
Submitted:
17 July 2023
Posted:
18 July 2023
You are already at the latest version
Abstract
Glaucoma, a group of diseases characterized by progressive retinal ganglion cell loss, cupping of the optic disc and by a typical pattern of visual field defects, is a leading cause of severe visual impairment and blindness worldwide. Elevated intraocular pressure (IOP) is the leading risk factor for glaucoma development. However, glaucoma can also develop at normal pressure levels. An increased susceptibility of retinal ganglion cells to IOP, systemic vascular dysregulation, endothelial dysfunction and autoimmune imbalances have been suggested to play a role in the pathophysiology of normal tension glaucoma. Since inflammation and oxidative stress play a role in all forms of glaucoma, the goal of this review article is to resent an overview on the inflammatory and prooxidant mechanisms in the pathophysiology of glaucoma and to discuss immunomodulatory and antioxidant treatment approaches.
Keywords:
autoimmune
; glaucoma
; retinal ganglion cell
; optic nerve
; inflammation
; ischemia
; oxidative stress
; vascular dysregulation
1. Introduction
The word glaucoma subsumes a spectrum of disorders which share a progressive optic nerve atrophy derived from a loss of retinal ganglion cells (RGCs), with concomitant optic disc cupping, retinal nerve fiber layer (RNFL) thinning and clinically detectable early visual field losses in form of arcuate defects that correspond to the fiber nerve bundle pattern [1,2,3,4]. Subsequently, in late disease stages, advanced optic nerve atrophy and perimetric defects can ultimately drive to blindness. Glaucoma is among the leading causes of irreversible visual loss worldwide [5,6,7,8]. Elevated intraocular pressure (IOP) is the major risk factor in this disorder [9,10]. Nonetheless, IOP alone appears not sufficient to properly account for all cases of glaucoma, since this disease can also occur without an elevation of IOP, such as in case of normal tension glaucoma (NTG) [11]. Relatively recent research shed light on the multifaceted pathophysiology of glaucoma, collecting evidence upon the involvement of vascular dysfunction, an altered redox status, neuroinflammation, and of autoimmunity as additional actors in the glaucomatous pathogenesis [11,12,13,14,15,16,17]. Considering the overall high prevalence and the severity of this disorder, various publications underlined the need for effective therapeutic strategies, exploring new pharmaceutical fields for glaucoma, with the purpose to prevent the severe visual impairment occurring in late stages [18,19,20].
A profound comprehension of the pathophysiological events in glaucoma is propaedeutic to eventually consider new alternative targets, which may finally hold additional benefits for patients. In this regard, this work aims to summarize the current understanding of the complex glaucomatous etiopathogenesis, highlighting alternative insights related to emerging pathomechanisms, such as inflammation and oxidative stress. Furthermore, we will explore immunomodulatory and antioxidant proposals as effective curative options for glaucoma.
2. General Characteristics of Glaucoma
2.1. Classification, Epidemiology and Economic Implications
Glaucoma is classified into primary and secondary forms based on the presence or absence of pre-existent pathological conditions such as uveitis, neoangiogenesis, traumas, lens abnormalities [2,21]. Glaucoma can also be categorized as open-angle or angle-closure, depending on the chamber angle located between the iris and the posterior surface of the cornea [22]. In a healthy state, this angle is physiologically open, allowing the outflow of aqueous humor (AH) through the trabecular meshwork (TM) to the uvea and conjunctiva, maintaining normal turnover [23,24]. Primary open-angle glaucoma (POAG) is the most common form of glaucoma [7], and is often associated with high IOP. However, it also includes a subtype known as normal-tension glaucoma (NTG), in which IOP is not elevated. NTG accounts for 30-90% of POAG cases and its prevalence varies significantly depending on geographical locations [11,25]. Remarkably, the prevalence of NTG among POAG cases in Asia is much higher (Japan 92% of cases [26], Singapore 84.6% [27], China 51.43-83.58% [28]), than in Western countries, where NTG accounts for 30-39% of cases [29,30,31]. Possible explanations for this significant difference were attributed another risk factor profile found in Asiatic populations, such as genetic components [32], long axial length [33], low intracranial pressure and vascular dysregulation [25]. Of note, elevated IOP can also be observed in individuals without glaucoma, as seen in cases of ocular hypertension [2].
In a context of primary angle-closure glaucoma (PACG), there is anatomical contact between the iris and the cornea, and in 90% of cases, between the iris and the lens, creating a pupillary block [34]. This condition leads to a significant increase in IOP, sometimes reaching levels as high as 80 mmHg [24]. Although PACG cases account for approximately 26% of the total glaucoma cases [35], they are responsible for approximatively half of the worldwide cases of glaucoma-related blindness [6,36]. Figure 1 illustrates the classification of various glaucoma variants.
From an epidemiological standpoint, it was estimated that in 2013, approximately 64.3 million people between the ages of 40 and 80 were affected by glaucoma. However, by the year 2040, it is projected that the number of individuals affected will exceed 110 million [7]. The high prevalence and severity of glaucoma have significant economic implications for both patients and healthcare systems. Direct costs associated with this condition are primarily linked to disease progression and the need for treatment adjustments when initial therapies are unsuccessful, contributing to cost escalation [37]. Indirect costs, such as the loss of well-being and visual disability experienced by patients, have been estimated to be the most impactful economic factors in Europe, surpassing three times the total healthcare costs. In late glaucoma stages, these indirect costs average €2,703 annually compared to total healthcare costs of €830 [38]. Additionally, a study conducted in a British clinic reported that approximately 11.7% of hospitalizations for falls were observed in patients with a secondary diagnosis of glaucoma [39].
Collectively, numerous studies on this subject emphasize the importance of halting disease progression and preventing late-stage glaucoma to minimize the loss of well-being for patients and prevent escalating costs.
2.2. Symptoms and Diagnostic Features
POAG and PACG present with different sets of symptoms. In POAG, the disease progression is often asymptomatic due to binocular compensation. As a result, patients typically experience the first noticeable symptoms only in advanced stages when significant damage to the visual field has already occurred [22,40]. Hence, an early detection through screening examinations such as fundoscopy, tonometry, or analysis of the retinal nerve fiber layer is fundamental to prevent the disease progression [22,41].
On the other hand, PACG manifests with rapid and painful symptoms. Affected individuals may experience a rock-hard sensation in the eye, corneal edema, reduced visual acuity, conjunctival hyperemia (redness), irradiating pain, and potentially accompanying nausea and vomiting [22]. PACG is considered an ophthalmologic emergency that necessitates immediate medical intervention to prevent severe visual loss [22].
The use of appropriate diagnostic tools is crucial for facilitating the detection of early signs of glaucoma and initiating prompt and appropriate therapy to prevent further damage. Tonometry, fundoscopy, and perimetry are valuable in enabling early diagnosis. Examination of the optic nerve head (ONH) helps evaluate the optic disc, neuroretinal rim, optic cup, and the cup-disc ratio (CDR), which represents the ratio between the optic cup and optic disc area [42]. Classic fundoscopic signs of glaucoma include an enlarged optic cup, resulting in an increased CDR, loss of neuroretinal rim, presence of disc hemorrhages, and parapapillary tissue atrophy [22,42]. Moreover, a spectral domain-optical coherence tomography (SD-OCT) and Heidelberg retina tomography can assist in detecting RNFL-thinning, decreased rim volume, or an increased CDR area [22,43,44].
Assessing the disease progression in glaucoma can be achieved through the examination of the neuroretinal rim of the ONH using fundoscopy or through perimetric evaluation [45]. Recently, SD-OCT has also been described as a suitable diagnostic tool for staging glaucoma [46]. However, despite the availability of various diagnostic features for assessing glaucomatous disease progression, there is currently no consensus on a singular criterion to determine specific disease stages [47].
2.3. Current Pharmacological Approches and Surgical Techniques
The primary objective of major established antiglaucoma drugs is to reduce intraocular pressure (IOP) to a personalized and acceptable range in order to halt the progression of the disorder [48]. These medications are typically administered topically via eye drops and can be categorized based on their pharmacological mechanisms into the following groups:
- Prostaglandin analogues: Examples include bimatoprost, which enhances both trabecular and uveoscleral outflow AH [22].
- β-blockers: Medications like levobunolol and timolol work by reducing the production of aqueous humor [22].
- α₂-adrenergic adrenoceptor agonists: Drugs such as apraclonidine and brimonidine lower IOP by decreasing aqueous humor production and augmenting trabecular outflow [22].
- Carbonic anhydrase inhibitors: Agents like brinzolamide act by reducing the production of aqueous humor [22].
- Miotic agents: Pilocarpine, for instance, increases the chamber angle by constricting the pupil and can also provide neuroprotective effects through the activation of muscarinic receptors [49].
- Rho-associated protein kinase (ROCK) inhibitors: Netarsudil is a ROCK inhibitor that targets the ROCK pathway, suppressing fibrotic events in the trabecular meshwork (TM) and optimizing aqueous humor flow, thereby reducing IOP [50]. This molecule has been approved for the treatment of glaucoma in the United States (2017) and Europe (2019) in the form of a 0.02% ophthalmic topical formulation for once-daily application [51].
In case of angle-closure glaucoma, the first-line therapy is typically a peripheral iridotomy performed using an ND-Yag laser. This procedure aims to create a full-thickness perforation in the iris, restoring the outflow of AH [4]. Simultaneously, carbonic anhydrase inhibitors, such as acetazolamide, can also be given intravenously to reduce AH production and the osmotic active molecule mannitol may be administered intravenously to facilitate the drainage of water from the eye [22]. Laser therapies are also employed for open-angle glaucoma. Selective laser trabeculoplasty, which utilizes pulsed ND-Yag laser, increases AH flow, while cyclophotocoagulation reduces AH production [22,52,53].
Surgical treatments in glaucoma are usually considered as a second-line therapy when conservative options fail to sufficiently lower IOP [22]. These surgical options can be categorized as follows [22]:
- Cyclocryocoagulation, which decreases AH production.
- Minimally invasive procedures, such as stent implantation, that reduce the outflow resistance of the trabecular meshwork (TM).
- Non-filtering procedures, such as deep sclerotomy, which enhance outflow pathways without incising the eye globe.
- Filtering procedures, like trabeculectomy, which create an additional drainage route for AH beneath the conjunctiva.
Due to the scarring processes that may affect the long-term efficacy of surgical techniques bypassing AH flow to subconjunctival spaces, medications are often employed postoperatively to inhibit excessive scar tissue growth [54]. Commonly used medications for this purpose in clinical practice include topical steroids and non-steroidal anti-inflammatory drugs. Off-label drugs, such as 5-fluorouracil and mitomycin C, are also utilized [54]. Additionally, there are ongoing investigations into the use of biologic drugs, such as bevacizumab (anti-vascular endothelial growth factor, VEGF), ranibizumab (anti-VEGF), infliximab (anti-tumor necrosis factor α, TNF-α), as well as molecules targeting the transforming growth factor (TGF-β) signaling pathway, like lerdelimumab (anti-TGF-β2) or decorin (a proteoglycan that also targets TGF-β signaling) [54].
3. Pathophysiology
3.1. Risk Factors
3.1.1. Elevated Intraocular Pressure
The main risk factor in both POAG and PACG is an elevated IOP, defined as a pressure value above the 97.5th percentile in the specific population under consideration, often considered to be higher than 21 mmHg [2,55]. In addition to IOP, other risk factors for POAG include myopia, advanced age, belonging to the black ethnic group, and a family history of the condition. For PACG, risk factors include being female, having a small corneal diameter, hyperopia, an anteriorly positioned lens, and shallower central and limbal anterior chamber depth [35]. However, IOP is recognized as the primary modifiable risk factor, making it the main target of current established antiglaucoma drugs [22].
Two major theories have been proposed to explain the pathogenesis of glaucoma, both emphasizing the association between elevated IOP and the development of the disease: the “vascular” and the “mechanical” theory. According to the vascular theory, high IOP leads to compression of the blood vessels supplying the ONH, resulting in reduced blood flow, hypoperfusion, and subsequent ischemia in RGCs [56,57]. On the other hand, the mechanical theory suggests that elevated IOP causes compressions and deformations in the lamina cribrosa and RGC axons, initiating a cascade of events that lead to cell death due to blocked axoplasmic traffic and inadequate cellular supply [58]. Figure 2 provides a summary of the events leading to mechanical damage in RGCs as a consequence of elevated IOP.
Elevated IOP is proposed to arise due to the pathological increase in resistance to AH flow within the TM [59]. The TM, located in the chamber angle, consists of three layers: the uveal TM, corneoscleral TM, and the juxtacanalicular TM (also known as the cribriform TM region), which borders the Schlemm's canal. The AH flows through the TM and reaches the episcleral veins of the conjunctiva via the Schlemm's canal [60]. The permeability of the TM to AH plays a crucial role in regulating IOP levels [60]. Structural alterations in the TM can lead to apoptosis of TM cells and disintegration of its structure [61]. Additionally, changes in the deposition of extracellular matrix within the TM can disrupt the adhesion of TM-endothelial cells [62]. The TGF-β2 appears to play a pivotal role in promoting the deposition of extracellular matrix within the human TM during glaucoma [63]. These events ultimately result in increased resistance to AH drainage within the TM, leading to elevated IOP [61].
3.1.2. Genetic Factors, Systemic Vascular Dysregulation and Endothelial Dysfunction
In the “Collaborative Normal-Tension Glaucoma Study”, a clinical trial, the effectiveness of IOP-lowering therapy in NTG was evaluated. The study revealed that although reducing IOP can have a positive impact, it alone cannot completely halt disease progression [12,64]. This suggests the involvement of additional factors in the development of NTG. The wide geographical variability in the prevalence of NTG and the relatively high percentage (approximately 21%) of patients reporting a family history of the condition [65] suggest a possible genetic predisposition. Individuals with NTG may have a lower tolerance for what are considered "normal" IOP levels [12]. The increased susceptibility of RGCs to IOP-induced damage is believed to contribute to mechanical injuries observed in NTG, similar to those seen in glaucomas associated with elevated IOP [12]. Numerous specific gene polymorphisms, resulting in altered functionality of corresponding proteins, have been associated with NTG [65]. For example, certain sequence variants of the optineurin (OPTN) gene, which encodes a neuroprotective and IOP-regulating protein, have been linked to NTG [66]. Mutations in the optic atrophy type 1 (OPA1) gene, which is crucial for mitochondrial function, have also been implicated in the pathogenesis of NTG. These mutations can lead to RGC apoptosis through mitochondrial dysfunction [65,67]. Furthermore, specific gene sequence variants of the endothelin-1 (ET-1) receptor A have been identified as being associated with NTG [68].
In addition to genetic factors, several alternative risk factors have been identified as potential contributors to the pathophysiology of NTG, including systemic vascular dysregulation, oxidative stress, and endothelial dysfunction [11,12,17]. A systemic vascular impairment, such as cerebral silent infarcts and nocturnal arterial hypotension, has been associated with NTG, potentially leading to a condition of hypoperfusion in the ONH [11,12,69,70,71]. Hypoperfusion-induced hypoxia may initiate the glaucomatous pathogenesis in NTG [72]. As a result of hypoxic insults, the hypoxia-inducible factor 1α (HIF-1α), a potent cytokine, triggers downstream transductions that activate glial cells, leading to neuroinflammation, similar to the events observed in glaucoma associated with high IOP [14,73,74]. Vascular endothelial dysfunction is another characteristic of NTG and may manifest through the impairment of vasoregulatory factors such as nitric oxide (NO) [75], and endothelin-1 (ET-1) [65]. Excessive reactive oxygen species (ROS) can reduce NO-dependent vasorelaxation due to impaired activity of endothelial nitric oxide synthase (eNOS). In a context of altered redox status, the fundamental cofactor of eNOS, tetrahydrobiopterin, undergoes oxidation to dihydrobiopterin, resulting in abnormal eNOS activity, the production of peroxynitrite (ONOO⁻), and a lower bioavailability of NO [75]. Consequently, dysfunctional vasoregulation occurs, leading to deficits in vasorelaxation [65,76,77]. Moreover, the vasoconstrictor peptide endothelin-1 (ET-1) has been reported to be increased in the plasma [78,79], and in AH [80], of NTG patients. The abnormal vasoconstriction induced by ET-1 may affect the blood vessels supplying the ONH in NTG, further contributing to reduced perfusion [81]. The combined processes of decreased NO-dependent vasodilation and increased ET-1-induced vasoconstriction in blood vessels may result in reduced perfusion of the ONH, forming the etiopathogenic basis for primary damage to RGCs in NTG [65,82].
In Figure 3, the pathomechanisms due to increased susceptibility to IOP in RGCs, to hypoperfusion and to endothelial dysfunction in NTG are illustrated.
3.2. Pathomechanisms
3.2.1. Inflammation, Chronic Oxidative Stress and Mitochondrial Dysfunction
High hydrostatic pressure and ischemia have been shown to trigger the release of the major proinflammatory cytokine, TNF-α, from glial cells, initiating inflammation and apoptosis in RGCs [73]. The role of TNF-α in the inflammatory and oxidative processes occurring in glaucoma is pivotal. This mediator is secreted by microglia, astrocytes and Müller cells and contributes to mitochondrial dysfunction, an overabundance of ROS, and the expression of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), a central transcription factor responsible for amplifying neuroinflammation and glial activation. NF-kB promotes the expression of proinflammatory cytokines and adhesion molecules [14,73]. Our studies in mice have demonstrated that elevated IOP leads to endothelial damage in the retina, accompanied by upregulation of nicotinamide adenine dinucleotide phosphate oxidase type 2 (NOX-2), one of the major prooxidative enzymes responsible for ROS generation through oxidation of oxygen (O2) in superoxide (O2•−) [83,84]. Furthermore, increased IOP has been shown to induce mitochondrial injuries, resulting in mitochondrial disruptions and expression abnormalities in the OPA1 gene [15,85]. Additionally, oxidized metabolites such as advanced glycation end products (AGEs) and oxidized low-density lipoproteins (oxLDLs) can act as "antigenic" stimuli, promoting ROS production, NF-kB activity, glial activation, and apoptosis [15,16]. In this context, other molecules such as damage-associated molecular patterns (DAMPs) and heat shock proteins (HSPs) have been extensively investigated as “highly antigenic molecules” [86] that associated with neuroinflammation in glaucoma [87,88,89]. Toll-like receptors (TLRs) are a relevant group of pattern recognition receptors, expressed in glial cells. They recognize bacterial components such as lipopolysaccharides, viral RNAs, and potentially oxidative stress products and upregulated HSPs. Activation of TLRs can lead to downstream activation of NF-kB and the amplification of neuroinflammation [87]. In addition to these pathomechanisms, also hypoxia has been observed to induce HIF-1α expression in the retina and optic nerve of patients with glaucoma [13]. HIF-1α has the ability to upregulate the expression ofNOX-2 and inducible nitric oxide synthase (iNOS), leading to the production of ROS [90,91,92,93]. In turn, ROS can trigger the expression of HIF-1α [94,95]. The activation of glial cells and the release of TNF-α follow, leading to the induction of NF-kB. This process amplifies glial activation, neuroinflammation, and ultimately apoptosis [14,73,74].
3.2.2. Endoplasmic Reticulum Stress, Apoptosis and Amplified Neuroinflammation
Elevated levels of ROS, mechanical and vascular insults in the pathogenesis of glaucoma disrupt the activities of mitochondria. Through calcium-dependent processes, mitochondria interact with the endoplasmic reticulum (ER), influencing each other and leading to energy deficiency, apoptosis, inflammation, and increased ROS production [96]. The ER is an intracellular organelle with a crucial role in protein processing and folding, ensuring their proper functionality [97,98]. Conditions such as oxidative stress, protein mutations, viral infections, nutritional deficits, and hypoxia can impact the ER, resulting in an accumulation of unfolded proteins [99,100,101]. This leads to ER stress, wherein the unfolded protein response (UPR) is initiated to restore cellular homeostasis [102]. chronic ER stress can paradoxically perpetuate UPR activation, leading to apoptosis, NF-kB activation, and further ROS formation [100,101]. The UPR consists of three main signalling pathways:
- The protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2α (eIF2α)/activating transcription factor 4 (ATF4)/CCAAT-enhancer-binding protein homologous protein (CHOP) pathway. This pathway reduces protein translation but can also increase ROS production and promote apoptosis [104].
NOX2-, mitochondrial and ER stress-generated ROS trigger activation of the NLRP3 inflammasome leading to the proteolytic processing of interleukin (IL)-1β and activation of NF-kB signaling [106]. ROS also activate proapoptotic signaling pathways, including the apoptosis signal-regulating kinase 1 (ASK-1)/p38 mitogen-activated protein kinase (MAPK)/JNK/extracellular-signal-regulated kinase (ERK) axis, which ultimately leads to caspase-3 activation and cellular membrane disassembly [107,108]. Chronic exposition to ROS activates the phosphoinositide 3-kinase (PI3K)/Akt axis while attenuating the mammalian target of rapamycin (mTOR) pathway, resulting in further stimulation of NF-kB and enhanced inflammatory events [109]. Through NF-kB-mediated abnormal neuroinflammation, infiltrations of activated T-cells and monocytes have been identified as fundamental factors contributing to RGC death [110,111]. Excessive ROS disrupts glutamate metabolism, leading to neurotoxic extracellular accumulation of glutamate [16]. Dysfunctional glial cells are unable to properly buffer the excess glutamate [16,96,112]. Additionally, aberrant immune responses include dysregulation of the complement system, which is implicated in synapse elimination and dendrite remodeling during glaucomatous neurodegeneration [86,113,114]. Consistent with this, various studies on murine glaucoma models have demonstrated that a transgenic lack of complement attenuates disease progression [115,116].
3.2.3. Autoimmune Imbalances
Evidence of autoimmune factors has been described in glaucoma [17,117,118]. Several publications have reported the presence of autoantibodies in sera and retina of patients with glaucoma [119,120,121,122,123,124,125,126]. Heat shock proteins (HSP) may play a critical role in this context. HSPs can be produced by bacteria or generated endogenously by cells at sites of inflammation, and they can activate specific HSP-induced T-regulatory cells [127]. High levels of HSP autoantibodies, including antibodies against HSP27, have been found in the sera of glaucoma patients. These autoantibodies have been shown to trigger neuronal apoptosis by interfering with the function of native HSPs in stabilizing the cytoskeleton [128,129,130]. Autoantibodies against HSP60 [131] and HSP70 [132] have also been detected in serum of glaucoma patients [133]. Furthermore, studies have demonstrated IgG autoantibody depositions in the glaucomatous retina, along with an increase in CD27+/IgG+ plasma cells and elevated levels of TNF-α, IL-6, and IL-8. These proinflammatory mediators were found to be released by activated microglia [117].
On the other hand, patients with glaucoma have shown a downregulation of protective naturally occurring autoantibodies, such as those against 14-3-3 and γ-synuclein. These autoantibodies have displayed protective effects by preserving against apoptosis and oxidative stress [133,134].
Taken together, considering the sequence of pathogenetic events, imbalances between pro-apoptotic and anti-apoptotic autoantibodies in autoimmune responses may contribute to secondary injuries in RGCs [133,135].
In Figure 4, processes leading to loss of RGCs in glaucoma are summarized.
4. Emerging Curative Strategies: Immunomodulatory and Antioxidants
4.1. Immunomodulatory Candidates for Glaucoma
4.1.1. Fas-Receptor Antagonists
The fragment apoptosis stimulator (Fas) ligand is a membrane-bound protein that has been described in the eye to have pro-inflammatory and pro-apoptotic activity when it binds its receptor [136]. However, when it is cleaved and released as a soluble isoform, Fas exhibits opposite functions [137]. A study conducted on mouse models of glaucoma demonstrated that an upregulation of the soluble form of Fas ligand, achieved through intravitreal adeno-associated virus-mediated gene treatment, can reduce the glial cell activation and prevent loss of RGCs [138]. Moreover, a small peptide inhibitor of the Fas receptor, known as ONL1204, has shown promising results in murine glaucoma models by suppressing RGC apoptosis, preserving axons, and inhibiting glial activation and neuroinflammation [139]. A dedicated clinical trial is currently underway to evaluate the effectiveness of an ophthalmic solution of ONL1204 on 25 patients with progressing open-angle glaucoma (NCT05160805). The estimated completion date of this study is September 2023.
4.1.2. Adenosine Receptor Modulators
By antagonizing the adenosine A2A receptor, caffeine has been shown to have the ability to protect against neuroinflammation and attenuate glial activation in neurodegenerative conditions [140]. Building on this evidence, studies have investigated the effectiveness of selective A2A receptor antagonists such as SCH 58261 in animal models of ischemia/reperfusion [141], as well as caffeine in rodent models of glaucoma [142]. In both cases, these interventions demonstrated a reduction in neuroinflammation through decreased glial activation, ultimately resulting in the preservation of RGCs. In addition, caffeic acid phenethyl ester, when administered in rodent models of glaucoma, has been shown to reduce the expression of pro-inflammatory cytokines such as IL-6 and IL-8, as well as inducible nitric oxide synthase (iNOS) and COX2. This leads to a decreased activation of NF-kB, thereby attenuating neuroinflammation and preventing RGC loss [143].
A modulator of the adenosine A3 receptor called FM101 has demonstrated its safety in rodent models of glaucoma [144], and a dedicated clinical trial is currently underway to evaluate its efficacy in patients with ocular hypertension (NCT04585100).
4.1.3. Biologic Perspectives
Etanercept is a monoclonal antibody that targets and antagonizes the human TNF-α receptor type 2 [18]. This biologic drug is approved for the treatment of autoimmune diseases such as rheumatoid arthritis and ankylosing spondylitis [145]. In murine glaucoma models, administration of etanercept has been shown to inhibit TNF-α signaling, leading to a reduction in glial activation and the preservation of RGCs [146]. In the context of biologic medications for the treatment of glaucoma, Geyer and Levo extensively reviewed the current literature upon the autoimmune aspects of glaucoma [17]. They suggest that immunomodulatory drugs approved for autoimmune diseases, such as Janus kinase inhibitors, anti-cytokines, and rituximab (an anti-CD20 monoclonal antibody), may be suitable for managing glaucoma [17]. However, an investigation designed to test intravitreal injections of rituximab for the treatment of retinal lymphomas reported that the procedure can inadvertently lead to an elevation in IOP, necessitating the use of antiglaucoma drugs postoperatively [147].
4.1.4. TGF-β2 and NF-kB Targeting
A recent study conducted on human TM cells found that baicalin, an extract from Scutellaria baicalensis Georgi, has potential in preventing fibrosis by reducing the deposition of TGF-β2-induced extracellular matrix. This effect was achieved through the modulation of the NF-kB pathway [148]. Another study utilizing an experimental mouse model of glaucoma, characterized by the transgenic inhibition of astroglial NF-kB, demonstrated a protective effect against neurodegeneration in RGC axons and somas. These findings suggest potential new approaches in immunomodulation for glaucoma by targeting NF-kB, a crucial mediator of neuroinflammation [149]. However, it is important to note that NF-kB targeting may be controversial due to its essential role in regulating physiological cell survival mechanisms [86,150,151]. Lack of cell-specific NF-kB targeting can lead to severe side effects, including RGC loss, as observed in transgenic mice lacking NF-kB [152].
4.1.5. Inhibition of the Complement System
As previously mentioned, abnormal activation of the complement system is a known event in the pathophysiology of glaucoma, and enhanced complement activity has been observed in glaucoma models [153,154]. Building upon this knowledge, a study investigated the protective effect of combined inhibition of the endothelin and complement systems in mouse models of glaucoma [155]. Additionally, a recent study on experimental autoimmune glaucoma models demonstrated that intravitreal treatment with an antibody against complement factor C5 suppressed complement activation, leading to reduced RGC loss and prevention of degenerative events associated with immune dysregulation in glaucoma [156].
4.1.6. cAMP Phosphodiesterase Inhibitors
Ibudilast is a non-selective 3′,5′-cyclic adenosine monophosphate (cAMP) phosphodiesterase (PDE) inhibitor with specific affinity for PDE type 4. It possesses important anti-inflammatory and vasodilator properties and is used in the treatment of stroke and asthma [157,158]. Ibudilast has been shown to suppress glial activation and the generation of inflammatory cytokines [159]. Ocular hypertension has been found to upregulate PDE type 4 in Müller cells, the major glial cell type in the retina [160]. In rodent models of glaucoma, Ibudilast has been found to mitigate neuroinflammation and improve RGC viability through the cAMP/Protein kinase A axis [161].
Interestingly, another PDE also expressed in the retina is PDE type 5 [162]. Sildenafil, a PDE type 5 inhibitor commonly used to treat erectile dysfunction due to its vasorelaxant effects, has been investigated in glaucoma rodent models. The study demonstrated that sildenafil promotes RGC survival by modulating the TNF-α pathway [163]. Furthermore, sildenafil was the subject of a dedicated clinical trial (NCT04052269) aimed at evaluating the effect of PDE inhibitors on blood circulation in the retina and choroid vessels of patients with glaucoma using OCT scans [164]. However, the trial was suspended due to the COVID-19 pandemic.
4.1.7. Antibiotics as Possible Immunomodulatory Approaches
Minocycline is a tetracycline antibiotic that has demonstrated anti-inflammatory and vasoregulatory activities in retinal ischemia/reperfusion models [165]. Administration of minocycline in glaucomatous rodent eyes and rodent eyes after optic nerve transection has been shown to prevent RGC loss by suppressing proapoptotic cascades [166]. Another study in murine glaucoma models demonstrated that minocycline can mitigate glial activation and improve RGC axonal transport and integrity [167]. Similarly, in an experimental model of glaucoma, minocycline antagonized microglial reactivity, preserving RGC axons and glia from degeneration [168]. However, in a recent investigation using glaucoma-like degenerative retinal models, minocycline was found to decrease inflammation and glial activation but did not provide complete protection for RGCs [169]. In rodent models of chronic OHT, intravitreal injections of minocycline induced Müller cell autophagy and increased RGC survival, confirming its role as a microglial inhibitor [170]. Mechanistically, it has been suggested that minocycline can upregulate genes associated with the antiapoptotic Bcl-2 family, as observed in optic nerve transection models, human TM cells, and optic nerve head astrocytes [171,172].
Azithromycin is a macrolide antibiotic with immunomodulatory properties that has been explored, for example, in the treatment of respiratory disorders [173]. In rodent models of ischemia/reperfusion, post-injury administration of azithromycin exhibited a neuroprotective effect by preventing RGC loss through the suppression of Bcl-2-associated death promoter (Bad) upregulation, inhibition of metalloproteinase (MMP)-2/-9 activity, and the ERK1/2 pathway [174]. Consistent with these findings, another recent investigation using rodent models of glaucoma found that azithromycin preserved RGCs from apoptosis and attenuated neuroinflammation by decreasing the Bcl-2 associated X-protein (Bax)/Bcl-2 ratio, TGF-β levels, and TNF-α levels [163].
4.1.8. Stem Cell Potential for Immunomodulation in Glaucoma
Stem-cell based treatments are well-known for their regenerative capabilities, however, they have also been suggested as an approach to modulate inflammatory events [86]. In this regard, several publications have highlighted the protective effect of mesenchymal stem cells (MSCs) on RGCs, inducing neuroprotection in terms of preserving RNFL thickness in rodent optic nerve crush models [175] and glaucoma models [176]. Nevertheless, a dedicated clinical trial involving two patients with advanced glaucoma (NCT02330978) showed that intravitreal injections of autologous bone marrow-derived MSCs did not result in changes in electroretinographic responses or improvements in visual acuity. In one of the patients, retinal detachment occurred two weeks after treatment [177]. These findings indicate the need for modified MSCs for glaucoma treatment [178]. A recent study examined the immunomodulatory features and safety of MSCs in an ex vivo neuroretina explant model. The study assessed the capabilities of MSCs to attenuate glial activation, TNF-α signaling, and IL-1β signaling. However, it also confirmed edema and gliosis as side effects of the stem cell treatment [179].
4.1.9. Toll-like Receptor Inhibitors and Modulation of the Microbiota
TLRs can interact with lipopolysaccharides as well as with DAMPs, playing a role in glial activation signaling and the amplification of neuroinflammation [86]. Interestingly, the commensal microbiota has been found to be partially involved in the pre-sensitization of T-cells, observed in murine infiltrates during glaucomatous neurodegeneration [180]. Astafurov et al. subcutaneously administered lipopolysaccharides, common bacterial constituents, in two different murine glaucoma models, resulting in increased axonal degeneration and RGC loss, along with microglial activation in the optic nerve and retina [181]. The study also demonstrated that lipopolysaccharide-induced TLR-4 activation was responsible for amplifying neuroinflammation and complement activation, thereby exacerbating glaucomatous degeneration. Naloxone, an opioid shown to inhibit TLR-4, partially attenuated these effects [181,182,183]. TAK-242, also known as resatorvid, is a small-molecule cyclohexene derivative that acts as a TLR-4 inhibitor. It has been shown to attenuate glial activation in RGCs of optic nerve crush models by reducing the p38 pathway and NF-kB activation [184]. In addition, TAK-242 has been demonstrated to block fibroblastic proliferation of the Tenon's capsule in rodent models, suggesting its potential as an anti-scarring drug after glaucoma surgery [185]. Mechanistically, TAK-242 decreases the TGF-β2 pathway in human TM through TLR-4 inhibition [186].
Furthermore, short-chain fatty acids (SCFAs), products of microbiota in fermentation, have been described as mediators of microglial homeostasis and are capable of binding to TLRs [187,188]. Chen et al. revealed that SCFAs can suppress inflammatory responses in retinal astrocytes by decreasing proinflammatory cytokines such as IL-6 [188]. Their potential role in modulating the microbiota and counteracting inflammation in the neurodegeneration of glaucoma has been suggested [17].
4.2. Promising Antioxidants for Glaucoma Treatment
4.2.1. Natural Antioxidants
Numerous naturally occurring molecules with antioxidant properties have been investigated in preclinical and clinical studies for their potential benefits in preserving RGCs in glaucoma:
Vitamin B3, or niacin, has been studied for its antioxidant features in the treatment of glaucoma [189]. An epidemiological study conducted in Korea found that patients with NTG had a lower dietary intake of niacin compared to other nutrients, suggesting a possible negative correlation between vitamin B3 intake and NTG risk [190]. Preclinical investigations in murine glaucoma models have shown that administration of nicotinamide (amide form of niacin) is effective in preventing and slowing down the progression of glaucoma by attenuating the age-related decline of nicotinamide adenine dinucleotide (NAD) [191]. A randomized controlled trial involving 57 patients with glaucoma demonstrated that a nicotinamide supplementation can improve the inner retinal function [192].
Astaxanthin (AST) is an antioxidant molecule found in microalgae and other sources [193,194]. In rat models of elevated IOP, AST has been shown to decrease apoptotic cascades [195]. In murine models of NTG, AST has demonstrated the ability to prevent RGC loss [196]. Mechanistically, AST appears to activate the nuclear factor erythroid-derived 2-related factor 2 (Nrf2), a transcription factor that upregulates several antioxidant genes, thus attenuating RGC loss in glaucoma [197].
Resveratrol is a polyphenol present in grapes, berries, and peanuts, known for its antioxidant properties [198]. This molecule has been shown to activate suirtin1 (SIRT1), a nuclear NAD⁺-dependent deacetylase that upregulates the Nrf2/ARE (antioxidant response elements) pathway [199,200]. In a rodent glaucoma model, resveratrol was reported to attenuate RGC loss [201]. Moreover, resveratrol was shown to preserve RGCs from ROS-triggered apoptosis by suppressing MAPK cascades (p38, JNK, ERK) [202]. Likewise, in a mouse model of retinal ischemia/reperfusion injury induced by elevated IOP, resveratrol promoted RGC survival by reducing oxidative stress possibly via downregulation of NOX2 expression [203].
The α-lipoic acid (ALA) is found in vegetables, fruits and the liver or heart of animals [204]. In glaucomatous mouse models, ALA decreased ROS formation and increased the activity of antioxidant enzymes like NOS and HO-1, possibly through the activation of Nrf-2 [204]. In a prospective case-control study, a formula containing ALA and other antioxidants, including vitamin C, enhanced systemic markers of antioxidative status such as total antioxidant status (TAS) and reduced systemic oxidative marker malondialdehyde (MDA), a marker of lipid peroxidation, in the blood of patients with POAG [205].
Curcumin is a constituent of the spice turmeric, traditionally used in medicine, and possesses antioxidant properties [206]. In rodent models of chronic elevated IOP, curcumin reduced ROS generation and inhibited apoptotic pathways by downregulating proapoptotic proteins such as caspase-3, Bax, and cytochrome c [207]. In murine models, curcumin prevented RGC loss by blocking MAPK, caspase-9 and caspase-3 activation [208].
Flavonoids are a class of molecules present in plants that possess antioxidant properties. Plant extracts from Gingko biloba L. contain over 70 diverse flavonoids, which have been shown to interfere with apoptotic pathways by binding proteins such as p53, Bax, Bcl-2, and caspase-3/-9 [209]. Flavonoids in Gingko biloba L. may attenuate RGC injuries in glaucoma by suppressing ROS-induced apoptosis [210]. However, a clinical study comparing oral antioxidants, including extracts of Ginkgo biloba and α-tocopherol, for the treatment of glaucoma (NCT01544192) did not show clear benefits associated with the use of Ginkgo biloba [211]. Coenzyme Q₁₀, another flavonoid, has been shown to reduce glutamate excitotoxicity and ROS formation in mice models of glaucoma, thus preserving RGCs from apoptosis by reducing Bax expression and enhancing Bad protein expression [212]. Currently, a clinical trial (NCT03611530) is underway to determine the effect of a formula containing coenzyme Q10 and vitamin E on patients with POAG [213]. Another trial (NCT04784234), is also ongoing, testing a mixture of Ginkgo biloba, α-lipoic acid, coenzyme Q10, curcumin, and other naturally occurring compounds in 100 patients with POAG. The expected completion date for this study is the end of 2023.
In a recent study from our laboratory, we found that mice devoid of the M1 muscarinic acetylcholine receptor subtype display a reduced RGC density and elevated retinal ROS levels at advanced age despite normal IOP [214]. Moreover, retinal mRNA levels for the prooxidant enzyme, NOX2, were elevated but mRNA levels for the antioxidant enzymes, SOD1, HO-1 and GPX1, reduced, suggesting that the M1 receptor may play an important role in regulating ROS levels in the retina and thus in neuroprotection [214]. In support of this concept, various other studies reported neuroprotective effects of cholinergic agents on retinal neurons pointing to the involvement of the M1 receptor [215,216,217]. Huperzine A, an alkaloid extracted and isolated from the plant Huperzia serrata, inhibits acetylcholinesterase activity, thus increasing acetylcholine levels. In a recent study, huperzine A was reported to produce neuroprotective effects in the rat retina subjected to ischemia/reperfusion injury via involvement of the M1/AKT/MAPK signaling pathway and by reducing oxidative stress [218]. Based on these promising studies, the role of the M1 signaling pathway on ROS generation and on neuroprotection in the retina should be pursued further.
4.2.2. Existing Drugs with Antioxidant Properties
Several existing drugs with antioxidant properties have been investigated for their potential benefits in glaucoma:
Valproic acid (VPA), an antiepileptic drug, has been shown in murine models of NTG to attenuate excessive ROS levels and improve RGC survival through a cascade associated with ERK [219]. In retina explant models, VPA has been found to decrease the expression of pro-inflammatory cytokines and reduce microglial activation [220]. A dedicated clinical trial demonstrated that VPA has benefits in patients in advanced stages of glaucoma, improving their visual acuity [221].
N-acetylcysteine, commonly used in cases of paracetamol overdose and as a mucolytic agent in respiratory diseases, possesses antioxidant capabilities [222]. It attenuates retinal oxidative stress caused by elevated IOP when combined with brimonidine in rodent models of OHT [223]. N-acetylcysteine has been shown to enhance concentrations of glutathione, a potent antioxidant, inhibiting oxidative stress and RGC autophagy in mouse models of NTG [224]. Another study demonstrated that this molecule can preserve RGCs from autophagy by interfering with the HIF-1α axis via BNIP3 (Bcl2 interacting protein 3) and the PI3K/Akt/mTOR cascade [225].
Edaravone, an anti-stroke drug, possesses free radical scavenging features [226]. It has been shown to inhibit the JNK/p38 proapoptotic pathways in glaucoma models, preventing RGC-loss [227,228,229].
Rapamycin, a macrolide antibiotic with anti-neurodegenerative capabilities reported in Alzheimer's and Parkinson's diseases, has been found to increase RGC survival in rat glaucoma models. It counters the release of TNF-α from microglia, regulates NF-kB activity, and retains Akt phosphorylation to antagonize RGC apoptosis [230,231,232].
Geranylgeranylacetone, a compound used in the case of gastric ulcers, possesses antioxidant properties. In the retina, it promotes the activity of thioredoxin and HSP-72, preserving against apoptosis [233]. In mouse models of NTG, geranylgeranylacetone counteracts RGC death by upregulating HSP-70 and reducing caspase-3 and -9 activities [234].
Metformin, a widely used antidiabetic medication, has shown in eye drop solution to prevent fibrosis after glaucoma surgeries in rat models by activating the AMP-activated protein kinase (AMPK)/Nrf2 signaling pathway [235].
Valdecoxib, a selective cyclooxygenase (COX)-2 inhibitor commonly used in osteoarthritis and rheumatoid arthritis, has been shown in an investigation to suppress apoptosis in ischemia/reperfusion-induced glaucoma-like damaged cells of rats by blocking the ATF4-CHOP axis [236], thereby preventing the CHOP-induced ROS-formation [104]. Another compound that antagonizes ER stress is 4-phenylbutyric acid (4-PBA). Traditionally employed in cystic fibrosis since the 1990s [237,238], 4-PBA has been found to mitigate ROS formation in activated microglia [239]. It can counteract ROS formation related to high-fat diet or acute ammonia challenge by opposing ER stress [240]. In mouse models of glaucoma, 4-PBA has demonstrated the ability to reduce ER stress and prevent disease phenotypes [241]. Another study revealed that 4-PBA can reduce IOP by activating matrix metalloproteinase -9 and subsequent extracellular matrix degradation [242].
4.2.3. Target-Specific Synthetic Compounds
Target-specific synthetic compounds, representing a new frontier in combating oxidative stress in glaucoma, focus on inhibiting specific molecular targets. One promising class of compounds is the NOX inhibitors, which aim to counteract the adverse effects of glial activation and supplement traditional IOP-reducing strategies [243]. GKT137831, also known as setanaxib, is a dual inhibitor of NOX1 and NOX4. It has demonstrated beneficial effects in mitigating retinal inflammation and ischemia by reducing hypoxia-related ROS formation [244]. Another notable compound in this class is GLX7013114, a specific NOX4 inhibitor. Intravitreal injections of GLX7013114 have been effective in mitigating glial activation in a rat model of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-induced retinal excitotoxicity [245].
NOX inhibitors offer new possibilities in the field of antioxidants for glaucoma treatment, as they act independently of IOP to counteract oxidative stress, prevent RGC loss, and attenuate neuroinflammatory events.
Another emerging class of molecules is ROCK inhibitors, as demonstrated by the approval of netarsudil. Among them, Y-27632 is a noteworthy ROCK inhibitor under investigation. This potential drug has been shown to upregulate antioxidant agents such as Catalase and partially reduce ROS formation [246]. Moreover, Y-27632 induces phagocytosis in glaucomatous TM cells, leading to IOP reduction [247]. Ripasudil, also known as K-115, is another ROCK inhibitor that promotes endothelium-independent relaxation in porcine retinal arterioles while suppressing ET-1 activity, suggesting potential as an antiglaucoma drug [248].
In summary, by targeting the ROCK pathway, these molecules hold significant potential for glaucoma treatment. They optimize TM functionality, reduce fibrotic processes, and potentially lower IOP.
5. Conclusion and Future Perspectives
Glaucoma, a highly prevalent neurodegenerative disease and a leading cause of irreversible visual loss globally, has a complex and multifactorial pathophysiology that requires in-depth exploration. Our review has shed light on key players and emerging factors in glaucomatous pathomechanisms, particularly neuroinflammation and oxidative stress. Immunomodulation and enhancement of antioxidant capabilities have shown promise in preclinical studies as effective strategies for promoting neuroprotection and RGC survival. These approaches offer new avenues for targeting glaucoma independently of IOP and present a horizon of potential curative possibilities [86,249,250,251,252]. However, the design of effective immunomodulatory and antioxidant therapies for glaucoma remains challenging on several fronts. Pivotal mediators such as NF-kB, involved in balancing pro- and anti-inflammatory events and pro- and anti-apoptotic processes, require careful consideration [152]. Immunomodulatory treatments need to address these complex factors to achieve a delicate balance and avoid potential side effects [86]. Similarly, antioxidant therapies should take into account the delicate redox status, maintaining basal levels of ROS that are physiologically essential for cellular homeostasis [253].
Continued efforts are needed to address challenges related to biomarker sensitivities, long-term follow-ups, drug bioavailability, and target-specific delivery systems for neuroprotective approaches. Advancements in these areas will help bridge the current translational gap [254,255,256]. The goal is to provide substantial benefits to patients, potentially supplementing current established antiglaucoma drugs with a new generation of molecules that offer alternative targeting strategies.
Author Contributions
Conceptualization, F.B. and A.G.; writing—original draft preparation, F.B.; writing—review and editing, A.G. and N.P.; visualization, F.B.; supervision, A.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anderson, D.R.; Patella, V.M. Automated static perimetry, 2nd ed.; Mosby: Saint Louis (Mo.), 1999. [Google Scholar]
- Casson, R.J.; Chidlow, G.; Wood, J.P.; Crowston, J.G.; Goldberg, I. Definition of glaucoma: clinical and experimental concepts. Clin Exp Ophthalmol 2012, 40, 341–349. [Google Scholar] [CrossRef]
- Burgoyne, C. The morphological difference between glaucoma and other optic neuropathies. J Neuroophthalmol 2015, 35 Suppl 1, S8–s21. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: a review. Jama 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990-2020: a systematic review and meta-analysis. Lancet Glob Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef]
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 2006, 90, 262–267. [Google Scholar] [CrossRef]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, J.; Li, Y.; Jiang, B. Prevalence of primary open angle glaucoma in the last 20 years: a meta-analysis and systematic review. Sci Rep 2021, 11, 13762. [Google Scholar] [CrossRef]
- Acott, T.S.; Keller, K.E.; Kelley, M.J. Role of Proteoglycans in the Trabecular Meshwork☆. In Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier, 2017. [Google Scholar]
- Cesareo, M.; Giannini, C.; Martucci, A.; Di Marino, M.; Pocobelli, G.; Aiello, F.; Mancino, R.; Nucci, C. Chapter 2 - Links between obstructive sleep apnea and glaucoma neurodegeneration. In Progress in Brain Research; Bagetta, G., Nucci, C., Eds.; Elsevier, 2020; Volume 257, pp. 19–36. [Google Scholar]
- Leung, D.Y.L.; Tham, C.C. Normal-tension glaucoma: Current concepts and approaches-A review. Clin Exp Ophthalmol 2022, 50, 247–259. [Google Scholar] [CrossRef]
- Killer, H.E.; Pircher, A. Normal tension glaucoma: review of current understanding and mechanisms of the pathogenesis. Eye (Lond) 2018, 32, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Wax, M.B. Hypoxia-inducible factor 1alpha in the glaucomatous retina and optic nerve head. Arch Ophthalmol 2004, 122, 1348–1356. [Google Scholar] [CrossRef]
- Tezel, G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog Brain Res 2008, 173, 409–421. [Google Scholar] [CrossRef]
- Tezel, G. Molecular regulation of neuroinflammation in glaucoma: Current knowledge and the ongoing search for new treatment targets. Prog Retin Eye Res 2022, 87, 100998. [Google Scholar] [CrossRef] [PubMed]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Current Opinion in Pharmacology 2013, 13, 12–15. [Google Scholar] [CrossRef]
- Geyer, O.; Levo, Y. Glaucoma is an autoimmune disease. Autoimmunity Reviews 2020, 19, 102535. [Google Scholar] [CrossRef]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Progress in Retinal and Eye Research 2021, 83, 100916. [Google Scholar] [CrossRef]
- Sim, R.H.; Sirasanagandla, S.R.; Das, S.; Teoh, S.L. Treatment of Glaucoma with Natural Products and Their Mechanism of Action: An Update. Nutrients 2022, 14. [Google Scholar] [CrossRef]
- Hsueh, Y.-J.; Chen, Y.-N.; Tsao, Y.-T.; Cheng, C.-M.; Wu, W.-C.; Chen, H.-C. The Pathomechanism, Antioxidant Biomarkers, and Treatment of Oxidative Stress-Related Eye Diseases. International Journal of Molecular Sciences 2022, 23, 1255. [Google Scholar] [CrossRef]
- Foster, P.J.; Buhrmann, R.; Quigley, H.A.; Johnson, G.J. The definition and classification of glaucoma in prevalence surveys. Br J Ophthalmol 2002, 86, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Schuster, A.K.; Erb, C.; Hoffmann, E.M.; Dietlein, T.; Pfeiffer, N. The Diagnosis and Treatment of Glaucoma. Dtsch Arztebl Int 2020, 117, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Flores-Sánchez, B.C.; Tatham, A.J. Acute angle closure glaucoma. Br J Hosp Med (Lond) 2019, 80, C174–c179. [Google Scholar] [CrossRef] [PubMed]
- Khazaeni, B.; Khazaeni, L. Acute Closed Angle Glaucoma. In StatPearls; StatPearls PublishingCopyright © 2022, StatPearls Publishing LLC.: Treasure Island (FL), 2022. [Google Scholar]
- Chen, M.-J. Normal tension glaucoma in Asia: Epidemiology, pathogenesis, diagnosis, and management. Taiwan Journal of Ophthalmology 2020, 10. [Google Scholar] [CrossRef]
- Iwase, A.; Suzuki, Y.; Araie, M.; Yamamoto, T.; Abe, H.; Shirato, S.; Kuwayama, Y.; Mishima, H.K.; Shimizu, H.; Tomita, G.; et al. The prevalence of primary open-angle glaucoma in Japanese: the Tajimi Study. Ophthalmology 2004, 111, 1641–1648. [Google Scholar] [CrossRef]
- Shen, S.Y.; Wong, T.Y.; Foster, P.J.; Loo, J.L.; Rosman, M.; Loon, S.C.; Wong, W.L.; Saw, S.M.; Aung, T. The prevalence and types of glaucoma in malay people: the Singapore Malay eye study. Invest Ophthalmol Vis Sci 2008, 49, 3846–3851. [Google Scholar] [CrossRef]
- Zhao, J.; Solano, M.M.; Oldenburg, C.E.; Liu, T.; Wang, Y.; Wang, N.; Lin, S.C. Prevalence of Normal-Tension Glaucoma in the Chinese Population: A Systematic Review and Meta-Analysis. Am J Ophthalmol 2019, 199, 101–110. [Google Scholar] [CrossRef]
- Bonomi, L.; Marchini, G.; Marraffa, M.; Bernardi, P.; De Franco, I.; Perfetti, S.; Varotto, A.; Tenna, V. Prevalence of glaucoma and intraocular pressure distribution in a defined population. The Egna-Neumarkt Study. Ophthalmology 1998, 105, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Dielemans, I.; Vingerling, J.R.; Wolfs, R.C.; Hofman, A.; Grobbee, D.E.; de Jong, P.T. The prevalence of primary open-angle glaucoma in a population-based study in The Netherlands. The Rotterdam Study. Ophthalmology 1994, 101, 1851–1855. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.E.; Klein, R.; Sponsel, W.E.; Franke, T.; Cantor, L.B.; Martone, J.; Menage, M.J. Prevalence of glaucoma. The Beaver Dam Eye Study. Ophthalmology 1992, 99, 1499–1504. [Google Scholar] [CrossRef]
- Belamkar, A.; Harris, A.; Oddone, F.; Verticchio Vercellin, A.; Fabczak-Kubicka, A.; Siesky, B. Asian Race and Primary Open-Angle Glaucoma: Where Do We Stand? Journal of Clinical Medicine 2022, 11, 2486. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-k.; Kee, C. Population-based glaucoma prevalence studies in Asians. Survey of Ophthalmology 2014, 59, 434–447. [Google Scholar] [CrossRef]
- Patel, K.; Patel, S. Angle-closure glaucoma. Disease-a-Month 2014, 60, 254–262. [Google Scholar] [CrossRef]
- Wright, C.; Tawfik, M.A.; Waisbourd, M.; Katz, L.J. Primary angle-closure glaucoma: an update. Acta Ophthalmol 2016, 94, 217–225. [Google Scholar] [CrossRef]
- Quigley, H.A. Number of people with glaucoma worldwide. Br J Ophthalmol 1996, 80, 389–393. [Google Scholar] [CrossRef]
- Lorenz, K.; Wolfram, C.; Breitscheidel, L.; Shlaen, M.; Verboven, Y.; Pfeiffer, N. Direct cost and predictive factors for treatment in patients with ocular hypertension or early, moderate and advanced primary open-angle glaucoma: the CoGIS study in Germany. Graefes Arch Clin Exp Ophthalmol 2013, 251, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, J.; Aagren, M.; Arnavielle, S.; Bron, A.; Fröhlich, S.J.; Baggesen, K.; Azuara-Blanco, A.; Buchholz, P.; Walt, J.G. Late-stage, primary open-angle glaucoma in Europe: social and health care maintenance costs and quality of life of patients from 4 countries. Curr Med Res Opin 2008, 24, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- McGinley, P.; Ansari, E.; Sandhu, H.; Dixon, T. The cost burden of falls in people with glaucoma in National Health Service Hospital Trusts in the UK. J Med Econ 2020, 23, 106–112. [Google Scholar] [CrossRef]
- Crabb, D.P.; Smith, N.D.; Glen, F.C.; Burton, R.; Garway-Heath, D.F. How does glaucoma look?: patient perception of visual field loss. Ophthalmology 2013, 120, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Aspberg, J.; Heijl, A.; Bengtsson, B. Screening for Open-Angle Glaucoma and Its Effect on Blindness. Am J Ophthalmol 2021, 228, 106–116. [Google Scholar] [CrossRef]
- Jonas, J.B.; Budde, W.M.; Panda-Jonas, S. Ophthalmoscopic evaluation of the optic nerve head. Surv Ophthalmol 1999, 43, 293–320. [Google Scholar] [CrossRef]
- Hoffmann, E.M.; Miglior, S.; Zeyen, T.; Torri, V.; Rulli, E.; Aliyeva, S.; Floriani, I.; Cunha-Vaz, J.; Pfeiffer, N. The Heidelberg retina tomograph ancillary study to the European glaucoma prevention study: study design and baseline factors. Acta Ophthalmol 2013, 91, e612–619. [Google Scholar] [CrossRef]
- Enders, P.; Adler, W.; Kiessling, D.; Weber, V.; Schaub, F.; Hermann, M.M.; Dietlein, T.; Cursiefen, C.; Heindl, L.M. Evaluation of two-dimensional Bruch's membrane opening minimum rim area for glaucoma diagnostics in a large patient cohort. Acta Ophthalmol 2019, 97, 60–67. [Google Scholar] [CrossRef]
- Sihota, R.; Angmo, D.; Ramaswamy, D.; Dada, T. Simplifying "target" intraocular pressure for different stages of primary open-angle glaucoma and primary angle-closure glaucoma. Indian J Ophthalmol 2018, 66, 495–505. [Google Scholar] [CrossRef]
- Brusini, P. OCT Glaucoma Staging System: a new method for retinal nerve fiber layer damage classification using spectral-domain OCT. Eye (Lond) 2018, 32, 113–119. [Google Scholar] [CrossRef]
- Vianna, J.R.; Chauhan, B.C. Chapter 7 - How to detect progression in glaucoma. In Progress in Brain Research, Bagetta, G., Nucci, C., Eds.; Elsevier: 2015; Volume 221, pp. 135–158.
- Prum, B.E., Jr.; Rosenberg, L.F.; Gedde, S.J.; Mansberger, S.L.; Stein, J.D.; Moroi, S.E.; Herndon, L.W., Jr.; Lim, M.C.; Williams, R.D. Primary Open-Angle Glaucoma Preferred Practice Pattern(®) Guidelines. Ophthalmology 2016, 123, P41–p111. [Google Scholar] [CrossRef]
- Tan, P.P.; Yuan, H.H.; Zhu, X.; Cui, Y.Y.; Li, H.; Feng, X.M.; Qiu, Y.; Chen, H.Z.; Zhou, W. Activation of muscarinic receptors protects against retinal neurons damage and optic nerve degeneration in vitro and in vivo models. CNS Neurosci Ther 2014, 20, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lee, C.; Read, A.T.; Wang, K.; Ha, J.; Kuhn, M.; Navarro, I.; Cui, J.; Young, K.; Gorijavolu, R.; et al. Anti-fibrotic activity of a rho-kinase inhibitor restores outflow function and intraocular pressure homeostasis. Elife 2021, 10. [Google Scholar] [CrossRef]
- Batra, M.; Gupta, S.; Nair, A.B.; Dhanawat, M.; Sandal, S.; Morsy, M.A. Netarsudil: A new ophthalmic drug in the treatment of chronic primary open angle glaucoma and ocular hypertension. Eur J Ophthalmol 2021, 31, 2237–2244. [Google Scholar] [CrossRef] [PubMed]
- Gulati, V.; Fan, S.; Gardner, B.J.; Havens, S.J.; Schaaf, M.T.; Neely, D.G.; Toris, C.B. Mechanism of Action of Selective Laser Trabeculoplasty and Predictors of Response. Invest Ophthalmol Vis Sci 2017, 58, 1462–1468. [Google Scholar] [CrossRef]
- Aquino, M.C.; Barton, K.; Tan, A.M.; Sng, C.; Li, X.; Loon, S.C.; Chew, P.T. Micropulse versus continuous wave transscleral diode cyclophotocoagulation in refractory glaucoma: a randomized exploratory study. Clin Exp Ophthalmol 2015, 43, 40–46. [Google Scholar] [CrossRef]
- Collotta, D.; Colletta, S.; Carlucci, V.; Fruttero, C.; Fea, A.M.; Collino, M. Pharmacological Approaches to Modulate the Scarring Process after Glaucoma Surgery. Pharmaceuticals 2023, 16, 898. [Google Scholar] [CrossRef] [PubMed]
- Kroese, M.; Burton, H. Primary open angle glaucoma. The need for a consensus case definition. J Epidemiol Community Health 2003, 57, 752–754. [Google Scholar] [CrossRef]
- McMonnies, C. Reactive oxygen species, oxidative stress, glaucoma and hyperbaric oxygen therapy. J Optom 2018, 11, 3–9. [Google Scholar] [CrossRef]
- Downs, J.C.; Roberts, M.D.; Burgoyne, C.F. Mechanical environment of the optic nerve head in glaucoma. Optom Vis Sci 2008, 85, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Flammer, J.; Orgül, S.; Costa, V.P.; Orzalesi, N.; Krieglstein, G.K.; Serra, L.M.; Renard, J.-P.; Stefánsson, E. The impact of ocular blood flow in glaucoma. Progress in Retinal and Eye Research 2002, 21, 359–393. [Google Scholar] [CrossRef] [PubMed]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid Med Cell Longev 2016, 2016, 3164734. [Google Scholar] [CrossRef] [PubMed]
- Llobet, A.; Gasull, X.; Gual, A. Understanding trabecular meshwork physiology: a key to the control of intraocular pressure? News Physiol Sci 2003, 18, 205–209. [Google Scholar] [CrossRef]
- Saccà, S.C.; Izzotti, A.; Rossi, P.; Traverso, C. Glaucomatous outflow pathway and oxidative stress. Exp Eye Res 2007, 84, 389–399. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; Yue, B.Y. Oxidative stress affects cytoskeletal structure and cell-matrix interactions in cells from an ocular tissue: the trabecular meshwork. J Cell Physiol 1999, 180, 182–189. [Google Scholar] [CrossRef]
- Pervan, C.L.; Lautz, J.D.; Blitzer, A.L.; Langert, K.A.; Stubbs, E.B. Rho GTPase signaling promotes constitutive expression and release of TGF-β2 by human trabecular meshwork cells. Experimental Eye Research 2016, 146, 95–102. [Google Scholar] [CrossRef]
- The effectiveness of intraocular pressure reduction in the treatment of normal-tension glaucoma. Collaborative Normal-Tension Glaucoma Study Group. Am J Ophthalmol 1998, 126, 498–505. [Google Scholar] [CrossRef]
- Trivli, A.; Koliarakis, I.; Terzidou, C.; Goulielmos, G.N.; Siganos, C.S.; Spandidos, D.A.; Dalianis, G.; Detorakis, E.T. Normal-tension glaucoma: Pathogenesis and genetics. Exp Ther Med 2019, 17, 563–574. [Google Scholar] [CrossRef]
- Tang, S.; Toda, Y.; Kashiwagi, K.; Mabuchi, F.; Iijima, H.; Tsukahara, S.; Yamagata, Z. The association between Japanese primary open-angle glaucoma and normal tension glaucoma patients and the optineurin gene. Hum Genet 2003, 113, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chen, X.; Zhang, H.; Li, N.; Yang, X.; Cheng, W.; Zhao, K. Association of OPA1 polymorphisms with NTG and HTG: a meta-analysis. PLoS One 2012, 7, e42387. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, J.Y.; Kim, D.M.; Ko, H.S.; Kim, S.Y.; Yoo, T.; Hwang, S.S.; Park, S.S. Investigations on the association between normal tension glaucoma and single nucleotide polymorphisms of the endothelin-1 and endothelin receptor genes. Mol Vis 2006, 12, 1016–1021. [Google Scholar]
- Stroman, G.A.; Stewart, W.C.; Golnik, K.C.; Curé, J.K.; Olinger, R.E. Magnetic resonance imaging in patients with low-tension glaucoma. Arch Ophthalmol 1995, 113, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.; Farinelli, A.; Billson, F.; Houang, M.; Stern, M. Comparative study of brain magnetic resonance imaging findings in patients with low-tension glaucoma and control subjects. Ophthalmology 1995, 102, 1632–1638. [Google Scholar] [CrossRef]
- Suzuki, J.; Tomidokoro, A.; Araie, M.; Tomita, G.; Yamagami, J.; Okubo, T.; Masumoto, T. Visual field damage in normal-tension glaucoma patients with or without ischemic changes in cerebral magnetic resonance imaging. Jpn J Ophthalmol 2004, 48, 340–344. [Google Scholar] [CrossRef]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog Retin Eye Res 2006, 25, 490–513. [Google Scholar] [CrossRef]
- Tezel, G.; Wax, M.B. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci 2000, 20, 8693–8700. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X. Caspase-independent component of retinal ganglion cell death, in vitro. Invest Ophthalmol Vis Sci 2004, 45, 4049–4059. [Google Scholar] [CrossRef]
- Nebbioso, M.; Franzone, F.; Lambiase, A.; Bonfiglio, V.; Limoli, P.G.; Artico, M.; Taurone, S.; Vingolo, E.M.; Greco, A.; Polimeni, A. Oxidative Stress Implication in Retinal Diseases-A Review. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Toda, N.; Nakanishi-Toda, M. Nitric oxide: ocular blood flow, glaucoma, and diabetic retinopathy. Prog Retin Eye Res 2007, 26, 205–238. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: regulation and function. Eur Heart J 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Moriya, S.; Oku, H.; Azuma, I. Association of endothelin-1 with normal tension glaucoma: clinical and fundamental studies. Surv Ophthalmol 1995, 39 Suppl 1, S49–56. [Google Scholar] [CrossRef]
- Cellini, M.; Possati, G.L.; Profazio, V.; Sbrocca, M.; Caramazza, N.; Caramazza, R. Color Doppler imaging and plasma levels of endothelin-1 in low-tension glaucoma. Acta Ophthalmol Scand Suppl 1997, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Sin, B.H.; Song, B.J.; Park, S.P. Aqueous vascular endothelial growth factor and endothelin-1 levels in branch retinal vein occlusion associated with normal tension glaucoma. J Glaucoma 2013, 22, 104–109. [Google Scholar] [CrossRef]
- Moore, D.; Harris, A.; Wudunn, D.; Kheradiya, N.; Siesky, B. Dysfunctional regulation of ocular blood flow: A risk factor for glaucoma? Clin Ophthalmol 2008, 2, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Galassi, F.; Giambene, B.; Varriale, R. Systemic vascular dysregulation and retrobulbar hemodynamics in normal-tension glaucoma. Invest Ophthalmol Vis Sci 2011, 52, 4467–4471. [Google Scholar] [CrossRef]
- Gericke, A.; Mann, C.; Zadeh, J.K.; Musayeva, A.; Wolff, I.; Wang, M.; Pfeiffer, N.; Daiber, A.; Li, H.; Xia, N.; et al. Elevated Intraocular Pressure Causes Abnormal Reactivity of Mouse Retinal Arterioles. Oxid Med Cell Longev 2019, 2019, 9736047. [Google Scholar] [CrossRef]
- Wang, M.; Liu, H.; Xia, N.; Li, H.; van Beers, T.; Gericke, A.; Prokosch, V. Intraocular Pressure-Induced Endothelial Dysfunction of Retinal Blood Vessels Is Persistent, but Does Not Trigger Retinal Ganglion Cell Loss. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Ju, W.K.; Kim, K.Y.; Lindsey, J.D.; Angert, M.; Duong-Polk, K.X.; Scott, R.T.; Kim, J.J.; Kukhmazov, I.; Ellisman, M.H.; Perkins, G.A.; et al. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Invest Ophthalmol Vis Sci 2008, 49, 4903–4911. [Google Scholar] [CrossRef]
- Bariş, M.; Tezel, G. Immunomodulation as a Neuroprotective Strategy for Glaucoma Treatment. Curr Ophthalmol Rep 2019, 7, 160–169. [Google Scholar] [CrossRef]
- Luo, C.; Yang, X.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Tezel, G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Invest Ophthalmol Vis Sci 2010, 51, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Luo, C.; Cai, J.; Powell, D.W. An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Invest Ophthalmol Vis Sci 2012, 53, 4220–4233. [Google Scholar] [CrossRef]
- Howell, G.R.; Macalinao, D.G.; Sousa, G.L.; Walden, M.; Soto, I.; Kneeland, S.C.; Barbay, J.M.; King, B.L.; Marchant, J.K.; Hibbs, M.; et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest 2011, 121, 1429–1444. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, K.; Krasnow, M.A. The hypoxic response: huffing and HIFing. Cell 1997, 89, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Rupin, A.; Paysant, J.; Sansilvestri-Morel, P.; Lembrez, N.; Lacoste, J.M.; Cordi, A.; Verbeuren, T.J. Role of NADPH oxidase-mediated superoxide production in the regulation of E-selectin expression by endothelial cells subjected to anoxia/reoxygenation. Cardiovasc Res 2004, 63, 323–330. [Google Scholar] [CrossRef]
- Mittal, M.; Roth, M.; König, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 2007, 101, 258–267. [Google Scholar] [CrossRef]
- Yokota, H.; Narayanan, S.P.; Zhang, W.; Liu, H.; Rojas, M.; Xu, Z.; Lemtalsi, T.; Nagaoka, T.; Yoshida, A.; Brooks, S.E.; et al. Neuroprotection from retinal ischemia/reperfusion injury by NOX2 NADPH oxidase deletion. Invest Ophthalmol Vis Sci 2011, 52, 8123–8131. [Google Scholar] [CrossRef]
- Kleikers, P.W.; Wingler, K.; Hermans, J.J.; Diebold, I.; Altenhöfer, S.; Radermacher, K.A.; Janssen, B.; Görlach, A.; Schmidt, H.H. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J Mol Med (Berl) 2012, 90, 1391–1406. [Google Scholar] [CrossRef]
- Kietzmann, T.; Görlach, A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol 2005, 16, 474–486. [Google Scholar] [CrossRef]
- Tezel, G.; Fourth, A.P.O.R.I.C.W.G. The role of glia, mitochondria, and the immune system in glaucoma. Invest Ophthalmol Vis Sci 2009, 50, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.-R.; Chae, H.-J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Experimental & Molecular Medicine 2021, 53, 151–167. [Google Scholar] [CrossRef]
- Kroeger, H.; Chiang, W.-C.; Felden, J.; Nguyen, A.; Lin, J.H. ER stress and unfolded protein response in ocular health and disease. The FEBS Journal 2019, 286, 399–412. [Google Scholar] [CrossRef]
- Hiramatsu, N.; Chiang, W.C.; Kurt, T.D.; Sigurdson, C.J.; Lin, J.H. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am J Pathol 2015, 185, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Hurley, D.J.; Normile, C.; Irnaten, M.; O'Brien, C. The Intertwined Roles of Oxidative Stress and Endoplasmic Reticulum Stress in Glaucoma. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annual Review of Pathology: Mechanisms of Disease 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem Sci 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Kaneko, M.; Niinuma, Y.; Nomura, Y. Activation Signal of Nuclear Factor-κB in Response to Endoplasmic Reticulum Stress is Transduced <i>via</i> IRE1 and Tumor Necrosis Factor Receptor-Associated Factor 2. Biological and Pharmaceutical Bulletin 2003, 26, 931–935. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Chen, A.C.-H.; Burr, L.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in respiratory disease. Clinical & Translational Immunology 2018, 7, e1019. [Google Scholar] [CrossRef]
- Martinon, F. Signaling by ROS drives inflammasome activation. European Journal of Immunology 2010, 40, 616–619. [Google Scholar] [CrossRef]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Kitsati, N.; Mantzaris, M.D.; Galaris, D. Hydroxytyrosol inhibits hydrogen peroxide-induced apoptotic signaling via labile iron chelation. Redox Biology 2016, 10, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-S.; Chang, Y.-C.; Chen, P.-H.; Li, C.-Y.; Wu, W.-C.; Kao, Y.-H. MicroRNA-100 Mediates Hydrogen Peroxide-Induced Apoptosis of Human Retinal Pigment Epithelium ARPE-19 Cells. Pharmaceuticals 2021, 14, 314. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Cho, K.S.; Thee, E.F.; Jager, M.J.; Chen, D.F. Neuroinflammation and microglia in glaucoma: time for a paradigm shift. J Neurosci Res 2019, 97, 70–76. [Google Scholar] [CrossRef]
- Mélik Parsadaniantz, S.; Réaux-le Goazigo, A.; Sapienza, A.; Habas, C.; Baudouin, C. Glaucoma: A Degenerative Optic Neuropathy Related to Neuroinflammation? Cells 2020, 9. [Google Scholar] [CrossRef]
- Martin, K.R.; Levkovitch-Verbin, H.; Valenta, D.; Baumrind, L.; Pease, M.E.; Quigley, H.A. Retinal glutamate transporter changes in experimental glaucoma and after optic nerve transection in the rat. Invest Ophthalmol Vis Sci 2002, 43, 2236–2243. [Google Scholar] [PubMed]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Williams, P.A.; Tribble, J.R.; Pepper, K.W.; Cross, S.D.; Morgan, B.P.; Morgan, J.E.; John, S.W.; Howell, G.R. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Molecular neurodegeneration 2016, 11, 1–13. [Google Scholar] [CrossRef]
- Howell, G.R.; Soto, I.; Ryan, M.; Graham, L.C.; Smith, R.S.; John, S.W. Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice. J Neuroinflammation 2013, 10, 76. [Google Scholar] [CrossRef]
- Bosco, A.; Anderson, S.R.; Breen, K.T.; Romero, C.O.; Steele, M.R.; Chiodo, V.A.; Boye, S.L.; Hauswirth, W.W.; Tomlinson, S.; Vetter, M.L. Complement C3-Targeted Gene Therapy Restricts Onset and Progression of Neurodegeneration in Chronic Mouse Glaucoma. Mol Ther 2018, 26, 2379–2396. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, O.W.; Beck, S.; von Thun und Hohenstein-Blaul, N.; Boehm, N.; Ziegler, A.; Vetter, J.M.; Pfeiffer, N.; Grus, F.H. Enhanced Insight into the Autoimmune Component of Glaucoma: IgG Autoantibody Accumulation and Pro-Inflammatory Conditions in Human Glaucomatous Retina. PLOS ONE 2013, 8, e57557. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.W.; Gülgün, T.; Junjie, Y.; Guanghua, P.; Rajkumar, V.P.; Neeraj, A.; Rebecca, M.S.; David, J.C. Induced Autoimmunity to Heat Shock Proteins Elicits Glaucomatous Loss of Retinal Ganglion Cell Neurons via Activated T-Cell-Derived Fas-Ligand. The Journal of Neuroscience 2008, 28, 12085. [Google Scholar] [CrossRef]
- Joachim, S.C.; Grus, F.H.; Pfeiffer, N. Analysis of Autoantibody Repertoires in Sera of Patients with Glaucoma. European Journal of Ophthalmology 2003, 13, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Joachim, S.C.; Reichelt, J.; Berneiser, S.; Pfeiffer, N.; Grus, F.H. Sera of glaucoma patients show autoantibodies against myelin basic protein and complex autoantibody profiles against human optic nerve antigens. Graefe's Archive for Clinical and Experimental Ophthalmology 2008, 246, 573–580. [Google Scholar] [CrossRef]
- Joachim, S.C.; Pfeiffer, N.; Grus, F.H. Autoantibodies in patients with glaucoma: a comparison of IgG serum antibodies against retinal, optic nerve, and optic nerve head antigens. Graefe's Archive for Clinical and Experimental Ophthalmology 2005, 243, 817–823. [Google Scholar] [CrossRef]
- Grus, F.H.; Joachim, S.C.; Hoffmann, E.M.; Pfeiffer, N. Complex autoantibody repertoires in patients with glaucoma. Mol Vis 2004, 10, 13–17. [Google Scholar]
- Joachim, S.C.; Wuenschig, D.; Pfeiffer, N.; Grus, F.H. IgG antibody patterns in aqueous humor of patients with primary open angle glaucoma and pseudoexfoliation glaucoma. Mol Vis 2007, 13, 1573–1579. [Google Scholar]
- Grus, F.; Joachim, S.; Wax, M.; Pfeiffer, N. Serum autoantibodies in glaucoma patients from Germany and the United States: further implications for autoimmune mechanisms in the neurodegenerative processes of glaucoma. Investigative Ophthalmology & Visual Science 2005, 46, 1285–1285. [Google Scholar]
- Wax, M.B.; Tezel, G.; Saito, I.; Gupta, R.S.; Harley, J.B.; Li, Z.; Romano, C. Anti-Ro/SS-A positivity and heat shock protein antibodies in patients with normal-pressure glaucoma. American journal of ophthalmology 1998, 125, 145–157. [Google Scholar] [CrossRef]
- Wax, M.B.; Barrett, D.A.; Pestronk, A. Increased incidence of paraproteinemia and autoantibodies in patients with normal-pressure glaucoma. American journal of ophthalmology 1994, 117, 561–568. [Google Scholar] [CrossRef]
- van Eden, W. Immune tolerance therapies for autoimmune diseases based on heat shock protein T-cell epitopes. Philosophical Transactions of the Royal Society B: Biological Sciences 2018, 373, 20160531. [Google Scholar] [CrossRef]
- Tezel, G.; Seigel, G.M.; Wax, M.B. Autoantibodies to small heat shock proteins in glaucoma. Investigative ophthalmology & visual science 1998, 39, 2277–2287. [Google Scholar]
- Tezel, G.; Wax, M.B. The Mechanisms of hsp27 Antibody-Mediated Apoptosis in Retinal Neuronal Cells. The Journal of Neuroscience 2000, 20, 3552–3562. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Hernandez, M.R.; Wax, M.B. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Archives of ophthalmology 2000, 118, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Wax, M.B.; Tezel, G.; Kawase, K.; Kitazawa, Y. Serum autoantibodies to heat shock proteins in glaucoma patients from Japan and the United States. Ophthalmology 2001, 108, 296–302. [Google Scholar] [CrossRef]
- Joachim, S.C.; Bruns, K.; Lackner, K.J.; Pfeiffer, N.; Grus, F.H. Antibodies to α B-crystallin, vimentin, and heat shock protein 70 in aqueous humor of patients with normal tension glaucoma and IgG antibody patterns against retinal antigen in aqueous humor. Current eye research 2007, 32, 501–509. [Google Scholar] [CrossRef]
- Von Thun Und Hohenstein-Blaul, N.; Bell, K.; Pfeiffer, N.; Grus, F.H. Autoimmune aspects in glaucoma. European Journal of Pharmacology 2016, 787, 105–118. [Google Scholar] [CrossRef]
- Bell, K.; Wilding, C.; Funke, S.; Pfeiffer, N.; Grus, F.H. Protective effect of 14-3-3 antibodies on stressed neuroretinal cells via the mitochondrial apoptosis pathway. BMC ophthalmology 2015, 15, 1–13. [Google Scholar] [CrossRef]
- Tezel, G. The immune response in glaucoma: A perspective on the roles of oxidative stress. Experimental Eye Research 2011, 93, 178–186. [Google Scholar] [CrossRef]
- Griffith, T.S.; Yu, X.; Herndon, J.M.; Green, D.R.; Ferguson, T.A. CD95-induced apoptosis of lymphocytes in an immune privileged site induces immunological tolerance. Immunity 1996, 5, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Murakami, Y.; Kataoka, K.; Notomi, S.; Mantopoulos, D.; Trichonas, G.; Miller, J.W.; Gregory, M.S.; Ksander, B.R.; Marshak-Rothstein, A.; et al. Membrane-bound and soluble Fas ligands have opposite functions in photoreceptor cell death following separation from the retinal pigment epithelium. Cell Death Dis 2015, 6, e1986. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Fei, F.; Jones, A.; Busto, P.; Marshak-Rothstein, A.; Ksander, B.R.; Gregory-Ksander, M. Overexpression of Soluble Fas Ligand following Adeno-Associated Virus Gene Therapy Prevents Retinal Ganglion Cell Death in Chronic and Acute Murine Models of Glaucoma. J Immunol 2016, 197, 4626–4638. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Kocab, A.J.; Zacks, D.N.; Marshak-Rothstein, A.; Gregory-Ksander, M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. Journal of Neuroinflammation 2019, 16, 184. [Google Scholar] [CrossRef]
- Brothers, H.M.; Marchalant, Y.; Wenk, G.L. Caffeine attenuates lipopolysaccharide-induced neuroinflammation. Neuroscience Letters 2010, 480, 97–100. [Google Scholar] [CrossRef]
- Madeira, M.H.; Boia, R.; Elvas, F.; Martins, T.; Cunha, R.A.; Ambrósio, A.F.; Santiago, A.R. Selective A2A receptor antagonist prevents microglia-mediated neuroinflammation and protects retinal ganglion cells from high intraocular pressure–induced transient ischemic injury. Translational Research 2016, 169, 112–128. [Google Scholar] [CrossRef]
- Madeira, M.H.; Ortin-Martinez, A.; Nadal-Nícolas, F.; Ambrósio, A.F.; Vidal-Sanz, M.; Agudo-Barriuso, M.; Santiago, A.R. Caffeine administration prevents retinal neuroinflammation and loss of retinal ganglion cells in an animal model of glaucoma. Scientific Reports 2016, 6, 27532. [Google Scholar] [CrossRef]
- Jia, Y.; Jiang, S.; Chen, C.; Lu, G.; Xie, Y.; Sun, X.; Huang, L. Caffeic acid phenethyl ester attenuates nuclear factor-κB-mediated inflammatory responses in Müller cells and protects against retinal ganglion cell death. Mol Med Rep 2019, 19, 4863–4871. [Google Scholar] [CrossRef]
- Park, C.W.; Han, C.T.; Sakaguchi, Y.; Lee, J.; Youn, H.Y. Safety evaluation of FM101, an A3 adenosine receptor modulator, in rat, for developing as therapeutics of glaucoma and hepatitis. Excli j 2020, 19, 187–200. [Google Scholar] [CrossRef]
- Ramiro, S.; Radner, H.; van der Heijde, D.; Van Tubergen, A.; Buchbinder, R.; Aletaha, D.; Landewé, R.B. Combination therapy for pain management in inflammatory arthritis (rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, other spondyloarthritis). Cochrane database of systematic reviews 2011. [Google Scholar] [CrossRef]
- Roh, M.; Zhang, Y.; Murakami, Y.; Thanos, A.; Lee, S.C.; Vavvas, D.G.; Benowitz, L.I.; Miller, J.W. Etanercept, a widely used inhibitor of tumor necrosis factor-α (TNF-α), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS One 2012, 7, e40065. [Google Scholar] [CrossRef]
- Hashida, N.; Ohguro, N.; Nishida, K. Efficacy and Complications of Intravitreal Rituximab Injection for Treating Primary Vitreoretinal Lymphoma. Translational Vision Science & Technology 2012, 1, 1–1. [Google Scholar] [CrossRef]
- Li, L.; Liu, Q.; Shi, L.; Zhou, X.; Wu, W.; Wang, X.; Wang, L.; Wu, Z. Baicalin prevents fibrosis of human trabecular meshwork cells via inhibiting the MyD88/NF-κB pathway. Eur J Pharmacol 2023, 938, 175425. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zeng, Q.; Barış, M.; Tezel, G. Transgenic inhibition of astroglial NF-κB restrains the neuroinflammatory and neurodegenerative outcomes of experimental mouse glaucoma. J Neuroinflammation 2020, 17, 252. [Google Scholar] [CrossRef] [PubMed]
- Harari, O.A.; Liao, J.K. NF-κB and innate immunity in ischemic stroke. Ann N Y Acad Sci 2010, 1207, 32–40. [Google Scholar] [CrossRef]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat Immunol 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Yanagidaira, T.; Takahashi, Y.; Sano, K.; Murata, T.; Hayashi, T. Development of spontaneous neuropathy in NF-κBp50-deficient mice by calcineurin-signal involving impaired NF-κB activation. Mol Vis 2011, 17, 2157–2170. [Google Scholar]
- Becker, S.; Reinehr, S.; Dick, H.B.; Joachim, S.C. [Complement activation after induction of ocular hypertension in an animal model]. Ophthalmologe 2015, 112, 41–48. [Google Scholar] [CrossRef]
- Kuehn, M.H.; Kim, C.Y.; Ostojic, J.; Bellin, M.; Alward, W.L.; Stone, E.M.; Sakaguchi, D.S.; Grozdanic, S.D.; Kwon, Y.H. Retinal synthesis and deposition of complement components induced by ocular hypertension. Exp Eye Res 2006, 83, 620–628. [Google Scholar] [CrossRef]
- Howell, G.R.; MacNicoll, K.H.; Braine, C.E.; Soto, I.; Macalinao, D.G.; Sousa, G.L.; John, S.W. Combinatorial targeting of early pathways profoundly inhibits neurodegeneration in a mouse model of glaucoma. Neurobiol Dis 2014, 71, 44–52. [Google Scholar] [CrossRef]
- Reinehr, S.; Gomes, S.C.; Gassel, C.J.; Asaad, M.A.; Stute, G.; Schargus, M.; Dick, H.B.; Joachim, S.C. Intravitreal Therapy Against the Complement Factor C5 Prevents Retinal Degeneration in an Experimental Autoimmune Glaucoma Model. Front Pharmacol 2019, 10, 1381. [Google Scholar] [CrossRef] [PubMed]
- Gibson, L.C.D.; Hastings, S.F.; McPhee, I.; Clayton, R.A.; Darroch, C.E.; Mackenzie, A.; MacKenzie, F.L.; Nagasawa, M.; Stevens, P.A.; MacKenzie, S.J. The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. European Journal of Pharmacology 2006, 538, 39–42. [Google Scholar] [CrossRef]
- Belforte, N.; Cueva-Vargas, J.L.; Di Polo, A. The phosphodiesterase inhibitor Ibudilast attenuates glial cell reactivity, production of proinflammatory cytokines and neuronal loss in experimental glaucoma. Investigative Ophthalmology & Visual Science 2014, 55, 2665–2665. [Google Scholar]
- Rodgers, K.M.; Deming, Y.K.; Bercum, F.M.; Chumachenko, S.Y.; Wieseler, J.L.; Johnson, K.W.; Watkins, L.R.; Barth, D.S. Reversal of established traumatic brain injury-induced, anxiety-like behavior in rats after delayed, post-injury neuroimmune suppression. J Neurotrauma 2014, 31, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Ruzafa, N.; Vecino, E. Efecto de las células de Müller en la supervivencia y neuritogénesis de las células ganglionares de la retina. Archivos de la Sociedad Española de Oftalmología 2015, 90, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Cueva Vargas, J.L.; Belforte, N.; Di Polo, A. The glial cell modulator ibudilast attenuates neuroinflammation and enhances retinal ganglion cell viability in glaucoma through protein kinase A signaling. Neurobiol Dis 2016, 93, 156–171. [Google Scholar] [CrossRef]
- Kerr, N.M.; Danesh-Meyer, H.V. Phosphodiesterase inhibitors and the eye. Clin Exp Ophthalmol 2009, 37, 514–523. [Google Scholar] [CrossRef]
- Sela, T.C.; Zahavi, A.; Friedman-Gohas, M.; Weiss, S.; Sternfeld, A.; Ilguisonis, A.; Badash, D.; Geffen, N.; Ofri, R.; BarKana, Y.; et al. Azithromycin and Sildenafil May Have Protective Effects on Retinal Ganglion Cells via Different Pathways: Study in a Rodent Microbead Model. Pharmaceuticals (Basel) 2023, 16. [Google Scholar] [CrossRef]
- Storgaard, L.; Tran, T.L.; Freiberg, J.C.; Hauser, A.S.; Kolko, M. Glaucoma Clinical Research: Trends in Treatment Strategies and Drug Development. Frontiers in Medicine 2021, 8. [Google Scholar] [CrossRef]
- Abcouwer, S.F.; Lin, C.-m.; Shanmugam, S.; Muthusamy, A.; Barber, A.J.; Antonetti, D.A. Minocycline prevents retinal inflammation and vascular permeability following ischemia-reperfusion injury. Journal of Neuroinflammation 2013, 10, 913. [Google Scholar] [CrossRef]
- Levkovitch-Verbin, H.; Kalev-Landoy, M.; Habot-Wilner, Z.; Melamed, S. Minocycline Delays Death of Retinal Ganglion Cells in Experimental Glaucoma and After Optic Nerve Transection. Archives of Ophthalmology 2006, 124, 520–526. [Google Scholar] [CrossRef]
- Bosco, A.; Inman, D.M.; Steele, M.R.; Wu, G.; Soto, I.; Marsh-Armstrong, N.; Hubbard, W.C.; Calkins, D.J.; Horner, P.J.; Vetter, M.L. Reduced Retina Microglial Activation and Improved Optic Nerve Integrity with Minocycline Treatment in the DBA/2J Mouse Model of Glaucoma. Investigative Ophthalmology & Visual Science 2008, 49, 1437–1446. [Google Scholar] [CrossRef]
- Bordone, M.P.; González Fleitas, M.F.; Pasquini, L.A.; Bosco, A.; Sande, P.H.; Rosenstein, R.E.; Dorfman, D. Involvement of microglia in early axoglial alterations of the optic nerve induced by experimental glaucoma. Journal of Neurochemistry 2017, 142, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Grotegut, P.; Perumal, N.; Kuehn, S.; Smit, A.; Dick, H.B.; Grus, F.H.; Joachim, S.C. Minocycline reduces inflammatory response and cell death in a S100B retina degeneration model. J Neuroinflammation 2020, 17, 375. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhong, H.; Sun, J.; Li, N.; Chen, J.; Shen, B.; Huang, P.; Shen, X.; Huang, S.; Zhong, Y. Molecular signaling from microglia impacts macroglia autophagy and neurons survival in glaucoma. iScience 2023, 26, 106839. [Google Scholar] [CrossRef]
- Levkovitch-Verbin, H.; Waserzoog, Y.; Vander, S.; Makarovsky, D.; Ilia, P. Minocycline mechanism of neuroprotection involves the Bcl-2 gene family in optic nerve transection. Int J Neurosci 2014, 124, 755–761. [Google Scholar] [CrossRef]
- Kernt, M.; Neubauer, A.S.; Eibl, K.H.; Wolf, A.; Ulbig, M.W.; Kampik, A.; Hirneiss, C. Minocycline is cytoprotective in human trabecular meshwork cells and optic nerve head astrocytes by increasing expression of XIAP, survivin, and Bcl-2. Clin Ophthalmol 2010, 4, 591–604. [Google Scholar] [CrossRef]
- Cramer, C.L.; Patterson, A.; Alchakaki, A.; Soubani, A.O. Immunomodulatory indications of azithromycin in respiratory disease: a concise review for the clinician. Postgrad Med 2017, 129, 493–499. [Google Scholar] [CrossRef]
- Varano, G.P.; Parisi, V.; Adornetto, A.; Cavaliere, F.; Amantea, D.; Nucci, C.; Corasaniti, M.T.; Morrone, L.A.; Bagetta, G.; Russo, R. Post-ischemic treatment with azithromycin protects ganglion cells against retinal ischemia/reperfusion injury in the rat. Mol Vis 2017, 23, 911–921. [Google Scholar]
- Mead, B.; Tomarev, S. Bone Marrow-Derived Mesenchymal Stem Cells-Derived Exosomes Promote Survival of Retinal Ganglion Cells Through miRNA-Dependent Mechanisms. Stem Cells Transl Med 2017, 6, 1273–1285. [Google Scholar] [CrossRef]
- Mead, B.; Amaral, J.; Tomarev, S. Mesenchymal Stem Cell-Derived Small Extracellular Vesicles Promote Neuroprotection in Rodent Models of Glaucoma. Invest Ophthalmol Vis Sci 2018, 59, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Jaganathan, B.G. Stem Cell Therapy for Retinal Degeneration: The Evidence to Date. Biologics 2021, 15, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Vilela, C.A.P.; Messias, A.; Calado, R.T.; Siqueira, R.C.; Silva, M.J.L.; Covas, D.T.; Paula, J.S. Retinal function after intravitreal injection of autologous bone marrow-derived mesenchymal stromal cells in advanced glaucoma. Doc Ophthalmol 2021, 143, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Reboussin, É.; Buffault, J.; Brignole-Baudouin, F.; Réaux-Le Goazigo, A.; Riancho, L.; Olmiere, C.; Sahel, J.-A.; Mélik Parsadaniantz, S.; Baudouin, C. Evaluation of neuroprotective and immunomodulatory properties of mesenchymal stem cells in an ex vivo retinal explant model. Journal of Neuroinflammation 2022, 19, 63. [Google Scholar] [CrossRef]
- Chen, H.; Cho, K.S.; Vu, T.H.K.; Shen, C.H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat Commun 2018, 9, 3209. [Google Scholar] [CrossRef]
- Astafurov, K.; Elhawy, E.; Ren, L.; Dong, C.Q.; Igboin, C.; Hyman, L.; Griffen, A.; Mittag, T.; Danias, J. Oral microbiome link to neurodegeneration in glaucoma. PLoS One 2014, 9, e104416. [Google Scholar] [CrossRef]
- Nayyar, A.; Gindina, S.; Barron, A.; Hu, Y.; Danias, J. Do epigenetic changes caused by commensal microbiota contribute to development of ocular disease? A review of evidence. Human Genomics 2020, 14, 11. [Google Scholar] [CrossRef]
- Lewis, S.S.; Loram, L.C.; Hutchinson, M.R.; Li, C.M.; Zhang, Y.; Maier, S.F.; Huang, Y.; Rice, K.C.; Watkins, L.R. (+)-naloxone, an opioid-inactive toll-like receptor 4 signaling inhibitor, reverses multiple models of chronic neuropathic pain in rats. J Pain 2012, 13, 498–506. [Google Scholar] [CrossRef]
- Nakano, Y.; Shimazawa, M.; Ojino, K.; Izawa, H.; Takeuchi, H.; Inoue, Y.; Tsuruma, K.; Hara, H. Toll-like receptor 4 inhibitor protects against retinal ganglion cell damage induced by optic nerve crush in mice. J Pharmacol Sci 2017, 133, 176–183. [Google Scholar] [CrossRef]
- Liang, L.; Zhu, M.N.; Chen, B.J.; Wang, Z.; He, L.Y.; Zhang, R. Inhibitive effect of TAK-242 on Tenon's capsule fibroblasts proliferation in rat eyes. Int J Ophthalmol 2019, 12, 1699–1707. [Google Scholar] [CrossRef]
- Sharma, T.P.; Curry, S.; McDowell, C.M. Effects of Toll-Like Receptor 4 Inhibition on Transforming Growth Factor-β2 Signaling in the Human Trabecular Meshwork. J Ocul Pharmacol Ther 2020, 36, 170–178. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wu, J.; Wang, J.; Piri, N.; Chen, F.; Xiao, T.; Zhao, Y.; Sun, D.; Kaplan, H.J.; Shao, H. Short chain fatty acids inhibit endotoxin-induced uveitis and inflammatory responses of retinal astrocytes. Exp Eye Res 2021, 206, 108520. [Google Scholar] [CrossRef]
- Kamat, J.P.; Devasagayam, T.P. Nicotinamide (vitamin B3) as an effective antioxidant against oxidative damage in rat brain mitochondria. Redox Rep 1999, 4, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.I.; Kim, Y.C.; Park, C.K. Dietary Niacin and Open-Angle Glaucoma: The Korean National Health and Nutrition Examination Survey. Nutrients 2018, 10. [Google Scholar] [CrossRef]
- Williams, P.A.; Harder, J.M.; John, S.W.M. Glaucoma as a Metabolic Optic Neuropathy: Making the Case for Nicotinamide Treatment in Glaucoma. J Glaucoma 2017, 26, 1161–1168. [Google Scholar] [CrossRef]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin Exp Ophthalmol 2020, 48, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health-promoting effects of astaxanthin: a high-value carotenoid mostly from microalgae. Mol Nutr Food Res 2011, 55, 150–165. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, R.; Kumari, A.; Panwar, A. Astaxanthin: A super antioxidant from microalgae and its therapeutic potential. J Basic Microbiol 2022, 62, 1064–1082. [Google Scholar] [CrossRef]
- Cort, A.; Ozturk, N.; Akpinar, D.; Unal, M.; Yucel, G.; Ciftcioglu, A.; Yargicoglu, P.; Aslan, M. Suppressive effect of astaxanthin on retinal injury induced by elevated intraocular pressure. Regul Toxicol Pharmacol 2010, 58, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Dong, Z.; Shinmei, Y.; Murata, M.; Kanda, A.; Noda, K.; Harada, T.; Ishida, S. Cytoprotective Effect of Astaxanthin in a Model of Normal Intraocular Pressure Glaucoma. J Ophthalmol 2020, 2020, 9539681. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Q.; Chu, C.; Liu, S. Astaxanthin protects retinal ganglion cells from acute glaucoma via the Nrf2/HO-1 pathway. J Chem Neuroanat 2020, 110, 101876. [Google Scholar] [CrossRef] [PubMed]
- Galiniak, S.; Aebisher, D.; Bartusik-Aebisher, D. Health benefits of resveratrol administration. Acta Biochim Pol 2019, 66, 13–21. [Google Scholar] [CrossRef]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of Human SIRT1 Activation by Resveratrol*. Journal of Biological Chemistry 2005, 280, 17187–17195. [Google Scholar] [CrossRef]
- Ciccone, L.; Piragine, E.; Brogi, S.; Camodeca, C.; Fucci, R.; Calderone, V.; Nencetti, S.; Martelli, A.; Orlandini, E. Resveratrol-like Compounds as SIRT1 Activators. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Pirhan, D.; Yüksel, N.; Emre, E.; Cengiz, A.; Kürşat Yıldız, D. Riluzole- and Resveratrol-Induced Delay of Retinal Ganglion Cell Death in an Experimental Model of Glaucoma. Curr Eye Res 2016, 41, 59–69. [Google Scholar] [CrossRef]
- Ye, M.J.; Meng, N. Resveratrol acts via the mitogen-activated protein kinase (MAPK) pathway to protect retinal ganglion cells from apoptosis induced by hydrogen peroxide. Bioengineered 2021, 12, 4878–4886. [Google Scholar] [CrossRef]
- Chronopoulos, P.; Manicam, C.; Zadeh, J.K.; Laspas, P.; Unkrig, J.C.; Göbel, M.L.; Musayeva, A.; Pfeiffer, N.; Oelze, M.; Daiber, A.; et al. Effects of Resveratrol on Vascular Function in Retinal Ischemia-Reperfusion Injury. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Inman, D.M.; Lambert, W.S.; Calkins, D.J.; Horner, P.J. α-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS One 2013, 8, e65389. [Google Scholar] [CrossRef]
- Sanz-González, S.M.; Raga-Cervera, J.; Aguirre Lipperheide, M.; Zanón-Moreno, V.; Chiner, V.; Ramírez, A.I.; Pinazo-Durán, M.D. Effect of an oral supplementation with a formula containing R-lipoic acid in glaucoma patients. Arch Soc Esp Oftalmol (Engl Ed) 2020, 95, 120–129. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J Med Chem 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.K.; Mo, B.; Zhao, J.; Yu, Y.J.; Liu, L.; Yue, C.L.; Liu, W. Neuroprotective effect of curcumin against oxidative damage in BV-2 microglia and high intraocular pressure animal model. J Ocul Pharmacol Ther 2014, 30, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Buccarello, L.; Dragotto, J.; Hassanzadeh, K.; Maccarone, R.; Corbo, M.; Feligioni, M. Retinal ganglion cell loss in an ex vivo mouse model of optic nerve cut is prevented by curcumin treatment. Cell Death Discov 2021, 7, 394. [Google Scholar] [CrossRef]
- Liu, X.G.; Wu, S.Q.; Li, P.; Yang, H. Advancement in the chemical analysis and quality control of flavonoid in Ginkgo biloba. J Pharm Biomed Anal 2015, 113, 212–225. [Google Scholar] [CrossRef]
- Yu, H.; Dong, L.-H.; Zhang, Y.; Liu, Q. A network pharmacology-based strategy for predicting the protective mechanism of Ginkgo biloba on damaged retinal ganglion cells. Chinese Journal of Natural Medicines 2022, 20, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Anne-Marie, B.; Beatriz, V.; Ana Isabel, J.; Covadonga, P. Managing Intraocular Pressure: Innovation in Glaucoma Management. In Glaucoma, Parul, I., Ed.; IntechOpen: Rijeka, 2016. [Google Scholar]
- Lee, D.; Shim, M.S.; Kim, K.Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Invest Ophthalmol Vis Sci 2014, 55, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Quaranta, L.; Riva, I.; Biagioli, E.; Rulli, E.; Rulli, E.; Poli, D.; Legramandi, L. Evaluating the Effects of an Ophthalmic Solution of Coenzyme Q10 and Vitamin E in Open-Angle Glaucoma Patients: A Study Protocol. Adv Ther 2019, 36, 2506–2514. [Google Scholar] [CrossRef]
- Laspas, P.; Zhutdieva, M.B.; Brochhausen, C.; Musayeva, A.; Zadeh, J.K.; Pfeiffer, N.; Xia, N.; Li, H.; Wess, J.; Gericke, A. The M(1) muscarinic acetylcholine receptor subtype is important for retinal neuron survival in aging mice. Sci Rep 2019, 9, 5222. [Google Scholar] [CrossRef]
- Pereira, S.P.; Medina, S.V.; Araujo, E.G. Cholinergic activity modulates the survival of retinal ganglion cells in culture: the role of M1 muscarinic receptors. Int J Dev Neurosci 2001, 19, 559–567. [Google Scholar] [CrossRef]
- Zhou, W.; Zhu, X.; Zhu, L.; Cui, Y.Y.; Wang, H.; Qi, H.; Ren, Q.S.; Chen, H.Z. Neuroprotection of muscarinic receptor agonist pilocarpine against glutamate-induced apoptosis in retinal neurons. Cell Mol Neurobiol 2008, 28, 263–275. [Google Scholar] [CrossRef]
- Almasieh, M.; Zhou, Y.; Kelly, M.E.; Casanova, C.; Di Polo, A. Structural and functional neuroprotection in glaucoma: role of galantamine-mediated activation of muscarinic acetylcholine receptors. Cell Death Dis 2010, 1, e27. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Dong, W.P.; Tang, Y.B.; Chen, H.Z.; Cui, Y.Y.; Bian, X.L. Huperzine A lowers intraocular pressure via the M3 mAChR and provides retinal neuroprotection via the M1 mAChR: a promising agent for the treatment of glaucoma. Ann Transl Med 2021, 9, 332. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Guo, X.; Noro, T.; Harada, C.; Tanaka, K.; Namekata, K.; Harada, T. Valproic acid prevents retinal degeneration in a murine model of normal tension glaucoma. Neurosci Lett 2015, 588, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Tribble, J.R.; Kastanaki, E.; Uslular, A.B.; Rutigliani, C.; Enz, T.J.; Williams, P.A. Valproic Acid Reduces Neuroinflammation to Provide Retinal Ganglion Cell Neuroprotection in the Retina Axotomy Model. Front Cell Dev Biol 2022, 10, 903436. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, K.; Chaurasia, A.K.; Gowtham, L.; Gupta, S.; Somarajan, B.I.; Velpandian, T.; Sihota, R.; Gupta, V. Therapeutic potential of valproic acid in advanced glaucoma: A pilot study. Indian J Ophthalmol 2018, 66, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Harada, C.; Noro, T.; Kimura, A.; Guo, X.; Namekata, K.; Nakano, T.; Harada, T. Suppression of Oxidative Stress as Potential Therapeutic Approach for Normal Tension Glaucoma. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, G.; Tolun, F.I.; Gul, M.; Imrek, S. Retinal Oxidative Stress Induced by Intraocular Hypertension in Rats May be Ameliorated by Brimonidine Treatment and N-acetyl Cysteine Supplementation. Journal of Glaucoma 2009, 18, 662–665. [Google Scholar] [CrossRef]
- Sano, H.; Namekata, K.; Kimura, A.; Shitara, H.; Guo, X.; Harada, C.; Mitamura, Y.; Harada, T. Differential effects of N-acetylcysteine on retinal degeneration in two mouse models of normal tension glaucoma. Cell Death Dis 2019, 10, 75. [Google Scholar] [CrossRef]
- Yang, L.; Tan, P.; Zhou, W.; Zhu, X.; Cui, Y.; Zhu, L.; Feng, X.; Qi, H.; Zheng, J.; Gu, P.; et al. N-acetylcysteine protects against hypoxia mimetic-induced autophagy by targeting the HIF-1α pathway in retinal ganglion cells. Cell Mol Neurobiol 2012, 32, 1275–1285. [Google Scholar] [CrossRef]
- Lapchak, P.A. A critical assessment of edaravone acute ischemic stroke efficacy trials: is edaravone an effective neuroprotective therapy? Expert Opin Pharmacother 2010, 11, 1753–1763. [Google Scholar] [CrossRef]
- Masuda, T.; Shimazawa, M.; Hara, H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxid Med Cell Longev 2017, 2017, 9208489. [Google Scholar] [CrossRef] [PubMed]
- Akaiwa, K.; Namekata, K.; Azuchi, Y.; Guo, X.; Kimura, A.; Harada, C.; Mitamura, Y.; Harada, T. Edaravone suppresses retinal ganglion cell death in a mouse model of normal tension glaucoma. Cell Death Dis 2017, 8, e2934. [Google Scholar] [CrossRef] [PubMed]
- Aksar, A.T.; Yuksel, N.; Gok, M.; Cekmen, M.; Caglar, Y. Neuroprotective effect of edaravone in experimental glaucoma model in rats: a immunofluorescence and biochemical analysis. Int J Ophthalmol 2015, 8, 239–244. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin Protects against Neuron Death in In Vitro andIn Vivo Models of Parkinson's Disease. Journal of Neuroscience 2010, 30, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Majumder, S.; Deng, J.J.; Bai, Y.; Thornton, F.B.; Oddo, S. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. Journal of Biological Chemistry 2009, 284, 27416–27424. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Li, Z.; Jia, Y.; Zhuo, Y. Rapamycin is neuroprotective in a rat chronic hypertensive glaucoma model. PLoS One 2014, 9, e99719. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Kwon, Y.W.; Kondo, N.; Bai, J.; Masutani, H.; Nakamura, H.; Fujii, J.; Ohira, A.; Yodoi, J. Cytoprotective effects of geranylgeranylacetone against retinal photooxidative damage. J Neurosci 2005, 25, 2396–2404. [Google Scholar] [CrossRef]
- Dong, Z.; Shinmei, Y.; Dong, Y.; Inafuku, S.; Fukuhara, J.; Ando, R.; Kitaichi, N.; Kanda, A.; Tanaka, K.; Noda, K.; et al. Effect of geranylgeranylacetone on the protection of retinal ganglion cells in a mouse model of normal tension glaucoma. Heliyon 2016, 2, e00191. [Google Scholar] [CrossRef]
- Li, X.; Leng, Y.; Jiang, Q.; Wang, Z.; Luo, P.; Zhang, C.; Chen, L.; Wang, Y.; Wang, H.; Yue, X.; et al. Eye Drops of Metformin Prevents Fibrosis After Glaucoma Filtration Surgery in Rats via Activating AMPK/Nrf2 Signaling Pathway. Front Pharmacol 2020, 11, 1038. [Google Scholar] [CrossRef]
- Gao, Z.; Li, M.; Yao, F.; Xia, X.; Duan, T.; Meng, J.; Huang, Y.; He, Y.; Saro, A.; Huang, J. Valdecoxib Protects against Cell Apoptosis Induced by Endoplasmic Reticulum Stress via the Inhibition of PERK-ATF4-CHOP Pathway in Experimental Glaucoma. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Feillet, F.; Leonard, J.V. Alternative pathway therapy for urea cycle disorders. J Inherit Metab Dis 1998, 21 Suppl 1, 101–111. [Google Scholar] [CrossRef]
- Brown, C.R.; Hong-Brown, L.Q.; Biwersi, J.; Verkman, A.S.; Welch, W.J. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1996, 1, 117–125. [Google Scholar] [CrossRef]
- Roy, A.; Ghosh, A.; Jana, A.; Liu, X.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Sodium phenylbutyrate controls neuroinflammatory and antioxidant activities and protects dopaminergic neurons in mouse models of Parkinson's disease. PLoS One 2012, 7, e38113. [Google Scholar] [CrossRef]
- Dong, Y.; Li, L.; Xia, T.; Wang, L.; Xiao, L.; Ding, N.; Wu, Y.; Lu, K. Oxidative Stress Can Be Attenuated by 4-PBA Caused by High-Fat or Ammonia Nitrogen in Cultured Spotted Seabass: The Mechanism Is Related to Endoplasmic Reticulum Stress. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Zode, G.S.; Kuehn, M.H.; Nishimura, D.Y.; Searby, C.C.; Mohan, K.; Grozdanic, S.D.; Bugge, K.; Anderson, M.G.; Clark, A.F.; Stone, E.M.; et al. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J Clin Invest 2011, 121, 3542–3553. [Google Scholar] [CrossRef] [PubMed]
- Maddineni, P.; Kasetti, R.B.; Kodati, B.; Yacoub, S.; Zode, G.S. Sodium 4-Phenylbutyrate Reduces Ocular Hypertension by Degrading Extracellular Matrix Deposition via Activation of MMP9. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Fan Gaskin, J.C.; Shah, M.H.; Chan, E.C. Oxidative Stress and the Role of NADPH Oxidase in Glaucoma. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Deliyanti, D.; Wilkinson-Berka, J.L. Inhibition of NOX1/4 with GKT137831: a potential novel treatment to attenuate neuroglial cell inflammation in the retina. J Neuroinflammation 2015, 12, 136. [Google Scholar] [CrossRef]
- Dionysopoulou, S.; Wikström, P.; Walum, E.; Thermos, K. Effect of NADPH oxidase inhibitors in an experimental retinal model of excitotoxicity. Exp Eye Res 2020, 200, 108232. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Inoue, T.; Ohira, S.; Awai-Kasaoka, N.; Kameda, T.; Inoue-Mochita, M.; Tanihara, H. Inhibition of Rho Kinase Induces Antioxidative Molecules and Suppresses Reactive Oxidative Species in Trabecular Meshwork Cells. Journal of Ophthalmology 2017, 2017, 7598140. [Google Scholar] [CrossRef]
- Chen, W.; Yang, X.; Fang, J.; Zhang, Y.; Zhu, W.; Yang, X. Rho-Associated Protein Kinase Inhibitor Treatment Promotes Proliferation and Phagocytosis in Trabecular Meshwork Cells. Front Pharmacol 2020, 11, 302. [Google Scholar] [CrossRef]
- Kamiya, T.; Omae, T.; Nakabayashi, S.; Takahashi, K.; Tanner, A.; Yoshida, A. Effect of Rho Kinase Inhibitor Ripasudil (K-115) on Isolated Porcine Retinal Arterioles. J Ocul Pharmacol Ther 2021, 37, 104–111. [Google Scholar] [CrossRef]
- Aslan, M.; Dogan, S.; Kucuksayan, E. Oxidative stress and potential applications of free radical scavengers in glaucoma. Redox Rep 2013, 18, 76–87. [Google Scholar] [CrossRef]
- Sanz-Morello, B.; Ahmadi, H.; Vohra, R.; Saruhanian, S.; Freude, K.K.; Hamann, S.; Kolko, M. Oxidative Stress in Optic Neuropathies. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Noro, T.; Harada, C.; Harada, T. Targeting Oxidative Stress for Treatment of Glaucoma and Optic Neuritis. Oxid Med Cell Longev 2017, 2017, 2817252. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.J.; Chen, Y.N.; Tsao, Y.T.; Cheng, C.M.; Wu, W.C.; Chen, H.C. The Pathomechanism, Antioxidant Biomarkers, and Treatment of Oxidative Stress-Related Eye Diseases. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Shu, D.Y.; Chaudhary, S.; Cho, K.-S.; Lennikov, A.; Miller, W.P.; Thorn, D.C.; Yang, M.; McKay, T.B. Role of Oxidative Stress in Ocular Diseases: A Balancing Act. Metabolites 2023, 13, 187. [Google Scholar] [CrossRef]
- Wubben, T.J.; Besirli, C.G.; Johnson, M.W.; Zacks, D.N. Retinal Neuroprotection: Overcoming the Translational Roadblocks. American Journal of Ophthalmology 2018, 192, xv. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.E.H.; Guo, L.; Hill, D.; Yap, T.E.; Cordeiro, M.F. Neuroprotective Strategies in Glaucoma - Translation to Clinical Trials. OBM Neurobiology 2020, 04, 062. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Mehta, C. Adaptive Designs for Clinical Trials. New England Journal of Medicine 2016, 375, 65–74. [Google Scholar] [CrossRef]
Figure 1.
Classification of glaucoma. Glaucoma suspect indicates suggestive, but without diagnostic evidence for glaucoma; OHT: ocular hypertension; OAG: open-angle glaucoma; PACG: primary angle-closure glaucoma; POAG: primary open-angle glaucoma; elevated IOP: intraocular pressure values ≥ 97.5th percentile for the population under consideration; normal IOP: intraocular pressure values < 97.5th percentile for the population under consideration; NTG: normal tension glaucoma; SOAG: secondary open-angle glaucoma; SACG: secondary angle-closure glaucoma.
Figure 1.
Classification of glaucoma. Glaucoma suspect indicates suggestive, but without diagnostic evidence for glaucoma; OHT: ocular hypertension; OAG: open-angle glaucoma; PACG: primary angle-closure glaucoma; POAG: primary open-angle glaucoma; elevated IOP: intraocular pressure values ≥ 97.5th percentile for the population under consideration; normal IOP: intraocular pressure values < 97.5th percentile for the population under consideration; NTG: normal tension glaucoma; SOAG: secondary open-angle glaucoma; SACG: secondary angle-closure glaucoma.
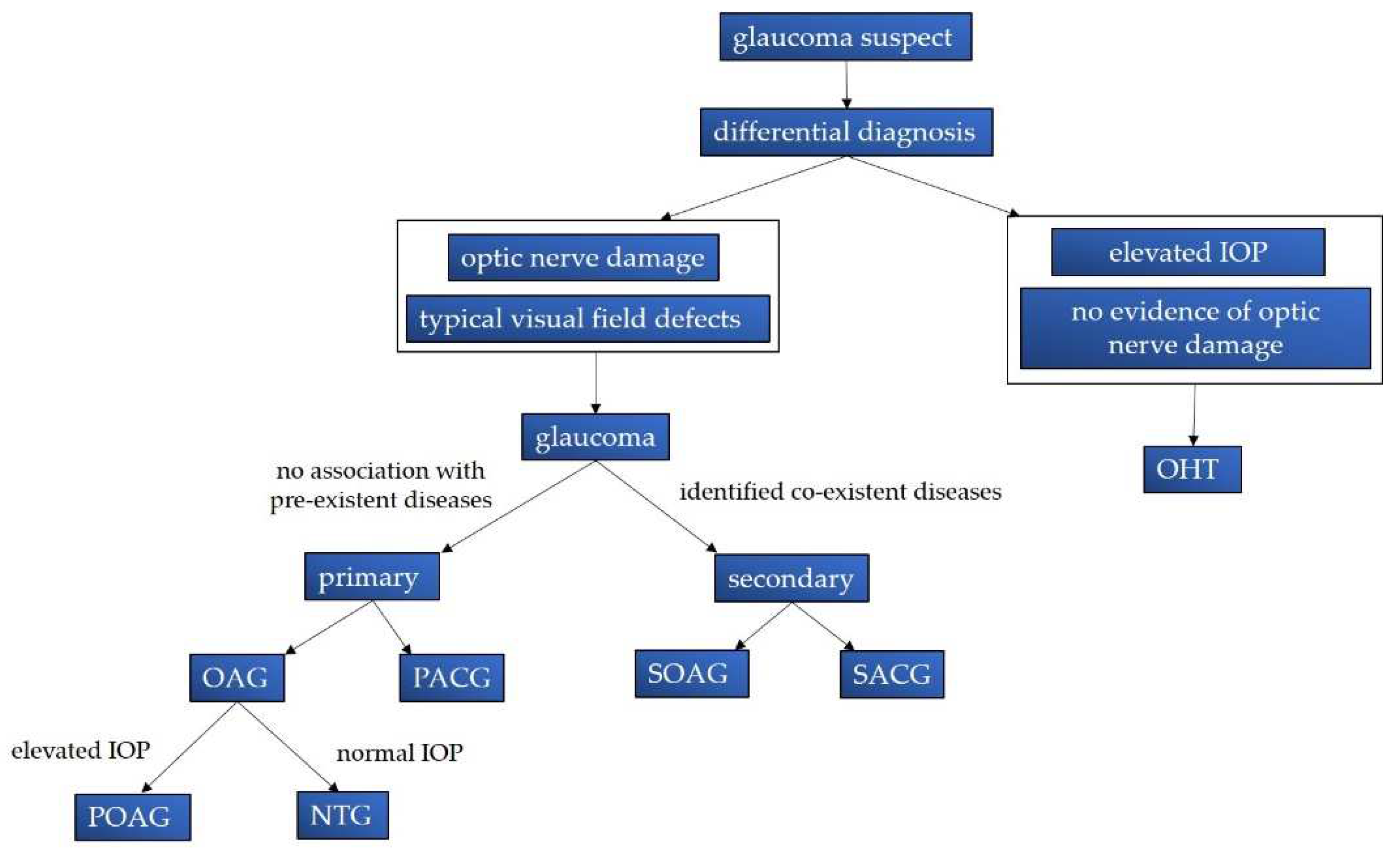
Figure 2.
RGC-degeneration due to elevated IOP in RGCs. IOP: intraocular pressure; RGC: retinal ganglion cell; RNFL: retinal nerve fiber layer.
Figure 2.
RGC-degeneration due to elevated IOP in RGCs. IOP: intraocular pressure; RGC: retinal ganglion cell; RNFL: retinal nerve fiber layer.
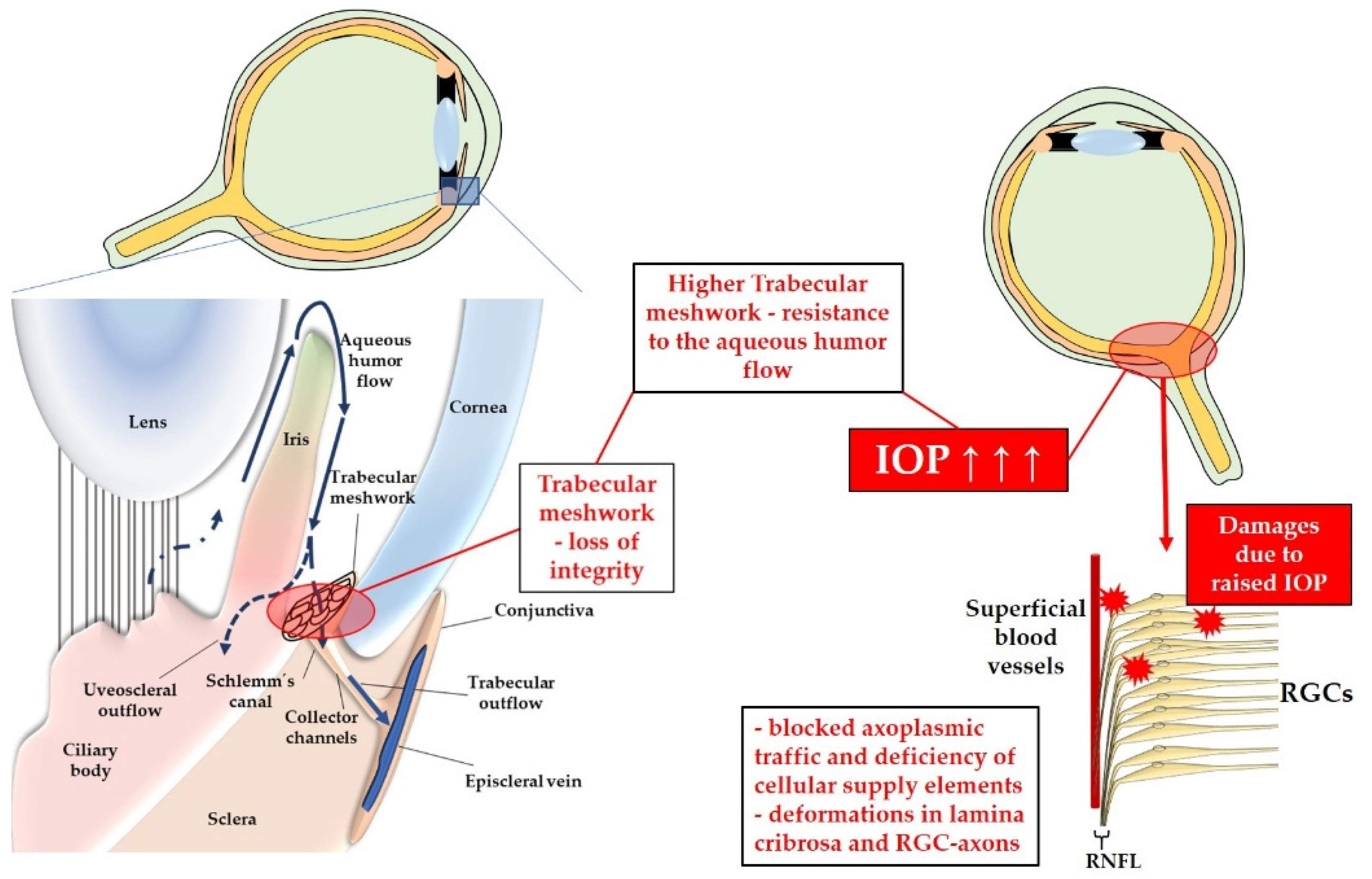
Figure 3.
Possible etiologic initiators in NTG. IOP: intraocular pressure; ATP: Adenosintriphosphat; ONH: optic nerve head; CSI: cerebral silent infarcts; NO: nitric oxide; ET-1: endothelin-1; ROS: reactive oxygen species.
Figure 3.
Possible etiologic initiators in NTG. IOP: intraocular pressure; ATP: Adenosintriphosphat; ONH: optic nerve head; CSI: cerebral silent infarcts; NO: nitric oxide; ET-1: endothelin-1; ROS: reactive oxygen species.
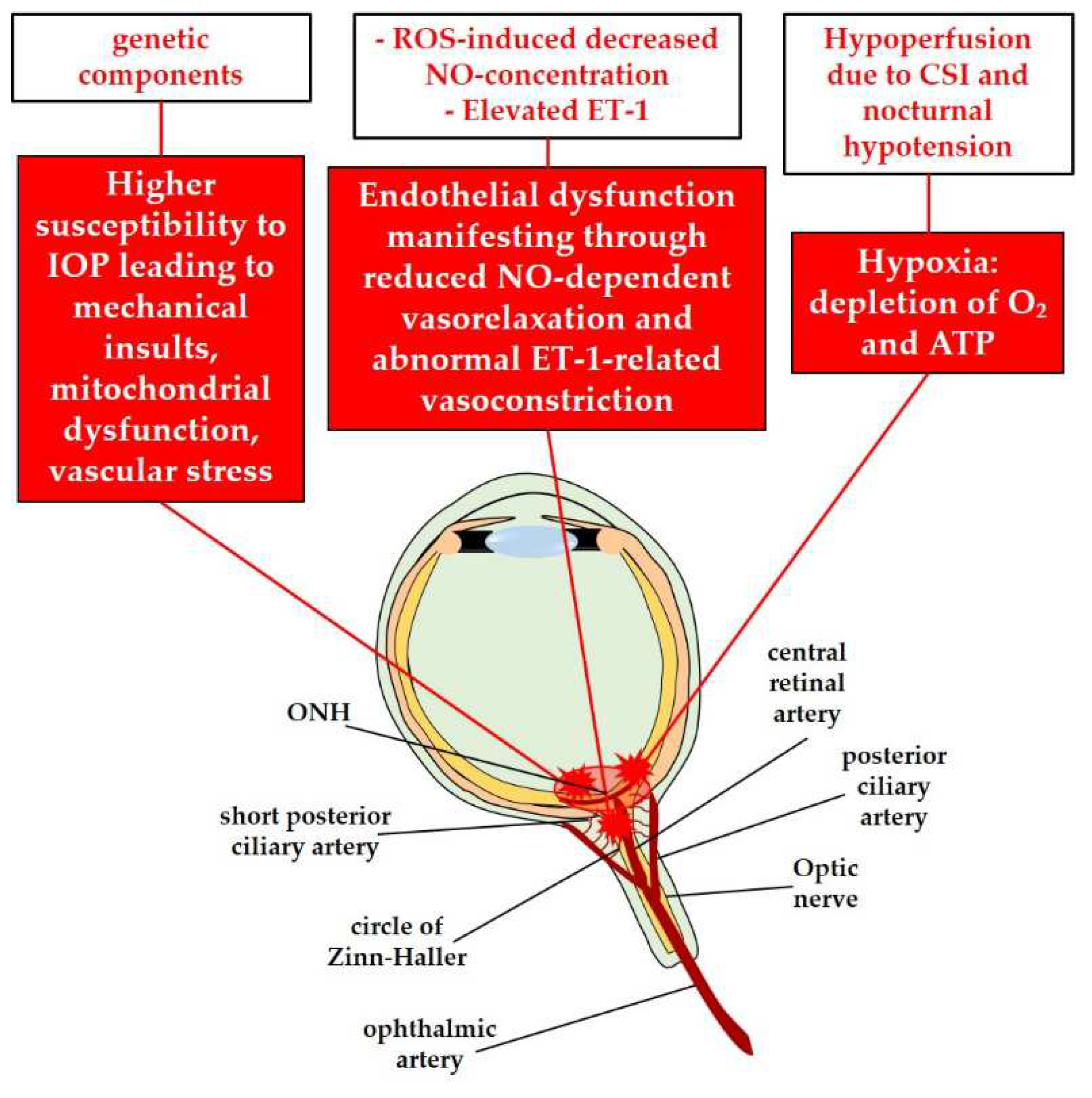
Figure 4.
RGC-death in glaucoma caused by ROS-excess, NF-kB activation, complement activation and autoimmune imbalances. IOP: intraocular pressure; NOX2: nicotinamide adenine dinucleotide phosphate oxidase type 2; NF-kB: nuclear factor 'kappa-light-chain-enhancer' of activated B-cells; RGC: retinal ganglion cell; ROS: reactive oxygen species; HIF-1α: hypoxia inducible factor 1α; TNF-α: tumor necrosis factor α; ER: endoplasmic reticulum; ASK-1: apoptosis signal-regulating kinase 1; MAPK: mitogen activated protein kinase; ERK: extracellular-signal-regulated kinase.; JNK: Janus kinase; PI3K: Phosphoinositide 3-kinase; Akt: Ak strain transforming ; mTor: mammalian target of rapamycin.
Figure 4.
RGC-death in glaucoma caused by ROS-excess, NF-kB activation, complement activation and autoimmune imbalances. IOP: intraocular pressure; NOX2: nicotinamide adenine dinucleotide phosphate oxidase type 2; NF-kB: nuclear factor 'kappa-light-chain-enhancer' of activated B-cells; RGC: retinal ganglion cell; ROS: reactive oxygen species; HIF-1α: hypoxia inducible factor 1α; TNF-α: tumor necrosis factor α; ER: endoplasmic reticulum; ASK-1: apoptosis signal-regulating kinase 1; MAPK: mitogen activated protein kinase; ERK: extracellular-signal-regulated kinase.; JNK: Janus kinase; PI3K: Phosphoinositide 3-kinase; Akt: Ak strain transforming ; mTor: mammalian target of rapamycin.
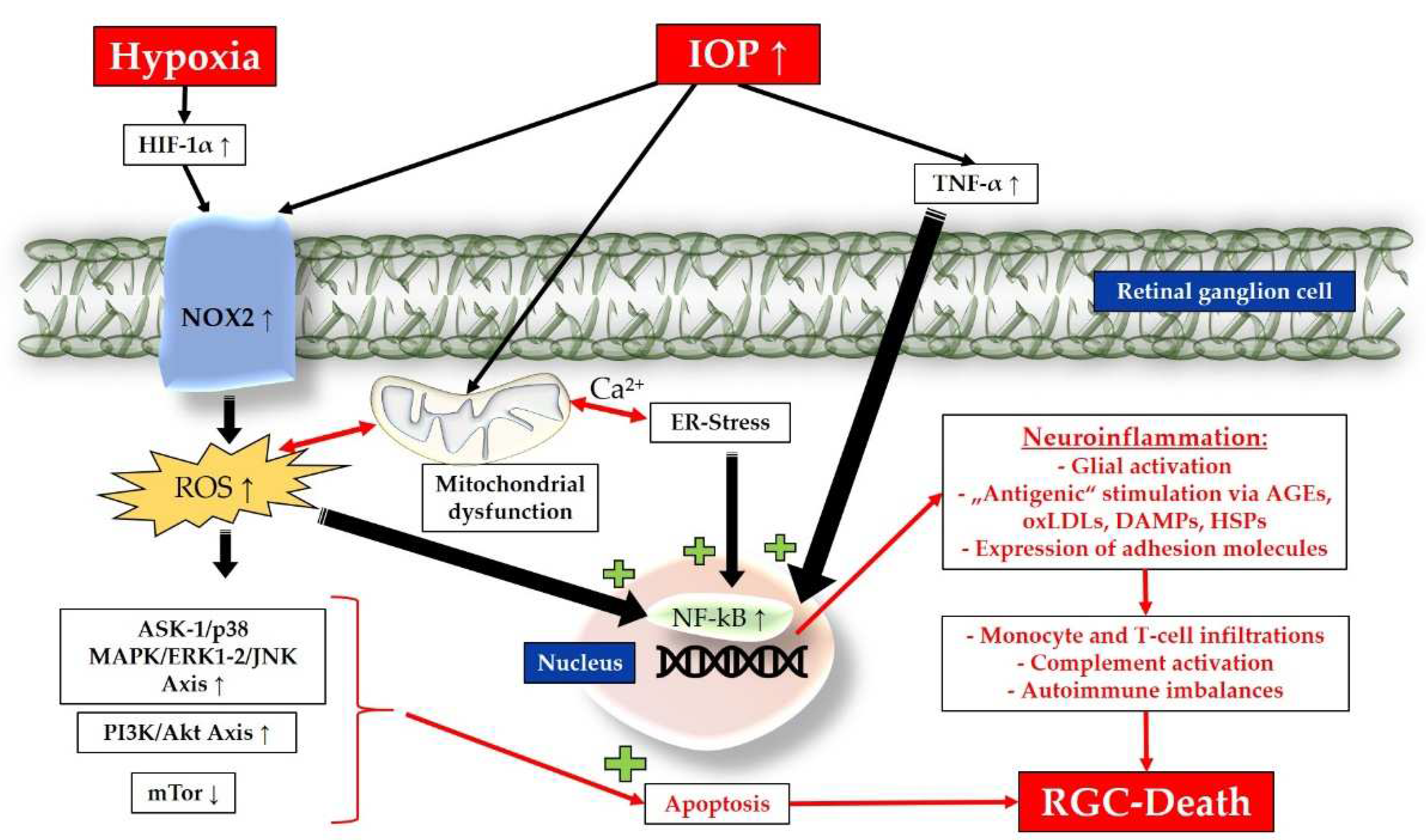
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



