
Submitted:
09 September 2023
Posted:
12 September 2023
You are already at the latest version
Abstract
Ebola virus is a zoonotic virus comprised of 6 different species designated within the family Filoviridae and genus Ebolavirus. The first recorded outbreak of an Ebola virus (EBOV) was in Yambuku, Zaire (ZEBOV) in 1976, followed by Sudan Ebola virus (SUBOV) later that year. Outbreaks have been increasing throughout the 21st century, and mortality rates can reach up to 90%. Such extraordinary virulence is evidenced with few pathogens, similarly with Marburg virus (MARV) that originated in Uganda and was first detected in Germany in 1967. The virulent nature of filovirus disease has established these related viruses as a formidable global concern. There are currently four types of Ebolaviridae species known to infect humans, with two more recently identified in other animals that are genomically different with respect to cellular pathogenesis or aetiology of disease. Recent advances into understanding the pathogenesis of filovirus disease infections have been remarkable, yet the immunological response to filovirus infection remains unknown. Scientific analysis of cellular mechanisms can provide insight into virulence factors utilised by other pathogenic viruses that also cause febrile illness with occasional haemorrhagic fever in humans. In this review, we aim to provide a brief summary of EBOV proteins and the role of innate and adaptive immune cells known since 2000. We will consider the relevance and implications of immunological proteins measured by CD marker, alongside cytokine, chemokine and other biologically relevant pathways, as well as genetic research. Thorough understanding of immunological correlates affecting host responses to Ebola viruses will facilitate both clinical and applied research knowledge, contributing towards protection against potential public health threats.
Keywords:
Adaptive
; Ebola
; Filoviridae
; Immunology
; Innate
; Molecular
Introduction
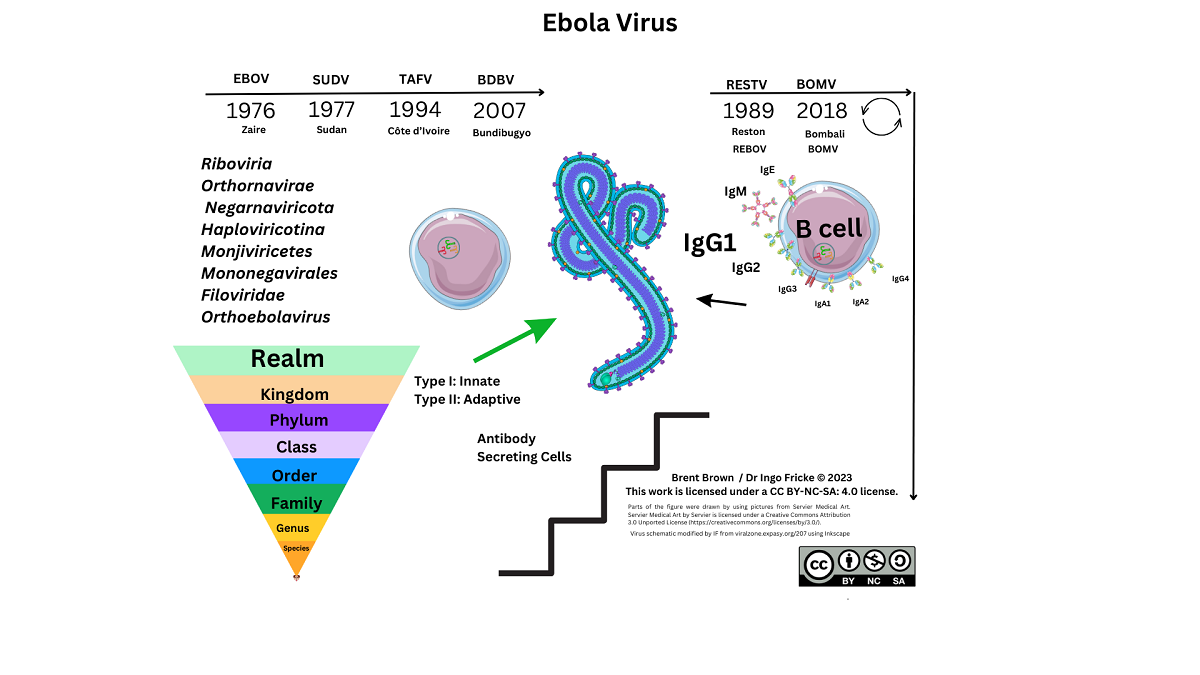
Ebola virus (EBOV) is the causal virion of a severe viral–induced pathology and disease affecting human hosts through cellular infection and across specific tissues, via transmission through host cell receptors or membranes and intracellular propagation. Total cases of EBOV have amounted to more than 34,626 infections since the virus was first isolated and characterised. Past outbreak mortality was at least 15,266 individuals which is indicative of an infection fatality rate (IFR) of around 50% (range 48%–74%) and the reproduction number (R0) between 2013–2014 was estimated within the ranges 1.51–2.53 1,2. Consideration of global incidence of disease or pathology of any description in all age groups is required to clarify research undertaken so far 3. Despite developments that occurred during the tragic outbreaks in the Republic of Guinea and surrounding areas between 2013–2016 much still remains unclear. However, transmission of zoonotic viruses occurs in other animal species 4. In 2017, the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (ICTV) was published with updates. Subsequently, another species of Filoviridae genus named Bombali virus (BOMV) was defined in 2018 not seen in humans currently 5.
Ebola virus is categorised as a Biosafety Level 4 (BSL4) pathogen only researched in designated laboratories (see Supplementary Data). Diagnostics are discussed elsewhere in other articles subject to criteria from the World Health Organization (WHO) protocol and criteria “ASSURED” in order to meet the benchmark for usage as point of care (PoC) products to test for any pathogen 6,7. From the research that has been conducted, EBOV virion particle size is estimated at a length of 1–2 µm with a diameter in the region of 80–100nm. Each is composed of a negative (–ve) sense single–stranded (ss) ribonucleic acid (RNA) genome of around 19kb in size encoding for at least seven predominant proteins. A number of enzymes are necessary for the cleavage of host or virion particles for transcription including polymerases, transferases but also a methyl–transferase. The interaction between Ebola viral proteins (VP) within a host cell is still being clarified. In general, this family of viruses utilise the RNA genome as a complementary strand to synthesise messenger RNA (mRNA) via the RNA–dependent polymerase (RdRp) within a host cell. Viral, bacterial and fungal pathogens utilise different mechanisms for adhesion, permeation, propagation and replication within a host. Other (–ve) ssRNA viruses include the Rabies lyssavirus (Rabies), Hantavirus, Mumps virus (MuV), Measles virus (MeV) and also Rift Valley fever (RVF) phlebovirus that infect different hosts. Currently, data on Uniprot suggests that EBOV proteins are observed to be synthesised in rats and mice. Therefore, here we discuss the structure and function of EBOV virion particles, including virus protein components, cellular entry, and cellular regulation, along with immune cellular receptor research in context with innate and adaptive immunological mechanisms during EBOV infection.
Method
We performed a comprehensive PubMed literature review encompassing the period from discovery of the Ebola virus, in 1976, until 1st July 2023, using the search terms “Ebola”, “Filovirus”, “Cluster of Differentiation”, “Immunology”, “Innate ” and “Adaptive” or in various combinations. Relevant articles to the manuscript title were included.
Structure and Function of Ebola Virus
Ebola Virus Protein Components and Cellular Entry
Other reviews summarise current knowledge of proteins known to have been sequenced from the EBOV genome in great depth 8. The method of transmission of EBOV may be considered to be through bodily fluid and similarly discussed in other reports with sequencing ongoing 9. Therefore, the scope of this review is to explore knowledge thus far with regard to the interplay between innate/adaptive immune system cells, cytokines (interleukins, IL), interferons (IFN) and chemokines (denoted by CC or CXC with receptor or ligand (R/L)) alongside cluster of differentiation (CD) molecules 8.
At least seven known EBOV proteins are characterised between the 3’ to 5’ EBOV genomic (–ve) ssRNA strand. In order, these include a helical nucleoprotein (NP), viral protein 35 (VP35), VP40, glycoprotein (GP), VP30, and VP24 with a polymerase (L) protein required for transcription. During 1998 the structure of EBOV GP was further elucidated after prior outbreaks with GP2 appearing to form a trimer with GP1 more important for receptor binding/viral entry requiring a comparatively conserved furin enzyme for cleavage differing between Filoviridae with soluble GP (sGP) traversing organelles including the Golgi apparatus and Endoplasmic Reticulum 8,10. Particle entry of EBOV occurs by forming a ribonucleoprotein (RNP) complex with VP35/VP30 interacting as a dimer, and then VP24 followed by the matrix (M) protein, VP40 8,11. Recently from 2022, reports suggest that VP35 forms a dimer with the polymerase L protein and may underpin the transcription process of the NP as well as replication thereafter 8,11. It is necessary to correlate the individual size, structure and function of protein composition as immune cell epitope recognition relies on antigen presentation of degraded viral peptides. The EBOV NP protein is estimated at 739 amino–acids, VP35 is 340 amino–acids, VP40 is 326 amino–acids, GP a total of 1,378 amino–acids, VP30 288 amino–acids, with VP24 251 amino–acids and the longer RNA polymerase (L protein) 2212 amino–acids at the 5’ end of the EBOV viral genome 8.
Characterisation of the (–ve) ssRNA EBOV GP occurred before 2009 and brought to light two other glycoproteins (GP1/GP2) together with a small–soluble glycoprotein (ssGP) 8. The ssGP is described as a di–sulphide linked homodimer and N–glycosylated protein produced through transcriptional RNA editing that can occur of GP1/GP2 with the addition of seven uridine (U) residues in GP1 and GP2 together with an adenine (A) amino–acid 8,12. It was previously considered that three biological processes contribute to EBOV virion particle cellular entry. Two methods of virion particle entry were described before 2016 being either clathrin–dependent and/or caveolin–dependent EBOV virion particle cellular uptake. Recently, between 2010 and 2011, this was clarified as remaining a grey area although logical suggestions were that EBOV utilises micropinocytosis as the main process modifying actin cytoskeletal elements for cellular vesicular entry 13,14. Thereafter, virion transport occurs intracellularly where GP1 undergoes proteolysis by one of two cathepsins (B/L) eventually leading to GP2–dependent host/virion endosome entry 8. Further detail is shown below (see Figure 1).
Cellular Regulation and Ebola Virus Proteins
Therefore, cellular EBOV virion–like particles (VLP) produce variable and complex functional proteins within host cells through transcription of viral genome elements affecting parts of the homeostatic cellular compartment with much remaining unknown. For example, EBOV VP35 has amino–acid residues considered to inactivate protein kinase R (PKR) whilst interacting with a protein activator of IFN–induced protein kinase (PACT) 8,15. Predominant functions of VP40 include those of maintaining virion particle structure with significance as a matrix (M) protein; while VP30 role is that of transcription, activation as well as initiation. In comparison, VP24 seemingly interacts with the un–unphosphorylated signal transduction activator of transcription 1 (STAT1), but also two types of karyopherin (KPNA1/5) downstream reducing nuclear accumulation of phosphorylated STAT1 8. Therefore, it can be considered that a key cellular pathway producing type I IFN (α/β) release is disrupted that trains the immune system response. Many authors concur that the mechanisms of viral EBOV cellular responses are unclear to date 16,17. Intriguingly, utilising co–immunoprecipitation assays in 2021, it was discovered that the EBOV VP24 protein may interact with emerin (EMD), lamin A (LMNA), lamin B (LNMB) as well as lamin C (LMNC). It was highlighted and seen that each protein was required to stabilise nuclear membranes that are disrupted 18–20.
Prior articles investigating EBOV GP mutations, specifically within A82V, are relevant and may or may not enhance cellular virion particle entry in vitro of the EBOV glycoprotein 21. Nevertheless, it was highlighted in vitro that an amino–acid substitution where alanine was substituted with valine, as both contain neutral non–polar R groups and a methyl (CH3) addition, that an increase in EBOV cellular infectivity was observed 21. Moreover, a change between threonine to isoleucine at residue 544 of the glycoprotein (GP) increased virion particle kinetics during viral host plasma membrane (PM) fusion utilising the intracellular Niemann–Pick type C1 (NPC1) receptor within endosomes 21. The crystallised protein structure of NPC1 was observed with clarity that a pocket exists within the cholesterol transport pathway where low–density lipoproteins (LDL) are processed 22. Thereafter, in vivo, NPC1 was observed in deficiency studies, where EBOV was non–contagious with EBOV GP appearing to form a complex with the NPC1 luminal domain. One enzyme, a serine cathepsin protease L, was considered to be more important in proteolytic cleavage enhanced by the virion EBOV GP around or within the endosome. In order to replicate, the RNA polymerase transcribes from the 3’ trailer to the 5’ leader genetic viral RNA sequence intracellularly. This occurs through capping and adenylation utilising the L protein RNA polymerase sequentially but also through RNA editing to express full–length glycoprotein (GP). Ebola virus replication through endosomal pathways remains under research investigation 23. The exact mechanism EBOV utilises to form intracellular late endosomes remains unknown. Recently VP40 is ascribed to be crucial in membrane fusion during endosomal disassembly within cells 24.
Immune Cellular Receptor Research during Ebola Infection and Disease
Viral entry occurs by attachment to host cell membrane receptors or disruption of the cell/ PM utilising either receptors, ion channels, adhesion mechanisms or attachment factors for permeation. It is considered that the EBOV GP utilises a C–type lectin DC–SIGN (CD209) receptor expressed by dendritic cells (DCs) and macrophages (Mϕ) as well as other host cells that facilitates virion particle entry. The corresponding receptor is a DC–specific intercellular adhesion molecule–3 (CD50/ICAM–3) receptor. Data on proteinatlas.org shows DC–SIGN (CD209) is differentially expressed by host cells but at increased levels throughout the gastrointestinal (GI) tract, bladder, heart muscle and within the lymphoid organs that are the thymus, spleen as well as lymph nodes. It is also present in smaller quantities within the brain (e.g. cortex, hypothalamus etc.), but specifically within the choroid plexus. The current understanding of DC–SIGN evolved from 2000 research when it was ascribed that DC–SIGN and a homologous protein L–SIGN (CD209L) receptor both had mannose–binding motifs and are calcium–dependent and bind with high affinity to mannose N–glycans present in pathogens 25. Shortly after, between 2001 and 2003, indications were that this receptor was required for Mycobacterium tuberculosis infection of DCs and CD209 may potentially be utilised with ICAM–3 (CD50) present on T cells as an immunological synapse necessary for priming naive T cells26,27. In addition, the DC–SIGN receptor (CD50) is suggested to have high avidity for EBOV GP with both binding to CD209 28.
From 2006, emerging in vitro research indicated a synthetic peptide that could affect (inhibit) two types of Filoviridiae (MARV/EBOV) by activation of neutrophils. Activation of neutrophils during EBOV is intertwined with the pro–inflammatory response occurring through triggering receptors expressed in myeloid cells (TREM–1) regulating viral–induced increases in intracellular calcium as well as reducing secretion of cytokine tumor necrosis factor–α (TNF–α) 29,30. Circulating neutrophils are central to both endothelial cell (EC) adhesion, and platelet aggregation as well as activating other immune cells and also secret serine proteases (e.g., cathepsin G, elastase) and are sensitive to IL–6 due to high expression of the IL–6Rα/gp130 receptor subunit domain 30–32. It was indicated then that TREM–1 could be shed from the PM whilst increasing phosphorylation of decay activating proteins (e.g. DAP–10/DAP–12) possessing a conserved immunoreceptor tyrosine–based activation motif (ITAM) 33,34. Since discovery, DAP–12 protein has been described as a signalling adaptor molecule containing aspartic acid within the PM forming stable non–covalent homodimers that may interact with a number of natural killers (NK) cell receptors like CD94/NK2GC/E but also CD158 and other immune cells 33. Gene transcripts and proteins of DAP–12 have been described as in abundance in plasmacytoid DCs (pDCs), monocytes, Mϕ as well as NK cells with lesser amounts present in as yet unknown subtypes of αβ T cells expressing a T cell receptor (TCR) known as CD4+ and CD8+ T cells 33. Signalling regulation occurs through c–Jun N–terminal kinases (JNKs) but also spleen tyrosine kinases (Syk) in myeloid cells 33. Further to this, a homologous zeta–chain–associated protein kinase of molecular weight 70kDa (ZAP70) was identified as a crucial CD3 TCR expressed during T cell maturation 35. Interestingly Syk is not expressed in T cells but ZAP70 is expressed in NK cell and T cell populations with research ongoing. The importance of ZAP70, a cytoplasmic tyrosine kinase, can be considered, as ITAM tyrosine residues can be doubly phosphorylated; thereafter resulting in signalling through ZAP70 with high affinity through –SH2 domains within the locality of the TCR/MHC class II complex required for antigen/peptide dependent TCR signalling 33,35–37.
Prior to 2011, reports emerged investigating other receptors further clarifying unknown mechanisms of EBOV cellular PM entry. The T–cell immunoglobulin–mucin–domain 1 (TIM–1) was of consideration, denoted as CD365 in vitro, that may facilitate EBOV cellular infection in overall Filoviridae research 38. Mucosal epithelial expression of TIM–1 was suggested to be a potential receptor for EBOV cellular infection and route of entry. Recently, TIM–1 has been suggested to also facilitate late viral virion particle egress. In 2017, adaptive T–cell studies investigating the immune response further clarified that TIM–1 is a relevant attachment factor for EBOV together with a further three attachment factors that are L–SIGN, folate receptor–α and Tyro3 receptor tyrosine kinases 39,40. Conundrums remain, as during 2019, TIM–1 would appear to in vivo not affect overall mortality, although as discussed further, EBOV stimulation affects T cell receptors with TIM–1 (CD365) affecting cytokine regulation. As research evolved, deletion of the mucin domain of EBOV GP1–5 has some effects unknown so far and may hold future promise with chemokines emerging that include CXCL10 and CCL2 and others of note in immunological responses detailed further below 40–42.
It is indicated that the EBOV VP40 protein requires phosphatidylinositol 4,5–bisphosphate (PI(4,5)P2) and phosphatidylserine (PS) forming oligomers at the PM surface also facilitating vesicular particle egress 43. Recently this was followed up to note VP40–membrane binding via electrostatic interactions with cationic VP40 residues. Therefore VP40 interaction with lipid residues was further clarified with phosphatidic acid (PA) as a key part of the formation of EBOV M proteins 44. It is of note that phosphatidylinositol (PI) is phosphorylated at the PM surface into PI(4,5)P2 and requires PA but also protein lipase C (PLC) and can hydrolyse phospholipids, whilst PI(4,5)P2 regulates actin formation required within epithelial layers an important EBOV method of transmission 45. Other receptors remain unclear, although considered prior were fibronectin receptors (denoted by integrin subunit domains) present within the extracellular matrix (ECM) (e.g., α5β1). As discussed, NPC1 protein is a 1278 amino–acid internal cellular receptor localised to endosomal and lysosomal membranes and in 2015 was confirmed to be essential for EBOV replication and pathogenesis in vivo 46. These were notable findings because NPC1 is uniformly expressed throughout the brain but preferentially within pDCs and endothelial cells (ECs) of the vascular system and possibly at decreased quantity within other immune cells. The unique receptor that EBOV utilises remains unknown, but viral EBOV GP is required for micropinocytosis and vesicular transport within cells 13,14. Furthermore, the ECM may contain other short signalling peptides unknown to date acting as cryptic pockets that differential EBOV proteins interact with triggering structural interaction and allowing differential EBOV binding to cellular receptors 47.
During 2011 it was further elucidated in vitro that VP24 disrupted a nuclear p38 mitogen–activated kinase (MAPK) gene transcript (MAPK14) and the resultant enzyme synthesis 48. It was distinguished that MAPK p38–α disruption was one key factor in type I IFN–β inhibition; but also that EBOV VP24 binds to karyopherin A1 (KPNA1) while inhibiting phosphorylated signal transducer and activator of transcription–1 (STAT1) nuclear accumulation. Therefore, as KPNAs would usually transport proteins through nuclear pores, the resultant STAT1 signalling stimulating IFN synthesis through interferon stimulatory genes (e.g. ISG15) could be affected by different EBOV virion particle subunit domains 48. Mitogen–activated protein kinases are serine/threonine–protein kinases regulating the p38 cell cycle progression. Disruption to p38–α regulation between the growth and mitosis cell cycle (G2/M) could therefore plausibly be considered; because p38 may regulate stability and translation of both cytokines, TNF–α as well as IL–6, affecting neutrophil migration and can be expressed within endothelial/epithelial cells 49. Furthermore, it is apparent that the deletion of p38–α within DCs in vivo has consequential effects on DC function. The p38–α deletion can result in changing viral antigen presentation to cytotoxic T (TC) cells expressing CD8 from conventional DCs (cDC) and reducing antigen presentation ability of CD8− cDCs accompanied with reduction of DC maturation cytokines IL–12p40 and IL–12p70 50. Therefore, viral EBOV mRNA may in effect dysregulate cytokine production as well as nuclear cell cycle regulation and transcriptional regulation affecting antigen presentation to T cells remaining unknown to date (see Figure 2).
Innate and Adaptive Immunological Mechanisms during Ebola Virus Infection
Overall Perspectives and Antigen Presenting Cells
During 2006, early research articles implicated the GP relevant to Filoviridae pathogenesis as development and results allowed more insight whilst genomic sequencing technology improvements occurred. At this time within the Marburg viruses whole–genome sequencing identified the existence of a Ravn virus (RAVV) characterised 51. Initial immunological research investigating Filoviridae was suggestive that an imbalance in helper T cell (TH), as well as cytotoxic T cell (TC) responses, may occur. The combined interaction of innate and adaptive regulation of the T cell response remains a topic requiring further detail. It is known that optimal immune responses require B cell antibody production; but also T cells alongside type I/II/III interferon (IFN) synthesis and requiring a homeostatic cytokine/chemokine balance to function in an autocrine/paracrine systemic manner. This in effect regulates antiviral, antibacterial and antifungal activity through cytolytic actions and phagocytosis by antigen–presenting cells (APCs), TC cells, and NK cells. In our last article, we discussed antigen–presenting cells (APCs) encompassing DCs, monocytes and macrophages (M1ϕ and/or M2ϕ) by CD molecule expression 32. These cells present and process pathogen–derived peptide epitopes to T cells utilising major histocompatibility complex (MHC) class I/II molecules. Dendritic cells are considered to have a unique tolerogenic profile. This cell population has the highest expression of MHC class II molecules and presents EBOV antigens 52. Monocytes are classified as non–classical, intermediate or classical with varying functions including pathogen phagocytosis. Macrophage categorisation is intertwined with cellular polarisation and the upregulation/downregulation of various CD markers. Recently these have been referred to as M1ϕ or M2ϕ that can have pro–inflammatory or anti–inflammatory functions. During 2017 it was designated that four EBOV proteins could potentially be key immunogenic proteins in positive innate and adaptive immunological responses, namely VP24, NP, GP, and sGP 53. Below is shown some of the known EBOV protein functions to this point with some of the relevant observed gene transcripts and cytokines observed during infection and within laboratory research hitherto that affect IFN synthesis and coagulation regulation pathways (see Table 1) 8,54.
Initial immunological laboratory observations are researched and measured by serum cytokine concentration changes during infection often referred to as a “cytokine storm” in an autocrine/paracrine dependent manner in conjunction with chemokines and Toll–like receptors (TLR) with a 2015 CD classification update on all relevant immune cellular markers 55. Studies of these can explain some of how the cellular/molecular immune system synchronises during health and disease with much remaining unclear 56–58. During 2003, the predominant APCs were further investigated with regards to Filoviridae infection. Dendritic cell research has evolved since 2017 59. Accordingly, the profile of DCs remains a mystery in EBOV infection amongst other viral diseases that is dependent on a multitude of transcription and activation factors. These contribute towards the tolerogenic and anergic profile of DCs. Since 2017, DCs are broadly classified into non–classical pDC, but also a further three classical/conventional subtypes (cDC1, cDC2 and cDC3) 32.
In 2003 it was indicated that EBOV VP35 could infect DCs of the monocytic lineage affecting the T cell response 60. It was then observed through in vitro culture that DCs maturing into a monocytic lineage secrete IL–1β, IL–6 together with IL–8 and express CCL5 (RANTES), with abrogation of type I IFN–α that is usually secreted in high quantities 60,61. More recently, it was confirmed that DCs obtained from EBOV peripheral blood mononuclear cell (PBMC) derived monocytes (moDCs) could permit EBOV replication but pDCs did not 60. Importantly a generic leukocyte–expressed marker, CD45+, was used to determine the cellular lineages as either MoDC (CD11c+CD16–CD14–HLA–DR+) or pDC (CD11c+CD14–CD16–CD123+HLA–DR+) expressing human leukocyte antigens (HLA–DR) that are required to present peptides to T cells (see Figure 3) 60. With research ongoing, it was confirmed that several key immune cell checkpoint gene transcripts (CD40, CD80, CD83) were potentially affected in DCs by EBOV during the 2013–2016 outbreak with clarity that VP35 could unblock antigen processing pathways and IFN transduction 48,62. Uniquely, it was seen in a key study that each of the type I/III IFN gene transcript changes in dendritic cells 52. These indicated that during EBOV infection VP24 may counteract earlier VP35 antagonism of DC IFN transcription of genes between 8–24 hours after infection and up to 5 days later 52,63. Type I IFNs are usually required to activate and regulate both Mϕ, NK cells and the T cell effector response 64. The significance of this still remains unclear. Below is depicted cytokine expression regulators during EBOV infection (see Table 2).
Seminal in vitro reports prior to 2013 also clarified some of the mystery investigating monocyte cell roles during EBOV infection. It was indicated that monocytes could be permissive to infection after a delay of up to 48 hours in effect allowing EBOV cellular replication. Key differences were clarified with preferential mRNA expression in monocytes of three IFN regulatory gene transcripts (IFITM1, IFITM2, IFITM3) that in effect regulate intracellular viral replication cycles through IFN synthesis. Whilst NPC1 was distinctively expressed in pDCs at higher concentrations than within monocyte–derived lineages usually presenting viral antigenic epitopes to T cells 65. It is therefore observed that EBOV can infect two APCs (Mϕ and DCs) with EBOV VP24 and VP35 having dual effects modulating the overall duration of leukocyte type I IFN synthesis and secretion of which the significance is unclear 48,52,62.
During EBOV infection, early indications were that a change in secretion of type II IFN–γ may occur usually secreted by T cells and NK cells. This was accompanied by reduced IL–12 and IL–2 synthesis with increased IL–10 and increased T cell apoptosis within 12 hours upon cellular stimulation 60,66. Synthesis of IL–12 is crucial (see Figure 4 below), as signalling occurs through p35/p40 subunits effecting DC responses in synergy with IL–23 and other cytokines via the heterodimer p70 formed with IL–12R (IL–12Rβ1/IL–12Rβ2) and a heterodimer shared with IL–23 67,68. In 2015, this was examined further to find exogenous stimulation could in vivo inhibit EBOV replication within Mϕ to find four chemokines expressed (CXCL9, CXCL10, CCL8 and CXCL11) together with complement proteins (C1s and C1r), both of which are usually regulated by IFNγ and three of these have a receptor (CXCR3 (CD183)) on DCs 69–71. Moreover, upregulation was noted of the Mϕ endocytic marker (CD163) shed from the PM localising with viral EBOV antigens around Mϕ and hepatocytes 72. The glycoprotein CD163 is also expressed by DCs during maturation. It is a haemoglobin scavenger receptor specific for both monocyte and M1ϕ/M2ϕ lineages and facilitates the uptake of haptoglobin/haemoglobin (Hp–Hb) complexes in the circulation where lysis of erythrocytes occurs during EBOV infection. It was further noted in 2006 that lipopolysaccharide (LPS) stimulation of TLR4 also causes CD163 shedding in monocyte lineages with TLR2 and TLR5 present at the cell PM surface 73–75. Recently (2017), TLR4 studies in vivo and in vitro in the absence of T cells are indicative that EBOV activates TLR4 with each of the CD11b+ and CD11c+ lineages (DCs, monocytes, M1ϕ and neutrophils) circulating in draining lymph nodes (dLNs) 76. Further to this observation, using GP–stimulated bone marrow–derived Mϕ (BMDM), it was observed in vivo that TNF–α, IL–1β, and IL–6 normalised within 24 hours; moreover CCL2 and CCL4 together with T cell–derived cytokines (IL–2, IL–4, IL–5, IFN–γ) and IL–10 remained elevated. Noteworthy was that CCL5 (RANTES) and IL–12 expression/synthesis remained unchanged. However, in vivo studies in detail indicated Mϕ cells continue to upregulate two co–stimulatory molecules (CD40/CD80) and potentially could still signal to three respective T cell ligands (CD40L/CD28/CTLA4) which are required for adaptive T cell responses.
From 2016, further detailed analysis occurred in vitro comparing EBOV and MARV of consideration as both are classified as Filoviridae 77. This laboratory utilised cellular transcriptome mapping in vitro to note the upregulation of an annexin gene transcript (ANXA3), with the potential protein encoded known to inhibit phospholipase 2 (PLA2) enzymes 77,78. These were key observations as PLA2 is crucial and metabolised by cyclooxygenases (COXs) into anti–inflammatory mediators including leukotrienes (LTs), prostaglandins (e.g., PGE2) as well as eicosanaoids. Also observed were IL–8 and IL–32 which could affect T cell apoptosis. Another gene transcript, CYR61, was observed known to encode a known cysteine–rich protein binding to heparin and is a growth factor (GF) affecting vascularisation. The dual specificity protein phosphatase 1 (DUSP1) gene transcript was highlighted encoding a protein processing intrinsic phosphatase activity that can inhibit the extracellular signal–regulated kinase (ERK1/2/MAPK) pathway together with phosphothreonine and phosphotyrosine residues present in STAT proteins 77. In longer–term studies, it is indicated that Mϕ expressing CD68+ together with leukocytes expressing CD45+ but also T cells (denoted by CD3 +) could also infiltrate heart ventricles and the choroid plexus 79. Historically, and since 2021 the blood–brain barrier (BBB) in terms of how immune cells permeate across semi–permeable membranes is unclear 80.
Antibody Responses to Ebola Virus
In the early 21st century between 2008–2009, reports appeared utilising an isolated neutralising antibody binding to EBOV GP to elucidate the crystalline structure of an antibody 81. It was then specified as in vivo research continued that a mouse Ig2a monoclonal antibody could bind specifically to the glycosylated mucin–like domain of the EBOV GP (EQHHRRTDN, amino–acids 405–413) 82. The overall structure of EBOV GP seemingly then was considered to be a GP1 trimer composed of base, head and glycan cap forming a chalice with a trimer of GP2 subunits forming a cradle around the GP1 domain when investigations began to indicate differential antibody responses against GP1/GP2 and the sGP cellular product 83. Furthermore, the existence of a smaller Δ–peptide clarity remains unknown to date with two theories which are that of a peptide that could be immuno–adhesive or act as a viroporin 84–87. To this effect, during 2011, observations were made in comparison with different Filoviridae between sGP and ssGP function with hints of differences in the immunological function of the Δ–peptide resulting from a C–terminal cleavage product of sGP 87. However longitudinal research studies of the 2013–2016 outbreak, indications were that a polyfunctional B cell and APC–induced CD8+ T cell phenotype remained crucial (n=206) for an effective immune response. This was measured as cells being able to synthesise and secrete type II IFN–γ alongside two other cytokines, TNF and IL–2, synthesised in 0.046% of total CD8+ T cells 88. Observations of immunological relevance were examined within a cohort (n=207), where 96% possessed antibodies after EBOV infection with sub–characterisation confirming that 9% of blood samples were considered to possess neutralising antibodies (nAb) 88. It was observed in Western Blot analysis that antibodies could be polyfunctional against four of the seven EBOV virion–encoded proteins (GP, NP, VP35 and VP40). Therefore, this would indicate characterised viral–specific epitopes within EBOV particles that may generate antigen/epitope–specific innate and/or adaptive immunological responses 88. In longer–term studies utilising the development of a cell–based reporter system, the affinity of EBOV–stimulated antibody binding via their respective Fc (constant) receptors to effector cell surface receptors was examined. It was confirmed that EBOV infection may generate IgG1 antibodies that display the highest affinity for the FcγR1 (CD64) receptor rather than FcγRIII (CD16) and FcγRII (CD32) over a time–frame of more than 10 years 89. These three receptors are utilised differentially by antibody Fc regions to effect B cell antibody–dependent responses on a variety of host immune cells like macrophages.
Serology reports in 2020 are indicative that EBOV protective antibodies can be synthesised in humans after infection with isotypes produced predominantly ten times higher in concentration of the IgG1 isotype compared to IgG2, IgG3 and IgG4 still produced 90. In addition, IgM is also produced which is historically seen to be a marker of natural infection in other viral pathologies. It is further indicated (n=4) in a longer kinetics study that IgG specific for GP remained over 2 years and up to 12 years with IgM declining and IgA remaining at increased concentrations alongside IgG responses to both EBOV NP and VP40 91. The characterisation of B cells producing antibodies then was indicative of antibody–secreting B cells (ASC) expressing CD27hi/CD38hi with less expression of other activation markers CD71+/CD20+. Notable at this point was that the removal of mucin and glycan caps of EBOV GP further increased the binding affinity of serum IgG to cleaved viral GP (sGP) 92.
Adaptive Immune Responses to Ebola Virus
A key report utilised a control group of non–pathogenic Filoviridae, (REBOV), in comparison with human pathogenic Filoviridae (EBOV, MARV) in both humans and macaques was performed in 2006 66. It was noted, utilising annexin V and propidium iodide staining, that T cell apoptotic rates at least doubled indicating cell cycle changes and apoptotic cells, but not in the non–pathogenic control virus group (REBOV). Furthermore, utilising in vitro viral peptide sequences stimulating peripheral blood mononuclear cells (PBMCs) and measuring hypodiploid DNA content that a three–fold DNA synthesis reduction occurred during the T cell synthesis (S) phase to each of the pathogenic Filoviridae (MARV/EBOV) 66. It was considered that pathogenic Filoviriade could alter CD4/C8 cell count ratios measured by TH1, TH2 and TC cell populations with reasons unknown as to cellular mechanisms. Indications may be suggestive that this could be caused by nuclear membrane changes, cell communication, intracellular signalling, or single nucleotide point mutations (SNP) that affect any of the proteins or immune cell populations. However, EBOV T cell research is evolving and continues with T cell phenotypes only further researched that include mucosal–associated invariant T cells (MAIT) and others 32.
It was noted in 2003 that EBOV may suppress TH1 cell phenotypes alongside IL–12p40 and type II IFN–γ production with inhibition of IL–2, TNF–α, as well as CCL2, but not CCL3 utilising monoclonal antibody reagents that are validated as specific in research 60. The reasons for this remain unclear. Further indications evidenced then were reduction in CD25 within T cell populations categorised into regulatory T cell (TREGS) amongst others known to express this soluble secreted PM cellular receptor. Shortly after in 2011, independently, T cell populations were further researched and it was noted that a memory T cell subset could possess regenerative–like properties (denoted by CD45RO–CCR7+CD45RA+CD62L+CD27+CD28+) and IL–7R+ T cell compartment characteristic of T cells 93. Importantly it indicated that T cells could be specific for viral and tumour–associated antigens (TAA) whilst specifically expressing high concentrations of IL–2Rβ, CXCR3, and LFA–1 with the CD95 activation marker 93. The role of T cell responses to EBOV in 2015 indicated that polyfunctional TC (CD8+) cells could express CCR7 and not CD45RO as phenotypical TEM cells developing EBOV–specific responses upregulating CD28 and CD95 in response 40,94. These were critical as CCR7 is expressed on the majority of the T cell population whereas CD45RO/RA is used to define T cell maturation, while CD28 is a required co–stimulatory molecule for TC cell stimulation and survival. Other investigations of the exact mechanism of action of EBOV on T cells are indicative of a “superagonist–like” effect. When EBOV–stimulated PBMCs were depleted of DCs/monocytes it could be seen that activation markers CD25/CD69 were significantly upregulated at 48 hours on T cells 40. The reasons for EBOV stimulation and its effects on host cells remain unknown. Below is shown an infographic of the overall immune system interactions based on prior research (see Figure 3).

Figure 3.
Immune Cellular Response Profile by Cluster of Differentiation (CD) Molecule during EBOV infection.
Figure 3.
Immune Cellular Response Profile by Cluster of Differentiation (CD) Molecule during EBOV infection.
With regards to the CD4+ T cell population, stimulation indicated preferential expression of CD45RO+ on T cells in survivors predominantly central memory (TCM) T cells expressing CXCR3+ that were EBOV GP antigen–specific with the overall TH (CD4hi) cell population expressing another activation (CD38) marker. Simultaneously in a 2018 report, it was noted in vivo that CD69 expression downregulation did not occur within myeloid–derived DCs (mDCs) in the spleen 95. Though the earlier report did indicate that CD4+ and CD8+ T cells significantly downregulated CD69 at 12 hours, it was recently confirmed that this occurred at 48 hours with CD25 upregulation in T cells 66. During 2019, annexin–A5 was suggested to potentially be a beneficial therapeutic during microvascular disease. Annexin proteins are both soluble and hydrophilic and bind to oppositely charged phospholipids reversibly dependent on calcium. Such research would be interesting to see as the above annexin proteins can aggregate vesicles but also phosphatidylserine (PS) does appear to be implicated in EBOV host–cell interaction mostly unknown 96,97.
Notable further investigations into EBOV host cell receptors showed that deficiency of TIM–1 in vitro cell line culture could regulate T cell secretion of crucial cytokines that were type II IFN–γ, IL–2, and TNF–α as well as IL–12p40 with the granulocyte–macrophage colony–stimulating factor (GM–CSF) 40. Much remains unknown regarding EBOV effect on T cells; however, it was evidenced that EBOV could both stimulate T cells and be internalised without replicating. Investigations showed the EBOV NP protein could be found within T cells expressing the TCR complex differentially affected. Specifically, the TCR CD3ε subunit domain could be found at increased frequency in EBOV–stimulated samples. This was accompanied by downregulation and degradation of the TCR CD3ζ with intracellular signalling maintained but CD3ζ co–localised with a monomeric guanosine–5'–triphosphate (GTP) enzyme (a GTPase, Rab7) and EBOV GP in late endosomes 98–100. The CD3/TCR complex predominantly utilises lymphocyte–specific protein tyrosine kinase (Lck) with phosphorylation of the TIM–1 cytoplasmic domain resulting in activating phosphatidylinositol 3–kinase (PI3K) representing a known cell survival and proliferation pathway. In other longitudinal kinetic studies (n=2), individuals underwent treatment, the CD95 marker was observed as transiently upregulated on both TC/TH cells with PD–1 transiently upregulated on TH cells indicative of cellular activation. Antigen presentation proteins denoted by human leukocyte antigens (HLA–DR/ MHC Class II) were present on CD8+ T cell PMs, but it was inferred that CD45RA and CCR7 may not be expressed on all T cell populations 101. During EBOV infection, each of the DCs, monocytes, Mϕ as well and T cell phenotypes of the immune system are predominantly affected similarly to other viral pathologies 32,102. Recent observations are that EBOV sequesters the interferon regulatory factor (IRF3) in viral inclusion bodies (VIB) blocking required type I IFN induction. This usually occurs through IRF3 binding to the TNFR–associated factor (TRAF) associated proteins usually activating a NF–κB kinase (TANK/TBK1) and the IκB kinase epsilon (IKKε) 103,104. It is plausible that some of the kinetic studies outlined above that many of the delayed onset of immune cell upregulation of CD molecules could be regulated by type I/II/III IFN unknown so far; because in conjunction with the tolerogenic profile of DCs that T and NK cell responses usually stimulated may be seemingly affected by EBOV subunit protein domains that are toxic with the appearance of agonist–like effects. However, the cytokine IL–8 (CXCL8) is known to differentially affect CD4+/– T cell activation of effector memory (TEM) CD4+/– T cells but not naive (TN) or central memory (TCM) CD4+/– T cells. In addition, IL–8 can upregulate IL–2 synthesis with no effect on type II IFN–γ and IL–4 production 105. These observations remain key as IL–8 effect on T cell apoptosis remains unclear.
Natural Killer Cells during Immune Responses to Ebola Virus
During 2017, authors indicated using digital cell quantification (DCQ) that NK cells showed an evidential increase after infection during recovery from EBOV disease. In a comparison (n=112) transcriptome analysis with non–human primates (NHP), clear significance was noted between non–fatal and fatal outcomes with potential pro–inflammatory chemokine mRNA gene transcripts (CXCL10, CCL2, CCL8, CXCL11) during severe EBOV infection of which two have affinity to a known receptor present on DCs that is CXCR3 106. Shortly after in 2019, laboratory research investigated the role of Mϕ and NK cells (sCD163) in comparison given the similarities that severe EBOV disease has led to Mϕ activation syndrome (MAS) and haemophagocytic lymphohistiocytosis (HLH). At this juncture, it was hypothesised that three cytokines (IL–6, IL–8, M–CSF), three chemokines (CCL2, CCL3 and CCL4), alongside two modulatory cytokines IL–10/IL–1RA remained pertinent to immunological cell function and migration during EBOV infection 72. The T cell marker expressed by NK cells denoted as an immune cell shed soluble receptor (sIL–2R) is present on TREG (CD25+) cells and was also found at increased levels during infection 72. As above, in vivo, TLR4 was considered to be an activation marker instigated by both pathogenic/non–pathogenic viruses. Authors noted that overall NK cell frequencies may be considered slightly reduced reflecting unknown NK cell activation and proliferation then. Furthermore, a decrease in CD56dim/bright NK cells, alongside increased frequencies of CD56negNK cells hold possibilities and may be attributed as positive regulators of immunological relevance unknown to date 72.
More recently during 2020, the role that NK cells perform indicated that CD14+ monocytes may be considered the source of pro–inflammatory cytokines. In vitro stimulation/inhibition studies using EBOV GP indicated that NK cell activation was dependent on IL–18 and IL–12 and could be enhanced by an antagonist that blocked the IL–10 receptor 107. Two NK cell activation markers, CD107a and CD25, were observed to be upregulated in response to EBOV GP but without secretion of type II IFN–γ usually synthesised by both NK and T cells 108. Simultaneously the EBOV VP40 protein subunit domain was found to stimulate IL–12/IL–18 production 109. However, it was determined that the degranulation and cytolytic actions of NK cells was dependent on IL–12 to activate CD56+ NK cells. These observations noted that NK cell function and maturation were crucial 109. Interleukin–18 is considered to act in concert with IL–12 as a regulator of TH1 differentiation along with IL–23 as well as regulating TH17 cell secretion of IL–17. Dual regulatory effects can occur with the activation of TC cells and NK cells 110. Activation markers like CD95 are used to examine NK cell maturation and observed to be upregulated without change during EBOV infection; but concurrently more recently subtypes of immune cells have been characterised that include CCR7neg effector Vδ2 cells that can be an antigen–stimulated source of TNF–α 111. The other activation marker, CD69, was expressed on each of the observed NK subtypes. As above type II IFN can inhibit EBOV infection and be produced by the above four cell types. Some of the unknown genetic mechanisms of type II IFN synthesis became slightly clearer with predominant ISG gene transcripts researched and observed that are relevant. Initially, guanylate binding protein 5 (GBP5) upregulation was observed that is a factor that can inhibit type II IFN in other viral infections like Respiratory Syncytial virus (RSV) 112; but also others were observed that may affect other immune cell types regulating retinoic acid receptor responder protein 3 (RARRES3) and vesicle–associated membrane protein (VAMP5) as yet unknown 70,113.
Very recently, developments occurring showed the neutrophil chemoattractant IL–8 (CXCL–8) together with serum IL–1RA, TNF–α, CCL2 and TGF–α as essential factors in beneficial immune responses 54. Metabolic enzymes were depicted as significant including high–density lipoproteins (HDL) that were apolipoproteins (E/L1/A4/C3/C4). Notably CSF1, GM–CSF, CCL3, CX3CL1, and a binding protein to IL–18 were associated with high EBOV viral loads but VP40 was not associated in severity with GP associated with PCR cycle threshold (CT) values over 50 54. These reports in combination indicate that M–CSF is a key cytokine required during Mϕ development; but also IL–18 can independently mediate type II IFN–γ secretion from helper T cells (TH1) (through IL–18 binding protein (BP)) and is usually produced by Mϕ that NK cells require for cytolytic function. The role of IL–18 BP in regulating IFN–γ during EBOV infection is worthy of exploring as it is produced by mononuclear cells but also IL–18 does signal between IL–1 receptor–associated kinases (IRAK1/4) and could plausibly affect T cell and NK cell responses 114,115. The synergism of IL–18 activity with other cytokines does stimulate NK cell proliferation and cytotoxicity via the production of perforins and granzyme B remaining unclear to date 116. The duality of NK cell function remains unclear as a multitude of activation and inhibitory receptors exist regulating NK cell–dependent cytotoxicity; however, activation/inhibition signalling was further researched in 2022. Researchers Jarahian et al. (Heidelberg, Germany) performed in vitro research to observe the EBOV GP may stimulate NK cell cytotoxic activation receptors (NKp44/NKp46) as well as selectin adhesion, receptors (CD62L/CD62P) and inhibitory sialylated Siglec–7 (CD328) receptors as well as Siglec–5 (CD170) receptor proteins 28. These were significant findings because CD62L is constitutively expressed and required by leukocytes for cellular migration across EC layers, with CD62P similarly on platelets; while short–chain CD328 (also CD170) contains immune–receptor tyrosine–based inhibitory motif (ITIM) cellular cytoplasmic domains and external domain receptors which bind to glycans containing sialic acid.
Recent developments during in vivo transcriptional research demonstrate that during EBOV infection (n=9), four genes are markedly upregulated at all stages of infection (IRF7, S100A8, S100A9, IFI44). These correspond to calprotectin and IRF proteins potentially encoded 117. A further seven mRNA transcripts were noted at early stages of infection (CD19, FCGR3, PRDM1, IRF1, CXCR2, IL13RA1, CD79A) which do encode proteins that all may regulate B cells, antibody receptors, cytokines, receptors and chemokine receptor synthesis 117. Importantly, one key microRNA (miRNA) regulating post–transcriptional gene expression was highlighted and is under investigation in sepsis research. Future research on miR–122–5p will be very interesting to see. Recently, miR–122–5p was observed to regulate coagulation where mimics of miR–122–5p (n=84) were observed to downregulate IL–1β, IL–6, CCL2, and TNF–α in sepsis models 118. Therefore, these observations may have direct relevance to research in the future given that miRNA modulation of cytokine levels could potentially be applicable to different pathologies after extensive further research. More recently, nine EBOV GP epitopes were examined against HLA alleles in silico to predict that thirteen HLA–A and HLA–B alleles may represent optimal CD8+ T cell responses that could generate broad immunological responses with two predicted to be within regions of the GP1.1 and GP2.2 domains 119. Below is shown interpretation of current immunological response factors so far based on research to date (see Figure 5)
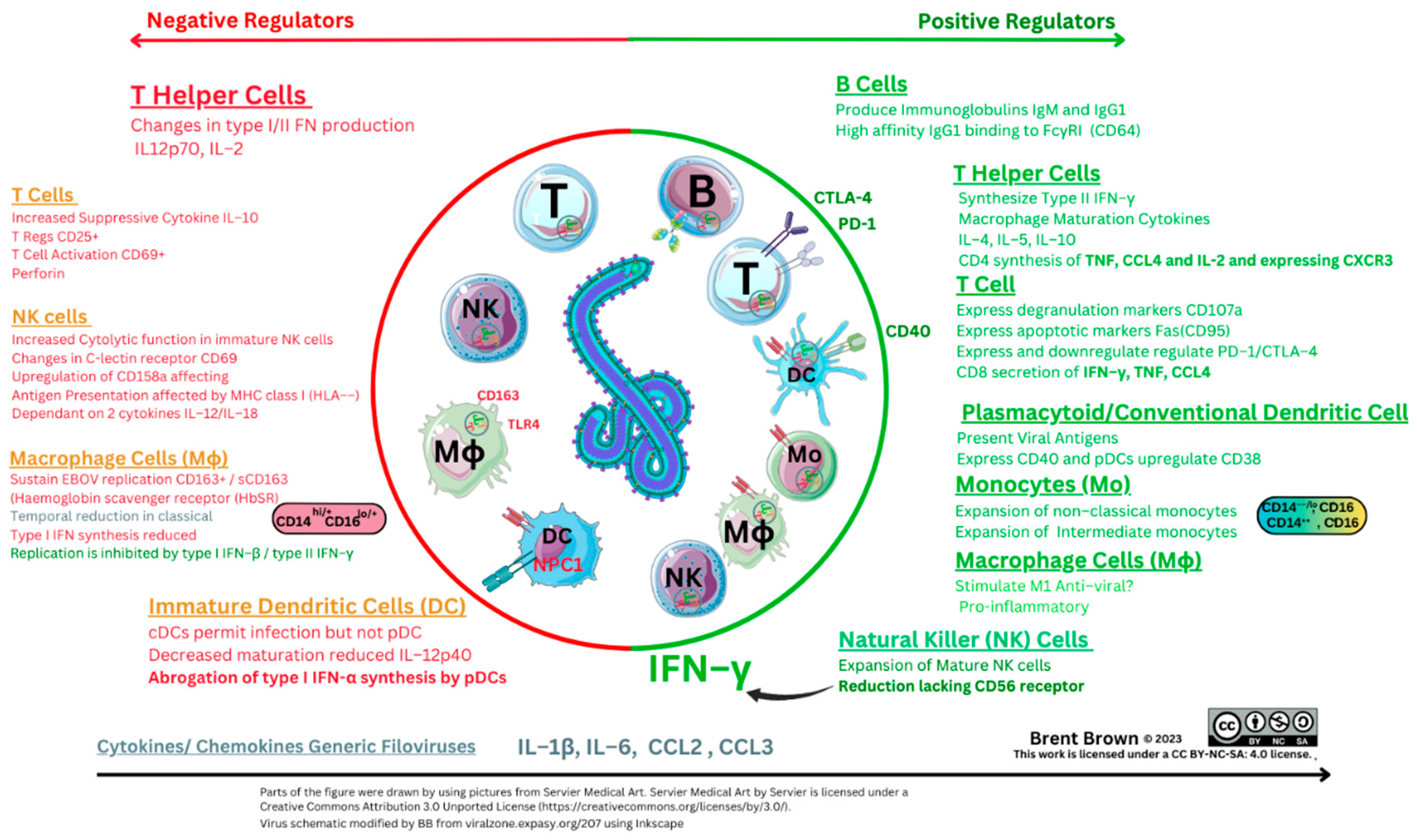
Figure 4.
Overall Immunological Factors during Ebola Virus Infection.
Limitations
Vaccine and clinical trial studies are documented in other reviews 120. In 2014, limitations of polymerase chain reaction (PCR) testing were noted with a report outlining potential options of lateral flow immunoassays (LFI) as a potential accurate viral antigen diagnostic 6,7,121. Other articles discuss monoclonal antibodies that have been identified and historically are undergoing further research as both diagnostic and therapeutic antibodies are expensive and require validation for sensitivity and specificity which can vary between proteins in different host species 122,123. Furthermore, single nucleotide point mutations (SNP) can occur in any cellular host gene encoding any protein 124. However much remains unclear with research ongoing.
Discussion
During individual host immune response to pathogens, overall host innate and adaptive immune response may remain consistent as viral genomes will differ upon transcription and translation between species. As a prerequisite, if it is considered that both DNA and RNA viruses evolve and mutate, with more or less severity regardless, host immune proteins can vary with errors that occur during gene transcription and translation into proteins. Previously we discussed the CD4+ T cell upregulation during EBOV infection. This is important in determining the T–cell response and contribution to disease phenotype. Therapeutic strategies used may vary and the differential role of TIM–1 in T–cell signalling is still being clarified 125. Recent suggestions are that EBOV is dependent on phosphosphatidylserine with caspase–dependent scramblases (XK–related protein (Xkr)) utilised further elucidating EBOV intracellular pathways 126. The role of STAT protein mutations and chemokines are still being discovered; but also type II IFN–γ was discovered some 50 years ago with errors and remains unknown within populations globally. Historically, errors in type II IFN–γ signalling are rarely documented in research or literature in mycobacterial infection and remains a key determinant of adaptive T cell function in pathogenic host immune cell responses 127,128. Interferon research began in 1965 with the discovery of type I IFNs with the key role of type II IFN–γ in T cell antiviral responses shortly after. More recently, the 2003 discovery of type III IFN–λ and four subtypes remains largely unknown as to cellular signalling and transduction mechanisms. It can be seen in the diagrams above that there is an outline as to overall EBOV host immune responses. However, chemokines were also only characterised between 2000–2014, to highlight atypical chemokine receptors (ACKRs) present within epithelial layers. In addition, TLR research during EBOV infection is comparatively recent 124. During any infection caused by viral, bacterial or mycobacterial pathogens, the overall risk is usually defined by R0 as the transmission rate. This is a measure denoting pathogen transmission referred to as the reproduction number or how many people could be affected during contagious infectious diseases. It is reported that in 2015 this was estimated for EBOV to be in the range of 1.37–2.53. Other reports indicate that the overall risk of EBOV infection estimate was around 45% estimated within community settings 129,130. The infection fatality rate (IFR) of EBOV between males/females is indicated with a slightly increased survival rate of EBOV–affected females 131. Given EBOV severity in disease it is prudent to consider that adaptive immune systems can vary by age with thymic T cell development mainly documented during and after cytomegalovirus (CMV) infection 132–134.
In the 21st century real–time polymerase chain reaction (PCR) has become regularly used for sequencing viral genomes. The Ebola virus has been investigated with regards to effects in different age–ranges. It was seen that a characterised marker, regulated upon activated T cell expressed and secreted molecule known as RANTES (CCL5) appears to correlate with survival outcome but also that two soluble adhesion molecules (sICAM, sVCAM) and plasminogen activator inhibitor–1 (PAI–1) released by endothelial cells were detrimental factors. These can vary in different age ranges and cellular mechanisms will require further clinical and laboratory research 135. Also of note given the complexities of EBOV infection and disease together with the immune system, there are immunological characteristics that appear consistent between studies. As recently as 2021, other authors agree that the complexities of IL–10 regulation of innate lymphoid cells including NK cells will require clarity 136. Furthermore, the role that pDCs play in immune systems similarly requires clarity as to mechanisms of IFN regulation in a cell population where cell viability and culture provides difficulty.
Further research on maturation between DCs, monocytes and macrophages will be required but also the mechanisms that Ebola virus proteins like VP24 may use to regulate CD38 and type I/III IFN synthesis are as yet unknown52. Questions remain given other recent research with similarities as outlined above with macrophage activation syndrome, but also sepsis appearing similar within systemic conditions of such severity. These observations will undoubtedly require further research in the future. Uniquely, a group in Seattle, USA, (Bruchez et al.) recently characterised a transcription factor (the major histocompatibility complex class II transactivator) that seemingly has 2–3 times proprietary antiviral activity against two viruses (both Ebola as well as SARS–CoV–2) and is induced by type II interferon whilst inducing resistance through a CD74/p41 isoform antagonising a known host cathepsin L protease required to allow endocytosis of viral proteins 137. However given the above, further research is required on the other immunologically relevant antibodies that include subtypes of IgG (1–4), and IgA (1–2) alongside relevant cellular functions of natural killer cells, mast cells, basophils and eosinophils. Therapeutic strategies that are under research investigation for Filoviridae (EBOV/MARV) include antivirals, polyclonal/monoclonal antibodies as well as proprietary small–interfering RNA (siRNA) molecules. It is currently indicated that there are at least 115 clinical trials at various stages globally on the National Institute of Health (NIH) website either completed or in progress. Of these three vaccines are at various stages of development and are viral–vector modified with two monoclonal antibody therapeutics recently recommended by the WHO (see Supplementary Materials) 120,138–140. We concur with other authors, as in 2023, in vitro research with recombinant shed GP further indicated the complexities and unknown EBOV immunological mechanisms. It was clarified that induction of B cell, T cell and NK cell apoptosis could occur either dependent or independent of caspase proteins with further research needed to ascertain these mechanisms further 141.
Conclusions
In conclusion, the immunological systemic innate and adaptive responses remain under–researched in many viral and pathological fields, and this is particularly true for Ebola virus. The factors we have outlined are immunological characteristics of cellular basis. Positive indicators of immunological relevance are the upregulation of T helper (CD4+) cells alongside the cytotoxic T (CD8+) cell response as illustrated in Figure 4. The T cell phenotype will remain crucial to elucidate the dynamics of Ebola virus immune responses and other pathologies as well in future. The role of pattern recognition receptors including Toll–like receptors and other immune cell types like mucosal associated invariant T cells will also require further investigation as will other receptors relevant to T cell signalling responses. There are currently 371 CD molecules classified that immune cells could express with variable cellular function. Additionally, there are 10 Toll–like Receptors classified within the CD nomenclature that remain unclear as to relevance within how or why the Ebola virus evokes such differential immunological characteristics. Although technological advancements and transcriptome studies allow further clarity and insights into mechanisms underlying scientific progress. Human leukocyte alleles may also vary between global populations encoding major histocompatibility complexes (type I/II) required to present viral peptide antigens to systemic immune cells.
Characterisation of the Ebola virus genome is ongoing, and combined with technological advancements and transcriptome studies will further advance clarity and insights into mechanisms. A holistic overview is of utility, however, for which we have provided known research to date. The authors therefore hope that the comprehensive information presented will be useful for clinicians, academics and researchers alike in the future.
Author Contributions
Original Manuscript Conceptualization–B.B.; methodology: –B.B.; formal analysis–B.B., I.F., C.I., A.G., P.M ; data curation–B.B, I.F, C.I.; writing—original draft preparation, B.B.; writing, review and editing B.B., C.I., I.F., A.G., P.M; software: B.B. and I.F.; visualization: B.B and I.F.; supervision by B.B.; All authors have signed, read and agreed to the published version of the manuscript according to IJCME guidelines.
Funding
This research received no external funding.
Conflicts of Interest
The authors have no conflict of interest to declare.
Ethical approval
Not required. Individual citations are subject to the Helsinki Declaration and individual regulatory authorities.
Consent to participate
Not applicable.
Consent to publication
Written signed consent has been obtained from the author(s) to publish this paper. Research contained within, refer to original citation(s) for further details. .
Availability of data and materials
Above.
Use of Artificial Intelligence (AI) Disclaimer
During the preparation of this work the author(s) did not use AI tools or service. Citations are referenced using Mendeleey. After using the above tool/service, each author(s) reviewed and edited the content.
List of Abbreviations
Lymphocyte function–associated antigen 1 (LFA–1), Intercellular Adhesion Molecule (ICAM), Vascular Cell Adhesion Molecule (VCAM). For other protein abbreviations not listed see www.reactome.org
References
- Barbiero, V.K. Ebola: A Hyperinflated Emergency. Glob. Heal. Sci. Pr. 2020, 8, 178–182. [Google Scholar] [CrossRef]
- Althaus, C.L. Estimating the Reproduction Number of Ebola Virus (EBOV) During the 2014 Outbreak in West Africa. PLoS Curr. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- James SL, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018, 392, 1789–1858. [CrossRef]
- Kallio-Kokko, H.; Uzcategui, N.; Vapalahti, O.; Vaheri, A. Viral zoonoses in Europe. FEMS Microbiol. Rev. 2005, 29, 1051–1077. [Google Scholar] [CrossRef] [PubMed]
- Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Com-mittee on Taxonomy of Viruses (2017). Arch Virol. 2017, 162, 2505–2538. [CrossRef]
- Bettini, A.; Lapa, D.; Garbuglia, A.R. Diagnostics of Ebola virus. Front. Public Heal. 2023, 11, 1123024. [Google Scholar] [CrossRef]
- Land, K.J.; Boeras, D.I.; Chen, X.-S.; Ramsay, A.R.; Peeling, R.W. REASSURED diagnostics to inform disease control strategies, strengthen health systems and improve patient outcomes. Nat. Microbiol. 2018, 4, 46–54. [Google Scholar] [CrossRef]
- Jain, S.; Martynova, E.; Rizvanov, A.; Khaiboullina, S.; Baranwal, M. Structural and Functional Aspects of Ebola Virus Proteins. Pathogens 2021, 10, 1330. [Google Scholar] [CrossRef]
- Vetter, P.; Fischer, W.A.; Schibler, M.; Jacobs, M.; Bausch, D.G.; Kaiser, L. Ebola Virus Shedding and Transmission: Review of Current Evidence. J. Infect. Dis. 2016, 214, S177–S184. [Google Scholar] [CrossRef]
- Sanchez, A.; Yang, Z.-Y.; Xu, L.; Nabel, G.J.; Crews, T.; Peters, C.J. Biochemical Analysis of the Secreted and Virion Glycoproteins of Ebola Virus. J. Virol. 1998, 72, 6442–6447. [Google Scholar] [CrossRef]
- Yuan, B.; Peng, Q.; Cheng, J.; Wang, M.; Zhong, J.; Qi, J.; Gao, G.F.; Shi, Y. Structure of the Ebola virus polymerase complex. Nature 2022, 610, 394–401. [Google Scholar] [CrossRef]
- Mehedi M, Falzarano D, Seebach J, Hu X, Carpenter MS, Schnittler HJ, et al A New Ebola Virus Non-structural Glycoprotein Expressed through RNA Editing. J Virol. 2011, 85, 5406–5414. [CrossRef] [PubMed]
- Lim, J.P.; A Gleeson, P. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol. Cell Biol. 2011, 89, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus Is Internalized into Host Cells via Macropinocytosis in a Viral Glycoprotein-Dependent Manner. PLOS Pathog. 2010, 6, e1001121. [Google Scholar] [CrossRef]
- Di Palma, F.; Daino, G.L.; Ramaswamy, V.K.; Corona, A.; Frau, A.; Fanunza, E.; Vargiu, A.V.; Tramontano, E.; Ruggerone, P. Relevance of Ebola virus VP35 homo-dimerization on the type I interferon cascade inhibition. Antivir. Chem. Chemother. 2019, 27. [Google Scholar] [CrossRef] [PubMed]
- Yu DS, Weng TH, Wu XX, Wang FXC, Lu XY, Wu HB, et al The lifecycle of the Ebola virus in host cells. Oncotarget. 2017, 8, 55750–55759.
- Di Paola, N.; Sanchez-Lockhart, M.; Zeng, X.; Kuhn, J.H.; Palacios, G. Viral genomics in Ebola virus research. Nat. Rev. Microbiol. 2020, 18, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Vidal S, Sánchez-Aparicio M, Seoane R, El Motiam A, Nelson E V., Bouzaher YH, et al. Expression of the Ebola Virus VP24 Protein Compromises the Integrity of the Nuclear Envelope and Induces a Laminopa-thy-Like Cellular Phenotype. mBio. 2021 Aug 31;12(4).
- Simon, T.; Bromberg, J.S. Regulation of the Immune System by Laminins. Trends Immunol. 2017, 38, 858–871. [Google Scholar] [CrossRef]
- Dubik, N.; Mai, S. Lamin A/C: Function in Normal and Tumor Cells. Cancers 2020, 12, 3688. [Google Scholar] [CrossRef] [PubMed]
- Fels, J.M.; Bortz, R.H.; Alkutkar, T.; Mittler, E.; Jangra, R.K.; Spence, J.S.; Chandran, K. A Glycoprotein Mutation That Emerged during the 2013–2016 Ebola Virus Epidemic Alters Proteolysis and Accelerates Membrane Fusion. Mbio 2021, 12. [Google Scholar] [CrossRef]
- Li X, Wang J, Coutavas E, Shi H, Hao Q, Blobel G Structure of human Niemann–Pick C1 protein. Pro-ceedings of the National Academy of Sciences. 2016, 113, 8212–8217. [CrossRef] [PubMed]
- Martin B, Hoenen T, Canard B, Decroly E. Filovirus proteins for antiviral drug discovery: A struc-ture/function analysis of surface glycoproteins and virus entry. Antiviral Res. 2016, 135, 1–14. [CrossRef] [PubMed]
- Winter SL, Golani G, Lolicato F, Vallbracht M, Thiyagarajah K, Ahmed SS, et al. The Ebola virus VP40 matrix layer undergoes endosomal disassembly essential for membrane fusion. EMBO J. 2023, 42. [Google Scholar]
- Soilleux EJ, Barten R, Trowsdale J.Cutting Edge: DC-SIGN; a Related Gene, DC-SIGNR; and CD23 Form a Cluster on 19p13 . The Journal of Immunology. 2000, 165, 2937–2942. [CrossRef]
- Tailleux L, Schwartz O, Herrmann JL, Pivert E, Jackson M, Amara A, et al DC-SIGN Is the Major My-cobacterium tuberculosis Receptor on Human Dendritic Cells. Journal of Experimental Medicine. 2003, 197, 121–127. [CrossRef] [PubMed]
- Bashirova AA, Geijtenbeek TBH, van Duijnhoven GCF, van Vliet SJ, Eilering JBG, Martin MP, et al. A Dendritic Cell–Specific Intercellular Adhesion Molecule 3–Grabbing Nonintegrin (Dc-Sign)–Related Pro-tein Is Highly Expressed on Human Liver Sinusoidal Endothelial Cells and Promotes HIV-1 Infection. Journal of Experimental Medicine. 2001, 193, 671–678.
- Jarahian M, Marstaller K, Banna N, Ahani R, Etemadzadeh MH, Boller LK, et al. Activating Natural Killer Cell Receptors, Selectins, and Inhibitory Siglecs Recognize Ebolavirus Glycoprotein. J Innate Immun. 2022, 14, 135–147. [Google Scholar] [CrossRef]
- Mohamadzadeh M, Coberley SS, Olinger GG, Kalina W V, Ruthel G, Fuller CL, et al. Activation of Trig-gering Receptor Expressed on Myeloid Cells-1 on Human Neutrophils by Marburg and Ebola Viruses. J Virol. 2006, 80, 7235–7244. [CrossRef]
- Herrero-Cervera, A.; Soehnlein, O.; Kenne, E. Neutrophils in chronic inflammatory diseases. Cell. Mol. Immunol. 2022, 19, 177–191. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Brown B, Ojha V, Fricke I, Al-Sheboul SA, Imarogbe C, Gravier T, et al. Innate and Adaptive Immunity during SARS-CoV-2 Infection: Biomolecular Cellular Markers and Mechanisms. Vaccines (Basel). 2023, 11, 408.
- Lanier, LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev. 2009, 227, 150–160. [Google Scholar] [CrossRef]
- Turnbull, I.R.; Colonna, M. Activating and inhibitory functions of DAP12. Nat. Rev. Immunol. 2007, 7, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kadlecek, T.A.; Au-Yeung, B.B.; Goodfellow, H.E.S.; Hsu, L.-Y.; Freedman, T.S.; Weiss, A. ZAP-70: An Essential Kinase in T-cell Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002279–a002279. [Google Scholar] [CrossRef] [PubMed]
- Mócsai A, Ruland J, Tybulewicz VLJ. The SYK tyrosine kinase: a crucial player in diverse biological func-tions. Nat Rev Immunol. 2010, 10, 387–402. [CrossRef]
- Zhong L, Zhang ZL, Li X, Liao C, Mou P, Wang T, et al. TREM2/DAP12 Complex Regulates Inflammatory Responses in Microglia via the JNK Signaling Pathway. Front Aging Neurosci. 2017 Jun 21;9.
- Kondratowicz AS, Lennemann NJ, Sinn PL, Davey RA, Hunt CL, Moller-Tank S, et al. T-cell immuno-globulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proceedings of the National Academy of Sciences. 2011, 108, 8426–8431. [CrossRef]
- Hsu, P.-L.; Jou, J.; Tsai, S.-J. TYRO3: A potential therapeutic target in cancer. Exp. Biol. Med. 2019, 244, 83–99. [Google Scholar] [CrossRef]
- Younan, P.; Iampietro, M.; Nishida, A.; Ramanathan, P.; Santos, R.I.; Dutta, M.; Lubaki, N.M.; Koup, R.A.; Katze, M.G.; Bukreyev, A. Ebola Virus Binding to Tim-1 on T Lymphocytes Induces a Cytokine Storm. Mbio 2017, 8, e00845–17. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, J.; Tillieux, S.; Guo, Z.; Natividade, R.d.S.; Koehler, M.; Petitjean, S.; Cui, Z.; Alsteens, D. Stepwise Enzymatic-Dependent Mechanism of Ebola Virus Binding to Cell Surface Receptors Monitored by AFM. Nano Lett. 2022, 22, 1641–1648. [Google Scholar] [CrossRef]
- Brunton, B.; Rogers, K.; Phillips, E.K.; Brouillette, R.B.; Bouls, R.; Butler, N.S.; Maury, W. TIM-1 serves as a receptor for Ebola virus in vivo, enhancing viremia and pathogenesis. PLOS Neglected Trop. Dis. 2019, 13, e0006983. [Google Scholar] [CrossRef]
- Johnson, K.A.; Taghon, G.J.F.; Scott, J.L.; Stahelin, R.V. The Ebola Virus matrix protein, VP40, requires phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) for extensive oligomerization at the plasma membrane and viral egress. Sci. Rep. 2016, 6, srep19125. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.D. Role of lipid composition on human plasma membrane interactions with the Ebola virus matrix protein VP40. Biophys. J. 2023, 122, 506a–507a. [Google Scholar] [CrossRef]
- Katan, M.; Cockcroft, S. Phosphatidylinositol(4,5)bisphosphate: diverse functions at the plasma membrane. Essays Biochem. 2020, 64, 513–531. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A.S.; Davidson, C.; Kuehne, A.I.; Bakken, R.; Braigen, S.Z.; Gunn, K.E.; Whelan, S.P.; Brummelkamp, T.R.; Twenhafel, N.A.; Chandran, K.; et al. Niemann-Pick C1 Is Essential for Ebolavirus Replication and Pathogenesis In Vivo. Mbio 2015, 6, e00565–15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shmidov, Y.; Harris, E.A.; Theus, M.H.; Bitton, R.; Matson, J.B. Activating hidden signals by mimicking cryptic sites in a synthetic extracellular matrix. Nat. Commun. 2023, 14, 3635. [Google Scholar] [CrossRef]
- Fanunza E, Frau A, Corona A, Tramontano E. Insights into Ebola Virus VP35 and VP24 Interferon In-hibitory Functions and their Initial Exploitation as Drug Targets. Infect Disord Drug Targets. 2019, 19, 362–374. [CrossRef]
- Coxon, P.; Rane, M.; Uriarte, S.; Powell, D.; Singh, S.; Butt, W.; Chen, Q.; McLeish, K. MAPK-activated protein kinase-2 participates in p38 MAPK-dependent and ERK-dependent functions in human neutrophils. Cell. Signal. 2003, 15, 993–1001. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, J.; Liu, C.; Guo, X.; Zhu, X.; Yao, Y.; Jiao, Y.; He, P.; Han, J.; Wu, L. p38α has an important role in antigen cross-presentation by dendritic cells. Cell. Mol. Immunol. 2016, 15, 246–259. [Google Scholar] [CrossRef]
- Carroll SA, Towner JS, Sealy TK, McMullan LK, Khristova ML, Burt FJ, et al. Molecular Evolution of Vi-ruses of the Family Filoviridae Based on 97 Whole-Genome Sequences. J Virol. 2013, 87, 2608–2616. [CrossRef]
- Ilinykh, P.A.; Lubaki, N.M.; Widen, S.G.; Renn, L.A.; Theisen, T.C.; Rabin, R.L.; Wood, T.G.; Bukreyev, A. Different Temporal Effects of Ebola Virus VP35 and VP24 Proteins on Global Gene Expression in Human Dendritic Cells. J. Virol. 2015, 89, 7567–7583. [Google Scholar] [CrossRef]
- LaVergne, S.M.; Sakabe, S.; Kanneh, L.; Momoh, M.; Al-Hassan, F.; Yilah, M.; Goba, A.; Sandi, J.D.; Gbakie, M.; Cubitt, B.; et al. Ebola-Specific CD8+ and CD4+ T-Cell Responses in Sierra Leonean Ebola Virus Survivors With or Without Post-Ebola Sequelae. J. Infect. Dis. 2020, 222, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Viodé A, Smolen KK, Fatou B, Wurie Z, Van Zalm P, Konde MK, et al. Plasma Proteomic Analysis Dis-tinguishes Severity Outcomes of Human Ebola Virus Disease. mBio. 2022 Jun 28;13(3).
- Engel P, Boumsell L, Balderas R, Bensussan A, Gattei V, Horejsi V, et al. CD Nomenclature 2015: Human Leukocyte Differentiation Antigen Workshops as a Driving Force in Immunology. The Journal of Immu-nology. 2015, 195, 4555–4563. [CrossRef] [PubMed]
- Xuan, W.; Qu, Q.; Zheng, B.; Xiong, S.; Fan, G.-H. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J. Leukoc. Biol. 2014, 97, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol [Internet]. 2015, 7, a016303.
- Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the Eye of the Cytokine Storm. Microbiology and Molecular Biology Reviews. 2012, 76, 16–32.
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef]
- Bosio, C.M.; Aman, M.J.; Grogan, C.; Hogan, R.; Ruthel, G.; Negley, D.; Mohamadzadeh, M.; Bavari, S.; Schmaljohn, A. Ebola and Marburg Viruses Replicate in Monocyte-Derived Dendritic Cells without Inducing the Production of Cytokines and Full Maturation. J. Infect. Dis. 2003, 188, 1630–1638. [Google Scholar] [CrossRef]
- Wittling MC, Cahalan SR, Levenson EA, Rabin RL. Shared and Unique Features of Human Interferon-Beta and Interferon-Alpha Subtypes. Front Immunol. 2021 Jan 19;11.
- Hammou, R.A.; Kasmi, Y.; Khataby, K.; Laasri, F.E.; Boughribil, S.; Ennaji, M.M. Roles of VP35, VP40 and VP24 Proteins of Ebola Virus in Pathogenic and Replication Mechanisms. Ebola 2016. [Google Scholar] [CrossRef]
- Wiedemann, A.; Foucat, E.; Hocini, H.; Lefebvre, C.; Hejblum, B.P.; Durand, M.; Krüger, M.; Keita, A.K.; Ayouba, A.; Mély, S.; et al. Long-lasting severe immune dysfunction in Ebola virus disease survivors. Nat. Commun. 2020, 11, 3730. [Google Scholar] [CrossRef]
- Gessani S, Conti L, Del Cornò M, Belardelli F. Type I Interferons as Regulators of Human Antigen Pre-senting Cell Functions. Toxins (Basel). 2014, 6, 1696–1723. [CrossRef]
- Martinez, O.; Johnson, J.C.; Honko, A.; Yen, B.; Shabman, R.S.; Hensley, L.E.; Olinger, G.G.; Basler, C.F. Ebola Virus Exploits a Monocyte Differentiation Program To Promote Its Entry. J. Virol. 2013, 87, 3801–3814. [Google Scholar] [CrossRef]
- Yaddanapudi, K.; Palacios, G.; Towner, J.S.; Chen, I.; Sariol, C.A.; Nichol, S.T.; Lipkin, W.I. Implication of a retrovirus-like glycoprotein peptide in the immunopathogenesis of Ebola and Marburg viruses. FASEB J. 2006, 20, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, K.A.-M.; Schulze, L.L.; Paap, E.-M.; Müller, T.M.; Neurath, M.F.; Zundler, S. Immunology of IL-12: An update on functional activities and implications for disease. EXCLI J. 2020, 19, 1563–1589. [Google Scholar] [PubMed]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Falasca, L.; Agrati, C.; Petrosillo, N.; Di Caro, A.; Capobianchi, M.R.; Ippolito, G.; Piacentini, M. Molecular mechanisms of Ebola virus pathogenesis: focus on cell death. Cell Death Differ. 2015, 22, 1250–1259. [Google Scholar] [CrossRef]
- Rhein, B.A.; Powers, L.S.; Rogers, K.; Anantpadma, M.; Singh, B.K.; Sakurai, Y.; Bair, T.; Miller-Hunt, C.; Sinn, P.; Davey, R.A.; et al. Interferon-γ Inhibits Ebola Virus Infection. PLOS Pathog. 2015, 11, e1005263. [Google Scholar] [CrossRef]
- Van Raemdonck, K.; Van den Steen, P.E.; Liekens, S.; Van Damme, J.; Struyf, S. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev. 2015, 26, 311–327. [Google Scholar] [CrossRef]
- McElroy AK, Shrivastava-Ranjan P, Harmon JR, Martines RB, Silva-Flannery L, Flietstra TD, et al. Mac-rophage Activation Marker Soluble CD163 Associated with Fatal and Severe Ebola Virus Disease in Humans1. Emerg Infect Dis. 2019, 25, 290–298. [CrossRef]
- Sharygin D, Koniaris LG, Wells C, Zimmers TA, Hamidi T. Role of CD14 in human disease. Immunology. 2023.
- Mass, E.; Nimmerjahn, F.; Kierdorf, K.; Schlitzer, A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat. Rev. Immunol. 2023, 23, 563–579. [Google Scholar] [CrossRef]
- Weaver, L.K.; A Hintz-Goldstein, K.; A Pioli, P.; Wardwell, K.; Qureshi, N.; Vogel, S.N.; Guyre, P.M. Pivotal Advance: Activation of cell surface Toll-like receptors causes shedding of the hemoglobin scavenger receptor CD163. J. Leukoc. Biol. 2006, 80, 26–35. [Google Scholar] [CrossRef]
- Lai, C.-Y.; Strange, D.P.; Wong, T.A.S.; Lehrer, A.T.; Verma, S. Ebola Virus Glycoprotein Induces an Innate Immune Response In vivo via TLR4. Front. Microbiol. 2017, 8, 1571–1571. [Google Scholar] [CrossRef] [PubMed]
- Hölzer M, Krähling V, Amman F, Barth E, Bernhart SH, Carmelo VAO, et al. Differential transcriptional responses to Ebola and Marburg virus infection in bat and human cells. Sci Rep. 2016 Oct 7;6(1):34589. [CrossRef]
- Purvis, G.S.D.; Solito, E.; Thiemermann, C. Annexin-A1: Therapeutic Potential in Microvascular Disease. Front. Immunol. 2019, 10, 938. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Trefry, J.C.; Babka, A.M.; Schellhase, C.W.; Coffin, K.M.; Williams, J.A.; Raymond, J.L.W.; Facemire, P.R.; Chance, T.B.; Davis, N.M.; et al. Ebola virus persistence and disease recrudescence in the brains of antibody-treated nonhuman primate survivors. Sci. Transl. Med. 2022, 14, eabi5229. [Google Scholar] [CrossRef] [PubMed]
- Rustenhoven, J.; Drieu, A.; Mamuladze, T.; de Lima, K.A.; Dykstra, T.; Wall, M.; Papadopoulos, Z.; Kanamori, M.; Salvador, A.F.; Baker, W.; et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell 2021, 184, 1000–1016. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Saphire, E.O. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009, 4, 621–635. [Google Scholar] [CrossRef]
- Lee, J.E.; Kuehne, A.; Abelson, D.M.; Fusco, M.L.; Hart, M.K.; Saphire, E.O. Complex of a Protective Antibody with Its Ebola Virus GP Peptide Epitope: Unusual Features of a Vλx Light Chain. J. Mol. Biol. 2008, 375, 202–216. [Google Scholar] [CrossRef] [PubMed]
- de La Vega MA, Wong G, Kobinger GP, Qiu X. The Multiple Roles of sGP in Ebola Pathogenesis. Viral Immunol. 2015, 28, 3–9. [CrossRef]
- Pokhrel, R.; Pavadai, E.; Gerstman, B.S.; Chapagain, P.P. Membrane pore formation and ion selectivity of the Ebola virus delta peptide. Phys. Chem. Chem. Phys. 2019, 21, 5578–5585. [Google Scholar] [CrossRef]
- He, J.; Melnik, L.I.; Komin, A.; Wiedman, G.; Fuselier, T.; Morris, C.F.; Starr, C.G.; Searson, P.C.; Gallaher, W.R.; Hristova, K.; et al. Ebola Virus Delta Peptide Is a Viroporin. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Melnik, L.I.; Guha, S.; Ghimire, J.; Smither, A.R.; Beddingfield, B.J.; Hoffmann, A.R.; Sun, L.; Ungerleider, N.A.; Baddoo, M.C.; Flemington, E.K.; et al. Ebola virus delta peptide is an enterotoxin. Cell Rep. 2022, 38, 110172. [Google Scholar] [CrossRef]
- Radoshitzky SR, Warfield KL, Chi X, Dong L, Kota K, Bradfute SB, et al. Ebolavirus Δ-Peptide Immuno-adhesins Inhibit Marburgvirus and Ebolavirus Cell Entry. J Virol. 2011, 85, 8502–8513. [CrossRef] [PubMed]
- Thom, R.; Tipton, T.; Strecker, T.; Hall, Y.; Bore, J.A.; Maes, P.; Koundouno, F.R.; Fehling, S.K.; Krähling, V.; Steeds, K.; et al. Longitudinal antibody and T cell responses in Ebola virus disease survivors and contacts: an observational cohort study. Lancet Infect. Dis. 2021, 21, 507–516. [Google Scholar] [CrossRef]
- Radinsky O, Edri A, Brusilovsky M, Fedida-Metula S, Sobarzo A, Gershoni-Yahalom O, et al. Sudan ebo-lavirus long recovered survivors produce GP-specific Abs that are of the IgG1 subclass and preferentially bind FcγRI. Sci Rep. 2017, 7, 6054. [CrossRef] [PubMed]
- Koch, T.; Rottstegge, M.; Ruibal, P.; Gomez-Medina, S.; Nelson, E.V.; Escudero-Pérez, B.; Pillny, M.; Ly, M.L.; Koundouno, F.R.; Bore, J.A.; et al. Ebola Virus Disease Survivors Show More Efficient Antibody Immunity than Vaccinees Despite Similar Levels of Circulating Immunoglobulins. Viruses 2020, 12, 915. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.W.; Jackson, K.J.; McElroy, A.K.; Halfmann, P.; Huang, J.; Chennareddy, C.; Piper, A.E.; Leung, Y.; Albariño, C.G.; Crozier, I.; et al. Longitudinal Analysis of the Human B Cell Response to Ebola Virus Infection. Cell 2019, 177, 1566–1582. [Google Scholar] [CrossRef] [PubMed]
- Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glyco-protein bound to an antibody from a human survivor. Nature. 2008, 454, 177–182. [CrossRef]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell–like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Tipton, T.R.W.; Hall, Y.; Bore, J.A.; White, A.; Sibley, L.S.; Sarfas, C.; Yuki, Y.; Martin, M.; Longet, S.; Mellors, J.; et al. Characterisation of the T-cell response to Ebola virus glycoprotein amongst survivors of the 2013–16 West Africa epidemic. Nat. Commun. 2021, 12, 1–9. [Google Scholar] [CrossRef]
- Lavender, K.J.; Williamson, B.N.; Saturday, G.; Martellaro, C.; Griffin, A.; Hasenkrug, K.J.; Feldmann, H.; Prescott, J. Pathogenicity of Ebola and Marburg Viruses Is Associated With Differential Activation of the Myeloid Compartment in Humanized Triple Knockout-Bone Marrow, Liver, and Thymus Mice. J. Infect. Dis. 2018, 218, S409–S417. [Google Scholar] [CrossRef]
- Kennedy, JR. Phosphatidylserine’s role in Ebola’s inflammatory cytokine storm and hemorrhagic con-sumptive coagulopathy and the therapeutic potential of annexin V. Med Hypotheses. 2020 Feb;135:109462.
- Bohan D, Van Ert H, Ruggio N, Rogers KJ, Badreddine M, Aguilar Briseño JA, et al. Phosphatidylserine receptors enhance SARS-CoV-2 infection. PLoS Pathog. 2021, 17, e1009743.
- Érsek, B.; Molnár, V.; Balogh, A.; Matkó, J.; Cope, A.P.; Buzás, E.I.; Falus, A.; Nagy, G. CD3ζ-Chain Expression of Human T Lymphocytes Is Regulated by TNF via Src-like Adaptor Protein-Dependent Proteasomal Degradation. J. Immunol. 2012, 189, 1602–1610. [Google Scholar] [CrossRef]
- Bozso, S.J.; Kang, J.J.H.; Nagendran, J. The role of competing mechanisms on Lck regulation. Immunol. Res. 2020, 68, 289–295. [Google Scholar] [CrossRef]
- Bhuin, T.; Roy, J.K. Rab proteins: The key regulators of intracellular vesicle transport. Exp. Cell Res. 2014, 328, 1–19. [Google Scholar] [CrossRef]
- Agrati C, Castilletti C, Casetti R, et al. Longitudinal characterization of dysfunctional T cell-activation during human acute Ebola infection. Cell Death Dis. 2016, 7, e2164. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.; Gravier, T.; Fricke, I.; Al-Sheboul, S.A.; Carp, T.-N.; Leow, C.Y.; Imarogbe, C.; Arabpour, J. Immunopathogenesis of Nipah Virus Infection and Associated Immune Responses. Immuno 2023, 3, 160–181. [Google Scholar] [CrossRef]
- AL Hamrashdi, M.; Brady, G. Regulation of IRF3 activation in human antiviral signaling pathways. Biochem. Pharmacol. 2022, 200, 115026. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qi, X.; Qu, B.; Zhang, Z.; Liang, M.; Li, C.; Cardona, C.J.; Li, D.; Xing, Z. Evasion of Antiviral Immunity through Sequestering of TBK1/IKK /IRF3 into Viral Inclusion Bodies. J. Virol. 2014, 88, 3067–3076. [Google Scholar] [CrossRef]
- Meniailo ME, Malashchenko VV, Shmarov VA, Gazatova ND, Melashchenko OB, Goncharov AG, et al. Direct effects of interleukin-8 on growth and functional activity of T lymphocytes. Int Immunopharmacol. 2017, 50, 178–185. [CrossRef] [PubMed]
- Liu, X.; Speranza, E.; Muñoz-Fontela, C.; Haldenby, S.; Rickett, N.Y.; Garcia-Dorival, I.; Fang, Y.; Hall, Y.; Zekeng, E.-G.; Lüdtke, A.; et al. Transcriptomic signatures differentiate survival from fatal outcomes in humans infected with Ebola virus. Genome Biol. 2017, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Wagstaffe, H.R.; Clutterbuck, E.A.; Bockstal, V.; Stoop, J.N.; Luhn, K.; Douoguih, M.; Shukarev, G.; Snape, M.D.; Pollard, A.J.; Riley, E.M.; et al. Ebola virus glycoprotein stimulates IL-18–dependent natural killer cell responses. J. Clin. Investig. 2020, 130, 3936–3946. [Google Scholar] [CrossRef] [PubMed]
- Wagstaffe HR, Anzala O, Kibuuka H, Anywaine Z, Sirima SB, Thiébaut R, et al. NK Cell Subset Redis-tribution and Antibody Dependent Activation after Ebola Vaccination in Africans. Vaccines (Basel). 2022, 10, 884. [CrossRef] [PubMed]
- Le, H.; Spearman, P.; Waggoner, S.N.; Singh, K. Ebola virus protein VP40 stimulates IL-12– and IL-18–dependent activation of human natural killer cells. J. Clin. Investig. 2022, 7. [Google Scholar] [CrossRef] [PubMed]
- Esmailbeig, M.; Ghaderi, A. Interleukin-18: a regulator of cancer and autoimmune diseases. Eur. Cytokine Netw. 2017, 28, 127–140. [Google Scholar] [CrossRef]
- Cimini, E.; Viola, D.; Cabeza-Cabrerizo, M.; Romanelli, A.; Tumino, N.; Sacchi, A.; Bordoni, V.; Casetti, R.; Turchi, F.; Martini, F.; et al. Different features of Vδ2 T and NK cells in fatal and non-fatal human Ebola infections. 2017. [CrossRef]
- Li, Z.; Qu, X.; Liu, X.; Huan, C.; Wang, H.; Zhao, Z.; Yang, X.; Hua, S.; Zhang, W. GBP5 Is an Interferon-Induced Inhibitor of Respiratory Syncytial Virus. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Anderson, A.M.; Kalimutho, M.; Harten, S.; Nanayakkara, D.M.; Khanna, K.K.; Ragan, M.A. The metastasis suppressor RARRES3 as an endogenous inhibitor of the immunoproteasome expression in breast cancer cells. Sci. Rep. 2017, 7, 39873. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Damsky, W.; Weizman, O.-E.; McGeary, M.K.; Hartmann, K.P.; Rosen, C.E.; Fischer, S.; Jackson, R.; Flavell, R.A.; Wang, J.; et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 2020, 583, 609–614. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 Binding Protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef]
- Choi YH, Lim EJ, Kim SW, Moon YW, Park KS, An HJ. IL-27 enhances IL-15/IL-18-mediated activation of human natural killer cells. J Immunother Cancer. 2019, 7, 168. [CrossRef]
- Stefan CP, Arnold CE, Shoemaker CJ, Zumbrun EE, Altamura LA, Douglas CE, et al. Transcriptomic Analysis Reveals Host miRNAs Correlated with Immune Gene Dysregulation during Fatal Disease Pro-gression in the Ebola Virus Cynomolgus Macaque Disease Model. Microorganisms. 2021, 9, 665. [CrossRef]
- Wang, H.; Zhang, C.; Zhang, C.; Wang, Y.; Zhai, K.; Tong, Z. MicroRNA-122-5p regulates coagulation and inflammation through MASP1 and HO-1 genes. Infect. Genet. Evol. 2022, 100, 105268. [Google Scholar] [CrossRef]
- Powlson, J.; Wright, D.; Zeltina, A.; Giza, M.; Nielsen, M.; Rampling, T.; Venkatrakaman, N.; Bowden, T.A.; Hill, A.V.; Ewer, K.J. Characterization of Antigenic MHC-Class-I-Restricted T Cell Epitopes in the Glycoprotein of Ebolavirus. Cell Rep. 2019, 29, 2537–2545. [Google Scholar] [CrossRef]
- Woolsey, C.; Geisbert, T.W. Current state of Ebola virus vaccines: A snapshot. PLOS Pathog. 2021, 17, e1010078. [Google Scholar] [CrossRef]
- Wonderly, B.; Jones, S.; Gatton, M.L.; Barber, J.; Killip, M.; Hudson, C.; Carter, L.; Brooks, T.; Simpson, A.J.H.; Semper, A.; et al. Comparative performance of four rapid Ebola antigen-detection lateral flow immunoassays during the 2014-2016 Ebola epidemic in West Africa. PLOS ONE 2019, 14, e0212113. [Google Scholar] [CrossRef]
- Tshiani Mbaya O, Mukumbayi P, Mulangu S. Review: Insights on Current FDA-Approved Monoclonal Antibodies Against Ebola Virus Infection. Front Immunol. 2021 Aug 30;12.
- Yamaoka, S.; Ebihara, H. Pathogenicity and Virulence of Ebolaviruses with Species- and Variant-specificity. Virulence 2021, 12, 885–901. [Google Scholar] [CrossRef]
- Su, S.; Tao, L.; Deng, Z.; Chen, W.; Qin, S.; Jiang, H. TLR10: Insights, controversies and potential utility as a therapeutic target. Scand. J. Immunol. 2020, 93, e12988. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, X.; Hu, L.; Zhang, Y.; Zheng, H.; Wu, H.; Wang, J.; Luo, L.; Xiao, H.; Qiao, C.; et al. TIM-1 Augments Cellular Entry of Ebola Virus Species and Mutants, Which Is Blocked by Recombinant TIM-1 Protein. Microbiol. Spectr. 2022, 10, e0221221. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Maruyama, J.; Imai, M.; Ujie, M.; Fujioka, Y.; Nishide, S.; Takada, A.; Ohba, Y.; Kawaoka, Y. Ebola virus requires a host scramblase for externalization of phosphatidylserine on the surface of viral particles. PLOS Pathog. 2018, 14, e1006848. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Kerner, G.; Rosain, J.; Guérin, A.; Al-Khabaz, A.; Oleaga-Quintas, C.; Rapaport, F.; Massaad, M.J.; Ding, J.-Y.; Khan, T.; Al Ali, F.; et al. Inherited human IFN-γ deficiency underlies mycobacterial disease. J. Clin. Investig. 2020, 130, 3158–3171. [Google Scholar] [CrossRef] [PubMed]
- Bower, H.; Johnson, S.; Bangura, M.S.; Kamara, A.J.; Kamara, O.; Mansaray, S.H.; Sesay, D.; Turay, C.; Checchi, F.; Glynn, J.R. Exposure-Specific and Age-Specific Attack Rates for Ebola Virus Disease in Ebola-Affected Households, Sierra Leone. Emerg. Infect. Dis. 2016, 22, 1403–1411. [Google Scholar] [CrossRef]
- Khan, A.; Naveed, M.; Dur-E-Ahmad, M.; Imran, M. Estimating the basic reproductive ratio for the Ebola outbreak in Liberia and Sierra Leone. Infect. Dis. Poverty 2015, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ebola Virus Disease among Male and Female Persons in West Africa. New England Journal of Medicine. 2016, 374, 96–98. [CrossRef] [PubMed]
- Hassouneh F, Goldeck D, Pera A, van Heemst D, Slagboom PE, Pawelec G, et al. Functional Changes of T-Cell Subsets with Age and CMV Infection. Int J Mol Sci. 2021, 22, 9973. [CrossRef]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Ebola Virus Disease among Children in West Africa. New England Journal of Medicine. 2015, 372, 1274–1277. [CrossRef]
- McElroy, A.K.; Erickson, B.R.; Flietstra, T.D.; Rollin, P.E.; Nichol, S.T.; Towner, J.S.; Spiropoulou, C.F. Biomarker Correlates of Survival in Pediatric Patients with Ebola Virus Disease. Emerg. Infect. Dis. 2014, 20, 1683–1690. [Google Scholar] [CrossRef]
- Sun, H.; Wu, Y.; Zhang, Y.; Ni, B. IL-10-Producing ILCs: Molecular Mechanisms and Disease Relevance. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Bruchez A, Sha K, Johnson J, Chen L, Stefani C, McConnell H, et al. Faculty Opinions recommendation of MHC class II transactivator CIITA induces cell resistance to Ebola virus and SARS-like coronaviruses. 2020, 370, 241–247. [CrossRef]
- Albakri, K.M.; Al-Hajali, M.M.; Saleh, O.M.; Alkhalil, A.M.M.; Mohd, A.B.M.; Samain, C.A.M.; Abuasad, N.N.M.; Hasan, H.M.; Khaity, A.M.; Farahat, R.A.M. Marburg virus disease treatments and vaccines: recent gaps and implications. Ann. Med. Surg. 2023, 85, 328–330. [Google Scholar] [CrossRef]
- Lee, A. Ansuvimab: First Approval. Drugs 2021, 81, 595–598. [Google Scholar] [CrossRef]
- Markham, A. REGN-EB3: First Approval. Drugs 2021, 81, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Perez-Valencia, L.J.; Vannella, K.M.; Ramos-Benitez, M.J.; Sun, J.; Abu-Asab, M.; Dorward, D.W.; Awad, K.S.; Platt, A.; Jacobson, E.; Kindrachuk, J.; et al. Ebola virus shed glycoprotein is toxic to human T, B, and natural killer lymphocytes. iScience 2023, 26, 107323. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Ebola Virus Genome.
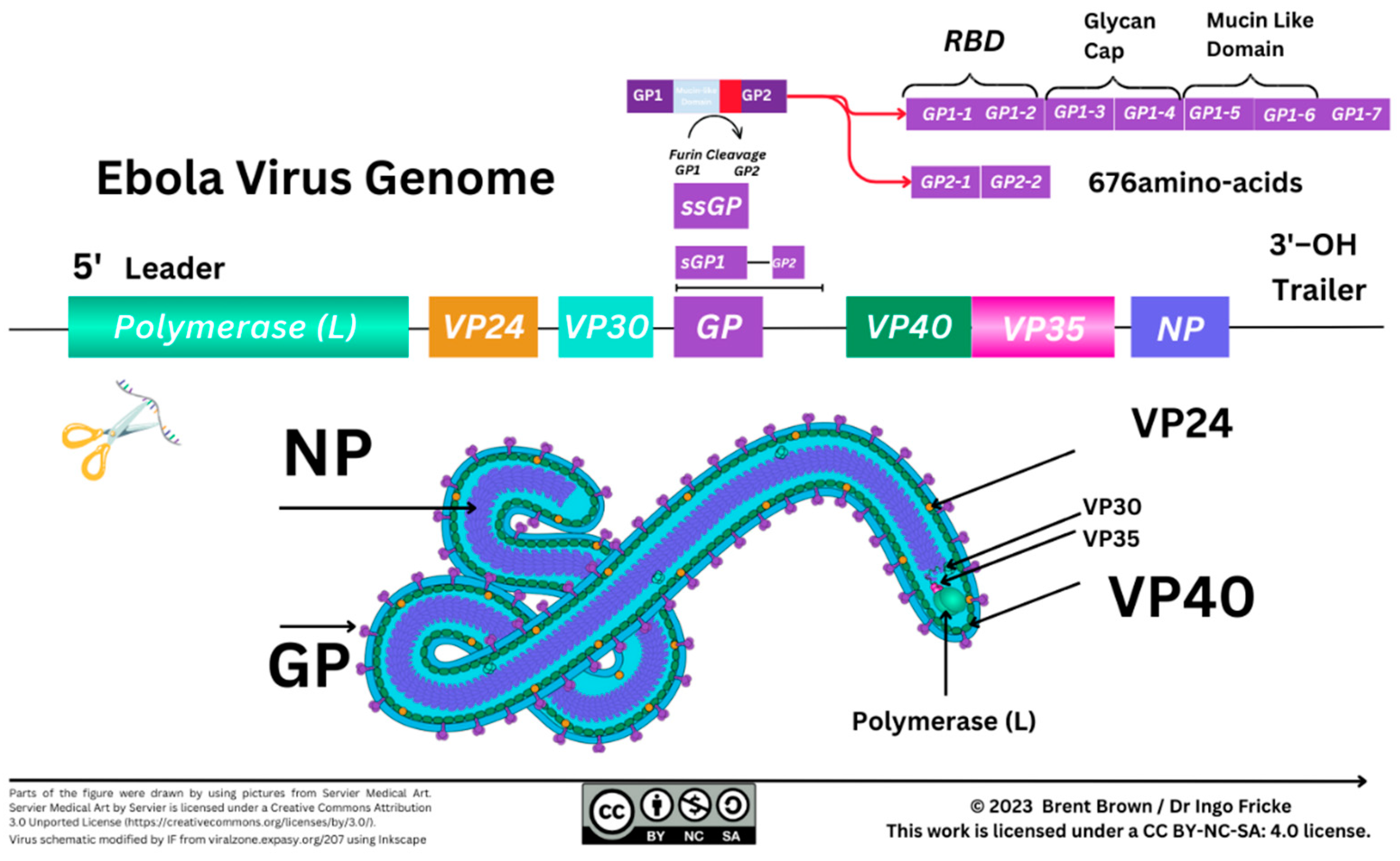
Figure 2.
Ebola Virus Pathogenesis.
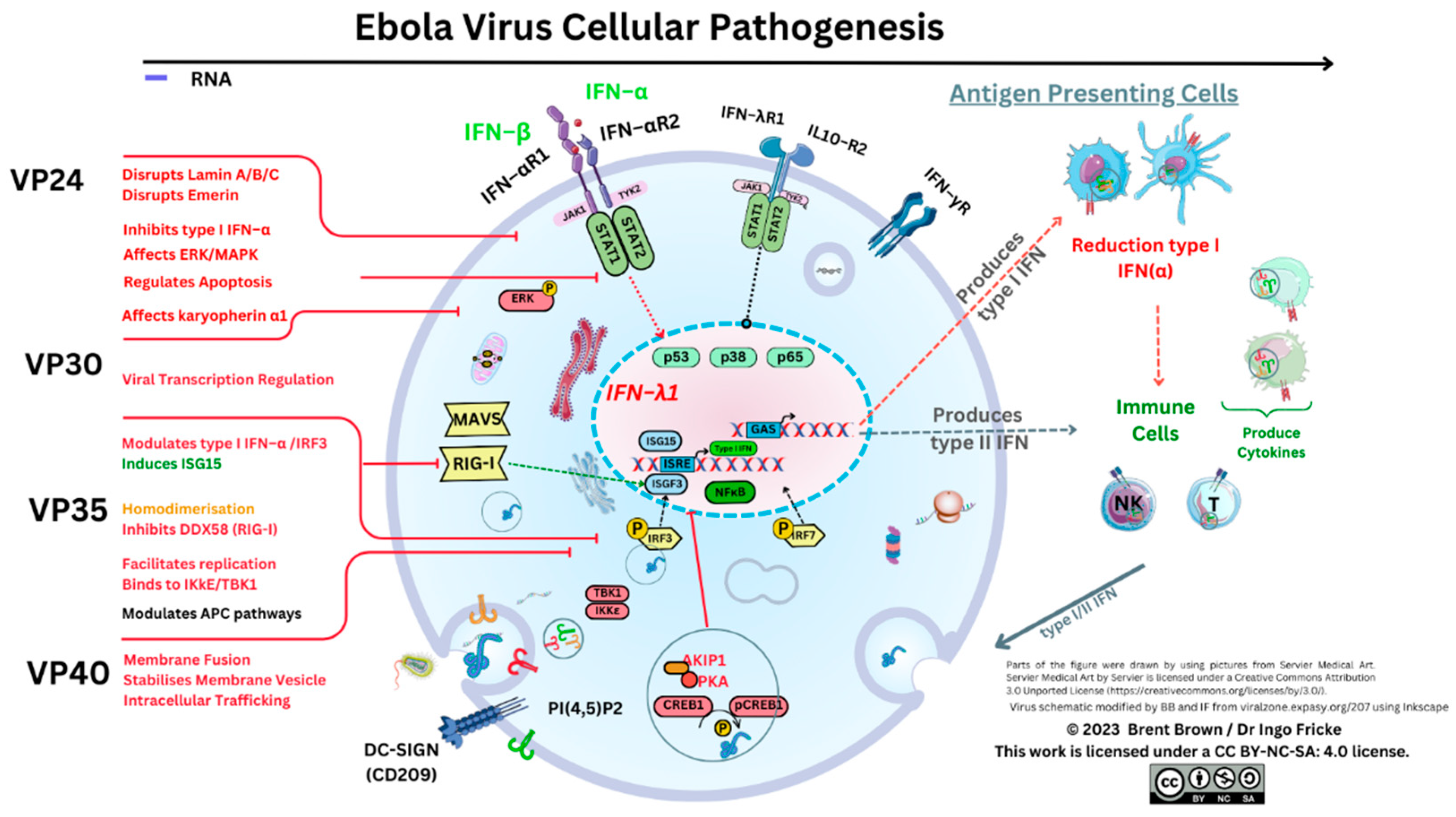

Table 1.
Ebola Virus Proteins and Gene Regulation Factors.
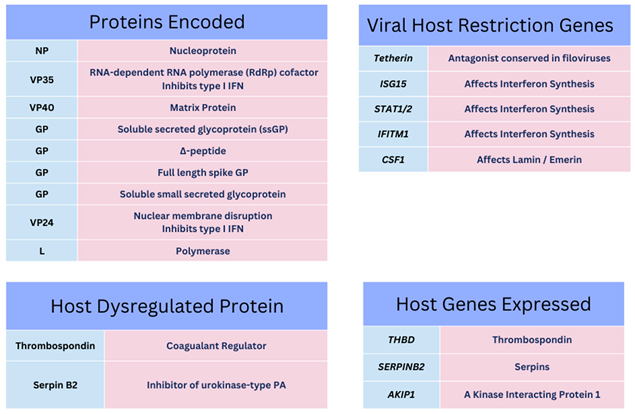 |
Table 2.
Cytokine Regulators during Ebola Virus Infection.
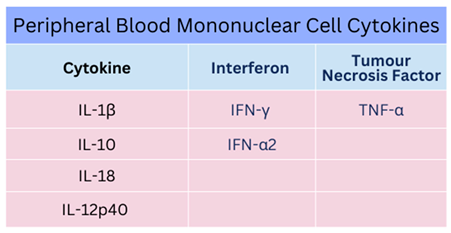 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



