Submitted:
13 July 2023
Posted:
14 July 2023
You are already at the latest version
Abstract
Life is supported by metabolism, a vital physiological activity. It guarantees enough energy available to power every cellular activity. The primary cellular substrate, glucose, is where the majority of the energy needed by the cell to support all other life activities and the growth of living creatures is derived. The quality of life is greatly impacted by any disease or flaw that alters the metabolism of glucose. There is no known cure for the metabolic illness known as diabetes. As diabetes is so varied, the only effective treatments would be based on precision medicine and a molecular strategy. This review covers a wide range of topics, including the ability to recognize the various forms of diabetes, the anthropogenic and genetic influence of the cause of diabetes, the severity of the various forms of diabetes, its potential existential risk, the disease burden, and clinical methods for diagnosing them. The most recent research goes on to recommend ways to ameliorate the disease, including diet-based management, biotherapies, and other individual lifestyle choices. Futuristic therapy using molecular biology approach was suggested. Recent research found a direct correlation between an individual's lifestyle and the rate of gene mutation in diabetes. There is an existential risk associated with the sickness as the rate of the spread of the disease among population globally is faster than the world's population growth rate. It is possible to think of diabetes as a silent pandemic.
Keywords:
Diabetes mellitus
; silent pandemic
; existential risk
; heterogeneity
; Insulin gene
; health
; genetic
; anthropogenics
; hyperglycemia
1. Introduction
Diabetes mellitus is a group of metabolic illnesses that come in diverse forms and are characterized by persistent hyperglycemia due to a lack of insulin, brought on by a reduction in insulin secretion and/or an increase in insulin resistance [1]. The ubiquity of fast-food culture and our way of life are the main causes of diabetes, which has risen to one of the most frequent diseases affecting individuals of all ages. Diabetes is a result of lifestyle activities with immediate and long-term impacts on the metabolism of glucose, lipids, and proteins [2].
The World Health Organization estimates that 537 million people globally have diabetes as of 2021. From 108 million in 1980 to 537 million in 2021, there has been a significant rise. There have been 6 times as many cases of diabetes worldwide. A 1.5 million deaths per year attributable rate is currently predicted, and by 2030, more than 600 million deaths are anticipated [3]. The ninth biggest cause of death worldwide is diabetes [4]. Diabetes prevalence is predicted to climb by more than two-thirds in low- and middle-income nations, including those in sub-Saharan Africa [5] . This may be explained by the fact that environmental influences can affect the genes involved in metabolic processes.
That is to say, since personal habits in developed and underdeveloped nations differ, having a healthy lifestyle reduces the risk of developing diabetes. From an estimated 7.1 million persons in the early 2000s to a projected 18.6 million by 2030, the illness burden in Africa alone has increased dramatically [6] . The overall prevalence of diabetes mellitus among adult Ghanaians was high in a developing nation like Ghana, coming in at 6.46 percent [7]. They understood that the statistics might not be worrisome because many people, particularly kids, go untreated in the adult population. The majority of people in the globe, including health professionals, only have a basic awareness of diabetes and believe that it exclusively affects people who are aged. This demonstrates why there is no glucose monitoring at the hospital's pediatric center.
Though occurrences of diabetes are increasingly occurring in youngsters, diabetes is still far more common among the elderly [8]. In children under the age of 15, type 1 diabetes is the most prevalent type of the disease, and there are more than 500,000 children living with it worldwide at the moment [9]. Globally, the prevalence of Type 1 diabetes is rising, with an estimated 90,000 children receiving diagnoses per year [10]
Endocrine disrupting substances have been found in the placenta, breast milk, and blood. This implies that babies as young as a few months old or those still in the womb who have genes and immune systems that are vulnerable to these chemicals are exposed to them at a young age, increasing their risk of developing diabetes in infancy. Furthermore, children may inherit faulty insulin genes from their parents, usually in an autosomal dominant manner, however incidences of the recessive mode of inheritance have also been recorded in the Donohue syndrome and other diabetes related conditions.
A child has a 50% chance of acquiring the mutant gene if one of their parents has it. Now that there is homozygous cases of mutant alleles, there is 100% chance of one parent passing the defective trait to an offspring [11]. This explains why the range of insulin gene mutations is expanding, including instances of homozygous mutations.
Patients with recessive (homozygous) mutations exhibit lower birth rates, earlier diagnosis between 1 week and 10 weeks, and premature death as a result of the transmission of defective genes from couples to offspring with mutations at the same loci [11]. Now that children are born with recessive mutation who die prematurely, what will happen to the survival of human on earth when all children are born with the severe cases of diabetes and die prematurely? According to Hanson and his associates [12], they described diabetes as a pandemic. The rate at which the diabetes is spreading faster than the world population growth. The sickness thus poses a threat to all future generations, creating the possibility of an existential risk. This review aims to serve as a wake-up call regarding the existential risk associated with diabetes and potential cures.
2. The Different Forms of Diabetes Mellitus
Diabetes is a complex condition, which manifest in different forms [13]. They are categorised based on the age of the onset and the cause of the disease. Seven different types of diabetes have gained attention by researchers lately. Aside the 7, other rare forms of diabetes exist whilst many are yet to be discovered. By the scope of this review, the 7 are gestational diabetes, diabetes type 1, type 2, [4,14], maturity-onset diabetes of the youth (MODY), Wolfram syndrome, neonatal diabetes, and latent autoimmune diabetes in adult [14]. Apart from these 7 types, there are other forms of diabetes that are also significant. They are barely caused by underlying illnesses as well as environmental factors. This type of diabetes is said to affect 2% of the population. They include type 3c, steroid-induced and cystic fibrosis various types of diabetes [14]. According to Tuomi and his associates (13), type 1, type 2, and LADA diabetes all have more than 60 susceptibility loci (polygenic), whereas MODY, Neonatal Diabetes Mellitus, and Wolfram syndrome are monogenic. The rampancy of monogenic diabetes is lower than the polygenic ones. However, monogenic forms are equally significant and are accounted for just 1% to 2% of cases overall [15].
Type 1 diabetes mellitus which is also called insulin-dependent diabetes mellitus is caused by a complete insulin deficiency as a result of loss of pancreatic β –cells [16]. Type 1 diabetes is solely characterized by autoimmunity and ketoacidosis which is clinically tested by the presence of antibodies and ketones in both older and young people whilst type 2 diabetes is known distinctively by its metabolic syndrome [13]. Metabolic syndrome describes a number of disorders that include glucose intolerance, hypertension, dyslipidemia and obesity [17]. In a genetically predisposed person, type 1 diabetes is linked to an autoimmune response against pancreatic beta cells. T-cells play a role in the autoimmune destruction of pancreatic beta cells [16]. Type 2 diabetes develops due to insulin defects and insulin resistance. Insulin resistance is mainly caused by an impairment in the insulin receptor, leading to Leprechaunism (Donohue syndrome) and Rabson–Mendenhall syndrome. They are very fatal and responsible for early death in diabetic patients [18]. With type 2, autoantibody and ketoacidosis is not detectable and that is the bottomline between type 1 and type 2. Type 1 and 2 can also be distinguished by an absolute loss of beta cell and a partial loss of mass of beta cell respectively. It was believed that type 1 diabetes was the only heterogenous diseases that occur in children but it is recently revealed than type 2 also occurs in children [13]. Type 1 diabetes occurs in children younger than 10 years whiles type 2 occurs after 10 years, usually after the onset of puberty. There is another subgroup within type 2 called latent autoimmune diabetes in the youth. This subgroup of diabetes is caused by autoimmunity as in type 1 but occurs in adolescence between the ages of 10 and 17. It is just an analogue form of latent autoimmunity diabetes in adults (LADA), a subgroup labelled diabetes 1/½ or autoimmune diabetes in an adult who are clinically diagnosed with type 2 diabetes yet carry antibodies [13,19].
Adolescents who have a family history that supports autosomal dominant inheritance of non-insulin-dependent diabetes with onset in the second or third decade of life are considered to have maturity-onset diabetes in youth (MODY) [14]. MODY is a kind of monogenic diabetes that does not occur during pregnancy and is inherited in an autosomal dominant manner. MODY is non-ketotic [20]. There are genetic subtypes of MODY which are denoted MODY 1, MODY 2 etc based on the type of monogenic mutation. Some of the monogenic mutations occur on mitochondrion, which allows for maternal inheritance of their variants, often associated with a particular syndrome such as deafness [21,22]. MODY is not caused by obesity or insulin resistance. Due to anomalies in the regulation of beta cell mass or function, hyperglycemia results even in the absence of insulin resistance [22]. Young individuals are prone to MODY. Unlike any other diabetes, MODY and type 2 is non-insulin-dependent diabetes mainly characterized either by a defective control of insulin secretion or a low mass of the beta cell. A decrease in the mass of beta cells produces normal insulin but their level of production and its response may not be enough to meet the high glucose level. The major difference between type 2 and MODY is that MODY involves only a single gene that is responsible for the defect whilst type 2 diabetes involves many defective genes.
The term "gestational diabetes mellitus" refers to glucose intolerance that begins or is first noticed during pregnancy [22]. It is characterized by insufficient pancreatic beta cell activity to supply the body with the insulin it requires. The range of causes for hyperglycemia in general, including autoimmune disorders, monogenic causes, and insulin resistance, are thought to contribute to the beta cell dysfunction, according to the evidence that is now available.
It is unknown if pregnancy can start or speed up islet-directed autoimmunity. Wolfram syndrome, also known as DIDMOAD syndrome and caused by recessive mutations in WFS1, is characterized by diabetes insipidus, diabetes mellitus, optic atrophy, and deafness [14]. Endoplasmic reticulum stress has been demonstrated to happen due to the WFS1 mutation (23). Diabetes that first manifests before the age of six months is referred to as neonatal diabetes mellitus (NDM). Most frequently, it is monogenic. NDM is frequently brought on by mutations in the INS gene, ABCC8, KCNJ11, and other genes [20].
3. The Anthropogenic Lifestyles Connected to Diabetes Mellitus
Persistent eating expecially high glycemic (GI) meals, keeps the blood glucose high all the time and reduces the insulin mRNA thereby predisposing a person to diabetes [24,25,26,27,28]. Eating too many high GI foods at regular intervals is enough to cause diabetes if there is little or no involvement in high energy demand activities after meals. Obesity has been linked with hyperglycemia as the condition is reported to accumulate fat in the skeletal muscles and liver. Accumulation of fat in these organs especially in the liver causes mutation in the insulin signalling pathway and affect β cell compensation, thereby resulting in insulin resistance [13,29]. This is a result of the abnormality in adipocyte cells that are influenced by the activation of P13K. However, there are instances children and lean patients contract this insulin resistance disorder due to a single gene defect [30]. Preproinsulin translation has been hypothesized to gradually reduce with aging, which may be related to the age-associated rise in type 2 diabetes incidence [31,32]. However, age here is not the cause but the lifestyle put up across ones entire life raises the risks of getting diabetes. Other forms of diabetes may be brought on by surgery, or hormonal imbalances of some drugs, including steroids and antipsychotics [31].
It has been suggested that a number of chemicals, dietary elements, and viral infections may act as environmental triggers for type 1 diabetes [33]. The frequently used compounds may have a significant impact on key elements governing glucose metabolism.
Endocrine disrupting chemicals (EDC) include a wide variety of chemicals including bisphenol A, dioxins, polychlorinated bisphenyls (PCB), organochlorine pesticides, hexachlorobenzene (HCB), β-HCB, dichlorodiphenyldichloroethylene (DDE), furans, phthalates [17,34] etc. They are industrial chemicals and products of burning waste. These chemicals are widespread and are found everywhere in the environment, making exposure continuous for humans and wildlife. These EDCs are used in making pesticides, pulp and paper bleaching, dielectric fluids in capacitors and transformers, plastics, epoxy resins, detergents, thermal paper, can foods, perfumes and other toiletries [17,33,34]. These materials have become inseparable part of humans. Long-term exposure to Bisphenol A (BPA) is known to impair pancreatic islet morphology and beta cell function and there is evidence of BPA association with type 2 diabetes [17,35]. Research has also discovered the ability of BPA in accelerating spontaneous insulitis and diabetes development [36]. BPA is one of the chemicals largely produced worldwide and used as additives for plastic, metal cans linings, resin linings, and reinforced water pipes [37,38]. BPA leaks into food and water and has been detected in human urine, serum, blood, sweat and milk [36,39]. Unlike BPA, due to their lipophilic nature, certain EDC are remarkably resistant to breakdown and are retained in adipose tissue. These EDCs are dioxins, polychlorinated bisphenyls (PCB), organochlorine pesticides and dichlorodiphenyldichloroethylene (DDE) and they wallow for years in human adipose tissue [17]. Through the ingestion of meals high in fat, humans are exposed to EDC. EDCs attach to many targets, including cellular receptors, to either mimic or disrupt hormonal responses. Many of them mimic the effects of oestrogens in beta cells and insulin-sensitive tissues, leading to a metabolic state resembling pregnancy marked by insulin resistance and hyperinsulinemia [17].
Another commonly used EDC is phthalates, a plasticizer used to make plastic products more flexible, are linked to a high prevalence of diabetes in the elderly. Numerous phthalate metabolites are connected to the occurrence of diabetes [34]. Phthalates may make about 40–50% of the weight of plastic [40]. Phthalate is also a component of personal care goods including cosmetic and medicinal additives. They can easily leach from the plastics because they are not covalently attached to them, which allows them to transfer to food [40]. The use of phthalates is linked to a number of health issues, including an increased risk for other unfavorable reproductive development, asthma, obesity, allergies and atherosclerosis are associated with diabetes. Humans are exposed to phthalates by inhalation, ingestion, and skin contact [41].
Thus, they serve as inhibitors that can bind to insulin receptors, disallowing insulin from its receptor, causing hyperinsulinemia and eventually resulting in hyperglycemia. These are all risk factors in the etiology of type 2 diabetes and other diseases related to insulin resistance. Ruiz et al.[42] in 2017 discovered in their review that the diabetes disparities among people which are believed to have a relation with race, ethnicity and socio-economic differences may not be true upon concrete findings and thorough analysis but it is a result of unequal exposure to diabetogenic endocrine disruptors.
High animal protein consumption has been linked with the prevalence of diabetes, whereas plant-based protein is relatively low in the risk. When plant-based protein is consumed in the appropriate amounts, they offer significant protection [8]. This could be explained by the widely used endocrine disrupting chemicals in the environment with animals consuming a lot of them. By this, animal proteins may be contaminated with the EDC metabolites harboured in the animal meat and poses humans at risk of diabetes when their meat are eaten. However, protein in itself, be it either from plants or animals may not necessarily be involved in the pathogenesis of diabetes.
Increased dietary intake of selenium above the recommended dietary allowance of 55µg/day has been found to increase the risk of the pathogenesis of type 2 diabetes [43]. Selenite mimics insulin activity in experimental models [44] and may bind the insulin receptors, hence causing hyperinsulinemia. This may be why the high intake of selenium diet may increase the risk of developing diabetes. Due to the fluctuation in selenium content, selenium differs significantly between nations and areas in the soil which ends up in plant foods and animal forages. In many Western countries, selenium is used widely to enrich foods, fertilizers and supplements owing to the perception that it can lower the risk of cancer and other chronic diseases. It was preceded by notion that, selenoproteins are rich in antioxidant [45]. They failed to realise that the two, selenoproteins and selenium are chemically different and may be involved in different biochemical pathway in the human body. Early life exposure of selenium is likely to cause type 1 diabetes as well [33].
Studies have shown that excessive intake of salt is associated with glucose regulation and insulin levels as salt play a major role in regulating blood pressure [46] which means too much salt will aggravate the progression of diabetes in people with hypertensive diseases. Asiwe et al.[47] in 2021 also discovered that high salt intake distorts the histomorphology of the pancreas and renal tissues, confirming the association of high salt intake with diabetes.
Excessive intake of caffeine has the tendency of reducing insulin sensitivity [48] which explains the rise in glucose levels after taking an energy drink. Energy drinks and coffee are products that contain caffeine. Energy drink contains high doses of caffeine coupled with the incorporation of the high glycemic index glucose and sucrose which raises consumers' risks of obesity, eventually leading to diabetes [49].
4. Genetic Role in Diabetes Mellitus
A two-chain structure with two disulfide bonds between the B and A chains and one internal disulfide bond within the A chain is necessary for mature, maximally bioactive insulin [31]. Insulin formation in response to glycemic control always follows a simple cycle, beginning with the transcription and translation of the insulin gene into preproinsulin. Then the preproinsulin undergoes translocation into the endoplasmic reticulum (ER) which gets processed and converted into proinsulin. Proinsulin then undergoes folding and is exported from the endoplasmic reticulum, delivered through the golgi complex to the secretory granules for proinsulin conversion to insulin. The creation and maintenance of the total insulin granule pool require all of these processes, and flaws in any of these processes may, weakly or strongly, disrupt glycemic control [50].
The chromosome 6 with the major histocompatibility complex, including the DR3-DR2 and DR4-DQ8 alleles increases one’s risk of getting type 1 diabetes whiles the DR2-DQ6 allele protects an individual from getting type 1 diabetes. The role of the genetic component of diabetes has increased the risk of relatives who have diabetes [16].
Diabetes mellitus is mainly a result of single-point mutations in insulin genes resulting in amino acid substitutions within either the preproinsulin, proinsulin or the insulin molecule itself [51]. The replacements occur at different sites within the insulin molecule. There are five possible stages in endogeneous insulin processing and production at which these single mutations could occur [52]. The transcription and translation of the insulin gene, the targeting and translocation of preproinsulin into the ER, the folding of proinsulin in the ER, the trafficking and processing of proinsulin are all possible effects of insulin gene mutations [50]. Liu et al. [50] in 2021 reviewed extensively human diabetes-related genes such as NEUROD1, MAFA, PDX1 that are associated with INS gene transcription. Human diabetes-related genes such as CDKAL1, EIF2B1, and SSR1 are also linked to preproinsulin translation and/or translocation. Other genes such as GCK, SLC2A2, ABCC8, CAMKK2 are associated with insulin granule packaging, processing and secretion. All these genes must be considered and explored for diagnosis and treatment of diabetes. Mutations in any of these genes can cause diabetes. Specific mutations in KCNJ11 or ABCC8 are discovered to cause either MODY or neonatal diabetes [20] depending on the time of start of the disease. All these are feasible ways of contracting diabetes mellitus. This means the insulin gene is not solely responsible for the etiology of diabetes.
Heterozygous individuals with diabetes mellitus co-express both healthy and unhealthy molecules. The genetic disorder can be brought on by one copy of a faulty gene from one parent. There is a relatively late onset of diabetes as compared to the homozygous or recessive mutations. This is a result of the coexistence of the normal allele with the defective allele, which is sufficient to produce insulin to maintain normoglycemia. A child has a 50% chance of acquiring the mutant gene if one of their parents has it. This explains why the range of insulin gene mutations is expanding, including instances of homozygous mutations.
Recessive (homozygous) mutations due to the passage of defective genes from couples to offsprings with mutations at the same loci lead to patients showing lower birth rate, earlier diagnosis between 1 week to 10 weeks and premature death [11].
One perturbing issue in the role of diabetes mellitus is factors that control the level of insulin mRNA and stability [53,54]. These factors include transcriptional networks involved in β cell development, differentiation, and dedifferentiation [52,55]. Additionally, Kir6.2-V59M, a mutant form of the ATP channel, hinders the release of insulin from beta cells, resulting in hyperglycemia [56].
Transposable elements (TE) are also responsible for the molecular etiology of several diseases including diabetes [57]. Both null and hypomorphic mutations were effectively produced using Sleeping Beauty (SB) transposon mutagenesis and they could be responsible for diabetes [58]. Transposons naturally set into genes and cause mutations that may result in diabetes. Despite the fact that total insulin deficiency is clinically fatal, impaired insulin action has become a major medical front in the early twenty-first century due to the rising prevalence of obesity-related "insulin resistance" and the wide range of pandemic diseases, such as type 2 diabetes [12].
4.1. Genetic Anomalies to Transcriptional and Translational Mechanisms of Preproinsulin and Preprotein of Insulin-Receptor
A frameshifted protein is produced when a mutation in the INS gene's intron 2 results in the creation of a preferential splice acceptor site, terminating the typical preproinsulin sequence in the same location as in INS-IGF2, and replacing the remaining C-peptide and A chain with a novel peptide sequence [59]. A typical splicing of the INS mRNA may also result in the development of the INS-IGF2 chimaera [60].
Cdkal1 is a tRNA methylthiotransferase necessary for the accuracy of reading AAA and AAG, and its deficit can lead to the misreading of Lys codons. Its abnormality has been associated to abnormal preproinsulin translation [61]. It has long been understood that Sel1L expression is necessary for the islet cells to secrete insulin in response to glucose [62]. Loss of Sel1L in cells seems to cause an increase in TGF- signaling, which is accompanied by a dedifferentiated cell phenotype [63]. Additionally, Cdkal1 deficiency can cause ER stress and have a negative impact on the proinsulin translation product's quality [64].
Mutations affect the biological potency of the insulin itself and are impaired in their receptor-binding affinities [65,66,67]. Mutations to the cysteine residue or the addition of a new cysteine tend to disrupt the normal disulfide bonding and conformation, ultimately causing the accumulation of misfolded proinsulin, endoplasmic reticulum stress, and pancreatic-cell death [1,68]. The C96Y [69,70], C95S [71] and C43G [72] are the likely mutations that affect cysteine residue and cause permanent neonatal diabetes.
Furthermore, translational initiation at an incorrect AUG start site may result in either the yield of preproinsulin with an N-terminally shortened signal peptide or a novel defective ribosomal initiation product which has absolutely no sequence similarity to preproinsulin. This may activate the production of neoantigens, recognized by the immune system and cause an autoimmune reaction to destroy beta cells for the pathogenesis of diabetes type 1 [73,74].
The most prevalent kind of monogenic diabetes, maturity onset diabetes of the young 3 (MODY3), is caused by HNF1A gene mutations. The mutation lowers the transcriptional activity of preproinsulin synthesis [75,76]. Reduced transcriptional activity results from the H126D mutation in the DNA-binding domain of HNF1A, which prevents target DNA complexes from forming hydrogen bonds with the HNF1A protein due to expected changes in POUS-POUH domain interactions [77]. It is distinguished by deficiencies in insulin secretion and decreased GLUT2 expression, which is connected to impaired glucose absorption and ATP synthesis in the MODY. It is challenging to reach the pancreatic cell for prognostic research since MODY3 patients are known to have progressive pancreatic cell failure and eventual loss of it [78].
In some cases, the insulin gene may be normally processed and have no defect in its function to metabolize glucose, however, the problem may stem from the insulin receptor gene (INSR). A study by Ardon et al. [79] showed that, there are 22 exons in this gene, which is mapped to the short arm of chromosome 19. Inherited insulin-resistant metabolic syndromes such as Leprechanism and Rabson-Mendenhall syndrome led to type 2 diabetes, which is brought on by mutations in the insulin receptor gene.
4.2. Genetic Anomalies to Proinsulin Processing
Before preproinsulin gets processed and converted into proinsulin, it must be translocated into the ER. In some cases, the ER encounters stress. That is, it carries enough preprotein (preproinsulin) to the extent that, it fails to allow the entry of newly formed preprotein. This mechanism initiates the preemptive quality control and the ER Associated Degradation (ERAD) mechanism to degrade the newly formed preproinsulin. When cells are under the ER stress of type 1 diabetes (T1D), type 2 diabetes (T2D), or certain kinds of monogenic diabetes, poor preproinsulin translocation can contribute to cell failure [80,81,82]. Proinsulin misfolding in the ER is a result of 30 out of 51 insulin gene mutations, or more than half of all insulin gene mutation [83].
Hyperproinsulinemia is caused by an amino acid substitution during the production of proinsulin. Proinsulin Providence (H34D), proinsulin Tokyo (R89H), proinsulin Kyoto (R89L), and proinsulin Oxford (R89P) are the four kinds that have been identified. There are three processing site mutations among these [84]. The R89 mutations affect how des-31,32 split proinsulin is processed, which determines how PC2 splits proinsulin into insulin and C-peptide. As a result, these individuals have significant levels of des-31,32 split proinsulin in their blood, which only allows PC1/3 to cleave the -chain-C-peptide junction. The H34D insulin gene mutation involves yet another pathway in the development of hyperproinsulinemia. The constitutive route, where no processing takes place, is where H34D proinsulin is released [85]. When compared to normal insulin, H34D insulin has receptor binding that is about five times stronger [86]. Hypoglycemia could develop from this circumstance.
The preproinsulin that successfully and efficiently gets translocated into the ER gets processed and converted into proinsulin. However, not all the proinsulin is properly folded into the biologically active insulin molecule. In the absence of iron-responsive element binding protein 2 (IRP2), which is necessary for Cdkal1 to continue functioning, proinsulin is susceptible to misfolding. Its absence is known to cause Lys codon misreading, which results in compromised proinsulin folding and reduced insulin production [87]. Thus, the secretory route is generally dysfunctional since the misfolded proinsulin is degraded into proteotoxic forms, and any natural selection of the proteotoxic forms damages cell mass in addition to impairing insulin synthesis [88,89]. Sometimes the creation of abnormal interactions between the wild-type (WT) and mutant alleles may lead to the misfolding [90,91,92]. Long before decreasing cell mass, these aberrant interactions seem to be the first step in the development of insulin-deficient diabetes [93,94,95]. The proinsulin's natural folding is affected by the ineffective signal peptide cleavage caused by the mutation A24D at the position of the signal peptide's cleavage [83].
Figure 1.
The various stages of getting a matured insulin.
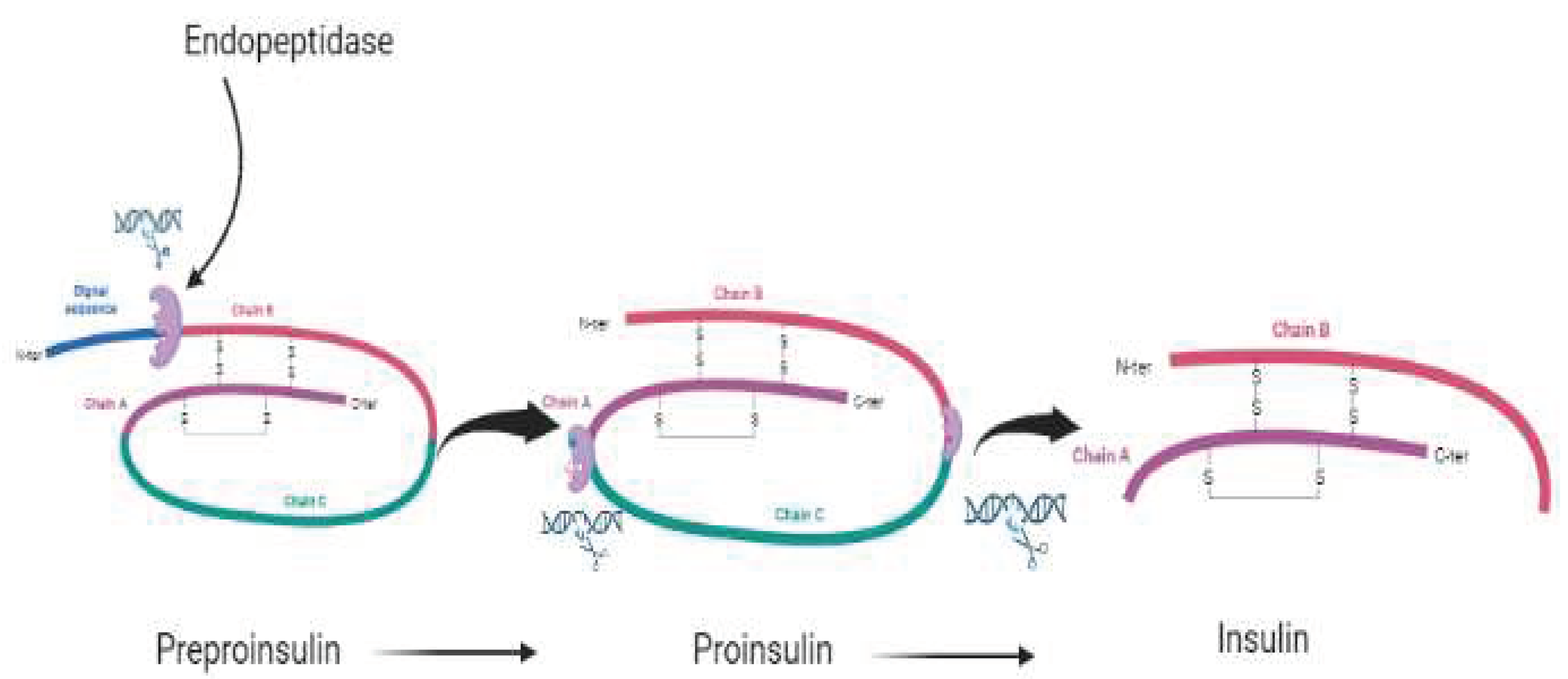
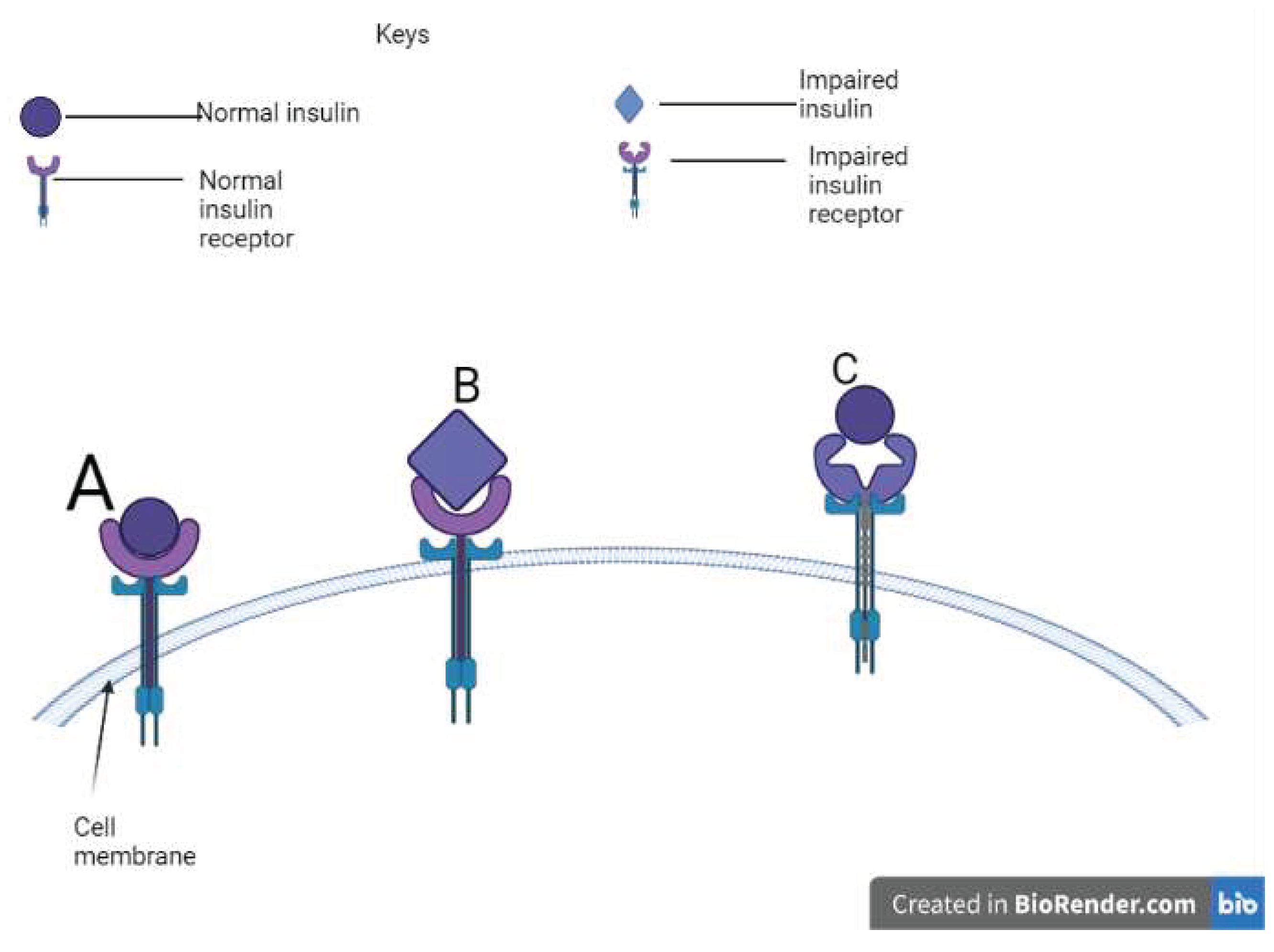
Figure 2.
A schematic view of insulin and receptor binding potency.
5. The Severity of the Disease
Mutations vary with the severity of the disease ranging from asymptomatic to very lethal stage, thus some mutations show no symptoms others show mild symptoms and some have dangerous health repercussions and lead to death. The fact that some mutations may not carry symptoms is not a guarantee of a healthy life throughout. Consequences may appear in later old age. A mutated human INS allele are heterozygote and recessive, supporting the idea that blood glucose levels can typically be kept within the normal range with just one functioning insulin allele, thus several mutations of the INS allele may be asymptomatic [8,11,96,97]. According to Raile et al. [97], the interaction between mutant and wild-type proinsulin alters the co-expressed wild-type proinsulin's ability to fold normally in the ER, which increases the chance of acquiring diabetes at maturity [91,98]. The intermolecular thiol assault, in which mutant proinsulin cysteine residues create intermolecular disulfide bonds with wild-type proinsulin, appears to be one of these aberrant interactions [99]. The mutant and wild-type proinsulin abnormal protein complexes keep the wild-type partner in the ER. Proinsulin is therefore continually produced by cells in normal levels [88,93]. The inability to export wild-type proinsulin from the ER is one mechanism by which the loss of a functioning insulin allele predisposes to decreased insulin production, which is a key determinant in the development of insulin-deficient diabetes [91,93].
Some mutations that show glucose intolerance or syndrome of mild diabetes are as a result of the substitution of B25-Phe with Leu (Chicago), A3-Val with Leu (Los Angeles) or B24- Phe with Ser (Wakayama). The four proinsulin mutations which are proinsulin Providence (H34D), proinsulin Tokyo (R89H), proinsulin Kyoto (R89L), and proinsulin Oxford (R89P) also show mild symptoms [100,101]. Such individuals with those mutations are diagnosed with the synthesis of biologically impaired insulin in their receptor binding potency due to changes of A or B chains. This abnormal insulin is secreted at higher levels and results in a condition known as hyperinsulinemia [67]. Providence (H34D) also show only glucose intolerance or mild diabetes as a result of hyperproinsulinemia since it enters the constitutive pathway where the processing does not exist [84]. The Providence (H34D) may show no symptoms as proinsulin has approximately fivefold enhanced receptor binding compared with normal insulin, thus exhibiting a strong bonding between the proinsulin and the receptor [85,86]. This situation may subject a person to hypoglycemia due to the abnormal strong affinity of insulin and its receptors, depleting blood glucose in lesser time than usual.
One kind of those mutations that yield no symptoms is a point mutation affecting proinsulin, which leads to a replacement of Arg-65 by His. The mutation inhibits the 3-cell processing protease from recognizing the C-peptide-A-chain dibasic cleavage site, which causes a type II proinsulin intermediate form known as des 64 and 65 HPI to circulate in the body. The replacement of B10-His with Asp results in proinsulin, which displays a negative change in the subcellular sorting behaviour, and is another mutation with no symptoms. A proportion of 15% of the newly produced Asp-10 proinsulin is released via an uncontrolled or constitutive secretory route in an unprocessed form and these two mutations result in a condition called hyperproinsulinemia due to a large build-up of the impaired proinsulin in the circulation [102]. However, the WT allele is enough to make insulin to stabilize the body’s glucose level and prevent the mutated proinsulin from progressing into diabetes symptoms.
By default, proinsulin enters the regulated secretory pathway so that it would undergo processing and transformation into insulin. It tends to cause problems when proinsulin enters the non-regulated constitutive pathway, which rapidly secretes the proinsulin unconverted and moves to the surfaces of cells within 60-90 min for insulin metabolism via receptor-mediated endocytosis [103]. Diabetes results from an excessive amount of proinsulin being secreted in an uncontrolled manner because it inhibits the release of regularly processed insulin through the granule pathway. This is where any culprits of this situation responding to no symptoms begin to show symptoms of diabetes. Thus, there is always a progression of the disease from mild to severe symptoms.
Mutations along the preproinsulin sequence are normally detrimental when they affect both alleles of the gene. It affects the biological potency of the insulin itself and is impaired in its receptor-binding affinities [65,66,67]. Its signs and symptoms are quick and very fatal. Particularly those changes at the cysteine residue or adding a new cysteine likely to affect the appropriate disulfide bonding and conformation, which ultimately causes an accumulation of misfolded proinsulin, endoplasmic reticulum stress, and pancreatic-cell death [1,68]. They are the C96Y, C95S, and C43G mutation. The C96S causes hypoinsulinemic hyperglycemia as early as 1 month. When mutant insulin molecules were observed under electron microscopy, they were improperly folded and decreased in number and size, with an expanded ER and inflated mitochondria [71]. The C43G mutation causes very severe diabetes at 43 weeks of age and is extremely deadly [72].
6. The Existential Risk Potential of Diabetes
The ever-increasing cases of diabetes recorded worldwide by the World Health Organization (WHO) are raising an alarm within our century and the projections put the future at great risk. According to estimates, 4 million deaths from diabetes were reported globally in 2017. Diabetes is one of the top 10 causes of death for people. Globally, 150 million persons had diabetes in 2002. In 2016, the number increased to 422 million adults only. The number has gone up to 563 in 2019 [4]. By 2030, the number of deaths that may be directly attributed to human activity is predicted to reach over 600 million [3]. By 2030 and 2045, respectively, it is predicted that there would be 578 million people and 700 million people [104].
Although only adults are included in these statistics, many people in the adult population go undetected, which further reduces the serious nature of the numbers [7]. Though children are not regarded for diabetes but research has shown how the diseases affect them. Not only adults, but children are also subject to diabetes. With all this consideration, the disease prevalence rate worldwide is expected to be greater than what is stated if everyone including those seen as normal is involved in the diagnosis since the disease could stay with a normal person for a while before signs and symptoms manifest. Diabetes was the ninth leading cause of death globally in 2019 and a significant factor in kidney failure, heart attacks, strokes, lower limb amputation, blindness, and other conditions [4].
Figure 3 can compare two decades from 1980-2000 (late 20th century) to 2000-2021 (early 21st century). Most synthetic products like plastics hit the world in the late 20th century and their production augmented from 60 million tonnes in 1980 to 368 million tonnes in 2019 [105]. The cases of diabetes have augmented by about 6-folds in the last four decades which seem to have a direct correlation with the rate of plastic production within the same timeframe with a similar 6-fold increase. Meanwhile, the world population has only doubled from 1980 to date [106]. This means diabetes is spreading faster than the growing population. The graph depicts how an anthropogenic lifestyle within the environment can pose humans with a higher risk of getting diabetes. A steady rise can be seen in the late 20th century whilst the 21st century is marked by a sharp rise, depicting a direct link with the over-reliance of synthetic materials such as plastics. This has accounted for the reason for reporting large numbers of diabetes in our time.
This is due to the fact that these synthetic materials harbours endocrine disrupting chemicals (EDC) such as BPA and they have been detected in blood, placenta and breast milk and are known to cause diabetes [36,39]. Children are very vulnerable to these chemicals. Exposing children to these chemicals at an early age can result in the onset of diabetes. Though the prevalence of children with diabetes is relatively low compared with the adult. Their cases are rising at a greater pace. These environmental factors from early EDC exposure can affect genes responsible for insulin production, hence altering or causing lifetime mutation and consequently into diabetes.
Moreover, children could be born with defective insulin and their related genes, inherited from parents in an autosomal dominant fashion. The onset of diabetes varies according to the type of mutation, from early months to years in adulthood. When a father with the same C43G mutation was 30, he was diagnosed with mild type 2 diabetes; however, when his son, who inherited the same gene, was 43 weeks old, he had very severe type 1 diabetes [72]. This demonstrates that environmental influences, rather not just only genetics, have a significant impact in how diabetes manifests. Now that children are inheriting diabetogenic mutations from parents, what happens to the survival of human on earth when all children are born with the severe cases of diabetes and die prematurely?
Thus, our discriminated use and over-reliance on synthetic and some dangerous natural chemicals not only cause diabetes in adults but in children and unborn generations. This has qualified diabetes for having existential risk characteristics with a faster rate of spread of the disease as compared to thess rate of population growth. Diabetes in our era is a silent pandemic that needs attention.
7. The Disease Burdens on Diabetic Patients
Many years before you are given a diabetes diagnosis, high blood sugar and high insulin levels begin to harm your body. Due to diabetes's impact on immune function and blood flow, a minor wound could not receive the care it requires to heal. Doctors may have to amputate a foot or other damaged regions in severe situations. Similar to how it harms the heart, it causes kidney blood vessels to thicken and lose their ability to filter blood [14]. As a result, the body struggles to eliminate waste and water, which causes it to retain additional fluid and toxins in the blood as well as lose vital proteins in the urine. The kidney could stop functioning. The immune system is impacted by diabetes because high blood glucose levels reduce the immunological activity of the white blood cells. Due to this, one is more prone to experience major flu complications that could necessitate hospitalization. High blood sugar can speed tooth decay and make treating gum infections more difficult. Dysbiosis can be brought on by diabetes and obesity, which disturb the balance of good and bad gut flora [107]. This is where some particular microbes are proliferated in larger numbers and get involved in the metabolic processes. This may result in the excessive release of glucose in the blood, thus worsening the diabetes condition and causing gut inflammation in the end through their activities of breaking down complex food materials to release energy.
The long-term detrimental complications include neuropathy, renal failure, cardiovascular diseases and retinopathy such as atherosclerotic vascular diseases, stroke, and heart attack. These are all related to hyperglycemia and are the most common causes of death. Kinsley [108] found 64 of 68 patients suffering from wolfram syndrome diagnosed with optic atrophy. Other symptoms such as neurological dysfunction, characterized by neurosensory hearing loss, seizures, and ataxia were also very common among wolfram syndromic patients. Partial or complete diabetes insipidus, impaired urinary tract disease characterised by dilated neurogenic bladders, impaired renal function, malignant hypertension, chronic bilateral upper urinary tract obstruction, chronic obstructive uropathy, erectile dysfuction, and autopsy evidence of mild chronic renal pyelitis. Also, cardiac anomalies are common with Wolfram syndrome, characterized by sinus tachycardia, atrial and ventricular arrhythmias, and tetralogy of falot and pulmonic valve stenosis [109,110].
8. The Clinical Manifestation of Diabetes Mellitus and Its Related Diseases
One of the most accurate clinical indicators of diabetes-related illnesses brought on by insulin resistance is acanthosis nigricans (AN). The skin in the axillae, nuchal region, groin, and other flexural areas develops a black, dark spot that is characteristic of the condition. Skin tags are frequently linked to it. As β-cells compensate, AN increasingly manifests [79]. Other symptoms such as hyperandrogenism, usually with oligomenorrhea or amenorrhoea are also very common in females with insulin resistance diabetes [111].
Donohue syndrome is the most severe diabetogenic disease. For the first year or so of life, there is no ketoacidosis even in the absence of insulin receptor function, only fasting hypoglycemia with postprandial hyperglycemia and very significantly raised plasma insulin. The condition progresses to a later stage that is marked by linear growth retardation with overgrowth of soft tissues, giving the face a coarse look, severe AN, hypertrichosis, and organomegaly, frequently with nephrocalcinosis [79]. The slightly less severe Rabson-Mendenhall syndrome frequently manifests as ketoacidosis, recurrent infections, pineal hyperplasia, and ovarian tumor [112]. It was first observed in three siblings who also had acanthosis nigricans, prognathism, thick fingernails, abdominal distension, penile enlargement, early dentition, coarse senile-looking facies, striking hirsutism, and dental and skin abnormalities. Children experience initial postprandial hyperglycemia and fasting hypoglycemia as a result of improperly raised fasting insulin levels [18,113]. Rabson-Mendenhall syndrome patients can live past the age of one and eventually develop chronic hyperglycemia, which is followed by diabetic ketoacidosis and death. This is accompanied by a gradual drop in insulin levels, which are insufficient to stop the production of liver glucose and the release of fatty acids by adipocytes [18].
The risk of developing type 1 diabetes is increased by the presence of antibodies in the serum of patients that target particular proteins like insulin, glutamic acid decarboxylase, insulinoma-associated protein-2, and a protein tyrosine phosphatase. These antibodies can be found months to two years prior to the onset of diabetes [16]. They are thought to harm the pancreatic beta cells necessary for producing insulin. Before glycemic control is compromised, more than 80% beta cells must be lost.
When blood glucose levels exceed 180 mg/dL (10.0 mmol/L), glucosuria develops, which causes osmotic diuresis and polyuria with a fruity odor because glucose is lost through urine. Polyuria is a symptom brought on by polydipsia, which makes one drink more water. The result of patients' frequent urine and bedwetting is polyuria. Further insulin shortage leads to an increase in protein oxidation and lipolysis from fat cells.
At this state, a diabetic patient begins to lose weight and profound insulin deficiency drives ketoacidosis to occur, leading to the accumulation of ketoacids [16]. which can be detected in urine. A fasting glucose level of 126 mg/dL (7.0 mmol/L) or more is also a sign of having diabetes.
9. Management of the Disease by Medications and Other Lifestyle
Diabetes has no direct medication for treatment. However, the symptoms are taken care of by already existing drugs which is administered together with artificially made insulin. Insulin secretory capacity and some of the rise of INS mRNA levels are preserved in interventions to control obesity and diabetes using diet or drugs that promote cell rest [114,115]. the early use of metformin at the greatest permissible dose, dietary modification, and the administration of sufficient amounts of insulin, often using concentrated formulations remain the mainstays of current treatment for milder receptoropathies [116].
Oral contraceptives may be used to inhibit gonadotrophins when treating hyperandrogenism. They are occasionally used with anti-androgens to achieve this. Use of potent GnRH agonists with hormone replacement therapy is a more extreme method that can regulate androgens even in the face of the most severe IR-related disease. This is a type of "block and replace" therapy that has been discovered to be successful in a small number of patients. Recombinant insulin analogues, insulin pumps, and more recent home monitoring gadgets have significantly improved diabetic individuals' capacity to regulate high blood glucose levels. Current diabetes therapies cannot make a full metabolic normalization possible. Mutations of KCNJ11 or ABCC8 which cause MODY or neonatal diabetes can be controlled by the usage of oral hypoglycemic drugs known as sulfonylurea which when binds to the sulfonylurea receptor, induce endogenous insulin production [16]. Sulfonylureas are a group of oral drugs that lower blood sugar levels in people with type 2 diabetes or MODY2 by boosting the pancreas to produce more insulin. Beta cells lost to immunological destruction can be replaced via pancreas transplantation or intraportal injection of isolated islets. Due to the need for immunosuppressions to avoid alloimmune rejection of the graft, the risk of transplantation is still too great for most patients to consider it as a treatment option.
Regular blood tests for ketones and blood sugar are recommended (at least every 3 to 4 hours). Excessive liquids should be administered to help patients stay hydrated, which also aids in the excretion of extra glucose and ketoacids. Glucagon must be available to treat hypoglycaemia. Physical activities help manage diabetes by lowering glucose levels. Being active makes the body more sensitive to insulin. Cells can take up glucose for energy when muscles contract during exercise and one must strive for at least 150 minutes of moderate to intense exercise per week [117].
Members of nuclear peroxisome proliferator-activated receptors (PPARs) are known to bind to phthalates, which aids in adipose tissue and lipid homeostasis as phthalate levels have been linked to obesity [34]. Pharmaceutical substances like fibrates and glitazones, which are known to affect fat distribution and change lipid status and are commercially accessible as PPAR-agonists, serve to lower the risk of diabetes brought on by obesity [118]. Additionally, it is understood that PPAR-antagonists are used to manage type 2 diabetes and have an impact on glucose homeostasis through the reduction of insulin resistance [118,119].
9.1. Diet Based Management
Diabetic patients should eat complex carbohydrate foods that are rich in fibre [120]. Fibre does not produce glucose directly to human cells, as the human digestive system cannot metabolize it. However, friendly microorganisms in the digestive system feed on this fibre to produce fatty acids that some of the cells used to make glucose. Thus, eating fibre-rich foods will help to regulate and manage glucose levels in the blood. Foods that contain simple carbohydrates should be avoided [117,121] as their pathway for gluconeogenesis is very short. Those foods keep our blood glucose always high. Some of the complex carbohydrates are whole grain foods such as brown rich rice, parboiled rice, barley, buckwheat, bulgur, millet, oatmeal and pasta. Beans, vegetables and fruits are equally healthy choices of foods [117, 122–124]. Limit the consumption of refined whole grains such as white rice and jasmine rice as they are low in fibre content and have lost most of the nutrients [124]. They have a higher glycemic index which means, they can quickly convert into blood glucose more easily than brown rice. Avoid eating canned foods and drinks since they contain added syrups such as the EDCs. Foods should have a low amount of saturated fat and cholesterol. The glycemic index (GI) is a number that indicates how much certain foods raise blood sugar levels [125]. Foods and their glycemic index must be known and determined as one of the parameters for all processed and packaged foods. This will inform a consumer to make the right choice, especially regarding victims of diabetes. A low GI falls in the range of 1 to 55, a medium is from 56-69 and a high GI is 70 and higher [125]. Avoid eating simple carbohydrates foods (sweaters) such as the common white sugar and even honey, reduce salt intake, potatoes. Monounsaturated fats, in particular, should be substituted for saturated fats as much as possible [117,121]. They have a relatively higher glycemic index than complex carbohydrate foods. Honey and white sugar are very similar as they have 73±15 and 65±4 glycemic indexes respectively [122], thus honey is not a choice sweeter for diabetic patients. Fructose has a relatively lower GI of 28 and can be considered for diabetic patients. Thus, fructose should also be processed and sold just as sucrose (white sugar) for diabetic patients to use as their sweeter in breakfast foods. Low GI foods decrease the risk of type 2 diabetes and its complications and must be encouraged to be taken [117]. They reduce the complications of type 1 diabetes as well. Table 1 has the list of all the foods that can be considered the best choice for consumers based on the ranking from low, moderate to high.
9.2. Common Naturally Occurring Biotherapies Used in the Prevention and Management of Diabetes
Some plants such as the leaves of Ginkgo biloba and Prunus mume fruits are edible and medicinal and they contain bioactive compounds that help in alleviating the risk of diabetic patients. In China, the edible and medicinal fungi known commonly as mushroom, Ganoderma lucidum have been proven efficient in adipogenesis, lipolysis, and adipocyte being/browning. In HPA-v and 3T3-L1, the medicinal herb Ganoderma lucidum inhibits adipogenesis and promotes lipolysis [126]. In 3T3-L1 adipocytes, the concentrated extract of Prunus mume fruit inhibits adipogenesis and induces beijing, or browning [127]. Ginkpo biloba is a herb that contain an essential phytochemical called bilobalide, known to prevent diabetes [128]. According to Bu et al. [129] bilobalide was able to inhibits adipogenesis in 3T3-L1 adipocytes through the AMPK signaling pathway. All this have given scientist a rationale for creating products that lower cholesterol and promote weight reduction through the utilization of bioactive components in fungi and plants. This will offer fresh perspectives on treating and preventing metabolic illnesses like type 2 diabetes and obesity.
As it is reported that, diabetes mellitus has a link with dysbiosis in the gut, some Chinese herbal medicine has proven efficient in ameliorating diabetes whilst regulating the gut microbiota. There is a correlation analysis of the gut microbiota being an active regulator of metabolic functions [130]. Plants such as Folium mori, Fructus mori L., Ganoderma atrum, Maydis stigma, Morus nigra, Radix pseudostellariae, Momordica charantia L. and Rhizoma dioscoreae all have hypoglycemic effect to regulate blood sugar level whilst regulating intestinal microecology [107]. Most of these plant based foods and herbs contain important chemicals such as anthocyanins, reported to have hypoglycemic effects to control type 2 diabetes [131]. All including those not mentioned in this review only helps to reduce blood glucose level to lower a person’s risk of getting type 2 diabetes or regulate the blood glucose level of diabetic patient but cannot reverse the disease as diabetes impairs genes of the metabolic processes which can be best treated via gene therapy.
10. Therapeutic Molecular Interventions so Far
Researchers and scientists were able to determine the origin of diabetes mellitus and develop effective interventions via the use of high-performance liquid chromatography (HPLC). When it was first used, it was to resolve plasma insulin, which helped identify aberrant insulins [132]. The HLPC's discovery of aberrant insulins led to the development of insulin biotherapies, produced via recombinant DNA technology [133,134,135]. The human insulin gene was cloned in 1980 [102,136], which made it possible to look for naturally occurring mutations that could change how the insulin molecule operates. It became feasible to produce native human insulin via the recombinant DNA technology. Recombinant DNA technology is used to create all insulin, and it is based on the amino acid sequence of the hormone. They are insulin mimics that work quickly and are absorbed and eliminated faster than naturally occurring insulin. Preproinsulin was harvested ex-vivo, chemically cleaved and folded into insulin [137] which is used on commercial basis for both insulin and non-insulin dependent diabetic patient. However, the main disadvantage of the common commercial crystalline insulin is its inability to undertake quick glucose stimulating insulin response [102]. This has led to the production of insulin analogues, produced using similar molecular approaches. With insulin analogues, insulin are modified by combining the expression of the A and B chains or altering some of the bases in the chains of insulin to make them more biologically active via molecular biology techniques and tools [138]. Miniproinsulins have also been designed with the linking peptide region removed and replaced with a brief bridge using similar strategies to enable in-vivo or in-vitro cleavage of the synthesized protein [139]. According to reports, modifying the connections between residues in the insulin molecule can result in the creation of dimers and hexamers that are more easily soluble and trigger a fast insulin response to glucose [140]. Additionally, changes such the substitution of aspartic acid for histidine at position B10 tend to produce insulins with much higher receptor-binding affinities as determined by in vitro binding assays and even somewhat higher biological potencies in vivo. This is feasible due to their quicker elimination from the circulation by receptor-mediated endocytosis [140,141,142]. However, patients have not yet been tested for their usefulness, and nothing is known about the potential benefits or drawbacks of such potent insulin molecules. Thus, human insulin molecule has undergone several advantageous modifications to change its properties, particularly its binding capabilities, solubility, and self-association to enhance its efficiency [102]. Other modifications have been made that have negative effects because they cause insulin crystals to accumulate in tissue depots, which are absorbed considerably more slowly [140,143]. The addition of new antigenic sites to the original human insulin molecule poses a risk that it may be recognized as a foreign protein, negating one of the key benefits of employing human insulin as a therapeutic approach [144].
The ability to express healthy or defective insulin genes through DNA transfection in cultured cells, injection of in vitro-produced mRNA into Xenopus oocytes, or translation of mRNA in reticulocyte cell-free systems allows for the evaluation of their changed properties. These and other molecular biological techniques have led to the development of novel insulins for the management of both insulin and non-insulin dependent diabetes and a deeper comprehension of the mechanisms underlying the biosynthesis, intracellular sorting, processing, and secretion of insulin under both normal and abnormal circumstances [102].
Thus, we can conclude that the data on insulin mutations only scratch the surface of insulin variability. Further examination of human populations will likely reveal more frequent low-level polymorphisms brought on by point mutations as well as other more uncommon genetic alterations (such as deletions and insertions) [102]. Here, we create a clinically validated sequencing technique to find INSR gene mutations. The INSR gene's coding regions and exon-intron boundaries were examined for alterations using bidirectional sequencing with BigDye terminator and M13 primers. An accurate diagnosis of insulin receptor insufficiency can be made using a combination of biochemical and DNA studies [79]. Insulin receptor mutations is a great problem in the pathogenesis of diabetes as insulin may be produced with no impairment yet the insulin receptor cannot interact with its insulin to ensure normoglycemia. In an experiment by Wang and his associate, they showed that a single dosage of an adeno-associated virus (AAV) vector could express human IR (hIR) in the liver of inducible IR-knockout mice and considerably ameliorate the diabetes phenotype brought on by adult IR deletion in ob/ob mice. They concluded that, AAV-hIR increased hepatic insulin signaling, induced liver-tropic IR expression, and improved systemic insulin sensitivity [145].To identify the genes that, when disrupted, affect T1D pathogenesis, a complementary approach to current techniques called the Sleeping Beauty (SB) transposon mutagenesis system are applied [58].
11. The Prospective Wayforward
Strategies that rescue wild-type proinsulin from blockade may therefore potentially restore sufficient insulin production to delay/prevent the onset of insulin-deficient diabetes because one normal INS gene allele is sufficient to produce enough insulin to maintain normoglycemia [11,96]. Two different strategies could be used to free mutant proinsulin from its inhibition of wild-type proinsulin. First off, one potential therapeutic approach for the MODY syndrome involves targeting the mutant proinsulin for degradation, which would make the stoichiometry of mutant and wild-type proinsulin more favorable to the wild-type proinsulin [90,91,94]. Targeting wild-type proinsulin for better oxidative folding in the ER is the second strategy. Accelerating the oxidative folding of wild-type proinsulin may promote its folding, limiting its interactions with co-expressed mutant proinsulin, and enabling it to escape from the ER before its retention caused by aberrant interaction with the mutant gene products. This is because abnormal inter-molecular disulfide bonds between mutant and wild-type proinsulins contribute to the dominant negative effect of mutant proinsulin [11,91]. In order to induce disulfide pairing, the oxidizing environment is kept in place by the actions of the enzymes protein disulfide isomerase (PDI) and ER oxidoreducin-1 (ERO1) [146,147]. The oxidative folding of wild-type proinsulin can be improved by overexpressing (ERO1), according to studies published recently, even when it is co-expressed with mutants [90,91]. As a result, the ER export of wild-type proinsulin will rise significantly, and insulin synthesis will rise as well.
Additionally, the essential intracellular components that control the breakdown of improperly folded proinsulin are unclear, thus finding those molecules may offer potential targets to control the breakdown of misfolded mutant proinsulin and it will go a long way to preserve the wild-type proinsulin as well.
The major histocompatibility complex on chromosome 6, the DR2-DQ6 allele has been found to protect an individual from getting type 1 diabetes [16]. With recombinant technology, these genes can be cloned and fixed in the human body to be immuned against type 1 diabetes whereas DR3-DR2 and DR4-DQ8 alleles can increase one’s risk of getting type 1 diabetes and can be subjected to early screening and detection for the silencing of those genes.
There should also be an effort of education to everybody including health care workers using one-health approach to bring the awareness of people on the environmental exposures of chemicals and personal lifestyles that increases the risks of the pathogenesis of diabetes [42]. This will reduce individual and the global burdens of diabetes.
12. Conclusions
Diabetes mellitus is a heterogenous disease with the etiology involving many complex ways that all arrive at one condition of hyperglycemia, though there are few instances of hypoglycaemia. The disease has no treatment or cannot be reversed currently, though a lot of molecular approaches have been used. However, personal lifestyle, surgery and oral therapy are used to manage the condition. Personal lifestyle correlates with the pathology of diabetes, where the use of plastics with 6-fold increase over the last 4 decades has resulted in a similar 6-fold increase in the cases of diabetes worldwide. This condition has also resulted in a lot of individuals harbouring mutant insulin genes that are passed onto offspring.
The rampancy of the disease today is alarming with the disease spreading at a faster rate with a 6-fold increase as compared to the rate of population growth with only a two-fold increase. In addition, the heterozygote mutants are spreading with a 50% chance of inheritance from parent to offspring. Currently, there are cases of homozygous mutant’s forms with 100% chance of inheritance and are diagnosed in the early weeks after birth with a short lifespan of patients. This proves that diabetes has the potency of spreading throughout the whole world into the future and consequently, affects the existence of man. It is therefore wise for one-health approach to be harnessed by reaching out to everyone including health workers to educate them on an appropriate lifestyle that would be good to avoid the incidence of diabetes. Foods glycemic index should be one of the required parameters that must be considered by Food and Drugs Board Authority. Foods that are known by their GI will inform consumer to make right choices at a particular time. Doing this will lower the risk of getting diabetes through diet. Also, in the treatment of diabetes, precision medicine is the only method combined with the molecular approach to help in reversing the gene defect.
Author Contributions
KM developed the original idea, organized periodic discussions to develop and consolidate materials in this review manuscript and did most of the write-up. SMA and BT contributed to the part related to the therapeutic molecular interventions and the prospective wayforward. EKA and AS contributed to the part related to management of the disease and personal lifestyle. DA contributed the part related to the clinical manifestation of the disease and its associated burden. CKSS contributed to the part related to the common naturally occurring biotherapies used in the prevention and management of diabetes. SB reviewed the manuscripts and supervised the work. All authors read and approved the final manuscript.
Funding
This paper received no funding.
Acknowledgments
Biorender.com was used to create the figures.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
AN- Acanthosis nigricans, CEL- Carboxylesterlipase, EDC- Endocrine disrupting chemicals, GI- Glycemic index, INSR-Insulin gene receptor, MODY- maturity-onset diabetes of the youth, WFS-Wolfram syndrome.
References
- Nishi M, Nanjo K. Insulin gene mutations and diabetes. J Diabetes Investig. 2011;2(2):92–100. [CrossRef]
- Feo P, De Loreto C, Di Ranchelli A, Fatone C, Gam G, Lucidi P, Santeusanio F. Exercise and diabetes. Acta Biomed. 2006;3:14–17.
- Rowley WR, Bezold C, Arikan Y et al. Diabetes 2030: insights from yesterday, today, and future trends. Popul Heal Manag. 2017;20(1):6–12. [CrossRef]
- World Health Organization. Diabetes. Accessed on 15th of July 2022. Available at https://www.who.int/health-topics/diabetes. 2021.
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. [CrossRef]
- Azevedo M, Alla S. Diabetes in Sub-Saharan Africa: Kenya, Mali, Mozambique, Nigeria, South Africa and Zambia. Int J Diabetes Dev Ctries. 2008;28(4):101–108. [CrossRef]
- Asamoah-Boaheng M, Sarfo-kantanka O, Tuffour AB, Eghan B, Mbanya JC. Prevalence and risk factors for diabetes mellitus among adults in Ghana : a systematic review and meta-analysis. Int Health. 2018;11:83–92. [CrossRef]
- Pounis GD, Tyrovolas S, Antonopoulou M, Zeimbekis A, Anastasiou F, Bountztiouka V, Metallinos G, Gotsis E, Lioliou E, Polychronopoulos E, Lionis C, Panagiotakos D. Long-term animal-protein consumption is associated with an increased prevalence of diabetes among the elderly: the Mediterranean islands (MEDIS) study G.D. Diabetes Metab. 2010;36:484–490.
- Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Publ Gr [Internet]. 2017;3:1–18. [CrossRef]
- Diaz-Valencia PA, Bougneres P, Valleron AJ. Global epidemiology of type 1 diabetes in young adults and adults: a systematic review. BMC Public Health. 2015;15:255. [CrossRef]
- Garin I, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci U S A. 2010;107(7):3105–3110. [CrossRef]
- Hanson RL, Imperatore G, Bennett PH, Knowler W. Components of the “metabolic syndrome” and incidence of type 2 diabetes. Diabetes. 2002;51(10):3120–7.
- Tuomi T, Santoro N, Caprio S, Cai M, Weng J, Groop L. The many faces of diabetes : a disease with increasing. Lancet [Internet]. 2014;383(9922):1084–94. [CrossRef]
- Diabetes, UK. Types of diabetes. Accessed on 15th of July 2022. Available at https://www.diabetes.org.uk/diabetes-the-basics/types-of-diabetes.
- Murphy R, Ellard S, Hattersley A. Clinical implications of a molecular genetic classifi cation of monogenic β-cell diabetes. Nat Clin Pr Endocrinol Metab. 2008;4:200–213.
- Cooke DW, Plotnick L. Type 1 Diabetes Mellitus in. Pediatr Rev 2008. 2008;29:374–85.
- Alonso-Magdalena P, Quesada I, Nadal A. Endocrine disruptors in the etiology of type 2 diabetes mellitus. Nat Publ Gr [Internet]. 2011;7(6):346–53. [CrossRef]
- Longo N, Wang Y. Progressive decline in insulin levels in Rabson–Mendenhall syndrome. J Clin Endocrinol Metab. 1999;84:2623–2629.
- Dabelea D, Bell RA, D’Agostino RB Jr, et al. Incidence of diabetes in youth in the United States. JAMA. 2007;297:2716–2724.
- Naylor RN, Greeley SAW, Bell GI, Philipson LH. Genetics and pathophysiology of neonatal diabetes mellitus. J Diabetes Investig. 2011;2(3):158–69. [CrossRef]
- Leibowitz G, et al. Mitochondrial regulation of insulin production in rat pancreatic islets. Diabetologia. 2005;48(8):1549–1559. [CrossRef]
- Buchanan TA, Kjos SI, Xiang A, Watanabe R. What is gestational diabetes? Diabetes Care. 2007;30:105–11.
- Bonnycastle LL, Chines PS, Hara T, et al. Autosomal dominant diabetes arising from a Wolfram syndrome 1 mutation. Diabetes. 2013;61:1974–1977. [CrossRef]
- Talchai C, Xuan S, Lin HV, Sussel L AD. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150(6):1223–1234.
- Cinti F, et al. Evidence of β-cell dedifferentia- tion in human type 2 diabetes. J Clin Endocrinol Metab. 2016;101(3):1044–1054.
- Sun J, et al. β-Cell dedifferentiation in patients with T2D with adequate glucose control and non- diabetic chronic pancreatitis. J Clin Endocrinol Metab. 2019;104(1):83–94.
- Perreira M, et al. “Reversine” and its 2-substitut- ed adenine derivatives as potent and selective A3 adenosine receptor antagonists. J Med Chem. 2005;48(15):4910–4918.
- Ojamaa K, Hedo JA, Roberts CT, Moncada VY, Gorden P, Ullrich A, et al. Defects in Human Insulin Receptor Gene Expression. Mol Endocrinol. 1988;2(3):242–7. [CrossRef]
- Alderete TL, Toledo-Corral CM, Desai P, Weigensberg MJ, Goran M. Liver fat has a stronger association with risk factors for type 2 diabetes in African-American compared with Hispanic adolescents. J Clin Endocrinol Metab. 2013;98:3748–3754. [CrossRef]
- Challis BG, K S and R. Genetic Disorders of Insulin Action : Far More than Diabetes. Curr Obes Rep. 2013;2:293–300.
- Wang SY, Halban PA, Rowe J. Effects of aging on insulin synthesis and secretion. Differential effects on preproinsulin messenger RNA levels, proinsulin biosynthesis, and secretion of newly made and preformed insulin in the rat. J Clin Invest. 1988;81(1):176–184. [CrossRef]
- Halter JB, et al. Diabetes and cardiovascular disease in older adults: current status and future directions. Diabetes. 2014;63(8):2578–2589. [CrossRef]
- Bodin J, Stene LC, Nygaard UC. Can Exposure to Environmental Chemicals Increase the Risk of Diabetes Type 1 Development ? BioMed Res Int. 2014;2015:1–9.
- Lind PM, Zethelius B, Lind L. Circulating Levels of Phthalate Metabolites Are Associated With Prevalent Diabetes in the Elderly. Diabetes Care. 2012;35:1519–24. [CrossRef]
- Song Y, Chou EL, Baecker, A, et al. Endocrinedisrupting chemicals, risk of type 2 diabetes, and diabetes-related metabolic traits: a systematic review and meta-analysis. J Diabetes; 2016;8:516–532. [CrossRef]
- Bodin J, Samuelsen M, Becher R, Nygaard UC. diabetes-prone NOD mice. Immunopharmacol Immunotoxicol. 2013;3973(3):349–58.
- Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol A (BPA). Reprod Toxicol. 2007;24:139–177. [CrossRef]
- Vandenberg LN, Chahoud I, Heindel JJ, et al. Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Env Heal Perspect. 2010;118:1055–1070.
- He Y, Miao M, Herrinton LJ, et al. Bisphenol A levels in blood and urine in a Chinese population and the personal factors affecting the levels. Env Res. 2009;109:629–633. [CrossRef]
- Heudorf U, Mersch-Sundermann V, Angerer J. Phthalates: toxicology and exposure. Int J Hyg Env Heal. 2007;210:623–634. [CrossRef]
- Hatch EE, Nelson JW, Qureshi MM, et al. Association of urinary phthalate metabolite concentrations with body mass index and waist circumference: a cross-sectional study of NHANES data, 1999-2002. Env Heal. 2008;7:27. [CrossRef]
- Ruiz D, Becerra M, Sargis RM. Disparities in Environmental Exposures to Endocrine- Disrupting Chemicals and Diabetes Risk in Vulnerable Populations. Diabetes Care. 2017;1–13. [CrossRef]
- Stranges S, Sieri S, Vinceti M, Grioni S, Guallar E, Laclaustra M, et al. A prospective study of dietary selenium intake and risk of type 2 diabetes. BMC Public Heal 2010. 2010;10:564–74. [CrossRef]
- Mueller AS, Pallauf J. Compendium of the antidiabetic effects of supranutritional selenate doses. In vivo and in vitro investigations with type II diabetic db/db mice. J Nutr Biochem. 2006;17:548–60. [CrossRef]
- Burk, R. Selenium, an antioxidant nutrient. Nutr Clin Care. 2002;5:75–9.
- Han S, Cheng D, Liu N, Kuang H. The relationship between diabetic risk factors, diabetic complications and salt intake. J Diabetes Complications [Internet]. 2018;32(5):531–7. [CrossRef]
- Asiwe JN, Anachuna KK, Moke EG, Sanusi KO, Okonofua DE, Omeru O, Fasanmade AA. High dietary salt intake alleviates fasting blood glucose in streptozotocin-induced diabetic male wilstar rats. Thai J Pharm Sci. 2021;45(3):172–7.
- Beaudoin M, Allen B, Mazzetti G, Sullivan PJ, Graham TE. Manner in Both Men and Women. Appl Physiol Nutr Metab. 2013;38:140–7. [CrossRef]
- Alsunni, AA. Energy drink consumption: Beneficial and adverse health effects. Int J Heal Sci. 2015;9(4):468–74. [CrossRef]
- Liu M, Qi L, Arvan P, Liu M, Huang Y, Xu X, et al. Normal and defective pathways in biogenesis and maintenance of the insulin storage pool Normal and defective pathways in biogenesis and maintenance of the insulin storage pool. J Clin Invest. 2021;131(2):1–14.
- Vasiljević J, Torkko JM, Knoch KP, et al. The making of insulin in health and disease. Diabetologia. 2020;63(10):1981–1989. [CrossRef]
- Vasiljević J, Torkko JM, Knoch KP SM. The role of insulin in health and disease. Int J Mol Sci. 2021;22:6403.
- Carrano AC, Mulas F, Zeng C SM. Interro- gating islets in health and disease with single-cell technologies. Mol Metab. 2017;6(9):991–1001.
- Zhu Y, Liu Q, Zhou Z IY. PDX1, Neurogen- in-3, and MAFA: critical transcription regulators for beta cell development and regeneration. Stem Cell Res Ther. 2017;8(1):240. [CrossRef]
- Haliyur R, et al. Human islets expressing HNF1A variant have defective β cell transcriptional regula- tory networks. J Clin Invest. 2019;129(1):246–251.
- Wang Z, York NW, Nichols CG RM. Pancreatic β cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014;19(5):872–882. [CrossRef]
- Chénais, B. Transposable Elements in Cancer and Other Human Benoît Chénais To cite this version : HAL Id : hal-01905441. 2018. 227–242 p.
- Elso CM, Chu EPF, Alsayb MA, Mackin L, Ivory ST, Ashton MP, et al. Sleeping Beauty Transposon Mutagenesis as a Tool for Gene Discovery in the NOD Mouse Model of Type 1 Diabetes. G3. 2015;5:2903–11. [CrossRef]
- Garin I, Perez de Nanclares G, Gastaldo E, Harries LW, Rubio-Cabezas O CL. Permanent neonatal diabetes caused by creation of an ectopic splice site within the INS gene. e29205. PLoS One. 2012;7(1). [CrossRef]
- Wernersson R, et al. Analysis artefacts of the INS- IGF2 fusion transcript. BMC Mol Biol. 2015;16:13. [CrossRef]
- Kaufman, R. Beta-cell failure, stress, and type 2 diabetes. N Engl J Med. 2011;365(20):1931–1933.
- Diaferia GR, Cirulli V, Biunno I. SEL1L regulates adhesion, proliferation and secretion of insulin by affecting integrin signaling. :e79458. PLoS One. 2013;8(11).
- Shrestha N, et al. Sel1L-Hrd1 ER-associated degradation maintains β cell identity via TGF-β signaling. J Clin Invest. 2020;130(7):3510. [CrossRef]
- Wei F, et al. Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. J Clin Invest. 2011;121(9):3598–3608.
- Wollmer A, Strassburger W, Glatter U, Dodson GG, McCall M, Gattner H-G, Danho W, Brandenburg DRW. Two mutant forms of human insulin—structural conse- quences of the substitution of variant B24- or B25-phen- ylalanine by leucine. Hoppe-Seyler’s Z Physiol Chem. 1981;362:581–91.
- Haneda M, Kobayashi M, Maegawa H, Watanabe N, Takata Y, Ishibashi O, Shigeta Y IK. Decreased bio- logic activity and degradation of human [SerB24]-insulin, a second mutant insulin. Diabetes. 1985;34:568–73.
- Shoelson SE, Polonsky KS, Zeidler A, Rubenstein AH, et al. Human insulin B24 (Phe-Ser): secretion and metabolic clearance of the abnormal insulin in man and in a dog model. J Clin Invest. 1984;73:1351–8. [CrossRef]
- Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress- mediated diabetes. J Clin Invest. 2002;109:525–532.
- Yoshioka M, Kayo T, Ikeda T, et al. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early- onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46:887–894.
- Wang J, Takeuchi T, Tanaka S, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic b-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103:27–37.
- Herbach N, Rathkolb B, Kemter E, et al. Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe b-cell loss in Munich Ins2 C95S mutant mice. Diabetes. 2007;56:1268–1276.
- krokStøy J, Edghill EL, Flanagan SE, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. PNAS. 2007;104:15040–15044. [CrossRef]
- Kracht M, et al. Autoimmunity against a defec- tive ribosomal insulin gene product in type 1 diabetes. Nat Med. 2017;23(4):501–507.
- Laban S, et al. Heterogeneity of circulating CD8 T-cells specific to islet, neo-antigen and virus in patients with type 1 diabetes mellitus. Laban S, al. 2018;13(8). [CrossRef]
- Byrne MM, et al. Altered insulin secretory responses to glucose in diabetic and nondiabetic subjects with mutations in the diabetes susceptibility gene MODY3 on chromosome. Diabetes. 1996;12(45):1503–1510.
- Glucksmann MA, et al. Novel mutations and a mutational hotspot in the MODY3 gene. Diabetes. 1997;46:1081–1086. [CrossRef]
- Chi YI, et al. Diabetes mutations delineate an atypical POU domain in HNF-1α. Mol Cell. 2002;10:1129–1137. [CrossRef]
- Su B, Low J, Lim CS, Suet S, Ding L, Tan YS, et al. Decreased GLUT2 and glucose uptake contribute to insulin secretion defects in MODY3/HNF1A hiPSC-derived mutant Î2 cells. Nat Commun [Internet]. 2021;12:3133–53. [CrossRef]
- Ardon O, Procter M, Tvrdik T, Longo N, Mao R. Molecular Genetics and Metabolism Reports Sequencing analysis of insulin receptor defects and detection of two novel mutations in INSR gene ☆. YMGMR [Internet]. 2014;1:71–84. [CrossRef]
- Liu M, et al. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol Metab. 2010;21(11):652–659. [CrossRef]
- Arunagiri A, et al. Misfolded proinsulin in the endoplasmic reticulum during development of beta cell failure in diabetes. Ann N Y Acad Sci. 2018;1418(1):5–19. [CrossRef]
- Tersey S, et al. Islet β-cell endoplasmic reticu- lum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes. 2012;61(4):818–827.
- Liu M, Sun J, Cui J, Chen W, Guo H, Barbetti F, et al. INS-gene mutations: From genetics and beta cell biology to clinical disease. Mol Asp Med. 2016;42:3–18. [CrossRef]
- Nishi M, Nanjo K. Insulin gene mutations and diabetes. J Diabetes Investig. 2011;2(2):92–100.
- Carroll RJ, Hammer RE, Chan SJ, et al. Amutanthuman proinsulin is secreted from islets of Langerhans in increased amounts via an unregulated pathway. Proc Natl Acd Sci USA. 1988;85:8943–8947.
- Schwartz GP, Burke SGT, Katsoyannis PA. Superactive insulin:[B10-aspartic acid] insulin (human). Proc Natl Acad Sci USA. 1987;84:6408–6411. [CrossRef]
- Santos M, et al. Irp2 regulates insulin production through iron-mediated Cdkal1- catalyzed tRNA modification. Nat Commun. 2020;11(1):296.
- Izumi T, Yokota-Hashimoto H, Zhao S, Wang J, Halban PA TT. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes. 2003;52(2):409–416. [CrossRef]
- Riahi Y, et al. Inhibition of mTORC1 by ER stress impairs neonatal β-cell expansion and predisposes to diabetes in the Akita mouse:e38472. Elife. 2018;7.
- Wright J, et al. Dominant protein interactions that influence the pathogenesis of conformational diseases. J Clin Invest. 2013;123(7):3124–3134. [CrossRef]
- Liu M, et al. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes. 2012;61(4):828–837. [CrossRef]
- Liu M, et al. Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on wild-type proin- sulin transport. e13333. 77. PLoS One. 2010;5(10):77.
- Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci U S A. 2007;104(40):15841–15846.
- Renner S, et al. Permanent neonatal diabe- tes in INS(C94Y) transgenic pigs. Diabetes. 2013;62(5):1505–1511.
- Gupta S, McGrath B, Cavener D. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes. 2010;59(8):1937–1947. [CrossRef]
- Leroux L, Desbois P, Lamotte L, Duvillié B, Cordonnier N, Jackerott M, et al. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes. 2001;50(90001):150–3.
- Raile K, O’Connell M, Galler A, Werther G, Khnen P, Krude H, et al. Diabetes caused by insulin gene (INS) deletion: clinical characteristics of homozygous and heterozygous individuals. Eur J Endocrinol. 2011;165(2):255–260. [CrossRef]
- Haataja L, Snapp E, Wright J, Liu M, Hardy AB, Wheeler MB, et al. Proinsulin intermolecular interactions during secretory trafficking in pancreatic β cells. J Biol Chem. 2013;288(3):1896– 1906. [CrossRef]
- Hodish I, Liu M, Rajpal G, Larkin D, Holz RW, Adams A, et al. Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. J Biol Chem. 2010;285(1):685–694. [CrossRef]
- Nanjo K, Miyano M, Kondo M, Sanke T, Nishimura S, Miyamura K, Inouye K, Given BD, Chan SJ, Polonsky KS, Tager HS, Steiner DF RA. Insulin Wakayama: familial mutant insulin syndrome in Japan. Diabetologia. 1987;30:87–92. [CrossRef]
- Iwamoto Y, Sakura H, Ishii Y, Yamamoto R, Kumakura S, Sakamoto Y, Matsuda A KT. A new case of ab- normal insulinemia with diabetes: reduced insulin values determined by radioreceptor assay. Diabetes. 1986;35:1237–42.
- Steiner DF, Tager HS, Chan SJ, Nanjo K, Sanke T, Rubenstein AH. Lessons Learned From Molecular Biology of Insulin-Gene Mutations. Diabetes Care. 1990;13(6):600–9. [CrossRef]
- Burgess TL, KR. Constitutive and regulated secretion of proteins. Annu Rev Cell Biol. 1987;3:243–93.
- Saeedi P, Petersohn I, Salpea P, Malanda B, Kaaruranga S, Unwin N, Colagiuri S, Guanguata L, Motala AA, Ogurtsova K, Shaw JE, Bright D, Williams R. Diabetes Research and Clinical Practise. JDiabres. 2019. [CrossRef]
- PlasticEurope. Plastics – the facts 2020. An analysis of European plastics production, demand and waste data. Available at http://www.plasticseurope.org/Document/plastics –the-facts-2020.aspx. 2020.
- Infoplease. Total world population by decade, 1950-2050. Accessed in 22nd July, 2022, Available at https://www.infoplease.com/world/population/total-population-world-decade-1950-2050.
- Zhang B, Yue R, Chen Y, Yang M, Huang X, Shui J, et al. Gut Microbiota, a Potential New Target for Chinese Herbal Medicines in Treating Diabetes Mellitus. Evidence-based Complement Altern Med. 2019;2019. [CrossRef]
- Kinsley, BT. Morbidity and Mortality. Diabetes Care. 1995;18(12):1566–1570.
- Krolewski AH, Warram JH, Rand LI, Kahn C. Epidemiological approach to the etiology of type 1 diabetes mellitus and its complications. N Engl Med. 1987;317:390–9. [CrossRef]
- Deckert T, Poulsen JE, Larsen M. Prognosis of diabetics with diabetes onset before age of thirty-one: survival causes of death and complications. Diabetologia. 1978;14.
- Kahn CR, Goldstein BJ. Molecular defects in insulin action. Science (80- ). 1989;245:13. [CrossRef]
- Rabson SM, Mendenhall EN. Familial hypertrophy of pineal body, hyperplasia of adrenal cortex and diabetes mellitus; report of 3 cases. Am J Clin Pathol. 1956;26:283–290.
- Longo N, Wang Y, Smith SA, Langley SD, DiMeglio LA, Giannella-Neto D. Genotype–phenotype correlation in inherited severe insulin resistance. Hum Mol Genet. 2002;11:1465–1475. [CrossRef]
- Boland BB, et al. Pancreatic β-cell rest replen- ishes insulin secretory capacity and attenuates diabetes in an extreme model of obese type 2 diabetes. Diabetes. 2019;68(1):131–140.
- Orland MJ, Permutt M. Quantitative analysis of pancreatic proinsulin mRNA in genetically diabetic (db/db) mice. Diabetes. 1987;36(3):341–347.
- van der Vorm ER, van der Zon GCW, Moller, HM, Krans, D, Lindhout JAM. An Arg for Gly substitution at position 31 in the insulin receptor, linked to insulin resistance, inhibits receptor processing and transport. J Biol Chem. 1992;267:66–71.
- Dyson PA, Twenefour D, Breen C, Duncan A, Elvin E, Goff L, et al. Diabetes UK evidence-based nutrition guidelines for the prevention and management of diabetes. Diabet Med. 2018;35(5):541–7. [CrossRef]
- Kim SK, Hur KY, Kim HJ et al. The increase in abdominal subcutaneous fat depot is an independent factor to determine the glycemic control after rosiglitazone treatment. Eur J Endocrinol. 2007;157:167–174. [CrossRef]
- Lebovitz HE, Banerji M. Insulin resistance and its treatment by thiazolidine- diones. Recent Prog Horm Res. 2001;56:265–294.
- Jenkins DJA, Axelsen M, Kendall CWC, Augustin LSA, Vuksan V, Smith U. Dietary fibre, lente carbohydrates and the insulin-resistant diseases. Br J ofNutrition. 2000;83(1):157–63. [CrossRef]
- Dyson PA, Kelly T, Deakin T, Duncan A, Frost G, Harrison Z, et al. Diabetes UK evidence-based nutrition guidelines for the prevention and management of diabetes. Diabet Med. 2011;28(11):1282–8. [CrossRef]
- Foster-Powell K, Miller J. International tables of glycemic index. Am J Clin Nuir. 1995;62:871–93. [CrossRef]
- Liu S, Manson JE, Stampfer MJ, Hu FB, Giovannucci E, Colditz GA, et al. A Prospective Study of Whole-Grain Intake and Risk of Type 2 Diabetes Mellitus in US Women. Am J Public Health. 2000;90(9):1409–15. [CrossRef]
- Liu, S. Journal of the American College of Nutrition Intake of Refined Carbohydrates and Whole Grain Foods in Relation to Risk of Type 2 Diabetes Mellitus and Coronary Heart Disease Intake of Refined Carbohydrates and Whole Grain Foods in Relation to Risk of Type. J Am Coll Nutr. 2013;21(4):298–306.
- BetterHealth. Carbohydrate and the glycemic index. Last reviewed 1st July 2022. Accessed 24th July, 2022. Available at https://www.betterhealth.vic.gov.au/health/healthyliving/carbohydrates-and-the-glycaemic-index.
- Bu S, Yuan C, Cao F, Xu Q, Zhang Y, Ju R., Chen L, Li Z. Lingzhi or reishi medicinal mushroom, Ganoderma lucidum (Agaricomycetes) polysaccharides suppressed adipogenesis and stimulated lipolysis in HPA-v and 3T3-L1 adipocytes.. Int J Med Mushroom. 2021.
- Bu S, Zheng H, Yuan C, Tiaz Z, Xu C, Zhao L, Zhu X, Chen H. Concentrated extract of Prunus mume fruits exert dual effect in 3T3-L1 adipocytes by inbiting adipogenesis and inducing beiging/browning. Food Nutr Res. 2020.
- Johnston GAR, Chebib M, Duke RK, Fernandez SP, Hanrahan JR, Hinton T, et al. Herbal Products and GABA Receptors. 2009;1095–101. [CrossRef]
- Bu S, Yuan CY, Xue Q, Chen Y, Cao F. Bilobalide suppresses adipogenesis in 3T3-L1 adipocytes via AMPK signaling patway. Molecules. 2019;24(19).
- Wei X, Tao J, Xiao S, et al. “XiexinTang improves the symptom of type 2 diabetic rats by modulation of the gut microbiota,” Article ID3685,. Sci Rep. 2018;8(1).
- Sancho RAS, Pastore G. “Evaluation of the effects of anthocyanins in type2 diabetes,.” Food Res Int. 2012;46(1):378–86. [CrossRef]
- Shoelson S, Haneda M, Blix P, Nanjo A, Sanke T, Steiner DF, Inouye K, Rubenstein, AH, Tager H. Three mutant insulins in man. Nat. 1983;302:540–3.
- Cousens LS, Shuster JR, Gallegos C, Ku L, Stempien MM, Urdea MS, Sanchez-Pescador R, Taylor A, Tekamp-Olson P. High level expression of proinsulin in yeast. Cene. 1987;61:265–75.
- Chan SJ, Weiss J, Konrad M, White T, Bahl C, Yu SD, Marks D, Steiner D. Biosynthesis and periplasmic segregation of human proinsulin in E. coli. Proc Natl Acad Sci USA. 1981;78:5401–5.
- Talmadge K, Brosius J, Gilbert W. An “internal” signal sequence directs secretion and processing of proinsulin in bacteria. Nat. 1981;294:176–8.
- Bell Gl, Pictet RL, Rutter WJ, Cordell B, Tischer E GH. Sequence of the human insulin gene. Nat. 1980;284:26–32. [CrossRef]
- Paul DC, VanFrank RM, Muth WL, Ross JW, Williams D. Immunocytochemical demonstration of human proinsulin chimeric polypeptide within cytoplasmic inclusion bodies of Escherichia coli. Eur J Cell Biol. 1983;31:171–4.
- Chance RE, Kroeff EP, Hoffman J. Chemical, physical, and biological properties of recombinant human insulin. In Insulins, Growth Hormone and Recombinant DNA Technology. Guerigian JL, Ed. New York, Raven. 1981. 71–86 p.
- Thim L, Hansen MT, Norris K, Hoegh I, Boel E, Forstrom J, Ammerer G, Fiil N. Secretion and processing of insulin precursors in yeast. Proc Natl Acad Sci USA. 1986;83:6766–70. [CrossRef]
- Brange J, Ribe U, Hansen JF, Dodson G, Hansen MT, Havelune S, Melberg SG, Norris F, Norris K, Snel L, Rensen AR, Voigt H. Monomeric insulins obtained by protein engineering and their medical implications. Na- ture. 1988;333:679–82. [CrossRef]
- Schwartz GP, Burke SGT KP. A superactive insulin: [B10-aspartic acid] insulin (human). Proc Natl Acad Sci USA. 1987;84:6408–11.
- Schwartz GP, Burke GT KP. A highly potent insulin: Des-(B26-B30)-[AspB10,TyrB25-NH2] insulin. Proc Natl Acad Sci USA. 1989;86(6):458–61. [CrossRef]
- Markussen J, Diers I, Hougaard P, Langkjaer Norris K, Snel L, Rensen, AR, Rensen, E, Voigt H. Soluble, prolonged-acting insulin derivatives. III. Degree of protrac- tion, crystallizability and chemical stability of insulins substituted in positions A21, B13, B23, B27 and B30. Protein Eng. 1988;2:157–66. [CrossRef]
- Konrad, M. Immunogenicity of proteins administered to humans for therapeutic purposes. Trends Biotechnol. 1989;7:175–9. [CrossRef]
- Wang Y, Zhou H, Palyha O, Mu J. Restoration of insulin receptor improves diabetic phenotype in T2DM mice. JCI Insight. 2019;4(15):1–11. [CrossRef]
- Bulleid, NJ. Disulfide bond formation in the mammalian endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2012;4(11). [CrossRef]
- Sevier, CS. New insights into oxidative folding. J Cell Biol. 2010;188(6):757–758. [CrossRef]
Figure 3.
Showing the trend in the increase of diabetes worldwide reported by WHO.
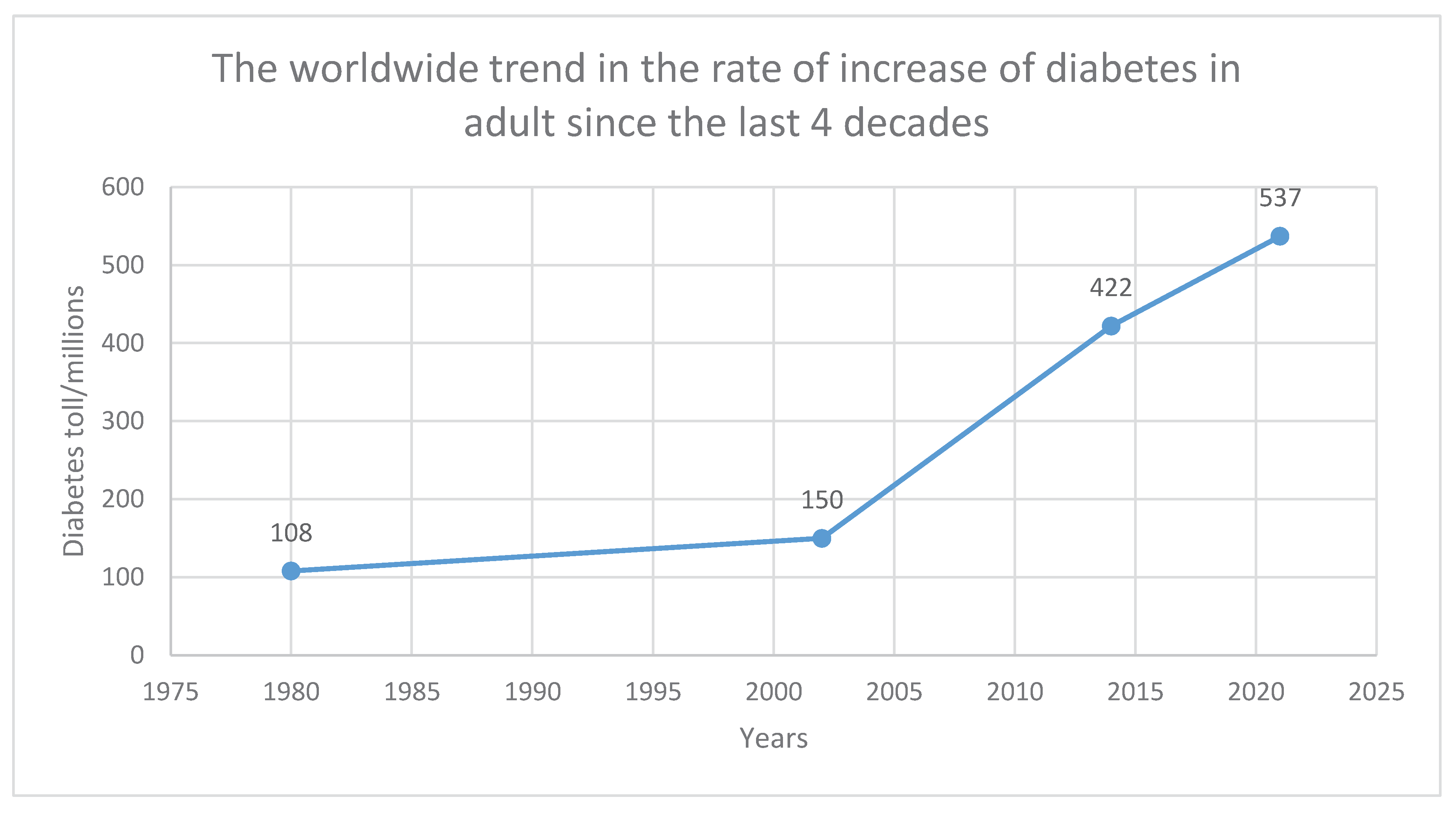

Table 1.
An illustration of the normal level of insulin resistance and insulin insufficiency in the various diabetes subgroups.
Table 1.
An illustration of the normal level of insulin resistance and insulin insufficiency in the various diabetes subgroups.
| The Forms of Diabetes | Disease Severity | |
|---|---|---|
| Insulin Deficiency | Insulin Resistance | |
| Autoimmunity, type 1 diabetes | From mild to severe | None/Mild |
| LADA, Autoimmunity | From mild to severe | From mild to noticeable |
| CEL-MODY2 | Noticeable | Mild |
| Glucokinase MODY | Mild | None/Mild |
| HNF1α-MODY | Noticeable | None/Mild |
| HNF4α-MODY | Noticeable | Mild |
| HNF1β-MODY3 | Mild to noticeable | None/Mild |
| PDX1-MODY heterozygote | Mild | None/Mild |
| PDX1-MODY homozygote | Severe | None/Mild |
| Other forms of MODY | Severe | None/Mild |
| Mitochondrial diabetes | From mild to severe | None/Mild |
| Type 2 Diabetes | From mild to noticeable | From noticeable to severe |
| WFS1, Wolfram syndrome | Noticeable | None/Mild |
| Ketosis-prone diabetes | Mild to severe | Noticeable to severe |
Source: [13], Keys: None/mild means the disease may show or not show any symptoms, CEL= carboxylesterlipase. The disease severity is depicted in the order from None<mild<noticeable<severe.
Table 2.
Some selected foods and their mean glycemic index from multiple sources.
| Foods | Mean Glycemic Index | Metabolic Effect |
|---|---|---|
| Cereal crops | ||
| Barley | 25±2 | Low |
| Buckwheat | 54±4 | Low |
| Maize | 65±5 | Moderate |
| Millet | 71±10 | High |
| White rice | 56±2 | Low/moderate |
| Low amylose white rice | 88±3 | High |
| High amylose white rice | 59±3 | Moderate |
| Brown rice | 55±5 | Low to moderate |
| Parboiled rice | 47±3 | Low |
| Wheat | 41±3 | Low |
| Diary foods | ||
| Ice cream | 61±7 | Moderate |
| Low-fat ice cream | 50±8 | Low/moderate |
| Full-fat milk | 27±7 | Low |
| Fruits and vegetables | ||
| Apple | 36±2 | Low |
| Banana | 53±6 | Low/moderate |
| Grapefruit | 25 | Low |
| Mango | 55±5 | Low/moderate |
| Orange | 43±4 | Low |
| Pawpaw | 58±2 | Moderate |
| Pear | 36±3 | Low |
| Pinapple | 66±7 | Moderate/high |
| Watermelon | 72±13 | Moderate/high |
| Tomato | 38±9 | Low |
| Carrot | 71±22 | High |
| Potato | 83±1 | High |
| Sweet potato | 54±8 | Low/moderate |
| Yam | 51±12 | Low/moderate |
| Sugars | ||
| Honey | 73±15 | High |
| Fructose | 20±5 | Low |
| Glucose | 97±3 | High |
| Sucrose | 65±4 | Moderate |
| Lactose | 46±3 | Low |
| Maltose | 105±12 | Extreme |
| Kidney beans | 29±8 | Low |
| Soyabeans | 18±3 | Low |
| Baked beans | 40±3 | Low |
| Indigenous processed foods | ||
| South Africa maize meal porridge | 71±6 | High |
| Spaghetti, boiled 15 min | 41±3 | Low |
| Spaghetti, boiled 5 min | 37±3 | Low |
| Spaghetti, boiled 20 min | 58±7 | Low/moderate |
| Wheat bread | 69±12 | Moderate/high |
| Barley kernel bread | 34±4 | Low |
| Barley flour bread | 66±1 | Moderate |
| Lucozade | 95±10 | High to extreme |
| Soft drink, coca cola, Fanta | 68±6 | Moderate/high |
Source: [122].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



