
Submitted:
11 July 2023
Posted:
11 July 2023
You are already at the latest version
Abstract
Treatment of the [AuCl(tetrahydrothiophene)] complex with 4-chloro-2-iodo-1-isocyanobenzene furnished the gold(I) compound [AuCl(CNC6H3-4-Cl-2-I)] (1). In the crystal structure of 1, the linear C–Au–Cl group is subject to the solid-state head-to-tail pairing which is determined by the aurophilic Au···Au and the rare π-holeCN···Cl interactions. These two types of structure-determining interactions are complementary to each other and the system of Au···Au and CCN···Cl contacts accomplishes 2D extended ladder-type architecture. In addition, the terminal I-atoms are involved in the three-center halogen bonding. Density functional theory calculations, employing a set of computational tools, verified the role of Au···Au and π-holeCN···Cl noncovalent bonds in the spectrum of noncovalent forces.
Keywords:
gold(I) complexes
; isocyanide ligands
; π-hole interactions
; aurophilic interactions
1. Introduction
Noncovalent interactions (for general reviews see refs. [1,2,3,4,5,6]) play key roles in various fields of modern science spanning from chemistry to molecular biology. Despite their low energy, in many instances they function collectively and the sum of their actions can significantly affect synthesis [7,8], catalytic transformations (including noncovalent organic catalysis [9,10]), and reactivity of chemical compounds [11]. Currently, a classification of these interactions includes their division into the σ- and π-hole interactions (for reviews on σ- and π-hole interactions see refs. [12,13,14,15,16,17,18]) – depending on the location of the regions with positive molecular electrostatic potential (abbreviated as MEP). If a σ-hole interaction is associated with the positive MEP on the extension of the covalent σ-bond, a π-hole interaction usually occurred in the direction perpendicular to the σ-framework of the molecular moiety [19].
In the overwhelming majority of cases, most popular π-hole donors, utilized as coformers of noncovalent interaction, include electron-deficient aromatics, while π-hole donor properties of triple-bonded small molecules were revealed in only a few cases, namely, for carbon monoxide [20,21], organic nitriles [22], and cyanamides [23]. Furthermore, it has been previously reported that coordinated isocyanides are chameleonic species [24,25] and they featuring either nucleophilic (low-oxidation state metal ions) [26] or electrophilic (high-oxidation state metal ions) character depending on identity of a metal site, in particular, on their donor/acceptor abilities. When the ligated isocyanides are bound to high-oxidation state metal ions, they could be involved in π-hole···dz2-[M], [27] π-hole···π, [28] and π-hole···lone pair (LP) [29] interactions.
We report herein on π-holeCN···Cl interactions between an isocyano group and a chlorido ligand from the neighboring complex; these contacts have no covalent analogs. The observed type of π-holeCN interaction is supported by and complementary to aurophilic forces (for reviews on aurophilicity see refs. [30,31,32,33,34]) occurred in the same compound on its association in the solid state. DFT calculations are used to closely interrogate the bonding in the RNC–Au–Cl linear units that in the solid state are assembled in head-to-tail (or, in other words, antiparallel) binding mode. The aurophilic Au···Au and π-hole···LP interactions were characterized using several computational tools.
2. Materials and Methods
2.1. Reagents, Instrumentation, and Methods
Solvents, tetrahydrothiophene (THT), and HAuCl4 were obtained from commercial sources and used as received. 4-Chloro-2-iodo-1-isocyanobenzene [35] and [AuCl(THT)] [36] were prepared by the published procedures.
The high-resolution mass spectra were obtained on a Bruker micrOTOF spectrometer equipped with electrospray ionization (ESI) source and MeOH was used as the solvent. The instrument was operated at positive ion mode using m/z range of 50–3000. The most intensive peak in the isotopic pattern is reported. Infrared spectra (4000–400 cm–1) were recorded on Shimadzu IRAffinity-1 FTIR spectrophotometer in KBr pellets. NMR spectra were measured on Bruker AVANCE III 400 spectrometers in CDCl3 at ambient temperature (at 400 and 100 MHz for 1H and 13C NMR, respectively). Chemical shifts are given in δ-values [ppm] referenced to the residual signals of undeuterated solvent (CHCl3): δ 7.27 (1H) and 77.0 (13C).
Synthesis of 1. 4-chloro-2-iodo-1-isocyanobenzene (42 mg, 0.16 mmol) was added to a solution of [AuCl(THT)] (50 mg, 0.16 mmol) in CH2Cl2 (5 mL), whereupon the reaction mixture was stirred for 40 min at 20 °С. The resulting suspension was evaporated under a reduced pressure (20 mbar) at room temperature to dryness and the colorless residue was washed with Et2O (two 2-mL portions) and then dried in air at room temperature. Yield is 77 mg, 98%. Colorless solid. Calcd (%) for C7H3NAuCl2I: C 16.96, H 0.61, N 2.82; Found: C 17.11, H 0.70, N 2.73. ESI HRMS+ (MeOH, m/z): 517.8251 ([1 + Na]+, calcd. for C7H3NAuCl2INa+ 517.8250). FTIR, νmax (KBr, cm–1): 2221 ν(C≡N). 1H NMR (CDCl3, δ): 7.47–7.51 (m, 1H), 7.51–7.55 (m, 1H), 7.97 (d, 1H, 3JH,H = 2 Hz). 13C{1H} NMR (CDCl3, δ): 95.28, 128.00, 128.71, 129.96, 138.32, 139.88, 144.60.
2.2. X-ray Diffraction Study
Crystals of 1 were obtained by slow evaporation of its CH2Cl2 solution at room temperature. Single-crystal X-ray diffraction experiment was carried out on SuperNova, Single source at offset/far, HyPix3000 diffractometer with monochromated CuKα radiation. Crystal was kept at 100(2) K during data collection. Structures were solved by ShelXT [37] (structure solution program using Intrinsic Phasing) and refined by means of the ShelXL [38] program incorporated in the OLEX2 program package [39]. Crystallographic details are summarized in Table S1. CCDC number 2280014 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
2.3. Computational Details
Turbomole 7.7 program and X-ray coordinates were used for the calculations reported herein [40]. The crystallographic coordinates were used because we are interested in evaluating the interactions as they stand in the solid state. The level of theory used for the calculations was PBE0-D3/def2-TZVP [41,42,43]. For iodine, effective core potentials (ECP) and relativistic were used for the inner electrons.[44] The 0.001 a.u. isovalue was used for the MEP surface plots. The quantum theory of atoms in molecules (QTAIM) and noncovalent interaction plot index (NCIplot) methods proposed by Bader [45] and W. Yang et al. [46], were used to analyze the topology of the electron density and were plotted using the VMD software [47]. NCIplot index settings: RDG = 0.5 a.u.; cut-off ρ = 0.04 a.u., and color scale –0.03 ≤ sign(λ2)ρ ≤ 0.03 a.u. The electron localization function (ELF) [48] analysis was performed using the MultiWFN program [49] at the PBE0-D3/def2-TZVP level of theory.
3. Results
3.1. X-ray Diffraction Study
Molecular structure of the gold(I) complex [AuCl(CNC6H3-4-Cl-2-I)] (1) (Figure 1) is analogous to those reported for other [AuCl(CNR)] complexes [50,51,52,53]. The gold(I) ion is in a linear coordination environment, which is formed by one isocyanide and one chlorido ligands. Bond distances and angles are typical for this type of complexes [50,51,52].
In the structure of 1, each gold(I) center forms several intermolecular contacts. The shortest separation, which involve a metal atom, is Au···Au (3.3050(8) Å; vs Bondi [54,55] ΣvdW 3.32 Å); this metallophilic bond is characteristic for (RNC)[AuI] species [20,50,52]. The Au···Au contact is involved in the system of noncovalent interactions which includes also two antiparallel CCN···Cl short separations. The linear C–Au–Cl group is subject to the solid-state head-to-tail pairing accordingly to the association mode predicted and studied computationally [20] for similar gold(I) species. Another Au···Au contact in the system of noncovalent interactions includes two CCN···Au and one Au···Au contact (Figure 1). In the latter aurophilic contact, the Au···Au distance is 3.5672(7) Å. It is still larger than the sum of the corresponding Bondi ΣvdWAu + Au = 3.32 Å, but lower than Alvarez [54,55] ΣvdWAu + Au = 4.64 Å.
Other unconventional contacts in the crystal structure are CCN···Cl (3.428(6) Å) and CCN···Au (3.495(5) Å). For the CCN···Cl separation, the C···Cl distance is smaller than Bondi ΣvdWC + Au 3.45 Å. Geometry considerations based on the data collected in Table 1 indicate that the Cl nucleophilic site is directed to the isocyano carbon. The availability of the π-holeCN···LPCl interaction was confirmed in appropriate theoretical study (see later).
ϕ is a torsion angle ClAuAuC.
To find our similarities with the crystal structure of our gold complex – which based on one Au···Au and two CCN···Cl contacts – we performed a Cambridge Structural Database (CSD) search using the following parameters: the structures of [AuX(CNR)] (X = Cl, Br, I) exhibiting one Au···Au contact with d(Au + Au)<Bondi ΣvdW(Au + Au) and two antiparallel CCN···X with d(C + X)<Bondi ΣvdW(C + X). By applying these criteria, we retrieved six structures of [AuCl(CNAr)] species, but only 4 out of 6 structures were of acceptable quality (Rw <5%). Examination of the geometrical parameters of the four structures (Table 1) allows the assignment of these contacts to aurophilic Au···Au and π-holeCN···LPCl interactions.
Another unconventional contact is CCN···Au, in which the distances between the C-atom of the isocyanide group and the gold(I) site are slightly larger than Bondi ΣvdWC + Au 3.36 Å, but are significantly less than Alvarez ΣvdWC + Au = 4.09 Å [56]. The CCN···Au contact exhibits a comparable length with those in the earlier reported structures of [AuCl(CNR)] (R = C6H4OMe-4; BUVCAX [57]; 3.662(5) Å and R = 2-naphthyl; TAHVIK;[58] 3.605(8) Å) and even in other (RNC)AuI species (see CSD search with brief analysis for CCN···M contacts in refs. [27,59]). In these reports, π-hole···[AuI] nature of the C···Au contact has not been analyzed, while analogous π-hole···[MII] (M = Pd, Pt) contacts were thoroughly investigated, including DFT studies of their nature [27,59].
Based on the angular parameters analysis (both ∠N≡C···Au and ∠C···Au–C angles are ca. 100°) one can conclude that the C and Au atoms are in contact but parallel displaced. Our theoretical calculations revealed no bond critical point and bond path and only a small value of energetic stabilization due to electron transfer from Au(5dx2-y2) to the π*(C≡N) orbital. As further explained below, the NCIplot analysis also suggests the presence of a weak interaction and confirms that both the CN group and Au-atoms are in contact. The existence of such π-holeCN···[AuI] interaction seems quite logical as gold(I) site, despite its positive charge, exhibits d-orbital nucleophilicity and can function as a nucleophilic coformer of noncovalent interactions, for instance, halogen bonding [60,61,62]. However, the results of our geometry considerations and the computations data give only a hint that such interaction likely exists and it is very weak.
The system of Au···Au and CCN···Cl contacts accomplishes 2D extended ladder-type architecture (Figure 2). Molecules from different 2D-ladder arrays are linked to each other via I···I contacts (3.7932(7) Å vs. Bondi ΣvdWI + I = 3.96 Å) (Figure 3). These contacts, as follows from consideration of the ∠C–I–I values (150.10(15) and 128.76(14)°), are close to Type-II halogen···halogen interactions (idealized angles θ1 ≈ 180 and θ2 ≈ 90°) [63,64]. In addition, molecules from the same 2D-ladder array exhibit short I···I contacts (4.0486(1) Å vs Bondi ΣvdWI + I = 3.96 Å) of Type-I (∠C–I–I values are 103.04(14) and 76.96(14)° that are close to 90°) halogen···halogen interactions [64]. To be conclusive regarding the nature of such contacts, an elaborated theoretical study is detailed below, where different computational tools were used to investigate the donor/acceptor role of the I-atoms.
3.2. Theoretical Study
First, to study the electron rich and electron poor parts of compound 1, the MEP surface was computed and corresponding surface is represented in Figure 4. It can be observed that the MEP minimum, as expected, is located at the chlorido ligand (–33.8 kcal/mol). The MEP is also negative at the Au and negative belts around the iodine and chlorine substituents (–5.0 kcal/mol). The MEP maximum is located at the iodine’s σ-hole (33.9 kcal/mol) and it is also large and positive at the aromatic H-atoms, ranging from 30 to 34 kcal/mol. The MEP is also positive over the center of the aromatic ring (16.9 kcal/mol), σ-hole of chlorine (18.8 kcal/mol), and over the isocyano group (11.9 over C and 18.5 over N, measured perpendicular to the molecular plane). This distribution of the electron density explains the formation of the 2D-ladder type architecture with the antiparallel arrangement of the molecules.
To characterize the 2D-ladder packing and to gain an insight into the nature of the noncovalent interactions, DFT calculations were performed using two independent dimers, denoted as “A” and “B” that exemplify the two binding modes observed in the infinite assembly depicted in Figure 2. The energetic features of the dimers and the QTAIM/NCIplot analyses are given in Figure 5. As can be inferred from consideration of our computational results, both binding modes exhibit very similar interaction energies (–13.6 kcal/mol and –13.4 kcal/mol for dimers A and B, respectively). Both dimers A and B are interconnected by five bond critical points (CPs, represented as small red spheres) and bond paths (orange lines). One bond CP interconnects both Au-atoms, thus disclosing the existence of aurophilic interactions. Moreover, in both dimers the Cl is connected to the isocyano group by a bond CP and bond path, in the case of dimer A to the C-atom and in the case of B to the N-atom. Finally, in dimer A, two symmetrically equivalent CPs and bond paths connect two aromatic CH bonds to the chlorido ligands. For dimer B, the additional bond CPs and bond paths connect the Cl to one C-atom of the aromatic ring, thus evidencing the occurrence of π···LP interactions.
Such combination of interactions explains the rather strong dimerization energies. The main difference between both dimers is observed in the NCIplot analysis and shape of the reduced density gradient (RDG) isosurfaces. That is, dimer A exhibits well defined disk-shape isosurfaces for each contact, revealing that the Au···Au interaction is the strongest one (blue RDG isosurface). In contrast, dimer B presents a continuous and green RDG isosurface, thus suggesting a more complicated (cooperative) binding and supporting the possibility of a partial contribution of π-holeCN···[AuI] interaction in combination with the aurophilic one and also π-holeCN···Cl contacts.
To shed light into the physical nature interactions in both dimers, we performed the natural bond orbital (NBO) analysis since it is useful to study charge-transfer effects from an orbital donor-acceptor viewpoint. The most important contributions (>1 kcal/mol) obtained from the NBO analysis of both dimers are provided in Figure 6. For dimer A, two orbital donor-acceptor contributions were found: one corresponds to the aurophilic interaction that is composed by an electron donation from the LP located at the filled 5dx2-y2 atomic orbital of gold to the antibonding C–Au orbital of the adjacent molecule, and vice versa. The associated stabilization energy is E(2) = 3.7 kcal/mol due to 5dx2-y2[Au]→σ*(C–Au) is the largest donor-acceptor orbital contribution. In addition, an electron donation from one LP of the chlorido ligand to one of the π*(C≡N) orbitals of the isocyano group is also observed, with a concomitant second order stabilization of E(2) = 1.4 kcal/mol.
For dimer B it is interesting to highlight that the most important charge transfer is from the LP of gold (5dx2-y2) to the π*(C≡N) orbital, thus confirming the existence of the interaction between the π-hole at the isocyano group and gold.
Echeverria[20] has also analyzed CCN···Cl contacts in AuI dimers similar to those observed in Dimer A (characterized by bond CPs and disk shaped RDG isosurfaces). The reported NBO stabilization energies are ranged from 0.05 to 0.2 kcal/mol for the LP(Cl)→π*(C≡N) donor-acceptor interactions, thus revealing that the CCN···Cl contacts studied herein are stronger than those previously studied.
Finally, the noncovalent three-center I···I contacts in the structure of 1 (Figure 3) were also studied using QTAIM analysis and the electron localization function (ELF) method to verify halogen bonding (for reviews on halogen bonding see refs. [1,2,3,4,5,65]) nature of these contacts. The results are shown in Figure 7 and it can be observed that the QTAIM analysis reveals the existence of three I···I contacts characterized by the corresponding bond CPs and bond paths. The trimerization energy is moderately strong (–10.3 kcal/mol) likely due to the contribution of the π-stacking of two out of the three molecules that form the I···I triangle. The ELF 2D-map is represented in Figure 7b, using the plane defined by the three I-atoms for the orientation. This ELF analysis reveals that the I···I contacts characterized by the bond CPs denoted as “a” and “b” can be defined as halogen bonds since the bond path that connects the I-atoms crosses LP of one I (electron donor) and the σ-hole of the other one (electron acceptor). In contrast, the bond CP denoted as “c” corresponds to a I···I interaction that cannot classified as halogen bond, since the bond path crosses the LPs of both iodine atoms that are, in fact, pointing to each other. The size of these electron rich regions is significantly smaller than other similar regions (LPs) that are not pointing to each other, indicating some polarization and/or conjugation of the LPs to the aromatic ring, likely reducing the electrostatic repulsion. This I···I van der Waals contact can be rationalized considering that other forces like dispersion or polarization compensate the electrostatics or that is a consequence of packing effects.
4. Discussion
By inspection of the X-ray structure of gold(I)-based complex 1, we recognized rare structure-determining π-holeCN···Cl contact that involve an isocyanide ligand acting as a π-hole donor. The observed type of π-holeCN···LPCl interaction is complementary to the Au···Au aurophilic forces occurred in the same compound on its head-to-tail association in the solid state. The interplay of these two types of interactions largely determines the crystal structure.
Results of DFT calculations revealed that both binding modes (antiparallel dimers) are energetically similar and that the Au···Au interaction in dimer A is the most favorable as shown by the NCIplot and NBO analyses. In the case of dimer B, the NBO analysis gives a hint for the existence of the rare π-hole···d-metal, namely π-holeCN···[AuI], interaction since a LP(Au)→π*(C≡N) charge transfer is observed. Moreover, the RDG isosurface in dimer B, that embraces the whole region between the NC–Au–Cl is also compatible with the contribution of the π-hole···[AuI] interaction. We hope that these results will stimulate further studies focused on π-holeCN···[AuI] systems and eventually lead to the discovery of a system, in which these contacts are unambiguously recognized.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Crystal data and structure refinement for 1.
Author Contributions
Conceptualization, V.Y.K. and N.A.B.; methodology, A.S.S., M.A.K., R.M.G. and A.F.; investigation, A.S.S., M.A.K., and R.M.G.; writing—original draft preparation, N.A.B., A.F. and A.S.S.; writing—review and editing, V.Y.K.; visualization, N.A.B. and R.M.G.; funding acquisition, A.S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Russian Science Foundation, project number 22-23-00307.
Data Availability Statement
The data that support the findings of this study are available upon reasonable request from the authors.
Acknowledgments
The article is in commemoration of the 300th anniversary of St. Petersburg State University’s founding. Physicochemical studies were performed at the Center for Magnetic Resonance, Center for X-ray Diffraction Studies, Center for Thermogravimetric and Calorimetric Research, and Center for Chemical Analysis and Materials Research (all belonging to Saint Petersburg State University).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shukla, R.; Chopra, D. Chalcogen and Pnictogen Bonds: Insights and Relevance. Curr. Sci. 2021, 120, 1848–1853. [Google Scholar] [CrossRef]
- Cornaton, Y.; Djukic, J.-P. Noncovalent Interactions in Organometallic Chemistry: From Cohesion to Reactivity, a New Chapter. Accounts Chem. Res. 2021, 54, 3828–3840. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. On the Importance of σ–Hole Interactions in Crystal Structures. Crystals 2021, 11, 1205. [Google Scholar] [CrossRef]
- Miller, D.K.; Chernyshov, I.Y.; Torubaev, Y.V.; Rosokha, S.V. From weak to strong interactions: structural and electron topology analysis of the continuum from the supramolecular chalcogen bonding to covalent bonds. Phys. Chem. Chem. Phys. 2022, 24, 8251–8259. [Google Scholar] [CrossRef]
- Schneider, H. Noncovalent interactions: A brief account of a long history. J. Phys. Org. Chem. 2022, 35, e4340. [Google Scholar] [CrossRef]
- Non-covalent Interactions in the Synthesis and Design of New Compounds; Abel M. Maharramov, K.T.M., Maximilian N. Kopylovich, Armando J. L. Pombeiro, Ed.; 2016.
- Loh, C.C.J. Exploiting non-covalent interactions in selective carbohydrate synthesis. Nat. Rev. Chem. 2021, 5, 792–815. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Guseinov, F.I.; da Silva, M.F.C.G. Noncovalent interactions in metal complex catalysis. Co-ord. Chem. Rev. 2019, 387, 32–46. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; da Silva, M.F.C.G.; Pombeiro, A.J. Noncovalent Interactions in Catalysis; Royal Society of Chemistry: 2019.
- Cornaton, Y.; Djukic, J.-P. Noncovalent Interactions in Organometallic Chemistry: From Cohesion to Reactivity, a New Chapter. Accounts Chem. Res. 2021, 54, 3828–3840. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional σ/π-Hole Interactions. Chem Phys Chem 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Electrostatics and Polarization in σ- and π-Hole Noncovalent Interactions: An Overview. Chem Phys Chem 2020, 21, 579–588. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: a perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.K.; Bauza, A.; Frontera, A. Quantitative analysis of weak non-covalent σ-hole and π-hole interactions. Monogr. Supramol. Chem. 2019, 26, 285–333. [Google Scholar]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. (Washington, DC, U. S.) 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Noncovalent Bonds through Sigma and Pi-Hole Located on the Same Molecule. Guiding Principles and Comparisons. Molecules 2021, 26, 1740. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chemical Reviews 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Echeverría, J. Intermolecular Carbonyl···Carbonyl Interactions in Transition-Metal Complexes. Inorganic Chemistry 2018, 57, 5429–5437. [Google Scholar] [CrossRef] [PubMed]
- Doppert, M.T.; van Overeem, H.; Mooibroek, T.J. Intermolecular π-hole/n→π* interactions with carbon monoxide ligands in crystal structures. Chemical Communications 2018, 54, 12049–12052. [Google Scholar] [CrossRef]
- Ruigrok van der Werve, A.; van Dijk, Y.R.; Mooibroek, T.J. π-Hole/n→π* interactions with acetonitrile in crystal structures. Chemical Communications 2018, 54, 10742–10745. [Google Scholar] [CrossRef] [PubMed]
- Toikka, Y.N.; Mikherdov, A.S.; Ivanov, D.M.; Mooibroek, T.J.; Bokach, N.A.; Kukushkin, V.Y. Cyanamides as π-Hole Donor Components of Structure-Directing (Cyanamide)···Arene Noncovalent Interactions. Crystal Growth & Design 2020, 20, 4783–4793. [Google Scholar] [CrossRef]
- Vatsadze, S.Z.; Loginova, Y.D.; Gomes, G.d.P.; Alabugin, I.V. Stereoelectronic Chameleons: The Donor–Acceptor Dichotomy of Functional Groups. Chem. – A Eur. J. 2016, 23, 3225–3245. [Google Scholar] [CrossRef] [PubMed]
- Gomes, G.d.P.; Loginova, Y.; Vatsadze, S.Z.; Alabugin, I.V. Isonitriles as Stereoelectronic Chameleons: The Donor–Acceptor Dichotomy in Radical Additions. J. Am. Chem. Soc. 2018, 140, 14272–14288. [Google Scholar] [CrossRef]
- Kinzhalov, M.A.; Ivanov, D.M.; Melekhova, A.A.; Bokach, N.A.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. Chameleonic metal-bound isocyanides: a π-donating CuI-center imparts nucleophilicity to the isocyanide carbon toward halogen bonding. Inorganic Chemistry Frontiers 2022, 9, 1655–1665. [Google Scholar] [CrossRef]
- Katkova, S.A.; Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Zolotarev, A.A.; Boyarskiy, V.P.; Kukushkin, V.Y. (Isocyano Group π-Hole)⋅⋅⋅[d -MII] Interactions of (Isocyanide)[MII] Complexes, in which Positively Charged Metal Centers (d8-M=Pt, Pd) Act as Nucleophiles. Chemistry – A European Journal 2019, 25, 8590–8598. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Boyarskiy, V.P.; Boyarskaya, I.A.; Avdontceva, M.S.; Kukushkin, V.Y. Ligation-Enhanced π-Hole···π Interactions Involving Isocyanides: Effect of π-Hole···π Noncovalent Bonding on Conformational Stabilization of Acyclic Diaminocarbene Ligands. Inorganic Chemistry 2018, 57, 6722–6733. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Katkova, S.A.; Novikov, A.S.; Efremova, M.M.; Reutskaya, E.Y.; Kinzhalov, M.A. (Isocyano group)⋯lone pair interactions involving coordinated isocyanides: experimental, theoretical and CSD studies. CrystEngComm 2020, 22, 1154–1159. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Schier, A. Aurophilic interactions as a subject of current research: an up-date. Chem. Soc. Rev. 2012, 41, 370–412. [Google Scholar] [CrossRef] [PubMed]
- Schmidbaur, H.; Schier, A. A briefing on aurophilicity. Chemical Society Reviews 2008, 37, 1931–1951. [Google Scholar] [CrossRef]
- Tiekink, E.R. Supramolecular assembly of molecular gold(I) compounds: An evaluation of the competition and complementarity between aurophilic (Au⋯Au) and conventional hydrogen bonding interactions. Co-ord. Chem. Rev. 2014, 275, 130–153. [Google Scholar] [CrossRef]
- Mirzadeh, N.; Privér, S.H.; Blake, A.J.; Schmidbaur, H.; Bhargava, S.K. Innovative Molecular Design Strategies in Materials Science Following the Aurophilicity Concept. Chem. Rev. 2020, 120, 7551–7591. [Google Scholar] [CrossRef]
- Weber, D.; Gagné, M.R. Aurophilicity in Gold(I) Catalysis: For Better or Worse. In Homogeneous Gold Catalysis, Slaughter, L.M., Ed.; Springer International Publishing: Cham, 2015; pp. 167–211. [Google Scholar]
- Min, H.; Xiao, G.; Liu, W.; Liang, Y. Copper-catalyzed synthesis of 2-aminobenzothiazoles from 2-iodophenyl isocyanides, potassium sulfide and amines. Org. Biomol. Chem. 2016, 14, 11088–11091. [Google Scholar] [CrossRef]
- Uson, R.; Laguna, A.; Laguna, M.; Briggs, D.A.; Murray, H.H.; Fackler Jr., J.P. (Tetrahydrothiophene)Gold(I) or Gold(III) Complexes. In Inorganic Syntheses; 1989; pp. 85–91.
- Sheldrick, G. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS-2008/1-Bruker AXS area detector scaling and absorption correction. Bruker AXS, Madison, Wisconsin, USA 2008.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Yamada, S.; Rokusha, Y.; Kawano, R.; Fujisawa, K.; Tsutsumi, O. Mesogenic gold complexes showing aggregation-induced enhancement of phosphorescence in both crystalline and liquid-crystalline phases. Faraday Discuss. 2016, 196, 269–283. [Google Scholar] [CrossRef]
- Ruch, A.A.; Ellison, M.C.; Nguyen, J.K.; Kong, F.; Handa, S.; Nesterov, V.N.; Slaughter, L.M. Highly Sterically Encumbered Gold Acyclic Diaminocarbene Complexes: Overriding Electronic Control in Regiodivergent Gold Catalysis. Organometallics 2021, 40, 1416–1433. [Google Scholar] [CrossRef]
- Ecken, H.; M. Olmstead, M.; C. Noll, B.; Attar, S.; Schlyer, B.; L. Balch, A. Effects of anions on the solid state structures of linear gold(I) complexes of the type (o-xylyl isocyanide)gold(I) (monoanion). Journal of the Chemical Society, Dalton Transactions 1998, 3715–3720. [CrossRef]
- Fujisawa, K.; Okuda, Y.; Izumi, Y.; Nagamatsu, A.; Rokusha, Y.; Sadaike, Y.; Tsutsumi, O. Reversible thermal-mode control of luminescence from liquid-crystalline gold(i) complexes. J. Mater. Chem. C 2014, 2, 3549–3555. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii of Metals in Covalent Compounds. J. Phys. Chem. 1966, 70, 3006–3007. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Hengst, T.; Lothschütz, C.; Rominger, F. New and Easily Accessible Nitrogen Acyclic Gold(I) Carbenes: Structure and Application in the Gold-Catalyzed Phenol Synthesis as well as the Hydration of Alkynes. Advanced Synthesis & Catalysis 2010, 352, 1315–1337. [Google Scholar] [CrossRef]
- Hobbollahi, E.; List, M.; Redhammer, G.; Zabel, M.; Monkowius, U. Structural and photophysical characterization of gold(I) complexes bearing naphthyl chromophores. Inorg. Chem. Commun. 2016, 65, 24–27. [Google Scholar] [CrossRef]
- Katkova, S.A.; Mikherdov, A.S.; Sokolova, E.V.; Novikov, A.S.; Starova, G.L.; Kinzhalov, M.A. Intermolecular (Isocyano group)···PtII interactions involving coordinated isocyanides in cyclometalated PtII complexes. Journal of Molecular Structure 2022, 1253, 132230. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Jin, M.; Ito, H. Exploring Au(i) involving halogen bonding with N-heterocyclic carbene Au(i) aryl complexes in crystalline media. Chem. Sci. 2023, 14, 4485–4494. [Google Scholar] [CrossRef]
- Aliyarova, I.S.; Tupikina, E.Y.; Ivanov, D.M.; Kukushkin, V.Y. Metal-Involving Halogen Bonding Including Gold(I) as a Nucleophilic Partner. The Case of Isomorphic Dichloroaurate(I)·Halomethane Cocrystals. Inorg. Chem. 2022, 61, 2558–2567. [Google Scholar] [CrossRef]
- Aliyarova, I.S.; Tupikina, E.Y.; Soldatova, N.S.; Ivanov, D.M.; Postnikov, P.S.; Yusubov, M.; Kukushkin, V.Y. Halogen Bonding Involving Gold Nucleophiles in Different Oxidation States. Inorg. Chem. 2022, 61, 15398–15407. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Saeed, R.R.A.; Shehata, M.N.I.; Ahmed, M.N.; Shawky, A.M.; Khowdiary, M.M.; Elkaeed, E.B.; Soliman, M.E.S.; Moussa, N.A.M. Type I-IV Halogen⋯Halogen Interactions: A Comparative Theoretical Study in Halobenzene⋯Halobenzene Homodimers. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, K.; Singh, H.B. Adaptive responses of sterically confined intramolecular chalcogen bonds. Chem. Sci. 2018, 9, 7027–7042. [Google Scholar] [CrossRef]
Figure 1.
Identified Au···Au and CCN···Cl contacts in the crystal structure of 1.
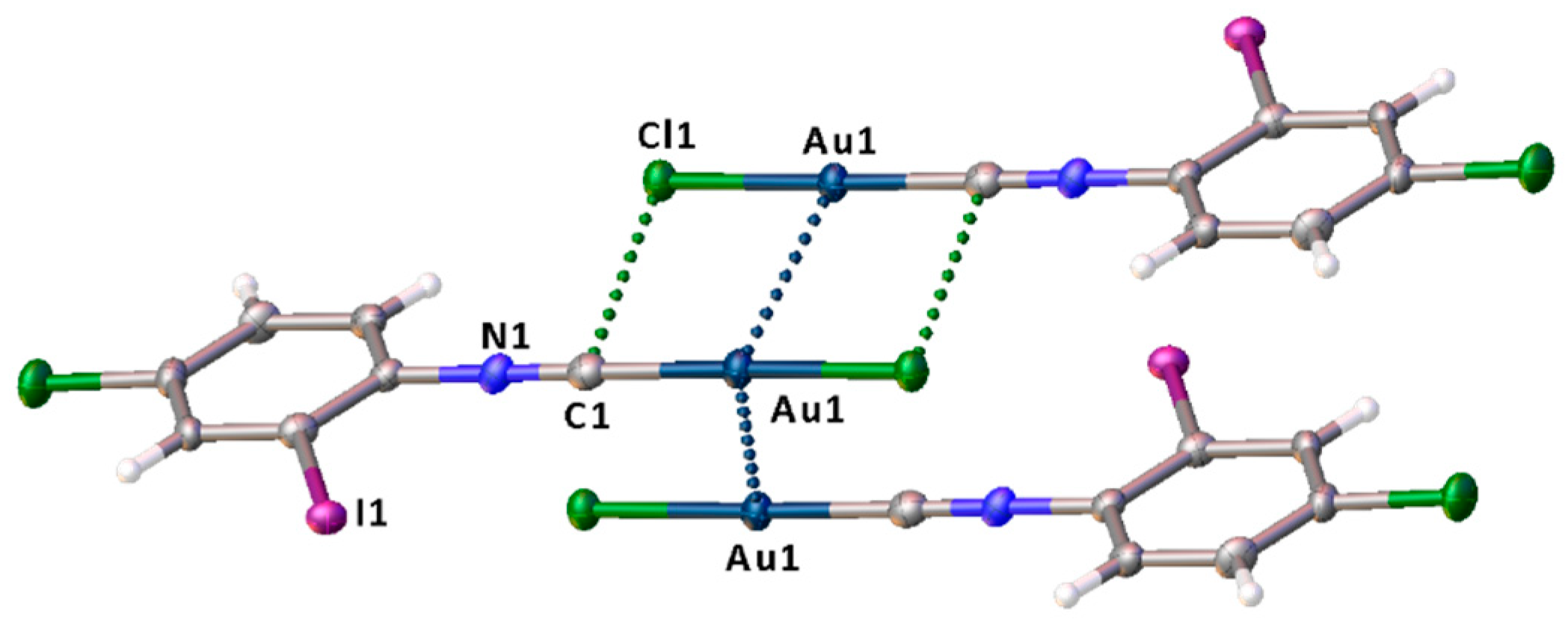
Figure 2.
Figure 2. 2D-ladder type architecture occurred via different type noncovalent interactions.
Figure 2.
Figure 2. 2D-ladder type architecture occurred via different type noncovalent interactions.
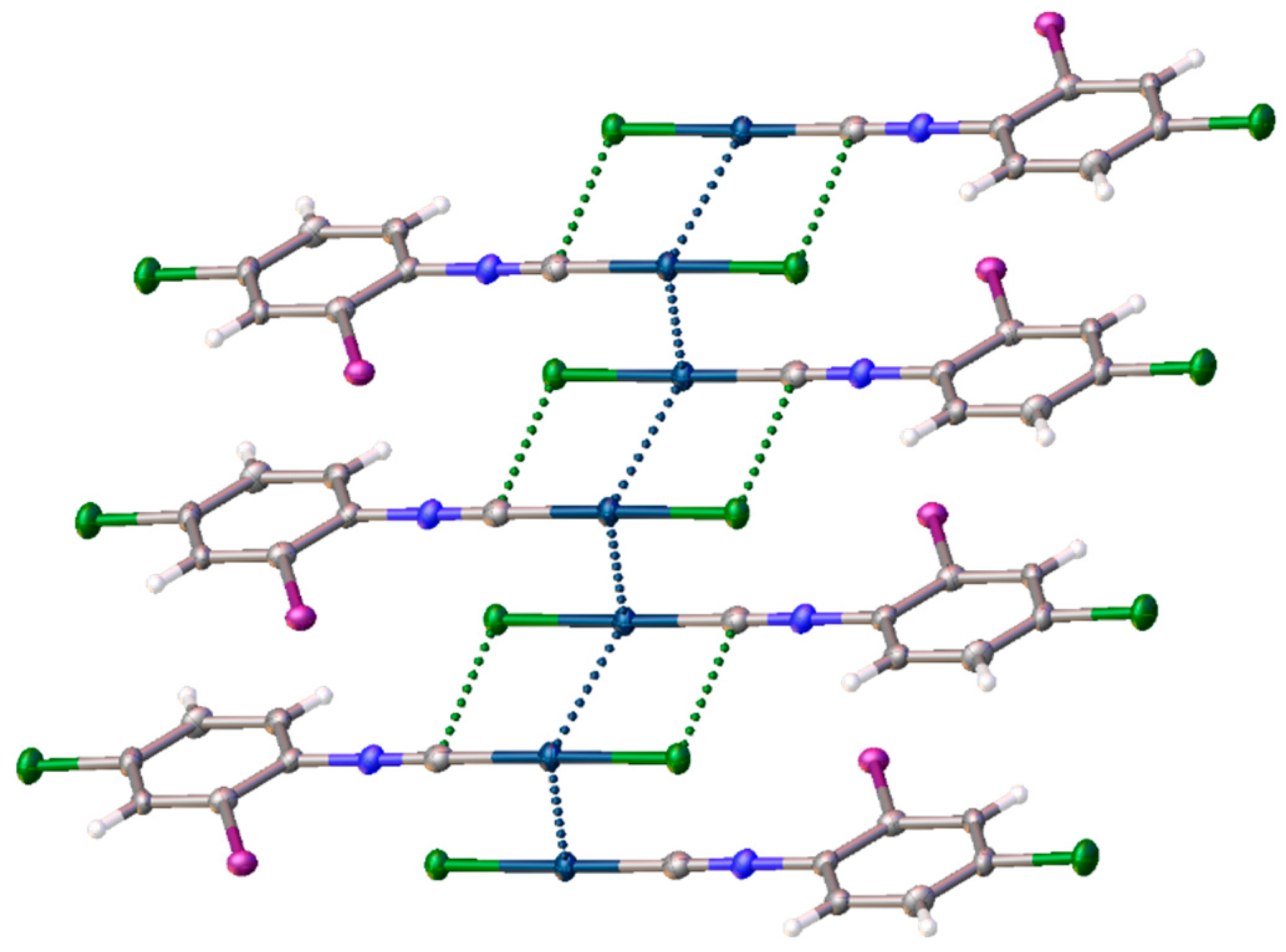
Figure 3.
I···I contacts in the structure of 1.
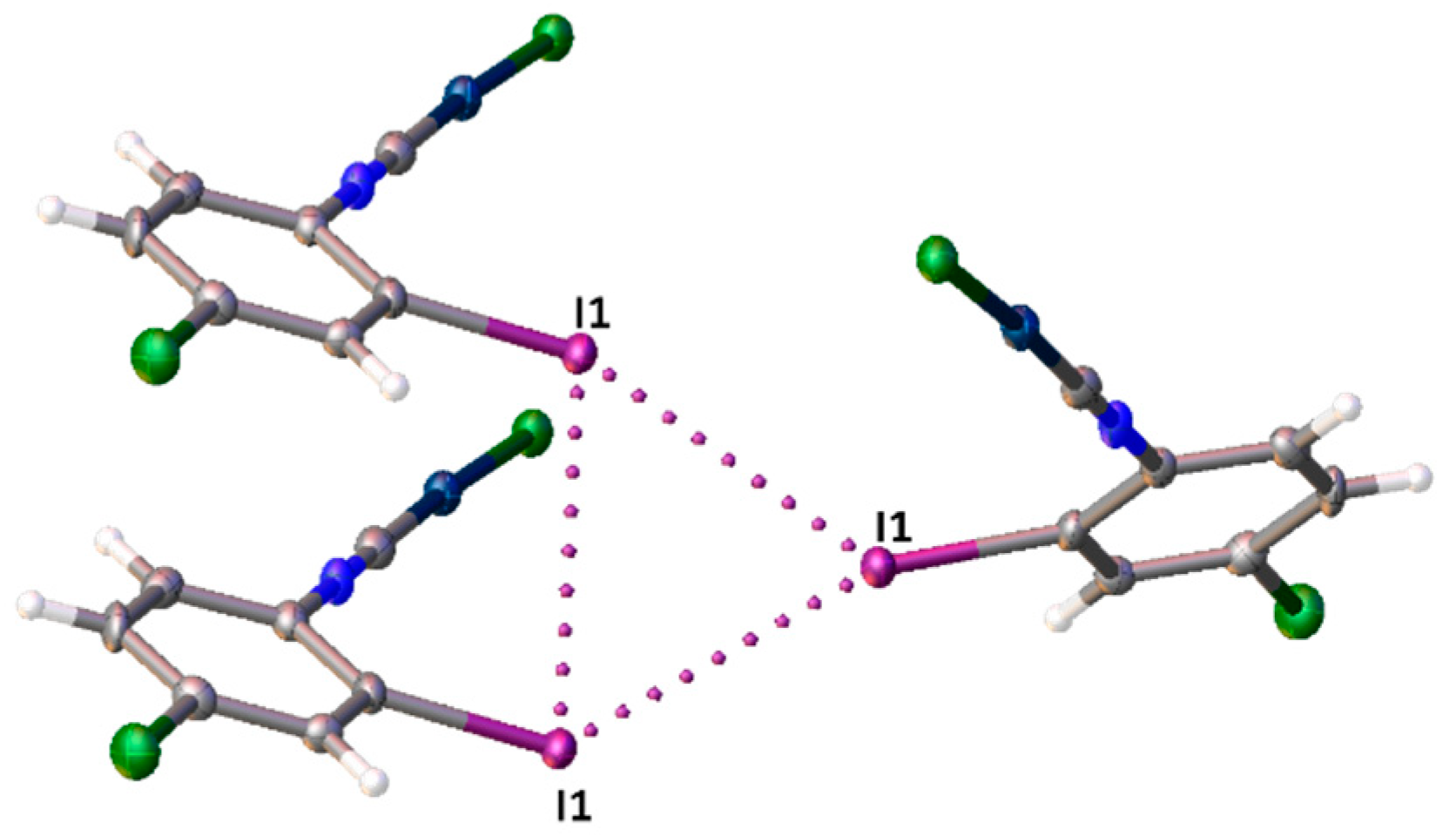
Figure 4.
MEP surface of 1. Energies at selected points are indicated in kcal/mol. Isovalue 0.001 a.u.
Figure 4.
MEP surface of 1. Energies at selected points are indicated in kcal/mol. Isovalue 0.001 a.u.
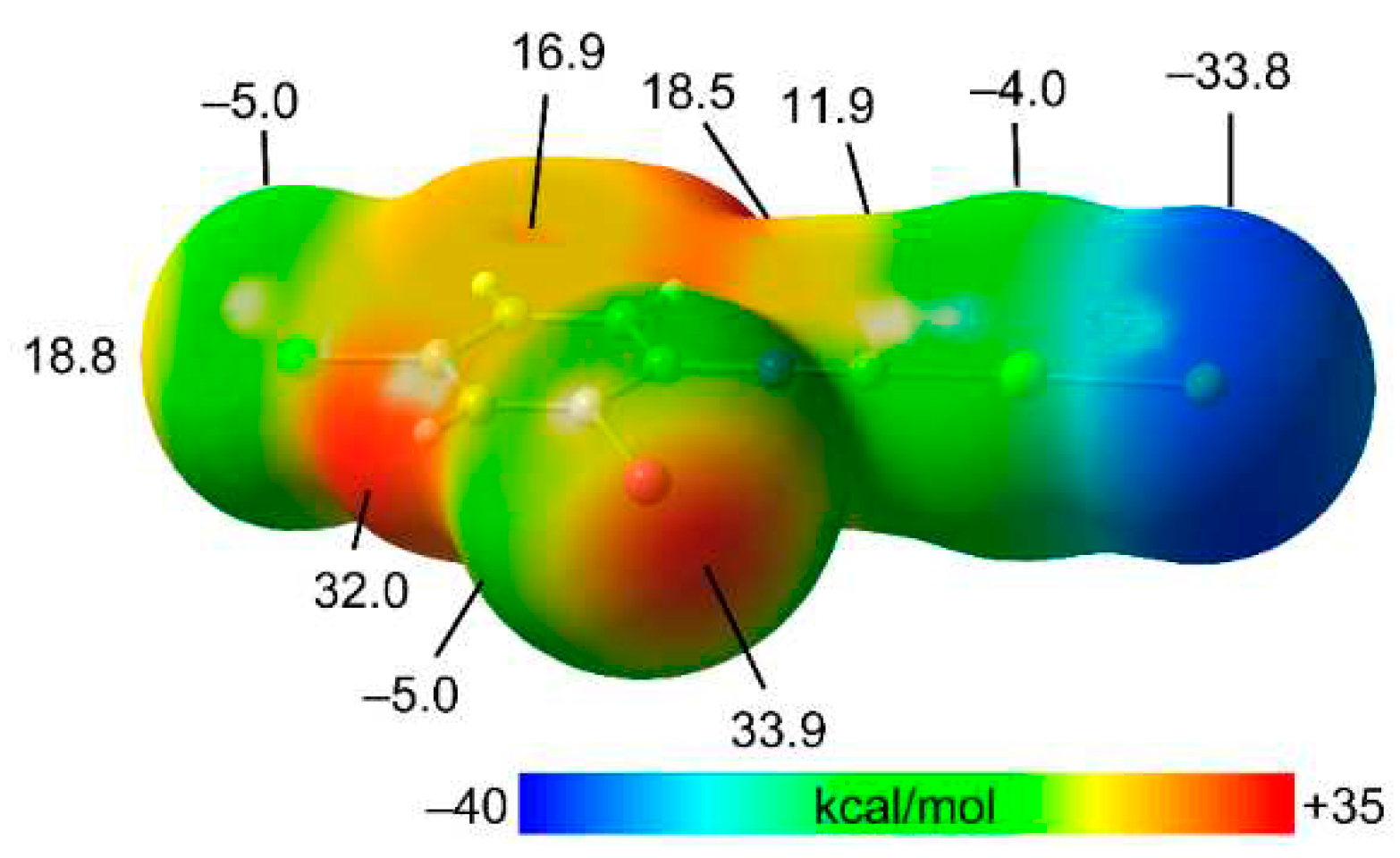
Figure 5.
Figure 5. QTAIM and NCIplot analyses of dimers A and B. The dimerization energies are indicated. See section 2.3 for the settings. Only intermolecular interactions are represented.
Figure 5.
Figure 5. QTAIM and NCIplot analyses of dimers A and B. The dimerization energies are indicated. See section 2.3 for the settings. Only intermolecular interactions are represented.
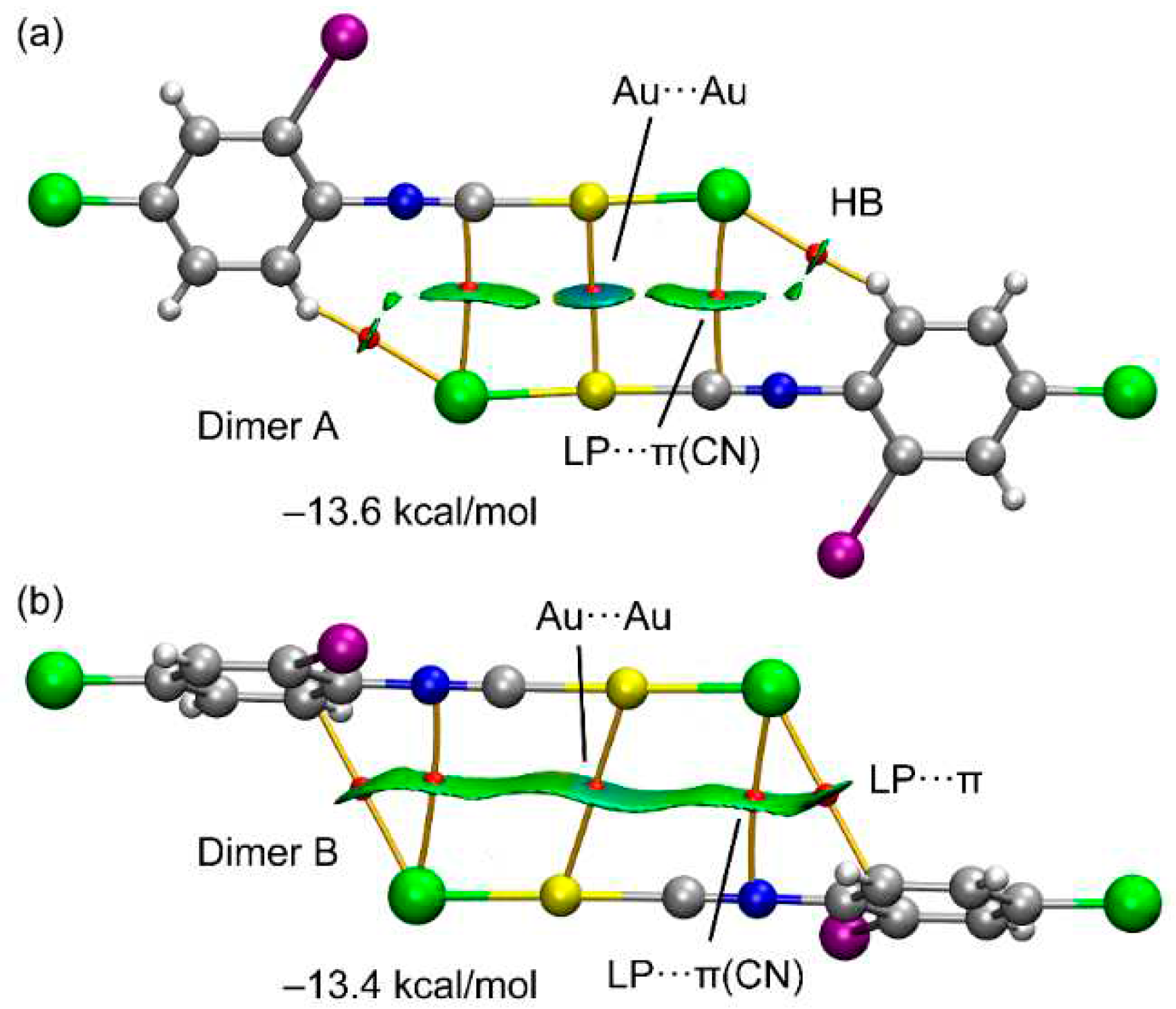
Figure 6.
NBOs involved in the charge transfer in dimers A (a) and B (b). The second order perturbation energies are also indicated.
Figure 6.
NBOs involved in the charge transfer in dimers A (a) and B (b). The second order perturbation energies are also indicated.
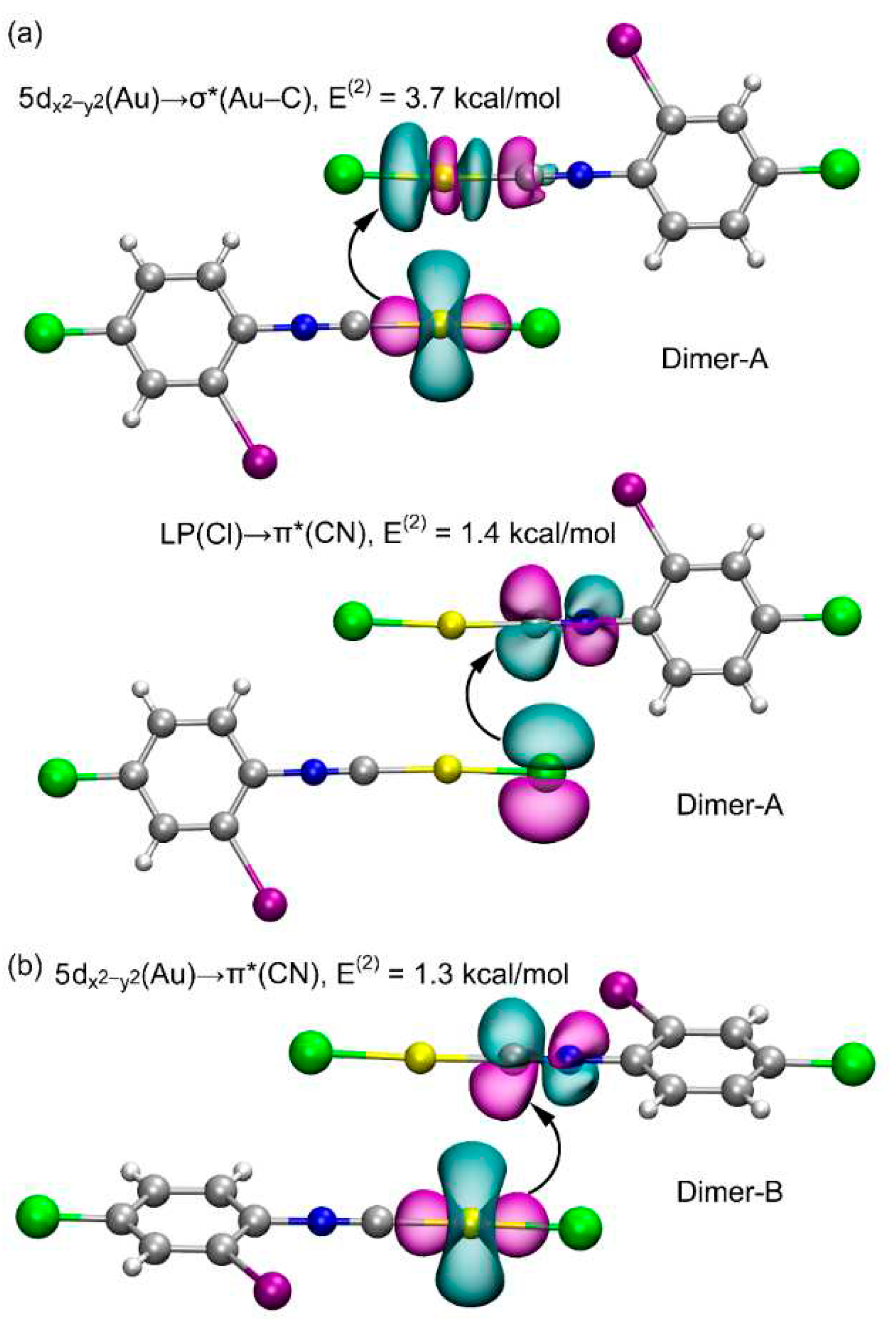
Figure 7.
(a) QTAIM of the trimeric assembly of compound 1. (b) ELF 2D of the trimer.
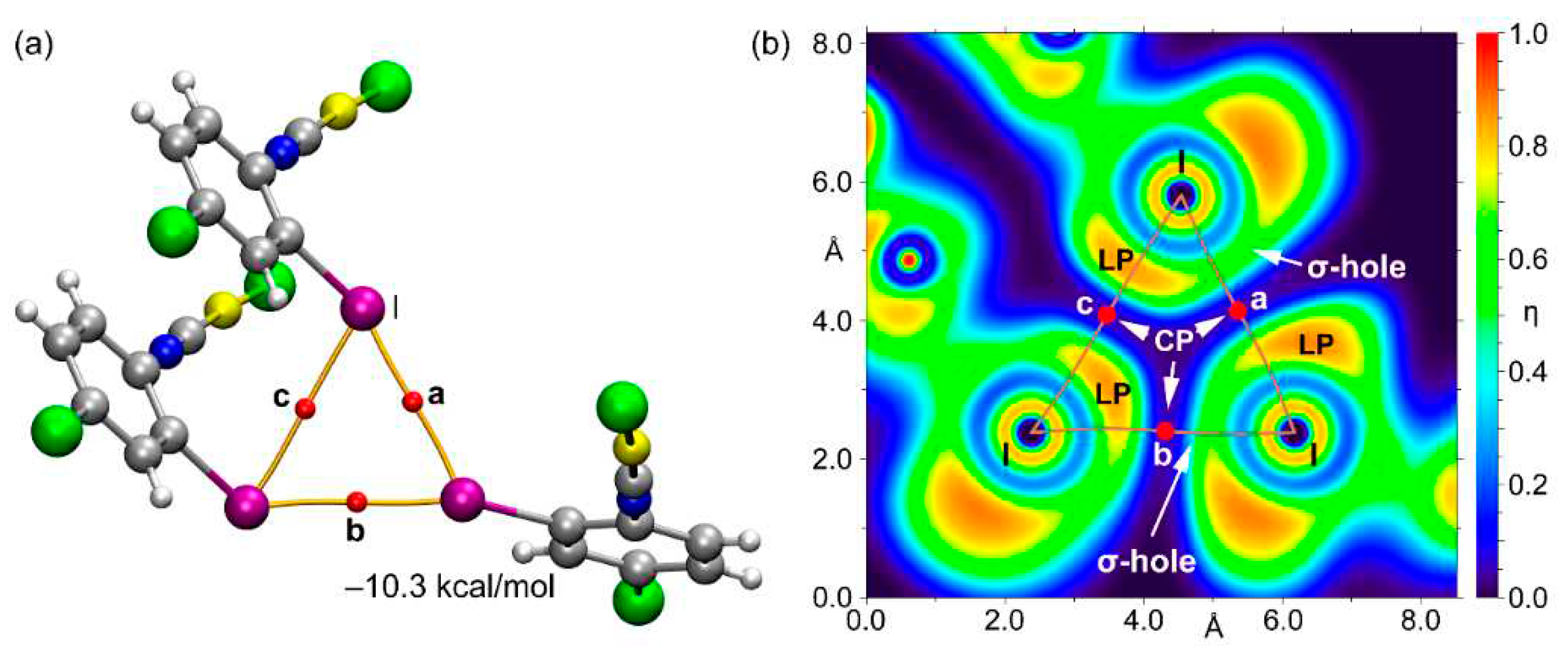

Table 1.
Geometrical parameters of short contacts in the structures of [AuCl(CNAr)].  ϕ is a torsion angle ClAuAuC.
ϕ is a torsion angle ClAuAuC.
ϕ is a torsion angle ClAuAuC.
Table 1.
Geometrical parameters of short contacts in the structures of [AuCl(CNAr)]. ϕ is a torsion angle ClAuAuC.
ϕ is a torsion angle ClAuAuC.| CSD code | d1 | d2 | d3 | d4 | ∠1 | ∠2 | ∠3 | ϕ |
|---|---|---|---|---|---|---|---|---|
| EBEZAP | 2.258(4) | 1.929(10) | 3.3159(8) | 3.373(11) | 94.0(3) | 86.7(2) | 91.6(4) | 0.1(3) |
| EQOGOJ | 2.2518(11) 2.2577(11) 2.2530(13) 2.2593(14) |
1.923(5) 1.919(4) 1.918(5) 1.935(5) |
3.2814(6) 3.2903(7) |
3.414(5) 3.436(5) 3.429(5) 3.448(4) |
87.34(13) 87.77(13) 87.63(13) 87.37(12) |
79.13(8) 79.27(8) 78.94(9) 79.29(9) |
97.32(16) 97.71(15) 97.36(15) 97.45(16) |
7.57(15) 8.88(14) 8.37(13) 9.57(14) |
| QIZTEA | 2.2564(15) 2.2584(14) |
1.946(6) 1.942(6) |
3.3092(6) | 3.433(6) | 90.64(16) 89.91(16) |
81.92(10) 82.57(10) |
95.24(19) 94.5(2) |
3.20(17) 2.88(17) |
| KEJRUQ | 2.255(2) 2.258(2) |
1.957(9) 1.939(9) |
3.3275(7) 3.2601(7) |
3.386(8) 3.410(8) |
101.5(2) 92.0(2) |
93.51(16) 82.92(18) |
83.8(2) 93.4(3) |
1.53(19) 1.12(18) |
| 1 | 2.2639(11) | 1.926(6) | 3.3050(8) | 3.428(6) | 92.53(16) | 83.77(10) | 93.15(19) | 0.90(17) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



