
Submitted:
12 July 2023
Posted:
13 July 2023
You are already at the latest version
Abstract
In organisms that use reduced sulfur compounds as alternative or additional electron donors to organic compounds, transcriptional regulation of genes for enzymes involved in sulfur oxidation is needed to adjust metabolic flux to environmental conditions. However, little is known about the sensing and response to inorganic sulfur compounds such as thiosulfate in sulfur-oxidizing bacteria. In the Alphaproteobacterium Hyphomicrobium denitrificans, one strategy is the use of the ArsR-SmtB-type transcriptional regulator SoxR. We show that this homodimeric repressor senses sulfane sulfur and that it is crucial for the expression not only of sox genes encoding the com-ponents of a truncated periplasmic thiosulfate-oxidizing enzyme system but also of several other sets of genes for enzymes of sulfur oxidation. DNA binding and transcriptional regulatory activity of SoxR are controlled by polysulfide-dependent cysteine modification. The repressor uses the formation of a sulfur bridge between two conserved cysteines as a trigger to bind and release DNA and can also form a vicinal disulfide bond to orchestrate a response to oxidizing conditions. The importance of the sulfur bridge forming cysteines was confirmed by site directed mutagen-esis, mass spectrometry and gel shift assays. In vivo, SoxR interacts directly or indirectly with a second closely related repressor, sHdrR.
Keywords:
Hyphomicrobium denitrificans
; sulfur oxidation
; thiosulfate
; SoxR
; transcriptional regulation
; reactive sulfur species
; repressor
1. Introduction
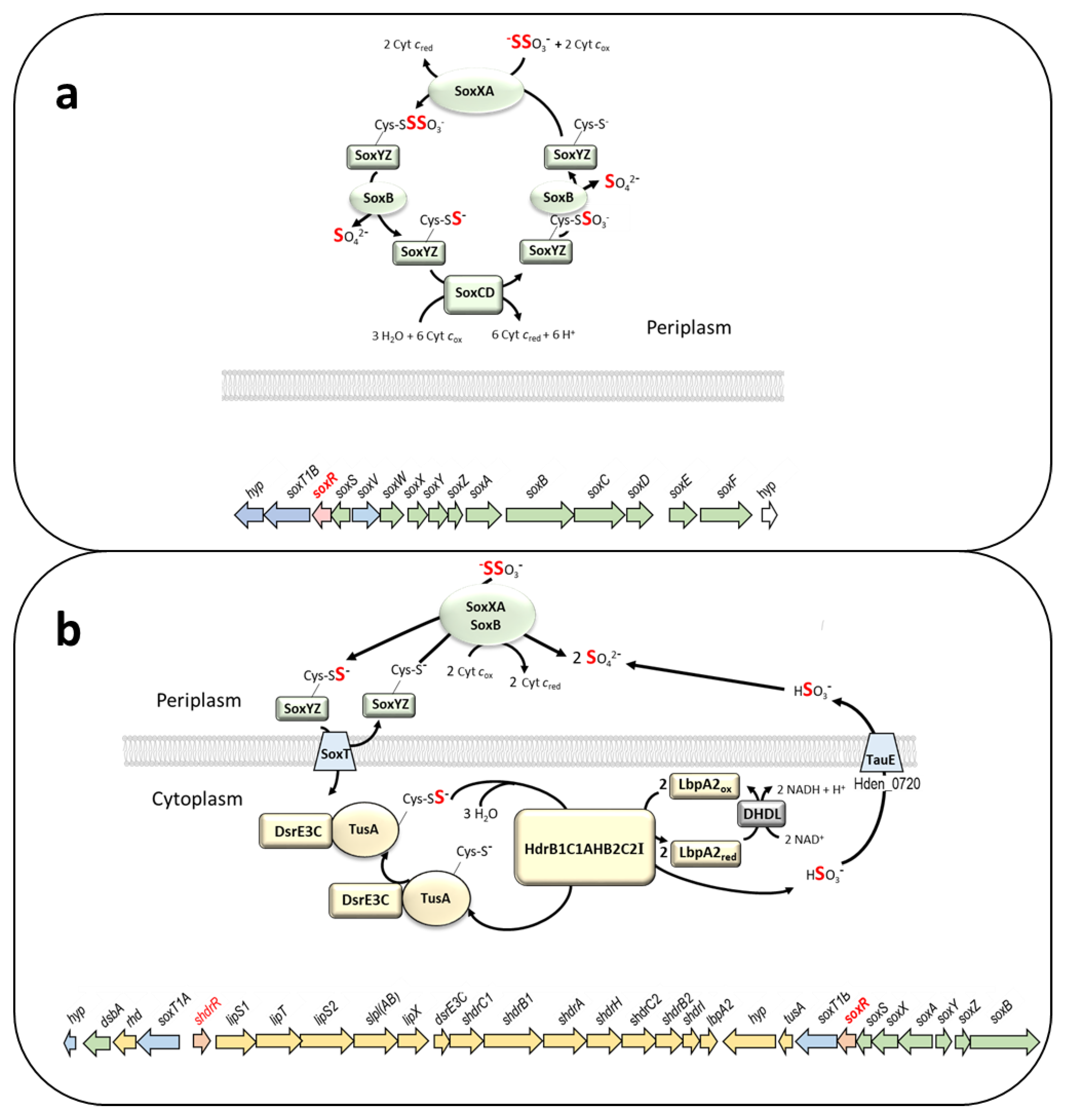
Thiosulfate (S2O32-) is a sulfur substrate that is oxidized by the majority of dissimilatory sulfur oxidizers. Its complete oxidation to sulfate is always initiated, and in many cases also completely performed, in the bacterial periplasm and involves the well-studied thiosulfate-oxidizing Sox multienzyme system [1,2,3] (Figure 1a). Three proteins, SoxYZ, SoxXA and SoxB are required for the initial steps. The c-type cytochrome SoxXA catalyzes the oxidative formation of a disulfide linkage between the sulfane sulfur of thiosulfate and the persulfurated active site cysteine residue of SoxY [4]. Then, SoxB catalyzes the hydrolytic release of the sulfone group as sulfate, leaving the original sulfane sulfur of thiosulfate bound to SoxY [5,6]. The reaction cycle can be fully completed in the periplasm in organisms containing the hemomolybdoprotein SoxCD, that catalyzes oxidation of SoxY-bound sulfane sulfur to a sulfone, followed again by SoxB-catalyzed hydrolytic release of sulfate [7].
Many sulfur oxidizers do not contain SoxCD and have a so-called “truncated” Sox system (Figure 1b) [2,8]. For complete oxidation to sulfate, truncated Sox systems can be combined with cytoplasmic sulfur oxidation systems. How the sulfur is transferred into the cytoplasm for further oxidation is still a mystery. The Alphaproteobacterium Hyphomicrobium denitrificans XT (DSM 1869T) is a representative of this group [9] (Figure 1b). In this organism, two genes encoding predicted sulfur compound transporters (SoxT1A and SoxT1B) are located in close proximity to the sox genes and the genes for the cytoplasmic sulfane sulfur-oxidizing heterodisulfide reductase-like (sHdr) system (Figure 1b). While the H. denitrificans Sox and sHdr proteins have been shown experimentally to be essential for thiosulfate oxidation [9,10,11], evidence for the proposed sulfur transport has not been provided so far.
Figure 1.
(a) Model of the complete periplasmic Sox pathway and exemplary sox gene cluster (A6W98_09510 to A6W98_09585) from the Alphaproteobacterium Rhodovulum sulfidophilum DSM 1374T (Rhodobacterales, Rhodobacteraceae) [12,13]. SoxS is neither part of the Sox enzyme system nor involved in its regulation [14]. This periplasmic thiol–disulfide oxidoreductase of the Dsb family prevents SoxYZ inactivation by reducing false mixed disulfides [15,16]. (b) Model of thiosulfate oxidation and a genetic island for sulfur oxidation (Hden_0678 to Hden_0706) in Hyphomicrobium denitrificans DSM 1869T (Hyphomicrobiales, Hyphomicrobiaceae) [9]. The lip genes encode proteins involved in posttranslational assembly of lipoate on the lipoate-binding LbpA2 protein. The truncated Sox system in the periplasm consists of SoxXY, SoxB and SoxYZ. The sulfane sulfur stemming from thiosulfate and bound to SoxY is transferred to the cytoplasm, possibly via one (or both) of the transporters SoxT1A and Soxt1B, and oxidized to sulfite by the sHdr-LbpA2 system. Sulfite is excreted, probably via TauE, and cannot be effectively oxidized. In panels (a) and (b), periplasmic, membrane-bound and cytoplasmic proteins and the encoding genes are shown in green, blue and yellow, respectively. Regulator genes are highlighted in red. The hyp and rhd genes encode a predicted cytochrome P450 and a rhodanese-like protein, respectively.
Figure 1.
(a) Model of the complete periplasmic Sox pathway and exemplary sox gene cluster (A6W98_09510 to A6W98_09585) from the Alphaproteobacterium Rhodovulum sulfidophilum DSM 1374T (Rhodobacterales, Rhodobacteraceae) [12,13]. SoxS is neither part of the Sox enzyme system nor involved in its regulation [14]. This periplasmic thiol–disulfide oxidoreductase of the Dsb family prevents SoxYZ inactivation by reducing false mixed disulfides [15,16]. (b) Model of thiosulfate oxidation and a genetic island for sulfur oxidation (Hden_0678 to Hden_0706) in Hyphomicrobium denitrificans DSM 1869T (Hyphomicrobiales, Hyphomicrobiaceae) [9]. The lip genes encode proteins involved in posttranslational assembly of lipoate on the lipoate-binding LbpA2 protein. The truncated Sox system in the periplasm consists of SoxXY, SoxB and SoxYZ. The sulfane sulfur stemming from thiosulfate and bound to SoxY is transferred to the cytoplasm, possibly via one (or both) of the transporters SoxT1A and Soxt1B, and oxidized to sulfite by the sHdr-LbpA2 system. Sulfite is excreted, probably via TauE, and cannot be effectively oxidized. In panels (a) and (b), periplasmic, membrane-bound and cytoplasmic proteins and the encoding genes are shown in green, blue and yellow, respectively. Regulator genes are highlighted in red. The hyp and rhd genes encode a predicted cytochrome P450 and a rhodanese-like protein, respectively.
The obligately heterotrophic H. denitrificans oxidizes thiosulfate as an additional electron donor during growth on compounds like methanol [10]. In batch culture, substantial amounts of sulfite are excreted as the product of sHdr-catalyzed sulfur oxidation and accumulate because an enzyme catalyzing efficient sulfite oxidation is not present [10]. Accumulation of sulfite as an intermediate has also been described for some facultatively autotrophic sulfur oxidizing Alphaproteobacteria, e.g., Rhodovulum (previously Rhodobacter) sulfidophilum [17].
(Bi)sulfite (HSO3-), SO32- is a highly reactive, strong nucleophile and has many toxic effects. Its strong reducing capacity (E0` for the sulfate/sulfite couple is -515 mV) contributes to its toxicity and antimicrobial action, which have led to its widespread use as food preservative [18,19]. Free sulfite can damage DNA through formation of adducts [20,21,22]. Its toxic effect on mammalian cells has been attributed to the formation of sulfur- and oxygen-based free radicals [23,24] which can in turn react with lipids and proteins [25,26]. The full Sox pathway or the truncated Sox/sHdr combination may be advantageous, despite the intermediate release of sulfite, for organisms such as H. denitrificans or R. sulfidophilum, at low thiosulfate concentrations if removal by other members of the community or chemical oxidation in oxygenated environments keeps sulfite concentrations below inhibitory levels. In any case, the formation of the toxic intermediate sulfite during the oxidation of sulfur compounds, as well as the switching between organic and inorganic electron donors requires fine-tuning to the environmental conditions.
Accordingly, complex regulatory patterns have been reported for facultative sulfur oxidizers, with upregulation usually occurring only in the presence of metabolizable sulfur substrates, whereas the corresponding genes are thought to be always highly expressed in chemolithoautotrophs restricted to the oxidation of sulfur compounds. In H. denitrificans, and other Alphaproteobacteria that are not restricted to sulfur oxidation, such as R. sulfidophilum, Paracoccus pantotrophus or Pseudaminobacter salicylatoxidans, the ability to oxidize thiosulfate and, depending on the organism, other reduced inorganic and organic sulfur compounds such as sulfide or dimethyl sulfide, is not constitutive but can be induced by the presence of oxidizable sulfur compounds [10,17,27,28]. While the transcriptional repressor sHdrR is involved in this process in H. denitrificans [10], genetic and biochemical studies have identified the related SoxR protein as a major regulator in P. pantotrophus and P. salicylatoxidans [27,28,29], both of which contain a complete Sox system and are unable to oxidize sulfane sulfur in the cytoplasm.
SoxR is a member of the arsenic repressor (ArsR-SmtB) family of prokaryotic repressors [30,31]. Members of the ArsR-SmtB family were originally recognized as metal-responsive transcriptional regulators, but there are also members in this family that have been shown to sense reactive oxygen or sulfur species [32]. SqrR from Rhodobacter capsulatus and BigR from Xylella fastidiosa belong to this group and control the transcription of genes involved in sulfide-dependent photosynthesis and the detoxification of H2S derived from associated host plants, respectively [33,34]. Knowledge about SoxR is comparatively sparse. While binding regions for the transcriptional repressor have been identified in promoter-operator segments within the sox gene clusters of P. denitrificans and P. salicylatoxidans [27,28], no information is available on factors that control its DNA-binding capacity. It is therefore completely unclear how SoxR senses the presence of oxidizable sulfur compounds and how it then triggers the transcription of sulfur oxidation genes.
Here, we start to close this knowledge gap by first providing information on the general distribution of complete and truncated Sox systems and their co-occurrence with SoxR. Furthermore, we present genetic information for SoxR function in H. denitrificans, identify target genes and map its binding sites. The DNA-binding properties of the homodimeric repressor and its response to bridging of the sulfur atoms of two conserved cysteine residues by one to three sulfur atoms are characterized via site-directed mutagenesis, mass spectrometry, MalPEG assays, and electrophoretic mobility shift assays (EMSA).
2. Materials and Methods
2.1. Bacterial strains, plasmids, primers, and growth conditions
Table S1 lists the bacterial strains, and plasmids that were used for this study. Escherichia coli strains were grown on complex lysogeny broth (LB) medium [35] under aerobic conditions at 37°C unless otherwise indicated. Escherichia coli. BL21 (DE3) was used for recombinant protein production. E. coli strains 10-beta and DH5α were used for molecular cloning. H. denitrificans strains were cultivated in minimal media containing 24.4 mM methanol kept at pH 7.2 with 100 mM 3-(N-Morpholino)propanesulfonic acid (MOPS) buffer as described before [9]. Thiosulfate was added as needed. Antibiotics for E. coli and H. denitrificans were used at the following concentrations (in μg ml-1): ampicillin, 100; kanamycin, 50; streptomycin, 200; chloramphenicol, 25.
2.2. Recombinant DNA techniques
Standard techniques for DNA manipulation and cloning were used unless otherwise indicated [36]. Restriction enzymes, T4 ligase and Q5 polymerase were obtained from New England Biolabs (Ipswich, UK) and used according to the manufacturer’s instructions. Oligonucleotides for cloning were obtained from Eurofins MWG (Ebersberg, Germany). Plasmid DNA from E. coli was purified using the GenJET Plasmid Miniprep kit (Thermo Scientific, Waltham, USA). Chromosomal DNA from H. denitrificans strains was prepared using the First-DNA all-tissue Kit (GEN-IAL GmbH, Troisdorf, Germany).
2.3. Construction of plasmid for deletion of soxR in H. denitrificans
For markerless deletion of the H. denitrificans soxR (Hden_0700) gene by splicing overlap extension (SOE) [37], PCR fragments were constructed using the primers P1 fwd up hden_0700, P2 rev up hden_0700, P3 fwd down hden 0700 and P4 rev down hden_0700 (Table S1). The resulting 1.04 kb SOE PCR fragment was cloned into the XbaI and PstI sites of pK18mobscaB-Tc [10]. The final construct pK18mobsacB_Tc_ΔsoxR was electroporated into H. denitrificans ΔtsdA and transformants were selected using previously published procedures [9,11]. Single crossover recombinants were Cmr and Tcr. Double crossover recombinants were Tcs and survived in the presence of sucrose due to loss of both, the vector-encoded levansucrase (SacB) and the tetracyclin resistance gene.
2.4. Characterization of phenotypes, quantification of sulfur compounds and protein content
Growth experiments with H. denitrificans were run in 200 ml medium with 24.4 mM methanol and varying concentrations of thiosulfate in 500-ml Erlenmeyer flasks as described in [10]. Thiosulfate concentrations, protein content and specific thiosulfate oxidation rates were determined by previously described methods [10,38]. All growth experiments were repeated three times. Representative experiments with two biological replicates for each strain are shown. All quantifications are based on at least three technical replicates.
2.5. RNA preparation
Total RNA of H. denitrificans was isolated from cells grown mid-log phase. H. denitrificans strains ΔtsdA and ΔtsdA ΔsoxR were grown in 50 ml methanol-containing medium at 30°C with shaking at 250 rpm in 100-ml Erlenmeyer flasks. Cells from 2 ml were harvested by centrifugation at 16,000 × g for 5 min. The cell pellet was incubated with 500 µl of 10% SDS containing 1 mg ml-1 lysozyme at room temperature for 5 min. Then 700 µl of TRIzol [39] was added and the mixture was incubated for another 5 min. This step was followed by addition of 1ml ROTI®Aqua-P/C/I reagent (Carl Roth GmbH, Karlsruhe, Germany), 10 min incubation and centrifugation at 13,000 × g for 5 min. RNA purification from the supernatant was achieved with the Monarch Total RNA Miniprep Kit (NEB, Frankfurt, Germany). gDNA was removed by treating 10-µl samples with an absorption at 260 nm corresponding to ~1 µg RNA with 1 U of RNase-free DNase I (ThermoFisher, Waltham, MA, USA) in the MgCl2-containing reaction buffer provided by the manufacturer. RNA concentrations were measured with an Eppendorf NanoDrop Biospectrometer. The absence of gDNA was verified using the primers rpoB-denitf and rpoB-denitr [40], which bind only to gDNA and not to the corresponding RNA.
2.6. Expression studies based on RT-qPCR
RNA samples of 100 ng were used for RT-qPCR analysis via the Luna Universal One-Step RT-qPCR Kit (NEB) and the CFX ConnectTM real-time detection system (Bio-Rad, Munich, Germany) according to the instructions of the manufacturers. The level of rpoB mRNA was used as internal standard [40]. Approximately 200-bp fragments were amplified (see Table S1 in the supplemental material) with an annealing temperature of 60°C. The RT-qPCR conditions were as follows: 10 min at 55°C (reverse transcription using random nonamer primers), 1 min at 95°C (inactivation of the reverse transcriptase and activation of the polymerase), 40 cycles of 15 s at 95°C, 30 s at 60°C, followed by melting curve analysis, in which the temperature was increased every 10 s by 1°C, from a start at 60°C to 95°C. Analyses of melting curves and calculation of Ct (calculated threshold) values were automatically quantified with The Bio-rad CFX Manager software. Ct values for each point in time were run in triplicate. Relative expression ratios were calculated by the 2-ΔΔCt method [41].
2.7. Cloning, site-directed mutagenesis, overproduction, and purification of recombinant SoxR proteins
The soxR gene was amplified from H. denitrificans genomic DNA with primers adding a sequence for an N-terminal Strep-tag and cloned between the NdeI and HindIII sites of pET-22b(+), resulting in pET22b-SoxR-Strep. Cysteine to serine exchanges were implemented with the Q5 Site-Directed Mutagenesis Kit (NEB) according to the manufacturer’s instructions and using the primers listed in Table S1. Recombinant SoxR proteins were overproduced in E. coli BL21(DE3) containing plasmids pET22b-SoxR-Strep, pET-22b-SoxR C50S, pET-22b-SoxR C116S, and pET-22b-SoxR C50S C116S. The cells were grown in 1-l Erlenmeyer flasks at 37°C in 400 ml LB medium containing ampicillin up to an OD600 of 0.5-0.6. Expression of soxR was induced by adding 0.5 mM IPTG. IPTG-induced E. coli cells were grown over night at 20 °C. Cells were harvested at 14,000 × g for 30 min. Three ml lysis buffer (100 mM Tris-HCl buffer pH 7.0, 5 mM EDTA containing a spatula tip of deoxyribonuclease I and protease inhibitor) were added per g wet weight for homogenization. Cell lysis was achieved by sonification and followed by centrifugation (16,100 × g, 30 min, and 4°C) and ultracentrifugation (145,000 × g, 1 h, 4°C). The supernatant was applied to a Strep-tactin affinity chromatography column equilibrated with buffer W (100 mM Tris-HCl, pH 8.0, 150 mM NaCl). The column was washed with six volumes of buffer W and eluted with buffer E (100 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2.5 mM D-desthiobiotin). The protein was assessed for its purity by 12.5 % SDS-PAGE. Pure SoxR proteins were stored on ice in buffer W. Buffer exchange was achieved with Amicon® Ultra-3K centrifugal filters.
2.8. Electrophoretic mobility shift assays (EMSA)
Gel electrophoretic mobility shift assays are used to detect interactions between proteins and nucleic acids. In the assay, solutions of protein and nucleic acid are combined and the resulting mixtures are subjected to polyacrylamide under native conditions. After electrophoresis, the distribution of nucleic acid species is determined. In general, protein-nucleic acid complexes migrate more slowly than the corresponding free nucleic acid [39]. The binding reaction mixture (15 μl final volume), contained purified SoxR wildtype or variant protein in various concentrations (up to 700 nM), 2 μl 50 % glycerol and 1.5 μl 10 × binding buffer (100 mM Tris-HCl, 500 mM KCl, 10 mM DTT, 5 % glycerol, pH 8.0). Reaction mixtures were pre-incubated for 20 min at room temperature followed by a further 30 min incubation at 30°C after adding the DNA probe to a final concentration of 17 nM. The DNA probes consisted of a 362-bp fragment covering the entire intergenic region between the shdrR (Hden_0682) and the soxT1A (Hden_0681) genes, a 180-bp fragment representing the central part of the first product (created with primers EMSA-Fr2-Fr and EMSA_Fr3-Rev), a 177-bp fragment situated between the shdrR and the lipS1 (Hden_0683) gene, a 173-bp fragment situated between the lipX (Hden_0687) and dsrE3C (Hden_0688) genes, a 176-bp fragment located between the tusA (Hden_0698) and hyp (Hden_0697) genes, and a 151-bp fragment situated between the soxA (Hden_0703) and soxY (Hden_0704) genes. All primers used are listed in Table S1. The reaction mixtures were loaded onto 6 % native polyacrylamide gels after these had been pre-run at 100 V for 1 h at 4 °C with 0.25 × TBE buffer (25 mM Tris-borate, 0.5 mM EDTA). The loaded gels were electrophoresed in 0.25 × TBE with 0.5 % glycerol at 180 V for 1h at 4 °C). Gels were subsequently stained for 20 min with SYBR green I. The bands corresponding to SoxR-bound and free DNAs were visualized with a ChemiDoc Imaging System (BioRad).
2.9. Gel permeation chromatography
The size exclusion chromatography column Superdex™ 75 Increase 10/300 GL (Cytiva, Freiburg, Germany) was calibrated using Blue dextran (2,000 kDa), conalbumin (75 kDa), bovine serum albumin (67 kDa), ovalbumin (43 kDa), lactoglobulin (35 kDa), carbonic anhydrase (29 kDa) chymotrypsin (23 kDa) and ribonuclease (13.7 kDa). The calibration curve was plotted using the gel-phase distribution coefficient (kav) versus the logarithm of molecular weight. kav = (Ve-V0/Vc-V0) where Ve = elution volume, V0 = column void volume (7.94 ml based on Blue dextran elution volume), Vc geometric column volume (24 ml). The column was run in 50 mM Tris-HCl, pH 8.0, 150 mM NaCl at a flow rate of 0.8 ml min-1 using an Äkta FPLC system.
2.10. Preparation of polysulfides
A polysulfide stock solution was prepared according to Ikeda et al. [42] by mixing 1.2 g NaHS × H20 and 0.16 g sulfur powder with 3 ml oxygen-free water in a closed 10 ml serum bottle under a nitrogen atmosphere for 1 h at room temperature. Then, the volume was filled up to 10 ml with oxygen-free water. Based on an average length for the resulting polysulfides of four sulfur atoms, their concentration is 0.5 M in the final solution that can be kept at room temperature for many months. If necessary, the polysulfide solution was diluted with oxygen-free water and immediately used for persulfuration reactions.
2.11. Redox treatments, persulfuration reactions, MalPEG gel-shift assays and mass spectrometry
5 µg protein was treated with DTT (1 mM and 5 mM for samples analyzed by mass spectrometry and EMSA, respectively) for reduction, 5 mM CuC12 for oxidation, 0.5 mM polysulfide for persulfuration and 1 mM MalPEG (methoxy-polyethylene glycol maleimide, MW 10000 g mol-1) for PEGylation or 5 mM iodoacetamide for carbamidomethylation in a final volume of 15 µl containing 100 mM Tris-HCl, pH 8.0, 150 mM NaCl. When polysulfide, MalPEG and DTT were applied consecutively, concentrations were 0.5 mM, 5 mM and 1 mM, respectively. When polysulfide and DTT were applied consecutively concentrations were 0.5 mM and 10 mM, respectively. Protein samples used in EMSA experiments were reacted with the reagents for 20 min at 25°C. Samples analyzed by SDS-PAGE were incubated with each reagent for 15 min at 30°C. Reactions were either stopped by addition of 5 µl 4 x non-reducing Roti®-Load2 (Carl Roth GmbH) and subjected to 15% SDS–PAGE without boiling the sample or analyzed by mass spectrometry. For MS, samples of 20 µl were desalted by ZiptipC4 Pipette tips (Merck Millipore, Darmstadt, Germany) and measured by MALDI-TOF at the Core Facility Protein synthesis & BioAnalytics, Pharmaceutical Institute, University of Bonn.
2.12. Distribution of Sox systems and SoxR: dataset generation and anaylsis
Archaeal and bacterial genomes were downloaded from Genome Taxonomy Database (GTDB, release R207). In GTDB, all genomes are sorted according to validly published taxonomies, they are pre-validated and have high quality (completeness minus 5*contamination must be higher than 50%). One representative of each of the current 65,703 species clusters was analyzed. It should be noted that GTDB is built on recently standardized bacterial and archaeal taxonomies derived by normalization of the evolutionary distance between taxonomic levels [43,44]. Among the bacteria, 148 phyla are currently distinguished. For the Archaea, GTDB lists 16 phyla. Open reading frames were determined using Prodigal [45] and subsequently annotated for SoxR, other Sox proteins and clustering of the respective genes via HMS-S-S using default conditions [46]. Chromatiaceae and Ectothiorhodospiraceae were treated as exceptions as they do not contain contiguous sox clusters but the thiosulfate-oxidizing capabilities and the functionality of the Sox proteins have been experimentally established for relevant species [47,48].
3. Results
3.1. Distribution of Sox systems and the SoxR regulator
We first asked how complete and truncated Sox systems (Figure 1) are distributed among the prokaryotes and analyzed the genomes available in the Genome Taxonomy Database (GTDB, release R207). In GTDB, all genomes are sorted according to validly published taxonomies. In addition, we asked which groups of these prokaryotes contain a soxR that is linked to the other sox genes. In order to accurately identify and discriminate the Sox components we used HMS-S-S, a tool that specifically finds sulfur metabolism-related proteins [46]. As shown in Figure 2 and Table S2, genes encoding Sox proteins are not found among the Archaea. They exclusively occur in 17 of the currently 169 bacterial phyla distinguished in GTDB. The highest proportion of species with Sox in a phylum is observed for the Aquificota (54%), followed by the Campylobacterota (30.7%), the Deinococcota (24.3%) and the Proteobacteria (19.3%) (Table S2). The SoxR regulator is strictly confined to the Proteobacteria (Figure 2).
The Aquificota contain exclusively organisms with a truncated Sox system (Table S2), which are strictly chemolithoautotrophic sulfur oxidizers, with a few having additional organoheterotrophic capacity [49]. Among the Sox-containing Campylobacteria about three quarters rely on a complete system. The type of Sox system varies within a family and even within a single genus. Many Campylobacterota species, e.g., members of the families Sulfurimonadaceae, Sulfurispirillaceae or Sulfurovoraceae are established chemolithoautotrophic sulfur oxidizers [50,51,52]. In the Deinococcota, the complete Sox system is much more abundant than the truncated version (Table S2) with occurrence in Thermus and Meiothermus species known as sulfur-oxidizing mixotrophs [53,54]. Among the Bacteroidota, the general abundance of Sox is low, but here we find the obligately photolithoautrophic sulfur oxidizers of the order Chlorobiales [55], all of which encode the truncated set of Sox proteins.
By far the highest absolute numbers of Sox-containing species are found among the Proteobacteria, here exclusively in the classes Alphaproteobacteria and Gammaproteobacteria. The complete Sox system appears more frequently than the truncated version in metabolically versatile members of the alphaproteobacterial families Rhizobiaceae [56] and Rhodobacteraceae [57], while the opposite is true for a number of gammaproteobacterial families, e.g., the Thioglobaceae, Chromatiaceae, Ectothiorhodospiraceae, Thiomicrospiraceae and Thiotrichaceae (Table S2), all of which contain members with established chemo- or photolithotrophic sulfur-oxidizing capabilities [58,59,60,61,62]. On the other hand, families like the alphaproteobacterial Xanthobacteraceae or the gammaproteobacterial Burkholderialaceae contain species encoding complete or incomplete Sox systems in almost equal numbers. The important general rule to emerge from our analysis is that the gene for the SoxR transcriptional regulator is more often linked to the genes for the complete than to those for the truncated Sox system (Figure 2).
3.2. Genetic evidence for SoxR function in H. denitrificans
Previously, we showed that the ArsR-type regulator encoded by the first gene of the H. denitrificans shdr-lbpA operon, sHdrR, functions as a repressor of shdr gene expression in the absence of oxidizable sulfur compounds [10]. Phenotypic characterization of a mutant strain lacking the shdrR gene indicated an additional regulator involved in the overall process. Indeed, a further candidate transcriptional repressor, SoxR, is encoded downstream of soxXA in H. denitrificans (Figure 1b). To assign a function for SoxR in transcriptional regulation of the hyphomicrobial sox and possibly also the shdr and associated genes, we constructed H. denitrificans ΔtsdA ΔsoxR, a mutant strain with a markerless deletion of soxR in a ΔtsdA background. The reference strain H. denitrificans ΔtsdA lacks thiosulfate dehydrogenase and oxidizes thiosulfate exclusively via the pathway combining Sox and sHdr-LbpA [9,10] (Figure 1b). When grown in the presence of methanol as a carbon source and thiosulfate as an additional electron source, the ΔtsdA reference strain excretes sulfite, which causes a growth retardation that is particularly impressive when cultures are inoculated with thiosulfate-induced cells ([10], also compare open and filled circles in the upper right panel of Figure 3). Just as the H. denitrificans strain lacking the shdrR gene, the soxR deficient strain exhibited a very strongly reduced growth rate and a high specific thiosulfate oxidation rate even without induction of pre-cultures (Figure 3). As soon as thiosulfate was consumed, the growth rate increased substantially. Both regulator-negative strains exhibited somewhat higher specific thiosulfate consumption rates when pre-cultures had been exposed to thiosulfate than for the non-induced case, fully in line with the finding that a second regulator is involved in the overall process.
3.3. Identification of genes controlled by SoxR by RT-qPCR for different H. denitrificans strains
To examine which genes are affected by the SoxR regulator protein, RT-qPCR experiments were performed and the transcription levels of twelve genes in the H. denitrificans sulfur oxidation island were compared in the absence and in the presence of thiosulfate for the ΔtsdA reference strain (Figure 4). In addition, transcription levels were determined for the same genes in the H. denitrificans ΔtsdA ΔsoxR mutant in the absence of thiosulfate. All cultures were harvested in the exponential growth phase. The studied genes included soxT1A, the first of a set of genes transcribed in the opposite direction of shdrR, the gene for the sHdrR regulator, and two of the genes encoding proteins involved in Lbp2 assembly (lipS1 and slpl(AB)). LbpA2 is a lipoate-binding protein essential for sulfur oxidation [11]. Four genes were chosen as examples for those encoding the shdr-lbpA cytoplasmic sulfane sulfur oxidation system (dsrE3C, shdrA, shdrB2 and lbpA). These genes are followed by genes transcribed in the opposite direction and encoding part of the Sox system (SoxXA), the SoxR regulator, SoxS, which is a periplasmic thiol–disulfide oxidoreductase, as well as a second potential sulfur transporter, SoxT1B, the cytoplasmic sulfurtransferase TusA and a predicted cytochrome P450 (Figure 1 and Figure 4b). Except of soxS, all of these genes were included in RT-qPCR analysis. Finally, the analysis was extended to soxY and soxB. These genes follow the previously described genes in the opposite direction in a soxYZB arrangement (Figure 1 and Figure 4b).
With the exception of the genes for the two transcriptional regulators, shdrR and soxR, and soxT1B, which is located just downstream of soxR, all the genes tested were upregulated at least five-fold when the reference strain was exposed to thiosulfate, with the strongest responses for lpl(AB), shdrA and shdrB2 (Figure 4a). In the strain lacking SoxR, transcription of various sox and shdr genes was much higher than in the reference strain even in the absence of thiosulfate. The lack of soxR most strongly affected transcription of soxXA and soxY but was also evident for shdr genes, soxT1A, lipS1 and lpl(AB) (Figure 4a). With the exception of the genuine sox genes tested, the reference strain showed a stronger response to the presence of thiosulfate than the ΔtsdA ΔsoxR mutant in its absence. This observation clearly points at the presence of at least one further regulatory element, most probably sHdrR [10]. On the other hand, the strong response of numerous genes in addition to those for the genuine Sox system, shows that their transcription is either directly or indirectly affected by SoxR.
3.3. Identification of SoxR target sites by EMSA
The finding that SoxR affects transcription of genes outside the sox operons was unexpected and afforded closer analysis. To that end, we inspected intergenic regions within the hyphomicrobial sulfur oxidation island and identified four with conspicuous inverted and direct repeats with the potential to serve as repressor binding sites and used them as probes for EMSA (Figure 4b). A 176-bp fragment located upstream of the hypothetical gene Hden_0697 served as a control (Figure 4b). Indeed, SoxR bound to four of the five tested probes (Figure 4c). Among these is the intergenic region between soxT1A and the gene for the SoxR-related repressor sHdrR. This region had before already been shown to serve as a binding site for sHdrR [10], further emphasizing the notion that the two repressors work intimately together.
3.4. Properties of the SoxR protein
The H. denitrificans SoxR protein has a length of 124 amino acids and a BlastP search identified R. capsulatus SqrR as the most similar structurally characterized protein. HdSoxR shows 53%, 52% and 43% and 42% amino acid identity to R. capsulatus SqrR, P. salicylatoxidans SoxR, Xylella fastidiosa BigR and H. denitrificans sHdrR, respectively. All of these regulators share two conserved cysteine residues, Cys50 and Cys116 in the hyphomicrobial protein (Figure 5). The equivalent residues in SqrR are required for sensing sulfide [34,63].
We sought to obtain information about the oligomerization state and conformation of SoxR as well as about the reactivity of the two cysteine residues. To this end, Strep-tagged SoxR as well as variants with serine in place of either one (SoxR Cys50Ser and SoxR Cys116Ser) or both cysteines (SoxR Cys50Ser Cys116Ser) were overexpressed in E. coli, purified by affinity chromatography and subjected to reducing and non-reducing SDS-PAGE analysis in the as-isolated state, after reduction with DTT and after oxidation with CuCl2. The same single 15-kDa band was obtained in all cases under reducing conditions (not shown). The band for the oxidized wildtype protein migrated slightly further than those for the as-isolate and reduced protein under non-reducing conditions (Figure 6a) indicating a more compact structure due to the formation of an intramolecular disulfide bond between Cys50 and Cys116. The oxidized SoxR Cys50Ser and SoxR Cys116Ser variants formed intermolecular dimers connected by the remaining cysteine on each of the monomers (Figure 6a). These observations indicated a homodimeric state for the native proteins that allows close contact between Cys50 and Cys116 residues, respectively, of the monomers and thus formation of disulfide bridges upon oxidizing conditions.
This conclusion was fully supported by size exclusion chromatography (Figure 6b). All variants as well as wildtype SoxR eluted with kav values corresponding to molecular masses between 37.6 and 41.6 kDa and indicating dimerization of the 15.2 kDa monomers. Tetramers were also observed with the highest abundance for the SoxR Cys116 variant. All proteins showed a tendency for formation of higher oligomers eluting in the void volume (Figure 6b). Notably, the Sox Cys50Ser single and the Cys50Ser Cys116Ser variant exchanges led to dimers eluting significantly earlier than those of wildtype SoxR and SoxR Cys116Ser, indicating that the loss of Cys50 but not that of Cys116 leads to a more open, space-demanding conformation of the regulator protein.
3.5. SoxR binding properties
In the next step, EMSA assays were performed that allowed more detailed insights into the binding capacity of SoxR to the intergenic region between the divergently oriented soxXA and soxYZB genes (Figure 7). SoxR binds to the DNA probe in a concentration-dependent manner and leads to appearance of two shifted bands indicating two different binding sites (Figure 7a, upper panel). As related proteins respond to persulfuration [34,63], we tested the answer of SoxR to treatment with polysulfide, oxidized and reduced glutathione (GSH and GSSG), tetrathionate, sulfite and thiosulfate in various molar ratios of protein and additive. Whereas GSH, GSSG, tetrathionate and sulfite had no effect even when present in 50-fold excess compared to the protein (not shown), treatment with polysulfide above a molar ratio of 1 completely prevented binding of SoxR to the target DNA and a shift was no longer observed (Figure 7a, lower panel and 7b, upper panel). Thiosulfate also had an effect, albeit a much milder one (Figure 7b, lower panel). The second shifted band disappeared at a ratio thiosulfate/SoxR of 5 and the first band still persisted at a ratio of 50, corresponding to a thiosulfate concentration of 0.2 mM. As outlined in the introduction, the initial steps of thiosulfate degradation occur in the periplasm and it is therefore unlikely that thiosulfate would ever reach concentrations in the cytoplasm that would be required to elicit a response of SoxR.
EMSA assays were also performed with the as-isolated, reduced, oxidized and polysulfide-treated SoxR variants and two different DNA probes (Figure 8). Reduction with DTT led to the same results as obtained for the untreated proteins indicating that they are fully reduced upon isolation and remain in this state during storage. Oxidation of wildtype SoxR prevented binding to both tested DNA probes. While the SoxR Cys50Ser variant completely lost its DNA binding ability, the Cys116Ser variant bound effectively to the DNA probes. When polysulfide-treated wildtype SoxR was reduced with DTT in a second step, the protein regained its DNA-binding capacity demonstrating that the modification caused by polysulfide was fully reversible by reduction. A response to oxidation or incubation with polysulfide was still observed for the Cys116Ser variant, albeit weaker than that of the wildtype protein. This behaviour differs significantly from that of the related SqrR from R. capsulatus, where variants lacking one of the two conserved cysteines bind to their target DNA but do not show a loss of affinity upon persulfuration [34]. The SoxR variant lacking both cysteines was unable to bind DNA, regardless of the treatments applied.
3.6. Redox state and modification of SoxR
To clarify the chemical nature of the SoxR modifications by polysulfide and oxidation, gel-shift assays were performed using MalPEG, which selectively labels free thiol groups covalently [66]. The modification can be detected by non-reducing SDS-PAGE, since the molecular mass of the protein is increased by ~10 kDa per SH group modified. Treatment of the recombinant wildtype SoxR protein with MalPEG resulted in a single 20 kDa band shift, indicating that it contains two free cysteine residues, as expected (Figure 9). In contrast, oxidized SoxR did not react with MalPEG, demonstrating the existence of a disulfide bridge between Cys50 and Cys116, as also suggested by non-reducing SDS-PAGE in the absence of MalPEG (Figure 6a). MalPEG labeling of the SoxR variants gave the expected results with the variants carrying one cysteine showing a single 10 kDa shift and the double mutant not reacting with MalPEG as predicted. After oxidation, the SoxR Cys50Ser variant produced exclusively dimers connected by Cys116-Cys116 disulfide bridges and not reacting with MalPEG, whereas the Cys116Ser variant showed only a minor dimer fraction. This observation is corroborated by the response of SoxR and its variants to polysulfide. While the wildtype protein stayed essentially monomeric, i.e., disulfide bonds between protein monomers were not formed, the SoxR Cys50Ser variant completely transformed into a dimer stable under denaturing conditions. The Cys116Ser variant behaved differently, with a substantial fraction staying monomeric. We note that the dimeric fraction of both variants obtained after treatment with polysulfide turned monomeric after incubation with MalPEG, possibly indicating a (poly)sulfur bridge between the remaining cysteine residues that is susceptible to cleavage by the thiol-binding agent. In conclusion, Cys50 residues appear less prone to reaction than Cys116 residues just as has been reported for the corresponding cysteines in R. capsulatus SqrR [67] and/or they reside further apart from each other in the native SoxR dimer than Cys116 residues.
The next set of reactions was most revealing. When wildtype polysulfide treated SoxR was reacted with MalPEG, it behaved just like the oxidized protein, i.e., MalPEG was not bound, indicating the absence of free cysteines (Figure 9). Instead, one MalPEG was bound to the polysulfide-treated single cysteine replacement variants and could be released upon reduction with DTT. We conclude that in the two latter cases, polysulfide led to persulfuration of the single remaining cysteines which then bound MalPEG. In the final step, MalPEG-sulfide conjugates were released by treatment with DTT and the single cysteine SoxR variants reappeared in their unmodified monomeric form. The situation for wildtype SoxR is completely different, either polysulfide merely leads to formation of a Cys50-Cys116 bridge or one or more sulfur atoms are enclosed by the two cysteines.
Mass spectrometric analyses finally allowed clear differentiation between these two possibilities (Table 1, Supplementary Figure S1). For these experiments, MalPEG was replaced by the thiol-modifying agent iodoacetamide, which leads to carbamidomethylation of free Cys sulfhydryl groups and thus adds a mass of 57 Da. As expected, wildtype SoxR gained twice 57 Da after iodoacetamide treatment, whereas the single Cys replacement mutants were modified with only one carbamido group. Notably, polysulfide treatment led to persulfuration of all SoxR proteins except of the cysteine-less double replacement variant, which was measured as a control. The SoxR wildtype protein was modified with up to three sulfur atoms (+ 32 Da each) and did not react with iodoacetamide, demonstrating the formation of an intramolecular tri-, tetra, or pentasulfide bond between Cys50 and Cys116. Although mass spectra do not provide exact quantitative information, peak heights indicate that bridges by two additional sulfur atoms are more abundant than one or three atom bridges for the SoxR wildtype protein, while the majority of the SoxR Cys50Ser and Cys116Ser variant polypeptide are modified by only one sulfur atom (Supplementary Figure S1)
4. Discussion
In this study, we collected a wealth of new information on the transcriptional repressor SoxR. We show that among the investigated more than 70,000 prokaryotic genomes bona-fide soxR (i.e., genetically linked to the genes for the SoxYZ sulfur-binding protein and/or catalytic Sox components) occurs exclusively among the bacterial phylum Proteobacteria, where it is more frequently found in gene clusters for complete than for truncated Sox systems. Based on the available data, it is difficult to draw general conclusions from this observation. However, it appears that a number of bacteria that operate the truncated Sox system, such as the green and purple sulfur bacteria or members of the Aquificota are dedicated sulfur-oxidizing chemolithoautrophs without much need for sophisticated transcriptional regulation of the sulfur oxidation machinery.
We show that in the model Alphaproteobacterium H. denitrificans SoxR is not only involved in the transcriptional regulation of true sox genes but that it also affects the transcription of a number of other genes. In particular, the shdr genes, which encode the cytoplasmic sulfur-oxidizing multi-enzyme system required for sulfane sulfur oxidation that cannot be achieved by the truncated hyphomicrobial Sox system, are co-controlled by SoxR. How it interacts with a second, related repressor, sHdrR, that affects the transcription of the same genes [10] is an important research question for the future.
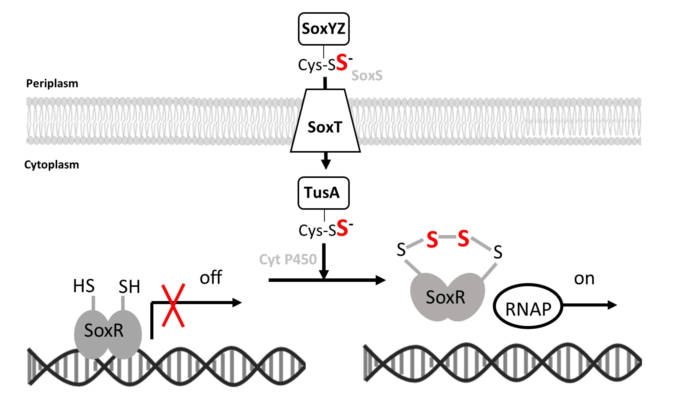
The expression levels of sox as well as of shdr and associated genes are increased by thiosulfate in wildtype cells and elevated in the soxR-deficient H. denitrificans mutant irrespective of the presence of thiosulfate (Figure 4). DNA binding in vitro and probably also transcriptional repression in living cells involve thiol modifications. This can be concluded from the observation that the DNA-binding activity of recombinant SoxR is strongly reduced upon incubation with polysulfide which leads to persulfuration of the regulator as proven by reaction with MalPEG (Figure 9) and mass spectrometry (Table 1, Supplementary Figure S1). In polysulfide-treated SoxR, the two conserved cysteine residues can neither be modified by MalPEG nor by iodoacetamide. In addition, polysulfide treatment increases the mass of wildtype SoxR by 32, 64 or 96 Da. These findings can be fully explained by the formation of an intramolecular tri-, tetra- or pentasulfide bond formed upon interaction with reactive sulfane sulfur species. Thus, SoxR clearly is not a simple redox sensor switching between dithiol and disulfide state but has been identified as a transcriptional regulator sensing reactive sulfane sulfur species (Figure 10), similar to the related SqrR protein from R. capsulatus [34,67]. Notably, the substitution of the two crucial conserved cysteine residues leads to a different outcome for SoxR as compared to SqrR: The lack of Cys50 causes complete loss of DNA binding in SoxR, whereas lack of Cys116 creates a variant that tightly binds to its target DNA and is insensitive to persulfuration. In SqrR, both equivalent Cys-Ser variants are DNA-binding competent and do not respond to persulfuration as a signal [34]. Clearly, this difference inspires future research that should also include detailed inspection of the conformational changes triggered by formation of a sulfur bridge and resulting in detachment of SoxR from its target DNA (Figure 10).
The physiological processes involving the various sulfane sulfur-responsive regulators characterized so far [33,34,68,69,70,71] differ fundamentally from that controlled by SoxR. The former mainly regulate stress responses, sense intracellular and extracellular reactive sulfur species and ensure upregulation of H2S oxidation genes for the purpose of detoxification, i.e., they control the removal of excess sulfide and sulfane sulfur thus contributing to cell survival in the presence of external reactive sulfur species. In contrast, SoxR regulates dissimilatory sulfur metabolism and enables the use of reduced sulfur compounds such as thiosulfate as electron donors for lithotrophic or mixotrophic growth.
As pointed out earlier, thiosulfate oxidation is initiated in the periplasm and it is highly unlikely that thiosulfate itself serves as the signalling molecule. Instead, SoxR responds to the presence of low concentrations of sulfane sulfur, which was provided as polysulfide in our in vitro assays. A working hypothesis for how this signal reaches its destination inspired by the arrangement of the respective genes in H. denitrificans (Figure 1b) is presented in Figure 10. It is conceivable that the sulfur bound to the sulfur carrier protein SoxYZ in the periplasm in the course of thiosulfate oxidation reaches the cytoplasm via a YedE-like SoxT transporter [72]. The periplasmic thiol-disulfide oxidoreductase SoxS [16] could be involved in the transfer of the sulfane sulfur to the transporter. Once in the cytoplasm, the sulfur transferase TusA [73] is a possible acceptor protein for the sulfur, which could be passed on from there to SoxR, possibly involving the cytochrome P450 encoded by gene Hden_0697.
In conclusion, our study shows that SoxR allows H. denitrificans to adapt to changes in thiosulfate availability via thiol persulfidation chemistry and the formation of an intramolecular sulfur bridge, which may involve transporters and sulfurtransferases encoded in the same genetic island. Clearly, much remains to be learned about this regulator not only in terms of signal transduction but also in terms of crosstalk with its counterpart sHdrR.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Strains, plasmids and primers.
Author Contributions
C.D. and J.L. conceptualized the study; J.L., K.T., J. K., T.S.T. and H.Y.H performed experiments; T.S.T. performed bioinformatic analyses. C.D. supervised the work and acquired funding. C.D. and J.L. wrote the manuscript.
Funding
This research was in part funded by German Science foundation, grant numbers Da 351/13-1 and Da 351/8-2. J.L. was financed by a Scholarship of the China Scholarship Council and T.S.T. received a scholarship from the Studienstiftung des Deutschen Volkes.
Institutional Review Board Statement
Not applicable.
Informed consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
We thank Laura Schiffer for help with protein production. We gratefully acknowledge the support of the Core Facility “Protein Synthesis and Bioanalytics” of the university of Bonn for performing mass spectrometry.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Friedrich, C.G.; Rother, D.; Bardischewsky, F.; Quentmeier, A.; Fischer, J. Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl. Environ. Microbiol. 2001, 67, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Dahl, C. A biochemical view on the biological sulfur cycle. In Environmental technologies to treat sulfur pollution: principles and engineering, 2 ed; Lens, P. (ed.), IWA Publishing: London, 2020; pp. 55–96. [Google Scholar]
- Appia-Ayme, C.; Little, P.J.; Matsumoto, Y.; Leech, A.P.; Berks, B.C. Cytochrome complex essential for photosynthetic oxidation of both thiosulfate and sulfide in Rhodovulum sulfidophilum. J. Bacteriol. 2001, 183, 6107–6118. [Google Scholar] [CrossRef] [PubMed]
- Grabarczyk, D.B.; Berks, B.C. Intermediates in the Sox sulfur oxidation pathway are bound to a sulfane conjugate of the carrier protein SoxYZ. PLoS One 2017, 12, e0173395. [Google Scholar] [CrossRef]
- Sauvé, V.; Roversi, P.; Leath, K.J.; Garman, E.F.; Antrobus, R.; Lea, S.M.; Berks, B.C. Mechanism for the hydrolysis of a sulfur-sulfur bond based on the crystal structure of the thiosulfohydrolase SoxB. J. Biol. Chem. 2009, 284, 21707–21718. [Google Scholar] [CrossRef] [PubMed]
- Grabarczyk, D.B.; Chappell, P.E.; Johnson, S.; Stelzl, L.S.; Lea, S.M.; Berks, B.C. Structural basis for specificity and promiscuity in a carrier protein/enzyme system from the sulfur cycle. Proc. Natl. Acad. Sci. USA 2015, 112, E7166–E7175. [Google Scholar] [CrossRef]
- Zander, U.; Faust, A.; Klink, B.U.; de Sanctis, D.; Panjikar, S.; Quentmeier, A.; Bardischewsky, F.; Friedrich, C.G.; Scheidig, A.J. Structural basis for the oxidation of protein-bound sulfur by the sulfur cycle molybdohemo-enzyme sulfane dehydrogenase SoxCD. J. Biol. Chem. 2010; 286, 8349. [Google Scholar]
- Dahl, C.; Friedrich, C.G.; Kletzin, A. Sulfur oxidation in prokaryotes. In Encyclopedia of Life Sciences, John Wiley & Sons, Ltd.: Chichester, 2008.
- Koch, T.; Dahl, C. A novel bacterial sulfur oxidation pathway provides a new link between the cycles of organic and inorganic sulfur compounds. ISME J. 2018, 12, 2479–2491. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Koch, J.; Flegler, W.; Garcia Ruiz, L.; Hager, N.; Ballas, A.; Tanabe, T.S.; Dahl, C. A metabolic puzzle: consumption of C1 compounds and thiosulfate in Hyphomicrobium denitrificans XT. Biochim. Biophys. Acta - Bioenergetics 2022, 1864, 148932. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Koch, T.; Steffens, L.; Finkensieper, J.; Zigann, R.; Cronan, J.E.; Dahl, C. Lipoate-binding proteins and specific lipoate-protein ligases in microbial sulfur oxidation reveal an atpyical role for an old cofactor. eLife 2018, 7, e37439. [Google Scholar] [CrossRef]
- Appia-Ayme, C.; Berks, B.C. SoxV, an orthologue of the CcdA disulfide transporter, is involved in thiosulfate oxidation in Rhodovulum sulfidophilum and reduces the periplasmic thioredoxin SoxW. Biochem. Biophys. Res. Commun. 2002, 296, 737–741. [Google Scholar] [CrossRef]
- Friedrich, C.G.; Quentmeier, A.; Bardischewsky, F.; Rother, D.; Orawski, G.; Hellwig, P.; Fischer, J. Redox control of chemotrophic sulfur oxidation of Paracoccus pantotrophus. In Microbial sulfur metabolism; Dahl, C., Ed.; Friedrich, C. G. (eds.), Springer: Berlin Heidelberg, 2008; pp. 139–150. [Google Scholar]
- Orawski, G.; Bardischewsky, F.; Quentmeier, A.; Rother, D.; Friedrich, C.G. The periplasmic thioredoxin SoxS plays a key role in activation in vivo of chemotrophic sulfur oxidation of Paracoccus pantotrophus. Microbiology 2007, 153, 1081–1086. [Google Scholar] [CrossRef]
- Carius, Y.; Rother, D.; Friedrich, C.G.; Scheidig, A.J. The structure of the periplasmic thiol-disulfide oxidoreductase SoxS from Paracoccus pantotrophus indicates a triple Trx/Grx/DsbC functionality in chemotrophic sulfur oxidation. Acta Crystallogr. D Biol. Cryst. 2009, 65, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Rother, D.; Ringk, J.; Friedrich, C.G. Sulfur oxidation of Paracoccus pantotrophus: The sulfur-binding protein SoxYZ is the target of the periplasmic thiol-disulfide oxidoreductase SoxS. Microbiology 2008, 154, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Neutzling, O.; Pfleiderer, C.; Trüper, H.G. Dissimilatory sulphur metabolism in phototrophic “non-sulphur” bacteria. J. Gen. Microbiol. 1985, 131, 791–798. [Google Scholar] [CrossRef]
- Kappler, U.; Enemark, J.H. Sulfite-oxidizing enzymes. J. Biol. Inorg. Chem. 2014, 20, 253–264. [Google Scholar] [CrossRef]
- Kappler, U.; Schwarz, G. The sulfite oxidase family of molybdenum enzymes. In Molybdenum and tungsten enzymes; Hille, R., Schulzke, C., Eds.; Kirk, M. L. (eds.), Royal Society of Chemistry: Cambridge, 2016; pp. 240–273. [Google Scholar]
- Meng, Z.; Qin, G.; Zhang, B.; Bai, J. DNA damaging effects of sulfur dioxide derivatives in cells from various organs of mice. Mutagenesis 2004, 19, 465–468. [Google Scholar] [CrossRef]
- Yi, H.; Liu, J.; Zheng, K. Effect of sulfur dioxide hydrates on cell cycle, sister chromatid exchange, and micronuclei in barley. Ecotoxicol. Environ. Saf. 2005, 62, 421–426. [Google Scholar] [CrossRef]
- Ozturk, O.H.; Kucukatay, V.; Yonden, Z.; Agar, A.; Bagci, H.; Delibas, N. Expressions of N-methyl-D-aspartate receptors NR2A and NR2B subunit proteins in normal and sulfite-oxidase deficient rat’s hippocampus: effect of exogenous sulfite ingestion. Arch. Toxicol. 2006, 80, 671–679. [Google Scholar] [CrossRef]
- Kao, Y.T.; Tan, C.; Song, S.H.; Ozturk, N.; Li, J.; Wang, L.; Sancar, A.; Zhong, D. Ultrafast dynamics and anionic active states of the flavin cofactor in cryptochrome and photolyase. J. Am. Chem. Soc. 2008, 130, 7695–7701. [Google Scholar] [CrossRef]
- Ozawa, T.; Hanaki, A. Spin-trapping of sulfite radical anion, SO3-., by a water-soluble, nitroso-aromatic spin-trap. Biochem. Biophys. Res. Commun. 1987, 142, 410–416. [Google Scholar] [CrossRef]
- Inouye, B.; Morita, K.; Ishida, T.; Ogata, M. Cooperative effect of sulfite and vanadium compounds on lipid peroxidation. Toxicol. Appl. Pharmacol. 1980, 53, 101–107. [Google Scholar] [CrossRef]
- Yang, S.F. Destruction of tryptophan during the aerobic oxidation of sulfite ions. Environmental Research 1973, 6, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Rother, D.; Orawski, G.; Bardischewsky, F.; Friedrich, C.G. SoxRS-mediated regulation of chemotrophic sulfur oxidation in Paracoccus pantotrophus. Microbiology 2005, 151, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Chatterjee, S.; Dam, B.; Roy, P.; Das Gupta, S.K. The dimeric repressor SoxR binds cooperatively to the promoter(s) regulating expression of the sulfur oxidation (sox) operon of Pseudaminobacter salicylatoxidans KCT001. Microbiology 2007, 153, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, C.; Mandal, S.; Ghosh, W.; Dam, B.; Roy, P. A novel gene cluster soxSRT is essential for the chemolithotrophic oxidation of thiosulfate and tetrathionate by Pseudaminobacter salicylatoxidans KCT001. Curr. Microbiol. 2006, 52, 267–273. [Google Scholar] [CrossRef]
- Ma, Z.; Jacobsen, F.E.; Giedroc, D.P. Coordination chemistry of bacterial metal transport and sensing. Chem Rev 2009, 109, 4644–4681. [Google Scholar] [CrossRef]
- Cook, W.J.; Kar, S.R.; Taylor, K.B.; Hall, L.M. Crystal structure of the cyanobacterial metallothionein repressor SmtB: a model for metalloregulatory proteins. J. Mol. Biol. 1998, 275, 337–346. [Google Scholar] [CrossRef]
- Capdevila, Daiana A. ; Edmonds, Katherine A.; Giedroc, David P. Metallochaperones and metalloregulation in bacteria. Essays Biochem. 2017, 61, 177–200. [Google Scholar] [CrossRef]
- Guimaraes, B.G.; Barbosa, R.L.; Soprano, A.S.; Campos, B.M.; de Souza, T.A.; Tonoli, C.C.; Leme, A.F.; Murakami, M.T.; Benedetti, C.E. Plant pathogenic bacteria utilize biofilm growth-associated repressor (BigR), a novel winged-helix redox switch, to control hydrogen sulfide detoxification under hypoxia. J. Biol. Chem. 2011, 286, 26148–26157. [Google Scholar] [CrossRef]
- Shimizu, T.; Shen, J.; Fang, M.; Zhang, Y.; Hori, K.; Trinidad, J.C.; Bauer, C.E.; Giedroc, D.P.; Masuda, S. Sulfide-responsive transcriptional repressor SqrR functions as a master regulator of sulfide-dependent photosynthesis. Proc. Natl. Acad. Sci. USA 2017, 114, 2355–2360. [Google Scholar] [CrossRef]
- Bertani, G. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 2004, 186, 595–600. [Google Scholar] [CrossRef]
- Ausubel, F.A.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidman, J.G.; Smith, J.A.; Struhl, K. Current protocols in molecular biology. John Wiley & Sons, New York, 1997.
- Horton, R.M. PCR mediated recombination and mutagenesis: SOEing together tailor-made genes. Mol. Biotechnol. 1995, 3, 93–99. [Google Scholar] [CrossRef]
- Dahl, C. Insertional gene inactivation in a phototrophic sulphur bacterium: APS-reductase-deficient mutants of Chromatium vinosum. Microbiology 1996, 142, 3363–3372. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Martineau, C.; Mauffrey, F.; Villemur, R. Comparative analysis of denitrifying activities of Hyphomicrobium nitrativorans, Hyphomicrobium denitrificans, and Hyphomicrobium zavarzinii. Appl. Environ. Microbiol. 2015, 81, 5003–5014. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-D D CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Satake, H.; Hisano, T.; Terazawa, T. Potentiometric argentimetric method for the successive titration of sulphide and dissolved sulphur in polysulphide solutions. Talanta 1972, 19, 1650–1654. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C.; Chuvochina, M.; Mussig, A.J.; Chaumeil, P.A.; Davin, A.A.; Waite, D.W.; Whitman, W.B.; Parks, D.H.; Hugenholtz, P. A standardized archaeal taxonomy for the Genome Taxonomy Database. Nat. Microbiol. 2021, 6, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11, 119. [Google Scholar] [CrossRef]
- Tanabe, T.S.; Dahl, C. HMS-S-S: a tool for the identification of sulphur metabolism-related genes and analysis of operon structures in genome and metagenome assemblies. Mol. Ecol. Resour. 2022, 22, 2758–2774. [Google Scholar] [CrossRef]
- Dahl, C. Sulfur metabolism in phototrophic bacteria. In Modern topics in the phototrophic prokaryotes: Metabolism, bioenergetics and omics; Hallenbeck, P. C. (ed.), Springer International Publishing: Cham, 2017; pp. 27–66. [Google Scholar]
- Frigaard, N.U.; Dahl, C. Sulfur metabolism in phototrophic sulfur bacteria. Adv. Microb. Physiol. 2009, 54, 103–200. [Google Scholar]
- Gupta, R.S. The phylum Aquificae. In The prokaryotes - other lineages of Bacteria and the Archaea; Rosenberg, E., DeLong, E. F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin Heidelberg, 2014. [Google Scholar]
- Kodama, Y.; Watanabe, K. Sulfuricurvum kujiense gen. nov., sp. nov., a facultatively anaerobic, chemolithoautotrophic, sulfur-oxidizing bacterium isolated from an underground crude-oil storage cavity. Int. J. Syst. Evol. Microbiol. 2004, 54, 2297–2300. [Google Scholar] [CrossRef]
- Xie, S.; Wang, S.; Li, D.; Shao, Z.; Lai, Q.; Wang, Y.; Wei, M.; Han, X.; Jiang, L. Sulfurovum indicum sp. nov., a novel hydrogen- and sulfur-oxidizing chemolithoautotroph isolated from a deep-sea hydrothermal plume in the Northwestern Indian Ocean. Int. J. Syst. Evol. Microbiol. 2019, 71. [Google Scholar] [CrossRef] [PubMed]
- Labrenz, M.; Grote, J.; Mammitzsch, K.; Boschker, H.T.S.; Laue, M.; Jost, G.; Glaubitz, S.; Jurgens, K. Sulfurimonas gotlandica sp. nov., a chemoautotrophic and psychrotolerant epsilonproteobacterium isolated from a pelagic redoxcline, and an emended description of the genus Sulfurimonas. Int. J. Syst. Evol. Microbiol. 2013, 63, 4141–4148. [Google Scholar] [CrossRef]
- Skirnisdottir, S.; Hreggvidsson, G.O.; Holst, O.; Kristjansson, J.K. Isolation and characterization of a mixotrophic sulfur-oxidizing Thermus scotoductus. Extremophiles 2001, 5, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Bjornsdottir, S.H.; Petursdottir, S.K.; Hreggvidsson, G.O.; Skirnisdottir, S.; Hjorleifsdottir, S.; Arnfinnsson, J.; Kristjansson, J.K. Thermus islandicus sp. nov., a mixotrophic sulfur-oxidizing bacterium isolated from the Torfajokull geothermal area. Int. J. Syst. Evol. Microbiol. 2009, 59, 2962–2966. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. Phylogenetic taxonomy of the family Chlorobiaceae on the basis of 16S rRNA and fmo (Fenna-Matthews-Olson protein) gene sequences. Int. J. Syst. Evol. Microbiol. 2003, 53, 941–951. [Google Scholar] [CrossRef]
- Carareto Alves, L.M.; de Souza, J.A.M.; Varani, A.d.M.; Lemos, E.G.d.M. The Family Rhizobiaceae. In The prokaryotes; Rosenberg, E., DeLong, E. F., Lory, S., Stackebrandt, E., Eds.; Thompson, F. (eds.), Springer: Berlin Heidelberg, 2014; pp. 419–437. [Google Scholar]
- Pujalte, M.J.; Lucena, T.; Ruvira, M.A.; Arahal, D.R.; Macián, M.C. The Family Rhodobacteraceae. In The prokaryotes; Rosenberg, E., DeLong, E. F., Lory, S., Stackebrandt, E., Eds.; Thompson, F. (eds.), Springer: Berlin, Heidelberg, 2014; pp. 439–512. [Google Scholar]
- Spietz, R.L.; Lundeen, R.A.; Zhao, X.; Nicastro, D.; Ingalls, A.E.; Morris, R.M. Heterotrophic carbon metabolism and energy acquisition in Candidatus Thioglobus singularis strain PS1, a member of the SUP05 clade of marine Gammaproteobacteria. Environ. Microbiol. 2019, 21, 2391–2401. [Google Scholar] [CrossRef]
- Imhoff, J.F. Family I. Chromatiaceae Bavendamm 1924, 125 AL emend. Imhoff 1984b, 339. In Bergey’s manual of systematic bacteriology; Brenner, D. J., Krieg, N. R., Staley, J. T., Eds.; Garrity, G. M. (eds.), Springer: New York, 2005. [Google Scholar]
- Imhoff, J.F. Family II. Ectothiorhodospiraceae Imhoff 1984b, 339 VP In Bergey’s manual of systematic bacteriology; Brenner, D. J., Krieg, N. R., Staley, J. T., Garrity, G. M., Eds.; Springer: New York, 2005; Vol. 2,pp. 41-57. [Google Scholar]
- Boden, R.; Scott, K.M.; Williams, J.; Russel, S.; Antonen, K.; Rae, A.W.; Hutt, L.P. An evaluation of Thiomicrospira, Hydrogenovibrio and Thioalkalimicrobium: reclassification of four species of Thiomicrospira to each Thiomicrorhabdus gen. nov. and Hydrogenovibrio, and reclassification of all four species of Thioalkalimicrobium to Thiomicrospira. Int. J. Syst. Evol. Microbiol. 2017, 67, 1140–1151. [Google Scholar]
- Boden, R.; Scott, K.M. Evaluation of the genus Thiothrix Winogradsky 1888 (Approved Lists 1980) emend. Aruga et al. 2002: reclassification of Thiothrix disciformis to Thiolinea disciformis gen. nov., comb. nov., and of Thiothrix flexilis to Thiofilum flexile gen. nov., comb nov., with emended description of Thiothrix. Int. J. Syst. Evol. Microbiol. 2018, 68, 2226–2239. [Google Scholar]
- Shimizu, T.; Masuda, S. Characterization of redox-active cysteine residues of persulfide-responsive transcriptional repressor SqrR. Commun. Integr. Biol. 2017, 10, e1329786. [Google Scholar] [CrossRef]
- Mukherjee, D.; Datta, A.B.; Chakrabarti, P. Crystal structure of HlyU, the hemolysin gene transcription activator, from Vibrio cholerae N16961 and functional implications. Biochim. Biophys. Acta 2014, 1844, 2346–2354. [Google Scholar] [CrossRef] [PubMed]
- Gueune, H.; Durand, M.J.; Thouand, G.; DuBow, M.S. The ygaVP genes of Escherichia coli form a tributyltin-inducible operon. Appl. Environ. Microbiol. 2008, 74, 1954–1958. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Deutsch, C. Pegylation: a method for assessing topological accessibilities in Kv1.3. Biochemistry 2001, 40, 13288–13301. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, D.A.; Walsh, B.J.C.; Zhang, Y.; Dietrich, C.; Gonzalez-Gutierrez, G.; Giedroc, D.P. Structural basis for persulfide-sensing specificity in a transcriptional regulator. Nat. Chem. Biol. 2021, 17, 65–70. [Google Scholar] [CrossRef]
- Barbosa, R.L.; Benedetti, C.E. BigR, a transcriptional repressor from plant-associated bacteria, regulates an operon implicated in biofilm growth. J. Bacteriol. 2007, 189, 6185–6194. [Google Scholar] [CrossRef]
- Grossoehme, N.; Kehl-Fie, T.E.; Ma, Z.; Adams, K.W.; Cowart, D.M.; Scott, R.A.; Skaar, E.P.; Giedroc, D.P. Control of copper resistance and inorganic sulfur metabolism by paralogous regulators in Staphylococcus aureus. J. Biol. Chem. 2011, 286, 13522–13531. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Hori, K.; Shimizu, T.; Kasamatsu, S.; Okamura, K.; Tanaka, K.; Ihara, H.; Masuda, S. The sulfide-responsive SqrR/BigR homologous regulator YgaV of Escherichia coli controls expression of anaerobic respiratory genes and antibiotic tolerance. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Tang, C.; Li, J.; Shen, Y.; Liu, M.; Liu, H.; Liu, H.; Xun, L.; Xia, Y. A sulfide-sensor and a sulfane sulfur-sensor collectively regulate sulfur-oxidation for feather degradation by Bacillus licheniformis. Commun. Biol. 2023, 6, 167. [Google Scholar] [CrossRef]
- Tanaka, Y.; Yoshikaie, K.; Takeuchi, A.; Ichikawa, M.; Mori, T.; Uchino, S.; Sugano, Y.; Hakoshima, T.; Takagi, H.; Nonaka, G.; Tsukazaki, T. Crystal structure of a YeeE/YedE family protein engaged in thiosulfate uptake. Science Advances 2020, 6, eaba7637. [Google Scholar] [CrossRef]
- Tanabe, T.S.; Leimkühler, S.; Dahl, C. The functional diversity of the prokaryotic sulfur carrier protein TusA. Adv. Microb. Physiol. 2019, 75, 233–277. [Google Scholar] [PubMed]
Figure 2.
Occurrence of complete and trunctated Sox systems among five bacterial phyla. Simultaneous presence of SoxR is indicated in black.
Figure 2.
Occurrence of complete and trunctated Sox systems among five bacterial phyla. Simultaneous presence of SoxR is indicated in black.
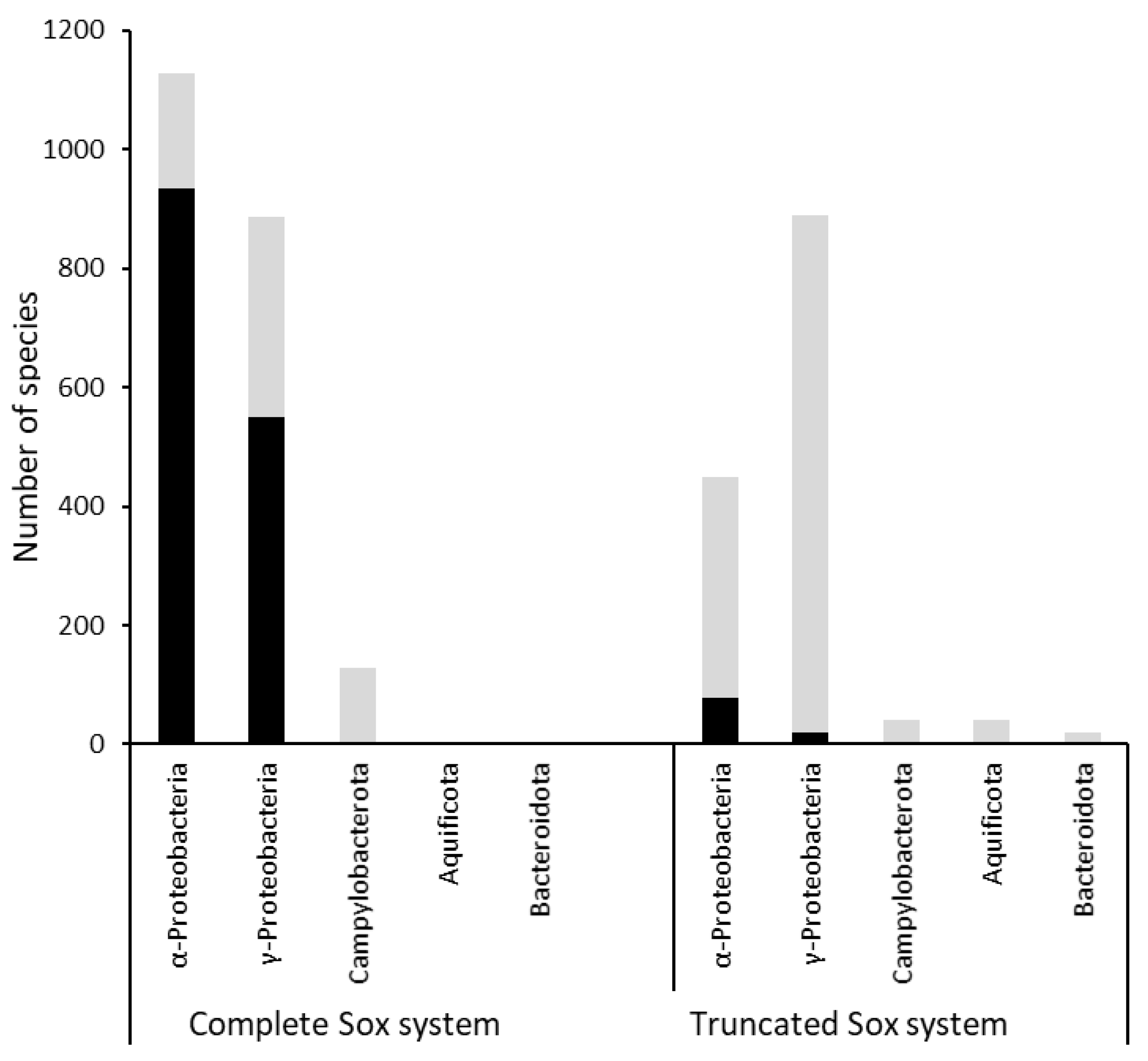
Figure 3.
Growth and thiosulfate consumption of H. denitrificans ΔtsdA (black circles and lines), ΔtsdA ΔshdrR (black triangles and lines) and ΔtsdA ΔsoxR (red boxes and lines). cultures were grown on methanol-containing medium (24.4 mM methanol) without (open symbols) or with 2 mM thiosulfate (filled symbols). Precultures contained either no thiosulfate (not induced, broken lines) or 2 mM thiosulfate (induced, solid lines). In the lower panels, thiosulfate concentrations for the different cultures are given. Symbol assignment is the same as in the upper panels. Specific thiosulfate (TS) oxidation rates are depicted in the same color code. Error bars indicating SD are too small to be visible for determination of biomass.
Figure 3.
Growth and thiosulfate consumption of H. denitrificans ΔtsdA (black circles and lines), ΔtsdA ΔshdrR (black triangles and lines) and ΔtsdA ΔsoxR (red boxes and lines). cultures were grown on methanol-containing medium (24.4 mM methanol) without (open symbols) or with 2 mM thiosulfate (filled symbols). Precultures contained either no thiosulfate (not induced, broken lines) or 2 mM thiosulfate (induced, solid lines). In the lower panels, thiosulfate concentrations for the different cultures are given. Symbol assignment is the same as in the upper panels. Specific thiosulfate (TS) oxidation rates are depicted in the same color code. Error bars indicating SD are too small to be visible for determination of biomass.
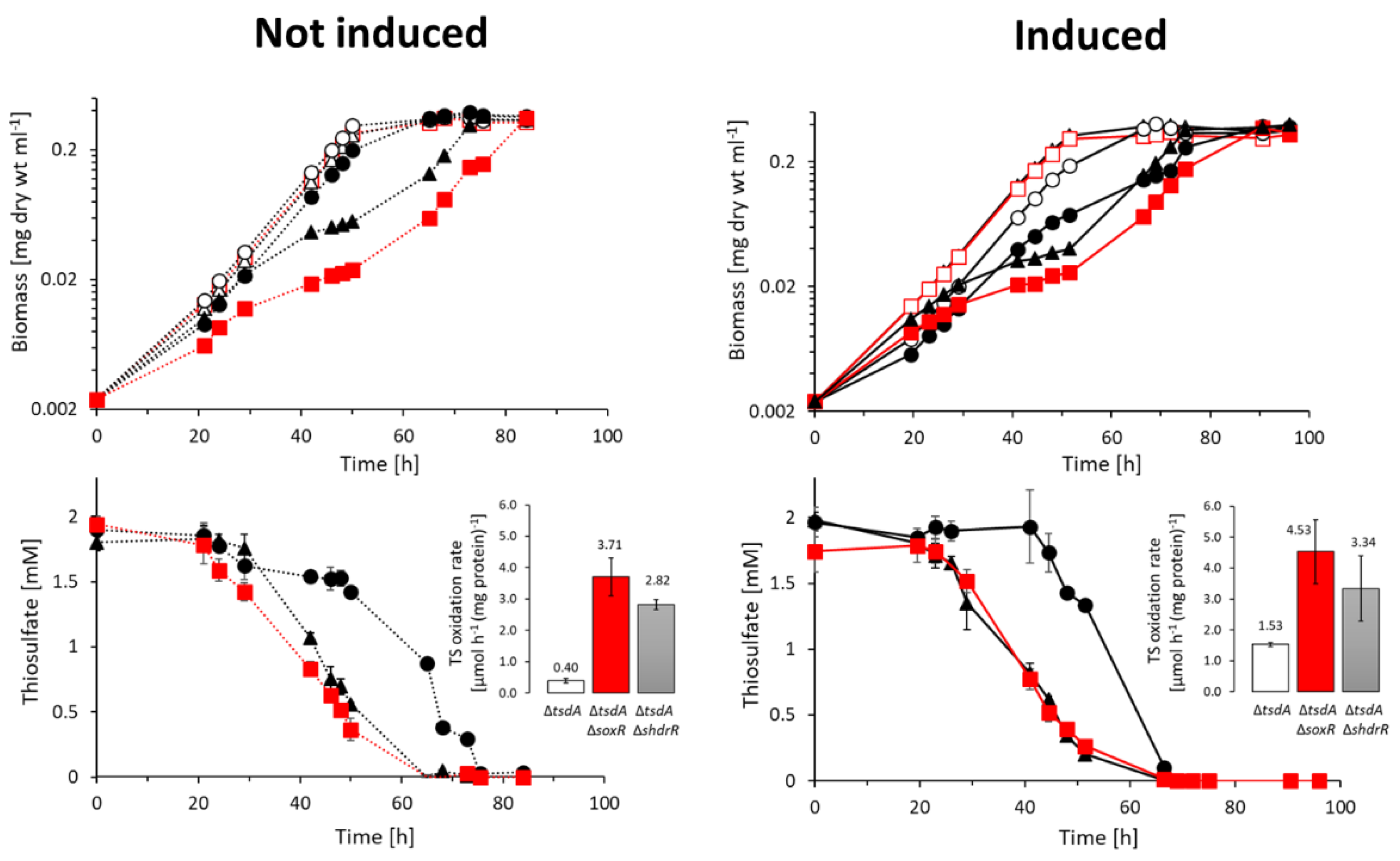
Figure 4.
(a) Relative mRNA levels of twelve genes located in the shdr-sox genetic island (depicted in panel (b)) from H. denitrificans for the ΔtsdA reference strain in the absence (gray columns) and presence of thiosulfate (white columns), as assessed by RT-qPCR. Results for H. denitrificans ΔtsdA ΔsoxR are shown by black columns. Results were adjusted using H. denitrifcans rpoB, which encodes the β-subunit of RNA polymerase, as an endogenous reference according to [40]. (b) DNA regions tested in EMSA assays for SoxR binding are indicated as black rectangles below the hyphomicrobial shdr-sox genetic island. Fragment sizes: 362 bp for the soxT1A-shdrR intergenic region, 177 bp and 173 bp for the regions upstream of lipS1 and dsrE3C, respectively. The fragments downstream of tusA and between soxA and soxY had sizes of 176 bp and 151 bp, respectively. (c) EMSA analysis of Strep-tagged SoxR with upstream promoter sequence probes of sulfur oxidation related genes as specified in (b). 17 nM DNA probes were incubated with different amounts of SoxR (300 and 700 nM).
Figure 4.
(a) Relative mRNA levels of twelve genes located in the shdr-sox genetic island (depicted in panel (b)) from H. denitrificans for the ΔtsdA reference strain in the absence (gray columns) and presence of thiosulfate (white columns), as assessed by RT-qPCR. Results for H. denitrificans ΔtsdA ΔsoxR are shown by black columns. Results were adjusted using H. denitrifcans rpoB, which encodes the β-subunit of RNA polymerase, as an endogenous reference according to [40]. (b) DNA regions tested in EMSA assays for SoxR binding are indicated as black rectangles below the hyphomicrobial shdr-sox genetic island. Fragment sizes: 362 bp for the soxT1A-shdrR intergenic region, 177 bp and 173 bp for the regions upstream of lipS1 and dsrE3C, respectively. The fragments downstream of tusA and between soxA and soxY had sizes of 176 bp and 151 bp, respectively. (c) EMSA analysis of Strep-tagged SoxR with upstream promoter sequence probes of sulfur oxidation related genes as specified in (b). 17 nM DNA probes were incubated with different amounts of SoxR (300 and 700 nM).
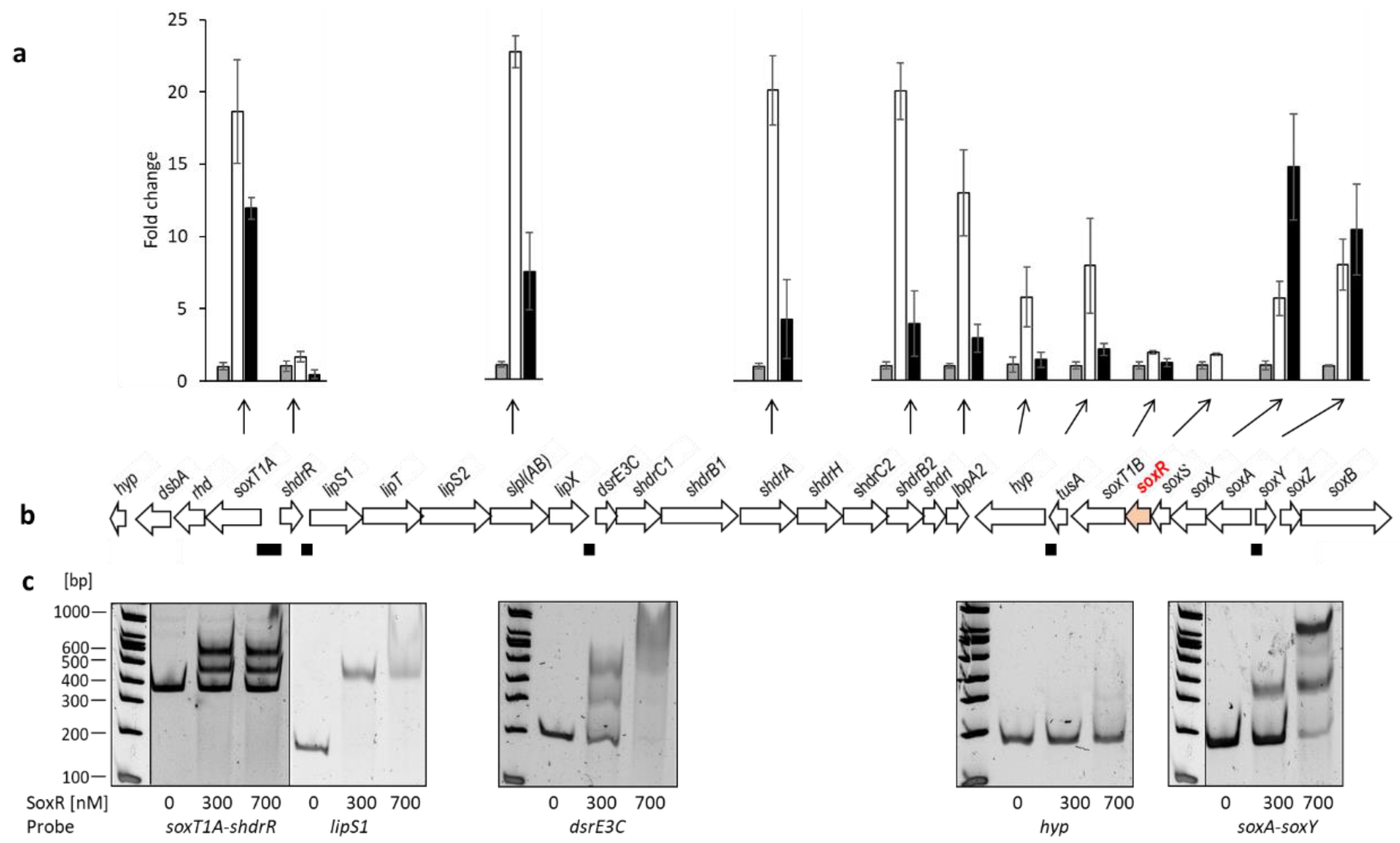
Figure 5.
Amino sequence alignment of SoxR homologs. Accession numbers/locus tags and references in the order of appearance: (Hden_0700 [9], Hden_0682, [9,10], WP_010893290 [33], HLYU_VIBCH [64], WP_019171658 [28], ADE85198 [34], b2667 [65].

Figure 6.
Conformation of SoxR and its variants as analyzed by (panels a and b) non-reducing SDS-PAGE analysis and (c) gel permeation chromatography. For the experiments shown in panels a and b 5 µg of SoxR or its variants were incubated in 15 µl 100 mM Tris-HCl, pH 8.0, 150 mM NaCl with either 1 mM DTT or 5 mM Cucl2 for 20 min at room temperature, mixed with 5 µl non-reducing Roti®-Load2 (Carl Roth GmbH) and run on 15% SDS polyacrylamide gels. The wildtype SoxR protein is shown in panels a and b to allow direct comparison with protein variants on different gels. In (c) the elution profiles upon gel filtration on Superdex 75 Increase 10/300 are depicted for SoxR, solid line; SoxR Cys50Ser, dotted line; SoxR Cys116Ser, dashed line; SoxR Cys50Ser Cys116Ser, dashed-dotted line. SoxR and SoxR Cys116Ser dimers elute at a kav of 0.2 corresponding to a molecular mass of 36.7 kDa, whereas SoxR Cys50Ser and SoxR Cys50Ser Cys116Ser elute earlier (kav = 0.174, 41.9 kDa) indicating a more open conformation. The resolution of the column does not allow clear separation of the different tetrameric conformations (kav 0.086 to 0.093, corresponding to 65.9 to 63.6 kDa).
Figure 6.
Conformation of SoxR and its variants as analyzed by (panels a and b) non-reducing SDS-PAGE analysis and (c) gel permeation chromatography. For the experiments shown in panels a and b 5 µg of SoxR or its variants were incubated in 15 µl 100 mM Tris-HCl, pH 8.0, 150 mM NaCl with either 1 mM DTT or 5 mM Cucl2 for 20 min at room temperature, mixed with 5 µl non-reducing Roti®-Load2 (Carl Roth GmbH) and run on 15% SDS polyacrylamide gels. The wildtype SoxR protein is shown in panels a and b to allow direct comparison with protein variants on different gels. In (c) the elution profiles upon gel filtration on Superdex 75 Increase 10/300 are depicted for SoxR, solid line; SoxR Cys50Ser, dotted line; SoxR Cys116Ser, dashed line; SoxR Cys50Ser Cys116Ser, dashed-dotted line. SoxR and SoxR Cys116Ser dimers elute at a kav of 0.2 corresponding to a molecular mass of 36.7 kDa, whereas SoxR Cys50Ser and SoxR Cys50Ser Cys116Ser elute earlier (kav = 0.174, 41.9 kDa) indicating a more open conformation. The resolution of the column does not allow clear separation of the different tetrameric conformations (kav 0.086 to 0.093, corresponding to 65.9 to 63.6 kDa).
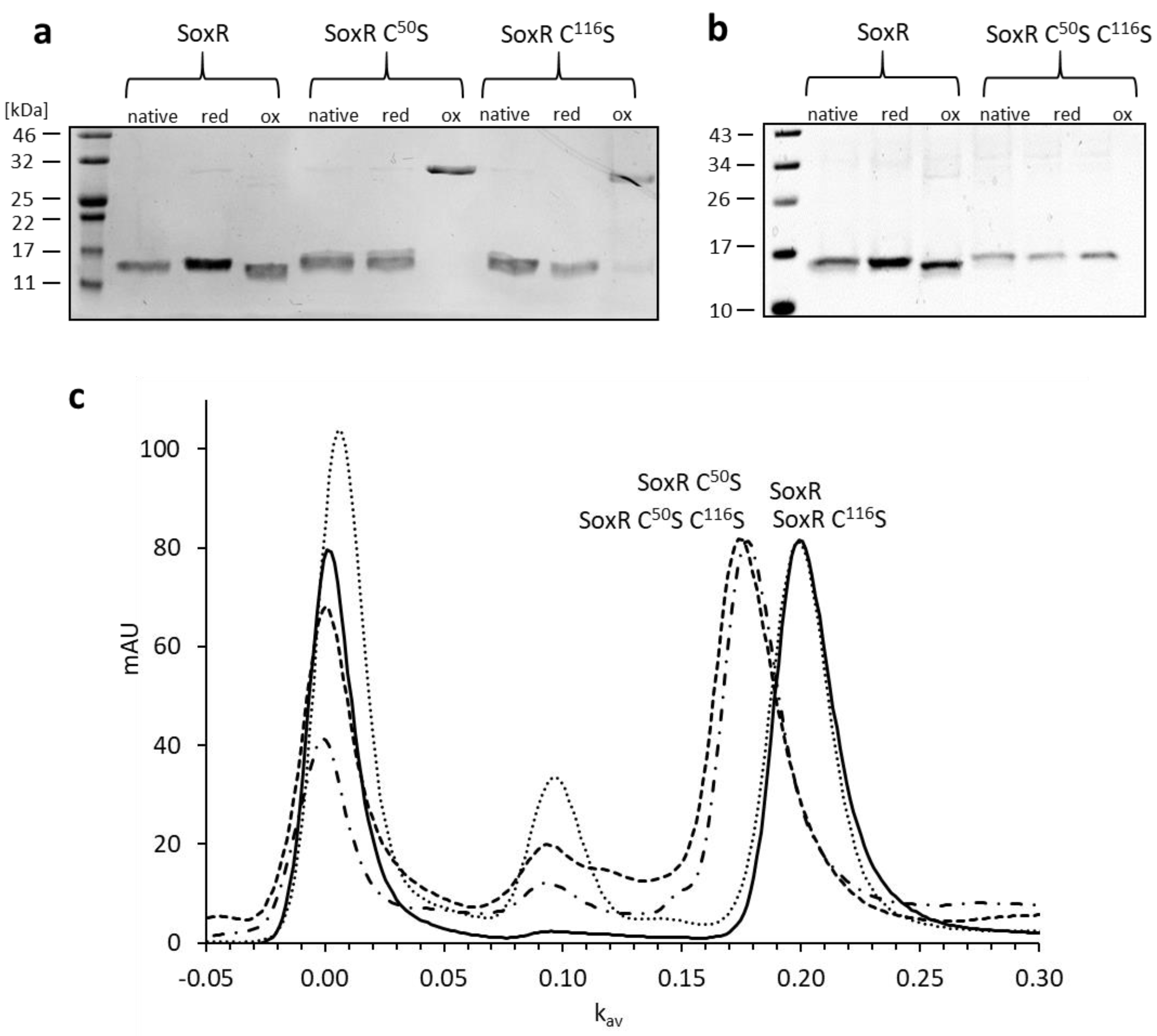
Figure 7.
(a) EMSA of the 151-bp soxA-soxY intergenic fragment with increasing amounts of untreated (upper panel) SoxR or SoxR pre-incubated with polysulfide in a molar ratio SoxR/polysulfide of 1:1 (lower panel), (b) EMSA of the 151-bp soxA-soxY intergenic fragment with SoxR pre-incubated with increasing amounts of polysulfide (upper panel) or thiosulfate (lower panel).
Figure 7.
(a) EMSA of the 151-bp soxA-soxY intergenic fragment with increasing amounts of untreated (upper panel) SoxR or SoxR pre-incubated with polysulfide in a molar ratio SoxR/polysulfide of 1:1 (lower panel), (b) EMSA of the 151-bp soxA-soxY intergenic fragment with SoxR pre-incubated with increasing amounts of polysulfide (upper panel) or thiosulfate (lower panel).
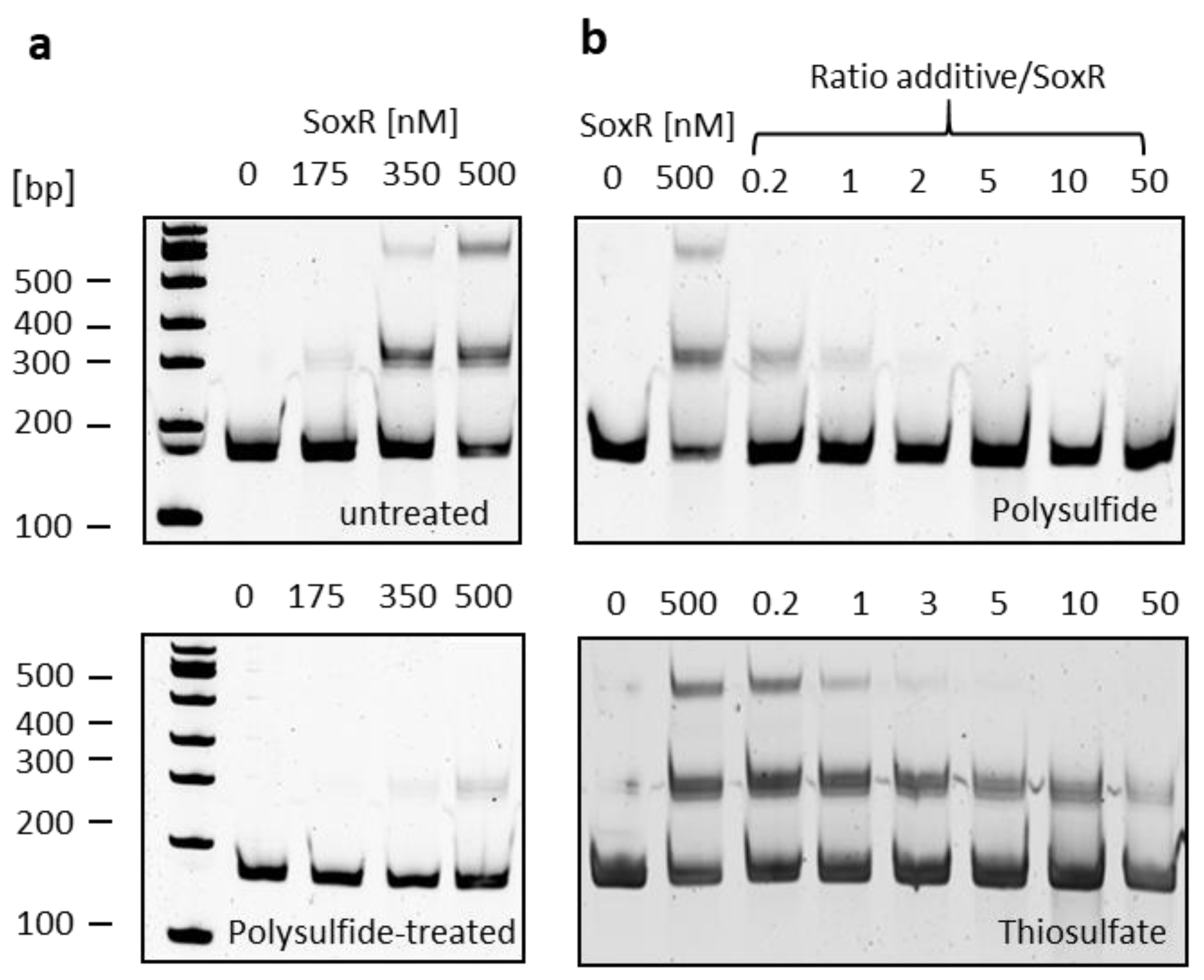
Figure 8.
(a) EMSA of the 151-bp soxA-soxY intergenic fragment (17 nM) with 700 nM SoxR wildtype and variant proteins as isolated, reduced with DTT, oxidized with CuCl2, treated with polysulfide and sequentially treated with polysulfide and DTT. (b) EMSA of the 180 bp central part of the soxT1A-shdrR intergenic fragment (17 nM) with 300 nM SoxR wildtype and variant proteins as isolated, oxidized with CuCl2 or treated with polysulfide.
Figure 8.
(a) EMSA of the 151-bp soxA-soxY intergenic fragment (17 nM) with 700 nM SoxR wildtype and variant proteins as isolated, reduced with DTT, oxidized with CuCl2, treated with polysulfide and sequentially treated with polysulfide and DTT. (b) EMSA of the 180 bp central part of the soxT1A-shdrR intergenic fragment (17 nM) with 300 nM SoxR wildtype and variant proteins as isolated, oxidized with CuCl2 or treated with polysulfide.
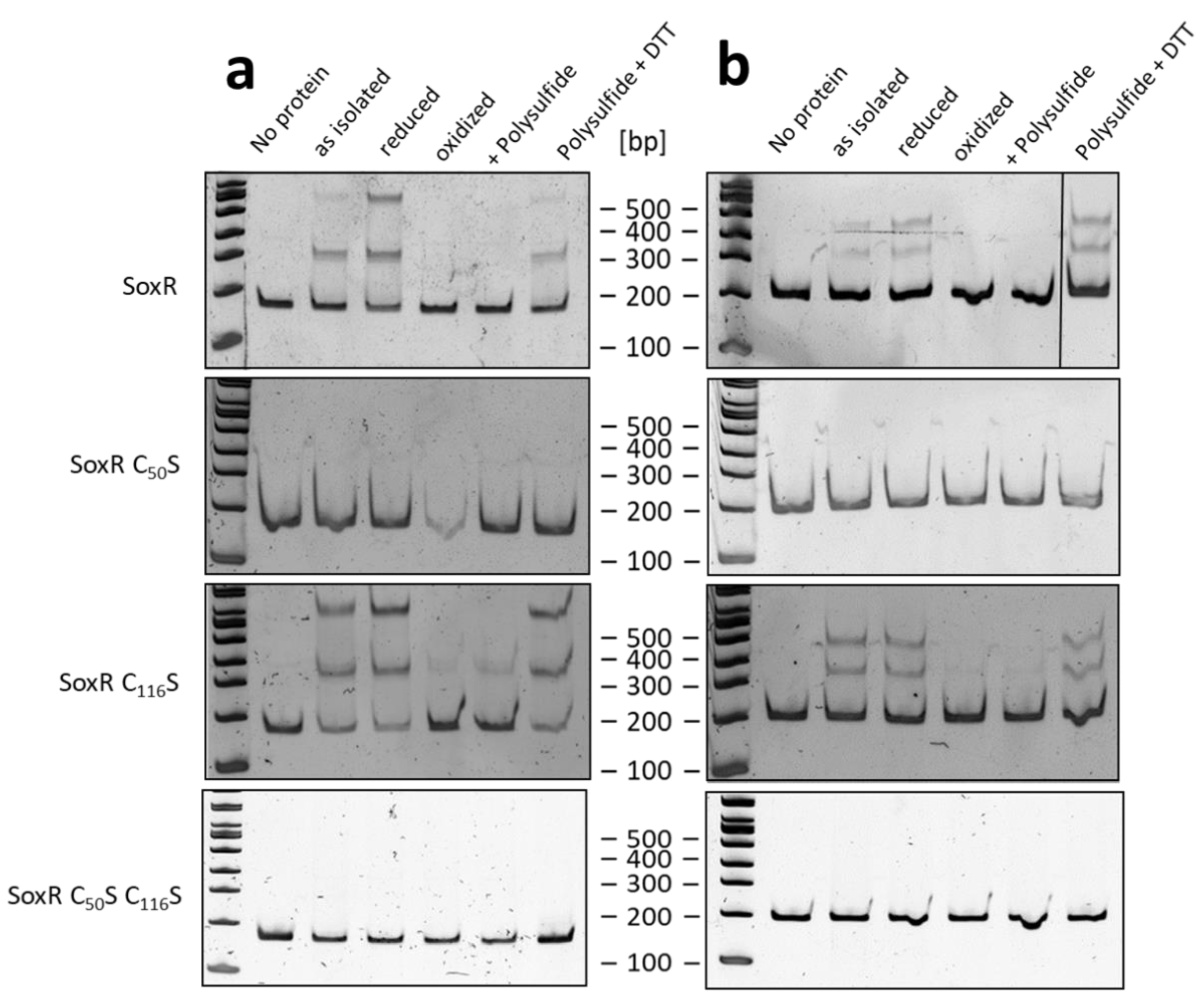
Figure 9.
Analysis of H. denitrificans SoxR cysteines with MalPEG gel-shift assays in non-reducing SDS-PAGE. Results are shown for the wildtype, single (Cys50Ser and Cys116Ser) and double (Cys50Ser Cys116Ser) mutants after MalPEG treatment of the as-isolated, reduced and oxidized states as well as after pre-incubation with polysulfide. Polysulfide and MalPEG reacted samples were furthermore reduced with DTT.
Figure 9.
Analysis of H. denitrificans SoxR cysteines with MalPEG gel-shift assays in non-reducing SDS-PAGE. Results are shown for the wildtype, single (Cys50Ser and Cys116Ser) and double (Cys50Ser Cys116Ser) mutants after MalPEG treatment of the as-isolated, reduced and oxidized states as well as after pre-incubation with polysulfide. Polysulfide and MalPEG reacted samples were furthermore reduced with DTT.
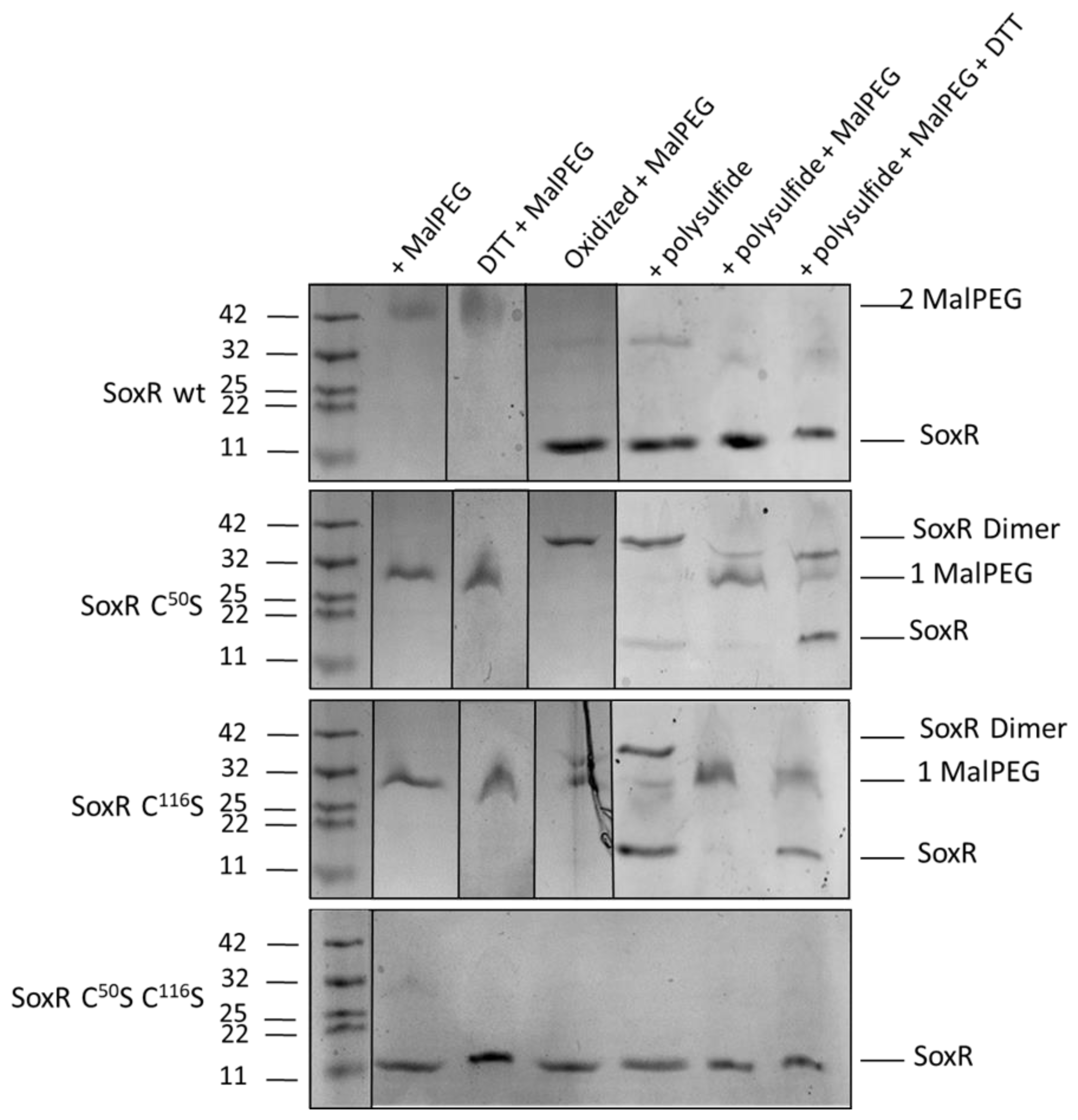
Figure 10.
Proposed signal transduction pathway and mode of action of the homodimeric SoxR repressor protein. Established sulfur-binding proteins are printed in black.
Figure 10.
Proposed signal transduction pathway and mode of action of the homodimeric SoxR repressor protein. Established sulfur-binding proteins are printed in black.


Table 1.
Mass spectrometry of SoxR and variants after treatment with modifying agents. CAM, carbamidomethylation; S, sulfur. Calculated masses for Strep-Tagged SoxR and SoxR Cys50Ser, SoxR Cys116Ser and SoxR Cys50Ser Cys116 without the initiator methionine are 15212.54 Da, 15197.54 Da, 15197.54 Da and 15182.54 Da, respectively.
Table 1.
Mass spectrometry of SoxR and variants after treatment with modifying agents. CAM, carbamidomethylation; S, sulfur. Calculated masses for Strep-Tagged SoxR and SoxR Cys50Ser, SoxR Cys116Ser and SoxR Cys50Ser Cys116 without the initiator methionine are 15212.54 Da, 15197.54 Da, 15197.54 Da and 15182.54 Da, respectively.
| Treatment | SoxR Mass [Da] (addition: [Da]) |
SoxR C50S Mass [Da] (addition: [Da]) |
SoxR C116S Mass [Da] (addition: [Da]) |
SoxR C50S C116S Mass [Da] (addition: [Da]) |
|---|---|---|---|---|
| Native | 15212.8 | 15197.3 | 15198.2 | 15182.3 |
| DTT reduced | 15212.5 | 15199.3 | 15198.8 | nd |
| CuCl2 oxidized | 15210.5 | 15196.7 | 15196.9 | 15182.0 |
| Iodoacetamide | 15328.0 (2 CAM: 2 × 57.07) |
15255.2 (1 CAM: 57.07) |
15255.22 (1 CAM: 57.07) |
nd |
| Polysulfide | 15212.5 15244.7 (1 S: 32) 15276.4 (2 S: 64) 15308.0 (3 S: 96) |
15198.9 15230.0 (1 S: 32) |
15198.0 15230.3 (1 S: 32) 15261.1 (2 S: 64) |
15182.0 |
| Polysulfide + Iodoacetamide | 15212.9 15244.3 (1 S: 32) 15275.2 (2 S: 64) 15306.0 (3 S: 96) |
15198.0 15286.0 (1 S + 1 CAM: 90) |
15197.6 15228.5 (1 S: 32) 15285.4 (1 S + 1 CAM: 90) |
15180.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



