
Submitted:
11 July 2023
Posted:
12 July 2023
You are already at the latest version
Abstract
Some p53 mutants lose their tumor suppressor activity and acquire new oncogenic functions, known as a gain of function. Recent studies have shown that p53 mutants can exert oncogenic effects through specific miRNAs. We identified the differentially expressed miRNA profiles of the three most frequent p53 mutants (p53R273C, p53R248Q, and p53R175H) in the Saos-2 cell line (null p53) transfected as compared with p53WT transfected cells. The association between these miRNAs and the signaling pathways in which they might participate was performed through the miRPath Software. QRT-PCR was employed to validate the miRNAs profiles. In addition, we explored if the induction of cell migration and invasion by the p53R48Q mutant was dependent on the miR-182-5p. We observed that p53 mutants have an overall negative effect on miRNA expression. Likewise, we found miRNAs differentially expressed associated with regulating Oncogenic signaling pathways. We found overexpression of miR-182-5p, which is associated with processes such as cell migration and invasion. Inhibition of mutant p53R248Q and miR-182-5p increased FOXF2-MTSS1 levels and decreased cell migration and invasion. Our results suggest that p53 mutants increase the expression of miR-182-5p, and this miRNA is also necessary to induce cell migration and invasion by the p53R248Q mutant in a cancer cell model.
Keywords:
miRNAs
; Mutant p53
; Gain of function
; Cancer
1. Introduction
p53WT is a tumor suppressor protein encoded by the TP53 gene located on chromosome 17p13.1. In response to cellular stress, p53 activates the expression of several genes associated with cell cycle arrest, apoptosis, and DNA repair; however, p53 is mutated in more than 50% of human cancer [1] TP53 missense mutations occur mainly in the DNA-binding domain and can be classified into two main categories. These two categories of mutations are commonly referred to as conformational or DNA contact mutations, or class I and II, respectively [2]. Class I involves the substitution of an amino acid residue that causes loss of contact with DNA affecting its transcriptional capacity; within this category are mutations at positions R248Q and R273C. Mutated class II proteins present structural changes (R175H) that arise from their affinity for DNA. Class I mutants of p53 usually have a native conformation, whereas class II are unable to acquire the native conformation and therefore misfold [3]. The effects of TP53 mutations can be classified into three. First, p53 mutations attenuate binding to its DNA response elements and block transcriptional activation of p53 target genes; so a partial or total loss function can define these mutations; Second, p53 mutant proteins exert a dominant-negative effect on the function of wild-type (WT) p53 protein, encoded by the second allele, through the formation of a heterotetramer deficient in its binding to specific DNA sequences, also known as dominant-negative mutations. Finally, some p53 mutants also acquire new functions independent of p53WT; this event is known as a gain of function (GOF) GOF p53 mutations are involved in critical oncogenic processes, such as increased cell migration and invasiveness [4,5]. The effects of TP53 mutations on function and cellular behavior depend on the cell type and environmental conditions [6]. Thus, mutant p53 proteins are able to interact with specific intracellular proteins and induce gene expression changes [7,8]. Moreover, GOF activities of p53 missense mutations vary depending on the mutation type, giving rise to phenotypic differences in vivo associated with developing different cancer types [9]. In summary, understanding how p53 mutants induce phenotypic differences may help cancer prevention and therapy strategies.
In the present study, we explored the possibility that p53 mutant proteins exert their gain of function activity by modulating the expression of miRNAs. The miRNAs are 20-24 nucleotides in length and are involved in the post-transcriptional control of genes [10]. Recently, it has been reported that some miRNAs also play an important role in the gain of function of mutant p53; it has been demonstrated how p53 mutants regulate gene expression and exert oncogenic effects by unbalancing specific microRNAs (miRNAs) levels, even disrupting its biogenesis which provokes epithelial-mesenchymal transition, chemoresistance, and cell survival, among others [11,12]. However, the details of how and which miRNAs are regulated by p53 mutants promoting tumorigenesis are not yet fully understood.
In this study, we observed that p53R273C, p53R248Q, and p53R175H mutants have an overall negative effect on the expression of miRNAs in cancer. Moreover, in Saos-2 cells transfected with the p53R175H mutant, we found 93 decreased and 10 increased miRNAs. On the other hand, in the p53R273C profile, we observed a decrease of 72 miRNAs and an increase of 35. Finally, in the expression profile regulated by the p53R248Q mutant, we found a decrease of 167 miRNAs and an increase of 6, compared to the control (p53WT). Furthermore, within the miRNAs over-expressed in the presence of p53 mutants, we found the miR-182-5p, which is associated with cell invasion and migration, and its expression was significantly higher in p53R248Q. Interestingly, we demonstrate that the p53R248Q mutant induces over-expression of miR-182-5p and that this miRNA is required for the p53R248Q mutant to induce cell invasion and migration capacity in Saos-2 and OVCAR-3 cell lines.
2. Materials and Methods
2.1. Cell Culture and Treatments.
Saos-2/Osteosarcoma (null-p53), SKBR3/Breast carcinoma (p53R175H), C33a/Cervical cancer (p53R273C), and OVCAR-3/Ovarian cancer (p53R248Q) cell lines were cultured in DMEM medium (Gibco BRL) supplemented with 10% Fetal Bovine Serum (SFB). The non-tumorigenic breast epithelial cell line MCF10a (p53WT) was cultured in DMEM-F12 medium supplemented with 10% SFB, 10 μg/ml insulin, 0.5 μg/ml hydrocortisone, and 20 ng/ml recombinant human epidermal growth factor (EGF). All cells were obtained from ATCC (American Type Cell Culture Collection) and were maintained at 37°C and 5% CO2.
For treatments with pifithrin-α hydrobromide (CAS 63208-82-2, Santa Cruz, cat # sc-126 HRP), OVCAR-3 cells were treated with or without the drug (30, 50, 75, and 100 μM) for 24 h. The vehicle was Dimethyl sulfoxide (DMSO). Subsequently, the cells were collected and used in the corresponding experiments.
2.2. Plasmids and transfections.
Saos-2 cells at 70% confluency were transfected with p53R273C [13], p53R175H [14], p53R248Q [13], p53WT [15] or empty vector. Transfections corresponding to p53 (R273C, R175H, or R248Q) and transient (p53WT) were performed using Lipofectamine 3000 Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Clone selection for transfections was performed using 800 µg/ml G418 (Sigma-Aldrich). The activity of miR-182-5p was inhibited using the Anti-miR™ miRNA Inhibitor (MH12369) Ambion®; for this purpose, Saos-2 cells were transfected with 10 µM negative control #1 (AM17010), and Saos-2 (p53R248Q) with 10 µM of the negative control and/or anti-miR, this was done using Lipofectamine 3000 following the manufacturing instructions (Invitrogen). Samples were collected within 24 hours for further experiments.
2.3. RNA extraction.
Total RNA was extracted with the Trizol method (Invitrogen, USA) according to the protocol provided by the manufacturer. RNA concentration was measured using NanoDrop ND-2000 (Thermo Scientific), and RNA quality was evaluated by Tape Station 2200 bioanalyzer (Agilent Technologies), with minimum quality requirements: A260/280 ≥ 1.8; RNA integrity number- RIN≥ 7.
2.4. Reverse transcription
Using the Applied Biosystems miScript RT II kit, cDNA was synthesized following the manufacturer’s recommendations. The RT reaction was performed on the GeneAmp System 7500 thermal cycler (Applied Biosystems).
Human miRNome PCR Array
Expression profiling was performed with miScript miRNA PCRNA Arrays Human miRNome (384-well plate) (MIHS-3216Z) QIAGEN, which is based on 1066 miRNAs reported in miRbase Release 16 (www.miRBase.org), plus controls. SYBR Green qPCR was performed as follows: 2.5 µL of the cDNA (p53WT, p53R275H, p53R248Q, or p53R273C) were mixed with 5 µL of RNAse-free water plus 12.5 µL of 2X QuantiTect SYBR Green PCR Master Mix and 2.5 µL of 10X miScript Universal primer. The reaction was carried out by programming one step at 95°C for 15 min, followed by 40 cycles of three temperatures 94°C for 15 s, 55°C for 30 s, and 70°C for 30 s. The amplification reactions were completed using the QuantStudio 6 Flex Real-Time PCR System (Life Technologies).
2.6. PCR Array Analysis
The data were analyzed through the "Web-based miScript miRNA PCR Array data analysis tool" software, which allowed us to generate the list of differentially expressed miRNAs using the comparative CT (2-∆∆ct) method. The expression resulting from the cell line transfected with p53WT was considered as a calibrator, and the expression of the small nucleolar RNAs RNU6 and RNU48 was considered as a normalizer. Differentially expressed miRNAs were considered to be those with a rate of change value of ≥2 or ≤-2 concerning the control and presenting statistical confidence or p-value < 0.05.
2.7. Bioinformatic Analysis and Visualization
All differentially expressed miRNAs from the three comparison analyses performed (p53R273C vs. p53WT, p53R248Q vs. p53WT, and p53R175H vs. p53WT) were used. Using a heatmap with an unsupervised hierarchical clustering approach with the gplots 2.14.1 library [16] of the Bioconductor library [17] of the R 3.1.1 statistical software [18].
Differential expression visualization was performed using circos plots with the Circos tool [19]. To identify similarities of differentially expressed miRNAs between comparisons, the online tool Venny 2.1 was used [20].
Enrichment results of differentially expressed miRNAs were processed through the online software miRPath 3.0 [21]. The microT-CDS 5.0 algorithm was used to determine hypothetical target genes and DIANA-TarBase 7.0 for experimentally validated targets. To identify signaling pathways altered by miRNAs, information from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was used [22]. Pathways were considered altered when p <0.05 and visualized by dotplot performed in R 3.1.1 statistical software.
2.8. Validation of miRNA expression by RT-qPCR
Taqman probes specific for hsa-miR-509-5p (ID: 002235), hsa-miR-3151 (ID: 243597 MAT), has-miR-27b (ID 002174), has-miR-200c* (ID: 002286), has-miR-517a (ID: 001151), has-miR-101 (ID: 002253), miR-885-3p (ID 002372) and hsa-miR-182 (ID: 002334), as well as the internal control RNU6 (ID: 001093) were purchased from Ambion (Applied Biosystems, Foster City, CA, USA) (P/N: 4427975). First, miRNA RT was performed using stem-loop primers (Applied Biosystems). For this, 5 µL (100 ng/µL) of total RNA were added to the mixture containing: 0.15 µL of dNTPs with dTTP (100 mM); 3 µL of miRNA RT primers;1. 0 µL of MultiScribe reverse transcriptase enzyme (50 U/µL); 1.5 µL of 10X RT buffer; 0.19 µL of RNase inhibitor (20 U/ µL), and 4.16 µL of RNase-free water, totaling 15 µL per reaction. The RT reaction was performed on a GeneAmp System 9700 thermal cycler (Applied Biosystems), programming three temperatures: at 16°C for 30 min, 42°C for 30 min, and 85°C for 5 min, plus a final step at 4°C. The real-time PCR reaction required for each miRNA 1.33 µL of the RT reaction product, which was mixed with 10 µL of TaqMan master mix (Universal PCR Master Mix, No 4 AmpErase UNG, 2X), plus 7.67 µL of RNAse-free water and 1.0 µL of PCR probe (specific for each miRNA), giving a total of 20 µL. The analysis was performed on an Applied Biosystems QuantStudio 3 Real-Time PCR. The reaction was carried out by programming a step at 95°C for 10 min; followed by 45 cycles of two temperatures: 95°C for 15 s and 60°C for 1 min. The results were analyzed by the 2-∆∆ct method, as described above in the PCR Array Analysis section.
2.9. Expression analysis of miR-182-5p target genes via RT-qPCR.
"High Capacity cDNA Reverse Transcription Kit" (Thermo Fisher Scientific, UK) was used for the retrotranscription reaction according to the manufacturer’s instructions. FOXF2 and MTSS1 expression was determined by QRT-PCR using SYBR Green/ROX Master Mix (Thermo Fisher Scientific, UK) following the manufacturer’s instructions. Quantitative data were normalized relative to HPRT. The sequences of the primers were: FOXF2-For: CAA GGT AGC GTT CCC CAA TC; FOXF2-Rev: GTC TGC TTT TTT CAC ACC CTG AT; MTSS1-For: TGA CCC GCT CTG TTG; MTSS1-Rev: GGT GCC CAC TAC GGA AAC G and for HPRT-For AGG GTG TTT ATT CCT CAT GG; HPRT-Rev CAC AGA GGG CTA CAA TGT. The reactions were performed in QuantStudio 3 (Thermo Fisher Scientific, UK). The cycling conditions were: 50°C for 2 min to activate the UNG, initial denaturation at 95°C for 10 min and 45 cycles at 95°C for 15 s, and finally 60°C for 1 min. Standard curves were analyzed to verify the amplification efficiency of each gene. The 2-ΔΔCT equation was applied to calculate the relative expression of the samples.
2.10. Western blot
Cells were collected and subsequently lysed using RIPA buffer (Beyotime Biotechnology Co., Ltd., Shanghai, China). Protein concentration was then determined via the Lowry method, and 50 µg were loaded onto 10% SDS-PAGE gels. After blocking, we used the specific antibodies against p53 (DO-1, Santa Cruz, cat # sc-126 HRP) (1:500) and β-actin (AC-15, Sigma Aldrich, cat # A3854) (1:50,000). Protein expression was detected by chemoluminescence using Supersignal West Pico (Thermo Scientific, Waltham MA).
2.11. Cell migration and invasion assays
For all cell migration assays, the cell lines (2x105) were resuspended in 300 µL of serum-free MEM medium and seeded on the top surface of Transwell® inserts (8 µm; Corning, USA) in 24-well box, to which 500 µL of 10% SFB (Chemoattractant) DMEM medium was previously added. Cell invasion was assessed using QCM ECMatrix Cell Invasion Assay, 24-well (8 µm) (ECM554) Chemicon (Millipore, Billerica, MA, USA); cells were seeded in ECMatrix Cell Invasion Assay QCM chambers (1x105) in the absence of Fetal Bovine Serum; the inserts were placed in 24-well boxes to which 500 µL of 10% SFB (Chemoattractant) DMEM medium was previously added. Subsequently, cells were incubated at 37°C for 24 h for the cell migration and invasion assay, cells on top of the insert were gently removed, and cells that migrated to the bottom of the insert were fixed with 4% paraformaldehyde for 15 min, stained with 0.1 % crystal violet (Sigma-Aldrich, St. Louis, MO, USA). The absorbance of stained cells was read on an ELISA plate reader (SofMaxPro USA) at 560 nm.
2.12. Statistical Analysis
The statistics used were ANOVA to observe the differences between groups of different treatments and incubation times, and the unpaired Student’s t-test to compare the differences between two groups, using the statistical program GraphPad Prism software 5.0 (GraphPad Software, Inc., San Diego, CA, USA). Each experiment was performed in triplicate and repeated at least three times; differences were considered significant when p<0.05.
3. Results
3.1. Global expression profiling of miRNAs in Saos-2 cells expressing p53R175H, p53R273C, or p53R248Q, as well as Signaling Pathway Enrichment (KEGG) analysis.
In order to identify novel miRNAs regulated by the most frequent p53 mutants, we performed PCR arrays of the Saos-2 cell line transfected with p53WT or with each of the p53R175H, p53R273C, and p53R248Q mutants. The p53 levels were verified by western blot (Supplementary Figure 1). Cells transfected with p53WT were used as control, and each experimental group had three biological replicates. In addition, to determine which miRNAs were differentially expressed, cutoff points ≥2 or ≤-2 Fold Change (FC) and a value of p<0.05 were used (Figure 1, Figure 2 and Figure 3).
The complete lists showing those miRNAs differentially expressed in the presence of p53R175H, p53R273C, or p53R248Q are in Supplementary Tables 1–3. The global expression profile of the Human miRNome regulated by the p53R273C mutant was represented by a Circos plot, in which 72 under-expressed (green) and 35 over-expressed (red) miRNAs are visible (Figure 1a). On the other hand, Pathways in Cancer (95 miRNAs/322 genes) presented a higher percentage of messenger RNAs (Hypothetical) compared to other signaling pathways, including, Adherens junction (75 miRNAs/65 genes) (Figure 1b, Supplementary Table 4). We found that both signaling pathways share 75 miRNAs. Interestingly, the cancer pathway has 21 unique miRNAs, and Adherens junction shares almost all of them with this pathway except for miR-3186-5p. Concerning the analysis of experimentally validated target mRNAs, we found a significant enrichment of the pathways: Cancer Proteoglycans (44 miRNAs / 137 genes), Adherens junction (40 miRNAs / 56 genes), and Cell cycle (45 miRNAs / 86 genes) (Figure 1C, Supplementary Table 5). Interestingly, the most miRNAs regulating hypothetical and validated target genes belong to the Hippo pathway and Adherens junction, with 31 miRNAs in common.
Concerning to p53R175H miRNAs profile, our results showed the downregulation of 93 and upregulation of 10 miRNAs (Figure 2a, Supplementary Table 2). In this case, hypothetical target gene analysis indicates that the Hippo signaling pathway (88 miRNAs / 116 genes), the signaling pathway regulating stem cell pluripotency (90 miRNAs / 112 genes), and Adherens junction (71 miRNAs / 62 genes) are shared with the miRNA profile of the p53R273C mutant. However, Estrogen Signaling Pathway (85 miRNAs / 79 genes) and Cancer Proteoglycans (92 miRNAs / 167 genes) were found exclusively in the p53R175H profile (Figure 2b, Supplementary Table 6). Whereas pathway analysis on validated target genes, we found miRNAs involved in Adherens junction (32 miRNAs / 55 genes), FoxO Signaling Pathway (33 miRNAs / 83 genes), Proteoglycans in Cancer (33 miRNAs / 116 genes), Pathways in Cancer (37 miRNAs / 211 genes), Focal Adhesion (36 miRNAs / 113 genes) and Cell Cycle (34 miRNAs / 88 genes). Similar to the p53R273C miRNAs profile, our results revealed that "Pathways in Cancer" was one of the pathways with a higher number of miRNAs and genes (Figure 2c, Supplementary Table 7).
On the other hand, in the miRNAs expression profile regulated by the p53R248Q mutant, we found 167 decreased and 6 increased miRNAs versus p53WT (Figure 3a, Supplementary Table 3). Furthermore, the analysis of signaling pathways regulated by hypothetical target genes shows Hippo signaling pathway (140 miRNAs / 134 genes), Signaling pathway regulating stem cell pluripotency (145 miRNAs / 123 genes), Wnt signaling pathway (83 miRNAs / 108 genes), TGF-beta signaling pathway (115 miRNAs / 71 genes), Adherens junction (114 miRNAs / 68 genes) and ErbB signaling pathway (132 miRNAs / 83 genes) (Figure 3b, Supplementary Table 8). Notably, the p53R273C, p53R175H, and p53R248Q miRNAs profiles share the Hippo Signaling Pathway, the pathway regulating stem cell pluripotency and Adherens junction. To further investigate the mechanisms by which mutant p53R248Q contributes to cancer development, we performed an analysis of validated target genes using the miRPath algorithm, which identified multiple genes involved, especially in pathways such as Cell cycle (58 miRNAs / 85 genes), Adherens junction (55 miRNAs / 61 genes), Pathways in Cancer (62 miRNAs / 228 genes), p53 signaling pathway (50 miRNAs / 49 genes), MAPK signaling pathway (54 miRNAs / 84 genes) and Focal adhesion (63 miRNAs / 126 genes) (Figure 3c, Supplementary Table 9). Remarkably, the "Cell cycle" pathway is shared among the three expression profiles. Specifically, we found the miRNAs miR-200b-3p, miR-431-3p, miR-508-5p, miR-509-5p, miR-520c-3p, miR-888-3p, miR-3140-3p, miR-3913-5p, miR-101-3p, have been reported experimentally validated and involved in the cell cycle shared among p53R175H, p53R248Q, and p53R273C. Finally, it is important to note that the three mutant profiles also share miRNAs (miR-200b-3p, miR-3140-3p, miR-508-5p, miR-509-5p, miR-888-3p, miR-101-3p) in the regulation of the Adherens junction pathway in both hypothetical and validated target analyses. These findings give us insight into the functions in which p53 mutants might be involved through the regulation of miRNAs in cancer.
3.2. Heat map and Venn diagram of differentially expressed miRNAs in p53R273C, p53R248Q, and p53R175H mutants.
After analyzing the signaling pathways of each expression profile, we performed hierarchical clustering of miRNAs. The results in the Heat map show that the expression profile of p53WT has a similar pattern to p53R273C and p53R248Q mutants but is different from p53R175H (Figure 4a). This is also reflected in the Venn diagram, where the p53R273C mutant shares 25 miRNAs with p53R248Q and only 5 with p53R175H (Figure 4b). In addition, the p53R248Q mutant shares 42 miRNAs with the p53R175H mutant, and p53R248Q mutant exclusively regulates 73 miRNAs (Figure 4b). Interestingly, the p53R175H mutant shows fewer shared miRNAs, but its expression profile is similar to p53R248Q (Figure 4a). Moreover, it is worth mentioning that the three mutants have 33 miRNAs in common, and 72 others are shared only by 2 mutants in any of their combinations (Figure 4b).
3.3. Validation of differentially expressed miRNAs in the presence of the p53R273C, p53R248Q, and p53R175H mutants.
After determining which miRNAs were differentially expressed in the presence of the p53R273C, p53R248Q, and p53R175H mutants; we selected some of them to validate our results as follows: We obtained 245 differentially expressed miRNAs using a cutoff of ≥2 or ≤-2 (Fold Change) and a p<0.05; 72 miRNAs were shared by at least 2 mutants and only 33 by the three mutants; then we search in the literature an association between these miRNAs differentially expressed in at least 2 p53 mutants with cancer. With this strategy, we selected and validated the downregulation of 6 tumor suppressor miRNAs: miR-509-5p [23,24], miR-200c-5p[25,26], miR-3151-5p [27], miR-885-3p[28,29,30], miR-517a-3p [31,32,33], miR-101-3p [34,35,36,37], as well as 2 oncomiRs: miR-182-5p [38,39,40,41,42,43] and miR-27b-5p [44,45,46] (Figure 5).
In the expression profile of the p53R273C mutant, we observed decreased tumor suppressors miR-509-5p (~ -2.77 fold), miR-101-3p (~ -48.74 fold), miR-517a-3p (~ -91.74 fold), miR-3151-5p (~ -2.56 fold), and miR-200c-5p (~-5.38 fold), and an increased of the oncomiR miR-27-5p (~ 2.66 fold) (Figure 1a, Supplementary Table 1). To validate these results, we performed real-time qRT-PCR with TaqMan probes, and the same tendency was obtained in the miR-509-5p (~ -10.93 fold), miR-101-3p (~ -4.41 fold), miR-517a-3p (~ -4.22 fold), miR-3151-5p (~ -6.51 fold), miR-200c-5p (~ -6.98 fold), and miR-27-5p ~ (6.25 fold) (Figure 6). Notably, in validations with TaqMan probes, we also found decreased miR-885-3p (~ -6.35 fold) and an increase in the miR-182-5p (~ 3.46 fold), which were not differentially expressed in the miRNAs expression profile (Figure 1a and Figure 6).
On the other hand, the p53R175H mutant miRNA profiling analysis shows negative regulation of miR-509-5p (~ -54.42 fold), miR-101-3p (~ -7.27 fold), miR-517a-3p (~ -67.41 fold), miR-3151-5p (~ -47.37 fold), and miR-885-3p (~ -4.71 fold); but a positive regulation of miR-182-5p (~ 8.54 fold) (Figure 2a, Supplementary Table 2). These miRNAs were validated by real-time PCR with Taqman probes and the results were consistent with the miRNA profiling analysis (miR-509-5p / ~ -16.12 fold), (miR-101-3p /~ -3.42 fold; miR-517a-3p/~ -8.17 fold; miR-3151-5p /~ -9.82 fold); miR-885-3p /~ -3. 20 fold); miR-182-5p /~ 1.95 fold) (Figure 2a and Figure 6). Additionally, in real-time validation with Taqman probes, we also found a decrease in miR-200c-5p expression (~ -4.98 fold) and an increase in the miR-27-5p (~ 9.95 fold) (Figure 6).
Finally, in the p53R248Q mutant expression profile of miRNAs, we observed the downregulation of miRNAs tumor suppressors miR-509-5p (~ -11. 99 fold), (miR-101-3p (~ -73.98 fold), miR-517a-3p (~ -845.16 fold), miR-3151-5p (~ -10.27 fold), miR-885-3p (~ -43.98 fold), and miR -200c-5p (~ -3.57 fold); in contrast, the miR-182-5p was positively regulated (~ 6.87 fold) (Figure 3a, Supplementary Table 3). Validations by qRT-PCR showed the following results: miR-509-5p (~ -11.63 fold), miR-101-3p (~ -2.47 fold), (miR-517a-3p (~ -5.47 fold), miR-3151-5p (~ -16.15 fold), miR-885-3p (~ -8.22 fold), miR -200c-5p (~ -3.04 fold), and miR-27-5-p (~ 6.86 fold) (Figure 6). The qRT-PCR analysis with TaqMan probes also showed overexpression of miR-182-5p in the presence of all three mutants (Figure 6).
3.4. Mutant p53R248Q is associated with overexpression of miR-185-5p and downregulation of FOXF2 and MTSS1.
The miR-182-5p is an oncomiR that is overexpressed in several cancer types; it has been reported to inhibit the expression of targets such as FOXF2 (Forkhead box F2) and MTSS1 (Metastasis suppressor-1). To determine whether the endogenous presence of the three most frequent p53 mutants could be associated with increased miR-182-5p expression, we performed QRT-PCR assays on cell lines displaying these mutants (OVCAR-3/p53R248Q, C33a/p53R273C, and SKBR3/p53R175H). Our results showed that the three cell lines endogenously expressing the 3 mutants significantly overexpress the miR-182-5p compared to the cell line with MCF10a (p53WT) or Saos-2 (null p53) (Figure 7a). In contrast, the miR-185-5p target genes, FOXF2 and MTSS1, exhibited significantly low expression in the Saos-2-p53R248Q and OVCAR-3 cell lines compared with their respective controls: Saos2 and empty vector (Saos-2 transfected with empty plasmid) (Figure 7b); which is consistent with the overexpression of miR-182-5p in these cell lines.
3.5. The p53R248Q mutant stimulates invasion and migration by overexpressing miR-182-5p in Saos-2 cells.
To assess whether the induction of migration and invasion by the p53R248Q mutant depends on miR-182-5p expression, we transfected the Saos-2 cell line with control-anti-miR (scramble control), transfected with p53R248Q and control-anti-miR or transfected with p53R248Q and antimiR-182. First, we evaluated the inhibition of the miR-182-5p by the siRNAs (Figure 7c), where we observed the decrease of mir-182 in the presence of the anti-miR-182 in cells transfected with the p53R248Q mutant and then evaluated their effect on cell migration and invasion (Figure 7d). We found that inhibition of miR-182-5p by shRNAs significantly decreased the migration and invasion ability of the Saos-2 cell line transfected with the p53R248Q mutant compared with scramble-transfected control cells (Figure 7d). Furthermore, we observed by inhibiting miR-182-5p in the presence of the p53R248Q mutant, the expression of MTSS1 and FOXF2 is restored (Figure 7e), which is related to the decreased effect on migration and invasion mentioned above (Figure 7d). Our results suggest that the p53R248Q mutant depends on miR-182-5p to induce cell migration and invasion in the Saos-2 cell line.
3.6. Inhibition of the p53R248Q mutant in the OVCAR-3 cell line induces decreased cell migration and invasion through miR-182-5p.
To corroborate that the p53R248Q mutant induces overexpression of miR-182-5p, we inhibited the expression of this mutant in the OVCAR-3/p53R248Q cell line, using pifithrin, a p53 inhibitor. First, we performed a dose-response curve of pifithrin from 30, 50, 75, and 100 µM and observed the decrease of p53R248Q protein at 75 and 100 µM concentrations. Subsequently, we selected the concentration (100 µM) where we observed a more significant effect on p53 knockdown and analyzed the expression of miR-182-5p. Our results showed that miR-182-5p significantly decreased with pifithrin treatment coinciding with the decrease in expression of the p53R248Q mutant (Figure 8a). In addition, we also evaluated the miR-182-5p target genes (FOXF2 and MTSS1) and observed an increase in FOXF2 and MTSS1 expression in pifithrin-treated cells (Figure 8b). Finally, we performed cell migration and invasion assays. Our results showed that pifithrin treatment decreased the ability of OVCAR3 cells to invade or migrate compared with vehicle-treated control cells (Figure 8c). These results suggest that the p53R248Q mutant positively regulates miR-182-5p expression, which results in the downregulation of FOXF2 and MTSS1 targets, and it is associated with an increase in cell migration and invasion.
4. Discussion
In this study, we observed most of the miRNAs differentially expressed downregulated in the presence of p53R175H, p53R273C, and p53R248Q. Our results agree with a previous study in which a miRNA profile was performed in the presence of the p53R282W mutant; they also observed an overall negative effect on miRNA expression [47]. In addition, Garibaldi et al. demonstrated that mutants bind and sequester the p72/82 RNA helicase of the microprocessor complex, interfering with the association between Drosha and pri-miRNA, inhibiting post-transcriptional maturation, which contributes to the negative regulation of miRNAs [33].
The association between the differentially expressed miRNAs in the three miRNAs profiles and the signaling pathways in which they might participate (target mRNAs hipotetic/mRNAs validates) led us to conclude that they have the "Adherens junction" pathway in common. This coincides with reports that p53 mutants can promote mesenchymal phenotype, inducing transcription factors such as TWIST-1 and SLUG, which promotes the loss of adherens junction, favoring cell motility [48]. In addition, up-regulation of some miRNAs involved in the regulation of EMT, metastasis, cell migration, and invasion (miR-130b, let-7i, miR-218, and miR-519a) has been previously reported in the presence of p53 mutants [33,49,50]. In another study, the exogenous expression of p53R248Q and p53R282W mutants in H1299 cell line (null p53) induce miR-155 overexpression in breast cancer. Moreover, knockdown of the R249S endogenous p53 mutant in BT-549 cells resulted in a significantly reduced level of miR-155, confirming a role for mutant p53 in the aberrant activation of miR-155 [51] In agreement with these reports, we also observed overexpression of miR-155 in the presence of the p53R248Q mutant. However, we also observed different pathways altered depending on which p53 mutant was expressed; This is in agreement with studies of GOF activities of p53 mutants, for example, in mice harboring a novel germline Trp53R245W allele (contact mutation), and compared them to existing with Trp53R172H (structural mutation) and Trp53R270H (contact mutation) alleles it was observed that Trp53R245W/+ and Trp53R270H/+ mice develop more frequently osteosarcomas and had a poor overall survival in contrast with Trp53R172H/+ mice [9].
On the other hand, previous research showed that p53R248Q and p53R248W mutants induce invasion and migration binding Stat3 and enhance activating Stat3 phosphorylation in colorectal and pancreatic cancer [52,53]. Interestingly, a positive correlation has been reported between miR-182-5p and Stat3 pathway in gliomas and breast cancer [41,54]. Recently, it has also been demonstrated that miR-182-5p target de PIAS1 (protein inhibitor of activated STAT) mRNA in endometrial cancer, and the overexpression of PIAS1 inhibited the Stat3 activity [42]. These reports suggest that p53 mutants could induce the activation of Stat3 pathway through miR-182-5p in addition to binding the protein directly; however, more experiments are necessary to prove this hypothesis.
Other signaling pathways shared in the three profiles based on target hypothetic mRNAs were the Hippo signaling pathway and the pluripotent stem cell regulation pathway, which in turn share several miRNAs (miR-101-3p , miR-3125, miR-3681-5p, miR-508-5p, miR-517a-3p, miR-888-3p, miR-200b-3p, miR-2052, miR-3122, miR-3140-3p, miR- 3151-5p, miR-3168, miR-3618, miR-3660, miR-3908, miR-3913-5p, miR-4267, miR-4275, miR-4280, miR-4288, miR-4311, miR-4323, miR-509-5p and miR-520c-3p). The Hippo signaling pathway is a key regulator of physiological processes such as: cell proliferation, differentiation, polarity and death [55,56]. Previously, it has been reported that the p53R280K and p53R175H mutants can physically interact with a modulator of this pathway known as YAP (YES-associated protein) and form a complex with NF-Y, increasing the transcription of genes involved in cell proliferation [57]. In addition, YAP/TAZ overexpression has been reported to induce cell proliferation and the acquisition of cancer stem cell characteristics [58]. It could be possible that p53R248Q, p53R175H, and p53R273C regulate the expression of some miRNAs through the Hippo pathway or induce this pathway by regulating some of these miRNAs; it would be interesting to analyze this possibility.
Moreover, analysis through DIANA-TarBase (validated mRNAs) shows that the Cell cycle is a shared pathway between the three miRNAs expression profiles from the three p53 mutants, with the following miRNAs in common: miR-200b-3p, miR-431-3p, miR-508-5p, miR-509-5p, miR-520c-3p, miR-888-3p, miR-3140-3p, miR-3913-5p. It is well known that activation of p53WT by DNA damage induces the expression of p21. However, the above does not occur in cells expressing p53 mutants, as p21 expression decreases [59]. Furthermore, it has been reported that p53 mutants can regulate the cell cycle through miR-128-2, miR-223 and miR-517a [33]. Interestingly, in our work, we found downregulation of miR-517a-3p, miR-509-5p, and miR-101-3p in the presence of all 3 mutants. In this sense, previous studies have shown that downregulation of miR-517a and miR-517c contribute to the development of hepatocellular carcinoma through post-transcriptional regulation of Pyk2 (protein tyrosine kinase 2 beta), which is associated with blockade of the G2/M transition [32]. In our study, we found decreased miR-517a and miR-517c in the profile of the p53R248Q mutant. miR-509-5p can also delay the G1/S transition in the cell cycle, as well as facilitate apoptosis in cervical cancer cells; because miR-509-5p negatively regulates MDM2, which increases p53WT levels, resulting in p21 overexpression [23]. Additionally, upregulation of miR-101-3p has been reported to suppress EZH2 and HDAC9 expression, thereby inhibiting cell cycle progression in Retinoblastoma cells [36]. HDAC9 expression is associated with cell proliferation in vitro, and its inhibition with cell arrest in the G1 phase is consistent with the reduction in Cyclin E2 and CDK2 expression in retinoblastoma cells [60]. On its part, EZH2 transcriptionally represses the cell cycle suppressor INK-ARF, driving cell cycle progression of cancer stem cells [61], so p53 mutants could induce cell cycle progression by inducing Cyclin E2, CDK2 expression and silencing INK-ARF expression through negative regulation of miR-101-3p.
On the other hand, it has been reported that miR-3151 is silenced by methylation of its promoter in chronic lymphocytic leukemia (CLL), favoring cell proliferation [27], although inactivation of this miRNA has not been associated with the presence of p53 mutations, it is known that mutations of this protein are frequent in patients with CLL and have been associated with resistance to chemotherapy and a poor prognosis [62], so it would be interesting to demonstrate whether there is an association between the presence of p53 mutants and miR-3151 expression, as well as its possible relationship with chemotherapy resistance and/or prognosis in patients with CLL.
The miRNAs are known to repress gene expression, but WT-induced overexpression of ΔNp63α (a dominant-negative isoform of p63) in cisplatin-treated cells has been reported to activate the MDM4 (MDM4 regulator of p53) expression [29]. Upregulation of miR-885-3p promotes p53-dependent cisplatin-induced mitochondrial pathway apoptosis in WT ΔNp63α-expressing head and neck squamous carcinoma cells through overexpression of MDM4 and downregulation of BCL2 (B-cell lymphoma 2). Interestingly, the decrease of MDM4 is related to resistance to cisplatin [29]. It has also been shown that mutant p53R273H and p53WT can interact with ΔNp63α, mediating its degradation [63]. Thus, it would be interesting to demonstrate whether there is an association with cisplatin resistance by p53 mutants dependent on inhibition of the ΔNp63α protein. On the other hand, miR-200c-5p suppresses proliferation and metastasis, inhibiting MAD2L1 (mitotic arrest deficient 2 like 1) in hepatocellular carcinoma [64]. It should be noted that several studies have shown that p53 is frequently mutated in this type of cancer [65,66]. Furthermore, the presence of p53 mutations correlates with tumor progression and survival in hepatocellular carcinoma [67], suggesting an important role of p53 mutations in hepatocellular carcinoma.
On the other hand, in our study, we observed overexpression of miR-27b-5p in the presence of p53 mutants. The miR-27b has been reported as an oncomiR and a tumor suppressor, which has also been observed for other miRNAs. It has been suggested that the cellular context is important to determine the expression and function of miRNAs, the balance between the targets of each miRNA present in a particular situation, and specific tissue. For example, miR-27b is overexpressed in breast, gastric, ovarian, and glioma cancers, where it has been associated with the induction of processes such as cell proliferation, metabolism, migration, and invasion [46,68]. In addition, miR-27b is overexpressed in the MDA-MB-231 breast cancer cell line. These levels increase in a subline selected for its high capacity to induce metastasis to the lung, called 4175. It was also demonstrated that inhibiting miR-27b expression decreases these cells’ migration and invasion capacity. Interestingly, the MDA-MB-231 cell line has a mutation in the p53 gene, so it would be interesting to analyze whether miR-27b expression is associated with the presence of p53 mutations in breast cancer [68].
Finally, miR-182-5p is also considered an oncomir because it is closely related to migration, invasion, and metastasis [38,39,40]. The active participation of adhesion molecules in the metastatic capacity of tumor cells is crucial since alteration in their expression generates a loss of function of the adhesion complex and gives rise to processes such as cell migration and invasion. This is consistent with the alteration of several miRNAs related to the signaling pathway "Adherens junction" in the expression profiles of p53 mutants, especially in the presence of the p53R248Q mutant. Among the differentially expressed miRNAs involved in regulating processes such as "Adherens junction" is the miR-182-5p. Some of the target genes of this miRNA have already been experimentally validated, such as FOXF2 and MTSS1. FOXF2 is a negative regulator of TWIST1 (Transcriptional repressor of E-cadherin) [38,69] and also negatively regulates the expression of matrix metalloproteinases such as MMP1 [70]. On the other hand, MTSS1 suppresses the formation of F-actin fibers, which is an important event in the rearrangement of the cytoskeleton in cell migration and invasion processes. Besides, MTSS1 accelerates the kinetics of Adherens junction assembly and makes cells more resistant to cell-cell junction disassembly [69,71]. In this study, we demonstrated that the three most frequent p53 mutants in cancer induce overexpression of miR-182-5p. In addition, we also observed miR-182-5p overexpression in the OVCAR-3/p53R248Q cell line, which coincides with low expression levels of the miRNA target genes FOXF2 and MTSS1. In agreement with our results, in high-grade serous ovarian carcinoma (HG-SOC), it has been reported that p53 mutations are frequent, and overexpression of miR-182 is common in the early stages [43]. Likewise, Xu et al., in 2014, observed that the OVCAR-3 cell line (p53R248Q) over-expressed miR-182-5p compared to the SKOV3 cell line (p53 null) [69]. Of note, these studies did not associate the overexpression of miR-182-5p with the presence of p53R248Q.
Additionally, we demonstrated that inhibiting miR-182-5p in the presence of mutant p53R248Q reestablished FOXF2 and MTSS1 expression, which correlated with decreased cell migration and invasion. Interestingly, the therapeutic potential of anti-miR-182 has been suggested in an orthotopic animal model to mimic human ovarian cancer, using the cell lines SKOV3 with transfection of miR-182 (intrabursal injection) and OVCAR-3 (intraperitoneal injection). In these models, they demonstrated that treatment with anti-miR-182-5p decreased tumor size, invasion, and distant metastasis compared to control [69]. On the other hand, Wang et al., in 2017, observed that miR-182 overexpression could promote the proliferation and migration of cancer cell lines from Head and Neck Squamous Cell Carcinoma (HNSCC), presenting TP53 mutations [72]. Notably, in this study, they only observed an association between miR-182-5p overexpression and the presence of p53 mutations in patients and cell lines from HNSCC but did not demonstrate whether p53 mutants induce miR-182-5p overexpression.
5. Conclusions
We found a direct relationship between the presence of p53 mutations and miR-182-5p overexpression because inhibition of the p53R248Q mutant in the OVCAR-3 cell line decreased miR-182-5p expression and correlated with decreased migration invasiveness in OVCAR-3 cells, as well as restoration of its target genes FOXF2 and MTSS1 expression. These results suggest that the ability of the p53R248Q mutant to induce cell migration and invasion is dependent on miR-182-5p expression. To acknowledge this is the first time that regulation of miR-182-5p expression by p53 mutants has been reported. However, further studies are needed to elucidate the mechanism by which p53 mutants induce miR-182-5p overexpression. In summary, our study provides a comprehensive overview of the regulation of miRNAs by the most frequent p53 mutants in cancer, contributing to the knowledge of how p53 mutants can induce cancer development through the regulation of miRNAs expression; it could also suggest new specific therapeutic strategies for cancer patients with p53 mutations.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1; Table S1 p53R273C; Table S2 p53175H; Table S3 p53248Q; Table S4 p53R273C; Table S5 p53R273C; Table S6 p53R175H; Table S7 p53R175H; Table S8 p53R248Q; Table S9 p53R248Q.
Author Contributions
JDC contributed to the conception and design of the review. TM, JDC, and DOB wrote the first draft of the manuscript. TM, DOB, GDG, and EAO developed the methodology, analysis, and interpretation of data; TM, JDC, LAH, and CHG wrote and reviewed the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
Grants from the CONACYT, México supported TM. The study was supported by grants from CONACyT (168896) and (261875) to JDC.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board and Ethics Committee of the Instituto Nacional de Cancerología (register number 017/039/IBI).
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pfister, N.T.; Prives, C. Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of P53. Cold Spring Harb Perspect Med 2017, 7, a026054. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant P53: One Name, Many Proteins. Genes Dev 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb Perspect Biol 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. Mutant P53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Yamamoto, S.; Iwakuma, T. Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers (Basel) 2018, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D.P. Therapeutic Targeting of P53: All Mutants Are Equal, but Some Mutants Are More Equal than Others. Nat Rev Clin Oncol 2018, 15, 13–30. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant P53 Partners in Crime. Cell Death Differ 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant P53: All the Roads Lead to Tumorigenesis. Int J Mol Sci 2019, 20, 6197. [Google Scholar] [CrossRef]
- Xiong, S.; Chachad, D.; Zhang, Y.; Gencel-Augusto, J.; Sirito, M.; Pant, V.; Yang, P.; Sun, C.; Chau, G.; Qi, Y.; et al. Differential Gain-of-Function Activity of Three P53 Hotspot Mutants In Vivo. Cancer Res 2022, 82, 1926–1936. [Google Scholar] [CrossRef]
- Annese, T.; Tamma, R.; De Giorgis, M.; Ribatti, D. MicroRNAs Biogenesis, Functions and Role in Tumor Angiogenesis. Front Oncol 2020, 10, 581007. [Google Scholar] [CrossRef]
- Madrigal, T.; Hernández-Monge, J.; Herrera, L.A.; González-De la Rosa, C.H.; Domínguez-Gómez, G.; Candelaria, M.; Luna-Maldonado, F.; Calderón González, K.G.; Díaz-Chávez, J. Regulation of MiRNAs Expression by Mutant P53 Gain of Function in Cancer. Front Cell Dev Biol 2021, 9, 695723. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant P53 Cancers Reprogram Macrophages to Tumor Supporting Macrophages via Exosomal MiR-1246. Nat Commun 2018, 9, 771. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-T.; Lung, M.L. Mutant P53 Expression Enhances Drug Resistance in a Hepatocellular Carcinoma Cell Line. Cancer Chemother Pharmacol 2004, 53, 519–526. [Google Scholar] [CrossRef]
- Baker, S.J.; Markowitz, S.; Fearon, E.R.; Willson, J.K.; Vogelstein, B. Suppression of Human Colorectal Carcinoma Cell Growth by Wild-Type P53. Science 1990, 249, 912–915. [Google Scholar] [CrossRef]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical Role for P53-Serine 15 Phosphorylation in Stimulating Transactivation at P53-Responsive Promoters. Nucleic Acids Res 2014, 42, 7666–7680. [Google Scholar] [CrossRef] [PubMed]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. Gplots: Various R Programming Tools for Plotting Data 2022.
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open Software Development for Computational Biology and Bioinformatics. Genome Biol 2004, 5, R80. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 7 July 2022).
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An Information Aesthetic for Comparative Genomics. Genome Res 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Venny 2.1.0. Available online: https://bioinfogp.cnb.csic.es/tools/venny/ (accessed on 7 July 2022).
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-MiRPath v3.0: Deciphering MicroRNA Function with Experimental Support. Nucleic Acids Res 2015, 43, W460–466. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ren, Z.-J.; Nong, X.-Y.; Lv, Y.-R.; Sun, H.-H.; An, P.-P.; Wang, F.; Li, X.; Liu, M.; Tang, H. Mir-509-5p Joins the Mdm2/P53 Feedback Loop and Regulates Cancer Cell Growth. Cell Death Dis 2014, 5, e1387. [Google Scholar] [CrossRef]
- Hiramoto, H.; Muramatsu, T.; Ichikawa, D.; Tanimoto, K.; Yasukawa, S.; Otsuji, E.; Inazawa, J. MiR-509-5p and MiR-1243 Increase the Sensitivity to Gemcitabine by Inhibiting Epithelial-Mesenchymal Transition in Pancreatic Cancer. Sci Rep 2017, 7, 4002. [Google Scholar] [CrossRef]
- Li, Y.; Bai, W.; Zhang, J. MiR-200c-5p Suppresses Proliferation and Metastasis of Human Hepatocellular Carcinoma (HCC) via Suppressing MAD2L1. Biomed Pharmacother 2017, 92, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Liang, H.; Zhou, J.; Wang, Y.; Liu, S.; Wang, X.; Su, L.; Kang, X. LncRNA Small Nucleolar RNA Host Gene 12 Promotes Renal Cell Carcinoma Progression by Modulating the MiR-200c-5p/Collagen Type XI A1 Chain Pathway. Mol Med Rep 2020, 22, 3677–3686. [Google Scholar] [CrossRef]
- Wang, L.Q.; Wong, K.Y.; Rosèn, A.; Chim, C.S. Epigenetic Silencing of Tumor Suppressor MiR-3151 Contributes to Chinese Chronic Lymphocytic Leukemia by Constitutive Activation of MADD/ERK and PIK3R2/AKT Signaling Pathways. Oncotarget 2015, 6, 44422–44436. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Qiu, H.; Cui, H.; Ni, X.; Li, J.; Liao, W.; Lu, L.; Ding, K. MicroRNA-885-3p Inhibits the Growth of HT-29 Colon Cancer Cell Xenografts by Disrupting Angiogenesis via Targeting BMPR1A and Blocking BMP/Smad/Id1 Signaling. Oncogene 2015, 34, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chuang, A.Y.; Ratovitski, E.A. Phospho-ΔNp63α/MiR-885-3p Axis in Tumor Cell Life and Cell Death upon Cisplatin Exposure. Cell Cycle 2011, 10, 3938–3947. [Google Scholar] [CrossRef]
- Vijayakumaran, R.; Tan, K.H.; Miranda, P.J.; Haupt, S.; Haupt, Y. Regulation of Mutant P53 Protein Expression. Front Oncol 2015, 5, 284. [Google Scholar] [CrossRef]
- Yoshitomi, T.; Kawakami, K.; Enokida, H.; Chiyomaru, T.; Kagara, I.; Tatarano, S.; Yoshino, H.; Arimura, H.; Nishiyama, K.; Seki, N.; et al. Restoration of MiR-517a Expression Induces Cell Apoptosis in Bladder Cancer Cell Lines. Oncol Rep 2011, 25, 1661–1668. [Google Scholar] [CrossRef]
- Liu, R.-F.; Xu, X.; Huang, J.; Fei, Q.-L.; Chen, F.; Li, Y.-D.; Han, Z.-G. Down-Regulation of MiR-517a and MiR-517c Promotes Proliferation of Hepatocellular Carcinoma Cells via Targeting Pyk2. Cancer Lett 2013, 329, 164–173. [Google Scholar] [CrossRef]
- Garibaldi, F.; Falcone, E.; Trisciuoglio, D.; Colombo, T.; Lisek, K.; Walerych, D.; Del Sal, G.; Paci, P.; Bossi, G.; Piaggio, G.; et al. Mutant P53 Inhibits MiRNA Biogenesis by Interfering with the Microprocessor Complex. Oncogene 2016, 35, 3760–3770. [Google Scholar] [CrossRef]
- Liu, P.; Ye, F.; Xie, X.; Li, X.; Tang, H.; Li, S.; Huang, X.; Song, C.; Wei, W.; Xie, X. Mir-101-3p Is a Key Regulator of Tumor Metabolism in Triple Negative Breast Cancer Targeting AMPK. Oncotarget 2016, 7, 35188–35198. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Liu, Z.-J.; He, H.; Zhang, C.; Wang, Y.-L. MicroRNA-101-3p Suppresses Cell Proliferation, Invasion and Enhances Chemotherapeutic Sensitivity in Salivary Gland Adenoid Cystic Carcinoma by Targeting Pim-1. Am J Cancer Res 2015, 5, 3015–3029. [Google Scholar]
- Hou, Y.; Li, L.; Ju, Y.; Lu, Y.; Chang, L.; Xiang, X. MiR-101-3p Regulates the Viability of Lung Squamous Carcinoma Cells via Targeting EZH2. J Cell Biochem 2017, 118, 3142–3149. [Google Scholar] [CrossRef] [PubMed]
- Luan, C.; Li, Y.; Liu, Z.; Zhao, C. Long Noncoding RNA MALAT1 Promotes the Development of Colon Cancer by Regulating MiR-101-3p/STC1 Axis. Onco Targets Ther 2020, 13, 3653–3665. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.T.; Byers, L.A.; Peng, D.H.; Roybal, J.D.; Diao, L.; Wang, J.; Tong, P.; Creighton, C.J.; Gibbons, D.L. The MiR-200 Family and the MiR-183~96~182 Cluster Target Foxf2 to Inhibit Invasion and Metastasis in Lung Cancers. Oncogene 2016, 35, 173–186. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Shen, J.; Wang, C.; Yang, L.; Zhang, X. MicroRNA-182 Downregulates Metastasis Suppressor 1 and Contributes to Metastasis of Hepatocellular Carcinoma. BMC Cancer 2012, 12, 227. [Google Scholar] [CrossRef]
- Lei, R.; Tang, J.; Zhuang, X.; Deng, R.; Li, G.; Yu, J.; Liang, Y.; Xiao, J.; Wang, H.-Y.; Yang, Q.; et al. Suppression of MIM by MicroRNA-182 Activates RhoA and Promotes Breast Cancer Metastasis. Oncogene 2014, 33, 1287–1296. [Google Scholar] [CrossRef]
- Xue, J.; Zhou, A.; Wu, Y.; Morris, S.-A.; Lin, K.; Amin, S.; Verhaak, R.; Fuller, G.; Xie, K.; Heimberger, A.B.; et al. MiR-182-5p Induced by STAT3 Activation Promotes Glioma Tumorigenesis. Cancer Res 2016, 76, 4293–4304. [Google Scholar] [CrossRef]
- Xiao, Y.; Huang, W.; Huang, H.; Wang, L.; Wang, M.; Zhang, T.; Fang, X.; Xia, X. MiR-182-5p and MiR-96-5p Target PIAS1 and Mediate the Negative Feedback Regulatory Loop between PIAS1 and STAT3 in Endometrial Cancer. DNA Cell Biol 2021, 40, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, J.; Segura, M.F.; Shao, C.; Lee, P.; Gong, Y.; Hernando, E.; Wei, J.-J. MiR-182 Overexpression in Tumourigenesis of High-Grade Serous Ovarian Carcinoma. J Pathol 2012, 228, 204–215. [Google Scholar] [CrossRef]
- Wang, Y.; Rathinam, R.; Walch, A.; Alahari, S.K. ST14 (Suppression of Tumorigenicity 14) Gene Is a Target for MiR-27b, and the Inhibitory Effect of ST14 on Cell Growth Is Independent of MiR-27b Regulation. J Biol Chem 2009, 284, 23094–23106. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, S.; Zhao, Z.; Mao, X.; Huang, J.; Wu, Z.; Zheng, L.; Wang, Q. MicroRNA-27b up-Regulated by Human Papillomavirus 16 E7 Promotes Proliferation and Suppresses Apoptosis by Targeting Polo-like Kinase2 in Cervical Cancer. Oncotarget 2016, 7, 19666–19679. [Google Scholar] [CrossRef] [PubMed]
- Eastlack, S.C.; Dong, S.; Ivan, C.; Alahari, S.K. Suppression of PDHX by MicroRNA-27b Deregulates Cell Metabolism and Promotes Growth in Breast Cancer. Mol Cancer 2018, 17, 100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, Y.; Fang, J.-Y.; Xu, J. Gain-of-Function MiRNA Signature by Mutant P53 Associates with Poor Cancer Outcome. Oncotarget 2016, 7, 11056–11066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, W.; Chen, X. Mutant P53 Disrupts MCF-10A Cell Polarity in Three-Dimensional Culture via Epithelial-to-Mesenchymal Transitions. J Biol Chem 2011, 286, 16218–16228. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant P53 Gain-of-Function Induces Epithelial-Mesenchymal Transition through Modulation of the MiR-130b-ZEB1 Axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef]
- Subramanian, M.; Francis, P.; Bilke, S.; Li, X.L.; Hara, T.; Lu, X.; Jones, M.F.; Walker, R.L.; Zhu, Y.; Pineda, M.; et al. A Mutant P53/Let-7i-Axis-Regulated Gene Network Drives Cell Migration, Invasion and Metastasis. Oncogene 2015, 34, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Neilsen, P.M.; Noll, J.E.; Mattiske, S.; Bracken, C.P.; Gregory, P.A.; Schulz, R.B.; Lim, S.P.; Kumar, R.; Suetani, R.J.; Goodall, G.J.; et al. Mutant P53 Drives Invasion in Breast Tumors through Up-Regulation of MiR-155. Oncogene 2013, 32, 2992–3000. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.-C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant P53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314.e7. [Google Scholar] [CrossRef]
- Klemke, L.; Fehlau, C.F.; Winkler, N.; Toboll, F.; Singh, S.K.; Moll, U.M.; Schulz-Heddergott, R. The Gain-of-Function P53 R248W Mutant Promotes Migration by STAT3 Deregulation in Human Pancreatic Cancer Cells. Front Oncol 2021, 11, 642603. [Google Scholar] [CrossRef]
- Zhang, W.; Qian, P.; Zhang, X.; Zhang, M.; Wang, H.; Wu, M.; Kong, X.; Tan, S.; Ding, K.; Perry, J.K.; et al. Autocrine/Paracrine Human Growth Hormone-Stimulated MicroRNA 96-182-183 Cluster Promotes Epithelial-Mesenchymal Transition and Invasion in Breast Cancer. J Biol Chem 2015, 290, 13812–13829. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Hu, Y.; Lan, T.; Guan, K.-L.; Luo, T.; Luo, M. The Hippo Signalling Pathway and Its Implications in Human Health and Diseases. Signal Transduct Target Ther 2022, 7, 376. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, J.; Feng, J.; Liu, Z.; Liu, S.; Zhang, M.; Zhang, Y.; Hou, Y.; Wu, D.; Li, C.; et al. Variation in the Life History Strategy Underlies Functional Diversity of Tumors. Natl Sci Rev 2021, 8, nwaa124. [Google Scholar] [CrossRef]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP Enhances the Pro-Proliferative Transcriptional Activity of Mutant P53 Proteins. EMBO Rep 2016, 17, 188–201. [Google Scholar] [CrossRef]
- Ferraiuolo, M.; Verduci, L.; Blandino, G.; Strano, S. Mutant P53 Protein and the Hippo Transducers YAP and TAZ: A Critical Oncogenic Node in Human Cancers. Int J Mol Sci 2017, 18, 961. [Google Scholar] [CrossRef] [PubMed]
- Elbendary, A.A.; Cirisano, F.D.; Evans, A.C.; Davis, P.L.; Iglehart, J.D.; Marks, J.R.; Berchuck, A. Relationship between P21 Expression and Mutation of the P53 Tumor Suppressor Gene in Normal and Malignant Ovarian Epithelial Cells. Clin Cancer Res 1996, 2, 1571–1575. [Google Scholar]
- Zhang, Y.; Wu, D.; Xia, F.; Xian, H.; Zhu, X.; Cui, H.; Huang, Z. Downregulation of HDAC9 Inhibits Cell Proliferation and Tumor Formation by Inducing Cell Cycle Arrest in Retinoblastoma. Biochem Biophys Res Commun 2016, 473, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-J.; Hung, M.-C. The Role of EZH2 in Tumour Progression. Br J Cancer 2012, 106, 243–247. [Google Scholar] [CrossRef]
- Zenz, T.; Eichhorst, B.; Busch, R.; Denzel, T.; Häbe, S.; Winkler, D.; Bühler, A.; Edelmann, J.; Bergmann, M.; Hopfinger, G.; et al. TP53 Mutation and Survival in Chronic Lymphocytic Leukemia. J Clin Oncol 2010, 28, 4473–4479. [Google Scholar] [CrossRef]
- Ratovitski, E.A.; Patturajan, M.; Hibi, K.; Trink, B.; Yamaguchi, K.; Sidransky, D. P53 Associates with and Targets Delta Np63 into a Protein Degradation Pathway. Proc Natl Acad Sci U S A 2001, 98, 1817–1822. [Google Scholar] [CrossRef]
- Li, Y.; Bai, W.; Zhang, J. MiR-200c-5p Suppresses Proliferation and Metastasis of Human Hepatocellular Carcinoma (HCC) via Suppressing MAD2L1. Biomed Pharmacother 2017, 92, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Sheen, I.S.; Chen, B.F.; Wu, J.Y. Is the P53 Gene Mutation of Prognostic Value in Hepatocellular Carcinoma after Resection? Arch Surg 2000, 135, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Sbisà, E.; Tullo, A.; Papeo, P.A.; Saccone, C.; Poole, S.; Pignatelli, M.; Mitry, R.R.; Ding, S.; Isla, A.; et al. P53 Mutation Is a Poor Prognostic Indicator for Survival in Patients with Hepatocellular Carcinoma Undergoing Surgical Tumour Ablation. Br J Cancer 1998, 77, 776–782. [Google Scholar] [CrossRef]
- Heinze, T.; Jonas, S.; Kärsten, A.; Neuhaus, P. Determination of the Oncogenes P53 and C-Erb B2 in the Tumour Cytosols of Advanced Hepatocellular Carcinoma (HCC) and Correlation to Survival Time. Anticancer Res 1999, 19, 2501–2503. [Google Scholar] [PubMed]
- Wang, Y.; Rathinam, R.; Walch, A.; Alahari, S.K. ST14 (Suppression of Tumorigenicity 14) Gene Is a Target for MiR-27b, and the Inhibitory Effect of ST14 on Cell Growth Is Independent of MiR-27b Regulation. J Biol Chem 2009, 284, 23094–23106. [Google Scholar] [CrossRef]
- Xu, X.; Ayub, B.; Liu, Z.; Serna, V.A.; Qiang, W.; Liu, Y.; Hernando, E.; Zabludoff, S.; Kurita, T.; Kong, B.; et al. Anti-MiR182 Reduces Ovarian Cancer Burden, Invasion, and Metastasis: An in Vivo Study in Orthotopic Xenografts of Nude Mice. Mol Cancer Ther 2014, 13, 1729–1739. [Google Scholar] [CrossRef]
- Wang, Q.-S.; Kong, P.-Z.; Li, X.-Q.; Yang, F.; Feng, Y.-M. FOXF2 Deficiency Promotes Epithelial-Mesenchymal Transition and Metastasis of Basal-like Breast Cancer. Breast Cancer Res 2015, 17, 30. [Google Scholar] [CrossRef]
- Saarikangas, J.; Mattila, P.K.; Varjosalo, M.; Bovellan, M.; Hakanen, J.; Calzada-Wack, J.; Tost, M.; Jennen, L.; Rathkolb, B.; Hans, W.; et al. Missing-in-Metastasis MIM/MTSS1 Promotes Actin Assembly at Intercellular Junctions and Is Required for Integrity of Kidney Epithelia. J Cell Sci 2011, 124, 1245–1255. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, H.; Li, W.; Jia, C.; Zhang, H.; Sun, Y.; Chen, X.; Song, X. Overexpression of TP53 Mutation-Associated MicroRNA-182 Promotes Tumor Cell Proliferation and Migration in Head and Neck Squamous Cell Carcinoma. Arch Oral Biol 2017, 73, 105–112. [Google Scholar] [CrossRef]
Figure 1.
Representation of the p53R273C-regulated miRNome and Signaling Pathway Enrichment (KEGG) analysis. a) Saos-2 cells were transfected with the p53R273C mutant, and p53WT was used as a control. To determine which miRNAs were differentially expressed, a p-value <0.05 and Fold Change ≥2 or ≤-2 were used. The Circos map distributes the differentially expressed miRNAs according to their chromosomal locations, within which "bar graphs" correspond to the Fold Change value of miRNAs that increased (red) or decreased (green) in the presence of the p53R273C vs. p53WT mutant are shown. Additionally, the names of those miRNAs that were selected for validation through TaqMan probe assays are highlighted in red-augmented (miR-27b-5p) and green-decreased (miR-509-5p, miR-101-3p, miR-517a-3p and miR-200c-5p). b) "KEGG" pathway enrichment analysis of hypothetical target genes, based on miRPath (DIANA-microT-CDS algorithm). c) "KEGG" pathway enrichment analysis of experimentally validated genes, based on miRPath (DIANA-microT-CDS algorithm). The scatter plot in b) and c) shows the significance level of each signaling Pathway on the "X" axis (p <0.05) and the pathway name on "Y". The color of the circles represents the number of miRNAs involved in the signaling pathways, and the size of the circles is the number of hypothetical and/or validated genes.
Figure 1.
Representation of the p53R273C-regulated miRNome and Signaling Pathway Enrichment (KEGG) analysis. a) Saos-2 cells were transfected with the p53R273C mutant, and p53WT was used as a control. To determine which miRNAs were differentially expressed, a p-value <0.05 and Fold Change ≥2 or ≤-2 were used. The Circos map distributes the differentially expressed miRNAs according to their chromosomal locations, within which "bar graphs" correspond to the Fold Change value of miRNAs that increased (red) or decreased (green) in the presence of the p53R273C vs. p53WT mutant are shown. Additionally, the names of those miRNAs that were selected for validation through TaqMan probe assays are highlighted in red-augmented (miR-27b-5p) and green-decreased (miR-509-5p, miR-101-3p, miR-517a-3p and miR-200c-5p). b) "KEGG" pathway enrichment analysis of hypothetical target genes, based on miRPath (DIANA-microT-CDS algorithm). c) "KEGG" pathway enrichment analysis of experimentally validated genes, based on miRPath (DIANA-microT-CDS algorithm). The scatter plot in b) and c) shows the significance level of each signaling Pathway on the "X" axis (p <0.05) and the pathway name on "Y". The color of the circles represents the number of miRNAs involved in the signaling pathways, and the size of the circles is the number of hypothetical and/or validated genes.
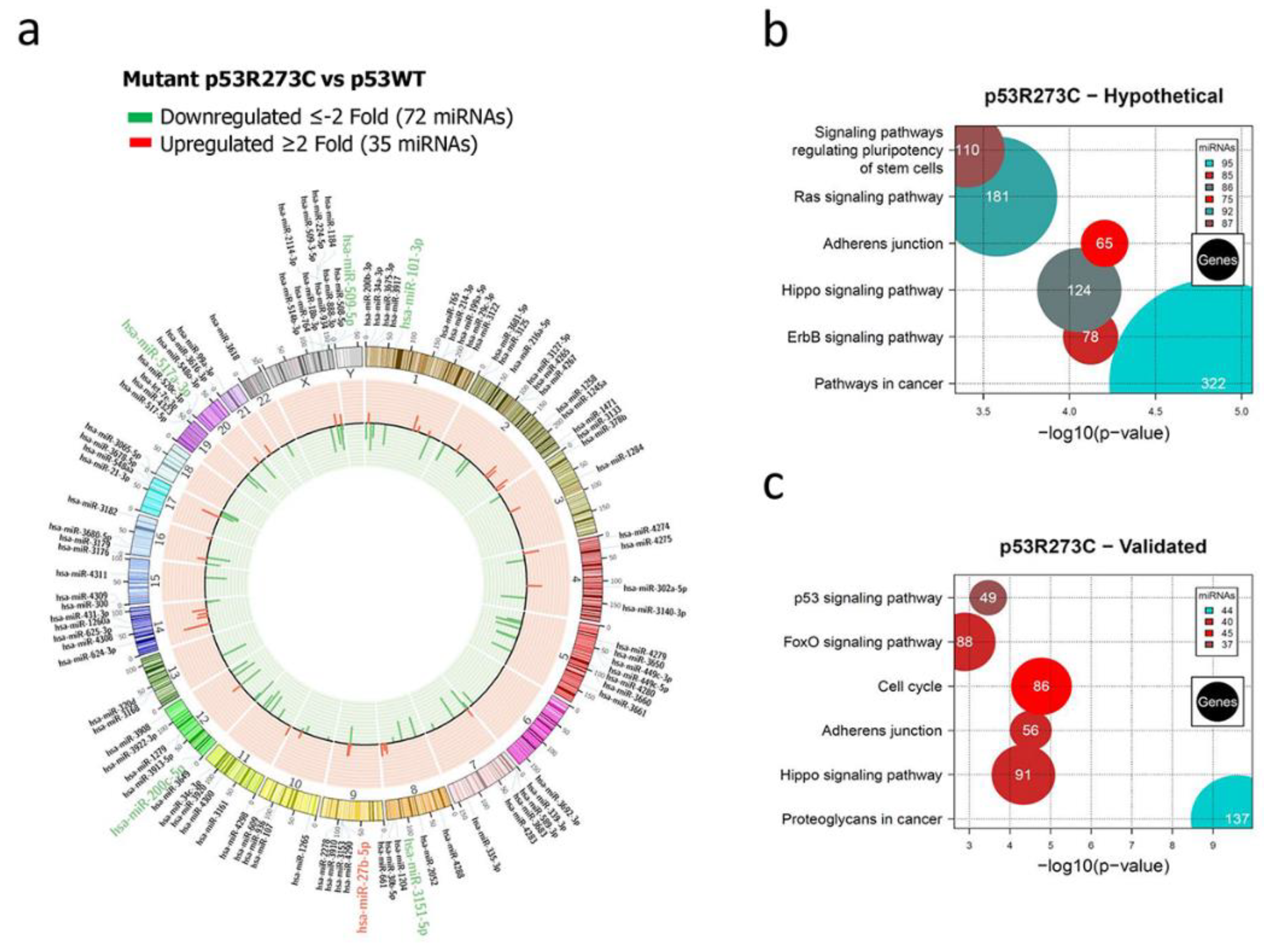
Figure 2.
Representation of the p53R175H-regulated miRNome and Signaling Pathway Enrichment (KEGG) analysis. a) Saos-2 cells were transfected with p53R175H, and p53WT was used as a control. To determine which miRNAs were differentially expressed, a value of p <0.05 and (Fold Change ≥2 or ≤-2) was used. The Circos map distributes the differentially expressed miRNAs according to their chromosomal locations, within which "bar graphs" correspond to the Fold Change value of miRNAs that increased (red) or decreased (green) in the presence of the p53R175H vs. p53WT mutant are shown. Additionally, the names of those miRNAs that were selected for validation through TaqMan probe assays are highlighted in red-increased (miR-182-5p) and green-decreased (miR-509-5p, miR-101-3p, miR-517a-3p, miR-885-3p, miR-3151-5p). (b) "KEGG" pathway enrichment analysis of hypothetical target genes, based on miRPath (DIANA-microT-CDS algorithm). C) "KEGG" pathway enrichment analysis of experimentally validated genes, based on miRPath (DIANA-microT-CDS algorithm). The scatter plot in b) and c) shows the significance level of each signaling Pathway on the "X" axis (p <0.05) and the pathway name on "Y". The color of the circles represents the number of miRNAs involved in signaling pathways, and the size of the circles the number of hypothetical and/or validated genes.
Figure 2.
Representation of the p53R175H-regulated miRNome and Signaling Pathway Enrichment (KEGG) analysis. a) Saos-2 cells were transfected with p53R175H, and p53WT was used as a control. To determine which miRNAs were differentially expressed, a value of p <0.05 and (Fold Change ≥2 or ≤-2) was used. The Circos map distributes the differentially expressed miRNAs according to their chromosomal locations, within which "bar graphs" correspond to the Fold Change value of miRNAs that increased (red) or decreased (green) in the presence of the p53R175H vs. p53WT mutant are shown. Additionally, the names of those miRNAs that were selected for validation through TaqMan probe assays are highlighted in red-increased (miR-182-5p) and green-decreased (miR-509-5p, miR-101-3p, miR-517a-3p, miR-885-3p, miR-3151-5p). (b) "KEGG" pathway enrichment analysis of hypothetical target genes, based on miRPath (DIANA-microT-CDS algorithm). C) "KEGG" pathway enrichment analysis of experimentally validated genes, based on miRPath (DIANA-microT-CDS algorithm). The scatter plot in b) and c) shows the significance level of each signaling Pathway on the "X" axis (p <0.05) and the pathway name on "Y". The color of the circles represents the number of miRNAs involved in signaling pathways, and the size of the circles the number of hypothetical and/or validated genes.
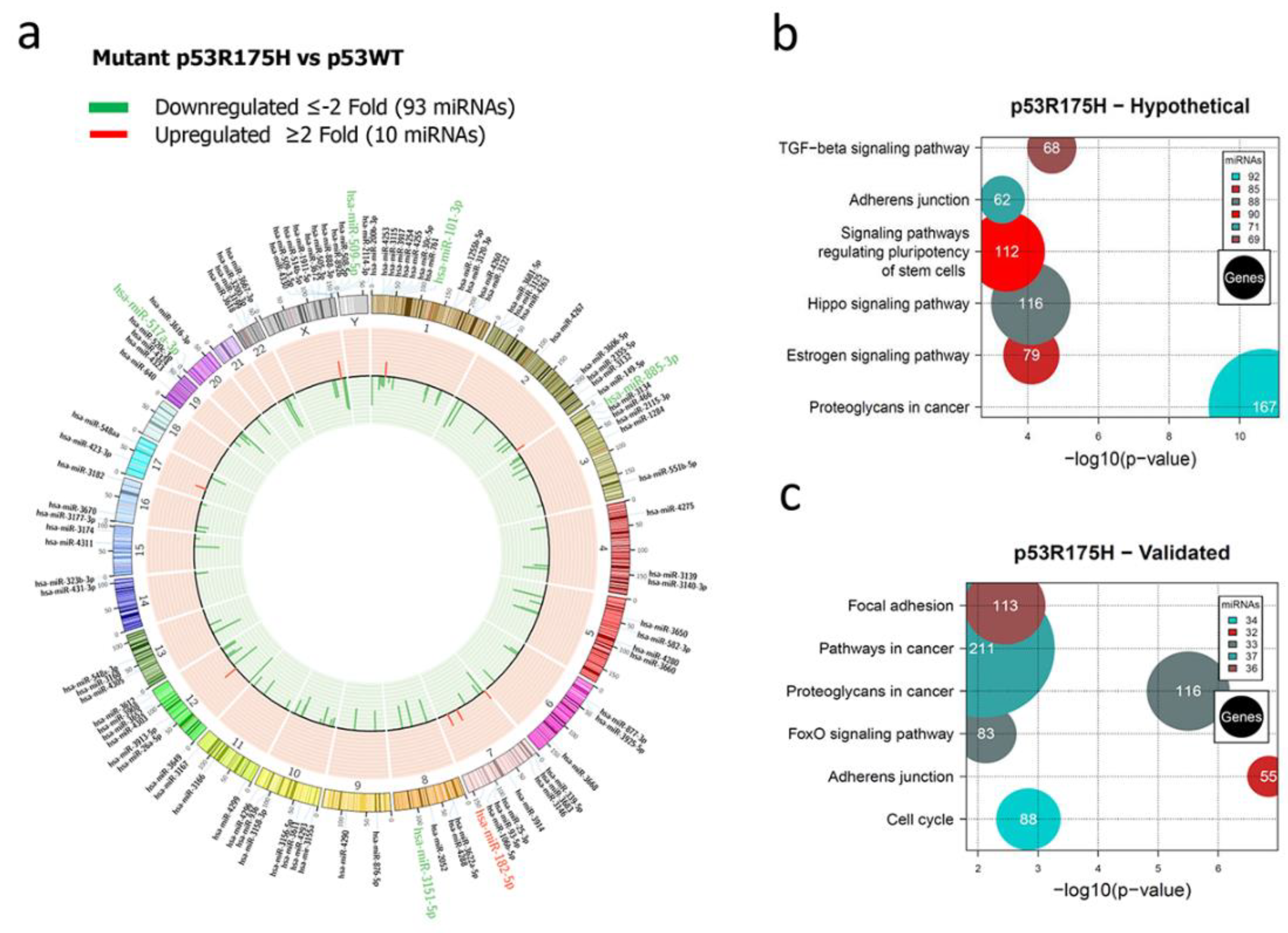
Figure 3.
Representation of the p53R248Q-regulated miRNome and Signaling Pathway Enrichment (KEGG) analysis. Saos-2 cells were transfected with p53R248Q, and p53WT was used as a control. To determine which miRNAs were differentially expressed, a value of p <0.05 and (Fold Change ≥2 or ≤-2 was used. The Circos map distributes the differentially expressed miRNAs according to their chromosomal locations, within which "bar graphs" correspond to the Fold Change value of miRNAs that increased (red) or decreased (green) in the presence of the p53R248Q vs. p53WT mutant are shown. Additionally, the names of those miRNAs that were selected for validation through TaqMan probe assays are highlighted in red-increased (miR-182-5p and miR-27b-5p) and green-decreased (miR-509-5p, miR-101-3p, miR-517a-3p, miR-885-3p, miR-3151-5p, miR-200c-5p) b). "KEGG" pathway enrichment analysis of hypothetical target genes, based on miRPath (DIANA-microT-CDS algorithm).c) "KEGG" pathway enrichment analysis of experimentally validated genes, based on miRPath (DIANA-microT-CDS algorithm). The scatter plot in b) and c) shows the significance level of each signaling Pathway on the "X" axis (p <0.05) and the pathway name on "Y". The color of the circles represents the number of miRNAs involved in signaling pathways, and the size of the circles the number of hypothetical and/or validated genes.
Figure 3.
Representation of the p53R248Q-regulated miRNome and Signaling Pathway Enrichment (KEGG) analysis. Saos-2 cells were transfected with p53R248Q, and p53WT was used as a control. To determine which miRNAs were differentially expressed, a value of p <0.05 and (Fold Change ≥2 or ≤-2 was used. The Circos map distributes the differentially expressed miRNAs according to their chromosomal locations, within which "bar graphs" correspond to the Fold Change value of miRNAs that increased (red) or decreased (green) in the presence of the p53R248Q vs. p53WT mutant are shown. Additionally, the names of those miRNAs that were selected for validation through TaqMan probe assays are highlighted in red-increased (miR-182-5p and miR-27b-5p) and green-decreased (miR-509-5p, miR-101-3p, miR-517a-3p, miR-885-3p, miR-3151-5p, miR-200c-5p) b). "KEGG" pathway enrichment analysis of hypothetical target genes, based on miRPath (DIANA-microT-CDS algorithm).c) "KEGG" pathway enrichment analysis of experimentally validated genes, based on miRPath (DIANA-microT-CDS algorithm). The scatter plot in b) and c) shows the significance level of each signaling Pathway on the "X" axis (p <0.05) and the pathway name on "Y". The color of the circles represents the number of miRNAs involved in signaling pathways, and the size of the circles the number of hypothetical and/or validated genes.
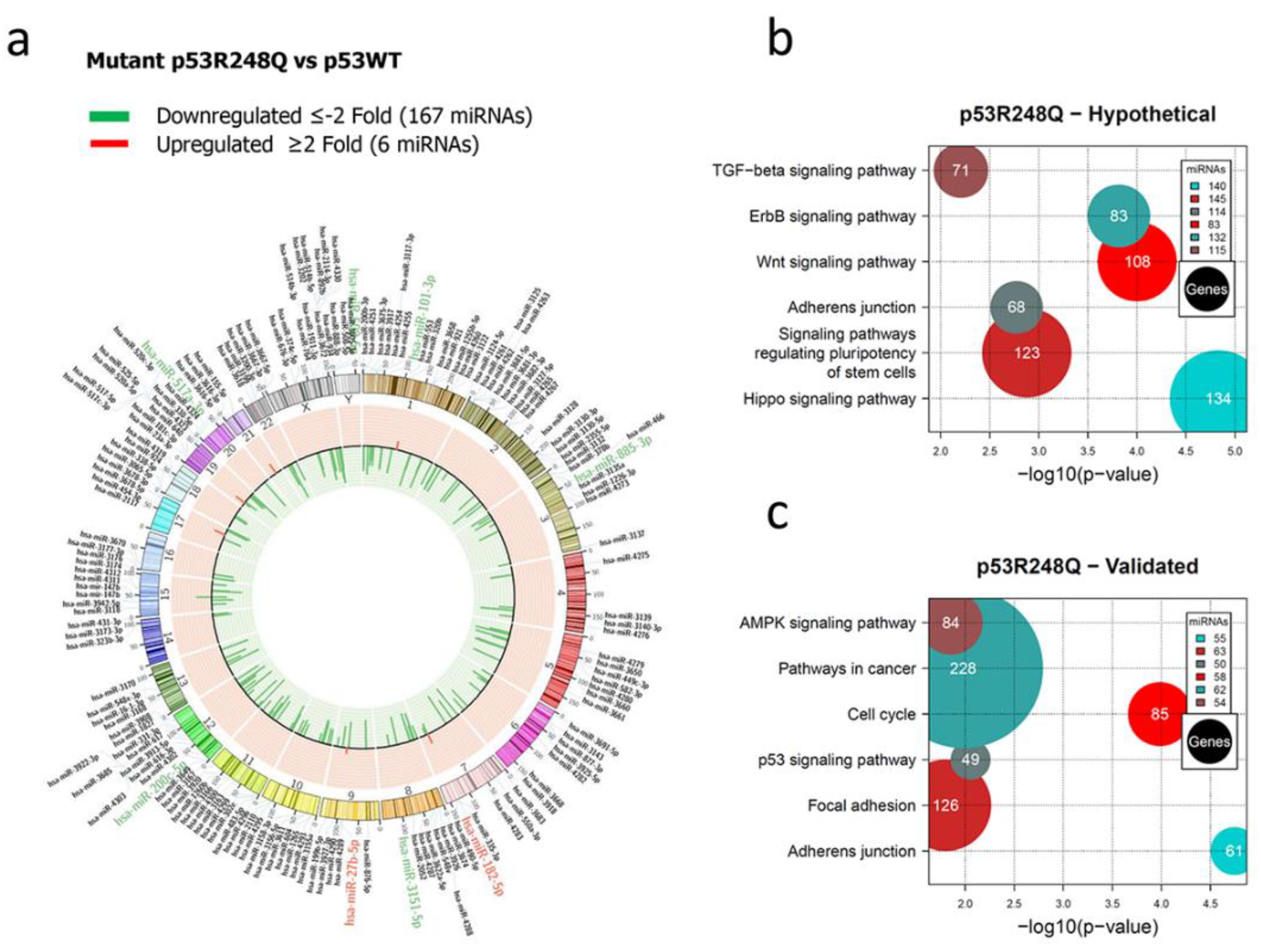
Figure 4.
Heat map and Venn diagram of differentially expressed miRNAs in Saos-2 cells transfected with p53R273C, p53R248Q, and p53R175H mutants. a) The heat map represents the color-coded expression levels; red indicates overexpression, and green, indicates underexpression. The expression of each miRNA is hierarchically grouped on the "Y" axis; furthermore, the p53 mutants are represented on the "X" axis. b) The Venn Diagram shows the overlap of miRNAs among the three p53 mutants (33 miRNAs). The p53R273C mutant shares 25 miRNAs with p53R248Q and only 5 with p53R175H. In addition, the p53R248Q mutant exclusively regulates 73 miRNAs, and 42 are shared with the p53R175H mutant.
Figure 4.
Heat map and Venn diagram of differentially expressed miRNAs in Saos-2 cells transfected with p53R273C, p53R248Q, and p53R175H mutants. a) The heat map represents the color-coded expression levels; red indicates overexpression, and green, indicates underexpression. The expression of each miRNA is hierarchically grouped on the "Y" axis; furthermore, the p53 mutants are represented on the "X" axis. b) The Venn Diagram shows the overlap of miRNAs among the three p53 mutants (33 miRNAs). The p53R273C mutant shares 25 miRNAs with p53R248Q and only 5 with p53R175H. In addition, the p53R248Q mutant exclusively regulates 73 miRNAs, and 42 are shared with the p53R175H mutant.
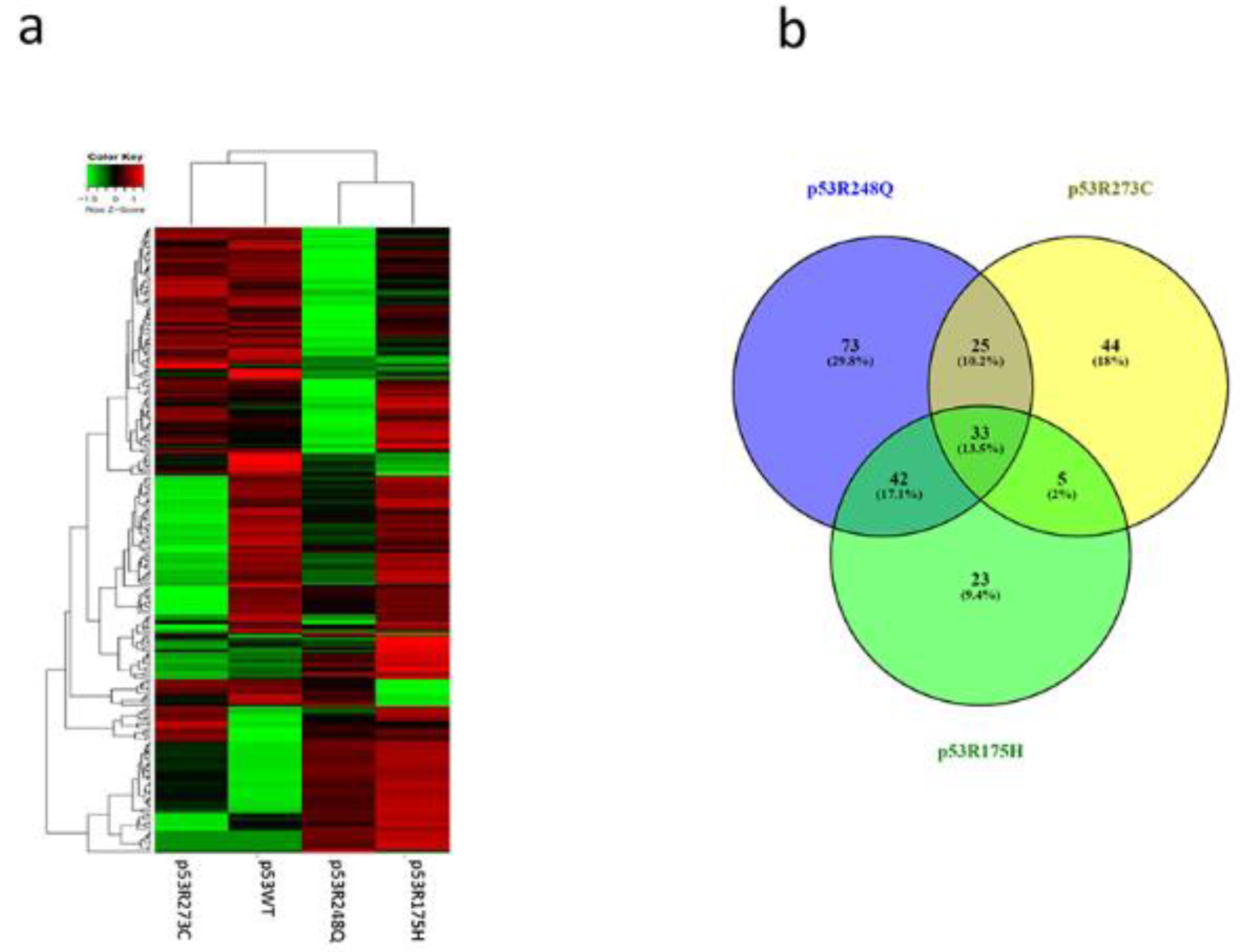
Figure 5.
Flowchart showing the selection mechanism of miRNAs validated by Taqman Probes assays. PCR arrays (miRBase version 16, 1066 miRNAs; Qiagen) of the mutants (p53R175H, p53R273C, and p53R248Q) were performed. 254 differentially expressed miRNAs were identified according to cutoff points of ≥2 or ≤-2 (Fold Change) and a value of p<0.05. Subsequently, we found that 72 miRNAs were shared by at least 2 p53 mutants and 33 by all three. We also identified these miRNAs’ association with cancer through a literature survey. With this strategy, we selected and validated the downregulation of 6 tumor suppressor miRNAs: miR-509-5p , miR-200c-5p, miR-3151-5p, miR-885-3p, miR-517a-3p , miR-101-3p , as well as 2 oncomiRs: miR-182-5p and miR-27b-5p.
Figure 5.
Flowchart showing the selection mechanism of miRNAs validated by Taqman Probes assays. PCR arrays (miRBase version 16, 1066 miRNAs; Qiagen) of the mutants (p53R175H, p53R273C, and p53R248Q) were performed. 254 differentially expressed miRNAs were identified according to cutoff points of ≥2 or ≤-2 (Fold Change) and a value of p<0.05. Subsequently, we found that 72 miRNAs were shared by at least 2 p53 mutants and 33 by all three. We also identified these miRNAs’ association with cancer through a literature survey. With this strategy, we selected and validated the downregulation of 6 tumor suppressor miRNAs: miR-509-5p , miR-200c-5p, miR-3151-5p, miR-885-3p, miR-517a-3p , miR-101-3p , as well as 2 oncomiRs: miR-182-5p and miR-27b-5p.
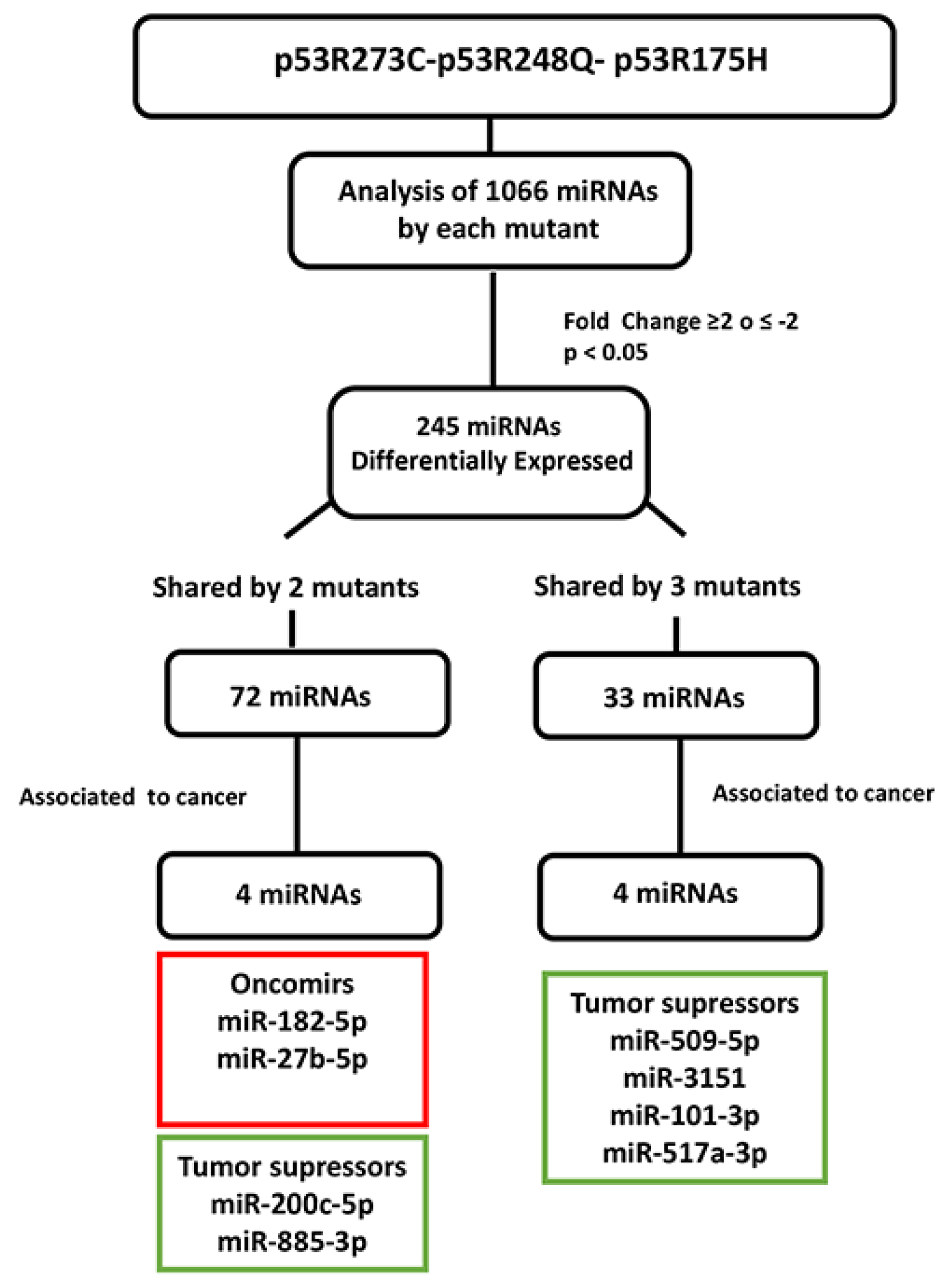
Figure 6.
Validation of miRNAs by TaqMan probe assays. Relative expression of miR-509-5p, miR-101-3p, miR-517a-3p, miR-3151-5p, miR-885-3p, miR-200c-5p, miR-182-5p and miR-27b-5p in cell line Saos-2 transfected with empty vector (Control) or with mutant p53 (Saos-2/p53R248Q, Saos-2/p53R273C and Saos-2/p53R175H) vs Saos-2/p53WT. Data are presented as the two-fold change in miRNA level normalized for U6 (endogenous control). Data represent mean ± SD (n = 3) and (*) Refers to expression changes was significant compared to cells transfected with p53WT (Saos-2/p53WT) (p<0.05).
Figure 6.
Validation of miRNAs by TaqMan probe assays. Relative expression of miR-509-5p, miR-101-3p, miR-517a-3p, miR-3151-5p, miR-885-3p, miR-200c-5p, miR-182-5p and miR-27b-5p in cell line Saos-2 transfected with empty vector (Control) or with mutant p53 (Saos-2/p53R248Q, Saos-2/p53R273C and Saos-2/p53R175H) vs Saos-2/p53WT. Data are presented as the two-fold change in miRNA level normalized for U6 (endogenous control). Data represent mean ± SD (n = 3) and (*) Refers to expression changes was significant compared to cells transfected with p53WT (Saos-2/p53WT) (p<0.05).
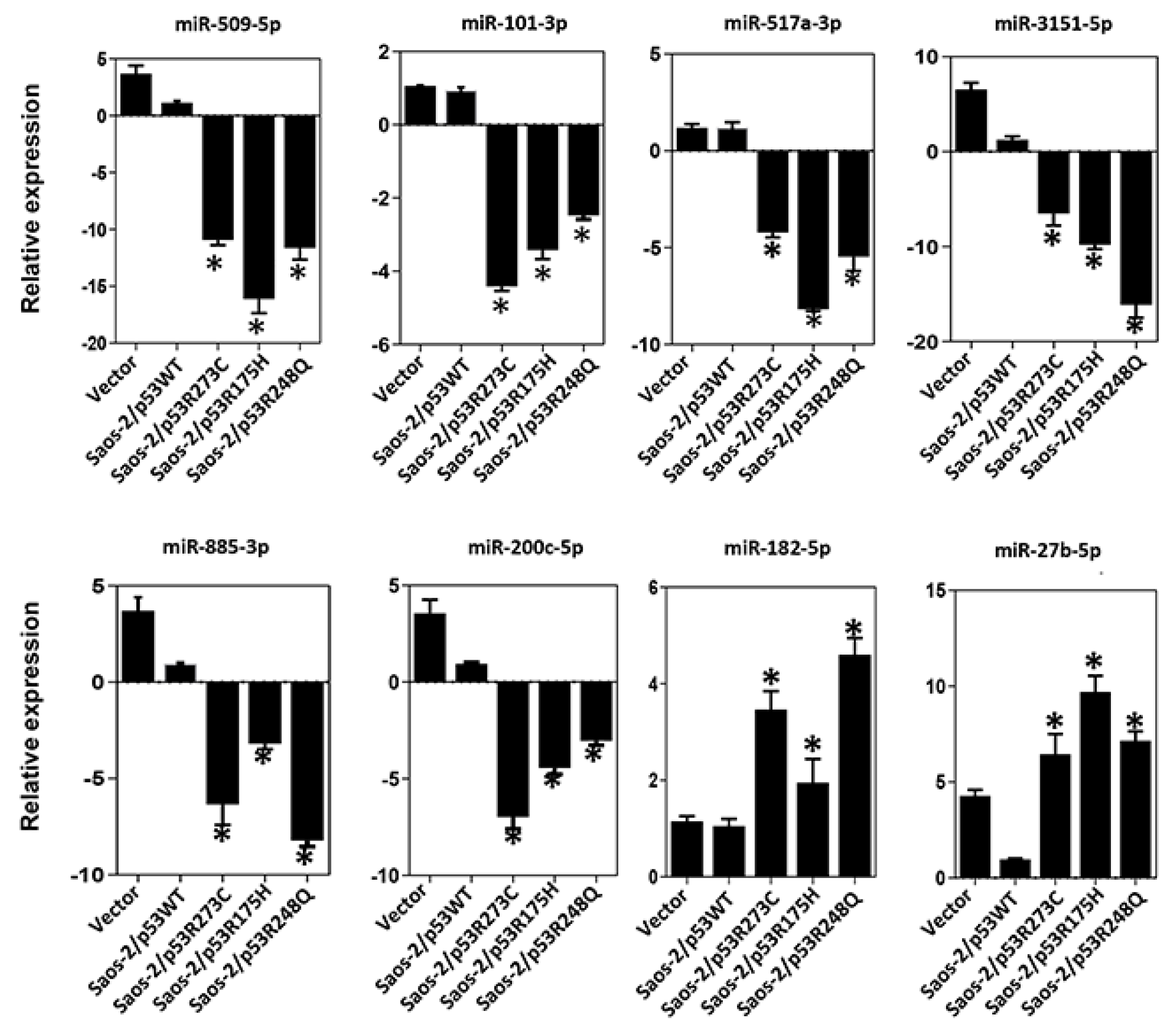
Figure 7.
Mutant p53R248Q stimulates invasion and migration through miR-182 upregulation in Saos-2 cells. a) Relative expression of miR-182-5p in Saos-2 cell line (Null p53) and cell lines with endogenous mutant p53 (OVCAR-3/p53R248Q, C33a/p53R273C and SKBR3/p53R175H) vs MCF10a/p53WT. b) Relative expression of FOXF2 and MTSS1 in Saos-2 (null p53) vs OVCAR-3/p53R248Q (endogenous p53R248Q) and in Saos-2 transfected with empty vector (Empty vector) vs Saos-2/p53R248Q (transfected with p53R248Q). c) Relative expression of miR-182-5p in Saos-2 cells transfected with Control-anti-miR/Vector; Saos-2 transfected with p53R248Q and control anti-miR (p53R248Q/control-anti-miR) or transfected with antimiR-182 (p53R248Q/antimiR-182). RT-PCRs were normalized with GAPDH and/or U6, respectively. d) Cell invasion and migration assays in Saos-2 cells transfected with Control-anti-miR/Vector; Saos-2 transfected with p53R248Q and control anti-miR (p53R248Q/control-anti-miR) or transfected with antimiR-182 (p53R248Q/antimiR-182). e) Relative expression of FOXF2 and MTSS1 in Saos-2 cells transfected with empty vector (Empty vector), transfected with p53WT (Saos-2/p53WT), transfected with p53R248Q and control anti-miR (Saos-2/p53R248Q) or transfected with antimiR-182 (antimiR-182). Error bars represent (mean ± SD) from three independent experiments (n = 3), * p<0.05.
Figure 7.
Mutant p53R248Q stimulates invasion and migration through miR-182 upregulation in Saos-2 cells. a) Relative expression of miR-182-5p in Saos-2 cell line (Null p53) and cell lines with endogenous mutant p53 (OVCAR-3/p53R248Q, C33a/p53R273C and SKBR3/p53R175H) vs MCF10a/p53WT. b) Relative expression of FOXF2 and MTSS1 in Saos-2 (null p53) vs OVCAR-3/p53R248Q (endogenous p53R248Q) and in Saos-2 transfected with empty vector (Empty vector) vs Saos-2/p53R248Q (transfected with p53R248Q). c) Relative expression of miR-182-5p in Saos-2 cells transfected with Control-anti-miR/Vector; Saos-2 transfected with p53R248Q and control anti-miR (p53R248Q/control-anti-miR) or transfected with antimiR-182 (p53R248Q/antimiR-182). RT-PCRs were normalized with GAPDH and/or U6, respectively. d) Cell invasion and migration assays in Saos-2 cells transfected with Control-anti-miR/Vector; Saos-2 transfected with p53R248Q and control anti-miR (p53R248Q/control-anti-miR) or transfected with antimiR-182 (p53R248Q/antimiR-182). e) Relative expression of FOXF2 and MTSS1 in Saos-2 cells transfected with empty vector (Empty vector), transfected with p53WT (Saos-2/p53WT), transfected with p53R248Q and control anti-miR (Saos-2/p53R248Q) or transfected with antimiR-182 (antimiR-182). Error bars represent (mean ± SD) from three independent experiments (n = 3), * p<0.05.
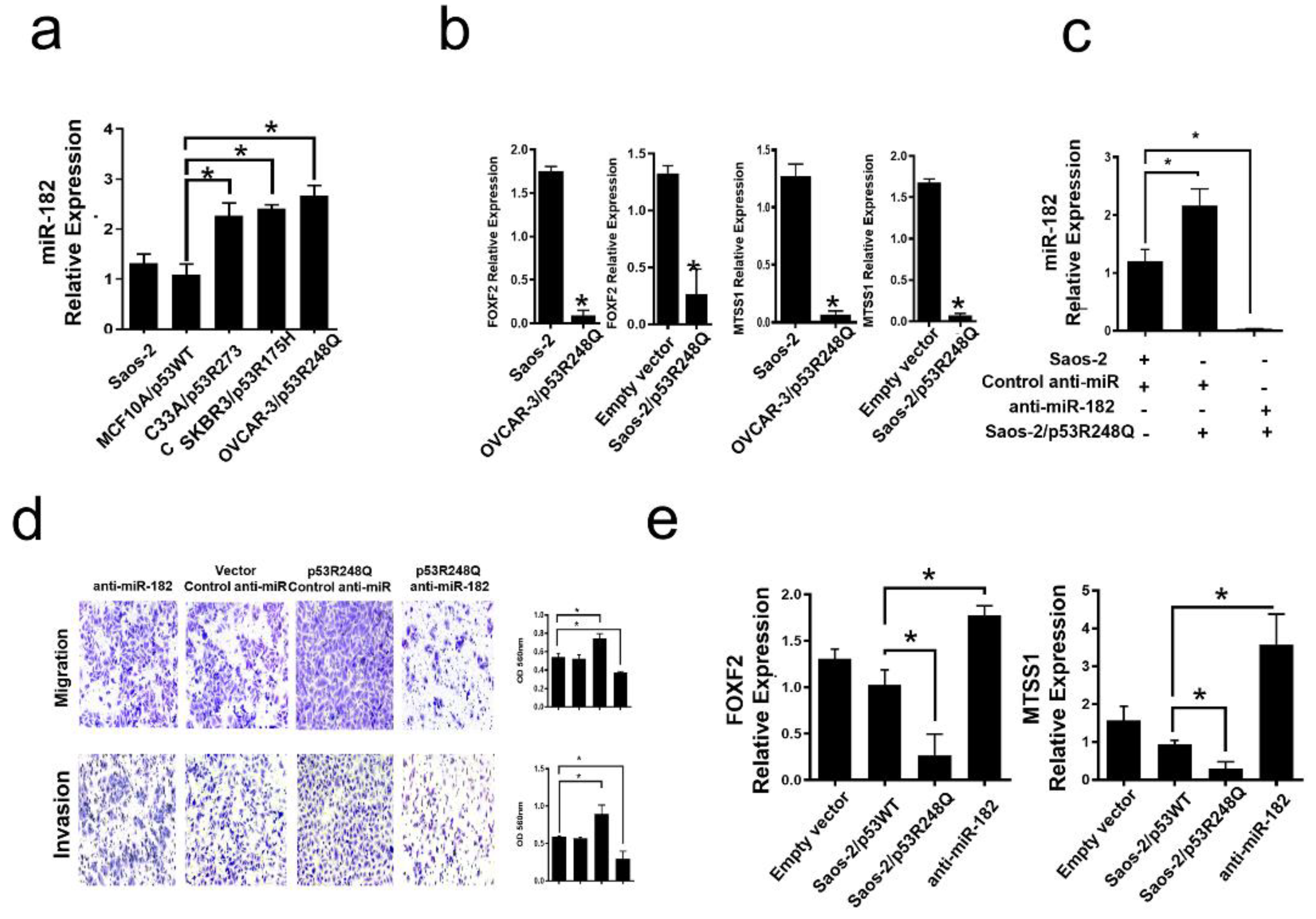
Figure 8.
Inhibition of mutant p53R248Q in OVCAR-3 cells promotes decreased cell invasion and migration through miR-182. a) Relative expression of miR-182-5p and Western Blot of p53R248Q in the cell line OVCAR-3 (endogenous p53R248Q) treated with pifithrin-α (30 μM, 50 μM, 75 μM, 100 μM) for 24 h. using as control cells without treatment (Control) and only vehicle (Vehicle). b) Relative expression levels of FOXF2 and MTSS1 in cells OVCAR-3 (endogenous p53R248Q) treated with pifithrin-α for 24 h compared with cells without treatment (Control) and only vehicle (Vehicle). c) Invasion and migration assays of OVCAR-3 cells with vehicle (Vehicle) or treated with pifithrin-α (Pifithrin 100 μM). Error bars represent (mean ± SD) of three independent experiments (n = 3), * p <0.05.
Figure 8.
Inhibition of mutant p53R248Q in OVCAR-3 cells promotes decreased cell invasion and migration through miR-182. a) Relative expression of miR-182-5p and Western Blot of p53R248Q in the cell line OVCAR-3 (endogenous p53R248Q) treated with pifithrin-α (30 μM, 50 μM, 75 μM, 100 μM) for 24 h. using as control cells without treatment (Control) and only vehicle (Vehicle). b) Relative expression levels of FOXF2 and MTSS1 in cells OVCAR-3 (endogenous p53R248Q) treated with pifithrin-α for 24 h compared with cells without treatment (Control) and only vehicle (Vehicle). c) Invasion and migration assays of OVCAR-3 cells with vehicle (Vehicle) or treated with pifithrin-α (Pifithrin 100 μM). Error bars represent (mean ± SD) of three independent experiments (n = 3), * p <0.05.
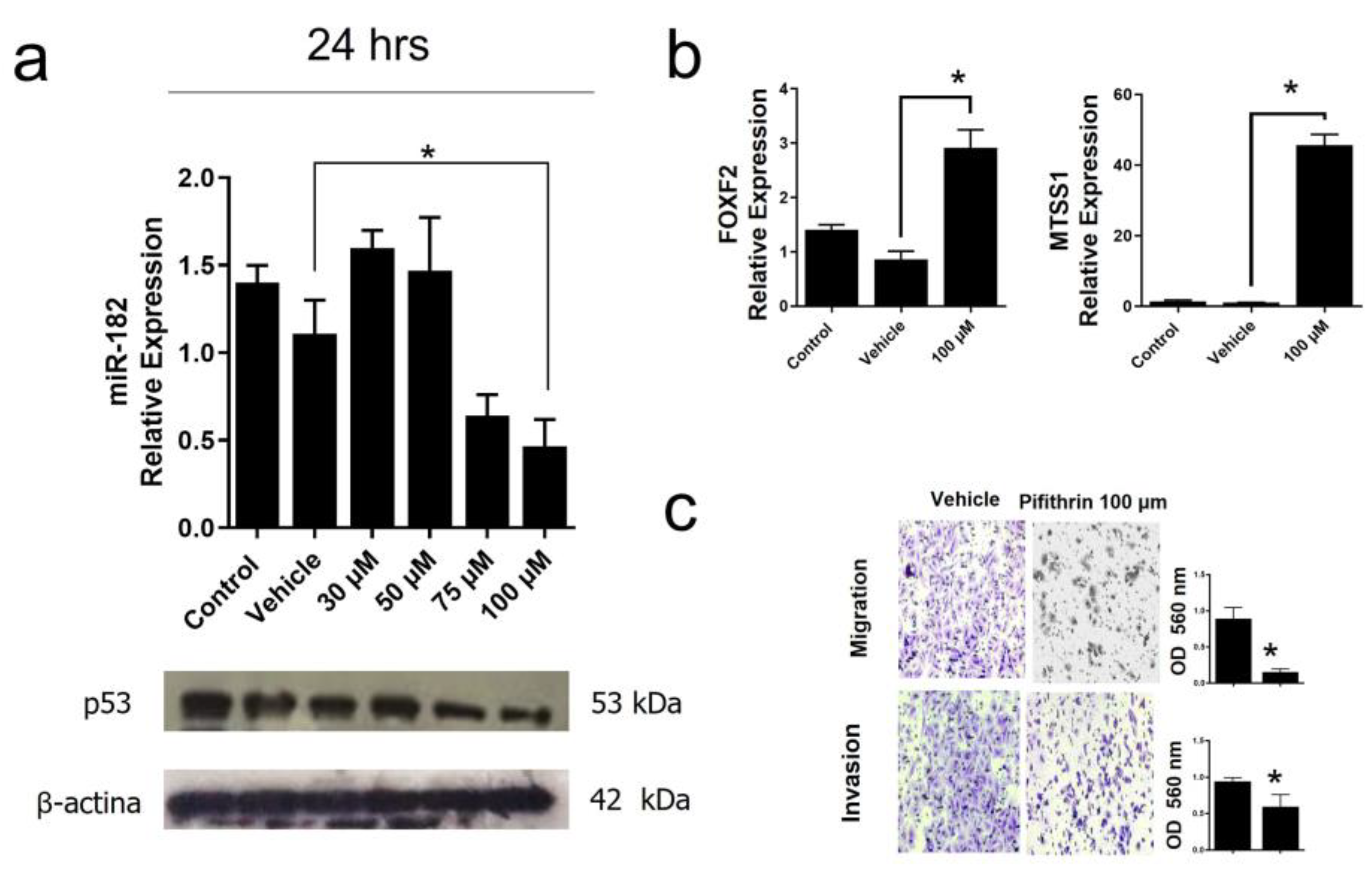
Figure 9.
p53R248Q induces cell migration and invasion through overexpression of miR-182-5p in cancer. The p53R248Q mutant induces overexpression of miR-182-5p, which promotes downregulation of MTSS1 and FOXF2 and increases cell migration and invasion. Mutant p53R248Q is required for the upregulation of miR-182-5p because inhibition of mutant p53 by pifithrin-α has a negative effect on the expression of miR-182-5p and its targets, leading to decreased cell migration and invasion.
Figure 9.
p53R248Q induces cell migration and invasion through overexpression of miR-182-5p in cancer. The p53R248Q mutant induces overexpression of miR-182-5p, which promotes downregulation of MTSS1 and FOXF2 and increases cell migration and invasion. Mutant p53R248Q is required for the upregulation of miR-182-5p because inhibition of mutant p53 by pifithrin-α has a negative effect on the expression of miR-182-5p and its targets, leading to decreased cell migration and invasion.
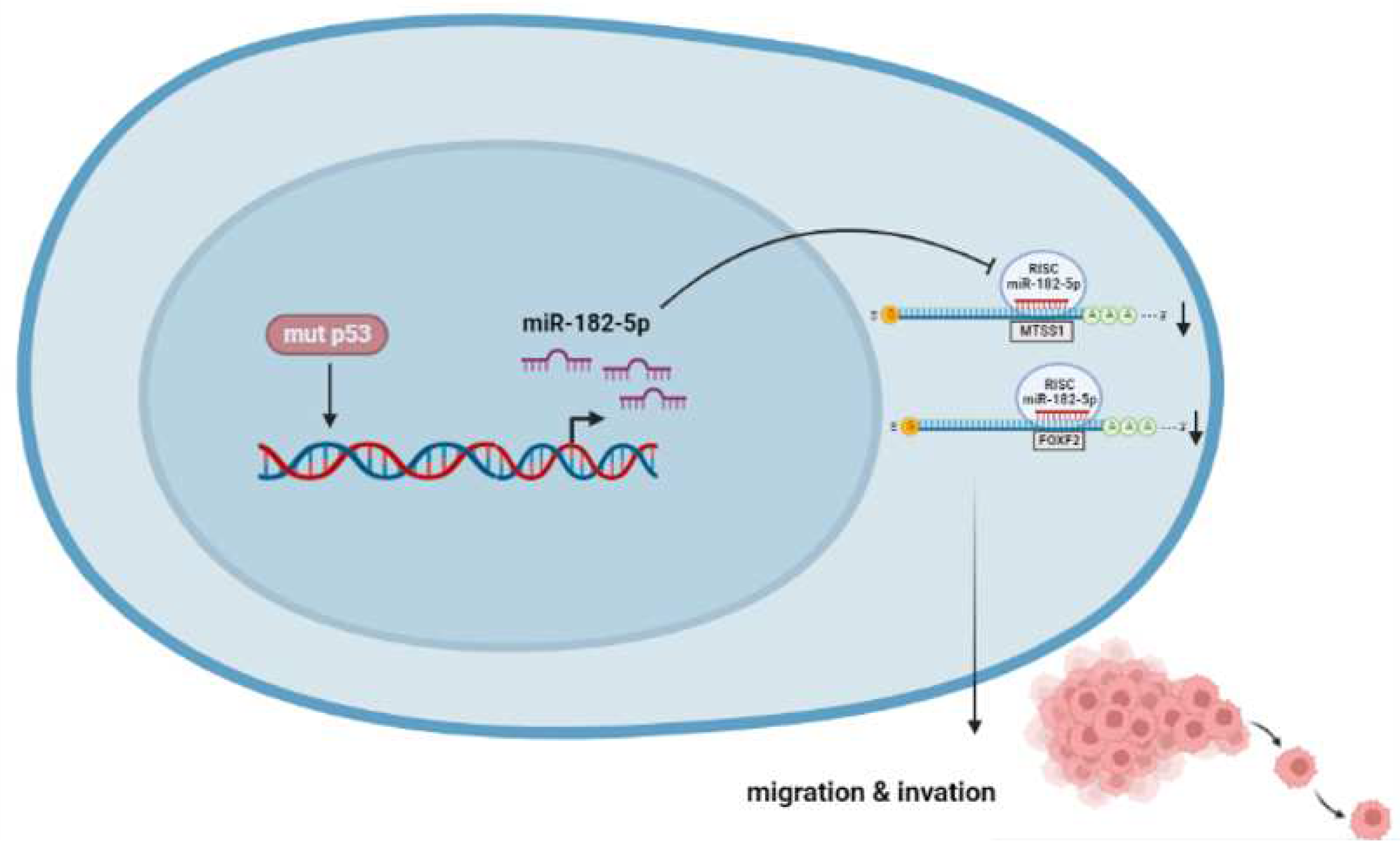
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



