
Submitted:
17 July 2023
Posted:
17 July 2023
You are already at the latest version
Abstract
Osteitis fibrosa cystica (OFC) and Brown Tumour are two related but distinct types of bone lesions that result from overactivity of osteoclasts most often associated with chronic kidney disease (CKD). Despite their potential consequences, these conditions are poorly understood because of their rare prevalence and variability in their clinical manifestation. Canonically, OFC and Brown Tumours are caused by secondary hyperparathyroidism in CKD. Recent literature showed that multiple factors such as hyperactivation of the renin-angiotensin-aldosterone system and chronic inflammation may also contribute to the occurrence of these diseases through osteoclast activation. Moreover, hotspot KRAS mutations were identified in these lesions placing them in the spectrum of RAS-MAPK-driven neoplasms, while until recently thought to be reactive lesions. Some risk factors contributed to the occurrence of OFC and Brown Tumour such as age, gender, comorbidities, and certain medications. The diagnosis of OFC and Brown Tumour includes clinical symptoms involving chronic bone pain and laboratory finding of hyperparathyroidism. In radiological imaging, the X-ray and Computed tomography (CT) scan could show lytic or multi-lobular cystic alterations. Histologically both lesions are characterized by clustered osteoclast in a fibrotic hemorrhagic background. Based on the latest understanding of the mechanism of OFC, this review elaborates on the manifestation, diagnosis, and available therapies that can be leveraged to prevent the occurrence of OFC and Brown Tumour.
Keywords:
Bone Tumour
; hyperparathyroidism
; giant cell
; osteoclast
1. Introduction
About 90% of patients undergoing dialysis are affected by renal osteodystrophy.1 Osteitis fibrosa cystica (OFC) also known as von Recklinghausen’s disease of bone and Brown Tumour are two related but distinct types of bone lesions that are associated with chronic kidney disease and hyperparathyroidism.2 OFC is characterized by excessive osteoclast activation/production, leading to the resorption of bone and cyst formation.2,3 OFC was first described by Engel in 1864 and von Recklinghausen in 1891, though the first recorded case was in the collection of Hunter who lived from 1718-1783.4,5Brown Tumour is a more aggressive tumour-forming lesion of OFC and is characterized by the presence of brown pigment in the lesion.6,7 The term was first coined as a tumour-like lesion by Henry Jaffe.8 Brown Tumour can present as a solitary lesion, but also multiple lesions within one patient are not uncommon. The pathophysiology of these conditions is not well understood.
Chronic kidney disease is a common condition that is characterized by a progressive loss of kidney function. The most common cause of chronic kidney disease is diabetes mellitus, followed by hypertension and glomerulonephritis.9 Haemodialysis is a common treatment for patients with advanced chronic kidney disease. These patients are at increased risk of developing OFC and Brown Tumours.1,2
Primary hyperparathyroidism is another important factor in the development of OFC and Brown Tumours.10–12 It is a condition in which the (hyperplastic) parathyroid glands or adenoma of the parathyroid produce excessive amounts of parathyroid hormone (PTH), leading to an imbalance in calcium and phosphorus metabolism.13 This can result in the hyperactivation of osteoclasts and the subsequent destruction of bone tissue leading to the development of OFC and Brown Tumours.13,14
The exact mechanisms by which chronic kidney disease and hyperparathyroidism lead to the development of OFC and Brown Tumour are not fully understood. Previous studies have suggested that PTH production and other factors, such as cytokines and growth factors, may play a role in the pathogenesis of these conditions.13,14 Numerous mediators such as NF-κB, receptor activator of NF-κB ligand (RANKL), and macrophage colony-stimulating factor (M-CSF) were involved in the downstream signalling of PTH and calcium homeostasis. 13,14
Here, we aim to provide a comprehensive overview of the current understanding of OFC and Brown Tumour in chronic kidney disease. We start by discussing the epidemiology of these conditions in patients with chronic kidney disease. We will then review the current understanding of the mechanisms by which chronic kidney disease and hyperparathyroidism can lead to the development of OFC and Brown Tumours. Some factors such as hyperparathyroidism, hyperstimulation of the renin-angiotensin-aldosterone system (RAAS), and chronic inflammation are discussed in relation to osteoclast activation. Finally, we will discuss the potential treatment options for these conditions and the challenges and gaps in current knowledge.
2. Epidemiology and Risk Factors
The exact prevalence of OFC and Brown Tumour is not well known, but previous studies have suggested that they are more common in patients with advanced chronic kidney disease who are on hemodialysis.1 Previous studies found that the prevalence of OFC in end-stage renal disease was 32%.15 In contrast, Brown Tumour is relatively rare with a prevalence of about 2% among patients with secondary hyperparathyroidism.16
There are several risk factors associated with an increased risk of developing these conditions, including age, gender, comorbidities, and certain medications. OFC and Brown Tumour are more common in older patients, with a peak incidence in the sixth and seventh decades of life.17–19 This may be due to age-related decline in renal function, which can affect the ability of the kidneys to regulate calcium and phosphorus metabolism.
Gender is also a significant risk factor. Specifically, females are more commonly affected, with a male-to-female ratio of 1:3 in ages below 30 years old.16 The exact reasons for this gender difference are not well understood, but hormonal and genetic factors have been suggested to play a role. The present comorbidities would influence the quality of life of patients with OFC and Brown Tumours. Spontaneous tendon ruptures, pruritus from calcium deposits in the skin, ocular calcification, and calcification of the joints also frequently accompany secondary hyperparathyroidism in CKD.20,21 Certain medications can also increase the risk of developing these conditions. Calcium supplementation, vitamin D deficiency, and the use of glucocorticoids have all been associated with an increased risk of OFC and Brown Tumour in patients with chronic kidney disease.20,21
In summary, age, sex, and certain medications are all significant risk factors for the development of OFC and Brown Tumours in patients with chronic kidney disease, however, the exact mechanisms of how they influence the occurrence and progression of the disease is as yet unknown.
3. The Mechanisms: Beyond Hyperparathyroidism
The exact mechanisms by which chronic kidney disease and hyperparathyroidism lead to the development of OFC and Brown Tumours are not fully understood. Previous studies have suggested that PTH production and other factors, such as renin-angiotensin-aldosterone system (RAAS) activation, cytokine production, and increased growth factors expression, may play a role in the pathogenesis of these conditions.13,14
Secondary hyperparathyroidism
Secondary hyperparathyroidism that classically occurred during CKD is central in the formation of Brown Tumour. The parathyroid glands are responsible to regulate calcium ion homeostasis by modulating bone metabolism, the synthesis of 1α, 25-dihydroxy vitamin D (1α,25(OH)2D) in proximal tubules, and the reabsorption of the calcium ion.
There are several factors involved in the pathogenesis of secondary HPT in CKD. Phosphate retention, hyperphosphatemia, low serum calcium ion, deficiency of 1α,25(OH)2D deficiency, elevated levels of PTH, intestinal calcium malabsorption, the reduction of vitamin D receptors (VDR) and calcium-sensing receptors (CaSR) in the parathyroid glands play a role in the development of secondary hyperparathyroidism.22–28 Furthermore, parathyroid hyperplasia is often present. Based on these observations concerning the pathogenesis, therapy for secondary HPT in the context of CKD and ESRD includes controlling serum phosphate concentrations, administering calcium and vitamin D analogs, and administering calcimimetics.29–32
In hyperparathyroidism, excessive production of PTH leads to an imbalance in calcium and phosphorus metabolism. 10 This can result in the activation of osteoclasts, which are cells that are responsible for the breakdown and removal of bone tissue.17Osteoclasts do not express functional PTH receptors. Therefore, PTH-induced increases in osteoclast activity and number result from non-cell autonomous pathways. The two main cytokines that promote osteoclast differentiation and function are M-CSF and RANKL (Figure 1). It has been established that PTH increases the expression of both of these cytokines.17 The bone microenvironment has numerous biological sources of both of these cytokines, including marrow stromal cells, osteoblasts, resident marrow lymphocytes, and osteocytes. Numerous cell types that express PTH receptors contain the well-researched PTH target gene RANKL. Osteocyte-derived RANKL is necessary for secondary hyperparathyroidism-induced increases in osteoclasts and bone loss. In OFC, excessive activation of osteoclasts leads to the destruction of bone tissue and the formation of cystic lesions.17
In Brown Tumours, the calcium imbalance induces vascular calcification and fragility. The excessive vascularization and focal haemorrhage become the hallmark of the lesion which differentiates them next to their size from OFC. The breakdown of red blood cells caused the accumulation of hemosiderin, appearing as a brown pigment.19,33
RAAS hyperactivity
Other than secondary hyperparathyroidism, individuals with CKD are at high risk of osteoclast activation (Figure 1). One of the leading comorbidities and cardiovascular risk factors in CKD is hypertension due to renin-angiotensin-aldosterone system (RAAS) activation.34,35 Previous studies reported that angiotensin II induces the expression of receptor activator of NFκB ligand (RANKL) in osteoblasts, which caused activation of osteoclast.36
Multinucleated cells of the monocyte/macrophage lineage, which can also be angiotensin II targets, are the source of osteoclasts.37 To direct the differentiation of osteoclasts from their precursors, osteoblasts and stromal cells express RANKL in response to many bone-resorbing agents, including vitamin D3.38 Through cell-to-cell contacts with osteoblasts and stromal cells, osteoclast precursors identify RANKL and develop into mature osteoclasts.36 Thus, targeted disruption of RANKL results in an osteoporotic phenotype and a lack of osteoclasts. Clinical studies have shown that angiotensin-converting enzyme (ACE) inhibitors decrease the risk of fractures and improve bone metabolism.39,40
Inflammatory factors induce osteoclast activation
Patients with CKD are at risk of developing chronic inflammation caused by infection, uraemic milieu, or tissue ischemia.41,42 Intrinsic damage-associated molecular patterns (DAMPs), which are generated by cells after cell death or tissue remodelling, frequently cause this inflammation.41,42 Additionally, various parenchymal cell types express Toll-like receptors (TLRs) and inflammasome components. Numerous pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, and Tumour necrosis factor (TNF)-α are consistently increasing across different stages of CKD.42,43 These trigger vascular dysfunction and the innate immune response, which cause microinflammation. 41,42
Inflammatory factors may also play a role in the development of OFC and Brown Tumours. Previous studies have suggested that immune cell-produced cytokines and growth factors can contribute to excessive osteoclast activation and subsequent destruction of bone tissue (Figure 1). A previous study reported that IL-1β activated osteoclast under the presence of macrophage colony-stimulating factor (M-CSF and RANKL).41Osteoclasts in healthy bone express both type 1 and type 2 IL-1 receptors at similar levels. However, pathologically activated osteoclasts preferentially express the stimulatory IL-1 receptor type 1 which is prone to induce osteoclast activation.41,44
Molecular drivers of tumour formation
Recently it became clear that in the majority of Brown Tumours hotspot somatic KRAS mutations are present.45 This sheds new light on these Tumours which were tilled and then considered being reactive instead of neoplastic lesions. As a hypothesis, the authors suggest that the regression of Brown Tumours after normalization of the hyperparathyroidism is the result of a second genetic hit mediated by endocrine stimulation.45
This mutation in Brown Tumours underpins its relation to non-ossifying fibroma and giant cell granuloma of the jaws which are also driven by RAS/MAPK signalling.46 It may also explain the morphological similarity between the lesions. Interestingly in non-ossifying fibroma, only a subset of the cells carries the mutation. Likewise, in Brown Tumours interplay between reactive and neoplastic cells may play a role in the sustaining of growth and it might be speculated that OFC is the non-neoplastic precursor lesion that can turn into a Brown Tumour following the induction of the KRAS mutation.47, 48
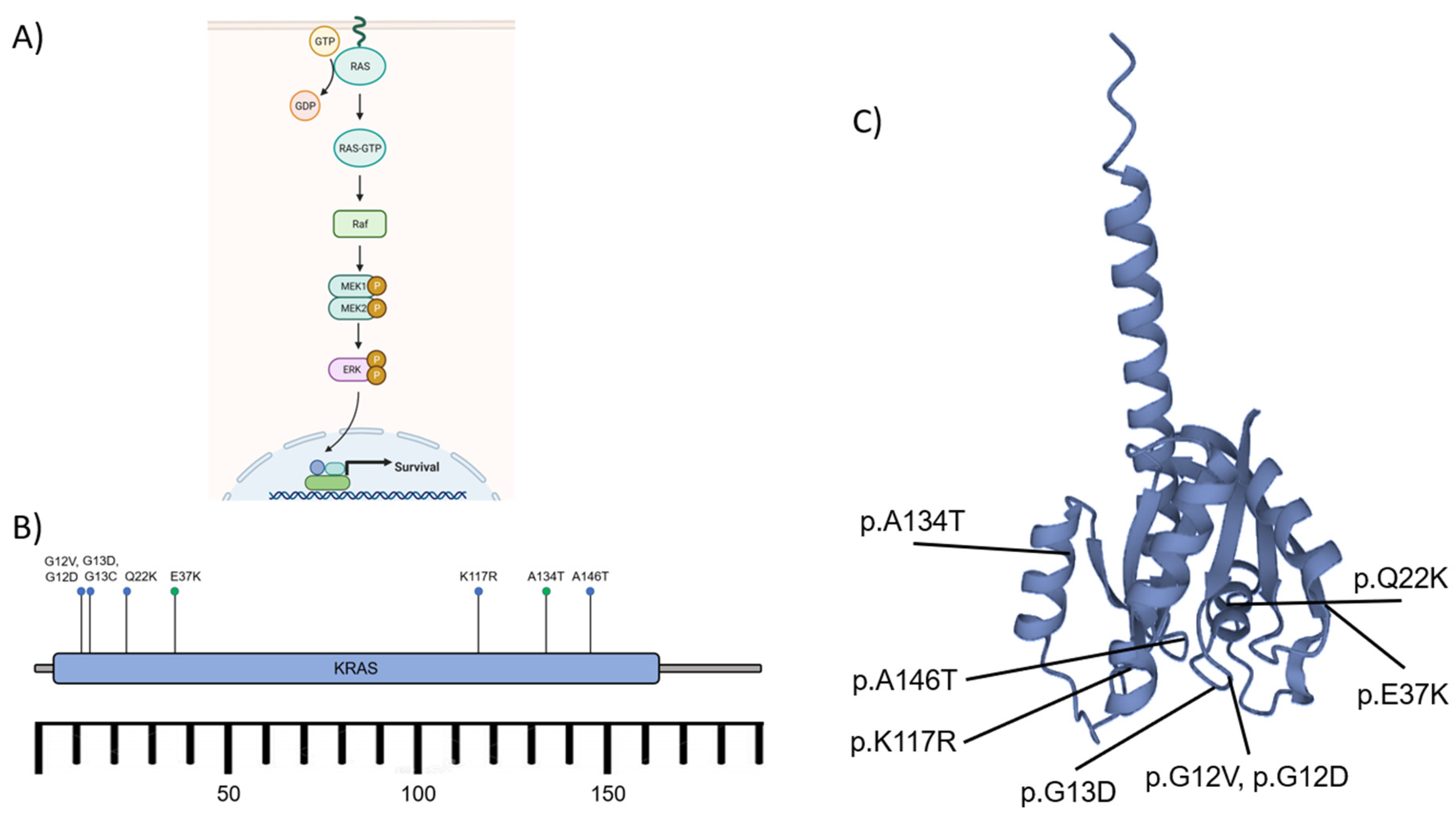
Similar to Giant Cell Lesions, mutations in KRAS have been identified in Brown Tumour which leads to MAPK/ERK activation.47 The most prevalent KRAS activating mutations found, were related to p.A146P/T/V, p.G12D/V, p.G13C/D, p.K117R, and p.Q22K affecting the BT development in both the axial and peripheral areas.45,47 Appealingly, the first two mutations mentioned accounted for more than 60% of frequencies and were seemingly related to colorectal cancer (codon 146) and numerous cancer (codon 12). 45,48,49
Other pathogenic activating mutations such as TRPV4 and FGFR1 have recently been reported in giant-cell lesions of the jaws and non-ossifying fibromas of the bones, which are histologically mimics of Brown Tumour.48 These mutations have been continuously triggering the activation of ERK1/2 in the MAPK pathway which resulted in cell survival.47,48
The involvement of genetic mutation shifted the prior paradigm of Brown Tumour, as non-neoplastic cell growth due to hyperparathyroidism turned into true neoplasms.47,50 However the cause of KRAS mutations in BT remains unclear. KRAS mutations have also been discovered under normal conditions and are associated with aging and smoking.51 Moreover, certain KRAS variants p.A134T and p.E37K also exhibited unknown significance.47
4. Diagnosis: The Role of Radiological Imaging and Lesion Biopsy
OFC and Brown Tumours can have significant consequences if left untreated, including bone pain, fractures, and deformities. Therefore, diagnosing these conditions as early as possible is important to initiate the appropriate treatment and prevent complications.
Radiological Imaging
The diagnosis of OFC and Brown Tumour typically begins with a thorough medical history and physical examination. The presence of chronic kidney disease and hyperparathyroidism should be evaluated, as these conditions are associated with an increased risk of developing these bone lesions.52,53 Imaging studies are also an important part of the diagnostic process. X-ray and computed tomography (CT) can help identify the presence of bone lesions and cysts. OFC is seen on X-rays or CT scans as lytic or multi-lobular cystic alterations. On a CT scan, numerous bony lesions that are Brown Tumours may be incorrectly identified as metastatic cancer, bone cysts, osteosarcoma, and particularly giant-cell tumour of bone.21,52,53 The concurrent measurement of the parathyroid hormone will help to distinguish between primary hyperparathyroidism and cancer.52 However, these imaging modalities may not be sensitive enough to detect early bone changes.
Magnetic resonance imaging (MRI) is a more sensitive imaging modality for the detection of bone lesions in patients with chronic kidney disease. The MRI of patients with OFC shows multiple bony osteolytic lesions and an enhanced mass-like change in the affected bone.12 The MRI can provide detailed information on the extent and severity of bone changes, and it can also help to differentiate between OFC and Brown Tumours.
Tissue Biopsy
A core needle biopsy is the gold standard for the diagnosis of fibrosa cystica osteitis and Brown Tumour.54 The fine needle biopsy (aspiration cytology) may suggest OFC appear as ossifying fibroma displaying spindled neoplastic cells producing trabecular bone and cementoid areas.55–57 Brown Tumours are highly vascularized lesions, histologically characterized by increased -often clustered - osteoclastic activity, haemorrhage, fibrosis, and reactive woven bone formation.55,56 In a Brown Tumour, the degradation of red blood cells will appear as brown pigment as the result of an accumulation of hemosiderin.55,56 The histological differential diagnosis of Brown Tumour includes giant cell Tumour of bone, giant cell reparative granuloma, or solid aneurysmal bone cyst. Clinical details include blood chemistry, radiological examination, and eventually FISH for USP6 rearrangements or immunohistochemistry for H3F3AG34 mutated protein.58
In summary, the diagnosis of OFC and Brown Tumour in chronic kidney disease requires a combination of medical history, physical examination, imaging studies, and biopsy. This approach can help to accurately diagnose these conditions and initiate the appropriate treatment to prevent complications.
5. Treatment: Beyond Removal of the Tumour
Pharmacological Prevention
There are potential pharmacological treatments for OFC and Brown Tumours in chronic kidney disease, ranging from Biphosphonate, Calcimimetics, and Vitamin D supplementation, to Denosumab.
Bisphosphonates are a class of medications that are commonly used to treat osteoporosis and other bone conditions. They work by inhibiting the activity of osteoclasts, which are the cells responsible for the breakdown and removal of bone tissue.59,60 In chronic kidney disease, bisphosphonates can help to prevent excessive osteoclast activation and the subsequent development of OFC and Brown Tumours.59,60
Calcimimetics are a relatively new class of medications that are designed to regulate calcium and phosphorus levels in the body.61 They work by activating the calcium-sensing receptor (CaSR) in the parathyroid glands, which can help reduce the production of PTH and restore the balance of calcium and phosphorus metabolism.61 In chronic kidney disease, calcimimetics can help prevent the development of hyperparathyroidism and the subsequent development of OFC and Brown Tumours.61,62
Vitamin D is an essential nutrient that is involved in the regulation of calcium and phosphorus metabolism. In chronic kidney disease, vitamin D deficiency can contribute to the development of hyperparathyroidism and the subsequent development of OFC and Brown Tumour.63–65 Supplementation with vitamin D can help restore normal levels of calcium and phosphorous.
Denosumab is a monoclonal antibody specifically designed to target the receptor activator of the nuclear factor kappa-B ligand (RANKL), which is a protein that plays a key role in osteoclast activation.66 By blocking the action of RANKL, denosumab can help to reduce osteoclast activity and prevent the development of OFC and Brown Tumours. However, there have not been any studies specifically looking at how well it prevents fractures.67 There have also been reports of denosumab-induced hypocalcemia, which disproportionately affects those with ESKD.67 Lower baseline blood calcium and 25-hydroxyvitamin D levels as well as both low and high bone turnover are risk factors for hypocalcemia with denosumab usage in CKD.67 If considering denosumab, it is crucial to choose the "appropriate patient," supplement with calcium and vitamin D, modify calcium dialysate, and undergo continuous clinical monitoring.
In summary, bisphosphonates, calcimimetics, vitamin D, and denosumab are potential pharmacological treatments for OFC and Brown Tumours in chronic kidney disease. These medications can help to prevent excessive osteoclast activation and restore the balance of calcium and phosphorus metabolism, which can prevent the development of these conditions. More clinical research is needed to determine the effectiveness and safety of these treatments in patients with chronic kidney disease.
Surgical treatment
A meta-analysis of the treatment options for OFC and Brown Tumour involves the removal of the parathyroid gland, cyst drainage, and bone grafting.10
Monoclonal proliferation with nodular hyperplasia and reduced expression of vitamin D and calcium-sensing receptors are likely to present when parathyroid hormone level continues above 800 pg/ml for more than 6 months despite extensive medical interventions.68 Therefore, a surgical parathyroidectomy should be taken into consideration, especially if other conditions like persistent hyperphosphatemia or hypercalcemia, vascular or tissue calcification, including calciphylaxis, and/or progressive osteodystrophy are present. 68
Parathyroidectomy is a surgical procedure involving removing one or more parathyroid glands.69 By removing the source of excess PTH production, parathyroidectomy can help restore the balance of calcium and phosphorus metabolism and prevent the development of these conditions.69 Furthermore, patients on dialysis who get a parathyroidectomy had a 15%–57% higher chance of surviving, as well as improved hypercalcemia, hyperphosphatemia, tissue calcification, bone mineral density, and health-related quality of life.68
In patients with OFC, cystic lesions may form in the affected bones. These cysts can cause pain and discomfortand can also increase the risk of fractures.16 Cyst drainage is a surgical procedure that involves the removal of fluid from these cysts to alleviate symptoms and prevent complications.16
In patients with advanced OFC or Brown Tumour, significant amounts of bone tissue may be lost. This can lead to deformities and a decreased ability to bear weight. Bone grafting is a surgical procedure involving bone tissue transplantation from a donor site to the affected area.16 This can help restore the structural integrity of the bone and improve function.16
6. Conclusions
OFC and Brown Tumours are bone lesion found in CKP patients. The risk factors for developing these two conditions include age, sex, a medication that disrupts calcium metabolism, and vitamin D deficiency. Secondary hyperparathyroidism is the main mechanism leading to the imbalance of calcium and phosphorous level which may cause osteoclast activation. However, other mechanisms such as RAAS hyperactivity and chronic inflammation also possibly contribute to the formation of OFC and Brown Tumours in CKD. Given those mechanisms, pharmacologic treatments such as bisphosphonate, calcimimetics, vitamin D supplementation, and denosumab could be utilized to attenuate hyperparathyroidism, restore the calcium level, and prevent the occurrence of OFC. The rare prevalence of Brown Tumour causes a lack of understanding about the manifestation and treatment of this disease. However, considering the life quality impact of this disease in CKD patients, it is important for nephrologists and medical practitioners working with dialysis patients to be aware of various options of diagnosis and treatment. Furthermore, more research is needed in terms of the comparative efficacy and safety between the treatment options in various populations of CKD patients.
Figure 1.
The schematic diagram of how CKD could induce Osteitis Fibrosa Cystica (OFC).
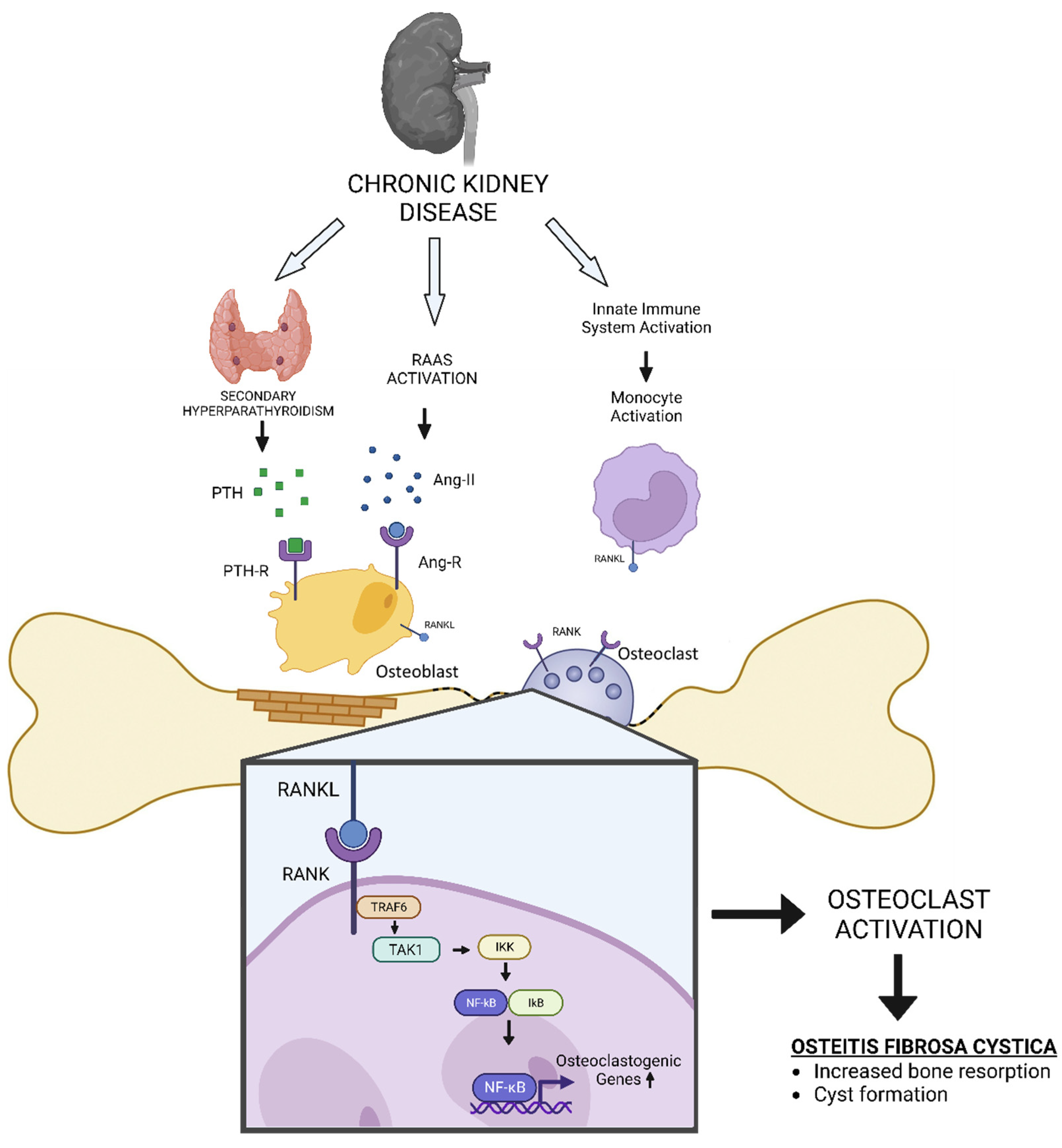
Figure 2.
X-ray of a 66-year-old female showing multiple radiolucency’s in the metacarpals and phalanges and subperiosteal erosions quite typical for hyperparathyroidism.
Figure 2.
X-ray of a 66-year-old female showing multiple radiolucency’s in the metacarpals and phalanges and subperiosteal erosions quite typical for hyperparathyroidism.
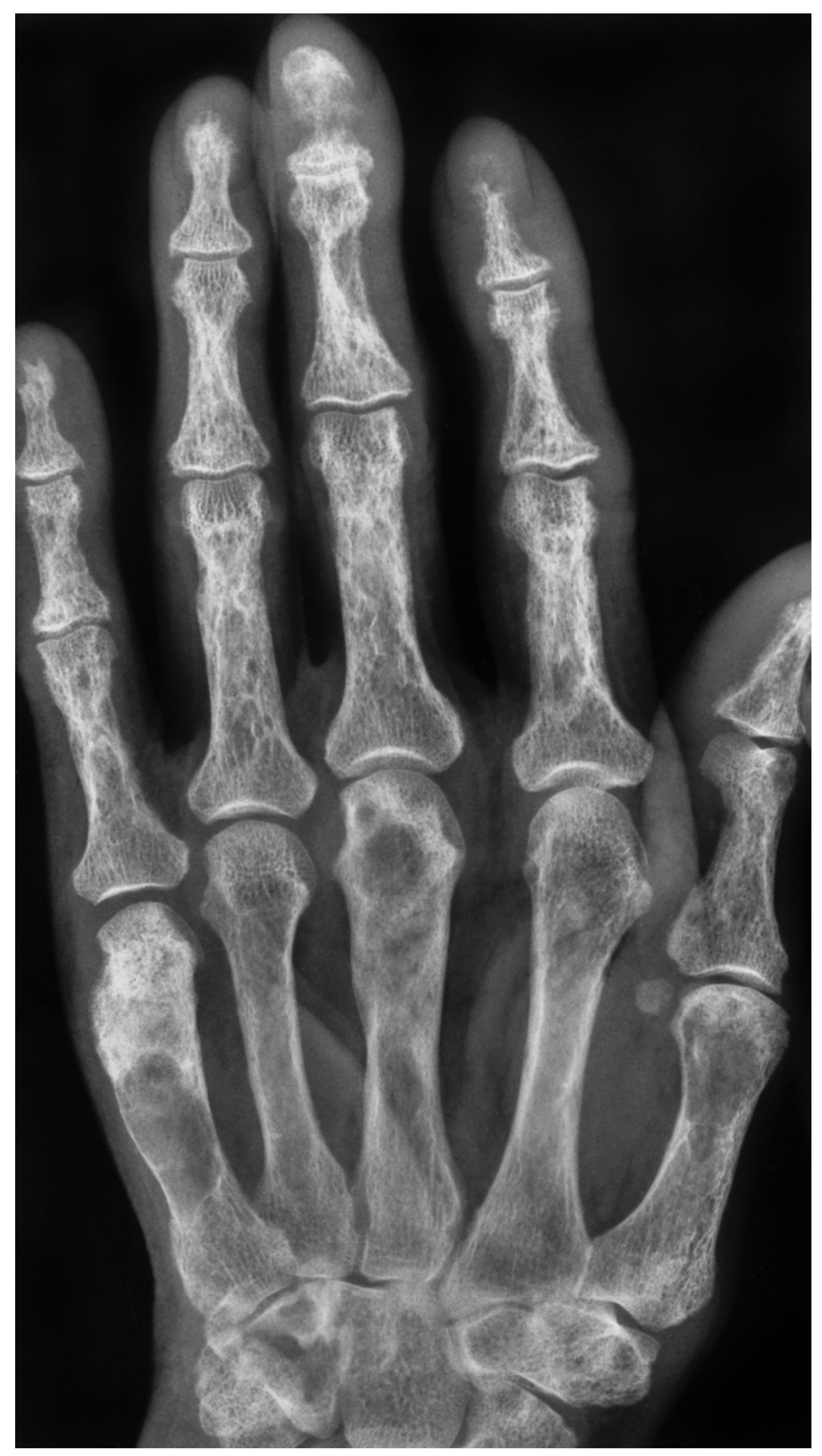
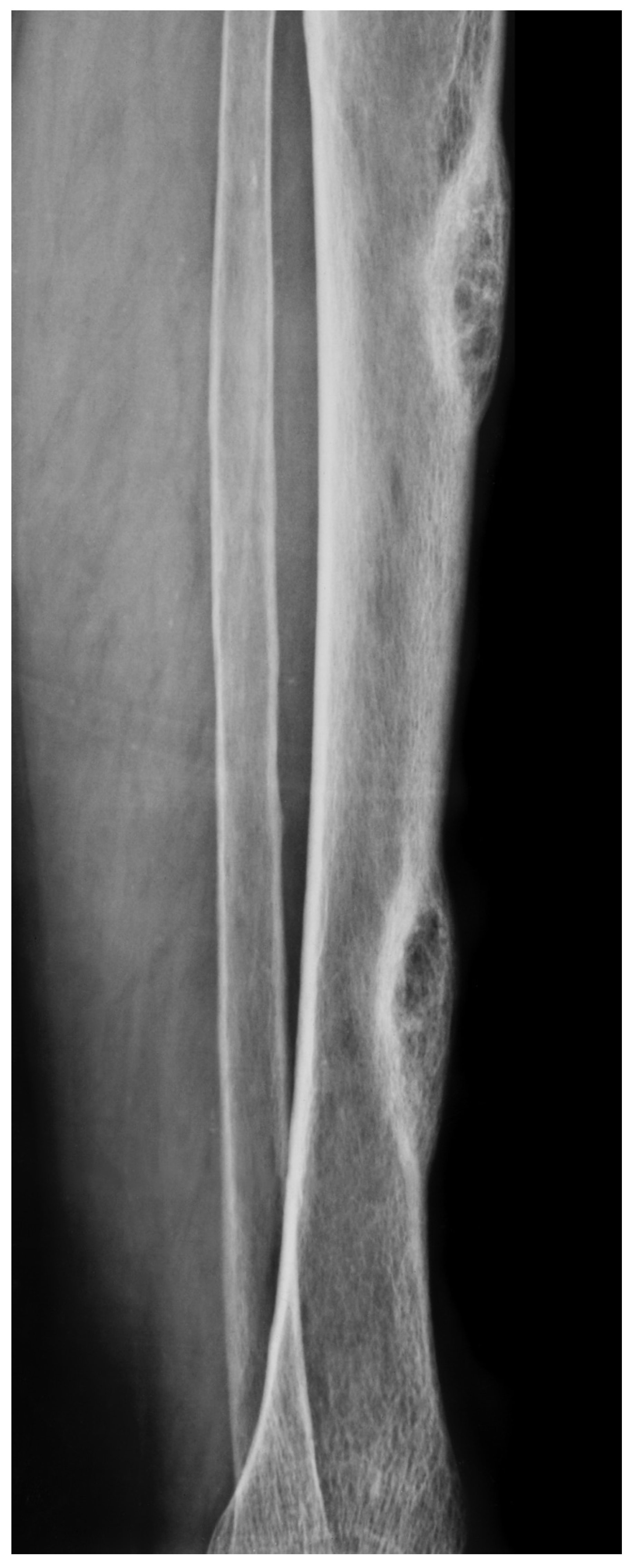
Figure 3.
A. X-ray of a 70-year-old female showing two diaphyseal cortical radiolucency’s with a thick radiopaque inner margin. B. Histology of a typical brown tumour associated with hyperparathyroidism showing scattered multinucleated osteoclasts in a slightly storiform arranged background of spindled and rounded pre-osteoclasts.
Figure 3.
A. X-ray of a 70-year-old female showing two diaphyseal cortical radiolucency’s with a thick radiopaque inner margin. B. Histology of a typical brown tumour associated with hyperparathyroidism showing scattered multinucleated osteoclasts in a slightly storiform arranged background of spindled and rounded pre-osteoclasts.
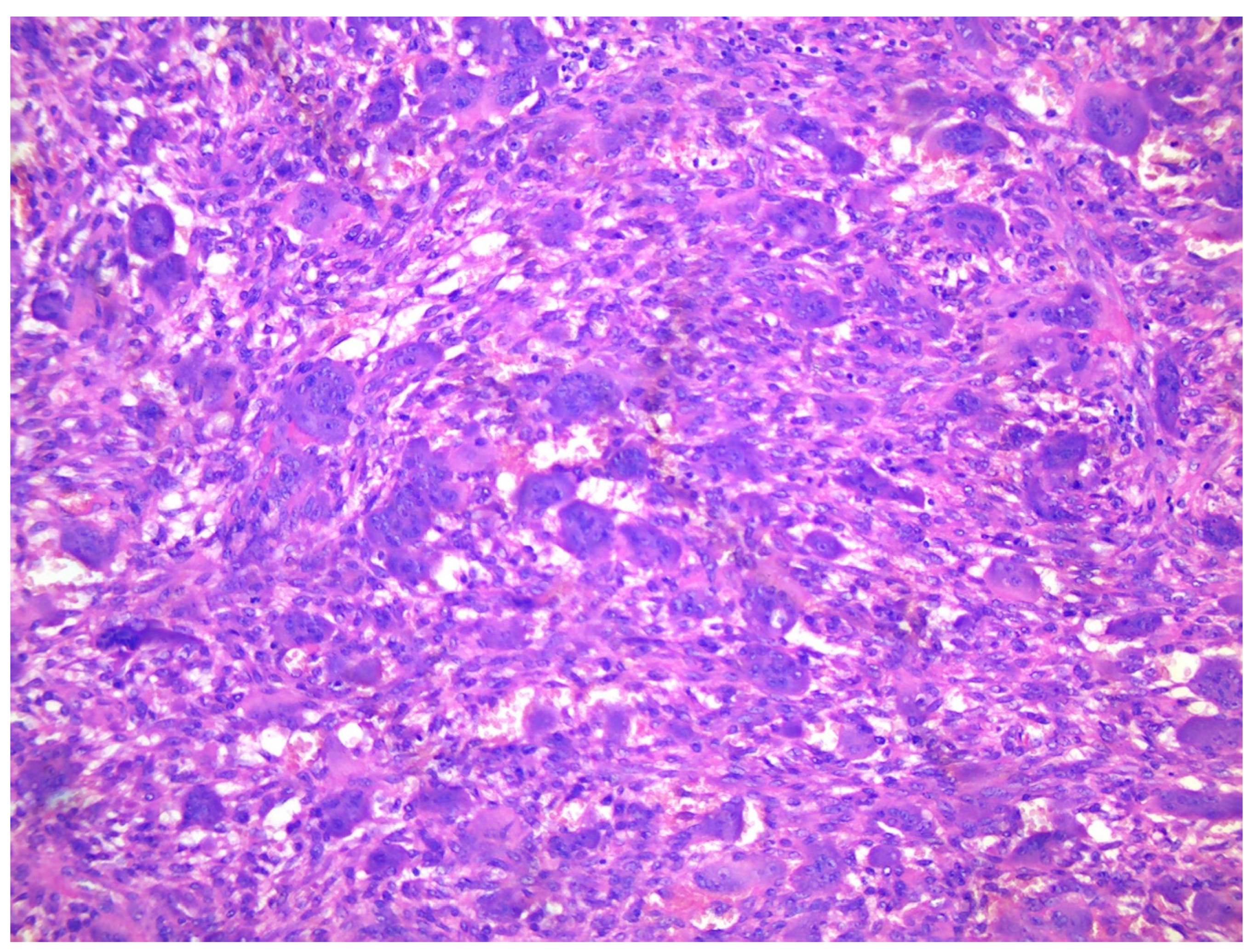
Figure 4.
A) The schematic diagram of the RAS activation to induce MAPK/ERK signaling pathway. The effect of cascade signaling of the pathway leads to nuclear transcription and cell survival. B) The KRAS gene, located on the short arm of chromosome 12, encodes protein K-ras consisting of ~188-189 amino acids. The activating mutations (blue dots) such as p.A146T, p.G12D/V, p.G13C/D, p.K117R, and p.Q22K affected Brown Tumor development. Some other known mutations (green dots) p.A134T and p.E37K exhibited no specific significance. C) K-ras protein and the mutation sites. GTP: Guanosine triphosphate, GDP: Guanosine diphosphate, Raf: rapidly accelerated fibrosarcoma kinases (related to Ser/Thr kinases), MEK1/2 (MAPK): Mitogen-activated protein kinase 1/2, ERK: extracellular signal-regulated kinase, P: phosphate (after phosphorylation), KRAS: Kirsten rat sarcoma viral oncogene homolog.45, 47, 48,70.
Figure 4.
A) The schematic diagram of the RAS activation to induce MAPK/ERK signaling pathway. The effect of cascade signaling of the pathway leads to nuclear transcription and cell survival. B) The KRAS gene, located on the short arm of chromosome 12, encodes protein K-ras consisting of ~188-189 amino acids. The activating mutations (blue dots) such as p.A146T, p.G12D/V, p.G13C/D, p.K117R, and p.Q22K affected Brown Tumor development. Some other known mutations (green dots) p.A134T and p.E37K exhibited no specific significance. C) K-ras protein and the mutation sites. GTP: Guanosine triphosphate, GDP: Guanosine diphosphate, Raf: rapidly accelerated fibrosarcoma kinases (related to Ser/Thr kinases), MEK1/2 (MAPK): Mitogen-activated protein kinase 1/2, ERK: extracellular signal-regulated kinase, P: phosphate (after phosphorylation), KRAS: Kirsten rat sarcoma viral oncogene homolog.45, 47, 48,70.
Acknowledgments
We would also like to express our sincere appreciation and gratitude to Jefferson Caesario, MD for his invaluable contribution to the editing of this manuscript.
References
- Antonelli JR, Hottei TL. Oral manifestations of renal osteodystrophy: case report and review of the literature. Spec care Dent. 2003;23(1):28–34. [CrossRef]
- Lehmann G, Ott U, Stein G, Steiner T, Wolf G. Renal osteodystrophy after successful renal transplantation: a histomorphometric analysis in 57 patients. In: Transplantation proceedings. Elsevier; 2007. p. 3153–8. [CrossRef]
- Tarrass F, Benjelloun M, Bensaha T. Severe jaw enlargement associated with uremic hyperparathyroidism. Hemodial Int. 2008;12(3):316–8. [CrossRef]
- Endel G. Ueber einen fall von cystoider entartung des ganzen skelettes. Giessen, Ger FC Pietsch. 1864;
- Von Recklinghausen F. Die fibrose oder deformierende Ostitis, die Osteomalacic und die osteoplastische Carcinose, in ihren gegenseitigen Beziehungen. Rudolf Virchow Festschriften. 1891;1–89.
- Fatma L Ben, Barbouch S, Fethi BH, Imen BA, Karima K, Imed H, et al. Brown tumors in patients with chronic renal failure and secondary hyperparathyroidism: report of 12 cases. Saudi J Kidney Dis Transplant. 2010;21(4):772.
- Morrone LF, Ettorre GC, Passavanti G, Tampoia M, Schiavone P, Coratelli P. Maxillary brown tumor in secondary hyperparathyroidism requiring urgent parathyroidectomy. J Nephrol. 2001;14(5):415–9.
- Jaffe HL, Lichtenstein L. Benign chondroblastoma of bone: a reinterpretation of the so-called calcifying or chondromatous giant cell tumor. Am J Pathol. 1942;18(6):969.
- Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, Jafar TH, Heerspink HJL, Mann JF, et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet. 2013;382(9889):339–52. [CrossRef]
- Baracaldo RM, Bao D, Iampornpipopchai P, Fogel J, Rubinstein S. Facial disfigurement due to osteitis fibrosa cystica or brown tumor from secondary hyperparathyroidism in patients on dialysis: A systematic review and an illustrative case report. Hemodial Int. 2015;19(4):583–92. [CrossRef]
- Nunes TB, Bologna SB, Witzel AL, Nico MMS, Lourenço SV. A rare case of concomitant maxilla and mandible Brown tumours, papillary thyroid carcinoma, parathyroid adenoma, and osteitis fibrosa cystica. Case Rep Dent. 2016;2016. [CrossRef]
- Lee JH, Chung SM, Kim HS. Osteitis fibrosa cystica mistaken for malignant disease. Clin Exp Otorhinolaryngol. 2013;6(2):110. [CrossRef]
- Kumar R, Thompson JR. The regulation of parathyroid hormone secretion and synthesis. J Am Soc Nephrol. 2011;22(2):216–24. [CrossRef]
- Quinn SJ, Thomsen ARB, Pang JL, Kantham L, Bräuner-Osborne H, Pollak M, et al. Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am J Physiol Metab. 2013;304(3):E310–20. [CrossRef]
- Jat JA, Mal P, Kumar D. Renal osteodystrophy in end stage renal failure patients on maintenance haemodialysis. J Clin Exp Nephrol. 2016;1(4):25. [CrossRef]
- Satpathy AS, Dasgupta A, Dutta C, Mohan NVK, Satpathy S. Osteitis fibrosa cystica of mandible in hyperparathyroidism-jaw tumor syndrome: A rare presentation and review of literature. Natl J Maxillofac Surg. 2017;8(2):162. [CrossRef]
- Jervis L, James M, Howe W, Richards S. Osteolytic lesions: osteitis fibrosa cystica in the setting of severe primary hyperparathyroidism. Case Reports. 2017;2017:bcr-2017. [CrossRef]
- Mellouli N, Belkacem Chebil R, Darej M, Hasni Y, Oualha L, Douki N. Mandibular osteitis fibrosa cystica as first sign of vitamin D deficiency. Case Rep Dent. 2018;2018. [CrossRef]
- Kemp AMC, Bukvic M, Sturgis CD. Fine Needle Aspiration Diagnosis of Osteitis Fibrosa Cystica (Brown Tumor of Bone). Acta Cytol. 2008;52(4):471–4.
- Crutchlow WP, David DS, Whitsell J. Multiple skeletal complications in a case of chronic renal failure treated by kidney homotransplantation. Am J Med. 1971;50(3):390–4. [CrossRef]
- Pfeifer M, Begerow B, Minne HW, Abrams C, Nachtigall D, Hansen C. Effects of a short-term vitamin D and calcium supplementation on body sway and secondary hyperparathyroidism in elderly women. J Bone Miner Res. 2000;15(6):1113–8. [CrossRef]
- Bricker NS, Slatopolsky E, Reiss E, Avioli L V. Calcium, Phosphorus, and Bone in Renal Disease and Transplantation. Arch Intern Med. 1969;123(5):543–53. [CrossRef]
- Reiss E, Canterbury JM, Kanter A. Circulating Parathyroid Hormone Concentration in Chronic Renal Insufficiency. Arch Intern Med. 1969;124(4):417–22. [CrossRef]
- Brown AJ, Dusso A, Lopez-Hilker S, Lewis-Finch J, Grooms P, Slatopolsky E. 1,25-(OH)2D receptors are decreased in parathyroid glands from chronically uremic dogs. Kidney Int. 1989;35(1):19–23. [CrossRef]
- Cantley LK, Russell J, Lettieri D, Sherwood LM. 1,25-Dihydroxyvitamin D3 Suppresses Parathyroid Hormone Secretion from Bovine Parathyroid Cells in Tissue Culture. Endocrinology. 1985;117(5):2114–9. [CrossRef]
- Naveh-Many T, Bell O, Silver J, Kilav R. Cis and trans acting factors in the regulation of parathyroid hormone (PTH) mRNA stability by calcium and phosphate. FEBS Lett. 2002 Oct 2;529(1):60–4.
- Kumar R, Thompson JR. The Regulation of Parathyroid Hormone Secretion and Synthesis. J Am Soc Nephrol [Internet]. 2011 Feb 1 [cited 2022 Dec 4];22(2):216–24. [CrossRef]
- Ritter CS, Martin DR, Lu Y, Slatopolsky E, Brown AJ. Reversal of secondary hyperparathyroidism by phosphate restriction restores parathyroid calcium-sensing receptor expression and function. J bone Miner Res Off J Am Soc Bone Miner Res. 2002 Dec 1;17(12):2206–13. [CrossRef]
- Shoben AB, Rudser KD, De Boer IH, Young B, Kestenbaum B. Association of oral calcitriol with improved survival in nondialyzed CKD. J Am Soc Nephrol. 2008;19(8):1613–9. [CrossRef]
- Block GA, Martin KJ, De Francisco ALM, Turner SA, Avram MM, Suranyi MG, et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med. 2004;350(15):1516–25. [CrossRef]
- Moe SM, Drüeke TB. Management of secondary hyperparathyroidism: the importance and the challenge of controlling parathyroid hormone levels without elevating calcium, phosphorus, and calcium-phosphorus product. Am J Nephrol. 2003;23(6):369–79. [CrossRef]
- Locatelli F, Cannata-Andía JB, Drüeke TB, Hörl WH, Fouque D, Heimburger O, et al. Management of disturbances of calcium and phosphate metabolism in chronic renal insufficiency, with emphasis on the control of hyperphosphataemia. Nephrol Dial Transplant. 2002;17(5):723–31.
- Fineman I, Johnson JP, Di-Patre P-L, Sandhu H. Chronic renal failure causing brown tumors and myelopathy: case report and review of pathophysiology and treatment. J Neurosurg Spine. 1999;90(2):242–6.
- Leelahavanichkul A, Yan Q, Hu X, Eisner C, Huang Y, Chen R, et al. Angiotensin II overcomes strain-dependent resistance of rapid CKD progression in a new remnant kidney mouse model. Kidney Int. 2010;78(11):1136–53. [CrossRef]
- Ruggenenti P, Cravedi P, Remuzzi G. Mechanisms and treatment of CKD. J Am Soc Nephrol. 2012;23(12):1917–28. [CrossRef]
- Shimizu H, Nakagami H, Osako MK, Hanayama R, Kunugiza Y, Kizawa T, et al. Angiotensin II accelerates osteoporosis by activating osteoclasts. FASEB J. 2008;22(7):2465–75. [CrossRef]
- Hatton R, Stimpel M, Chambers TJ. Angiotensin II is generated from angiotensin I by bone cells and stimulates osteoclastic bone resorption in vitro. J Endocrinol. 1997;152(1):5–10. [CrossRef]
- Hiruma Y, Inoue A, Hirose S, Hagiwara H. Angiotensin II stimulates the proliferation of osteoblast-rich populations of cells from rat calvariae. Biochem Biophys Res Commun. 1997;230(1):176–8. [CrossRef]
- Rejnmark L, Vestergaard P, Mosekilde L. Treatment with beta-blockers, ACE inhibitors, and calcium-channel blockers is associated with a reduced fracture risk: a nationwide case–control study. J Hypertens. 2006;24(3):581–9.
- Lynn H, Kwok T, Wong SYS, Woo J, Leung P. Angiotensin converting enzyme inhibitor use is associated with higher bone mineral density in elderly Chinese. Bone. 2006;38(4):584–8. [CrossRef]
- Shiratori T, Kyumoto-Nakamura Y, Kukita A, Uehara N, Zhang J, Koda K, et al. IL-1β induces pathologically activated osteoclasts bearing extremely high levels of resorbing activity: a possible pathological subpopulation of osteoclasts, accompanied by suppressed expression of Kindlin-3 and Talin-1. J Immunol. 2018;200(1):218–28.
- Zewinger S, Schumann T, Fliser D, Speer T. Innate immunity in CKD-associated vascular diseases. Nephrol Dial Transplant. 2016;31(11):1813–21. [CrossRef]
- Boswell JM, Yui MA, Burt DW, Kelley VE. Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. J Immunol. 1988;141(9):3050–4. [CrossRef]
- Xu LX, Kukita T, Nakano Y, Yu H, Hotokebuchi T, Kuratani T, et al. Osteoclasts in normal and adjuvant arthritis bone tissues express the mRNA for both type I and II interleukin-1 receptors. Lab Invest. 1996;75(5):677–87.
- Turek D, Haefliger S, Ameline B, Alborelli I, Calgua B, Hartmann W, Harder D, Flanagan AM, Amary F, Baumhoer D. Brown Tumors Belong to the Spectrum of KRAS -driven Neoplasms. Am J Surg Pathol. 2022 Nov 1;46(11):1577-1582. [CrossRef]
- Bovée JV, Hogendoorn PC. Non-ossifying fibroma: A RAS-MAPK driven benign bone neoplasm. J Pathol. 2019 Jun;248(2):127-130. [CrossRef]
- Guimarães LM, Gomes IP, Pereira Tdos, Andrade BA, Romañach MJ, Lacerda JC, et al. Kras mutations in brown tumor of the jaws in hyperparathyroidism. Journal of Oral Pathology & Medicine. 2020;49(8):796–802. [CrossRef]
- Gomes CC, Gayden T, Bajic A, Harraz OF, Pratt J, Nikbakht H, et al. TRPV4 and KRAS and FGFR1 gain-of-function mutations drive giant cell lesions of the jaw. Nature Communications. 2018;9(1). [CrossRef]
- Haigis KM. Kras alleles: The Devil is in the detail. Trends in Cancer. 2017;3(10):686–97.
- Baumhoer D, Kovac M, Sperveslage J, Ameline B, Strobl AC, Krause A, et al. Activating mutations in the map-kinase pathway define non-ossifying fibroma of Bone. The Journal of Pathology. 2019;248(1):116–22.
- Risques RA, Kennedy SR. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 2018;14:e1007108.
- Misiorowski W, Czajka-Oraniec I, Kochman M, Zgliczyński W, Bilezikian JP. Osteitis fibrosa cystica—a forgotten radiological feature of primary hyperparathyroidism. Endocrine. 2017;58(2):380–5.
- Hsieh M-C, Ko J-Y, Eng H-L. Pathologic fracture of the distal femur in osteitis fibrosa cystica simulating metastatic disease. Arch Orthop Trauma Surg. 2004;124(7):498–501. [CrossRef]
- van der Bijl AE, Taminiau AH, Hermans J, Beerman H, Hogendoorn PC. Accuracy of the Jamshidi trocar biopsy in the diagnosis of bone tumors. Clin Orthop Relat Res. 1997 Jan;(334):233-43. [CrossRef]
- Parfitt J, Harris M, Wright JM, Kalamchi S. Tumor suppressor gene mutation in a patient with a history of hyperparathyroidism–jaw tumor syndrome and healed generalized osteitis fibrosa cystica: a case report and genetic pathophysiology review. J Oral Maxillofac Surg. 2015;73(1):194-e1. [CrossRef]
- Kashkari S, Kelly TR, Bethem D, Pepe RG. Osteitis fibrosa cystica (brown tumor) of the spine with cord compression: report of a case with needle aspiration biopsy findings. Diagn Cytopathol. 1990;6(5):349–53. [CrossRef]
- Watson CW, Unger P, Kaneko M, Gabrilove JL. Fine needle aspiration of osteitis fibrosa cystica. Diagn Cytopathol. 1985;1(2):157–60. [CrossRef]
- Yang L, Zhang H, Zhang X, Tang Y, Wu Z, Wang Y, Huang H, Fu X, Liu J, Hogendoorn PCW, Cheng H. Clinicopathologic and molecular features of denosumab-treated giant cell tumour of bone (GCTB): Analysis of 21 cases. Ann Diagn Pathol. 2022 Apr;57:151882. [CrossRef]
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol. 2009;4(1):221–33. [CrossRef]
- Cremers S, Drake MT, Ebetino FH, Bilezikian JP, Russell RGG. Pharmacology of bisphosphonates. Br J Clin Pharmacol. 2019;85(6):1052–62.
- Evenepoel P. Calcimimetics in chronic kidney disease: evidence, opportunities and challenges. Kidney Int. 2008;74(3):265–75. [CrossRef]
- Ballinger AE, Palmer SC, Nistor I, Craig JC, Strippoli GFM. Calcimimetics for secondary hyperparathyroidism in chronic kidney disease patients. Cochrane Database Syst Rev. 2014;(12). [CrossRef]
- Jean G, Souberbielle J, Chazot C. Vitamin D in Chronic Kidney Disease and Dialysis Patients. Nutrients [Internet]. 2017 Mar 25;9(4):328. [CrossRef]
- González EA, Sachdeva A, Oliver DA, Martin KJ. Vitamin D Insufficiency and Deficiency in Chronic Kidney Disease. Am J Nephrol [Internet]. 2004;24(5):503–10. [CrossRef]
- Bosworth C, de Boer IH. Impaired Vitamin D Metabolism in CKD. Semin Nephrol [Internet]. 2013;33(2):158–68.
- Zaheer S, LeBoff M, Lewiecki EM. Denosumab for the treatment of osteoporosis. Expert Opin Drug Metab Toxicol [Internet]. 2015 Mar 4;11(3):461–70. [CrossRef]
- Gopaul A, Kanagalingam T, Thain J, Khan T, Cowan A, Sultan N, et al. Denosumab in chronic kidney disease: a narrative review of treatment efficacy and safety. Arch Osteoporos [Internet]. 2021 Dec 28;16(1):116. [CrossRef]
- Lau WL, Obi Y, Kalantar-Zadeh K. Parathyroidectomy in the Management of Secondary Hyperparathyroidism. Clin J Am Soc Nephrol [Internet]. 2018 Jun 7;13(6):952 LP – 961. [CrossRef]
- Apetrii M, Goldsmith D, Nistor I, Siriopol D, Voroneanu L, Scripcariu D, et al. Impact of surgical parathyroidectomy on chronic kidney disease-mineral and bone disorder (CKD-MBD) – A systematic review and meta-analysis. Shimosawa T, editor. PLoS One [Internet]. 2017 Nov 6;12(11):e0187025. [CrossRef]
- The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Research. 2023 Jan 6; 51(1): 523–31. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



