
Submitted:
11 July 2023
Posted:
12 July 2023
You are already at the latest version
Abstract
Alzheimer's disease (AD), the most common neurodegenerative disease and the first cause of dementia worldwide, has no effective treatment yet, and AD's pathological mechanisms are not yet fully understood. We conducted this study to explore the proteomic differences associated with Familial Alzheimer's Disease (FAD) in olfactory ecto-mesenchymal stem cells (MSCs) derived from PSEN1 (A431E) mutation carriers compared with healthy donors paired by age and gender through two label-free liquid chromatography-mass spectrometry approaches. The first analysis compared carrier 1 (patient with symptoms, P1) and its control (healthy donor, C1), and the second the Carrier 2 (patient with pre-symptoms, P2) with its respective control cells (C2) to evaluate whether the protein alterations presented in the symptomatic carrier were also present in pre-symptoms stages. Finally, we analyzed the differential expressed proteins (DEPs) for biological and functional enrichment. These proteins showed an impaired expression in a stage-dependent manner and are involved in energy metabolism, vesicle transport, actin cytoskeleton, cell proliferation, and proteostasis pathways resembling previous AD reports. Our study is the first proteomic analysis in MSCs from FAD patients in two stages of the disease (symptomatic and presymptomatic), showing these cells as a new and excellent in vitro model for future AD studies.
Keywords:
Proteome
; Alzheimer's Disease
; Familial Alzheimer's disease
; PSEN1
; A431E
; Mesenchymal Stem Cells
; Proteostasis
; Olfactory
; Neurodegeneration
; FAD
1. Introduction
Alzheimer's (AD) is the most common neurodegenerative disease, the first cause of dementia, and one of the seven causes of death globally. Due to the accelerated epidemiological growth of aging people, cases will triple over the next 30 years [1,2]. Currently, there are no curative treatments nor adequate diagnostic tools exist. AD is characterized by the formation of extracellular amyloid β (Aβ) aggregates derived from proteolytic cleavage of the Amyloid Precursor Protein (APP), known as amyloid plaques, and by intraneural neurofibrillary tangles (NFT), of hyperphosphorylated Tau in the entorhinal cortex, hippocampus (Hp), limbic system, and temporal cortex principally [3]. These pathological changes start at least 10-20 years before the first symptoms manifest by progressive memory loss and decline of cognitive functions, leading to dementia [4,5].
AD can be classified into Late-Onset Alzheimer's disease (LOAD) or Sporadic Alzheimer's disease (SAD) when it occurs over 65 years old and is of unknown etiology, comprising 95% of all cases, and Early-Onset Alzheimer's disease (EOAD) if it occurs under 65 years of age [6]. Approximately 80% of EOAD relates to an autosomal dominant gene defect in three principal genes: Presenilin-1 (PSEN1), presenilin-2 (PSEN2), and the APP. This inherited subgroup of AD is called Familial Alzheimer's Disease (FAD) [7]. FAD has been a model for AD study due to histopathological and clinical similarities with SAD. However, over 20% of FAD patients develop atypical clinical symptoms such as spastic paraparesis [8], dysarthria, schizophrenia, Parkinsonism, and depression [9,10]. Within the FAD, it is worth noting that a particular mutation has been reported in Mexico named Jalisco mutation A431E of the PSEN1. Almost half of these patients present spastic paraparesis, language impairments, and psychiatric and motor manifestations. Due to the genetic characteristics of FAD and its complete penetrance, it is crucial to study the early stages of the disease and develop new therapies [11,12].
To accomplish this, label-free UPLC-HDMSE quantitative proteomics, which uses proteome analysis with the highest coverage to look at changes in the proteomics profile, can be used to find novel pathways, molecular mechanisms, and therapeutic targets that may significantly advance our understanding of the disease [13]. Although recently, many proteomics studies have screened dysregulated proteins in various biological samples derived from patients with AD [14] like some postmortem brain tissues [15,16,17,18] cerebrospinal fluid (CSF) [19,20], blood (serum/plasma) [21,22] and even saliva [23]. Unfortunately, these samples have abundant proteins that can hide the identification of essential proteins or are not directly related to the brain's disease. The most recent research models, such as patient-derived mesenchymal cells, aid in understanding and tracking the physiopathology of the illness, discovering novel biomarkers, and creating novel therapeutics and diagnostics for the prevention or treatment of AD [24,25].
Olfactory ecto-mesenchymal stem cells (MSCs) have recently been discovered [26,27], and because of their ectoderm origin and mesenchymal characteristics, they have the benefit of having high neurogenic potential. Additionally, these cells are crucial to the disease because most AD patients also have hyposmia years before symptoms appear [28,29].
In the early stages of the disease, studies have shown the existence of Aβ and Tau protein aggregates in the olfactory pathway, olfactory bulb (OB), and neuroepithelium [30,31], as well as altered neurogenesis in AD with lower viability of neurons in comparison to controls [32]. Because of these factors, olfactory MSCs might provide a fresh and trustworthy research model for understanding AD. This work evaluates two stages of the disease (Presymptomatic and Symptomatic) in MSCs generated from human carriers of the PSEN1 (A431E) mutation. It is the first quantitative proteome investigation of its kind. Two Label-Free proteomic techniques were used to compare MSCs from mutation carriers and MSCs from healthy donors to identify changes in their overall protein expression linked to FAD and better understand the molecular pathways behind the progression of the disease.
2. Results
Two healthy adult donors and two PSEN1 (A431E) mutation carriers, whose clinical histories, neurological and cognitive information were gathered and described by Santos-Mandujano [33] (See Material and methods), were used to obtain and employ olfactory MSCs at pass 9. Siblings who carry the mutated gene have a three-generational history of lower limb paraparesis that has progressed. Figure 1 depicts the family's pedigree. Cells from symptomatic (P1) and presymptomatic (P2) patients were employed in this study.
We use label-free quantitative proteomic analysis to assess the DEPs between presymptomatic, symptomatic, and its control of MSCs. Figure 2 depicts the experimental process used in proteomics investigations, thoroughly described in the procedures. We gathered and characterized olfactory MSCs using flow cytometry and collected protein data using LC-MS/MS on a Synapt G2-Si in MSE. The raw mass spectrometry data were analyzed with Progenesis QI software and searched in WebGestalt, UniProt, Panther, Reactome, STRING, and GeneCards for biological and functional analysis.
2.1. Protein expression in symptomatic and presymptomatic PSEN1(A431E) carriers (P1, P2) and controls (C1, C2).
The first patient with the PSEN1(A431E) mutation is symptomatic (P1). He is a 54-year-old man with a 7-year history of lower extremity motor impairment, minor cognitive impairment, typical upper motor neuron disease symptoms, and anosmia. The second carrier is at the presymptomatic stage (P2). P2 is a 44-year-old lady with normal cognitive function, little leg weariness, generalized hyperreflexia, and hyposmia. The presymptomatic control (C2) and the symptomatic control (C1) were represented by healthy 42-year-old women and 55-year-old men, respectively.
2.1.1. Isolation and characterization of olfactory MSCs
Nasal exfoliates from research participants displayed varied cellular morphologies in the initial passes. However, a uniform population of MSCs emerged following the 5 passes [26]. Cells from nine passes of flow cytometry were used to confirm the expression of specific markers for mesenchymal stem cells (CD105, CD90, and CD73) and the absence of hematological markers and differentiated cells (CD34, CD45, CD14, CD19, and CD166) [34]. Figure 3 displays histograms contrasting P1 and P2 with C1 and C2, respectively.
2.1.2. Label-free UPLC-HDMSE analysis for P1 vs. C1.
We conducted a quantitative label-free proteome analysis after confirming that the isolated cells are homogenous MSCs to uncover the changed protein expression profile in MSCs from the pre and symptomatic carrier (P1 and P2) in comparison to the MSCs from its controls (C1 and C2) respectively, Figure 4.
2.1.3. Peptide and protein reliability and confidence.
At the peptide and protein levels, data quality control criteria were used. Our peptide analysis used ion mobility mass spectrometry (IM-MS) to consider signals with [M + 2H]2+ or more charges. There were 257,971 peptides detected in the symptomatic patient and 185,976 peptides in the presymptomatic patient; accuracy was 74.72% and 83.15%, respectively, with a maximum of 5 ppm (Figure 4A, E). Based on peptide-matched type, peptides were categorized (Figure 4C, G). The peptides with the highest quality (PepFrag1) in P1 were 75.9% and 76.1% in P2, those with lower reliability (PepFrag2) were 3.7% and 2.7%, missing cleavage peptides were 13.7% and 15.8%, variable modification peptides were 6.1% and 5.3% respectively. Both analyses showed that the Peptides fragmented at the ion source were less than 1%. The PepFrag1 did not exceed an error of ±10 ppm, consistent with the full range m/z 400−1600 (Figure 4B, F). These results show a proper mass spectrometer calibration, a good quality of peptides, and efficient enzymatic digestion.
At the protein level, 4750 proteins were identified in P1 and 3971 in P2, averaging 5.7 and 5.67 peptides per protein. 2870 and 2361 proteins were not quantified, and 330 and 246 were reversed sequences. The pattern of the significant values in P1 vs. P2 was maintained. Finally, 1553 proteins in P1 and 1277 in P2 were tested, with an average of 10.34 and 9.69 peptides per protein and a dynamic range of 6.5 and 6 orders of magnitude, respectively. These results revealed that the normalization of injection had been done correctly, with both conditions being highly sensitively comparable (Figure 4D, H). Showing that our analysis has proper reliability and confidence at the peptide/protein level based on Figures of Merit (FOMs) for label-free proteomic studies by Souza et al. [35].
Additionally, we identified 11 and 12 unique proteins for P1 and P2 (Table 1), 3 for C1, and 20 for C2. Tables S1 and S4 have a summary of all these findings.
2.1.2.2. Data Filtering and differentially expressed proteins in P1 and P2 MSCs
It considered only proteins with more than two identified peptides per protein and at least one unique peptide, an ANOVA p-value ≤ 0.05, and a coefficient of variation (CV) ≤ 0.3. Remove reversed proteins that were false positives and proteins in each condition in one of the three duplicates. Fold change (FC) levels of downregulated and upregulated proteins, respectively, are 0.6 and >1.5 in the differential expression. With these restrictive filters, 703 and 517 highly reliable quantified proteins remained in P1/C1 and P2/C2. Of these, 359 and 195 differentially expressed proteins (DEPs) resulted. For P1/C1, 118 proteins were upregulated, and 241 were downregulated. For P2/C2, 75 were upregulated, and 120 were downregulated, scattered in volcano plots (Figure 5A, C). Contrary to P2/C2, which had less than 50% of the proteome modified, P1 MSCs' proteome was altered by more than 50% compared to their C1 control (Figure 5B, D). The top 20 proteins up- and down-regulated in MSCs from P1 to C1 are shown in Table 2 and Table 3. P1 had a false discovery rate (FDR) of 2.8%, C1 had an FDR of 2.5%, P2 was 1.72%, and C2 was 1.75%. Tables S2 and S5 overview all differentially expressed proteins in both carriers.
HBB protein was the most down-regulated protein with -189.5 FC, followed by KIAA1551 protein with -52 FC. The most over-regulated proteins with these stringent filters were GUCY2C and PRPF4B, with 34.9 and 25.9 FC. For P2, the two genes that were most negatively regulated were SCIN and EEF1E1-BLOC1S5, with -14.9 and -9.7, and the two genes that were most positively regulated were DNM1L and COPS8, with 94.5 and 35 of FC.
2.1.3. Functional and biological analysis of DEPs in P1/C1 and P2/C2 analysis
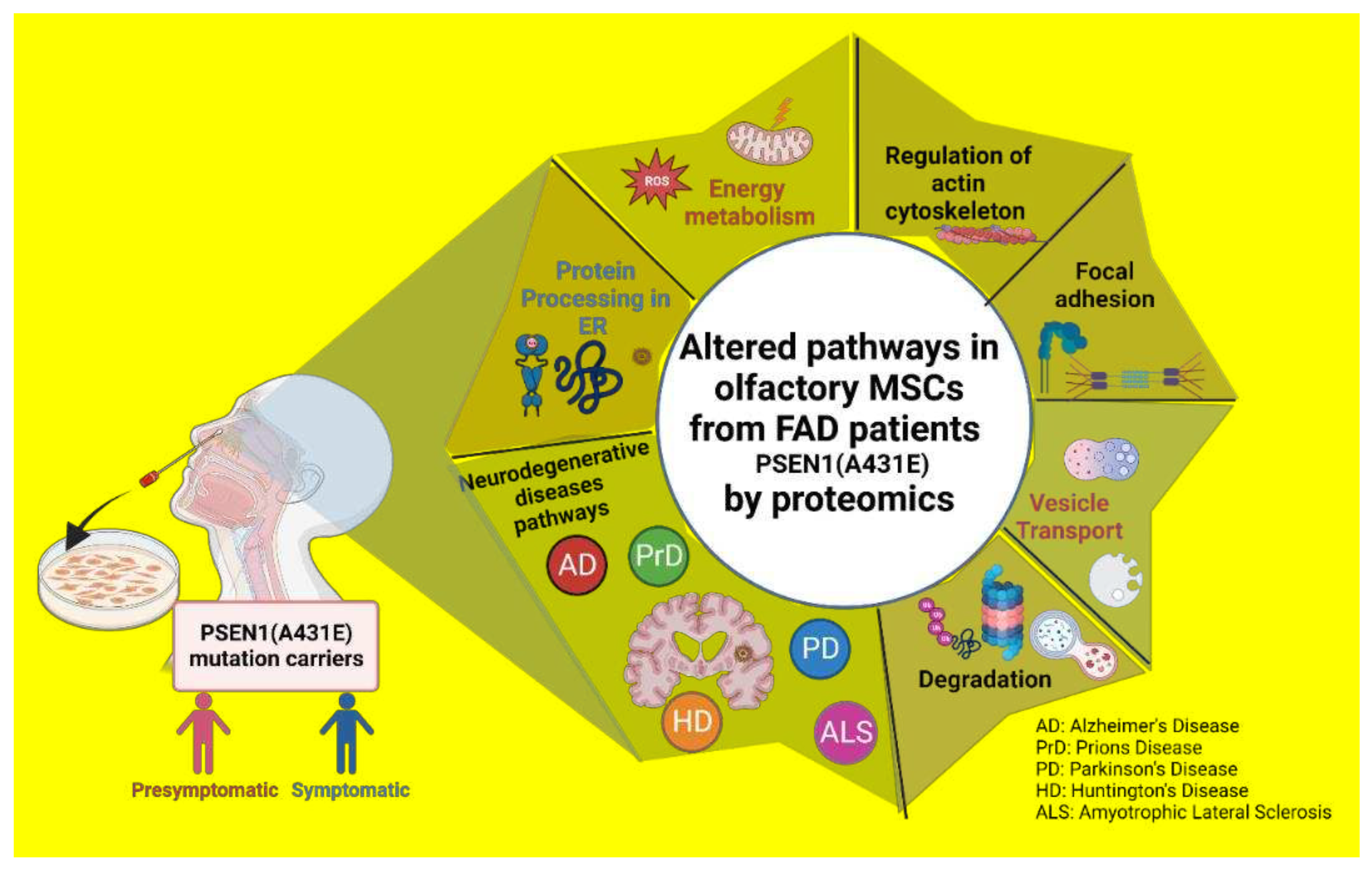
We performed a functional analysis of proteins with differential expression. GO and KEGG enrichment analyses were carried out using WebGestalt. Tables S3 and S6 display these outcomes and all functional enrichment analyses performed on WEBGESTALT and Gene Analytics. In contrast to P2/C2 biological processes, predominantly linked to leukocyte immunity, degranulation, and exocytosis, DEPs of P1/C1 are involved in protein folding and the endoplasmic reticulum stress response. (Figure 6A,D). In P1/C1 and P2/C2, the cell cortex and focal adhesions in both mutation carriers and cell junction substrate were the key cellular components of DEPs (Figure 6B, E). Similar to this, both carriers had the primary altered molecular activities of binding to cadherins and binding to cell adhesion molecules, whereas P1/C1 had changed cytoskeleton binding proteins and P2/C2 had altered proteins binding to actin filaments (Figure 6C, F).
The analysis of KEGG enrichment pathways with DEPs (Table 4 and Table 5 and Figure 7) revealed enrichment in pathways involved in energy metabolism, including the pentose phosphate pathway, amino acid biosynthesis, glutathione metabolism, carbon metabolism, and citrate cycle, as well as focal adhesion pathways and actin cytoskeleton regulation, even though the altered genes were different between the P1 and P2 mutation carriers. However, distinct pathways are enriched for each carrier, including phagosome or endocytosis pathways (Table 5) in P2 and neurodegenerative disease (NDD) pathways, such as Huntington's disease in P1(Table 4).
We also conducted pathway enrichment studies using WebGestalt with PANTHER and REACTOME databases (Table S3, S6). The sole pathway presented across all databases is the pentose phosphate route, an essential step in glucose metabolism that produces ribose-5-phosphate and NADPH. These compounds are crucial for maintaining redox equilibrium and nucleotide biosynthesis.
STRING was used to predict and show Protein-Protein Interactions (PPIs) between DEPs in MSCs from the symptomatic carrier compared to its control using the highest confidence score (0.9). According to KEGG-enriched pathways, proteins were highlighted, then categorized into clusters based on the overall biological processes they are engaged in. The leading group, in the middle of the network, with the highest number of proteins and interactions, are proteins related to protein processing in ER (red nodes), transport and degradation mechanisms (pink cluster), and focal adhesion (purple cluster). The biological processes were: energy metabolism pathways (green group), ECM-receptor interaction, and adhesion (purple cluster). We also discovered proteins (yellow group) involved in Interferon signaling (Figure 8A). However, because there were more DEPs in P1. MSCs than in the P2 carrier, the P2 MSC interactome was less complicated (Figure 8B). In addition, interferon signaling pathways and energy metabolism were also grouped. In contrast to P1 DEPs, protein, and translation-related pathways were grouped, and the core cluster was connected to vesicle secretion and transport.
Intriguingly, KEGG and STRING data showed enrichment in the pathways of the significant and most prevalent NDDs and proteinopathies, including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), prion disease (PrD), and Amyotrophic Lateral Sclerosis (ALS) (Table 6).
Since protein processing in the ER is the pathway with the most proteins engaged and a substantial number of interactions in P1, we conducted an individual interactome using these proteins and their direct protein connections. The majority were chaperones, STIP1, an adaptor protein that controls the ATPase cycles of some chaperones like HSP70 and HSP90. SOD1, a protein implicated in ROS detoxification, and three prolyl hydroxylases (P4HA1, P4HA2, and CRTAP) were down-regulated. The only elevated proteins (Figure 9A) included the stress-related proteins DCTN1, NUP214, and MAPK1, as well as CRYAB, SAR1A, and ERO1A. The physiological decline of chaperones and change of proteostasis in older people may have something to do with the early proteostasis imbalance, aggregation of misfolded proteins, and increased vulnerability to stress response in these patients.
As opposed to the P2 interactome (Figure 9B) of vesicle secretion and transport, where 13 proteins were elevated, and 12 proteins were downregulated with proteins also associated with phagosome and lysosome pathways (TUBB3, HLA-A, ITGA2, ITGB1, THBS1, CD63, PSAP)
2.3. Comparision of DEPs between PSEN(A431E) mutation carriers and with other NDD and AD DEPs previously reported
To compare the pathways between the DEPs involved in mutation carriers, Table 6 highlights the most critical pathways in both proteomic investigations.
2.3.1. Correlation between the proteomes of olfactory MSCs from PSEN1(A431E) mutant carriers with their clinical histories.
We assessed the conditions that might be involved using GeneAnalytics and WEBGESTALT with OMIM and DISGENET databases (Table S3 and S6), which allow us to compare the proteins that have been differentially expressed and reported as biomarkers in other diseases because our results contain proteins involved in some signaling pathways that play a crucial role in the biology of NDDs. Weirdly, using the WEBGESTALT tool, Spinocerebellar Ataxia, distal limb weakness, and muscular weakness were the pathogenic factors that led to symptomatic DEPs, which coincides with the spastic paraparesis displayed by the symptomatic carrier. Bulbar palsy is a condition where the region controlling the lower motor neurons accountable for speaking, chewing, and swallowing is altered. This finding is noteworthy because Santos-Mandujano noted that the patient's speech fluency, shortness of breath, and difficulty eating and drinking worsened [33].
Using GeneAnalytics, we obtained similar results for muscular dystrophy, ALS, FAD with 32 proteins matched (3 DE in brain PI4KA, S100A4, and DES, 4 in blood; DNAH6, CEP152, CRMP1, ITGA11 and 25 GeneCards inferred as ADAM10, ALB, HSPA1A, HSPA5, MAPK1, NOS2, SOD1, and STAT1), HD and surprisingly hereditary spastic paraplegia with 24 matched genes, ALDH18A1 as a causative mutation, 13 differentially expressed in skeletal muscle as BAG2, CANX, SCARB2, and UBR5 and 12 inferred genes (ALDH18A1, CANX, DCTN1, FUS, ITPR1, KIF5B, KIF5C, MATR3, REEP5, SETX, SOD1, and VPS13D). In the presymptomatic case, the analysis also revealed muscle degeneration and atrophy, particularly in the lower limbs, and abnormality in upper motor neuron disease, in which weakness, spasticity, and hyperreflexia are classic symptoms. The presymptomatic patient presented hyperreflexia. GeneAnalytics shows 30 proteins matched with FAD (4 DE in blood: CSPG4, DNAH6, TPM1, and TRIO. CSF1R as causative mutation and 26 inferred genes) and 24 with Hereditary Spastic Paraplegia (10 DE in skeletal muscle: as ACSL3, CFL2, GOLGA4, and ITGB1, 3 causative mutation ALD18A1, SACS, and WASHC5 and 14 inferred genes). These studies imply that progenitor cells of PSEN1 (A431E) mutation carriers may be connected with the clinic in these individuals, but further investigation is necessary to confirm these findings.
3. Discussion
Our understanding of the genesis of AD is mainly focused on the brain and neuronal cells. The molecular changes in olfactory Mesenchymal Stem cells are still unclear, though. It is generally known that these cells' epigenetic characteristics, cellular capabilities, and gene expression were preserved during in vitro culture. In order to compare the differences in protein expression between olfactory MSCs from patients with the PSEN1(A431E) mutation and healthy donors, two proteome profiles from various phases of FAD were examined. The significance of these DEPs was investigated further using bioinformatics and bibliographic techniques, including gene ontology analysis, KEGG pathway analysis, PPI network creation, and comparison with other AD databases.
3.1. Exclusive proteins of PSEN1(A431E) mutation carriers
The symptomatic carrier (P1) had eleven distinct proteins. Principally concerned with viability, proliferation, migration, and inflammation, which may account for the carrier's olfactory MSCs' observed difficulties in growth and proliferation.
The proteins involved in neurogenesis, brain development, self-renewal, survival, and plasticity include WDR62, MCPH1, and CDK5RAP2. Numerous neurological disorders, including Alzheimer's disease and microcephaly, have been linked to mutations in these proteins [36,37]. Because JNK and CDK5 kinases control tau phosphorylation, WDR62 and CDK5RAP2 may indirectly influence the development of neurofibrillary tangles (NFTs) [37,38].
Genes linked to the pathology of Alzheimer's disease may be affected by the expression of histone H1.5, encoded by the HIST1H1B gene and UTY. H1.5 affects the accessibility and structure of chromatin, and UTY is a histone demethylase involved in the epigenetic control of gene expression and cell differentiation. It has been reported that HIST1H1B can create amyloid-like fibers after it was discovered in amyloid plaques [39,40].
KIF5C is a kinesin that controls mitochondrial transport as well as the movement of organelles across microtubules. KIF5C decreased in mitochondria but rose in synaptosomal-enriched fractions in PS1-E280A mutant carriers [41]. SCUBE2 participates in SHH signaling and, when overexpressed, decreases glioma cell proliferation, migration, and invasion [42]. Epileptic encephalopathy and MCAHS1 (multiple congenital anomalies-hypotonia-seizures syndrome) are both brought on by PIGN mutations. Phosphatidylethanolamine (PE) is transferred from the ER to the first mannose of glycosylphosphatidylinositol (GPI) by the protein PIGN. Increased PE levels have been seen in mitochondria, plasma membranes, and ER-associated mitochondrial membranes (MAMs) in fibroblasts from patients with PSEN1 and PSEN2 mutations. These alterations could lead to an increase in γ-secretase activity and the generation of amyloid−β [43].
Furthermore, MCAHS1 mutations created by CRISPR/Cas9 in C. elegans result in protein aggregation and the activation of the UPR pathway in the ER [44] ATP5PF, a component of the mitochondrial ATP synthase and is in charge of producing ATP, has been linked to Alzheimer's disease, neurodegeneration, and oxidative stress as early manifestations of this pathology [45,46]. The functions of KIAA0825 and ANKRD36C are unknown; however, they may be connected to several activities, including inflammation [47].
The unique proteins found in the presymptomatic PSEN1 (A431E) mutation carrier are primarily involved in transcriptional regulation (RTRAF, DIP2C, and PROX1), mitochondrial function (ATP5H and TIMM13), proliferation control (CNBP, PROX1), neuronal development (PROX1, DIP2C), and cellular transport (FCHO1 DYNLRB1, BIN3, and CUL9), suggesting a potential compensatory or resistance process in the cell to maintain proper cell function. Interestingly, neurological illnesses such as autism, neurodevelopmental disorders, deafness dystonia syndrome, microcephaly, Alzheimer's disease, and several cancer types have been associated with RTRAF, DIP2C, YARS, CNBP, BIN3, DYNLRB1, and TIMM13. In addition, DIP2 paralogues and spastic hemiplegia are connected [48].
Tyrosyl-tRNA synthetase (YARS) mutations have been linked to Charcot-Marie-Tooth disease. This neuropathy causes progressive loss of muscle tissue and touch sensitivity in different body parts [49]. Additionally, elevated nuclear levels of YARS have been observed in a cell model of TDP-43 aggregation, a protein that builds up in ALS, and tau-negative frontotemporal lobar degeneration (FTLD) [50]. Type 2 myotonic dystrophy and sporadic inclusion body myositis disease (sIBM), an inflammatory muscle condition defined by the buildup of intramuscular Amyloid-β aggregates similar to those found in Alzheimer's disease, increasing muscle weakening, and atrophy [51], are both associated with CNBP [52]. These all resemble the spastic paraparesis experienced by carriers of this mutation and may be caused by proteins connected to the pathophysiology of these patients.
In sum, these specific proteins in presymptomatic PSEN1 (A431E) mutant carriers may indicate a complex interaction of cellular processes that offset the consequences of the mutation in MSCs at the early stages of the disease.
3.2. Downregulated proteins in PSEN1(A431E) mutation carriers
Most downregulated proteins in symptomatic carriers are distinct proteins that appear to be connected to changes in cell proliferation, cycle arrest, differentiation, and susceptibility to damage and ROS generation.
Unexpectedly, the α and β chains of hemoglobin, the protein that transports oxygen and carbon dioxide in cells of the erythroid lineage (FC-27.6 and -189.5, respectively), are two of the most downregulated proteins in symptomatic mutation carriers compared to the control. Hb chains, however, have been found in non-erythroid cells [53], in primary cell cultures of glial cells and neurons from the murine brain, and even in amyloid-β deposits [54,55,56]. Although mesenchymal cells have not been shown to express Hb, two Hb chains were discovered in a proteomic analysis of the olfactory bulb in 2012 [57].
Our findings corroborate those of Ferrer I. et al. [58], who found that nearly all neurons in senile plaques with hyperphosphorylated tau deposits and amyloid-β-core decreased in both hemoglobin chains. The downregulation of Hb chains indicates a change in oxygen homeostasis and reactive oxygen species detoxification.
The KIAA1551 (RESF1-retroelement silencing factor 1) is a protein necessary to maintain the repressive gene state in undifferentiated embryonic stem cells [59]. Downregulation of RASA2, CEP192, ZGRF1, and ITGA2 reduces cell migration, adhesion, and proliferation while also causing cell cycle arrest, which may have an impact on the phenotypic and course of the disease as it is now understood to exist. The maturation of the mitotic centrosome and the formation of the bipolar spindle both depend on the protein CEP192 (Centrosomal Protein 192). Cytosolic microtubule organizing centers (MTOCs) was in excess in CEP192 knockdown cells [60]. Several DNA-damaging substances, including PARPi and radiation, boosted the sensitivity of ZGRF1 null cells [61].
Integrin alpha 2 (ITGA2), an extracellular matrix receptor for laminin, collagen, fibronectin, and E-cadherin, plays a role in promoting cell proliferation and invasion in different types of cancer [62]. Cancer cells lost their ability to differentiate into epithelial cells and experienced a motility limitation due to the loss of the 2-integrin subunit [63,64]. With an FC of -3.8, ITGA2 is also one of the most downregulated proteins in the presymptomatic carrier.
Interferon induces several proteins, including MX1 (Interferon-induced GTP-binding protein Mx1), OAS3 (2'5'-oligoadenylate synthetase 3), and IFIT3 (Interferon-induced protein containing tetratricopeptide repeats 3), whose downregulation may prevent cell migration and proliferation. It is interesting to note that, in contrast to our findings, MxA expression has been found in senile plaques and reactive microglia in AD brains [65], and MxA polymorphisms have been linked to an increased risk of Alzheimer's disease, faster cognitive decline, and Multiple Sclerosis [66]. Apoptosis results from the knockdown of IFIT3 in lung epithelial cells [67].
Due to their involvement in myofibrillar myopathies and muscular dystrophy, which are diseases marked by progressive skeletal muscle weakness, atrophy, autolysis/proteolysis, and autophagy of muscle cells, Desmin and Dysferlin are among the top 20 most downregulated proteins that are noteworthy to mention [68].
Numerous oncogenes downregulated in the presymptomatic carrier are found among the downregulated proteins, which may also indicate changes in protein trafficking, apoptosis, and proliferation. Intriguingly, 4 of the proteins downregulated the greatest in the presymptomatic carrier also changed the symptomatic carrier. SCIN and LRP1B were downregulated, in contrast to UTY, a unique symptomatic carrier protein.
Cancer cell migration, proliferation, apoptosis, and invasion are correlated with SCIN, LRP1B, CPNE3, TICRR, and PPP1R7. A calcium-dependent actin filament protein called SCIN controls exocytosis and vesicle transit by arranging the cytoskeleton [69]. SCIN knockdown reduced proliferation, migration, and morphology in dental pulp stem cells (DPCs) [70].
Since LDL receptors are ApoE receptors and control both the clearance of Aβ and the amyloidogenic processing of APP, they are connected to the pathogenesis of AD [71,72]. Both PSEN1(A431E) mutant carriers exhibit downregulation of the tumor suppressor gene LRP1B (Low-density lipoprotein receptor-related protein 1B). LRP1B and APP interact, preventing APP from being internally processed and favoring not amyloidogenic processing [73]. Additionally, complement C1q protein promotes LRP1B expression and protects immature and adult neurons against fibrillar and oligomeric Aβ toxicity [74]. However, our findings indicate a decreased neuroprotective impact against Aβ toxicity in MSCs.
The protein-coding gene EEF1E1-BLOC1S5, or AIMP3, is an accessory part of the macromolecular tRNA synthase complex. When it was lacking in mouse embryonic stem cells (mESCs), an accumulation of DNA damage occurred, which caused p53 to become transcriptionally active and cause a loss of stemness and differentiation potential [75]. However, Kim et al. discovered that AIMP3 is inhibited under hypoxia, slowing cell senescence, lowering mitochondrial respiration, and activating autophagy. Additionally, it is favorably controlled by Notch3 in human placenta amnion-derived mesenchymal stem cells (hpMSCs) [76].
Non-muscle myosin II heavy chain (MYH10) inhibits the Wnt/-Catenin pathway and decreases glioma cell migration and invasion [77].
NTRK2, which codes for the tropomyosin receptor kinase TrkB, a neurotrophin receptor with a high affinity for BDNF, is another protein among the most downregulated that merits mentioning because it has been suggested as a potential candidate gene for AD [78].
Heat shock proteins (HSP27, HSP90, and HSP70) and mtp53 are downregulated by the knockdown of ribophorin II (RPN2), an essential subunit of the N-oligosaccharyl transferase (OST) complex in breast cancer stem cells [79]. RNF6 is an E3 ubiquitin ligase protein participating in IFN-antiviral response and proteasome degradation [80]. Additionally, RNF6 is silenced, which increases GSK3 activity, decreases p-GSK3 inhibition of the Wnt/-catenin pathway in cancer cells, and suppresses MAPK/ERK signaling, which in turn reduces cell growth and proliferation [81,82].
3.3. Upregulated proteins in PSEN1(A431E) mutation carriers
Many of the most upregulated proteins in symptomatic carriers are overexpressed in various cancer types and linked to cell proliferation, migration, and cell cycle progression via Notch1, -catenin, NF-B, AKT, and ERK1 signaling, pathways closely associated with AD, like PRPF4B, S100A16, PRPS2, PIK3C2B, MAPRE1, and SVEP1. There is evidence for both the opposite effects and suppression of cell proliferation, such as the overexpression of PRP4 in hepatocarcinoma cells (HCC), which caused cell growth retardation by inducing cell cycle arrest at the G1/S checkpoint [83], and S100A16, which inhibits proliferation, migration, and invasion via the JNK/p38 MAPK pathway [84].
Also, Guanylate Cyclase 2C or Heat Stable Enterotoxin Receptor (GUCY2C or GC-C), the most upregulated protein in the symptomatic carrier, inhibits phosphodiesterase 3 (PDE3) inducing cell cycle arrest cytostasis and senescence via activation of p21, PKGII, and p38 MAPK [85], inhibits proliferation by decreasing AkT signaling, β-catenin and promotes cell differentiation and migration in Colorectal Carcinoma Cells [86,87]. Consequently, the overexpression of these proteins may be a factor in the symptomatic mutation carrier's diminished migration and proliferation.
The S100 calcium-binding protein A16 (S100A16) has been linked to psychiatric disorders, depression, and neurodegeneration [88]. S100A16 promotes the Wnt/-catenin signaling pathway and the HRD1-induced ubiquitination and degradation of GSK3 and CK1 in cases of acute renal injury [89]. ER stress indicators like GRP78, p-IRE1, and XBP1s were upregulated in HK-2 cells after S100A16 overexpression [90].
A key player in the production of purines and pyrimidines, phosphoribosyl pyrophosphate synthetase 2 (PRPS2) acts as an oncogene by controlling the activities of matrix metalloproteinase 9 and the expression of E-cadherin [91,92]. Tumor suppressor gene TSC2 (Tuberous Sclerosis Complex subunit 2 or Tuberin) negatively controls mTORC1 signaling and is crucial for autophagy and dendritic formation [93,94]. While the frontal cortex of people with Alzheimer's disease or Down syndrome has decreased tuberin levels [95], other studies have found a significant upregulation of phospho-tuberin (Thr1462) in the post-mortem frontal cortex of people with AD and PD/DLB, which is mediated by the Akt-PTEN pathway [96]. TSC2 is linked to neurological deficits like epilepsy, autism, and intellectual disabilities.
EB1 or MAPRE1 controls the development of spindles, the stability of chromosomes, and microtubule architecture. Recently, Hahn et al. [97] reported that Eb1, XMAP215, and Tau cooperate to promote the polymerization of microtubules (MT), are also involved in the maintenance of mitochondrial morphology and dynamics, and interact with Bax and Bak to promote cell apoptosis via reactive oxygen species [98] form heterodimers with EB2 to promote inner mitochondrial membrane degradation [99], besides can regulate autophagosome biogenesis interacting with Beclin-1 to enhance PI3KC3 [100].
The one transmembrane immunoglobulin superfamily member DSCAML1 plays a role in several neurodevelopmental processes, including branching, migration, synaptogenesis, and dendritic self-avoidance. Dscam2 also inhibits the deposition of synaptic vesicles through an endosomal route dependent on phosphatidylinositol-3 kinase (PI3K) [101].
NAGA was proposed as a genetic risk for schizophrenia [106] because it encodes the lysosomal enzyme alpha-N-acetylgalactosaminidase, which is necessary for the breakdown of glycolipids and helps to maintain and regulate the dendritic spine [102].
The endosomal protein STAM2 (Signal Transducing Adaptor Molecule 2), a component of the ESCRT-0 complex that controls receptor signaling and trafficking, is another intriguing molecule among the most downregulated proteins. It plays a regulatory role in the endosomal sorting of ubiquitinated membrane proteins [103]. It is strongly expressed in stomach cancer, neurons, and olfactory epithelium [104]. In HeLa cells, STAM2b overexpression results in the expansion of early endosomes and the accumulation of ubiquitinated proteins and ligand-activated EGF receptors. This behavior has already been observed in AD [105]. The scaffold protein SRRM2 is overexpressed in the brain and blood of people with Parkinson's disease [106] and is necessary to establish nuclear speckles and mRNA splicing [107]. Dysfunction of SRRM2 has been connected to neurodevelopmental disorders.
Additionally, it accumulates and mislocalizes from cytosolic tau NFTs to nuclear splicing speckles in cells, mice, and the brains of AD patients [108]. In both the tricarboxylic acid (TCA) cycle and the mitochondrial electron transport chain (ETC), SDHA takes part. Inflammation-promoting fumarate buildup and enhanced mitochondrial respiration have been linked to increased SDHA function [109].
Phosphoglycerate dehydrogenase (PHGDH) upregulated in the subventricular zone (SVZ), a neural stem/progenitor niche, may play a part in neurogenesis. Severe neurological abnormalities in Phgdh knockout mice include congenital microcephaly and psychomotor impairment [110,111]. PHGDH and the astrocytic l-serine biosynthesis pathway are diminished in the AD brain and 3xTg-AD mice, according to a recent study by Le Douce et al. [112]. In contrast, Chen et al. later quoted Le Douce and provided results that concur with our findings, showing that PHGDH mRNA and protein levels rise as AD pathology and symptoms worsen in 3xTg-AD and P301S tau transgenic mice (PS19) and human AD brains [113]. Interestingly, in the presymptomatic carrier data (FC -2.3), this protein is downregulated rather than increased.
A GTPase crucial for mitochondrial fission is one of the most elevated proteins in the presymptomatic carrier, Dynamin-related protein 1 (DNM1L or Drp1). Abnormal mitochondrial dynamics, including increased fragmentation and decreased fusion, have been seen in Alzheimer's disease (AD). In AD neurons, elevated DNM1L activity led to mitochondrial fragmentation and dysfunction [114], while in AD mice models, inhibiting DNM1L prevents cell death, enhances synaptic function, and increases neuronal activity [115].
The regulation of cellular functions such as protein degradation, DNA damage response, and cell cycle progression is carried out by COPS8, also known as COP9 signalosome subunit 8. In murine livers, COP9 deficiency results in UPS dysfunction and apoptosis [116]. In a mouse model of cardiac proteinopathy, the COP9 signalosome regulates the breakdown of cytosolic misfolded proteins and guards against proteotoxicity [117]. Colony-stimulating factor 1 receptor (CSF1R) is a transmembrane protein that aids in the differentiation and survival of microglia. It is elevated in post-mortem samples from AD patients [118,119], and selective deletion of CSF1R in forebrain neurons in mice accelerated excitotoxin-induced death and neurodegeneration [120].
However, other studies showed that pharmacological inhibition in APP/PS1 mice improved performance in memory and behavioral tasks and prevented synaptic degeneration [119].
FKBP1A, a member of the immunophilin protein family involved in protein folding and trafficking, was protective against the cytotoxicity caused by Aβ in cultured cells when overexpressed [121]. Cognitive deficits have been associated with the increased expression of other members of this family, such as FKBP51 [122].
OLA1 (Obg-like ATPase 1) reduces the antioxidant response, which in turn enhances the integrated stress response and works as a regulator of protein synthesis. SAMHD1 is likewise elevated in the symptomatic carrier (both with FC of 1.7). Its overexpression makes cells more vulnerable to cytotoxicity from thiol-depleting and peroxide antioxidants [123]. But according to R-F Mao et al., OLA1 binds to and stabilizes HSP70 to shield cells from heat shock [124]. Overexpression of CPT1C lowered cell viability, oxidative stress, and apoptosis in Aβ25-35-induced neurons and decreased APP, p-Tau, and Bace-1 levels. CPT1A is an enzyme that transports fatty acids into the mitochondria to produce energy [125].
3.5. Signaling pathways and biological processes altered in PSEN1(A431E) mutation carriers.
The term "proteostasis" (PN) describes the interaction of a complex network of processes that allow the maintenance of the conformation, concentration, and localization of proteins for their correct function. This network comprises about 2,500 genes (> 10% of the total protein-coding genes) and multiple signaling pathways that control protein biogenesis, folding trafficking, and degradation. Any disruption of Protein Quality Control (PQC), changes in glucose uptake and metabolism, oxidative stress, transport, and abnormal autophagy or degradation system as a result of stress can result in misfolding or accumulation of proteins that interfere with various cellular functions and disrupt the entire proteostasis network, as happens in aging and the main NDDs including AD [126], causing oxidative stress, neuroinflammation and toxicity leading to dysfunction and cell death [127,128].
Our findings demonstrate that the primary categories that are altered in MSCs derived from carriers of the PSEN1 (A431E) mutation include Proteostasis network functions like energy metabolism, processing, synthesis, and folding of proteins, cytoskeleton organization, and intracellular and vesicular transport.
According to some studies, bioenergetic changes, including mitochondrial malfunction, changes in glycolysis and PPP, reduced ATP generation, and oxidative damage, are early symptoms of the disease [129,130,131].
One of the characteristics of AD is a decline in cerebral glucose absorption. The changes in glucose hypometabolism in pre-symptomatic AD are now being studied using MRI and 18F-FDG PET (Brain Fluorodeoxyglucose Positron Emission Tomography) imaging investigations [132,133]. A group of individuals from FAD families with mutations in PSEN1 who were presymptomatic had a glucose hypometabolism, according to Mosconi et al.'s findings [134].
Most DEPs participating in energy metabolism pathways were downregulated, pointing to disordered energy metabolism and a potential rise in ROS generation. Although elevated in P1, PHGDH, and ALDH18A1 are downregulated in P2. It's interesting to note that the gene for ALDH18A1 (Delta-1-Pyrroline-5-Carboxylate Synthase), a condition with symptoms comparable to those of people with PSEN1(A431E) mutation, is associated with spastic paraplegias (SPG9A and SP9B) [135]. AD and Down syndrome were also linked to ALDH18A1 mutations [136]. PGD, MAT2A, and PKM were the three proteins whose expression levels were downregulated in both mutation carriers. The pentose phosphate pathway (PPP) has three enzymes. The third enzyme is PGD (phosphogluconate dehydrogenase). In contrast to our results, an old work reported that G6PD and PGD activity increased in AD patients' inferior temporal cortex [137]. However, Tiwari and Patel said that the PPP flux is decreased in AβPP-PS1mice [138]. MAT2A (Methionine Adenosyl transferase 2A) is essential for the methylation of neurotransmitters, DNA, proteins, and lipids. As our results, Schrötter et al. [139] reported a MAT2A lower expression in frontal cortex samples from AD patients compared with controls and that KD of the APP protein family resulted in its downregulation at the RNA and protein level but with a subsequent SAM elevation. To produce ATP and pyruvate, the pyruvate kinase (PKM) catalyzes the transfer of a phosphoryl group from phosphoenolpyruvate to ADP. PKM2 controls the activity of -secretase and is necessary for cell proliferation. Contrary to our findings, Han et al. claimed that its expression is increased in brain samples from AD patients and mouse models and that its silencing led to lower levels of Aβ1-40 and Aβ1-42 and γ-secretase activity [140].
Regarding the mechanisms of degradation, there are numerous signs that AD plays a part in the endo-lysosomal, autophagic, and ubiquitin-proteasome systems, including the marked enlargement of endosomal compartments, the gradual accumulation of autophagic vacuoles (AVs), lysosome dyshomeostasis, the accumulation of ubiquitinated proteins, and changes in proteasome subunits that reduce the proteasomal activity [141].
The function and destiny of critical molecules involved in the genesis of Alzheimer's disease (AD) depend on endocytosis. For instance, early endosomes are an important location for producing Aβ peptides, which are noticeably increased within neurons in the brains of patients with early-stage AD [142,143]. These results aligned with our presymptomatic findings. The proteins Rab11, RabGDI (downregulated in our findings), phospholipase D1, syntaxins, and annexin A2 are all targets of interactions between Presenilins and intracellular vesicular trafficking [144]. PS1 loss also harms endocytosis and receptor-mediated transcytosis in neurons produced from iPSCs that have mutations in APP and PSEN1 [145]. Exosomes are intraluminal microvesicles that are made outside of the cell, and it has been demonstrated that exosomes can transfer pathogenic proteins like hyperphosphorylated tau, toxic amyloid beta, and other pathogens between cells that are engaged with other NDDs [146]. Defective lysosomal activity can also produce toxic Aβ and tau species oligomers, contributing to neuronal malfunction and death [143].
3.5.3. Protein Processing in Endoplasmic Reticulum.
Most of these events occur in the endoplasmic reticulum (ER), where most non-cytosolic proteins (secretory or membrane) are generated. Approximately 30% of all proteins misfold physiologically, yet there is tight quality control, where the first line of defense is given by chaperones, also known as heat shock proteins (HSPs). To keep the balance between protein synthesis and degradation, they bind to unfolded proteins and keep them in a properly folded state while preventing aggregation. These proteins are grouped into various families based on their molecular weight and functions, such as cotranslational folding, refolding or degradation of misfolded proteins, and the inactivation of the Unfolded Protein Response (UPR) [147]. 16 molecular chaperones (CRYAB, ERO1A, CALR, CANX, ERP29, PDIA3, PDIA4, PDIA6, BAG2, HSPA1A, HSPA5, DNAJB11, HSP90AB1, HSP90B1, HYOU1, and P4HB) were identified in the clinical data of 22 DEPs in Protein Processing in the Endoplasmic Reticulum pathway.
Almost all chaperone protein families are altered in AD brain tissue and animal models. Many have been overexpressed due to chronic inflammation, but some reports show HSP is downregulated in AD [148,149,150]. The altered regulation of HSP can increase oxidative stress or the response to the misfolded protein, potentiate the chronic overload, and functionally decrease the degradation systems that eventually lead to the development of the disease. There are conflicting reports from a few research suggesting PSEN1 mutations in transgenic cells or animals reduce the production of chaperone proteins [151].
Nevertheless, the results showing the downregulation of different molecular chaperones in non-differentiated cells are intriguing because they may be a factor in the earlier clinical presentation of FAD compared to SAD.
3.5.4. Regulation of Actin Cytoskeleton
Most excitatory synapses within the brain occur on small dendritic protrusions called dendritic spines. The quantity and size of dendritic spines, whose stability is provided by the actin cytoskeleton, significantly impact synaptic strength and neuronal function [152]. Actin dynamics are regulated by many molecules, particularly small GTPases like Rac and Rap. Glutamate receptors are lost from synaptic locations as a result of actin depolymerization. Similarly, spine and synapse loss are caused by disruption of the expression or activity of regulators upstream of the actin cytoskeleton, such as Rac-GEF, Rac, and Rac targets like PAK. Animal models of AD that include mutations in the susceptibility genes that cause familial Alzheimer's disease recapitulate the loss of the dendritic spine [153,154].
This decrease may be brought on directly by the toxicity of Aβ oligomers or indirectly by the change of several synaptic proteins in these patients [155]. In this line, Xia et al. [156] demonstrated that AD patients and animal models of the disease have higher levels of calcineurin activation, a calcium-sensitive phosphatase. Additionally, GSK-3, a downstream effector molecule of calcineurin, has enhanced activity in response to AD-related pathologies, as demonstrated by Li et al. [157].
In addition, p21-activated kinases (PAKs), essential regulators of actin assembly and subsequent spine modulation in neurons, decrease in the hippocampus of AD patients and animal models of the disease [156]. Kalirin, a crucial regulator of spine morphogenesis and an upstream activator of PAK in dendritic spines, is also consistently underexpressed in the hippocampus of AD patients regarding protein and mRNA levels. Similar to how PAKs are abnormally translocated from the cytosol to the membrane in cells from AD patients as opposed to cells from control subjects free of the disease, according to research [158]. The hippocampus of AD patients exhibits lower expression and altered location of RhoA, a small RhoGTPase that modifies cytoskeleton dynamics to modulate synaptic plasticity [159]. On the other hand, Liu et al. [160] demonstrated that the hippocampus of old mouse models and AD patients has less drebrin protein (DBN1), which accumulates in dendritic spines and creates a stable pool of slowly replenishing F-actin and bundles filaments by cross-linking them together.
In contrast to aging wild-type mice, APPPS1 mice have lower levels of activated CFL1, an actin-binding protein that depolymerizes and cleaves actin filaments to produce free G-actin monomers that are recruited for filament elongation and branching. This reduction is more significant in APPPS1 mice than in aging wild-type mice. Additionally, it has been noted that AD patients' brains had lower levels of active CFL1 [161].
3.5.5. Focal Adhesion and Extracellular Matrix-Receptor Interactions.
Regarding cell adhesion, several molecules involved in cell adhesion processes, including neuronal synapses, have also been reported to be dysregulated in animal models and subjects with AD [162]. For example, isoform NCAM180 levels of the neural cell adhesion molecule, but not total NCAM levels, are elevated in the frontal cortex of AD patients compared to healthy subjects of the same age [163].
On the other hand, contactin-2 levels are decreased in AD patients' temporal lobes [164], and N-cadherin levels are likewise reduced in their temporal cortex [165].
According to Bao et al. [162], who found significantly altered expression of cell adhesion pathway (CAM) genes in AD in cerebellar and temporal cortex samples from AD-affected individuals, several genome-wide association studies (GWAS) suggest the involvement of CAMs in AD. Meanwhile, single nucleotide polymorphisms (SNPs) in NCAM2 are associated as a risk factor related to AD progression in the Japanese population, according to Kimura et al. [166]. At the same time, another large GWAS encompassed more than 16 000 participants. SNPs in contactin-5, another synaptic CAM that localizes to presynaptic membranes, were strongly linked with disease development [167], similar to what was seen with SNPs in JAM2 [168].
3.5.6. Neurodegenerative diseases pathways
NDD are primarily distinguished by a failure of Protein Quality Control (PQC) to remove or degrade misfolded proteins accumulating in various brain regions. NDD share multiple pathophysiological processes that result in neuronal injury. In the current study, KEGG and STRING data for both carriers were enriched for the major NDDs pathways, including those for ALS, Parkinson's disease, prion disease, Huntington's disease, and Alzheimer's disease (Table 10). These proteins play a role in protein folding, processing, and stress in the ER, disruption of the ubiquitin-proteasome system, change in calcium signaling, oxidative stress and mitochondrial dysfunction, poor autophagy, and deficiencies in microtubule transport. Only 7 of these proteins were present in both PSEN1 (A431E) mutant carriers, and four (CLTA, AP2B1, DNAH6, and FUS) were downregulated. In both, RYR1 was elevated, and TUBB3 and MATR3 expression varied.
3.6. Comparison with AD studies previously reported.
The primary categories affected in MSCs produced from PSEN1 (A431E) mutation carriers are energy metabolism, the structure of the cytoskeleton, and intracellular and vesicular transport. These processes are related to the Proteostasis network and involve protein processing, synthesis, and folding.
Some AD investigations have reported proteostasis network change, with findings similar to ours. Vesicular transport, post-translational protein modifications, trafficking, and proteostasis were among the five classes of functionally related molecular pathways linked to AD that Rosenthal et al. [169] proposed as part of their recent identification of an AD network integrating multi-omics data with the most recent genome-wide association studies (GWAS). Shokhirev and Johnson used machine learning and bioinformatic techniques in a massive multi-omic dataset of 4089 blood and brain human samples from microarray, RNA-Seq, proteomics, and miRNA-accessible data generated from AD and control participants [170]. Their findings revealed aging-related characteristics, such as cell death in the youngest patients, cellular senescence, immunological system, and oxidative stress in the middle-aged group.
A thorough proteomic study of more than 2000 human brain tissues from 453 brains and 400 cerebrospinal fluid samples was carried out by Johnson and colleagues [171]. They included control, asymptomatic AD (AsymAD), and were connected to disease and reflected biological processes of synapses, mitochondria, RNA binding/splicing, and astrocyte/microglial metabolism.
Additionally, Pedrero-Prieto et al. [172] created a database of CSF from AD patients from 47 different proteomic studies that were compared. 36 proteins were also in our results: CDH6, CAMK2D, CUTA, MARCKS, SRCAP, TXN TPI1, WARS, BASP1, ALCAM, CYCS, HNRNPU, UCHL1, CHGB, HLA-B, some related to PI3k-Akt signaling pathway (COL1A1, COL1A2, FN1, NTRK2, and YWHAB) with the lysosome (CTSD, IGF2R, and PSAP) and with exocytosis, secretion and vesicle transport (A2M, ADAM10, ALB, CANX, CTSD, FN1, FTL, GOLGB1, GSN, HBB, IGF2R, PCLO, PKM, PSAP, and SOD1), protein processing in ER (PDIA3, PRKCSH, and CANX).
Furthermore, Higginbotham et al. [173] applied an integrative proteomics approach to AD brain and CSF of healthy controls, asymptomatic AD, and AD, finding progressive disease-specific changes in 5 main panels: immunological dysfunction, metabolic dysfunction, synaptic dysfunction, vascular dysfunction, and myelin dysfunction. This suggests that disruptions of energy and redox pathways may be necessary during the preclinical stages of the disease. Another redox proteomic investigation discovered the "triangle of death" in AD brains, which comprises abnormal interactions between protein homeostasis, mTOR signaling, and energy metabolism [174].
Recent research found that compared to fibroblasts from healthy donors, skin fibroblasts from FAD patients with PS1 (M146L or A246E) mutations expressed higher levels of HSPs and autophagic-lysosomal pathway proteins [175]. Our findings demonstrated that the present work was carried out in undifferentiated olfactory MSC cells, highlighting that the pathogenic pathways were comparable to those previously documented in AD.
4. Conclusion
Although many pathways, including protein misfolding and aggregation, mitochondrial dysfunction, autophagy, and oxidative stress, have been implicated in the etiology of AD, there is currently no effective treatment for the disease. Therefore, more research into disease processes and signaling pathways is required to identify new biomarkers and therapeutic targets for early diagnosis. Most pathogenic pathways have been tested in cell lines and animal models carrying FAD-causing gene mutations. Few investigations have been done using patient-derived cells like fibroblasts or astrocytes, even though several proteomics studies have recently screened dysregulated proteins in various biological samples obtained from individuals with AD, such as brain tissues and bodily fluids. This study evaluates two stages of the disease (presymptomatic and symptomatic) using quantitative proteome analysis in olfactory MSCs generated from human carriers of the PSEN1 (A431E) mutation. The MSCs are directly linked to the disease, have a high neurogenic potential, and possess mesenchymal characteristics that allow them to develop into diverse cell lineages. They are also simple to get and barely invasive. These fresh MSCs from patients are fantastic in vitro models for upcoming research on Alzheimer's disease.
Our findings may indicate a protein homeostasis (proteostasis) failure in undifferentiated cells, contributing to protein misfolding and aggregation. They may also point to an increased susceptibility to stress brought on by a disruption in energy metabolism, which can increase ROS production and symptoms earlier in FAD patients.
Our findings present a thorough proteomic analysis of MSCs from FAD patients in the early and late stages of the disease that resemble reported pathways in Sporadic Alzheimer's disease. Although more research and validation are required to understand some altered mechanisms reported in this work, our results provide guidelines for the study and identification of new therapeutic targets and the basis for the presymptomatic diagnosis of AD.
5. Materials and Methods
5.1. Subjects
Exfoliates. Two healthy adult donors and two PSEN1 (A431E) mutation carriers from a Mexican-Mestizo family provided the olfactory neuroepithelium. The family carriers are siblings affected with lower limb paraparesis for three generations (Figure 1). Santos-Mandujano [33]. gathered information on these PSEN1 (A431E) carriers, including mutation confirmation, clinical history, and neurological and cognitive data. The symptomatic (P1) and presymptomatic (P2) patients' cells were employed in this study. Family carriers are siblings with a three-generation history of progressive paraparesis of the lower limbs (Figure 1). Confirmation of the mutation, clinical history, and neurological and cognitive information of these PSEN1 (A431E) carriers were obtained and detailed by Santos-Mandujano [33]. In this work, we used cells from the symptomatic (P1) and presymptomatic subjects (P2).
Our patient (P1) is a 54-year-old guy with progressive motor dysfunction in the lower limbs, slight cognitive impairment, typical symptoms of upper motor neuron disease, and anosmia that has been present for seven years. The presymptomatic carrier (P2) is a 44-year-old asymptomatic female with normal cognitive function, modest hyperreflexia throughout her body, and slight hyposmia. In addition, control subjects consist of two healthy donors (without the mutation) matched by age and gender, with P1 and P2, a male 54-year-old serving as the control for the symptomatic subject and a female 42-year-old serving as the control for the presymptomatic subject.
These participants gave their informed consent following the guidelines of the Bioethics Committee on Human Research (COBISH, folio # 038/2016 of the Center for Research and Advanced Studies, CINVESTAV-IPN) by signing a form authorizing the collection of their medical history and biological samples.
5.2. Sample collection and cell culture conditions
Nasal epithelium cells were exfoliated from the anterior region of the medial and lateral turbinate using a special toothbrush according to the methodology reported by Benítez-King et al. [26] and harvested in Dulbecco's modified Eagle and F-12 media (DMEM/F-12) supplemented with 10% fetal bovine serum(Gibco, NY, EU), 4 mM l-Glutamine(Gibco, NY, EU), 100 g/ml Streptomycin, 100 IU/ml Penicillin (Gibco, NY, EU) at 37 °C in a humidified atmosphere with 5% CO2. Before obtaining the sample, we treated the dishes for two hours with poly-D-lysine (diluted in Milli-Q water at 50 mg/ml). The cells were transferred to a new plate and cultured under the same conditions when they reached 80% confluence.
5.3. Flow cytometry
Culture plates were trypsinized, inactivated, and supplied with DMEM/D-12 before being centrifuged at 1,300 rpm for 5 minutes, and the supernatant was discarded. Conjugated primary antibodies were added to the pellet after it had been resuspended in PBS with 1X106 cells per tube. Mesenchymal cells were identified using the markers CD105+ (Biolegend 323217), CD73+ (BD Biosciences 563198), CD90+ (BD Biosciences 555597), CD45- (MACSMylteny Biotec 130-080-202), CD14- (BD Biosciences 561712), CD19- (BD Biosciences 560911), and CD34- (BD Biosciences 562577). The information was recorded using a Fortessa flow cytometer (BD) and examined using the Flowjo 7.6 application.
5.4. Proteomic sample
At 80% confluence, MSCs cells were grown, and proteins were removed from the culture media with three cold PBS washes. Reconstituted cells were incubated at 95° C for 5 minutes in SDT lysis solution (4% SDS (w/v), 0.1 M DTT, 100 mM Tris/HCl pH 7.6). The samples were sonicated ten times (PEAK Bioruptor, pulse ON 30 sec and OFF 30 sec) at 20°C. The supernatant was then transferred to another tube after the undisturbed cells and cell debris were separated by centrifugation at 16000 x g for 5 min at -20°C. Following the manufacturer procedure, the protein concentration was assessed using a 2D Quant kit (GE Healthcare, Life Sciences).
5.5. Filter-aided sample preparation (FASP)
FASP digestion of the protein extract was done according to a modified protocol described by Wiśniewski et al. [176]. We used 50 μg of protein input, and protein digestion was performed with trypsin (enzyme: protein ratio of 1: 100), ON at 37 ◦C. The filtrates were transferred to new tubes and centrifuged at 14000 x g at 20°C for 40 minutes. Subsequently, they were rinsed with 50 μl of 0.5 M NaCl and centrifuged again (twice); finally, the sample was desalted in a C18 resin column and stored at -80°C until further use. Digestion of the samples was according to the FASP protocol described by Wiśniewski et al. [176] and modified by the LaNSE CINVESTAV proteomics team [177].
5.6. Liquid Chromatography-Tandem Mass Spectrometry
The resulting peptides were injected into the mass spectrometer Synapt G2-Si (Waters, Milford, MA) in MSE mode to calculate the area under the curve (AUC) of the Total Ion Chromatogram and thus normalize the injection in the HPLC. A Symmetry C18 Trap V/M precolumn with dimensions of 180 mm x 20 mm, a pore size of 100 °, and a particle size of 5 μm was loaded precisely with tryptic peptides in each condition. The precolumn was desalted using mobile phase A, which contained 0.1% formic acid (FA), in water, and mobile phase B, which had 0.1% FA in acetonitrile, under the following isocratic gradient: 99.9% mobile phase A and 0.1 % of mobile phase B at a flow of 5 μL min-1 during 3 min. Afterward, peptides were loaded and separated on HSS T3 C18 Column; 75 μm X 150 mm, 100 A° pore size, 1.8 μm particle size, using a UPLC ACQUITY M-Class with the same mobile phases under the following gradient: 0 min 7% B, 121.49 min 40% B, 123.15 to 126.46 min 85% B, 129 to 130 min 7% B, at a flow of 400 nL min-1 and 45 °C. Data-independent acquisition technique by HDMSE mode was used to obtain the spectra in a mass spectrometer equipped with electrospray ionization and ion mobility separation Synapt G2-Si (Waters, Milford, MA). The tune page for the ionization source was set with the following parameters: 2.75 kV in the sampler capillary tube, 30 V in the sampling cone, 30 V in the source offset, 70°C for the source temperature, 0.5 Bar for the nanoflow gas, and 150 L hr-1 for the purge gas flow. Two chromatograms (low and high energy) were acquired in a positive mode in a range of m/z 50-2000 with a velocity of 0.5 scans s-1. For the high-energy chromatograms, the precursor ions were fragmented in the transfer using a collision energy ramp of 19-55 V. We analyzed the generated raw files in the DriftScope v2.8 software (Waters, Milford, MA) to eliminate ions with z=1+ and to get the DrifTime from each peptide detected in the mass spectrometer to generate a .rul file to calculate specific collision energy for every peptide caught in the UDMSE mode (three technical replications were carried out).
5.7. Data Analysis
The .raw files containing the MS and MS/MS intensities were normalized, aligned, compared, and relatively quantified using Progenesis QI for Proteomics software v3.0.3 (Waters, Milford, MA) against a reversed Homo Sapiens database. The results generated from the software were exported to .xmls files to verify two levels of data quality control for label-free experiments (peptide and protein level) following the figures of merit described by Souza et al., 2017(19). (Downloaded from UniProt, 19229 protein sequences, last modified on May 16, 2016). The parameters used for the protein identification were: trypsin as a cut enzyme and one missed cleavage allowed; carbamidomethyl (C) as a fixed modification and oxidation (M), phosphoryl (S, T, Y) as variable modifications; besides automatic peptide and fragment tolerance, minimum fragment ion matches per peptide: 2, minimum fragment ion matches per protein: 5, minimum peptide matches per protein: 1, and false discovery rate ≤ 4%. Synapt G2-Si was calibrated with [Glu1]-Fibrinopeptide, [M+2H]2+= 785.84261 at ≤ 1 ppm. All the plots generated during the quality control were made using Spotfire software v7.0 (TIBCO, Palo Alto, CA). Differential expression was considered with P/C absolute ratios >1.5 and <0.66 for upregulated and downregulated proteins, respectively. The remaining proteins with ratios between 1.5 and 0.66 were considered unchanged. The ratio was calculated by dividing the average MS signal response of the three most intense tryptic peptides (Top3) of each well-characterized protein in the infected sample by the Top3 protein in the control sample. The images were created with BioRender.com.
5.8. Bioinformatic Analysis
For Gene Ontology (GO) terms, as well as a pathway enrichment analysis, we used WEB-based Gene SeT AnaLysis Toolkit (WEBGESTALT) http://www.webgestalt.org/ using the Kyoto Encyclopedia of Genes and Genomes (KEGG), REACTOME https://reactome.org/ and Protein ANalysis THrough Evolutionary Relationships (PANTHER) http://pantherdb.org/ databases. The protein lists were submitted with their respective gene symbol, and a comparison was made with the existing list of the human genome. For pathways enrichment, we used the top 15 significance level. KEGG Mapper-Color https://www.genome.jp/kegg/mapper/color.html was used for scheme pathways.
Interactomes of differentially expressed proteins were predicted using Search Tool for the Retrieval of Interacting Genes/Proteins STRING ver. 11.5 (https://string-db.org/) with the following settings: Homo Sapiens database; text mining, experiments, database, co-expression, neighborhood, gene fusion, and co-occurrence as active interaction source with the highest confidence score (0.90).
Additionally, we used GenAnalytics (http://geneanalytics.genecards.org) and WEBGESTALT with OMIM and DISGENET databases to find related diseases. UniProt (https://www.uniprot.org/) and GeneCards were used to know protein characteristics and function.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1. Measurements by Mass spectrometry P1-C1 (symptomatic), Table S2. Differentially expressed proteins (P1-C1), Table S3. WEBGESTALT analysis (P1-C1) GO, pathways and diseases, Table S4. Measurements by Mass spectrometry P2-C2 (presymptomatic), Table S5. Differentially expressed proteins (P2-C2), Table S6. WEBGESTALT Analysis (P2-C2), GO, pathways and diseases, Table S7. Comparison between both analyses, Table S8. Comparison with previously reported AD datasets. Figure S1. Volcano plot of P1/C1 DEPs with top 20 down and upregulated proteins labeled. Figure S2. Volcano plot of P2/C2 DEPs with top 20 down and upregulated proteins labeled.
Author Contributions
L.J.R.H and M.A.M.R developed the project, L.J.R.H analyzed the data and wrote the manuscript design, made the images, and Graphical Abstract. M.A.M.R. supervised the project, and M.A.M.R. and M.A.J.A. revised, examined, wrote, and edited the manuscript. L.R.R. made the sample process for/and Mass Spectrometry analysis. M.P.F.C. help with technical support and cell culture. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by CONACYT, grant number CF-2023-2206-. Student’s scholarships: 1008755 and 854550.
Institutional Review Board Statement
This study complied with the statutes of the Bioethics Committee on Human Research (COBISH, folio # 038/2016) of the Center for Research and Advanced Studies, CINVESTAV-IPN. The subjects signed an informed consent form.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study to collect their clinical history, biological samples and to publish this paper.
Data Availability Statement
Not applicable
Acknowledgments
We would like to thank GCSE of CINVESTAV, especially Lorena Ramírez Reyes, Emmanuel Ríos Castro (Genomic, Proteomic, and Metabolomic Service), and Gustavo Martins De Souza for their technical support in sample processing and fruitful comments. Also, to Lory S. Rochín Hernández and Erika Alejandra Cabrera Reyes for image design, edition support, and words.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Haque, R.U.; Levey, A.I. Alzheimer’s disease: A clinical perspective and future nonhuman primate research opportunities. Proc. Natl. Acad. Sci. 2019, 116, 26224–26229. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.J. World Alzheimer Report 2015: The Global Impact of Dementia Available online:. Available online: https://www.alz.co.uk/research/world-report-2015 (accessed on 23 July 2020).
- Nisbet, R.M.; Götz, J. Amyloid-β and Tau in Alzheimer’s Disease: Novel Pathomechanisms and Non-Pharmacological Treatment Strategies. J. Alzheimers Dis. JAD 2018, 64, S517–S527. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.M.; Götz, J. Amyloid-β and Tau in Alzheimer’s Disease: Novel Pathomechanisms and Non-Pharmacological Treatment Strategies. J. Alzheimers Dis. 2018, 64, S517–S527. [Google Scholar] [CrossRef]
- Tellechea, P.; Pujol, N.; Esteve-Belloch, P.; Echeveste, B.; García-Eulate, M.R.; Arbizu, J.; Riverol, M. Enfermedad de Alzheimer de inicio precoz y de inicio tardío: ¿son la misma entidad? Neurología 2018, 33, 244–253. [Google Scholar] [CrossRef]
- Chouraki, V.; Seshadri, S. Genetics of Alzheimer’s Disease. In Advances in Genetics; Elsevier, 2014; Vol. 87, pp. 245–294 ISBN 978-0-12-800149-3.
- Soosman, S.K.; Joseph-Mathurin, N.; Braskie, M.N.; Bordelon, Y.M.; Wharton, D.; Casado, M.; Coppola, G.; McCallum, H.; Nuwer, M.; Coutin-Churchman, P.; et al. Widespread white matter and conduction defects in PSEN1- related spastic paraparesis. Neurobiol. Aging 2016, 47, 201–209. [Google Scholar] [CrossRef]
- Ringman, J.M.; Casado, M.; Van Berlo, V.; Pa, J.; Joseph-Mathurin, N.; Fagan, A.M.; Benzinger, T.; Bateman, R.J.; Morris, J.C. A novel PSEN1 (S230N) mutation causing early-onset Alzheimer’s Disease associated with prosopagnosia, hoarding, and Parkinsonism. Neurosci. Lett. 2017, 657, 11–15. [Google Scholar] [CrossRef]
- Rudzinski, L.A.; Fletcher, R.M.; Dickson, D.W.; Crook, R.; Hutton, M.L.; Adamson, J.; Graff-Radford, N.R. Early Onset Familial Alzheimer Disease With Spastic Paraparesis, Dysarthria, and Seizures and N135S Mutation in PSEN1: Alzheimer Dis. Assoc. Disord. 2008, 22, 299–307. [Google Scholar] [CrossRef]
- Yescas, P.; Huertas-Vazquez, A.; Villarreal-Molina, M.T.; Rasmussen, A.; Tusié-Luna, M.T.; López, M.; Canizales-Quinteros, S.; Alonso, M.E. Founder effect for the Ala431Glu mutation of the presenilin 1 gene causing early-onset Alzheimer’s disease in Mexican families. Neurogenetics 2006, 7, 195–200. [Google Scholar] [CrossRef]
- Orozco-Barajas, M.; Oropeza-Ruvalcaba, Y.; Canales-Aguirre, A.A.; Sánchez-González, V.J. PSEN1 c.1292C<A Variant and Early-Onset Alzheimer’s Disease: A Scoping Review. Front. Aging Neurosci. 2022, 14, 860529. [Google Scholar] [CrossRef]
- Souza, G.H.M.F.; Guest, P.C.; Martins-de-Souza, D. LC-MSE, Multiplex MS/MS, Ion Mobility, and Label-Free Quantitation in Clinical Proteomics. In Multiplex Biomarker Techniques; Guest, P.C., Ed.; Methods in Molecular Biology; Springer New York: New York, NY, 2017; ISBN 978-1-4939-6729-2. [Google Scholar]
- Jain, A.P.; Sathe, G. Proteomics Landscape of Alzheimer’s Disease. Proteomes 2021, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Craft, G.E.; Chen, A.; Nairn, A.C. Recent advances in quantitative neuroproteomics. Methods San Diego Calif 2013, 61, 186–218. [Google Scholar] [CrossRef] [PubMed]
- Hondius, D.C.; van Nierop, P.; Li, K.W.; Hoozemans, J.J.M.; van der Schors, R.C.; van Haastert, E.S.; van der Vies, S.M.; Rozemuller, A.J.M.; Smit, A.B. Profiling the human hippocampal proteome at all pathologic stages of Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2016, 12, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Andreev, V.P.; Petyuk, V.A.; Brewer, H.M.; Karpievitch, Y.V.; Xie, F.; Clarke, J.; Camp, D.; Smith, R.D.; Lieberman, A.P.; Albin, R.L.; et al. Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J. Proteome Res. 2012, 11, 3053–3067. [Google Scholar] [CrossRef] [PubMed]
- Lachén-Montes, M.; Íñigo-Marco, I.; Cartas-Cejudo, P.; Fernández-Irigoyen, J.; Santamaría, E. Olfactory Bulb Proteomics Reveals Widespread Proteostatic Disturbances in Mixed Dementia and Guides for Potential Serum Biomarkers to Discriminate Alzheimer Disease and Mixed Dementia Phenotypes. J. Pers. Med. 2021, 11, 503. [Google Scholar] [CrossRef]
- Dayon, L.; Núñez Galindo, A.; Wojcik, J.; Cominetti, O.; Corthésy, J.; Oikonomidi, A.; Henry, H.; Kussmann, M.; Migliavacca, E.; Severin, I.; et al. Alzheimer disease pathology and the cerebrospinal fluid proteome. Alzheimers Res. Ther. 2018, 10. [Google Scholar] [CrossRef]
- Khoonsari, P.E.; Häggmark, A.; Lönnberg, M.; Mikus, M.; Kilander, L.; Lannfelt, L.; Bergquist, J.; Ingelsson, M.; Nilsson, P.; Kultima, K.; et al. Analysis of the Cerebrospinal Fluid Proteome in Alzheimer’s Disease. PloS One 2016, 11, e0150672. [Google Scholar] [CrossRef]
- Dey, K.K.; Wang, H.; Niu, M.; Bai, B.; Wang, X.; Li, Y.; Cho, J.-H.; Tan, H.; Mishra, A.; High, A.A.; et al. Deep undepleted human serum proteome profiling toward biomarker discovery for Alzheimer’s disease. Clin. Proteomics 2019, 16, 16. [Google Scholar] [CrossRef]
- Hye, A.; Lynham, S.; Thambisetty, M.; Causevic, M.; Campbell, J.; Byers, H.L.; Hooper, C.; Rijsdijk, F.; Tabrizi, S.J.; Banner, S. Proteome-based plasma biomarkers for Alzheimer’s disease. Brain 2006, 129, 3042–3050. [Google Scholar] [CrossRef]
- François, M.; Bull, C.F.; Fenech, M.F.; Leifert, W.R. Current State of Saliva Biomarkers for Aging and Alzheimer’s Disease Available online:. Available online: https://www.ingentaconnect.com/contentone/ben/car/2019/00000016/00000001/art00007 (accessed on Sep 24, 2019).
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: experimental models and reality. Acta Neuropathol. (Berl.) 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Jiménez-Acosta, M.A.; Hernández, L.J.R.; Cristerna, M.L.P.; Tapia-Ramírez, J.; Meraz-Ríos, M.A. Review: Neuronal Differentiation Protocols of Mesenchymal Stem Cells. Adv. Biosci. Biotechnol. 2022, 13, 15–71. [Google Scholar] [CrossRef]
- Benítez-King, G.; Riquelme, A.; Ortíz-López, L.; Berlanga, C.; Rodríguez-Verdugo, M.S.; Romo, F.; Calixto, E.; Solís-Chagoyán, H.; Jímenez, M.; Montaño, L.M.; et al. A non-invasive method to isolate the neuronal linage from the nasal epithelium from schizophrenic and bipolar diseases. J. Neurosci. Methods 2011, 201, 35–45. [Google Scholar] [CrossRef]
- Jiménez-Acosta, M.A.; Hernández, L.J.R.; Cristerna, M.L.P.; Meraz-Ríos, M.A. Mesenchymal Stem Cells: New Alternatives for Nervous System Disorders. Curr. Stem Cell Res. Ther. 2023, 18, 299–321. [Google Scholar] [CrossRef] [PubMed]
- Dan, X.; Wechter, N.; Gray, S.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. Olfactory dysfunction in aging and neurodegenerative diseases. Ageing Res. Rev. 2021, 70, 101416. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, A.; Corrêa, A.; Fisher, K.; Rushworth, J. Olfactory Dysfunction as a Global Biomarker for Sniffing out Alzheimer’s Disease: A Meta-Analysis. Biosensors 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Talamo, B.R.; Rudel, R.; Kosik, K.S.; Lee, V.M.-Y.; Neff, S.; Adelman, L.; Kauer, J.S. Pathological changes in olfactory neurons in patients with Alzheimer’s disease. Nature 1989, 337, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Kovács, T.; Cairns, N.J.; Lantos, P.L. Olfactory centres in Alzheimer’s disease: olfactory bulb is involved in early Braak’s stages. Neuroreport 2001, 12, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Scopa, C.; Marrocco, F.; Latina, V.; Ruggeri, F.; Corvaglia, V.; La Regina, F.; Ammassari-Teule, M.; Middei, S.; Amadoro, G.; Meli, G.; et al. Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Aβ oligomers. Cell Death Differ. 2020, 27, 934–948. [Google Scholar] [CrossRef]
- Santos-Mandujano, R.A.; Ryan, N.S.; Chávez-Gutiérrez, L.; Sánchez-Torres, C.; Meraz-Ríos, M.A. Clinical Association of White Matter Hyperintensities Localization in a Mexican Family with Spastic Paraparesis Carrying the PSEN1 A431E Mutation. J. Alzheimers Dis. JAD 2020, 73, 1075–1083. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Souza, G.H.M.F.; Guest, P.C.; Martins-de-Souza, D. LC-MSE, Multiplex MS/MS, Ion Mobility, and Label-Free Quantitation in Clinical Proteomics. In Multiplex Biomarker Techniques; Guest, P.C., Ed.; Methods in Molecular Biology; Springer New York: New York, NY, 2017; ISBN 978-1-4939-6729-2. [Google Scholar]
- Erten-Lyons, D.; Wilmot, B.; Anur, P.; McWeeney, S.; Westaway, S.K.; Silbert, L.; Kramer, P.; Kaye, J. Microcephaly genes and risk of late-onset Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2011, 25, 276–282. [Google Scholar] [CrossRef]
- Miron, J.; Picard, C.; Nilsson, N.; Frappier, J.; Dea, D.; Théroux, L.; Alzheimer’s Disease Neuroimaging Initiative; United Kingdom Brain Expression Consortium; Poirier, J. CDK5RAP2 gene and tau pathophysiology in late-onset sporadic Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2018, 14, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, T.; Katsenelson, K.; Daniliuc, S.; Hasin, T.; Choder, M.; Aronheim, A. A Novel c-Jun N-terminal Kinase (JNK)-binding Protein WDR62 Is Recruited to Stress Granules and Mediates a Nonclassical JNK Activation. Mol. Biol. Cell 2010, 21, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Duce, J.A.; Smith, D.P.; Blake, R.E.; Crouch, P.J.; Li, Q.-X.; Masters, C.L.; Trounce, I.A. Linker Histone H1 Binds to Disease Associated Amyloid-like Fibrils. J. Mol. Biol. 2006, 361, 493–505. [Google Scholar] [CrossRef]
- Roque, A.; Sortino, R.; Ventura, S.; Ponte, I.; Suau, P. Histone H1 Favors Folding and Parallel Fibrillar Aggregation of the 1–42 Amyloid-β Peptide. Langmuir 2015, 31, 6782–6790. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Falla, D.; Barrera-Ocampo, A.; Hagel, C.; Korwitz, A.; Vinueza-Veloz, M.F.; Zhou, K.; Schonewille, M.; Zhou, H.; Velazquez-Perez, L.; Rodriguez-Labrada, R.; et al. Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J. Clin. Invest. 2014, 124, 1552–1567. [Google Scholar] [CrossRef]
- Guo, E.; Liu, H.; Liu, X. Overexpression of SCUBE2 Inhibits Proliferation, Migration, and Invasion in Glioma Cells. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2017, 25, 437–444. [Google Scholar] [CrossRef]
- Area-Gomez, E.; del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease: Upregulated function of MAM in AD. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef]
- Ihara, S.; Nakayama, S.; Murakami, Y.; Suzuki, E.; Asakawa, M.; Kinoshita, T.; Sawa, H. PIGN prevents protein aggregation in the endoplasmic reticulum independently of its function in the GPI synthesis. J. Cell Sci. 2017, 130, 602–613. [Google Scholar] [CrossRef]
- Ebanks, B.; Ingram, T.L.; Chakrabarti, L. ATP synthase and Alzheimer’s disease: putting a spin on the mitochondrial hypothesis. Aging 2020, 12, 16647–16662. [Google Scholar] [CrossRef]
- Patro, S.; Ratna, S.; Yamamoto, H.A.; Ebenezer, A.T.; Ferguson, D.S.; Kaur, A.; McIntyre, B.C.; Snow, R.; Solesio, M.E. ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11185. [Google Scholar] [CrossRef]
- Lin, Q.; Liang, Q.; Qin, C.; Li, Y. CircANKRD36 Knockdown Suppressed Cell Viability and Migration of LPS-Stimulated RAW264.7 Cells by Sponging MiR-330. Inflammation 2021, 44, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, L.-Q.; He, Z.-X.; He, X.-X.; Wang, Y.-J.; Jian, Y.-L.; Wang, X.; Zhang, B.-B.; Su, C.; Lu, J.; et al. Autism candidate gene DIP2A regulates spine morphogenesis via acetylation of cortactin. PLOS Biol. 2019, 17, e3000461. [Google Scholar] [CrossRef] [PubMed]
- Jordanova, A.; Irobi, J.; Thomas, F.P.; Van Dijck, P.; Meerschaert, K.; Dewil, M.; Dierick, I.; Jacobs, A.; De Vriendt, E.; Guergueltcheva, V.; et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet. 2006, 38, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Prpar Mihevc, S.; Baralle, M.; Buratti, E.; Rogelj, B. TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci. Rep. 2016, 6, 33996. [Google Scholar] [CrossRef] [PubMed]
- Niedowicz, D.M.; Beckett, T.L.; Holler, C.J.; Weidner, A.M.; Murphy, M.P. APP(DeltaNL695) expression in murine tissue downregulates CNBP expression. Neurosci. Lett. 2010, 482, 57–61. [Google Scholar] [CrossRef]
- Sun, C.; Van Ghelue, M.; Tranebjærg, L.; Thyssen, F.; Nilssen, Ø.; Torbergsen, T. Myotonia congenita and myotonic dystrophy in the same family: coexistence of a CLCN1 mutation and expansion in the CNBP (ZNF9) gene. Clin. Genet. 2011, 80, 574–580. [Google Scholar] [CrossRef]
- Liu, L.; Zeng, M.; Stamler, J.S. Hemoglobin induction in mouse macrophages. Proc. Natl. Acad. Sci. 1999, 96, 6643–6647. [Google Scholar] [CrossRef]
- Biagioli, M.; Pinto, M.; Cesselli, D.; Zaninello, M.; Lazarevic, D.; Roncaglia, P.; Simone, R.; Vlachouli, C.; Plessy, C.; Bertin, N.; et al. Unexpected expression of α- and β-globin in mesencephalic dopaminergic neurons and glial cells. Proc. Natl. Acad. Sci. 2009, 106, 15454–15459. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Meurers, B.H.; Zhu, C.; Medvedeva, V.P.; Chesselet, M.-F. Neurons express hemoglobin α- and β-chains in rat and human brains. J. Comp. Neurol. 2009, 515, 538–547. [Google Scholar] [CrossRef]
- Wu, C.; Liao, P.; Yu, L.; Wang, S.; Chen, S.; Wu, C.; Kuo, Y. Hemoglobin promotes A? oligomer formation and localizes in neurons and amyloid deposits. Neurobiol. Dis. 2004, 17, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Irigoyen, J.; Corrales, F.J.; Santamaría, E. Proteomic atlas of the human olfactory bulb. J. Proteomics 2012, 75, 4005–4016. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Gómez, A.; Carmona, M.; Huesa, G.; Porta, S.; Riera-Codina, M.; Biagioli, M.; Gustincich, S.; Aso, E. Neuronal hemoglobin is reduced in Alzheimer’s disease, argyrophilic grain disease, Parkinson’s disease, and dementia with Lewy bodies. J. Alzheimers Dis. JAD 2011, 23, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Vojtek, M.; Chambers, I. Loss of Resf1 reduces the efficiency of embryonic stem cell self-renewal and germline entry. Life Sci. Alliance 2021, 4, e202101190. [Google Scholar] [CrossRef]
- Pitzen, V.; Sander, S.; Baumann, O.; Gräf, R.; Meyer, I. Cep192, a Novel Missing Link between the Centrosomal Core and Corona in Dictyostelium Amoebae. Cells 2021, 10, 2384. [Google Scholar] [CrossRef]
- Yan, S.; Song, M.; Ping, J.; Lai, S.; Cao, X.; Bai, C.-J.; Xie, D.-F.; Guan, H.; Gao, S.; Zhou, P.-K. ZGRF1 promotes end resection of DNA homologous recombination via forming complex with BRCA1/EXO1. Cell Death Discov. 2021, 7, 260. [Google Scholar] [CrossRef]
- Tirilomi, A.; Elakad, O.; Yao, S.; Li, Y.; Hinterthaner, M.; Danner, B.C.; Ströbel, P.; Tirilomis, T.; Bohnenberger, H.; von Hammerstein-Equord, A. Expression and prognostic impact of CD49b in human lung cancer. Medicine (Baltimore) 2022, 101, e28814. [Google Scholar] [CrossRef]
- Liu, H.; Al-aidaroos, A.Q.O.; Wang, H.; Guo, K.; Li, J.; Zhang, H.F.; Zeng, Q. PRL-3 suppresses c-Fos and integrin α2 expression in ovarian cancer cells. BMC Cancer 2013, 13, 80. [Google Scholar] [CrossRef]
- Zutter, M.M.; Santoro, S.A.; Staatz, W.D.; Tsung, Y.L. Re-expression of the alpha 2 beta 1 integrin abrogates the malignant phenotype of breast carcinoma cells. Proc. Natl. Acad. Sci. 1995, 92, 7411–7415. [Google Scholar] [CrossRef]
- Yamada, T.; Horisberger, M.A.; Kawaguchi, N.; Moroo, I.; Toyoda, T. Immunohistochemistry using antibodies to alpha-interferon and its induced protein, MxA, in Alzheimer’s and Parkinson’s disease brain tissues. Neurosci. Lett. 1994, 181, 61–64. [Google Scholar] [CrossRef]
- Furuyama, H.; Chiba, S.; Okabayashi, T.; Yokota, S.; Nonaka, M.; Imai, T.; Fujii, N.; Matsumoto, H. Single nucleotide polymorphisms and functional analysis of MxA promoter region in multiple sclerosis. J. Neurol. Sci. 2006, 249, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-L.; Shi, S.-F.; Wu, W.-L.; Ho, L.-J.; Lai, J.-H. Protective roles of interferon-induced protein with tetratricopeptide repeats 3 (IFIT3) in dengue virus infection of human lung epithelial cells. PloS One 2013, 8, e79518. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, E.M.; Xu, H.; Murphy, R.M.; Grounds, M.D.; Pinniger, G.J. Dysferlin-deficiency has greater impact on function of slow muscles, compared with fast, in aged BLAJ mice. PloS One 2019, 14, e0214908. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu Pene, T.; Rosé, S.D.; Lejen, T.; Marcu, M.G.; Trifaró, J.-M. Expression of various scinderin domains in chromaffin cells indicates that this protein acts as a molecular switch in the control of actin filament dynamics and exocytosis. J. Neurochem. 2005, 92, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, H.; Huang, Y.; Gong, Q.; Wang, J.; Ling, J. Expression and Function of the Actin-severing Protein Adseverin in the Proliferation, Migration, and Differentiation of Dental Pulp Cells. J. Endod. 2015, 41, 493–500. [Google Scholar] [CrossRef]
- Pohlkamp, T.; Wasser, C.R.; Herz, J. Functional Roles of the Interaction of APP and Lipoprotein Receptors. Front. Mol. Neurosci. 2017, 10, 54. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef]
- Bu, G.; Cam, J.; Zerbinatti, C. LRP in amyloid-beta production and metabolism. In Integrated Molecular Medicine for Neuronal and Neoplastic Disorders; Sobue, G., Takahashi, M., Yoshida, J., Kaibuchi, K., Naoe, T., Lahiri, D.K., Eds.; Blackwell Publishing: Oxford, 2006; ISBN 978-1-57331-655-2. [Google Scholar]
- Benoit, M.E.; Hernandez, M.X.; Dinh, M.L.; Benavente, F.; Vasquez, O.; Tenner, A.J. C1q-induced LRP1B and GPR6 proteins expressed early in Alzheimer disease mouse models, are essential for the C1q-mediated protection against amyloid-β neurotoxicity. J. Biol. Chem. 2013, 288, 654–665. [Google Scholar] [CrossRef]
- Kim, S.M.; Jeon, Y.; Kim, D.; Jang, H.; Bae, J.S.; Park, M.K.; Kim, H.; Kim, S.; Lee, H. AIMP3 depletion causes genome instability and loss of stemness in mouse embryonic stem cells. Cell Death Dis. 2018, 9, 972. [Google Scholar] [CrossRef]
- Kim, C.; Park, J.; Song, Y.; Kim, S.; Moon, J. HIF1α-mediated AIMP3 suppression delays stem cell aging via the induction of autophagy. Aging Cell 2019, 18, e12909. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Q.; Cheng, Y.; Gao, M.; Kuang, L.; Wang, C. Myosin Heavy Chain 10 (MYH10) Gene Silencing Reduces Cell Migration and Invasion in the Glioma Cell Lines U251, T98G, and SHG44 by Inhibiting the Wnt/β-Catenin Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 9110–9119. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Zou, H.-Q.; Zhou, H.-D.; Li, J.; Wang, L.; Cao, H.-Y.; Yi, X.; Wang, X.; Liang, C.-R.; Wang, Y.-R.; et al. The relationship between single nucleotide polymorphisms of the NTRK2 gene and sporadic Alzheimer’s disease in the Chinese Han population. Neurosci. Lett. 2013, 550, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Takeshita, F.; Honma, K.; Ono, M.; Kato, K.; Ochiya, T. Ribophorin II regulates breast tumor initiation and metastasis through the functional suppression of GSK3β. Sci. Rep. 2013, 3, 2474. [Google Scholar] [CrossRef]
- Sun, W.; Hua, X.; Gu, Y.; Xu, Q.; Zhu, S.; Lv, T.; Song, Y. E3 ubiquitin ligase RNF6 promotes antiviral immune responses through enhancing the expression of interferon stimulated genes in myeloid cells. Clin. Immunol. 2022, 242, 109099. [Google Scholar] [CrossRef]
- Li, Q.; Wang, G.; Tao, J.; Chen, W. RNF6 promotes colorectal cancer invasion and migration via the Wnt/β-catenin pathway by inhibiting GSK3β activity. Pathol. - Res. Pract. 2021, 225, 153545. [Google Scholar] [CrossRef]
- Zhu, K.; Bai, H.; Mu, M.; Xue, Y.; Duan, Z. Knockdown of RNF6 inhibits HeLa cervical cancer cell growth via suppression of MAPK/ERK signaling. FEBS Open Bio 2021, 11, 2041–2049. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-Y.; Diao, C.-F.; Zhang, Y.; Wu, N.; Wan, H.-Y.; Nong, X.-Y.; Liu, M.; Tang, H. miR-371-5p down-regulates pre mRNA processing factor 4 homolog B (PRPF4B) and facilitates the G1/S transition in human hepatocellular carcinoma cells. Cancer Lett. 2013, 335, 351–360. [Google Scholar] [CrossRef]
- Ou, S.; Liao, Y.; Shi, J.; Tang, J.; Ye, Y.; Wu, F.; Wang, W.; Fei, J.; Xie, F.; Bai, L. S100A16 suppresses the proliferation, migration and invasion of colorectal cancer cells in part via the JNK/p38 MAPK pathway. Mol. Med. Rep. 2021, 23, 164. [Google Scholar] [CrossRef]
- Bose, A.; Banerjee, S.; Visweswariah, S.S. Mutational landscape of receptor guanylyl cyclase C: Functional analysis and disease-related mutations. IUBMB Life 2020, 72, 1145–1159. [Google Scholar] [CrossRef]
- Li, P.; Schulz, S.; Bombonati, A.; Palazzo, J.P.; Hyslop, T.M.; Xu, Y.; Baran, A.A.; Siracusa, L.D.; Pitari, G.M.; Waldman, S.A. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology 2007, 133, 599–607. [Google Scholar] [CrossRef]
- Basu, N.; Saha, S.; Khan, I.; Ramachandra, S.G.; Visweswariah, S.S. Intestinal Cell Proliferation and Senescence Are Regulated by Receptor Guanylyl Cyclase C and p21. J. Biol. Chem. 2014, 289, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Rescher, U.; Gerke, V. S100A10/p11: family, friends and functions. Pflüg. Arch. - Eur. J. Physiol. 2007, 455, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Fan, Y.; Wang, Z.; Li, M.; Su, D.; Liu, Y.; Liang, X. S100A16 promotes acute kidney injury by activating HRD1-induced ubiquitination and degradation of GSK3β and CK1α. Cell. Mol. Life Sci. CMLS 2022, 79, 184. [Google Scholar] [CrossRef]
- Jin, R.; Zhao, A.; Han, S.; Zhang, D.; Sun, H.; Li, M.; Su, D.; Liang, X. The interaction of S100A16 and GRP78 actives endoplasmic reticulum stress-mediated through the IRE1α/XBP1 pathway in renal tubulointerstitial fibrosis. Cell Death Dis. 2021, 12, 942. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; Tan, X.; Lv, D.-J.; Xing, R.-W.; Shu, F.-P.; Zhong, C.-F.; Li, C.; Zou, Y.-G.; Mao, X.-M. Phosphoribosyl pyrophosphate synthetases 2 knockdown inhibits prostate cancer progression by suppressing cell cycle and inducing cell apoptosis. J. Cancer 2020, 11, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Wang, Y. Targeted Quantitative Kinome Analysis Identifies PRPS2 as a Promoter for Colorectal Cancer Metastasis. J. Proteome Res. 2019, 18, 2279–2286. [Google Scholar] [CrossRef]
- Han, J.M.; Sahin, M. TSC1/TSC2 signaling in the CNS. FEBS Lett. 2011, 585, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Ferrando-Miguel, R.; Rosner, M.; Freilinger, A.; Lubec, G.; Hengstschläger, M. Tuberin – A New Molecular Target in Alzheimer’s Disease? Neurochem. Res. 2005, 30, 1413–1419. [Google Scholar] [CrossRef]
- Habib, S.L.; Michel, D.; Masliah, E.; Thomas, B.; Ko, H.S.; Dawson, T.M.; Abboud, H.; Clark, R.A.; Imam, S.Z. Role of Tuberin in Neuronal Degeneration. Neurochem. Res. 2008, 33, 1113–1116. [Google Scholar] [CrossRef]
- Hahn, I.; Voelzmann, A.; Parkin, J.; Fülle, J.B.; Slater, P.G.; Lowery, L.A.; Sanchez-Soriano, N.; Prokop, A. Tau, XMAP215/Msps and Eb1 co-operate interdependently to regulate microtubule polymerisation and bundle formation in axons. PLoS Genet. 2021, 17, e1009647. [Google Scholar] [CrossRef]
- Kim, M.-J.; Yun, H.S.; Hong, E.-H.; Lee, S.-J.; Baek, J.-H.; Lee, C.-W.; Yim, J.-H.; Kim, J.-S.; Park, J.K.; Um, H.-D.; et al. Depletion of end-binding protein 1 (EB1) promotes apoptosis of human non-small-cell lung cancer cells via reactive oxygen species and Bax-mediated mitochondrial dysfunction. Cancer Lett. 2013, 339, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Jeong, S.-Y.; Youle, R.J. Endophilin B1 is required for the maintenance of mitochondrial morphology. J. Cell Biol. 2004, 166, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Odierna, G.L.; Kerwin, S.K.; Harris, L.E.; Shin, G.J.-E.; Lavidis, N.A.; Noakes, P.G.; Millard, S.S. Dscam2 suppresses synaptic strength through a PI3K-dependent endosomal pathway. J. Cell Biol. 2020, 219, e201909143. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, S.; Liu, J.; Huo, Y.; Luo, X.-J. The schizophrenia susceptibility gene NAGA regulates dendritic spine density: further evidence for the dendritic spine pathology of schizophrenia. Mol. Psychiatry 2021, 26, 7102–7104. [Google Scholar] [CrossRef] [PubMed]
- Kapuralin, K.; Ćurlin, M.; Mitrečić, D.; Kosi, N.; Schwarzer, C.; Glavan, G.; Gajović, S. STAM2, a member of the endosome-associated complex ESCRT-0 is highly expressed in neurons. Mol. Cell. Neurosci. 2015, 67, 104–115. [Google Scholar] [CrossRef]
- Furić Cunko, V.; Mitrecić, D.; Mavrić, S.; Gajović, S. Expression pattern and functional analysis of mouse Stam2 in the olfactory system. Coll. Antropol. 2008, 32 Suppl 1, 59–63. [Google Scholar]
- Mizuno, E.; Kawahata, K.; Kato, M.; Kitamura, N.; Komada, M. STAM Proteins Bind Ubiquitinated Proteins on the Early Endosome via the VHS Domain and Ubiquitin-interacting Motif. Mol. Biol. Cell 2003, 14, 3675–3689. [Google Scholar] [CrossRef]
- Shehadeh, L.A.; Yu, K.; Wang, L.; Guevara, A.; Singer, C.; Vance, J.; Papapetropoulos, S. SRRM2, a potential blood biomarker revealing high alternative splicing in Parkinson’s disease. PloS One 2010, 5, e9104. [Google Scholar] [CrossRef]
- Ilik, İ.A.; Malszycki, M.; Lübke, A.K.; Schade, C.; Meierhofer, D.; Aktaş, T. SON and SRRM2 are essential for nuclear speckle formation. eLife 2020, 9, e60579. [Google Scholar] [CrossRef] [PubMed]
- McMillan, P.J.; Strovas, T.J.; Baum, M.; Mitchell, B.K.; Eck, R.J.; Hendricks, N.; Wheeler, J.M.; Latimer, C.S.; Keene, C.D.; Kraemer, B.C. Pathological tau drives ectopic nuclear speckle scaffold protein SRRM2 accumulation in neuron cytoplasm in Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Burgener, A.-V.; Bantug, G.R.; Meyer, B.J.; Higgins, R.; Ghosh, A.; Bignucolo, O.; Ma, E.H.; Loeliger, J.; Unterstab, G.; Geigges, M.; et al. SDHA gain-of-function engages inflammatory mitochondrial retrograde signaling via KEAP1-Nrf2. Nat. Immunol. 2019, 20, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.O.; Shinoda, Y.; Sakai, K.; Hashikawa, T.; Watanabe, M.; Machida, T.; Hirabayashi, Y.; Furuya, S. Selective upregulation of 3-phosphoglycerate dehydrogenase (Phgdh) expression in adult subventricular zone neurogenic niche. Neurosci. Lett. 2009, 453, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Y.; Yoshida, K.; Yang, J.H.; Suzuki, T.; Azuma, N.; Sakai, K.; Hashikawa, T.; Watanabe, M.; Yasuda, K.; Kuhara, S.; et al. Impaired neurogenesis in embryonic spinal cord of Phgdh knockout mice, a serine deficiency disorder model. Neurosci. Res. 2009, 63, 184–193. [Google Scholar] [CrossRef]
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jégo, P.; Vigneron, P.-A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer’s Disease. Cell Metab. 2020, 31, 503–517. [Google Scholar] [CrossRef]
- Chen, X.; Calandrelli, R.; Girardini, J.; Yan, Z.; Tan, Z.; Xu, X.; Hiniker, A.; Zhong, S. PHGDH expression increases with progression of Alzheimer’s disease pathology and symptoms. Cell Metab. 2022, 34, 651–653. [Google Scholar] [CrossRef]
- Castora, F.J.; Kerns, K.A.; Pflanzer, H.K.; Hitefield, N.L.; Gershon, B.; Shugoll, J.; Shelton, M.; Coleman, R.A. Expression Changes in Mitochondrial Genes Affecting Mitochondrial Morphology, Transmembrane Potential, Fragmentation, Amyloidosis, and Neuronal Cell Death Found in Brains of Alzheimer’s Disease Patients. J. Alzheimers Dis. JAD 2022, 90, 119–137. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Pradeepkiran, J.A.; Morton, H.; Reddy, P.H. A partial reduction of Drp1 improves cognitive behavior and enhances mitophagy, autophagy and dendritic spines in a transgenic Tau mouse model of Alzheimer disease. Hum. Mol. Genet. 2022, 31, 1788–1805. [Google Scholar] [CrossRef]
- Lei, D.; Li, F.; Su, H.; Liu, J.; Wei, N.; Wang, X. Hepatic deficiency of COP9 signalosome subunit 8 induces ubiquitin-proteasome system impairment and Bim-mediated apoptosis in murine livers. PloS One 2013, 8, e67793. [Google Scholar] [CrossRef]
- Su, H.; Li, J.; Zhang, H.; Ma, W.; Wei, N.; Liu, J.; Wang, X. COP9 signalosome controls the degradation of cytosolic misfolded proteins and protects against cardiac proteotoxicity. Circ. Res. 2015, 117, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Nishimura, T.; Kondo, H.; Ikeda, K.; Hayashi, Y.; McGeer, P.L. Expression of the receptor for macrophage colony stimulating factor by brain microglia and its upregulation in brains of patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Brain Res. 1994, 639, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Alonso, A.; Schetters, S.T.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain J. Neurol. 2016, 139, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Elwood, F.; Britschgi, M.; Villeda, S.; Zhang, H.; Ding, Z.; Zhu, L.; Alabsi, H.; Getachew, R.; Narasimhan, R.; et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J. Exp. Med. 2013, 210, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-L.; Liu, T.-Y.; Kung, F.-L. FKBP12 regulates the localization and processing of amyloid precursor protein in human cell lines. J. Biosci. 2014, 39, 85–95. [Google Scholar] [CrossRef]
- Blair, L.J.; Criado-Marrero, M.; Zheng, D.; Wang, X.; Kamath, S.; Nordhues, B.A.; Weeber, E.J.; Dickey, C.A. The Disease-Associated Chaperone FKBP51 Impairs Cognitive Function by Accelerating AMPA Receptor Recycling. eNeuro 2019, 6, ENEURO–0242. [Google Scholar] [CrossRef]
- Zhang, J.; Rubio, V.; Lieberman, M.W.; Shi, Z.-Z. OLA1, an Obg-like ATPase, suppresses antioxidant response via nontranscriptional mechanisms. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 15356–15361. [Google Scholar] [CrossRef]
- Mao, R.-F.; Rubio, V.; Chen, H.; Bai, L.; Mansour, O.C.; Shi, Z.-Z. OLA1 protects cells in heat shock by stabilizing HSP70. Cell Death Dis. 2013, 4, e491. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, H.; Liu, Z.; Li, Q.; Guo, Y.; Chen, Y.; Chang, Y.; Cui, H. Carnitine palmitoyltransferase 1 (CPT1) alleviates oxidative stress and apoptosis of hippocampal neuron in response to beta-Amyloid peptide fragment Aβ25-35. Bioengineered 12, 5440–5449. [CrossRef]
- Yerbury, J.J.; Ooi, L.; Dillin, A.; Saunders, D.N.; Hatters, D.M.; Beart, P.M.; Cashman, N.R.; Wilson, M.R.; Ecroyd, H. Walking the tightrope: proteostasis and neurodegenerative disease. J. Neurochem. 2016, 137, 489–505. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Kabir, Md.T.; Uddin, Md.S.; Abdeen, A.; Ashraf, G.M.; Perveen, A.; Hafeez, A.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Evidence Linking Protein Misfolding to Quality Control in Progressive Neurodegenerative Diseases. Curr. Top. Med. Chem. 2020, 20, 2025–2043. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Barnes, K.; De Marco, M.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Venneri, A.; Mortiboys, H. Mitochondrial Dysfunction in Alzheimer’s Disease: A Biomarker of the Future? Biomedicines 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis. 2016, 7, 201–214. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain Glucose Hypometabolism and Oxidative Stress in Preclinical Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.-N.; Xu, W.; Li, J.-Q.; Guo, Y.; Cui, M.; Chen, K.-L.; Huang, Y.-Y.; Dong, Q.; Tan, L.; Yu, J.-T.; et al. FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis: a longitudinal study. Alzheimers Res. Ther. 2019, 11, 57. [Google Scholar] [CrossRef]
- Mosconi, L.; Sorbi, S.; de Leon, M.J.; Li, Y.; Nacmias, B.; Myoung, P.S.; Tsui, W.; Ginestroni, A.; Bessi, V.; Fayyazz, M.; et al. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2006, 47, 1778–1786. [Google Scholar]
- Koh, K.; Ishiura, H.; Beppu, M.; Shimazaki, H.; Ichinose, Y.; Mitsui, J.; Kuwabara, S.; Tsuji, S.; Takiyama, Y. ; Japan Spastic Paraplegia Research Consortium Novel mutations in the ALDH18A1 gene in complicated hereditary spastic paraplegia with cerebellar ataxia and cognitive impairment. J. Hum. Genet. 2018, 63, 1009–1013. [Google Scholar] [CrossRef]
- Patel, A.; Rees, S.D.; Kelly, M.A.; Bain, S.C.; Barnett, A.H.; Thalitaya, D.; Prasher, V.P. Association of variants within APOE, SORL1, RUNX1, BACE1 and ALDH18A1 with dementia in Alzheimer’s disease in subjects with Down syndrome. Neurosci. Lett. 2011, 487, 144–148. [Google Scholar] [CrossRef]
- Palmer, A.M. The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer’s disease. J. Neural Transm. 1999, 106, 317–328. [Google Scholar] [CrossRef]
- Tiwari, V.; Patel, A.B. Pyruvate Carboxylase and Pentose Phosphate Fluxes are Reduced in AβPP-PS1 Mouse Model of Alzheimer’s Disease: A 13C NMR Study. J. Alzheimers Dis. 2014, 41, 387–399. [Google Scholar] [CrossRef]
- Schrötter, A.; Pfeiffer, K.; El Magraoui, F.; Platta, H.W.; Erdmann, R.; Meyer, H.E.; Egensperger, R.; Marcus, K.; Müller, T. The Amyloid Precursor Protein (APP) Family Members are Key Players in S-adenosylmethionine Formation by MAT2A and Modify BACE1 and PSEN1 Gene Expression-Relevance for Alzheimer’s Disease. Mol. Cell. Proteomics MCP 2012, 11, 1274–1288. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Hyun, J.; Park, J.; Jung, S.; Oh, Y.; Kim, Y.; Ryu, S.-H.; Kim, S.-H.; Jeong, E.I.; Jo, D.-G.; et al. Aberrant role of pyruvate kinase M2 in the regulation of gamma-secretase and memory deficits in Alzheimer’s disease. Cell Rep. 2021, 37. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhong, M.B.; Toro, C.A.; Zhang, L.; Cai, D. Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer’s disease pathogenesis. Neurosci. Lett. 2019, 703, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Mathews, P.M.; Boiteau, A.B.; Hassinger, L.C.; Peterhoff, C.M.; Jiang, Y.; Mullaney, K.; Neve, R.L.; Gruenberg, J.; Nixon, R.A. Down syndrome fibroblast model of Alzheimer-related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am. J. Pathol. 2008, 173, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Van Acker, Z.P.; Bretou, M.; Annaert, W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol. Neurodegener. 2019, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Zwart, R.; van der Sluijs, P.; Annaert, W.; Gool, W.A.; Baas, F. Alzheimer’s presenilin 1 is a putative membrane receptor for rab GDP dissociation inhibitor. Hum. Mol. Genet. 2000, 9, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, G.; Reyna, S.M.; Dunlap, M.; Van Der Kant, R.; Callender, J.A.; Young, J.E.; Roberts, E.A.; Goldstein, L.S.B. Defective Transcytosis of APP and Lipoproteins in Human iPSC-Derived Neurons with Familial Alzheimer’s Disease Mutations. Cell Rep. 2016, 17, 759–773. [Google Scholar] [CrossRef]
- Malm, T.; Loppi, S.; Kanninen, K.M. Exosomes in Alzheimer’s disease. Neurochem. Int. 2016, 97, 193–199. [Google Scholar] [CrossRef]
- Lee, S.; Tsai, F.T.F. Molecular chaperones in protein quality control. J. Biochem. Mol. Biol. 2005, 38, 259–265. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.M.; Boelens, W.C.; Otte-Höller, I.; Kamps, B.; de Waal, R.M.W.; Verbeek, M.M. Small heat shock proteins inhibit amyloid-beta protein aggregation and cerebrovascular amyloid-beta protein toxicity. Brain Res. 2006, 1089, 67–78. [Google Scholar] [CrossRef]
- Gammazza, A.M.; Bavisotto, C.C.; Barone, R.; Macario, E.C.; de M. and A.J.L. Alzheimer’s Disease and Molecular Chaperones: Current Knowledge and the Future of Chaperonotherapy Available online:. Available online: https://www.eurekaselect.com/142309/article (accessed on Jul 24, 2020).
- Singh, R.; Kaur, N.; Dhingra, N.; Kaur, T. Protein misfolding, ER stress and chaperones: an approach to develop chaperone-based therapeutics for Alzheimer’s disease. Int. J. Neurosci. 2022, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1999, 1, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; VanLeeuwen, J.-E. Impaired regulation of synaptic actin cytoskeleton in Alzheimer’s disease. Brain Res. Rev. 2011, 67, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Knafo, S.; Alonso-Nanclares, L.; Gonzalez-Soriano, J.; Merino-Serrais, P.; Fernaud-Espinosa, I.; Ferrer, I.; DeFelipe, J. Widespread Changes in Dendritic Spines in a Model of Alzheimer’s Disease. Cereb. Cortex 2009, 19, 586–592. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–129. [Google Scholar] [CrossRef]
- Xie, Z.; Srivastava, D.P.; Photowala, H.; Kai, L.; Cahill, M.E.; Woolfrey, K.M.; Shum, C.Y.; Surmeier, D.J.; Penzes, P. Kalirin-7 Controls Activity-Dependent Structural and Functional Plasticity of Dendritic Spines. Neuron 2007, 56, 640–656. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble Oligomers of Amyloid β Protein Facilitate Hippocampal Long-Term Depression by Disrupting Neuronal Glutamate Uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef]
- Ma, Q.-L.; Yang, F.; Calon, F.; Ubeda, O.J.; Hansen, J.E.; Weisbart, R.H.; Beech, W.; Frautschy, S.A.; Cole, G.M. p21-activated Kinase-aberrant Activation and Translocation in Alzheimer Disease Pathogenesis. J. Biol. Chem. 2008, 283, 14132–14143. [Google Scholar] [CrossRef]
- Huesa, G.; Baltrons, M.A.; Gómez-Ramos, P.; Morán, A.; García, A.; Hidalgo, J.; Francés, S.; Santpere, G.; Ferrer, I.; Galea, E. Altered Distribution of RhoA in Alzheimer’s Disease and AβPP Overexpressing Mice. J. Alzheimers Dis. 2010, 19, 37–56. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, Y.-F.; Zhang, L.; Huang, L.; Yu, P.; Zhu, H.; Deng, W.; Qin, C. Effective expression of Drebrin in hippocampus improves cognitive function and alleviates lesions of Alzheimer’s disease in APP (swe)/PS1 (ΔE9) mice. CNS Neurosci. Ther. 2017, 23, 590–604. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Mosser, S.; Fraering, P.C. Inactivation of brain Cofilin-1 by age, Alzheimer’s disease and γ-secretase. Biochim. Biophys. Acta BBA - Mol. Basis Dis. 2014, 1842, 2500–2509. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Liu, G.; Jiang, Y.; Jiang, Q.; Liao, M.; Feng, R.; Zhang, L.; Ma, G.; Zhang, S.; Chen, Z.; et al. Cell adhesion molecule pathway genes are regulated by cis-regulatory SNPs and show significantly altered expression in Alzheimer’s disease brains. Neurobiol. Aging 2015, 36, 2904–e1. [Google Scholar] [CrossRef] [PubMed]
- Yew, D.T.; Li, W.P.; Webb, S.E.; Lai, H.W.L.; Zhang, L. Neurotransmitters, peptides, and neural cell adhesion molecules in the cortices of normal elderly humans and alzheimer patients: a comparison. Exp. Gerontol. 1999, 34, 117–133. [Google Scholar] [CrossRef]
- Gautam, V.; D’Avanzo, C.; Hebisch, M.; Kovacs, D.M.; Kim, D.Y. BACE1 activity regulates cell surface contactin-2 levels. Mol. Neurodegener. 2014, 9, 4. [Google Scholar] [CrossRef]
- Ando, K.; Uemura, K.; Kuzuya, A.; Maesako, M.; Asada-Utsugi, M.; Kubota, M.; Aoyagi, N.; Yoshioka, K.; Okawa, K.; Inoue, H.; et al. N-cadherin Regulates p38 MAPK Signaling via Association with JNK-associated Leucine Zipper Protein. J. Biol. Chem. 2011, 286, 7619–7628. [Google Scholar] [CrossRef]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between β-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2007, 16, 15–23. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Khondoker, M.; Newhouse, S.; Westman, E.; Muehlboeck, J.-S.; Mecocci, P.; Vellas, B.; Tsolaki, M.; Kłoszewska, I.; Soininen, H.; et al.; for the AddNeuroMed consortium and for the Alzheimer’s Disease Neuroimaging Initiative Linking Genetics of Brain Changes to Alzheimer’s Disease: Sparse Whole Genome Association Scan of Regional MRI Volumes in the ADNI and AddNeuroMed Cohorts. J. Alzheimers Dis. 2015, 45, 851–864. [Google Scholar] [CrossRef]
- Rosenthal, S.B.; Wang, H.; Shi, D.; Liu, C.; Abagyan, R.; McEvoy, L.K.; Chen, C.-H. Mapping the gene network landscape of Alzheimer’s disease through integrating genomics and transcriptomics. PLoS Comput. Biol. 2022, 18, e1009903. [Google Scholar] [CrossRef] [PubMed]
- Shokhirev, M.N.; Johnson, A.A. An integrative machine-learning meta-analysis of high-throughput omics data identifies age-specific hallmarks of Alzheimer’s disease. Ageing Res. Rev. 2022, 81, 101721. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Pedrero-Prieto, C.M.; García-Carpintero, S.; Frontiñán-Rubio, J.; Llanos-González, E.; Aguilera García, C.; Alcaín, F.J.; Lindberg, I.; Durán-Prado, M.; Peinado, J.R.; Rabanal-Ruiz, Y. A comprehensive systematic review of CSF proteins and peptides that define Alzheimer’s disease. Clin. Proteomics 2020, 17, 21. [Google Scholar] [CrossRef] [PubMed]
- Higginbotham, L.; Ping, L.; Dammer, E.B.; Duong, D.M.; Zhou, M.; Gearing, M.; Hurst, C.; Glass, J.D.; Factor, S.A.; Johnson, E.C.B.; et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci. Adv. 2020, 6, eaaz9360. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.A. The Triangle of Death in Alzheimer’s Disease Brain: The Aberrant Cross-Talk Among Energy Metabolism, Mammalian Target of Rapamycin Signaling, and Protein Homeostasis Revealed by Redox Proteomics. Antioxid. Redox Signal. 2017, 26, 364–387. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Toledo, G.; Silva-Lucero, M.-D.-C.; Herrera-Díaz, J.; García, D.-E.; Arias-Montaño, J.-A.; Cardenas-Aguayo, M.-D.-C. Patient-Derived Fibroblasts With Presenilin-1 Mutations, That Model Aspects of Alzheimer’s Disease Pathology, Constitute a Potential Object for Early Diagnosis. Front. Aging Neurosci. 2022, 14, 921573. [Google Scholar] [CrossRef]
- Wiśniewski, J.R. Filter-Aided Sample Preparation for Proteome Analysis. In Microbial Proteomics; Becher, D., Ed.; Methods in Molecular Biology; Springer New York: New York, NY, 2018; ISBN 978-1-4939-8693-4. [Google Scholar]
- Ríos-Castro, E.; Souza, G.H.M.F.; Delgadillo-Álvarez, D.M.; Ramírez-Reyes, L.; Torres-Huerta, A.L.; Velasco-Suárez, A.; Cruz-Cruz, C.; Hernández-Hernández, J.M.; Tapia-Ramírez, J. Quantitative Proteomic Analysis of MARC-145 Cells Infected with a Mexican Porcine Reproductive and Respiratory Syndrome Virus Strain Using a Label-Free Based DIA approach. J. Am. Soc. Mass Spectrom. 2020, 31, 1302–1312. [Google Scholar] [CrossRef]
Figure 1.
Family pedigree of the study subjects. Males are illustrated as squares and females as circles. Crossed figures depict deceased individuals. PSEN1 (A431E) mutation carriers genetically verified are shown with a dotted line [33].
Figure 1.
Family pedigree of the study subjects. Males are illustrated as squares and females as circles. Crossed figures depict deceased individuals. PSEN1 (A431E) mutation carriers genetically verified are shown with a dotted line [33].
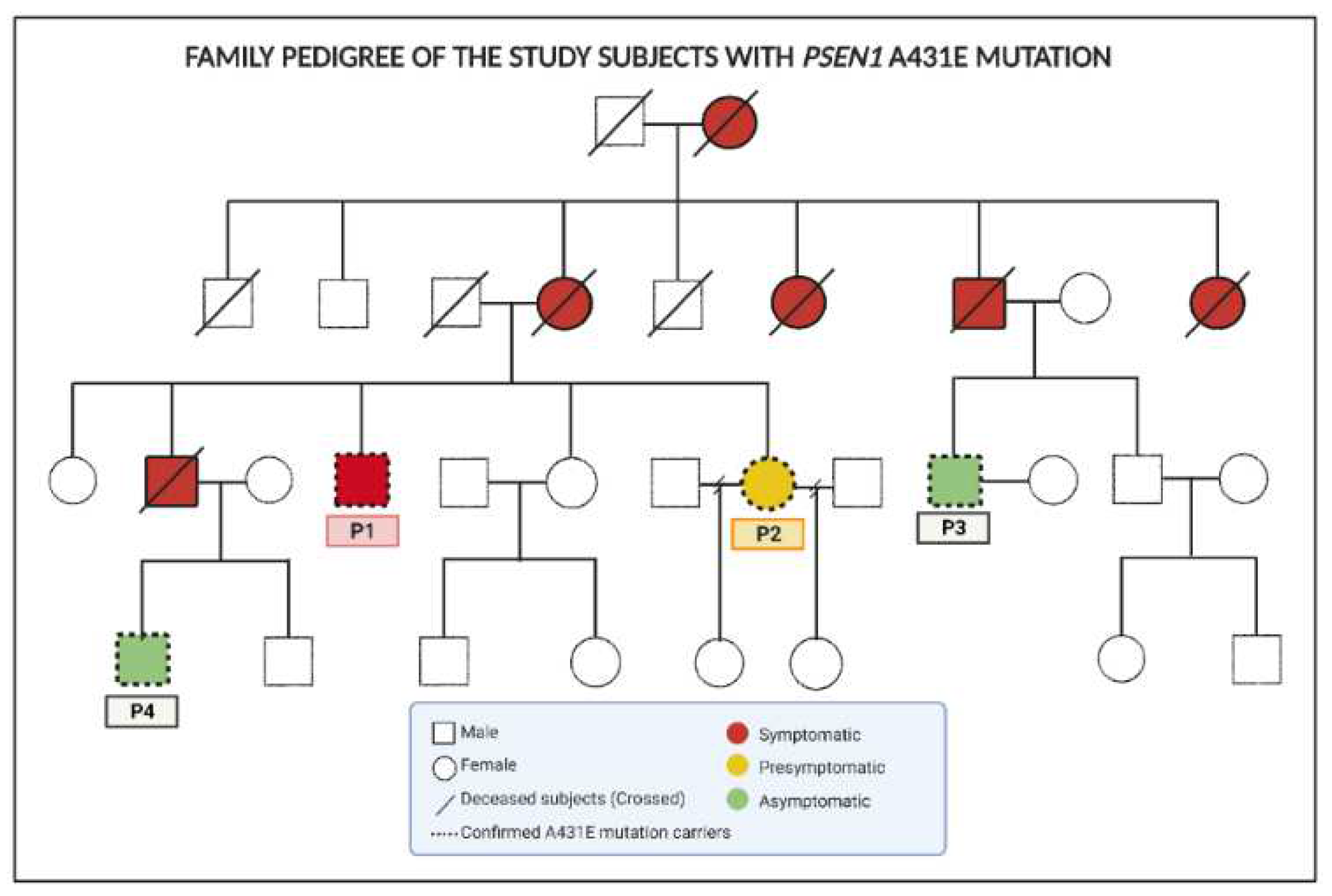
Figure 2.
Flowchart for the study's process. Olfactory ecto-mesenchymal stem cells were gathered and cultivated from two PSEN1 (A431E) mutant carriers and two healthy donors. MSC markers were then determined by flow cytometry. Protein was isolated and digested to identify DEPs by stage (symptomatic and presymptomatic) using two label-free proteomic techniques (P1/C1 and P2/C2). Data quality control and filtering were made to demonstrate proper reliability and confidence in the outcomes. Each protein group was subjected to functional analysis and comparisons using various bioinformatics techniques.
Figure 2.
Flowchart for the study's process. Olfactory ecto-mesenchymal stem cells were gathered and cultivated from two PSEN1 (A431E) mutant carriers and two healthy donors. MSC markers were then determined by flow cytometry. Protein was isolated and digested to identify DEPs by stage (symptomatic and presymptomatic) using two label-free proteomic techniques (P1/C1 and P2/C2). Data quality control and filtering were made to demonstrate proper reliability and confidence in the outcomes. Each protein group was subjected to functional analysis and comparisons using various bioinformatics techniques.
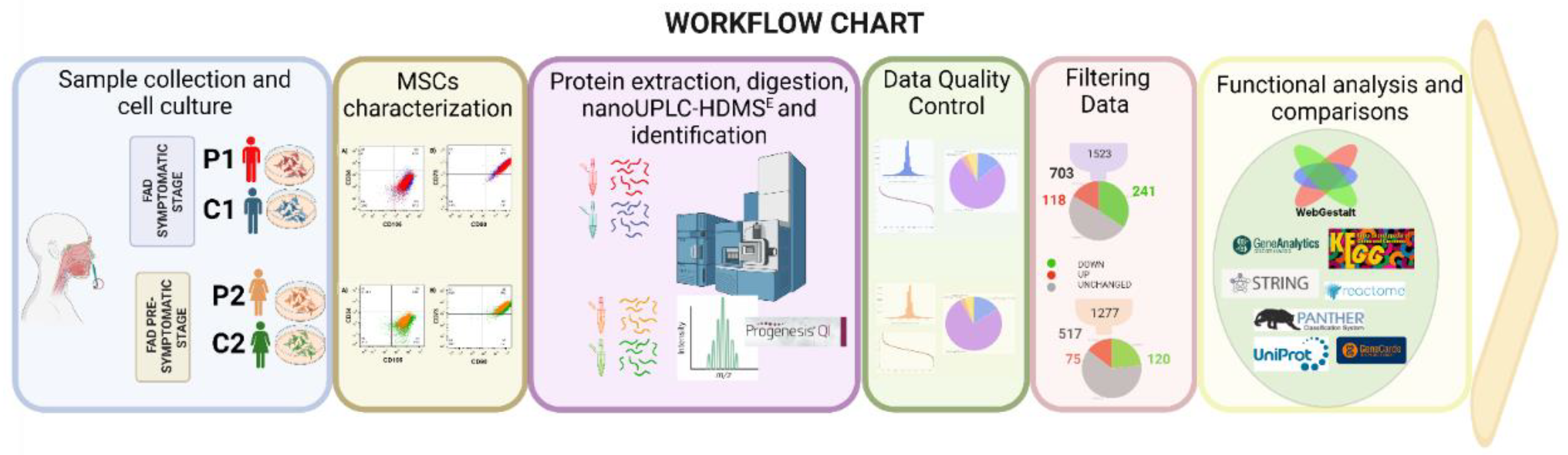
Figure 3.
Nine passes of nasal exfoliation result in cells expressing mesenchymal markers. A) The symptomatic mutation carrier (P1) showed in red. Its control (C1) is in blue, B) dot plots of cells from presymptomatic mutation carrier (P2) are shown in orange, and its control (C2) is in green. labeled with markers for CD34 and CD105, CD73 and CD90, CD45 and CD14, CD166 and CD19.
Figure 3.
Nine passes of nasal exfoliation result in cells expressing mesenchymal markers. A) The symptomatic mutation carrier (P1) showed in red. Its control (C1) is in blue, B) dot plots of cells from presymptomatic mutation carrier (P2) are shown in orange, and its control (C2) is in green. labeled with markers for CD34 and CD105, CD73 and CD90, CD45 and CD14, CD166 and CD19.
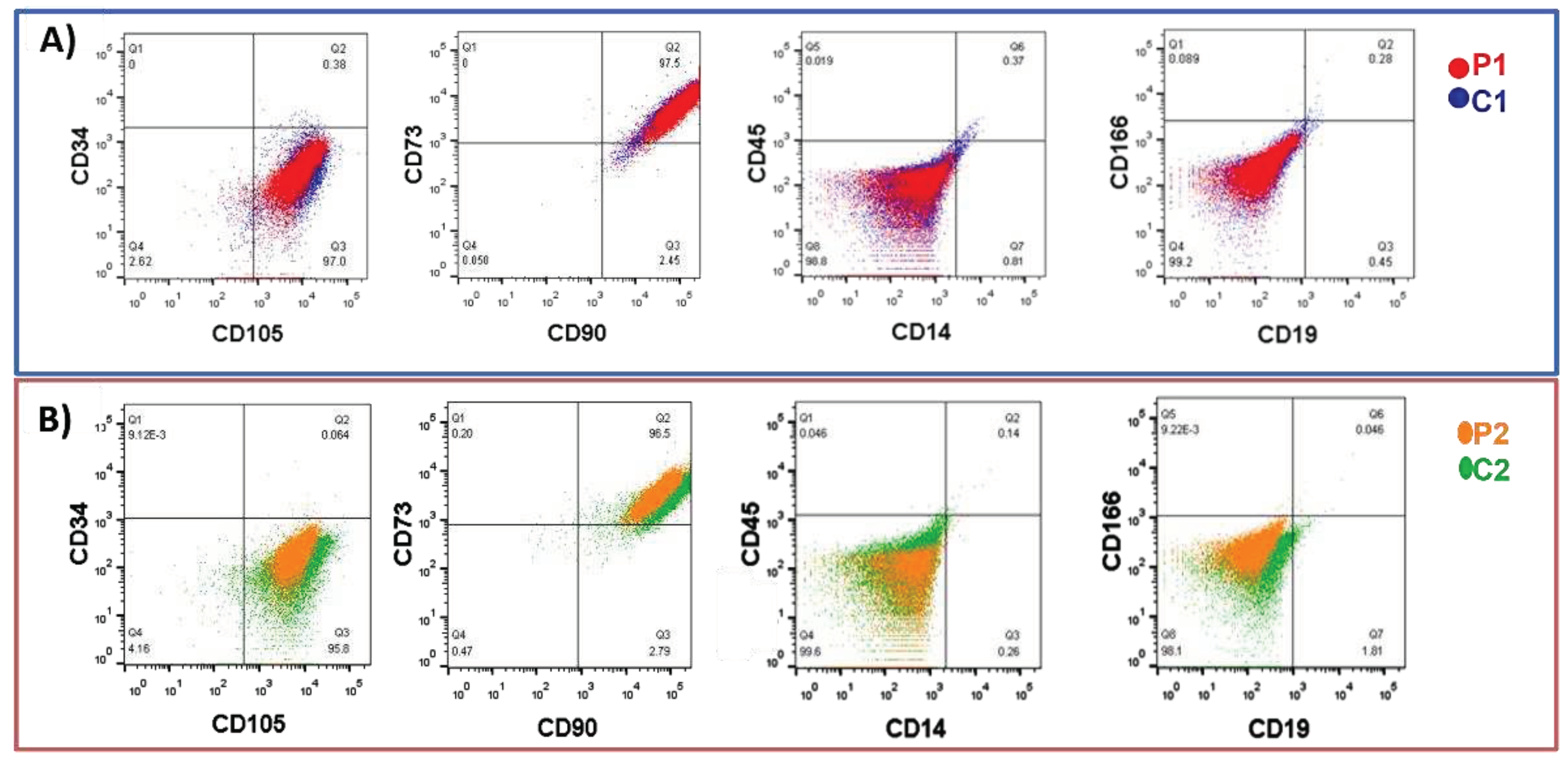
Figure 4.
Dependability and confidence of peptides and proteins (P1/C1, P2/C2). A and E) histograms show the total number of peptides with an inaccuracy of no more than 5 ppm (dark blue/orange). B and F). Dot plot of PepFrag1 peptides concentrated at a maximum of 18 ppm in case of B and 16 ppm in case of F, across the examined m/z range. C and G) Pie graph illustrating the different types of peptides The dynamic range of measured proteins in D and H. The Y-axis represents the average intensities for each quantified protein (expressed as log10), and the X-axis represents the number of quantified proteins (IDS).
Figure 4.
Dependability and confidence of peptides and proteins (P1/C1, P2/C2). A and E) histograms show the total number of peptides with an inaccuracy of no more than 5 ppm (dark blue/orange). B and F). Dot plot of PepFrag1 peptides concentrated at a maximum of 18 ppm in case of B and 16 ppm in case of F, across the examined m/z range. C and G) Pie graph illustrating the different types of peptides The dynamic range of measured proteins in D and H. The Y-axis represents the average intensities for each quantified protein (expressed as log10), and the X-axis represents the number of quantified proteins (IDS).
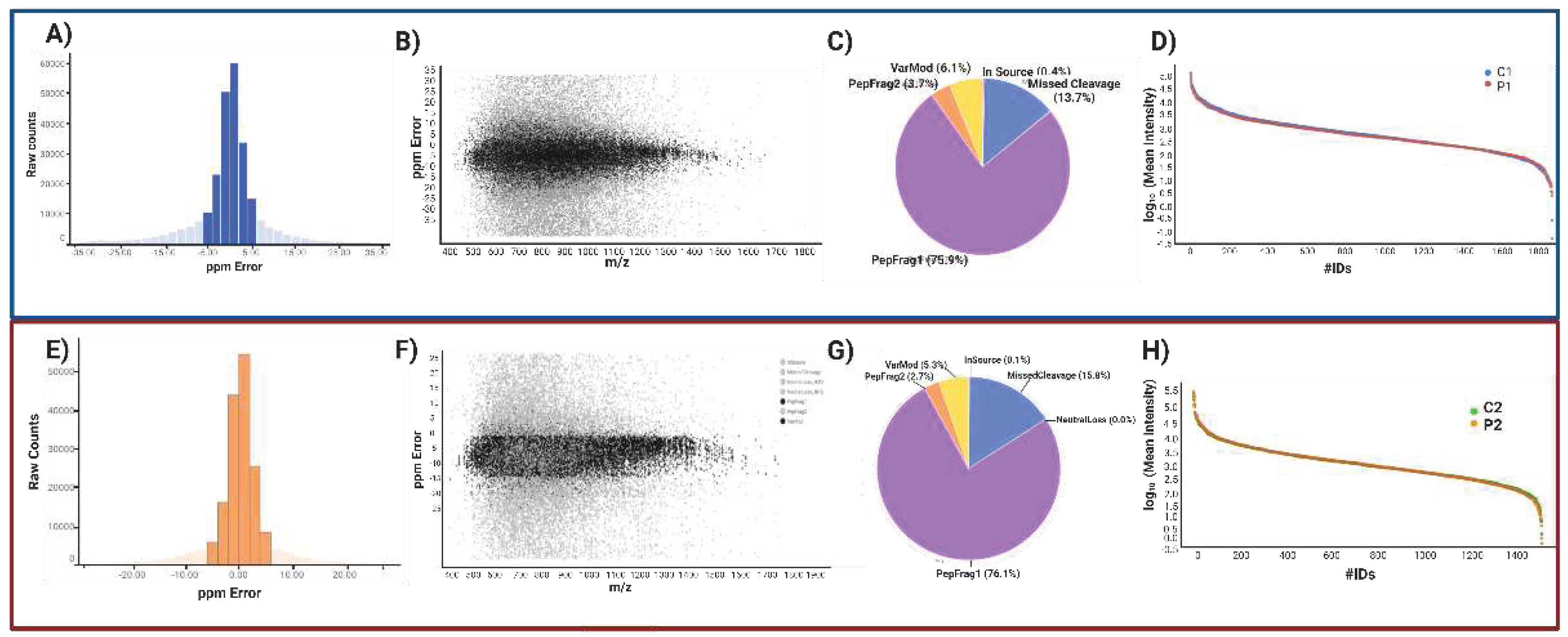
Figure 5.
Differentially expressed proteins in symptomatic and presymptomatic carriers related to controls. Both proteins that are up-regulated (red) and down-regulated (green) in all pairwise comparisons are depicted in the volcano plot (A, C). B) and D) A pie chart showing the different protein classes based on differential expression.
Figure 5.
Differentially expressed proteins in symptomatic and presymptomatic carriers related to controls. Both proteins that are up-regulated (red) and down-regulated (green) in all pairwise comparisons are depicted in the volcano plot (A, C). B) and D) A pie chart showing the different protein classes based on differential expression.
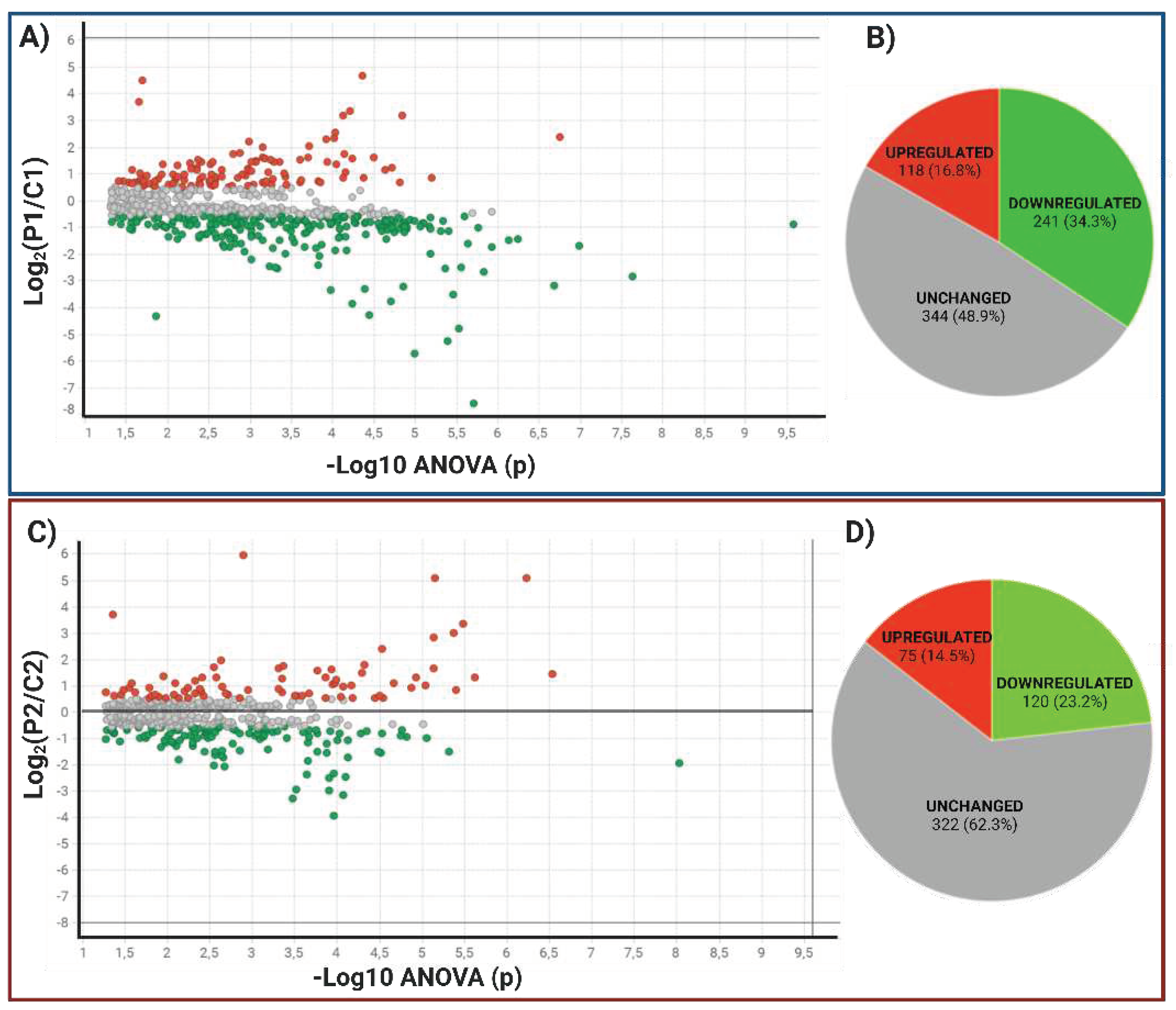
Figure 6.
Gene Ontology Annotation of proteins produced differently in symptomatic (A-C) and presymptomatic (D-E) carrier cells compared to C1 and C2. Biological Processes A and D) cellular component (B and E) and molecular function (C and F).
Figure 6.
Gene Ontology Annotation of proteins produced differently in symptomatic (A-C) and presymptomatic (D-E) carrier cells compared to C1 and C2. Biological Processes A and D) cellular component (B and E) and molecular function (C and F).
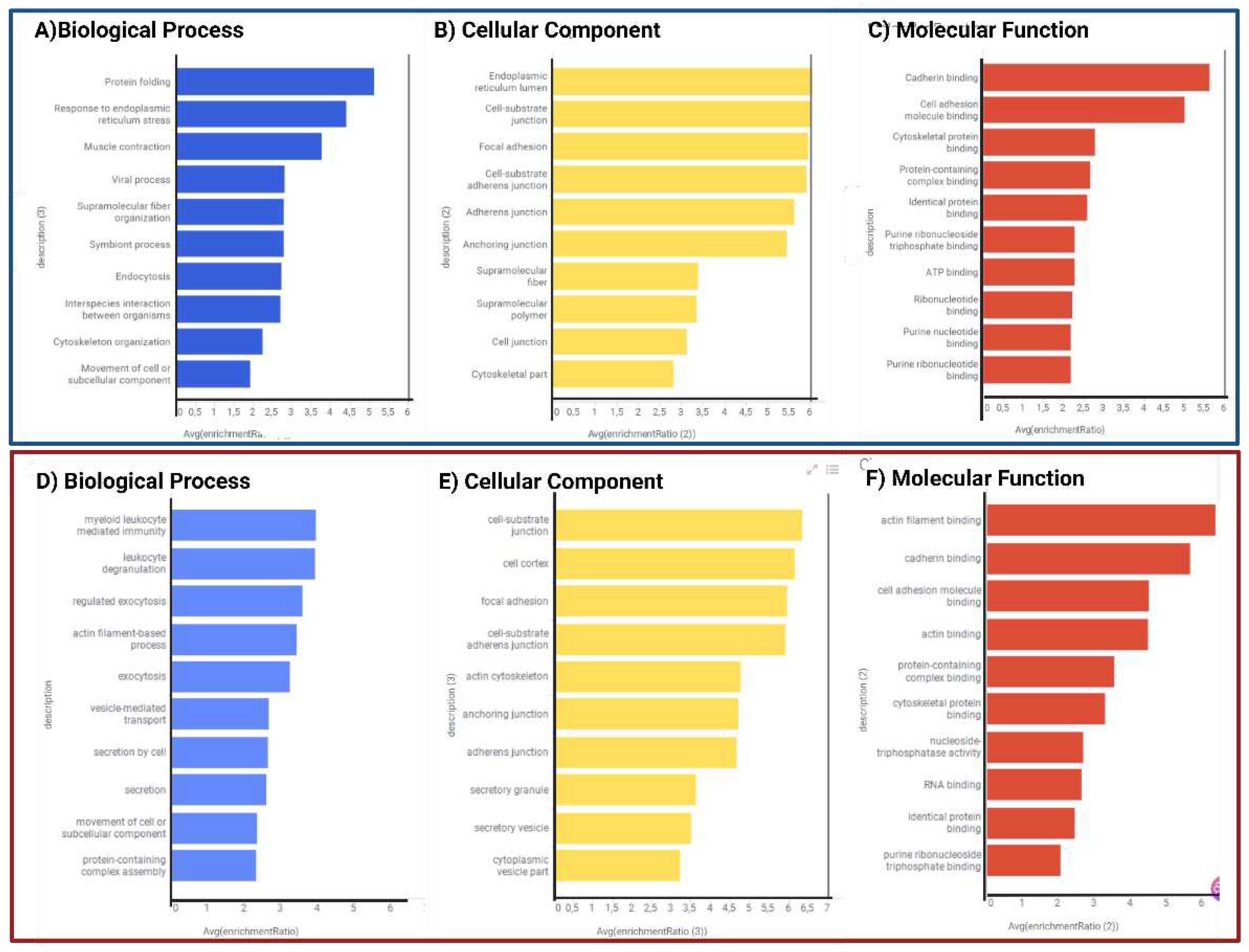
Figure 7.
Top 15 enriched pathways in P1/C1 (A) and P2/C2 (B) using WebGestalt with KEGG Database.
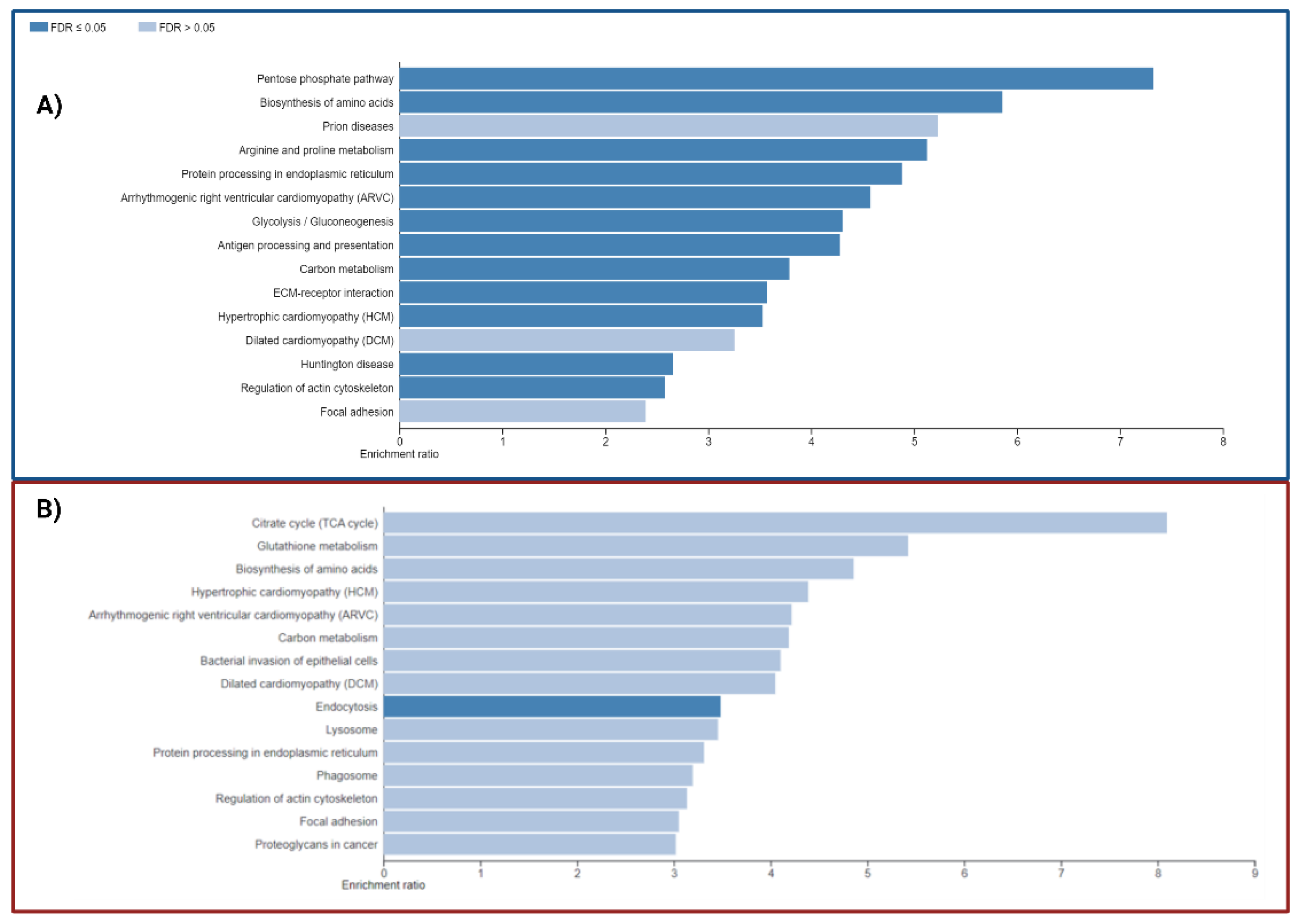
Figure 8.
Protein-protein interaction (PPI) network of P1/C1 (A) and P2/C2 (B) DEPs constructed on STRING with the KEGG-enriched pathways underlined. The upper-right corner displays disconnected nodes connected to the indicated paths. (0.9) Confidence rating.
Figure 8.
Protein-protein interaction (PPI) network of P1/C1 (A) and P2/C2 (B) DEPs constructed on STRING with the KEGG-enriched pathways underlined. The upper-right corner displays disconnected nodes connected to the indicated paths. (0.9) Confidence rating.
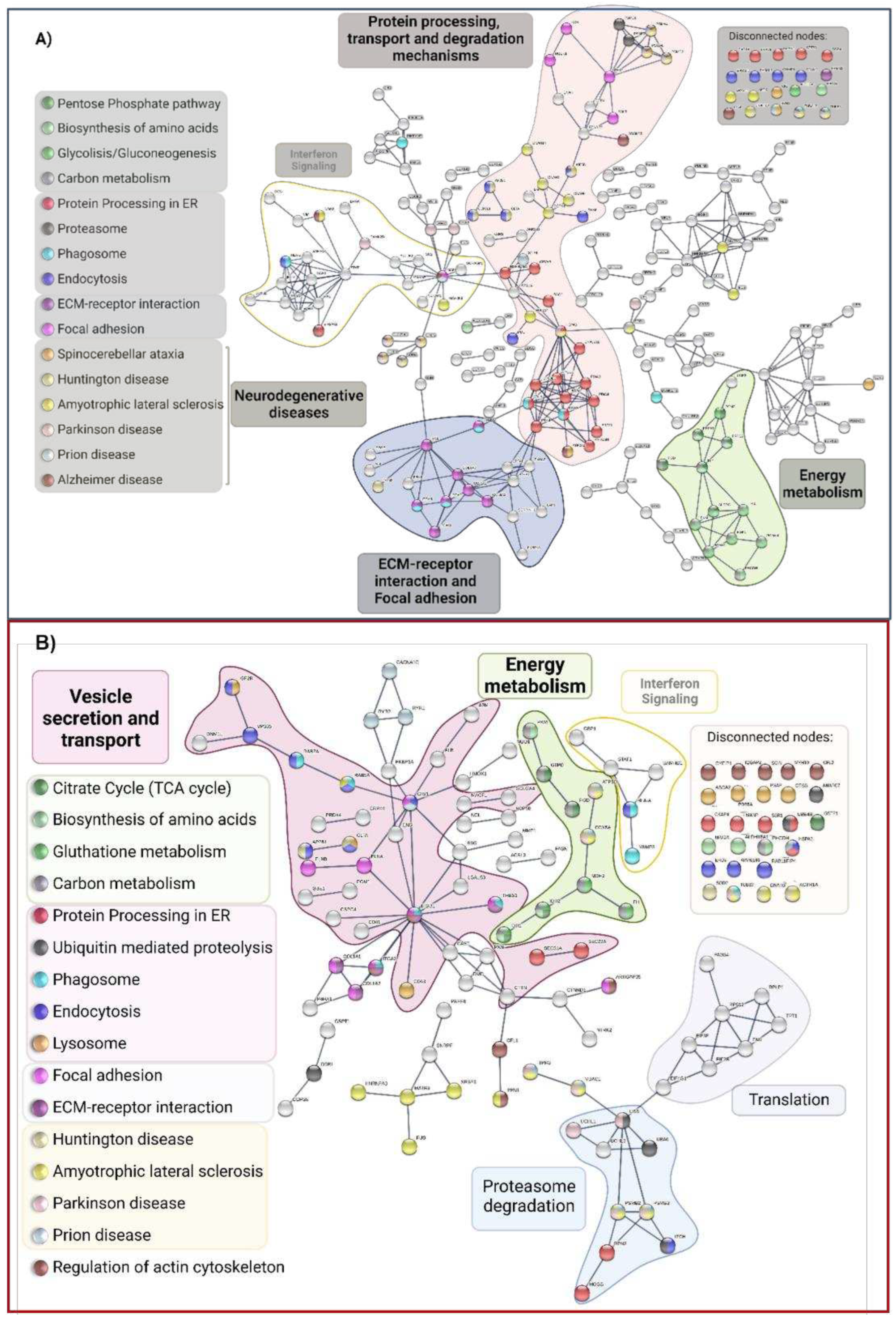
Figure 9.
PPIs of differentially expressed proteins of P1 and P2 involved in A) Protein Processing in ER and B) Vesicle secretion and transport.
Figure 9.
PPIs of differentially expressed proteins of P1 and P2 involved in A) Protein Processing in ER and B) Vesicle secretion and transport.
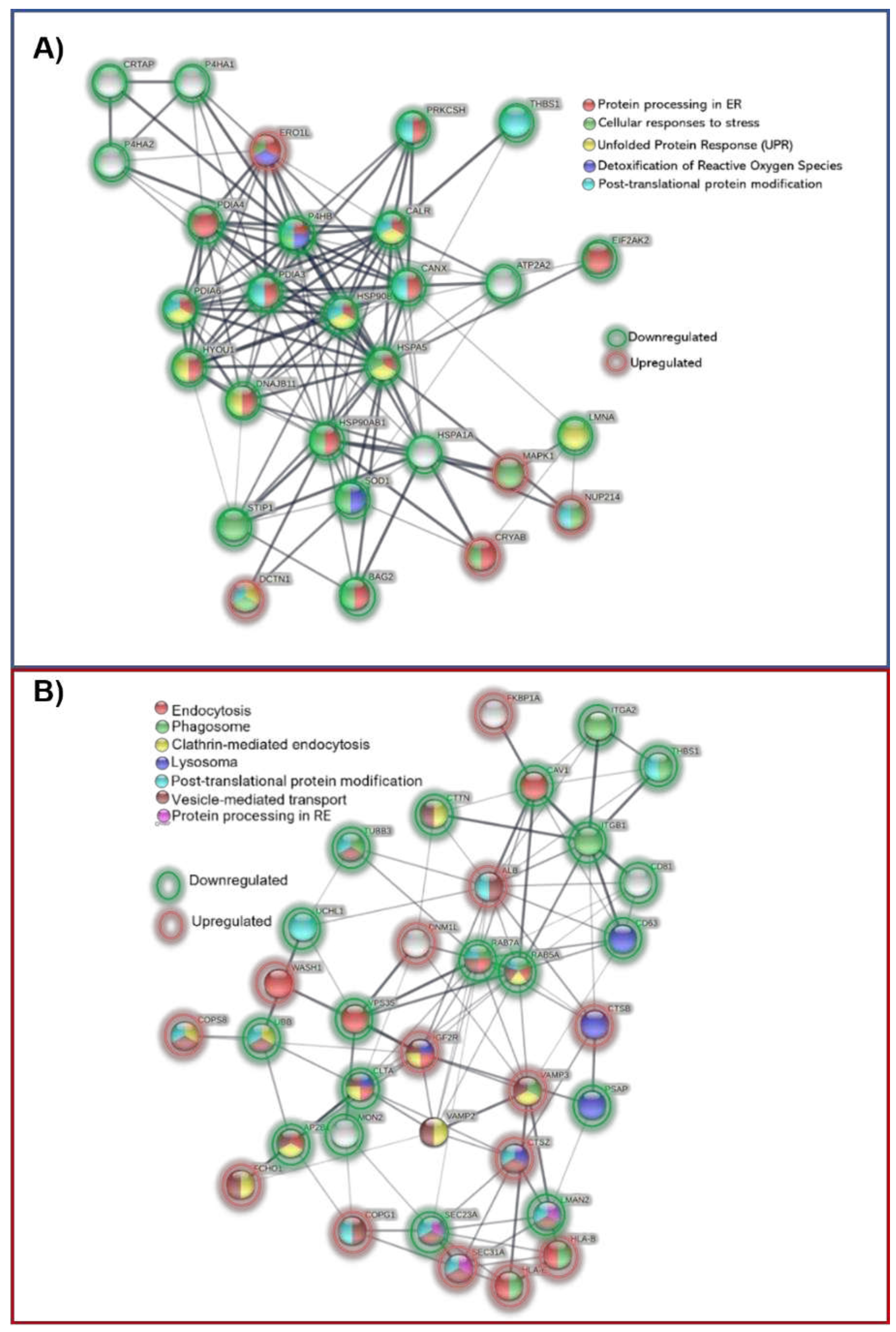
Figure 10.
Summary of dysproteostasis network in PSEN1 (A431E) mutation carriers.
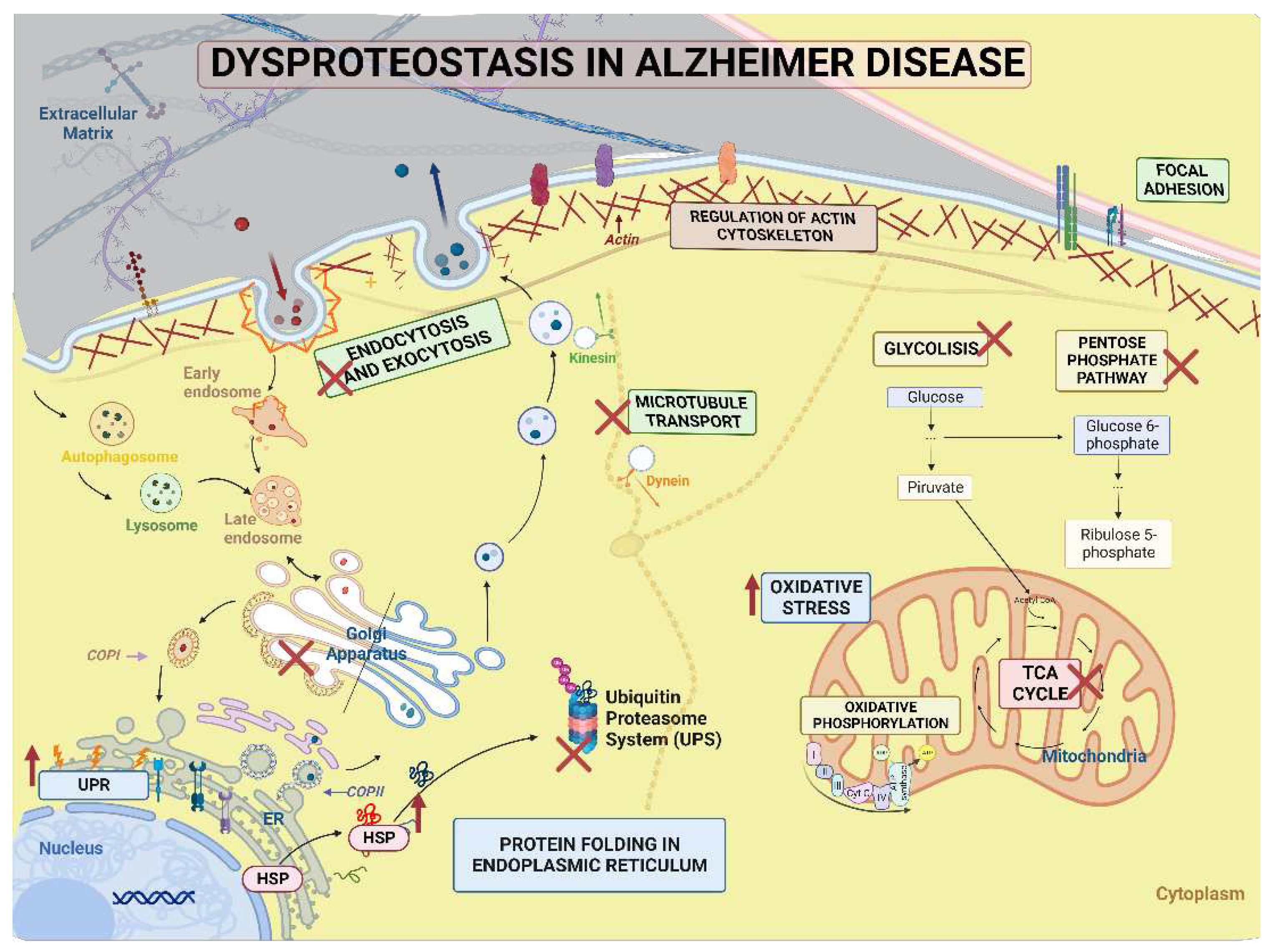

Table 1.
Unique proteins for P1 and P2.
| Uniprot ID | Gene Symbol | Description | ANOVA (p) | |
|---|---|---|---|---|
| P1 | O60282-2 | KIF5C | Kinesin heavy chain isoform 5C | 5.92E-09 |
| Q8IV33 | KIAA0825 | Uncharacterized protein KIAA0825 | 7.01E-08 | |
| A6NCS7 | UTY | Histone demethylase UTY | 1.23E-07 | |
| P16401 | HIST1H1B | Histone H1.5 | 3.42E-07 | |
| Q5JPF3-3 | ANKRD36C | Ankyrin repeat domain-containing protein 36C | 4.84E-07 | |
| Q9NQ36-3 | SCUBE2 | Signal peptide_ CUB and EGF-like domain-containing protein 2 | 8.00E-07 | |
| F8WBJ0 | CDK5RAP2 | CDK5 regulatory subunit-associated protein 2 | 1.87E-05 | |
| O43379-3 | WDR62 | WD repeat-containing protein 62 | 2.44E-05 | |
| Q8NEM0-3 | MCPH1 | Microcephalin | 3.85E-05 | |
| P18859-2 | ATP5PF | ATP synthase-coupling factor 6_ mitochondrial | 1.75E-03 | |
| K7ESH9 | PIGN | GPI ethanolamine phosphate transferase 1 | 2.50E-03 | |
| P2 | P62633 | CNBP | Cellular nucleic acid-binding protein | 1.6801E-09 |
| Q92786 | PROX1 | Prospero Homeobox protein 1 | 2.733E-09 | |
| H7BYV6 | BIN3 | Bridging integrator 3 | 3.9233E-09 | |
| G3V4C6 | RTRAF | RNA transcription_ translation, and transport factor protein | 2.3644E-07 | |
| O75947 | ATP5H | ATP synthase subunit d_ mitochondrial | 6.5903E-07 | |
| B1AKR6 | DYNLRB1 | Dynein light chain roadblock-type 1 | 8.1222E-07 | |
| Q95365 | HLA-B | HLA class I histocompatibility antigen_ B-38 alpha chain | 1.348E-05 | |
| A0A0C3SFZ9 | FCHO1 | F-BAR domain only protein 1 | 1.5595E-05 | |
| Q8IWT3 | CUL9 | Cullin-9 | 2.1129E-05 | |
| P54577 | YARS | Tyrosine--tRNA ligase_ cytoplasmic | 3.6024E-05 | |
| Q9Y5L4 | TIMM13 | Mitochondrial import inner membrane translocase subunit Tim13 | 0.0020643 | |
| Q9Y2E4 | DIP2C | Disco-interacting protein 2 homolog C | 0.00959067 |
Table 2.
Top 10 downregulated proteins in PSEN(A431E) mutation carriers.
| UNIPROT ID | Gene Symbol (GS) |
Description | FC | Anova (p) | |
|---|---|---|---|---|---|
| Symptomatic carrier (P1) | P68871 | HBB | Hemoglobin subunit beta | -189.5 | 1.99E-06 |
| Q9HCM1 | KIAA1551 | Uncharacterized protein KIAA1551 | -52.0 | 1.05E-05 | |
| P20591 | MX1 | Interferon-induced GTP-binding protein Mx1 | -37.1 | 4.19E-06 | |
| A0A2R8Y7C0 | HBA2 | Hemoglobin subunit alpha (Fragment) | -27.6 | 3.04E-06 | |
| E7EMF1 | ITGA2 | Integrin alpha-2 | -26.9 | 7.13E-05 | |
| Q86YA3 | ZGRF1 | Protein ZGRF1 | -29.7 | 1.42E-02 | |
| Q8TEP8 | CEP192 | Centrosomal protein of 192 kDa | -19.2 | 3.72E-05 | |
| Q15283 | RASA2 | Ras GTPase-activating protein 2 | -14.3 | 5.99E-05 | |
| Q9Y6K5 | OAS3 | 2'-5'-oligoadenylate synthase 3 | -13.5 | 2.02E-05 | |
| O14879 | IFIT3 | Interferon-induced protein with tetratricopeptide repeats 3 | -11.2 | 3.56E-06 | |
| Presymptomatic Carrier (P2) | Q9Y6U3 | SCIN | Adseverin | -14.9 | 1.10E-04 |
| C9J1V9 | EEF1E1-BLOC1S5 | EEF1E1-BLOC1S5 readthrough (NMD candidate) | -9.7 | 3.39E-04 | |
| Q7Z2Z1 | TICRR | Treslin | -8.7 | 8.58E-05 | |
| P35580 | MYH10 | Myosin-10 | -7.8 | 1.28E-04 | |
| Q16620 | NTRK2 | BDNF/NT-3 growth factors receptor | -7.6 | 3.14E-04 | |
| P32455 | GBP1 | Guanylate-binding protein 1 | -5.6 | 1.28E-04 | |
| Q9NZR2 | LRP1B | Low-density lipoprotein receptor-related protein 1B | -5.5 | 8.13E-05 | |
| O75131 | CPNE3 | Copine-3 | -5.2 | 2.32E-04 | |
| C9JD73 | PPP1R7 | Protein phosphatase 1 regulatory subunit 7 | -5.0 | 1.13E-04 | |
| A0A087X0Y2 | UTY | Histone demethylase UTY | -4.1 | 2.15E-03 |
Table 3.
Top 10 upregulated proteins in symptomatic carrier (P1) compared with control (C1) MSCs.
| UNIPROT ID | GS | Description | FC | Anova (p) | |
|---|---|---|---|---|---|
| Symptomatic carrier (P1) | P25092 | GUCY2C | Heat-stable enterotoxin receptor | 34.9 | 2.07E-02 |
| Q13523 | PRPF4B | Serine/threonine-protein kinase PRP4 homolog | 25.9 | 4.42E-05 | |
| Q15691 | MAPRE1 | Microtubule-associated protein RP/EB family member 1 | 19.8 | 2.28E-02 | |
| Q8TD84 | DSCAML1 | Down syndrome cell adhesion molecule-like protein 1 | 10.4 | 6.39E-05 | |
| Q4LDE5 | SVEP1 | Sushi_ von Willebrand factor type A_ EGF and pentraxin domain-containing protein 1 | 9.4 | 7.57E-05 | |
| Q96FQ6 | S100A16 | Protein S100-A16 | 9.3 | 1.45E-05 | |
| P11908 | PRPS2 | Ribose-phosphate pyrophosphokinase 2 | 5.9 | 9.41E-05 | |
| P17050 | NAGA | Alpha-N-acetylgalactosaminidase | 5.4 | 1.82E-07 | |
| O00750 | PIK3C2B | Phosphatidylinositol 4-phosphate 3-kinase C2 domain-containing subunit beta | 5.2 | 9.90E-05 | |
| A0A2R8Y5F1 | TSC2 | Tuberin | 5.0 | 1.22E-04 | |
| Presymptomatic Carrier (P2) | O00429 | DNM1L | Dynamin-1-like protein | 94.5 | 1.29E-03 |
| Q99627 | COPS8 | COP9 signalosome complex subunit 8 | 35.0 | 6.07E-07 | |
| P07333 | CSF1R | Macrophage colony-stimulating factor 1 receptor | 34.3 | 7.27E-06 | |
| P05386 | RPLP1 | 60S acidic ribosomal protein P1 | 19.8 | 4.54E-02 | |
| P62942 | FKBP1A | Peptidyl-prolyl cis-trans isomerase FKBP1A | 10.3 | 3.36E-06 | |
| P02768 | ALB | Serum albumin | 8.2 | 4.31E-06 | |
| J3KQ32 | OLA1 | Obg-like ATPase 1 | 7.3 | 7.48E-06 | |
| Q92817 | EVPL | Envoplakin | 5.3 | 3.00E-05 | |
| P50416 | CPT1A | Carnitine O-palmitoyltransferase 1_ liver isoform | 4.0 | 2.37E-03 | |
| Q9Y2X3 | NOP58 | Nucleolar protein 58 | 3.5 | 4.94E-05 |
Table 4.
KEGG pathways enrichment analysis in Symptomatic carrier compared to C1.
| GENESET | PATHWAY | SIZE | OVERLAPPED PROTEINS | Enrich Ratio |
pValue | FDR |
|---|---|---|---|---|---|---|
| hsa00030 | Pentose phosphate pathway | 30 | 6 (↑: PRPS1, PRPS2. ↓: ALDOC, PGD, PGM2, , TKT) | 7.32 | 1.32E-04 | 8.86E-03 |
| hsa01230 | Biosynthesis of amino acids | 75 | 12 (↑: ALDH18A1, PRPS1, PRPS2, PHGDH. ↓: ALDOC, MAT2A, PGAM1, PGAM4, PGK1, PKM, TKT, TPI1) |
5.86 | 7.19E-07 | 1.17E-04 |
| hsa05020 | Prion diseases | 35 | 5 (↑: MAPK1. ↓: HSPA1A, HSPA5, SOD1, STIP1) | 5.23 | 2.41E-03 | 6.04E-02 |
| hsa00330 | Arginine and proline metabolism | 50 | 7 (↑: ALDH18A1, OAT, ↓: ALDH7A1, LAP3, NOS2, P4HA1. P4HA2) | 5.13 | 3.77E-04 | 1.76E-02 |
| hsa04141 | Protein processing in the endoplasmic reticulum | 165 | 22 (↑: SAR1A, CRYAB. ERO1A. ↓: BAG2, CALR, CANX, DNAJB11, HSP90AB1, HSP90B1, HSPA1A, HSPA5, PDIA3, PDIA4, PDIA6, ERP29, P4HB, EIF2AK2, HYOU1, LMAN2, PRKCSH, SSR4, STT3A) | 4.88 | 5.29E-10 | 1.73E-07 |
| hsa05412 | Arrhythmogenic right ventricular cardiomyopathy | 72 | 9 (↓: ACTN2, ATP2A2, CTNNA2, DES, DSP, ITGA11, ITGA2, ITGA5, LMNA) | 4.58 | 1.36E-04 | 8.86E-03 |
| hsa00010 | Glycolysis / Gluconeogenesis | 68 | 8 (↓: ALDH7A1, ALDOC, PGAM1, PGAM4, PGK1, PGM2, PKM, TPI1) | 4.31 | 4.89E-04 | 1.99E-02 |
| hsa04612 | Antigen processing and presentation | 77 | 9 (↓: CANX, CALR, HLA-E, HSP90AB1, HSPA1A, HSPA5, PDIA3, PSME1, PSME2) | 4.28 | 2.29E-04 | 1.24E-02 |
| hsa01200 | Carbon metabolism | 116 | 12 (↑: PHGDH, PRPS1, PRPS2, SDHA, ↓: PKM, PGAM1, PGAM4, PGD, PGK1, TKT, ALDOC, TPI1.) |
3.79 | 7.08E-05 | 7.70E-03 |
| hsa04512 | ECM-receptor interaction | 82 | 8 (↑: COL6A3, ↓: COL1A2, COL1A1, FN1, ITGA11, ITGA2, ITGA5, THBS1) | 3.57 | 1.70E-03 | 5.00E-02 |
| hsa05410 | Hypertrophic cardiomyopathy (HCM) | 83 | 8 (↓: ATP2A2, DES, ITGA11, ITGA5, ITGA2, LMNA, TPM2, TPM3) | 3.53 | 1.84E-03 | 5.00E-02 |
| hsa05414 | Dilated cardiomyopathy (DCM) | 90 | 8 (↓: ATP2A2, DES, ITGA11, ITGA5, ITGA2, LMNA, TPM2, TPM3) | 3.25 | 3.07E-03 | 6.68E-02 |
| hsa05016 | Huntington disease | 193 | 14 (↑: SLC25A5, DCTN1, CLTCL1, CYCS, SDHA. ↓: AP2B1, CLTA, DNAH1, DNAH10, DNAH14, DNAH6, ITPR1, SOD1, TGM2) |
2.66 | 7.83E-04 | 2.55E-02 |
| hsa04810 | Regulation of actin cytoskeleton | 213 | 15 (↑: ENAH, MAPK1, NCKAP1, PAK2, ROCK1. ↓: FN1, GSN, IQGAP3, ITGA11, ITGA2, ITGA5, PAK3, PIKFYVE, RRAS, SCIN) |
2.58 | 6.95E-04 | 2.52E-02 |
| hsa04510 | Focal adhesion | 199 | 13 (↑: COL6A3, PAK2, MAPK1, ROCK1.↓: COL1A2, COL1A1, FN1, ITGA11, ITGA2, ITGA5, KDR, PAK3, THBS1) | 2.39 | 3.06E-03 | 6.68E-02 |
* ↑: upregulated protein, ↓: downregulated protein.
Table 5.
KEGG pathways in Presymptomatic carrier compared to C2.
| geneSet | Description | size | overlap | enrRatio | pValue | FDR |
|---|---|---|---|---|---|---|
| hsa00020 | Citrate cycle (TCA cycle) | 30 | 4 (↑: FH, MDH2, ↓: IDH1, IDH2) | 8.10 | 1.38E-03 | 6.93E-02 |
| hsa00480 | Glutathione metabolism | 56 | 5 (↓: G6PD, GSTP1, IDH1, IDH2,PGD) | 5.42 | 2.18E-03 | 7.73E-02 |
| hsa01230 | Biosynthesis of amino acids | 75 | 6 (↓: ALDH18A1, IDH1, IDH2, MAT2A, PHGDH, PKM) | 4.86 | 1.41E-03 | 6.93E-02 |
| hsa05410 | Hypertrophic cardiomyopathy (HCM) | 83 | 6 (↓: CACNA1C, DMD, ITGA2, ITGB1, RYR2, TPM1) | 4.39 | 2.37E-03 | 7.73E-02 |
| hsa05412 | Arrhythmogenic right ventricular cardiomyopathy | 72 | 5 (↓: CACNA1C, DMD, ITGA2, ITGB1, RYR2) | 4.22 | 6.48E-03 | 1.51E-01 |
| hsa01200 | Carbon metabolism | 116 | 8 (↑: FH, MDH2, ↓: G6PD, IDH1, IDH2, PGD, PHGDH, PKM) | 4.19 | 6.20E-04 | 6.93E-02 |
| hsa05100 | Bacterial invasion of epithelial cells | 74 | 5 (↓: CAV1, CLTA, CTTN, ITGB1, PXN) | 4.10 | 7.27E-03 | 1.58E-01 |
| hsa05414 | Dilated cardiomyopathy (DCM) | 90 | 6 (↓: CACNA1C, DMD, ITGA2, ITGB1, RYR2, TPM1) | 4.05 | 3.56E-03 | 9.67E-02 |
| hsa04144 | Endocytosis | 244 | 14 (↑: IGF2R, WASHC5 ↓: AP2B1, CAV1, CLTA, EHD3, HLA-A, HSPA2, ITCH, RAB11FIP4, RAB5A, RAB7A, UBB, VPS35) | 3.48 | 4.38E-05 | 1.43E-02 |
| hsa04142 | Lysosome | 123 | 7 (↑: ABCA2, CTSB, CTSZ, IGF2R ↓: CD63, CLTA, PSAP) |
3.46 | 4.06E-03 | 1.02E-01 |
| hsa04141 | Protein processing in the endoplasmic reticulum | 165 | 9 (↑: SEC31A, SSR1, UBE4B ↓: CKAP4, HSPA2, LMAN2, MOGS, RPN2, SEC23A) | 3.31 | 1.54E-03 | 6.93E-02 |
| hsa04145 | Phagosome | 152 | 8 (↑: VAMP3. ↓: HLA-A, ITGA2, ITGB1, RAB5A, RAB7A, THBS1, TUBB3,) | 3.20 | 3.50E-03 | 9.67E-02 |
| hsa04810 | Regulation of actin cytoskeleton | 213 | 11 (↑: ARHGAP35, CFL1, CFL2, CYFIP1, IQGAP2, PFN1, ↓: ITGA2, ITGB1, MYH10, PXN, SCIN) | 3.14 | 7.35E-04 | 6.93E-02 |
| hsa04510 | Focal adhesion | 199 | 10 (↑: ARHGAP35, COL1A1, COL1A2, FLNA, FLNB, ↓: CAV1, ITGA2, ITGB1, PXN, THBS1) | 3.05 | 1.58E-03 | 6.93E-02 |
| hsa05205 | Proteoglycans in cancer | 201 | 10 (↑: FLNA, FLNB, ↓: CAV1, CD63, CTTN, ITGA2, ITGB1, ITPR3, PXN, THBS1) | 3.02 | 1.70E-03 | 6.93E-02 |
* ↑: upregulated protein, ↓: downregulated protein.
Table 6.
Summary of essential pathways altered in PSEN1(A431E) mutation carriers.
| Pathways | DEPs in Symptomatic carrier | DEPs in Presymptomatic carrier |
|---|---|---|
| ENERGY METABOLISM | ↑: PRPS11,2,3, PRPS21,2,3, ALDH18A12,,6, PHGDH2,3, SDHA3,5,8, OAT6, CYCS8. ↓: ALDOC1,2,3,4, PGD1,3,7, PGM21,4, TKT1,2,3, MAT2A2, PGAM12,3,4, PGAM42,3,4, PGK12,3,4, PKM2,3,4, TPI12,3,4, ALDH7A14,6, LAP36,7, P4HA16, P4HA26, NOS26, GSTO17, PRDX67. | ↑: FH3,5, MDH23,5, GSTP17, COX5A8. ↓: G6PD1,3,7, PGD1,3,7, PGM11,4, ALDH18A12,6, IDH12,3,5,7, IDH22,3,5,7, MAT2A2, PHGDH2,3, PKM2,3,4, P4HA16, ATP5PO8 |
| Pentose Phosphate Pathway: 1, Biosynthesis of amino acids: 2, Carbon metabolism: 3, Glycolysis and Gluconeogenesis: 4, TCA: 5, Arginine and proline metabolism: 6, Glutathione metabolism: 7, Oxidative phosphorylation8 | ||
| VESICLE TRANSPORT AND DEGRADATION | ↑: KIF5B, STAM2a, CLTCL1a,b, NAGAb, TUBB3c, DYNC2H1c, TUBAL3c, PSMD2d, UBRSe, MAPK1f,g,TSC2g, IRS4g, ATG4Cg ↓: AP2B1a, CLTAa,b,, HLA-E a,c, HSPA1Aa, KIF5Ca, CHMPSa, PMLa, EPS15L1a, SNX6a, SH3GL1a, CTSDb,g,, SCARB2b, PIKFYVEc, ITGA2c, ITGA5c, THBS1c, CALRc, CANXc, PSMA4d, PSMA6d, PSME1d, PSME2d, PMLe, HERC1e, GSNf, SCINf, ITPR1g, RRASg |
↑: IGF2Ra,b,, WASHC5a,, ABCA2b, CTSBb, CTSZb, IGF2Ra,b, VAMP3c, UBE4Be, DDB1e, CFL1f, CFL2f, PP2CBg ↓: AP2B1a, CAV1a,, CLTAa,b, EHD3a,, HLA-Aa,c, HSPA2a,, ITCHa,e,, RAB11FIP4a, RAB5Aa,c, RAB7Aa,c,g, UBBa,e, VPS35a,, CD63b, PSAPb, ITGA2c, ITGB1c, THBS1c, TUBB3c, PSMB2d, PSMB3d, ANAPC7e, UBA6e, MARCKSf, SCINf |
| Endocytosis: a, Lysosome: b, Phagosome: c, Proteasome: d, Ubiquitin mediated proteolysise, Fc gamma R-mediated phagocytosis, Autophagyg | ||
| PROTEIN PROCESSING IN THE ENDOPLASMIC RETICULUM | 22 (↑: CRYAB, SAR1A ↓: BAG2, CALR, CANX, DNAJB11, HSP90AB1, HSP90B1, HSPA1A, HSPA5, PDIA3, PDIA4, PDIA6, ERP29, ERO1A, P4HB, EIF2AK2, HYOU1, LMAN2, PRKCSH, , SSR4, STT3A) | 9 (↑: SEC31A, SSR1, UBE4B ↓: CKAP4, HSPA2, LMAN2, MOGS, RPN2, SEC23A) |
| REGULATION OF ACTIN CYTOSKELETON | 15 (↑: ENAH, MAPK1, NCKAP1, PAK2, ROCK1 ↓: FN1, GSN, IQGAP3, ITGA11, ITGA2, ITGA5, PAK3, PIKFYVE, , RRAS, SCIN) | 11 (↑: ARHGAP35, CFL1. CFL2, CYFIP1, IQGAP2, PFN1, ↓: ITGA2, ITGB1, MYH10, PXN, SCIN) |
| FOCAL ADHESION | 13 (↑: COL6A3, MAPK1, PAK2, ROCK1, ↓: COL1A1, COL1A2, FN1, ITGA11, ITGA2, ITGA5, KDR, PAK3, THBS1, FREM2) | 10 (↑: ARHGAP35, COL1A1, COL1A2, FLNA, FLNB, ↓: CAV1, ITGA2, ITGB1, PXN, THBS1) |
| ARRHYTHMOGENIC CARDIOMYOPATHY | ↓: ACTN2, ATP2A2, CTNNA2, DES, DSP, ITGA11, ITGA2, ITGA5, LMNA TPM2, TPM3 | ↓: DMD, ITGA2, ITGB1, RYR2, TPM1, CACNA1C |
| NEURODEGENERATIVE DISEASES (NDD) | ↑: TUBB3A,B,C,D,E, TUBAL3A,B,C,D,E, KIF5BA,B,C,D,E, SLC25A5A,B,C,D, CYCSA,B,C,D,E, PSMD2A,B,C,D,E, MAPK1A, SDHAA,B,C,D,E, IRS4A , CAMK2DB RYR1C, DCTN1D,E, CLTCL1D, NUP214E ↓: ITPR1A,B,C,D, PSMA4A,B,C,D,E, PSMA6A,B,C,D,E, ADAM10A, NOS2A,E, ATP2A2A, EIF2AK2A HSPA5B, SOD1B,C,D,E, TXNB, GNAI1B, GNAI2B ,HSPA5C,E, HSPA1AC, STIP1C, CLTAH, DNAH14D,E, AP2B1D, DNAH6D,E, DNAH10D,E, DNAH1D,E, TGM2D , U: KIF5CA,B,D,E, ATP5PFA,B,D,E, SETXE, FUSE, ANXA11E, MAP2K3E, MATR3E |
↑: VDAC1A,B,C,D,E, COX5AA,B,C,D,E, MMEA, RYR1C, PFN1E, SRSF3E, HNRNPA3E ↓: TUBB3A,B,C,D,E, ITPR3A,B,C,E, ATP5POA,B,C,D,E, PSMB2A,B,C,D,E, PSMB3A,B,C,D,E, CACNA1CA,C, UBBB, UCHL1B CAV1C, RYR2C, HSPA2C, ACTR1AD,E, CLTAD, AP2B1D, DNAH6D,E, SOD2D, FUSE, MATR3E,RAB5AE |
| Alzheimer’s Disease: A, Parkinson’s Disease: B, Prions Disease: C, Huntington’s Disease: D, Amyotrophic Lateral Sclerosis: E | ||
* ↑: upregulated protein, ↓: downregulated protein, U: unique protein, Bold proteins: shared DEP in both mutation carriers.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



