
Submitted:
13 July 2023
Posted:
14 July 2023
You are already at the latest version
Abstract
Pancreatic cancer (PC) is the seventh leading cause of cancer-related death. PC incidence has con-tinued to increase by about 1% each year in both men and women. Although the 5‐year relative survival rate of PC has increased from 3% to 12%, it is still the lowest among cancers. Hence, novel therapeutic strategies are urgently needed. Challenges in PC-targeted therapeutic strate-gies stem from the high PC heterogeneity and from the poorly understood interplay between cancer cells and the surrounding microenvironment. Signaling pathways that drive PC cell growth have been the subject of intense scrutiny and interest has been attracted by necroptosis, a distinct type of programmed cell death. In this review, we provide a historical background on necroptosis and a detailed analysis of the ongoing debate on the role of necroptosis in PC malignant progression.
Keywords:
pancreatic cancer
; necroptosis
; tumor microenvironment
; cell death
1. Introduction
Pancreatic Cancer (PC) currently is the seventh leading cause of cancer-related death [1] and is expected to become the second one by 2030 [2].
PC incidence differs widely among countries: the highest age-standardized incidence is seen in Europe and North America, the lowest is recorded in Africa and South-Central Asia [3].
Late diagnosis, together with chemoresistance, and aggressive biology have contributed to its poor prognosis and to the concept of PC as a “silent killer”.
Early clinical manifestations are often vague, depending on the site of origin of the tumor. Approximately 70% of pancreatic tumors arise in the head of the pancreas and cause biliary obstruction leading to dark urine (49%) and jaundice (49%). Patients with PC arising from the pancreas body or tail present other symptoms, such as abdominal pain, back pain, and cachexia-related symptoms (appetite loss, weight loss, fatigue) [4].
Pancreatic Ductal Adenocarcinoma (PDAC) is the most prevalent type of PC, representing more than 90% of cases [5]. It shows an overall 5-year survival rate of less than 5% and a median survival time of less than 6 months if untreated [6].
Still today there is a common and keen interest in filling the gaps of our knowledge about PC, due to recent interesting findings that have encouraged researchers to go deeply into the complexity of this lethal disease.
The characteristic aggressiveness of PC should probably be assigned to several undisclosed strategies developed by neoplastic cells, that could be enhanced by the same pancreatic tissue context, particularly hostile but also crucial to cause a more powerful response by the tumor itself.
In this perspective, we discuss the recent hypothesis that necroptosis, a caspase-independent mechanism of programmed cell death, may play an unexpected role in PDAC malignant progression, through a peculiar interaction between the tumor cell and the extra-epithelial compartment. Here, we review opposing views on the contribution of programmed cell death in tumor progression and on the role of tumor microenvironment as a final “tiebreaker”.
2. Pancreatic Ductal Adenocarcinoma (PDAC)
After colorectal cancer, PDAC is the second most common cancer of the digestive system in the United States [7]. Only 10% of PDAC cases appear related to family history. Inherited cancer syndromes with known germline mutations, such as the Lynch syndrome (MLH1, MLH2, MLH6, PMS2), familiar breast and ovarian cancer (BRCA1 and BRCA2), familial adenomatous polyposis (FAP), familial atypical multiple mole melanoma (CDKN2A), Peutz–Jeghers (STK11/LKB1), show an increased risk of PDAC [8].
95% of PDAC show activation of K-RAS [9]. This gene encodes a small GTPase that regulates cellular signaling downstream of growth factor receptors. The most common K-RAS mutations are the GAT (aspartic acid; G12D), GTT (valine; G12V), and TGT (cysteine; G12C) mutations at codon 12 [10]. Instead, among the suppressor genes that are commonly mutated in PDAC there are P16/CDKN2A, TP53, and SMAD4/DPC4 [11].
However, subsequent mutational changes are necessary and take place gradually over time, exacerbating the genomic instability.
Besides genetic background, risk factors for PDAC are strongly related to age [12], obesity, diabetes [13], chronic pancreatitis [14] and lifestyle, especially cigarette smoking [15].
Macroscopically, PDAC appears as a solid and firm white yellowish poorly defined mass [5]. It is rarely diagnosed at early stages and presents at diagnosis as an advanced lesion (pT2 or higher), with infiltration of surrounding structures (peripancreatic adipose tissue, duodenum, stomach, portal vein). The reasons for this include the frequent lack of symptoms and the proximity of major blood vessels, which increases chances of early invasion by the tumor [16].
Several PDAC variants can be recognized based on distinctive histological characteristics, associated with specific genetic signatures (Supplementary, Table S1). Distinct non-invasive precursor lesions of PDAC have also been identified, providing further insight into PDAC carcinogenesis (Supplementary, Table S2). Key components of PDAC are invasive and mucin-producing epithelial lesions that microscopically appear structured as irregular tubular glands, most frequently when PDAC is in the proximal pancreas.
Improved diagnosis of PDAC precursor lesions would allow timely intervention before progression to invasive PDAC.
3. The vital crosstalk between PDAC and Tumor Microenvironment
Cancer is described as a hetero-cellular system containing both neoplastic and stromal cells that progressively form a surrounding protective microenvironment (tumor microenvironment, TME) where tumor development is facilitated.
A characteristic feature of PDAC is the so called “desmoplastic reaction”, an abundant fibrotic response whereby atypical tumor glands are embedded within a prominent desmoplastic stroma. This fibrotic reaction creates a barrier that can prevent the penetration of chemotherapeutic agents and promote cancer growth.
Different pancreatic stromal cells, such as pancreatic stellate cells (PSCs), cancer associated-fibroblasts (CAFs), immune cells such as tumor-associated macrophages (TAMs), regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) cooperate to create a typical highly immunosuppressive stroma [17]. Consequently, the survival of cancer becomes strongly related to the intense crosstalk with the TME, which is now established as a dynamic entity (Figure 1) [18].
The TME has an active role also in metastasis. PC is often considered a metastatic disease from the time of clinical diagnosis. The most common metastatic site for PC is the liver, followed by the lungs, peritoneum, and bones [19]. Several components of tumor microenvironment, such as PSCs and CAFs, can participate to the process of escaping of cancer cells from the primary site: they start to generate additional extracellular matrix, thereby increasing tumor pressure and diminishing vascularization [20].
Consequently, this hypoxic condition may exacerbate the malignant phenotype, suggesting that a hostile niche could favor specific cell-stroma interactions through paracrine signals and extracellular vesicle (EVs) trafficking. The latter, including exosomes, microvesicles (MVs), and apoptotic bodies, are secreted by cells in the extracellular matrix as cellular messengers to “educate” the recipient cells by delivering their specific cargo [21].
In this context, Leca et al. have shown that, under stressful conditions, CAFs within the PDAC microenvironment could manipulate cancer cell aggressive behavior through the secretion of specific ANXA6+-extracellular vesicles [22]. In turn, PDAC cells may uptake those EVs carrying the annexin A6 and so increase their spreading and metastatic potential.
Also, PSC-derived exosomes can promote neoplastic cell proliferation by delivering specific nuclear material. Li et al. have reported that PSC-derived exosomes deliver miR-5703 into PC cells that in turn binds and suppresses CMTM4 [23]. They also proposed that miR-5703 serum levels could be a promising diagnostic biomarker in PC.
On the other hand, the tumor itself can shape the surrounding environment to prepare the so called “soil” in secondary sites through the formation of pre-metastatic niches [24].
Finding new undiscovered ways of communication between stroma and cancer may provide novel therapeutic strategies for tumor tracking.
4. The “dark side” of cell death in PC cells
4.1. Necroptosis: a highly specific way of cell death
Cell death resistance is a recurrent feature of most types of cancer, including PDAC [25]. The different ways of cell dying can be broadly classified in two major categories: programmed (apoptosis) and non-programmed cell death (necrosis). Apoptosis acts both as a homeostatic and a restraining mechanism. It controls cellular turnover and the maintenance of cell populations in normal tissues. It also plays an important role during immune reactions or when cells are damaged by diseases or harmful agents. In contrast, necrosis is considered as a form of disruption of the cytoplasmic membrane and of internal cell structures, ending up with a characteristic inflammatory reaction that specifically differentiates it from apoptosis [26]. Necrosis includes swelling of mitochondria, followed by the explosive release of molecules from the dying cells into the extracellular space, upon loss of plasma membrane integrity.
Resistance to apoptosis induction results in enhanced cell viability and insensitivity to chemo- and radiation therapies [27]. Furthermore, cancer cells can respond to stresses, such as hypoxia and nutrient deficiency, by undergoing necrosis, thus considered as a form of “reparative cell death” [28].
Despite necrosis being usually defined as an accidental and uncontrolled event, segments of this response were recently recognized to be regulated by defined molecular pathways, leading to coin the new term “necroptosis”.
Necroptosis has been described as a novel form of programmed cell death, a sort of “chimera” born from apoptosis and necrosis [29] and characterized by a necrotic cell death morphology in response to activation of a defined and programmable mechanism. Cells generally undergo swelling and membrane rupture, with a subsequent release of intracellular content.
The triggering of necroptosis depends on the formation of the necrosome which requires co-activation of receptor-interacting serine/threonine protein kinase (RIPK)1/3 and Fas-associated protein with death domain (FADD) leading to the activation of the pseudokinase mixed lineage kinase like (MLKL) [30] followed by a rapid plasma membrane rupture and inflammatory response through the release of damage-associated molecular patterns (DAMPs) and cytokines. The process can be triggered by extracellular o intracellular activators; however, the TNFα-induced necroptosis is the best characterized necroptotic pathway [31]. It includes the initial recognition between the extracellular ligand, TNFα, and its receptor tumor necrosis factor receptor type I (TNFR1), present on the plasma membrane. Alternative extracellular signaling pathways involve Toll-like receptor 3 or 4 (TLR3/4) and the interferon receptors. Instead, the activator ZBP1 is responsible for the intracellular signaling of necroptosis [32].
Generally, necrosome formation is allowed only when caspase-8 activity is inhibited: this event determines the shift of the RIPK1-mediated death process from apoptosis to necroptosis, through the assembling of the RIPK1-RIPK3 complex [33]. Then, RIPK3 and MLKL act as downstream regulators of necroptosis, after undergoing phosphorylation. The activated MLKL finally migrates toward the plasma membrane, to generate membrane pores, accompanied by “ballooning” of the cell and massive calcium ions (Ca2+) influx, thus causing the rupture of the plasma membrane [33].
4.2. Necroptosis is a “double-edged sword” in cancer
Necrotic foci at tumor sites are a frequent occurrence resulting from inadequate vascularization and subsequent metabolic stresses, such as glucose deprivation and hypoxia.
On the other hand, necroptosis is not a simple passive consequence of triggering events. It is a finely regulated way of dying that neoplastic cells can choose to activate or switch off, according to induction pathways.
The role of necroptosis in oncology remains controversial, due to heterogeneous data derived from studies conducted across several distinct human cancers. An analysis of the results to date highlights context-dependent opposite functions, i.e tumor suppression and tumor-promotion, for this cell death program.
Several cases of decreased expression involving many key modulators of necroptosis have been found in different tumors, suggesting that cancer cells may evade necroptosis as it happens within apoptosis. Nugues et al. have identified a significant reduction of RIPK3 expression in acute myeloid leukemia samples [34], while in colon cancer tissues both RIPK1 and RIPK3 appeared to be downregulated versus adjacent normal tissue [35]. RIP1 and RIP3 switching off was proven to be associated with worse prognosis and disease progression both in patients with breast cancer [36] and in those with head and neck squamous cell carcinoma [37].
Some studies have also suggested the possibility that pro-necroptotic genes could be under epigenetic control such as DNA methylation-driven [38].
Key to this analysis is the inflammatory background to which necroptosis actively participates by secreting several pro-inflammatory cytokines and chemokines. During this process, necroptotic cells also liberate a plethora of damage-associated molecular patterns (DAMPs), promoting a final immunogenic outcome that in turn facilitates tumor-suppression. Yang et al. reported that TNF- and chemotherapy-driven necroptosis in cancer cells can release specific DAMPs such as ATP and HMGB1, thereby promoting the activation of tumor-regressive anticancer immunity [39]. The release of DAMPs from dead cells was found not sufficient by itself to trigger robust cross-priming of cytotoxic CD8+ T cells, which requires RIPK1 signaling and NF-κB-induced transcription in the dying cells [40], consistent with a model of anticancer action associated with pro-necroptotic processes.
However, the downregulation of necroptotic factors may not occur in all cancers: high expression of RIPK1 or MLKL has been reported to predict worse prognosis for some cancer patients, both in melanoma [41] and breast tumors [42]. Consistent with these observations, RIPK1 is commonly found overexpressed in glioblastoma, correlating with a poorer outcome [43].
These findings challenged the idea of necroptosis as an only fail-safe mechanism to prevent cancer when apoptosis is inhibited, highlighting a potential alternative tumor-promoting role. It should be noted that depending on the cell type, necroptotic cells can release a variety of chemokines, such as CCL2, CXCL8/IL8, CXCL1/2, CSF2. These molecules can facilitate the recruitment of myeloid cells and granulocytes and consequently promote a tumor-associated immunosuppression [33].
Thus, the modulation of anticancer immunity driven by necroptosis could take a specific direction according to several factors such as the tumor type, the microenvironment and the immune cell types involved: in this perspective, immunosurveillance mediated by DAMPs may more easily occur in “hot tumors” as compared to “cold tumors”, that are characterized by the lack of T cell infiltration.
These “two faces” of necroptosis can be observed in the same tumor. He et al. have classified colon cancer patients into two necroptosis-related molecular subtypes with diverse clinical outcomes and tumor microenvironment infiltration characteristics. Based on the differentially expressed genes between the two molecular subtypes, they found a high necroptosis risk signature score (NRS-score) associated with poor prognosis and especially with an immunosuppressive microenvironment, whereas colon cancer patients with low NRS-score showed a better overall survival [44]. These data support the idea that necroptosis may also shield tumors from antitumor immune responses by fostering an immune escape mechanism [45].
Indeed, when blocking necroptosis of tumor cells through MLKL deletion, lung metastasis was significantly inhibited [40]. Consistent with this, tumor cells were discovered to induce programmed necrosis of endothelial cells, with the consequence of facilitating neoplastic cell extravasation [46].
Taken together, these findings raise the issue that the outcome of necroptosis in cancer may be tissue-context dependent. In this perspective, the immunosuppressive TME of PC may turn out to be a decisive element to consider necroptosis as a tumor-promoting factor.
4.3. Necroptosis and PDAC: friends or foes?
Due to the desmoplastic reaction, PDAC belongs to the category of “cold tumors” which are characterized by a low infiltration of immunogenic cells, especially effector T cells, in contrast to the extensive presence of immune-suppressive cells, such as CD4+, CD25+ Tregs, MDSCs and TAMs. This immuno-evasive background can have a relevant impact on the behavior of pancreatic cancer cells: they may jealously preserve the immunosuppressive nature of the milieu by activating many pathways and, in this perspective, it cannot be excluded that they also undergo necroptosis. All these events would occur in a significant inflammatory scenario that is characteristic of PC. Here, necroptotic cells could give their contribution by producing additional inflammation-inducing factors, helping to establish a chronic inflammatory state that consequently may stimulate tumor growth.
Interestingly, a dual function of necroptosis may reflect that attributed to inflammation: it is known that the activity of inflammatory cells combined with the type and level of inflammation-modulating factors control the balance between their pro- and antitumor effects. Thus, while in the initial stage of cancer growth the acute phase responses may show anticancer action, in a chronic inflammatory state the presence of inflammatory cells, especially in the TME, acts for the benefit of cancer cells, stimulating their survival and proliferation [47]. In the same way, when massive acute necroptosis happens, following chemotherapy or irradiation treatment, the anti-tumor immunity is enhanced through the activation of cytotoxic CD8+ T cells; in contrast, a chronic necroptosis would have the opposite effect of an improved immune-suppression by producing molecules that modulate MDSC and M2-like macrophages [48].
Both scenarios have been observed in PC [49]. Considering the advantageous effects of chemotherapy in promoting cancer cells apoptosis, PDAC-bearing mice were initially treated with gemcitabine for a similar impact on necroptosis, thereby confirming that chemotherapy increased expression of RIPK1/3 in vivo. The second step was that of studying the effects of RIPK3 deletion both in vitro and in vivo: surprisingly, while in vitro K-RASG12D transformed PDAC cells increased their proliferative rate upon RIP3 deletion, in vivo findings were totally in contrast: p48Cre;KrasG12D;Rip3−/− pancreases showed a diminished rate of acinar replacement by dysplastic ducts, and a slower PanIN progression when compared to p48Cre;KrasG12D(KC);Rip3+/+ pancreases, indicating that RIPK3 deletion protects against oncogenesis.
To confirm the hypothesis of tumor-promotion by necroptosis, it has been shown that RIPK3 deletion inhibits infiltration by TAMs and MDSCs. This occurred via a concomitant decrease in the expression of chemokine attractant CXCL1 and macrophage inducible Ca2+-dependent lectin receptor (Mincle), proposed to mediate the pro-tumorigenic immune suppression associated with RIPK3 signaling [49]. Thus, we can suppose that the opposite outcome of RIPK1/3 expression in in vivo and in vitro assays must be function of the presence and the type of the microenvironment, well defined in PDAC.
Taken together these data support the idea of necroptosis as a “friend” of PDAC, as it tends to boost its growth and development by maintaining an inflammation-related immunosuppressive microenvironment (Figure 2).
However, there is a need to evaluate these findings especially in clinical trials since studies have been conducted only in vitro and through animal models.
Questions remain also about the “timing” of this programmed process. Jiao et al. indicate that necroptosis only happens in the late stage of tumor development in breast cancer [40]. Under stressful conditions, the expression of key necroptosis mediators seems to be reprogrammed by cancer cells to restore the necroptotic machinery [45].
It is also important to consider the phenotype of tumor associated macrophages (TAMs) found in pancreatic TME: while MHCIIhiTNFα+ M1-like macrophages promote anti-tumor cytotoxic T lymphocyte (CTL) activity by recruiting Th1 cells [50], CD206+IL-10+ M2-like macrophages enhance the expansion of Th2 cells and Tregs in PDAC [49]. Interestingly, high levels of RIP1 expression were found in M2-like TAMs [51]. Wang et al. have also showed that RIP1 inhibition reprogrammed TAMs towards the anti-tumor M1-like phenotype [51].
As changes in TME can have a critical effect in metastasis, necroptosis may have an additional role in remodeling the surrounding environment to promote neoplastic seeding. The idea that necroptosis could facilitate metastasis of PC is embraced by Ando et al. [52], after observing that conditioned media derived from necroptotic PC cells induced PC cell migration and invasion. The CXCL5 chemokine was upregulated in conditioned media, suggesting its possible implication in promoting colonization via the receptor CXCR2 [52].
Recently it has been found that also necroptotic cells can produce EVs that may play roles in the necroptosis-induced immune responses [53]. This phenomenon could additionally improve the communication with the TME, inducing stromal cells to create a more favorable environment for the survival of cancer.
Thus, given the variety of molecular mechanisms and events supported by experimental evidence, it can be argued that necroptosis may not have a pre-defined role [33], but rather a tissue specific one, where the stress degree as well as the amplitude and duration of inflammation may be the decisive tiebreaker of necroptosis functional fate.
5. Future Perspectives and Applications of Necroptosis
5.1. Pro-necroptotic markers: a novel focus of PDAC research?
The lack of specific biomarkers for early detection of PDAC is a critical problem in terms of diagnosis. Although many overexpressed mRNAs or proteins in PC have been reported [54], a critical step remains that of finding potential markers that can be easily detected in liquid biopsies with high sensitivity and specificity.
To date, the carbohydrate antigen CA19.9 represents the main serological marker associated with PC. Also known as Sialyl Lewis A, CA19.9 is a cell surface glycoprotein complex, synthesized in the normal pancreatic parenchyma and biliary tract. CA19.9 increased expression is seen not only in PC but also in stomach, colorectal, lung, thyroid, and biliary cancer [55].
High CA19.9 serum level is reported both in malignant and benign conditions such as pancreatitis, pancreatic cysts, diabetes mellitus, liver fibrosis, benign cholestatic diseases, and other urological, pulmonary, and gynecological diseases [56]. Thus CA19.9 serum level cannot be used for initial diagnosis but rather for post-therapy monitoring [57].
The need of innovative design for PDAC clinical trials is growing, due to the high degree of diversity emerging from several PDAC cases, which differ not only in histopathological features but also in genetic landscape [58].
Aharon and colleagues discovered heterogeneity in mutation rates between early- and average-age-onset PDAC patients: SMAD4 and PIK3CA were more frequently mutated in the early-onset compared with the average-age-onset disease, suggesting the possibility to delineate a unique profile for younger patients [59].
Of particular interest, increased risk of PDAC has been associated with germline loss-of-function mutations in BRCA1 and BRCA2, as 4 to 7% of patients with PC have a family history of BRCA mutations [60]. BRCA1/2 mutations have two types of relevance. The first one is in terms of prediction and diagnosis, because their identification as predictive biomarkers allows family members to have genetic counseling [61]. The second one is linked to the field of personalized medicine, because BRCA-related tumors have been observed to have higher sensitivity to poly (ADP-ribose) polymerase (PARP) inhibitors and platinum-based chemotherapies [62].
In this perspective of implementation of new targets for PC, the idea of monitoring the expression profile of the major necroptotic biomarkers could be an additional strategy to differentiate the large number of cases.
Immunohistochemistry assays for in vivo necroptosis detection rely on the use of antibodies that recognize the main actors of the process, RIP1, RIP3 and MLKL in the activated form that includes phosphorylation in specific sites [63].
As regards to markers in liquid biopsies, necroptotic EVs should be involved in this analysis, with the aim to clarify their role in tumor context.
It would be also interesting translating findings obtained in colon cancer by He et al., as mentioned above, to PC [44]. The goal should be that of trying to classify PDAC patients into different necroptosis-related molecular subtypes according to diverse clinical outcomes and tumor microenvironment infiltration characteristics. Expression data for the main pro-necroptotic markers, RIPK1 and RIPK3, could help us to understand more about the stage of tumor, allowing for more individualized and effective anti-cancer treatment strategies. This research is currently undergoing in our laboratories (unpublished data).
5.2. Immunotherapy: is there still a chance?
The principal basis of cancer immunotherapy is to activate patient’s T cells to kill cancer cells, by the identification of tumor-associated antigens. Among the different strategies of immunotherapy there are the use of monoclonal antibodies, cancer vaccines, immune checkpoint inhibitors and immune modulators.
The last frontier of immunotherapy is currently the chimeric antigen receptor (CAR) therapy, an emerging and promising approach to redirect T cells from cancer patients into tumor-specific killer cells [64]. Once removed from the patient’s blood, T cells are engineered in vitro to express artificial receptors, called CAR, which can recognize a specific tumor-associated antigen [65].
Despite validation for hematologic malignancies [66], CAR-T therapy in solid tumors has to face some additional obstacles that involve the immunosuppressive TME, the minimal migration and persistence of CAR-T cells within the tumors and the paucity of ideal targets [67]. Their ineffectiveness is linked in part to CAR-T cell exhaustion in TME [68], a phenomenon that happens also in pancreatic cancer, as it was studied by the group of Good et al. [69].
In the general perspective of immunotherapy, it can be expected that necroptosis of cancer cells could facilitate the activation of immune responses. However, as discussed above, the necroptotic immunobiology is contextually variable and depends on the tumor immune landscape. Desmoplastic reaction surrounding PDAC cells promotes T-cell capture, preventing them from reaching cancer cells [70]. As an effect, activated necroptosis may become an additional enemy to fight, given its potential in exacerbating the infiltration of immune-suppressive cells.
Thus, to validate immunotherapeutic approaches in PC the current mission is that of exploring alternative strategies in the search for new targets in tumor microenvironment that may contribute to disrupt this physical barrier for immune actors.
A consistent strategy may be that of the “normalization of the stroma”: the idea is to turn abnormalities in the tumor stroma, which are involved in tumor resistance to immunotherapy, into a normal-like context, by targeting some drivers of chronic hypoxia and chronic inflammation [71]. For example, retrospective observational cohort studies found that patients with PC, who were already taking angiotensin-converting enzyme inhibitors because of preexisting cardiovascular disease, showed longer survival [72]. This group of blockers, such as losartan, acts by inhibiting the TGF pathway, which is a key activator of fibroblasts associated with the development of fibrosis in cancer [73].
An additional goal would be that of reprogramming activated CAFs into a dormant state [74], as in the case of Froeling et al. who used the all-trans retinoic acid to induce quiescence in fibroblasts, obtaining reduced expansion and increased apoptosis of PDAC cells in murine models [75].
Looking for several strategies to disrupt the communication between cancer cells and stroma, that is hypothesized to be mediated also by necroptotic cells released in PDAC, is still one of the main attractive challenges [76].
6. Conclusions
PC remains one of the most devasting neoplastic diseases with poor prognosis and limited options for successful therapy. The complex genetics, the metastatic potential and the extensive TME that grows around the tumor primary site, combined with the lack of early diagnostic markers and specific symptoms, have made this cancer an unstoppable, terrible force.
A way to better reduce PDAC high resistance to therapy is by trying to penetrate its desmoplastic reaction/TME. An improved understanding of the mechanisms that contribute to maintain secure the PDAC, among which necroptosis, will be fundamental to achieve this goal.
By collecting data from recent works, necroptosis is increasingly emerging as an unexpected source for the tumor to improve its interaction with the surrounding microenvironment, especially with cellular components that may help neoplastic cells to suppress the immune system. Our review supports that this revolutionary tumor-promoting function attributed to neoplastic cells should be explored in the context of pancreatic cancer, where the support given by the TME is decisive for cancer survival, development and spreading. Recent advances in immune and targeted therapy may inspire novel strategies that could in turn increase the efficacy of precision medicine, thereby improving the outcome of different subtype-specific patients. In this perspective, a multidisciplinary approach arises as the one and only way for oncology research to translate findings into concrete and innovative clinical tools and to try to become stronger in this constant battle against PC.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, R.D.P. and V.G.; investigation, V.G. and G.S.; resources, G.S. and S.S.; data curation, V.G. and E.G.; writing—original draft preparation, V.G.; writing—review and editing, R.D.P.; visualization, S.S. and E.G.; supervision, S.A. and R.D.P.; project administration, S.A. and R.D.P.; funding acquisition, R.D.P. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research did not receive external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors wish to thank the medical student Luca Morello for the artwork and usage of BioRender Premium software.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, RL.; Miller, KD.; Wagle, NS.; Jemal, A. Cancer statistics, 2023. CA Cancer J Clin. 2023 Jan;73(1):17-48. [CrossRef]
- Rahib, L.; Smith, BD.; Aizenberg, R.; Rosenzweig, AB.; Fleshman, JM.; Matrisian, LM.; Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913-21. [CrossRef]
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J Gastroenterol. 2016;22(44):9694-9705. [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, EM. Pancreatic Cancer: A Review. JAMA 2021;326(9):851-862. [CrossRef]
- Cascinu, S.; Falconi, M.; Valentini, V.; Jelic, S; ESMO Guidelines Working Group. Pancreatic cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21 Suppl 5:v55-8. [CrossRef]
- Haeberle, L.; Esposito, I. Pathology of pancreatic cancer. Transl Gastroenterol Hepatol. 2019;4:50. [CrossRef]
- Zins, M.; Matos C. and Cassinotto C. Pancreatic adenocarcinoma staging in the era of preoperative chemotherapy and radiation therapy. Radiology 2018; 287(2): 374-390. [CrossRef]
- Bekkali, NLH.; Oppong, KW. Pancreatic ductal adenocarcinoma epidemiology and risk assessment: Could we prevent? Possibility for an early diagnosis. Endosc Ultrasound. 2017;6(Suppl 3):S58-S61. [CrossRef]
- Qian, Y.; Gong, Y.; Fan, Z. et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J Hematol Oncol 2020, 13, 130. [CrossRef]
- Scarpa, A.; Capelli, P.; Mukai K.; Zamboni, G.; Oda, T.; Iacono, C.; Hirohashi, S. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol. 1993;142(5):1534-43.
- Hahn, SA.; Schutte, M.; Hoque, AT.; Moskaluk, CA.; da Costa, LT.; Rozenblum, E.; Weinstein, CL.; Fischer, A.; Yeo, CJ.; Hruban, RH.; Kern, SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996 ;271(5247):350-3. [CrossRef]
- Midha, S.; Chawla, S.; Garg, PK. Modifiable and non-modifiable risk factors for pancreatic cancer: A review. Cancer Lett. 2016;381:269–277. [CrossRef]
- Pothuraju, R.; Rachagani, S.; Junker, WM.; Chaudhary, S.; Saraswathi, V.; Kaur, S.; Batra, SK. Pancreatic cancer associated with obesity and diabetes: An alternative approach for its targeting. J Exp Clin Cancer Res. 2018;37(1):319. [CrossRef]
- Raimondi, S.; Lowenfels, AB.; Morselli-Labate, AM.; Maisonneuve, P.; Pezzilli, R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358 . [CrossRef]
- Bosetti, C.; Lucenteforte, E.; Silverman, DT.; Petersen, G.; Bracci PM, Ji BT, Negri E, Li D, Risch HA, Olson SH; et al. Cigarette smoking and pancreatic cancer: An analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4) Ann Oncol. 2012;23:1880–1888. [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, RC.; Jones, C.; Coleman, HG.; McCain, RS. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol. 2018; 24(43):4846-4861. [CrossRef]
- Ren, B.; Cui, M.; Yang, G.; Wang, H.; Feng, M.; You, L.; Zhao, Y. Tumor microenvironment participates in metastasis of pancreatic cancer. Molecular cancer, 2018; 17(1), 1-15. [CrossRef]
- Hamada, S.; Masamune, A.; Shimosegawa, T. Alteration of pancreatic cancer cell functions by tumor-stromal cell interaction. Front Physiol. 2013;4:318.7. [CrossRef]
- Wang, S.; Zheng, Y.; Yang, F.; Zhu, L.; Zhu, XQ.; Wang, ZF.; Wu, XL.; Zhou, CH.; Yan, JY.; Hu, BY.; Kong, B.; Fu, DL.; Bruns, C.; Zhao, Y.; Qin, LX.; Dong, QZ. The molecular biology of pancreatic adenocarcinoma: Translational challenges and clinical perspectives. Signal Transduct Target Ther. 2021;6(1):249. [CrossRef]
- Sarantis, P.; Koustas, E.; Papadimitropoulou, A.; Papavassiliou, AG.; Karamouzis, MV. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J Gastrointest Oncol. 2020;12(2):173-181. [CrossRef]
- Komuro, H.; Kawai-Harada, Y.; Aminova, S.; Pascual, N.; Malik, A.; Contag, CH.; Harada, M. Engineering Extracellular Vesicles to Target Pancreatic Tissue In Vivo. Nanotheranostics. 2021; 5(4):378-390. [CrossRef]
- Leca, J.; Martinez, S.; Lac, S.; Nigri, J.; Secq, V.; Rubis, M.; Bressy, C.; Sergé, A.; Lavaut, MN.; Dusetti, N.; Loncle, C.; Roques, J.; Pietrasz, D.; Bousquet, C.; Garcia, S.; Granjeaud, S.; Ouaissi, M.; Bachet, JB.; Brun, C.; Iovanna, JL.; Zimmermann, P., Vasseur, S.; Tomasini, R. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J Clin Invest. 2016;126(11):4140-4156. [CrossRef]
- Li, M.; Guo, H.; Wang, Q.; Chen, K.; Marko, K.; Tian, X.; Yang, Y. Pancreatic stellate cells derived exosomal miR-5703 promotes pancreatic cancer by downregulating CMTM4 and activating PI3K/Akt pathway. Cancer Lett. 2020; 490:20-30. [CrossRef]
- Liu, Y.; Cao, X. Characteristics and significance of the pre-metastatic niche. Cancer Cell, 2016; 30:668–681. [CrossRef]
- Hanahan, D.; Weinberg, R. A. The hallmarks of cancer. Cell,2000;100(1), 57-70. [CrossRef]
- Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35(4):495-516. [CrossRef]
- Westphal, S., Kalthoff, H. Apoptosis: Targets in Pancreatic Cancer. Mol Cancer, 2003; 2,6. [CrossRef]
- Lee, SY.; Ju, MK.; Jeon, HM.; Jeong, EK.; Lee, YJ.; Kim, CH.; Park, HG.; Han, SI.; Kang, HS. Regulation of Tumor Progression by Programmed Necrosis. Oxid Med Cell Longev. 2018;2018:3537471. [CrossRef]
- Hanson, B. Necroptosis: A new way of dying? Cancer Biol Ther. 2016;17(9):899-910. [CrossRef]
- Vanlangenakker, N.; Vanden Berghe, T.; Vandenabeele, P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012; 19: 75–86. [CrossRef]
- Di Pietro, R; Zaulia G. Emerging non-apoptotic functions of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)/Apo2L. J Cell Physiol. 2004; 201(3):331-40. [CrossRef]
- Chen, Y.; Ren, W.; Wang, Q.; He, Y.; Ma, D.; Cai, Z. The regulation of necroptosis by ubiquitylation. Apoptosis. 2022;27(9-10):668-684. [CrossRef]
- Sprooten, J.; De Wijngaert, P.; Vanmeerbeerk, I.; Martin, S.; Vangheluwe, P.; Schlenner, S.; Krysko, DV.; Parys, JB.; Bultynck, G.; Vandenabeele, P.; Garg, AD. Necroptosis in Immuno-Oncology and Cancer Immunotherapy. Cells, 2020;9(8):1823. [CrossRef]
- Nugues, AL.; El Bouazzati, H.; He’tuin, D.; Berthon, C.; Loyens, A.; Bertrand. E.; Jouy, N.; Idziorek, T.; Quesnel, B. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis. 2014;5(8):e1384. [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, PJ.; Orlowski, GM.; Chan, FKM. Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis. 2015; 6: e1636–11. [CrossRef]
- Koo, GB.; Morgan, MJ.; Lee, DG.; Kim, WJ.; Yoon, JH.; Koo, JS.; Kim, SI.; Kim, SJ.; Son, MK.; Hong, SS.; Levy, JM.; Pollyea, DA.; Jordan, CT.; Yan, P.; Frankhouser, D.; Nicolet, D.; Maharry, K.; Marcucci, G.; Choi, KS.; Cho, H.; Thorburn, A.; Kim, YS. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25(6):707-25. [CrossRef]
- McCormick, KD.; Ghosh, A.; Trivedi, S.; Wang, L.; Coyne, CB.; Ferris, RL.; Sarkar, SN. Innate immune signaling through differential RIPK1 expression promote tumor progression in head and neck squamous cell carcinoma. Carcinogenesis. 2016;37(5):522-9. [CrossRef]
- Yang, C.; Li, J.; Yu, L,.; Zhang, Z.; Xu, F.; Jiang, L.; Zhou, X.; He, S. Regulation of RIP3 by the transcription factor Sp1 and the epigenetic regulator UHRF1 modulates cancer cell necroptosis. Cell Death Dis. 2017;8(10):e3084. [CrossRef]
- Yang, H.; Ma, Y.; Chen, G.; Zhou, H.; Yamazaki, T.; Klein, C.; Pietrocola, F.; Vacchelli, E.; Souquere, S.; Sauvat, A.; Zitvogel, L.; Kepp, O.; Kroemer, G. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016, 5, e1149673. [CrossRef]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O; Barreira, da Silva, R.; Reis e Sousa, C.; Green, DR.; Oberst, A.; Albert, ML. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8⁺ T cells. Science, 2015;350(6258):328-34. [CrossRef]
- Jin, L.; Chen, J.; Liu, X. Y.; Jiang, C. C.; Zhang, X. D. The double life of RIPK1. Molecular & cellular oncology, 2016; 3(1), e1035690. [CrossRef]
- Jiao, D.; Cai, Z.; Choksi, S.; Ma, D.; Choe, M.; Kwon, HJ.; Baik, JY.; Rowan, BG.; Liu, C.; Liu, ZG. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res. 2018;28(8):868-870. [CrossRef]
- Park, S.; Hatanpaa, KJ.; Xie, Y.; Mickey, BE.; Madden, CJ.; Raisanen, JM:; Ramnarair, DB.; Xiao, G.; Saha, D.; Boothman, RM.; Pieper, RO.; Habib, AA. The receptor interacting protein 1 inhibits p53 induction through NF-κB activation and confers a worse prognosis in glioblastoma. Cancer Res. 2009;69(7):2809–2816. [CrossRef]
- He, R.; Zhang, M.; He, L.; Huang, J.; Man, C.; Wang, X.; Lang. Y.; Fan, Y. Integrated Analysis of Necroptosis-Related Genes for Prognosis, Immune Microenvironment Infiltration, and Drug Sensitivity in Colon Cancer. Front Med (Lausanne), 2022;9:845271. [CrossRef]
- Tong, X.; Tang, R.; Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Targeting cell death pathways for cancer therapy: Recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J Hematol Oncol. 2022;15(1):174. [CrossRef]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, UC.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature, 2016;536(7615):215-8. [CrossRef]
- Korniluk, A.; Koper, O.; Kemona, H.; Dymicka-Piekarska, V. From inflammation to cancer. Ir J Med Sci. 2017;186(1):57-62. [CrossRef]
- Liu, ZG.; Jiao, D. Necroptosis, tumor necrosis and tumorigenesis. Cell Stress, 2019;4(1):1-8. [CrossRef]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, NN.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla ,R.; Daley, D.; Greco, SH.; Torres-Hernandez, A.; Pergamo, M.; Ochi, A.; Zambirinis, CP.; Pansari, M.; Rendon, M.; Tippens, D.; Hundeyin, M.; Mani, VR.; Hajdu, C.; Engle, D.; Miller, G. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature, 2016;532(7598):245-9. [CrossRef]
- Daley, D.; Zambirinis, CP.; Seifert, L.; Akkad, N.; Mohan, N.; Werba, G.; Barilla, R.; Torres-Hernandez, A.; Hundeyin, M.; Mani, VRK.; Avanzi,A.; Tippens, D.; Narayanan, R.; Jang, JE.;Newman, E.; Pillarisetty, VG.; Dustin, ML.; Bar-Sagi, D.; Hajdu, C.; Miller, G. γδ T Cells Support Pancreatic Oncogenesis by Restraining αβ T Cell Activation. Cell. 2016;166(6):1485-1499.e15. [CrossRef]
- Wang, W.; Marinis, JM.; Beal, AM.; Savadkar, S.; Wu,Y.; Khan, M.; Taunk, PS.; Wu, N.; Su, W.; Wu, J.; Ahsan, A.; Kurz, E.; Chen, T.; Yaboh, I.; Li, F.; Gutierrez, J.; Diskin, B.; Hundeyin, M.; Reilly, M.; Lich, JD.; Harris, PA.; Mahajan, MK.; Thorpe, JH.; Nassau, P.; Mosley, JE.; Leinwand, J.; Kochen Rossi, JA.; Mishra, A.; Aykut, B.; Glacken, M.; Ochi, A.; Verma, N.; Kim, JI.; Vasudevaraja, V.; Adeegbe, D.; Almonte, C.; Bagdatlioglu, E.; Cohen, DJ.; Wong, KK.; Bertin, J.; Miller, G. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell. 2018;34(5):757-774.e7. [CrossRef]
- Ando, Y.; Ohuchida, K.; Otsubo, Y.; Kibe, S.; Takesue, S.; Abe, T.; Iwamoto, C.; Shindo, K.; Moriyama, T.; Nakata, K.; Miyasaka, Y.; Ohtsuka, T.; Oda, Y.; Nakamura, M. Necroptosis in pancreatic cancer promotes cancer cell migration and invasion by release of CXCL5. PLoS ONE, 2020;15(1):e0228015. [CrossRef]
- Raden, Y.; Shlomovitz, I.; Gerlic, M. Necroptotic extracellular vesicles—Present and future. Semin Cell Dev Biol. 2021;109:106-113. [CrossRef]
- Harsha, HC.; Kandasamy, K.; Ranganathan, P.; Rani, S.; Ramabadran, S.; Gollapudi, S.; Balakrishnan, L.; Dwivedi, SB.; Telikicherla, D.; Selvan, LD.; Goel, R.; Mathivanan, S.; Marimuthu, A.; Kashyap, M.; Vizza, RF.; Mayer, RJ.; Decaprio, JA.; Srivastava, S.; Hanash, SM.; Hruban, RH.; Pandey, A. A compendium of potential biomarkers of pancreatic cancer. PLoS Med. 2009;6(4):e1000046. [CrossRef]
- Steinberg, W. The clinical utility of the CA 19-9 tumor-associated antigen. Am J Gastroenterol. 1990; 85(4):350-5.
- Lee, T.; Teng, TZJ.; Shelat, VG. Carbohydrate antigen 19-9—Tumor marker: Past, present, and future. World J Gastrointest Surg. 2020 ;12(12):468-490. [CrossRef]
- Micke, O.; Bruns, F.; Kurowski, R.; Horst, E.; deVries, AF.; Hausler, JW.; Willich. N., Schäfer, U. Predictive value of carbohydrate antigen 19-9 in pancreatic cancer treated with radiochemotherapy. Int J Radiat Oncol Biol Phys. 2003;57(1):90-7. [CrossRef]
- Bazzichetto, C.; Luchini, C.; Conciatori, F.; Vaccaro, V.; Di Cello, I.; Mattiolo, P.; Falcone, I.; Ferretti, G.; Scarpa, A.; Cognetti, F.; Milella, M. Morphologic and Molecular Landscape of Pancreatic Cancer Variants as the Basis of New Therapeutic Strategies for Precision Oncology. Int J Mol Sci. 2020;21(22):8841. [CrossRef]
- Ben Aharon, I.; Elkabets, M.; Pelossof, R.; Yu, KH.; Iacubuzio-Donahue, CA.; Leach, SD.; Lowery, MA.; Goodman, KA.; O’Reilly, EM. Genomic Landscape of Pancreatic Adenocarcinoma in Younger versus Older Patients: Does Age Matter? Clin Cancer Res. 2019;25(7):2185-2193. [CrossRef]
- Ghiorzo, P. Genetic predisposition to pancreatic cancer. World J Gastroenterol. 2014;20(31):10778-89. [CrossRef]
- Macchini, M.; Centonze, F.; Peretti, U.; Orsi, G.; Militello, AM.; Valente, MM.; Cascinu, S.; Reni, M. Treatment opportunities and future perspectives for pancreatic cancer patients with germline BRCA1-2 pathogenic variants. Cancer Treat Rev. 2021;100:102262. [CrossRef]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; Gallinger, S. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J Clin Oncol. 2015;33(28):3124-9. [CrossRef]
- He, S.; Huang, S.; Shen, Z. Biomarkers for the detection of necroptosis. Cell Mol Life Sci. 2016;73(11-12):2177-81. [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, AY.; Na, M.; Dyson, KA.; Grippin, AJ.; Deleyrolle, LP.; Zhang, W.; Rajon, DA.; Wang, QJ.; Yang, JC.; Kresak, JL.; Sayour, EJ.; Rahman, M.; Bova, FJ.; Lin, Z.; Mitchell, DA.; Huang, J. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10(1):4016. [CrossRef]
- Guerra, E.; Di Pietro, R.; Basile, M.; Trerotola, M.; Alberti, S. Cancer-Homing CAR-T Cells and Endogenous Immune Population Dynamics. Int J Mol Sci. 2021;23(1):405. [CrossRef]
- Kalos, M.; Levine, BL.; Porter, DL.; Katz, S.; Grupp, SA.; Bagg, A.; June, CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. [CrossRef]
- Ueda, T.; Shiina, S.; Iriguchi, S.; Terakura, S.; Kawai, Y.; Kabai, R.; Sakamoto, S.; Watanabe, A.; Ohara, K.; Wang, B.; Xu, H.; Minagawa, A.; Hotta, A.; Woltjen, K.; Uemura, Y.; Kodama, Y.; Seno, H.; Nakatsura, T.; Tamada, K.; Kaneko, S. Optimization of the proliferation and persistency of CAR T cells derived from human induced pluripotent stem cells. Nat Biomed Eng. 2023;7(1):24-37. [CrossRef]
- Blank, CU.; Haining, WN.; Held, W.; Hogan, PG.; Kallies, A.; Lugli, E.; Lynn, RC.; Philip, M.; Rao, A.; Restifo, NP.; Schietinger, A.; Schumacher, TN.; Schwartzberg, PL.; Sharpe, AH.; Speiser, DE.; Wherry, EJ.; Youngblood, BA.; Zehn, D. Defining ‘T cell exhaustion’. Nat Rev Immunol. 2019;19(11):665-674.
- Good, CR.; Aznar, MA.; Kuramitsu, S.; Samareh, P.; Agarwal, S.; Donahue, G.; Ishiyama, K.; Wellhausen, N.; Rennels, AK.; Ma, Y.; Tian, L.; Guedan, S.; Alexander, KA.; Zhang, Z.; Rommel, PC.; Singh, N.; Glastad, KM.; Richardson, MW.; Watanabe, K.; Tanyi, JL.; O’Hara, MH.; Ruella, M.; Lacey, SF.; Moon, EK.; Schuster, SJ.; Albelda, SM.; Lanier, LL.; Young, RM.; Berger, SL.; June, CH. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell, 2021;184(25):6081-6100.e26. [CrossRef]
- Mortezaee, K. Enriched cancer stem cells, dense stroma, and cold immunity: Interrelated events in pancreatic cancer. J Biochem Mol Toxicol. 2021;35(4):e22708. [CrossRef]
- Mortezaee K. Normalization in tumor ecosystem: Opportunities and challenges. Cell Biol Int. 2021;45(10):2017-2030. [CrossRef]
- Liu, H.; Naxerova, K.; Pinter, M.; Incio, J.; Lee, H.; Shigeta, K.; Ho, WW.; Crain, JA.; Jacobson, A.; Michelakos, T.; Dias-Santos, D.; Zanconato, A.; Hong, TS.; Clark, JW.; Murphy, JE.; Ryan, DP.; Deshpande, V.; Lillemoe, KD.; Fernandez-Del Castillo, C.; Downes, M.; Evans, RM.; Michaelson, J.; Ferrone, CR.; Boucher, Y.; Jain, RK. Use of Angiotensin System Inhibitors Is Associated with Immune Activation and Longer Survival in Nonmetastatic Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2017 ;23(19):5959-5969. [CrossRef]
- Kasi, A.; Allen, J.; Mehta, K.; Dandawate, P.; Saha, S.; Bossmann, S.; Anant, S.; Sun, W. Association of losartan with outcomes in metastatic pancreatic cancer patients treated with chemotherapy. J Clin Transl Res. 2021;7(2):257-262.
- Zhang, T.; Ren, Y.; Yang, P.; Wang, J.; Zhou, H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022;13(10):897. [CrossRef]
- Froeling, FE.; Feig, C.; Chelala, C.; Dobson, R.; Mein, CE.; Tuveson, DA.; Clevers, H.; Hart, IR.; Kocher, HM. Retinoicacid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-β-catenin signaling to slow tumor progression. Gastroenterology, 2011;141(4):1486-97, 1497.e1-14. [CrossRef]
- He. R.; Wang, Z.; Dong, S.; Chen, Z.; Zhou, W. Understanding Necroptosis in Pancreatic Diseases. Biomolecules. 2022;12(6):828. [CrossRef]
Figure 1.
The role of TME in the progression of PDAC. As shown in the figure, once PC arises, it enhances aggressiveness and malignancy by inducing a series of modifications of the surrounding microenvironment. A reciprocal interplay is established between cancer cells and other TME cellular types (CAFs, PSCs, Tregs, TAMs, and MDSCs) through the release of extracellular vesicles (EVs) and pro-inflammatory and immunomodulatory paracrine factors. As a result, the pancreatic stroma becomes denser and more compact (desmoplastic reaction), the degree of inflammation intensifies, and the normal immune response of the body is weakened, thus creating a “protective barrier” that promotes tumor immune-evasion and drug-resistance, and provides a perfect niche for growth, vascularization and metastatic spreading. Furthermore, it is worth noting that cancer cells undergo metabolic reprogramming (Warburg effect) and acquire the ability to replicate indefinitely and to resist to pro-apoptotic stimuli thanks to the progressive accumulation of DNA mutations (genomic instability), especially of oncogenes and tumor suppressor genes.
Figure 1.
The role of TME in the progression of PDAC. As shown in the figure, once PC arises, it enhances aggressiveness and malignancy by inducing a series of modifications of the surrounding microenvironment. A reciprocal interplay is established between cancer cells and other TME cellular types (CAFs, PSCs, Tregs, TAMs, and MDSCs) through the release of extracellular vesicles (EVs) and pro-inflammatory and immunomodulatory paracrine factors. As a result, the pancreatic stroma becomes denser and more compact (desmoplastic reaction), the degree of inflammation intensifies, and the normal immune response of the body is weakened, thus creating a “protective barrier” that promotes tumor immune-evasion and drug-resistance, and provides a perfect niche for growth, vascularization and metastatic spreading. Furthermore, it is worth noting that cancer cells undergo metabolic reprogramming (Warburg effect) and acquire the ability to replicate indefinitely and to resist to pro-apoptotic stimuli thanks to the progressive accumulation of DNA mutations (genomic instability), especially of oncogenes and tumor suppressor genes.
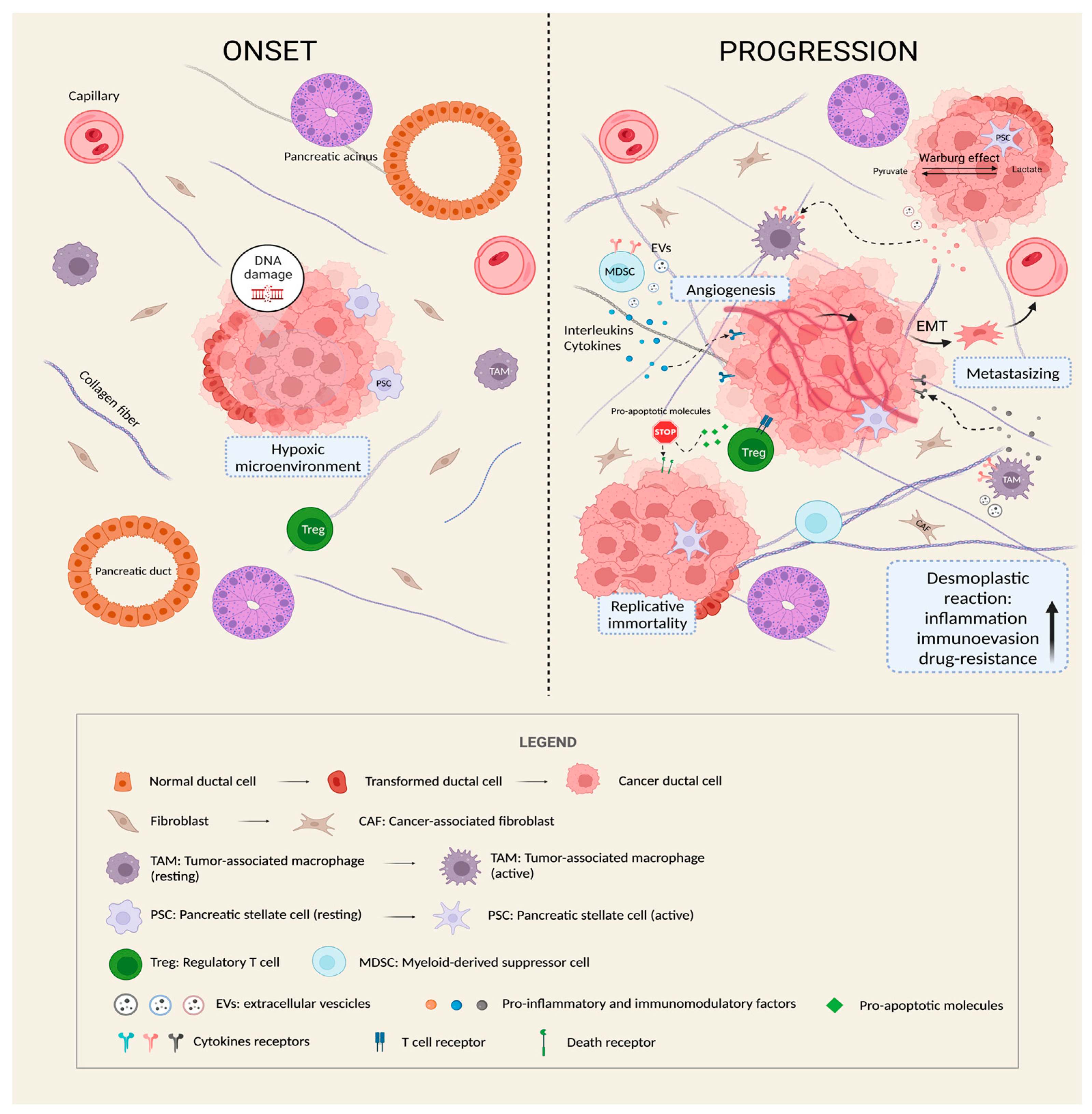
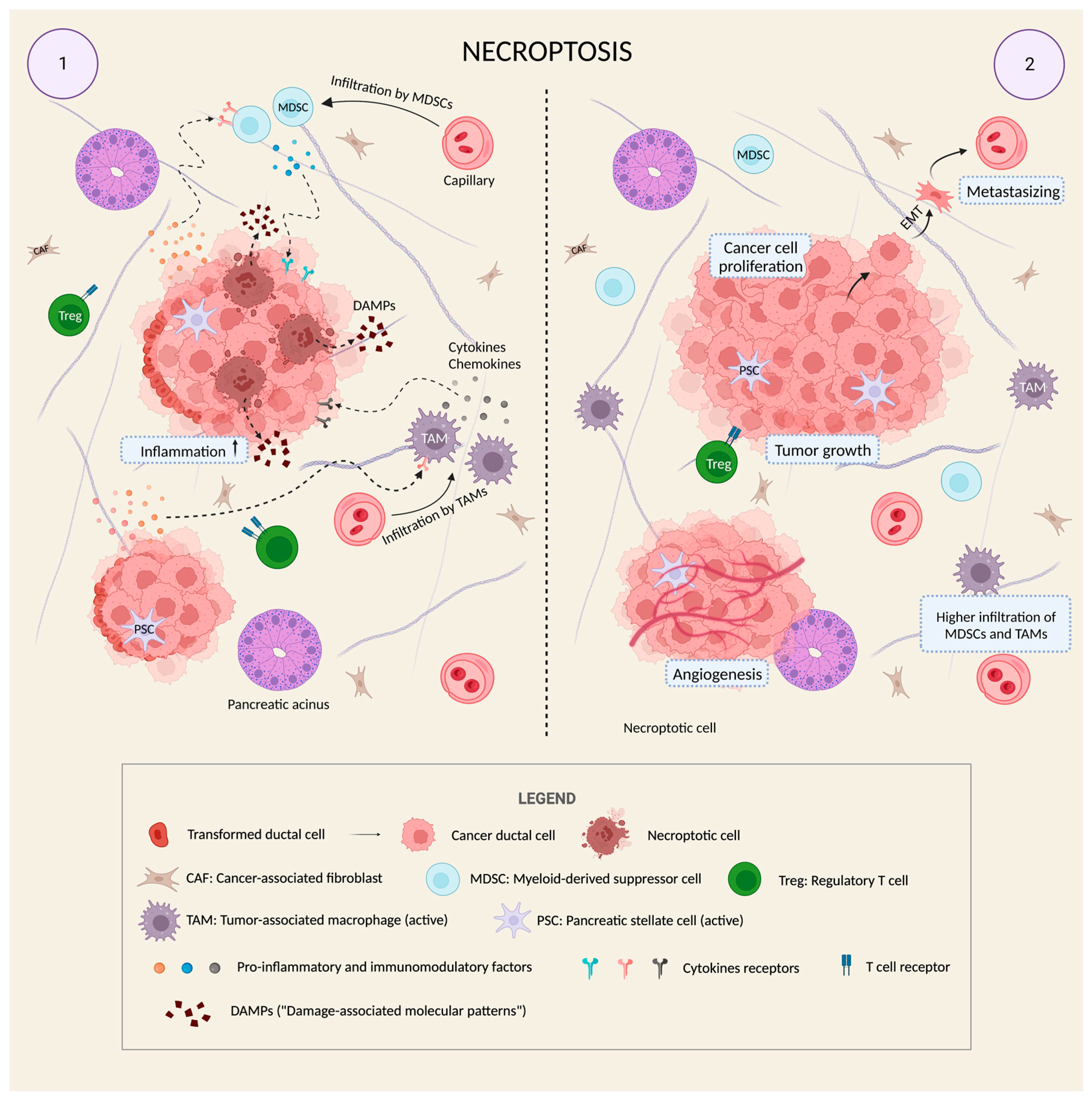
Figure 2.
Necroptosis as an additional means of progression in PDAC. PC may take advantage of a particular cell death mechanism that is able to activate the tumor microenvironment to enhance tumor malignancy. Panel 1: Within an already present desmoplastic reaction that is evident from the fibrous and dense stroma and from the activation of the typical cellular types of the tumor microenvironment (PSCs, CAFs, MDSCs, and TAMs), some cancer cells differ for a characteristic morphological change: these are necroptotic cells (brown cells in panel 1). They have a completely altered internal architecture, but their distinctive feature is the disruption of the plasma membrane which occurs in the form of pores in the membrane itself. Through these pores, the cytoplasm is violently released in the extra-cellular space. Among the wide variety of released material, the DAMPs (“Damage-associated molecular patterns”), which include ATP, heat-shock proteins, and other molecules, can increase the degree of inflammation in the microenvironment and make this inflammation chronic. Cytokines and chemokines released by cancer cells are particularly important, due to their ability to target some of the cellular types of the microenvironment (especially MDSCs and TAMs) thereby regulating their activity from anti-tumoral to pro-tumoral. Panel 2: As a result, thanks to the combination of microenvironment activation and necroptotic death, PDAC creates the perfect conditions to grow, promote angiogenesis, metastasize, and escape the immune response of the body, also generating a self-feeding cycle which supports its inflammatory background and its viability (the increasing infiltration of MDSCs and TAMs because of necroptotic stimuli is made evident in the cartoon).
Figure 2.
Necroptosis as an additional means of progression in PDAC. PC may take advantage of a particular cell death mechanism that is able to activate the tumor microenvironment to enhance tumor malignancy. Panel 1: Within an already present desmoplastic reaction that is evident from the fibrous and dense stroma and from the activation of the typical cellular types of the tumor microenvironment (PSCs, CAFs, MDSCs, and TAMs), some cancer cells differ for a characteristic morphological change: these are necroptotic cells (brown cells in panel 1). They have a completely altered internal architecture, but their distinctive feature is the disruption of the plasma membrane which occurs in the form of pores in the membrane itself. Through these pores, the cytoplasm is violently released in the extra-cellular space. Among the wide variety of released material, the DAMPs (“Damage-associated molecular patterns”), which include ATP, heat-shock proteins, and other molecules, can increase the degree of inflammation in the microenvironment and make this inflammation chronic. Cytokines and chemokines released by cancer cells are particularly important, due to their ability to target some of the cellular types of the microenvironment (especially MDSCs and TAMs) thereby regulating their activity from anti-tumoral to pro-tumoral. Panel 2: As a result, thanks to the combination of microenvironment activation and necroptotic death, PDAC creates the perfect conditions to grow, promote angiogenesis, metastasize, and escape the immune response of the body, also generating a self-feeding cycle which supports its inflammatory background and its viability (the increasing infiltration of MDSCs and TAMs because of necroptotic stimuli is made evident in the cartoon).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



