
Submitted:
11 July 2023
Posted:
12 July 2023
You are already at the latest version
Abstract
Polymer electrolyte membrane water electrolyser suffers mainly from slow kinetics regarding oxygen evolution reaction (OER). Noble metal oxides, like IrO2, RuO2, are generally more active for OER than metal electrodes, exhibiting low anodic overpotentials and high catalytic activity. However, issues like electrocatalyst stability under continuous operation and cost minimization through reduction of the catalyst loading are of great importance for the research community. In the present study, unsupported IrO2 of various particle sizes (different calcination temperatures) were evaluated for the OER and as anode electrodes for PEM water electrolyzers [1]. The elec-trocatalysts were synthesized by the modified Adams method [2][3] and the effect of calcination temperature on the properties of IrO2 electrocatalysts is investigated. Physicochemical charac-terization was conducted using X-ray Diffraction, BET, High-Resolution TEM and XPS analyses. For the electrochemical performance of synthesized electrocatalysts in the OER, cyclic voltammetry and linear sweep voltammetry were conducted in a typical 3-cell electrode configuration, using glassy carbon as working electrode, where the synthesized electrocatalysts were casted on, in 0.5 M H2SO4 solution. The materials, as anode PEM water electrolysis electrodes, were further evaluated in a typical electrolytic cell using Nafion®115 membrane as electrolyte and Pt/C as cathode elec-trocatalyst. The IrO2 electrocatalyst calcined at 400°C shows high crystallinity with 1.24 nm particle size, high specific surface area (185 m2 g-1) and a high OER activity of 177 mA cm-2 at 1.8 V as anode electrocatalyst for PEM water electrolyzer.
Keywords:
iridium oxide
; oxygen evolution
; PEM water electrolyzer
1. Introduction
Hydrogen is proposed to play a key role in forming a sustainable energy carrier in the future. Water electrolysis using renewable energy, such as wind, solar and tidal energy is supposed to be one of the green and sustainable methods for hydrogen production. Τhe Proton Exchange Membrane Water Electrolyzers (PEMWEs) are one of the most attractive and efficient technologies for water electrolysis due to the advantages of high energy efficiency (40-50%), high-purity hydrogen production, safety, non-pollution and long life time [1,4].
Clean hydrogen is produced through the electrolysis of water using PEMWE in a process that splits water into hydrogen (H2) and oxygen (O2) through the cathodic hydrogen evolution reaction (HER) and the anodic evolution reaction (OER), respectively. At the cathode, HER takes place with low overpotentional values when platinum is used as the electrocatalyst. In contrast, the OER involves much more complex mechanism, resulting in slow kinetics and high electrode overpotentials [5].
Ruthenium and iridium catalysts are the state-of-the-art choices for PEMWE anodes. IrO2 was shown to be more stable than RuO2, with a metal-dissolution rate 30 times smaller than that of RuO2. The higher stability of IrO2 has also been reported in other papers, justifying the choice of iridium oxide as an OER catalyst in acidic media [6]. However, despite the high activity and excellent stability of IrO2, iridium is a scarce and expensive metal. Therefore its use should be optimized in order to reduce PEMWE prices [5]. Therefore, synthesis of electrocatalysts with high activity and excellent stability for the oxygen evolution reaction (OER) in a facile and cost effective way is key to the development of PEMWE [4].
Several preparation methods, e.g. Adams method [2], sol-gel method [7], sulfite complex method [8], magnetron sputtering method [9], sonochemical method [10] and template method [11] have been reported to synthesis the IrO2 electrocatalyst. Puthiyapura et al. [12] synthesized IrO2 (d = 4 – 6 nm) electrocatalyst with the BET surface area of 112.8 m2 g-1 by Adams method. Hao et al. [13] also used Adams method to prepared IrO2 catalyst which BET surface area was about 68.10 m2 g-1. Nanosized IrO2 (d = 7-9 nm) electrocatalyst with specific surface area up to 100 m2 g-1 were synthesized by Cruz through sol-gel method, which was calcined at 500 °C [14]. Among them, the Adams method was firstly found by Adams and Shriner [2] which had the advantages of simple synthesized process, operational capability, widely adaptive and so on [4].
Generally, iridium oxide is obtained electrochemically by the anodic oxidation of metallic iridium, through the thermal decomposition of an iridium precursor, or chemically, by the Adams fusion method. Some studies have shown that changing the structural properties of IrO2 is a satisfactory way of improving the activity and stability of the electrocatalyst. Regarding the activity for the OER, usually amorphous iridium oxide is regarded to be more active than the crystalline version, while the latter is considered to be more stable. Ouattara et al. [15] compared the OER activity of an amorphous IrO2 film prepared by the anodic oxidation of metallic iridium, also reffered as a “hydrous iridium oxide film”, with that of a crystalline film prepared by the thermal decomposition of an iridium precursor. Both the specific electrocatalytic activity and Tafel slope for the OER in these films were similar, but no stability comparison studies were performed.
Reier et al. [16] investigated the OER performance of IrO2 thin films prepared by the thermal decomposition of iridium acetate at various temperatures. The electrocatalytic activities of the films prepared at 250 and 350 °C were almost identical, but superior compared to films calcined at 450 and 550 °C. The better performance of the former films was attributed to the presence of a mixture of amorphous and crystalline IrO2. In another study, the authors used on-line ICP-MS experiments to reveal that both calcination temperature and the crystallinity of the IrO2 film affect the Ir dissolution rate, with the stability essentially increasing with increasing calcination temperature [5,17].
In this study, nano-sized unsupported IrO2 electrocatalysts with high specific surface area and high OER activity have been synthesized by the modified Adams method [2]. IrO2 electrocatalysts were calcined at 400, 500 and 600°C and were systematically studied the effect of calcination temperature on the morphology, structure, and performance. The catalysts were characterized using several techniques such a specific surface area (BET) analysis, energy dispersive X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and transmission electron microscopy (TEM). The prepared materials were electrochemical characterized and their OER activities as well as stability evaluated in half-cell configurations in an acidic electrolyte. The synthesized materials were used as anode electrocatalysts to fabricate membrane electrode assemblies (MEAs), which were studied in a single cell electrolysis configuration regarding their activity and short-term stability.
2. Experimental Section
2.1. Catalysts Synthesis
A series of unsupported nanoparticle iridium oxide electrocatalysts were synthesized using a modified Adams method [2,3]. Firstly, the metal precursor H2IrCl6.xH2O (99 wt%, Alfa Aesar) was dissolved in isopropanol with excess of NaNO3 (Alfa Aesar, fine powder) and a homogeneous mixture was prepared. The metal concentration in the solution was 0.07 M. The salt mixture was heated at 60 °C under continuous stirring until isopropanol evaporated and the mixture was rather dry. Following, the mixture was allowed to completely dry for 30 min in an oven at 80 °C until a dry salt mixture was obtained. This dry salt mixture was then finally introduced into a ceramic furnace and sintered at 400, 500 and 600 °C, respectively, for 30 min. After cooling to room temperature, the mixture was washed with distilled water in order to remove all Cl- ions and remaining solvable salts, and finally dried in air at 80 °C overnight [3]. In the end, 0.2 g of catalyst was prepared.
2.2. Physicochemical Characterization
The crystalline structures of the synthesized electrocatalysts were analyzed by X-ray diffraction (XRD) using a Siemens D500 X-ray diffractometer with auto divergent slit and graphite monochromator using KaCu radiation (1.5418 Å) having a scanning speed 1.2° min-1. Τhe data were collected for 2θ values between 20 and 80°. The characteristic reflection peaks (d-values) were matched with JCPDS data files, and the crystalline phases were identified. The average crystallite size was calculated by means of the Scherrer formula after Warren’ s correction for instrumental broadening. The unit cell parameters were refined with the CrystalSleuth software.
The specific surface areas and pore volumes of the catalysts were determined using a Autosorb-1, Quantachrome instrument. The specific surface area was evaluated using the Brunauer-Emmett-Teller (BET) method.
The morphology, particles’ size and microstructure of the metal oxides were studied with Transmission Electron Microscopy (TEM). TEM images were obtained on a JEOL JEM 2010 High Resolution Transmission Electron Microscopy coupled with Oxford INCA X-ray EDS for elemental analysis and surface mapping of the catalyst surface. Initial TEM images of the catalysts were collected at different regions.
The surface composition and metals oxidation state of the electrocatalysts were evaluated by X-ray Photoelectron Spectroscopy (XPS). The photoemission experiments were carried out in an ultra-high vacuum system (UHV) which consists of a fast entry specimen assembly, a sample preparation and an analysis chamber. The base pressure in both chambers was 1 x 10-9 mbar. Unmonochromatized MgKa line at 1253.6 eV and an analyzer pass energy of 97 eV, giving a full width at half maximum of 1.7 eV for the Au 4f7/2 peak, were used in all XPS measurements. The XPS core level spectra were analyzed using a fitting routine, which can decompose each spectrum into individual mixed Gaussian-Lorentzian peaks after a Shirley background subtraction. The binding energy scale was calibrated by assigning the main C1s peak at 284.7 eV. Regarding measurement errors, for the XPS core level peaks for a good signal to noise ratio, errors in peak positions are of about ±0.2 eV while errors in the quantitative data are found in the range of ~10%.
2.3 Electrochemical Experiments
Electrochemical experiments were performed using a standard three-electrode electrochemical cell, in helium-purged 0.5 M H2SO4 solution to ensure the removal of diluted oxygen at 25 °C, using an Autolab PGSTAT302N potentiostat. A 3 mol/l KCl Ag/AgCl and Pt net were used as reference and counter electrode, respectively, while the working electrode substrate was a glassy carbon disk (rotating disk electrode, disc area: 0.07065 cm2). The working electrode consisted of a catalyst layer loading 3.25 mg cm-2. This layer was prepared by dispersing 10 mg of the catalyst powder in 1 mL of ethanol and 80 μL of Nafion solution (5 wt%, Sigma Aldrich) was also added to the catalyst ink solution as the binder. After that, the catalyst ink solution was sonicated for 30 min at room temperature to ensure uniform dispersion and consequently an amount of 25 μL was casted on the clean glassy carbon substrate using a micropipette. The formatted electrode was dried in air for 1 h.
Ιnitially, the prepared ΙrO2 catalysts were characterized by repetitive cyclic voltammetry (CV) in the three-electrode cell between -0.2 and 1.25 V vs Ag/AgCl with scan rates 20, 50, 75 and 100 mV s-1. The electrochemically active surface areas (ECSAs) were assumed to be proportional to the anodic redox charge of the CVs, as proposed by Ardizone et al. [18]. Here the results were obtained by integration of the anodic votlammetric profiles at various sweep rates.
The OER activities of the catalysts were evaluated by linear sweep voltammetry (LSV) within the potential range between 1.1 and 1.5 VAg/AgCl at a scan rate of 1 mV s-1 (near-steady-state). To avoid the accumulation of oxygen bubbles, a rotation speed of 2500 rpm was applied using a Metrohm rotator system. The electrolyte was deaerated via bubbling with He prior to and during all measurements. All reported measurements were repeated at least three times to ensure the reproducibility of the results.
2.4. Membrane Electrode Assemblies (MEAs) Preparation
A Nafion®115 (DuPontTM) membrane with a typical thickness of 127 μm was used as solid polymer electrolyte. IrO2 oxides powders, prepared as described above, were used as OER electrocatalyst while Pt/C catalyst (BASF, 20% Pt σε Vulcan XC-72 A65TDV 2.1 ELAT® V2.1) was used as hydrogen evolution electrocatalyst at the cathode. Electrodes were prepared as follows: catalysts and Nafion ionomer (5 wt%, Sigma Aldrich) mixtures were first ultrasonically suspended in a mixture of deionized water and isopropanol. Catalytic suspensions thus obtained were then brushed directly onto the Nafion®115 membrane (Catalyst Coated Membrane, CCM). The catalyst loadings were 0.5 mg cm-2 Pt/C for the hydrogen side and 1.5 mg cm-2 IrO2 for the oxygen side. Electrode layers (with 25 cm2 active area) were bonded by placing the catalyst coated membrane between carbon paper for oxygen side and carbon cloth for hydrogen side and then by hot pressing at 130 °C and 10 MPa for 3 min. The Nafion content of the electrocatalytic layers was set to 30 wt% for the anode. Finally, the single cell tests were carried out with a PEM water electrolysis cell. MEAs were clamped between a porous carbon paper at anode and a carbon cloth as gas diffusion layer at the cathode side. The cell body was made of end plates (stainless steel), graphite bipolar plates with serpentine flow fields (suitable for electrolysis) and the MEA between them. The cell was tightened between two stainless steel plates, using a dynamometric wrench to set the fastening screws to 40 cNm.
3. Results
3.1. Structural Characterization of the Catalysts
XRD analysis was used to investigate the evolution in the crystalline structure of the unsupported catalysts with increasing calcination temperature. The XRD analysis was performed on the calcined catalysts (referred to as “IrO2-x °C”, where x is the sintering temperature) and on the commercial catalyst (“IrO2-c”). The corresponding diffraction patterns are shown in Figure 1. In the commercial and the IrO2-400°C catalyst, two broad peaks (2θ=35 and 59°) are clearly visible in both XRD patterns. The XRD results for these catalysts are characteristic of materials that are amorphous and/or have small-sized crystallites, while in case of IrO2-c some additional sharp peaks appear (2θ=40.64, 47.36 and 69°), which are assigned to the metallic Ir. The characteristic peaks of the rutile tetragonal IrO2 started to develop in the diffraction patterns of the catalysts calcined at 500°C and the peaks were fully developed in the diffraction patterns of catalyst calcined at 600°C. In addition, by increasing the calcination temperature to 500°C, the amorphicity of the electrocatalyst was observed to decrease. After raising the temperature to 600°C, these reflection peaks became thinner and sharper, indicating a higher degree of crystallinity for this material. The XRD pattern also confirmed the absence of other phases, such metallic iridium, which is present in the commercial IrO2 catalyst.
The Scherrer equation was used to roughly estimate the mean crystal size and the lattice parameter for each IrO2 electrocatalyst. The IrO2 reflection peak (101) at 2θ=36° was used to define the full width at half maximum (FWHM) intensity and to calculate crystallite size of IrO2.The mean crystallite size determined for IrO2-500 °C is 5 nm, while that for the material obtained at 600 °C is 7 nm. For the amorphous catalysts, the absence of defined diffraction peaks makes it impossible to use the analysis Debye-Scherrer, which meant that the mean crystallite sizes of the materials treated at temperatures at or below 400°C could not be determined. The lattice parameters for the IrO2 catalysts were determined to be: a=b=4.467 Å and c=3.110 Å for IrO2-500 °C and a= b=4.457 Å and c=3.140 Å for IrO2-600 °C material (Table 1), which are in accordance with the JCPDS database for tetragonal IrO2-rutile crystallographic pattern (JCPDS No. 15-871).
Transmission electron micrographs for the IrO2 catalysts are presented in Figure 2 following the synthesis of these catalysts and before the OER activity evaluation. The presence of aggregates composed of small particles, which are characteristic of unsupported catalysts, are observed in all images. The TEM micrograph of the commercial and IrO2-400°C catalysts reveal spherical particles with sizes in the 1-2 nm range, but the mean particle size could not be determined due to the agglomeration of these nanoparticles. For the catalysts obtained at IrO2-500°C and IrO2-600°C (Figure 2a–c) spherical morphologies are no longer evident. Furthermore, the particle sizes increase due to coalescence resulting as a consequence of the thermal treatment. Some boundary zones observed for the IrO2-600°C catalyst are evidence of the majority presence of particles with sizes in the 5-7 nm range, in agreement with the XRD results (Table 1).
Figure 3a displays the XPS spectra of the Ir4f orbital of all electrocatalysts. Each Ir4f spectrum can be deconvoluted into two sets of doublets, which can be assigned to iridium in different oxidation states. The less intense doublet (the red curve), with 7/2 and 5/2 spin-orbit components located at 61.9 and 65.1 eV, respectively, corresponds to Ir4+ (IrO2), while the main doublet, with main components located at 63.8 and 66.9 eV is attributed to IrOx (x>2). These energy values are consistent with other results reported in the literature [19,20] and confirm the absence of metallic iridium (Ir0), in agreement with in the XRD data. The % percentages of Ir4+ are 41, 55 and 52% for IrO2-400 °C, IrO2-500 °C and IrO2-600 °C catalysts, respectively. It should be noted that, since XPS is a surface-sensitive technique, the atomic percentages of the Ir3+ and Ir4+ components are more accurate for IrO2-400oC, because of their smaller particle sizes. For materials with larger particle sizes, the bulk compositions may be somewhat different from those indicated by XPS, because the XPS results do not provide the bulk of these materials. Nevertheless, as electrochemical reactions take place on the surface, it is particularly important to consider the surface state of the catalysts in relation to their electrochemical behavior.
High-resolution oxygen 1s-orbital spectra were also acquired and are displayed in Figure 3b. Three species are identified at ~530.4 eV, 532.2 eV and 534.2 eV bond energies, attributed to Ir-O and Ir-OH bonds as well as weakly adsorbed molecules of water on their surface, respectively. The latter species do not participate in the stoichiometry of the catalyst and are related to surface adsorbates due to exposure to the atmosphere. Furthermore, the spectra reveals that the O1s orbital shifts to lower energy with increasing calcination temperature. This shift reflects an enhancement in the oxygen content of the oxide form compared to the oxy-hydroxide [21,22].
The oxygen to iridium atomic ratios were calculated using the signal intensity from O1s and Ir4f spectra, normalized for the different element sensitivity factor. It was found that the O:Ir ratio is between 2.41 to 2.82 for all three samples, which is slightly higher than the theoretical value of 2 for IrO2, revealing again the co-existence of iridium oxides at higher oxidation states. Furthermore, the O:Ir ratio did not exhibit a specific trend with sintering temperature, indicating that thermal treatment did not result in an oxygen loss on the surface due to the conversion of Ir4+ into Ir3+.
The BET surface areas of the calcined catalysts were determined using nitrogen physisorption isotherms and the BET equation [19]. The measured BET surface area is characterized by high values and is reduced with calcination temperature. The BET surface area was 185 m2 g-1 for IrO2-400°C, 127 m2 g-1 for IrO2-500°C and reduced to 50% of its initial value (66 m2 g-1) when the catalyst was calcined at 600°C. Despite calcination temperature, these values are higher than the reported values in the literature [14,20] (32 m2 g-1) for commercial IrO2. It is observed that the modified Adams methods results in the formation of nanosized particles with high surface area. In TEM images, it was shown that the iridium based species grew with calcination temperature. Therefore, the reduction in the BET surface area was sharper when the calcination temperature was increased from 400 to 600°C. This is in agreement with TEM images, where the increase in size of iridium based species had been more evident when the calcination temperature was increased from 400 to 600°C.
3.2. Surface Electrochemistry
Cyclic voltammetry was performed on the calcined catalysts and the commercial IrO2. During the potential scan, the oxidation states of iridium may change, and thus well-defined current peaks can usually be identified on the studied electrodes. Figure 4 shows the last cycle of a series of cyclic voltammograms (CVs) for IrO2-400°C, IrO2-500°C, IrO2-600°C and IrO2-c catalysts recorded at 20mV s-1 scan rate in 0.5 M aqueous H2SO4 at room temperature. One well defined oxidation reduction peak was observed at about 0.65 V vs Ag/AgCl (more pronounced for the IrO2-400°C). This redox peak corresponds to the redox reaction of iridium where the oxidation state changes between Ir3+ and Ir4+. Additionally, one reduction peak was observed at 1.1 V vs Ag/AgCl which is attributed to the change in the Ir4+ to Ir5+ transition. The oxidation peaks for IrO2-500°C, IrO2-600°C and IrO2-c catalysts corresponding to the above oxidation state transitions were not clearly observed. This was most likely due to an overlap with the OER. The OER starts around 1.23 V vs Ag/AgCl where the oxidation peak was supposed to be seen. The recorded current for the CVs decreased with calcination temperature, and it was accompanied by less pronounced redox peaks and smaller surface area under the voltammograms.
The surface area under the voltammograms divided by the scan rate corresponds to the voltametric charge. The oxidation voltametric charges were obtained from CVs over the whole anodic potential window. The charge transferred during the iridium redox reaction is proportional to the number of iridium active sites. It is frequently used as an estimation of electrochemically active surface area (ECSA). The ECSA reduced with the increase of calcination temperature (Table 2). This was due to the growth of iridium particles with calcination temperature. This result is comparable with the reported BET surface area results, where a similar trend was observed. According to the TEM images, 400-600°C is the temperature range where a major increase in the size of iridium species takes place [23].
3.3. Activity for the Oxygen Evolution Reaction (OER)
The catalysts’ activities towards the oxygen evolution reaction were evaluated through the polarization curves (LSV measurements) as shown in Figure 5. The currents in the curves are presented in the form of mass activity (mA/mg Ir). As depicted from this figure, the catalysts‘ performance decreased as the calcination temperature increased. The performance of the commercial IrO2 is also measured and added to the figure for comparison. The mass activities at 1.5 V vs Ag/AgCl are summarized on the inset table (Figure 5).
It is evident that the performance of the calcined catalyst (especially IrO2-400°C) synthesized by Adams method is superior to the commercial IrO2. The amorphous IrO2-400°C catalyst is the most active, and its corresponding starting potential of oxygen evolution is the lowest. With the increase of calcination temperature, the starting potential of oxygen evolution increases, since the IrO2-400°C is more electrocatalytically active (more active sites) than IrO2-500°C and IrO2-600°C in OER, resulting in lower current densities at any potential. This decrease to the current (in the performance) with the increase of calcination temperature was due to the reduction in the available ECSA, as previously discussed. This is comparable with the result discussed in surface electrochemistry section, regarding cyclic voltammograms. Moreover, it could be concluded that the amorphous iridium based species have higher activity toward OER compared to crystalline IrO2 [16,19].
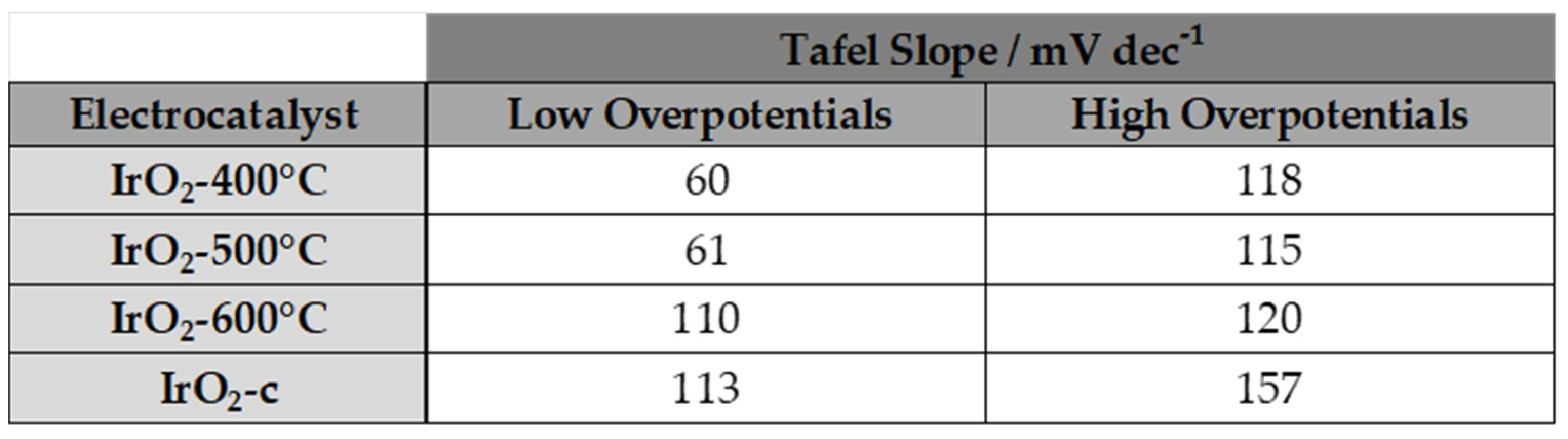
To further investigate the OER activity of the catalysts, the OER Tafel slopes for the prepared IrO2 samples were evaluated in 0.5 M H2SO4. For this purpose, the corresponding Tafel plots are shown in Figure 6. in the voltage range of 1.2-1.45 V vs Ag/AgCl and the calculated Tafel slope for each sample is listed in Table 3. Tafel slope is an important kinetic parameter to reveal changes in the apparent OER mechanism.
Two Tafel linear regions can be distinguished in these plots, the first one corresponds to slopes close to 60 mV dec-1 in the low current density regions, and the second with slopes close to 115-118 mV dec-1 in the higher current density regions for ΙrO2 400oC, ΙrO2 500oC. Tafel slopes of 40 and 120 mV dec-1 have been reported for sputtered iridium oxide films and thermally prepared iridium oxides [16]. The resulting Tafel slopes at low overpotentials deviated slightly from 55 mV dec-1 obtained for anodically oxidized Ir nanoparticles and are in good accordance with 61 mV dec-1 obtained for thermally prepared IrO2 [24,25]. However, at higher current densities, a Tafel slope of 120 mV dec-1 indicates that the rate determining step moves to the oxidative adsorption of water [5]. Finally, Tafel slopes for ΙrO2 600oC, ΙrO2-c are observed to be higher both of low and high overpotentials and are attributed to the formation of oxides on the surface which is in agreement with the results of XPS analysis.
A single cell PEM water electrolysis cell was used to assess the performance of the synthesized IrO2 oxides under typical water electrolysis conditions. Each experiment was repeated three times and then characterized using polarization curves. All parameters (ionomer content, cathode, membrane) were kept constant except the calcination temperature of IrO2 catalyst. Electrolysis performances were measured at a constant temperature of 50°C, at atmospheric pressure (1 bar). Liquid water was fed at the anode with a mass flow rate of 300 g h-1, while He saturated with vapor (58 kPa H2O) was supplied at the cathode. The corresponding steady-state polarization curves for all catalyst formulations are shown in Figure 7. The cell with IrO2-400°C as anodic electrocatalyst exhibited the highest performance, among all samples, achieving 177 mA cm-2 at 1.8 V. It should be noted that the reported values are not corrected for the ohmic losses. The superior performance of the MEA with the IrO2 calcined at 400oC is in agreement with the findings of the intrinsic OER activity measurements, performed in RDE configuration.
In order to evaluate the stability of the MEAs, short-term testing was carried out for 8 h continuous operation under constant applied potential (1.8 V) and the results are summarized in Figure 8.
The current density for all MEAs decreased quickly during the first minutes (~50 min) of potential application and then reached a steady-state values during the remaining time of testing. The best-performing MEA, with IrO2-400°C as anode electrocatalyst, showed remarkable stability reaching 200 mA cm-2 after the initial stabilization period. These results are very promising regarding the stability of the synthesized electrocatalysts and MEAs.
4. Conclusions
IrO2 catalysts were synthesized by the modified Adams method. The effect of the calcination temperature, in the range between 400 and 600oC, on the morphological/surface characteristics and the electrochemical performance of the catalysts for the oxygen evolution reaction (OER) was studied.
The application of a modified Adams method resulted in the synthesis of unsupported nanostructured IrO2 electrocatalysts with small, nano-scale particle size (1.24-7.77 nm) and high BET surface area (185-66 m2g-1). The structures, activities, and stabilities of Adams’ fusion method prepared iridium oxide nanomaterials as catalysts for the OER were studied. Heat treatment at different temperatures provided IrO2 catalysts with varying degrees of crystallinity. The unsupported prepared materials exhibit outstanding activity for oxygen evolution reaction (OER), compared to commercial IrO2. To achieve both high OER activity and stability three key factors have been identified: i) high anodic charge, ii) high surface area due to nano-size IrOx particles well dispersed in the Nafion ionomer electrolyte and iii) homogeneous layer morphology. The best performing material was the IrO2 calcined at 400°C.
The performances of the unsupported catalysts were also studied in MEA configuration under practical water electrolysis conditions. In agreement with the OER activity measurements, the best performing MEA was the one employing the 400oC calcined IrO2 catalyst. In terms of stability, the tests of 4 MEAs at steady operation stability (1.8 V) after 8 h continuous operation perform excellent stability. Further studies are required to examine the long term (in the range of 1,000-10,000 h) performance of the catalyst in electrolyzer conditions.
Author Contributions
Investigation, A.B. and K.M.P.; writing—original draft preparation, A.B.; writing—review and editing, K.M.P., S.B. and D.T. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
Thanks are due to O. Orfanou, A. Mavridou and P. Papazoglou for performing the ΧRD, BET measurements and Dr. A. Delimitis for HRTEM analysis. We also thank Lamprini Syggelou for XPS analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Carmo, M.; Fritz, D. L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrog. Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Adams, R.; Shriner, R. L. Platinum oxide as a catalyst in the reduction of organic compounds. III. Preparation and properties of the oxide of platinum obtained by the fusion of chloroplatinic acid with sodium nitrate. J. Am. Chem. Soc. 1923, 45, 2171–2179. [Google Scholar] [CrossRef]
- Papazisi, K. M.; Siokou, A.; Balomenou, S.; Tsiplakides, D. Preparation and characterization of IrxPt1−xO2 anode electrocatalysts for the oxygen evolution reaction. Int. J. Hydrog. Energy 2012, 37, 16642–16648. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, C.; Lei, Y.; Liu, F.; Tian, B.; Wang, J. Investigation of high-performance IrO2 electrocatalysts prepared by Adams method. Int. J. Hydrog. Energy 2018, 43, 19460–19467. [Google Scholar] [CrossRef]
- Silva, G. C.; Perini, N.; Ticianelli, E. A. Effect of temperature on the activities and stabilities of hydrothermally prepared IrOx nanocatalyst layers for the oxygen evolution reaction. Appl. Catal. B-Environ. 2017, 218, 287–297. [Google Scholar] [CrossRef]
- Antolini, E. Iridium as Catalyst and Cocatalyst for Oxygen Evolution/Reduction in Acidic Polymer Electrolyte Membrane Electrolyzers and Fuel Cells. ACS Catalysis 2014, 4, 1426–1440. [Google Scholar] [CrossRef]
- Hu, W.; Chen, S.; Xia, Q. IrO2/Nb–TiO2 electrocatalyst for oxygen evolution reaction in acidic medium. Int. J. Hydrog. Energy 2014, 39, 6967–6976. [Google Scholar] [CrossRef]
- Siracusano, S.; Baglio, V.; Stassi, A.; Ornelas, R.; Antonucci, V.; Aricò, A. S. Investigation of IrO2 electrocatalysts prepared by a sulfite-couplex route for the O2 evolution reaction in solid polymer electrolyte water electrolyzers. Int. J. Hydrog. Energy 2011, 36, 7822–7831. [Google Scholar] [CrossRef]
- Slavcheva, E.; Radev, I.; Bliznakov, S.; Topalov, G.; Andreev, P.; Budevski, E. Sputtered iridium oxide films as electrocatalysts for water splitting via PEM electrolysis. Electrochim. Acta 2007, 52, 3889–3894. [Google Scholar] [CrossRef]
- Angelucci, C. A.; D’Villa Silva, M.; Nart, F. C. Preparation of platinum–ruthenium alloys supported on carbon by a sonochemical method. Electrochim. Acta 2007, 52, 7293–7299. [Google Scholar] [CrossRef]
- Li, G.; Yu, H.; Song, W.; Dou, M.; Li, Y.; Shao, Z.; Yi, B. A Hard-Template Method for the Preparation of IrO2, and Its Performance in a Solid-Polymer-Electrolyte Water Electrolyzer. ChemSusChem 2012, 5, 858–861. [Google Scholar] [CrossRef]
- Puthiyapura, V. K.; Pasupathi, S.; Su, H.; Liu, X.; Pollet, B.; Scott, K. Investigation of supported IrO2 as electrocatalyst for the oxygen evolution reaction in proton exchange membrane water electrolyser. Int. J. Hydrog. Energy 2014, 39, 1905–1913. [Google Scholar] [CrossRef]
- Li, G.; Yu, H.; Yang, D.; Chi, J.; Wang, X.; Sun, S.; Shao, Z.; Yi, B. Iridium-Tin oxide solid-solution nanocatalysts with enhanced activity and stability for oxygen evolution. J. Power Sources 2016, 325, 15–24. [Google Scholar] [CrossRef]
- Cruz, J. C.; Baglio, V.; Siracusano, S.; Ornelas, R.; Ortiz-Frade, L.; Arriaga, L. G.; Antonucci, V.; Aricò, A.S. Nanosized IrO2 elec trocatalysts for oxygen evolution reaction in an SPE electrolyzer. J. Nanopart. Res. 2011, 13, 1639–166. [Google Scholar] [CrossRef]
- Ouattara, L.; Fierro, S.; Frey, O.; Koudelka, M.; Comninellis, C. Electrochemical comparison of IrO2 prepared by anodic oxidation of pure iridium and IrO2 prepared by thermal decomposition of H2IrCl6 precursor solution. J. Appl. Electrochem. 2009, 39, 1361–1367. [Google Scholar] [CrossRef]
- Reier, T.; Teschner, D.; Lunkenbein, T.; Bergmann, A.; Selve, S.; Kraehnert, R.; Schlogl, R.; Strasser, P. Electrocatalytic Oxygen Evolution on Iridium Oxide: Uncovering Catalyst-Substrate Interactions and Active Iridium Oxide Species. J. Electrochem. Soc. 2014, 161, F876–F882. [Google Scholar] [CrossRef]
- Cherevko, S.; Reier, T.; Zeradjanin, A. R.; Pawolek, Z.; Strasser, P.; Mayrhofer, K. J. J. Stability of nanostructured iridium oxide electrocatalysts during oxygen evolution reaction in acidic environment. Electrochem. Commun. 2014, 48, 81–85. [Google Scholar] [CrossRef]
- Ardizzone, S.; Fregonara, G.; Trasatti, S. ‘Inner’ and ‘outer’ active surface of RuO2 electrodes. Electrochim. Acta 1990, 35, 263–267. [Google Scholar] [CrossRef]
- Karimi, F.; Bazylak, A.; Peppleya, B. A. Effect of Calcination Temperature on the Morphological and Electrochemical Characteristics of Supported Iridium Hydroxyoxide Electrocatalysts for the PEM Electrolyzer Anode. J. Electrochem. Soc. 2017, 164, F464–474. [Google Scholar] [CrossRef]
- Karimi, F.; Peppley, B. A. Metal Carbide and Oxide Supports for Iridium-Based Oxygen Evolution Reaction Electrocatalysts for Polymer-Electrolyte-Membrane Water Electrolysis. Electrochim. Acta 2017, 246, 654–670. [Google Scholar] [CrossRef]
- Peuckert, M. XPS study on thermally and electrochemically prepared oxidic adlayers on iridium. Surface Science 1984, 144, 451–464. [Google Scholar] [CrossRef]
- Sanjinés, R.; Aruchamy, A.; Lévy, F. Thermal Stability of Sputtered Iridium Oxide Films. J. Electrochem. Soc. 1989, 136, 1740–1743. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, H.; Chen, G.; Zhang, Y. Study of IrxRu1−xO2 oxides as anodic electrocatalysts for solid polymer electrolyte water electrolysis. Electrochim. Acta 2009, 54, 6250–6256. [Google Scholar] [CrossRef]
- Reier, T.; Weidinger, I.; Hildebrandt, P.; Kraehnert, R.; Strasser, P. Electrocatalytic Oxygen Evolution Reaction on Iridium Oxide Model Film Catalysts: Influence of Oxide Type and Catalyst Substrate Interactions. ECS transactions 2013, 58, 39–51. [Google Scholar] [CrossRef]
- Fierro, S.; Kapałka, A.; Comninellis, C. Electrochemical comparison between IrO2 prepared by thermal treatment of iridium metal and IrO2 prepared by thermal decomposition of H2IrCl6 solution. Electrochem. Commun. 2010, 12, 172–174. [Google Scholar] [CrossRef]
Figure 1.
XRD patterns of IrO2-400°C, IrO2-500°C, IrO2-600°C and IrO2-c powder electrocatalysts.

Figure 2.
HRTEM images of IrO2 powders prepared under different calcination temperatures a. 400°C, b. 500°C and c. 600°C.
Figure 2.
HRTEM images of IrO2 powders prepared under different calcination temperatures a. 400°C, b. 500°C and c. 600°C.

Figure 3.
High resolution XPS spectra corresponding to (a) Ir4f and (b) O1s orbital of the IrOx electrocatalysts.
Figure 3.
High resolution XPS spectra corresponding to (a) Ir4f and (b) O1s orbital of the IrOx electrocatalysts.
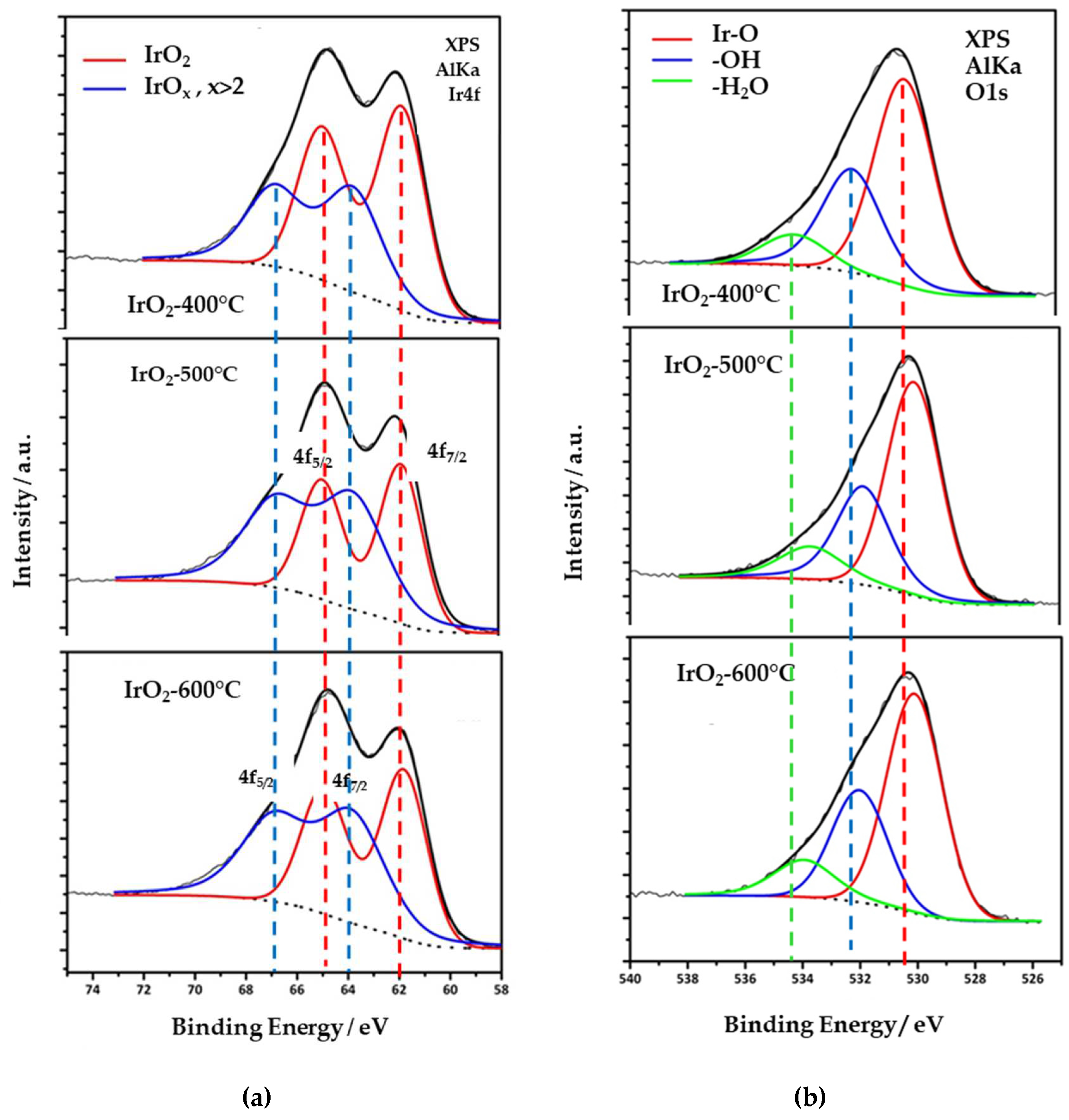
Figure 4.
Cyclic voltammograms of IrO2-400°C, IrO2-500°C, IrO2-600°C and IrO2-c electrocatalysts at a scan rate of 20 mV s-1 in 0.5 M H2SO4 at 25 °C.
Figure 4.
Cyclic voltammograms of IrO2-400°C, IrO2-500°C, IrO2-600°C and IrO2-c electrocatalysts at a scan rate of 20 mV s-1 in 0.5 M H2SO4 at 25 °C.
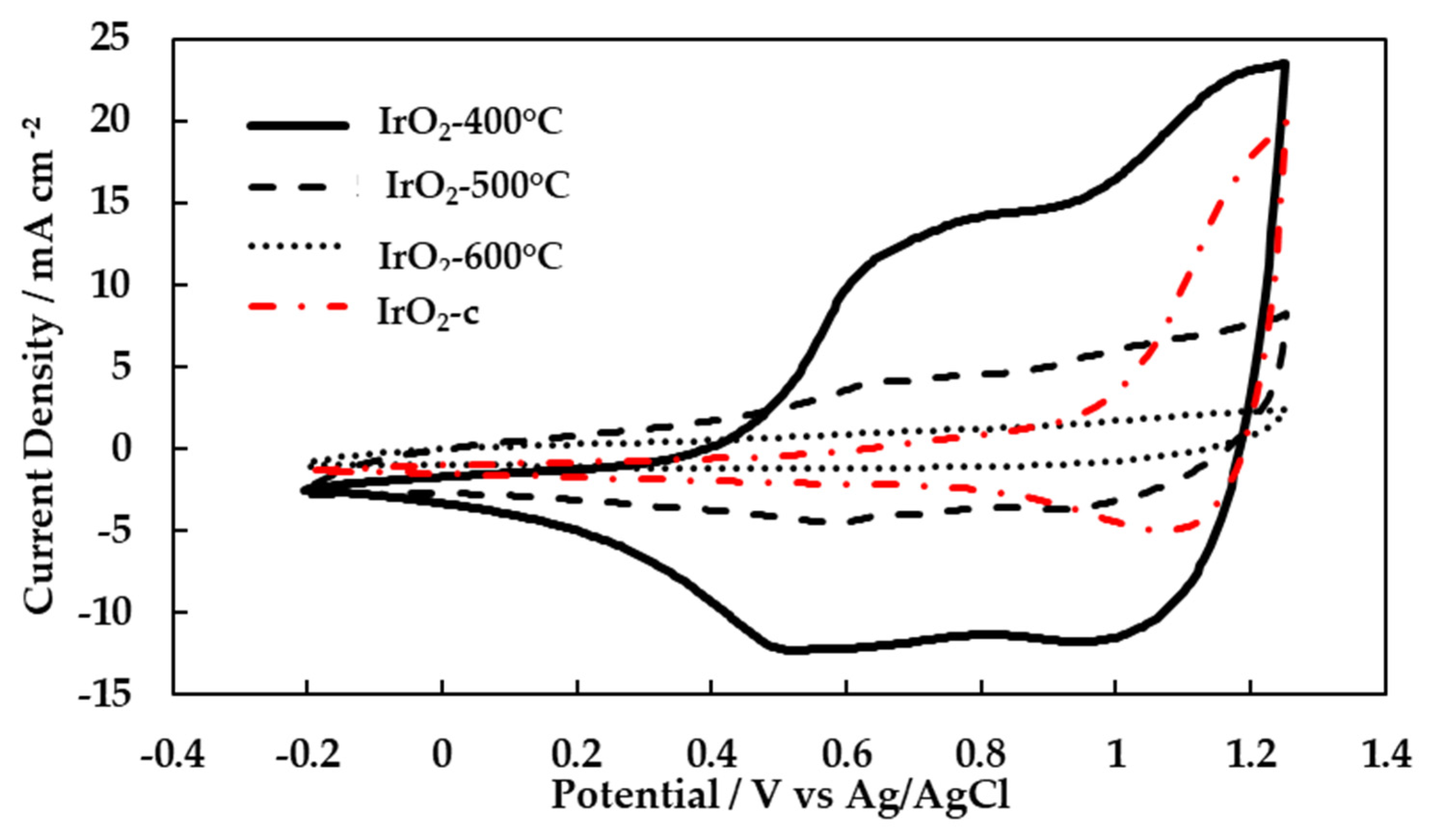
Figure 5.
OER polarization curves at 1 mV s-1 of the IrOx electrocatalysts in the potential range 1.1 to 1.25 V in 0.5 M H2SO4. The currents have been normalized to express mass activity.
Figure 5.
OER polarization curves at 1 mV s-1 of the IrOx electrocatalysts in the potential range 1.1 to 1.25 V in 0.5 M H2SO4. The currents have been normalized to express mass activity.
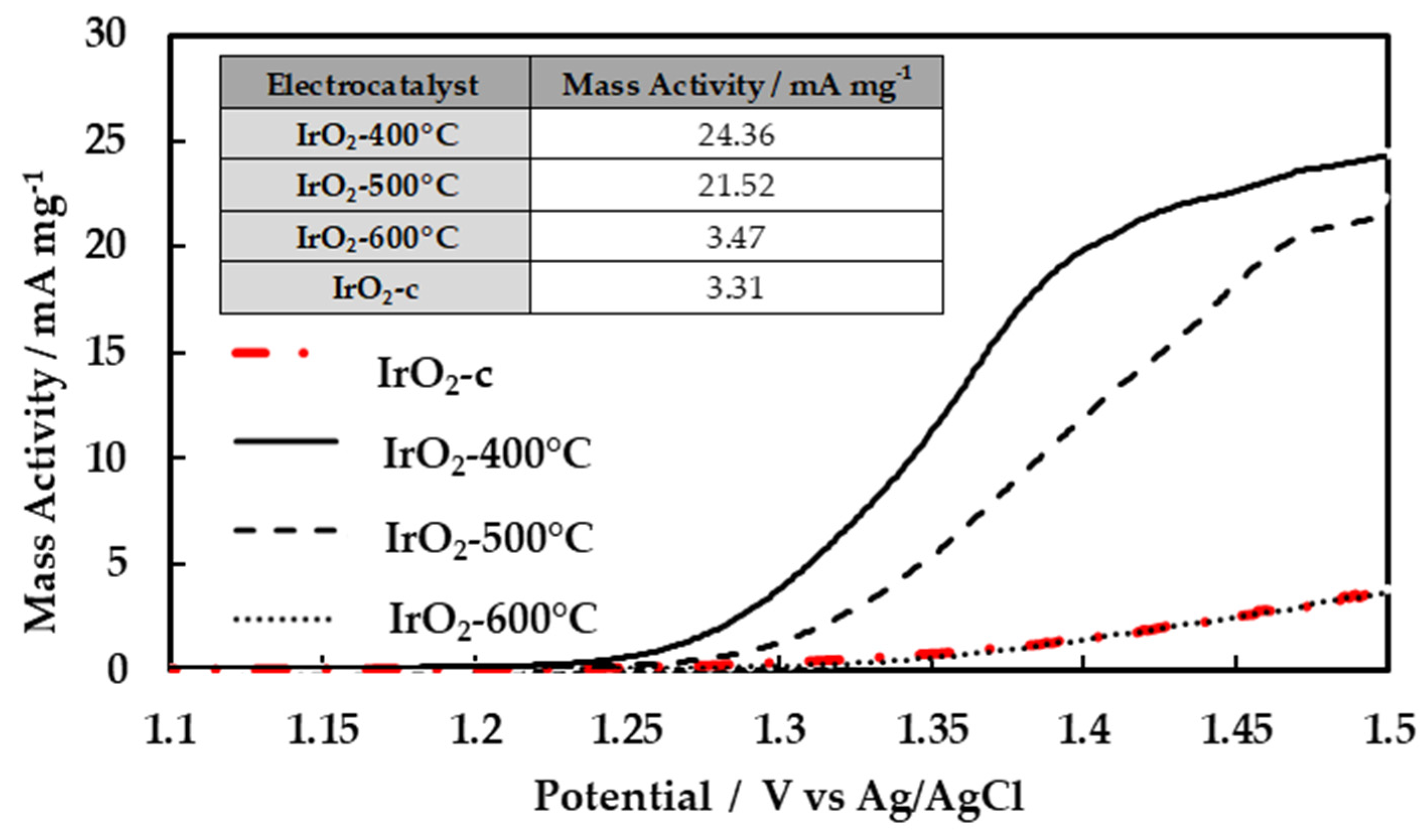
Figure 6.
Tafel plots of ΙrO2 400oC, ΙrO2 500oC, ΙrO2 600oC and ΙrO2 -c electrocatalysts.
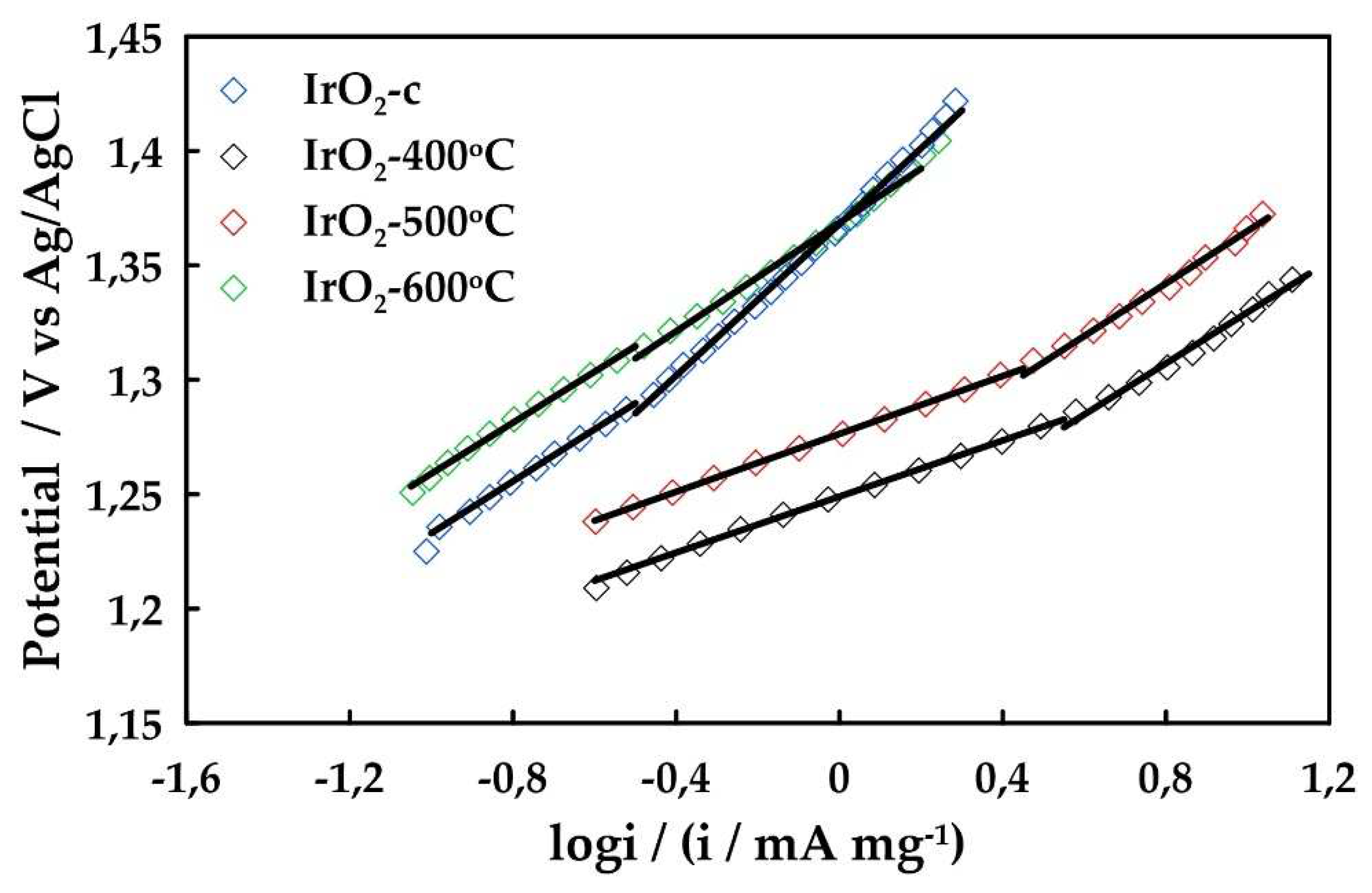
Figure 7.
Performance curves (potential vs. current density) for the MEAs employing the four different IrO2 catalysts as anode electrode. Inset table: Current densities at 1.8V.
Figure 7.
Performance curves (potential vs. current density) for the MEAs employing the four different IrO2 catalysts as anode electrode. Inset table: Current densities at 1.8V.
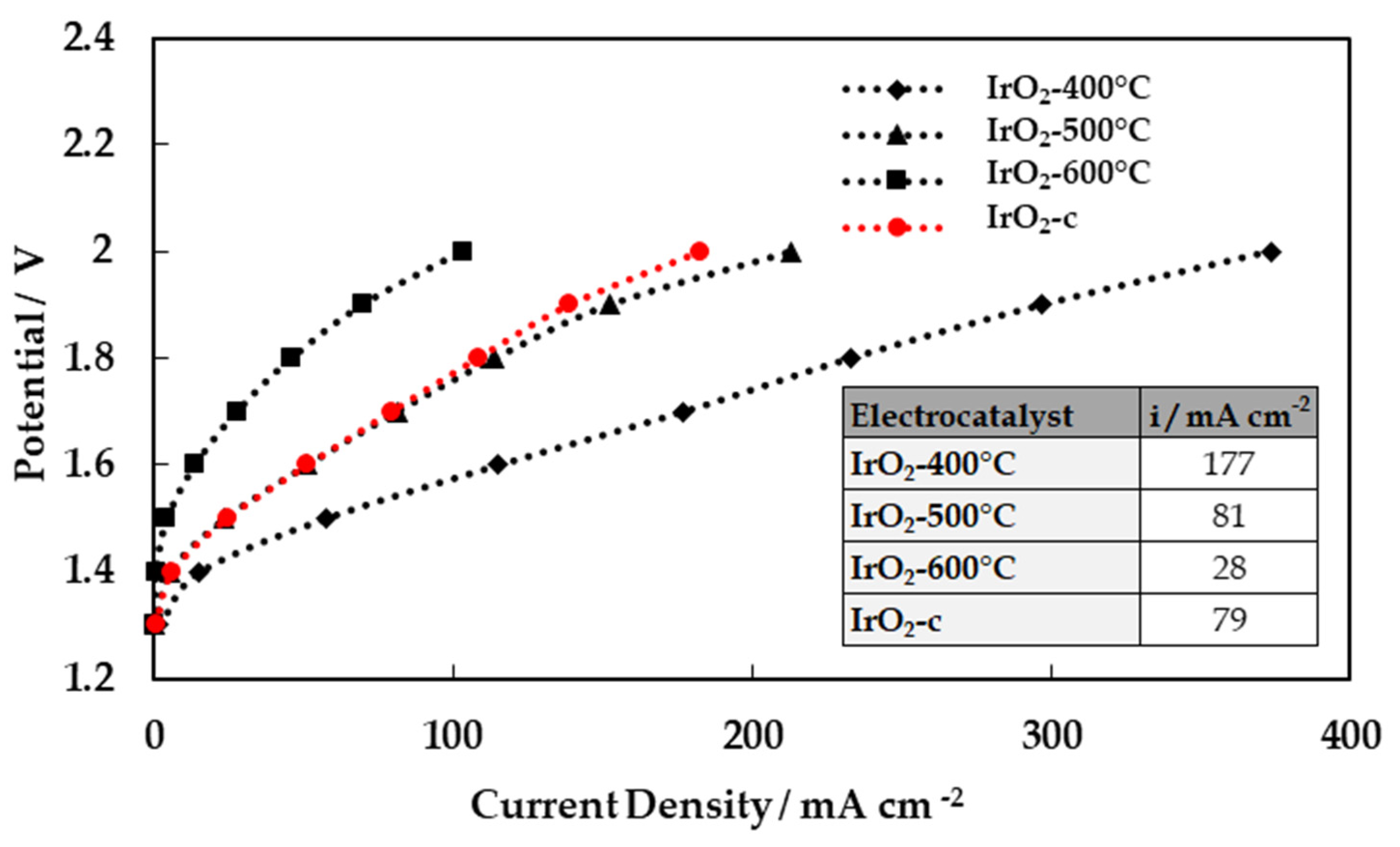
Figure 8.
Voltage of single cell test in 8 h with IrO2-400°C, IrO2-500°C, IrO2-600°C, IrO2-c as anodic electrocatalysts at 1.8 V.
Figure 8.
Voltage of single cell test in 8 h with IrO2-400°C, IrO2-500°C, IrO2-600°C, IrO2-c as anodic electrocatalysts at 1.8 V.
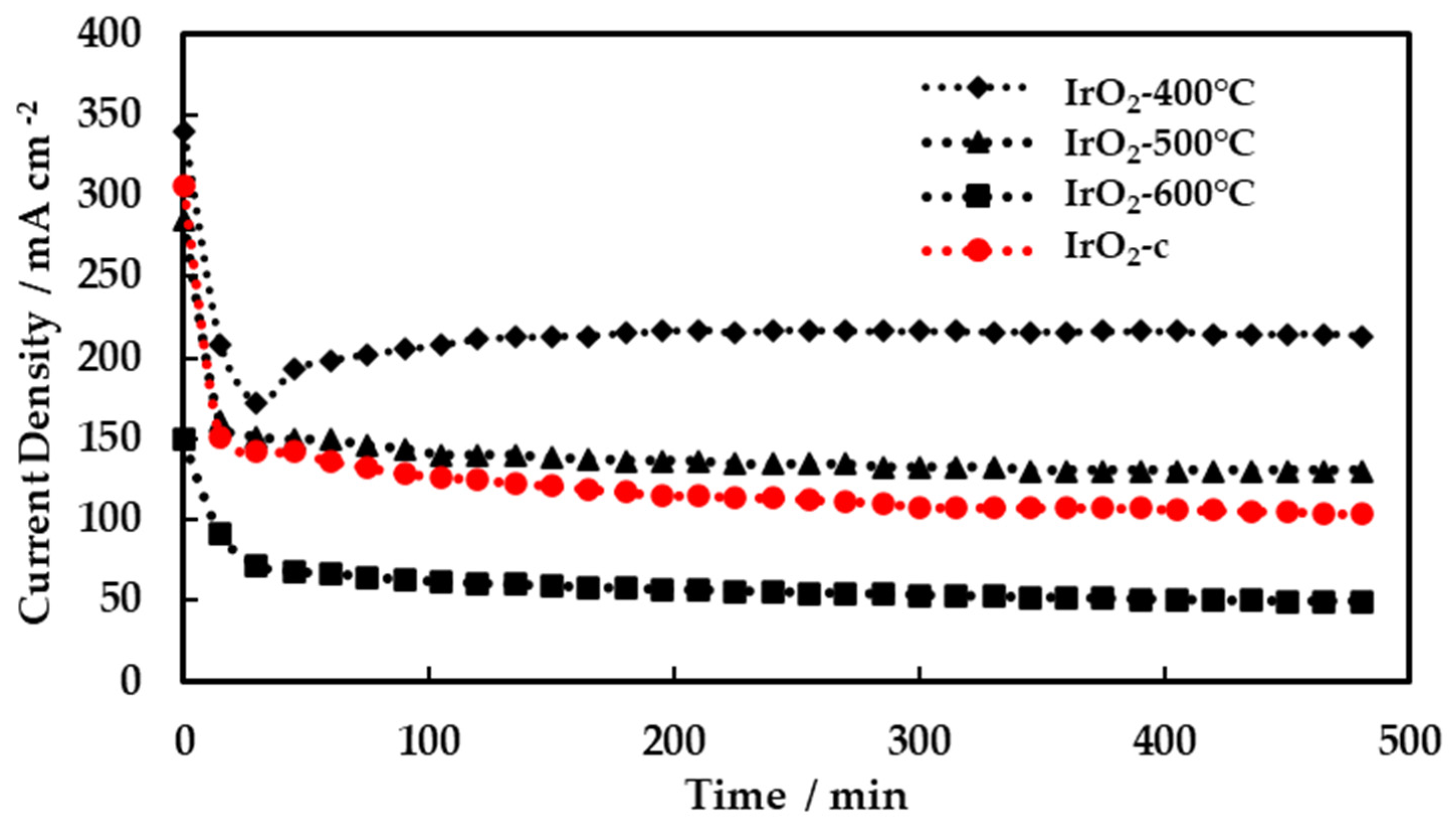

Table 1.
Results of the XRD analysis.
| Electrocatalyst | Lattice Parameters | Crystallite Size | Unit Cell Volume | |
|---|---|---|---|---|
| nm | Å3 | |||
| IrO2-c | - | - | 1.24 | - |
| IrO2-400°C | - | - | 1.40 | - |
| IrO2-500°C | a=b=4.467 | c=3.110 | 5.18 | 62.0 |
| IrO2-600°C | a=b=4.457 | c=3.140 | 7.77 | 62.4 |
Table 2.
BET surface, pore volume and total ESCA obtained for IrO2 electrocatalysts calcined at different temperatures.
Table 2.
BET surface, pore volume and total ESCA obtained for IrO2 electrocatalysts calcined at different temperatures.
| Electrocatalyst | Surface Area | Pore Volume | ESCA |
|---|---|---|---|
| m2 g-1 | cm3 g-1 | m2 g-1 | |
| IrO2-c | 32 | 0.130 | 11 |
| IrO2-400°C | 185 | 0.200 | 198 |
| IrO2-500°C | 127 | 0.167 | 27 |
| IrO2-600°C | 66 | 0.145 | 21 |
Table 3.
Tafel slops of ΙrO2 400oC, ΙrO2 500oC, ΙrO2 600oC and ΙrO2 -c electrocatalysts.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



