
Submitted:
18 July 2023
Posted:
18 July 2023
You are already at the latest version
Abstract
Peripheral arterial diseases (PAD) are complex cardiovascular conditions influenced by environmental factors and somatic mutations in multiple genes involved in hematopoiesis and inflammation. While traditional risk factors such as smoking, hypercholesterolemia, and hypertension have been extensively studied, the role of somatic mutations in PAD progression remains underexplored. This article aims to provide a comprehensive review of the molecular mechanisms, genetic landscape, prognostic significance, and clinical implications of somatic mutations in PAD. The expansion of clonal hematopoiesis of indeterminate potential (CHIP) clones in the circulating blood, named clonal hematopoiesis (CH), leads to the infiltration of these clones into atherosclerotic plaques and the production of inflammatory cytokines, increasing the risk of cardiovascular diseases including PAD. Furthermore, recent experimental evidence has demonstrated the involvement of somatically mutated TP53 genes with a high variant allele frequency (VAF) in PAD development and prognosis. The review delves into the relationship between CH and PAD, elucidating the prevalence, impact, and underlying mechanisms of this association. This understanding paves the way for novel therapeutic approaches targeting CHIP to promote tissue regeneration and improve outcomes in PAD patients. It emphasizes the need for further re-search to fully unravel the genetic footprint of the disease and highlights potential clinical implications. The findings presented in this article lay the foundation for personalized medicine approaches and open avenues for the development of targeted therapies based on somatic mutation profiling.
Keywords:
Peripheral arterial diseases
; Somatic mutation
; Thrombosis
; Atherosclerosis
; Inflammation
; Genetic disorders
1. Introduction
Peripheral arterial diseases (PAD), which encompass a group of disorders characterized by a narrowing or occlusion of the arteries supplying blood to the lower extremities, result in reduced blood flow, claudication, non-healing ulcers, and, in severe cases, tissue necrosis or gangrene, posing significant morbidity and mortality risks that make them a global healthcare burden [1]. The complex interplay of genetic and environmental factors contributes to the development and progression of PAD [2]. Thus, understanding the underlying impaired mechanisms and pathophysiology of this condition is crucial for early detection, appropriate management, and the development of novel therapeutic approaches.
Somatic mutations are genetic alterations that occur in the DNA of non-germ cells, including the cells of the arterial walls in PAD. These mutations can arise from a variety of factors, including exposure to environmental toxins, chronic inflammation, and DNA replication errors [3]. The relevance of somatic mutations in PAD progression lies in their potential to disrupt critical cellular processes involved in maintaining vascular health, promoting atherosclerotic plaque formation, altering of vascular tone and remodeling, and modulating angiogenesis [2]. Somatic mutations can affect genes involved in lipid metabolism, extracellular matrix remodeling, and endothelial cell function, thereby influencing plaque stability and arterial remodeling [4]. Additionally, mutations in genes regulating angiogenesis can impact the development of collateral vessels, which are essential compensatory mechanisms in PAD [5]. For example, mutations in genes that regulate vascular smooth muscle cell function, endothelial cell integrity, and inflammation can influence the development and progression of atherosclerosis [6]. These mutations may alter the behavior of vascular cells, leading to increased cell proliferation, impaired cell death (apoptosis), enhanced inflammation, and aberrant responses to oxidative stress [7]. Furthermore, somatic mutations can disrupt lipid metabolism, affecting the stability and function of genes involved in this process, which in turn plays a crucial role in atherosclerotic plaque formation. Disruptions in lipid metabolism can result in the accumulation of cholesterol and other lipids within the arterial walls, further exacerbating the occlusion of the arteries in PAD and may contribute to the pathogenesis of the disease [8].
First of all, there are several genetic terms in this field that are deemed to be defined: As mentioned above, a somatic mutation is a genetic alteration that occurs in non-germline cells during a person's lifetime, rather than being inherited from their parents. Of note, clonal hematopoiesis (CH) refers to the expansion of a single mutated clone of blood cells, usually in the bone marrow (BM), and the presence of a dominant population of blood cells with a specific somatic mutation. Aging-related clonal hematopoiesis (ARCH) is benign CH defined by the presence of somatic mutations in the blood or BM whose incidence increases with age, with a variant allele frequency (VAF) below 2% [9]. Also, clonal hematopoiesis of indeterminate potential (CHIP) is given to a condition characterized by the presence of CH without associated hematologic malignancy [10]. The current diagnostic criteria for CHIP encompass the following: (1) the absence of evident hematological malignancy, (2) a normal blood count, and (3) the presence of mutant cells carrying significant driver mutations in at least 2% of peripheral white blood cells, indicated by VAF [11]. CHIP is often associated with an increased risk of cardiovascular disease (CVD) and other age-related conditions [12]. Furthermore, clonal hematopoiesis of oncogenic potential (CHOP) refers to clonal hematopoiesis occurring in a clinical context where there is a notable probability of evolving into an overt malignancy [9]. Moreover, VAF points out the proportion of sequencing reads that contain a specific variant allele compared to the total number of reads at that genomic position, which is used to estimate the abundance of a particular mutation within a sample [13] (Figure 1).
Understanding the role of somatic mutations in PAD is important for several reasons. Firstly, it helps to elucidate the underlying molecular mechanisms involved in disease development, providing insights into potential therapeutic targets. By identifying specific mutations associated with PAD, researchers can develop personalized treatment approaches tailored to individual patients' genetic profiles. Secondly, the detection of somatic mutations in PAD patients may serve as a diagnostic tool for assessing disease severity and progression, enabling clinicians to monitor patients more effectively and adjust treatment strategies accordingly.
2. Molecular Mechanisms of Somatic Mutations in PAD
The etiology of somatic mutations in PAD is multifactorial. Several risk factors, including aging, exposure to environmental toxins, chronic inflammation, radiation, and oxidative stress, can induce DNA damage and the subsequent accumulation of somatic mutations [14]. Additionally, genetic predisposition and underlying vascular diseases, such as atherosclerosis, can increase the susceptibility to somatic mutations in PAD. Epidemiological studies have provided valuable insights into the prevalence and distribution of somatic mutations in PAD. Large-scale genomic sequencing efforts have identified specific somatic mutations in genes associated with vascular biology, inflammation, and DNA repair pathways [13]. CHIP-PAD and CHIP pan-arterial atherosclerosis mutations most frequently occur in genes responsible for regulating epigenetics (DNMT3A and TET2), DNA damage repair (DDR) genes (PPM1D, TP53, and BRCA1/2), cell cycle and transcriptional regulator genes (JAK2 and ASXL1) [15], and mutations that specifically disrupt splicing factor genes (LUC7L2, PRPF8, SF3B1, SRSF2, U2AF1, and ZRSR2) [16]. These studies have revealed the heterogeneity of somatic mutations across PAD patients, highlighting the importance of understanding their impact on vascular biology. Various types of somatic mutations can occur in PAD, each with distinct effects on vascular biology. Point mutations, insertions, deletions, and structural rearrangements can disrupt critical genes involved in vascular homeostasis, cellular proliferation, differentiation, and apoptosis, leading to PAD progression [17,18]. These mutations can affect key regulators of vascular function, including endothelial nitric oxide synthase (eNOS) [19], vascular endothelial growth factor (VEGF) [20], and components of the renin-angiotensin system [21], contributing to the development and progression of PAD. Large-scale chromosomal rearrangements, including copy number variations and chromosomal translocations, can occur as somatic mutations in PAD, disrupting critical genes and regulatory regions and resulting in dysregulation of cellular processes involved in vascular biology, such as angiogenesis, extracellular matrix remodeling, and smooth muscle cell function [22,23]. Denny et al. [24] utilized electronic health record (EHR) definitions of diseases to investigate the connection between CHIP and various types of atherosclerotic disease affecting different vascular beds, including the mesenteric, coronary, cerebral, and aneurysmal vessels. The study found significant associations between CHIP and coronary artery disease (CAD), any aortic aneurysm, and chronic mesenteric ischemia. Notably, these associations were consistently more pronounced when large CHIP clones were present. Furthermore, CHIP was linked to the development of pan-arterial atherosclerosis, and once again, the effects were stronger in the presence of large CHIP clones. Of note, Zekavat et al. [25] classified the CHIP-PAD and CHIP pan-arterial atherosclerosis assessments by putative driver genes and specific mutations, centering on DNMT3A, TET2, ASXL1, JAK2, the DDR genes (PPM1D and TP53), and mutations that specifically disrupt splicing factor genes (LUC7L2, PRPF8, SF3B1, SRSF2, U2AF1, and ZRSR2). They discovered an association of CHIP with PAD involving the four frequently occurring CHIP genes (DNMT3A, TET2, ASXL1, and JAK2), with drastic heterogeneity of incident PAD effect sizes across the CHIP genes. These data also revealed the new outcome that DDR-TP53 and PPM1D CHIP associate with incident PAD/CAD, with a greater effect on PAD conferred by TP53. Among the mutations associated with CHIP, the role of TET2 is well-established in vascular disease. TET2's normal function plays a significant role in regulating important processes in both macro- and microcirculation [26]. These processes include preventing the transformation of vascular smooth muscle cells, offering protection to endothelial cells, and exerting anti-inflammatory and anti-atherogenic effects [27,28].
Maintenance of genomic integrity is crucial for proper vascular function, and impairments in DNA damage response can contribute to the accumulation of somatic mutations in PAD [29]. Cells have intricate DNA repair pathways, including base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), and homologous recombination (HR), which counteract DNA damage and maintain genomic stability [30]. Chronic exposure to risk factors like oxidative stress and chronic inflammation can overwhelm the DNA repair capacity, leading to increased DNA damage and the accumulation of somatic mutations [31,32]. Defects in specific DNA repair genes, such as those involved in the BER or NER pathways, can further exacerbate DNA damage accumulation in PAD. Dysfunctional DNA repair and response pathways can perpetuate the accumulation of somatic mutations, promote genomic instability, and contribute to the progression of PAD [25,29]. In this respect, Zekavat et al. identified 338 and 419 incident PAD cases in UK biobank (UKB) and Mass General Brigham Biobank (MGBB), respectively. Based on their results, CHIP was positively associated with an increased risk of PAD incidents in the UKB and MGBB [25]. More interestingly, Bick et al. revealed that those with larger CHIP clone sizes had greater risk for PAD, which is associate more strongly with unfavorable clinical manifestations [33]. Specific genes affected by somatic mutations in PAD have been identified, and understanding their functional consequences is critical for unraveling disease mechanisms [2].
3. Genetic Landscape of Peripheral Arterial Diseases
During the past decade, with the availability of high throughput and relatively low-cost genotyping, genome-wide association studies (GWAS) became the dominant methodology for studying the genetics of PAD. In 2019, Klarin and colleagues [13] conducted the largest GWAS study on PAD. They utilized EHR data and significantly increased the sample size by nearly 10 times through the analysis of the genetic biorepositories of the Million Veteran Program (MVP) and the UKB. The researchers examined approximately 32 million DNA sequence variants for their association with PAD. As a result, they identified 19 genomic regions that were significantly associated with PAD, 18 of them had not been reported previously. These genomic regions included genes such as CELSR2/SORT1, F5, LPA, HLA-B, HDAC9, IL-6, LPL, ABO, CDKN2B-AS1/9p21, TCF7L2, MMP3, CREB3L1, PTPN11, RP11-359M6.3, COL4A1, SMOC1, CHRNA3, LOC732538, and LDLR. Further analysis revealed that the identified genetic variants were also associated with several known risk factors for PAD, such as abnormal lipid levels, type 2 diabetes, smoking, thrombosis, and hypertension. Additionally, the researchers investigated the contribution of these genetic variants to peripheral, coronary, and cerebral artery atherosclerosis. They found that 14 of the PAD risk variants showed at least a nominal association with CAD, and 12 variants were associated with large artery stroke (LAS). Interestingly, when considering the presence of concomitant CAD or LAS, the effects of some genetic variants related to lipids (SORT1, LPA, and LDLR) on PAD risk were significantly reduced. This suggests that the shared causal pathways or comorbidities for atherosclerosis in different vascular beds may drive some of the risk for PAD. In contrast, four of the significant PAD variants in the RP11-359M6.3, HLA-B, CHRNA3, and F5 (Leiden variant, p.R506Q) loci were uniquely associated with PAD, while they were not associated with other types of atherosclerosis. Additionally, the COL4A1 locus, previously linked to CAD [34] and small vessel disease of the brain [35], was found to be associated with both PAD and CAD. Overall, this study provides valuable insights into the genetic factors underlying PAD and their relationships with various risk factors and atherosclerosis. These mutations disrupt the balance of cell proliferation and apoptosis, contributing to the progression of vascular lesions and PAD pathogenesis. Understanding the specific molecular alterations induced by these mutations provides valuable insights into the dysregulated pathways and cellular processes driving disease progression in PAD.
Somatic mutations do not act in isolation but interact with other molecular alterations in PAD progression. The interplay between somatic mutations and factors such as chronic inflammation, oxidative stress, and epigenetic modifications can influence disease severity and outcomes. For instance, somatic mutations can synergize with chronic inflammation to promote sustained immune activation and cytokine release, contributing to vascular damage and remodeling [36]. In turn, the inflammatory microenvironment can further enhance the accumulation of somatic mutations through increased DNA damage and impaired repair processes [37]. Additionally, epigenetic modifications, such as DNA methylation and histone modifications, can modulate the impact of somatic mutations in PAD [38]. Aberrant epigenetic changes can affect gene expression patterns, including genes involved in vascular biology and DNA repair, amplifying the effects of somatic mutations on disease progression [39]. Understanding the intricate interplay between somatic mutations and other molecular alterations provides a comprehensive view of the complex mechanisms underlying PAD. Integration of these diverse factors is crucial for developing targeted therapeutic strategies that address the multifaceted nature of PAD and improve patient outcomes.
4. CHIP as Prognostic Markers in PAD
The studies revealed that somatic mutation accumulates in almost all of the tissues, and depending on the mutation variant, type, and gene region, it may cause disease. Zekavat et al. [40] demonstrated that PAD-CHIP carriers tended to be older, male, previous smokers, and have a history of CAD, hypertension, and hyperlipidemia, indicating a common vascular bed damage pathway as reported earlier [41,42]. Notably, larger CHIP clone sizes (as measured by VAF) have a significant strong effect on peripheral blood counts by increasing of pro-inflammatory total white blood cells, monocytes, neutrophils, and platelets than smaller CHIP clone sizes. For example, TET2 deficiency in hematopoietic cells promotes the aberrant production of inflammatory cytokines/chemokines, such as IL-6 and IL-1β, in macrophages. This provides a direct link between TET2 loss and microenvironmental changes within the BM niche [43,44]. Moreover, a recent large cross-sectional analysis of (n = 750, 249) patients with MDS revealed that patients with TET2, DNMT3A, and AXSL1 mutations were associated with a high prevalence of PAD (prevalence among individuals aged < 65 and > 65 years was 14.5% and 43.2%, respectively; P < 0.0001) [45]. These findings indicate that CHIP carrier-patients with PAD aged > 65 were at a 3 – 4-fold higher risk of developing PAD than non-carriers. In another study, authors also demonstrated a graded association between CHIP VAF and PAD, as those with a VAF < 10% and a VAF >10% increased the risk of PAD incidence up to 58% and 100%, respectively [40]. These findings verify that CHIP is an independent risk factor that increases PAD events in a VAF-grade dependent manner. Further studies are needed to elucidate the association between specific somatic mutations and clinical outcomes and the precise predictive value of somatic mutations in disease progression and recurrence.
4.1. Accumulation of Mutant Clones in Ischemic Tissues of PAD Patients:
The emerging evidence reveals a new insight into the infiltration of circulating mutant cells into ischemic tissue and the para-vascular area, as well as their contribution to the development of atherosclerotic lesions. Büttner et al. [46] conducted a study sequencing 31 consecutive patients with PAD who underwent open surgical procedures, demonstrating that 45% of these patients had various CHIP gene mutations, including DNMT3A, TET2, ASXL1, and JAK2. Furthermore, almost 90% of the mutations detected in peripheral blood were also found in atherosclerotic lesions, perivascular fat, or subcutaneous tissues [46]. This data confirms that the circulating mutant clones infiltration into the ischemia tissues and also the involvement of in the development of atherosclerosis in PAD patients. The contribution of circulating CHIP-mutated clone to atherosclerosis lesion was also shown in sample from carotid endarterectomy patients. The fraction of CHIP clones in circulating cells in peripheral blood correlated with the plaque fractions of the corresponding clones. In several cases, CHIP clones entering from the circulation contributed to more than 25% of the cell population in individual plaque segments [47]. In conclusion, somatic mutations play a significant role in the development of atherosclerosis in PAD patients. Accurate evaluation of the size of circulating cell somatic clones is necessary to predict atherosclerotic lesion progression. Future studies should focus on developing prognostic tools based on circulating cell somatic mutations to assess PAD development and management.
5. Diabetes mellitus and atherosclerosis as a one of the main drivers of somatic mutation accumulation in patients PAD
An earlier investigation revealed a connection between type 2 diabetic mellitus (T2DM) and blood CHIP incidents. [48]. There was a significant association between T2DM and CHIP incidents, especially in non-obese individuals who had T2DM. The association suggests that aging, long-term T2DM history, atherosclerosis, vessel formation, and revascularization may all play a role in mechanisms that lead to persistent glucotoxicity, oxidative stress, and inflammatory damage, which can then reduce the capacity of vascular cell lineages to regenerate. [49,50]. The self-renewal of hematopoietic stem cells (HSCs) within the endosteal niche can also be disrupted by chronic inflammation and increased reactive oxygen species (ROS), which results in ongoing premature mobilization of HSCs into the peripheral vessels and exhaustion of the reservoir of early myeloid progenitor cells with proangiogenic secretory function [51,52]. These observations can be attributed to the genetic background or CH events in T2DM patients, which impair vascular regeneration and promote inflammation through the above-mentioned mechanisms. Patients with T2DM who have clonal mosaic events (71.4%) have a higher prevalence of vascular problems including microvascular and macrovascular lesions than patients with T2DM who do not have clonal events. [48]. In a study by Heyde et al. [53], the investigation of hematopoiesis in atherosclerotic animals and humans demonstrated that the complex trait of atherosclerosis enhances HSC proliferation. Additionally, they determine whether mild hypercholesterolemia, in the absence of atherosclerosis, is sufficient to increase HSC proliferation and promote the expansion of mutant cells or not. The results indicate that mild hypercholesterolemia, without atherosclerosis, does not trigger increased HSC proliferation, nor lead to an altered expansion rate of Tet2−/− myeloid cells. Taking together, multiple risk factors such as T2DM-related impairments in stem/progenitor cell quality, atherosclerosis, metabolites and hypercholesterinemia in the presence of atherosclerosis significantly contribute to the accumulation of somatic mutations (Table 1).
6. Somatic Mutation Search in PAD-related Thrombosis
Recent meta-analysis has revealed that JAK2V617F and CALR mutations are associated with alterations of blood counts and thrombosis. Even at very low VAF, JAK2V617F poses a thrombosis risk factor in the general population and is overrepresented in populations with thrombosis. In each thrombosis group, the prevalence of JAK2V617F in thrombosis patients was higher compared to the control population, suggesting the JAK2V617F mutation may contribute to increased risk of both arterial and venous thrombosis, particularly within the context of the population with clonal hematopoiesis. The subgroup analysis revealed that patients with splanchnic vein thrombosis had the highest prevalence of the JAK2V617F mutation (18.7%), followed by patients with ischemic stroke (8.5%), cerebral vein thrombosis (6.0%), deep vein thrombosis/pulmonary embolism, and PAD was 1.6% and 3.1%, respectively [67]. Muendlein et al. reported that the prevalence of JAK2V617F mutation was higher in a cohort of 287 patients with sonographically confirmed PAD compared to a group of 997 healthy subjects [68]. The acquired JAK2V617F mutation frequency in patients with PAD was 5-fold higher than that in healthy individuals (P < 0.001). Interestingly, the frequency of mutations in patients with PAD markedly decreased in patients who received aspirin (P < 0.003) [68]. Moreover, JAK2 mutations are reported to play an essential role in systemic inflammation, coagulation, decreased proliferation and angiogenesis, and thrombosis through the activation of the downstream STAT1,6, MAPK, and PI3K/AKT signaling pathways [69]. Mechanistically, patients with JAK2V617F mutation exhibit decreased blood endothelial cell outgrowth and enhanced expression of interferon-related genes, including serine protease inhibitor B2, early growth response protein 1, and chemokine ligand 2 [70]. Platelets from JAK2V617F-positive patients demonstrated an enhanced activation status and procoagulant potential. In addition, the fraction of immature platelets, which can be more active than mature platelets, was higher in carriers of the JAK2V617F mutation versus non-carriers [71]. Somatic mutation-related articles comprehensively summarized elsewhere [72,73].
7. Molecular Aspects of Somatic Mutation and Inflammation
While atherosclerosis is recognized as a major contributor to PAD pathogenesis, emerging evidence suggests that somatic mutations and associated inflammatory processes play a significant role in disease progression [12]. The inflammatory effects caused by specific mutations associated with CHIP have not been fully characterized; however, a common feature appears to be the induction of a pro-inflammatory state. Individuals with evidence of CHIP exhibit elevated levels of pro-inflammatory markers such as IL-6, TNF-α, and MCP-1 compared to those without CHIP [41,74]. It is believed that CHIP can trigger inflammation in immune cells that have genetic mutations via the rise in circulating inflammatory markers [74], as well as the presence of specific subsets of inflammatory cells [75]. Experiments conducted on bone marrow cells with mutations in TET2 or DNMT3A have further confirmed the heightened inflammatory capacity of CHIP [76]. Recent advancements in single-cell RNA sequencing of human peripheral blood cells have also verified that the presence of CHIP mutations promotes an inflammatory characteristic in both monocytes and T cells [77,78]. Analysis focusing on specific driver genes in a large cohort of CHIP individuals revealed that TET2 mutations were associated with increased IL-1β, while JAK2 and SF3B1 mutations were linked to higher circulating levels of IL-18 [41]. As mentioned earlier, Among the mutations associated with CHIP, TET2, DNMT3A, JAK2V617F, and ASXL1 are the most frequently observed. The following is an overview of the underlying cascade resulting from CHIP with these mutations.
TET2: TET2 catalyzes the first reaction in cytosine demethylation, which is critical for maintaining the normal development of HSCs [79]. TET2 plays a crucial role in regulating the immune system, and evidence suggests that CH driven by TET2 mutations contributes to the pathophysiology and progression of vascular diseases by promoting a pro-inflammatory state. TET2 controls the release of pro-inflammatory cytokines by modulating histone acetylation [80,81]. Studies on TET2-deficient mice have shown that stimulation of macrophages with lipopolysaccharide (LPS) and IFN-γ leads to excessive activation of pro-inflammatory cytokines and chemokines like IL-1β and IL-6 [80]. Moreover, mice with TET2 deficiency in myeloid-derived cells exhibit increased expression of IL-1β through induction of the NLRP3 inflammasome [26], IL-6 [81], and IL-8 [82]. In an unselected cohort of patients without vascular disease, the presence of TET2 mutations was associated with more than a two-fold increase in circulating levels of IL-8 compared to those without these mutations [82].
DNMT3A: DNMT3A is a gene that regulates gene transcription by catalyzing DNA methylation and is the most commonly mutated gene in individuals with CHIP. DNMT3A plays multiple roles in inflammation regulation. Specifically, it controls the expression of cytokines through the regulation of IQ motif-containing GTPase Activating Protein 2 (IQGAP2), a scaffold protein found in mast cells [83]. In patients with osteoarthritis, the activity of the IL-6 gene is associated with DNMT3A expression, and those with increased DNMT3A expression exhibit significantly lower levels of IL-6 secretion [84]. Additionally, in patients with severe aortic stenosis, the presence of DNMT3A mutations is linked to a significantly higher ratio of T helper 17 cells (TH17) to regulatory T cells (Tregs), indicating a pro-inflammatory polarization of T cells [75].
JAK2V617F: Among the genetic abnormalities associated with CHIP, the JAK2V617F mutation is notably linked to inflammatory processes. In humans, this mutation acts as a downstream signal transmitter for major cytokine receptors, leading to the activation of granulocytes, T cells, increased inflammation in macrophages, and the formation of neutrophil extracellular traps [85]. JAK2V617F mutations are commonly found in myeloproliferative neoplasms like essential thrombocythemia and polycythemia vera [86]. These conditions are associated with a higher risk of stroke, myocardial infarction, and deep vein thrombosis primarily due to elevated blood viscosity and a pro-coagulant state. However, JAK2V617F mutations are increasingly identified in individuals with normal peripheral blood counts and remain associated with an elevated risk of vascular mortality [87,88].
ASXL1: The impact of ASXL1 mutations in CHIP on inflammation is not fully understood. However, studies have suggested that ASXL1 mutations may play a role in promoting an inflammatory state. Mutation of ASXL1 is common in patients with atherosclerosis and chronic ischemic heart failure but the mechanisms contributing to this increased CV risk are not defined [82,89]. ASXL1 is involved in regulating gene expression and chromatin structure, and its mutations have been associated with dysregulation of inflammatory pathways. In particular, ASXL1 mutations have been linked to increased levels of pro-inflammatory cytokines, such as IL-6, in certain contexts [90]. Further research is needed to elucidate the specific mechanisms by which ASXL1 mutations contribute to inflammation in CHIP.
Understanding the molecular mechanisms linking somatic mutations and inflammation in PAD is of paramount importance. Recent studies have identified specific somatic mutations in PAD patients that directly contribute to the activation of inflammatory pathways. For instance, mutations in genes encoding toll-like receptors (TLRs), which play a crucial role in recognizing and responding to pathogen-associated molecular patterns, have been observed in PAD patients [91,92]. These mutations can result in enhanced TLR signaling, leading to increased production of pro-inflammatory cytokines and chemokines. Moreover, somatic mutations in genes associated with nuclear factor-kappa B (NF-κB) signaling have been implicated in PAD progression [93]. NF-κB is a key transcription factor that regulates the expression of numerous inflammatory mediators. Somatic mutations in genes encoding components of the NF-κB pathway can dysregulate its activity, resulting in sustained inflammation and vascular dysfunction. Within this framework, the discovery that a genetic variation, specifically the EE genotype polymorphism of intercellular adhesion molecule-1, significantly amplifies the susceptibility to PAD, underscores the notion that inflammation indeed contributes significantly to the development of PAD [94]. Additionally, Flex and colleagues [95] conducted a study involving 157 individuals with PAD and 206 control participants. They discovered that genetic variations in various molecules, including intercellular adhesion molecule-1, IL-6, E-selectin, monocyte chemoattractant protein-1 (MCP-1), as well as matrix metalloproteinases 1 and 3 (MMP1 & 3), were individually linked to PAD [95]. Of note, these mutations can disrupt the delicate balance between pro-inflammatory and anti-inflammatory signals, leading to chronic inflammation and the promotion of atherosclerotic processes. One notable consequence of somatic mutations in PAD is the activation of inflammatory transcription factors such as NF-κB and activator protein-1 (AP-1) [89]. Mutated genes can aberrantly activate these transcription factors, leading to the upregulation of inflammatory cytokines, adhesion molecules, and matrix metalloproteinases [96]. This dysregulated inflammatory response promotes leukocyte recruitment, vascular smooth muscle cell proliferation, and extracellular matrix remodeling, all of which contribute to the development and progression of atherosclerotic lesions in PAD [97]. Furthermore, somatic mutations can also affect other inflammatory signaling pathways, including the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. Mutations in JAK or STAT genes can result in enhanced signaling and dysregulated cytokine responses, further perpetuating the inflammatory milieu in PAD [98].
Given the intricate interplay between CHIP and inflammation in PAD, targeting inflammation has emerged as a promising therapeutic strategy. By specifically addressing the molecular aspects associated with somatic mutations and inflammation, novel therapeutic interventions can be developed to attenuate disease progression. One potential approach is to target the dysregulated inflammatory signaling pathways directly linked to somatic mutations. These targeted therapies aim to restore the balance between pro-inflammatory and anti-inflammatory signals, mitigating the detrimental effects of somatic mutations on inflammation and disease progression. Another therapeutic avenue lies in the development of immunomodulatory agents that can regulate the overall inflammatory response in PAD. By targeting common downstream mediators of inflammation, such as cytokines or chemokines, these agents could alleviate inflammation irrespective
8. Somatic Mutation and Regeneration, Unraveling the Connection
Precision medicine approaches have revolutionized the field of healthcare by tailoring treatment strategies based on individual genetic profiles [99]. In the context of PAD, targeting somatic mutations has emerged as a promising avenue for precision medicine interventions [100]. Somatic mutations can have a profound impact on tissue regeneration and angiogenesis in PAD. By identifying specific somatic mutations present in individual patients, clinicians can develop personalized treatment plans that directly target the underlying genetic abnormalities contributing to disease progression. Studies examining specific genes as potential candidates have employed case-control methodology to investigate allele frequency differences between individuals with and without PAD. These studies have identified several biological pathways implicated in atherosclerosis and PAD, such as leukocyte adhesion, coagulation, and inflammation [101,102,103]. A small study focused on symptomatic PAD patients revealed the involvement of the Notch signaling pathway, which plays a crucial role in regulating vascular smooth muscle cells and macrophages. Activation of this pathway, specifically through the Delta-like 4 ligands, was associated with gene expression related to "inflamed plaque," contributing to the unfavorable progression of PAD [104]. The largest candidate gene study on PAD involved the Candidate Gene Association Resource consortium, which examined approximately 50,000 gene variants in over 29,000 individuals [105]. In European Americans, two variants, rs2171209 in SYTL3 and rs290481 in TCF7L2, showed significant associations with ABI (ankle-brachial index). However, these associations were not observed in African Americans. The significance of these two single-nucleotide polymorphisms (SNPs) for ABI in European Americans could not be replicated in an additional study involving 13,524 Europeans and Americans of European ancestry. Overall, candidate gene studies have faced challenges, including the arbitrary selection of candidate genes and frequent failure to replicate results in other cohorts [2]. For instance, certain NURR1 variants (rs13428968 and rs12803) were associated with restenosis/re-occlusion rates following femoropopliteal percutaneous angioplasty in one population [106], but this association was not observed in another population, and there was no association with either PAD or adverse cardiovascular events [107]. Understanding the mutation profiles of individual patients can guide the selection of targeted therapeutic strategies in PAD. Advances in genomic sequencing technologies have facilitated the identification of specific mutations associated with impaired tissue healing and angiogenesis. Based on the identified mutation profiles, therapeutic interventions can be tailored to address the specific genetic aberrations. For instance, small molecule inhibitors or gene therapies targeting genes with somatic mutations can be employed to modulate the downstream signaling pathways involved in tissue regeneration and angiogenesis. These targeted therapies hold the potential to restore normal cellular functions, promote tissue repair, and improve outcomes in PAD patients with specific mutation profiles.
Genetic alterations occurring in key genes involved in angiogenic and regenerative processes can disrupt the normal cellular mechanisms required for efficient tissue repair. Somatic mutations affecting genes encoding growth factors, receptors, or signaling molecules involved in angiogenesis can impair the formation of new blood vessels, leading to inadequate tissue perfusion and compromised healing in PAD [108]. For instance, it has been reported that mutations in the JAK2 gene play a crucial role in various physiological processes such as systemic inflammation, coagulation, reduced cell proliferation, impaired angiogenesis, and thrombosis, which are mediated through the activation of downstream signaling pathways including STAT1,6, MAPK, and PI3K/AKT [109]. Mechanistically, individuals with the JAK2V617F mutation exhibit decreased outgrowth of blood ECs and increased expression of interferon-related genes, such as SERPIN-B2, EGR1, and chemokine ligand 2 [110], suggesting that individuals with the JAK2V617F mutation experience persistent inflammation and increased permeability, limited cell growth (or angiogenesis), and accelerated senescence of EPCs compared to healthy individuals [110]. Investigating the influence of CHIP on tissue regeneration processes in PAD is a crucial area of research. By studying the functional consequences of specific mutations, researchers can uncover the molecular mechanisms by which genetic alterations impact tissue regeneration. Experimental models and in vitro studies can elucidate the effects of somatic mutations on cellular processes such as angiogenesis, cell proliferation, migration, and extracellular matrix remodeling. These investigations can provide valuable insights into the specific pathways and molecular targets affected by somatic mutations, aiding in the development of novel therapeutic strategies aimed at enhancing tissue regeneration in PAD patients.
9. Conclusion
Somatic mutation of multiple genes affecting hematopoiesis and mutant clone size expand in circulating blood, the latter infiltrates into the ischemic tissue/atherosclerosis plaque and produce variety of inflammatory cytokines which contribute risk factor to development of cardiovascular diseases, including PAD. The second, there are solid experimental evidences that hypercholesterinemia enhances HSC proliferation and leads to chronic HSC proliferation process which also significantly increases inflammatory-thrombosis risk in patients with PAD. The patients with blood count abnormalities or elevated pro-inflammatory cytokines or interleukins, and elderly age should be screened and father exposure with various risk factors which promoting CHIP mutation such as hypercholesterinemia, smoking, atherosclerosis, diabetes, and hypertension are needed adequate care. The third, not all the mutated CHIP genes promote pro-inflammatory cytokines production such as IL-1β and IL-6. For instance, TP53 and PPM1D genes mutation do not produce pro-inflammatory cytokine [40,41] while in patients with epigenetic genes (DNMT3A, TET2, and ASXL1) CHIP mutation circulating levels of pro-inflammatory cytokines are significantly increased [82,111,112]. These mechanistic differences between TP53/PPM1D- CHIP and DNMT3A/TET2/ASXL1/JAK2-CHIP require consideration when designing preventive care strategies targeting the effects of CHIP on atherosclerosis. Thus, while the pathogenic effects of TET2- CHIP may be prevented by targeting IL-1b-driven inflammation as shown in CANTOS interim data analysis [113].
The large VAF somatic mutated TP53 genes in PAD development and prognosis was recently experimentally demonstrated in large more than 50000 patient’s exome sequence data [40]. Further future studies require to establish PAD patients’ disease prognostic calculator based on somatic mutated genes type, variant, and VAF grade, etc., to boost personalized therapy.
Author Contributions
A.A.S., and M.H., collected data, designed figures, drafted manuscript, and finally approved for publication.
Funding
This research was funded by the Science Committee of the Ministry of Science and Higher Education of the Republic of Kazakhstan grant # AP14872543 to AAS.
Acknowledgments
We gratefully acknowledge the Science Committee of the Ministry of Science and Higher Education of the Republic of Kazakhstan for providing grant (# AP14872543 to AAS) and unwavering support provided by Qazaq Institute of Innovative Medicine in the completion of this review article. The contributions and guidance from our colleagues and mentors at Qazaq Institute of Innovative Medicine have been invaluable in shaping the content and direction of this work. We would like to express our sincere appreciation for their expertise, feedback, and assistance throughout the research process.
References
- Hinchliffe, R.J.; Forsythe, R.O.; Apelqvist, J.; Boyko, E.J.; Fitridge, R.; Hong, J.P.; Katsanos, K.; Mills, J.L.; Nikol, S.; Reekers, J.; et al. Guidelines on diagnosis, prognosis, and management of peripheral artery disease in patients with foot ulcers and diabetes (IWGDF 2019 update). Diabetes/Metabolism Res. Rev. 2020, 36, e3276. [Google Scholar] [CrossRef] [PubMed]
- Klarin, D.; Tsao, P.S.; Damrauer, S.M. Genetic Determinants of Peripheral Artery Disease. Circ. Res. 2021, 128, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Amancherla, K.W.J.; Bick, A.G. Clonal hematopoiesis and vascular disease. Seminars in immunopathology 2022, 44. [Google Scholar] [CrossRef] [PubMed]
- Bonafiglia, Q.A.; Bendeck, M.; Gotlieb, A.I. Vascular Pathobiology: Atherosclerosis and Large Vessel Disease. Cardiovasc. Pathol. 2022, 265–306. [Google Scholar] [CrossRef]
- Salybekov, A.A.; Wolfien, M.; Kobayashi, S.; Steinhoff, G.; Asahara, T. Personalized Cell Therapy for Patients with Peripheral Arterial Diseases in the Context of Genetic Alterations: Artificial Intelligence-Based Responder and Non-Responder Prediction. Cells 2021, 10, 3266. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Qian, M.; Kyler, K.; Xu, J. Endothelial–Vascular Smooth Muscle Cells Interactions in Atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Du, W.; Ren, L.; Hamblin, M.H.; Becker, R.C.; Chen, Y.E.; Fan, Y. Vascular Smooth Muscle Cells in Aortic Aneurysm: From Genetics to Mechanisms. J. Am. Hear. Assoc. 2021, 10, e023601. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, L.B.; et al. Somatic mutations reveal clonal cell populations in atherosclerotic plaques. medRxiv 2022. [Google Scholar] [CrossRef]
- Bejar, R. CHIP, ICUS, CCUS and other four-letter words. Leukemia 2017, 31, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.d.M.; et al. Clonal hematopoiesis of indeterminate potential, DNA methylation, and risk for coronary artery disease. Nature Communications 2022, 13, 5350. [Google Scholar] [CrossRef]
- DeZern, A.E.; Malcovati, L.; Ebert, B.L. CHIP, CCUS, and Other Acronyms: Definition, Implications, and Impact on Practice. American Society of Clinical Oncology Educational Book, 2019(39): p. 400-410.
- Marnell, C.S.; Bick, A.; Natarajan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cell. Cardiol. 2021, 161, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Klarin, D.; Program, V.M.V.; Lynch, J.; Aragam, K.; Chaffin, M.; Assimes, T.L.; Huang, J.; Lee, K.M.; Shao, Q.; Huffman, J.E.; et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat. Med. 2019, 25, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Heimlich, J.B.; Bick, A.G. Somatic Mutations in Cardiovascular Disease. Circ. Res. 2022, 130, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Visconte, V.; Nakashima, M.O.; Rogers, H.J. Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features. Cancers 2019, 11, 1844. [Google Scholar] [CrossRef] [PubMed]
- Haring, B.; Wissel, S.; Manson, J.E. Somatic Mutations and Clonal Hematopoiesis as Drivers of Age-Related Cardiovascular Risk. Curr. Cardiol. Rep. 2022, 24, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Feldkamp, J.D.; Vetter, V.M.; Arends, C.M.; Lang, T.J.L.; Bullinger, L.; Damm, F.; Demuth, I.; Frick, M. Clonal hematopoiesis of indeterminate potential-related epigenetic age acceleration correlates with clonal hematopoiesis of indeterminate potential clone size in patients with high morbidity. Haematologica 2022, 107, 1703–1708. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef]
- Newman, J.D.; Cornwell, M.G.; Zhou, H.; Rockman, C.; Heguy, A.; Suarez, Y.; Cheng, H.S.; Feinberg, M.W.; Hochman, J.S.; Ruggles, K.V.; et al. Gene Expression Signature in Patients With Symptomatic Peripheral Artery Disease. Arter. Thromb. Vasc. Biol. 2021, 41, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Koller, A.; Fazzini, F.; Lamina, C.; Rantner, B.; Kollerits, B.; Stadler, M.; Klein-Weigel, P.; Fraedrich, G.; Kronenberg, F. Mitochondrial DNA copy number is associated with all-cause mortality and cardiovascular events in patients with peripheral arterial disease. J. Intern. Med. 2020, 287, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Denny, J.C.; Bastarache, L.; Ritchie, M.D.; Carroll, R.J.; Zink, R.; Mosley, J.D.; Field, J.R.; Pulley, J.M.; Ramirez, A.H.; Bowton, E.; et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 2013, 31, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Zekavat, S.M.; et al. TP53-mediated clonal hematopoiesis confers increased risk for incident peripheral artery disease. medRxiv 2021. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jin, Y.; Tang, W.H.; Qin, L.; Zhang, X.; Tellides, G.; Hwa, J.; Yu, J.; Martin, K.A.; Y, X.; et al. Ten-Eleven Translocation-2 (TET2) Is a Master Regulator of Smooth Muscle Cell Plasticity. Circulation 2013, 128, 2047–2057. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Yang, Q.; Li, A.-F.; Li, R.-Q.; Wang, Z.; Liu, L.-S.; Ren, Z.; Zheng, X.-L.; Tang, X.-Q.; Li, G.-H.; et al. Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE−/− mice. Oncotarget 2016, 7, 76423–76436. [Google Scholar] [CrossRef]
- Saini, S.K.; McDermott, M.M.; Picca, A.; Li, L.; Wohlgemuth, S.E.; Kosmac, K.; Peterson, C.A.; Tian, L.; Ferrucci, L.; Guralnik, J.M.; et al. Mitochondrial DNA damage in calf skeletal muscle and walking performance in people with peripheral artery disease. Free. Radic. Biol. Med. 2020, 160, 680–689. [Google Scholar] [CrossRef]
- Sirbu, B.M.; Cortez, D. DNA Damage Response: Three Levels of DNA Repair Regulation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012724. [Google Scholar] [CrossRef]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kröller-Schön, S.; Steven, S.; Schulz, E.; Münzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2016, 174, 1670–1689. [Google Scholar] [CrossRef]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef] [PubMed]
- Bick, A.G.; Pirruccello, J.P.; Griffin, G.K.; Gupta, N.; Gabriel, S.; Saleheen, D.; Libby, P.; Kathiresan, S.; Natarajan, P. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 2020, 141, 124–131. [Google Scholar] [CrossRef] [PubMed]
- A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nature genetics 2015, 47, 1121–1130. [CrossRef] [PubMed]
- Sibon, I.; Coupry, I.; Menegon, P.; Bouchet, J.-P.; Gorry, P.; Burgelin, I.; Calvas, P.; Orignac, I.; Dousset, V.; Lacombe, D.; et al. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann. Neurol. 2007, 62, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; An, H.; Lim, J.; Park, I.S.; Kim, M.H.; Kim, J.H.; Kim, S.W.; Koh, Y.I.; Lee, E.Y.; Cheon, J.H. Interplay between chronic inflammation and clonal haematopoiesis of indeterminate potential in Behçet’s disease. Thromb. Haemost. 2023, 25, 33. [Google Scholar] [CrossRef]
- Li, J.; Wang, C.; Liu, J.; Yu, Y.; Liu, Y.; Peng, Q.; Liu, H.; Guan, X. A feedback loop: Interactions between Inflammatory Signals and Clonal Hematopoiesis in Cardiovascular Disease. Mol. Biol. Rep. 2021, 48, 3785–3798. [Google Scholar] [CrossRef]
- Golledge, J.; Biros, E.; Bingley, J.; Iyer, V.; Krishna, S.M. Epigenetics and Peripheral Artery Disease. Curr. Atheroscler. Rep. 2016, 18, 15. [Google Scholar] [CrossRef]
- Krishna, S.M.; Trollope, A.F.; Golledge, J. The relevance of epigenetics to occlusive cerebral and peripheral arterial disease. Clin. Sci. 2015, 128, 537–558. [Google Scholar] [CrossRef]
- Zekavat, S.M.; Viana-Huete, V.; Matesanz, N.; Jorshery, S.D.; Zuriaga, M.A.; Uddin, M.; Trinder, M.; Paruchuri, K.; Zorita, V.; Ferrer-Pérez, A.; et al. TP53-mediated clonal hematopoiesis confers increased risk for incident atherosclerotic disease. Nat. Cardiovasc. Res. 2023, 2, 144–158. [Google Scholar] [CrossRef]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- AH, C.; et al. Tet2 Restrains Inflammatory Gene Expression in Macrophages. Experimental hematology 2017, 55. [Google Scholar]
- SO, A.; et al. An Inflammatory Environment Containing TNFα Favors Tet2-mutant Clonal Hematopoiesis. Experimental hematology 2018, 59. [Google Scholar]
- Rivero, G.A.; Perli, E.; Moreno, S.; Salemi, J.L. Excess in Atherosclerotic and Inflammametabolic Diseases Are Differentially Expressed in Myelodysplasia and Are Highly Dependent on Age, R-IPSS and Ethnicity. Blood 2018, 132, 4855–4855. [Google Scholar] [CrossRef]
- Büttner, P.; Böttner, J.; Krohn, K.; Baber, R.; Platzbecker, U.; Cross, M.; Desch, S.; Thiele, H.; Steiner, S.; Scheinert, D.; et al. Clonal Hematopoiesis Mutations Are Present in Atherosclerotic Lesions in Peripheral Artery Disease. Int. J. Mol. Sci. 2023, 24, 3962. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, L.B.; et al. Somatic mutations reveal clonal cell populations in atherosclerotic plaques. medRxiv 2022. [Google Scholar] [CrossRef]
- A, B.; et al. Association Between Large Detectable Clonal Mosaicism and Type 2 Diabetes With Vascular Complications. Nature genetics 2013, 45. [Google Scholar]
- Albiero, M.; Poncina, N.; Tjwa, M.; Ciciliot, S.; Menegazzo, L.; Ceolotto, G.; de Kreutzenberg, S.V.; Moura, R.; Giorgio, M.; Pelicci, P.; et al. Diabetes Causes Bone Marrow Autonomic Neuropathy and Impairs Stem Cell Mobilization via Dysregulated p66Shc and Sirt1. Diabetes 2014, 63, 1353–1365. [Google Scholar] [CrossRef]
- Fadini, G.P.; Ciciliot, S.; Albiero, M. Concise Review: Perspectives and Clinical Implications of Bone Marrow and Circulating Stem Cell Defects in Diabetes. STEM CELLS 2016, 35, 106–116. [Google Scholar] [CrossRef]
- Kojima, H.; Kim, J.; Chan, L. Emerging roles of hematopoietic cells in the pathobiology of diabetic complications. Trends Endocrinol. Metab. 2014, 25, 178–187. [Google Scholar] [CrossRef]
- Orlandi, A.; Chavakis, E.; Seeger, F.; Tjwa, M.; Zeiher, A.M.; Dimmeler, S. Long-term diabetes impairs repopulation of hematopoietic progenitor cells and dysregulates the cytokine expression in the bone marrow microenvironment in mice. Basic Res. Cardiol. 2010, 105, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P.; A, H.; D, R.; CS, M.; S, Z.; Ff, H.; Jm, G.; D, C.; Y, I.; Mj, S.; et al. Faculty Opinions recommendation of Increased stem cell proliferation in atherosclerosis accelerates clonal hematopoiesis. Cell 2021, 184, 1348–1361. [Google Scholar] [CrossRef]
- Dezfulian, C.; N, S.; Cb, R.; Ah, S.; Y, G.; Ma, A.; T, R.; Js, B. Faculty Opinions recommendation of Association between advanced age and vascular disease in different arterial territories: a population database of over 3. 6 million subjects. Journal of the American College of Cardiology 2017, 61. [Google Scholar] [CrossRef]
- Fuster, J.J.; Walsh, K. Somatic mutations and clonal hematopoiesis: unexpected potential new drivers of age-related cardiovascular disease. Circ Res. 2018, 122, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Santulli, G. Update on peripheral artery disease: Epidemiology and evidence-based facts. Atherosclerosis 2018, 275, 379–381. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Shah, A.D.; Langenberg, C.; Rapsomaniki, E.; Denaxas, S.; Pujades-Rodriguez, M.; Gale, C.P.; Deanfield, J.; Smeeth, L.; Timmis, A.; Hemingway, H. Type 2 diabetes and incidence of cardiovascular diseases: a cohort study in 1·9 million people. Lancet Diabetes Endocrinol. 2015, 3, 105–113. [Google Scholar] [CrossRef]
- Brevetti, G.; Giugliano, G.; Brevetti, L.; Hiatt, W.R.; W, H.; E, A.; C, L.; E, B.; R, R.; F, F.; et al. Inflammation in Peripheral Artery Disease. Circulation 2010, 122, 1862–1875. [Google Scholar] [CrossRef]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef] [PubMed]
- Desai, U.; Kharat, A.; Hess, C.N.; Milentijevic, D.; Laliberté, F.; Zuckerman, P.; Benson, J.; Lefebvre, P.; Hiatt, W.R.; Bonaca, M.P. Incidence of Major Atherothrombotic Vascular Events among Patients with Peripheral Artery Disease after Revascularization. Ann. Vasc. Surg. 2021, 75, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Kaizer, H.; Naik, R.P.; Lobner, K.; Moliterno, A.R. JAK2 V617F Prevalence Study: Associations in the General Population and Vascular Disease Populations. Blood 2020, 136, 36–36. [Google Scholar] [CrossRef]
- Michael, H.; Criqui, V.A. Epidemiology of Peripheral Artery Disease | Circulation Research. Circulation Research 2015, 116. [Google Scholar]
- Yoshida, K.; Gowers, K.H.C.; Lee-Six, H.; Chandrasekharan, D.P.; Coorens, T.; Maughan, E.F.; Beal, K.; Menzies, A.; Millar, F.R.; Anderson, E.; et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 2020, 578, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Kourie, H.R.; Ameye, L.; Paesmans, M.; Bron, D. Re: Prognostic value of CALR vs. JAK2V617F mutations on splenomegaly, leukemic transformation, thrombosis, and overall survival in patients with primary fibrosis: a meta-analysis. Ann. Hematol. 2016, 95, 2105–2106. [Google Scholar] [CrossRef] [PubMed]
- A, M.; et al. Occurrence of the JAK2 V617F mutation in patients with peripheral arterial disease. American journal of hematology 2015, 90. [Google Scholar]
- F, P.; et al. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8. [Google Scholar]
- Megan E Cosgrove, R.S.; Harrison, H.J.; Jackson, G.E.; Howard, M.R.; Hitchcock, I.S. Endothelial JAK2V617F Expression Drives Inflammation and Cellular Senescence; New Evidence for the Roles of Endothelial Cells in MPN-Related Clotting Abnormalities? Blood 2016, 128. [Google Scholar]
- Barbui, T.; Finazzi, G.; Falanga, A. Myeloproliferative neoplasms and thrombosis. Blood 2013, 122, 2176–2184. [Google Scholar] [CrossRef]
- Veninga, A.; De Simone, I.; Heemskerk, J.W.; Cate, H.T.; van der Meijden, P.E. Clonal hematopoietic mutations linked to platelet traits and the risk of thrombosis or bleeding. Haematologica 2020, 105, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Misaka, T.; Kimishima, Y.; Yokokawa, T.; Ikeda, K.; Takeishi, Y. Clonal hematopoiesis and cardiovascular diseases: role of JAK2V617F. J. Cardiol. 2022, 81, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Cook, E.K.; Izukawa, T.; Young, S.; Rosen, G.; Jamali, M.; Zhang, L.; Johnson, D.; Bain, E.; Hilland, J.; Ferrone, C.K.; et al. Comorbid and inflammatory characteristics of genetic subtypes of clonal hematopoiesis. Blood Adv. 2019, 3, 2482–2486. [Google Scholar] [CrossRef] [PubMed]
- Mas-Peiro, S.; Hoffmann, J.; Fichtlscherer, S.; Dorsheimer, L.; A Rieger, M.; Dimmeler, S.; Vasa-Nicotera, M.; Zeiher, A.M. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Heart J. 2020, 41, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef]
- Abplanalp, W.T.; Mas-Peiro, S.; Cremer, S.; John, D.; Dimmeler, S.; Zeiher, A.M. Association of Clonal Hematopoiesis of Indeterminate Potential with Inflammatory Gene Expression in Patients with Severe Degenerative Aortic Valve Stenosis or Chronic Postischemic Heart Failure. JAMA Cardiol. 2020, 5, 1170–1175. [Google Scholar] [CrossRef]
- Abplanalp, W.T.; Cremer, S.; John, D.; Hoffmann, J.; Schuhmacher, B.; Merten, M.; Rieger, M.A.; Vasa-Nicotera, M.; Zeiher, A.M.; Dimmeler, S. Clonal Hematopoiesis–Driver DNMT3A Mutations Alter Immune Cells in Heart Failure. Circ. Res. 2021, 128, 216–228. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017, 55, 56–70. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef]
- Jaiswal, S.; et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. New England Journal of Medicine 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Leoni, C.; Montagner, S.; Rinaldi, A.; Bertoni, F.; Polletti, S.; Balestrieri, C.; Monticelli, S. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. 2017, 114, E1490–E1499. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhou, S.; Wang, C.; Huang, Y.; Li, H.; Wang, Y.; Zhu, Z.; Tang, J.; Yan, M. Epigenetic modifications of interleukin-6 in synovial fibroblasts from osteoarthritis patients. Sci. Rep. 2017, 7, 43592. [Google Scholar] [CrossRef]
- O'Shea, J.J.; Kontzias, A.; Yamaoka, K.; Tanaka, Y.; Laurence, A. Janus kinase inhibitors in autoimmune diseases. Rheumatol. 2013, 72, ii111–ii115. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Sidon, P.; El Housni, H.; Dessars, B.; Heimann, P. The JAK2V617F mutation is detectable at very low level in peripheral blood of healthy donors. Leukemia 2006, 20, 1622–1622. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.; Birgens, H.S.; Nordestgaard, B.G.; Kjaer, L.; Bojesen, S.E. The JAK2 V617F somatic mutation, mortality and cancer risk in the general population. Haematologica 2010, 96, 450–453. [Google Scholar] [CrossRef]
- Dorsheimer, L.; Assmus, B.; Rasper, T.; Ortmann, C.A.; Ecke, A.; Abou-El-Ardat, K.; Schmid, T.; Brüne, B.; Wagner, S.; Serve, H.; et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019, 4, 25–33. [Google Scholar] [CrossRef]
- Mooney, L.; Goodyear, C.S.; Chandra, T.; Kirschner, K.; Copland, M.; Petrie, M.C.; Lang, N.N. Clonal haematopoiesis of indeterminate potential: intersections between inflammation, vascular disease and heart failure. Clin. Sci. 2021, 135, 991–1007. [Google Scholar] [CrossRef]
- Varela, C.; De Haro, J.; Bleda, S.; Ferruelo, A.; Acin, F. Serum of peripheral arterial disease patients with poor flow-mediated-arterial-dilation values triggers a genomic over-expression of toll-like-receptor 4 by endothelial cells. Atherosclerosis 2014, 235, e42. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Chen, Y.; Wang, Y.; Zhang, S.; Liu, P.; Chen, Z.; Song, P.; Luo, L.; Luo, Y.; et al. Corilagin Ameliorates Atherosclerosis in Peripheral Artery Disease via the Toll-Like Receptor-4 Signaling Pathway in vitro and in vivo. Front. Immunol. 2020, 11, 1611. [Google Scholar] [CrossRef]
- Hu, Y.; et al. Correlation of mimecan with nuclear factor kappa B and P53 in peripheral arterial disease and peripheral arterial disease combined with type 2 diabetes in the elderly. Chinese Journal of Geriatrics 2014, 26–28. [Google Scholar]
- Gaetani, E.; Flex, A.; Pola, R.; Papaleo, P.; De Martini, D.; Pola, E.; Aloi, F.; Flore, R.; Serricchio, M.; Gasbarrini, A.; et al. The K469E polymorphism of the ICAM-1 gene is a risk factor for peripheral arterial occlusive disease. Blood Coagul. Fibrinolysis 2002, 13, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Flex, A.; Gaetani, E.; Angelini, F.; Sabusco, A.; Chillà, C.; Straface, G.; Biscetti, F.; Pola, P.; Castellot, J.J.; Pola, R. Pro-inflammatory genetic profiles in subjects with peripheral arterial occlusive disease and critical limb ischemia. J. Intern. Med. 2007, 262, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Alleboina, S.; Singh, M.V.; Wong, T.; Dokun, A. OR14-06 Inhibition of Protein Kinase C-beta2 Phosphorylation Restores Nuclear Factor-Kappa B Activation and Improves Peripheral Arterial Disease in Diabetes. J. Endocr. Soc. 2020, 4, OR14–06. [Google Scholar] [CrossRef]
- Golledge, J. Update on the pathophysiology and medical treatment of peripheral artery disease. Nat. Rev. Cardiol. 2022, 19, 456–474. [Google Scholar] [CrossRef]
- Ott, N.; Faletti, L.; Heeg, M.; Andreani, V.; Grimbacher, B. JAKs and STATs from a Clinical Perspective: Loss-of-Function Mutations, Gain-of-Function Mutations, and Their Multidimensional Consequences. J. Clin. Immunol. 2023, 43, 1326–1359. [Google Scholar] [CrossRef]
- Mathur, S.; Sutton, J. Personalized medicine could transform healthcare. Biomed. Rep. 2017, 7, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Poredoš, P.; Šabovič, M.; Mijovski, M.B.; Nikolajević, J.; Antignani, P.L.; Paraskevas, K.I.; Mikhailidis, D.P.; Blinc, A. Inflammatory and Prothrombotic Biomarkers, DNA Polymorphisms, MicroRNAs and Personalized Medicine for Patients with Peripheral Arterial Disease. Int. J. Mol. Sci. 2022, 23, 12054. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.B.; Connor, J.; Lee, A.J.; Cooke, A.; Lowe, G.D.; Rumley, A.; Fowkes, F. Relationship of the platelet glycoprotein PlA and fibrinogen T/G+1689 polymorphisms with peripheral arterial disease and ischaemic heart disease. Thromb. Res. 2003, 112, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Reny, J.L.; et al. The factor II G20210A gene polymorphism, but not factor V Arg506Gln, is associated with peripheral arterial disease: results of a case–control study. Journal of Thrombosis and Haemostasis 2004, 2, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Flex, A.; Gaetani, E.; Pola, R.; Santoliquido, A.; Aloi, F.; Papaleo, P.; Lago, A.D.; Pola, E.; Serricchio, M.; Tondi, P.; et al. The −174 G/C Polymorphism of the Interleukin-6 Gene Promoter is Associated with Peripheral Artery Occlusive Disease. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Aquila, G.; Fortini, C.; Pannuti, A.; Delbue, S.; Pannella, M.; Morelli, M.B.; Caliceti, C.; Castriota, F.; de Mattei, M.; Ongaro, A.; et al. Distinct gene expression profiles associated with Notch ligands Delta-like 4 and Jagged1 in plaque material from peripheral artery disease patients: a pilot study. J. Transl. Med. 2017, 15, 1–14. [Google Scholar] [CrossRef]
- Wassel, C.L.; Lamina, C.; Nambi, V.; Coassin, S.; Mukamal, K.J.; Ganesh, S.K.; Jacobs, D.R.; Franceschini, N.; Papanicolaou, G.J.; Gibson, Q.; et al. Genetic determinants of the ankle-brachial index: A meta-analysis of a cardiovascular candidate gene 50K SNP panel in the candidate gene association resource (CARe) consortium. Atherosclerosis 2012, 222, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Božič-Mijovski, M.; Bedenčič, M.; Stegnar, M.; Salapura, V.; Ježovnik, M.; Kozak, M.; Blinc, A. Nurr1 Haplotypes are Associated with Femoropopliteal Restenosis/Re-occlusion after Percutaneous Transluminal Angioplasty. Eur. J. Vasc. Endovasc. Surg. 2012, 43, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Boc, V.; Mijovski, M.B.; Perme, M.P.; Blinc, A. Diabetes and smoking are more important for prognosis of patients with peripheral arterial disease than some genetic polymorphisms. Vasa 2019, 48, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Perner, F.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef]
- Cosgrove, M.E.; et al. Endothelial JAK2V617F Expression Drives Inflammation and Cellular Senescence; New Evidence for the Roles of Endothelial Cells in MPN-Related Clotting Abnormalities? Blood 2016, 128, 3134. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef]
- Min, K.; Polizio, A.H.; Kour, A.; Thel, M.C.; Walsh, K. Experimental ASXL1 -Mediated Clonal Hematopoiesis Promotes Inflammation and Accelerates Heart Failure. J. Am. Hear. Assoc. 2022, 11, e026154. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.C.; et al. TET2-driven clonal hematopoiesis and response to canakinumab: an exploratory analysis of the CANTOS randomized clinical trial. JAMA cardiology 2022, 7, 521–528. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
CHIP mutation and its impact on the cardiovascular system. A) Clonal hematopoiesis (CH) occurs when somatic mutations in hematopoietic stem cells (HSCs) start producing the same clone of cells, leading to the expansion of mutant blood cells. B) It is well-documented that the somatic mutation with a variant allele frequency (VAF) less than 2% are known as age-related clonal hematopoiesis (ARCH). However, clonal hematopoiesis of indeterminate potential (CHIP) is defined as the presence of myeloid cancer-associated somatic mutations with a VAF of ≥2% in the hematopoietic cells of individuals without hematologic malignancy or it may transform into an oncogenic status, also known as clonal hematopoiesis with oncogenic potential (CHOP). C) CHIP mutated cells infiltrate into the atherosclerotic plaque and secrete enormous pro-inflammatory cytokines such as IL-1β, IL-6, etc., initiating immune-thrombosis and occlusion of the vessel.
Figure 1.
CHIP mutation and its impact on the cardiovascular system. A) Clonal hematopoiesis (CH) occurs when somatic mutations in hematopoietic stem cells (HSCs) start producing the same clone of cells, leading to the expansion of mutant blood cells. B) It is well-documented that the somatic mutation with a variant allele frequency (VAF) less than 2% are known as age-related clonal hematopoiesis (ARCH). However, clonal hematopoiesis of indeterminate potential (CHIP) is defined as the presence of myeloid cancer-associated somatic mutations with a VAF of ≥2% in the hematopoietic cells of individuals without hematologic malignancy or it may transform into an oncogenic status, also known as clonal hematopoiesis with oncogenic potential (CHOP). C) CHIP mutated cells infiltrate into the atherosclerotic plaque and secrete enormous pro-inflammatory cytokines such as IL-1β, IL-6, etc., initiating immune-thrombosis and occlusion of the vessel.
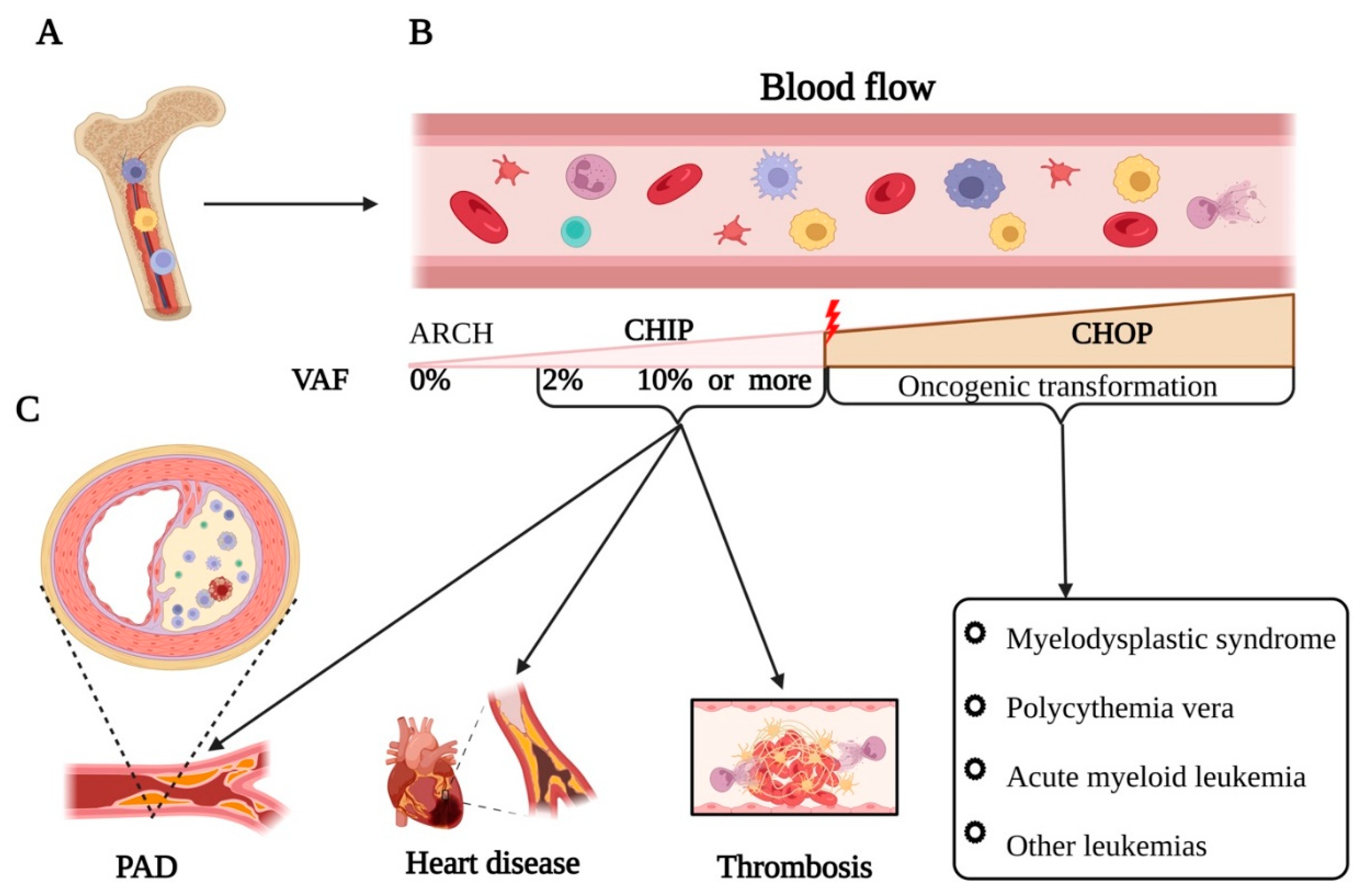

Table 1.
Peripheral artery disease and somatic mutations; Shared points and similarities.
| PAD | Shared points | Somatic mutation |
|---|---|---|
| The risk of PAD markedly increases with age. Prevalence of PAD among individuals aged 80–100 years is 22 to 33% [54] | Age | somatic mutations generally increase by around 2-6 mutations per cell division. Somatic mutation incidence increases in the ageing population by 10 to 20 % at the age of 70 years [55,56]. |
| Atherosclerosis incidence among PAD cases is more than 90% [57] | Atherosclerosis | Atherosclerosis patients is sufficient to produce a 3.5-fold increased risk of clonal hematopoiesis by age 70 [53]. Presence of somatic mutation in TET2 increases atherosclerotic lesions of vessels by 3- to 4-fold [58,59]. |
| Diabetic patients have 2- to 4-fold increased risk of developing PAD, CAD, and ischemic stroke [57] | Diabetes mellitus | Clonal mosaic event carriers with T2DM were associated with 2-fold increased prevalence of vascular complications [48,60]. |
| Approximately 70-80% of PAD patients exhibited elevated levels of inflammatory markers, suggesting a high incidence of inflammation in this population [61]. | Inflammation | Inflammation, by generating reactive oxygen and nitrogen species that damage DNA, contributes to mutagenesis and can result in a 2- to 4-fold higher accumulation of somatic mutations compared to non-inflammatory conditions [62]. |
| 15-20% of PAD patients experienced a thrombotic event over a five-year follow-up period [63]. | Thrombosis | The presence of JAK2 V617F mutation in PAD patients increased the risk of thrombosis by 3.1% [64]. |
| Smoking is the most common risk factor for PAD occurrence with a population attributable fraction of 44% [57,65]. | Tobacco smoking | Tobacco smoking dramatically increase the occurrence of somatic mutation from 1,000 to 10,000 mutations per cell [66]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



