
Submitted:
17 July 2023
Posted:
18 July 2023
You are already at the latest version
Abstract
The microenvironment of most tumors is complex, comprising numerous aspects of immunosuppression. Several studies have indicated that the adrenergic system is vital for controlling immunological responses. In the context of tumor microenvironment, Nor-Adrenaline (NA) is poured-in by innervating nerves and tumor tissues itself. The receptors for nor-adrenaline are present on the surface of cancer and immune cells and are often involved in activation of pro-tumoral signaling pathways. β2-adrenergic receptor (β2-AR) is an emerging class of receptors that are capable of modulating the functioning of immune cells. β2-AR is reported to activate the regulatory immune cells and inhibit the effector immune cells. Blocking β2-AR increases activation, proliferation and cytokine release of T lymphocytes. Moreover β2-AR deficiency during metabolic reprograming of T cells increases mitochondrial membrane potential and biogenesis. In the view of available research data, immunosuppressive role of β2-AR in T cells presents it a targetable checkpoint in CAR-T cell therapies. In this review, we have abridged contemporary knowledge about adrenergic stress mediated β2-AR activation on T lymphocytes inside tumor milieu.
Keywords:
CAR-T therapy
; Immunosuppression
; Tumor microenvironment
; adrenergic stress
; immunotherapy
1. Introduction
Neurotransmitters play a bridging role between nervous system and body. Catecholamines are important set of neurotransmitters released by supra-renal glands and comprise of adrenaline/epinephrine (A/E) and nor-adrenaline/nor-epinephrine (NA/NE). Adrenergic receptors are activated in response to stimulation by adrenaline and nor-adrenaline to chemically coordinate the signals/messages from nervous system to the target tissues [1]. Adrenergic receptors (ARs) belong to G-protein coupled, seven transmembrane receptor (GPCR) family and constitute alpha-adrenergic receptor (α-AR) and beta-adrenergic receptor (β-AR) subtypes, which further has been classified as Alpha-1 adrenergic receptor (α1-AR), Alpha-2 adrenergic receptor (α2-AR) and Beta-1 adrenergic receptor (β1-AR), Beta-2 adrenergic receptor (β2-AR), Beta-3 adrenergic receptor (β3-AR) respectively. In short term or acute stress condition like fear or exercise, body releases catecholamines, via activation of sympathetic nervous system (SNS) that binds with adrenergic receptors on various organs and results in increased heart rate, dilation of pupils, mobilizes the energy and divert the blood flow from other body organs to skeletal muscles to cope with the said stress [2].
However, in chronic stress conditions body remains in persistently higher concentration of catecholamines and sustained activation of SNS, leads to initiation and progression of cancer [3]. Many studies demonstrate that this cancer progression is because of catecholamines that triggers β-adrenergic receptors more specifically β2-Adrenergic receptors downstream signaling in cancer cells [4]. Studies of β2-AR influence on tumor progression were further motivated when psychological factors and depressive disorders acted as a main cause of incidence of cancer incidence. Persistent elevated catecholamines induces cancer by compromising genomic integrity of the cells, and making the cells hyper-responsive to the environmental carcinogens [5]. Recently the role of β2-AR in immunity is the focus of attention of a bunch of published data. β2-Adrenergic receptors can modulate the functions of various immune cells as the corresponding receptors are displayed on the surface of T cells, natural killer (NK) cells and dendritic cells (DCs) [6, 7]. The ligand for β2-AR is released inside tumor microenvironment (TME), and leads to inhibitory signaling in T lymphocytes [8, 9].
The immune system plays a critical role in identifying and eliminating cells that undergo malignant transformation, in addition to its primary function of eradicating infectious agents. If the inflammatory response remains unresolved, the affected cells undergo transformation and become cancerous [10]. However, at this stage, the immune system identifies the tumor cells by recognizing the tumor-specific antigens displayed on their surface as foreign particles [11], which facilitates their elimination by effector immune responses primarily mediated by CD8+ T cells and NK cells. The activation of these immune responses form the basis of immunotherapy, as it provides a brake on tumorigenesis [12, 13].
Since the first clinical trial of Chimeric antigen receptor (CAR)-T therapy has achieved enormous success in hematological malignancies, especially in infants, where the clinical response to CAR-T therapy has been reported up to 90% in leukemia patients [14, 15]. As a result, number of successful clinical trials for CAR-T therapy targeting hematological malignancies directing numerous antigens has upsurged significantly, yet the parallel success of CAR-T therapy in solid tumors is still awaited [16]. The number of ongoing clinical trials in solid tumors is far less than liquid tumors, courtesy to toxic side effects and suboptimal therapeutic outcomes, achieved almost every time. There can be several explanations to this including following. Hematological cancers often display analogous antigens and their distribution on different types of hematological cancers apart from some exceptions, is often alike [17]. On contrary antigens referred to solid tumors vary greatly not just in tumor to tumor but also in between different forms of a similar tumor i.e., primary and metastatic forms [18]. Moreover, hematological cancers are widespread in the circulatory system that makes them less dense and easily targetable as compared to solid tumors. The solid tumors are denser, concentrated on a single site creating a physical barrier for CAR-T cells, by developing extensive vasculature in order to supply ample of nutrients to fast growing tumor, thus inhibits chemo attractive signals that are necessary for CAR-T cells to reach at the tumor site [19]. Immune suppressive behavior of solid tumors as a result of displaying check point inhibitory ligands, metabolites of different metabolic pathways as well as presence of inhibitory immune cells creates an immune inhibitory environment often termed as immunosuppressive tumor microenvironment (TME); creates such a scenario that it becomes almost impossible for CAR-T cells to penetrate and successfully eradicate solid tumors [20]. Adrenergic stress among others is one of the culprits responsible for immunosuppression inside TME. Previously we have attempted to target immunosuppressive factors inside TME in an attempt to increase the efficacy of CAR-T therapy in prostate and pancreatic tumor microenvironments [21, 22].
The role of adrenergic stress in cancer and immunity has been reviewed previously [23-25]. In this review we will specifically discuss consequences of adrenergic stress mediated β2-AR activation in T lymphocytes and its implications for CAR-T cell therapy. To the best of our knowledge the inhibitory role of β2-AR in T lymphocytes and in the context of CAR-T therapy has not been published before. Digging deep down into immunosuppression inside TME, and how it negatively regulates CAR-T signaling is the topic of interest recently.
2. Nor-Adrenaline in Tumor Microenvironment
The production of adrenaline and nor-adrenaline within TME can arise from various sources, including the tumor cells themselves, the nerve fibers that innervate the tumor, and the surrounding stromal cells such as fibroblasts and immune cells. It seems that the NA mainly from the innervating nerves plays part in the incidence and initial progression of the cancer as TME is absent initially. After TME is established, it is mainly the TME that pours sufficient amounts of NA in tumor surroundings that carries its immunosuppressive roles in CD8+ T cells. This NA comes from two sources mainly. Firstly, the tumor itself contains all the necessary machinery to synthesize NA itself. Secondly many tumor types are innervated by sympathetic nerve fibers [26]. Neuronal progenitor cells migrate towards the tumor tissue during the process of neurogenesis and adapt the sympathetic tone while innervating tumor tissue. Moreover, the sensory neurons innervating tumor tissues are also reprogrammed to sympathetic nerve phenotype [27]. This altogether creates hyperactive SNS signaling inside TME. Growing number of evidences suggest the immunosuppressive role of adrenergic system inside TME could possibly hinder antitumor functions of immunotherapy [5, 25].
3. Adrenergic Stress Endorses Immunosuppressive Tumor Milieu Development
The catecholamines bind to adrenergic receptors on target cells, including immune cells, and trigger a range of biological responses. In the context of the immune system, adrenergic stress can change the activity of different immune cell subsets due to its wide expression on diverse immune cells subsets. One of the key mechanisms by which adrenergic stress promotes immunosuppression is through the recruitment and activation of myeloid-derived suppressor cells (MDSCs) [28]. MDSCs are a heterogeneous population of immature myeloid cells that have the ability to suppress T cell responses and promote tumor growth. Adrenergic stress has been shown to promote the expansion and activation of MDSCs in the tumor microenvironment, through the activation of adrenergic receptors on MDSCs themselves and on other cells that produce cytokines and chemokines that promote MDSC recruitment and activation [29]. β2-AR activation on MDSCs has been shown to enhance the expression of arginase-1, an enzyme that can deplete arginine, an amino acid that is important for T cell function. Additionally adrenergic stress inhibits T cell functions by upregulating interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β) [30, 31]. The production of these anti-inflammatory cytokines owes to the activation of β2-AR on the surface of tumor associated macrophages (TAMS) also demonstrated as M2-phenotype. Adrenergic signaling in cancer can promote the release of vascular endothelial growth factor (VEGF), a vital mediator of angiogenesis, from tumor cells and M2-macrophages in the tumor microenvironment [32]. VEGF promotes the recruitment of endothelial cells, the formation of new blood vessels, and the delivery of oxygen and nutrients to the tumor [25]. β2-AR can also increase the activity of Tregs and thus increases the immunosuppression in TME [33, 34].
According to literature, adrenergic stress has been shown to inhibit T cell activation, proliferation, and promote apoptosis. Similarly, it can inhibit NK cell and B cell activation. These effects are mediated by the activation of adrenergic receptors and the production of cytokines and chemokines that promote immune cell dysfunction [35]. In conclusion, adrenergic stress plays a key role in the development of TME by promoting the recruitment and activation of immune regulatory cells, inhibiting the function of pro-inflammatory immune cells (Figure 1), endorsing the production of immunosuppressive cytokines and chemokines, and promoting tumor angiogenesis [36]. In this review, we are discussing the role of adrenergic stress specifically in T lymphocytes.
4. Eminent Role of β2-AR in T Cells
Catecholamines can activate five distinct types of adrenergic receptors. Some of them have been reported to be expressed in immune cells. T lymphocytes express both α-AR and β-AR receptors [37, 38]. Expression of α-AR on T lymphocytes has remained controversial and there is a bunch of conflicting data on this issue. There are some studies which have completely declined the possibility of presence of α-AR in T cell populations, while others have reported the expression of α-AR in lymphocytes population upon LPS stimulation [39, 40]. Conflict in these studies might be due to lack of availability of specific antibodies. The data regarding α-AR expression mostly relies on RT-PCR analysis which is prone to contamination. Whatever the reason might be, the fact is that evidence regarding the role of α-AR in T lymphocytes is insufficient and less documented [41]. On the other hand, role of β2-AR, in T lymphocytes is well documented and has been the focus of many recent research [19, 42, 43].
The effects of NA on CD8+ T cells are primarily mediated by β2-adrenergic receptors (β2-AR) rather than the other receptors of the same class. It was found that the α-AR antagonist phentolamine and the β1-AR-specific antagonist atenolol were unable to reverse the effects of NA in CD8+ T cells. This was demonstrated by the fact that phentolamine did not alter the expression of IL-1 and IL-6 genes under the influence of NA. Therefore, the study suggests that β2-AR plays a more exclusive role in the suppression of T cell receptor (TCR)-mediated human and mouse CD8+ T cell effector functions [44, 45]. Moreover β2-AR specific blocker nadolol can abolish NA mediated effects on CD8+ T cells. On the other hand, β2-AR specific agonist terbutaline mimics inhibitory actions of NA [45]. In a model of influenza virus, role of beta-adrenergic receptors was assessed by pharmacological inhibition of β2-AR as well as chemical inhibition of SNS. Both the aforementioned systems mimicked each other signifying the importance and vitality of β2-AR signaling in T cell population [46].
Pharmacological inhibition or genetic deletion of β2-AR in mice can improve anti-tumor immunity in colon and breast cancer mice model under stress condition. Inhibition of β2-AR signaling led to improved number and function of CD8+ T cells owing to better tumor control as compared to control [47]. The beneficial effects of β2-AR blockade (pharmacological or genetic) in case of cancer depends upon CD8+ T cells [47, 48]. CD8+ T cells being the backbone of CAR-T therapy are vital for anti-tumor immune responses. Certain studies have demonstrated that blocking β2-AR signaling can halt the growth of tumors. The mechanism of this phenomenon is relatively complex and often cancer cell intrinsic signaling pathways are held responsible for adrenergic blockade mediated anti-tumor responses. Poor prognosis of cancer might be due to adrenergic stress driven loss of T cell activity. In the absence of CD8+ T cells, propranolol and β2-AR Knockout (KO) mice does not exert its anti-tumor effects which signifies that β2-AR blockade apart from affecting tumor intrinsic pathways, improves the status of anti-tumor immunity and hence better anti-tumor control [49]. Overall, the evidence suggests that the inhibitory effects of NA on T cells are mainly due to the exclusive involvement of β2-AR, highlighting the importance of this receptor subtype in regulating immune responses.
5. β2-AR and T Cell Signaling
Molecular mechanism of β2-AR is elusive as it works with different transducers (Gs, Gi and β-Arrestin) and exhibited different intracellular signaling pathway. Continuous stimulation of nor-adrenaline in TME triggers the canonical pathway of β2-AR expressed on T lymphocytes [27]. Once nor-adrenaline binds to its receptor, it leads to uncoupling to Gα subunit from other two (β and γ) subunits along with conversion of GDP to GTP. Gαs-GTP complex is recruited to lipid rafts where activation of adenyl cyclase (AC) takes place. Adenyl cyclase catalyzes the ATP and convert it into cyclic-AMP (cAMP) which further activates Protein Kinase A (PKA). cAMP can activate both isoforms of PKA i.e., PKA-1 and PKA-2 but only PKA-1 has a recognized role in T cell signaling [50]. In the context, of CAR-T, cAMP dependent PKA-1 phosphorylation activates C-terminal SRC kinase (CSK), which phosphorylates and inactivate Lck. Lymphocyte–specific protein tyrosine kinase (LcK) is important for the activation and phosphorylation of CD3-zeta chain associated protein kinase 70 (ZAP70) and other substrates that initiate the downstream signaling such as AP-1, NFAT, Erk and NF-kB [51]. These transcription factors play an important role in T and CAR-T cell activation, proliferation, survival and cytotoxic ability. β2-AR inhibits ZAP70 activation, CD-3ζ tonnic signaling by activating cAMP and PKA (Figure 2). On the other hand, β2-AR upregulate the expression of checkpoint receptor PD-1, which further inhibits CD28 mediated T cell activation signaling [52]. Overall β2-AR signaling in T cells is complex, and more studies are needed to unveil molecular mechanisms in detail.
6. β2-AR as a Potential Target for Enhancing Cancer Immunotherapy Efficacy
6.1. β2-AR and T Cell Co-Stimulation
Co-stimulation is a critical step in the activation of T cells, which requires not only the binding of the T cell receptor (TCR) to antigen presented by DCs but also the engagement of co-stimulatory molecules such as CD28, 41-BB etc on T cell surface [53]. In many solid tumors, abnormal cell activation has been observed in both mice and human populations. This is because tumor infiltrating lymphocytes (TILs) demonstrate decrease in CD-3ζ tonic signaling. Defects in TCR signaling in turn affects the proliferative as well cytotoxic ability [54]. Some studies have shown that β2-AR signaling can inhibit co-stimulation by downregulating the expression of CD28 on the surface of T cells. For example, by employing human T cells that have undergone through purification, experiment was conducted for evaluation of the proliferative potential following stimulation with immobilized anti-CD3 monoclonal antibody (mAb) while under the influence of the beta-adrenergic agonist isoproterenol (ISO). The proliferative capacity of T cells, including their CD4+, CD8+, or CD45RO+ subsets, in response to anti-CD3 mAb, was suppressed in a manner that was dependent on the dosage of ISO [49, 55].
For CAR-T therapy to magnificently eliminate existing tumors, novel strategies are being employed to boost co-stimulatory signals specifically within tumor microenvironment. Introduction of ‘switch receptors’ and ‘inverted cytokine receptors’ (ICRs) are examples of countless efforts to boost T cell co-stimulation and hence augment immunotherapeutic response to CAR-T cell therapy [56]. The mechanism by which β2-AR signaling regulates T cell co-stimulation has been demonstrated in Figure 2. One proposed mechanism is that β2-AR signaling can inhibit the activation of NF-κB, a transcription factor that is critical for the expression of co-stimulatory molecules [36, 57]. One potential strategy to boost CAR-T cell co-stimulation could be to inhibit β2-AR.
6.2. β2-AR and T Cell Activation Signals
T cells requires two-three signals for exhibiting complete activation. Antigen presenting cells or cancer cells present antigen (MHC-1) on their surface which binds with T cell receptor TCR and give 1st signal for activation. Co-stimulatory molecules, such as CD28 and 4-1BB binds with its ligands, B7.1 or B7.2, on APCs provides a crucial “second signal” (Figure 3) [58, 59]. Soon after activation, cells undergo extensive proliferation and release pro-inflammatory cytokines such as IL-2, IL-7, IL-15, IL-21 which bind to their receptors on T cells surface and in a positive feedback loop manner providing 3rd signal. Extensive proliferation is important as significantly greater number of T cells are required to successfully inhibit the rapidly growing tumor tissue [60]. Although CAR-T is getting enormous success in many tumors with excellent remission rates, still there are reports of relapse. There can be several explanations to this failure of treatment by CAR-T therapy, including adrenergic stress mediated inhibition of T cell activation and subsequently proliferation at tumor site [42, 61, 62].
CD69 is an early activation marker expressed on various immune cells, including T cells. Its expression is rapidly induced upon T cell activation and is involved in the regulation of T cell activation, differentiation, and effector functions [63, 64]. Some studies have suggested that β2-AR can inhibit the activation marker CD69 in T cells [47, 49, 65, 66]. For example, when exposed to isoproterenol (ISO), a beta-2 adrenergic receptor agonist, CD8+ T-cells showed a significant reduction in CD69 expression both 24 hours and 48 hours after activation [49, 66]. We have found that downregulation of β2-AR via shRNA in NKG2D-CAR-T cells led to a significant increase in CD69 marker when co-cultured with prostate cancer cell line PC3 (data not published). Many recent clinical trials in CAR-T therapy have also demonstrated failure to achieve optimum proliferation of T cells in different tumor models with complex TME. Inhibition of proliferation by β2-AR might be due to the fact that it does not let T cells to activate properly, and subsequently results in decreased proliferation.
6.3. β2-AR Self-Regulates T Cell Activation
DCs are professional antigen presenting cells that are a bridge between innate and adaptive immune system [67]. Certain studies have demonstrated that DCs have an excellent skill to penetrate tumor sites, arrest tumor antigens and subsequently present those antigens to CD8+ T cells after processing thus play vital role in anti-tumor immunity [68]. β2-AR interfere with anti-presenting ability of DCs, that has been demonstrated to be critical for proliferation of CD8+ T cells under adrenergic stress in mice model of influenza virus [7, 46, 69]. As DCs have extensive ability to prime T lymphocytes, so it possibly has broader implications in immunotherapy. β2-AR inhibits TLR-4 mediated maturation of DCs and antigen presenting ability yet, the direct effects of adrenergic signaling in T cells is sufficient to suppress the activation as well as proliferation in CD8+ T cells independent of DCs [70]. Recently Daher C lara et al., 2019 demonstrated that NA mediated inhibition of proliferation in T cells was not due to involvements of DCs. Rather the proliferative capacity and responsiveness of T cells to adrenaline depends upon the activation status [71]. That is why Tn are not susceptible to NA affects but the opposite scenario occurs in effector T cells. This can further be explained by another study where it was reported that the β2-AR expression is related to the activation status of T cells [44]. Moreover β2-AR mediated inhibitory signals are enough to suppress T cell functions independent of their presence and signaling in another important set of immune cells that is DCs.
6.4. β2-AR and IL-2 Signaling
Ex-vivo expansion of CAR-T cells is an important step before administration of therapy to the patients. T cell cultures are supplemented with exogenous cytokines in order to provide proliferative and growth signals. IL-2 is commonly used cytokine for ex-vivo CAR-T expansion [72]. IL-2 resides upstream as well as downstream of Akt molecule in a positive feedback cascade. IL-2 activates JAK/STAT, and JUN pathway both of which are involved in production of growth factors required for T cell proliferation [73]. β2-AR has been shown to inhibit IL-2 signaling in T cells [74]. β2-AR signaling pathway appears to have a multifaceted role in the regulation of lymphoid cell proliferation and survival. Additionally, β2-AR activation induced threonine phosphorylation of the IL-2Rβ, indicating a potential mechanism for ISO-induced changes in lymphoid cell function. In contrast, when cells were treated with ISO prior to IL-2 stimulation, an inhibitory signal was generated, which hindered IL-2-induced activation of the JAK/STAT, MEK/ERK, and PI3K pathways. This inhibition was attributed to the disruption of the IL-2R beta-gamma chain complex, which is necessary for downstream signaling and subsequent cell proliferation [75].
6.5. Metabolic Profiling of T Lymphocytes
During the resting phase, T cells don’t have fastidious requirements as the demand for nutrients is low. So, glucose is being metabolized through oxidative phosphorylation. Metabolic reprogramming in T cells refers to the changes in the metabolic pathways that occur when T cells are activated to perform their immune functions [76]. Activated T cells undergo a shift from oxidative phosphorylation to glycolysis, a process known as the Warburg effect. This shift in metabolism allows T cells to rapidly produce energy and biosynthetic intermediates required for the production of cytokines and the proliferation of effector T cells. This metabolic reprogramming is regulated by a variety of signaling pathways and transcription factors, including mTOR, HIF-1α, and c-Myc [77, 78].
6.5.1. Role of β2-AR in T Cell Glycolysis
Interestingly, different subsets of T cells exhibit distinct metabolic profiles. For example, effector CD8+ T cells primarily rely on glycolysis. Activated T cells increase their glucose uptake and upregulate the expression of glycolytic enzymes, such as hexokinase and pyruvate kinase, to support glycolysis. This increased glycolytic activity is crucial for T cell activation and function. β2-AR has been reported to downregulate the expression of GLUT-1 gene and glycolytic enzymes in T cells during T cell activation phase [49]. The authors demonstrated that ISO treatment leads to a reduced expression of glucose transporter 1 (GLUT-1) following activation and a subsequent decrease in glucose uptake and glycolysis in comparison to CD8+ T-cells activated in the absence of ISO. Importantly, this effect was specific to β2-AR signaling, as it was not observed in CD8+ T cells deficient in β2-AR and could be blocked by the β-AR antagonist propranolol. That is why, in another study knockdown of β2-AR in mice led to increased glycolysis and oxidative phosphorylation in T cells [49, 79]. It means that the adrenergic stress inside tumor microenvironment can negatively impact the metabolic profile of CAR-T cells and any intervention for blocking β2-AR in T lymphocytes might be a potential strategy to boost the efficacy of CAR-T cell therapy. Increased glycolytic activity by inhibiting β2-AR signaling might be due to the increased expression of co-stimulatory molecules and decreased expression of co-inhibitory molecules on the surface of tumor infiltrating lymphocytes [50, 51]. Inhibiting glycolysis might lead to the reduced T cell proliferation and cytokine release rendering T cell dysfunctional against cancers [80].
Hypoxia-inducible factor 1 alpha (HIF-1α) is a transcription factor that regulates the expression of several genes involved in glucose metabolism, including GLUT-1. β2-AR signaling has been shown to inhibit HIF-1α expression in CD8+ T cells, which could further lead to reduced GLUT-1 expression [81]. This may have important implications in determining immunotherapeutic efficacy.
6.5.2. Role of β2-AR in Mitochondrial Metabolism of T Cells
During the process of T cell activation, the cells upregulate glycolysis but at the same time mitochondrial metabolism remains vibrant part of T cell metabolism. The very reason for this might be that T cells also upregulate mitochondrial respiration during the process of activation, proliferation and differentiation [82]. 4I-BB co-stimulatory domain of CAR-T cells owe to their improved mitochondrial respiration and biogenesis while on the other hand, CD28 increases glycolysis and hence effector function of CAR-T cells. Third generation CAR-T may contain two co-stimulatory domains which suggests the vitality of mitochondrial metabolism in CAR-T functioning while dealing solid tumors [76]. The TME of many solid tumors render T cells exhausted and dysfunctional attributed to loss of mitochondrial function [83]. In mitochondria, β2-AR can modulate mitochondrial biogenesis, respiration capacity as well as membrane potential [49, 65, 84]. β2-AR leads to an exhausted phenotype of T cells expressing greater amounts of checkpoint inhibitors for example PD-1 [47].
Upregulation of checkpoint inhibitors especially PD-1 has been shown to negatively regulate metabolic fitness of T lymphocytes. PD-1 carries its immunosuppressive function by inhibiting PGC1-α expression which is the master regulator of T cell metabolism. As a result, T lymphocytes are unable to metabolic reprogram themselves in-vivo during their infiltration into tumor milieu in PD-1/mTOR dependent manner [85, 86]. Blocking β2-AR signaling results in a tumor-infiltrating T cell phenotype that is more activated and less exhausted [65, 87]. In a study, propranolol treated mice had increased mitochondrial mass in CD8+ T cells and CD4+ T cells in-vivo. Moreover, blocking β2-AR signaling led to an increased mitochondrial mass and spare respiratory capacity of propranolol treated tumor bearing mice by blocking checkpoint inhibitors expression on T cells [65]. Overall, the inhibition of mitochondrial metabolism by β2-AR signaling in CD8+ T cells is thought to contribute to the suppression of T cell activation and function (Figure 4).
6.6. β2-AR Mediated Redox Signaling in T Cells
Within body’s very own cells, a treacherous threat lurks in the form of highly reactive and unstable molecules known as ROS. These harmful agents possess a lone, unpaired electron in their outermost shell, rendering them volatile and prone to uncontrolled behavior. They are not mere invaders from without, but rather insidious byproducts of metabolic processes, waiting to wreak havoc on T lymphocytes [88]. CAR-T therapy has been designed to inhibit oxidative stress previously. The study demonstrated that CAR-T cells co-expressing catalase had better anti-tumor control and free radical scavenging capacity than control [89].
As discussed above, β2-AR can cause metabolic insufficiencies in T cells, and these changes especially in mitochondrial bioenergetics can lead to increased ROS production and signaling in T cells [90]. This was demonstrated in study, where it was shown that Nor-epinephrine can lead to the production of excess ROS in T lymphocytes, by disrupting mitochondrial metabolism and hence depicting mitochondria as a major source of ROS in T cells [91]. Some recent researches have highlighted that most of the NA mediated affects in T cells are via β2-AR receptors, so one of the proposed mechanisms by which β2-AR can alter T cell proliferation and cytokine profile might be due to their ability to produce excess ROS by disrupting mitochondrial metabolism [49, 65].
7. Perspective for CAR-T Cell Therapy
CAR-T cell therapy has gained excellent success for treating cancers and has evolved as a promising new strategy to control tumor growth in patients especially in blood cancers. Yet many cancers especially the solid tumors are mostly resistant to beneficial affects resulting in lower remission rates and relapse [92, 93]. Immunotherapies including CAR-T cell therapy rely on patient’s immune system in order to fight back with cancer displaying robust anti-tumor response, which is hindered by immunosuppressive factors within tumor milieu. Therefore, the need to develop novel therapeutic strategies to reverse immunosuppression never ceases.
Adrenergic stress is one of the several culprits behind immunosuppression inside TME, and has evolved recently as an important regulator of inflammation, immunity and cancer [94]. Chronic release of NA inside TME affluences tumor growth by impeding immune responses as evident in preclinical models of some cancers [9, 95]. β2-AR among adrenergic receptors is most studied receptor for its immunosuppressive function in T lymphocytes. β2-AR has been shown to decreases T cell activation, proliferation and cytokine release in several studies. The receptor performs this function by distorting mitochondrial fitness of the cell [6, 42]. That is why β-antagonists have demonstrated reduced tumor growth in many cancers by increasing immune response. This has been implicated in checkpoint inhibitor therapies, where β-blockers mediated inhibition of β2 signaling led to significant improvement in response to checkpoint inhibitor therapies. Blocking β2 signaling has been shown to increase tumor infiltrating CD8+ T cells, and hence can potentiate immunotherapy [96]. We have got some promising results in CAR-T cell therapy, where β2 inhibition has led to increased efficacy of CAR-T cell therapy by increasing activation, degranulation, cytokine release of CAR-T cells and at the same time decreasing exhaustion (data not published). Hence, we propose in the light of present literature and our own findings that blocking β2-adrenergic signaling in T cells can be an applicable strategy to improve CAR-T cell functioning in complex tumor microenvironments. Inhibitory role of β2 in T lymphocytes is often demonstrated which gives sufficient rationale to translate this research into CAR-T research by interfering adrenergic signaling for the benefit of immunotherapeutic response. Implying current knowledge about the role of β2-AR in tumor microenvironments and especially how does it impact T cell functions can synergize anti-tumor efficacy of CAR-T cell therapies in future clinical trials. More studies unearthing the molecular mechanisms of β2-AR mediated T cell inhibition will pave the way for designing novel immunotherapies for cancer treatment.
Author Contributions
Conceptualization: M.A.F., I.A., W.J; Literature search: M.A.F., I.A., X.H., Y.C., R.Y; Writing: M.A.F., I.A., W.J; Art-work: I.A; Review and editing: X.H., Y.C., R.Y; Supervision and Guidance: W.J. All the authors have contributed in gathering the literature and contributed significantly in the manuscript. All the authors have read and agree to information presented here.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This work was supported by Science and Technology Commission of Shanghai Municipality (21S11906200), National Natural Science Foundation of China (81771306), National Key Research and Development Program of China (2021YFF0702400).
Conflicts of Interest
Authors declare no conflict of interests.
References
- Lymperopoulos, A. Physiology and pharmacology of the cardiovascular adrenergic system. Front. Physiol. 2013, 4, 240. [Google Scholar] [CrossRef] [PubMed]
- Molina, P.E. Neurobiology of the stress response: contribution of the sympathetic nervous system to the neuroimmune axis in traumatic injury. Shock 2005, 24, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Chrousos, G.P. Stress and disorders of the stress system. Nat. Rev. Endocrinol. 2009, 5, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Zhen, Z.; Chao, Y.; Li, W.; Gang, D.; Feng, J. Epinephrine increases malignancy of breast cancer through p38 MAPK signaling pathway in depressive disorders. Int. J. Clin. Exp. Pathol. 2019, 12, 1932. [Google Scholar] [PubMed]
- Irwin, M.R.; Miller, A.H. Depressive disorders and immunity: 20 years of progress and discovery. Brain, Behav. Immun. 2007, 21, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Graff, R.M.; Kunz, H.E.; Agha, N.H.; Baker, F.L.; Laughlin, M.; Bigley, A.B.; et al. β2-Adrenergic receptor signaling mediates the preferential mobilization of differentiated subsets of CD8+ T-cells, NK-cells and non-classical monocytes in response to acute exercise in humans. Brain Behav. Immun. 2018, 74, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.C.; Araujo, L.P.; Maricato, J.T.; Nascimento, V.M.; Guereschi, M.G.; Rezende, R.M.; et al. Norepinephrine Controls Effector T Cell Differentiation through β2-Adrenergic Receptor–Mediated Inhibition of NF-κB and AP-1 in Dendritic Cells. J. Immunol. 2016, 196, 637–644. [Google Scholar] [CrossRef]
- Xiao, L.; Li, X.; Fang, C.; Yu, J.; Chen, T. Neurotransmitters: promising immune modulators in the tumor microenvironment. Front. Immunol. 2023, 14, 1118637. [Google Scholar] [CrossRef]
- Silva, D.; Quintas, C.; Gonçalves, J.; Fresco, P. Contribution of adrenergic mechanisms for the stress-induced breast cancer carcinogenesis. J. Cell. Physiol. 2022, 237, 2107–2127. [Google Scholar] [CrossRef]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. , PPARγ and the innate immune system mediate the resolution of inflammation. PPAR Res. 2015, 2015. [Google Scholar]
- Apavaloaei, A.; Hardy, M.-P.; Thibault, P.; Perreault, C. The Origin and Immune Recognition of Tumor-Specific Antigens. Cancers 2020, 12, 2607. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Ellis, G.I.; Sheppard, N.C.; Riley, J.L. Genetic engineering of T cells for immunotherapy. Nat. Rev. Genet. 2021, 22, 427–447. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; He, J.; Liu, L.; Wang, J.; Wang, S.; Liu, L.; Gao, L.; Liu, Y.; Kong, P.; Liu, J.; et al. CD19-Directed Fast CART Therapy for Relapsed/Refractory Acute Lymphoblastic Leukemia: From Bench to Bedside. Blood 2019, 134, 1340–1340. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dogan, A.; Siegel, D.; Tran, N.; Fu, A.; Fowler, J.; Belani, R.; Landgren, O. B-cell maturation antigen expression across hematologic cancers: a systematic literature review. Blood Cancer J. 2020, 10, 73. [Google Scholar] [CrossRef]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef]
- El-Sayes, N.; Vito, A.; Mossman, K. Tumor Heterogeneity: A Great Barrier in the Age of Cancer Immunotherapy. Cancers 2021, 13, 806. [Google Scholar] [CrossRef]
- Li, W.; Song, X.; Jin, Y.; Li, F.; Yu, H.; Cao, C.; et al. Carts for solid tumors: feasible or infeasible? Oncol Res Treat. 2017, 40, 540–546. [Google Scholar] [CrossRef]
- He, C.; Zhou, Y.; Li, Z.; Farooq, M.A.; Ajmal, I.; Zhang, H.; Zhang, L.; Tao, L.; Yao, J.; Du, B.; et al. Co-Expression of IL-7 Improves NKG2D-Based CAR T Cell Therapy on Prostate Cancer by Enhancing the Expansion and Inhibiting the Apoptosis and Exhaustion. Cancers 2020, 12, 1969. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lin, H.; Guo, D.; Cheng, S.; Zhou, Y.; Zhang, L.; Yao, J.; Farooq, M.A.; Ajmal, I.; Duan, Y.; et al. Suppression of 4.1R enhances the potency of NKG2D-CAR T cells against pancreatic carcinoma via activating ERK signaling pathway. Oncogenesis 2021, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sanders, V.M.; Straub, R.H. , Norepinephrine, the β-adrenergic receptor, and immunity. Brain Behav. Immun. 2002, 16, 290–332. [Google Scholar] [CrossRef] [PubMed]
- Sanders, V.M. The beta2-adrenergic receptor on T and B lymphocytes: do we understand it yet? Brain Behav. Immun. 2012, 26, 195–200. [Google Scholar] [CrossRef]
- Wang, W.; Cao, X. Beta-Adrenergic Signaling in Tumor Immunology and Immunotherapy. Crit. Rev. Immunol. 2019, 39, 93–103. [Google Scholar] [CrossRef]
- Wrobel, L.J.; Gayet-Ageron, A.; Le Gal, F.-A. Effects of Beta-Blockers on Melanoma Microenvironment and Disease Survival in Human. Cancers 2020, 12, 1094. [Google Scholar] [CrossRef]
- Lorton, D.; Bellinger, D.L. Molecular Mechanisms Underlying β-Adrenergic Receptor-Mediated Cross-Talk between Sympathetic Neurons and Immune Cells. Int. J. Mol. Sci. 2015, 16, 5635–5665. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Ammons, D.T.; MacDonald, C.R.; Chow, L.; Repasky, E.A.; Dow, S. Chronic adrenergic stress and generation of myeloid-derived suppressor cells: Implications for cancer immunotherapy in dogs. Veter- Comp. Oncol. 2023, 21, 159–165. [Google Scholar] [CrossRef]
- Koinis, F.; Xagara, A.; Chantzara, E.; Leontopoulou, V.; Aidarinis, C.; Kotsakis, A. Myeloid-Derived Suppressor Cells in Prostate Cancer: Present Knowledge and Future Perspectives. Cells 2021, 11, 20. [Google Scholar] [CrossRef]
- Ağaç, D.; Estrada, L.D.; Maples, R.; Hooper, L.V.; Farrar, J.D. The β2-adrenergic receptor controls inflammation by driving rapid IL-10 secretion. Brain, Behav. Immun. 2018, 74, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Zahalka, A.H.; Arnal-Estapé, A.; Maryanovich, M.; Nakahara, F.; Cruz, C.D.; Finley, L.W.S.; Frenette, P.S. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017, 358, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Gardner, K.P.; Cristofanilli, M.; Chumsri, S.; Lapidus, R.; Tang, C.-M.; Raghavakaimal, A.; Adams, D.L. Beta 2-Adrenergic Receptor in Circulating Cancer-Associated Cells Predicts for Increases in Stromal Macrophages in Circulation and Patient Survival in Metastatic Breast Cancer. Int. J. Mol. Sci. 2022, 23, 7299. [Google Scholar] [CrossRef] [PubMed]
- Lamkin, D.M.; Ho, H.-Y.; Ong, T.H.; Kawanishi, C.K.; Stoffers, V.L.; Ahlawat, N.; Ma, J.C.; Arevalo, J.M.; Cole, S.W.; Sloan, E.K. β-Adrenergic-stimulated macrophages: Comprehensive localization in the M1-M2 spectrum. Brain, Behav. Immun. 2016, 57, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Thapa, S.; Cao, X. Nervous regulation: beta-2-adrenergic signaling in immune homeostasis, cancer immunotherapy, and autoimmune diseases. Cancer Immunol. Immunother. 2023, 72, 2549–2556. [Google Scholar] [CrossRef]
- Qiao, G. Understanding the Mechanisms by Which β-Adrenergic Stress Signaling Impairs T-Cell Immune Responses. Doctoral Dissertation State University of New York at Buffalo, 2021.
- Kavelaars, A. Regulated expression of α-1 adrenergic receptors in the immune system. Brain, Behav. Immun. 2002, 16, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Kalinichenko, V.V.; Mokyr, M.B.; Graf, L.H.; Cohen, R.L.; Chambers, D.A. Norepinephrine-Mediated Inhibition of Antitumor Cytotoxic T Lymphocyte Generation Involves a β-Adrenergic Receptor Mechanism and Decreased TNF-α Gene Expression. PEDIATRICS 1999, 163, 2492–2499. [Google Scholar] [CrossRef]
- Casale, T.; Kaliner, M. Demonstration that circulating human blood cells have no detectable alpha1-adrenergic receptors by radioligand binding analysis. J. Allergy Clin. Immunol. 1984, 74, 812–818. [Google Scholar] [CrossRef]
- van der Voort, C.R.; Kavelaars, A.; van de Pol, M.; Heijnen, C.J. Noradrenaline induces phosphorylation of ERK-2 in human peripheral blood mononuclear cells after induction of α1-adrenergic receptors. J. Neuroimmunol. 2000, 108, 82–91. [Google Scholar] [CrossRef]
- Grisanti, L.A.; Perez, D.M.; Porter, J.E. Modulation of Immune Cell Function by α1-Adrenergic Receptor Activation. Curr Top Membr. 2011, 67, 113–138. [Google Scholar] [CrossRef]
- Nissen, M.D.; Sloan, E.K.; Mattarollo, S.R. β-Adrenergic Signaling Impairs Antitumor CD8+ T-cell Responses to B-cell Lymphoma ImmunotherapyβAR Signaling Impairs T cell–Mediated Immunotherapy. Cancer Immunol. Res. 2018, 6, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; He, Y.; Chen, F.; Zhang, F.; Shao, D.; Wang, Z. Leveraging β-Adrenergic Receptor Signaling Blockade for Improved Cancer Immunotherapy Through Biomimetic Nanovaccine. Small 2023, 19, e2207029. [Google Scholar] [CrossRef] [PubMed]
- Estrada, L.D.; Ağaç, D.; Farrar, J.D. Sympathetic neural signaling via the β2-adrenergic receptor suppresses T-cell receptor-mediated human and mouse CD8+ T-cell effector function. Eur. J. Immunol. 2016, 46, 1948–1958. [Google Scholar] [CrossRef] [PubMed]
- Slota, C.; Shi, A.; Chen, G.; Bevans, M.; Weng, N.-P. Norepinephrine preferentially modulates memory CD8 T cell function inducing inflammatory cytokine production and reducing proliferation in response to activation. Brain, Behav. Immun. 2015, 46, 168–179. [Google Scholar] [CrossRef]
- Grebe, K.M.; Hickman, H.D.; Irvine, K.R.; Takeda, K.; Bennink, J.R.; Yewdell, J.W. Sympathetic nervous system control of anti-influenza CD8 + T cell responses. Proc. Natl. Acad. Sci. 2009, 106, 5300–5305. [Google Scholar] [CrossRef]
- Chen, M.; Qiao, G.; Hylander, B.L.; Mohammadpour, H.; Wang, X.-Y.; Subjeck, J.R.; Singh, A.K.; Repasky, E.A. Adrenergic stress constrains the development of anti-tumor immunity and abscopal responses following local radiation. Nat. Commun. 2020, 11, 1821. [Google Scholar] [CrossRef] [PubMed]
- Bucsek, M.J.; Qiao, G.; MacDonald, C.R.; Giridharan, T.; Evans, L.; Niedzwecki, B.; et al. b-Adrenergic Signaling in Mice Housed at Standard Temperatures Suppresses an Effector Phenotype in CD8 þ T Cells and Undermines Checkpoint Inhibitor Therapy. Cancer Res. 2017, 77, 5639–5651. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.; Bucsek, M.J.; Winder, N.M.; Chen, M.; Giridharan, T.; Olejniczak, S.H.; Hylander, B.L.; Repasky, E.A. β-Adrenergic signaling blocks murine CD8+ T-cell metabolic reprogramming during activation: a mechanism for immunosuppression by adrenergic stress. Cancer Immunol. Immunother. 2018, 68, 11–22. [Google Scholar] [CrossRef]
- Wehbi, V.L.; Taskén, K. Molecular mechanisms for cAMP-mediated immunoregulation in T cells–role of anchored protein kinase a signaling units. Front Immunol. 2016, 7, 222. [Google Scholar] [CrossRef]
- Zheng, W.; Jones, L.L.; Geiger, T.L. Modulation of PI3K signaling to improve CAR T cell function. Oncotarget 2018, 9, 35807–35808. [Google Scholar] [CrossRef]
- Bucsek, M.J.; Qiao, G.; MacDonald, C.R.; Giridharan, T.; Evans, L.; Niedzwecki, B.; et al. β-Adrenergic Signaling in Mice Housed at Standard Temperatures Suppresses an Effector Phenotype in CD8+ T Cells and Undermines Checkpoint Inhibitor Therapyβ-Blockers Increase T-cell Effectors and Anti-PD-1 Efficacy. Cancer Res. 2017, 77, 5639–5651. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive Strategies that are Mediated by Tumor Cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Bartik, M.M.; Brooks, W.H.; Roszman, T.L. Modulation of T Cell Proliferation by Stimulation of the β-Adrenergic Receptor: Lack of Correlation between Inhibition of T Cell Proliferation and cAMP Accumulation. Cell. Immunol. 1993, 148, 408–421. [Google Scholar] [CrossRef] [PubMed]
- AHarrison, A.J.; Du, X.; von Scheidt, B.; Kershaw, M.H.; Slaney, C.Y. Enhancing co-stimulation of CAR T cells to improve treatment outcomes in solid cancers. Immunother. Adv. 2021, 1, ltab016. [Google Scholar] [CrossRef]
- Torchia, M.L.G.; Conze, D.B.; Jankovic, D.; Ashwell, J.D. Balance between NF-κB p100 and p52 regulates T cell costimulation dependence. J. Immunol. 2013, 190, 549–555. [Google Scholar] [CrossRef]
- Boucher, J.C.; Li, G.; Kotani, H.; Cabral, M.L.; Morrissey, D.; Lee, S.B.; et al. CD28 Costimulatory Domain–Targeted Mutations Enhance Chimeric Antigen Receptor T-cell FunctionCD28 Mutation Enhances CAR T-cell Function. Cancer Immunol. Res. 2021, 9, 62–74. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2020, 18, 842–859. [Google Scholar] [CrossRef]
- Pettitt, D.; Arshad, Z.; Smith, J.; Stanic, T.; Holländer, G.; Brindley, D. CAR-T cells: a systematic review and mixed methods analysis of the clinical trial landscape. Mol. Ther. 2018, 26, 342–353. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, W.; Li, Y.; Yi, Y.; Yu, Z.; Liu, X.; Zhang, L.; Zheng, Y.; Niu, T. A systematic review on performance analysis of critical time points in multiple myeloma treated by CAR-T cell immunotherapy. Int. Immunopharmacol. 2023, 114, 109592. [Google Scholar] [CrossRef] [PubMed]
- González-Amaro, R.; Cortes, J.R.; Sánchez-Madrid, F.; Martín, P. Is CD69 an effective brake to control inflammatory diseases? Trends Mol. Med. 2013, 19, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Fernández, M.; de la Fuente, H.; Martín, P.; Cibrián, D.; Sánchez-Madrid, F. Unraveling CD69 signaling pathways, ligands and laterally associated molecules. EXCLI J. 2023, 22, 334. [Google Scholar] [PubMed]
- Qiao, G.; Chen, M.; Mohammadpour, H.; MacDonald, C.R.; Bucsek, M.J.; Hylander, B.L.; et al. Chronic Adrenergic Stress Contributes to Metabolic Dysfunction and an Exhausted Phenotype in T Cells in the Tumor Microenvironment Chronic Stress Drives T-cell Dysfunction in Tumors. Cancer Immunol. Res. 2021, 9, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Alexandre, Y.O.; Loi, J.K.; Gillis, R.; Ghazanfari, N.; Creed, S.J.; Holz, L.E.; Shackleford, D.; Mackay, L.K.; Heath, W.R.; et al. Adrenergic regulation of the vasculature impairs leukocyte interstitial migration and suppresses immune responses. Immunity 2021, 54, 1219–1230. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hemmi, H. Dendritic Cells: Translating Innate to Adaptive Immunity. Curr. Top. Microbiol. Immunol. 2006, 311, 17–58. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Burga, R.A.; Powell, A.B.; Chorvinsky, E.A.; Hoq, N.; McCormack, S.E.; Van Pelt, S.N.; Hanley, P.J.; Cruz, C.R.Y. Beyond CAR T Cells: Other Cell-Based Immunotherapeutic Strategies Against Cancer. Front. Oncol. 2019, 9, 196. [Google Scholar] [CrossRef]
- Hervé, J.; Dubreil, L.; Tardif, V.; Terme, M.; Pogu, S.; Anegon, I.; Rozec, B.; Gauthier, C.; Bach, J.-M.; Blancou, P. β2-Adrenoreceptor Agonist Inhibits Antigen Cross-Presentation by Dendritic Cells. Perspect. Surg. 2013, 190, 3163–3171. [Google Scholar] [CrossRef]
- Nissen, M. The impact of β-adrenergic signalling on immune function and immunotherapy in lymphoma. 2017. [CrossRef]
- Daher, C.; Vimeux, L.; Stoeva, R.; Peranzoni, E.; Bismuth, G.; Wieduwild, E.; et al. Blockade of β-Adrenergic Receptors Improves CD8+ T-cell Priming and Cancer Vaccine Efficacyβ-Blockers Boost T-cell Priming and Cancer Vaccine Efficacy. Cancer Immunol. Res. 2019, 7, 1849–1863. [Google Scholar] [CrossRef]
- Egri, N.; de Landazuri, I.O.; Bartolomé, C.S.; Ortega, J.R.; Español-Rego, M.; Juan, M. CART manufacturing process and reasons for academy-pharma collaboration. Immunol. Lett. 2019, 217, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Won, A.; Refaeli, Y.; Van Parijs, L. IL-2 and Related Cytokines Can Promote T Cell Survival by Activating AKT. Heal. Educ. 2002, 168, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.D.; Hunninghake, G.W.; McArdle, W.L. Beta-adrenergic-receptor-mediated suppression of interleukin 2 receptors in human lymphocytes. PEDIATRICS 1987, 139, 3355–3359. [Google Scholar] [CrossRef]
- Ruiz-Medina, B.E.; Cadena-Medina, D.A.; Esparza, E.; Arrieta, A.J.; Kirken, R.A. Isoproterenol-induced beta-2 adrenergic receptor activation negatively regulates interleukin-2 signaling. Biochem. J. 2018, 475, 2907–2923. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Yang, R.; Tu, J.; Xi, Y.; Yang, S.; Lv, L.; Zhai, X.; Zhu, Y.; Dong, D.; Tao, X. Metabolic reprogramming of immune cells in pancreatic cancer progression. BioMedicine 2023, 157, 113992. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Rathmell, J.C.; Macintyre, A.N. Metabolic Reprogramming towards Aerobic Glycolysis Correlates with Greater Proliferative Ability and Resistance to Metabolic Inhibition in CD8 versus CD4 T Cells. PLOS ONE 2014, 9, e104104. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, S.E.; O’neill, L.A. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef]
- Kokolus, K.M.; Capitano, M.L.; Lee, C.-T.; Eng, J.W.-L.; Waight, J.D.; Hylander, B.L.; Sexton, S.; Hong, C.-C.; Gordon, C.J.; Abrams, S.I.; et al. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc. Natl. Acad. Sci. 2013, 110, 20176–20181. [Google Scholar] [CrossRef]
- Chen, Z.; Vaeth, M.; Eckstein, M.; Delgobo, M.; Ramos, G.; Frantz, S.; Hofmann, U.; Gladow, N. Characterization of the effect of the GLUT-1 inhibitor BAY-876 on T cells and macrophages. Eur. J. Pharmacol. 2023, 945, 175552. [Google Scholar] [CrossRef]
- Park, S.Y.; Kang, J.H.; Jeong, K.J.; Lee, J.; Han, J.W.; Choi, W.S.; et al. Retracted: Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1α protein-dependent mechanism. Int. J. Cancer. 2011, 128, 2306–2316. [Google Scholar] [CrossRef]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B., Jr; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.W.P.; Simões, A.M.C.; Straten, P.T.; Olofsson, G.H. Adrenergic Signaling in Immunotherapy of Cancer: Friend or Foe? Cancers 2021, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692–6692. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8 + T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef]
- Yu, Y.-R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.-C.; et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor–Redirected T Cells as well as Bystander Cells from Oxidative Stress–Induced Loss of Antitumor Activity. Heal. Educ. 2016, 196, 759–766. [Google Scholar] [CrossRef]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid. Med. Cell. Longev. 2016, 2016, 1580967. [Google Scholar] [CrossRef]
- Case, A.J.; Roessner, C.T.; Tian, J.; Zimmerman, M.C. Mitochondrial superoxide signaling contributes to norepinephrine-mediated T-lymphocyte cytokine profiles. PloS one 2016, 11, e0164609. [Google Scholar] [CrossRef]
- Ogbodo, E.J.; Ruff, M.W.; Sakemura, R.L.; ManriquezRoman, C.; Huynh, T.; Girsch, J.H.; Sirpilla, O.L.; Yun, K.; Stewart, C.M.; Can, I.; et al. Abstract 5017: Simultaneous targeting of EphA3 on glioblastoma and tumor microenvironment to overcome resistance to CART cell therapy in brain cancer. Cancer Res 2023, 83, 5017–5017. [Google Scholar] [CrossRef]
- He, J.; Munir, F.; Ragoonanan, D.; Zaky, W.; Khazal, S.J.; Tewari, P.; Fueyo, J.; Gomez-Manzano, C.; Jiang, H. Combining CAR T Cell Therapy and Oncolytic Virotherapy for Pediatric Solid Tumors: A Promising Option. Immuno 2023, 3, 37–56. [Google Scholar] [CrossRef]
- Dünser, M.W.; Hasibeder, W.R. Sympathetic Overstimulation During Critical Illness: Adverse Effects of Adrenergic Stress. J. Intensiv. Care Med. 2009, 24, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.-F.; Jin, F.-J.; Li, N.; Guan, H.-T.; Lan, L.; Ni, H.; Wang, Y. Adrenergic receptor β2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB Rep. 2015, 48, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.S.; Guzner, A.; Wainwright, D.A.; Mohindra, N.A.; Chae, Y.K.; Behdad, A.; Villaflor, V.M. The Impact of Beta Blockers on Survival Outcomes in Patients With Non–small-cell Lung Cancer Treated With Immune Checkpoint Inhibitors. Clin. Lung Cancer 2020, 22, e57–e62. [Google Scholar] [CrossRef]
Figure 1.
Nor-adrenaline (NA) mediated immunosuppression inside tumor microenvironment. Nor-adrenaline released in tumor microenvironment activates β2-AR on the surface of various immune cells. The activation of these adrenergic receptors activates Myeloid Derived Suppresser Cells (MDSCs), recruits T-Regulatory cells (Tregs) and M2 Tumor-associated Macrophage (TAMS). On the other hand, the activity of Natural killer (NK) cells, T cells and Dendritic cells (DCs) is inhibited by β2-AR signaling. .
Figure 1.
Nor-adrenaline (NA) mediated immunosuppression inside tumor microenvironment. Nor-adrenaline released in tumor microenvironment activates β2-AR on the surface of various immune cells. The activation of these adrenergic receptors activates Myeloid Derived Suppresser Cells (MDSCs), recruits T-Regulatory cells (Tregs) and M2 Tumor-associated Macrophage (TAMS). On the other hand, the activity of Natural killer (NK) cells, T cells and Dendritic cells (DCs) is inhibited by β2-AR signaling. .
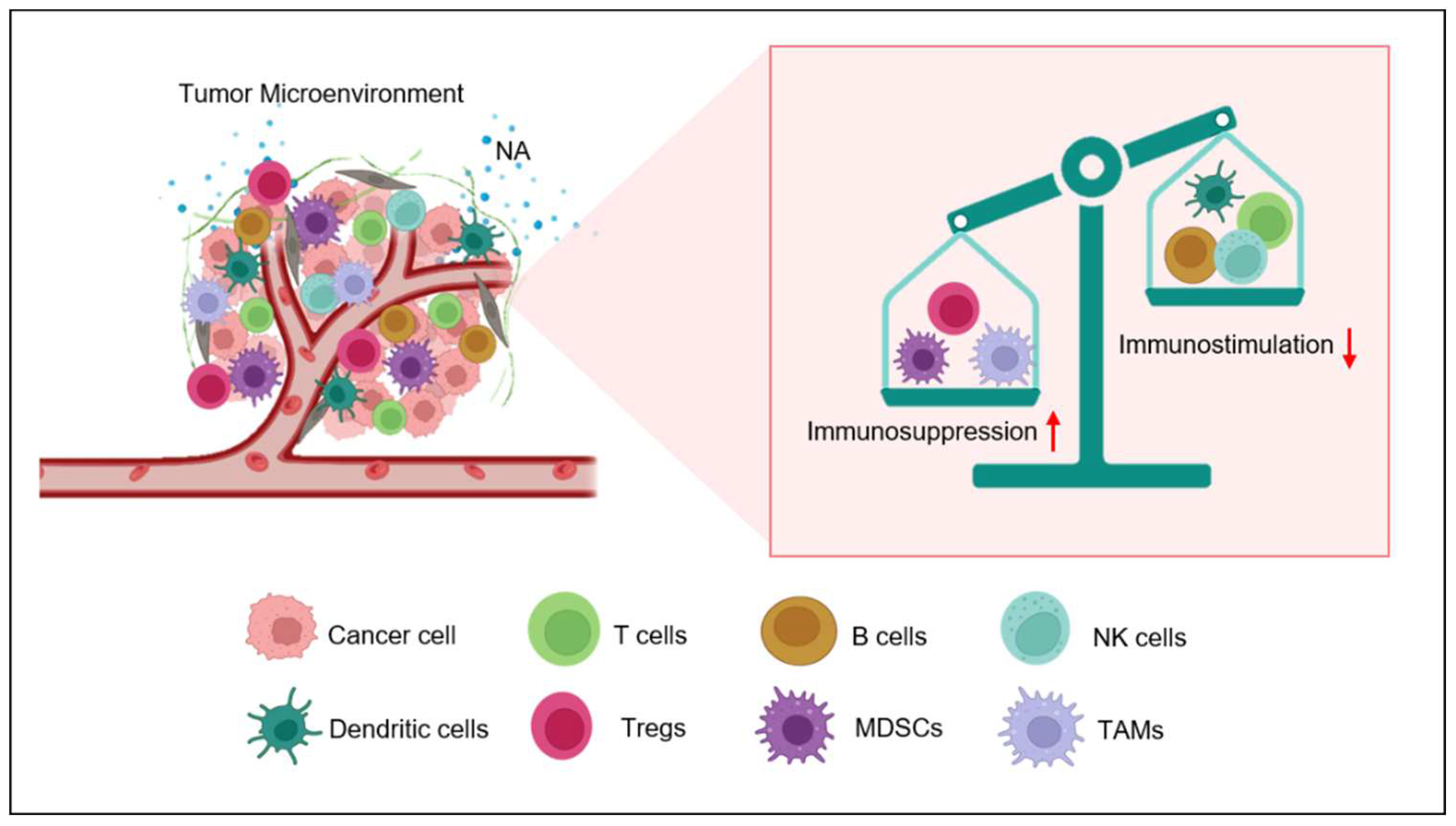
Figure 2.
β2-AR mediated CAR-T cell signaling. Panel A indicated the signaling pathways of CAR-T cell after activation. Panel B indicated that activation of β2-AR hinders the normal functioning of CAR-T cells by inhibiting the phosphorylation of ZAP70 in a PKA dependent manner. As a result, various downstream signaling pathways (NFAT, ERK, ATF2, and NF-κB) that regulate T cell function are also inhibited.
Figure 2.
β2-AR mediated CAR-T cell signaling. Panel A indicated the signaling pathways of CAR-T cell after activation. Panel B indicated that activation of β2-AR hinders the normal functioning of CAR-T cells by inhibiting the phosphorylation of ZAP70 in a PKA dependent manner. As a result, various downstream signaling pathways (NFAT, ERK, ATF2, and NF-κB) that regulate T cell function are also inhibited.
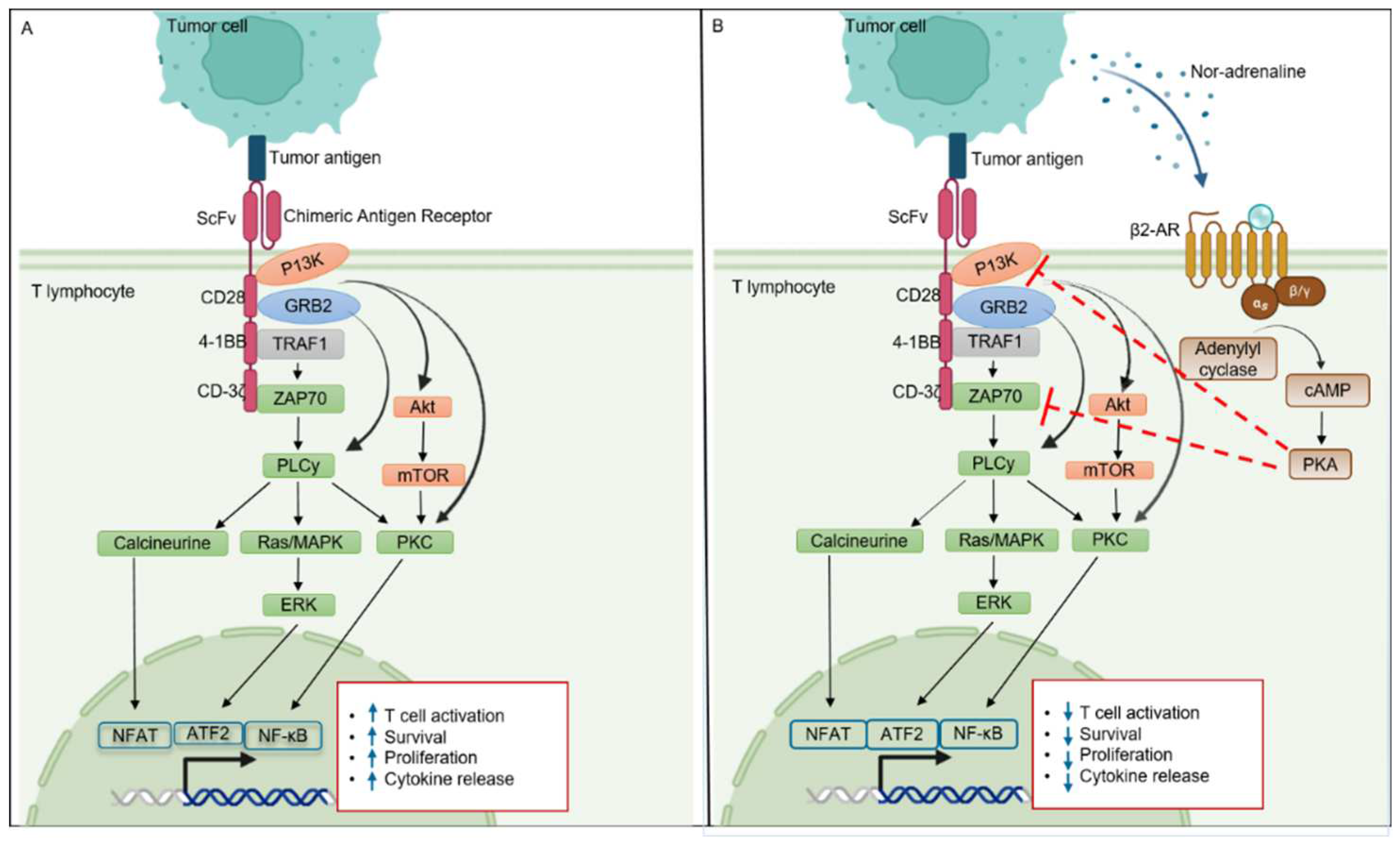
Figure 3.
Schematic representation of β2-AR mediated inhibition of CAR-T cell function. Optimum CAR-T activation requires signaling via all the three signals (Signal 1, Signal 2 and Signal 3). Adrenergic signaling via β2-AR activation on the surface of T cells can block CAR-T cell activation signaling by inhibiting stimulatory domain, co-stimulatory domain and the activity of IL-2 Receptor.
Figure 3.
Schematic representation of β2-AR mediated inhibition of CAR-T cell function. Optimum CAR-T activation requires signaling via all the three signals (Signal 1, Signal 2 and Signal 3). Adrenergic signaling via β2-AR activation on the surface of T cells can block CAR-T cell activation signaling by inhibiting stimulatory domain, co-stimulatory domain and the activity of IL-2 Receptor.
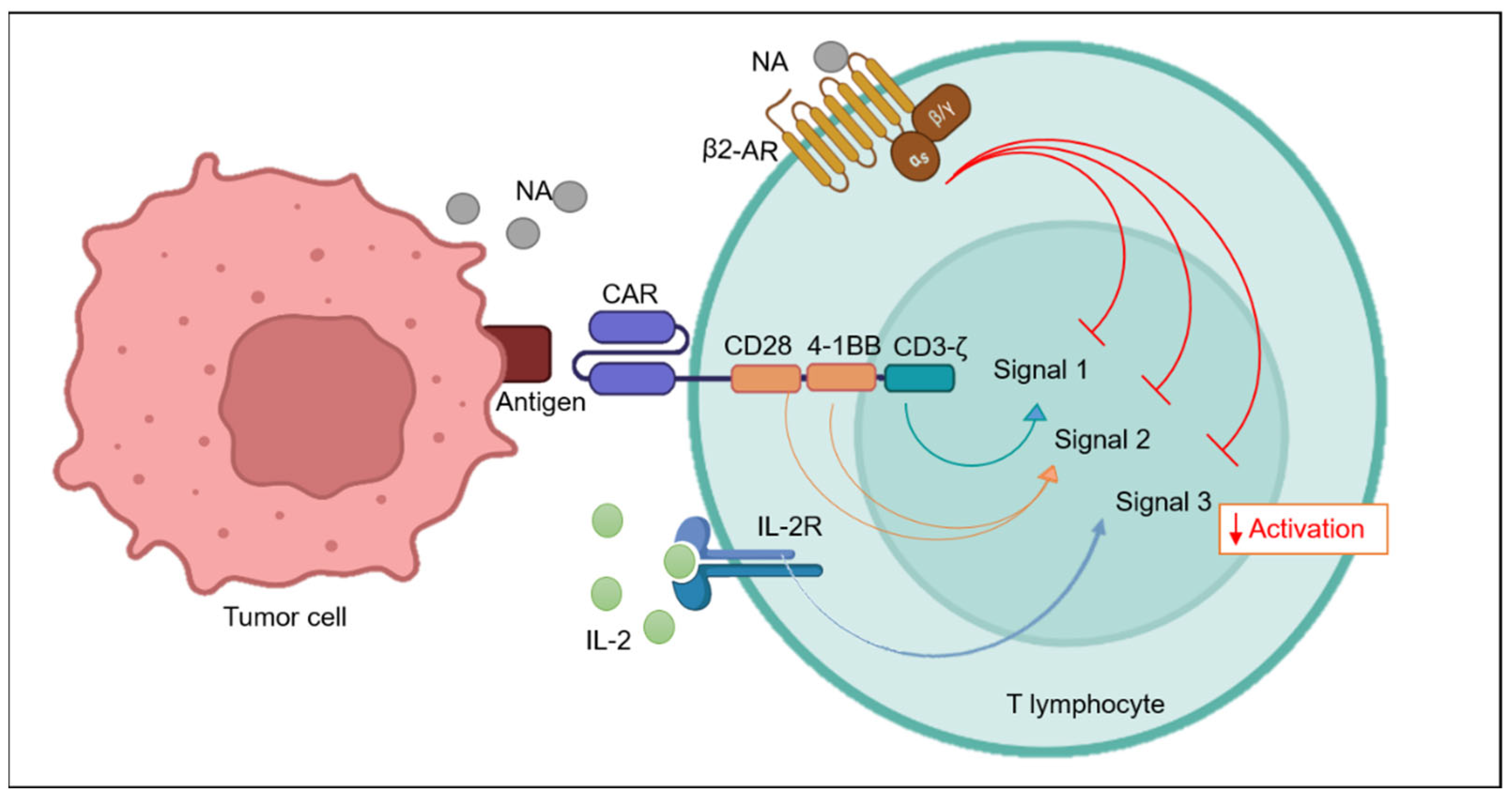
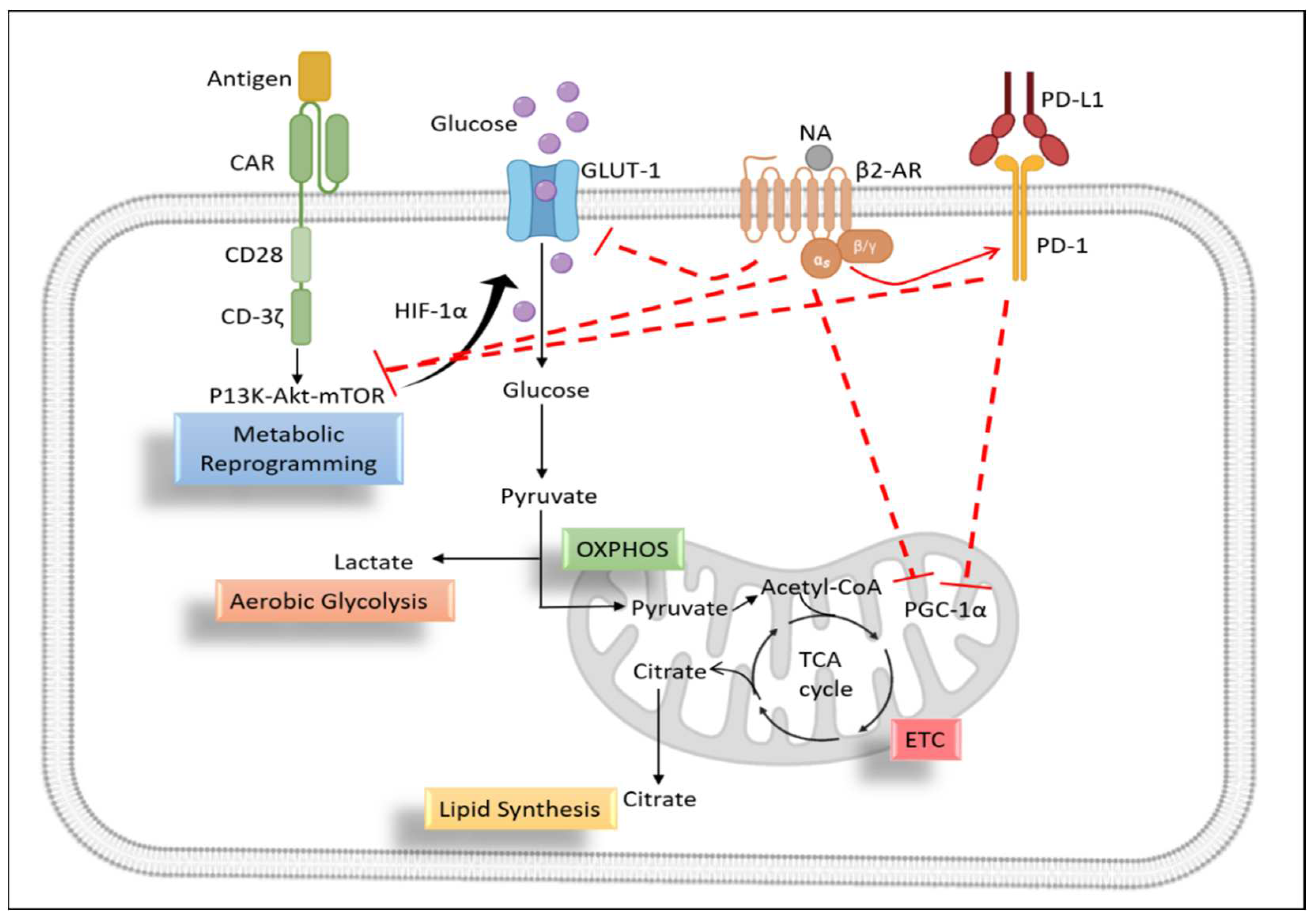
Figure 4.
Proposed Role of β2-Adrenergic signaling in metabolic reprogramming of CAR-T cells. CAR-T cells have been activated after recognizing their specific antigen on tumor cells. CD3-ζ and co-stimulatory molecules undergoes metabolic reprogramming by activating P13K-Akt-mTOR pathway. This pathway increases the expression of GLUT-1 and release pyruvate as a by-product of aerobic glycolysis. Pyruvate enter the mitochondria and starts TCA cycle which in turn produce high amount of energy for T cell functioning. Nor-adrenaline in TME binds with β2-AR and releases PKA which can inhibit GLUT-1 and CD-3ζ, CD28, 4-1BB mediated activation signaling and therefore blocks the metabolic reprogramming. On the other hand, β2-AR increases the inhibitors (PD-1) signaling in T cells which also hinders the CAR signaling and let to mitochondrial dysfunction.
Figure 4.
Proposed Role of β2-Adrenergic signaling in metabolic reprogramming of CAR-T cells. CAR-T cells have been activated after recognizing their specific antigen on tumor cells. CD3-ζ and co-stimulatory molecules undergoes metabolic reprogramming by activating P13K-Akt-mTOR pathway. This pathway increases the expression of GLUT-1 and release pyruvate as a by-product of aerobic glycolysis. Pyruvate enter the mitochondria and starts TCA cycle which in turn produce high amount of energy for T cell functioning. Nor-adrenaline in TME binds with β2-AR and releases PKA which can inhibit GLUT-1 and CD-3ζ, CD28, 4-1BB mediated activation signaling and therefore blocks the metabolic reprogramming. On the other hand, β2-AR increases the inhibitors (PD-1) signaling in T cells which also hinders the CAR signaling and let to mitochondrial dysfunction.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



