T.A.; Jessop, L.; Delattre, O.; Yu, K.; Chanock, S.J. Landscape of Combination Immunotherapy and Targeted Therapy to Improve Cancer Management. Cancer Res. 2017 77(13),3666-3671. [CrossRef]
- Gupta, J.; Ahuja, A.; Gupta, R. Green Approaches for Cancers Management: An Effective Tool for Health Care. Anticancer Agents Med. Chem. 2022, 22(1),101-114. [CrossRef]
- Raju, S.K.; Sekar, P.; Kumar, S.; Murugesan, M.; Karthikeyan, M.; Elampulakkadu, A. Plant secondary metabolites for the prevention and treatment of colorectal cancer: A review. J. Pharmacogn. Phytochem. 2022, 11 (2), 229–246. [CrossRef]
- Levin, B. Nutrition and colorectal cancer. Cancer 1992, 70 (6 Suppl),1723-6. [CrossRef]
- Esmeeta, A.; Adhikary, S.; Dharshnaa, V.; Swarnamughi, P.; Ummul Maqsummiya, Z.; Banerjee, A.; Pathak, S.; Duttaroy, A.K. Plant-derived bioactive compounds in colon cancer treatment: An updated review. Biomed. Pharmacother. 2022, 53,113384. [CrossRef]
- Lee, C.J.; Chen, L.G.; Liang, W.L, Wang, C.C. Anti-inflammatory effects of Punica granatum Linne in vitro and in vivo. Food. Chem. 2010, 118 (2), 315-322. [CrossRef]
- Fischer, U.A.; Carle, R.; Kammerer, D.R. Identification and quantification of phenolic compounds from pomegranate (Punica granatum L.) peel, mesocarp, aril and differently produced juices by HPLC-DAD-ESI/MSn. Food Chem. 2011, 127, 807–821. [CrossRef]
- Akhtar, S.; Ismail, T.; Fraternale, D.; Sestili, P. Pomegranate peel and peel extracts: Chemistry and food features. Food Chem. 2015, 174, 417–425. [CrossRef]
- Eghbali, S.; Askari,S.F.; Avan, R.; Sahebkar, A. Therapeutic Effects of Punica granatum(Pomegranate): An Updated Review of Clinical Trials. J Nutr Metab. 2021, 16, 5297162. [CrossRef]
- Ranjha, M. M. A. N., Shafique, B., Wang, L., Irfan, S., Safdar, M. N., Murtaza, M. A., Nadeem, M.; Mahmood, S.; Mueen-ud-Din, G.; Nadeem, H.R. A comprehensive review on phytochemistry, bioactivity and medicinal value of bioactive compounds of pomegranate (Punica granatum). Adv in Trad Med. 2023, 23, 37–57. [CrossRef]
- Dikmen, M.; Ozturk, N.; Ozturk, Y. The antioxidant potency of Punica granatum L. Fruit peel reduces cell proliferation and induces apoptosis on breast cancer. J Med Food. 2011, 14(12),1638-46. [CrossRef]
- Kim, N.D.; Mehta, R.; Yu, W.; Neeman, I.; Livney, T.; Amichay, A.; Poirier, D.; Nicholls, P.; Kirby, A.; Jiang, W.; et al. Chemopreventive and adjuvant therapeutic potential of pomegranate (Punica granatum) for human breast cancer. Breast Cancer Res Treat. 2002, 71(3),203-17. [CrossRef]
- Panda, D.; Saharan, V.; Manickam, S. Controlled hydrodynamic cavitation: A review of recent advances and perspectives for greener processing. Processes 2020, 8, 220. [CrossRef]
- Meneguzzo, F.; Albanese, L.; Zabini, F. 3.21 - Hydrodynamic Cavitation in Beer and Other Beverage Processing. Innovative Food Processing Technologies; Knoerzer, K., Muthukumarappan, K. Elsevier, 2021, 369-394. [CrossRef]
- Benedetti, G.; Zabini, F.; Tagliavento, L.; Meneguzzo, F.; Calderone, V.; Testai, L. An Overview of the Health Benefits, Extraction Methods and Improving the Properties of Pomegranate. Antioxidants 2023, 12, 1351. [CrossRef]
- Meneguzzo, F.; Brunetti, C.; Fidalgo, A.; Ciriminna, R.; Delisi, R.; Albanese, L.; Zabini, F.; Gori, A.; dos Santos Nascimento, L.B.; De Carlo, A.; et al. Real-Scale Integral Valorization of Waste Orange Peel via Hydrodynamic Cavitation. Processes 2019, 7, 581. [CrossRef]
- Impei, S.; Gismondi, A.; Canuti, L.; Canini, A. Metabolic and biological profile of autochthonous Vitis vinifera L. ecotypes. Food Funct 2015, 6(5), 1526–1538. [CrossRef]
- Di Marco, G.; Gismondi, A.; Canuti, L.; Scimeca, M.; Volpe, A.; Canini, A. Tetracycline accumulates in Iberis sempervirens L. through apoplastic transport inducing oxidative stress and growth inhibition. Plant Biol. 2014, 16(4), 792–800. [CrossRef]
- Weidner, S.; Karolak, M.; Karamac, M.; Kosinska, A.; Amarowicz, R. Phenolic compounds and properties of antioxidants in grapevine roots [Vitis vinifera L.] under drought stress followed by recovery. Acta Soc Bot Pol. 2009, 78(2), 97-103. [CrossRef]
- Giusti, M.M.; Wrolstad, R.E. Characterization and measurement of anthocyanins by UV-visible spectroscopy. Curr protoc food anal chem. 2001, 1, F1-2. [CrossRef]
- Benzie, I. F.; Strain, J. J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Analytical Biochemistry 1996, 239(1), 70–76. [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Radical Biology and Medicine 1999, 26(9–10), 1231–1237. [CrossRef]
- Gismondi, A.; De Rossi, S.; Canuti, L.; Novelli, S.; Di Marco, G.; Fattorini, L.; Canini, A. From Robinia pseudoacacia L. nectar to Acacia monofloral honey: biochemical changes and variation of biological properties. J Sci Food Agric. 2018, 98(11), 4312-4322. [CrossRef]
- Sharma, A.; Thakur, N.S. Influence of active packaging on quality attributes of dried wild pomegranate (Punica granatum L.) arils during storage. J Appl Nat Sci. 2016, 8(1), 398–404. [CrossRef]
- Bule, M.; Khan, F.; Nisar, M.F.; Niaz, K.; Nabavi, S.; Saeedi, M.; et al. Tannins (hydrolysable tannins, condensed tannins, phlorotannins, flavono-ellagitannins). Recent advances in natural products analysis 2020, 132-146.
- Zhao, X.; Yuan, Z. Anthocyanins from pomegranate (Punica granatum L.) and their role in antioxidant capacities in vitro. Chem biodivers. 2021, 18(10), e2100399. [CrossRef]
- Wang, Y.; Lei, Z.; Lan, W.; Tan, B.; Chen, C.; Zeng, X. Theoretical study on the spectroscopic properties of anthocyanin molecules. In 2nd International Conference on Signal Image Processing and Communication 2022, 12246, 479-484. [CrossRef]
- Kennas, A.; Amellal-Chibane, H. Comparison of five solvents in the extraction of phenolic antioxidants from pomegranate (Punica granatum L.) peel. Nor Afr J Food Nutr Res. 2019, 3(5),140–147. [CrossRef]
- Sharma, P.; McClees, S.F.; Afaq, F. Pomegranate for Prevention and Treatment of Cancer: An Update. Molecules 2017, 22(1),177. [CrossRef]
- Khan, N.; Syed, D.N.; Pal, H.C.; Mukhtar, H.; Afaq, F. Pomegranate fruit extract inhibits UVB-induced inflammation and proliferation by modulating NF-κB and MAPK signaling pathways in mouse skin. Photochem Photobiol. 2012, 88(5), 1126-34. [CrossRef]
- Wang, Y.; Zhang, S.; Iqbal, S.; Chen, Z.; Wang, X.; Wang, Y.A.; Liu, D.; Bai, K.; Ritenour, C.; Kucuk, O.; et al. Pomegranate extract inhibits the bone metastatic growth of human prostate cancer cells and enhances the in vivo efficacy of docetaxel chemotherapy. Prostate 2013. Epub ahead of print. [CrossRef]
- Zahin, M.; Ahmad, I.; Gupta, R.C.; Aqil, F. Punicalagin and ellagic acid demonstrate antimutagenic activity and inhibition of benzo[a]pyrene induced DNA adducts. Biomed Res Int. 2014, 2014, 467465. [CrossRef]
- Adams, L.S.; Seeram, N.P.; Aggarwal, B.B.; Takada, Y.; Sand, D.; Heber, D. Pomegranate juice, total pomegranate ellagitannins, and punicalagin suppress inflammatory cell signaling in colon cancer cells. J Agric Food Chem. 2006, 54(3), 980-5. [CrossRef]
- Potestà, M.; Roglia, V.; Fanelli, M.; Pietrobono, E.; Gismondi, A.; Vumbaca, S.; Nguedia Tsangueu, R.G.; Canini, A.; Colizzi, V.; Grelli, S.; Minutolo, A.; et al. Effect of microvesicles from Moringa oleifera containing miRNA on proliferation and apoptosis in tumor cell lines. Cell Death Discov. 2020, 6, 43. [CrossRef]
- Shirode, A.B.; Kovvuru, P.; Chittur, S.V.; Henning, S.M.; Heber, D.; Reliene, R. Antiproliferative effects of pomegranate extract in MCF-7 breast cancer cells are associated with reduced DNA repair gene expression and induction of double strand breaks. Mol Carcinog. 2014, 53(6), 458-70. [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us?. Oxid Med Cell Longev. 2016. [CrossRef]
- Rodrigo, R.; Miranda, A.; Vergara, L. Modulation of endogenous antioxidant system by wine polyphenols in human disease. Clin Chim Acta 2011, 412(5-6),410-24. [CrossRef]
- Seeram, N.P.; Aronson, W.J.; Zhang, Y.; Henning, S.M.; Moro, A.; Lee, R.P.; Sartippour, M.; Harris, D.M.; Rettig, M.; Suchard, M.A.; et al. Pomegranate ellagitannin-derived metabolites inhibit prostate cancer growth and localize to the mouse prostate gland. J Agric Food Chem. 2007, 55(19),7732-7. [CrossRef]
Figure 1.
Content of secondary metabolites in pomegranate samples. (a) Total content of phenolics; (b) Total content of flavonoids; (c) Total content of condensed tannins. These quantities were measured by spectrophotometric assays in the fractions (M1-M5) of crude, lyophilized and aqueous extracts. The unit of measure was indicated in the y-axis of each graph. Only in (c), the significance of the results showed a p-value <0.05 for all the aqueous samples vs the respective crude and lyophilized ones.
Figure 1.
Content of secondary metabolites in pomegranate samples. (a) Total content of phenolics; (b) Total content of flavonoids; (c) Total content of condensed tannins. These quantities were measured by spectrophotometric assays in the fractions (M1-M5) of crude, lyophilized and aqueous extracts. The unit of measure was indicated in the y-axis of each graph. Only in (c), the significance of the results showed a p-value <0.05 for all the aqueous samples vs the respective crude and lyophilized ones.
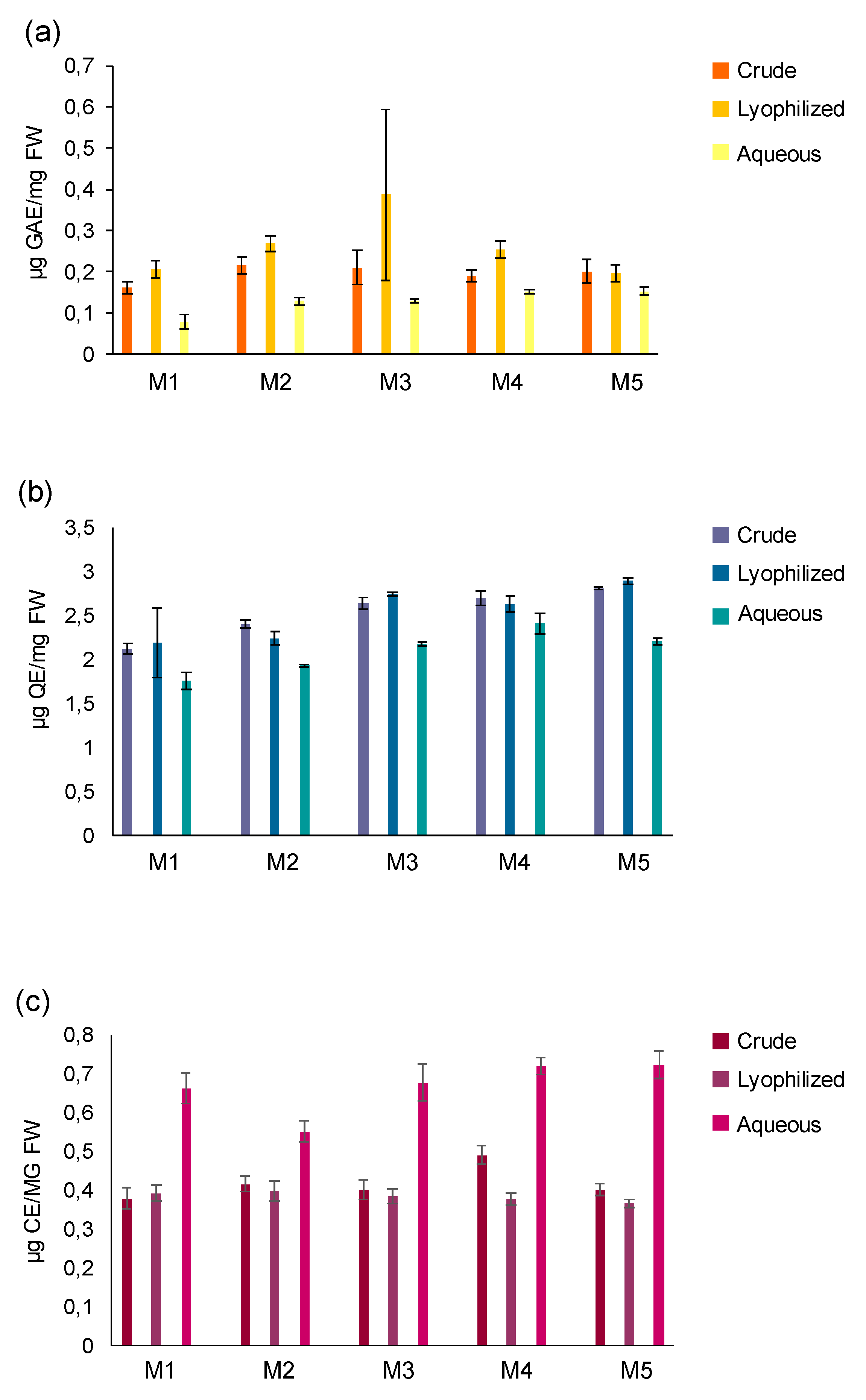
Figure 2.
Anthocyanin quantitation in pomegranate samples in the fractions (M1-M5) of crude, lyophilized and aqueous extracts. Line charts showing the level of (a) Cyanidin; (b) Delphinidin; (c) Pelargonidin 3-glucoside; (d) Peonidin; (e) Malvidin; (f) Pelargonidin; (g) Petunidin 3-glucoside. Results were expressed as µg per mg of plant fresh weight.
Figure 2.
Anthocyanin quantitation in pomegranate samples in the fractions (M1-M5) of crude, lyophilized and aqueous extracts. Line charts showing the level of (a) Cyanidin; (b) Delphinidin; (c) Pelargonidin 3-glucoside; (d) Peonidin; (e) Malvidin; (f) Pelargonidin; (g) Petunidin 3-glucoside. Results were expressed as µg per mg of plant fresh weight.
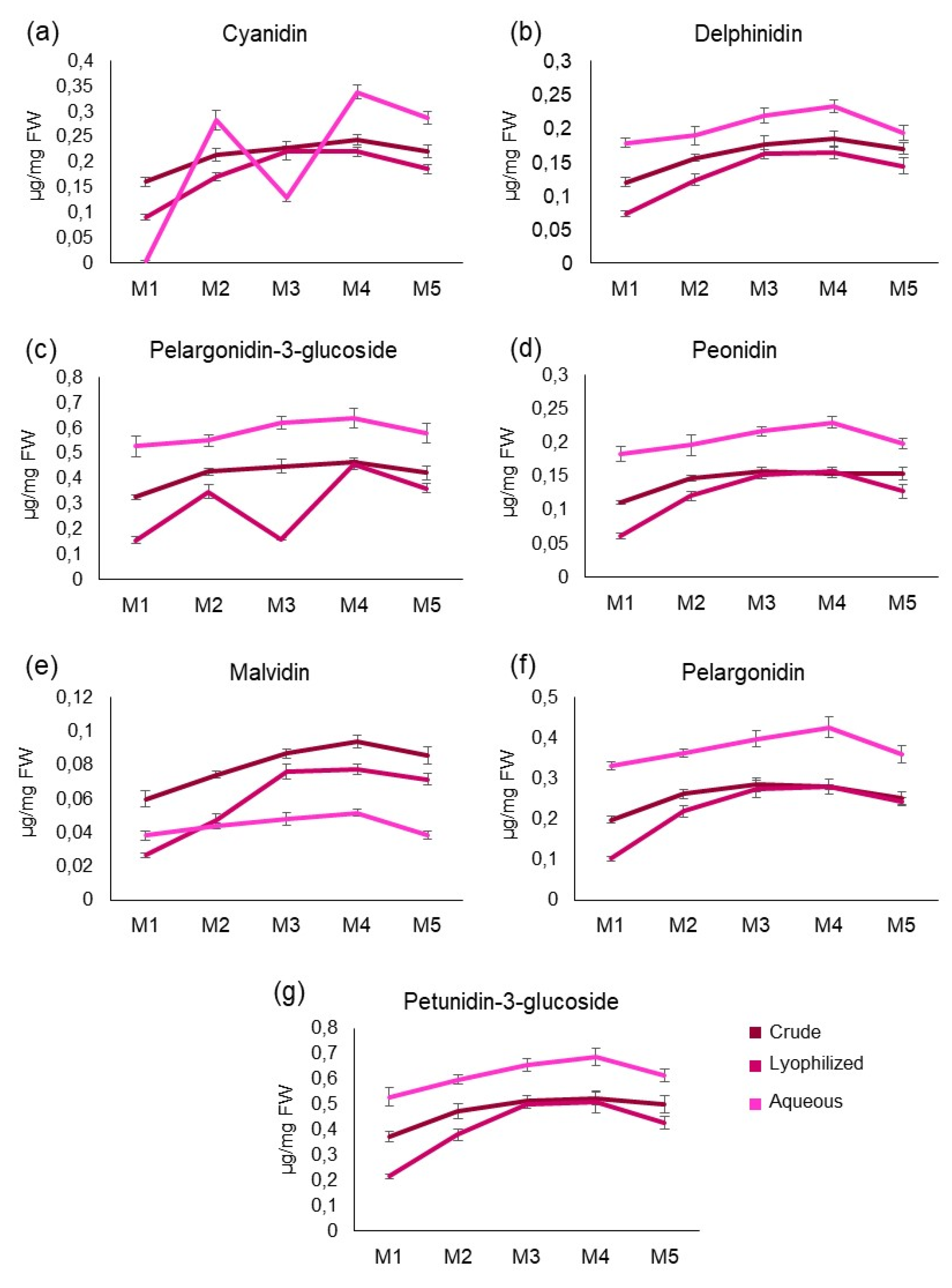
Figure 3.
Antioxidant activity of pomegranate samples. (a) Antiradical power measured by ABTS and FRAP assays in the fractions (M1-M5) of crude, lyophilized and aqueous extracts. Results were expressed as µg AAE and µmol AAE, respectively for FRAP and ABTS, per mg of plant fresh weight. (b) Pearson’s correlation between content of plant secondary metabolites and antioxidant potentials (p<0.05 boxed). Quantities were phenols (P), flavonoids (F), condensed tannins (CT).
Figure 3.
Antioxidant activity of pomegranate samples. (a) Antiradical power measured by ABTS and FRAP assays in the fractions (M1-M5) of crude, lyophilized and aqueous extracts. Results were expressed as µg AAE and µmol AAE, respectively for FRAP and ABTS, per mg of plant fresh weight. (b) Pearson’s correlation between content of plant secondary metabolites and antioxidant potentials (p<0.05 boxed). Quantities were phenols (P), flavonoids (F), condensed tannins (CT).
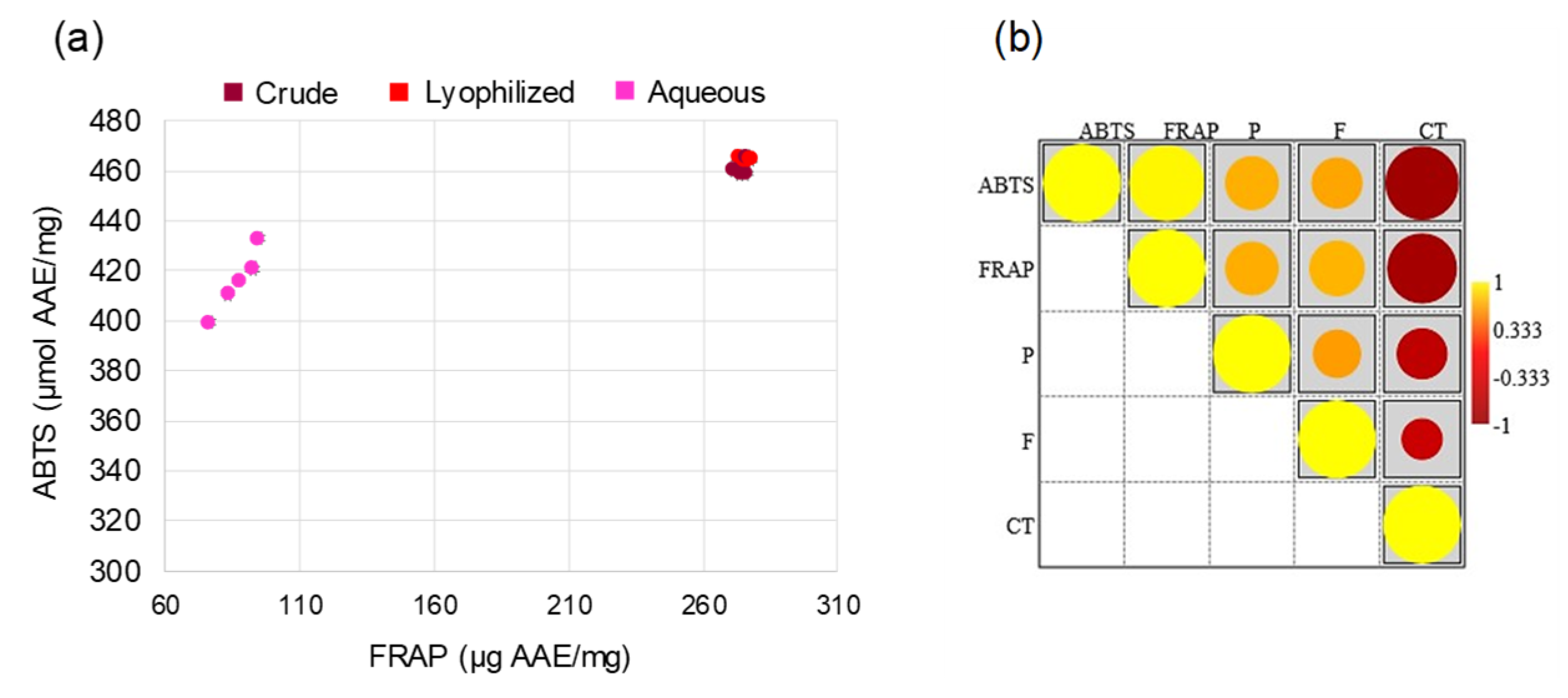
Figure 4.
Viability analysis following treatment with increasing doses of M3. (a) PBMC from healthy donors; (b) AU565 cells. Results are expressed as the ratio between the number of alive cells after treatments and that measured in the control condition (fold change). (*) p≤0.050, (**) p≤0.010, (***) p<0.001. Data are shown as mean ± SD of at least three experiments performed.
Figure 4.
Viability analysis following treatment with increasing doses of M3. (a) PBMC from healthy donors; (b) AU565 cells. Results are expressed as the ratio between the number of alive cells after treatments and that measured in the control condition (fold change). (*) p≤0.050, (**) p≤0.010, (***) p<0.001. Data are shown as mean ± SD of at least three experiments performed.
Figure 5.
Evaluation of the apoptosis expressed as mean fluorescence intensity in (a,b) PBMC; (c,d) AU565 cells. The results in the histograms are expressed as the ratio between the percentage of hypodiploid nuclei measured in the treatment and control conditions (fold change). (*) p≤0.050, (**) p≤0.010, (***) p≤0.001. Data are shown as mean ± SD of at least three independent experiments.
Figure 5.
Evaluation of the apoptosis expressed as mean fluorescence intensity in (a,b) PBMC; (c,d) AU565 cells. The results in the histograms are expressed as the ratio between the percentage of hypodiploid nuclei measured in the treatment and control conditions (fold change). (*) p≤0.050, (**) p≤0.010, (***) p≤0.001. Data are shown as mean ± SD of at least three independent experiments.
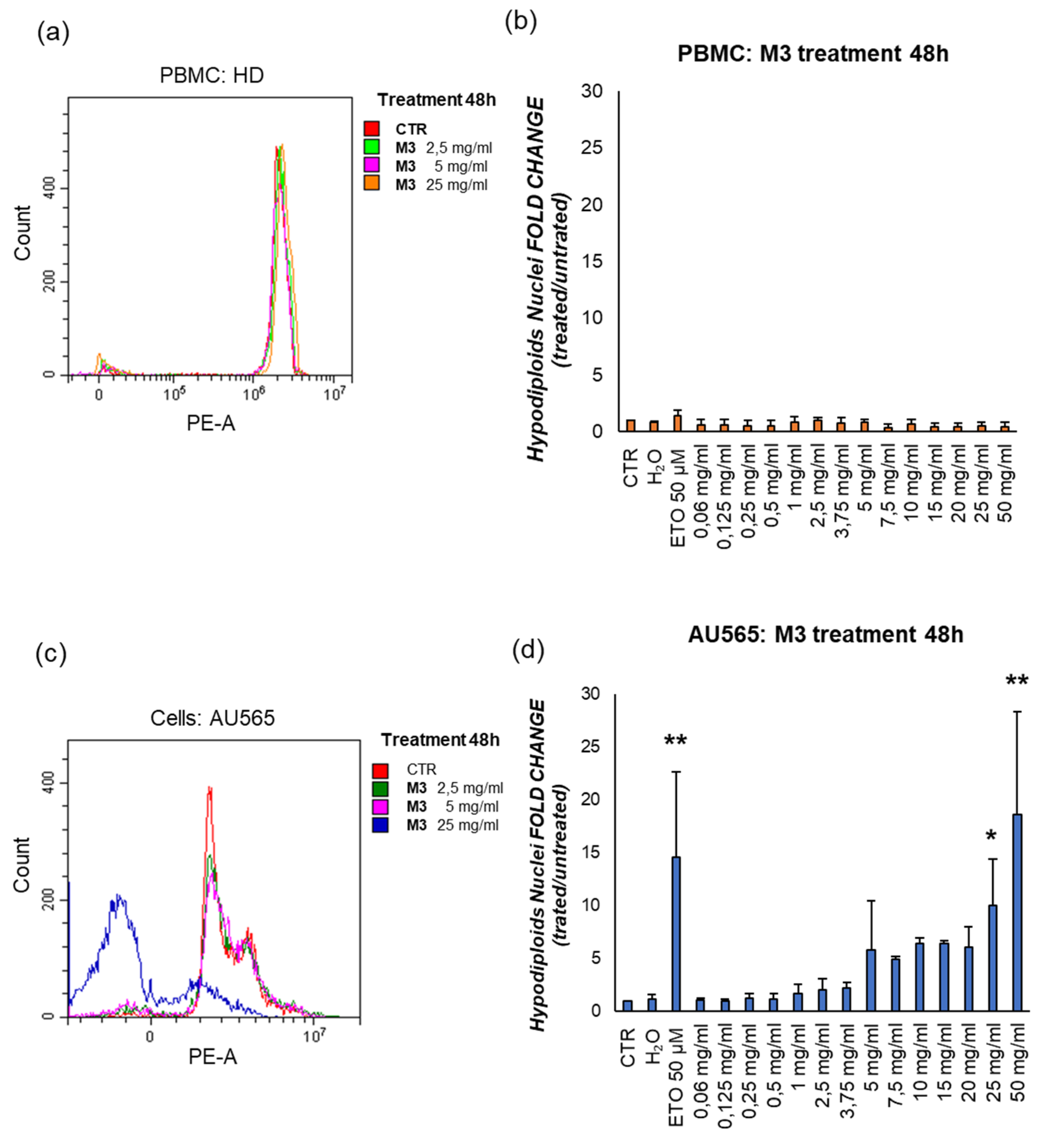
Figure 6.
Representative flow cytometric analysis of the effect of M3 25 mg/mL on early apoptosis (ANX-V+/7AAD-) and late apoptosis/necrosis (ANX-V+/7AAD+) in (b) AU565; (f) PBMC, with respect to their control conditions (a,e). The results in the histograms represent the fold change of the ratio between the percentage of cells ANX-V+/7AAD- and ANX-V+/7AAD+, respectively in (c,d) AU565; (g,h) PMBCs. (*) p≤0.050, (**) p≤0.010, (***) p<0.001. Data are shown as mean ± SD.
Figure 6.
Representative flow cytometric analysis of the effect of M3 25 mg/mL on early apoptosis (ANX-V+/7AAD-) and late apoptosis/necrosis (ANX-V+/7AAD+) in (b) AU565; (f) PBMC, with respect to their control conditions (a,e). The results in the histograms represent the fold change of the ratio between the percentage of cells ANX-V+/7AAD- and ANX-V+/7AAD+, respectively in (c,d) AU565; (g,h) PMBCs. (*) p≤0.050, (**) p≤0.010, (***) p<0.001. Data are shown as mean ± SD.
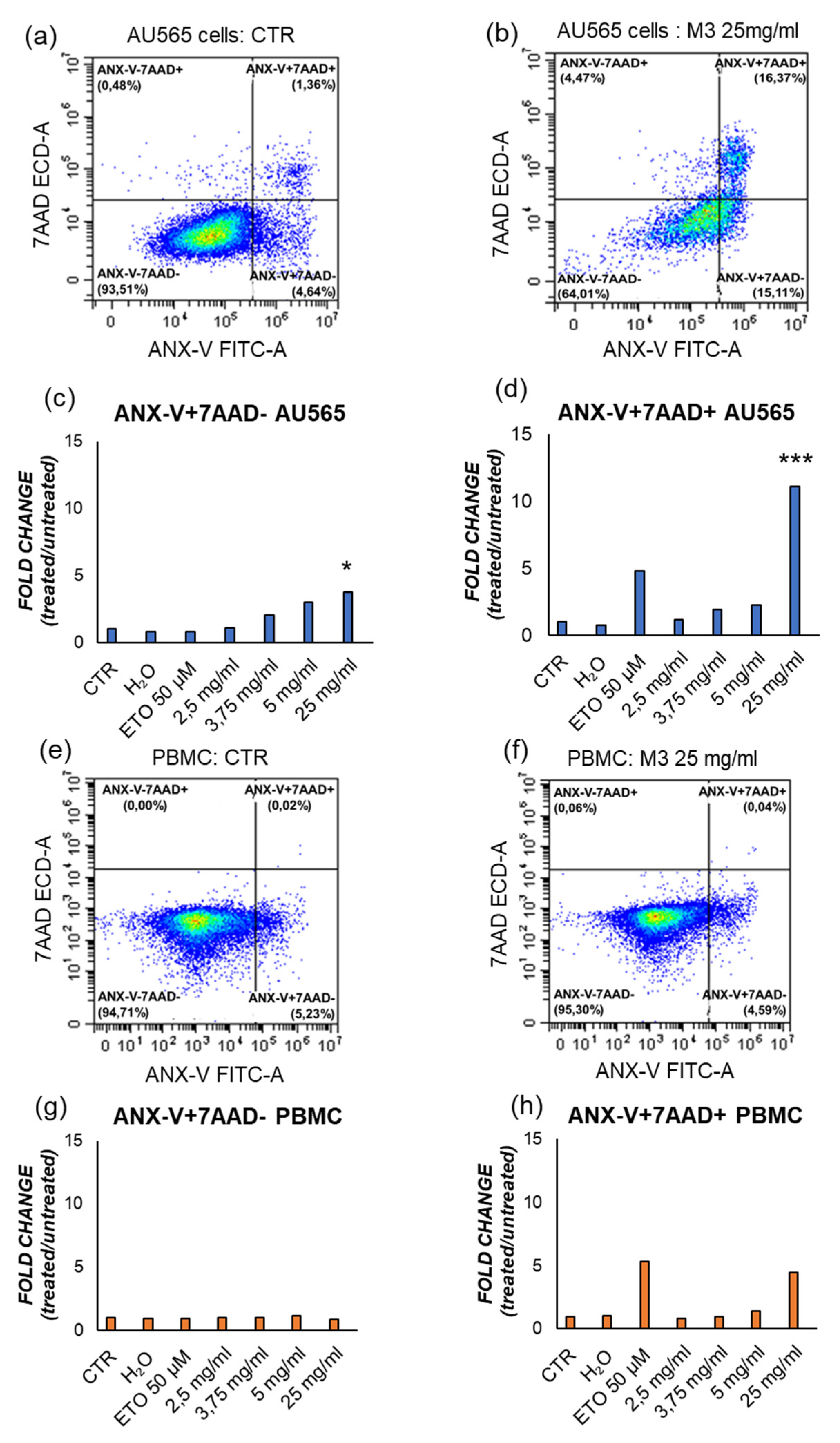
Figure 7.
Evaluation of effects of M3 treatment on the modulation of ROS production in AU565. (a) Results are expressed as cell counts in relation to the fluorescence intensity of the DCFDA probe (FITC-A) following treatment with M3. (b) In the histogram, the results are expressed as mean fluorescence intensity in AU565. (**) p≤0.010, (***) p<0.001. Data are shown as mean ± SD of at least three experiments performed.
Figure 7.
Evaluation of effects of M3 treatment on the modulation of ROS production in AU565. (a) Results are expressed as cell counts in relation to the fluorescence intensity of the DCFDA probe (FITC-A) following treatment with M3. (b) In the histogram, the results are expressed as mean fluorescence intensity in AU565. (**) p≤0.010, (***) p<0.001. Data are shown as mean ± SD of at least three experiments performed.
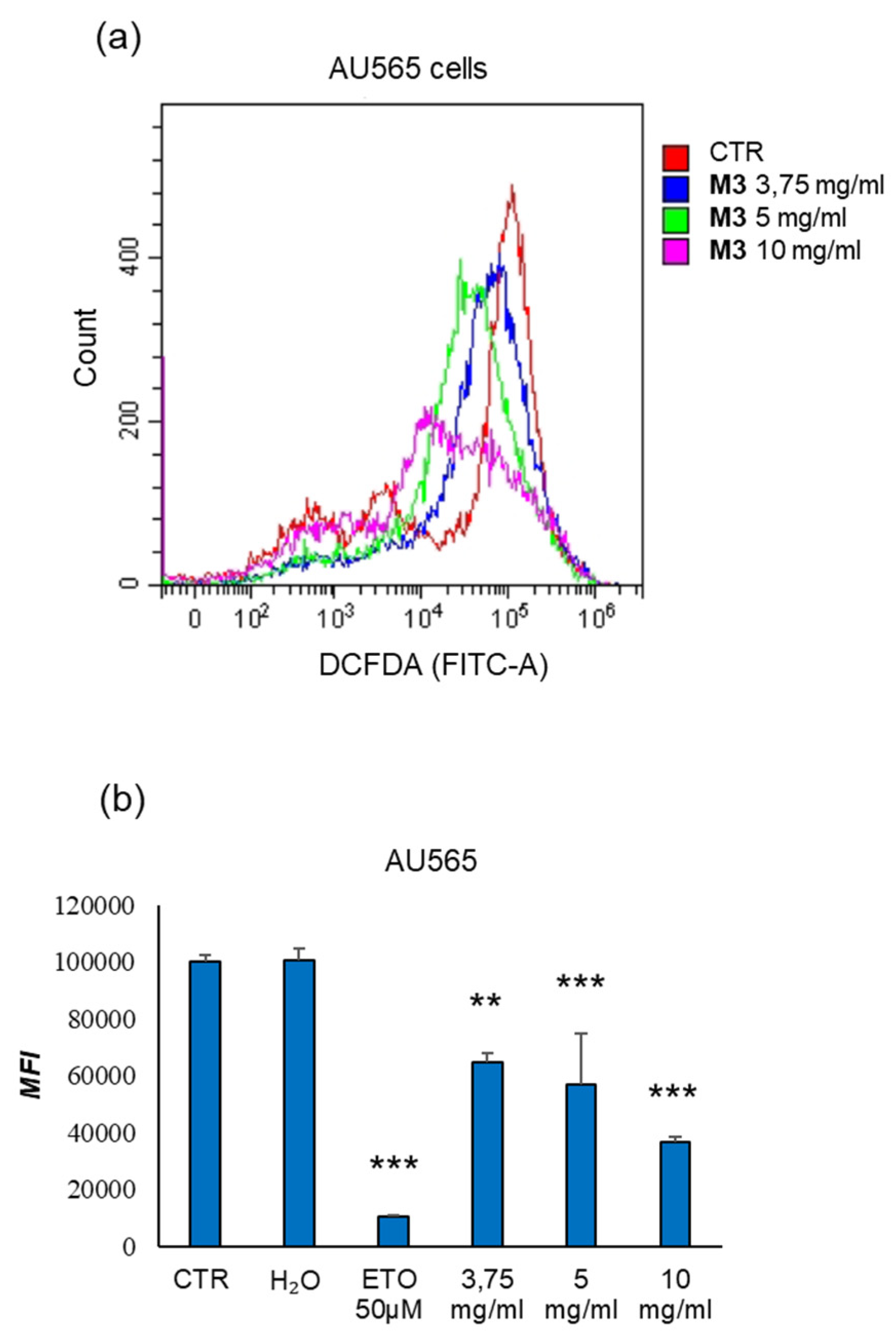

Table 1.
Samples collected at different extraction times (minutes), temperatures (°C), and specific energy (Wh per kg of fresh raw material).
Table 1.
Samples collected at different extraction times (minutes), temperatures (°C), and specific energy (Wh per kg of fresh raw material).
| Fractions | Process Time (minutes) |
Process Temperature (°C) |
Specific Energy (Wh/kg) |
|---|---|---|---|
| M1 | 5 | 24.0 | 13 |
| M2 | 10 | 27.5 | 25 |
| M3 | 15 | 30.5 | 38 |
| M4 | 25 | 36.5 | 62 |
| M5 | 45 | 47 | 108 |
Table 2.
Cytotoxic effect of M3 extract treatments on AU565 and PBMCs from healthy donors (n = 3). Mean ± SD of three independent measurements of Trypan blue assay.
Table 2.
Cytotoxic effect of M3 extract treatments on AU565 and PBMCs from healthy donors (n = 3). Mean ± SD of three independent measurements of Trypan blue assay.
| Cells Type | EC30 (mg/mL) | LD30 (mg/mL) |
|---|---|---|
| AU565 | 2.44 ± 0.01 | >50 |
| PBMCs | >50 | >50 |
Table 3.
Percentage of Annexin V (ANX-V) and propidium iodide (PI) negative and positive AU565 cells treated with M3-L extract for 48 hours. Results are reported in percentage, as mean ± SD. (*) p≤0.050; (**) p≤0.010.
Table 3.
Percentage of Annexin V (ANX-V) and propidium iodide (PI) negative and positive AU565 cells treated with M3-L extract for 48 hours. Results are reported in percentage, as mean ± SD. (*) p≤0.050; (**) p≤0.010.
| AU565 | ANX+/7AAD+ (%) | ANX+/7AAD- (%) |
| CTR | 1.28±0.30 | 4.74±0.25 |
| H₂O | 0.94±0.02 | 3.66±0.18 |
| ETO 50 µM | 6.16±0.10 | 3.86±0.28 |
| 2,5 mg/mL | 1.5±0.15 | 4.98±0.01 |
| 3,75 mg/mL | 2.47±0.25 | 9.74±0.7 |
| 5 mg/mL | 2.92±0.23 | 14.28±1.14* |
| 25 mg/mL | 14.15±1.35** | 17.6±2.56** |
| PBMCs | ANX-V+/7AAD+ (%) | ANX-V+/7AAD- (%) |
| CTR | 4.73±0.30 | 0.84±0.15 |
| H₂O | 0.92±0.10 | 0.75±0.35 |
| ETO 50 µM | 0.81±0.36 | 3.42±0.25 |
| 2,5 mg/mL | 2.45±0.45 | 0.65±0.28 |
| 3,75 mg/mL | 3.16±0.12 | 0.79±0.14 |
| 5 mg/mL | 9.03±0.18 | 1.24±0.32 |
| 25 mg/mL | 4.72±0.45 | 4.06±0.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



