Submitted:
14 July 2023
Posted:
17 July 2023
You are already at the latest version
Abstract
Multiple myeloma (MM) is an incurable, diverse cancer in which abnormal plasma cells produce and release monoclonal immunoglobulin (Ig), also known as monoclonal protein or M protein. MM is always preceded by monoclonal gammaglobulinemia of unknown significance (MGUS) and smoldering multiple myeloma (SMM). Diagnosis of MM is made from a bone marrow sample, which involves analysis of the appearance of plasma cells. Quantification of CD138+ plasma cells in core biopsies is performed using techniques such as immunohistochemistry, flow cytometry, fluorescence in situ hybridization (FISH) and conventional cytogenetics. Urinalysis includes total protein by immunofixation and 24-hour urine by serum protein electrophoresis. FISH probes should detect the presence of at least del 13, del 17p, t(4;14), t(11;14), t(14;16), and 1q21 amplifications. Indeed, next-generation sequencing (NGS) has become an increasingly important tool in the diagnosis and treatment of MM. It can be used to identify genetic mutations, copy number changes, and clonal structures to help determine disease prognosis and treatment strategies. NGS can identify subclonal populations of cancer cells that are resistant to therapy and may lead to disease relapse, and can be used to identify potential targets for personalized therapy. For example, NGS can identify mutations in genes involved in drug resistance, enabling the selection of drugs that may be more effective in treating a patient's cancer. This systematic review analyzes 182 papers to determine the position and role of NGS in the diagnosis of MM. We first analyze the platforms and sample types used in the selected studies, and then start discussing the results.
Keywords:
multiple myeloma
; next generation sequencing
; whole genome sequencing
; whole exome se-quencing
; myTYPE panel
; minimal residual disease.
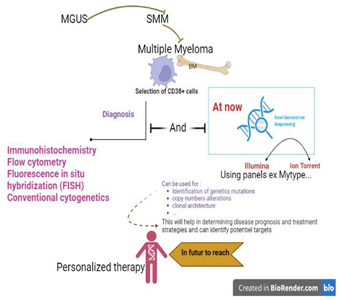
1. Introduction and Etiology of Multiple Myeloma
Multiple myeloma is an incurable and heterogeneous malignancy. It is distinguished by the abnormal plasma cells producing and secreting a monoclonal immunoglobulin (Ig), referred to as monoclonal protein or M-protein [1]. Monoclonal gammaglobulinemia of unknown significance (MGUS) and smoldering multiple myeloma (SMM) precede MM, representing clinically detectable but asymptomatic premalignant conditions. The progression from these conditions to malignant MM in certain patients is not well understood [2]. MM represents approximately 1% of the total cancer cases [3] and constitutes approximately 13-15% of all hematologic malignancies, making it the second most prevalent cancer following non-Hodgkin lymphoma [4]. MM is considered rare among young patients, with less than 2% of MM cases occurring before age 40 years at diagnosis [1].
The etiology of multiple myeloma is poorly understood. It includes a lot of factors like: life style (obesity, diet, use of hormonal therapy, use of alcohol and tobacco…), the occupation and environment of the patient (agriculture and farming, pesticides, organic solvents, radiation, hair coloring products exposure, working in the cosmetic or hair-dressers industry…), chronic immune stimulation or autoimmune pathology, family history, genetic variation, monoclonal gammopathy of undetermined significance… [2].
2. Epidemiology of Multiple Myeloma
2.1. Incidence
The occurrence of MM varies depending on the geographic location [4]. The regions showing the highest standardized incidence rates (ASIR) for MM were Australasia (5.8; 95% uncertainty interval [UI], 4.4-6.5), high-income North America (5.2; 95% UI, 4.7-6.5), and Western Europe (4.6; 95% UI, 3.7-5.5) [6].
In 2016, the global number of newly reported MM cases was 138,509 (95% uncertainty interval [UI], 121,000-155,480), and the age-standardized incidence rate (ASIR) was 2.1 per 100,000 individuals (95% UI, 1.8-2.3) [3].
In 2018, the Global Cancer Observatory data estimated approximately 160,000 fresh instances of MM, representing 0.9% of all cancer diagnoses. Out of the total number of cases, around 90,000 were among males, while approximately 70,000 were among females, resulting in age-standardized incidence rates of 2.1/100,000 and 1.4/100,000, respectively. The cumulative risk of being diagnosed with MM from birth to the age of 74 was 0.24% for men and 0.17% for women [7].
According to the most recent statistics provided by the Global Cancer Observatory, (GLOBOCAN) in 2020, an approximate estimation of 176,404 new cases of MM was recorded, representing 0.9% of all cancer diagnoses. Out of these cases, approximately 98,000 were male and 77,000 were female, resulting in age-standardized incidence rates of 2.2/100,000 and 1.5/100,000, respectively. The cumulative risk of being diagnosed with MM from birth to the age of 74 was 0.25% for men and 0.17% for women, indicating that men are 1.5 times more likely to develop the condition compared to women [8].
It is worth noting that worldwide, annually, there are more than 120,000 newly diagnosed MM (NDMM) patients [4].
2.2. Mortality
In 2016, there were 98,437 global deaths attributed to multiple myeloma, with an age-standardized death rate (ASDR) of 1.5/100,000 individuals (95% uncertainty interval [UI], 1.3-1.7) [6].
In 2018, it was estimated that there were approximately 106,000 deaths from multiple myeloma, comprising 1.1% of the total number of deaths caused by cancer. Among these deaths, around 59,000 of the individuals were male, while approximately 47,000 were female, this led to age-standardized mortality rates of 1.3 per 100,000 for males and 0.9 per 100,000 for females, respectively. For men, the risk of death from MM was 0.15%, while for women, it was 0.10% [7].
According to the statistics for 2020, it is estimated that there were approximately 117,000 deaths due to multiple myeloma, accounting for 1.2% of all cancer-related deaths during that year. Out of these deaths, 65,197 were males and 51,880 were females, resulting in age-standardized mortality rates of 1.4/100,000 and 0.9/100,000, respectively. The risk of death from MM was 0.15% for men and 0.10% for women [5].
2.3. Risk factors
2.3.1. Age
The majority of multiple myeloma cases occur in older adults, with a median age of onset typically around 69 years. Among those diagnosed, 34.8% are above the age of 75, and 9.6% are above 85 years old [9]. At the time of death, the median age for individuals with MM is 74 years, and the interquartile range (the range between the 25th and 75th percentiles) is from 65 to 81 years [10].
The concept of clonal hematopoiesis (CH) was introduced 25 years ago by Busque et al. In their study, they observed an increase in acquired skewing of X-chromosome inactivation as individuals aged, particularly after the age of 60. Additionally, they discovered that age-dependent acquired mutations in the TET2 gene contribute to non-random skewing of X-chromosome inactivation. Following this, several age-related somatic mutations have been identified in the blood cells of healthy individuals who do not meet the diagnostic criteria for a hematologic neoplasm. Initially, this condition was referred to as “age-related clonal hematopoiesis” (ARCH) [6].
2.3.2. Sex
2.3.3. Race
In the United States: When compared to White individuals, Black individuals face twice the risk of developing MM. The differences in MM incidence rates for other races in the US are not as significant. Asians have a noticeably lower incidence rate of MM compared to non-Hispanic Whites (3.8 per 100,000 versus 6.2 per 100,000) and Hispanics have a slightly higher incidence rate of multiple myeloma compared to Whites, with a rate of 6.7 cases per 100,000 population. However, multiple studies have shown that Black individuals in the US have a significantly higher prevalence of MGUS compared to Whites, particularly at younger ages [7].
3. Diagnosis Multiple Myeloma
3.1. Clinical Features
Multiple myeloma is a malignant condition that specifically impacts the plasma cells. (B lymphocytes), a type of white blood cell found in the bone marrow. B cells produce antibodies that help fight infections and diseases. In multiple myeloma, cancerous B cells multiply and build up in the bone marrow, leading to a decrease in the production of normal blood cells and an increase in abnormal plasma cells and as result, an excessive production of dysfunctional intact immunoglobulins or immunoglobulin chains.
Multiple myeloma symptoms may include bone pain, weakness, fatigue, and anemia. Other possible symptoms include increased risk of infections, kidney problems, and nerve damage.
The primary complication observed in individuals with MM is infection, surpassing all other causes in terms of both morbidity and mortality. The most prevalent infections are bacterial pneumonia, arising from a complex interplay of factors including the patient’s overall health, environmental conditions, the nature of the disease itself, and the efficacy of treatment. Diagnostic assessment for MM involves conducting laboratory tests, urinalysis, bone marrow biopsy (utilizing the IMWG updated criteria for multiple myeloma diagnosis), and radiological examinations [9].
3.2. Laboratory Evaluation in MM
The first step in conducting laboratory tests for multiple myeloma involves a comprehensive analysis of the blood. This includes examining various factors such as the complete blood count. Additionally, it is crucial to measure certain substances in the blood, namely serum creatinine, serum calcium, albumin, lactate dehydrogenase, serum free light chain, β2-microglobulin levels, and 24-hour urine protein electrophoresis by immunofixation. Among patients diagnosed with multiple myeloma, around 86% exhibit monoclonal proteins in their blood, which refers to the presence of abnormal antibodies.
To assess the impact of elevated kappa and lambda free light chains on organ damage, it is recommended to conduct serum-free light chain assays to quantify their levels. It is crucial to initially measure the levels of monoclonal protein and free light chains in the bloodstream to establish the extent of the disease and evaluate the response to treatment. Monitoring these levels over time serves as an indicator of treatment effectiveness. A small percentage, approximately 1% to 2% of patients, may not exhibit any detectable serum or urine markers, referred to as “non-secretory” multiple myeloma.
Flow cytometry is utilized to identify abnormal circulating B cells, which is particularly important when considering a diagnosis of B cell leukemia.
In all patients suspected of having multiple myeloma, it is recommended to perform a bone marrow aspiration and biopsy on one side. The analysis of the bone marrow includes examining the morphology of plasma cells, quantifying CD138+ plasma cells in core biopsies using immunohistochemistry, as well as employing techniques such as flow cytometry, fluorescence in situ hybridization (FISH), and conventional cytogenetics.
For urinalysis, it is essential to assess total protein levels using immunofixation and to collect a 24-hour urine sample for serum protein electrophoresis.
FISH probes should be capable of detecting specific abnormalities, such as the presence of deletions (del) in chromosomes 13 and 17p, translocations (t) such as t(4;14), t(11;14), and t(14;16), as well as amplifications in chromosome 1q21. The identification of loss and rearrangement can also be valuable for assessing the risk associated with the disease [3].
3.3. Imaging Investigation
X-Rays: Skeletal lesions can be classified into four types. Single lesion (plasmacytoma), diffuse skeletal lesion (myeloma), diffuse skeletal osteopenia, and sclerosing myeloma.
Computed tomography (CT) scans are more sensitive than X-rays and can detect lesions with less than 5% trabecular destruction. In addition, it helps assess the involvement of surrounding soft tissue.
Whole-body magnetic resonance imaging (MRI) is a highly sensitive and specific imaging technique used to detect bone disease [3].
4. NGS
4.1. Definition and Brief History of NGS
Next-Generation Sequencing (NGS) is an advanced technology known for its exceptional capacity and rapidity. It is utilized to decipher the sequence of nucleotides in entire genomes or specific regions of DNA or RNA. NGS has completely transformed and enhanced the field of life sciences, enabling laboratories to explore a wide range of applications and study biological systems at an unprecedented level.
The study of intricate cellular instructions in today’s complex scientific inquiries requires a depth of information that traditional DNA sequencing technologies are unable to provide. NGS has successfully bridged that gap and has now become a commonplace tool for addressing and understanding these complex questions [10].
NGS technology became accessible in the early 21st century, bringing with it significant advancements. One of the most notable breakthroughs was its capability to generate vast quantities of data, surpassing the limitations of traditional Sanger methods. In addition, NGS introduced a highly efficient, rapid, cost-effective, and accurate approach to DNA sequencing.
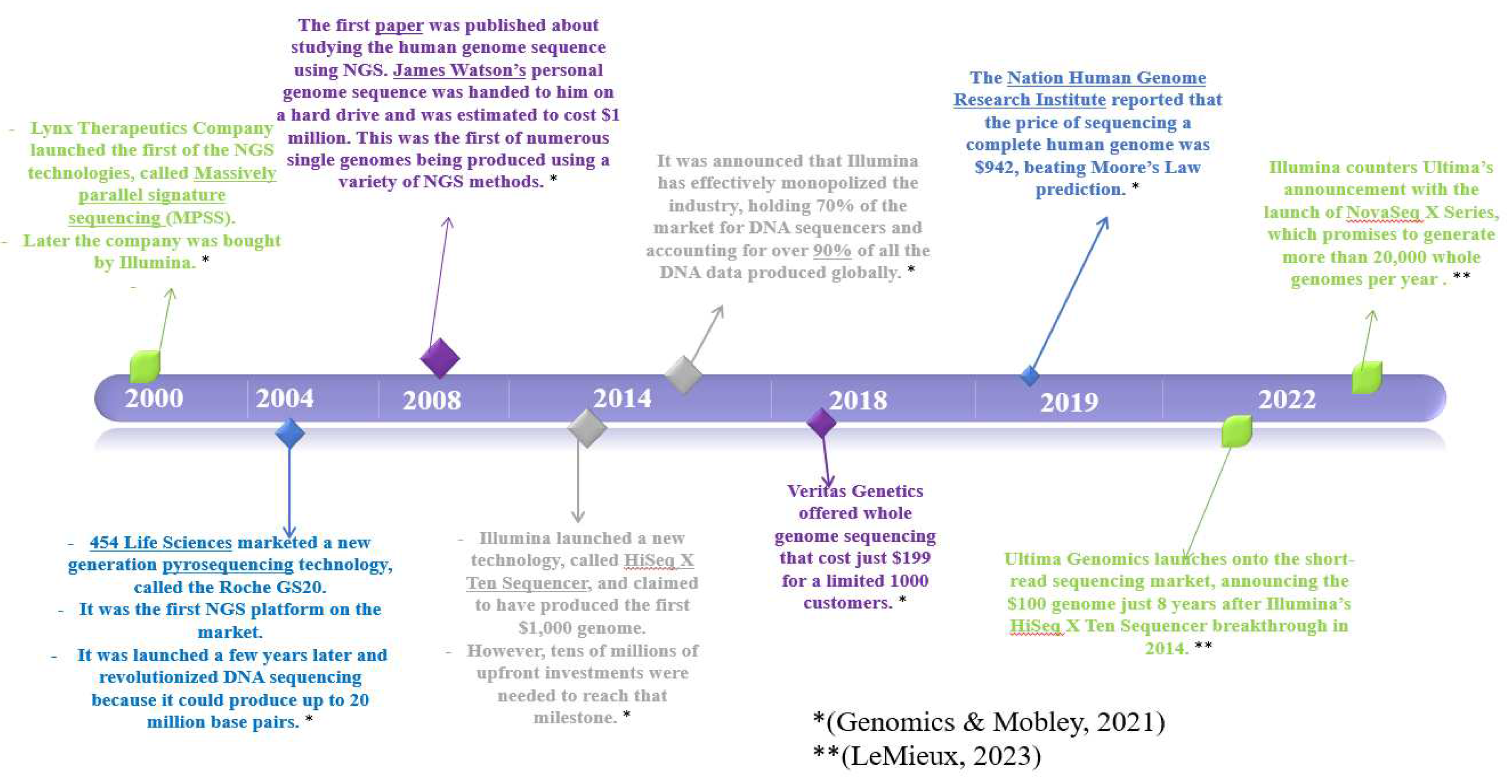
4.2. Key Moments in Evolution of NGS Technology
4.3. NGS Platforms and Techniques for MM
4.3.1. Introduction
Next-generation sequencing (NGS), refers to technology of the high-throughput sequencing which enables a large number of DNA templates(millions to billions) to be sequenced at the same time, and to generate a considerable amount of genetic information in a single run [13]. Two NGS platforms (Ion Torrent and Illumina) are to mention, including NGS library preparation, data analysis, and validation. Nowadays, the choice of NGS platform is one of the first decisions a laboratory must make. In terms of installation costs, chemistry, read lengths, sequencing capacities, instrument footprints, and sequencing time.
4.3.2. Illumina Sequencing Platform
The Illumina sequencing platform, also known as Illumina sequencing or Illumina technology, is a widely used next-generation sequencing (NGS) technology that has revolutionized genomic research and applications. It is based on the principle of sequencing-by-synthesis, where DNA fragments are amplified and sequenced in parallel.
The Illumina sequencing platform utilizes a massively parallel sequencing approach, allowing millions of DNA fragments to be sequenced simultaneously on a single run. The process involves several key steps:
Library Preparation: DNA samples are fragmented and adapters are added to the DNA fragments. These adapters contain sequences that allow the DNA fragments to bind to the surface of the flow cell.
Cluster Generation: The adapter-ligated DNA fragments are immobilized on a flow cell surface, with each fragment occupying a distinct location known as a cluster. Through bridge amplification, the DNA clusters are amplified to create clonal clusters, where each cluster contains many identical DNA fragments.
Sequencing: The sequencing process begins by using reversible terminators. Fluorescently labeled nucleotides are added one at a time, and the incorporation of each nucleotide is detected by imaging the emitted fluorescence. After detection, the fluorescent dye and blocking group are chemically removed, allowing the next nucleotide to be added.
Image Processing and Base Calling: The fluorescent images captured during sequencing are processed to generate raw data. The intensities of the fluorescence signals are translated into DNA base calls, resulting in a sequence of DNA fragments for each cluster.
Data Analysis: The generated sequences are then aligned to a reference genome or assembled de novo to obtain the complete DNA sequence. Various bioinformatics tools and software are used for data analysis, including read mapping, variant calling, and identification of structural variations [14].
The Illumina sequencing platform offers high throughput, generating large amounts of sequencing data with high accuracy and quality. It has been widely adopted in various fields of genomics research, including whole-genome sequencing, exome sequencing, transcriptome profiling, epigenetics, metagenomics, and many other applications. The platform’s flexibility, scalability, and cost-effectiveness have made it a popular choice for diverse genomic studies.
4.3.3. Ion Torrent sequencing platform
The Ion Torrent sequencing platform, developed by Thermo Fisher Scientific, is a next-generation sequencing (NGS) technology that utilizes semiconductor-based sequencing to determine DNA or RNA sequences. It is based on the principle of detecting hydrogen ions (protons) released during DNA synthesis, enabling the identification of nucleotides.
The Ion Torrent sequencing platform operates in the following steps:
Library Preparation: Similar to other NGS technologies, the first step involves preparing the DNA or RNA sample by fragmenting it and attaching adapter sequences. These adapters facilitate the attachment of the DNA or RNA fragments to beads or microparticles.
Template Preparation: The DNA or RNA fragments are amplified through emulsion polymerase chain reaction (PCR) or clonal amplification. Each bead or microparticle carries multiple copies of the same DNA or RNA fragment, resulting in clonal populations.
Sequencing: The prepared templates are loaded onto a semiconductor chip, with each bead or microparticle occupying a separate well or ion-sensitive field-effect transistor (ISFET). During sequencing, the addition of nucleotides triggers DNA synthesis. If the added nucleotide matches the template DNA or RNA strand, a hydrogen ion is released as a byproduct. The released ions generate a detectable change in pH, which is measured by the ISFET.
Data Acquisition and Analysis: The changes in pH are converted into electrical signals and interpreted as DNA sequences. The data is processed and analyzed using bioinformatics tools to align the reads to a reference genome, detect variants, and perform other downstream analyses [15].
The Ion Torrent sequencing platform offers advantages such as speed, simplicity, and scalability. It has been used in various applications, including targeted sequencing, whole-exome sequencing, microbial genomics, cancer research, and genetic disease studies. The technology is known for its relatively low instrument cost and flexibility, making it accessible for a wide range of research laboratories and applications.
4.5. Minimal Residual Disease (MRD) in Multiple Myeloma
Minimal residual disease (MRD) denotes the presence of a limited quantity of cancer cells within a patient’s body following treatment, even if these cells are undetectable using conventional tests or imaging techniques. In the context of multiple myeloma, MRD testing is a way to monitor the effectiveness of treatment and predict the likelihood of relapse.
MRD testing in multiple myeloma typically involves analyzing bone marrow samples for the presence of clonal cells using highly sensitive techniques, such as next-generation sequencing or flow cytometry. The goal of MRD testing is to determine whether a patient has achieved a deep response to treatment, meaning that there are very few or no detectable cancer cells remaining.
Patients who achieve a negative MRD status (i.e., no detectable cancer cells) have been shown to have significantly better outcomes, including longer progression-free survival and overall survival, compared to those who still have MRD-positive disease. As such, MRD testing is becoming an increasingly important tool in the management of multiple myeloma.
It’s worth noting that MRD testing is not yet widely available or routinely used in all clinical settings, but it is a field of study that is currently undergoing extensive research. and is likely to become more widely adopted in the future as the technology and understanding of the disease continue to advance [17,18,19,20].
4.6. Types of MRD Tests
In case of multiple myeloma (MM), there are several types of MRD tests that can be used to detect and measure the disease. The most commonly used tests for MRD in MM are:
Next-generation sequencing (NGS)-based MRD: This method uses high-throughput DNA sequencing to detect and quantify cancer cells in the blood or bone marrow. NGS-based MRD tests are highly sensitive and can detect very low levels of residual disease, making them useful for monitoring treatment response and predicting patient outcomes [18,21,22,23].
Flow cytometry-based MRD: analysis involves examining blood or bone marrow cells using a flow cytometer to identify any remaining myeloma cells. This technique is known for its efficiency and simplicity, allowing for convenient monitoring of disease progression and response to treatment [24,25,26].
While qPCR is commonly used for monitoring MRD in lymphoproliferative disorders, it has a notable drawback in that it depends on a serial dilution standard curve for the quantification of the target. However, the latest iteration of PCR, called droplet digital PCR (ddPCR), has introduced several practical advantages over qPCR. Importantly, ddPCR enables direct and precise quantification of the target without requiring a reference standard curve. Furthermore, recent research has demonstrated that ddPCR exhibits comparable sensitivity, accuracy, and reproducibility to qPCR when assessing MRD in mature lymphoproliferative disorders. Given its broader applicability and reduced labor intensity compared to qPCR, ddPCR shows promise as an alternative method for evaluating MRD [27].
PCR-based MRD: This technique amplifies specific DNA sequences that are unique to the myeloma cells, allowing for the detection of small numbers of residual cancer cells. PCR-based MRD tests are highly sensitive and specific, and can be used to monitor treatment response and predict patient outcomes [28,29].
The choice of MRD method may depend on factors such as the stage of the disease, the availability of technology in the healthcare facility, and the patient’s individual circumstances.
5. Methods
5.1. Search Strategy
We conducted a search in accordance with the guidelines outlined by the Preferred Reporting Items for Systematic Reviews and Meta-analysis (PRISMA) reporting guideline. PubMed and Web of Science were searched using relevant terms such as “Multiple myeloma” and “Next Generation Sequencing.” There were no limitations imposed on language, time period, study design or publication location, from 2013 to January 07, 2023. Similarly, ScienceDirect from 2013 to January 10, 2023.
5.2. Eligibility Criteria
They are eligible all articles that deal with the importance or the role of next generation sequencing in the diagnosis, treatment or prognosis of multiple myeloma and the follow-up of minimal residual disease. Articles not dealing with multiple myeloma or not using the NGS technique and literature reviews were excluded.
5.3. Data Extraction
Data extraction from the open access selected articles was done by drawing a table including data about the country and/or city of the study, type of study, number of patients included in the study, type of samples used in the study, the NGS panels used, etc.
6. Results and Discussion
6.1. Platforms Used
As far as the NGS platforms used in our articles are concerned, 72% of the studies use the ion torrent platform, probably because of its speed and simplicity and also its low cost.
6.2. Type of Samples
Among all the publications we studied, different types of samples were used in multiple myeloma diagnosis and it is clear that the commonly used sample was BM (44.55%). This can be explained by the high concentration of tumor plasma cells in the bone marrow.
The use of peripheral blood samples is also possible in studies with NGS because of the high sensitivity of this technique to evaluate tumor cells even at low quantities and this is what made 6.02% of our articles choose peripheral blood as the sample type for their studies.

Table 1.
The different types of samples used in our studied articles.
| Type of samples | Percentage of use |
|---|---|
| Bone Marrow (BM) | 44,55% |
| Unsited | 35,26% |
| Peripheral Blood (PB) | 6,02% |
| BM and PB | 5,46% |
| BM, PB and Plasma Samples | 2,18% |
| Plasma Samples | 2,18% |
| BM and Plasma Samples | 1,09% |
| Serum Samples | 1,09% |
| Plasmacytoid Dendritic cells (PDCs) | 1,09% |
| BM and Biopsies | 0,54% |
| PB, BM and Formalin Fixed Paraffin Embedded Samples (FFPE) | 0,54% |
6.3. Studies Focusing on the Study of WES in Myeloma Patients by the NGS
Whole-exome sequencing (WES) by next-generation sequencing (NGS) has been utilized in several ways to study MM: identification of somatic mutations, characterization of clonal architecture, prognostic biomarker discovery or drug target discovery.
Overall, WES by NGS is a powerful tool for studying MM and has the potential to help researchers better understand the disease and develop more effective treatments.
15.38% of the studies selected in our review are interested in the study of the exome “Whole Exome Sequencing” WES data. Those studies are reported in the table below and an analysis and discussion of the results of those studies is quoted directly after the table.
Table 2.
Studies focusing on the study of Whole Exome Sequencing.
| Study | Year of the study | Country | Number of patients used in the study | Type of patients | Type of samples used | |
|---|---|---|---|---|---|---|
| 1 | [30] | 2022 | Erbil Iraq | - | - | PB |
| 2 | [31] | 2021 | New delhi India | 62 | - | - |
| 3 | [32] | 2021 | AIIMS, New Delhi | 71 | NDMM | PCs |
| 4 | [33] | 2021 | Helsinki, Finland | 8 | - | BM |
| 5 | [34] | 2021 | New York, NY | 1154 | - | - |
| 6 | [35] | 2020 | Galway, Ireland | 291 | RRMM | BM |
| 7 | [36] | 2020 | Tampa, FL | 196 | - | - |
| 8 | [37] | 2019 | Turin, Italy | 42 | RRMM | - |
| 9 | [38] | 2019 | Milano, Italy | 40 | RRMM | - |
| 10 | [39] | 2018 | Arkansas USA | 1141 | - | |
| 11 | [40] | 2018 | Wurzburg Germany | 1 | - | - |
| 12 | [41] | 2018 | Boston, MA | 629, 1144, 205 | MM, NDMM, NDMM | PB |
| 13 | [42] | 2017 | Boston MA | 186 | - | - |
| 14 | [43] | 2017 | Adelaide Australia | 10 | - | - |
| 15 | [44] | 2017 | New York USA | 19 | - | - |
| 16 | [45] | 2017 | Boston, MA | 151 | MM, SMM, MGUS | PB |
| 17 | [46] | 2017 | Liège, Belgium | 10 | - | BM |
| 18 | [47] | 2016 | Cambridge UK | 14 | - | - |
| 19 | [48] | 2016 | Southampton UK | 25 | - | - |
| 20 | [49] | 2016 | Boston, MA | 63 | NDMM, RRMM, SMM, MGUS | PB |
| 21 | [50] | 2016 | - | 1000 | NDMM | - |
| 22 | [51] | 2016 | Little Rock, AR | 2161 | - | - |
| 23 | [52] | 2016 | Little Rock, AR | 33 | - | - |
| 24 | [53] | 2015 | Boston, MA, USA | 29 | - | - |
| 25 | [54] | 2015 | Little Rock, AR | - | - | BM |
| 26 | [55] | 2014 | Cambridge UK | 23 | - | - |
| 27 | [56] | 2012 | London, United Kingdom | - | - | BM |
| 28 | [57] | 2011 | Boston, MA, USA | - | - | - |
Some of these studies aim to investigate the genetic landscape of multiple myeloma, like the study carried out by Kakoo et al. which included 22 MM patients from Iranian population, the WES analysis revealed 6,959 somatic mutations across all patients and the most commonly mutated genes in MM patients were TP53, KRAS, NRAS, DIS3, and FAM46C.
This study recognized potential novel driver genes in MM, including ARID1A, RYR2, and ARID2 and also highlighted the potential clinical implications of the identified mutations, such as the association between TP53 mutations and worse overall survival in MM patients.
The authors also found that the mutational landscape of MM patients in the Iranian population was distinct from other populations, with some specific mutations being more prevalent in Iranian MM patients [58]. The study conducted by Munshi et al. identified recurrent mutations commonly seen in multiple myeloma cells. These recurring mutations included: the TP53, KRAS and NRAS genes which were found in up to 20% of multiple myeloma cases studied in this study, the DIS3 gene: which is involved in the degradation of messenger RNA and which has been identified in up to 13% of cases of multiple myeloma, the FAM46C which is involved in the regulation of protein synthesis and which has been mutated in up to 15% of cases, finally, the IRF4 gene, which plays a role in regulating immune responses, has been found to be mutated in approximately 20% of cases [57]. In the study conducted by Bolli et al., these genes were investigated as recurring mutations. KRAS and NRAS mutations were detected in 36% of patients, TP53 mutations were found in 28% of patients, which is a common mutation observed in various types of cancer, including MM. DIS3 mutations were identified in 20% of patients, FAM46C mutations were observed in 16% of patients, and TRAF3 mutations were found in 12% of patients. TRAF3 is a gene that has also been associated with the development of MM [47].
KRAS, NRAS, TP53, DIS3 and FAM46C are also the most frequently mutated genes in the study carried by Manier et al., this study which involved 45 patients with MM and sequenced their cfDNA samples using both WES and targeted deep sequencing panels. The research detected a total of 4714 somatic mutations in all patients, with an average of 104 mutations per patient [49]. In the study conducted by Miller et al., specific gene mutations were linked to the duration of progression-free survival (PFS) in multiple myeloma (MM) patients. KRAS and NRAS mutations were associated with longer PFS, while TP53 mutations were associated with shorter PFS. This particular study focused on analyzing exome sequencing data from 765 MM patients with the aim of investigating the relationship between the burden of somatic mutations, neoantigen load, and progression-free survival (PFS) in MM patients [50].
The study by Walker, Samur, et al. and that performed by Munshi et al. joins the set of previous studies in identifying TP53, KRAS and NRAS as genes frequently mutated in affected patients and which are associated with a poor prognosis and higher-risk molecular subtypes. The DIS3, FAM46C and CYLD genes also recurrently mutated and which are involved in the pathogenesis of MM [51], [59].
The study conducted by Zielinska et al. study developed a high-throughput pipeline for NGS analysis of multiple myeloma samples. The study found that the NextSeq 500 platform performed well for identifying known gene variants in multiple myeloma samples. The high-throughput pipeline developed in the study was also found to identify various gene variants in multiple myeloma samples, including mutations in the TP53, KRAS, NRAS, and BRAF genes [54].
Another study conducted by Bolli et al. sequenced the exomes of tumor cells from 50 MM patients and matched germline DNA from 44 of them. Additionally, they sequenced the IGH locus and performed copy number analysis on the tumor cells.
The study identified a range of mutations and copy number changes in the MM tumor cells, including mutations in genes such as KRAS, NRAS, TP53, and DIS3, as well as copy number gains or losses in chromosomes 1, 6, 8, 9, 11, 12, 13, 15, 16, 17, and 19. The study also detected IGH translocations in 35 out of the 50 MM patients.
The authors also compared their NGS results with those obtained using traditional techniques, for example fluorescence in situ hybridization (FISH) and Sanger sequencing. They found that NGS provided a more comprehensive analysis of genomic abnormalities and enabled the detection of mutations that would have been missed by other methods.
Overall, the study demonstrates the potential of NGS for identifying clinically relevant gene variants in MM and suggests that this approach could be used to guide personalized treatment strategies for MM patients [55].
So, these studies performed on myeloma patients highlight the importance of the KRAS, NRAS, BRAF, TP53, DIS3 and FAM46C as regularly mutated genes in the pathology of MM and suggest their potential as therapeutic targets or prognostic biomarkers, and in studies investigating smoldering multiple myeloma SMM, mutations in the TP53, BRAF, DIS3, and ATM genes, among others. provide high-risk abnormalities in these patients and can help identify patients who might benefit from early intervention as mentioned in the study by Bustoros et al. for example [42].
6.4. Studies Involving MRD Assessment by NGS
The World Health Organization (WHO) defines minimal residual disease (MRD) as “the presence of residual disease at levels below the threshold of detection by conventional morphological and cytogenetic techniques” [60].
MRD testing involve sensitive methods such as flow cytometry, polymerase chain reaction (PCR), or next-generation sequencing (NGS). The detection of MRD is important because it can help to predict a patient’s risk of relapse and guide treatment decisions.
For the assessment of MRD some from our selection studies used the LymphoTrack® IGH panel which is a molecular diagnostic tool utilizes next-generation sequencing (NGS) technology to detect and quantify clonal immunoglobulin heavy chain (IGH) gene rearrangements in the patient’s blood or bone marrow.
The LymphoTrack® IGH panel works in the context of MRD assessment as following:
A sample of the patient’s blood or bone marrow is collected.
DNA is extracted from the sample and sequenced using NGS technology to identify clonal IGH gene rearrangements.
And MRD detection and monitoring how the sequences obtained are compared to those from the patient’s baseline sample to identify any remaining cancer cells that may indicate minimal residual disease (MRD). The level of MRD is then quantified to monitor treatment response and disease progression over time.
There are some studies aimed to compare next-generation sequencing (NGS) and next-generation flow cytometry (NGF) for detecting minimal residual disease (MRD) in multiple myeloma (MM) patients like the one made by Medina et al., (2020) this found that NGS and NGF had similar levels of sensitivity for detecting MRD in MM patients, with NGS detecting MRD in 92% of cases and NGF detecting MRD in 89% of cases. However, NGS was able to detect MRD at lower levels than NGF, with a limit of detection of 0.01% for NGS compared to 0.1% for NGF.
Moreover, the study suggests that both NGS and NGF are effective methods for detecting MRD in MM patients, but NGS may have some advantages in terms of its ability to detect MRD at lower levels. Nevertheless, additional research is required to obtain a more comprehensive understanding which method is more clinically useful in guiding treatment decisions for MM patients [61].
Another study made by Ha et al. (2022) found that Ig gene clonality analysis using NGS was a highly sensitive and specific method for MRD detection in MM patients. In addition, the study found that MRD negativity by Ig gene clonality analysis using NGS was associated with improved progression-free survival (PFS) and overall survival (OS).
The study also found that Ig gene clonality analysis using NGS was able to detect MRD in a higher percentage of patients than standard flow cytometry-based methods, and that Ig gene clonality analysis was particularly useful in patients with low levels of MRD [62].
Evaluation of the utility of the NGS-based assay for the detection of MRD was also performed by Ho et al. (2018), where he revealed through his study that the NGS-based test had a high success rate for the clonal characterization of plasma cell neoplasms, with a success rate of 95.7% in the MM cohort. The test was also able to detect MRD in a high percentage of patients, with a sensitivity of 89.5% for detecting MRD in the MM cohort.
The study also found a high degree of concordance between the NGS-based assay and flow cytometry for MRD detection, with a kappa coefficient of 0.68 indicating substantial agreement between the two methods.
Global, the findings of the study indicate that the NGS-based assay may be a useful tool for identifying MRD in plasma cell neoplasms and that it has a high success rate for clonal characterization. The study also suggests that the NGS-based assay has a high degree of concordance with flow cytometry for MRD detection, indicating that it may be a reliable alternative to flow cytometry-based methods for MRD detection [63].
Similarly, the study realized by M. Kim et al. (2019) on Korean multiple myeloma (MM) patients found that NGS-based analysis of IGH and IGK rearrangements and somatic hypermutation (SHM) status was able to detect clonality in a large proportion of individuals at high-risk MM, with a success rate of 91.5%. The study also found that SHM analysis was able to identify clonality in some patients where rearrangement analysis was not successful, suggesting that SHM analysis may be a useful complementary approach for clonality detection in MM.
In addition, the study found that the clonality status determined by NGS was highly concordant with that determined by conventional methods such as PCR and flow cytometry, indicating that NGS may be a reliable alternative to these methods for clonality detection in MM.
Overall, the study suggests that NGS-based analysis of IGH and IGK rearrangements and SHM status may be a useful tool to identify clonality in MM patients at a high risk, and that it has a high success rate and concordance with conventional methods for clonality detection [64].
In the same direction of all these studies Rustad and Boyle (2020) study found that NGS was able to detect MRD in a high proportion of patients, with a sensitivity of 95.2% and a specificity of 100% for MRD detection in bone marrow samples. The study also found a high degree of concordance between NGS and standard flow cytometry for MRD detection, with a kappa coefficient of 0.85 indicating almost perfect agreement between the two methods.
In addition, this study found that NGS was able to detect clonal evolution in some patients, which may have implications for treatment decisions and patient outcomes.
Overall, the study suggests that NGS may be a useful tool for monitoring MRD in the bone marrow of multiple myeloma patients, and that it has a high sensitivity and concordance with standard flow cytometry for MRD detection. The study also suggests that NGS may be useful for detecting clonal evolution in multiple myeloma patients (Rustad & Boyle, 2020).
Other: the study realized by Cho et al. (2022) compare the efficacity of the fragment analysis (FA) technique and next-generation sequencing (NGS) in evaluation of the MRD prognostic value in MM assessment and the both techniques had confirmed that the MRD negativity had being associated with improved progression-free survival (PFS) and overall survival (OS). However, the study also found that NGS was more sensitive than FA in detecting of MRD, and that NGS had a stronger association with improved PFS and OS than FA.
Overall, the study suggests that NGS may be a more sensitive method than FA for detecting MRD in MM patients, and that MRD negativity by NGS is associated with improved outcomes regardless of the type of treatment received [22].
Other studies have compared the performance of NGS and real-time quantitative PCR (qPCR) in detecting of MRD like the study by Yao et al. (2021) which has been included 50 MM patients who had successfully attained complete or partial response after treatment and had undergone both NGS and qPCR for MRD assessment. This study found that both NGS and qPCR were highly sensitive in detecting MRD in MM patients, with NGS detecting MRD in 45 out of 50 patients (90%) and qPCR detecting MRD in 43 out of 50 patients (86%) and found a strong agreement between NGS and qPCR results, with a correlation coefficient of 0.92.
The study also found that NGS had a higher specificity compared to qPCR, with no false-positive results observed in the NGS analysis. Moreover, the study found that NGS was able to detect MRD in a higher proportion of patients with low-level disease compared to qPCR, indicating a higher sensitivity of NGS in detecting low-level disease.
Finally, the study showed that NGS was able to detect clonal evolution and the emergence of new mutations in some patients, which could have implications for treatment decisions and monitoring of disease progression.
Overall, the study suggests that NGS is a highly sensitive and specific method for MRD detection in MM and may offer advantages over qPCR in detecting low-level disease and identifying clonal evolution [65].
In 2019 Yao et al. aimed to develop a standardized NGS-based MRD assay for multiple myeloma and evaluate its sensitivity and specificity. They analyzed bone marrow samples from 40 multiple myeloma patients using an NGS-based MRD assay targeting patient-specific somatic mutations and found that the NGS-based MRD assay had a sensitivity of 0.001% and a specificity of 100%. MRD was detected in 22 out of 40 (55%) patients using the NGS-based assay, compared to 16 out of 40 (40%) patients using traditional methods such as flow cytometry or ASO-qPCR. The study also demonstrated that the NGS-based MRD assay was reproducible and had a fast turnaround time. The authors concluded that the NGS-based MRD assay could be a reliable tool for MRD detection in multiple myeloma patients [66].
And in 2020 Yao et al. aimed to develop an upgraded version of the previously developed standardized NGS-based MRD assay for multiple myeloma and evaluate its sensitivity and specificity. The authors analyzed bone marrow samples from 166 multiple myeloma patients using the upgraded NGS-based MRD assay targeting patient-specific somatic mutations. The study found that the upgraded NGS-based MRD assay had a sensitivity of 0.001% and a specificity of 100%. MRD was detected in 103 out of 166 (62%) patients using the upgraded NGS-based assay, compared to 51 out of 166 (31%) patients using traditional methods such as flow cytometry or ASO-qPCR. The study also showed that the upgraded NGS-based MRD assay was reproducible and had a fast turnaround time.
The authors concluded that the upgraded NGS-based MRD assay could be a reliable tool for MRD detection in multiple myeloma patients and has the potential to guide treatment decisions and predict outcomes in clinical practice [67].
6.5. The Use of WGS and In-House Panels in the Diagnosis of MM
Whole genome sequencing allows for the comprehensive analysis of an individual’s complete genetic code, including mutations and variations in the DNA sequence. In multiple myeloma, WGS has been used to identify somatic mutations and chromosomal aberrations that are associated with the disease. These genetic changes can be used to develop personalized treatment plans and improve patient outcomes.
In addition to WGS, Gene panels are a targeted sequencing approach that focuses on a panel of genes that are known to be associated with MM or other hematologic malignancies. Gene panels typically include genes involved in oncogenesis, DNA repair, cell cycle regulation, and other biological processes that are relevant to cancer development and progression.
A large cohort of 2161 was studied by Walker, Samur, et al. (2016) and focused on the use of WGS, WES and RNAseq to establish a strategy for the Clinical Classification of Multiple Myeloma to segment the disease into therapeutically meaningful subgroups. Actually 5 translocation groups effecting prognosis have been elucidated: t(4;14), t(6;14), t(11;14), t(14;16) and t(14;20) but minor translocation and mutational groups need a sensitive technology as WGS to be detected [51].
Another study of Ashby et al. (2018) included a cohort of 1141 NDMM and used WGS as well as WES based on previous karyotype information in order to study the presence of hyperhaploidy related to poor prognosis (Double-Hit Bi-Allelic Inactivation of TP53). [39].
One study made by Raab et al. (2020) about the phase II clinical trial investigated the effectiveness of a combination of BRAF/MEK inhibitors in patients with relapsed/refractory multiple myeloma (RRMM) who had activating BRAF V600E mutations. This study used WGS and phospho-IHC to explore the therapeutic efficacy of the combination between Encorafenib and Binimetinib. The results showed that phospho-IHC was more effective in proving that pharmacodynamic markers reveal suppression of BRAF/MEK signaling et cycle 1/day 28 and restoration of expression at the time of the relapse [68].
Table 3.
Studies that have used WGS for Diagnosis of MM.
| Study | Year | Country | Number of cases | Type of patients | Type of samples | Type of NGS investigation |
|---|---|---|---|---|---|---|
| [69] | 2021 | New York, NY | 1154 | - | - | WGS and WES |
| [68] | 2020 | Heidelberg, Germany | 15 | RRMM | - | Exploratory biomarker assessments include cytogenetics, genomic analysis (WGS, RNAseq) and phospho-IHC. |
| [70] | 2019 | New York, USA | 154 | - | BM | WGS with myTYPE panel |
| [71] | 2018 | New York, USA | - | - | BM | WGS using myTYPE panel |
| [39] | 2018 | Arkansas USA | 1141 | WGS, WES, and targeted panel sequencing | ||
| [72] | 2018 | Little Rock, AR | 439 | NDMM | - | WGS, WES or targeted panel (TP) modalities. |
| [73] | 2017 | St. Louis MO | 995 | - | - | WGS (were identified using custom Seq-FISH software on long-insert whole genome sequencing data.) |
| [42] | 2017 | Boston MA | 186 | - | - | WES and WGS libraries were constructed with Agilent SureSelect XT2 library prep kit, |
| [51] | 2016 | Little Rock, AR | 2161 | - | - | WGS, WES, targeted panel sequencing, expression data from RNA-Seq and Gene Expression array |
| [74] | 2016 | New York, USA | - | - | - | WGS |
| [59] | 2015 | Boston, MA, USA | 29 | - | - | WGS, (22 patients) WES (17 patients) |
| [75] | 2014 | Phoenix, AZ | - | - | - | WGS |
| [76] | 2012 | London, UK | 13 | MM, SMM | BM | WGS |
| [56] | 2012 | London, UK | - | - | BM | WGS, WES, SNVs |
The studies identified several genes that were frequently mutated in multiple myeloma, including TP53, KRAS, NRAS, and BRAF.
Overall, practice of WGS in the diagnosis and management of multiple myeloma has shown great promise. As the technology continues to advance, it is likely that WGS will become an increasingly important tool in the diagnosis and treatment of this complex disease.
Table 4.
Studies that have used myTYPE Panel for Diagnosis of MM.
| Study | Year | Country | Number of cases | type of samples | Type of NGS investigations |
|---|---|---|---|---|---|
| [77] | 2022 | Stockholm Sweden | 159 | - | NGS MRD assay with myTYPE panel |
| [78] | 2020 | New York USA | 74 | - | NGS-based assay with myTYPE panel |
| [70] | 2019 | New York, USA | 154 | BM | WGS with myTYPE panel |
| [71] | 2018 | New York, USA | - | BM | WGS using myTYPE panel |
| [79] | 2018 | New York USA | 147 | BM | NGS assay based myTYPE panel |
| [80] | 2018 | Trondheim, Norway | 177 | BM | LymphoTrack® VDJ assay NGS based myTYPE panel assay |
Three studies were leaded by Hultcrantz et Al in 2018, 2020 and 2022 about usage of MyType Panel multiple myeloma diagnosis (Table 5). The first study of 2018 published in Blood Journal [79] compared the adaptive NGS VDJ assay and the internal NGS panel myTYPE and concluded that the assay was less sensitive in samples with insufficient DNA content. The last study of 2022 published in Clinical Cancer Research journal [77] has been conducted at Memorial Sloan Kettering Cancer Center (MSK) between 2010 and 2017 and has also focused on the comparison of MyType Panel with Adaptive next generation sequencing (NGS) MRD. The results were similar to the first study( MyType panel had significantly higher V(D)J clonotype detection rates in univariate and multivariate analysis.
Two studies published in the Blood Cancer Journal in 2019 and 2018 by Yellapantula et Al used both whole genome sequencing and MyType Panel. The first study of 2018 [71] compared in one hand MyType Panel and WGS especially in deletions of 1p, 13p, 16q, 17p and gains of 1q, 11q and concluded a total concordance of these aberrations with a percentage of 100% identified by both assays. In The second hand, this study compared MyTYPE panel with FISH and the results were additional t(11;14) translocations identified uniquely by myTYPE. FISH was also used in deletions of 17q, 13q, 1p and 1q gain. All aberrations identified by FISH were identified in myTYPE but 13q- in four samples and 1p- in one sample were uniquely identified by myTYPE and concluded that evaluation of specificity and sensitivity require larger clinical cohort. This team leaded then a larger.
Therefore, considering the findings of these investigations collectively, it can be inferred that NGS is a remarkably accurate and precise method for identifying minimal residual disease (MRD) in individuals with multiple myeloma.
Some studies have shown that NGS-based assays have a high success rate in characterizing clonality and detecting MRD in plasma cell neoplasms (C. Ho et al., 2018; Rustad et al., 2018) and others demonstrated that NGS can achieve a sensitivity of 10-6 or better, which is superior to other techniques such as PCR and flow cytometry (Yao et al., 2019).
Furthermore, several studies have compared the performance of NGS with other MRD detection methods, such as flow cytometry and PCR, and found that NGS provides higher sensitivity and specificity for MRD detection (Cho et al., 2022b; Medina et al., 2020b).
Overall, these studies suggest that NGS is a highly accurate and reliable method for MRD detection in multiple myeloma patients, and may be used to guide treatment decisions and predict patient outcomes.
7. Conclusion
In conclusion, the use of whole-exome sequencing (WES) by next-generation sequencing (NGS) in MM studies has been shown to be a powerful tool for identifying somatic mutations, characterizing clonal architecture, prognostic biomarker discovery, and drug target discovery. Several studies highlighted the potential clinical implications of the identified mutations, such as the association between TP53 mutations and worse overall survival in MM patients. The TP53, KRAS, NRAS, DIS3, and FAM46C genes were found to be the most commonly mutated genes in MM patients across various studies. The study by Kakoo et al. identified potential novel driver genes in MM, including ARID1A, RYR2, and ARID2. These studies demonstrate the potential of WES by NGS for more understanding MM and developing more effective treatments.
Moreover NGS, is a highly sensitive and specific method for detecting MRD in MM patients. Because it has been found to detect MRD at lower levels than standard flow cytometry-based methods, with a limit of detection of 0.01% compared to 0.1%. NGS has a high success rate for clonal characterization, and its clonality status determination has been found to be highly concordant with that determined by conventional methods such as PCR and flow cytometry. The detection of MRD through NGS can help predict a patient’s risk of relapse and guide treatment decisions. However, further studies are needed to determine which method is more clinically useful in guiding treatment decisions for MM patients.
Author Contributions
Conceptualization: I.E.F., H.I., L.B. and M.E.E.; methodology: L.B.; software: S.N.; validation: M.A. and M.A.; formal analysis: R.B.; resources: I.E.F. and H.I.; data curation: M.E.E. and L.B.; writing—original draft preparation, I.E.E. and H.I.; writing—review and editing: I.E.E. and H.I.; visualization, K.O.; supervision: M.E.E. and L.B.; project administration: M.E.E and L.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Data Availability Statement
The datasets utilized in the present study are openly accessible to the public.
Conflicts of Interest
The researchers state that this study was carried out without any commercial or financial affiliations that could be perceived as potential conflicts of interest.
References
- Caulier et al., « Epidemiological landscape of young patients with multiple myeloma diagnosed before 40 years of age: the French experience », Blood, vol. 138, no 25, p. 2686-2695, déc. 2021. [CrossRef]
- Veres et I. A. Cardos, « Multiple myeloma: focus on international epidemiology literature », Acta Medica Transilv., vol. 27, no 1, 2022.
- J. Cowan et al., « Global Burden of Multiple Myeloma: A Systematic Analysis for the Global Burden of Disease Study 2016 », JAMA Oncol., vol. 4, no 9, p. 1221-1227, sept. 2018. [CrossRef]
- E. M. Boyle et al., « Improving prognostic assignment in older adults with multiple myeloma using acquired genetic features, clonal hemopoiesis and telomere length », Leukemia, vol. 36, no 1, Art. no 1, janv. 2022. [CrossRef]
- H. Sung et al., « Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries », CA. Cancer J. Clin., vol. 71, no 3, p. 209-249, 2021. [CrossRef]
- DeStefano, S. J. Gibson, A. S. Sperling, P. G. Richardson, I. Ghobrial, et C. C. Mo, « The emerging importance and evolving understanding of clonal hematopoiesis in multiple myeloma », Semin. Oncol., vol. 49, no 1, p. 19-26, févr. 2022. [CrossRef]
- R. Marinac, I. M. Ghobrial, B. M. Birmann, J. Soiffer, et T. R. Rebbeck, « Dissecting racial disparities in multiple myeloma », Blood Cancer J., vol. 10, no 2, p. 19, févr. 2020. [CrossRef]
- L. H. Schinasi et al., « Multiple myeloma and family history of lymphohaematopoietic cancers: Results from the International Multiple Myeloma Consortium », Br. J. Haematol., vol. 175, no 1, p. 87-101, oct. 2016. [CrossRef]
- K. Brigle et B. Rogers, « Pathobiology and Diagnosis of Multiple Myeloma », Semin. Oncol. Nurs., vol. 33, no 3, p. 225-236, août 2017. [CrossRef]
- « Next-Generation Sequencing (NGS) | Explore the technology ». https://www.illumina.com/science/technology/next-generation-sequencing.html (consulté le 16 janvier 2023).
- LeMieux, « The NGS Race Is On Souped-Up Sequencers Vie for Frontrunner Status », Genet. Eng. Biotechnol. News, vol. 43, no 1, p. 44-48, janv. 2023. [CrossRef]
- F. L. Genomics et I. Mobley, « A brief history of Next Generation Sequencing (NGS) », Front Line Genomics, 26 juillet 2021. https://frontlinegenomics.com/a-brief-history-of-next-generation-sequencing-ngs/ (consulté le 16 janvier 2023).
- S. Yohe et B. Thyagarajan, « Review of Clinical Next-Generation Sequencing », Arch. Pathol. Lab. Med., vol. 141, no 11, p. 1544-1557, nov. 2017. [CrossRef]
- « NGS Workflow Steps | Illumina sequencing workflow ». https://www.illumina.com/science/technology/next-generation-sequencing/beginners/ngs-workflow.html (consulté le 3 juin 2023).
- R. Pereira, J. Oliveira, et M. Sousa, « Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics », J. Clin. Med., vol. 9, no 1, p. 132, janv. 2020. [CrossRef]
- Y. Yin, C. Butler, et Q. Zhang, « Challenges in the application of NGS in the clinical laboratory », Hum. Immunol., vol. 82, no 11, p. 812-819, nov. 2021. [CrossRef]
- Charalampous et T. Kourelis, « Minimal Residual Disease Assessment in Multiple Myeloma Patients: Minimal Disease With Maximal Implications », Front. Oncol., vol. 11, 2022, Consulté le: 23 février 2023. [En ligne]. Disponible sur: https://www.frontiersin.org/articles/10.3389/fonc.2021.801851.
- J. Hillengass et al., « Disease Monitoring In Multiple Myeloma », Clin. Lymphoma Myeloma Leuk., janv. 2023. [CrossRef]
- M. Ho et T. Kourelis, « The burden of myeloma: novel approaches to disease assessment », Hematology, vol. 2022, no 1, p. 356-362, déc. 2022. [CrossRef]
- Paiva, J. San-Miguel, et H. Avet-Loiseau, « MRD in multiple myeloma: does CR really matter? », Blood, vol. 140, no 23, p. 2423-2428, déc. 2022. [CrossRef]
- Bal et al., « Impact of autologous hematopoietic cell transplantation on disease burden quantified by next-generation sequencing in multiple myeloma treated with quadruplet therapy », Am. J. Hematol., vol. 97, no 9, p. 1170-1177, 2022. [CrossRef]
- H. Cho et al., « Real-world data on prognostic value of measurable residual disease assessment by fragment analysis or next-generation sequencing in multiple myeloma », Br. J. Haematol., vol. 198, no 3, p. 503-514, 2022. [CrossRef]
- R. Fonseca et al., « Integrated analysis of next generation sequencing minimal residual disease (MRD) and PET scan in transplant eligible myeloma patients », In Review, preprint, sept. 2022. [CrossRef]
- K. Kriegsmann et al., « Comparison of bone marrow and peripheral blood aberrant plasma cell assessment by NGF in patients with MM », Blood Adv., vol. 7, no 3, p. 379-383, janv. 2023. [CrossRef]
- R. Urushihara et al., « Eight-color multiparameter flow cytometry (EuroFlow-NGF) is as sensitive as next-generation sequencing in detecting minimal/measurable residual disease in autografts of patients with multiple myeloma », eJHaem, vol. 4, no 1, p. 184-191, 2023. [CrossRef]
- T. Yoroidaka et al., « Measurable Residual Disease Assessment Using Next-Generation Flow in Patients With Relapsed and Refractory Multiple Myeloma Treated With a Combination of Carfilzomib, Lenalidomide, and Dexamethasone », Anticancer Res., vol. 43, no 1, p. 157-165, janv. 2023. [CrossRef]
- Drandi, S. Ferrero, et M. Ladetto, « Droplet Digital PCR for Minimal Residual Disease Detection in Mature Lymphoproliferative Disorders », in Digital PCR: Methods and Protocols, G. Karlin-Neumann et F. Bizouarn, Éd., in Methods in Molecular Biology. New York, NY: Springer, 2018, p. 229-256. [CrossRef]
- S. Galimberti, S. Balducci, F. Guerrini, M. Del Re, et R. Cacciola, « Digital Droplet PCR in Hematologic Malignancies: A New Useful Molecular Tool », Diagnostics, vol. 12, no 6, Art. no 6, juin 2022. [CrossRef]
- V. V. Subhash et al., « Whole-genome sequencing facilitates patient-specific quantitative PCR-based minimal residual disease monitoring in acute lymphoblastic leukaemia, neuroblastoma and Ewing sarcoma », Br. J. Cancer, vol. 126, no 3, Art. no 3, févr. 2022. [CrossRef]
- Kakoo, M. Al-Attar, et T. Rasheed, « Exonic variants in multiple myeloma patients associated with relapsed/ refractory and response to bortezomib regimens », Saudi J. Biol. Sci., vol. 29, no 1, p. 610-614, janv. 2022. [CrossRef]
- Farswan et al., « Branching clonal evolution patterns predominate mutational landscape in multiple myeloma », Am. J. Cancer Res., vol. 11, no 11, p. 5659-+, 2021.
- G. Kaur et al., « P-050: Whole Exome Sequencing provides novel insights in synonymous and non-synonymous mutational landscapes of Multiple Myeloma », Clin. Lymphoma Myeloma Leuk., vol. 21, p. S65-S66, oct. 2021. [CrossRef]
- Kumar, S. Adhikari, M. Kankainen, et C. A. Heckman, « Comparison of Structural and Short Variants Detected by Linked-Read and Whole-Exome Sequencing in Multiple Myeloma », Cancers, vol. 13, no 6, p. 1212, mars 2021. [CrossRef]
- L. Williams et al., « Hispanic or Latin American Ancestry Is Associated with a Similar Genomic Profile and a Trend Toward Inferior Outcomes in Newly Diagnosed Multiple Myeloma As Compared to Non-Hispanic White Patients in the Multiple Myeloma Research Foundation (MMRF) CoMMpassstudy », Blood, vol. 138, p. 4117, nov. 2021. [CrossRef]
- M. E. O’Dwyer et al., « Integrative Analysis of the Genomic and Transcriptomic Landscape of Relapsed/Refractory Multiple Myeloma Patients Treated With Venetoclax in Combination With Bortezomib and Dexamethasone: Biomarker Analyses From the Phase 3 BELLINI Study », Blood, vol. 136, p. 40-41, nov. 2020. [CrossRef]
- J. Song et al., « Molecular Insights into Clonal Hematopoiesis and Therapy-Related Myeloid Neoplasm in Patients with Multiple Myeloma and Cytopenia(s) », Blood, vol. 136, p. 6-7, nov. 2020. [CrossRef]
- Ziccheddu et al., « The Genomic and Transcriptomic Landscape of Double-Refractory Multiple Myeloma », Blood, vol. 134, p. 3056, nov. 2019. [CrossRef]
- Ziccheddu et al., « Analysis of the genomic and transcriptomic landscape of chemoresistant multiple myeloma », Clin. Lymphoma Myeloma Leuk., vol. 19, no 10, Supplement, p. e58-e59, oct. 2019. [CrossRef]
- Ashby et al., « Da », Blood, vol. 132, p. 4441, nov. 2018. [CrossRef]
- M. C. Da Via’ et al., « mou », Blood, vol. 132, p. 3181, nov. 2018. [CrossRef]
- T. H. Mouhieddine et al., « The Role of Clonal Hematopoiesis of Indeterminate Potential (CHIP) in Multiple Myeloma: Immunomodulator Maintenance Post Autologous Stem Cell Transplant (ASCT) Predicts Better Outcome », Blood, vol. 132, p. 749, nov. 2018. [CrossRef]
- M. Bustoros et al., « Next Generation Sequencing Identifies Smoldering Multiple Myeloma Patients with a High Risk of Disease Progression », Blood, vol. 130, p. 392, déc. 2017.
- K. Dutta, J. P. Grady, D. R. Hewett, L. B. To, L. Fink, et A. C. W. Zannettino, « laga », Blood, vol. 130, p. 391, déc. 2017.
- Lagana et al., « Clonal Evolution in Newly Diagnosed Multiple Myeloma Patients: A Follow-up Study from the Mmrf Commpass Genomics Project », Blood, vol. 130, p. 325, déc. 2017.
- S. Manier, X. Leleu, et H. Avet-Loiseau, « Rôle pronostique des microARN des exosomes circulants dans le myélome multiple », médecine/sciences, vol. 33, no 11, p. 939-941, nov. 2017. [CrossRef]
- S. Wenric et al., « Exome copy number variation detection: Use of a pool of unrelated healthy tissue as reference sample », Genet. Epidemiol., vol. 41, no 1, p. 35-40, janv. 2017. [CrossRef]
- N. Bolli et al., « A DNA target-enrichment approach to detect mutations, copy number changes and immunoglobulin translocations in multiple myeloma », Blood Cancer J., vol. 6, no 9, p. e467, sept. 2016. [CrossRef]
- Bryant et al., « Single Cell Whole Exome Sequencing in an Index Case of Amp1q21 Multiple Myeloma to Define Intraclonal Variation », Blood, vol. 128, no 22, p. 5651, janv. 2016. [CrossRef]
- S. Manier, K. Salem, S. V. Glavey, A. M. Roccaro, et I. M. Ghobrial, « Genomic Aberrations in Multiple Myeloma », Cancer Treat. Res., vol. 169, p. 23-34, 2016. [CrossRef]
- Miller et al., « Correlation Between Somatic Mutation Burden, Neoantigen Load and Progression Free Survival in Multiple Myeloma: Analysis of MMRF CoMMpass Study », Blood, vol. 128, no 22, p. 193, janv. 2016. [CrossRef]
- Walker et al., « The Multiple Myeloma Genome Project: Development of a Molecular Segmentation Strategy for the Clinical Classification of Multiple Myeloma », Blood, vol. 128, no 22, p. 196, janv. 2016. [CrossRef]
- N. Weinhold et al., « Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma », Blood, vol. 128, no 13, p. 1735-1744, sept. 2016. [CrossRef]
- N. C. Munshi et al., « Next Generation Sequencing in Multiple Myeloma », Clin. Lymphoma Myeloma Leuk., vol. 15, p. e2-e3, sept. 2015. [CrossRef]
- K. Zielinska, K. Leigh, H. Gomez, et R. K. Van Laar, « Validation of the Nextseq 500 and Development of a High-Throughput NGS Pipeline for Identifying Clinically-Relevant Gene Variants in Multiple Myeloma Specimens », Blood, vol. 126, no 23, déc. 2015, Consulté le: 7 juin 2022. [En ligne]. Disponible sur: https://www.webofscience.com/wos/woscc/summary/8e9f0957-98a2-4fc0-b446-ccf7a6255ce7-3ca6fb9a/relevance/1.
- N. Bolli et al., « A Next Generation Sequencing-Based Approach to Detect Gene Mutations, Copy Number Changes and IGH Translocations in Multiple Myeloma », Blood, vol. 124, no 21, p. 3364, déc. 2014. [CrossRef]
- Walker et al., « mun », Blood, vol. 120, no 5, p. 1077-1086, août 2012. [CrossRef]
- N. C. Munshi et al., « Whole Genome Sequencing Defines the Clonal Architecture and Genomic Evolution in Myeloma: Tumor Heterogeneity with Continued Acquisition of New Mutational Change », Blood, vol. 118, no 21, p. 297, nov. 2011. [CrossRef]
- Kakoo, M. Al-Attar, et T. Rasheed, « Exonic variants in multiple myeloma patients associated with relapsed/ refractory and response to bortezomib regimens », Saudi J. Biol. Sci., vol. 29, no 1, p. 610-614, janv. 2022. [CrossRef]
- N. C. Munshi et al., « Next Generation Sequencing in Multiple Myeloma », Clin. Lymphoma Myeloma Leuk., vol. 15, p. e2-e3, sept. 2015. [CrossRef]
- J. D. Khoury et al., « The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms », Leukemia, vol. 36, no 7, Art. no 7, juill. 2022. [CrossRef]
- Medina et al., « Comparison of next-generation sequencing (NGS) and next-generation flow (NGF) for minimal residual disease (MRD) assessment in multiple myeloma », Blood Cancer J., vol. 10, no 10, p. 108, oct. 2020. [CrossRef]
- J. Ha et al., « Ig Gene Clonality Analysis Using Next-Generation Sequencing for Improved Minimal Residual Disease Detection with Significant Prognostic Value in Multiple Myeloma Patients », J. Mol. Diagn., vol. 24, no 1, p. 48-56, janv. 2022. [CrossRef]
- Ho et al., « Next-Generation Sequencing-Based Assay Shows High Clonal Characterization Success Rate for Plasma Cell Neoplasms, and Concordance with Flow Cytometry in Minimal Residual Disease Detection », Blood, vol. 132, p. 4475, nov. 2018. [CrossRef]
- M. Kim, H. J. Kim, et Y. K. Lee, « IGH and IGK Rearrangement and IGH Somatic Hypermutation Analysis Using Next-generation Sequencing for the Detection of Clonality in High-risk Korean Multiple Myeloma Patients », Clin. Lymphoma Myeloma Leuk., vol. 19, no 10, Supplement, p. e68-e69, oct. 2019. [CrossRef]
- Q. Yao, Y. Bai, S. Kumar, E. Au, A. Orfao, et C. S. Chim, « Minimal Residual Disease Detection by Next-Generation Sequencing in Multiple Myeloma: A Comparison With Real-Time Quantitative PCR », Front. Oncol., vol. 10, p. 611021, janv. 2021. [CrossRef]
- Q. Yao, Y. Bai, A. Orfao, et C. S. Chim, « Standardized Minimal Residual Disease Detection by Next-Generation Sequencing in Multiple Myeloma », Front. Oncol., vol. 9, 2019, Consulté le: 17 mai 2022. [En ligne]. Disponible sur: https://www.frontiersin.org/article/10.3389/fonc.2019.00449.
- Q. Yao, Y. Bai, S. Kumar, E. Au, A. Orfao, et C. S. Chim, « Minimal Residual Disease Detection by Next-Generation Sequencing in Multiple Myeloma: A Comparison With Real-Time Quantitative PCR », Front. Oncol., vol. 10, p. 611021, 2020. [CrossRef]
- M. S. Raab et al., « Safety and Preliminary Efficacy Results from a Phase II Study Evaluating Combined BRAF and MEK Inhibition in Relapsed/Refractory Multiple Myeloma (rrMM) Patients with Activating BRAF V600E Mutations: The GMMG-Birma Trial », Blood, vol. 136, p. 44-45, nov. 2020. [CrossRef]
- L. Williams et al., « Hispanic or Latin American Ancestry Is Associated with a Similar Genomic Profile and a Trend Toward Inferior Outcomes in Newly Diagnosed Multiple Myeloma As Compared to Non-Hispanic White Patients in the Multiple Myeloma Research Foundation (MMRF) CoMMpassstudy », Blood, vol. 138, p. 4117, nov. 2021. [CrossRef]
- V. Yellapantula et al., « Comprehensive detection of recurring genomic abnormalities: a targeted sequencing approach for multiple myeloma », Blood Cancer J., vol. 9, p. 101, déc. 2019. [CrossRef]
- V. Yellapantula et al., « Mytype: A Capture Based Sequencing Approach to Detect Somatic Mutations, Copy Number Changes and IGH Translocations in Multiple Myeloma », Blood, vol. 132, p. 5588, nov. 2018. [CrossRef]
- Wardell et al., « Extracting Prognostic Molecular Information from PET-CT Imaging of Multiple Myeloma Using Radiomic Approaches », Blood, vol. 132, p. 1906, nov. 2018. [CrossRef]
- M. Fiala et al., « Next Generation Sequencing Based Revised International Staging System (R-ISS) for Multiple Myeloma », Clin. Lymphoma Myeloma Leuk., vol. 17, no 1, Supplement, p. e18, févr. 2017. [CrossRef]
- Perumal et al., « Network Modeling Reveals CDC42BPA and CLEC11A As Novel Driver Genes of t(4; 14) Multiple Myeloma », Blood, vol. 128, no 22, p. 802, janv. 2016. [CrossRef]
- K. Stephenson, B. Benard, M. Klug, A. Christofferson, et J. J. Keats, « Clonal Diversity of a Myeloma Tumor: From Generations of Cells to Treatment Implications », Blood, vol. 124, no 21, p. 3424, déc. 2014. [CrossRef]
- L. Melchor et al., « Intra-Clonal Heterogeneity Is a Critical Early Event in the Preclinical Stages of Multiple Myeloma and Is Subject to Darwinian Fluctuation throughout the Disease », Blood, vol. 120, no 21, p. 3941, nov. 2012. [CrossRef]
- M. Hultcrantz et al., « Capture Rate of V(D)J Sequencing for Minimal Residual Disease Detection in Multiple Myeloma », Clin. Cancer Res., vol. 28, no 10, p. 2160-2166, mai 2022. [CrossRef]
- M. Hultcrantz et al., « Baseline VDJ clonotype detection using a targeted sequencing NGS assay: allowing for subsequent MRD assessment », Blood Cancer J., vol. 10, no 7, p. 76, juill. 2020. [CrossRef]
- M. Hultcrantz et al., « Capture Rate of the Adaptive Next Generation Sequencing VDJ Assay in Multiple Myeloma », Blood, vol. 132, p. 3184, nov. 2018. [CrossRef]
- H. Rustad et al., « V(D)J Sequence Capture for DNA-Based Minimal Residual Disease Detection in Multiple Myeloma », Blood, vol. 132, p. 4444, nov. 2018. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



