
Submitted:
18 July 2023
Posted:
19 July 2023
You are already at the latest version
Abstract
Antimicrobial resistance (AMR) has a significant global impact on human, animal, and environmental health. Misuse and overuse of antibiotics in clinical and animal production settings are the main drivers behind the emergence of antimicrobial resistant bacteria. However, other compounds with antimicrobial activity may also contribute to this global public health problem. The aim of this comprehensive review is to provide detailed insights into the impact of metals and organic acids on the emergence and spread of AMR in the food chain, for which their role is not fully understood. The review examines the widespread use of organic acids in the food industry as feed additives or disinfectants, the crucial role of copper in animal growth and the harmful effects of mercury and arsenic as pollutants in food-producing environments. Additionally, it explores the antimicrobial mechanisms of metals and organic acids, the tolerance mechanisms developed by bacteria, and the interplay between genes responsible for metal tolerance and AMR. The comprehensive and integrated data presented highlights the need to further explore and understand the role of metals and organic acids as drivers of AMR to develop well-defined strategies effectively mitigating the AMR crisis within the food chain context.
Keywords:
Copper
; Mercury
; Arsenic
; Organic Acids
; Antibiotic Resistance
; Food Safety
1. Introduction
Antimicrobial resistance (AMR) is a critical global health challenge, ranked among the top ten to public health threats worldwide [1]. This biological process occurs when microorganisms change over time and no longer respond to antimicrobials, becoming resistant and making infections harder to treat, which consequently increases the risk of disease spread, serious illness and death [2]. Often referred to as the “silent pandemic” of the 21st century, the true global impact of AMR is difficult to assess, but estimates point to 700,000 deaths each year, globally [3]. More recent estimates indicate that this number could be significantly higher, with 4.95 million human deaths associated with bacterial AMR in 2019, including 1.27 million directly linked to it [4], representing a much greater threat to public health than some infectious diseases such as malaria or HIV [4]. If no action plans are taken, projections indicate that the number of deaths due to AMR could rise to 10 million per year by 2050 [5]. In the European Union (EU) alone, bacterial AMR is estimated to be responsible for 33.000 deaths per year, with an economic impact of 1.5 billion/year in healthcare costs and productivity losses [6].
Since the discovery of antibiotics in the 1940s, the global threat of AMR has evolved dramatically over the past century [7]. While antibiotics are recognized as the greatest advance in the history of medicine [8], revolutionizing medical practice and saving millions of lives, their misuse and overuse, particularly in the medical, veterinary, and agricultural sectors, triggered the emergence, escalation and spread of AMR on a local and global scale (Figure 1) [9,10]. Such use creates selective pressure on bacteria, leading to the survival and proliferation of antimicrobial resistant strains while eliminating susceptible ones, and promoting the exchange and spread of antibiotic resistance genes (ARGs) among multiple bacterial species through horizontal transfer events or bacteria vertical heritage [11].
Although the link between human or animals antimicrobial use and AMR seems clear cut, this association is a complex process involving multiple events, including pathogen-drug and pathogen-host interactions, high mutation rates of particular strains, emergence and expansion of successful antimicrobial resistant clones and/or mobile genetic elements, co-selection events by unrelated antimicrobials (e.g., different antibiotics or biocides), and variable transmission rates of pathogens between humans, animals and the environment [11]. Diverse pathogenic bacterial species, as well as the microbiota of humans, animals and the environment are active participants in these events and can act as important reservoirs and disseminators of ARGs in different settings [12,13].
Figure 1.
Drivers of antimicrobial resistance and its impact at different levels: humans, terrestrial and aquatic animals, food and feed, plants and crops, water sanitation and hygiene, environment (Adapted from [14]).
Figure 1.
Drivers of antimicrobial resistance and its impact at different levels: humans, terrestrial and aquatic animals, food and feed, plants and crops, water sanitation and hygiene, environment (Adapted from [14]).
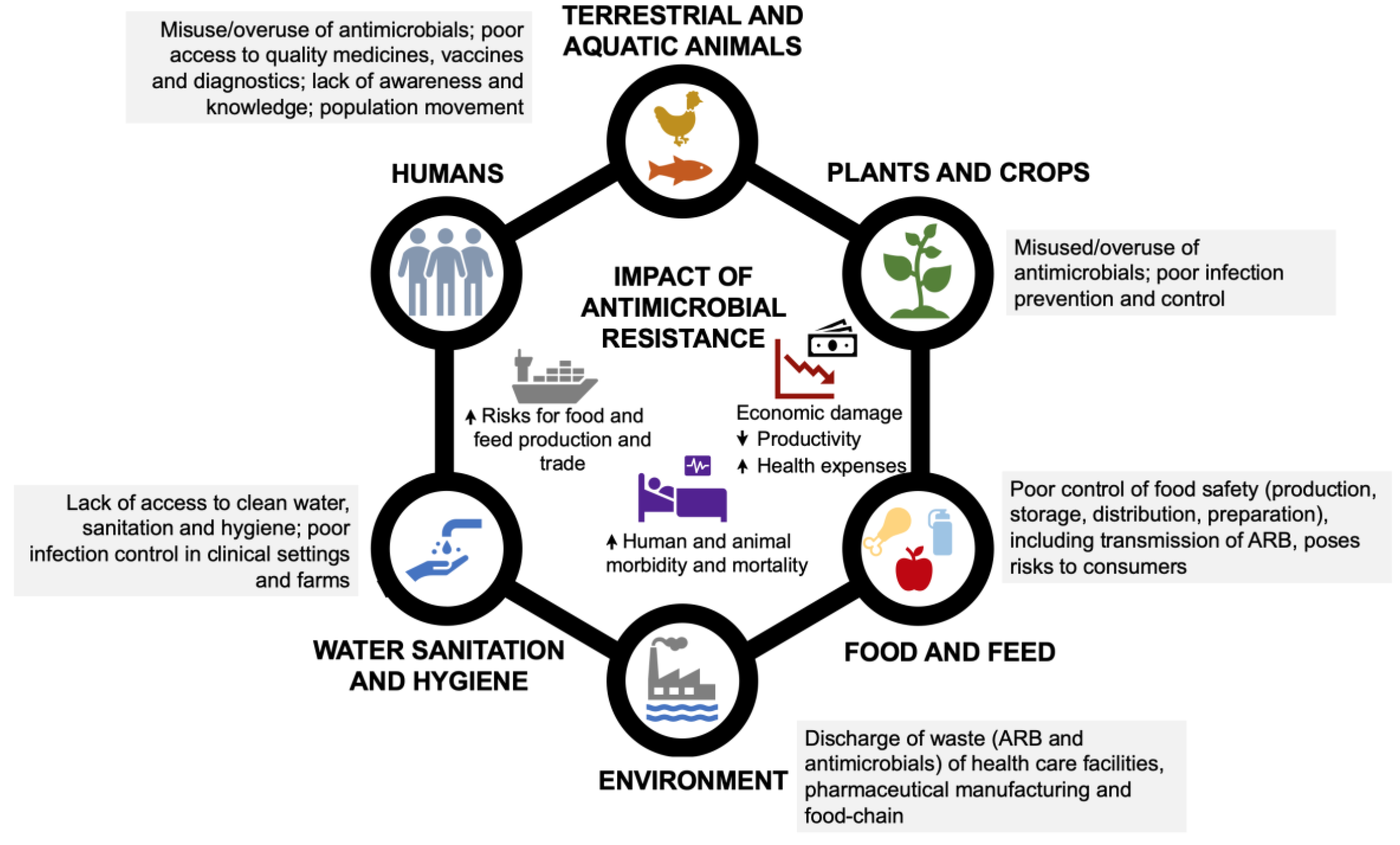
In addition to antimicrobials misuse, other factors are also important drivers of AMR spread, including poor infection control practices (e.g., vaccination), hygiene or biosecurity measures in healthcare facilities or animal production settings, limited access to clean water and sanitation, environmental waste discharges and poor food hygiene and safety practices (Figure 1). Also, the globalization of human, animal and food products, as well as variable policies in different countries regarding antibiotics use and AMR surveillance in food-production and other sectors, contribute to this threat [15,16,17]. All these events that facilitate the spread of AMR have significant multi-layered implications, including in human and animal morbidity and mortality, food and feed trade and economy in general (Figure 1).
The food production sector has long been recognized as a significant contributor to the selection and evolution of antibiotic-resistant bacteria (ARB) [18], which can be introduced at any stage along the farm-to-fork continuum [19] and pose a potential risk to consumers. In fact, ARB from food and animals are important causes of human infections, highlighting the importance of global measures related to food hygiene and safety [20].
Changes in consumption trends as a result of rapid human population growth has led to the increase and globalization of the food supply [11], with the animal-food production industry accounting for approximately 70% of global antimicrobials sales worldwide [12,21]. Antimicrobials are used in different agri-food sectors and at different stages of production, both in intensive food-producing animals (terrestrial and aquatic) and crop productions [22]. However, while antimicrobials play a vital role in preserving animal health and welfare, as well as ensuring food safety and security, most of their use worldwide is to prevent rather than to treat infections (e.g., to compensate for poor farming practices) or, in specific countries, to promote animal growth [5]. Antibiotics use, including as veterinary agents, at subtherapeutic doses to increase the feed-to-weight ratio in animals or as pesticides in crop production, leads to the emergence of ARB in the food-chain [13,18]. However, beside antibiotic use, AMR transmission routes are intricate and involve the participation of different players external to the food-producing animals, which can also promote the spread of AMR in the food-chain. These include feed, workers, air/dust, equipment, water, soil, crops, rodents and other wildlife and visitors, which can be vehicles or vectors of ARB into and out of farms or food processing plants. Ultimately, ARB can be transmitted to humans through contaminated food and water consumption, direct contact with animals, or exposure to water sources contaminated by agricultural and farm wastes [10].
Over time, numerous studies have identified ARB and/or ARGs of higher public health priority in food-producing settings where animal or non-animal foods are produced or processed, including both pre-harvest (primary production) and post-harvest levels (such as slaughterhouses and processing plants) [13]. Among them, methicillin-resistant Staphylococcus aureus has been identified in livestock and poultry meat [23,24,25], vancomycin-resistant or linezolid-resistant Enterococcus in poultry and pork [26,27,28,29], mcr-1 colistin-resistant Escherichia coli or Salmonella in vegetables, unprocessed meat, livestock and farm environments [30,31,32,33], and carbapenem-resistant Enterobacteriaceae in vegetables and livestock [34,35], including Salmonella enterica serovar Typhimurium in pork [36,37], all bacteria that may pose serious risks to human and animal health. Despite growing concerns about the role of the food chain in the emergence and spread of AMR and currently available surveillance data on animals, food, human and the environment, there is still limited information on the proportion of ARGs or clones transferred and spread from the food chain to humans for most bacteria [38,39]. This knowledge gaps makes it difficult to accurately assess the extent to which the food chain contributes to AMR transmission to humans [38]. Thus, effective AMR control requires a coordinated effort within and across countries to identify targeted interventions, improving surveillance and monitoring systems, raising stakeholder awareness, implementing good practices to prevent and control AMR spread, using antibiotics responsibly and strengthening governance [40].
Implementing restrictions on the use of antibiotic in food-producing animals is an important measure to curb the spread of AMR through the food chain, with numerous studies demonstrating a positive impact of limiting the use of antibiotics in reducing the prevalence of AMR in animal bacteria [41,42,43,44]. Global efforts and effective actions have been debated and implemented worldwide to address the issue of AMR in the food chain sector, with the EU taking a leading role in this commitment [6]. One of the main efforts is to reduce the use of antibiotics in food-producing animals by setting national reduction targets [e.g., reduction of colistin in veterinary medicine to 5 mg/PCU (Population Correction unit) by 2021 in Portugal] [45], restriction of antimicrobials drugs only for the treatment of certain human infections (e.g., carbapenems, glycopeptides, oxazolidinones) [46], benchmarking antibiotic use at the farm level and promoting rational antibiotic stewardship, such as requiring susceptibility testing before use of some high-priority antibiotics [47]. Over the past few years, EU/EEA (European Economic Area) countries have made important progress in reducing the use of antibiotics in food-producing animals, resulting in a 47% decrease in sales between 2011 and 2021 [48]. This achievement is partly due to the actions taken in the early 1980s by some European countries such as Sweden, Norway and Denmark, which were pioneers in restricting or banning the use of antibiotics as growth promoters in animal farms [49], leading to an EU-wide ban in 2006 through Regulation (EC) No. 1831/2003. More recently, a new milestone was reached with the interdiction of all forms of routine use of antibiotics in farm animals, including for prophylactic use [Regulation (EU) 2019/6 on veterinary medicinal products and Regulation (EU) 2019/4 on medicated feed]. With these actions, the EU aims to reduce by 50% the sale of antibiotics for farm animals and aquaculture by 2030 [50]. This paradigm creates new expectations regarding AMR reduction, but also new challenges for the animal-farming sector to ensure animals’ safety, health and welfare, and at the same time to obtain the desired production level [51]. Apart from the reduction on antimicrobials use, other measures are essential to mitigate AMR, including the effective implementation of good hygiene practices and biosecurity measures [13,52]. Also, improving animal nutrition contributes to a good level of animal yield by reducing vulnerability to bacterial infections and, consequently, the need for antimicrobials in animal husbandry practices [53].
In-feed supplementation with probiotics, enzymes, phytochemicals, antimicrobial peptides, metals and organic acids are among the available alternatives to antibiotics, with an important contribution to animal growth and disease prevention [54,55]. Some metals are essential nutrients for most animal species and are widely incorporated into animal feeds to contribute to meet nutritional requirements [56]. Some metals, such as copper and zinc, are even added to feed in higher concentrations to act as growth promoters [56]. In addition to their use as feed additives, some metals have for decades been important antimicrobials in veterinary medicine, including arsenic (as coccidiostat) [57], copper (as fungicide and bactericide) [58], mercury (as preservative of veterinary drugs/bacteriostatic) [59], silver (as bacteriostatic/bactericide) and zinc (to treatment and prevention of diarrhea and skin infections) [60]. Currently some metals (e.g., copper and zinc) continue to be promoted by official bodies [e.g., European Medicines Agency (EMA) and European Food Safety Authority (EFSA)] as alternatives to antibiotics due to their antimicrobial properties [47,61,62]. Thus, food production, as well as other anthropogenic activities, promote the release of metals into the environment, beyond their natural occurrence through biogeochemical processes [63,64,65]. The persistence of metals in the environment, due to their limited biodegradability, can lead to their accumulation in soil, water and sediments, resulting in significant environmental contamination and selective pressure for ARB [56]. Therefore, new rules on metals use (e.g., zinc, copper) as feed additives or growth promoters have been implemented by the EU to control such events [66,67]. Other compounds, such as some organic acids (e.g., peracetic acid) have been used as disinfectants for equipment and surfaces in food production environments [68,69,70], decontaminants on carcass surfaces following slaughter (e.g., lactic acid on bovines) [71] or feed additives (e.g., lactic and citric acids as preservatives) [72], with less environmental impact than other biocides [55].
Regardless of their importance in food production environments, metals or other compounds (e.g., biocides, organic acids) have been suggested to be potentially associated with the co-selection and dissemination of ARB [73,74]. This association stems from the fact that many genes that confer tolerance to these chemical agents are frequently located in the same genetic elements as ARGs (co-resistance) (Figure 2). Also, other less frequently described co-selection mechanisms might be involved, including the occurrence of a single mechanism that may confer resistance to metals and antibiotics simultaneously (e.g., efflux pumps) (cross-resistance) or the presence of a common regulator responsible for controlling the expression of metal and antibiotic resistance systems (co-regulation/co-expression) (Figure 2) [75].
Additionally, exposure to low antimicrobial concentrations have been described to increase horizontal transfer events or the occurrence of bacteria genome mutations with and impact on AMR [73,76,77,78,79]. To better understand the bacterial response to various antimicrobials widely used in food production environments (such as copper and organic acids) or commonly present as environmental contaminants from anthropogenic activities (such as arsenic or mercury), and their contribution to the selection of multidrug-resistant (MDR) bacteria, this topic will be discussed in more detail in the following sections, with a focus on the food chain.
2. Metals
2.1. Copper
Copper (Cu) is an essential mineral for all living organisms [80], participating in various biological processes. In bacteria it is found as a cofactor in proteins and enzymes due to its redox potential, acting as an electron donor/acceptor by alternating between the reduced cuprous form [Cu(I) or Cu+] and the oxidized cupric form [Cu(II) or Cu2+], critical for a wide range of cellular metabolic and regulatory functions [81,82,83] (e.g., electron transport, oxidative respiration, denitrification, etc.) [84,85]. However, in certain forms and concentrations, it can be toxic and inhibit or kill bacteria [86,87].
The antimicrobial properties of copper are well described [87] and its use dates back to ancient Egypt for the preservation of water and food, as well as for medical applications [88]. In the agri-food sector, copper-based compounds have been used as antimicrobial since the end of the 19th century, when its activity as fungicide was first described, being used as the “Bordeaux mixture” in vineyards [89]. Since then, it has been widely used in pesticides and fertilizers [90,91]. Although the role of copper as an antimicrobial agent was widely recognized in the past, it lost significance with the advent of antibiotics [92]. However, the biocidal properties of copper against a wide range of pathogens have made it regain importance as a promising alternative in the fight against the spread of MDR bacteria [92]. Among the currently authorized copper applications in the EU are several copper-based biocidal products not intended for direct application to humans or animals [93]. In recent years, the use of copper plating of surfaces, including in the food and medical sectors [94,95,96,97], has been proposed as a more effective measure to limit bacterial adhesion than stainless steel [87], being the first solid antimicrobial material registered with the U.S Environmental Protection Agency [92]. Other antimicrobial applications of copper have been made, most in clinical settings (e.g., medical devices such as copper-impregnated fabrics) [98,99,100,101].
Although copper is commonly known for its antimicrobial properties, it also plays a crucial role in human and veterinary medicine in the treatment of nutritional deficiencies [58]. In food-producing animals, feed is routinely supplemented with copper not only to meet the animals’ nutritional needs but also to improve their growth performance by modulating the gastrointestinal tract microbiota, leading to improved nutrient absorption [102]. Varying concentrations of copper are used, depending on the species, age group and feed composition, as copper can interact with other nutrients, including other metals (e.g., zinc, iron, calcium, molybdenum) and phytates [103]. As an example, the maximum concentration allowed in poultry feed is 25 mg Cu/kg, while in piglets up to 4 weeks after weaning it is 150 mg Cu/kg and from the 5th to the 8th week after weaning it is 100 mg Cu/kg [66]. Traditionally, feed supplementation with inorganic trace mineral (ITM) copper has been used as a cost-effective solution ([104,105], but the use of other forms, mainly organic species (organic trace mineral, OTM) and copper nanoparticles, has been increasing, as they present higher bioavailability, improving animals’ growth performance, with a less environmental impact [105,106,107]. The application of copper nanoparticles has also been exploited in the food industry and agriculture sectors, mainly to prevent microorganism spoilage (e.g., in food packaging) [108] and as agro-nanochemicals (e.g., fertilizers and pesticides) with a larger specific surface area than conventional forms [109]. However, the widespread use of copper-based compounds in many anthropogenic activities has led to its accumulation in different ecosystems, making it a pollutant and potentially toxic to many organisms, including bacteria.
Copper poses a unique challenge to bacteria due to its dual nature – it is an essential trace mineral, but it can also be cytotoxic when present in excess. This ambivalence highlights the importance of strict regulation of cellular copper levels [110]. Maintaining copper homeostasis requires a delicate balance between providing the required dose of the micronutrient while avoiding toxic excess [56,111]. Although the mechanisms of how copper ions affect bacteria are still not fully understood, it seems that the cycling between the cupric [Cu(II)] and the cuprous [Cu(I)] states can disturb the intracellular redox potential, being the main cause of cytotoxicity. In particular, the intracellular soluble fraction of copper [Cu(I)], via a Fenton-like reaction, catalyzes the formation of superoxide (O2-) and other reactive oxygen species [hydroxyl radicals (OH⋅) and hydrogen peroxide (H2O2)], which are responsible for lipid peroxidation, protein oxidation and DNA damage [112]. Under low oxygen conditions, the reduced ionic species Cu(I) is prevalent and is highly toxic, showing great affinity for thiolates and other sulfur-containing compounds, disrupting the binding of iron-sulfur (Fe-S) clusters, leading to poor protein metallation, protein inactivation and ultimately to dysfunctional cell metabolism [112,113,114]. In human macrophages, copper is pumped to their phagosomes after engulfing pathogenic bacteria to induce bacteria death by oxidative stress [115].
Copper can often enter the bacterial cells in an unspecific manner by using other metal uptake systems, making it difficult for bacteria to limit the amount of copper entering the cytoplasm [56]. Bacteria have evolved a number of mechanisms implicated in the uptake, internal traffic, storage and efflux of copper from the cell, including the extracellular sequestration of copper ions, the relative impermeability of outer and inner bacterial membranes to copper ions, the presence of metallothionein-like copper-scavenging proteins in the cytoplasm and periplasm, and the active extrusion of copper from the cell [92].
The extrusion of excess cytoplasmic copper by homeostatic mechanisms appears to be the main defense mechanism in bacteria, a process that has been extensively studied in both Gram-positive and Gram-negative bacteria [92]. Specifically, copper efflux occurs through transporters, members of the P1B-1-ATPase subfamily [Cu(I) transporters] of P1B-ATPases [116]. The first copper-transporting ATPases were described in Enterococcus hirae [117,118], represented by the cop operon (copYZAB), which formed by four genes coding for the following proteins: CopA and CopB, responsible for the uptake and removal of excess Cu(I) from the cytoplasm, respectively [119]; CopZ, a chaperone responsible for intracellular copper transport; and CopY, a promoter regulator [120,121]. Unlike to Gram-positive bacteria which lack a periplasmic space and an outer membrane, Gram-negative bacteria require additional mechanisms to deal with the presence of copper in the periplasm. In the most studied Gram-negative bacterium, E. coli, in addition to the presence of the Cu(I)-translocating P-type ATPase CopA in the cytoplasmic membrane, responsible for pumping excess Cu(I) from the cytoplasm to the periplasm [122], there is also the CusCBA multicomponent copper efflux system and the CueO multicopper oxidase. These two systems are chromosomally encoded and play important roles in controlling copper level and redox state, respectively [56]. Since CueO acts only in the presence of oxygen, presumably oxidizing Cu(I) into the less toxic Cu(II) [56], the CusCBA transport complex is important to copper detoxification from the periplasm in the absence of CueO [123]. In Salmonella, copper defense determinants are quite similar to those of wild-type E. coli, also containing CopA and CueO. However, most Salmonella strains do not contain the CusCBA system, instead having the periplasmic copper-binding protein CueP [112].
In environments with high copper concentrations, which would overwhelm chromosomally encoded copper metabolic systems, some bacteria have acquired copper tolerance mechanisms, regulated mainly by extrachromosomal loci [124]. The first mechanism described in Gram-negative bacteria was identified in the pRJ1004 plasmid of an Australian pig E. coli isolate [125], linked to the presence of the pco (plasmid-borne copper resistance) system. This system includes different structural proteins, including PcoA, a periplasmic multicopper oxidase, PcoB and PcoD, outer and inner membrane proteins, respectively, and PcoC and PcoE, two periplasmic proteins [125,126,127,128]. While PcoE is responsible for temporarily sequestering excess copper [128], PcoC is also capable of transferring it to the membrane-bound PcoD [56]. In turn, PcoD catalyzes the uptake of Cu(I) into the cell, which is incorporated into PcoA and exported to the periplasm, where it will be detoxified either by sequestration or oxidation and removed via PcoB (Figure 3) [129]. A two-component regulatory system, PcoRS, seems to be responsible for the transcription of PcoABCD proteins [126], while the chromosomally encoded CusRS system regulates the transcription of PcoE protein [128]. Two additional proteins, PcoF and PcoG, corresponding to a putative copper-binding protein and a putative metallopeptidase, respectively, may be present, but their role has yet to be determined [130]. The pco gene cluster encodes proteins responsible for periplasmic copper management, being dependent on the supply of copper by the cytoplasmic CopA protein to confer copper tolerance to bacteria [110]. Contiguous to the pco system in pRJ1004 is the sil gene, first described in the S. Typhimurium plasmid pMG101, and initially linked to silver tolerance [131]. The Sil system includes a SilCBA efflux complex responsible for exporting Cu(I) and Cu(II) from the periplasm, three periplasmic proteins, SilE [homolog to PcoE, presumably to bind Cu(I) and Cu(II)], SilF and SilG, the first two acting as chaperones of the SilCBA complex and the last one with unknown function, as well as a P-type ATPase SilP that transports copper and silver ions from the cytoplasm to the periplasm [132]. The two-component membrane sensor and transcriptional responder SilRS appear to be involved in silCFBAGP expression [130]. The occurrence of sil efflux systems is associated with a CuSO4 tolerance phenotype in several Enterobacteriaceae under anaerobic conditions, where the more toxic form Cu(I) is predominant, a distinct feature of isolates carrying sil±pco genes in comparison with those without it [44,133,134,135]. A minimum inhibitory concentration (MIC) for CuSO4 between 16-36 mM has been described in isolates with sil±pco, contrasting with a MICCuSO4 between 2-12 mM in isolates without these genes [44,133,134], being proposed a CuSO4 tolerance cut-off ≥ 16 mM to differentiate isolates with and without sil±pco gene clusters, under anaerobioses [44,134].
Since the entire sil determinant confers copper tolerance, the contiguous 20-gene clusters of pco+sil have been referred to as copper-pathogenicity island [130]. Although the pco+sil determinants were initially identified in plasmids, it is worth noting that this gene cluster may also be located on chromosome [133,134], due to the bacteria genetic plasticity, which is often facilitated by the presence of Tn7-like transposons [134,136,137]. Several studies have been describing the wide occurrence and distribution of sil-pco clusters in diverse species and multiple environments, including food and food-producing animals [134,138], hospitals and urban wastewaters [139], freshwaters [140], veterinary clinical settings [141] and human clinic [134].
Gram-positive bacteria with high acquired tolerance to copper have also been described, namely in several species of Enterococcus genus. The most characterized gene is the plasmid encoded tcrB (transferable copper resistance gene B) initially identified in an E. faecium isolate from pigs in Denmark [142]. The tcrB gene codes for an efflux pump, presumably belonging to the P1B-3-ATPase subfamily of copper transporters P1B-ATPases, which is activated mainly by Cu(II) and to a lesser extent by Cu(I) [129,143]. This gene is part of the tcrYAZB operon (homologous to the copYZAB copper-homeostasis gene cluster of E. hirae) [144], together with the tcrA gene, an additional P1B-ATPase of the P1B-1-ATPase subfamily and responsible for Cu(I) export, the tcrZ gene, which encodes a cytoplasmic copper chaperone (TcrZ) responsible for Cu(I) transport, and the tcrY gene, a copper-dependent regulator (TcrY) involved in controlling operon expression (Figure 4) [142,144]. These copper tolerant determinants are often flanked by insertion sequences, allowing their transferability [145,146,147].
As the sil efflux systems, the acquisition of the tcrYAZB operon represents a clear advantage for bacteria in anaerobic environments, allowing them to survive in higher Cu concentrations [148]. Enterococcus spp. carrying tcrYAZB operon have shown a MICCuSO4 between 16-36 mM, while in isolates without these genes the MICCuSO4 ranged between 4-12 mM [148,149,150]. Thus, a CuSO4 tolerance cut-off ≥ 16 mM was proposed to differentiate isolates with and without tcrB gene, under anaerobic conditions [148]. In the vicinity of the tcrYAZB operon is often a multicopper oxidase (CueO), potentially involved in the oxidation of Cu(I) to Cu(II) [145].
As in Gram-negative bacteria, the tcrYAZB operon genes are located mainly in plasmids [142,146,149], unlike chromosomal genes related to copper homeostasis [151]. Since the first description of the tcrYAZB operon in the pA17sv1 plasmid of an E. faecium from a healthy pig [144], the presence of the tcrB gene has been mainly associated with Enterococcus genus isolates from food-animal production environments [145,146] and foodstuffs [145,149,152], with few studies describing its occurrence in humans (clinical and community isolates) and aquatic environments [145,148].
A major issue is that copper tolerance has been strongly associated with antibiotic- resistant bacteria in different environments (e.g., aquatic, animal-food production, agri-food, clinical settings) [153,154,155], including those without antimicrobial pressure (e.g., pristine environments) [75]. Co-selection of copper tolerance genes and ARGs often occurs because they all share the same genetic elements [146,150,156]. Shortly after the first description of the tcrB gene, a link to macrolide and glycopeptide resistance was established by the co-occurrence of such resistance determinants on the same conjugative plasmid of porcine E. faecium [142,156]. More recently, other ARGs (e.g., vanA- vancomycin; tet(M) or tet(L)-tetracycline; aadE-streptomycin; aac(6’)-Ie-aph(2’’)-Ia-gentamycin) have also been described in the same Enterococcus plasmids as the tcrYAZB operon and other metals in Enterococcus spp. of the food-chain and other niches [149,150]. A single description of tcrYAZB on the chromosome is available for E. faecalis from poultry meat alongside mercury (merA) tolerance genes [149]. Plasmids carrying sil±pco genes (and other metal tolerance genes, including to mercury – mer genes) and ARGs for beta-lactams (blaTEM-1, blaCTX-M), aminoglycosides [aac(3), aadA], sulfonamides (sul), trimethoprim (dfrA), chloramphenicol (cmlA) and tetracyclines (tet) have also been described in E. coli, Klebsiella pneumoniae and S. enterica isolates from food-producing environments and human sources [133,134,157,158]. In addition, chromosomal co-localization of copper (pco+sil) with other metal tolerance genes (e.g., mer) and ARGs for beta-lactams (blaTEM-1), aminoglycosides (aadA, str) sulfonamides (sul), trimethoprim (dfrA) and tetracyclines (tet) was described in S. enterica isolates from various sources (animal-food production; food; human) [133,134]. Cross-resistance and co-regulation mechanisms have been poorly described, with some studies suggesting the role of efflux systems (e.g., membrane transporters belonging to the RND family) in the extrusion of both copper and antibiotics (e.g., cefotaxime) in some Gammaproteobacteria [159,160], and overexpression of some binding proteins (e.g., Rob encoded by robA gene) associated with increased resistance to metals (including copper) and multiple antibiotics (e.g., tetracycline, chloramphenicol) in E. coli [161].
2.2. Arsenic
Arsenic (As) is a metalloid naturally present in the earth’s crust and widely distributed in soil, sediments, water, air and living organisms [162,163]. Unlike other elements (e.g., copper, zinc), arsenic is not required for biological functions in most bacteria, exerting a toxic effect on the cell [164,165]. The toxicity of arsenic greatly depends on its oxidation state, and it can occur in four valence states: As3- (arsine gas, AsH3), As0 (elemental arsenic), As3+ (trivalent arsenic or arsenite) and As5+ (pentavalent arsenic or arsenate) [166]. Arsenite and arsenate are the predominant species under reduced and oxygenated conditions, respectively, the former being 100 times more toxic than the pentavalent form [166].
Regardless of its ubiquitous distribution and the contribution of natural processes to increasing environmental arsenic contamination (e.g., mineralized and mined areas, volcanogenic activity, thermal springs and Holocene alluvial sediments) [167], it is human activity that has greatly contributed to increase arsenic concentrations in different environments [163]. Arsenic or arsenic-based compounds have historically been used in a range of applications, including pharmaceuticals, wood preservatives, agricultural chemicals (e.g., pesticides, cotton desiccants, defoliants and soil sterilant) and in industry (e.g., mining, and metallurgy) [162]. Inorganic arsenic compounds have been used in medicine since 2000 BC, when arsenic trioxide (As2O3, commonly referred to as ATO) was used as both a drug and a poison [168]. Over time, the use and development of arsenicals in medicine has evolved, with important milestones including its use by Hippocrates to treat skin cancers (using orpiment – As2S3, and realgar – As4S4) and its recommendation by Paracelsus for use in medicine [168]. After the 17th century, ATO became widely used as a drug to cure headaches and specifically to treat trypanosomiasis, syphilis and leukemia [168]. Currently, ATO is still used as an anticancer chemotherapeutic agent for hematological diseases, listed as one of the essential medicines by the World Health Organizations [169]. Although arsenic has this history of use in medicine, it is the agricultural and industrial sectors that have contributed the most to environmental pollution by arsenic. In agriculture and animal-farming, arsenic-based compounds have been extensively in pesticides [e.g., sodium arsenite or sodium arsenate, Na2HAsO3/Na2HAsO4; calcium arsenite or calcium arsenate, Ca(AsO2)2/Ca3(AsO4)2], as coccidiostats and as a feed additive, mainly in the poultry and swine industries [57,168,170]. Roxarsone, a pentavalent nitroaromatic arsenical, has been used exclusively for animal husbandry, particularly poultry, to promote growth, treat coccidiosis and prevent gastrointestinal infections [57]. Despite possible accumulation in animals’ meat [57], most of the roxarsone ingested by animals is excreted in feces and urine, which might contribute to its accumulation in and around the animal production environment (e.g., manure, waste lagoons, amended soils) [171,172]. For this reason, roxarsone is now banned in several countries around the world (e.g., EU countries, USA and China) [173,174].
Although many arsenic compounds are no longer used, their residues persist from past activities. A recent study showed that arsenic concentrations in more than half of European agricultural soils exceeded the threshold of 5 mg/kg [175], posing a threat to the environment, food safety and human health. Moreover, concentrations found in animal-production environments (e.g., total arsenic in manure: ~0.016-2.5 mM; sludge: ~0.15 mM; feed: ~0.0003-0.174 mM) [176,177,178,179], suggest that arsenic may create a selective pressure on bacteria in these environments, favoring the selection of those with tolerance to arsenic (and other metals), with particular concern for MDR zoonotic bacteria [180].
Throughout Earth’s evolutionary history, bacteria have always been exposed to arsenic in different environments and have evolved numerous mechanisms to deal with it, either through detoxification or metabolic pathways [181,182]. Several arsenic biotransformation systems have been identified in bacteria, most of which are associated with detoxification processes. These include the arsenic resistance efflux system (ars), arsenic methylation and associated pathways (e.g., arsM), as well as metabolic processes such as arsenite oxidation (aio/arx) and reduction (arr) systems (Figure 5) [181].
Arsenic metabolic pathways involving biotransformation between As3+ and As5+ (aio/arx and arr systems), represent an important energy-generating process in the respiratory process of some bacteria [182,184]. However, for most bacteria, arsenic is not essential, which explains the absence of specific arsenic uptake systems [165]. In fact, the analogy of some arsenic species with other molecules allows arsenic entrance into bacterial cell via non-specific intrinsic transporters [185]. For example, arsenate is a phosphate analogue, entering to cell through phosphate transporters (Pit or Pst) (Figure 5) and inhibiting phosphorylation reactions (such as glycolysis and ATP production) [186]. However, it is unstable and can rapidly dissociate into the more toxic trivalent arsenite (As3+) [187]. Arsenite has a structural similarity to glycerol and enters the cell via aqua-glycerolporins (GlpF), the glycerol transport system (Figure 5) [165,181]. The greater toxicity of arsenite is related to its ability to bind strongly with sulfhydryl groups in proteins, impairing the function of many proteins important for biochemical processes, and to bind weakly to other small thiol molecules (glutathione, lipoic acid, and cysteine), affecting respiration [184,186].
To cope with continued exposure to arsenic toxicity, most bacteria have evolved and acquired genes for arsenic detoxification, mostly encoded by ars operons (Figure 5), often found among prokaryotic genomes, either on chromosomes or on plasmids of Gram-positive and Gram-negative bacteria [165,181,184,188], which reflects its ubiquitous presence in nature. The first description of arsenic tolerance genes occurred more than 50 years ago, when a clinical strain of S. aureus was identified as carrying a plasmid (pI258) conferring tolerance to arsenate, arsenite and other metals and resistance to antibiotics [189]. Shortly thereafter, another plasmid (R773) identified in a clinical strain of E. coli also revealed the occurrence of arsenic tolerance genes [190]. In both cases, ars operons involved in the arsenic tolerance phenotype were identified, encoding homologous proteins, but with different configurations: the three-gene arsRBC operon in the Staphylococcus pI258 plasmid and the extended five-gene arsRDABC operon in the E. coli R773 plasmid [184]. In fact, several genomic configurations of ars operons have been described and suggested to be strain-specific [165,184]. Most of ars operons are involved in inorganic arsenic detoxification, although coupling with other ars-related genes also allows for organoarsenicals detoxification (Figure 5) [181]. In both types of ars operons, the core genes include a trans-acting transcriptional repressor protein (ArsR) that binds to the promoter region of the ars operons, an arsenite efflux pump (ArsB) and an arsenate reductase (ArsC) (Figure 5) [184]. ArsR interacts with arsenite, dissociating the repressor protein from DNA, thereby downregulating transcription of other ars operon genes [184,191]. ArsB is an integral membrane protein responsible for the extrusion of arsenite [As(OH)3/H+ antiporter) from the cell cytoplasm, representing the basic mechanism of arsenite detoxification by decreasing its accumulation [192]. ArsB activity can involve two types of energy sources: acting independently on the arsenite transport channel, using the membrane potential to catalyze the extrusion of As3+ from the cell; or acting in conjugation with ArsA (in the case of operons arsRDABC), to potentiate arsenic tolerance to a higher degree [181]. Specifically, the ArsA ATPase protein catalyzes the hydrolysis of ATP, which energizes the arsenite efflux pump, forming the ArsA-ArsB membrane-bound complex (Figure 5). The ArsC protein is an arsenate reductase enzyme, capable of reducing intracellular arsenate to arsenite, which will then be extruded out of the cell through the ArsB pump [193]. Finally, the ArsD protein, which occurs in the extended ars operons (arsRDABC), is a metallochaperone responsible for sequestering cytosolic arsenite and transferring it to the ArsA subunit of the efflux pump, increasing the efficiency of arsenic extrusion (Figure 5) [192].
Genomic analysis has been contributed to identify the existence of atypical ars clusters [194,195] or the occurrence of additional genes associated with these clusters and involved in arsenic tolerance genes, including the acr3 gene [196,197]. Acr3 (also known as ACR3 or ArsY) is a member of the BART (bile/arsenite/riboflavin transporter) superfamily, first reported in the arsRBC operon of B. subtilis as a typical ArsB membrane protein (Figure 5) [184]. In fact, the literature often describes members of the Acr3 family as ArsB-type, even though they do not exhibit significant sequence similarity to ArsB [198]. While the ArsB-type is mostly restricted to bacteria, including Bacillota (formerly Firmicutes) and Pseudomonadota (formerly Proteobacteria) [180,199,200], the Acr3-type family has a wide distribution, also being found in archaea and eukaryotes (mainly fungi and some plants) [187,201,202]. Interestingly, a predominance of acr3 over arsB genes was found in arsenic tolerant bacterial isolates from arsenic-contaminated soils, and in some cases, concurrently with the arsB gene [203]. However, no evidence of the coexistence of the two transporters encoded in the same operon has been reported so far [202]. As with the ArsB-type, Acr3 can also couple with ArsA to form a more efficient arsenite efflux system [201]. A phenotype of increased arsenate (sodium arsenate Na2AsO4) tolerance was observed in Gram-positive (Enterococcus spp.) and Gram-negative (Salmonella enterica) bacteria with arsenic tolerance genes (arsA, arsB or acr3) compared to those without these genes, with MICs ranging between 8 and ≥ 128 mM and between 0.5 and 4 mM, respectively, regardless of the atmosphere used (aerobic or anaerobic) [150,180].
The wide distribution of arsenic tolerance genes in bacteria from diverse sources (environment, food, clinical) reflects not only the ubiquitous nature of this metal, but also bacteria adaptive characteristics. In particular, arsenic tolerance genes (arsA/arsB/acr3) have been predominantly found in bacteria from natural environmental sites, regardless of whether they had a history of arsenic contamination, including soils (from forests or close to gold mining activities or geothermal effluents), creek water and sewage [200,203,204,205]. Additionally, other contexts have been associated with the occurrence of arsenic tolerance genes, such as clinical (e.g., human samples, clinical settings) [141,206] and food-associated environments (e.g., food-producing animals, processing plants, food products) [206,207]. In animal-food production environments, arsenic can accumulate and persist in sublethal concentrations, leading to long-term selective pressure on bacteria, which favors those with reduced susceptibility to arsenic and other antimicrobials (metals and antibiotics) [154]. In fact, there is growing evidence of the wide dispersion of arsenic tolerance genes in these environments, ranging from animals to other variable stages in food production, including raw, processed, and ready-to-eat animal products (e.g., swine, poultry, cattle), associated or not with foodborne pathogens [207,208,209].
The co-localization of arsenic and other metal tolerance operons (e.g., mercury and copper) in the same genetic context have been described, either in plasmids or in chromosomal regions. These genetic regions have been pointed as potential hotspots for the accretion of metal tolerance genes, either in bacteria with an environmental lifestyle (e.g., Alteromonas sp.) or food-chain associated bacteria (e.g., Listeria sp., Salmonella sp.) [206,210]. Furthermore, arsenic tolerance genes have been described as being on the same mobile genetic elements as other metal tolerance genes or ARGs, including in plasmids (e.g., E. coli, Klebsiella, Listeria monocytogenes, E. faecalis) [44,188,211], or ICEs (Integrative Conjugative Elements) (e.g., S. Typhimurium) [212]. The variability of mobile genetic elements carrying arsenic tolerance genes may favor their horizontal transfer between bacterial hosts. Also, when integrated and fixed in the chromosome, arsenic tolerance genes can confer a lower fitness cost to bacteria and be spread by vertical transmission. In all cases, there is a selective advantage for bacterial survival, particularly in food-animal production or other metal polluted environments. In fact, arsenic-polluted environments (e.g., water reservoirs, urban soils) have been described as contributing to the co-selection of ARGs [e.g., for aminoglycosides – aadA/aacC, beta-lactams – blaCMY/ampC, MLSB – erm(F) tetracyclines – tet(B)] and mobile genetic elements (e.g., integron – intI-1, transposon – Tn21/Tn22/Tn24/Tn614) [213,214]. The occurrence of arsenic and other metals (e.g., copper, zinc, cadmium, lead) in a Chinese poultry production environment has also recently been found to have a greater impact on MeT and ARGs gene composition than some antibiotics, showing a positive correlation between arsenic concentrations and resistance genes to aminoglycosides [aac(6’)-Ia], macrolides (erm35), bacitracin (bacA) and, in particular, with resistance genes to tetracycline (tet genes), probably promoted by co-selection events [154].
2.3. Mercury
Mercury (Hg) is a highly toxic heavy metal widely dispersed in nature [215]. Like arsenic and other heavy metals, mercury is a non-essential element for living organisms, with no known beneficial function for cells [216]. The toxicological properties of mercury depend on the different chemical forms in which it can occur [217]. In the environment and in biological systems, mercury can be present in three oxidation states, namely, elemental mercury (Hg0) (known as metallic mercury, a highly volatile liquid, at room temperature), and the mercuric [Hg2+ /Hg(II)] and mercurous [Hg+/Hg(I)] forms [218]. It can also occur as organic (or organomercuric) forms, such as the methylmercury (MeHg) ion (HgCH3+) and its compounds methylmercury chloride (CH3HgCl), methylmercury hydroxide (CH3HgOH), dimethylmercury and phenylmercury, identified as the most toxic forms of Hg [219,220]. The occurrence of these different chemical species depends on the environmental physicochemical features and how they are metabolized by different biological processes that occur in the local microbiota [217]. While Hg0 occurs mainly in the atmosphere, mercuric species [Hg(II)] are dominant in water, soil and sediments and methylmercury (MeHg) in biota [221].
Mercury is a natural component of the Earth’s crust, often found as salts such as mercury sulfide (HgS, known as cinnabar) and other sulfate minerals (e.g., HgSO4), mercury oxide (HgO), mercury chloride (HgCl2) or as elemental mercury [222]. It can be released into atmosphere through natural events such as volcanic activity, geothermal sources, biomass burning, and soil-water-air exchanges [223]. Both biotic (including bacteria) and abiotic (e.g., meteorological conditions, human activity) processes are involved in the transformation of mercury (geochemistry mercury cycle) into different inorganic and organic forms, as well as the gaseous element that returns to the atmosphere and contributes to its wide dispersion [224]. Nonetheless, 75% of the global mercury input and distribution to the environment is caused by extensive anthropogenic use [225], making it one of the most prevalent and persistent environmental pollutants [215].
Historical records reveal the use of quicksilver (liquid metallic mercury) in ancient Greek, Indian, Persian, Arabic and Chinese medicine and alchemy [226,227]. In fact, it has been employed in traditional Chinese medicine for over 3000 years [226]. Additionally, evidence suggests that this metal was used as a preservative in Egyptian tombs [226]. Mercury compounds gained significant importance in medical applications during the late 15th century in Europe, particularly in the treatment of syphilis [228]. Moreover, the use of mercury became common in the 20th century in many applications (e.g., dental amalgam fillings; drug preservative; antiseptics) [217,229,230]. Currently, it is still used in very small amounts as a preservative in some human and animal vaccines and pharmaceuticals, in the form of ethylmercury (known as thiomersal) [59]. In the agri-food sector, mercury was also used for decades, until the mid/late 20th century, in pesticides, mainly insecticides and fungicides, in the form of mercurous chloride and ethylmercury [230,231,232]. Although mercury contamination from industrial sources has declined globally in recent years due to stricter regulations (mainly as a result of Minamata Convention on Mercury involving several countries worldwide) [232,233], anthropogenic processes are still responsible for a significant input of mercury into the environment [221,233]. Among the main activities that have been contributing to environmental contamination with mercury are cinnabar mining, coal combustion for energy production (an important source of atmospheric mercury), cement production, metal processing (gold, silver), waste incineration (from urban, medical and industrial sources), chlor-alkali and steel industry and the production of electric equipment, paints and wood [223,232,234].
The extensive use of mercury in different applications has led to severe pollution in aquatic and terrestrial ecosystems. In recent years, a wide range of mercury concentrations have been found in soil (topsoil/agricultural land: 0 – 8 889 mg/kg) water (marine sediments: 0.0023 – 5 330 mg/kg; marine water: 0.5 – 27 060 ng/L; surface freshwater: 1.6 – 28.7 ng/L) [175,235,236,237], and across food webs, particularly in aquatic ecosystems where predatory fish (e.g., dusky grouper, barracuda, porbeagle) bioaccumulate mercury (sea fish: 0.001-3.1 mg/kg; estuarine/freshwater fish: 0.04 – 1.74 mg/kg) [238,239]. Given the wide distribution of mercury in the environment and the abundance of bacteria on Earth, microorganisms are commonly exposed to and affected by toxic levels of mercury [240]. As a result, there is a widespread prevalence of genetic determinants of mercury tolerance among bacterial populations, which allows their survival and adaptation in the presence of this toxic element in diverse environments. However, the mechanisms underlying mercury toxicity in bacterial cells are still not fully understood and continue to be the subject of study. Mercury exhibits a similar chemical reactivity to other metals (e.g., cadmium, lead, arsenic) within cells, where it binds to sulfhydryl groups of enzymes and proteins [241], causing changes in protein structure and often loss of function [242]. Recently, the affinity of mercury for the low molecular weight thiol molecules cysteine and glutathione (the most prevalent) and for proteins was described as involved in the replacement of essential zinc cofactors in DNA-binding proteins, which are involved in the transcription of tRNA genes and DNA repair, vital for many cellular functions [240].
Bacterial tolerance to mercury has been described in various Gram-positive and Gram-negative species from diverse sources (e.g., natural environments such as water, soil, and glaciers) or in human commensal/pathogenic bacteria [243,244,245,246], but mainly associated with environments contaminated by mercury [247]. In fact, the first description of bacterial mercury tolerance (phenotypic feature) occurred at a time when mercurial compounds were widely used as topical disinfectants and antiseptics in hospitals, community and food-producing animals [248,249], and it was observed in a clinical isolate of S. aureus also resistant to penicillin [250]. At the same time the role of some anaerobic bacteria in the geochemistry of mercury, participating in the production of the most toxic form, methylmercury, was recognized in aquatic bottom sediments and fish [251]. To cope with mercury toxicity, bacteria have evolved the ability to convert toxic forms of mercury into nontoxic or relatively less harmful species, including the reduction of the highly reactive Hg2+ to metallic Hg0 (relatively inert, water insoluble, and volatile) [252,253], or the degradation of organomercury compounds to inorganic mercury [248]. The mer operon is the most extensively studied cluster of genes that lead to mercury tolerance. It is highly variable among bacteria [248,254] and allows them to resist both inorganic and organic forms of mercury, known as narrow- or broad-spectrum mercury tolerance operons, respectively [215]. They typically consist of a combination of operators, regulators, promoter genes, and functional genes (e.g., merT, merP, merE, merC, merA, merG, merB and merD), all or part of which are present, which contribute to the proper functioning of the operon system [247] (Figure 6).
The central enzyme in the mercury detoxification system is the mercuric reductase – MerA (encoded by the merA gene) [252], a cytosolic flavin disulfide oxidoreductase, which uses NAD(P)H as a reducing agent [248]. This protein is responsible for the volatilization of mercury, catalyzing the conversion of Hg2+ to Hg0 [255], and it is present both in narrow- and broad-spectrum mer operons [215]. While exhibiting a similar function role, variations in MerA amino acid sequences have been observed among Gram-positive and Gram-negative bacteria [255], suggesting a distinct ancestral origin of the mer operon for these two bacterial groups during the course of evolution [255]. In addition to MerA, a cytoplasmatic organomercury lyase – MerB (encoded by the merB gene) might also occur, allowing bacteria to resist organomercurials [215], catalyzing the demethylation of organic mercury compounds by lysing the carbon-Hg bond, transforming it into relatively less toxic Hg2+, which is then reduced by MerA to form Hg0 [215]. Therefore, the merB gene is associated only with the broad-spectrum mer operon [215]. The presence of merB gene is more common in Gram-negative mer than in Gram-positive operons [248].
Other functional genes are primarily related to mercury transport and may include: merT, encoding an inner cytoplasmic membrane (MerT) protein responsible for accepting organic and inorganic mercury from MerP and transporting it to the cytoplasmic side of the membrane [248]; merP, which encodes a periplasmic scavenger protein that aids in the exchange of Hg2+ in the early transmembrane domain of MerP to MerT [215,248]; merE, which encodes a transport protein (MerE) that helps transport both inorganic and organic mercury compounds across the bacterial cell membrane into the cytoplasm [215,248]; and merC, which encodes an inner membrane-spanning transporter protein (MerC), which helps transport inorganic (Hg2+) and organic (C6H5Hg) mercury from the periplasm to the cytosol [215]. Additionally, merG is responsible for decreasing cell membrane permeability to phenylmercury (since it and other organomercurials can potentially undergo simple diffusion [248]), contributing along with merB to broad-spectrum resistance against mercurial compounds [256]. The merR gene is associated with mercury tolerance expression, as it encodes an Hg2+-dependent transacting activator-repressor protein (MerR), which activates the mer genes in the presence of Hg2+ or represses it when a deficiency in Hg2+ occurs [257]. Other genes are also involved in the regulation of the mer operon, including the merD gene, which encodes a regulatory protein (MerD), responsible for downregulation of the mercury tolerance system [215] and the merO gene, which is the operator region linked to the merR gene, responsible to upregulating and downregulating the expression of the mer operon genes [215]. A mercury tolerance phenotype associated with the presence of only the merR and merA genes was recently described in Enterococcus spp., with MICs to HgCl2 ranging between 16 and 64 μM, contrasting with those of 4-8 μM among isolates without such genes [150].
Mercury tolerance determinants are often located on the chromosome or plasmids of Gram-positive and Gram-negative bacteria, usually as components of transposable elements, in a striking diversity of arrangements [248]. The mer operon was first described in Gram-negative bacteria associated with Tn501 and related transposons [246] and since then several associations with plasmids and transposons have been identified in bacteria from natural environments [258] or with clinical relevance, including pathogenic strains of E. coli (e.g., genomic island GI-3) [259], and S. Typhimurium (e.g, GI-DT12 containing a Tn21-like transposon) [260]. In Gram-positive bacteria, mer operons have been found in diverse MGEs, including in S. aureus [e.g., plasmid pTW20_1 borne SCCmec (beta-lactamase) cassette] [261] and in E. faecalis and E. faecium (e.g., chromosomal Tn5385-like, pPPM1000) isolated from human (clinical) and animal samples, respectively [253,262]. The same mercury tolerance-associated transposons or plasmids often carry ARGs genes, which makes them potential vectors of multiple genes involved in co-resistance and co-selection events. Shortly after the first description of mercury tolerance in S. aureus resistant to penicillin, a plasmid (pI285) carrying both mercury tolerance and penicillin resistance genes was identified [189,263], along with other metal tolerance genes (arsenic/antimony, lead/zinc, cadmium) [189]. In recent years, several reports have been published on the co-occurrence of mercury tolerance, ARGs and biocide tolerance genes in the same MGEs, including in conjugative plasmids [253,264,265,266]. Specific associations of mercury tolerance genes with aminoglycosides [e.g., aac(3)-IV, aadA], sulfonamides (e.g., sul) or tetracycline [e.g., tet(A)] were described in plasmids of Klebsiella, Escherichia, Salmonella, and Enterobacter isolated from diverse sources (human, animal, wastewater and sludge) [133,134,267]. Additionally, co-location of mer operon genes with β-lactams genes (blaCTX-M, blaOXA, or blaTEM) has also been described in plasmids of K. pneumoniae, E. coli and Salmonella from clinical, surveillance, food and environmental samples [133,134,268,269,270]. In Gram-positive bacteria, particularly Enterococcus spp. from different sources (e.g., animal, healthy human, clinical, hospital sewage), mercury tolerance genes have been co-localized on plasmids with ARGs, mainly for erythromycin [erm(B)], tetracycline [tet(M), tet(L)], aminoglycosides [aadE, aadK, aac(6’)-aph(2’)] and vancomycin (vanA) [150,253]. The distribution of mercury tolerance genes in MGEs along with ARGs genes highlights the potential impact of mercury on the co-transfer and dissemination of such determinants among bacteria of different sources.
3. Organic acids
Organic acids are organic compounds with acidic properties [271], widely distributed in nature, either as natural constituents of plants and animals or metabolites of the activity of microorganisms (e.g., microbial fermentation) [55,272,273]. The most common organic acids comprise the carboxylic acids, distinguished from other acids by the presence of the –COOH functional group, to which an organic group or a hydrogen atom may be attached [274]. Among this group of compounds are the straight-chain saturated monocarboxylic acids and their derivates such as unsaturated (e.g., cinnamic, sorbic), hydroxylic (e.g., citric, lactic), phenolic (e.g., benzoic, cinnamic, salicylic) and multi-carboxylic (e.g., azelaic, citric, succinic) acids [274]. Chemically, organic acids are classified based on the number of hydroxy or carboxy functional groups and double bonds of carbon-carbon in their structures [271,275]. Other features, such as the nature of carbon chain (aromatic, aliphatic, alicyclic, and heterocyclic) and saturation properties are important to categorize these compounds [275]. The number of carboxyl groups or other functional groups (e.g., alcohol, phenol, thiol, enol, and OSO3H) determines the compounds’ acidity [271]. In general, organic acids are weak acids not dissociating completely in the presence of water [271].
Organic acids are suspected to have been used in their natural form since prehistoric times [272,276], having a long tradition in the preservation of food products [277]. Acting mainly in the inhibition of microbial growth, these compounds prevent the deterioration of food products and extend their shelf life, especially the most perishable ones [278,279]. Originally, they began to be used as fungistats in animal feed [279] and, with the discovery of their potential microbiocidal activity, they soon became widely applied in many products [279]. Currently, several organic acids and their salts are listed as food and feed additives in European legislation, most acting as preservatives and acidifiers (e.g., acetic, citric, formic, malic, fumaric, lactic, propionic, phosphoric, sorbic) [280,281]. In food-producing animals, organic acids have been suggested as alternatives to other antimicrobials for use in non-clinical animal management practices [55,279]. Thus, dietary supplementation with organic acids (e.g., fumaric, lactic, citric, formic, malic, sorbic, tartaric) in the feed and drinking water of animals for food production has become a common practice, given the benefits associated with weight gain and feed efficiency improvement [55,282]. In particular, the use of blends of various acids or their salts has been shown to enhance the beneficial effects of organic acids, improving feed conversion ratio [283,284]. Additionally, general recognition of the safety of organic acids in food products has led to their wider application as sanitizers, not just in the food production setting (e.g., disinfection of surfaces and equipment in food production settings, including slaughterhouses), but also in food products (e.g., disinfection of fruits and vegetables or animal carcasses) [285,286]. In Europe, the application of organic acid solutions (e.g., lactic, acetic, peroxyacetic acids) to reduce microbial surface contamination of animal carcasses and meat has been evaluated by EFSA [287,288], and the use of lactic acid is currently authorized in bovine carcasses [71]. Also, the application of organic acids (e.g., citric acid, succinic acid) has been tested for plant protection against phytopathogens (as bactericide, fungicide, nematicide) [289], although only acetic acid is currently authorized as an herbicide by some EU countries [290]. In recent years, promising new approaches have been explored in the food industry, including the use of organic acid-based antimicrobial packaging, which combined with different preservation technologies contribute to increasing the shelf life of products [278].
The effectiveness of organic acids as antimicrobial agents relies on their ability to penetrate cell membranes as protonated acids [291]. Organic acids show a great ability to penetrate the cell wall, which makes them compounds with higher antimicrobial activity than the highly dissociated inorganic acids at the same pH level [278]. This feature is related to the ability of the organic acid to exist in a pH-dependent equilibrium between the undissociated and dissociated state [292]. The undissociated form is predominant at low pH and is primarily responsible for antimicrobial activity as it can freely diffuse across the cell membrane into the cytoplasm [293]. Once inside the cell, the higher pH will promote acid dissociation, resulting in the release of charged anions and protons and their accumulation in the cytoplasm. This creates not only an intracellular pH shift out of the optimal range for enzyme activity, affecting protein and DNA/RNA synthesis [273,294,295,296], but also hinders the proton motive force and inhibits the cell’s ability to re-alkalinize its cytoplasm [297]. In fact, pH homeostasis is a critical factor for cell growth and metabolism, influencing nutrient uptake and utilization, substrate degradation, and protein and nucleic acid synthesis [273]. Since the undissociated form of the acid is responsible for the antimicrobial effect, the pKa dissociation constant is an important factor, representing the pH at which 50% of the acid is dissociated. Thus, the higher the pKa of an organic acid, the more effective it will be, a factor potentiated by other variables, including increasing the carbon chain length and the degree of unsaturation of the acid [293].
In contrast to other acids, peracetic acid (also known as peroxyacetic acid – PAA), widely used in the food and healthcare industries [298], also acts as a strong oxidant [299]. This organic peroxide (synthetic chemical) is available in the form of a quaternary equilibrium mixture containing acetic acid, hydrogen peroxide, PAA and water [300]. Thus, PAA combines the active oxygen characteristics of a peroxide within an acetic acid molecule [301] with the PAA, showing the highest biocidal activity [302]. Although there are few descriptions of PAA’s mode of action as an antimicrobial compound, its activity is assumed to be similar to other peroxides and oxygen agents [299,303], causing oxidative stress in the cell by oxidizing and disrupting sulfhydryl and sulfur bonds in proteins, enzymes and other metabolites [299,301]. It can also act on the lipoprotein cytoplasmic membrane, disrupting its chemiosmotic function [301]. Intracellular PAA can also oxidize essential enzymes and impair vital biochemical pathways, active transport across membranes and intracellular solute levels [301]. pH is one of the most important factors of PAA activity, affecting the acid-base balance of PAA, which in turn affects the generation of free radicals [300]. The pKa value of PAA is 8.2, which means that under acidic conditions the predominant species is the undissociated acid form. At acidic-neutral pH (3-7), reactive radicals (e.g., OH⋅) increase [300], which contributes to the oxidizing properties of PAA. Additionally, in acidic environments (pH < 5.5), the decomposition of PAA to acetic acid by protonation and the release of protons during this process [304] may also contribute to the antimicrobial activity of PAA.
Bacteria are often exposed to both strong and mild acidic environments, either within the human/animal host (e.g., dental plaque, gastrointestinal tract, macrophage phagosome) or outside in other human-associated niches, such as food processing and preservation [305], which creates a major challenge for the cell in maintaining pH homeostasis. In general, neutralophilic bacteria can grow at external pH values between ~5.5-9.0, while maintaining a cytoplasmic pH between ~7.2-7.8 (data reported for E. coli) [306]. However, when exposed to acid stress (pH 2.5-3.0), neutralophilic bacteria have evolved multiple tolerance or resistance mechanisms, responsible for increasing bacterial survival [305]. Cytoplasmic pH is buffered by small molecules (e.g., amino acids, proteins, polyamines, polyphosphate and inorganic phosphate), representing a passive system in regulating pH homeostasis [307]. However, to counteract acid stress, active systems involving physiological, metabolic and proton-consuming mechanisms are essential [307]. Common mechanisms involved in bacterial acid tolerance and part of the active systems include the decarboxylation of amino acids (e.g., glutamate, arginine or lysine), F1-F0-ATPase proton pump, and alkali production [308].
The decarboxylation of amino acids are enzyme-catalyzed reactions that consume protons [291]. Often called amino acid-dependent acid resistance systems, four distinct systems may be involved in bacterial defense against acid damage: a) the glutamic acid-depended acid resistance (GDAR) system; b) the arginine-dependent acid resistance (ADAR) system; c) the lysine-dependent acid resistance (LDAR) system; and d) the ornithine-dependent acid resistance (ODAR) system [307]. The GDAR system is present in several bacteria such as E. coli, Shigella flexnerii, L. monocytogenes, Lactobacillus reuteri and Enterococcus avium [308,309], and provides robust protection against extreme acid stress [310,311]. This system is responsible for catalyzing the conversion of protonated glutamate (Glu) to Glu/γ-aminobutyrate acid (GABA) and carbon dioxide, followed by the export of GABA through the GadC antiporter in exchange for a new extracellular Glu molecule (Figure 7) [308]. Recently, the gad gene (glutamate decarboxylase) was described in isolates of E. coli from chicken meat [43], suggesting an important feature for bacterial survival in food-producing animal environments, particularly poultry, where acidic pH can occur in different contexts (e.g., feed with organic acids additives, gastrointestinal tract of animals, processing plants using acidic disinfectants). In Salmonella enterica, the presence of genes associated with the ADAR (adiA - arginine decarboxylase and adiC – arginine-agmatine antiporter) and LDAR (cadA - lysine decarboxylase and cadB - lysine-cadaverine antiporter) systems has also been described as an important feature for neutralizing and surviving acid stress [312], allowing bacterial survival in harsh acidic environments (e.g., stomach, phago-lysosomes), determinant for the dissemination capacity and virulence of this food-borne pathogen. Additional decarboxylation pathways have been less studied in other bacteria, including tyrosine decarboxylation associated to the acid response mechanism in several lactic acid bacteria, such as Enterococcus spp., given them a competitive advantage in acidic environments [313].
Deamination of amine-containing amino acids [e.g., arginine (Arg), agmatine (Agm) or glutamine (Gln)] and the urease system are also important acid response mechanisms, being associated with the production of basic compounds such as ammonia (NH3), important to avoid a critical drop in internal pH (Figure 7) [291]. In the urease system, urea is hydrolyzed to NH3 and carbon dioxide (CO2) by ureases [308]. Furthermore, the conversion of Gln to Glu by acid-activated glutaminase (YbaS), of Arg to ornithine (Orn) by arginine deaminase (ADI system) and of Agm to putrescine (Putr) by agmatine deiminase (AgDI system) releases NH3 and CO2 (Figure 7) [305,308]. NH3 directly neutralizes protons and regulates the cytoplasmic pH [314].
Another important mechanism relies on the activity of proton pumps (e.g., H+-ATPase, symporter, antiporter) that promote proton efflux in a proton motive force (PMF) dependence system [273]. The efflux of protons out of the cell is an ATP-consuming process (Figure 7), which leads to a depletion of the energy available to cells and, consequently, affects their survival [273]. The F1-F0-ATPase is a bifunctional proton pump, catalyzing the synthesis and hydrolysis of ATP [315]. This multi-subunit enzyme uses the energy released from the movement of protons across cell membranes to generate ATP and, in a reverse reaction, hydrolyzes ATP to export protons across the membrane, thereby maintaining pH homeostasis particularly in acidic environments [308]. In fact, induction of the F1-F0 operon by exposure to acidic pH suggests that this enzyme plays a critical role in acid resistance in several bacteria [308].
In contrast to inorganic acids (e.g., hydrochloric acid), which primarily lower cytoplasmatic pH, organic acids have the additional ability to accumulate as intracellular anions [312]. When these anions accumulate in high concentrations within bacterial cells, they can exert inhibitory effects. As a result, bacteria have evolved mechanisms to efflux these anions using membrane pumps [316]. Consequently, the mechanisms involved in the acid stress response that are induced by organic acids appear to differ from those triggered by inorganic acids [317]. However, it is important to note that cells adapted to withstand inorganic acids also acquire resistance to acid stress induced by organic acids and vice versa [312]. Some organic acid tolerance mechanisms have been explored, mainly in organic acid producing bacteria (e.g., Acetobacter, Lactobacillus) [318,319]. In addition to those previously described (e.g., amino acid decarboxylation, proton pumps and neutralization processes), additional mechanisms have been reported, for example, the PQQ-ADH (pyrroloquinoline quinine dependent alcohol dehydrogenase) system, known to be involved in tolerance to acetic acid in acetic acid bacteria [320]. Interestingly, in acetic acid bacteria, the GDAR acid-resistance system is absent, and the urea degradation was down-regulated after acetic acid production [317]. In the case of PAA, certain bacteria, including pathogenic strains such as S. enterica [312], can induce the expression of genes associated with oxidative stress (e.g., SoxRS, OxyR and PerR regulon), with such induction being associated with a protective response against the activity of PAA [321]. In fact, in-use concentrations of PAA for food and feed area disinfection (20-3000 mg/L for Product Type-PT4) have recently been described as being, in some cases, lower than MIC (60-70 mg/L) and MBC (70-90 mg/L) shown by poultry associated S. enterica strains [322].
Unlike metals, limited information on the co-selection of antibiotic resistance and acid tolerance is available. A recent study using metagenomic approaches reveal the co-occurrence of the pmrA/B/C polymyxin resistance genes and actP acid resistance gene [323]. Furthermore, other acid resistance genes (e.g., gadE, hdeA, mdtE, mdtF, gadW, gadX, gadA) were co-located with metal tolerance genes (mainly arsenic – arsA/B/R) in the same contig [323]. In the case of PAA, the literature suggests the absence of a strong pattern between tolerance to PAA and resistance to antibiotics. For example, some studies have shown that exposure of S. enterica strains to sub-inhibitory concentrations of PAA (MIC/2; ~0.040 mg/mL) [324] resulted in increased resistance to streptomycin and neomycin [325], but this association appears to be strain-specific [325]. On the other hand, a study involving more than 500 S. enterica isolates from Danish pig slaughterhouses found little evidence of an association between increased MIC for PAA and antibiotic resistance [326]. Likewise, E. faecium exposed to low doses of PAA did not show changes in the abundance of ARGs [327]. In addition, no antibiotic cross-resistance was observed in L. monocytogenes from food production plants exposed to PAA [328]. Indeed, according to the EFSA there is no evidence to suggest that PAA can lead to acquired antibiotic resistance [329]. However, a recent study has suggested that reactive oxygen species may promote antibiotic resistance by increasing expression of the MDR efflux pump via activation of the SoxRS redox regulon [330], a mechanism that should not be ruled out due to the oxidative stress created by PAA.
4. Conclusions
While antibiotic overuse remains the main driving force behind the emergence of ARB in the agri-food sector, there is growing recognition of the potential role of other antimicrobial compounds for this problem. Metals, including copper, commonly found in feed, as well as pollutants such as arsenic and mercury that enter the food chain, can potentially contribute to the co-selection of ARB, often sharing metal tolerance genes and ARGs in diverse mobile genetic contexts. In contrast, the impact of widely used organic acids on the emergence of ARB and ARGs is still not fully understood, although there is limited evidence suggesting the need for further investigation in this area.
To effectively address the challenge of AMR, it is crucial to conduct comprehensive research focused on understanding the mechanisms of action and bacterial tolerance to both metals and organic acids, as well as their potential ecological impacts on microbiota diversity and the promotion of AMR emergence. It remains to be determined the minimum selective concentrations of metals and organic acids for particular MDR bacterial clones and mobile genetic elements, as well as those that promote horizontal gene transfer events or ARGs expression, especially on bacteria of clinical relevance to human and animals. By knowing so, we can optimize metals and organic acids use to mitigate the microbial risks associated with food production but also prevent pollution and develop holistic approaches to combating AMR in the food chain and in other environments beyond. These efforts will contribute to protecting human, animal and environmental health and ensure the long-term effectiveness of antimicrobial treatments for all.
Funding
This study was financed by national funds from FCT - Fundação para a Ciência e a Tecnologia, I.P., in the scope of the project UIDP/04378/2020 and UIDB/04378/2020 of the Research Unit on Applied Molecular Biosciences - UCIBIO, the project LA/P/0140/2020 of the Associate Laboratory Institute for Health and Bioeconomy - i4HB, by the AgriFood XXI I&D&I project (NORTE-01-0145-FEDER-000041) co-financed by European Regional Development Fund (ERDF) and through the NORTE 2020 (Programa Operacional Regional do Norte 2014/2020). Andreia Rebelo was supported by a PhD fellowship from FCT (SFRH/BD/137100/2018), co-financed by European Social Fund through Norte Portugal Regional Operational Program (NORTE 2020).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the writing of the manuscript; or in the decision to publish the results.
References
- World Health Organization (WHO). 10 global health issues to track in 2021. Available online: https://www.who.int/news-room/spotlight/10-global-health-issues-to-track-in-2021 (accessed on 9 May 2023).
- World Health Organization (WHO). Antimicrobial resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 9 May 2023).
- European Commission. AMR: A Major European and Global Challenge. 2017. Available online: https://health.ec.europa.eu/system/files/2020-01/amr_2017_factsheet_0.pdf (accessed on 24 May 2023).
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. The Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations, 2016.
- European Commission. Communication from the Commission to the council and the European Parliament: A European One Health Action Plan against Antimicrobial Resistance (AMR). 2017. Available online: https://health.ec.europa.eu/system/files/2020-01/amr_2017_action-plan_0.pdf.
- Levy, S.B.; Marshall, B. Antibacterial Resistance Worldwide: Causes, Challenges and Responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Rigol, S. A Brief History of Antibiotics and Select Advances in Their Synthesis. J. Antibiot. (Tokyo) 2018, 71, 153–184. [Google Scholar] [CrossRef]
- Bell, B.G.; Schellevis, F.; Stobberingh, E.; Goossens, H.; Pringle, M. A Systematic Review and Meta-Analysis of the Effects of Antibiotic Consumption on Antibiotic Resistance. BMC Infect. Dis. 2014, 14, 13. [Google Scholar] [CrossRef]
- Landers, T.F.; Cohen, B.; Wittum, T.E.; Larson, E.L. A Review of Antibiotic Use in Food Animals: Perspective, Policy, and Potential. Public Health Rep. 2012, 127, 4–22. [Google Scholar] [CrossRef]
- Holmes, A.H.; Moore, L.S.P.; Sundsfjord, A.; Steinbakk, M.; Regmi, S.; Karkey, A.; Guerin, P.J.; Piddock, L.J.V. Understanding the Mechanisms and Drivers of Antimicrobial Resistance. The Lancet 2016, 387, 176–187. [Google Scholar] [CrossRef]
- Marshall, B.M.; Levy, S.B. Food Animals and Antimicrobials: Impacts on Human Health. Clin. Microbiol. Rev. 2011, 24, 718–733. [Google Scholar] [CrossRef]
- EFSA Panel on Biological Hazards (BIOHAZ); Koutsoumanis, K.; Allende, A.; Álvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Herman, L.; et al. Role Played by the Environment in the Emergence and Spread of Antimicrobial Resistance (AMR) through the Food Chain. EFSA J. 2021, 19. [Google Scholar] [CrossRef]
- Interagency Coordination Group on Antimicrobial Resistance (IACG). No Time to Wait: Securing the Future from Drug-Resistant Infections - Report to the Secretary-General of the United Nations. 2019. Available online: https://www.who.int/docs/default-source/documents/no-time-to-wait-securing-the-future-from-drug-resistant-infections-en.pdf.
- McEwen, S.A.; Collignon, P.J. Antimicrobial Resistance: A One Health Perspective. Microbiol. Spectr. 2018, 6, 6210. [Google Scholar] [CrossRef]
- White, A.; Hughes, J.M. Critical Importance of a One Health Approach to Antimicrobial Resistance. EcoHealth 2019, 16, 404–409. [Google Scholar] [CrossRef]
- Thakur, S.; Gray, G.C. The Mandate for a Global “One Health” Approach to Antimicrobial Resistance Surveillance. Am. J. Trop. Med. Hyg. 2019, 100, 227–228. [Google Scholar] [CrossRef]
- Irfan, M.; Almotiri, A.; AlZeyadi, Z.A. Antimicrobial Resistance and Its Drivers—A Review. Antibiotics 2022, 11, 1362. [Google Scholar] [CrossRef]
- Samtiya, M.; Matthews, K.R.; Dhewa, T.; Puniya, A.K. Antimicrobial Resistance in the Food Chain: Trends, Mechanisms, Pathways, and Possible Regulation Strategies. Foods 2022, 11, 2966. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Antibiotic Resistance Threats in the United States, 2019; Centers for Disease Control and Prevention: United States, 2019. [Google Scholar] [CrossRef]
- Van Boeckel, T.P.; Pires, J.; Silvester, R.; Zhao, C.; Song, J.; Criscuolo, N.G.; Gilbert, M.; Bonhoeffer, S.; Laxminarayan, R. Global Trends in Antimicrobial Resistance in Animals in Low- and Middle-Income Countries. Science 2019, 365, eaaw1944. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations (FAO); World Health Organization (WHO). Foodborne Antimicrobial Resistance – Compendium of Codex standards; First revision; Codex Alimentarius Commission: Rome, 2022. [Google Scholar] [CrossRef]
- Ariza-Miguel, J.; Hernández, M.; Fernández-Natal, I.; Rodríguez-Lázaro, D. Methicillin-Resistant Staphylococcus aureus Harboring mecC in Livestock in Spain. J. Clin. Microbiol. 2014, 52, 4067–4069. [Google Scholar] [CrossRef]
- Quddoumi, S.S.; Bdour, S.M.; Mahasneh, A.M. Isolation and Characterization of Methicillin-resistant Staphylococcus aureus from Livestock and Poultry Meat. Ann. Microbiol. 2006, 56, 155–161. [Google Scholar] [CrossRef]
- Cui, S.; Li, J.; Hu, C.; Jin, S.; Li, F.; Guo, Y.; Ran, L.; Ma, Y. Isolation and Characterization of Methicillin-Resistant Staphylococcus aureus from Swine and Workers in China. J. Antimicrob. Chemother. 2009, 64, 680–683. [Google Scholar] [CrossRef]
- Pantosti, A.; Del Grosso, M.; Tagliabue, S.; Macri, A.; Caprioli, A. Decrease of Vancomycin-Resistant Enterococci in Poultry Meat after Avoparcin Ban. The Lancet 1999, 354, 741–742. [Google Scholar] [CrossRef]
- Lemcke, R.; Bülte, M. Occurrence of the Vancomycin-Resistant Genes vanA, vanB, vanC1, vanC2 and vanC3 in Enterococcus Strains Isolated from Poultry and Pork. Int. J. Food Microbiol. 2000, 60, 185–194. [Google Scholar] [CrossRef]
- Eisner, A.; Feierl, G.; Gorkiewicz, G.; Dieber, F.; Kessler, H.H.; Marth, E.; Köfer, J. High Prevalence of VanA-Type Vancomycin-Resistant Enterococci in Austrian Poultry. Appl. Environ. Microbiol. 2005, 71, 6407–6409. [Google Scholar] [CrossRef]
- Bates, J.; Jordens, J.Z.; Griffiths, D.T. Farm Animals as a Putative Reservoir for Vancomycin-Resistant Enterococcal Infection in Man. J. Antimicrob. Chemother. 1994, 34, 507–514. [Google Scholar] [CrossRef]
- Suzuki, S.; Ohnishi, M.; Kawanishi, M.; Akiba, M.; Kuroda, M. Investigation of a Plasmid Genome Database for Colistin-Resistance Gene mcr-1. Lancet Infect. Dis. 2016, 16, 284–285. [Google Scholar] [CrossRef]
- Tse, H.; Yuen, K.-Y. Dissemination of the mcr-1 Colistin Resistance Gene. Lancet Infect. Dis. 2016, 16, 145–146. [Google Scholar] [CrossRef]
- Webb, H.E.; Granier, S.A.; Marault, M.; Millemann, Y.; Den Bakker, H.C.; Nightingale, K.K.; Bugarel, M.; Ison, S.A.; Scott, H.M.; Loneragan, G.H. Dissemination of the mcr-1 Colistin Resistance Gene. Lancet Infect. Dis. 2016, 16, 144–145. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, P.; Du, P.; Zhang, X.; Wang, J.; Yang, Y.; Sun, H.; Wang, Z.; Cui, S.; Li, R.; et al. Prevalence and Genomic Characteristics of Mcr -Positive Escherichia coli Strains Isolated from Humans, Pigs, and Foods in China. Microbiol. Spectr. 2023, 11, e04569-22. [Google Scholar] [CrossRef]
- Liu, B.-T.; Zhang, X.-Y.; Wan, S.-W.; Hao, J.-J.; Jiang, R.-D.; Song, F.-J. Characteristics of Carbapenem-Resistant Enterobacteriaceae in Ready-to-Eat Vegetables in China. Front. Microbiol. 2018, 9, 1147. [Google Scholar] [CrossRef]
- Köck, R.; Daniels-Haardt, I.; Becker, K.; Mellmann, A.; Friedrich, A.W.; Mevius, D.; Schwarz, S.; Jurke, A. Carbapenem-Resistant Enterobacteriaceae in Wildlife, Food-Producing, and Companion Animals: A Systematic Review. Clin. Microbiol. Infect. 2018, 24, 1241–1250. [Google Scholar] [CrossRef]
- Gao, Y.; Wen, J.; Wang, S.; Xu, X.; Zhan, Z.; Chen, Z.; Bai, J.; Qu, X.; Zhang, H.; Zhang, J.; et al. Plasmid-Encoded blaNDM-5 Gene That Confers High-Level Carbapenem Resistance in Salmonella Typhimurium of Pork Origin. Infect. Drug Resist. 2020, Volume 13, 1485–1490. [Google Scholar] [CrossRef]
- Wang, Z.; He, J.; Li, Q.; Tang, Y.; Wang, J.; Pan, Z.; Chen, X.; Jiao, X. First Detection of NDM-5-Positive Salmonella enterica Serovar Typhimurium Isolated from Retail Pork in China. Microb. Drug Resist. 2020, 26, 434–437. [Google Scholar] [CrossRef]
- Bennani, H.; Mateus, A.; Mays, N.; Eastmure, E.; Stärk, K.D.C.; Häsler, B. Overview of Evidence of Antimicrobial Use and Antimicrobial Resistance in the Food Chain. Antibiotics 2020, 9, 49. [Google Scholar] [CrossRef]
- European Food Safety Authority; European Centre for Disease Prevention and Control. The European Union Summary Report on Antimicrobial Resistance in Zoonotic and Indicator Bacteria from Humans, Animals and Food in 2019–2020. EFSA J. 2022, 20. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations (FAO). The FAO Action Plan on Antimicrobial Resistance 2021–2025. 2021. [Google Scholar] [CrossRef]
- Tang, K.L.; Caffrey, N.P.; Nóbrega, D.B.; Cork, S.C.; Ronksley, P.E.; Barkema, H.W.; Polachek, A.J.; Ganshorn, H.; Sharma, N.; Kellner, J.D.; et al. Restricting the Use of Antibiotics in Food-Producing Animals and Its Associations with Antibiotic Resistance in Food-Producing Animals and Human Beings: A Systematic Review and Meta-Analysis. Lancet Planet. Health 2017, 1, e316–e327. [Google Scholar] [CrossRef]
- Postma, M.; Vanderhaeghen, W.; Sarrazin, S.; Maes, D.; Dewulf, J. Reducing Antimicrobial Usage in Pig Production without Jeopardizing Production Parameters. Zoonoses Public Health 2017, 64, 63–74. [Google Scholar] [CrossRef]
- Ribeiro, S.; Mourão, J.; Novais, Â.; Campos, J.; Peixe, L.; Antunes, P. From Farm to Fork: Colistin Voluntary Withdrawal in Portuguese Farms Reflected in Decreasing Occurrence of Mcr-1- Carrying Enterobacteriaceae from Chicken Meat. Environ. Microbiol. 2021, 23, 7563–7577. [Google Scholar] [CrossRef]
- Mourão, J.; Ribeiro-Almeida, M.; Novais, C.; Magalhães, M.; Rebelo, A.; Ribeiro, S.; Peixe, L.; Novais, Â.; Antunes, P. From Farm to Fork: Persistence of Clinically-Relevant Multidrug-Resistant and Copper-Tolerant Klebsiella pneumoniae Long after Colistin Withdrawal in Poultry Production. Microbiol. Spectr. 2023. [CrossRef]
- Direção Geral da Saúde. Plano Nacional de Combate à Resistência Aos Antimicrobianos 2019–2023. Âmbito Do Conceito “Uma Só Saúde,” 2019.
- European Commission. Commission Implementing Regulation (EU) on Designating Antimicrobials or Groups of Antimicrobials Reserved for Treatment of Certain Infections in Humans, in Accordance with Regulation (EU) 2019/6 of the European Parliament and of the Council; OJ L191/58 2022. 2022. [Google Scholar]
- European Medicines Agency (EMA) Committee for Medicinal Products for Veterinary Use (CVMP) and EFSA Panel on Biological Hazards (BIOHAZ); Murphy, D.; Ricci, A.; Auce, Z.; Beechinor, J.G.; Bergendahl, H.; Breathnach, R.; Bureš, J.; Duarte Da Silva, J.P.; Hederová, J.; et al. EMA and EFSA Joint Scientific Opinion on Measures to Reduce the Need to Use Antimicrobial Agents in Animal Husbandry in the European Union, and the Resulting Impacts on Food Safety (RONAFA). EFSA J. 2017, 15. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA), European Surveillance of Veterinary Antimicrobial Consumption. “Sales of Veterinary Antimicrobial Agents in 31 European Countries in 2021,” 2022.
- Kirchhelle, C. Pharming Animals: A Global History of Antibiotics in Food Production (1935–2017). Palgrave Commun. 2018, 4, 96. [Google Scholar] [CrossRef]
- European Commission. A Farm to Fork Strategy for a Fair, Healthy and Environmentally-Friendly DG SANTE/Unit ‘Food Information and Composition, Food Waste’. 2020. Available online: https://food.ec.europa.eu/system/files/2020-05/f2f_action-plan_2020_strategy-info_en.pdf.
- Cheng, G.; Hao, H.; Xie, S.; Wang, X.; Dai, M.; Huang, L.; Yuan, Z. Antibiotic Alternatives: The Substitution of Antibiotics in Animal Husbandry? Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- World Health Organization, Regional Office for Europe. Tackling Antibiotic Resistance from a Food Safety Perspective in Europe. 2021. Available online: https://apps.who.int/iris/handle/10665/326398.
- Mehdi, Y.; Létourneau-Montminy, M.-P.; Gaucher, M.-L.; Chorfi, Y.; Suresh, G.; Rouissi, T.; Brar, S.K.; Côté, C.; Ramirez, A.A.; Godbout, S. Use of Antibiotics in Broiler Production: Global Impacts and Alternatives. Anim. Nutr. 2018, 4, 170–178. [Google Scholar] [CrossRef]
- Shannon, M.C.; Hill, G.M. Trace Mineral Supplementation for the Intestinal Health of Young Monogastric Animals. Front. Vet. Sci. 2019, 6, 73. [Google Scholar] [CrossRef]
- Gadde, U.; Kim, W.H.; Oh, S.T.; Lillehoj, H.S. Alternatives to Antibiotics for Maximizing Growth Performance and Feed Efficiency in Poultry: A Review. Anim. Health Res. Rev. 2017, 18, 26–45. [Google Scholar] [CrossRef]
- Rensing, C.; Moodley, A.; Cavaco, L.M.; McDevitt, S.F. Resistance to Metals Used in Agricultural Production. In Antimicrobial Resistance in Bacteria from Livestock and Companion Animals; Schwarz, S., Cavaco, L.M., Shen, J., Eds.; ASM Press: Washington, DC, USA, 2018; pp. 83–107. [Google Scholar] [CrossRef]
- Nachman, K.E.; Baron, P.A.; Raber, G.; Francesconi, K.A.; Navas-Acien, A.; Love, D.C. Roxarsone, Inorganic Arsenic, and Other Arsenic Species in Chicken: A U.S.-Based Market Basket Sample. Environ. Health Perspect. 2013, 121, 818–824. [Google Scholar] [CrossRef]
- European Medicines Agency (EMA), Committee for Veterinary Medicinal Products (CVMP). Copper Chloride, Copper Gluconate, Copper Heptanoate, Copper Oxide, Copper Methionate, Copper Sulfate and Dicopper Oxide: Summary Report, 1998. Available online: https://www.ema.europa.eu/en/documents/mrl-report/copper-chloride-copper-gluconate-copper-heptanoate-copper-oxide-copper-methionate-copper-sulphate_en.pdf (accessed on 10 May 2023).
- European Medicines Agency (EMA), Committee for Veterinary Medicinal Products (CVMP). Thiomersal and Timerfonate Summary Report, 1996. Available online: https://www.ema.europa.eu/en/documents/mrl-report/thiomersal-timerfonate-summary-report-committee-veterinary-medicinal-products_en.pdf (accessed on 10 May 2023).
- European Medicines Agency (EMA), Committee for Veterinary Medicinal Products (CVMP). Zinc Salts Summary Report, 1996. Available online: https://www.ema.europa.eu/en/documents/mrl-report/zinc-salts-summary-report-committee-veterinary-medicinal-products_en.pdf (accessed on 10 May 2023).
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.; Dowson, C.G.; Dujardin, G.; Jung, N.; et al. Metal Complexes as a Promising Source for New Antibiotics. Chem. Sci. 2020, 11, 2627–2639. [Google Scholar] [CrossRef]
- Lemire, J.A.; Harrison, J.J.; Turner, R.J. Antimicrobial Activity of Metals: Mechanisms, Molecular Targets and Applications. Nat. Rev. Microbiol. 2013, 11, 371–384. [Google Scholar] [CrossRef]
- Puschenreiter, M.; Horak, O.; Friesl, W.; Hartl, W. Low-Cost Agricultural Measures to Reduce Heavy Metal Transfer into the Food Chain - a Review. Plant Soil Environ. 2005, 51, 1–11. [Google Scholar] [CrossRef]
- Vareda, J.P.; Valente, A.J.M.; Durães, L. Assessment of Heavy Metal Pollution from Anthropogenic Activities and Remediation Strategies: A Review. J. Environ. Manage. 2019, 246, 101–118. [Google Scholar] [CrossRef]
- Hu, Y.; Cheng, H.; Tao, S. Environmental and Human Health Challenges of Industrial Livestock and Poultry Farming in China and Their Mitigation. Environ. Int. 2017, 107, 111–130. [Google Scholar] [CrossRef]
- European Commission. Commission Implementing Regulation (EU) 2018/1039 of 23 July 2018 concerning the authorization of Copper(II) diacetate monohydrate, Copper(II) carbonate dihydroxy monohydrate, Copper(II) chloride dihydrate, Copper(II) oxide, Copper(II) sulphate pentahydrate, Copper(II) chelate of amino acids hydrate, Copper(II) chelate of protein hydrolysates, Copper(II) chelate of glycine hydrate (solid) and Copper(II) chelate of glycine hydrate (liquid) as feed additives for all animal species and amending Regulations (EC) No 1334/2003, (EC) No 479/2006 and (EU) No 349/2010 and Implementing Regulations (EU) No 269/2012, (EU) No 1230/2014 and (EU) 2016/2261. 2018, OJ L186/3.
- European Commission. Commission Implementing Regulation (EU) 2016/1095 of 6 July 2016—Concerning the authorisation of Zinc acetate dihydrate, Zinc chloride anhydrous, Zinc oxide, Zinc sulphate heptahydrate, Zinc sulphate monohydrate, Zinc chelate of amino acids hydrate, Zinc chelate of protein hydrolysates, Zinc chelate of glycine hydrate (solid) and Zinc chelate of glycine hydrate (liquid) as feed additives for all animal species and amending Regulations (EC) No 1334/2003, (EC) No 479/2006, (EU) No 335/2010 and Implementing Regulations (EU) No 991 / 2012 and (EU) No 636/2013. 2016, OJ L182/7.
- Ölmez, H.; Kretzschmar, U. Potential Alternative Disinfection Methods for Organic Fresh-Cut Industry for Minimizing Water Consumption and Environmental Impact. LWT - Food Sci. Technol. 2009, 42, 686–693. [Google Scholar] [CrossRef]
- Kim, J.; Huang, C.-H. Reactivity of Peracetic Acid with Organic Compounds: A Critical Review. ACS EST Water 2021, 1, 15–33. [Google Scholar] [CrossRef]
- European Chemicals Agency (ECHA). Peracetic acid. Available online: https://echa.europa.eu/es/substance-information/-/substanceinfo/100.001.079.
- European Commission. Commission Regulation (EU) No 101/2013 of 4 February 2013 Concerning the Use of Lactic Acid to Reduce Microbiological Surface Contamination on Bovine carcases. 2013, OJ L3471.
- European Commission. Commission Directive of 8 July 1985 Amending the Annexes to Council Directive 70/524/EEC Concerning Additives in Feedingstuffs (85/429/EEC); 1985, OJ L 245.
- Davies, R.; Wales, A. Antimicrobial Resistance on Farms: A Review Including Biosecurity and the Potential Role of Disinfectants in Resistance Selection. Compr. Rev. Food Sci. Food Saf. 2019, 18, 753–774. [Google Scholar] [CrossRef]
- Yazdankhah, S.; Rudi, K.; Bernhoft, A. Zinc and Copper in Animal Feed – Development of Resistance and Co-Resistance to Antimicrobial Agents in Bacteria of Animal Origin. Microb. Ecol. Health Dis. 2014, 25. [Google Scholar] [CrossRef]
- Pal, C.; Asiani, K.; Arya, S.; Rensing, C.; Stekel, D.J.; Larsson, D.G.J.; Hobman, J.L. Metal Resistance and Its Association With Antibiotic Resistance. Advances in Microbial Physiology 2017, 70, 261–313. [Google Scholar] [CrossRef]
- Jutkina, J.; Marathe, N.P.; Flach, C.-F.; Larsson, D.G.J. Antibiotics and Common Antibacterial Biocides Stimulate Horizontal Transfer of Resistance at Low Concentrations. Sci. Total Environ. 2018, 616–617, 172–178. [Google Scholar] [CrossRef]
- Li, X.; Gu, A.Z.; Zhang, Y.; Xie, B.; Li, D.; Chen, J. Sub-Lethal Concentrations of Heavy Metals Induce Antibiotic Resistance via Mutagenesis. J. Hazard. Mater. 2019, 369, 9–16. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y.; Song, H.; Lu, J.; Yuan, Z.; Guo, J. Copper Nanoparticles and Copper Ions Promote Horizontal Transfer of Plasmid-Mediated Multi-Antibiotic Resistance Genes across Bacterial Genera. Environ. Int. 2019, 129, 478–487. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, A.Z.; Cen, T.; Li, X.; He, M.; Li, D.; Chen, J. Sub-Inhibitory Concentrations of Heavy Metals Facilitate the Horizontal Transfer of Plasmid-Mediated Antibiotic Resistance Genes in Water Environment. Environ. Pollut. 2018, 237, 74–82. [Google Scholar] [CrossRef]
- Dalecki, A.G.; Crawford, C.L.; Wolschendorf, F. Copper and Antibiotics. Advances in Microbial Physiology 2017, 70, 193–260. [Google Scholar] [CrossRef]
- Samanovic, M.I.; Ding, C.; Thiele, D.J.; Darwin, K.H. Copper in Microbial Pathogenesis: Meddling with the Metal. Cell Host Microbe 2012, 11, 106–115. [Google Scholar] [CrossRef]
- Ladomersky, E.; Petris, M.J. Copper Tolerance and Virulence in Bacteria. Metallomics 2015, 7, 957–964. [Google Scholar] [CrossRef]
- Kim, B.-E.; Nevitt, T.; Thiele, D.J. Mechanisms for Copper Acquisition, Distribution and Regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef]
- Festa, R.A.; Thiele, D.J. Copper: An Essential Metal in Biology. Curr. Biol. 2011, 21, R877–R883. [Google Scholar] [CrossRef]
- Tavares, P.; Pereira, A.S.; Moura, J.J.G.; Moura, I. Metalloenzymes of the Denitrification Pathway. J. Inorg. Biochem. 2006, 100, 2087–2100. [Google Scholar] [CrossRef]
- Frei, A.; Verderosa, A.D.; Elliott, A.G.; Zuegg, J.; Blaskovich, M.A.T. Metals to Combat Antimicrobial Resistance. Nat. Rev. Chem. 2023, 7, 202–224. [Google Scholar] [CrossRef]
- Vincent, M.; Hartemann, P.; Engels-Deutsch, M. Antimicrobial Applications of Copper. Int. J. Hyg. Environ. Health 2016, 219, 585–591. [Google Scholar] [CrossRef]
- Dollwet, H.H.A.; Sorenson, J.R.J. Historic Uses of Copper Compounds in Medicine. Trace Elem. Med. 1985, 2, 80–87. [Google Scholar]
- Lamichhane, J.R.; Osdaghi, E.; Behlau, F.; Köhl, J.; Jones, J.B.; Aubertot, J.-N. Thirteen Decades of Antimicrobial Copper Compounds Applied in Agriculture. A Review. Agron. Sustain. Dev. 2018, 38, 28. [Google Scholar] [CrossRef]
- Kuehne, S.; Roßberg, D.; Röhrig, P.; Von Mehring, F.; Weihrauch, F.; Kanthak, S.; Kienzle, J.; Patzwahl, W.; Reiners, E.; Gitzel, J. The Use of Copper Pesticides in Germany and the Search for Minimization and Replacement Strategies. Org. Farming 2017, 3, 66–75. [Google Scholar] [CrossRef]
- Adrees, M.; Ali, S.; Rizwan, M.; Ibrahim, M.; Abbas, F.; Farid, M.; Zia-ur-Rehman, M.; Irshad, M.K.; Bharwana, S.A. The Effect of Excess Copper on Growth and Physiology of Important Food Crops: A Review. Environ. Sci. Pollut. Res. 2015, 22, 8148–8162. [Google Scholar] [CrossRef]
- Grass, G.; Rensing, C.; Solioz, M. Metallic Copper as an Antimicrobial Surface. Appl. Environ. Microbiol. 2011, 77, 1541–1547. [Google Scholar] [CrossRef]
- European Chemicals Agency (ECHA). Information on biocides. Available online: https://echa.europa.eu/pt/information-on-chemicals/biocidal-active-substances?p_p_id=dissactivesubstances_WAR_dissactivesubstancesportlet&p_p_lifecycle=1&p_p_state=normal&p_p_mode=view&_dissactivesubstances_WAR_dissactivesubstancesportlet_javax.portlet.a (accessed on 13 May 2023).
- Montero, D.A.; Arellano, C.; Pardo, M.; Vera, R.; Gálvez, R.; Cifuentes, M.; Berasain, M.A.; Gómez, M.; Ramírez, C.; Vidal, R.M. Antimicrobial Properties of a Novel Copper-Based Composite Coating with Potential for Use in Healthcare Facilities. Antimicrob. Resist. Infect. Control 2019, 8, 3. [Google Scholar] [CrossRef]
- Noyce, J.O.; Michels, H.; Keevil, C.W. Use of Copper Cast Alloys To Control Escherichia coli O157 Cross-Contamination during Food Processing. Appl. Environ. Microbiol. 2006, 72, 4239–4244. [Google Scholar] [CrossRef]
- Bharadishettar, N.; Bhat K, U.; Bhat Panemangalore, D. Coating Technologies for Copper Based Antimicrobial Active Surfaces: A Perspective Review. Metals 2021, 11, 711. [Google Scholar] [CrossRef]
- DeFlorio, W.; Liu, S.; White, A.R.; Taylor, T.M.; Cisneros-Zevallos, L.; Min, Y.; Scholar, E.M.A. Recent Developments in Antimicrobial and Antifouling Coatings to Reduce or Prevent Contamination and Cross-contamination of Food Contact Surfaces by Bacteria. Compr. Rev. Food Sci. Food Saf. 2021, 20, 3093–3134. [Google Scholar] [CrossRef]
- Marcus, E.-L.; Yosef, H.; Borkow, G.; Caine, Y.; Sasson, A.; Moses, A.E. Reduction of Health Care–Associated Infection Indicators by Copper Oxide–Impregnated Textiles: Crossover, Double-Blind Controlled Study in Chronic Ventilator-Dependent Patients. Am. J. Infect. Control 2017, 45, 401–403. [Google Scholar] [CrossRef]
- Hewawaduge, C.; Senevirathne, A.; Jawalagatti, V.; Kim, J.W.; Lee, J.H. Copper-Impregnated Three-Layer Mask Efficiently Inactivates SARS-CoV2. Environ. Res. 2021, 196, 110947. [Google Scholar] [CrossRef]
- Liu, H.; Tang, Y.; Zhang, S.; Liu, H.; Wang, Z.; Li, Y.; Wang, X.; Ren, L.; Yang, K.; Qin, L. Anti-Infection Mechanism of a Novel Dental Implant Made of Titanium-Copper (TiCu) Alloy and Its Mechanism Associated with Oral Microbiology. Bioact. Mater. 2022, 8, 381–395. [Google Scholar] [CrossRef]
- Melamed, E.; Kiambi, P.; Okoth, D.; Honigber, I.; Tamir, E.; Borkow, G. Healing of Chronic Wounds by Copper Oxide-Impregnated Wound Dressings—Case Series. Medicina (Mex.) 2021, 57, 296. [Google Scholar] [CrossRef]
- Forouzandeh, A.; Blavi, L.; Abdelli, N.; Melo-Duran, D.; Vidal, A.; Rodríguez, M.; Monteiro, A.N.T.R.; Pérez, J.F.; Darwich, L.; Solà-Oriol, D. Effects of Dicopper Oxide and Copper Sulfate on Growth Performance and Gut Microbiota in Broilers. Poult. Sci. 2021, 100, 101224. [Google Scholar] [CrossRef]
- Espinosa, C.D.; Stein, H.H. Digestibility and Metabolism of Copper in Diets for Pigs and Influence of Dietary Copper on Growth Performance, Intestinal Health, and Overall Immune Status: A Review. J. Anim. Sci. Biotechnol. 2021, 12, 13. [Google Scholar] [CrossRef]
- da Cruz Ferreira Júnior, H.; da Silva, D.L.; de Carvalho, B.R.; de Oliveira, H.C.; Cunha Lima Muniz, J.; Junior Alves, W.; Eugene Pettigrew, J.; Eliza Facione Guimarães, S.; da Silva Viana, G.; Hannas, M.I. Broiler Responses to Copper Levels and Sources: Growth, Tissue Mineral Content, Antioxidant Status and mRNA Expression of Genes Involved in Lipid and Protein Metabolism. BMC Vet. Res. 2022, 18, 223. [Google Scholar] [CrossRef]
- Lu, W.B.; Kuang, Y.G.; Ma, Z.X.; Liu, Y.G. The Effect of Feeding Broiler with Inorganic, Organic, and Coated Trace Minerals on Performance, Economics, and Retention of Copper and Zinc. J. Appl. Poult. Res. 2020, 29, 1084–1090. [Google Scholar] [CrossRef]
- De Marco, M.; Zoon, M.V.; Margetyal, C.; Picart, C.; Ionescu, C. Dietary Administration of Glycine Complexed Trace Minerals Can Improve Performance and Slaughter Yield in Broilers and Reduces Mineral Excretion. Anim. Feed Sci. Technol. 2017, 232, 182–189. [Google Scholar] [CrossRef]
- Scott, A.; Vadalasetty, K.P.; Chwalibog, A.; Sawosz, E. Copper Nanoparticles as an Alternative Feed Additive in Poultry Diet: A Review. Nanotechnol. Rev. 2018, 7, 69–93. [Google Scholar] [CrossRef]
- Pathakoti, K.; Manubolu, M.; Hwang, H.-M. Nanostructures: Current Uses and Future Applications in Food Science. J. Food Drug Anal. 2017, 25, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elsalam, K.A. Copper-Based Nanomaterials: Next-Generation Agrochemicals: A Note from the Editor. In Copper Nanostructures: Next-Generation of Agrochemicals for Sustainable Agroecosystems; Elsevier, 2022; pp. 1–14. [Google Scholar] [CrossRef]
- Bondarczuk, K.; Piotrowska-Seget, Z. Molecular Basis of Active Copper Resistance Mechanisms in Gram-Negative Bacteria. Cell Biol. Toxicol. 2013, 29, 397–405. [Google Scholar] [CrossRef]
- Puig, S.; Rees, E.M.; Thiele, D.J. The ABCDs of Periplasmic Copper Trafficking. Structure 2002, 10, 1292–1295. [Google Scholar] [CrossRef]
- Dupont, C.L.; Grass, G.; Rensing, C. Copper Toxicity and the Origin of Bacterial Resistance—New Insights and Applications. Metallomics 2011, 3, 1109. [Google Scholar] [CrossRef]
- Banci, L. Metallomics and the Cell; Metal ions in life sciences; Springer: Dordrecht Heidelberg New York London, 2013. [Google Scholar] [CrossRef]
- Macomber, L.; Imlay, J.A. The Iron-Sulfur Clusters of Dehydratases Are Primary Intracellular Targets of Copper Toxicity. Proc. Natl. Acad. Sci. 2009, 106, 8344–8349. [Google Scholar] [CrossRef]
- Besold, A.N.; Culbertson, E.M.; Culotta, V.C. The Yin and Yang of Copper during Infection. JBIC J. Biol. Inorg. Chem. 2016, 21, 137–144. [Google Scholar] [CrossRef]
- Purohit, R.; Ross, M.O.; Batelu, S.; Kusowski, A.; Stemmler, T.L.; Hoffman, B.M.; Rosenzweig, A.C. Cu + -Specific CopB Transporter: Revising P 1B -Type ATPase Classification. Proc. Natl. Acad. Sci. 2018, 115, 2108–2113. [Google Scholar] [CrossRef]
- Odermatt, A.; Suter, H.; Krapf, R.; Solioz, M. An ATPase Operon Involved in Copper Resistance by Enterococcus hirae. Ann. N. Y. Acad. Sci. 1992, 671, 484–486. [Google Scholar] [CrossRef]
- Solioz, M.; Stoyanov, J.V. Copper Homeostasis in Enterococcus hirae. FEMS Microbiol. Rev. 2003, 27, 183–195. [Google Scholar] [CrossRef]
- Solioz, M.; Odermatt, A. Copper and Silver Transport by CopB-ATPase in Membrane Vesicles of Enterococcus hirae. J. Biol. Chem. 1995, 270, 9217–9221. [Google Scholar] [CrossRef]
- Cobine, P.; Wickramasinghe, W.A.; Harrison, M.D.; Weber, T.; Solioz, M.; Dameron, C.T. The Enterococcus hirae Copper Chaperone CopZ Delivers Copper(I) to the CopY Repressor. FEBS Lett. 1999, 445, 27–30. [Google Scholar] [CrossRef]
- Strausak, D.; Solioz, M. CopY Is a Copper-Inducible Repressor of the Enterococcus hirae Copper ATPases. J. Biol. Chem. 1997, 272, 8932–8936. [Google Scholar] [CrossRef]
- Rensing, C.; Grass, G. Escherichia coli Mechanisms of Copper Homeostasis in a Changing Environment. FEMS Microbiol. Rev. 2003, 27, 197–213. [Google Scholar] [CrossRef]
- Outten, F.W.; Huffman, D.L.; Hale, J.A.; O’Halloran, T.V. The Independent Cue and cus Systems Confer Copper Tolerance during Aerobic and Anaerobic Growth in Escherichia coli. J. Biol. Chem. 2001, 276, 30670–30677. [Google Scholar] [CrossRef]
- Chaturvedi, K.S.; Henderson, J.P. Pathogenic Adaptations to Host-Derived Antibacterial Copper. Front. Cell. Infect. Microbiol. 2014, 4. [Google Scholar] [CrossRef]
- Brown, N.L.; Barrett, S.R.; Camakaris, J.; Rouch, D.A. Molecular Genetics and Transport Analysis of the Copper-Resistance Determinant (Pco) from Escherichia coli Plasmid pRJ1004. Mol. Microbiol. 1995, 17, 1153–1166. [Google Scholar] [CrossRef]
- Rouch, D.A.; Brown, N.L. Copper-Inducible Transcriptional Regulation at Two Promoters in the Escherichia coli Copper Resistance Determinant Pco. Microbiology 1997, 143, 1191–1202. [Google Scholar] [CrossRef]
- Lee, S.M.; Grass, G.; Rensing, C.; Barrett, S.R.; Yates, C.J.D.; Stoyanov, J.V.; Brown, N.L. The Pco Proteins Are Involved in Periplasmic Copper Handling in Escherichia coli. Biochem. Biophys. Res. Commun. 2002, 295, 616–620. [Google Scholar] [CrossRef]
- Zimmermann, M.; Udagedara, S.R.; Sze, C.M.; Ryan, T.M.; Howlett, G.J.; Xiao, Z.; Wedd, A.G. PcoE — A Metal Sponge Expressed to the Periplasm of Copper Resistance Escherichia coli. Implication of Its Function Role in Copper Resistance. J. Inorg. Biochem. 2012, 115, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Rensing, C.; Alwathnani, H.A.; McDevitt, S.F. The Copper Metallome in Prokaryotic Cells. In Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria; De Bruijn, F.J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar] [CrossRef]
- Hao, X.; Lüthje, F.L.; Qin, Y.; McDevitt, S.F.; Lutay, N.; Hobman, J.L.; Asiani, K.; Soncini, F.C.; German, N.; Zhang, S.; et al. Survival in Amoeba—a Major Selection Pressure on the Presence of Bacterial Copper and Zinc Resistance Determinants? Identification of a “Copper Pathogenicity Island.”. Appl. Microbiol. Biotechnol. 2015, 99, 5817–5824. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Matsui, K.; Lo, J.-F.; Silver, S. Molecular Basis for Resistance to Silver Cations in Salmonella. Nat. Med. 1999, 5, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Silver, S. Bacterial Silver Resistance: Molecular Biology and Uses and Misuses of Silver Compounds. FEMS Microbiol. Rev. 2003, 27, 341–353. [Google Scholar] [CrossRef]
- Mourão, J. Metal Tolerance in Emerging Clinically Relevant Multidrug-Resistant Salmonella enterica Serotype 4,[5],12:I:− Clones Circulating in Europe. Int. J. of Antimicrob. Agents 2015, 45, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Mourão, J.; Marçal, S.; Ramos, P.; Campos, J.; Machado, J.; Peixe, L.; Novais, C.; Antunes, P. Tolerance to Multiple Metal Stressors in Emerging Non-Typhoidal MDR Salmonella Serotypes: A Relevant Role for Copper in Anaerobic Conditions. J. Antimicrob. Chemother. 2016, 71, 2147–2157. [Google Scholar] [CrossRef]
- Andrade, L.N.; Siqueira, T.E.S.; Martinez, R.; Darini, A.L.C. Multidrug-Resistant CTX-M-(15, 9, 2)- and KPC-2-Producing Enterobacter hormaechei and Enterobacter asburiae Isolates Possessed a Set of Acquired Heavy Metal Tolerance Genes Including a Chromosomal Sil Operon (for Acquired Silver Resistance). Front. Microbiol. 2018, 9, 539. [Google Scholar] [CrossRef]
- Peters, J.E.; Fricker, A.D.; Kapili, B.J.; Petassi, M.T. Heteromeric Transposase Elements: Generators of Genomic Islands across Diverse Bacteria: Heteromeric Transposase Elements. Mol. Microbiol. 2014. [Google Scholar] [CrossRef]
- Moreno Switt, A.I.; Den Bakker, H.C.; Cummings, C.A.; Rodriguez-Rivera, L.D.; Govoni, G.; Raneiri, M.L.; Degoricija, L.; Brown, S.; Hoelzer, K.; Peters, J.E.; et al. Identification and Characterization of Novel Salmonella Mobile Elements Involved in the Dissemination of Genes Linked to Virulence and Transmission. PLoS ONE 2012, 7, e41247. [Google Scholar] [CrossRef]
- Chalmers, G.; Rozas, K.; Amachawadi, R.; Scott, H.; Norman, K.; Nagaraja, T.; Tokach, M.; Boerlin, P. Distribution of the pco Gene Cluster and Associated Genetic Determinants among Swine Escherichia coli from a Controlled Feeding Trial. Genes 2018, 9, 504. [Google Scholar] [CrossRef]
- Zagui, G.S.; Moreira, N.C.; Santos, D.V.; Darini, A.L.C.; Domingo, J.L.; Segura-Muñoz, S.I.; Andrade, L.N. High Occurrence of Heavy Metal Tolerance Genes in Bacteria Isolated from Wastewater: A New Concern? Environ. Res. 2021, 196, 110352. [Google Scholar] [CrossRef] [PubMed]
- Furlan, J.P.R.; Ramos, M.S.; Rosa, R.D.S.; Savazzi, E.A.; Stehling, E.G. Occurrence and Genetic Characteristics of Multidrug-Resistant Escherichia coli Isolates Co-Harboring Antimicrobial Resistance Genes and Metal Tolerance Genes in Aquatic Ecosystems. Int. J. Hyg. Environ. Health 2022, 244, 114003. [Google Scholar] [CrossRef] [PubMed]
- Kamathewatta, K.; Bushell, R.; Rafa, F.; Browning, G.; Billman-Jacobe, H.; Marenda, M. Colonization of a Hand Washing Sink in a Veterinary Hospital by an Enterobacter hormaechei Strain Carrying Multiple Resistances to High Importance Antimicrobials. Antimicrob. Resist. Infect. Control 2020, 9, 163. [Google Scholar] [CrossRef]
- Hasman, H.; Aarestrup, F.M. tcrB, a Gene Conferring Transferable Copper Resistance in Enterococcus faecium : Occurrence, Transferability, and Linkage to Macrolide and Glycopeptide Resistance. Antimicrob. Agents Chemother. 2002, 46, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Arguello, J.M. Identification of Ion-Selectivity Determinants in Heavy-Metal Transport P1B -Type ATPases. J. Membr. Biol. 2003, 195, 93–108. [Google Scholar] [CrossRef]
- Hasman, H. The tcrB Gene Is Part of the tcrYAZB Operon Conferring Copper Resistance in Enterococcus faecium and Enterococcus faecalis. Microbiology 2005, 151, 3019–3025. [Google Scholar] [CrossRef] [PubMed]
- Silveira, E.; Freitas, A.R.; Antunes, P.; Barros, M.; Campos, J.; Coque, T.M.; Peixe, L.; Novais, C. Co-Transfer of Resistance to High Concentrations of Copper and First-Line Antibiotics among Enterococcus from Different Origins (Humans, Animals, the Environment and Foods) and Clonal Lineages. J. Antimicrob. Chemother. 2014, 69, 899–906. [Google Scholar] [CrossRef]
- Amachawadi, R.G.; Scott, H.M.; Alvarado, C.A.; Mainini, T.R.; Vinasco, J.; Drouillard, J.S.; Nagaraja, T.G. Occurrence of the Transferable Copper Resistance Gene tcrB among Fecal Enterococci of U.S. Feedlot Cattle Fed Copper-Supplemented Diets. Appl. Environ. Microbiol. 2013, 79, 4369–4375. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, D.; Wang, Y.; Hasman, H.; Aarestrup, F.M.; Alwathnani, H.A.; Zhu, Y.-G.; Rensing, C. Genome Sequences of Copper Resistant and Sensitive Enterococcus faecalis Strains Isolated from Copper-Fed Pigs in Denmark. Stand. Genomic Sci. 2015, 10, 35. [Google Scholar] [CrossRef]
- Mourão, J.; Rae, J.; Silveira, E.; Freitas, A.R.; Coque, T.M.; Peixe, L.; Antunes, P.; Novais, C. Relevance of tcrYAZB Operon Acquisition for Enterococcus Survival at High Copper Concentrations under Anaerobic Conditions. J. Antimicrob. Chemother. 2016, 71, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, A.; Duarte, B.; Ferreira, C.; Mourão, J.; Ribeiro, S.; Freitas, A.R.; Coque, T.M.; Willems, R.; Corander, J.; Peixe, L.; et al. Enterococcus spp. from Chicken Meat Collected 20 Years Apart Overcome Multiple Stresses Occurring in the Poultry Production Chain: Antibiotics, Copper and Acids. Int. J. Food Microbiol. 2023, 384, 109981. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, A.; Mourão, J.; Freitas, A.R.; Duarte, B.; Silveira, E.; Sanchez-Valenzuela, A.; Almeida, A.; Baquero, F.; Coque, T.M.; Peixe, L.; et al. Diversity of Metal and Antibiotic Resistance Genes in Enterococcus spp. from the Last Century Reflects Multiple Pollution and Genetic Exchange among Phyla from Overlapping Ecosystems. Sci. Total Environ. 2021, 787, 147548. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, C.; Gutierrez-Corona, F. Copper Resistance Mechanisms in Bacteria and Fungi. FEMS Microbiol. Rev. 1994, 14, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Choi, S. Copper Resistance and Its Relationship to Erythromycin Resistance in Enterococcus Isolates from Bovine Milk Samples in Korea. J. Microbiol. 2012, 50, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, L.; Ye, C.; Yu, X. Co-Selection of Antibiotic Resistance via Copper Shock Loading on Bacteria from a Drinking Water Bio-Filter. Environ. Pollut. 2018, 233, 132–141. [Google Scholar] [CrossRef]
- Mazhar, S.H.; Li, X.; Rashid, A.; Su, J.; Xu, J.; Brejnrod, A.D.; Su, J.-Q.; Wu, Y.; Zhu, Y.-G.; Zhou, S.G.; et al. Co-Selection of Antibiotic Resistance Genes, and Mobile Genetic Elements in the Presence of Heavy Metals in Poultry Farm Environments. Sci. Total Environ. 2021, 755, 142702. [Google Scholar] [CrossRef]
- Hu, H.; Wang, J.; Li, J.; Li, J.; Ma, Y.; Chen, D.; He, J. Field-based Evidence for Copper Contamination Induced Changes of Antibiotic Resistance in Agricultural Soils. Environ. Microbiol. 2016, 18, 3896–3909. [Google Scholar] [CrossRef]
- Hasman, H.; Aarestrup, F.M. Relationship between Copper, Glycopeptide, and Macrolide Resistance among Enterococcus faecium Strains Isolated from Pigs in Denmark between 1997 and 2003. Antimicrob. Agents Chemother. 2005, 49, 454–456. [Google Scholar] [CrossRef]
- Fang, L.; Li, X.; Li, L.; Li, S.; Liao, X.; Sun, J.; Liu, Y. Co-Spread of Metal and Antibiotic Resistance within ST3-IncHI2 Plasmids from E. coli Isolates of Food-Producing Animals. Sci. Rep. 2016, 6, 25312. [Google Scholar] [CrossRef]
- Zhai, Y.; He, Z.; Kang, Y.; Yu, H.; Wang, J.; Du, P.; Zhang, Z.; Hu, S.; Gao, Z. Complete Nucleotide Sequence of pH11, an IncHI2 Plasmid Conferring Multi-Antibiotic Resistance and Multi-Heavy Metal Resistance Genes in a Clinical Klebsiella pneumoniae Isolate. Plasmid 2016, 86, 26–31. [Google Scholar] [CrossRef]
- Teixeira, P.; Tacão, M.; Alves, A.; Henriques, I. Antibiotic and Metal Resistance in a ST395 Pseudomonas aeruginosa Environmental Isolate: A Genomics Approach. Mar. Pollut. Bull. 2016, 110, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Flach, C.-F.; Pal, C.; Svensson, C.J.; Kristiansson, E.; Östman, M.; Bengtsson-Palme, J.; Tysklind, M.; Larsson, D.G.J. Does Antifouling Paint Select for Antibiotic Resistance? Sci. Total Environ. 2017, 590–591, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Kobayashi, K.; Kobayashi, M.; Asako, H.; Aono, R. Overexpression of the robA Gene Increases Organic Solvent Tolerance and Multiple Antibiotic and Heavy Metal Ion Resistance in Escherichia coli. Appl. Env. Microbiol. 1995, 61, 2302–2307. [Google Scholar] [CrossRef]
- Centre international de recherche sur le cancer. A Review of Human Carcinogens; IARC monographs on the evaluation of carcinogenic risks to humans; International agency for research on cancer: Lyon, 2012. [Google Scholar]
- Mandal, B.K.; Suzuki, K.T. Arsenic Round the World: A Review. Talanta 2002, 58, 201–235. [Google Scholar] [CrossRef]
- Cervantes, C.; Ji, G.; Ramirez, J.; Silver, S. Resistance to Arsenic Compounds in Microorganisms. FEMS Microbiol. Rev. 1994, 15, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Páez-Espino, D.; Tamames, J.; De Lorenzo, V.; Cánovas, D. Microbial Responses to Environmental Arsenic. BioMetals 2009, 22, 117–130. [Google Scholar] [CrossRef]
- Gomez-Caminero, A.; Howe; Paul, D.; Hughes, M.; Kenyon, E.; Lewis, D.R.; et al. Arsenic and Arsenic Compounds, 2nd ed.; World Health Organization: Geneva, 2001; Available online: https://apps.who.int/iris/handle/10665/42366.
- Nordstrom, D.K. Worldwide Occurrences of Arsenic in Ground Water. Science 2002, 296, 2143–2145. [Google Scholar] [CrossRef]
- Paul, N.P.; Galván, A.E.; Yoshinaga-Sakurai, K.; Rosen, B.P.; Yoshinaga, M. Arsenic in Medicine: Past, Present and Future. BioMetals 2023, 36, 283–301. [Google Scholar] [CrossRef]
- World Health Organization. Model List of Essential Medicines – 22nd List, 2021. World Health Organization; Geneva WHO/MHP/HPS/EML/2021.02.
- Argudín, M.A.; Hoefer, A.; Butaye, P. Heavy Metal Resistance in Bacteria from Animals. Res. Vet. Sci. 2019, 122, 132–147. [Google Scholar] [CrossRef]
- Nachman, K.E.; Graham, J.P.; Price, L.B.; Silbergeld, E.K. Arsenic: A Roadblock to Potential Animal Waste Management Solutions. Environ. Health Perspect. 2005, 113, 1123–1124. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wen, M.; Wu, L.; Cao, S.; Li, Y. Environmental Behavior and Remediation Methods of Roxarsone. Appl. Sci. 2022, 12, 7591. [Google Scholar] [CrossRef]
- Food and Drug Administration. Withdrawal of Approval of New Animal Drug Applications for Combination Drug Medicated Feeds Containing an Arsenical Drug, 2014, Vol. 79, No. 39; FDA–2014–N–0002. Available online: https://public4.pagefreezer.com/content/FDA/27-04-2023T11:46/http://www.gpo.gov/fdsys/pkg/FR-2014-02-27/pdf/2014-02617.pdf (accessed on 29 May 2023).
- Hu, Y.; Cheng, H.; Tao, S.; Schnoor, J.L. China’s Ban on Phenylarsonic Feed Additives, A Major Step toward Reducing the Human and Ecosystem Health Risk from Arsenic. Environ. Sci. Technol. 2019, 53, 12177–12187. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Hermann, T.; Silva, M.R.D.; Montanarella, L. Heavy Metals in Agricultural Soils of the European Union with Implications for Food Safety. Environ. Int. 2016, 88, 299–309. [Google Scholar] [CrossRef]
- Sager, M. Trace and Nutrient Elements in Manure, Dung and Compost Samples in Austria. Soil Biol. 2007, 39, 1383–1390. [Google Scholar] [CrossRef]
- Nachman, K.E.; Raber, G.; Francesconi, K.A.; Navas-Acien, A.; Love, D.C. Arsenic Species in Poultry Feather Meal. Sci. Total Environ. 2012, 417-418, 183–188. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Y.; Yang, M.; Li, W. Content of Heavy Metals in Animal Feeds and Manures from Farms of Different Scales in Northeast China. Int J Env. Res Public Health 2012, 9, 2658–2668. [Google Scholar] [CrossRef]
- Cui, E. The Behavior of Antibiotic Resistance Genes and Arsenic Influenced by Biochar during Different Manure Composting. Env. Sci Pollut Res 2017, 24, 14484–14490. [Google Scholar] [CrossRef]
- Mourão, J.; Rebelo, A.; Ribeiro, S.; Peixe, L.; Novais, C.; Antunes, P. Tolerance to Arsenic Contaminant among Multidrug-resistant and Copper-tolerant Salmonella Successful Clones Is Associated with Diverse ars Operons and Genetic Contexts. Environ. Microbiol. 2020, 22, 2829–2842. [Google Scholar] [CrossRef]
- Yan, G.; Chen, X.; Du, S.; Deng, Z.; Wang, L.; Chen, S. Genetic Mechanisms of Arsenic Detoxification and Metabolism in Bacteria. Curr. Genet. 2019, 65, 329–338. [Google Scholar] [CrossRef]
- Andres, J.; Bertin, P.N. The Microbial Genomics of Arsenic. FEMS Microbiol. Rev. 2016, 40, 299–322. [Google Scholar] [CrossRef] [PubMed]
- Kruger, M.C.; Bertin, P.N.; Heipieper, H.J.; Arsène-Ploetze, F. Bacterial Metabolism of Environmental Arsenic—Mechanisms and Biotechnological Applications. Appl. Microbiol. Biotechnol. 2013, 97, 3827–3841. [Google Scholar] [CrossRef]
- Ben Fekih, I.; Zhang, C.; Li, Y.P.; Zhao, Y.; Alwathnani, H.A.; Saquib, Q.; Rensing, C.; Cervantes, C. Distribution of Arsenic Resistance Genes in Prokaryotes. Front. Microbiol. 2018, 9, 2473. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.P.; Liu, Z. Transport Pathways for Arsenic and Selenium: A Minireview. Environ. Int. 2009, 35, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Oremland, R.S.; Stolz, J.F. The Ecology of Arsenic. Science 2003, 300, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.P. Biochemistry of Arsenic Detoxification. FEBS Lett. 2002, 529, 86–92. [Google Scholar] [CrossRef]
- Parsons, C.; Lee, S.; Kathariou, S. Dissemination and Conservation of Cadmium and Arsenic Resistance Determinants in Listeria and Other Gram-positive Bacteria. Mol. Microbiol. 2020, 113, 560–569. [Google Scholar] [CrossRef]
- Novick, R.P.; Roth, C. Plasmid-Linked Resistance to Inorganic Salts in Staphylococcus aureus. J Bacteriol. 1968, 95, 1335–1342. [Google Scholar] [CrossRef]
- Hedges, R.W.; Baumberg, S. Resistance to Arsenic Compounds Conferred by a Plasmid Transmissible Between Strains of Escherichia coli. J. Bacteriol. 1973, 115, 459–460. [Google Scholar] [CrossRef]
- Xu, C.; Zhou, T.; Kuroda, M.; Rosen, B.P. Metalloid Resistance Mechanisms in Prokaryotes. J Biochem 1998, 123, 16–23. [Google Scholar] [CrossRef]
- Yang, H.-C.; Fu, H.-L.; Lin, Y.-F.; Rosen, B.P. Pathways of Arsenic Uptake and Efflux. Curr. Top. Membr. 2012, 69, 325–358. [Google Scholar] [CrossRef]
- Ji, G.; Silver, S. Reduction of Arsenate to Arsenite by the ArsC Protein of the Arsenic Resistance Operon of Staphylococcus aureus Plasmid P1258. Proc Natl Acad Sci USA 1992, 89, 9474–9478. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.N.; Saltikov, C.W. The ArsR Repressor Mediates Arsenite-Dependent Regulation of Arsenate Respiration and Detoxification Operons of Shewanella Sp. Strain ANA-3ᰔ. J. Bacteriol 2009, 191, 6722–6731. [Google Scholar] [CrossRef] [PubMed]
- Kleerebezem, M.; Siezen, R.J. Functional Analysis of Three Plasmids from Lactobacillus plantarum. Appl. Env. Microbiol. 2005, 71, 1223–1230. [Google Scholar] [CrossRef]
- Sato, T.; Kobayashi, Y. The Ars Operon in the Skin Element of Bacillus subtilis Confers Resistance to Arsenate and Arsenite. J. Bacteriol 1998, 180, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, E.K.J.; Silow, M. Transmembrane Topology of the Acr3 Family Arsenite Transporter From. Biochim. Biophys. Acta. 2008, 1778, 963–973. [Google Scholar] [CrossRef]
- Fu, H.-L.; Meng, Y.; Gil, J.A.; Mateos, L.M.; Rosen, B.P. Properties of Arsenite Efflux Permeases (Acr3) from Alkaliphilus metalliredigens and Corynebacterium glutamicum. J. Biol. Chem. 2009, 284, 19887–19895. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G.M. Valid Publication of the Names of Forty two Phyla of Prokaryotes. Int J Syst Evol Microbiol 2021, 71. [Google Scholar] [CrossRef]
- Achour, A.R.; Bauda, P.; Billard, P. Diversity of Arsenite Transporter Genes from Arsenic-Resistant Soil Bacteria. Res. Microbiol. 2007, 158, 128–137. [Google Scholar] [CrossRef]
- Castillo, R.; Saier, M.H. Functional Promiscuity of Homologues of the Bacterial ArsA ATPases. Int. J. Microbiol. 2010, 1–21. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, S.; Lilley, R.M.; Zhang, R. The Diversity of Membrane Transporters Encoded in Bacterial Arsenic-Resistance Operons. PeerJ 2015, 3, e943. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Liu, G.; Rensing, C.; Wang, G. Genes Involved in Arsenic Transformation and Resistance Associated with Different Levels of Arsenic-Contaminated Soils. BMC Microbiol. 2009, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.R.; Cook, G.M. Isolation and Characterization of Arsenate-Reducing Bacteria from Arsenic-Contaminated Sites in New Zealand. Curr. Microbiol. 2004, 48, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Saltikov, C.W.; Olson, B.H. Homology of Escherichia coli R773 arsA, arsB, and arsC Genes in Arsenic-Resistant Bacteria Isolated from Raw Sewage and Arsenic-Enriched Creek Waters. Appl. Environ. Microbiol. 2002, 68, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Ward, T.J.; Jima, D.D.; Parsons, C.; Kathariou, S. The Arsenic Resistance-Associated Listeria Genomic Island LGI2 Exhibits Sequence and Integration Site Diversity and a Propensity for Three Listeria monocytogenes Clones with Enhanced Virulence. Appl. Environ. Microbiol. 2017, 83, e01189-17. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.; Card, R.M.; Nunez-Garcia, J.; Mendonça, N.; Da Silva, G.J.; Anjum, M.F. Multidrug-Resistant Salmonella enterica Isolated from Food Animal and Foodstuff May Also Be Less Susceptible to Heavy Metals. Foodborne Pathog. Dis. 2019, 16, 166–172. [Google Scholar] [CrossRef]
- Noormohamed, A.; Fakhr, M. Arsenic Resistance and Prevalence of Arsenic Resistance Genes in Campylobacter jejuni and Campylobacter coli Isolated from Retail Meats. Int. J. Environ. Res. Public. Health 2013, 10, 3453–3464. [Google Scholar] [CrossRef]
- Argudín, M.A.; Lauzat, B.; Kraushaar, B.; Alba, P.; Agerso, Y.; Cavaco, L.; Butaye, P.; Porrero, M.C.; Battisti, A.; Tenhagen, B.-A.; et al. Heavy Metal and Disinfectant Resistance Genes among Livestock-Associated Methicillin-Resistant Staphylococcus aureus Isolates. Vet. Microbiol. 2016, 191, 88–95. [Google Scholar] [CrossRef]
- Cusick, K.D.; Polson, S.W.; Duran, G.; Hill, R.T. Multiple Megaplasmids Confer Extremely High Levels of Metal Tolerance in Alteromonas Strains. Appl. Environ. Microbiol. 2019, 86, e01831-19. [Google Scholar] [CrossRef]
- Sandegren, L.; Linkevicius, M.; Lytsy, B.; Melhus, Å.; Andersson, D.I. Transfer of an Escherichia coli ST131 Multiresistance Cassette Has Created a Klebsiella pneumoniae-Specific Plasmid Associated with a Major Nosocomial Outbreak. J. Antimicrob. Chemother. 2012, 67, 74–83. [Google Scholar] [CrossRef]
- Branchu, P.; Charity, O.J.; Bawn, M.; Thilliez, G.; Dallman, T.J.; Petrovska, L.; Kingsley, R.A. SGI-4 in Monophasic Salmonella Typhimurium ST34 Is a Novel ICE That Enhances Resistance to Copper. Front. Microbiol. 2019, 10, 1118. [Google Scholar] [CrossRef]
- Zhang, M.; Wan, K.; Zeng, J.; Lin, W.; Ye, C.; Yu, X. Co-Selection and Stability of Bacterial Antibiotic Resistance by Arsenic Pollution Accidents in Source Water. Environ. Int. 2020, 135, 105351. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cocerva, T.; Cox, S.; Tardif, S.; Su, J.-Q.; Zhu, Y.-G.; Brandt, K.K. Evidence for Co-Selection of Antibiotic Resistance Genes and Mobile Genetic Elements in Metal Polluted Urban Soils. Sci. Total Environ. 2019, 656, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshanee, M.; Chatterjee, S.; Rath, S.; Dash, H.R.; Das, S. Cellular and Genetic Mechanism of Bacterial Mercury Resistance and Their Role in Biogeochemistry and Bioremediation. J. Hazard. Mater. 2022, 423, 126985. [Google Scholar] [CrossRef] [PubMed]
- Nies, D.H. Microbial Heavy-Metal Resistance. Appl. Microbiol. Biotechnol. 1999, 51, 730–750. [Google Scholar] [CrossRef]
- Syversen, T.; Kaur, P. The Toxicology of Mercury and Its Compounds. J. Trace Elem. Med. Biol. 2012, 26, 215–226. [Google Scholar] [CrossRef]
- O’Connor, D.; Hou, D.; Ok, Y.S.; Mulder, J.; Duan, L.; Wu, Q.; Wang, S.; Tack, F.M.G.; Rinklebe, J. Mercury Speciation, Transformation, and Transportation in Soils, Atmospheric Flux, and Implications for Risk Management: A Critical Review. Environ. Int. 2019, 126, 747–761. [Google Scholar] [CrossRef]
- Mahbub, K.R.; Krishnan, K.; Naidu, R.; Andrews, S.; Megharaj, M. Mercury Toxicity to Terrestrial Biota. Ecol. Indic. 2017, 74, 451–462. [Google Scholar] [CrossRef]
- Bernhoft, R.A. Mercury Toxicity and Treatment: A Review of the Literature. J. Environ. Public Health 2012, 2012, 1–10. [Google Scholar] [CrossRef]
- Liu, G.; Cai, Y.; O’Driscoll, N.; Feng, X.; Jiang, G. Overview of Mercury in the Environment. In Environmental Chemistry and Toxicology of Mercury; Liu, G., Cai, Y., O’Driscoll, N., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Beryllium, Cadmium, Mercury and Exposures in the Glass Manufacturing Industry: This Publication Represents the Views and Expert Opinions of an IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Which Met in Lyon, 9-16 February 1993; International Agency for Research on Cancer, Ed.; IARC monographs on the evaluation of carcinogenic risks to humans; World Health Organization: Geneva, 1993. [Google Scholar]
- Pirrone, N.; Cinnirella, S.; Feng, X.; Finkelman, R.B.; Friedli, H.R.; Leaner, J.; Mason, R.; Mukherjee, A.B.; Stracher, G.B.; Streets, D.G.; et al. Global Mercury Emissions to the Atmosphere from Anthropogenic and Natural Sources. Atmospheric Chem. Phys. 2010, 10, 5951–5964. [Google Scholar] [CrossRef]
- Mason, R.P. Mercury Emissions from Natural Processes and Their Importance in the Global Mercury Cycle. In Mercury Fate and Transport in the Global Atmosphere; Mason, R., Pirrone, N., Eds.; Springer US: Boston, MA, 2009; pp. 173–191. [Google Scholar] [CrossRef]
- Fitzgerald, W.F.; Mason, R.P. The Global Mercury Cycle: Oceanic and Anthropogenic Aspects. In Global and Regional Mercury Cycles: Sources, Fluxes and Mass Balances; Baeyens, W., Ebinghaus, R., Vasiliev, O., Eds.; Springer Netherlands: Dordrecht, 1996. [Google Scholar] [CrossRef]
- Zhao, M.; Li, Y.; Wang, Z. Mercury and Mercury-Containing Preparations: History of Use, Clinical Applications, Pharmacology, Toxicology, and Pharmacokinetics in Traditional Chinese Medicine. Front. Pharmacol. 2022, 13, 807807. [Google Scholar] [CrossRef] [PubMed]
- Wujastyk, D. Histories of Mercury in Medicine across Asia and Beyond. Asiat. Stud. - Études Asiat. 2015, 69, 819–830. [Google Scholar] [CrossRef]
- O’Shea, J.G. ‘Two Minutes with Venus, Two Years with Mercury’-Mercury as an Antisyphilitic Chemotherapeutic Agent. J. R. Soc. Med. 1990, 83, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Dadar, M.; Mutter, J.; Aaseth, J. The Toxicology of Mercury: Current Research and Emerging Trends. Environ. Res. 2017, 159, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Tchounwou, P.B.; Ayensu, W.K.; Ninashvili, N.; Sutton, D. Review: Environmental Exposure to Mercury and Its Toxicopathologic Implications for Public Health. Environ. Toxicol. 2003, 18, 149–175. [Google Scholar] [CrossRef]
- Jepson, P.C. Pesticides, Uses and Effects Of. In Encyclopedia of Biodiversity; Elsevier, 2001; pp. 692–702. [Google Scholar] [CrossRef]
- Boening, D.W. Ecological Effects, Transport, and Fate of Mercury: A General Review. Chemosphere 2000, 40, 1335–1351. [Google Scholar] [CrossRef]
- Marnane, I. Mercury in Europe’s Environment: A Priority for European and Global Action; Publications Office of the European Union: Luxembourg, 2018. [Google Scholar]
- Teng, H.; Altaf, A.R. Elemental Mercury (Hg0) Emission, Hazards, and Control: A Brief Review. J. Hazard. Mater. Adv. 2022, 5, 100049. [Google Scholar] [CrossRef]
- Gworek, B.; Dmuchowski, W.; Baczewska-Dąbrowska, A.H. Mercury in the Terrestrial Environment: A Review. Environ. Sci. Eur. 2020, 32, 128. [Google Scholar] [CrossRef]
- Gworek, B.; Bemowska-Kałabun, O.; Kijeńska, M.; Wrzosek-Jakubowska, J. Mercury in Marine and Oceanic Waters—a Review. Water. Air. Soil Pollut. 2016, 227, 371. [Google Scholar] [CrossRef]
- Tóth, G.; Hermann, T.; Szatmári, G.; Pásztor, L. Maps of Heavy Metals in the Soils of the European Union and Proposed Priority Areas for Detailed Assessment. Sci. Total Environ. 2016, 565, 1054–1062. [Google Scholar] [CrossRef]
- Bettoso, N.; Pittaluga, F.; Predonzani, S.; Zanello, A.; Acquavita, A. Mercury Levels in Sediment, Water and Selected Organisms Collected in a Coastal Contaminated Environment: The Marano and Grado Lagoon (Northern Adriatic Sea, Italy). Appl. Sci. 2023, 13, 3064. [Google Scholar] [CrossRef]
- Llull, R.M.; Garí, M.; Canals, M.; Rey-Maquieira, T.; Grimalt, J.O. Mercury Concentrations in Lean Fish from the Western Mediterranean Sea: Dietary Exposure and Risk Assessment in the Population of the Balearic Islands. Environ. Res. 2017, 158, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Manceau, A.; Nagy, K.L.; Glatzel, P.; Bourdineaud, J.-P. Acute Toxicity of Divalent Mercury to Bacteria Explained by the Formation of Dicysteinate and Tetracysteinate Complexes Bound to Proteins in Escherichia coli and Bacillus subtilis. Environ. Sci. Technol. 2021, 55, 3612–3623. [Google Scholar] [CrossRef] [PubMed]
- Quig, D. Cysteine Metabolism and Metal Toxicity. Altern. Med. Rev. J. Clin. Ther. 1998, 3, 262–270. [Google Scholar]
- Nies, D.H. Efflux-Mediated Heavy Metal Resistance in Prokaryotes. FEMS Microbiol. Rev. 2003, 27, 313–339. [Google Scholar] [CrossRef]
- Møller, A.K.; Barkay, T.; Hansen, M.A.; Norman, A.; Hansen, L.H.; Sørensen, S.J.; Boyd, E.S.; Kroer, N. Mercuric Reductase Genes ( merA ) and Mercury Resistance Plasmids in High Arctic Snow, Freshwater and Sea-Ice Brine. FEMS Microbiol. Ecol. 2014, 87, 52–63. [Google Scholar] [CrossRef]
- Gionfriddo, C.M.; Tate, M.T.; Wick, R.R.; Schultz, M.B.; Zemla, A.; Thelen, M.P.; Schofield, R.; Krabbenhoft, D.P.; Holt, K.E.; Moreau, J.W. Microbial Mercury Methylation in Antarctic Sea Ice. Nat. Microbiol. 2016, 1, 16127. [Google Scholar] [CrossRef]
- Oregaard, G.; Sørensen, S.J. High Diversity of Bacterial Mercuric Reductase Genes from Surface and Sub-Surface Floodplain Soil (Oak Ridge, USA). ISME J. 2007, 1, 453–467. [Google Scholar] [CrossRef]
- Hobman, J.L.; Crossman, L.C. Bacterial Antimicrobial Metal Ion Resistance. J. Med. Microbiol. 2015, 64, 471–497. [Google Scholar] [CrossRef]
- Dash, H.R.; Das, S. Bioremediation of Mercury and the Importance of Bacterial mer Genes. Int. Biodeterior. Biodegrad. 2012, 75, 207–213. [Google Scholar] [CrossRef]
- Barkay, T.; Miller, S.M.; Summers, A.O. Bacterial Mercury Resistance from Atoms to Ecosystems. FEMS Microbiol. Rev. 2003, 27, 355–384. [Google Scholar] [CrossRef]
- Addie, D.D. Metals as Antiseptics and Disinfectants for Use With Animals. MDS Manual, Veterinary Manual. 2022. Available online: https://www.msdvetmanual.com/pharmacology/antiseptics-and-disinfectants/metals-as-antiseptics-and-disinfectants-for-use-with-animals.
- Moore, B. A new screen test and selective medium for the rapid detection of epidemic strains of Staph. aureus. The Lancet 1960, 276, 453–458. [Google Scholar] [CrossRef]
- Jensen, S.; Jernelöv, A. Biological Methylation of Mercury in Aquatic Organisms. Nature 1969, 223, 753–754. [Google Scholar] [CrossRef]
- Boyd, E.S.; Barkay, T. The Mercury Resistance Operon: From an Origin in a Geothermal Environment to an Efficient Detoxification Machine. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.J.; Roberts, A.P.; Ready, D.; Richards, H.; Wilson, M.; Mullany, P. Linkage of a Novel Mercury Resistance Operon with Streptomycin Resistance on a Conjugative Plasmid in Enterococcus faecium. Plasmid 2005, 54, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Chiba, K.; Nishizawa, H.; Ishii, H.; Huang, C.-C.; Kawabata, Z.; Silver, S.; Endo, G. Diversity of Mercury Resistance Determinants among Bacillus Strains Isolated from Sediment of Minamata Bay. FEMS Microbiol. Lett. 2003, 223, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Dash, H.R.; Sahu, M.; Mallick, B.; Das, S. Functional Efficiency of MerA Protein among Diverse Mercury Resistant Bacteria for Efficient Use in Bioremediation of Inorganic Mercury. Biochimie 2017, 142, 207–215. [Google Scholar] [CrossRef]
- Nascimento, A.M.A.; Chartone-Souza, E. Operon Mer: Bacterial Resistance to Mercury and Potential for Bioremediation of Contaminated Environments. Genet. Mol. Res. 2003.
- Lund, P.A.; Brown, N.L. Regulation of Transcription in Escherichia coli from the Mer and merR Promoters in the Transposon Tn501. J. Mol. Biol. 1989, 205, 343–353. [Google Scholar] [CrossRef]
- Osborn, A.M.; Bruce, K.D.; Strike, P.; Ritchie, D.A. Sequence Conservation between Regulatory Mercury Resistance Genes in Bacteria from Mercury Polluted and Pristine Environments. Syst. Appl. Microbiol. 1995, 18, 1–6. [Google Scholar] [CrossRef]
- Grad, Y.H.; Godfrey, P.; Cerquiera, G.C.; Mariani-Kurkdjian, P.; Gouali, M.; Bingen, E.; Shea, T.P.; Haas, B.J.; Griggs, A.; Young, S.; et al. Comparative Genomics of Recent Shiga Toxin-Producing Escherichia coli O104:H4: Short-Term Evolution of an Emerging Pathogen. mBio 2013, 4, e00452-12. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, H.; Sekizuka, T.; Nakaya, H.; Taguchi, M.; Oguchi, A.; Ichikawa, N.; Nishiko, R.; Yamazaki, S.; Fujita, N.; Watanabe, H.; et al. Whole-Genome Analysis of Salmonella enterica Serovar Typhimurium T000240 Reveals the Acquisition of a Genomic Island Involved in Multidrug Resistance via IS 1 Derivatives on the Chromosome. Antimicrob. Agents Chemother. 2011, 55, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Holden, M.T.G.; Feil, E.J.; Lindsay, J.A.; Peacock, S.J.; Day, N.P.J.; Enright, M.C.; Foster, T.J.; Moore, C.E.; Hurst, L.; Atkin, R.; et al. Complete Genomes of Two Clinical Staphylococcus aureus Strains: Evidence for the Rapid Evolution of Virulence and Drug Resistance. Proc. Natl. Acad. Sci. 2004, 101, 9786–9791. [Google Scholar] [CrossRef]
- Rice, L.B.; Carias, L.L. Transfer of Tn5385, a Composite, Multiresistance Chromosomal Element from Enterococcus faecalis. J. Bacteriol. 1998, 180, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Richmond, M.H.; John, M. Co-Transduction by a Staphylococcal Phage of the Genes Responsible for Penicillinase Synthesis and Resistance to Mercury Salts. Nature 1964, 202, 1360–1361. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G.J. Co-Occurrence of Resistance Genes to Antibiotics, Biocides and Metals Reveals Novel Insights into Their Co-Selection Potential. BMC Genomics 2015, 16, 964. [Google Scholar] [CrossRef]
- McIntosh, D.; Cunningham, M.; Ji, B.; Fekete, F.A.; Parry, E.M.; Clark, S.E.; Zalinger, Z.B.; Gilg, I.C.; Danner, G.R.; Johnson, K.A.; et al. Transferable, Multiple Antibiotic and Mercury Resistance in Atlantic Canadian Isolates of Aeromonas salmonicida Subsp. salmonicida Is Associated with Carriage of an IncA/C Plasmid Similar to the Salmonella enterica Plasmid pSN254. J. Antimicrob. Chemother. 2008, 61, 1221–1228. [Google Scholar] [CrossRef]
- Rodríguez-Blanco, A.; Lemos, M.L.; Osorio, C.R. Integrating Conjugative Elements as Vectors of Antibiotic, Mercury, and Quaternary Ammonium Compound Resistance in Marine Aquaculture Environments. Antimicrob. Agents Chemother. 2012, 56, 2619–2626. [Google Scholar] [CrossRef]
- Li, X.; Yang, Z.; Zhang, G.; Si, S.; Wu, X.; Cai, L. Plasmid Genomes Reveal the Distribution, Abundance, and Organization of Mercury-Related Genes and Their Co-Distribution with Antibiotic Resistant Genes in Gammaproteobacteria. Genes 2022, 13, 2149. [Google Scholar] [CrossRef]
- Gaeta, N.C.; De Carvalho, D.U.; Fontana, H.; Sano, E.; Moura, Q.; Fuga, B.; Munoz, P.M.; Gregory, L.; Lincopan, N. Genomic Features of a Multidrug-Resistant and Mercury-Tolerant Environmental Escherichia coli Recovered after a Mining Dam Disaster in South America. Sci. Total Environ. 2022, 823, 153590. [Google Scholar] [CrossRef]
- Perez-Palacios, P.; Delgado-Valverde, M.; Gual-de-Torrella, A.; Oteo-Iglesias, J.; Pascual, Á.; Fernández-Cuenca, F. Co-Transfer of Plasmid-Encoded bla Carbapenemases Genes and Mercury Resistance Operon in High-Risk Clones of Klebsiella pneumoniae. Appl. Microbiol. Biotechnol. 2021, 105, 9231–9242. [Google Scholar] [CrossRef] [PubMed]
- Novais, Â.; Cantón, R.; Valverde, A.; Machado, E.; Galán, J.-C.; Peixe, L.; Carattoli, A.; Baquero, F.; Coque, T.M. Dissemination and Persistence of bla CTX-M-9 Are Linked to Class 1 Integrons Containing CR1 Associated with Defective Transposon Derivatives from Tn 402 Located in Early Antibiotic Resistance Plasmids of IncHI2, IncP1-α, and IncFI Groups. Antimicrob. Agents Chemother. 2006, 50, 2741–2750. [Google Scholar] [CrossRef] [PubMed]
- Chahardoli, A.; Jalilian, F.; Memariani, Z.; Farzaei, M.H.; Shokoohinia, Y. Analysis of Organic Acids. In Recent Advances in Natural Products Analysis; Elsevier, 2020; pp. 767–823. [Google Scholar] [CrossRef]
- Yadav, P.; Chauhan, A.K.; Singh, R.B.; Khan, S.; Halabi, G. Organic Acids: Microbial Sources, Production, and Applications. In Functional Foods and Nutraceuticals in Metabolic and Non-Communicable Diseases; Elsevier, 2022; pp. 325–337. [Google Scholar] [CrossRef]
- Guan, N.; Liu, L. Microbial Response to Acid Stress: Mechanisms and Applications. Appl. Microbiol. Biotechnol. 2020, 104, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Cherrington, C.A.; Hinton, M.; Mead, G.C.; Chopra, I. Organic Acids: Chemistry, Antibacterial Activity and Practical Applications. Advances in Microbial Physiology 1991, 32, 87–108. [Google Scholar] [CrossRef]
- Nollet, L.M.L. Food Analysis by HPLC, Food science and technology, 2nd ed.; Marcel Dekker: New York, 2000. [Google Scholar]
- Anyasi, T.A.; Jideani, A.I.O.; Edokpayi, J.N. Application of Organic Acids in Food Preservation. In Organic Acids: Characteristics, Properties and Synthesis; Vargas, C., Ed.; Nova Publishers: New York, 2017. [Google Scholar]
- Theron, M.M.; Lues, J.F.R. Organic Acids and Food Preservation; CRC Press, Taylor & Francis Group: Boca Raton, 2011. [Google Scholar] [CrossRef]
- Hauser, C.; Thielmann, J.; Muranyi, P. Organic Acids. In Antimicrobial Food Packaging; Elsevier, 2016; pp. 563–580. [Google Scholar] [CrossRef]
- Ricke, S. Perspectives on the Use of Organic Acids and Short Chain Fatty Acids as Antimicrobials. Poult. Sci. 2003, 82, 632–639. [Google Scholar] [CrossRef]
- European Commission. Regulation (EC) No 1831/2003 of the European Parliament and of the Council of 22 September 2003 on Additives for Use in Animal Nutrition; 2003; OJ L268. 18.10.2003, pp. 29-3. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2003:268:0029:0043:EN:PDF.
- European Commission. Regulation (EC) No 1333/2008 of the European Parliament and of the Council of 16 December 2008 on Food Additives; 2008; OJ L 354, 31.12.2008, pp. 16–33. Available online: https://eur-lex.europa.eu/eli/reg/2008/1333/oj (accessed on 23 August 2019).
- Saleem, K.; Saima; Rahman, A.; Pasha, T.N.; Mahmud, A.; Hayat, Z. Effects of Dietary Organic Acids on Performance, Cecal Microbiota, and Gut Morphology in Broilers. Trop. Anim. Health Prod. 2020, 52, 3589–3596. [Google Scholar] [CrossRef]
- Samanta, S.; Haldar, S.; Ghosh, T.K. Comparative Efficacy of an Organic Acid Blend and Bacitracin Methylene Disalicylate as Growth Promoters in Broiler Chickens: Effects on Performance, Gut Histology, and Small Intestinal Milieu. Vet. Med. Int. 2010, 1–8. [Google Scholar] [CrossRef]
- Ma, J.; Wang, J.; Mahfuz, S.; Long, S.; Wu, D.; Gao, J.; Piao, X. Supplementation of Mixed Organic Acids Improves Growth Performance, Meat Quality, Gut Morphology and Volatile Fatty Acids of Broiler Chicken. Animals 2021, 11, 3020. [Google Scholar] [CrossRef]
- Park, S.-H.; Choi, M.-R.; Park, J.-W.; Park, K.-H.; Chung, M.-S.; Ryu, S.; Kang, D.-H. Use of Organic Acids to Inactivate Escherichia coli O157:H7, Salmonella Typhimurium, and Listeria monocytogenes on Organic Fresh Apples and Lettuce. J. Food Sci. 2011, 76, M293–M298. [Google Scholar] [CrossRef]
- Thomas, C.; Schönknecht, A.; Püning, C.; Alter, T.; Martin, A.; Bandick, N. Effect of Peracetic Acid Solutions and Lactic Acid on Microorganisms in On-Line Reprocessing Systems for Chicken Slaughter Plants. J. Food Prot. 2020, 83, 615–620. [Google Scholar] [CrossRef]
- EFSA Panel on Biological Hazards (BIOHAZ). Scientific Opinion on the Evaluation of the Safety and Efficacy of Peroxyacetic Acid Solutions for Reduction of Pathogens on Poultry Carcasses and Meat. EFSA J. 2014, 12. [Google Scholar] [CrossRef]
- EFSA Panel on Food Contact Materials, Enzymes and Processing Aids (CEP); Silano, V.; Barat Baviera, J.M.; Bolognesi, C.; Brüschweiler, B.J.; Chesson, A.; Cocconcelli, P.S.; Crebelli, R.; Gott, D.M.; Grob, K.; et al. Evaluation of the Safety and Efficacy of the Organic Acids Lactic and Acetic Acids to Reduce Microbiological Surface Contamination on Pork Carcasses and Pork Cuts. EFSA J. 2018, 16. [Google Scholar] [CrossRef]
- Morgunov, I.G.; Kamzolova, S.V.; Dedyukhina, E.G.; Chistyakova, T.I.; Lunina, J.N.; Mironov, A.A.; Stepanova, N.N.; Shemshura, O.N.; Vainshtein, M.B. Application of Organic Acids for Plant Protection against Phytopathogens. Appl. Microbiol. Biotechnol. 2017, 101, 921–932. [Google Scholar] [CrossRef] [PubMed]
- European Commission, Directorate-General for Health and Food Safety. Active substance: Acetic acid. EU Pesticides Database. Available online: https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/start/screen/active-substances/details/1051.
- Lund, P.A.; De Biase, D.; Liran, O.; Scheler, O.; Mira, N.P.; Cetecioglu, Z.; Fernández, E.N.; Bover-Cid, S.; Hall, R.; Sauer, M.; et al. Understanding How Microorganisms Respond to Acid pH Is Central to Their Control and Successful Exploitation. Front. Microbiol. 2020, 11, 556140. [Google Scholar] [CrossRef] [PubMed]
- Theron, M.M.; Lues, J.F.R. Organic Acids and Meat Preservation: A Review. Food Rev. Int. 2007, 23, 141–158. [Google Scholar] [CrossRef]
- Huyghebaert, G.; Ducatelle, R.; Immerseel, F.V. An Update on Alternatives to Antimicrobial Growth Promoters for Broilers. Vet. J. 2011, 187, 182–188. [Google Scholar] [CrossRef]
- Mani-López, E.; García, H.S.; López-Malo, A. Organic Acids as Antimicrobials to Control Salmonella in Meat and Poultry Products. Food Res. Int. 2012, 45, 713–721. [Google Scholar] [CrossRef]
- Cherrington, C.A.; Hinton, M.; Chopra, I. Effect of Short-Chain Organic Acids on Macromolecular Synthesis in Escherichia coli. J. Appl. Bacteriol. 1990, 68, 69–74. [Google Scholar] [CrossRef]
- Trček, J.; Mira, N.P.; Jarboe, L.R. Adaptation and Tolerance of Bacteria against Acetic Acid. Appl. Microbiol. Biotechnol. 2015, 99, 6215–6229. [Google Scholar] [CrossRef]
- Russell, J.B. Another Explanation for the Toxicity of Fermentation Acids at Low pH: Anion Accumulation versus Uncoupling. J. Appl. Bacteriol. 1992, 73, 363–370. [Google Scholar] [CrossRef]
- Kampf, G. Antiseptic Stewardship: Biocide Resistance and Clinical Implications; Springer International Publishing: Cham, 2018. [Google Scholar] [CrossRef]
- Kitis, M. Disinfection of Wastewater with Peracetic Acid: A Review. Environ. Int. 2004, 30, 47–55. [Google Scholar] [CrossRef]
- Shi, C.; Li, C.; Wang, Y.; Guo, J.; Barry, S.; Zhang, Y.; Marmier, N. Review of Advanced Oxidation Processes Based on Peracetic Acid for Organic Pollutants. Water 2022, 14, 2309. [Google Scholar] [CrossRef]
- Fraser, J.A.L.; Godfree, A.F.; Jones, F. Use of Peracetic Acid in Operational Sewage Sludge Disposal to Pasture. Water Sci. Technol. 1985, 17, 451–466. [Google Scholar] [CrossRef]
- Wagner, M.; Brumelis, D.; Gehr, R. Disinfection of Wastewater by Hydrogen Peroxide or Peracetic Acid: Development of Procedures for Measurement of Residual Disinfectant and Application to a Physicochemically Treated Municipal Effluent. Water Environ. Res. 2002, 74, 33–50. [Google Scholar] [CrossRef]
- Chauret, C.P. Sanitization. In Encyclopedia of Food Microbiology; Elsevier, 2014; pp. 360–364. [Google Scholar] [CrossRef]
- Da Silva, W.P.; Carlos, T.D.; Cavallini, G.S.; Pereira, D.H. Peracetic Acid: Structural Elucidation for Applications in Wastewater Treatment. Water Res. 2020, 168, 115143. [Google Scholar] [CrossRef] [PubMed]
- Lund, P.; Tramonti, A.; De Biase, D. Coping with Low pH: Molecular Strategies in Neutralophilic Bacteria. FEMS Microbiol. Rev. 2014, 38, 1091–1125. [Google Scholar] [CrossRef]
- Slonczewski, J.L.; Fujisawa, M.; Dopson, M.; Krulwich, T.A. Cytoplasmic pH Measurement and Homeostasis in Bacteria and Archaea. Advances in Microbial Physiology 2009, 55, 1–317. [Google Scholar] [CrossRef] [PubMed]
- Kanjee, U.; Houry, W.A. Mechanisms of Acid Resistance in Escherichia coli. Annu. Rev. Microbiol. 2013, 67, 65–81. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, H.; Lin, Z.; Xu, P. Mechanisms of Acid Tolerance in Bacteria and Prospects in Biotechnology and Bioremediation. Biotechnol. Adv. 2015, 33, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhao, J.; Zhang, R.; Yu, R.; Guo, T.; Kong, J. Molecular Analysis of Glutamate Decarboxylases in Enterococcus avium. Front. Microbiol. 2021, 12, 691968. [Google Scholar] [CrossRef]
- Castanie-Cornet, M.-P.; Penfound, T.A.; Smith, D.; Elliott, J.F.; Foster, J.W. Control of Acid Resistance in Escherichia coli. J. Bacteriol. 1999, 181, 3525–3535. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Ma, D.; Chen, Y.; Guo, Y.; Chen, G.-Q.; Deng, H.; Shi, Y. L-Glutamine Provides Acid Resistance for Escherichia coli through Enzymatic Release of Ammonia. Cell Res. 2013, 23, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Spector, M.P.; Kenyon, W.J. Resistance and Survival Strategies of Salmonella enterica to Environmental Stresses. Food Res. Int. 2012, 45, 455–481. [Google Scholar] [CrossRef]
- Pereira, C.I.; Matos, D.; San Romão, M.V.; Barreto Crespo, M.T. Dual Role for the Tyrosine Decarboxylation Pathway in Enterococcus faecium E17: Response to an Acid Challenge and Generation of a Proton Motive Force. Appl. Environ. Microbiol. 2009, 75, 345–352. [Google Scholar] [CrossRef]
- Miller, E.F.; Maier, R.J. Ammonium Metabolism Enzymes Aid Helicobacter pylori Acid Resistance. J. Bacteriol. 2014, 196, 3074–3081. [Google Scholar] [CrossRef]
- Sun, Y. F1F0-ATPase Functions Under Markedly Acidic Conditions in Bacteria. In Regulation of Ca2+-ATPases,V-ATPases and F-ATPases; Chakraborti, S., Dhalla, N.S., Eds.; Springer International Publishing: Cham, 2016. [Google Scholar] [CrossRef]
- Du, D.; Wang-Kan, X.; Neuberger, A.; Van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug Efflux Pumps: Structure, Function and Regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef]
- Yang, H.; Yu, Y.; Fu, C.; Chen, F. Bacterial Acid Resistance Toward Organic Weak Acid Revealed by RNA-Seq Transcriptomic Analysis in Acetobacter pasteurianus. Front. Microbiol. 2019, 10, 1616. [Google Scholar] [CrossRef]
- Wang, B.; Shao, Y.; Chen, F. Overview on Mechanisms of Acetic Acid Resistance in Acetic Acid Bacteria. World J. Microbiol. Biotechnol. 2015, 31, 255–263. [Google Scholar] [CrossRef]
- Wang, C.; Cui, Y.; Qu, X. Mechanisms and Improvement of Acid Resistance in Lactic Acid Bacteria. Arch. Microbiol. 2018, 200, 195–201. [Google Scholar] [CrossRef]
- Wang, B.; Shao, Y.; Chen, T.; Chen, W.; Chen, F. Global Insights into Acetic Acid Resistance Mechanisms and Genetic Stability of Acetobacter pasteurianus Strains by Comparative Genomics. Sci. Rep. 2015, 5, 18330. [Google Scholar] [CrossRef]
- Seixas, A.F.; Quendera, A.P.; Sousa, J.P.; Silva, A.F.Q.; Arraiano, C.M.; Andrade, J.M. Bacterial Response to Oxidative Stress and RNA Oxidation. Front. Genet. 2022, 12, 821535. [Google Scholar] [CrossRef]
- Mourão, J.; Rebelo, A.; Ribeiro, S.; Peixe, L.; Novais, C.; Antunes, P. Atypical Non-H2S-Producing Monophasic Salmonella Typhimurium ST3478 Strains from Chicken Meat at Processing Stage Are Adapted to Diverse Stresses. Pathogens 2020, 9, 701. [Google Scholar] [CrossRef]
- Li, X.; Rensing, C.; Vestergaard, G.; Arumugam, M.; Nesme, J.; Gupta, S.; Brejnrod, A.D.; Sørensen, S.J. Metagenomic Evidence for Co-Occurrence of Antibiotic, Biocide and Metal Resistance Genes in Pigs. Environ. Int. 2022, 158, 106899. [Google Scholar] [CrossRef]
- Alonso-Hernando, A.; Capita, R.; Prieto, M.; Alonso-Calleja, C. Adaptation and Cross-Adaptation of Listeria monocytogenes and Salmonella enterica to Poultry Decontaminants. J. Microbiol. 2009, 47, 142–146. [Google Scholar] [CrossRef]
- Alonso-Hernando, A.; Capita, R.; Prieto, M.; Alonso-Calleja, C. Comparison of Antibiotic Resistance Patterns in Listeria monocytogenes and Salmonella enterica Strains Pre-Exposed and Exposed to Poultry Decontaminants. Food Control 2009, 20, 1108–1111. [Google Scholar] [CrossRef]
- Gantzhorn, M.R.; Pedersen, K.; Olsen, J.E.; Thomsen, L.E. Biocide and Antibiotic Susceptibility of Salmonella Isolates Obtained before and after Cleaning at Six Danish Pig Slaughterhouses. Int. J. Food Microbiol. 2014, 181, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Turolla, A.; Sabatino, R.; Fontaneto, D.; Eckert, E.M.; Colinas, N.; Corno, G.; Citterio, B.; Biavasco, F.; Antonelli, M.; Mauro, A.; et al. Defence Strategies and Antibiotic Resistance Gene Abundance in Enterococci under Stress by Exposure to Low Doses of Peracetic Acid. Chemosphere 2017, 185, 480–488. [Google Scholar] [CrossRef]
- Roedel, A.; Dieckmann, R.; Brendebach, H.; Hammerl, J.A.; Kleta, S.; Noll, M.; Al Dahouk, S.; Vincze, S. Biocide-Tolerant Listeria monocytogenes Isolates from German Food Production Plants Do Not Show Cross-Resistance to Clinically Relevant Antibiotics. Appl. Environ. Microbiol. 2019, 85, e01253-19. [Google Scholar] [CrossRef]
- EFSA Panel on Biological Hazards (BIOHAZ). Assessment of the Possible Effect of the Four Antimicrobial Treatment Substances on the Emergence of Antimicrobial Resistance - Scientific Opinion of the Panel on Biological Hazards. EFSA J. 2008. [Google Scholar] [CrossRef]
- Li, H.; Zhou, X.; Huang, Y.; Liao, B.; Cheng, L.; Ren, B. Reactive Oxygen Species in Pathogen Clearance: The Killing Mechanisms, the Adaption Response, and the Side Effects. Front. Microbiol. 2021, 11, 622534. [Google Scholar] [CrossRef]
Figure 2.
Mechanisms of metal/biocide and antibiotic co-selection: cross-resistance, co-resistance and co-regulation/co-expression (Adapted from [75]). Abbreviations: R - Resistance, T - Tolerance.
Figure 2.
Mechanisms of metal/biocide and antibiotic co-selection: cross-resistance, co-resistance and co-regulation/co-expression (Adapted from [75]). Abbreviations: R - Resistance, T - Tolerance.
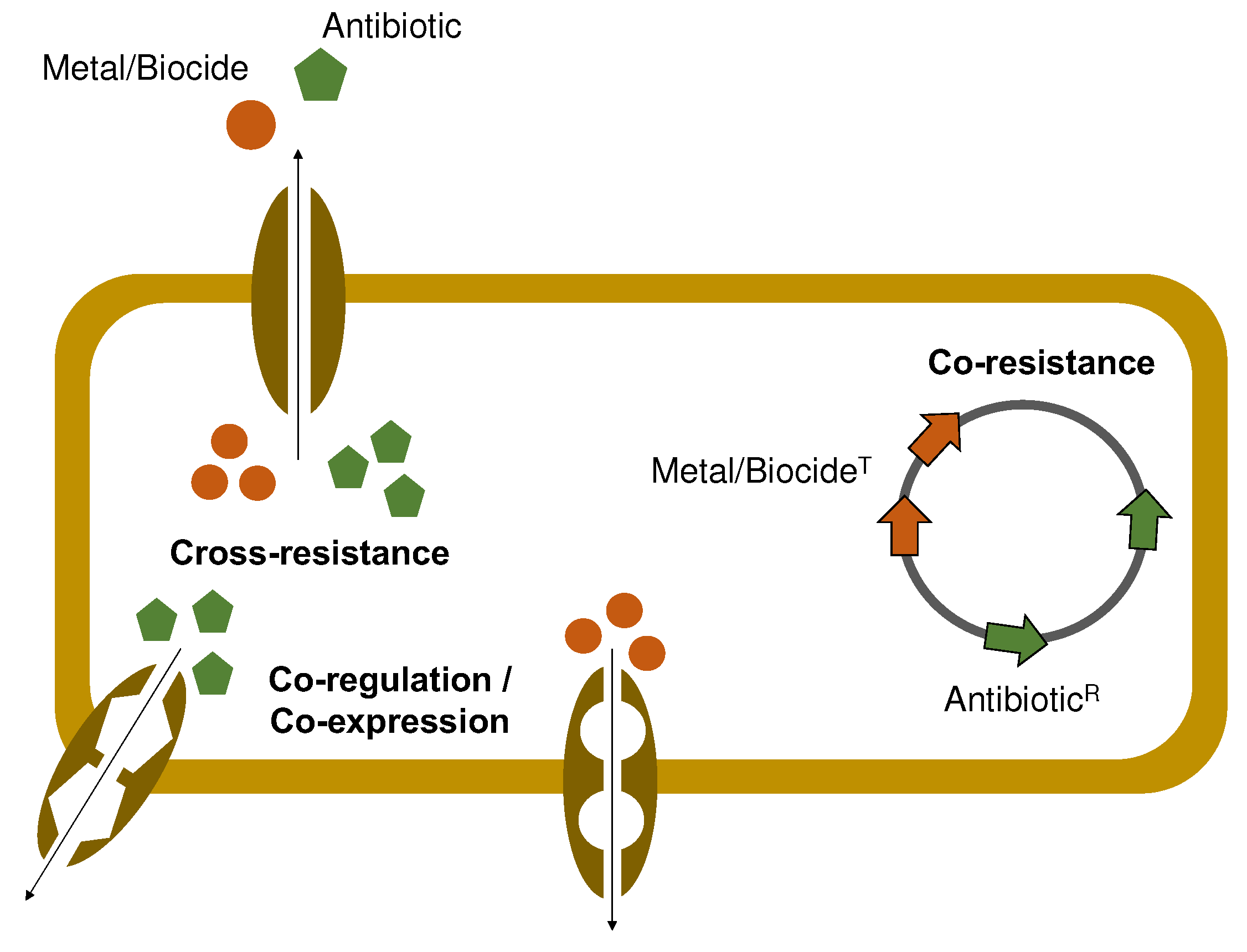
Figure 3.
Mechanisms of copper tolerance associated with the pco (figure-A) and sil (figure-B) genes clusters. The genes and their transcriptional and translation directions are indicated below the illustration. Genes with unknown functions are not represented (Adapted from [130]). The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Figure 3.
Mechanisms of copper tolerance associated with the pco (figure-A) and sil (figure-B) genes clusters. The genes and their transcriptional and translation directions are indicated below the illustration. Genes with unknown functions are not represented (Adapted from [130]). The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
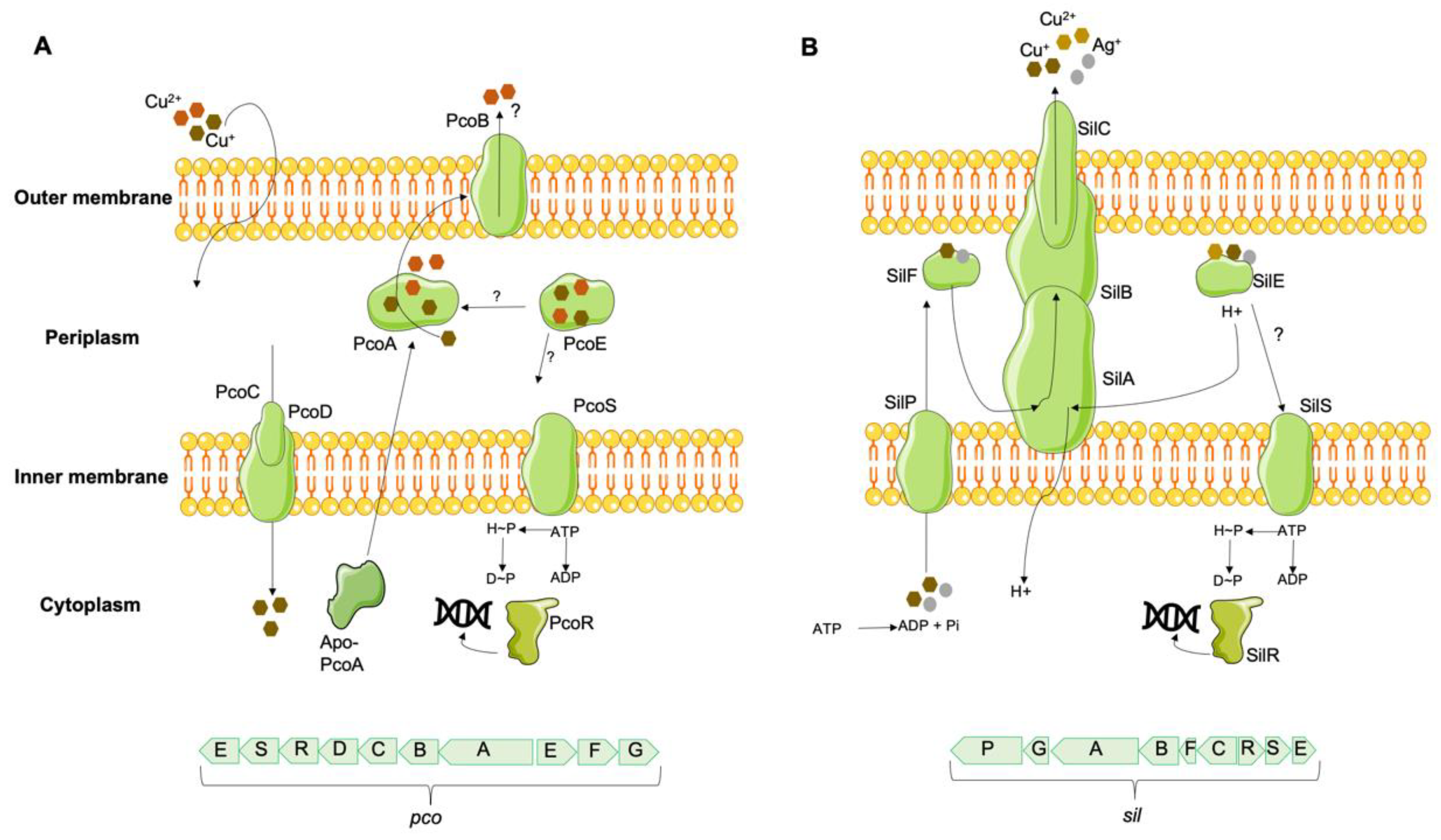
Figure 4.
Representation of genes and protein products associated with the TcrYAZB operon and CueO multicopper oxidase protein in Enterococcus spp. The tcrYAZB operon genes and their transcriptional and translation directions are indicated below the illustration (Adapted from [129]). The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attibution 3.0 unported license.
Figure 4.
Representation of genes and protein products associated with the TcrYAZB operon and CueO multicopper oxidase protein in Enterococcus spp. The tcrYAZB operon genes and their transcriptional and translation directions are indicated below the illustration (Adapted from [129]). The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attibution 3.0 unported license.
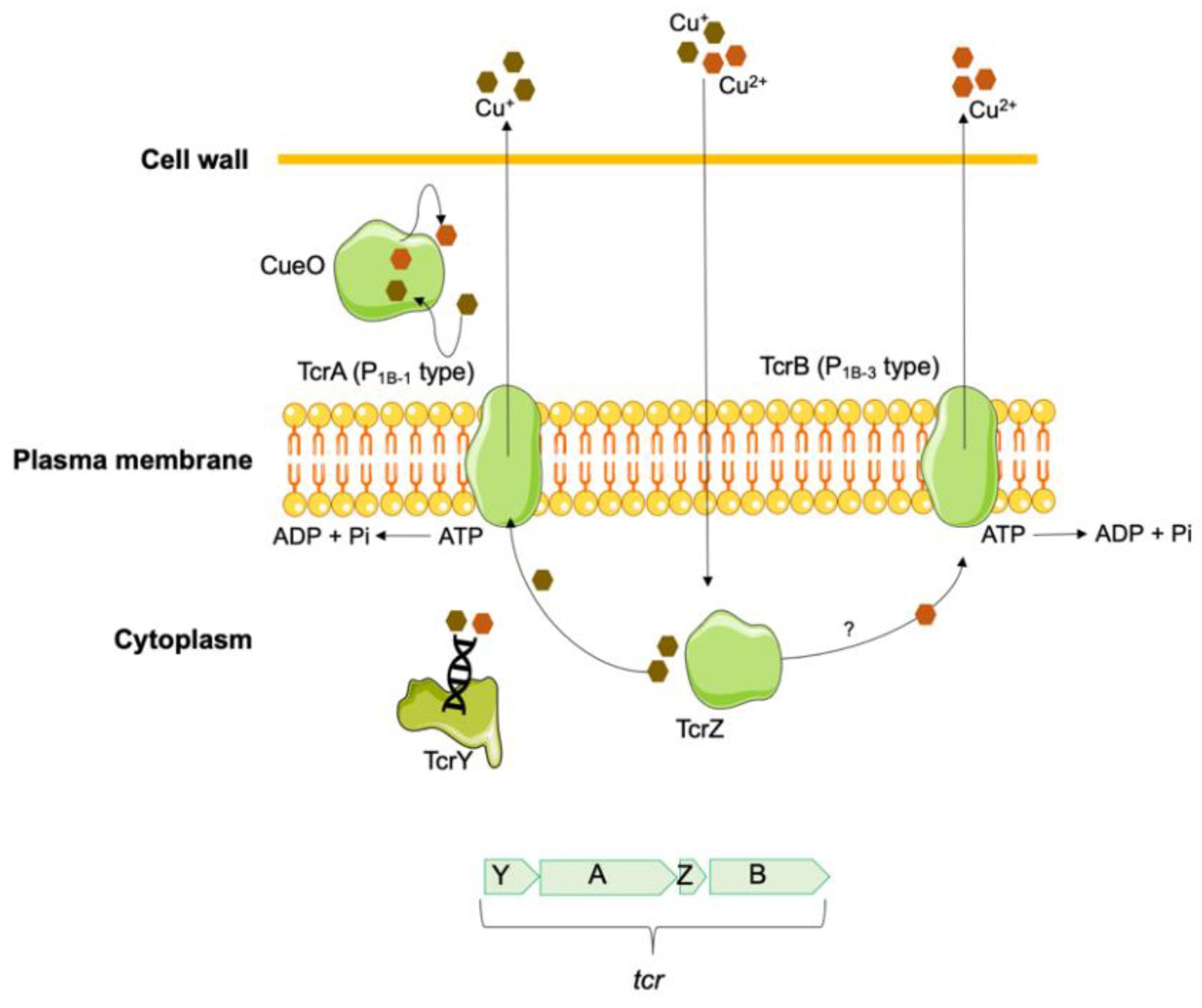
Figure 5.
Arsenic detoxification and respiratory metabolic pathways in bacteria. Four different systems might be involved in arsenic biotransformation pathways. Inorganic and organic arsenic detoxification pathways include arsenic resistance efflux system (ars) (yellow) and arsenic methylation (green), respectively. The respiratory oxidization of As(III) to As(V) and the reduction of As(V) to As(III) are represented by the aio/arx (light pink) and arr systems (dark pink), respectively. Uptake of As(V) and As(III) is represented by phosphate transporters (Pit or Pst) and by aqua-glycerolporins (GlpF) (brown) (Adapted from [183]). The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Figure 5.
Arsenic detoxification and respiratory metabolic pathways in bacteria. Four different systems might be involved in arsenic biotransformation pathways. Inorganic and organic arsenic detoxification pathways include arsenic resistance efflux system (ars) (yellow) and arsenic methylation (green), respectively. The respiratory oxidization of As(III) to As(V) and the reduction of As(V) to As(III) are represented by the aio/arx (light pink) and arr systems (dark pink), respectively. Uptake of As(V) and As(III) is represented by phosphate transporters (Pit or Pst) and by aqua-glycerolporins (GlpF) (brown) (Adapted from [183]). The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
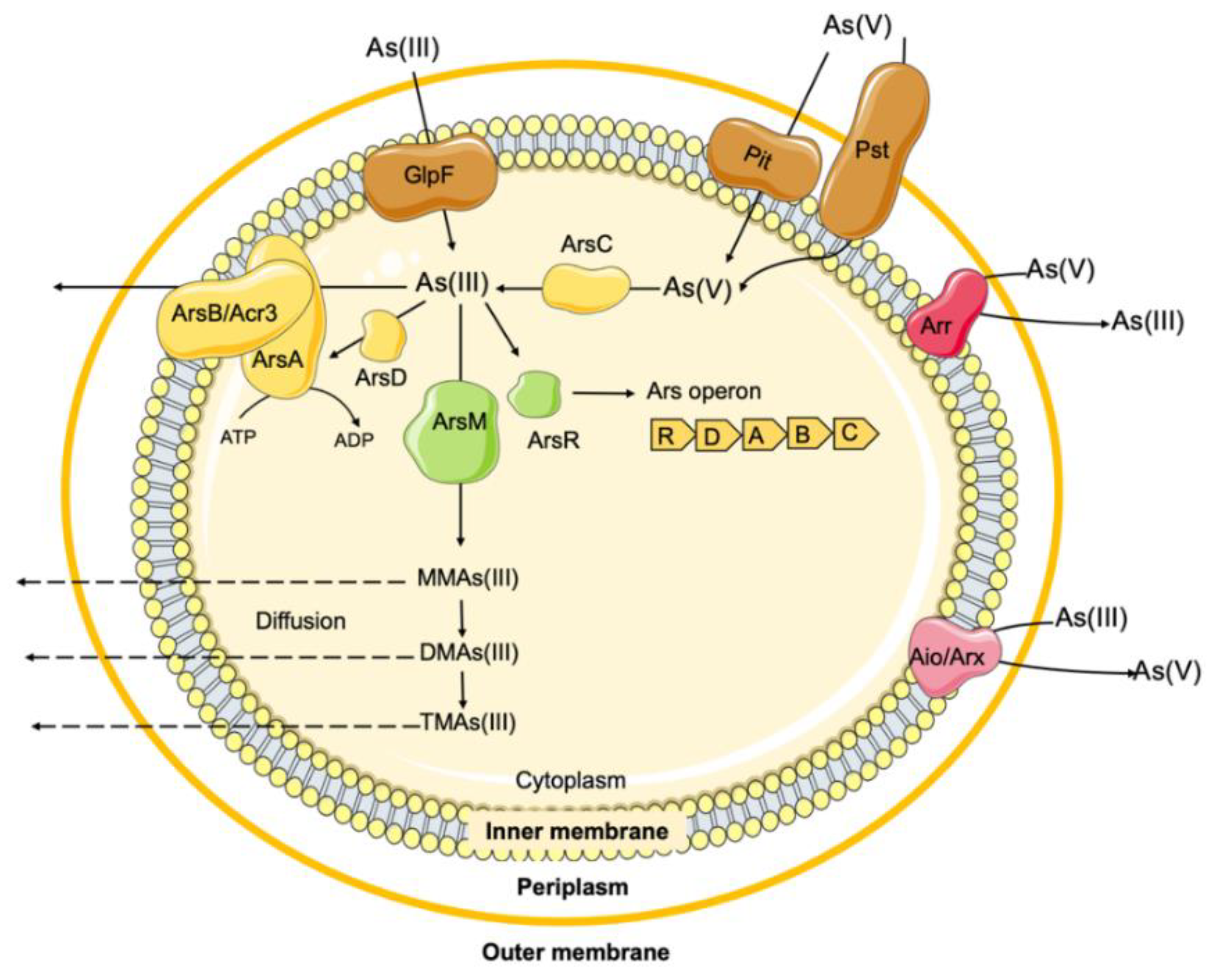
Figure 6.
Generic model of bacterial mer operon system. The mer operon genes are indicated below the illustration, with those in parentheses representing genes with variable presence in mer operons. Despite the variability of mer determinants in both Gram-positive and Gram-negative bacteria, overall mer expression is regulated by the MerR protein. The red diamonds represent the different types of Hg (MeHg - methylated Hg, Hg2+ - inorganic Hg, Hg0 – elemental Hg) (Adapted from [215]). The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Figure 6.
Generic model of bacterial mer operon system. The mer operon genes are indicated below the illustration, with those in parentheses representing genes with variable presence in mer operons. Despite the variability of mer determinants in both Gram-positive and Gram-negative bacteria, overall mer expression is regulated by the MerR protein. The red diamonds represent the different types of Hg (MeHg - methylated Hg, Hg2+ - inorganic Hg, Hg0 – elemental Hg) (Adapted from [215]). The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
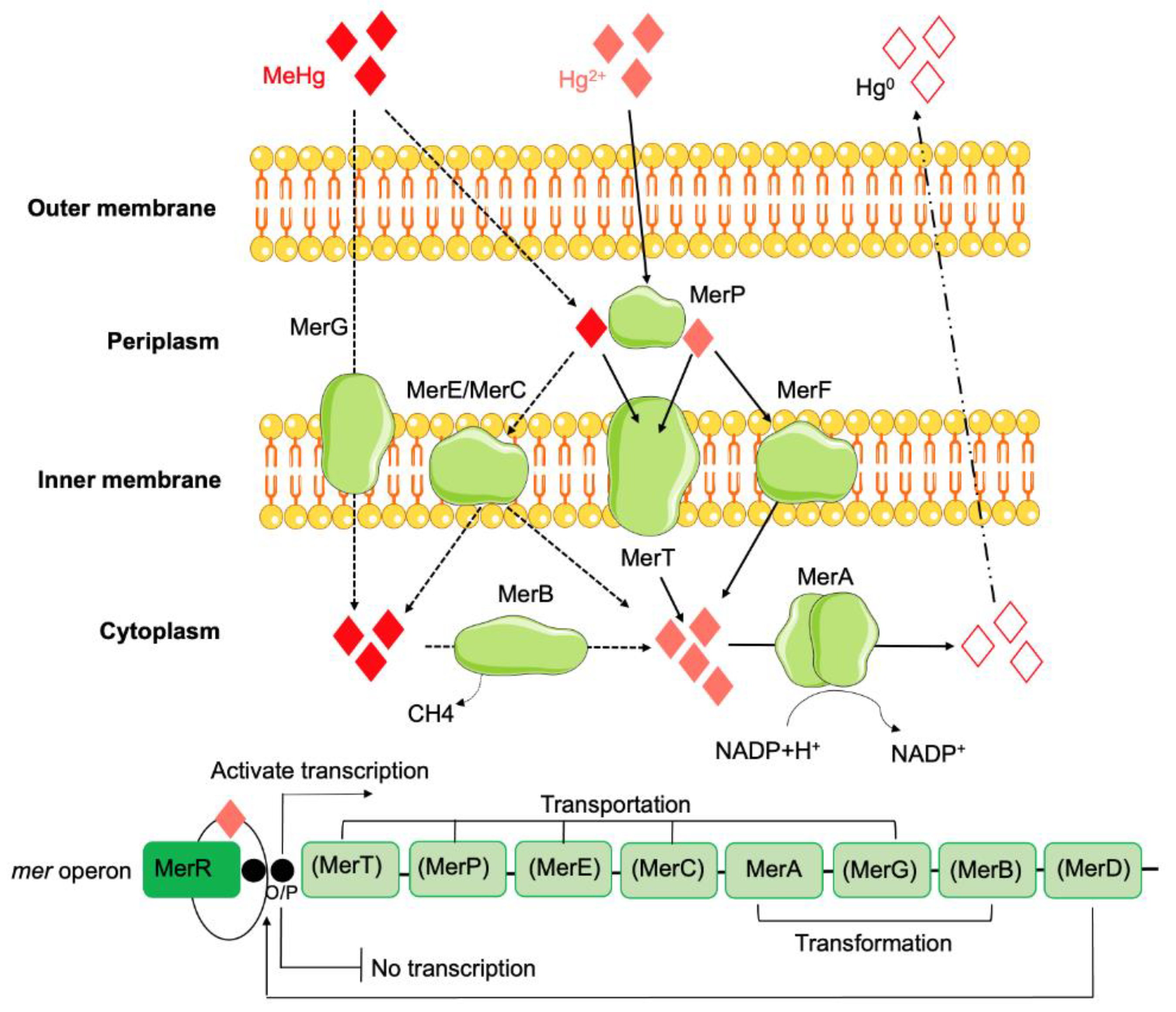
Figure 7.
Bacterial mechanisms for responding to acidic pH stress. The image illustrates some of the best-studied acid response mechanisms used by Gram-negative (figure A) and Gram-positive (figure B) bacteria (adapted from [291]). Abbreviations: ADP – adenosine diphosphate, ATP – adenosine triphosphate, Arg – arginine, Agm – agmatine, Cad – cadaverine, GABA - Glu/γ-aminobutyrate acid, Gln – glutamine, Glu – glutamate, Lys – lysine, Orn – ornithine, Putr – putrescine. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Figure 7.
Bacterial mechanisms for responding to acidic pH stress. The image illustrates some of the best-studied acid response mechanisms used by Gram-negative (figure A) and Gram-positive (figure B) bacteria (adapted from [291]). Abbreviations: ADP – adenosine diphosphate, ATP – adenosine triphosphate, Arg – arginine, Agm – agmatine, Cad – cadaverine, GABA - Glu/γ-aminobutyrate acid, Gln – glutamine, Glu – glutamate, Lys – lysine, Orn – ornithine, Putr – putrescine. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
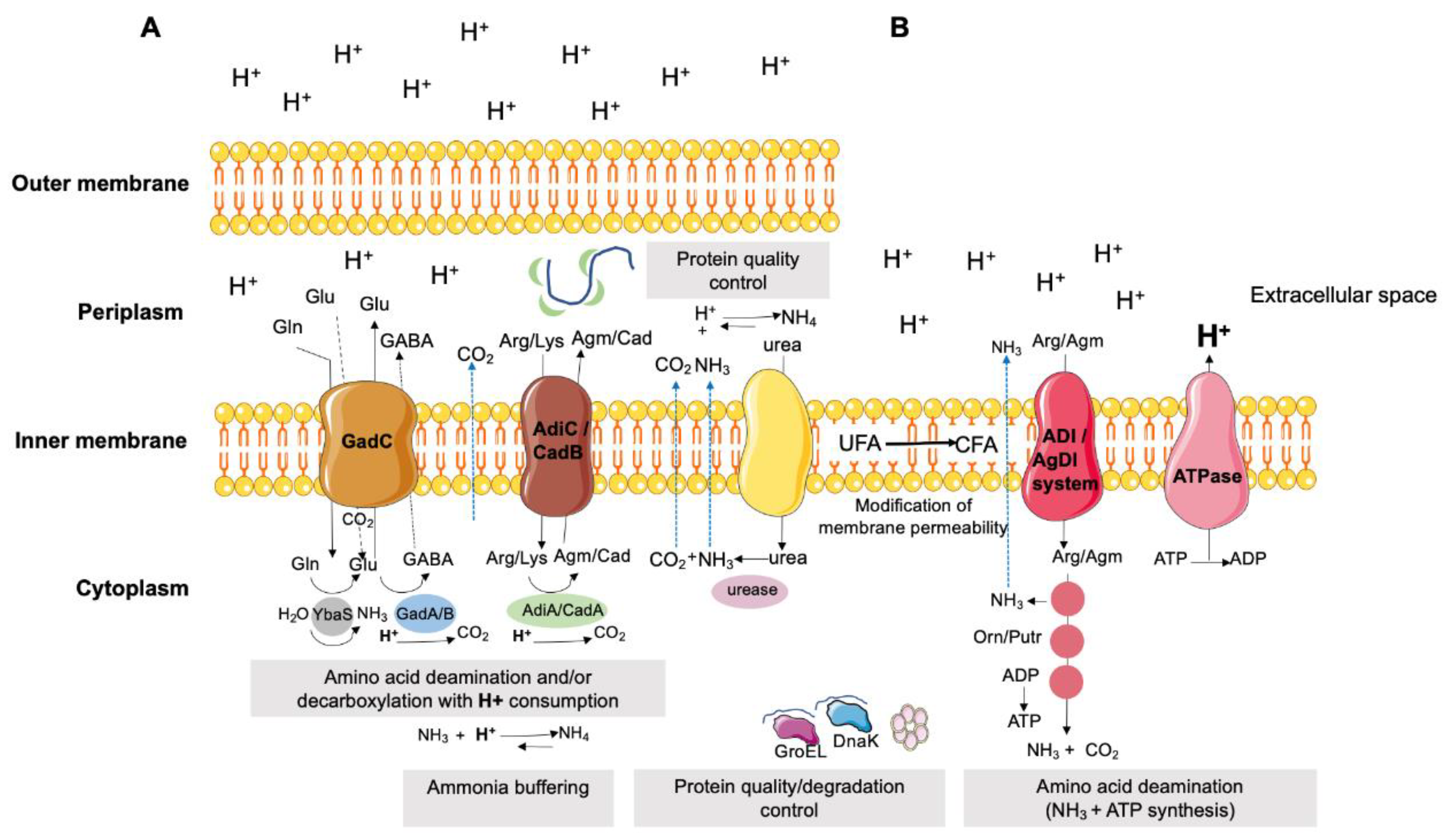
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



