
Submitted:
11 July 2023
Posted:
11 July 2023
You are already at the latest version
Abstract
We compared the performance of matrix-assisted laser desorption/ionization followed by a time of flight (MALDI-TOF) mass spectrometry and genomic DNA extraction followed by sequencing, assembly, and alignment for phylogenetic assessment (Genomics method). We collected the samples from four contaminated rivers in the Dominican Republic and analyzed MALDI-TOF efficacy and accuracy for identifying bacteria in the samples. We evaluated the results for both methods (MALDI-TOF and Genomics) and reported a similarity percentage between each method's results. The MALDI-TOF method had a 72.41\% of coincidence with the Genomics method. This could have been sequence contamination found in the Genomics method. When it was later filtered, the result's coincidence rate went up to 90\%.
Keywords:
MALDI-TOF
; enviromental
; Rivers
; Genomics method
; Dominican Republic
1. Introduction
When it comes to analytic methods, humanity has always moved from the classic chemical and physical analysis to instrumental and more accurate methods, and for the latter ones, new updates have been coming out to facilitate them while maintaining or upgrading the accuracy of results [1]. In the case of the identification of species in environmental samples, sequencing became the standard due to the gradual increase in its availability, as well as its high accuracy on this topic [2]. However, in the last decade, the Matrix-Assisted Laser Desorption Ionization-Time of Flight mass spectrometry (MALDI-TOF) has been explored as a possible alternative method with lower cost and time [3]. This technique has been used to profile bacterial proteins from cell extracts [2]. The procedure provides a unique spectral mass (a signature graph for each microorganism, also called their fingerprint) of the microorganisms that can identify their species with an accuracy higher than 90%, making this procedure ideal for clinical microbiology [4]. It is important to mention the latter because bacterial identification is essential to develop effective antimicrobial decisions. Also, this method of identification has great potential in water samples analysis due to the capacity to process greater amounts of samples at lower cost and time, which is a major advantage when it comes to characterizing a microbiome [5]. For this reason, since the last decade, studies regarding surface water in rivers have been published [6,7].
On the other hand, genomic analysis of complex environmental samples is becoming an essential tool for understanding the evolutionary history and functional and ecological biodiversity, the only downside being a high-cost and long-time procedure [8,9]. In addition, the widespread use of DNA sequencing in the last decades has played a pivotal role in accurately identifying bacterial isolates and discovering novel bacteria [2]. DNA barcoding utilizes standardized species-specific genomic regions (DNA barcodes) to generate vast DNA libraries to identify unknown specimens [10]. This methodology is essential, particularly in bacteria with unusual phenotypic profiles, slow-growing, uncultivable bacteria, and culture-negative infections [11]. An excellent example of this in bacteria is the 16S ribosomal RNA [12].
The main objective of this article will be to compare the viability of the MALDI-TOF technique as an alternative to the Whole Genome Sequencing technique in surface water river samples. The objective is to determine how close the accuracy of mass spectrometry is to the accuracy of a high-precision and high-cost method in DNA sequencing, as the funds destined to research in the Dominican Republic are rather limited, and the need for studying bacterial profiles in the environment is crucial for the public health, as many pathogenic bacteria have been identified in its rivers [6].
2. Materials and Methods
2.1. Sampling
The sampling was done in 2019 for four rivers, the Isabela River and the Ozama River from Santo Domingo, in the Dominican Republic. The points sampled for these rivers were, for Isabela: 18.510111, −69.913111 (Point A), 18.513805, −69.898666 (Point B) and 18.696638, −70.158861 (Point C) see Figure 1; for the Ozama: 18.526683, −69.860283 (Point A), 18.518502, −69.895071 (Point B) and 18.552950, −69.822415 (Point C) see Figure 2; for the Yaque del Norte river: 19.067232, −70.864517 (Point A), 19.153600, −70.644250 (Point B), 19.454433, −70.716350 (Point C), 19.589842, −71.059632 (Point D), 19.706249, −71.499045 (Point E), 19.829313, −71.647867 (Point F) see Figure 3; for the Yaque del Sur river: 18.911396, −71.014936 (Point A), 18.398545, −71.185798 (Point B), 18.299459, −71.172602 (Point C), 18.252541, −71.140495 (Point D) see Figure 4. For the sampling method, we decided to take 1 sample and two replicas of each point in a time lapse of six hours, one replica every two hours. These waters are not treated, all samples were taken directly from the river, and proper sewage and wastewater systems are yet to be implemented.
2.2. Bacterial Isolation
In accordance with the recommendations from [13], 1 mL, 10 mL, and 50 mL aliquots were subjected to vacuum filtration in triplicate using a nitrocellulose membrane (0.22 m pore size) from Simsii, INC. Bacteria retained on the membranes were subsequently cultured in two different media: (a) MacConkey agar supplemented with imipenem (4 g/mL); and (b) MacConkey agar supplemented with cefotaxime (8 g/mL). Then, the samples were then incubated at 37°C for a duration ranging from 16 to 48 hours. In addition, isolates were obtained by streaking the samples onto chromogenic culture media (ChromAgar™, France) using the streak plate method. Finally, Pure isolates were preserved at -70°C in 25% glycerol.
2.3. Bacterial Classification and Identification
Microbial identification followed the CLSI guidelines with some adaptations, using MALDI-TOF (matrix-assisted laser desorption ionization-time of flight) technology [12,14]. The BioTyper® 3.1 software from Bruker Daltonics, Germany, equipped with the MBT 6903 MPS library released in 2019, was utilized to ensure reproducible results. The manufacturer’s recommended MALDI BioTyper Preprocessing Standard Method and the MALDI Biotyper MSP Identification Standard Method were adjusted and employed. Isolated colonies were cultured on blood agar at 35°C for 24 hours. Approximately 0.1 mg was inoculated onto a sample carrier for each new culture using the complete cell transfer protocol. Subsequently, the samples were coated with 1 L of a matrix solution (10 mg/mL) composed of -cyano-4-hydroxycinnamic acid in a mixture of 50% acetonitrile and 2.5% trichloroacetic acid. The coated samples were then allowed to dry at 25°C for 20 minutes. Identification was performed in triplicate for each sample.
2.4. Genomic DNA extraction from Isolates
Genomic DNA was extracted from colonies that were incubated in TSB at 35°C for 24 hours. A 4 mL aliquot of the culture was centrifuged at 8,000× g for 2 minutes to initiate the extraction process. The resulting cell pellet was then subjected to the QIAamp DNA extraction kit (Qiagen, Germany) with the following adaptations: the bacterial pellet was resuspended in a modified lysis buffer consisting of 20 L proteinase K, 200 L of TSB, and 100 L of Qiagen’s ATL buffer. This suspension was incubated at 56°C for 10 minutes. Next, 50 L of absolute ethanol was added, followed by a 3-minute incubation at room temperature. Subsequently, the extraction protocol continued according to the manufacturer’s recommendations. The obtained DNA was suspended in 50 L of Qiagen’s TE buffer. The integrity of the extracted DNA was assessed by running it on a 1% agarose gel stained with SYBR Green, and the gel was electrophoresed at 100 V for 60 minutes.
2.5. Genome Sequencing, Assembly, and Analysis
The construction of sequencing libraries involved the following steps:
(I) The genomic DNA was fragmented randomly through sonication.
(II) The fragmented DNA was subjected to end polishing, A-tailing, and ligation with Illumina sequencing full-length adapters. This was followed by PCR amplification using P5 and indexed P7 oligos.
(III) The PCR products, which represented the final construction of the libraries, were purified using the AMPure XP system from Beckman Coulter Inc., located in Indianapolis, IN, USA.
To ensure the quality and size distribution of the sequencing libraries, a quality control assessment was performed using an Agilent 2100 Bioanalyzer from Agilent Technologies, based in CA, USA. Additionally, the libraries were quantified by real-time PCR to meet the criteria of 3 nM.
An Illumina NovaSeq 600 platform was utilized for sequencing the whole genomes, employing the PE 150bp strategy. The sequencing process was conducted at the America Novogene Bioinformatics Technology facility Co., Ltd.
Whole genomes were sequenced using the Illumina NovaSeq 600 platform, employing the PE 150 strategy. The sequencing process was performed at the America Novogene Bioinformatics Technology Co., Ltd., located in Sacramento, California, United States. The genomes were assembled using the Assembly HiSeq Pipeline [13], which utilizes various quality control tools such as FastQC for read quality analysis and visualization [15], AdapterRemoval v2 for adapter removal [16], and KmerStream for k-mer distribution computation [17]. The assemblers Edena V3 [18]and Spades 3.9.1 [19] were used for genome graph construction, and Unicycler [20] were employed to optimize and integrate the assemblies. Whole-genome annotation was performed using RAST [21]and Prokka [22]. Individual plasmid sequences within the genome assemblies were predicted and reconstructed using MOB-recon [23]. Assembly quality metrics were computed using QUAST [24], and genome phylogenetic affiliation was confirmed using JSpeciesWS web tools [25]. In addition, tools for filtering fasta sequences according to taxonomy were used for the low identity samples, Autometa 1.0 [26] and MaxBin 2.0 [27] were used. The genome shotgun projects have been deposited in the DDBJ/ENA/GenBank databases.
To gain insights into the genomes, the RAST annotation server [21] was used to identify the subsystem of each genome, providing information on genes associated with various functions, including virulence, pathogenicity, plasmids, and antibiotic resistance. Pathogenicity and virulence analyses were conducted using the PathogenFinder [28] and VirulenceFinder [29] tools from the Center for Genomic Epidemiology. Plasmid identification was performed using the PlasmidFinder [30] tool from the same center. Resistomes were identified using the CARD database Resistance Gene Identifier tool [31].
For the analysis of multi-resistant genomes, Resistance Genes Identifier (RGI) with the CARD protein database and ResFinder-4.0 were employed to predict resistance genes [31]. Plasmid detection was carried out using the MOB-suite and PlasmidFinder-2.1 [30]. Pathogenicity classification was performed using PathogenFinder-1.1 [28], and virulence factors were determined using VirulenceFinder-2.0 [29]. The serotypes of the E. coli genomes were determined using SerotypeFinder-2.0 [32], and the number of mobile elements was assessed using MobileElementFinder [33].
3. Results
There was found some similarity between some results, to be concrete, a similarity of 72.41%, in Table 1 it is shown each sample with their results for the MALDI-TOF and Sequencing techniques. The results that differed between both methods were highlighted, and the reasons for these discordances were discussed. It was found that some of the sequences in the Genomics method didn’t have enough similarity to be assigned to a concrete species correctly, as they had a similarity lower than 98.9% in the phylogenetic analysis performed through JspeciesWS (which is the minimum similarity this tool catalogs as acceptable). Because of this, these sequences were filtrated by taxonomy utilizing MaxBin 2.0 [27] and Autometa 1.0 [26] following the authors’ recommendations.
After filtrating the sequences, only YNP1-2, YSP3-2, YSP4-2, YSP6-2 were still different, as the rest of the results came out to coincide with the MALDI-TOF results and upgrading the similarity percent to 86.2%, which is a much more respectable percent considering the difference in price and time per sample in the Dominican Republic (See Table 2).
The contamination in the sequences might have been a manipulation mistake. After the filtering with MaxBin 2.0 and Autometa 1.0, we could see that half of the results were fixed and coincided with MALDI-TOF. The filtered sequences had over 99% similitude with the database sequences.
4. Discussion
This research was conducted for three years, using different techniques to identify the bacteria: required culture-based (MALDI-TOF) and Whole genome sequencing (WGS). It is essential to mention the number of organisms that each technique covers per sample as MALDI-TOF does one per sample and WGS does one per sample as well; hence MALDI-TOF is a more efficient and cost-effective option for bacteria identification compared to WGS as it was described by [2], many new packages also allow you to scan for particular antimicrobial resistance proteins inside bacteria using MALDI-TOF [4]. This is very limited at the moment since MALDI-TOF is still a culture-based technique and requires at least eighteen hours for microbe isolation and eighteen to twenty-four more hours for growth to produce enough proteins to be read by MALDI-TOF. However, this technology will eventually allow fast identification of ARGs based on expressed proteins in the cytoplasm [34].
The samples were retrieved and processed successfully. It was possible to correctly identify most of the bacteria through MALDI-TOF methodology. There was a contamination problem that might be related to a manipulation mistake detected in the phylogenetics assessments, causing some results to differ from the MALDI-TOF results, but it was possible to detect the contamination due to JspeciesWS giving a range of acceptable similitude percentages between the submitted fasta files (the samples) and the sequences from the database (reference sequences), as well as other calculated features like the size and the GC% from the files being too high.
The final results of the MALDI-TOF technique were found to have 86.2% of similarity with the results obtained from the whole genomes phylogenetic analysis from JspeciesWS, which is very close to the results described by [35]. We hypothesize that the four results that differed between them could’ve been caused by the MALDI-TOF database needing some improvement [12]. It was stated this way because the data analyzed by the JspeciesWS tool is still continuously updated and improved [25]. Due to this, it is important to mention that the high cost of the Genomics methodologies justifies itself with its frequent updating and high specificity.
The post-filtering results prove the high efficacy of the MALDI-TOF technique in river water bacterial identification, and that it can be used as a low cost alternative to whole genome sequencing in the Dominican Republic, also, we expect more improvements in the future that could make possible to characterize whole microbiomes without the need to invest in nucleic acid sequencing.
5. Conclusions
It was proved that MALDI-TOF could be a low cost alternative to Whole Genome Sequencing when it comes to identification of bacteria sampled from surface river water. This finding is of great importance for developing countries as the Dominican Republic, in which hiring an external company for the sequencing of samples represents a lower efficiency when it comes to price and time, making it possible for these countries to make related studies with this major resource facility.
Finally, it is important to state that more uses are being standardized for the MALDI-TOF technique, like identifying proteins related to antibiotic resistance, and the used databases are being improved, expanding MALDI-TOF scope in microbiology even more.
Author Contributions
Conceptualization, E.F. and L.M.; methodology, E.F., O.C., R.R., L.M.; software, V.C. and R.B.; Investigation R.B., W.L., I.O., A.G., S.M. and C.P.; validation, A.G., I.O., R.B., V.C., Y.R. and O.C.; formal analysis, R.B, Y.R. and V.C.; resources, E.F. and L.M.; data curation, R.B. and V. C.; writing—original draft preparation, E.F., R.B. and V.C.; writing—review and editing, E.F. and R.R and L.M.; visualization, R.B. and V.C.; supervision, R.R., O.C., E.F., Y.R. and L.M.; project administration, E.F. and L.M.; funding acquisition, E.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fondo Nacional de Innovación y Desarrollo Científico y Tecnológico (FONDOCYT) of Miniterio de Eduacion Superior Ciencia y Tecnología (MESCyT) grant number 2018-2019-2B4-157.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Acknowledgments
This research project was successfully conducted thanks to the support provided by the Research Vice-Rectory and the Deanship of Basic and Environmental Sciences at Instituto Tecnologico de Santo Domingo. The Federal University of Para team was support by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES, Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq, Pró-reitoria de Pesquisa e Pós-graduação(PROPESP)-UFPA and Pró-Reitora de Relações Internacionais (PROINTER)-UFPA.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Skoog, D.; Holler, F.; Crouch, S. Principles of Instrumental Analysis, 6 ed.; Cengage Learning; 2008; pp. 1–2. [Google Scholar]
- Carbonnelle, E.; Mesquita, C.; Bille, E.; Day, N.; Dauphin, B.; Beretti, J.L.; Ferroni, A.; Gutmann, L.; Nassif, X. MALDI-TOF mass spectrometry tools for bacterial identification in clinical microbiology laboratory. Clinical Biochemistry 2011, 44, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Buylla, A.; Culebras, E.; Picazo, J.J. Identification of Acinetobacter species: is Bruker biotyper MALDI-TOF mass spectrometry a good alternative to molecular techniques? Infection, genetics and evolution 2012, 12, 345–349. [Google Scholar] [CrossRef]
- Shannon, S.; Kronemann, D.; Patel, R.; Schuetz, A.N. Routine use of MALDI-TOF MS for anaerobic bacterial identification in clinical microbiology. Anaerobe 2018, 54, 191–196. [Google Scholar] [CrossRef]
- Sala-Comorera, L.; Caudet-Segarra, L.; Galofré, B.; Lucena, F.; Blanch, A.R.; García-Aljaro, C. Unravelling the composition of tap and mineral water microbiota: Divergences between next-generation sequencing techniques and culture-based methods. International Journal of Food Microbiology 2020, 334, 108850. [Google Scholar] [CrossRef]
- Bonnelly, R.; Queiroz Cavalcante, A.L.; Calderon, V.V.; Baraúna, R.A.; Jucá Ramos, R.T.; Rodríguez-Rodríguez, Y.; De Francisco, L.E.R.; Maroto Martín, L.O.; Perdomo, O.P.; Franco De Los Santos, E.F. Beta-lactam susceptibility profiles of bacteria isolated from the Ozama river in Santo Domingo, Dominican Republic. Sustainability 2023, 15, 5109. [Google Scholar] [CrossRef]
- Ashfaq, M.Y.; Da’na, D.A.; Al-Ghouti, M.A. Application of MALDI-TOF MS for identification of environmental bacteria: A review. Journal of Environmental Management 2022, 305, 114359. [Google Scholar] [CrossRef] [PubMed]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Molecular ecology 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Hassan, F.M.; Salih, W.Y.; Al-Haideri, H.H. Next-Generation Sequencing Technologies for Environmental DNA as an Efficient Bio Indicator for Bacterial Biodiversity in Tigris River, Iraq. Systematic Reviews in Pharmacy 2020, 11, 1107–1114. [Google Scholar]
- Valentini, A.; Pompanon, F.; Taberlet, P. DNA barcoding for ecologists. Trends in ecology & evolution 2009, 24, 110–117. [Google Scholar]
- Purty, R.; Chatterjee, S. DNA barcoding: an effective technique in molecular taxonomy. Austin J Biotechnol Bioeng 2016, 3, 1059. [Google Scholar]
- Strejcek, M.; Smrhova, T.; Junkova, P.; Uhlik, O. Whole-Cell MALDI-TOF MS Versus 16S rRNA Gene Analysis for Identification and Dereplication of Recurrent Bacterial Isolates. Frontiers in microbiology 2018, 9, 1294. [Google Scholar] [CrossRef]
- Freitas, D.Y.; Araújo, S.; Folador, A.R.; Ramos, R.T.; Azevedo, J.S.; Tacão, M.; Silva, A.; Henriques, I.; Baraúna, R.A. Extended spectrum beta-lactamase-producing gram-negative bacteria recovered from an Amazonian lake near the city of Belém, Brazil. Frontiers in microbiology 2019, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, M.P.; Lewis, J.S. The Clinical and Laboratory Standards Institute Subcommittee on Antimicrobial Susceptibility Testing: Background, Organization, Functions, and Processes. Journal of Clinical Microbiology 2020, 58, e01864–19. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.; others. FastQC: a quality control tool for high throughput sequence data. 2010. [Google Scholar]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC research notes 2016, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Melsted, P.; Halldórsson, B.V. KmerStream: streaming algorithms for k-mer abundance estimation. Bioinformatics 2014, 30, 3541–3547. [Google Scholar] [CrossRef]
- Hernandez, D.; François, P.; Farinelli, L.; sterås, M.; Schrenzel, J. De novo bacterial genome sequencing: millions of very short reads assembled on a desktop computer. Genome research 2008, 18, 802–809. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; others. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS computational biology 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; others. The RAST Server: rapid annotations using subsystems technology. BMC genomics 2008, 9, 1–15. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Robertson, J.; Nash, J.H. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microbial genomics 2018, 4. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Miller, I.J.; Rees, E.R.; Ross, J.; Miller, I.; Baxa, J.; Lopera, J.; Kerby, R.L.; Rey, F.E.; Kwan, J.C. Autometa: automated extraction of microbial genomes from individual shotgun metagenomes. Nucleic acids research 2019, 47, e57–e57. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder-distinguishing friend from foe using bacterial whole genome sequence data. PloS one 2013, 8, e77302. [Google Scholar] [CrossRef]
- Kleinheinz, K.A.; Joensen, K.G.; Larsen, M.V. Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 2014, 4, e27943. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Hasman, H. PlasmidFinder and in silico pMLST: identification and typing of plasmid replicons in whole-genome sequencing (WGS). In Horizontal gene transfer; Springer, 2020; pp. 285–294. [Google Scholar]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S.; others. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic acids research 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Joensen, K.G.; Tetzschner, A.M.; Iguchi, A.; Aarestrup, F.M.; Scheutz, F. Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. Journal of clinical microbiology 2015, 53, 2410–2426. [Google Scholar] [CrossRef]
- Johansson, M.H.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. Journal of Antimicrobial Chemotherapy 2021, 76, 101–109. [Google Scholar] [CrossRef]
- Murray, P.R. What is new in clinical microbiology—microbial identification by MALDI-TOF mass spectrometry: a paper from the 2011 William Beaumont Hospital Symposium on molecular pathology. The journal of molecular diagnostics 2012, 14, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Carbonnelle, E.; Grohs, P.; Jacquier, H.; Day, N.; Tenza, S.; Dewailly, A.; Vissouarn, O.; Rottman, M.; Herrmann, J.L.; Podglajen, I.; others. Robustness of two MALDI-TOF mass spectrometry systems for bacterial identification. Journal of microbiological methods 2012, 89, 133–136. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Sampling points corresponding to the Isabela river. Point A corresponds to a region surrounded by multiple communities, Point B corresponds to the delta of the river, where it interacts with the Ozama river, and Point C corresponds to a pristine zone close to the river source.
Figure 1.
Sampling points corresponding to the Isabela river. Point A corresponds to a region surrounded by multiple communities, Point B corresponds to the delta of the river, where it interacts with the Ozama river, and Point C corresponds to a pristine zone close to the river source.
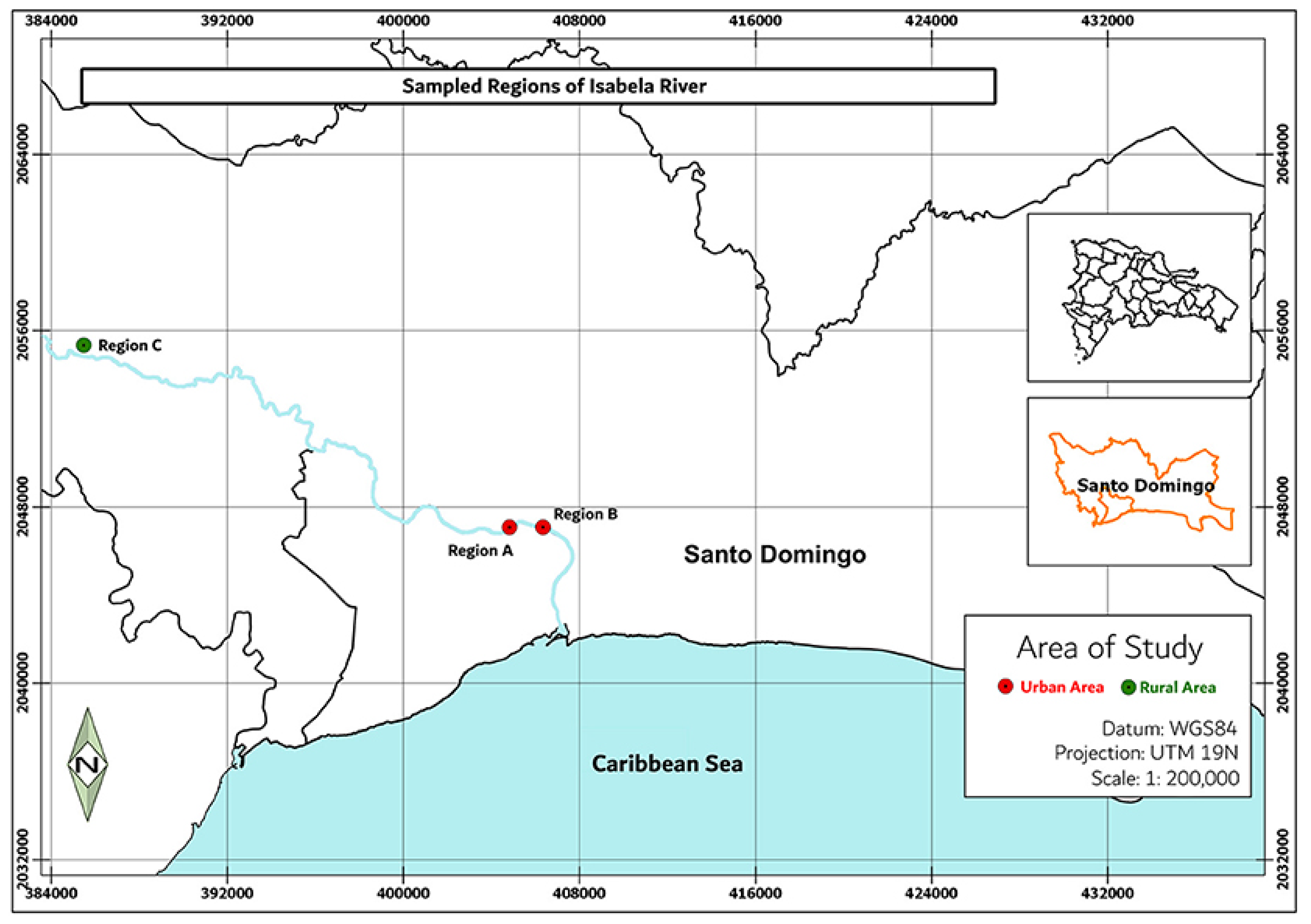
Figure 2.
Sampling points corresponding to the Ozama river. Like in the Isabela description, Point A corresponds to a region surrounded by multiple communities, Point B corresponds to the delta of the river, where it interacts with the Isabela river, and Point C corresponds to a pristine zone close to the river source.
Figure 2.
Sampling points corresponding to the Ozama river. Like in the Isabela description, Point A corresponds to a region surrounded by multiple communities, Point B corresponds to the delta of the river, where it interacts with the Isabela river, and Point C corresponds to a pristine zone close to the river source.
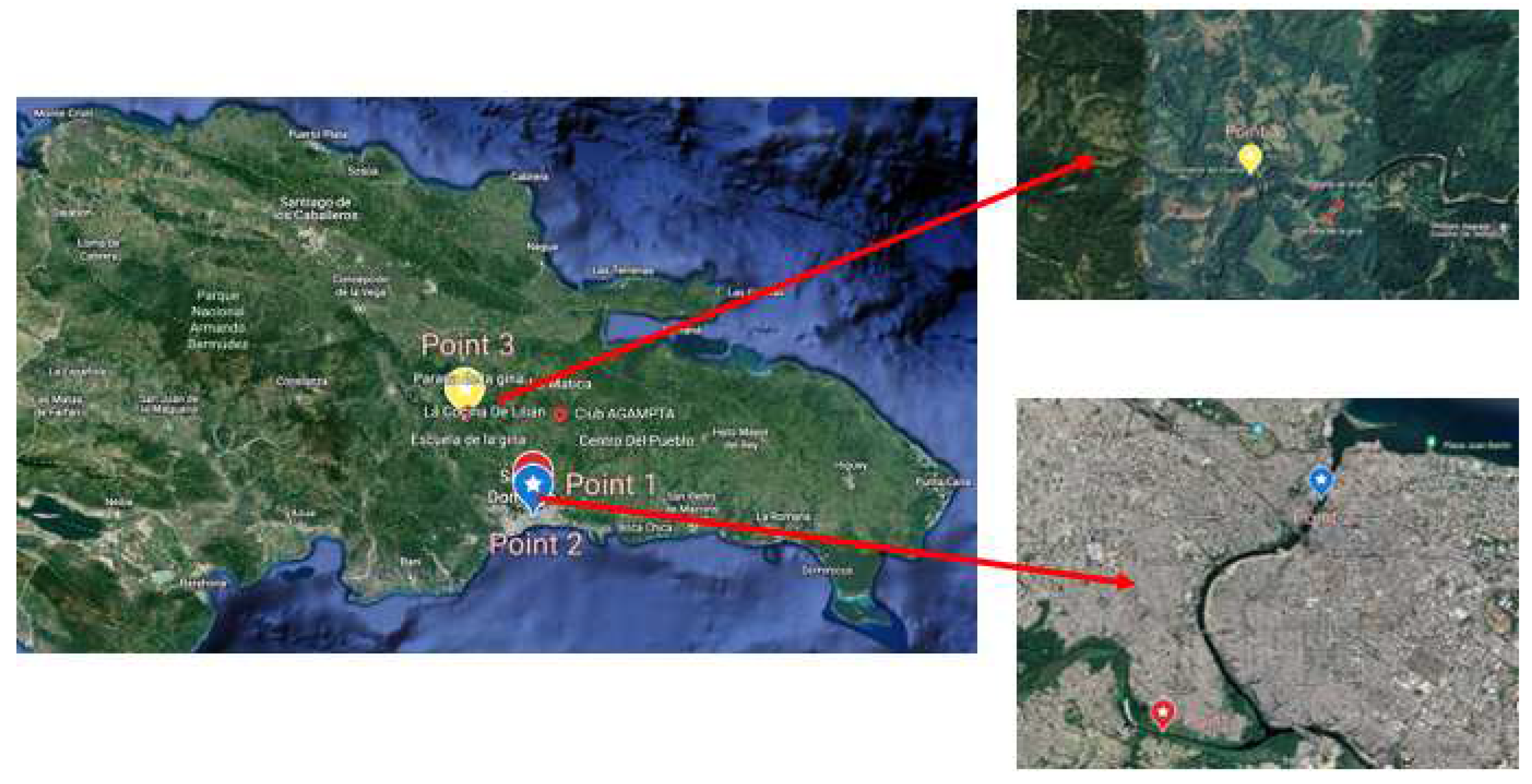
Figure 3.
Sampling points corresponding to the Yaque del Norte river. In this map, the points go from the coordinates with less demographic impact to a highly populated city.
Figure 3.
Sampling points corresponding to the Yaque del Norte river. In this map, the points go from the coordinates with less demographic impact to a highly populated city.
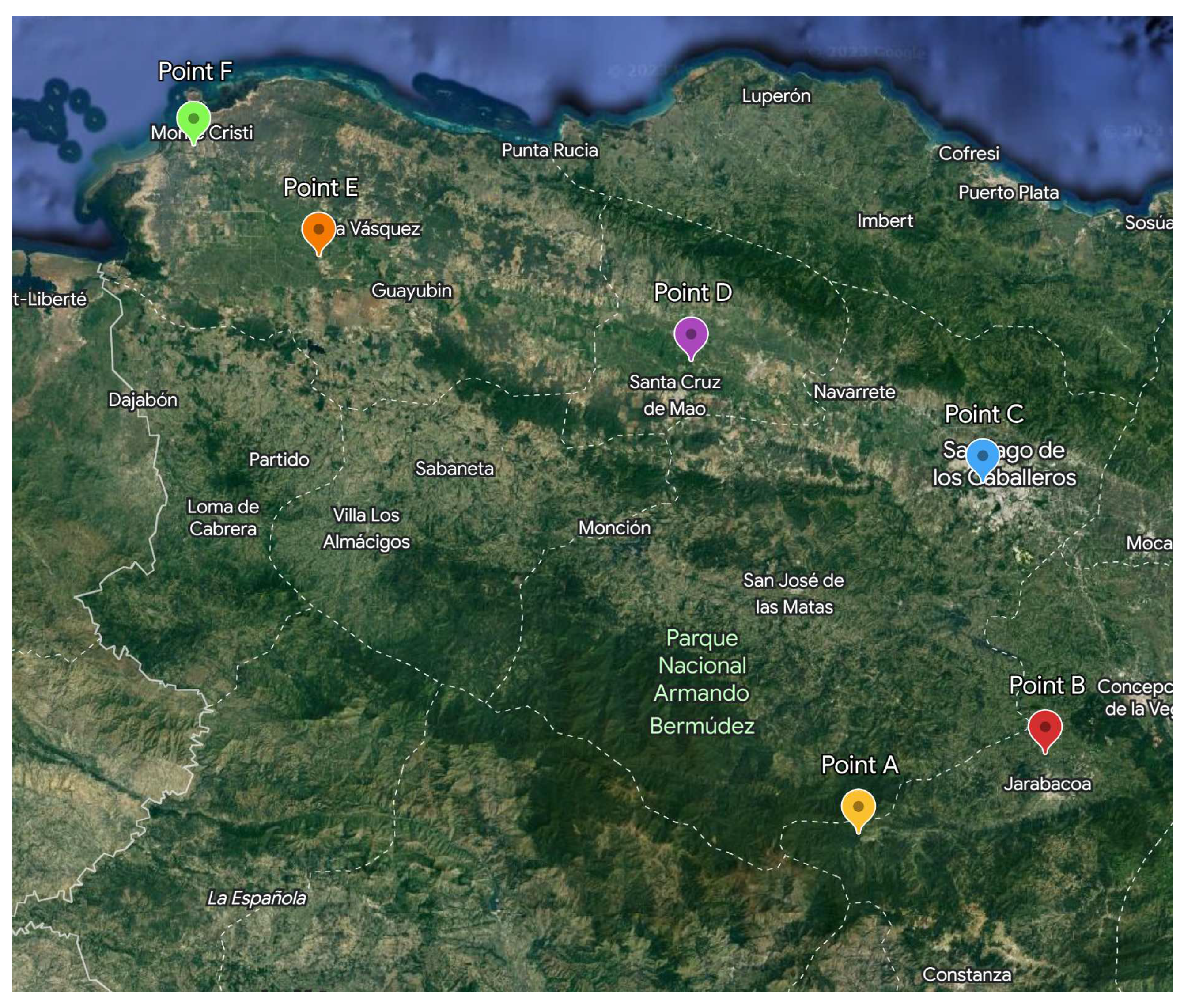
Figure 4.
Sampling points corresponding to the Yaque del Sur river. Similar to the Yaque del Norte river, it was sampled from a pristine zone to a more populated zone.
Figure 4.
Sampling points corresponding to the Yaque del Sur river. Similar to the Yaque del Norte river, it was sampled from a pristine zone to a more populated zone.
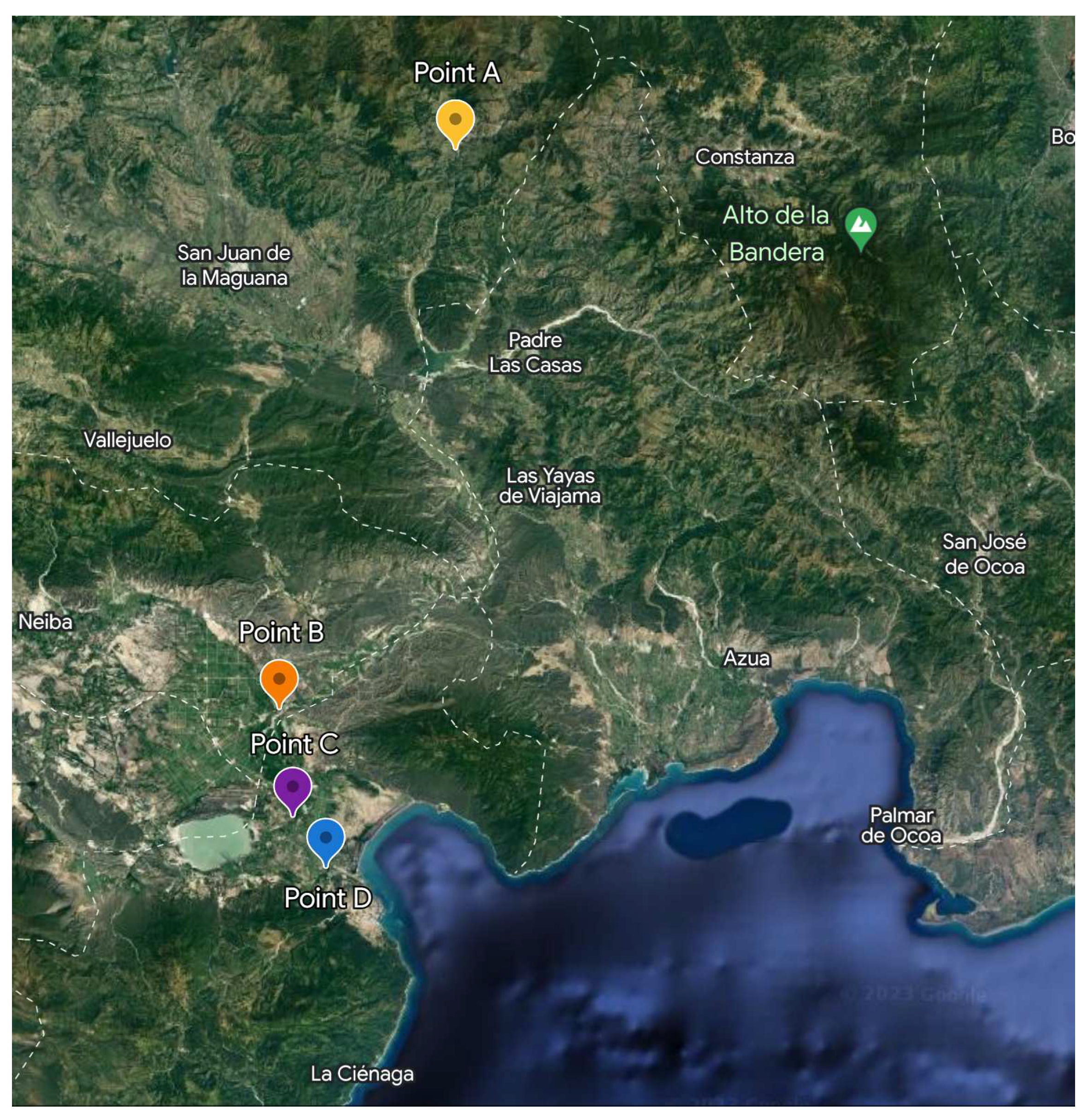

Table 1.
In this table, the samples’ Identification Codes with their respective results for both methods are presented. It is important to mention that with the sequencing technique, it was also possible to detect the subspecies of some samples.
Table 1.
In this table, the samples’ Identification Codes with their respective results for both methods are presented. It is important to mention that with the sequencing technique, it was also possible to detect the subspecies of some samples.
| Sample Code | MALDI-TOF Result | JSpeciesWS Result |
|---|---|---|
| INTEC BC5 1.1 | Enterobacter cloacae | Enterobacter cloacae subsp. Cloacae SMART_901 |
| INTEC BI4 1.1 | Enterobacter kobei | Enterobacter kobei 35730 |
| INTEC AC6 1.1 | Escherichia coli | Escherichia coli SQ2203 |
| INTEC BI10 1.1 | Escherichia coli | Escherichia coli 50816743 |
| INTEC BC4 | Escherichia coli | Escherichia coli KOEGE 40 (102a) |
| INTEC BC8 | Escherichia coli | Eascherichia coli O32:H37 str. P4 |
| INTEC AI11 1.1 | Acinetobacter baumannii | Acinetobacter baumannii ABBL129 |
| INTEC BI5 | Acinetobacter baumannii | Acinetobacter baumannii BR097 |
| INTEC BI9 | Acinetobacter baumannii | Acinetobacter baumannii NIPH 67 |
| INTEC AI6 | Acinetobacter baumannii | Acinetobacter baumannii UH6507 |
| INTEC AI12 | Acinetobacter baumannii | Acinetobacter baumannii BR097 |
| INTEC AI10 | Acinetobacter baumannii | Acinetobacter baumannii BR097 |
| INTEC BI15 | Acinetobacter baumannii | Acinetobacter baumannii NIPH 67 |
| DC2 | Escherichia coli | Pseudomonas monteilii |
| DC8 | Escherichia coli | Escherichia coli |
| DC10 | Acinetobacter pitii | Achromobacter xylosoxidans |
| EC4 | Klebsiella pneumoniae | Klebsiella pneumoniae |
| EC7 | Klebsiella pneumoniae | Klebsiella pneumoniae |
| FC5 | Acinetobacter baumanii | Acinetobacter baumanii |
| FC7 | Acinetobacter baumanii | Acinetobacter baumanii |
| YNP1-2 | Bacillus licheniformis | Acinetobacter pittii |
| YNP2-2 | Klebsiella pneumoniae | Klebsiella pneumoniae |
| YNP5-3 | Pseudomonas aeruginosa | Pseudomonas aeruginosa |
| YSP2-1 | Enterobacter bugandensis | Enterobacter mori |
| YSP3-2 | Serratia marcescens | Salmonella enterica |
| YSP4 | Raoultella ornithinolytica | Raoultella ornithinolytica |
| YSP4-2 | Salmonella spp. | Klebsiella pneumoniae |
| YSP5 | Escherichia coli | Klebsiella pneumoniae |
| YSP6-2 | Klebsiella variicola | Salmonella enterica |
Table 2.
Table indicating cost and time needed for identifying the bacteria per sample for the Dominican Republic.
Table 2.
Table indicating cost and time needed for identifying the bacteria per sample for the Dominican Republic.
| Technique | Cost per Sample | Time per Sample |
|---|---|---|
| MALDI-TOF | $7.5USD | 2 days |
| WGS | $120.00USD | 21 days |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



