Submitted:
18 July 2023
Posted:
19 July 2023
You are already at the latest version
Abstract
One of the leading causes of death worldwide, in both man and woman, is cancer. Despite the significant development in therapeutic strategies, the inevitable emergence of drug resistance limits the success and impedes the curative outcome. Intrinsic and acquired resistance are common mechanisms responsible for cancer relapse. Several factors crucially regulate tumourigenesis and resistance, including physical barriers, tumour microenvironment (TME), heterogeneity, genetic and epigenetic alterations, the immune system, tumour burden, growth kinetics and undruggable targets. Moreover, transforming growth factor-beta (TGF-β), Notch, epidermal growth factor receptor (EGFR), integrin-extracellular matrix (ECM), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), phosphoinositol-3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR), wingless-related integration site (Wnt)/β-catenin), Janus kinase/signal transducers and activators of transcription (JAK/STAT) and RAS/RAF/mitogen-activated protein kinase (MAPK) signalling pathways are some of the key players that have a pivotal role in drug resistance mechanisms. To guide future cancer treatments and improve results, a deeper comprehension of drug resistance pathways is necessary. This review will cover both intrinsic and acquired resistance and give a comprehensive overview of recent research on mechanisms that enable cancer cells to bypass barriers put by treatments, and like “satellite navigation”, find alternative routes to carry on their “journey” to cancer progression.
Keywords:
Cancer
; tumourigenesis
; drug resistance
; signalling pathways
; Wnt/β-catenin pathway
; JAK/STAT pathway
; PI3K/Akt/mTOR pathway
; RAS/RAF/MAPK/ERK signalling
1. Introduction
Over many years, cancer has consistently been a significant global public health burden. According to GLOBOCAN, in 2020 there were estimated around 19.3 million new cancer cases and 10.0 million cancer-related deaths (all cancers combined excluding non-melanoma skin cancer) worldwide [1,2]. Cancer continues to be the greatest cause of mortality in the world despite the enormous progress that has been made in developing more effective treatment options, partially due to drug resistance that is challenging to bypass. The heterogeneous, versatile, and adaptable nature of cancer to overcome “obstacles” put by treatments is some of the key reasons why it has been proven difficult to combat drug resistance, and therefore, reduce effectively and significantly relapse and mortality.
Carcinomas can be thought of as an organ-like structure made up of both transformed cancer cells and nontransformed stroma [3,4]. Tumour stroma is a complex milieu and a crucial component of the tumour microenvironment (TME) which is highly active, heterogeneous, and frequently tumour-type specific. It consists of noncellular components like the extracellular matrix (ECM; including secreted exosomes, metabolites, chemokines and cytokines) and the distinctive vascular system associated with cancer as well as a wide range of cellular elements like activated cancer-associated fibroblasts (CAFs), pericytes and mesenchymal stromal cells (MSCs). It also includes immune cells (such as natural killer (NK) cells, T and B lymphocytes and tumour-associated macrophages (TAMs)) and lymphatics [5,6,7].
Although there are many different types of disorders that go under the umbrella term "cancer," the formation of abnormal cells that proliferate beyond their natural bounds serves as both a defining characteristic and a common denominator. Normal cells gradually transition into the neoplastic stage because tumour formation is a multi-step process, and along the way they gain specific abilities that make them tumourigenic. These fundamental signature traits, which are both unique and supplementary, include continuing proliferative signalling, eluding growth inhibitors, allowing replicative immortality, avoiding cell death, generating angiogenesis, reprogramming of energy metabolism, avoiding immune destruction and triggering invasion and metastasis as well as relative autonomy of cancer cells in relation to the stroma. These traits are characterised by genomic instability, which constructs the genetic variety that expedites their inflammation and acquisition, which supports many hallmark functions. These distinguishing traits and enabling qualities describe essential components for cellular transformation [8,9].
Throughout the past few decades, chemotherapy, radiation, hormone therapy, targeted therapy, and immunotherapy have all undergone remarkable advancements in their development and therapeutic use for cancer [10]. However, the occurrence of intrinsic and acquired resistance to treatments in cancer patients, has been a significant barrier that limits the efficacy of cancer treatments and has an impact on patient survival [11,12]. A complete understanding of the interactions between tumour cells and their microenvironment is required to fully comprehend the acquirement of growth advantage and drug resistance in tumour cells. This review will aim to address the complexity of interactions within a tumour, recent findings on distinct drug resistance signalling pathways and strategies for combating anticancer drug resistance and enhancing its effectiveness.
2. Evolution of cancer
Depending on the type of tumour, where it is located, and what stage of the disease it is in, the TME's composition and structure changes. The following criteria are used to categorise the stages: (i) development of the primary tumour; (ii) invasion of cancer cells into nearby tissue; (iii) spread of cancer cells to distant organs via the blood or lymphatic system; (iv) extravasation into a secondary organ’ stroma; and (v) development of secondary tumours and metastasis [12,13].
The four stages of carcinogenesis are initiation, promotion, progression, and metastasis (Figure 1). Initiation is the first stage in which mutation of genes can occur naturally or because of a carcinogen exposure. The molecular signalling pathways involved in cellular proliferation, differentiation and survival can become dysregulated due to genetic alterations. Numerous factors, including the type and rate of carcinogenic metabolism as well as the response of the DNA repair function, have an impact on these pathways. During the protracted and reversible promotion stage, a buildup of actively proliferating preneoplastic cells takes place. Chemopreventive medicines may alter the mechanism at this point and impact growth rates. The transitional period between the emergence of a premalignant lesion and the beginning of an invasive malignancy is referred to as progression. This latter is the last stage of neoplastic transformation which involves genetic and phenotypic alterations as well as cell proliferation. This entails a sharp increase in tumour size and the possibility of new mutations in the cells, which could lead to invasion and metastatic dissemination. Chemopreventive medications should have the ability to work more effectively at the stages of carcinogenesis' initiation and promotion. Cancer cells spread to different parts of the body through the process of metastasis, which involves the lymphatic or circulatory systems. Chemopreventive drugs have been shown to prevent angiogenesis and invasion of primary tumours; as a result, they may be used to limit cancer metastasis [14].
3. Drug resistance in cancer
Cancer therapy, in its most basic form, consists of a treatment that goes against a group of cancer cells located in a particular host environment. The range of clinical responses is caused by the pharmacological qualities of the therapy, as well as the intrinsic and acquired molecular and physical features of cancer cells and external environmental factors [15]. Many studies have focused on the differences between intrinsic and acquired cancer drug resistance, however, a variety of overlapping mechanisms can also be contributing factors. By comprehending the underpinnings of resistance, to both current and upcoming treatments, we can develop a paradigm of mechanisms that takes into consideration all variables of cancer and therapeutic science that can influence it (Figure 2).
Therefore, development of drug resistance is a complex phenomenon that involves several different components and numerous interconnected signalling pathways. To our understanding, altered drug targets, modified drug metabolism, enhanced drug efflux, repair of DNA damage, epigenetics modifications, dormancy, undruggable targets, TME and epithelial-mesenchymal transition (EMT) are some of the fundamental molecular mechanisms by which cancer cells develop chemoresistance. The molecular mechanisms linked to drug resistance in cancer are explained below, and a schematic sketch is shown in Figure 2.
3.1. Intrinsic Drug Resistance
The natural resistance that occurs before the patient is exposed to medications is typically referred to as intrinsic resistance and can influence treatment effectiveness. The latter can be brought on by several factors, such as increased DNA repair capacity, altered drug metabolism, mutated or altered drug targets, reduced drug accumulation and deactivated cell death signals [11,12].
Cancer stem cells (CSCs) exhibit drug resistance because they overexpress adenosine triphosphate (ATP)-binding cassette (ABC) transporters [16]. Through certain regulatory genes, FOXM1, a transcription factor specifically for cell proliferation, controls the transition between the G1/S and G2/M cell cycle phases. Additionally, it is an oncogene that promotes the expansion and multiplication of cancer cells [17]. Through the expression of ABCC5 (ATP binding cassette subfamily C member 5), FOXM1 overexpression causes paclitaxel resistance in nasopharyngeal carcinoma [18]. Growth differentiation factor-15 (GDF-15) is a member of the superfamily of transforming growth factor-beta (TGF-β). Proliferation, angiogenesis, stemness, metastasis, drug resistance, and immunological modulation are all associated with overexpression of GDF-15 in cancer. It was demonstrated that stemness and indicators of treatment resistance were significantly positively correlated with GDF-15 expression in breast cancer patients. This suggests that the p-Akt/FOXM1 axis mediates the relationship between increased GDF-15 expression and enhanced stemness and treatment resistance in breast cancer [19]. Oestrogen receptor positive (ER+)/ human epidermal growth factor receptor 2 positive (HER2+) breast cancer is strongly influenced by the HER2-E subtype and erbB2, which results in resistance to endocrine therapy and a higher probability of recurrence [20]. About 20–30% of metastatic breast tumours overexpress the human epidermal growth factor receptor 2 (HER2/erbB2), which is associated with a poor prognosis [21]. In studies using trastuzumab as the sole treatment, over two thirds of patients showed intrinsic resistance to the drug [22,23]. High levels of GDF15 may be a factor in trastuzumab resistance in HER2 overexpressing breast cancer cells through activation of TGF-β receptor-Src-HER2 signalling crosstalk [24]. Furthermore, the aberrant activation of the phosphoinositol-3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) signalling pathway is closely related to resistance to anti-HER2 treatment [25]. Moreover, due to the genetic mutation(s) of genes involved in cancer cell proliferation and/or death, intrinsic drug resistance may develop in cancer cells before therapy. For instance, HER2 overexpression induced EMT and promoted resistance to cisplatin in gastric cancer cells [26]. CSCs and EMT are both associated to intrinsic drug resistance via these concurrent alterations mentioned above [27,28].
Intercellular genetic heterogeneity in cancer can result from genomic instability, which is characterised by mutations, gene amplifications, chromosomal rearrangements, gene deletions, gene translocations and alterations in microRNA [29]. Moreover, genotypic changes can have an impact on epigenetic variables affecting the heterogeneity of the mRNA, transcriptome, and proteome [30].
3.2. Acquired Drug Resistance
Gradual declines in a drug's ability to treat cancer after treatment can indicate acquired resistance. A number of factors can contribute to acquired resistance, including changes in the TME following therapy through various mechanisms, such as low pH, hypoxia, shifts and polarisations in the immune cell population, exosomes, various secretomes, vascular abnormalities, and soluble factors derived from stromal cells [11,12,31]. Paracrine signalling connections between stromal and tumour cells, mutations or altered levels of drug target expression and activation of a second proto-oncogene that develops into the driver gene, can also contribute to acquired dug resistance [11,12,32].
Targeted medicines cause subtler alterations that can be classified as acquired resistance after repeated exposures or early adaptive responses. Adaptive responses may be the cause of transient clinical reactions because they might happen so quickly that no response is ever clinically evident. Adaptive processes are frequently the result of epigenetic modification and/or non-genetic relief of negative feedback of signalling pathways, which activates parallel pathways or reactivates the initial one [33,34]. For example, due to the reactivation of upstream receptor tyrosine kinases (RTKs), such as epidermal growth factor receptor (EGFR), BRAF-mutant colorectal tumours are resistant to BRAF inhibitors, while low level of EGFR expression in BRAF-mutant melanomas were not affected by the negative feedback relief [35,36].
New genetic mutations can cause resistance and regeneration in cancers that had previously shrunk. Whole-genome sequencing comparing the genetic profiles of eight patients with acute myeloid leukaemia before and after relapse revealed novel gene mutations (e.g., DAXX, DDX41, DIS3, SMC3 and WAC) responsible for tumour resistance and regeneration [37]. Chemotherapeutic medications disrupt malignant cells' DNA, which probably accelerates the occurrence of new mutations. Also linked to acquired chemoresistance is the crosstalk that occurs between tumour cells and their microenvironment as the disease progresses [38]. This will be discussed later in the TME section below (section 3.10).
Some non-small-cell lung cancer (NSCLC) patients experience acquired resistance due to circumstances that can interfere with EGFR signalling, such as the upregulation of other RTKs like MET, the downstream activation of specific pathway elements, or phenotypic and histological changes [39]. Recently it was demonstrated that EGFR signalling pathways was activated by autocrine EGF and TGF-α and withstand c-Met and anaplastic lymphoma kinase (ALK) inhibition leading to primary and acquired resistance to TAE684/SGX-523 (ALK/c-Met inhibitors) in NSCLC [40]. In hepatocellular carcinoma (HCC) cells that heavily express c-MET, hepatocyte growth factor (HGF) activated the downstream PI3K/Akt and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathways through c-MET and concurrently reduced the anticancer effects of lenvatinib (a tyrosine kinase inhibitor) and promoted EMT [41]. Activating PIK3CA mutations in HER2+ breast cancer will unable a favourable response to pyrotinib plus trastuzumab neoadjuvant therapy [42]. By encouraging FOXD1 translation through PIK3CA/PI3K/Akt/mTOR signalling, FOXD1-AS1 (an oncogenic long non-coding RNAs (lncRNA)) exacerbates gastric cancer development and chemoresistance [43]. Moreover, one of the primary causes of medication resistance is the point mutation in the c-ros oncogene 1 (ROS1) gene. ROS1 is a receptor of the insulin family of tyrosine kinases. Recently it was demonstrated that the point mutations CD74-ROS1 D2033N and CD74-ROS1 S1986F render NSCLC cells resistant to crizotinib via the FAK/PI3K/Akt signalling pathway activation [44].
Activation of hypoxia-inducible pathways, EMT, interaction between the PI3K/Akt and Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathways, and enrichment of tumour-initiating cell population are some of the processes that cause acquired resistance to sorafenib [45]. Furthermore, potential mechanisms were revealed that underlie acquired resistance to gemcitabine in gallbladder cancer such as disruption of drug metabolism, and activation of receptor and non RTK (i.e., PDGFRA, ABL1 and LYN) as well as the increased expression of EMT-related markers, FN1, CDA and LAMC2 [46].
A significant increase was observed in single-nucleotide variants in the genes ATM, ATR, BRCA1, LRP1B, MAP2K1, PIK3CG and ZNF217 in addition to BRAF, KRAS, NRAS and EGFR among tumours receiving prior anti-EGFR [47]. Genes with possible signalling implications and those involved in DNA repair pathways made up the two main categories of these changes. ZNF217 and PIK3CG converge on Akt1 signalling which may encourage anti-EGFR acquired resistance. Protein kinase MAP2K1, whose acquisition occurs following anti-EGFR therapy, enhances the translation of signalling from MEK to ERK [48,49]. LRP1B inhibits β-catenin signalling [50]. β-catenin activation might be a different route to get around EGFR suppression, similar to Akt1 bypass signalling. It is interesting to note that after exposure to anti-EGFR, a greater frequency of mutations in BRCA1, ATR and ATM was found. These modifications could aid in the evolution of a response to targeted therapy and could also account for increases in relative tumour mutation burden (rTMB) in patients exposed to anti-EGFR leading to acquired resistance [47].
3.3. Altered drug targets
One of the main causes of drug resistance is drug targeting alteration, which occurs when drug targets' expression and functionality are altered. Di(2-ethylhexyl) phthalate (DEHP) is a chemical that is frequently found in everyday items and polyvinylchloride medical equipment. As a result, phthalates can enter the human body through eating, inhalation, and medical procedures. Phthalates induce cancer progression and chemotherapeutic resistance [53]. Recently it was demonstrated that DEHP increased trefoil factor 3 (TFF3) expression through the vinculin/aryl hydrocarbon receptor (AhR)/ERK signalling pathway which induced EMT [54]. In human breast cancer, the expression of the oncogene TFF3 is favourably connected with both ER+ and negative cells, and it increases cell metastasis, invasion, proliferation and treatment resistance [55]. Through the ubiquitination pathway, DEHP promoted AhR-related changes in oestrogen receptor expression, which reduced tamoxifen's effects in AhR knockout mice [54].
T315I point mutation that arises in BCR-ABL kinase domain is the most frequent mutation in BCR-ABL that causes resistance to first-generation (imatinib) or second-generation tyrosine kinase inhibitors (TKIs) that target the BCR-ABL protein, leading to a poor clinical prognosis in chronic myeloid leukaemia [56,57]. Due to the activation of intrinsic signalling pathways such as the RAS/RAF/MAPK/ERK, GSK3 β and JAK/STAT5 pathways, imatinib intolerance or initial resistance arises, and many leukaemic patients acquire secondary resistance [58,59,60,61]. Most cellular intrinsic mechanisms play a role in the development of resistance; either directly through BCR-ABL1 point mutations, which predominate in primary resistance, or indirectly through activation of signalling pathways independent of BCR-ABL1, which frequently lead to disease recurrence and therapy relapse [59,62]. Typically, such activation frequently happens in a BCR-ABL1-independent manner; as a result, those oncogenic pathways continue to be active even after treating leukaemic cells with imatinib, including nonmutated BCR-ABL1 cells.
Moreover, osimertinib is a third generation powerful EGFR-TKI used to treat NSCLC patients with EGFR mutations. The therapeutic use of osimertinib is nonetheless restricted by the emergence of acquired resistance associated with the triple mutation Del19/T790M/C797S in EGFR [63]. Furthermore, mutations at either V550 (a gatekeeper residue) or C552 (hinge-1 residue) in the kinase domain of fibroblast growth factor receptor 4 (FGFR4) prevent fisogatinib (a potent and selective FGFR4 inhibitor) from interacting with the FGFR4's ATP binding site resulting in acquired clinical resistance to fisogatinib in patients with HCC [64].
TKIs can prevent downstream pathways from being activated improperly by aberrant protein tyrosine kinases (PTKs). PI3K/Akt, RAS/MAPK/ERK, and JAK/STAT are examples of key signalling pathways that control a variety of cellular processes by stimulating proliferation, encouraging angiogenesis, preventing apoptosis, and promoting drug resistance [65]. Therefore, due to mutations at the drug binding sites, TKIs lose the ability to inhibit PTKs (e.g., FGFRs, EGFRs, ALK, platelet-derived growth factor receptor (PDGFRs), Insulin-like growth factor receptor (IGFRs), vascular endothelial growth factor receptor (VEGFRs)), resulting in constant activation of downstream signalling pathways.
3.4. Modified drug metabolism
Once ingested, drugs are transformed biochemically by drug metabolism enzymes. Metabolic activation is necessary for many anticancer drugs to carry out their mechanism of action. These enzymes have been linked to drug activation and inactivation in cancer cells, including the uridine diphosphoglucuronosyltransferase (UGT) superfamily, glutathione S-transferase (GST) superfamily and cytochrome P450 (CYP) system [66,67]. A modification in CYP may alter these proteins' capacity for drug metabolism, leading to both a large increase in drug release and a change in how the drug is broken down. As a result, patients' intratumoural medication concentrations drop, and the treatment loses its effectiveness. Recently it was shown that docetaxel's resistance in HCC cells may be significantly mediated by the metabolic deactivation of the CYP isoforms 3A4 (CYP3A4) [68].
Key metabolic enzymes of 5-fluorouracil (5-FU) include thymidylate synthase (TYMS). 5-FU is a chemotherapy drug and TYMS is one of its target enzymes. It was established that SNHG15 (lncRNA) increased 5-FU chemoresistance in colorectal cancer (CRC) by controlling TYMS expression [69]. Moreover, the activation of detoxification systems that serve as a defence against contaminants can limit the therapeutic efficiency of anticancer medicines. A compromised detoxification mechanism in cancer cells makes medication responses inefficient and encourages resistance. One of the primary contributing causes to the development of treatment resistance in cancer is the exclusion of medicines by GST [70]. Numerous biological activities, including cell differentiation, proliferation, and death, depend on GST. Drug resistance is influenced by an increase of glutathione. For instance, upregulation of GST-π production and activation of PI3K/Akt/mTOR signalling pathway were promoted by regenerating gene 4 (REG4) overexpression which contributed to an invasive phenotype and induced cisplatin and paclitaxel resistance in ovarian cancer. [71]. Furthermore, by blocking the MAPK pathway, GSTs contribute to the emergence of drug resistance [70].
3.5. Enhanced drug efflux
The term "drug efflux" refers to the rise in the efflux of cytotoxic medications by active ATP-binding cassette (ABC) transporter proteins, which is one mechanism of drug resistance. Chemotherapy can only be used successfully to a limited extent because of these drug efflux transporters, which lower intracellular drug concentration and inhibit therapeutic response [72,73,74,75]. Humans have been found to have 48 members of the ABC transporter family. There are only 13 different types of ABC transporters that have been found to play a part in drug resistance in cancer (ABCC1/2/3/4/5/6/10, ABCB1/2/5, ABCA2/3 and ABCG2) [76]. Breast cancer resistance protein (BCRP/ABCG2), P-glycoprotein (P-gp/MDR1/ABCB1), and multidrug resistance-associated protein 1 (MRP1/ABCC1) are three major ABC transporters that have recently undergone substantial research to better understand how multiple drug resistant (MDR) works [77,78].
The role of proteins and signalling pathways in the regulation of ABC transporters in cancer cells has been extensively documented in recent years. Through interactions with upstream and downstream targets, the PI3K/Akt pathway which is elevated in many human malignancies, has been found as a critical elusive link in MDR. This signalling pathway promotes the progression of cancer progression and confers resistance to chemotherapy treatments by increasing the expression of the ABC transporters BCRP, MRP1 and P-gp [79,80].
In the human acute lymphoblastic leukaemia cell lines, activation of the MAPK/ERK and JNK pathways upregulated the expression of the ABCB1 and ABCG2 genes respectively [81]. Other research revealed that the ABCG2-mediated multidrug resistance in colon cancer cells was caused by the JNK1/c-jun signalling pathway [82]. Interestingly it was recently found that activation of the RhoB/PI3K/Akt pathway-mediated overexpression of ABC transporters by hsa-miR-3178 is an intriguing mechanism which promotes gemcitabine resistance in pancreatic cancer cells [83].
Through the SIRT1/CREB/ABCG2 signalling pathway, miR-132 has also been shown to increase cisplatin resistance in LGR5+ gastric cancer stem cell-like cells [84]. Noting that ABC transporter proteins are highly expressed on the cell surfaces of CSCs which have been found to have a significant role in drug resistance and play a role as indicators for CSC isolation and identification [85,86,87,88,89,90]. Furthermore, LGR5 is a receptor that enhances the wingless-related integration site (Wnt)/β-catenin signalling pathway, promoting the growth and self-renewal of CSCs. It was recently discovered that a strong cooperation between the expression of LGR5 and LRP6 (mediators of Wnt/β-catenin signalling) was enhanced in neuroblastoma resistant cells [91]. As well as the higher β-catenin expression in those neuroblastoma cell lines with acquired resistance to vincristine or doxorubicin is an indicator of β-catenin-dependent Wnt signalling [91]. Moreover, a correlation between elevated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation/phosphorylation and upregulated MDR efflux transporter expression was demonstrated in doxorubicin resistant breast cancer cells [92].
3.6. Repair of DNA damage
Numerous chemotherapeutic drugs focus heavily on targeting DNA [93]. Resistance to drugs that target DNA, however, might be brought on by increased DNA repair and tolerance to DNA damage [94]. By causing DNA damage, several chemotherapy medicines, including 5-FU and cisplatin, destroy cancer cells. Drug resistance may occur from DNA lesion repairs caused by the DNA damage response of damaged cells to anti-cancer medications [95]. Upregulation of p53-target genes on DNA damage response and repair was brought about by 5-FU therapy. When the damage was successfully repaired, the resistant colon cancer cell lines experienced decreased cell cycle arrest and apoptosis than the wild type ones [96]. Moreover, it was discovered that 5-FU resistant human colon cancer cell lines have increased levels of the DNA repair genes FANCG, FEN1 and RAD23B [96,97].
Checkpoint kinase 1 (CHK1) plays a vital role in DNA damage and response and is a crucial effector in the control of replication. The mechanism of resistance to prexasertib, a CHK1 inhibitor was investigated in sarcoma xenografts [98]. BCL-xL, an anti-apoptotic protein, was found in higher concentrations and the PI3K and MAPK signalling pathways were phosphorylated more frequently in prexasertib-resistant tumours. Akt, MEK1/2, and ERK1/2 were found to be substantially active in resistant tumours [98]. Other cell lines have also shown increased RAS/MEK/ERK activity in response to CHK1 inhibition [99,100]. Combining prexasertib with MAPK or PI3K inhibitors, were not enough to overcome developed resistance in sarcoma xenografts [98]. The stimulation of RAS/MAPK and, to a lesser extent, PI3K/Akt/mTOR signalling by EGFR overexpression or activation promotes cell proliferation and may avoid the replication stress caused by prexasertib. About 50% of triple negative breast cancer (TNBC) show overexpression of the EGFR, which is linked to poor overall survival [101,102,103]. It was suggested that EGFR activation or overexpression is a factor in the innate resistance of TNBC to prexasertib and may also be responsible for the drug's low clinical efficacy [104].
3.7. Epigenetics modifications
Cell destiny and pathogenic provenience are greatly influenced by epigenetics. It appears that non-genetic heterogeneity contributes to the development of cancer-causing cells and/or resistance to treatment. Impairment in gene expression is caused by epigenetic alterations, which last for several cell divisions and finally result in non-genetic heterogeneity and treatment resistance [105]. The development of chemoresistance in cancer is fuelled by epigenetic changes that are linked to histone modification, DNA methylation, chromatin remodelling, and changes associated to non-coding RNA (ncRNAs) [106]. Accumulating evidence show that epigenetic changes contribute to the development of various resistance mechanisms, such as improved DNA repair, enhanced drug efflux, and defective apoptosis. For example, the chromodomain helicase DNA-binding protein 4 (CHD4), which modulates chromatin remodelling, specifically causes drug resistance in breast cancer gene1/2 (BRCA1/2) deficient cells through aiding DNA damage repair [107]. Recently, it was demonstrated that by interacting with major vault protein (MVP), CHD4 encourages gastric cancer cell proliferation and chemoresistance. As well as by stimulating drug efflux, CHD4 promotes the reduction in the intracellular concentration of cisplatin. It also enhances the protein interaction between ERK1/2 and MEK1/2 leading to the activation of MVP/MEK/ERK signalling axis [108].
Moreover, DNA methylation and gene expression profiles of fulvestrant- and tamoxifen-resistant MCF7 derivatives with oestrogen-responsive MCF7 human breast cancer cells were analysed. Resistance to tamoxifen is developed by significant alterations in downstream ER target gene networks, whereas acquired resistance to fulvestrant revealed a general up-regulation of growth-stimulatory pathways including cytokines and cytokine receptors, the EGFR, ErbB2 and related proteins, the Notch pathway, the Wnt/β-catenin pathway and the interferons (IFN) signalling pathway/IFN-inducible genes were among the prominently altered pathways in MCF7 cells resistant to fulvestrant [109]. For instance, demethylation of DNA near an oncogene's promoter region would increase the gene's expression, leading to treatment resistance. In a resistant HCC cell line, thymosin 4 (Tβ4), a G-actin monomer binding protein, was shown to be enhanced through DNA demethylation and active modification of histone H3 at the promoter region [110]. In the HCC cell line, overexpression of Tβ4 caused stem cell-like capabilities to develop, as well as in vivo resistance to the VEGFR inhibitor sorafenib [110].
A study shows that sorafenib resistance develops because of the histone demethylase KDM1A also known as Lysine demethylase 1A (LSD1). They found that cells resistant to sorafenib (a TKIs) had a higher capacity for self-renewal. KDM1A's importance for the stemness of liver CSCs by epigenetic alteration was previously discovered by the same team [111]. They found a potential mechanism by which KDM1A causes resistance to sorafenib through the control of important β-catenin signalling pathway antagonists. They also showed that KDM1A is necessary for maintaining the stemness of resistant HCC cells to sorafenib [111].
The first histone modification enzyme shown to be linked to drug resistance to several anticancer drugs is a lysine demethylase called Lysine demethylase 5A (KDM5A) [112,113]. Erlotinib (an EGFR inhibitor) was less effective against breast cancer cells with amplifications of the KDM5A gene due to increased expression of a set of genes associated with apoptosis/cell cycle, including the apoptosis effector BCL2 antagonist/killer1 (BAK1) and the tumour suppressor p21 [113]. Lewis lung carcinoma and renal cell carcinoma become resistant to sunitinib (a RTK inhibitor) due to KDM5C which was discovered to be a significant epigenetic regulator in this process. In patients with NSCLC, KDM1A is crucial for inducing gefitinib resistance by the development of hypoxia through generating EMT [114].
Moreover, by changing gene expression and the structure of regulatory proteins, N6-methyladenosine (m6A), a particular type of RNA alteration, influences the development of tumours [115]. KIAA1429 plays a vital role in m6A methylation or controls ncRNAs, including microRNAs (miRNAs) and lncRNA to promote the growth and metastasis of many cancers [116,117]. Recently it was demonstrated that the activation of the JNK/MAPK signalling pathway results in m6A KIAA1429-mediated gefitinib resistance in lung adenocarcinoma cells [118].
The effects of mucin 17 (MUC17) on the epigenome of EGFR-TKI-acquired drug resistance was examined in NSCLC cells. Gefitinib/osimertinib-resistant (GR/OR) cells were found to increase genome-wide DNA hypermethylation, notably in 5-UTR related to several oncogenic pathways, where GR/OR cells had a pro-oncogenic effect by decreasing MUC17 expression. The downregulation of MUC17 caused by acquired GR/OR was triggered by a methylation promoter dependent on the DNA methyltransferases1/ Ubiquitin-like containing PHD Ring Finger 1 (DNMT1/UHRF1) complex, which in turn stimulated NF-κB activity [119].
3.8. Slow growing cells
Tumour cells may have transcriptional plasticity, due to epigenetic reprogramming, which will change them into persister cells. These "persisters" are a collection of cells that are growing slowly and have the potential to either re-grow when therapy is stopped or develop enduring resistance. KDM5B, a member of the KDM5A family, designates a small subset of slow-cycling cells in melanomas that are necessary for ongoing tumour maintenance and are dynamically triggered depending on the microenvironmental situation. These KDM5B-positive cells cycle slowly and have increased self-renewal. They are intrinsically resistant to many cytotoxic therapies, and through a dysfunctional Jagged 1/Notch 1-signalling pathway, they can produce an offspring that are extremely proliferative [120].
Recent research demonstrated that abnormal expression of nerve growth factor receptor (NGFR), SRY-Box transcription factor 2 (SOX2), AXL RTK and melanocyte inducing transcription factor (MITF) in melanoma cells make them more susceptible to shift into a persister state in response to RAF and MAPK inhibition [121,122].
In response to targeted kinase inhibitors, the histone H3 lysine 27 trimethylation (H3K27me3) specific demethylases, KDM6A/B, are activated and crucial for the transformation of naive glioblastoma stem cells into the slow-cycling drug-tolerant persisters (DTPs). Pervasive acetylation (H3K27ac) of cis-regulatory components occurs in conjunction with the transition to the persister state and is made possible by a widespread redistribution of the repressive mark H3K27me3. These persisting cells display primitive neurodevelopmental hallmarks because of this modified chromatin state and heavily rely on Notch signalling [123]. Sharma et al. consistently identified a small fraction of reversibly "drug-tolerant" cells while simulating the acute response to several anticancer drugs in drug-sensitive human tumour cell lines. These cells exhibit a >100-fold decrease in drug sensitivity and continue to exist due to activation of the insulin-like growth factor 1 receptor (IGF-1R) signalling and a modified chromatin state that needs the histone demethylase RBP2/KDM5A/Jarid1A. Individual cells within the population transiently acquire this drug-tolerant phenotype at low frequency, suggesting that drug tolerance is dynamically regulated by phenotypic heterogeneity [112]. In addition, KDM5A is necessary to create a transient chromatin state in EGFR-mutant lung cancer cell lines with elevated expression driven by IGF-1 signalling pathway in both DTPs and drug-tolerant expanded persisters (DTEPs). This will mediate the development of EGFR inhibitor resistance [112].
The irreversible stop of cell growth known as cellular senescence is what causes tumour-suppressive pathways regulated by p16 and/or p53 to be activated. As a tumour suppressor, the protein p16INK4a (also known as p16) inhibits the activity of cyclin-dependent kinases (CDKs) and slows cell division by delaying the transition from the G1 to the S phases of the cell cycle [124]. Both endogenous and external factors can promote cellular senescence. The three main factors are shortening of telomere, increased mitogenic signalling created by oncogene activation, and non-telomeric DNA damage brought on by chemotherapeutic medicines. Senescence can begin, for instance, when chemotherapy drugs like doxorubicin and cisplatin cause cell death [125,126,127]. In part via inhibiting apoptosis, p53 and INK4a/ARF mutations encourage carcinogenesis and treatment resistance [126]. Drug resistance and tumour progression/recurrence have been linked to a mechanism known as escape from therapy-induced senescence (TIS) [128]. The ability of cancer cells with TIS to acquire stem-cell characteristics explains how they can avoid senescence and relapse [129,130].
Moreover, metastasis, chemoresistance, and cancer recurrence are all influenced by tumour dormancy. CSCs frequently exist in a quiescent state where they might stay in the G0/G1 stage and proliferate at a slow rate [131,132]. Quiescence (reversible cell cycle arrest) features help CSCs develop resistance to radiation and chemotherapy because most traditional chemotherapeutic agents target proliferating cells [87,133,134]. For example, the majority of 5-FU-resistant gastric cancer cells with CSC characteristics were quiescent cells that stayed in the G0/1 phase [135]. In response to chemotherapies, CSCs enter quiescence by initiating a complex array of intracellular molecular and epigenetic programmes [131]. The three signalling pathways most frequently engaged in CSC quiescence are Notch, Wnt, and p38-MAPK. Active p38 mitogen-activated protein kinase 1 (MAPK1) can cause CSC to enter a dormant state in prostate cancer [136]. It is noteworthy that the Wnt canonical pathway component c-Myc can speed up the CSC cell cycle and encourage CSC reawakening, whereas their inactivation was directly linked to the onset of reversible quiescence [137,138,139,140].
3.9. Undruggable targets
Several of the most powerful oncogenes and tumour suppressor genes, such as MYC, RAS, and TP53, remain intractable despite increasing progress in efforts to target oncogenic driver mutations. Ras proteins were discovered to be oncogenes in the early 1980s, but despite extensive research over more than three decades to identify particular inhibitors, they were thought to be unreachable targets.
In up to 90% of human melanoma, mutated BRAF or mutated NRAS hyperactivate the kinase ERK, according to the examination of genetic changes [141,142]. The rationale for developing targeted inhibitors of mutant BRAF and MEK, the kinase that functions downstream of BRAF to activate ERK, as treatments for advanced melanoma was supplied by these findings [143]. The overall survival of patients significantly increased as a result of the introduction of targeted medicines (MAPK pathway inhibitors such as BRAF and MEK inhibitors) and immunotherapies (immune checkpoint inhibitors). However, a lack of clinical effects, side effects, and the rapidly escalating treatment resistance limit the long-term efficacy of such treatments. This resistant phenotype is supported by several molecular pathways [144]. Moreover, resistance may also be caused by target indifference, in which the effects of focusing on an oncogenic driver are mitigated by changes made to the pathway later on or concurrently. This is demonstrated by the fact that resistance to anti-EGFR therapy in colon cancer can be caused by downstream mutations that activate NRAS or KRAS [145]. Recently it was shown that JAK/STAT pathway activation occurs as BRAFV600E thyroid cancer cells become resistant to BRAF inhibitors [146]. Interestingly, insensitivity to inhibition of the MAPK/ERK pathway in advanced melanoma tumours harbouring the BRAFV600E mutation resulted from the activation of compensatory signalling cascades. Particularly in mesenchymal-like cells, the PI3K/Akt/mTOR axis displayed increased activity, resulting in a decreased MAPK/ERK signalling dependency and promoting stem-like features making the latter pathway's inhibitors ineffective [147].
Cancer frequently harbours mutations in the p53 pathway. In fact, the TP53 gene exhibits mutations or deletions in around 50% of human malignancies, which predominantly cause decreased tumour suppressor activity [148]. Damaged cells may multiply after losing their p53 functioning, passing on changes to the following generation [149]. Deregulation of p53 frequently causes tumour development and mutant p53 cancers are frequently characterised by genomic instability promoting proliferation, migration, invasion, angiogenesis and increased drug resistance [149,150].
In NSCLC, mutated p53 increases binding to the nuclear factor erythroid 2 –related factor 2 (Nrf2) promoter, supported by an activation of the NF-κB signalling pathway, which further increases Nrf2 expression. Nrf2 is a transcription factor that codes for detoxification enzymes and confers resistance to anticancer drugs. In addition, in p53 mutant colon cancer cells, absence of DNA mismatch repair trigger resistance to cisplatin [151]. Furthermore, mutant p53 affects the ERK-mediated transcription of early growth response-1 (Egr-1) and ERK pathway which enhance the production of EGFR ligands and stimulates EGFR signalling, rendering therapy to EGFR-inhibitor ineffective [152]. Moreover, mutations in PI3K and MAPK pathways are common in metastatic CRC and accelerate tumour growth in conjunction with other prevalent mutations in the p53, TGF-β and Wnt signalling pathways [153] . Mutations in the MAPK pathway are present in these CRC patients (0.8% in MAP2K1, 1.7% in MAP2K4, 3.9% in NRAS, 8.5% in BRAF and 44% in KRAS). The PI3K/Akt/mTOR pathway is mutated in CRC patients (1% in AKT1, 2.4% in PIK3R1, 2.5% in PIK3CG, 2.8% in PTEN and 18% in PIK3CA) [5]. Additionally, 11% of the remaining patients exhibit mutations in RTKs which are upstream of both pathways triggering the emergence of resistance mechanisms to chemotherapy or targeted therapies [15,154].
Breast, colorectal, liver and other cancers are all mostly driven by the MYC oncogene. More than 70% of human malignancies exhibit high and/or abnormal Myc expression, which is associated with aggressive diseases and a bad prognosis [155,156]. Myc is a difficult oncoprotein to target due to its high frequency of overexpression in malignancies and its pervasive function in transcriptional control. There are presently no specific medications that can be used to target Myc, primarily due to its "undruggable" characteristics: Myc is primarily localised in the nucleus, making it inaccessible to antibodies and lacking an enzyme site where typical small molecules can bind [157]. BRD4 is a crucial epigenetic regulator (a chromatin regulator) and a member of the BET family. The human genome contains regulatory components including silencers (repressors), enhancers/super-enhancers and promoters that are used to dynamically modulate the regulation of transcription. In BET inhibitor-sensitive leukaemia cells, the classic enhancer or super-enhancer controls MYC expression through BRD4 binding. The expression of MYC is inhibited and cell proliferation is suppressed as a result of BET inhibitor’s blocking of BRD4's ability to bind to its genomic targets. However, by various mechanisms, long-term drug therapy may restore MYC expression. One of those mechanism is maintaining MYC expression by activating Wnt/β-catenin signalling pathways which result in enhanced β-catenin binding to the sites that were initially occupied by BRD4 leading to drug resistance [158].
The assessment of tumour heterogeneity is a crucial clinical concern. Genomic sequencing is being used to assess heterogeneity in cancer samples that were either archived at the time of diagnosis or later biopsied upon recurrence. This method has significant limitations because it is unlikely to adequately capture tumour heterogeneity, which has clear consequences for cancer therapy despite its utility in some circumstances for therapy selection [159]. Targeting an 'actionable' driver mutation, for instance, might only be successful if the mutation is truncal (i.e., clonal and present in the majority of subclones and parts of the tumour during the course of its lifetime) [160]. In other situations, the presence of a particular mutation may not indicate that it is clonal, and vice versa, the scarcity of a mutation does not indicate that it is accidental. In fact, resistance to targeted medications can be brought on by subclonal driver mutations in the PI3K pathway genes and ESR1. A list of the 'clonality' of driver mutations might be helpful in this case [161,162].
3.10. Tumour Microenvironment
Cancer cells, stromal cells, ECM, blood and lymphatic vessels, immune cells, nerve fibres, signalling molecules and related acellular components make up the TME. This latter is sculpted and instructed by cancer cells to support the emergence of cancer hallmarks, react to stimulation, internal or external stress and therapy, and eventually support the survival, growth, angiogenesis, migration, invasion, and immune evasion as well as drug resistance of these cells [10].
TME consists of myeloid-derived suppressor cells (MDSCs), mast cells, CAFs, TAMs, vascular endothelial cells, adipocytes, pericytes, tumour-associated neutrophils, dendritic cells, and granulocytes. It also includes malignant cells, NK cells, T and B cells. Cancer is protected from immunological eradication by the suppressive immune microenvironment [4,163]. Regulatory T (Treg) cells, neutrophils, macrophages, MDSCs, CD4+, FOXP3+, and CD25+ assist in establishing an immunosuppressive pre-metastatic microenvironment [164,165]. The activation of MDSCs, TAMs, and CAFs by reactive oxygen species (ROS) was demonstrated to be crucial in strengthening their immunosuppressive functions [166,167]. Immune cell recruitment into the TME can be affected by the ECM. For example, the ECM can activate the pro-survival pathway PI3K/Akt, which makes it easier for CSCs to evade the immune system [168]. The recruitment of immunosuppressive cells like Tregs and TAMs by ECM proteins has also been demonstrated to support CSC survival while inhibiting the recruitment of cytotoxic T cells, which are anti-tumourigenic immune cells [169,170,171]. Moreover, lipid metabolism has been associated with tumour progression, recurrence, and exhaustion of CD8 T cell through activation of programmed-cell death protein-1 (PD-1), which results in escaping the immune surveillance after treatment [172,173].
Key aspects that define cancer stemness, the recruitment of non-malignant cells that support tumour cells and ECM remodelling are coordinated by cellular crosstalk via several signalling network such as juxtracrine and paracrine pathways [174]. The suppression or modification of interferon-gamma (IFN-γ) signalling, activation of the MAPK and Wnt/β-catenin pathways, a decreased T-cell response and tumour antigen production are a few often found pathways that inhibit the immunotherapy response leading to treatment resistance [175].
Avoiding detection and eradication by the immune system results in multidrug resistance [176]. PD-1 are frequently expressed on the membranes of immune cells such as macrophages, T and B cells. While various tumour cells express programmed death ligand 1 (PD-L1). It has been demonstrated that the interaction of PD-1 and PD-L1 on T cell surfaces can inhibit the activity of killer T cells by promoting apoptosis, which causes tumour cells to escape the immune system [177]. Through the IL-6/STAT3/PD-L1 axis, CAFs modulated neutrophil activation, survival, and function in tumour tissues in HCC to promote immune suppression [178].
MSCs can produce a wide range of cells that engage in paracrine signalling, including IL-6 and IL-8, advancing the development of cancer and enhancing chemoresistance [179]. When exposed to cisplatin, instead of going through apoptosis, a subpopulation of cisplatin-resistant MSCs activate a phenotype linked to senescence [179]. As a result, various proteins (such as PLC-y1, RSK1/2/3, WNK1, c-Jun and p53) get phosphorylated, activating signalling pathways resulting in secretion of IL-6 and IL-8 into the TME. When breast cancer cells and MSCs were co-cultured simultaneously, the therapeutic impact was diminished in vivo due to the upregulation of resistance-related genes (such as MUC1, MYC and BRCA1) in the breast cancer cells after cisplatin pre-treatment [179].
MSCs can differentiate into CAFs. Recently, it was revealed that CAF triggered TKI resistance in HCC via the activation of PI3K/Akt/mTOR and RAF/ERK/STAT3 pathways [180]. Moreover, it was determined that the major signalling pathway activated by CAF is STAT3 driving everolimus resistance in neuroendocrine tumours cells [181].
In oesophageal squamous cell carcinoma, PAI-1 secreted by CAF activate the MAPK and Akt pathways in a paracrine manner resulting in production of ROS, induction of DNA damage and cell death leading to chemoresistance [182]. Additionally, drug resistance was promoted in tumour cells via NF-κB pathway induction by CAF-derived paracrine signals, such as exosomes, metabolites and chemoattractant cytokines [183,184]. CAFs can also enhance stemness through NF-κB signalling activation in gastric cancer [185]. Moreover, CAF enhanced the stemness of HCC by activating the Notch1 signalling pathway [186]. Furthermore, recently it was discovered INF-γ/STAT1/Notch3 as a molecular connection between CSCs and CAF using a bioinformatics strategy in TNBC cell lines resistant to doxorubicin [187].
The cellular composition and functional state of the TME will differ depending on the organ in which the tumour is located, as well as on cancer type and stage which will affect the delivery of treatment leading to a heterogeneous exposure to anticancer drugs [13,188,189]. TME can be divided into six different types of specialised microenvironments: the hypoxic, immunological, innervated, metabolic, mechanical and acidity niches. All these niches interact together and facilitate the progression and drug resistance of cancer [190].
Depending on their location within the cancer tissue, the cells in the tumour mass grow in a 3D tissue structure and are exposed to oxygen unevenly. As opposed to the tumour core, which is poorly vascularized, blood vessels in tumour tissues are typically randomly arranged and only cover the outer portion of the tumour mass [189]. A hypoxic microenvironment is created within the tumour core as a result of increased tumour cell proliferation, which places the cells there further away from the supporting blood vessels than the cells outside the tumour. This can result in varied treatment responses. By increasing the expression of genes linked to cell survival, angiogenesis, and anti-apoptotic pathways, tumour cells respond to hypoxic circumstances and the modified TME leading to the progression of cancer and the development of treatment resistance [191,192,193].
Interestingly, cancer cells may proliferate and colonise in anatomical areas which are sanctuary sites where medications administered systemically are unable to reach the therapeutic window. The brain's blood-brain barrier (BBB) and the central nervous system (CNS) are the two most typical examples [194]. Additionally, the peritoneum is another sanctuary site in severe paediatric leukaemia that may be treated with intra-peritoneal chemotherapy and tests that result in the management of preventative emission. Among these sanctuaries, the CNS is conceivably the most resentful therapeutic necessity. The extent of CNS tropism is higher in some types of diseases including melanoma, lung, breast, and kidney cancers. Those sanctuaries are physical barriers that lead to devastating clinical outcome [195].
The TME causes chemotherapeutic resistance via intrinsic or acquired mechanisms. Cancer dormancy, stemness and progression as well as intercellular communication, redox adaptability and drug resistance are reprogrammed by hypoxia [196]. Hypoxia affects the TME and treatment efficacy by encouraging cancer cells greater production of hypoxia-inducing factors (HIFs), most frequently HIF-1α. This latter stimulates the transcription of numerous genes, including vascular endothelial growth factor (VEGF) which enhance angiogenesis and as a result, cancer cells are better able to sustain their oxygen supply and metabolism, improving their chances of surviving [197,198]. Increased somatic mutational burden of oncogenes and tumour suppressors, such as TP53, MYC and PTEN is also linked to the hypoxic niche [199]. Cancer cells with p53 mutations or suppressed p53 transcription have the ability to avoid p53-mediated apoptosis pathways under hypoxic conditions, leading to the selection of cancer cell clones and the production of apoptosis-resistant cells [200]. Under hypoxic conditions, it has been demonstrated that p53 transcriptional activity is inhibited and the expression of efflux pumps, ABCB1 and ABCB5 is increased once HIF-1α binds to p53 in ovarian cancer cells, promoting their resistance to commonly used chemotherapeutics [201].
One of the characteristics of cancer is metabolic reprogramming, which is a modification in metabolism or nutrition supply. Increased metabolism of glutamine, glucose, amino acids, lipids, addiction to ROS and accumulation of lactate are common characteristics of cancer [202,203,204]. The synthesis of brain-derived neurotrophic factor by CAFs was driven by lactate in cancer cells in an NF-κB -dependent way, which in turn activated TrkB/Nrf2 signalling in cancer cells to lessen their susceptibility to anlotinib [205]. These results support the connection between drug resistance, metabolism, and NF-κB signalling.
Cancer is characterized by dysregulated pH, which is one of the TME variables. Extracellular pH (pHe 7.3–7.5) is often higher than intracellular pH (pHi 6.8–7.2) in healthy tissues and cells, while cancer cells generate a "reversed pH gradient" with increased internal pH and decreased external pH [206,207,208]. This reversed pH gradient makes it difficult for cancer cells to undergo apoptosis and prevents them from dying off [209,210]. Cancer cells' acidic extracellular environment (pH 6.5–7.1) plays a role in their chemotherapy resistance [211]. Recent research showed that an acidic tumour environment promotes cellular stemness and increases radio- and chemoresistance in oral cancer cells by causing increased cancer cell migration [212]. Acidic environments are extremely stressful for cells triggering many signalling pathways and likely activating powerful survival signalling pathways, such as those linked to cell stemness and undifferentiation leading to an increase in treatment resistance. Melanoma, neuroblastoma and breast cancer cells become more invasive and undergo an increase in oxidative phosphorylation and EMT in an acidic niche [213,214,215]. The development of acidic niches is also influenced by the activation of oncogenes like Ras and Myc and the inactivation of tumour suppressors like p53. Acidic pHe produces resistance to daunorubicin by inducing the activation of P-gp and the subsequent activation of p38 MAPK [216,217]. Inhibition of apoptosis in colon cancer cells is also associated with tumour acidity and p53 function loss [218]. Moreover, the absorption and resistance to cisplatin in melanoma cells are influenced by an acidified TME [219].
Neurology and cancer science are closely related, with neurotransmitters and neuropeptides generated from the nerve creating an "innervated niche" [220,221]. The neuroligin-3 (NLGN3)-stimulated PI3K/mTOR pathway, which is activated by active neurons, aids in the formation of high-grade gliomas [222]. Paracrine stimulations of cGAMP to astrocytes, cytokines production, activation of STING pathway and NF-κB and STAT1 signalling are triggered in brain metastatic cells via gap junctions between astrocytes and lung/breast cancer, which promotes cancer growth and resistance to chemotherapy [223,224].
The creation of a mechanical niche depends on stromal cells, extracellular and intracellular components, and intercellular signalling [225]. There are various structural proteins in the ECM such as collagen, laminins, fibronectin, elastin, proteoglycans and glycoproteins. The ECM is a 3D network of macromolecules that provides the biochemical and biophysical characteristics of the non-cellular bulk surrounding the cells. Additionally, non-malignant tumour-associated stroma cells are a crucial component of the TME, altering tumour characteristics, illness prognosis, and therapeutic response. Cell surface proteoglycans, cell adhesion molecules such as integrins, and hyaluronic acid receptors such as CD44, mediate biochemical and biophysical signalling as well as cell anchoring to the ECM [189,226]. For instance, in breast cancer increased laminin-mediated signalling and overexpression have been connected to diminished treatment responsiveness and improved tumour cell invasion and metastasis [227]. Fibronectin-integrin β1 interactions activate the PI3K/Akt and MAPK/ERK 1/2 pathways leading to chemotherapy resistance [228]. The integrin β1 downstream kinases FAK and Src are activated in HER2+ breast cancer cells that are resistant to lapatinib (a HER2-targeted therapy), resulting in these overcoming HER2 inhibition [229].
Matrix cells in the TME communicate with cancer cells through exosomes. Exosomes are small, bilayered molecules involved in autocrine, paracrine and endocrine signalling that are released by stromal and cancer cells in the TME. Altering vital survival signal transduction pathways, inducing EMT, activating anti-apoptotic pathways, and modifying the immune system are just a few of the ways that exosomes can make tumour cells resistant to treatment [230]. Exosome-mediated transfer of different ncRNAs, such as lncRNAs and miRNAs, may be a way for cancer cells to develop treatment resistance by causing genetic and epigenetic changes [230,231]. Recently it was shown that miR-1228-3p carried by CAF-derived extracellular vesicles increases HCC's chemoresistance by activating the PI3K/Akt pathway [232]. It was also revealed that Wnt/β-catenin and BMP signalling diminish the susceptibility of hepatoma cells to sorafenib and promote EMT in CAFs-derived Gremlin-1-rich exosomes [233]. Moreover, it was demonstrated that CAF-derived exosomes harbouring miR-20a can encourage chemoresistance and aggressive growth in NSCLC cells via the PTEN/PI3K/Akt signalling pathway [234]. Exosomal miR-21 and IL-6 produced from CAFs together increased MDSC formation in oesophageal squamous cell carcinoma by activating STAT3, which made tumour cells resistant to cisplatin [235]. Furthermore, SOX2 and PD-L1 expression was mediated by PI3K/Akt signalling pathway activation and showed to be a mechanism by which exosomes from CRC/MDR cells may increase cetuximab resistance in KRAS wild type cells [236].
3.11. Epithelial-Mesenchymal Transition
The phenotypic change from epithelial to mesenchymal cells, or epithelial-mesenchymal transition (EMT), occurs when epithelial cells lose their cell identity and take on mesenchymal traits altering the cell's shape and expression of surface markers in the process [237]. Epithelial cells, in the EMT process, experience depolarization, lose their cell-cell contact, adherent property, and develop elongated fibroblast-like morphology, is known to be triggered by ncRNAs, growth factors, cytokines, and hypoxia. These occurrences are accompanied by a concurrent increase in mesenchymal markers (integrin, laminin 5, N-cadherin, fibronectin, vimentin and type I collagen) and a concurrent decrease in epithelial markers (laminin 1, desmoplakin, E-cadherin and type IV collagen) expression. EMT is typically seen under healthy conditions, but tumour cells have the ability to carry out the same process while cancer is developing. Recent evidence suggests that pathological hyperactivated EMT is closely linked to elevated therapeutic resistance of cancer cells. Intracellular regulatory miRNA, exogenous inducers, epigenetic modulators, and cellular signalling pathways such as SMADs, PI3K, MAPK, ERK, TGF-β, Notch and Wnt/β-catenin are only a few of the molecular players involved in the regulation of EMT [238,239]. For instance, through the Wnt/β-catenin pathway, tongue squamous cell carcinoma cells gained cisplatin resistance and stem cell-like properties resulting in an enhanced EMT [240]. Moreover, in oral cancer, Notch signalling increases the population of CSCs, improves angiogenesis and EMT, and responds strongly to the DNA damage response induced by cisplatin [241]. TGF-β is the primary substance released by CAFs; it causes EMT and encourages the acquisition of gastric CSC features both of which eventually result in drug resistance [242]. Furthermore, miR-155 is overexpressed in oral squamous cell carcinoma, which results in resistance to cisplatin by inhibiting the expression of FOXO3a and promoting the EMT pathway [243].
Recently, it was demonstrated that cancer cells treated with chemotherapy release IL-1β triggering the release of integrin-αvβ1 and matrix metalloproteinase 9 causing the activation of TGF-β which in turn promote EMT in breast cancer cells [244]. Moreover, family with sequence similarity 46, member A (FAM46A) activated TGF-β pathways promoting chemoresistance in ovarian cancer cells [245]. TGF-β signalling promoted EMT and resistance to doxorubicin in breast cancer cells by upregulating lncRNA urothelial carcinoma-associated 1(lncRNA UCA1) [246]. HIF-1α/TGF-β2/GLI2 signalling is responsible for chemoresistance in CRC cells [247].
It was shown that hexokinase domain containing protein-1 (HKDC1) is essential for gastric cancer cell glycolysis, carcinogenesis, and EMT by activating the NF-κB pathway resulting in resistance to 5-FU, oxaliplatin and cisplatin in gastric cancer patients [248]. For instance, epithelial ovarian cancer (EOC) cells resistant to paclitaxel, cisplatin, erlotinib and carboplatin displayed high NF-κB activity [249]. The Notch signalling pathway is upregulated in breast cancer patients that are resistant to tamoxifen which can promote CSCs and EMT [250]. Furthermore, as a result of the activation of PI3K/Akt/mTOR signalling, the expression of EMT and CSC markers was considerably increased in cisplatin-resistant EOC cells [251].
4. Altered signalling pathways involved in drug resistance in cancer
Cellular signalling is an intracellular network of related crosstalking molecules ensuring cellular homeostasis. A potent stimulus activates molecular receptor promoting downstream signalling cascade that will determine the cellular function. As discussed above, genetic and epigenetic modifications of certain molecular components of these signalling pathways (e.g., RTK) can lead to their dysregulation which causes cancer progression and drug resistance.
In cancer, oncogenic pathways are abnormally active whilst tumour suppressor pathways are inhibited. The abnormal activation or inhibition of one or more signalling pathways can have a pivotal role in cancer drug resistance. Below we will discuss some of the key downstream pathways involved in drug resistance in response to altered upstream receptors.
4.1. Wnt/β-catenin pathway
It has been discovered that EMT resistance to chemotherapeutic drugs in cancer cells depend on the zinc-finger transcription factor pleomorphic adenoma gene like-2 (PLAGL2). Recently it was demonstrated that through the activation of the Wnt/β-catenin signalling pathway, PLAGL2 encourages adriamycin resistance and aggressiveness of cells in breast cancer [252]. A recent study on squamous transitioned lung cancer suggests that Wnt signalling may have a role in increasing adeno-to-squamous transdifferentiation (AST) [253]. For instance, upregulation of Wnt pathway was detected in transitioned lung cancer following osimertinib resistance. Various investigations have shown the significant role that AST has played in the development of resistance to molecular targeted therapy in lung cancer. Recently it was found that the ROS-Wnt axis as the AST tipping point and plays a crucial role in dynamically managing the homeostasis between the adeno- and squamous-specific transcription factors networks [254]. Wnt signalling is the primary regulator of the CSC gene expression program. Wnt3a has been reported to be able to activate p38 MAPK. This latter has been demonstrated to interact with Wnt/β-catenin pathway and functions as a β-catenin chromatin-related kinase, which is essential for controlling the signalling system involved in tumour growth, metastasis, and chemoresistance in CRCs [255]. Moreover, recent research suggests that SNORD1C (small nucleolar RNAs) promotes cancer cell stemness and drug resistance in CRC via the Wnt/β-catenin pathway and may serve as a biomarker that predicts the prognosis and aggressiveness of this cancer [256]. It was demonstrated how IL-6/STAT3 signalling is activated by Hsp90 (heat shock protein) inhibitor therapy through the actions of ERK and Akt which subsequently activate the Wnt signalling pathway allowing NSCLC cells to develop CSC characteristics and resistance to Hsp90 inhibitor [257].
4.2. The JAK/STAT signalling pathway
It was discovered that miR-106a-3p is an oncomiR in gastric cancer that trigger apatinib resistance due to the overexpression of JAK2/STAT3 proteins and their signalling [258]. Moreover, through the PTEN/Akt/SMAD2 and RAS/MEK/FOS MAPK/Akt pathways, the apurinic/apyrimidinic endodeoxyribonuclease 1 (APEX1)/miR-27a-5p axis contributed to the resistance to doxorubicin in gastric cancer cells [259]. Recently it was discovered that chronic myeloid leukaemia (CML) cells with a high amount of intracellular angiopoietin-1 (iANG-1) were resistant to dasatinib, nilotinib, imatinib and other TKIs. Furthermore, a unique drug-resistant mechanism in CML is revealed by the substantial upregulation of the p-SRC/p-STAT5 axis by iANG-1 [260].
4.3. PI3K/Akt/mTOR pathway
Proprotein convertase subtilisin/kexin type 9 (PCSK9), a crucial enzyme for antitumour immune responses, also activated Akt by suppressing PTEN, which caused HCC to develop sorafenib resistance [261]. Furthermore, nuclear paraspeckle assembly transcript 1 (NEAT1) which is a lncRNA activated the c-MET/Akt pathway via miR-335 in HCC cells, resulting in sorafenib resistance [262]. Recently it was elucidated that in male HCC and hepatoma cell lines, the NEAT1v1-superoxide dismutase 2 (SOD2) axis confers lenvatinib and sorafenib resistance and shifts the growth mode from MEK/ERK-dependent to Akt-dependent mode [263]. Lenvatinib's lethal effects were amplified by the MEK inhibitor selumetinib, which implies that thyroid cancer cells convert from Akt-dependent to MEK/ERK-dependent cell growth modes to develop resistance to lenvatinib [264]. Acid sensing ion channel 1a (ASIC1a) is an H+-gated cation channel that promotes tumour cell invasion and migration. ASIC1a is highly expressed in resistant HCC cells. ASIC1a-induced calcium influx activates the PI3K/Akt pathway leading to drug resistance in resistant HCC cells [265].
4.4. MAPK pathway
Recently, it was demonstrated that mitochondrial fusion dramatically decreases the susceptibility of breast cancer cells to tamoxifen under metabolic stress and likely contributes to the development of acquired drug resistance through AMPK and MAPK signalling [266]. Through p44/42 MAPK-Drp1 (a dynamin-related GTPase) signalling, membrane-bounded G-protein coupled oestrogen receptor (GPER) causes mitochondria fission, which is essential for GPER-induced cell apoptosis in breast cancer cells [267]. According to pertinent studies, one of the key mechanisms by which CRC cells develop resistance to cetuximab is activation of the RAS/RAF/MEK/MAPK pathway [268]. Fucosyltransferase VI (FUT6) modulates the EGFR/ERK/STAT signalling pathway to control head and neck squamous cell carcinoma invasion, migration, proliferation and EGF-induced EMT [269]. Moreover, it was recently identified that resistance to gefitinib and osimertinib in NSCLC cells is driven through the cholesterol/EGFR/Src/ERK/SP1 axis [270].
5. Strategies to overcome drug resistance in cancer
It will be very challenging to choose the optimal approach to combat drug resistance due to the high complexity and heterogeneity of tumours. Here we state strategies used to manage drug resistance and present how the deployment of cutting-edge diagnostic and therapeutic technologies are used to prevent its emergence (Figure 3).
Liquid biopsy has received a lot of interest in oncology diagnosis over the past several years. It is a biological sample approach that offers details on the real time dynamics of tumour biomarkers in a quick, cheap, easy to access, minimally invasive, and patient-friendly way. Several soluble components associated to tumour genetics, including exosomes, circulating tumour cells, cell-free DNA (cfDNA) and circulating tumour DNA (ctDNA). In this latter, genetic modifications associated with cancer can be detected such as amplification, point mutations, aneuploidy, rearrangements and patterns of fusion and methylation. By using platforms based on the polymerase chain reaction (PCR) and next-generation sequencing (NGS), liquid biopsy can be analysed to depict the current complexity of the patient's overall tumour mass [271,272]. ctDNA amount may serve as a prognostic indicator as its analysis may reveal the factors affecting prognosis. Following surgical resection, ctDNA is sensitive enough to detect minimal residual disease (MRD). Following surgery, ctDNA analysis offers a good prognostic evaluation and can help identify patients who have a very high risk of recurrence potentially avoiding unnecessary chemotherapy. Moreover, according to genetic changes, tailored treatments can be created using ctDNA. Patients whose BRAF V600E was missed in tissue analysis due to spatial heterogeneity can have their BRAF V600E found in their plasma using ctDNA. Therefore, offering the opportunity of BRAF inhibitors administration in combination with anti-EGFR monoclonal antibodies for example in CRC patients with a BRAF V600E mutation [271]. This offers evidence that ctDNA screening is useful and equivalent to tissue-based biomarker screening for choosing treatments. Therefore, it may be possible to use ctDNA as a surveillance tool to spot clones that are developing resistance to current treatments and provide a chance to convert therapy early on before the disease progresses.
Moreover, immunotherapy, which includes cancer vaccines, monoclonal antibodies, and inhibitors of immune checkpoints like anti-PD-1/PD-L1, anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA4) [273,274], is promising in that it may revolutionise the treatment of cancer by inducing, enhancing, or suppressing immune responses against cancer cells. Recently anti-CD47 agents have been gaining attention. Numerous tumour cell surface membranes have high levels of CD47 expression, which controls macrophage phagocytosis by binding SIRPα to prevent the eradication of host cells. Inhibiting the interaction between cancer cells and macrophages and inducing phagocytosis may be achieved by CD47 blocking drugs, such as monoclonal antibodies that target CD47/SIRPα [275]. Thus, combining immunotherapy and chemotherapy can be an effective approach to overcome drug resistance. For example, recent clinical trial results for unresectable HCC have shown that combination therapies, such as tremelimumab (anti-CTLA4 Ab) (HIMALAYA) + durvalumab (anti-PD-L1 Ab) + bevacizumab (anti-VEGF Ab) (IMbrave 150) + atezolizumab (anti-PD-L1 Ab) outperform monotherapy in terms of clinical outcomes [276,277]. Interestingly, it was recently shown that DTPs cells and EGFR TKI-resistant cells are effectively eliminated by CD70-targeting chimeric antigen receptor (CAR) T and NK cells and anti-CD70 antibody drug conjugates. These findings point to CD70, a cell surface protein, as a promising therapeutic target for EGFR mutant tumours that have developed acquired resistance to EGFR TKI [278]. Moreover, Akt inhibition specifically can result in a favourable immunological profile in the TME of breast, including an enhanced density of CD3+CD8+ cells and improved IFN genes expression, offering a justification for utilising Akt inhibition and immunotherapy in combination [279]. In PTEN-deficient xenografts, AZD8186 (a PI3Kβ inhibitor) improved anticancer activity by combination with anti-PD-1 Ab [280].
Furthermore, in cancer treatment, the combination of cytotoxic medicines and autophagy inhibitors like chloroquine (CQ) and its derivative is gaining more attention. Recently, it was demonstrated that CQ promotes colon cancer cells to become more sensitive to 5-FU via inhibiting ataxia telangiectasia and Rad3-related (ATR) kinase-mediated HIF-1α translation and interfering with HIF-1α's hypoxic function [281]. Moreover, recent data implies that inhibition of autophagy with CQ could circumvent in TNBC a therapeutic resistance mechanism to PI3K/Akt inhibitors with paclitaxel making the assessment of such combinations in clinical trials justified [282]. Dong et al. in 2023 present a drug loading system that combines CQ and a CD47 antibody (aCD47) and with a bionic lipoprotein (BLP) carrier (BLP-CQ-aCD47) to improve drug delivery, cancer immunotherapy and potentially helping to overcome drug resistance [283].
Nanoparticle-based medications have effectively decreased side effects, eliminated drug resistance, and increased medicinal efficacy [284]. The selectivity of the target gives nano-based medications an edge over traditional therapy [285]. Numerous nanoparticles, including mesoporous silica, metal, and polymeric nanoparticles as well as micelles, liposomes, dendrimers and nanostructured lipid carriers have been created and investigated over time and have significantly reduced chemoresistance in cancer [286]. The newly created doxorubicin-melittin polymersome (Dox-Mel PL) drug delivery system was capable of controlling MDR cancer cells and offered the following benefits: (1) biocompatible polymersome (a poly lactic acid-hyaluronic acid (20k-10k) di-block copolymer) promote synergistic effect of the simultaneous administration of Dox (anticancer agent) and Mel (a major component of bee venom); (2) reduction of Dox and Mel side effects; and (3) downregulation of P-gp by Mel prevent drug resistance. Dox-Mel PL overcomes MDR through P-gp inhibition and PI3K/Akt/NF-κB pathway downregulation [287]. Moreover, by inhibiting the NF-κB expression and activation and downregulating PD-L1 level, Ab-G/S-NP (nanoparticles that are loaded with sorafenib and GSK1059615) controlled the activation of cellular signalling pathways in HCC resistant cells to overcome their drug resistance to sorafenib. For the purpose of creating a more potent treatment for sorafenib-resistant malignancies, these findings call for additional research on the combination of treating HCC resistant cells with GSK1059615 (a PI3K/mTOR inhibitor) in vivo [288]. Furthermore, an innovative multifunctional medication delivery system based on targeted gold nanoparticles has been created as a useful approach for highly focused and EGFR-TKI-resistant reversal therapy. The nanoparticle (cRGD-GIPG) ensures that the treatment gets delivered successfully to the EGFR-TKI-resistant NSCLC by inhibiting the activation of the TGF-β/PDLIM5/SMAD resistance pathway and triggering drug-resistant cells to die by mitochondrial apoptosis. Thus, cRGD-GIPG exhibits strong anticancer effects against EGFR-TKI-resistant NSCLC cells both in vitro and in vivo [289]. Extensive work has also been put into finding novel compounds (like allosteric modulators) [290], creating new biotechnology (like PROTAC) [291,292,293] and suggesting effective drug combinations that successfully combat drug resistance.
It is common practise to utilise high-throughput forward genetic screening methods to investigate the molecular processes behind particular cellular phenotypes, such as treatment resistance in malignancies. To undertake loss-of-function screening across a variety of signalling pathways and biological processes, CRISPR-associated nuclease Cas9 (CRISPR/Cas9) is a particularly successful method. Single or double knockouts or modification of genes responsible for drug resistance can now be produced using the genome-wide CRISPR/Cas9 gene editing method [294,295]. For instance, lung cancer cell proliferation and EMT were suppressed by PNO1 (RNA-binding protein)/CRISPR/Cas9 through inhibiting the Notch signalling pathway in lung adenocarcinoma. Using CRISPR/Cas9 technology may be a beneficial technique [296].
Moreover, the emergence of deep learning, the vast amount of digital data, and powerful computing resources can offer an effective pipeline for enhanced drug discovery, help us understand how drugs become resistant to them, and help us make the best decisions possible when treating patients with EGFR-mutated NSCLC [297,298]. Furthermore, Fröhlich et al. recently discussed the second-generation MAPK Adaptive Resistance Model (MARM2.0), which aims to explain how drug adapted BRAFV600E melanoma cells rewire EGFR/MAPK signalling. MARM2.0 is developed using rule-based modelling in PySB (python program) with thermodynamic balance and builds on an extensive body of theoretical, biochemical and structural work on EFGR/MAPK signalling and feedback regulation [299]. Also, a unique phosphoproteomic-based machine learning technology called VESPA (Virtual Enrichment-based Signalling Protein-activity Analysis) is used to analyse enzyme-substrate connections and measure the activity of signalling proteins. Scientists have used it to investigate the mechanisms of post-translational cell adaptation that cause CRC to be resistant to or insensitive to clinically useful targeted therapies [300]. Interestingly, 'DRESIS', a comprehensive database describing information on drug resistance, was recently created and is anticipated to have significant effects on clinical treatment optimisation and the discovery of novel drugs in the future [301].
miRNAs can also control drug sensitivity and modulate resistance by post transcriptional gene regulation. Therefore, they do not only serve as biomarkers, but also as drug targets for overcoming drug resistance. For instance, exosomal miR-107 modulated the HMGA2/mTOR/P-gp axis, drastically increasing the susceptibility of resistant gastric cancer cells to cisplatin, indicating that exosomal miR-107 may be a promising target in the therapy of gastric cancer [302]. Recently, the tumour suppressor miR-4486 was used to increase the chemo-sensitivity of cisplatin in gastric cancer. The JAK3/STAT3 pathway was the target of this activity [303]. Moreover, by inhibiting CD44-induced CSC-like features via EGFR-mediated MAPK and Akt signalling pathways, miR-302a has also been discovered to restore the response to cetuximab [304].
PTK overexpression, including HER2, EGFR, FGFR, PDGFR, VEGFR and IGFR, activates numerous cell signalling pathways, including STAT3, NF-κB, PI3K/Akt and ERK1/2. It also results in aberrant expression of proteins associated with apoptosis in cancer cells, which is a major contributor to the chemotherapy resistance in tumour cells. Target therapy directed at specific tyrosine kinases will therefore overcome this resistance. There are various instances where platelet-derived growth factor (PDGF) ligands and receptors are both expressed in malignant cells; nevertheless, PDGF expression and function typically involve the tumour stroma. The pursuit of PDGFR inhibitors represents a successful strategy given the significance of the TME and the critical part that PDGF signalling plays in creating and maintaining that milieu [305]. A reversible and ATP-competitive PDGFR inhibitor called CP-673451 inhibits both PDGFRα and PDGFRβ kinase and effectively suppresses the downstream phosphorylation of PI3K/Akt [306]. Although CP-673451 reduces PDGFR-β expression and tumour growth in Lewis lung carcinoma-carrying mice, it did not increase overall survival when compared to radiation and Endostar combined therapy [307]. In a previous study it was shown that in soft tissue cancers, PDGFRα loss and bypass of the Akt-signalling pathway are one cause of acquired resistance to PDGFR inhibitor via activation of alternative compensatory signalling pathways. This study also outlines that by blocking FGFR1 in vitro this resistance was eliminated. Therefore, as an alternative strategy, combination therapy that simultaneously targets several compensatory signalling pathways can be developed to overcome resistance [308] (Figure 4).
It is interesting to note that in some cases, resistance to target therapy may develop over time and may be brought on by several translocation break points, various mutations at the target site or abnormalities in the phosphorylation of protein substrates. Therefore, discovering novel combination therapies might reduce drug resistance development. For instance, inhibition of both PI3K/Akt/mTOR and MAPK pathways presents a viable approach to treat the majority of CRC patients and circumvent potential resistance mechanisms that result from single target treatment, as revealed for the MEK inhibitor pathway [309]. Moreover, MEK1/2 inhibitor (BAY 86-9766) therapy of multi-drug resistant human CRC cell lines showed a considerable effect. Cetuximab (EGFR inhibitor)-resistant CRC cells also experienced synergistic apoptotic and antiproliferative effects from combination therapy with BAY 86-9766 and cetuximab through inhibition of the Akt and MAPK pathways [268]. Furthermore, combining BEZ235 (a dual PI3K/mTOR inhibitor) and cisplatin treatment dramatically reduced the capacity of chemoresistant EOC cells to form colonies, increased ROS levels, and increased apoptosis when compared to cisplatin treatment alone. In addition, compared to cisplatin mono-treatment, the combination method efficiently inhibited the PI3K/Akt/mTOR signalling pathway, reversed EMT, and lowered the expression of CSC marker and re-sensitized chemoresistant EOC cells to cisplatin [251]. It was found that imatinib (a TKI) sensitivity is increased, blastic phase of chronic myeloid leukaemia (CML-BP) cells resistant to TKIs are eliminated, and leukaemia engraftment is decreased when the integrated stress response (ISR) signalling is inhibited by the small molecule ISRIB in combination with imatinib. It was demonstrated how this dual therapy precisely alters the profile of gene expression and inhibits oncogenic JAK/STAT5 and RAS/RAF/MAPK/ERK signalling. To combat TKI-resistant leukaemic cells that demonstrate RAS/RAF/MAPK and STAT5 signalling hyperactivation due to driver mutations such as PTPN11 (SHP2) that can be detected by NGS analysis, it was suggested the combination of ISRIB and imatinib as a potential treatment approach [310].
Inhibiting several signalling targets with a single drug is an alternate strategy for overcoming resistance. For instance, sorafenib is a multitarget kinase inhibitor that targets several RTKs in the cell membranes, such as fibroblast growth factor receptor 1 (FGFR1), PDGFR, VEGFR 1, 2, and 3, FMS-related tyrosine kinase 3 receptor (FLT3), stem cell factor receptor (KIT) and RET proto-oncogene (RET), as well as downstream intracellular serine/threonine kinases such as B-Raf, Raf-1, and kinases in the Ras/Raf/MEK/ERK signalling pathway. By inhibiting these kinases and the downstream signalling molecules in a variety of oncogenic pathways, tumour cell apoptosis, proliferation and angiogenesis are all markedly reduced [311,312,313,314]. However, within 5 years of surgery, 70% of HCC patients who received adjuvant sorafenib treatment following surgical resection or local ablation (or both) experienced tumour recurrence, and the majority of these recurrent HCCs were sorafenib-resistant [315]. There is growing evidence that acquired sorafenib resistance is significantly influenced by the IGF/FGF axis, an upstream Akt regulator, such as in HCC [316]. It was discovered that ceritinib, an IGFR inhibitor first used to treat NSCLC, could sensitise HCC cells to sorafenib both in vitro and in xenograft and HCC mice models by inhibiting the IGF-1R/Akt pathway [317]. Comparatively to when it is taken alone, ceritinib significantly inhibits the proliferation of HCC cells when combined with sorafenib. Additionally, it was discovered that the linsitinib (IGFR inhibitor) and the brigatinib (FGFR inhibitor) were successful in reducing the viability of sorafenib-resistant HCC cells via the Akt pathway [316]. Recently a study showed that niclosamide, through modulation of IGF-1R/p-IGF-1R/stemness and metabolic alterations, can boost sorafenib sensitivity in sorafenib-resistant HCC cells/organoids. For sorafenib-resistant HCC cells, combining sorafenib and niclosamide can result in a synergistic combination index that lowers IGF-1R/p-IGF-1R/OCT4 expression. Niclosamide significantly increased the capacity of sorafenib to decrease the mitochondrial membrane potential in vitro by substantially downregulating the sorafenib-induced gene expression of stemness (OCT4), drug resistance (ABCG2), and glycolysis (GLUT1, HK2, LDHA, and PEPCK) [318]. Growing data suggests that niclosamide, a antihelminthic drug, has the potential to be a novel treatment for diseases such as cancer other than helminthic disease since it is a multifunctional medication that may interfere with a variety of biological processes and signalling pathways [319]. The growth of tumours in several cancers, including drug-resistant HCC, prostate and oesophageal cancer has been shown to be inhibited by niclosamide [320,321,322]. Moreover, lenvatinib and MEK inhibitors were evaluated in vitro and in vivo for the treatment of anaplastic thyroid cancer. Based on decreased tumour proliferation and increased apoptosis, which are caused by the Akt and ERK signal pathways, they discovered that the combination revealed synergistic effects and strengthened the anticancer impact [264]. Furthermore, immense effort has gone into several ongoing clinical trials (Table 1) aiming to evaluate the clinical benefits of various novel combination therapy to overcome drug resistance.
6. Conclusions
Each tumour has a unique collection of traits that govern how it progresses. Despite the challenges, it is still possible to sketch out a strategy for combating the issue of cancer drug resistance by employing our knowledge of its biological constituents and moving to a more personalised approach to treatment. This informative and comprehensive review highlights the importance of understanding the tumour properties, its drivers and dependencies to effectively block one or more signaling pathways to stop cancer progression and prevent resistance to treatment. Real-time monitoring of cancer progression, checkpoint blockade immunotherapy, multimodality therapy and systematically identifying cancer addictions are progressive steps towards effectively eradicating cancer without giving it a chance to adapt and utilise compensatory mechanisms. Combination of drugs targeting multiple pathways along with the utilisation of computational strategies that foresee cancer growth and signalling adaptability will be a promising approach to reduce the likelihood of developing drug resistance in the future.
Author Contributions
Conceptualization, N.B.A. and A.-M.C.; writing—original draft preparation, N.B.A; writing— review and editing, A-M C.; visualization, A.-M.C. All authors have read and agreed to the published version of the manuscript.
Funding
This literature research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ABC | ATP-binding cassette |
| ABCC5 | ATP binding cassette subfamily C member 5 |
| aCD47 | CD47 antibody |
| AhR | Aryl hydrocarbon receptor |
| ALK | Anaplastic lymphoma kinase |
| APEX1 | Apurinic/apyrimidinic endodeoxyribonuclease 1 |
| ASIC1a | Acid sensing ion channel 1a |
| AST | Adeno-to-squamous transdifferentiation |
| ATR | Ataxia telangiectasia and Rad3-related |
| ATP | Adenosine triphosphate |
| BAK1 | BCL2 antagonist/killer1 |
| BBB | Blood-brain barrier |
| BCRP/ABCG2 | Breast cancer resistance protein |
| BLP | Bionic lipoprotein |
| BRCA1/2 | Breast cancer gene1/2 |
| CAFs | Cancer-associated fibroblasts |
| CAR | Chimeric antigen receptor |
| CDKs | Cyclin-dependent kinases |
| cfDNA | Cell-free DNA |
| CHD4 | Chromodomain helicase DNA-binding protein 4 |
| CHK1 | Checkpoint kinase 1 |
| CML | Chronic myeloid leukaemia |
| CML-BP | Blastic phase of chronic myeloid leukaemia |
| CNS | Central nervous system |
| CQ | Chloroquine |
| CRC | Colorectal cancer |
| CSCs | Cancer stem cells |
| ctDNA | Circulating tumour DNA |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CYP | Cytochrome P450 |
| CYP3A4 | CYP isoforms 3A4 |
| DEHP | Di(2-ethylhexyl) phthalate |
| DNMT1/UHRF1 | DNA methyltransferases1/ Ubiquitin-like containing PHD Ring Finger 1 |
| Dox | Doxorubicin |
| Dox-Mel PL | Doxorubicin-melittin polymersome |
| DTEPs | Drug-tolerant expanded persisters |
| DTPs | Drug-tolerant persisters |
| ECM | Extracellular matrix |
| EGFL7 | EGF like domain multiple 7 |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| Egr-1 | Early growth response-1 |
| EMT | Epithelial-mesenchymal transition |
| EOC | Epithelial ovarian cancer |
| ER+ | Oestrogen receptor positive |
| 5-FU | 5-fluorouracil |
| FAM46A | Family with sequence similarity 46, member A |
| FGFR4 | Fibroblast growth factor receptor 4 |
| FGFR1 | Fibroblast growth factor receptor 1 |
| FLT3 | FMS-related tyrosine kinase 3 receptor |
| FUT6 | Fucosyltransferase VI |
| GDF-15 | Growth differentiation factor-15 |
| GPER | G-protein coupled oestrogen receptor |
| GR/OR | Gefitinib/osimertinib-resistant |
| GST | Glutathione S-transferase |
| H3K27ac | Histone H3 lysine 27 trimethylation acetylation |
| H3K27me3 | Histone H3 lysine 27 trimethylation |
| HCC | Hepatocellular carcinoma |
| HGF | Hepatocyte growth factor |
| HER2/erbB2 | Human epidermal growth factor receptor 2 |
| HER2+ | Human epidermal growth factor receptor 2 positive |
| HKDC1 | Hexokinase domain containing protein-1 |
| iANG-1 | Intracellular angiopoietin-1 |
| IFN | Interferons |
| IFN-γ | Interferon-gamma |
| IGFR | Insulin-like growth factor receptor |
| IGF-1R | Insulin-like growth factor 1 receptor |
| ISR | Integrated stress response |
| JAK/STAT | Janus kinase/signal transducers and activators of transcription |
| KDM5A | Lysine demethylase 5A |
| lncRNA | Long non-coding RNAs |
| lncRNA UCA1 | LncRNA urothelial carcinoma-associated 1 |
| LSD1/KDM1A | Lysine demethylase 1A |
| m6A | N6-methyladenosine |
| MAPK/ERK | Mitogen-activated protein kinase/extracellular signal-regulated kinase |
| MAPK1 | Mitogen-activated protein kinase 1 |
| MARM2.0 | Second-generation MAPK Adaptive Resistance Model |
| MDR | Multiple drug resistant |
| MDSCs | Myeloid-derived suppressor cells |
| Mel | Melittin |
| miRNAs | MicroRNAs |
| MITF | Melanocyte inducing transcription factor |
| MRD | Minimal residual disease |
| MRP1/ABCC1 | Multidrug resistance-associated protein 1 |
| MSCs | Mesenchymal stromal cells |
| MUC17 | Mucin 17 |
| MVP | Major vault protein |
| ncRNAs | Non-coding RNAs |
| NEAT1 | Nuclear paraspeckle assembly transcript 1 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGFR | Nerve growth factor receptor |
| NGS | Next-generation sequencing |
| NK | Natural killer |
| NLGN3 | Neuroligin-3 |
| Nrf2 | Nuclear factor erythroid 2 –related factor 2 |
| NSCLC | Non-small-cell lung cancer |
| PCR | Polymerase chain reaction |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| PD-1 | Programmed-cell death protein-1 |
| PDGF | Platelet-derived growth factor |
| PDGFR | Platelet-derived growth factor receptor |
| PD-L1 | Programmed death ligand 1 |
| P-gp/MDR1/ABCB1 | P-glycoprotein |
| pHe | Extracellular pH |
| pHi | Intracellular pH |
| PI3K/Akt/mTOR | Phosphoinositol-3-kinase/protein kinase B/mammalian target of rapamycin |
| PTKs | Protein tyrosine kinases |
| REG4 | Regenerating gene 4 |
| ROS | Reactive oxygen species |
| ROS1 | Ros oncogene 1 |
| RTKs | Receptor tyrosine kinases |
| rTMB | Relative tumour mutation burden |
| SOD2 | Superoxide dismutase 2 |
| SOX2 | SRY-Box transcription factor 2 |
| TAMs | Tumour-associated macrophages |
| Tβ4 | Thymosin 4 |
| TFF3 | Trefoil factor 3 |
| TGF-β | Transforming growth factor-beta |
| TIS | Therapy-induced senescence |
| TKIs | Tyrosine kinase inhibitors |
| TME | Tumour microenvironment |
| TNBC | Triple negative breast cancer |
| Treg | Regulatory T |
| TYMS | Thymidylate synthase |
| UGT | Uridine diphosphoglucuronosyltransferase |
| Wnt | Wingless-related integration site |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| VESPA | Virtual Enrichment-based Signalling Protein-activity Analysis |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int J Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther 2020, 5, 166. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J Cell Sci 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Kaemmerer, E.; Loessner, D.; Avery, V.M. Addressing the tumour microenvironment in early drug discovery: a strategy to overcome drug resistance and identify novel targets for cancer therapy. Drug Discov Today 2021, 26, 663–676. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordao, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov 2021, 11, 933–959. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Assaraf, Y.G.; Brozovic, A.; Goncalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist Updat 2019, 46, 100645. [Google Scholar] [CrossRef]
- Rafaeva, M.; Erler, J.T. Framing cancer progression: influence of the organ- and tumour-specific matrisome. FEBS J 2020, 287, 1454–1477. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.A.; Sanna, V.; Ahmad, N.; Sechi, M.; Mukhtar, H. Resveratrol nanoformulation for cancer prevention and therapy. Ann N Y Acad Sci 2015, 1348, 20–31. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, Y.K. Cancer Stem Cells as a Potential Target to Overcome Multidrug Resistance. Front Oncol 2020, 10, 764. [Google Scholar] [CrossRef]
- Liu, Y.; Hock, J.M.; Van Beneden, R.J.; Li, X. Aberrant overexpression of FOXM1 transcription factor plays a critical role in lung carcinogenesis induced by low doses of arsenic. Mol Carcinog 2014, 53, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhu, Q.; Li, Z.; Peng, Y.; Yu, X.; Yuan, B.; Liu, Y.; Liu, Y.; Yin, L.; Peng, Y.; et al. The FOXM1-ABCC5 axis contributes to paclitaxel resistance in nasopharyngeal carcinoma cells. Cell Death Dis 2017, 8, e2659. [Google Scholar] [CrossRef]
- Modi, A.; Purohit, P.; Roy, D.; Vishnoi, J.R.; Pareek, P.; Elhence, P.; Singh, P.; Sharma, S.; Sharma, P.; Misra, S. FOXM1 mediates GDF-15 dependent stemness and intrinsic drug resistance in breast cancer. Mol Biol Rep 2022, 49, 2877–2888. [Google Scholar] [CrossRef]
- Bergamino, M.A.; Lopez-Knowles, E.; Morani, G.; Tovey, H.; Kilburn, L.; Schuster, E.F.; Alataki, A.; Hills, M.; Xiao, H.; Holcombe, C.; et al. HER2-enriched subtype and novel molecular subgroups drive aromatase inhibitor resistance and an increased risk of relapse in early ER+/HER2+ breast cancer. EBioMedicine 2022, 83, 104205. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Esteva, F.J.; Valero, V.; Booser, D.; Guerra, L.T.; Murray, J.L.; Pusztai, L.; Cristofanilli, M.; Arun, B.; Esmaeli, B.; Fritsche, H.A.; et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J Clin Oncol 2002, 20, 1800–1808. [Google Scholar] [CrossRef]
- Joshi, J.P.; Brown, N.E.; Griner, S.E.; Nahta, R. Growth differentiation factor 15 (GDF15)-mediated HER2 phosphorylation reduces trastuzumab sensitivity of HER2-overexpressing breast cancer cells. Biochem Pharmacol 2011, 82, 1090–1099. [Google Scholar] [CrossRef]
- Pernas, S.; Tolaney, S.M. HER2-positive breast cancer: new therapeutic frontiers and overcoming resistance. Ther Adv Med Oncol 2019, 11, 1758835919833519. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Duan, H.; Huang, H.; Tong, X.; Han, Y.; Ru, G.; Qu, L.; Shou, C.; Zhao, Z. Cisplatin resistance in gastric cancer cells is associated with HER2 upregulation-induced epithelial-mesenchymal transition. Sci Rep 2016, 6, 20502. [Google Scholar] [CrossRef]
- Witta, S.E.; Gemmill, R.M.; Hirsch, F.R.; Coldren, C.D.; Hedman, K.; Ravdel, L.; Helfrich, B.; Dziadziuszko, R.; Chan, D.C.; Sugita, M.; et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 2006, 66, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Sayan, A.E.; Griffiths, T.R.; Pal, R.; Browne, G.J.; Ruddick, A.; Yagci, T.; Edwards, R.; Mayer, N.J.; Qazi, H.; Goyal, S.; et al. SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc Natl Acad Sci U S A 2009, 106, 14884–14889. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef]
- Milman, N.; Ginini, L.; Gil, Z. Exosomes and their role in tumorigenesis and anticancer drug resistance. Drug Resist Updat 2019, 45, 1–12. [Google Scholar] [CrossRef]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat Med 2013, 19, 1401–1409. [Google Scholar] [CrossRef]
- Sun, C.; Bernards, R. Feedback and redundancy in receptor tyrosine kinase signaling: relevance to cancer therapies. Trends Biochem Sci 2014, 39, 465–474. [Google Scholar] [CrossRef]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Challagundla, K.B.; Wise, P.M.; Neviani, P.; Chava, H.; Murtadha, M.; Xu, T.; Kennedy, R.; Ivan, C.; Zhang, X.; Vannini, I.; et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst 2015, 107. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Chen, R.; Tian, X. Autocrine EGF and TGF-α promote primary and acquired resistance to ALK/c-Met kinase inhibitors in non-small-cell lung cancer. Pharmacol Res Perspect 2023, 11, e01047. [Google Scholar] [CrossRef]
- Fu, R.; Jiang, S.; Li, J.; Chen, H.; Zhang, X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Medical Oncology 2020, 37, 24. [Google Scholar] [CrossRef]
- Shi, Q.; Xuhong, J.; Luo, T.; Ge, J.; Liu, F.; Lan, Y.; Chen, Q.; Tang, P.; Fan, L.; Chen, L.; et al. PIK3CA mutations are associated with pathologic complete response rate to neoadjuvant pyrotinib and trastuzumab plus chemotherapy for HER2-positive breast cancer. Br J Cancer 2023, 128, 121–129. [Google Scholar] [CrossRef]
- Wu, Q.; Ma, J.; Wei, J.; Meng, W.; Wang, Y.; Shi, M. FOXD1-AS1 regulates FOXD1 translation and promotes gastric cancer progression and chemoresistance by activating the PI3K/AKT/mTOR pathway. Mol Oncol 2021, 15, 299–316. [Google Scholar] [CrossRef]
- Sun, R.; Meng, Y.; Xu, R.; Li, Y.; Xu, X.; Li, Z.; Zuo, D. Construction of crizotinib resistant models with CD74-ROS1 D2033N and CD74-ROS1 S1986F point mutations to explore resistance mechanism and treatment strategy. Cell Signal 2023, 101, 110497. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.J.; Zheng, B.; Wang, H.Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin 2017, 38, 614–622. [Google Scholar] [CrossRef]
- Vergara-Gomez, L.; Bizama, C.; Zhong, J.; Buchegger, K.; Suarez, F.; Rosa, L.; Ili, C.; Weber, H.; Obreque, J.; Espinoza, K.; et al. A Novel Gemcitabine-Resistant Gallbladder Cancer Model Provides Insights into Molecular Changes Occurring during Acquired Resistance. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Topham, J.T.; O'Callaghan, C.J.; Feilotter, H.; Kennecke, H.F.; Lee, Y.S.; Li, W.; Banks, K.C.; Quinn, K.; Renouf, D.J.; Jonker, D.J.; et al. Circulating Tumor DNA Identifies Diverse Landscape of Acquired Resistance to Anti-Epidermal Growth Factor Receptor Therapy in Metastatic Colorectal Cancer. J Clin Oncol 2023, 41, 485–496. [Google Scholar] [CrossRef]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med 2015, 21, 795–801. [Google Scholar] [CrossRef]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med 2019, 25, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, P.; Gao, C.; Chen, J.; Li, J.; Chen, Z.; Xu, M.; Shao, J.; Zhang, Y.; Xie, J. Down-regulation of LRP1B in colon cancer promoted the growth and migration of cancer cells. Exp Cell Res 2017, 357, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, P.; Zhao, M.; Cao, H.; Zhao, Y.; Ji, M.; Hou, P.; Chen, M. EGFL7 drives the evolution of resistance to EGFR inhibitors in lung cancer by activating NOTCH signaling. Cell Death Dis 2022, 13, 910. [Google Scholar] [CrossRef]
- Cheng, D.; Fan, J.; Qin, K.; Zhou, Y.; Yang, J.; Ma, Y.; Shi, M.; Jin, J. LncRNA SNHG7 Regulates Mesenchymal Stem Cell Through the Notch1/Jagged1/Hes-1 Signaling Pathway and Influences Folfirinox Resistance in Pancreatic Cancer. Front Oncol 2021, 11, 719855. [Google Scholar] [CrossRef]
- Hsieh, T.H.; Tsai, C.F.; Hsu, C.Y.; Kuo, P.L.; Lee, J.N.; Chai, C.Y.; Hou, M.F.; Chang, C.C.; Long, C.Y.; Ko, Y.C.; et al. Phthalates stimulate the epithelial to mesenchymal transition through an HDAC6-dependent mechanism in human breast epithelial stem cells. Toxicol Sci 2012, 128, 365–376. [Google Scholar] [CrossRef]
- Hsieh, T.H.; Hsu, C.Y.; Yang, P.J.; Chiu, C.C.; Liang, S.S.; Ou-Yang, F.; Kan, J.Y.; Hou, M.F.; Wang, T.N.; Tsai, E.M. DEHP mediates drug resistance by directly targeting AhR in human breast cancer. Biomed Pharmacother 2022, 145, 112400. [Google Scholar] [CrossRef]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W.L. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef]
- Gao, C.; Zhang, L.; Xu, Y.; Ma, X.; Chen, P.; Chen, Z.S.; Wei, L. I13 overrides resistance mediated by the T315I mutation in chronic myeloid leukemia by direct BCR-ABL inhibition. Front Pharmacol 2023, 14, 1183052. [Google Scholar] [CrossRef] [PubMed]
- El-Damasy, A.K.; Jin, H.; Park, J.W.; Kim, H.J.; Khojah, H.; Seo, S.H.; Lee, J.H.; Bang, E.K.; Keum, G. Overcoming the imatinib-resistant BCR-ABL mutants with new ureidobenzothiazole chemotypes endowed with potent and broad-spectrum anticancer activity. J Enzyme Inhib Med Chem 2023, 38, 2189097. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef]
- Soverini, S.; De Benedittis, C.; Papayannidis, C.; Paolini, S.; Venturi, C.; Iacobucci, I.; Luppi, M.; Bresciani, P.; Salvucci, M.; Russo, D.; et al. Drug resistance and BCR-ABL kinase domain mutations in Philadelphia chromosome-positive acute lymphoblastic leukemia from the imatinib to the second-generation tyrosine kinase inhibitor era: The main changes are in the type of mutations, but not in the frequency of mutation involvement. Cancer 2014, 120, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Pohnert, S.C.; Shelton, J.G.; Franklin, R.A.; Bertrand, F.E.; McCubrey, J.A. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004, 18, 189–218. [Google Scholar] [CrossRef] [PubMed]
- Warsch, W.; Grundschober, E.; Sexl, V. Adding a new facet to STAT5 in CML: Multitasking for leukemic cells. Cell Cycle 2013, 12, 1813–1814. [Google Scholar] [CrossRef]
- Chien, S.H.; Liu, H.M.; Chen, P.M.; Ko, P.S.; Lin, J.S.; Chen, Y.J.; Lee, L.H.; Hsiao, L.T.; Chiou, T.J.; Gau, J.P.; et al. The landscape of BCR-ABL mutations in patients with Philadelphia chromosome-positive leukaemias in the era of second-generation tyrosine kinase inhibitors. Hematol Oncol 2020, 38, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Hao, S.; Liu, X.; Li, Y.; Zheng, X. Feiyiliu Mixture sensitizes EGFR(Del19/T790M/C797S) mutant non-small cell lung cancer to osimertinib by attenuating the PRC1/Wnt/EGFR pathway. Front Pharmacol 2023, 14, 1093017. [Google Scholar] [CrossRef]
- Hatlen, M.A.; Schmidt-Kittler, O.; Sherwin, C.A.; Rozsahegyi, E.; Rubin, N.; Sheets, M.P.; Kim, J.L.; Miduturu, C.; Bifulco, N.; Brooijmans, N.; et al. Acquired On-Target Clinical Resistance Validates FGFR4 as a Driver of Hepatocellular Carcinoma. Cancer Discov 2019, 9, 1686–1695. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective. Signal Transduct Target Ther 2022, 7, 329. [Google Scholar] [CrossRef]
- Michael, M.; Doherty, M.M. Tumoral Drug Metabolism: Overview and Its Implications for Cancer Therapy. J Clin Oncol 2005, 23, 205–229. [Google Scholar] [CrossRef]
- Kawahara, B.; Mascharak, P.K. Inhibition of Cytochrome P450 by Carbon Monoxide: Relevance to Drug Resistance in Human Breast Cancer Therapy. Med Res Arch 2023, 11. [Google Scholar] [CrossRef]
- Hofman, J.; Vagiannis, D.; Chen, S.; Guo, L. Roles of CYP3A4, CYP3A5 and CYP2C8 drug-metabolizing enzymes in cellular cytostatic resistance. Chem Biol Interact 2021, 340, 109448. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, S.; Bian, Z.; Yao, S.; Liu, M.; You, X.; Li, M. SNHG15 promotes chemoresistance and glycolysis in colorectal cancer. Pathol Res Pract 2023, 246, 154480. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.W.; Xue, H.; Ha, M.W.; Yu, D.Y.; Xiao, L.J.; Zheng, H.C. The effects of REG4 expression on chemoresistance of ovarian cancer. J Obstet Gynaecol 2022, 42, 3149–3157. [Google Scholar] [CrossRef] [PubMed]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R., Jr.; Chen, Z.S. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat 2015, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep 2015, 7, 14. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Pastan, I.H. The Role of Multidrug Resistance Efflux Pumps in Cancer: Revisiting a JNCI Publication Exploring Expression of the MDR1 (P-glycoprotein) Gene. J Natl Cancer Inst 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Liu, Y.; Gao, Z.; Huang, W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharm Sin B 2021, 11, 55–70. [Google Scholar] [CrossRef]
- Wang, J.Q.; Wu, Z.X.; Yang, Y.; Teng, Q.X.; Li, Y.D.; Lei, Z.N.; Jani, K.A.; Kaushal, N.; Chen, Z.S. ATP-binding cassette (ABC) transporters in cancer: A review of recent updates. J Evid Based Med 2021, 14, 232–256. [Google Scholar] [CrossRef]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat 2016, 27, 14–29. [Google Scholar] [CrossRef]
- Xue, X.; Liang, X.J. Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology. Chin J Cancer 2012, 31, 100–109. [Google Scholar] [CrossRef]
- Lampada, A.; O'Prey, J.; Szabadkai, G.; Ryan, K.M.; Hochhauser, D.; Salomoni, P. mTORC1-independent autophagy regulates receptor tyrosine kinase phosphorylation in colorectal cancer cells via an mTORC2-mediated mechanism. Cell Death Differ 2017, 24, 1045–1062. [Google Scholar] [CrossRef]
- Tazzari, P.L.; Cappellini, A.; Ricci, F.; Evangelisti, C.; Papa, V.; Grafone, T.; Martinelli, G.; Conte, R.; Cocco, L.; McCubrey, J.A.; et al. Multidrug resistance-associated protein 1 expression is under the control of the phosphoinositide 3 kinase/Akt signal transduction network in human acute myelogenous leukemia blasts. Leukemia 2007, 21, 427–438. [Google Scholar] [CrossRef]
- Tomiyasu, H.; Watanabe, M.; Sugita, K.; Goto-Koshino, Y.; Fujino, Y.; Ohno, K.; Sugano, S.; Tsujimoto, H. Regulations of ABCB1 and ABCG2 expression through MAPK pathways in acute lymphoblastic leukemia cell lines. Anticancer Res 2013, 33, 5317–5323. [Google Scholar] [PubMed]
- Zhu, M.M.; Tong, J.L.; Xu, Q.; Nie, F.; Xu, X.T.; Xiao, S.D.; Ran, Z.H. Increased JNK1 signaling pathway is responsible for ABCG2-mediated multidrug resistance in human colon cancer. PLoS One 2012, 7, e41763. [Google Scholar] [CrossRef]
- Gu, J.; Huang, W.; Wang, X.; Zhang, J.; Tao, T.; Zheng, Y.; Liu, S.; Yang, J.; Chen, Z.-S.; Cai, C.-Y.; et al. Hsa-miR-3178/RhoB/PI3K/Akt, a novel signaling pathway regulates ABC transporters to reverse gemcitabine resistance in pancreatic cancer. Mol Cancer 2022, 21, 112. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, X.; Zhang, D.; Fan, Y.; Qin, L.; Dong, S.; Zhang, L. Upregulated miR-132 in Lgr5(+) gastric cancer stem cell-like cells contributes to cisplatin-resistance via SIRT1/CREB/ABCG2 signaling pathway. Mol Carcinog 2017, 56, 2022–2034. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Ajani, J.A.; Song, S. Drug resistance and Cancer stem cells. Cell Commun Signal 2021, 19, 19. [Google Scholar] [CrossRef]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; ME, L.L. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin Cancer Biol 2020, 60, 166–180. [Google Scholar] [CrossRef]
- Cojoc, M.; Mäbert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol 2015, 31, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jeon, H.Y.; Kim, H. The molecular mechanisms underlying the therapeutic resistance of cancer stem cells. Arch Pharm Res 2015, 38, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Moitra, K.; Lou, H.; Dean, M. Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin Pharmacol Ther 2011, 89, 491–502. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther 2020, 5, 8. [Google Scholar] [CrossRef]
- Clark-Corrigall, J.; Myssina, S.; Michaelis, M.; Cinatl, J., Jr.; Ahmed, S.; Carr-Wilkinson, J. Elevated Expression of LGR5 and WNT Signaling Factors in Neuroblastoma Cells With Acquired Drug Resistance. Cancer Invest 2023, 41, 173–182. [Google Scholar] [CrossRef]
- Abdin, S.M.; Tolba, M.F.; Zaher, D.M.; Omar, H.A. Nuclear factor-κB signaling inhibitors revert multidrug-resistance in breast cancer cells. Chem Biol Interact 2021, 340, 109450. [Google Scholar] [CrossRef]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat Rev Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- Salehan, M.R.; Morse, H.R. DNA damage repair and tolerance: a role in chemotherapeutic drug resistance. Br J Biomed Sci 2013, 70, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, P.M.; Svendsrud, D.H.; Kravik, K.L.; Stokke, T. Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol Cancer 2006, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- de Angelis, P.M.; Fjell, B.; Kravik, K.L.; Haug, T.; Tunheim, S.H.; Reichelt, W.; Beigi, M.; Clausen, O.P.; Galteland, E.; Stokke, T. Molecular characterizations of derivatives of HCT116 colorectal cancer cells that are resistant to the chemotherapeutic agent 5-fluorouracil. Int J Oncol 2004, 24, 1279–1288. [Google Scholar] [CrossRef]
- Lowery, C.D.; Dowless, M.; Renschler, M.; Blosser, W.; VanWye, A.B.; Stephens, J.R.; Iversen, P.W.; Lin, A.B.; Beckmann, R.P.; Krytska, K.; et al. Broad Spectrum Activity of the Checkpoint Kinase 1 Inhibitor Prexasertib as a Single Agent or Chemopotentiator Across a Range of Preclinical Pediatric Tumor Models. Clin Cancer Res 2019, 25, 2278–2289. [Google Scholar] [CrossRef]
- Booth, L.; Cruickshanks, N.; Ridder, T.; Dai, Y.; Grant, S.; Dent, P. PARP and CHK inhibitors interact to cause DNA damage and cell death in mammary carcinoma cells. Cancer Biol Ther 2013, 14, 458–465. [Google Scholar] [CrossRef]
- Lee, H.J.; Cao, Y.; Pham, V.; Blackwood, E.; Wilson, C.; Evangelista, M.; Klijn, C.; Stokoe, D.; Settleman, J. Ras-MEK Signaling Mediates a Critical Chk1-Dependent DNA Damage Response in Cancer Cells. Mol Cancer Ther 2017, 16, 694–704. [Google Scholar] [CrossRef]
- Gonzalez-Conchas, G.A.; Rodriguez-Romo, L.; Hernandez-Barajas, D.; Gonzalez-Guerrero, J.F.; Rodriguez-Fernandez, I.A.; Verdines-Perez, A.; Templeton, A.J.; Ocana, A.; Seruga, B.; Tannock, I.F.; et al. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: A systematic review and a meta-analysis. Cancer Treat Rev 2018, 62, 1–8. [Google Scholar] [CrossRef]
- Liu, D.; He, J.; Yuan, Z.; Wang, S.; Peng, R.; Shi, Y.; Teng, X.; Qin, T. EGFR expression correlates with decreased disease-free survival in triple-negative breast cancer: a retrospective analysis based on a tissue microarray. Med Oncol 2012, 29, 401–405. [Google Scholar] [CrossRef]
- Rimawi, M.F.; Shetty, P.B.; Weiss, H.L.; Schiff, R.; Osborne, C.K.; Chamness, G.C.; Elledge, R.M. Epidermal growth factor receptor expression in breast cancer association with biologic phenotype and clinical outcomes. Cancer 2010, 116, 1234–1242. [Google Scholar] [CrossRef]
- Lee, K.J.; Wright, G.; Bryant, H.; Wiggins, L.A.; Schuler, M.; Gassman, N.R. EGFR signaling promotes resistance to CHK1 inhibitor prexasertib in triple negative breast cancer. Cancer Drug Resist 2020, 3, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Wilting, R.H.; Dannenberg, J.H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist Updat 2012, 15, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Zeller, C.; Brown, R. Therapeutic modulation of epigenetic drivers of drug resistance in ovarian cancer. Ther Adv Med Oncol 2010, 2, 319–329. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, Z.; Li, J.; Liu, H.; Zhang, H.; Zhang, J.; Huang, W.; He, Y.; Zhu, S.; Huo, M.; et al. CHD4 promotes acquired chemoresistance and tumor progression by activating the MEK/ERK axis. Drug Resist Updat 2023, 66, 100913. [Google Scholar] [CrossRef]
- Fan, M.; Yan, P.S.; Hartman-Frey, C.; Chen, L.; Paik, H.; Oyer, S.L.; Salisbury, J.D.; Cheng, A.S.; Li, L.; Abbosh, P.H.; et al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res 2006, 66, 11954–11966. [Google Scholar] [CrossRef]
- Ohata, Y.; Shimada, S.; Akiyama, Y.; Mogushi, K.; Nakao, K.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; Ochiai, T.; et al. Acquired Resistance with Epigenetic Alterations Under Long-Term Antiangiogenic Therapy for Hepatocellular Carcinoma. Mol Cancer Ther 2017, 16, 1155–1165. [Google Scholar] [CrossRef]
- Huang, M.; Chen, C.; Geng, J.; Han, D.; Wang, T.; Xie, T.; Wang, L.; Wang, Y.; Wang, C.; Lei, Z.; et al. Targeting KDM1A attenuates Wnt/β-catenin signaling pathway to eliminate sorafenib-resistant stem-like cells in hepatocellular carcinoma. Cancer Lett 2017, 398, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wu, J.; Dombkowski, A.; Zhang, K.; Holowatyj, A.; Boerner, J.L.; Yang, Z.Q. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am J Transl Res 2012, 4, 247–256. [Google Scholar] [PubMed]
- Lu, Y.; Liu, Y.; Oeck, S.; Glazer, P.M. Hypoxia Promotes Resistance to EGFR Inhibition in NSCLC Cells via the Histone Demethylases, LSD1 and PLU-1. Mol Cancer Res 2018, 16, 1458–1469. [Google Scholar] [CrossRef] [PubMed]
- Tuncel, G.; Kalkan, R. Importance of m N 6-methyladenosine (m 6 A) RNA modification in cancer. Med Oncol 2019, 36, 1–6. [Google Scholar] [CrossRef]
- Lan, T.; Li, H.; Zhang, D.; Xu, L.; Liu, H.; Hao, X.; Yan, X.; Liao, H.; Chen, X.; Xie, K.; et al. KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol Cancer 2019, 18, 186. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov 2018, 4, 10. [Google Scholar] [CrossRef]
- Lin, X.; Ye, R.; Li, Z.; Zhang, B.; Huang, Y.; Du, J.; Wang, B.; Meng, H.; Xian, H.; Yang, X.; et al. KIAA1429 promotes tumorigenesis and gefitinib resistance in lung adenocarcinoma by activating the JNK/ MAPK pathway in an m(6)A-dependent manner. Drug Resist Updat 2023, 66, 100908. [Google Scholar] [CrossRef]
- Lin, S.; Ruan, H.; Qin, L.; Zhao, C.; Gu, M.; Wang, Z.; Liu, B.; Wang, H.; Wang, J. Acquired resistance to EGFR-TKIs in NSCLC mediates epigenetic downregulation of MUC17 by facilitating NF-κB activity via UHRF1/DNMT1 complex. Int J Biol Sci 2023, 19, 832–851. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e819. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e237. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu Rev Physiol 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.R.; Nelson, P.S. Cellular senescence and cancer chemotherapy resistance. Drug Resistance Updates 2012, 15, 123–131. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef]
- Demaria, M.; O'Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.Y.; Wu, D.Y. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res 2005, 65, 2795–2803. [Google Scholar] [CrossRef]
- Sabisz, M.; Skladanowski, A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: implications for drug resistance and in vitro drug screening models. Cell Cycle 2009, 8, 3208–3217. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv Cancer Res 2019, 141, 43–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, C.; Ling, S.; Wei, R.; Wang, J.; Xu, X. The metabolic flexibility of quiescent CSC: implications for chemotherapy resistance. Cell Death Dis 2021, 12, 835. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat Med 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Takeishi, S.; Nakayama, K.I. To wake up cancer stem cells, or to let them sleep, that is the question. Cancer Sci 2016, 107, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.Y.; Tang, J.N.; Xie, H.X.; Du, Y.A.; Huang, L.; Yu, P.F.; Cheng, X.D. 5-Fluorouracil chemotherapy of gastric cancer generates residual cells with properties of cancer stem cells. Int J Biol Sci 2015, 11, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med 2011, 208, 2641–2655. [Google Scholar] [CrossRef]
- Yang, A.; Qin, S.; Schulte, B.A.; Ethier, S.P.; Tew, K.D.; Wang, G.Y. MYC Inhibition Depletes Cancer Stem-like Cells in Triple-Negative Breast Cancer. Cancer Res 2017, 77, 6641–6650. [Google Scholar] [CrossRef]
- Civenni, G.; Malek, A.; Albino, D.; Garcia-Escudero, R.; Napoli, S.; Di Marco, S.; Pinton, S.; Sarti, M.; Carbone, G.M.; Catapano, C.V. RNAi-mediated silencing of Myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer Res 2013, 73, 6816–6827. [Google Scholar] [CrossRef]
- Wu, C.H.; van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A 2007, 104, 13028–13033. [Google Scholar] [CrossRef]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1117. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef]
- Henriques, V.; Martins, T.; Link, W.; Ferreira, B.I. The Emerging Therapeutic Landscape of Advanced Melanoma. Curr Pharm Des 2018, 24, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim Biophys Acta Rev Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O'Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Limberg, J.; Egan, C.E.; Gray, K.D.; Singh, M.; Loewenstein, Z.; Yang, Y.; Riascos, M.C.; Al Asadi, H.; Safe, P.; El Eshaky, S.; et al. Activation of the JAK/STAT Pathway Leads to BRAF Inhibitor Resistance in BRAFV600E Positive Thyroid Carcinoma. Mol Cancer Res 2023, 21, 397–410. [Google Scholar] [CrossRef]
- Corrales, E.; Levit-Zerdoun, E.; Metzger, P.; Mertes, R.; Lehmann, A.; Münch, J.; Lemke, S.; Kowar, S.; Boerries, M. PI3K/AKT signaling allows for MAPK/ERK pathway independency mediating dedifferentiation-driven treatment resistance in melanoma. Cell Commun Signal 2022, 20, 187. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.Y. Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb Perspect Med 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Khoury, K.; Dömling, A. P53 mdm2 inhibitors. Curr Pharm Des 2012, 18, 4668–4678. [Google Scholar] [CrossRef]
- Liu, D.P.; Song, H.; Xu, Y. A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 2010, 29, 949–956. [Google Scholar] [CrossRef]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef]
- Sauer, L.; Gitenay, D.; Vo, C.; Baron, V.T. Mutant p53 initiates a feedback loop that involves Egr-1/EGF receptor/ERK in prostate cancer cells. Oncogene 2010, 29, 2628–2637. [Google Scholar] [CrossRef]
- Zaidi, S.H.; Harrison, T.A.; Phipps, A.I.; Steinfelder, R.; Trinh, Q.M.; Qu, C.; Banbury, B.L.; Georgeson, P.; Grasso, C.S.; Giannakis, M.; et al. Landscape of somatic single nucleotide variants and indels in colorectal cancer and impact on survival. Nat Commun 2020, 11, 3644. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef]
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol 2006, 16, 318–330. [Google Scholar] [CrossRef]
- Ben-David, E.; Bester, A.C.; Shifman, S.; Kerem, B. Transcriptional dynamics in colorectal carcinogenesis: new insights into the role of c-Myc and miR17 in benign to cancer transformation. Cancer Res 2014, 74, 5532–5540. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative approaches to target Myc for cancer treatment. Signal Transduct Target Ther 2021, 6, 117. [Google Scholar] [CrossRef]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.N.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Gerlinger, M.; Futreal, P.A.; Pusztai, L.; Swanton, C. Intratumor heterogeneity: seeing the wood for the trees. Sci Transl Med 2012, 4, 127ps110. [Google Scholar] [CrossRef]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 2015, 7, 283ra254. [Google Scholar] [CrossRef]
- Schiavon, G.; Hrebien, S.; Garcia-Murillas, I.; Cutts, R.J.; Pearson, A.; Tarazona, N.; Fenwick, K.; Kozarewa, I.; Lopez-Knowles, E.; Ribas, R.; et al. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med 2015, 7, 313ra182. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Laughney, A.M.; Hu, J.; Campbell, N.R.; Bakhoum, S.F.; Setty, M.; Lavallée, V.P.; Xie, Y.; Masilionis, I.; Carr, A.J.; Kottapalli, S.; et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med 2020, 26, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e717. [Google Scholar] [CrossRef]
- Costa, N.L.; Valadares, M.C.; Souza, P.P.; Mendonça, E.F.; Oliveira, J.C.; Silva, T.A.; Batista, A.C. Tumor-associated macrophages and the profile of inflammatory cytokines in oral squamous cell carcinoma. Oral Oncol 2013, 49, 216–223. [Google Scholar] [CrossRef]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res 2013, 23, 898–914. [Google Scholar] [CrossRef] [PubMed]
- Dituri, F.; Mazzocca, A.; Giannelli, G.; Antonaci, S. PI3K functions in cancer progression, anticancer immunity and immune evasion by tumors. Clin Dev Immunol 2011, 2011, 947858. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Fröse, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol 2014, 16, 1105–1117. [Google Scholar] [CrossRef]
- Stahl, M.; Schupp, J.; Jäger, B.; Schmid, M.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PLoS One 2013, 8, e81382. [Google Scholar] [CrossRef]
- Bollyky, P.L.; Wu, R.P.; Falk, B.A.; Lord, J.D.; Long, S.A.; Preisinger, A.; Teng, B.; Holt, G.E.; Standifer, N.E.; Braun, K.R.; et al. ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc Natl Acad Sci U S A 2011, 108, 7938–7943. [Google Scholar] [CrossRef]
- Long, J.; Zhang, C.J.; Zhu, N.; Du, K.; Yin, Y.F.; Tan, X.; Liao, D.F.; Qin, L. Lipid metabolism and carcinogenesis, cancer development. Am J Cancer Res 2018, 8, 778–791. [Google Scholar] [PubMed]
- Ma, L.; Hernandez, M.O.; Zhao, Y.; Mehta, M.; Tran, B.; Kelly, M.; Rae, Z.; Hernandez, J.M.; Davis, J.L.; Martin, S.P.; et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 2019, 36, 418–430.e416. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat Med 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pascual, J.; Ayuso-Sacido, A.; Belda-Iniesta, C. Drug resistance in cancer immunotherapy: new strategies to improve checkpoint inhibitor therapies. Cancer Drug Resist 2019, 2, 980–993. [Google Scholar] [CrossRef]
- Mandal, R.; Şenbabaoğlu, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. [Google Scholar] [CrossRef]
- Liu, C.; Wang, M.; Zhang, H.; Li, C.; Zhang, T.; Liu, H.; Zhu, S.; Chen, J. Tumor microenvironment and immunotherapy of oral cancer. Eur J Med Res 2022, 27, 198. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis 2018, 9, 422. [Google Scholar] [CrossRef]
- Skolekova, S.; Matuskova, M.; Bohac, M.; Toro, L.; Durinikova, E.; Tyciakova, S.; Demkova, L.; Gursky, J.; Kucerova, L. Cisplatin-induced mesenchymal stromal cells-mediated mechanism contributing to decreased antitumor effect in breast cancer cells. Cell Commun Signal 2016, 14, 4. [Google Scholar] [CrossRef]
- Eun, J.W.; Yoon, J.H.; Ahn, H.R.; Kim, S.; Kim, Y.B.; Lim, S.B.; Park, W.; Kang, T.W.; Baek, G.O.; Yoon, M.G.; et al. Cancer-associated fibroblast-derived secreted phosphoprotein 1 contributes to resistance of hepatocellular carcinoma to sorafenib and lenvatinib. Cancer Commun (Lond) 2023, 43, 455–479. [Google Scholar] [CrossRef]
- Amin, T.; Viol, F.; Krause, J.; Fahl, M.; Eggers, C.; Awwad, F.; Schmidt, B.; Benten, D.; Ungefroren, H.; Fraune, C.; et al. Cancer-Associated Fibroblasts Induce Proliferation and Therapeutic Resistance to Everolimus in Neuroendocrine Tumors through STAT3 Activation. Neuroendocrinology 2023, 113, 501–518. [Google Scholar] [CrossRef]
- Che, Y.; Wang, J.; Li, Y.; Lu, Z.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Zang, R.; Sun, N.; et al. Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis 2018, 9, 759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.H. Tumor-Stroma IL1β-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res 2018, 78, 1700–1712. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e816. [Google Scholar] [CrossRef]
- Han, M.E.; Kim, H.J.; Shin, D.H.; Hwang, S.H.; Kang, C.D.; Oh, S.O. Overexpression of NRG1 promotes progression of gastric cancer by regulating the self-renewal of cancer stem cells. J Gastroenterol 2015, 50, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Zhao, Y.; Chen, W.; Peng, W.; Wang, Y.; Xiong, S.; Aruna; Li, Y. ; Yang, Y.; Chen, S.; et al. The stromal-tumor amplifying STC1-Notch1 feedforward signal promotes the stemness of hepatocellular carcinoma. J Transl Med 2023, 21, 236. [Google Scholar] [CrossRef]
- Lawal, B.; Wu, A.T.; Chen, C.H.; T, A.G.; Wu, S.Y. Identification of INFG/STAT1/NOTCH3 as γ-Mangostin's potential targets for overcoming doxorubicin resistance and reducing cancer-associated fibroblasts in triple-negative breast cancer. Biomed Pharmacother 2023, 163, 114800. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat 2012, 15, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Sermeus, A.; Cosse, J.P.; Crespin, M.; Mainfroid, V.; de Longueville, F.; Ninane, N.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia induces protection against etoposide-induced apoptosis: molecular profiling of changes in gene expression and transcription factor activity. Mol Cancer 2008, 7, 27. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Magnussen, A.L.; Mills, I.G. Vascular normalisation as the stepping stone into tumour microenvironment transformation. Br J Cancer 2021, 125, 324–336. [Google Scholar] [CrossRef]
- Sharma, M.; Bakshi, A.K.; Mittapelly, N.; Gautam, S.; Marwaha, D.; Rai, N.; Singh, N.; Tiwari, P.; Agarwal, N.; Kumar, A.; et al. Recent updates on innovative approaches to overcome drug resistance for better outcomes in cancer. J Control Release 2022, 346, 43–70. [Google Scholar] [CrossRef]
- Qiu, G.Z.; Jin, M.Z.; Dai, J.X.; Sun, W.; Feng, J.H.; Jin, W.L. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol Sci 2017, 38, 669–686. [Google Scholar] [CrossRef]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683.e665. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, S.; Gao, L.; Zhi, K.; Ren, W. The Molecular Basis and Therapeutic Aspects of Cisplatin Resistance in Oral Squamous Cell Carcinoma. Front Oncol 2021, 11, 761379. [Google Scholar] [CrossRef]
- Bhandari, V.; Li, C.H.; Bristow, R.G.; Boutros, P.C.; Aaltonen, L.A.; Abascal, F.; Abeshouse, A.; Aburatani, H.; Adams, D.J.; Agrawal, N.; et al. Divergent mutational processes distinguish hypoxic and normoxic tumours. Nat Commun 2020, 11, 737. [Google Scholar] [CrossRef]
- Deben, C.; Deschoolmeester, V.; De Waele, J.; Jacobs, J.; Van den Bossche, J.; Wouters, A.; Peeters, M.; Rolfo, C.; Smits, E.; Lardon, F.; et al. Hypoxia-Induced Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Mediated by HIF-1α and Mutant p53 and Can Be Overcome by Induction of Oxidative Stress. Cancers (Basel) 2018, 10. [Google Scholar] [CrossRef]
- Parmakhtiar, B.; Burger, R.A.; Kim, J.H.; Fruehauf, J.P. HIF Inactivation of p53 in Ovarian Cancer Can Be Reversed by Topotecan, Restoring Cisplatin and Paclitaxel Sensitivity. Mol Cancer Res 2019, 17, 1675–1686. [Google Scholar] [CrossRef]
- Mao, X.Y.; Jin, M.Z.; Chen, J.F.; Zhou, H.H.; Jin, W.L. Live or let die: Neuroprotective and anti-cancer effects of nutraceutical antioxidants. Pharmacol Ther 2018, 183, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Schulze, A. Lipid Metabolism at the Nexus of Diet and Tumor Microenvironment. Trends Cancer 2019, 5, 693–703. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front Oncol 2019, 9, 1143. [Google Scholar] [CrossRef]
- Jin, Z.; Lu, Y.; Wu, X.; Pan, T.; Yu, Z.; Hou, J.; Wu, A.; Li, J.; Yang, Z.; Li, C.; et al. The cross-talk between tumor cells and activated fibroblasts mediated by lactate/BDNF/TrkB signaling promotes acquired resistance to anlotinib in human gastric cancer. Redox Biol 2021, 46, 102076. [Google Scholar] [CrossRef]
- Sharma, M.; Astekar, M.; Soi, S.; Manjunatha, B.S.; Shetty, D.C.; Radhakrishnan, R. pH Gradient Reversal: An Emerging Hallmark of Cancers. Recent Pat Anticancer Drug Discov 2015, 10, 244–258. [Google Scholar] [CrossRef]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos Trans R Soc Lond B Biol Sci 2014, 369, 20130099. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 2010, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol Pharm 2011, 8, 2032–2038. [Google Scholar] [CrossRef]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist Updat 2015, 23, 69–78. [Google Scholar] [CrossRef]
- de Bem Prunes, B.; Nunes, J.S.; da Silva, V.P.; Laureano, N.K.; Gonçalves, D.R.; Machado, I.S.; Barbosa, S.; Lamers, M.L.; Rados, P.V.; Kurth, I.; et al. The role of tumor acidification in aggressiveness, cell dissemination and treatment resistance of oral squamous cell carcinoma. Life Sci 2022, 288, 120163. [Google Scholar] [CrossRef]
- Peppicelli, S.; Toti, A.; Giannoni, E.; Bianchini, F.; Margheri, F.; Del Rosso, M.; Calorini, L. Metformin is also effective on lactic acidosis-exposed melanoma cells switched to oxidative phosphorylation. Cell Cycle 2016, 15, 1908–1918. [Google Scholar] [CrossRef]
- LaMonte, G.; Tang, X.; Chen, J.L.-Y.; Wu, J.; Ding, C.-K.C.; Keenan, M.M.; Sangokoya, C.; Kung, H.-N.; Ilkayeva, O.; Boros, L.G.; et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab 2013, 1, 23. [Google Scholar] [CrossRef]
- Mazzio, E.A.; Boukli, N.; Rivera, N.; Soliman, K.F. Pericellular pH homeostasis is a primary function of the Warburg effect: inversion of metabolic systems to control lactate steady state in tumor cells. Cancer Sci 2012, 103, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med 2010, 14, 771–794. [Google Scholar] [CrossRef] [PubMed]
- Sauvant, C.; Nowak, M.; Wirth, C.; Schneider, B.; Riemann, A.; Gekle, M.; Thews, O. Acidosis induces multi-drug resistance in rat prostate cancer cells (AT1) in vitro and in vivo by increasing the activity of the p-glycoprotein via activation of p38. Int J Cancer 2008, 123, 2532–2542. [Google Scholar] [CrossRef]
- Williams, A.C.; Collard, T.J.; Paraskeva, C. An acidic environment leads to p53 dependent induction of apoptosis in human adenoma and carcinoma cell lines: implications for clonal selection during colorectal carcinogenesis. Oncogene 1999, 18, 3199–3204. [Google Scholar] [CrossRef] [PubMed]
- Federici, C.; Petrucci, F.; Caimi, S.; Cesolini, A.; Logozzi, M.; Borghi, M.; D'Ilio, S.; Lugini, L.; Violante, N.; Azzarito, T.; et al. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS One 2014, 9, e88193. [Google Scholar] [CrossRef]
- Hanoun, M.; Maryanovich, M.; Arnal-Estapé, A.; Frenette, P.S. Neural regulation of hematopoiesis, inflammation, and cancer. Neuron 2015, 86, 360–373. [Google Scholar] [CrossRef]
- Amit, M.; Na'ara, S.; Gil, Z. Mechanisms of cancer dissemination along nerves. Nat Rev Cancer 2016, 16, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Mao, X.Y.; Li, Q.Q.; Gao, Y.F.; Zhou, H.H.; Liu, Z.Q.; Jin, W.L. Gap junction as an intercellular glue: Emerging roles in cancer EMT and metastasis. Cancer Lett 2016, 381, 133–137. [Google Scholar] [CrossRef]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; S. Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; Rowan, A.E.; Span, P.N. The mechanical microenvironment in cancer: How physics affects tumours. Semin Cancer Biol 2015, 35, 62–70. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv Drug Deliv Rev 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Yang, X.H.; Flores, L.M.; Li, Q.; Zhou, P.; Xu, F.; Krop, I.E.; Hemler, M.E. Disruption of laminin-integrin-CD151-focal adhesion kinase axis sensitizes breast cancer cells to ErbB2 antagonists. Cancer Res 2010, 70, 2256–2263. [Google Scholar] [CrossRef]
- Wang, J.P.; Hielscher, A. Fibronectin: How Its Aberrant Expression in Tumors May Improve Therapeutic Targeting. J Cancer 2017, 8, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Park, C.C.; Hilsenbeck, S.G.; Ward, R.; Rimawi, M.F.; Wang, Y.C.; Shou, J.; Bissell, M.J.; Osborne, C.K.; Schiff, R. β1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res 2011, 13, R84. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther 2020, 5, 145. [Google Scholar] [CrossRef]
- Vaidya, F.U.; Sufiyan Chhipa, A.; Mishra, V.; Gupta, V.K.; Rawat, S.G.; Kumar, A.; Pathak, C. Molecular and cellular paradigms of multidrug resistance in cancer. Cancer Rep (Hoboken) 2022, 5, e1291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pan, Q.; Shao, Z. Extracellular vesicles derived from cancer-associated fibroblasts carry tumor-promotive microRNA-1228-3p to enhance the resistance of hepatocellular carcinoma cells to sorafenib. Hum Cell 2023, 36, 296–311. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Wang, L.; Tian, H.; Wu, X.; Xiao, C.; Pan, Y.; Fan, M.; Tai, Y.; Liu, W.; Zhang, Q.; et al. CAF-derived exosomes transmitted Gremlin-1 promotes cancer progression and decreases the sensitivity of hepatoma cells to sorafenib. Mol Carcinog 2022, 61, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhu, W.; Huang, Y.; Zhuo, L.; Wang, S.; Chen, S.; Zhang, B.; Ke, B. Cancer-associated fibroblast-derived exosomal microRNA-20a suppresses the PTEN/PI3K-AKT pathway to promote the progression and chemoresistance of non-small cell lung cancer. Clin Transl Med 2022, 12, e989. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Huang, L.; Qin, G.; Qiao, Y.; Ren, F.; Shen, C.; Wang, S.; Liu, S.; Lian, J.; Wang, D.; et al. Cancer-associated fibroblasts induce monocytic myeloid-derived suppressor cell generation via IL-6/exosomal miR-21-activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett 2021, 518, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Wang, Z.; Chai, Q.; Li, Z.; Zhang, M.; Zhang, Y.; Zhang, L.; Tang, Q.; Zhu, H.; Sui, H. Exosomes derived from MDR cells induce cetuximab resistance in CRC via PI3K/AKT signaling-mediated Sox2 and PD-L1 expression. Exp Ther Med 2023, 25, 86. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J Clin Invest 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.L.; Fan, S.; Zhang, S.Y.; Chen, W.X.; Li, Q.X.; Pan, G.K.; Zhang, H.Q.; Wang, W.W.; Weng, B.; Zhang, Z.; et al. SOX8 regulates cancer stem-like properties and cisplatin-induced EMT in tongue squamous cell carcinoma by acting on the Wnt/β-catenin pathway. Int J Cancer 2018, 142, 1252–1265. [Google Scholar] [CrossRef]
- Usman, S.; Jamal, A.; Teh, M.T.; Waseem, A. Major Molecular Signaling Pathways in Oral Cancer Associated With Therapeutic Resistance. Front Oral Health 2020, 1, 603160. [Google Scholar] [CrossRef]
- Huang, L.; Wu, R.L.; Xu, A.M. Epithelial-mesenchymal transition in gastric cancer. Am J Transl Res 2015, 7, 2141–2158. [Google Scholar]
- Kirave, P.; Gondaliya, P.; Kulkarni, B.; Rawal, R.; Garg, R.; Jain, A.; Kalia, K. Exosome mediated miR-155 delivery confers cisplatin chemoresistance in oral cancer cells via epithelial-mesenchymal transition. Oncotarget 2020, 11, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Mousset, A.; Lecorgne, E.; Bourget, I.; Lopez, P.; Jenovai, K.; Cherfils-Vicini, J.; Dominici, C.; Rios, G.; Girard-Riboulleau, C.; Liu, B.; et al. Neutrophil extracellular traps formed during chemotherapy confer treatment resistance via TGF-β activation. Cancer Cell 2023, 41, 757–775.e710. [Google Scholar] [CrossRef]
- Liang, S.; Liu, Y.; He, J.; Gao, T.; Li, L.; He, S. Family with sequence similarity 46 member a confers chemo-resistance to ovarian carcinoma via TGF-β/Smad2 signaling. Bioengineered 2022, 13, 10629–10639. [Google Scholar] [CrossRef] [PubMed]
- Wo, L.; Zhang, B.; You, X.; Hu, Y.; Gu, Z.; Zhang, M.; Wang, Q.; Lv, Z.; Zhao, H. Up-regulation of LncRNA UCA1 by TGF-β promotes doxorubicin resistance in breast cancer cells. Immunopharmacol Immunotoxicol 2022, 44, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.A.; Chen, Y.F.; Bao, Y.; Mahara, S.; Yatim, S.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci U S A 2018, 115, E5990–e5999. [Google Scholar] [CrossRef]
- Wang, M.Q.; Chen, Y.R.; Xu, H.W.; Zhan, J.R.; Suo, D.Q.; Wang, J.J.; Ma, Y.Z.; Guan, X.Y.; Li, Y.; Zhu, S.L. HKDC1 upregulation promotes glycolysis and disease progression, and confers chemoresistance onto gastric cancer. Cancer Sci 2023, 114, 1365–1377. [Google Scholar] [CrossRef]
- Momeny, M.; Yousefi, H.; Eyvani, H.; Moghaddaskho, F.; Salehi, A.; Esmaeili, F.; Alishahi, Z.; Barghi, F.; Vaezijoze, S.; Shamsaiegahkani, S.; et al. Blockade of nuclear factor-κB (NF-κB) pathway inhibits growth and induces apoptosis in chemoresistant ovarian carcinoma cells. Int J Biochem Cell Biol 2018, 99, 1–9. [Google Scholar] [CrossRef]
- Boustan, A.; Jahangiri, R.; Ghalehno, A.D.; Khorsandi, M.; Mosaffa, F.; Jamialahmadi, K. Expression analysis elucidates the roles of Nicastrin, Notch4, and Hes1 in prognosis and endocrine-therapy resistance in ER-positive breast cancer patients. Res Pharm Sci 2023, 18, 78–88. [Google Scholar] [CrossRef]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, R.; Han, X.; Xu, W.; Liu, Y. PLAGL2 increases adriamycin resistance and EMT in breast cancer cells by activating the Wnt pathway. Genes Genomics 2023, 45, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Quintanal-Villalonga, A.; Taniguchi, H.; Zhan, Y.A.; Hasan, M.M.; Chavan, S.S.; Meng, F.; Uddin, F.; Allaj, V.; Manoj, P.; Shah, N.S.; et al. Comprehensive molecular characterization of lung tumors implicates AKT and MYC signaling in adenocarcinoma to squamous cell transdifferentiation. J Hematol Oncol 2021, 14, 170. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Han, X.; Chen, Y.; Tong, X.; Xue, Y.; Yao, S.; Tang, S.; Pan, Y.; Sun, Y.; Wang, X.; et al. Oxidative stress-triggered Wnt signaling perturbation characterizes the tipping point of lung adeno-to-squamous transdifferentiation. Signal Transduct Target Ther 2023, 8, 16. [Google Scholar] [CrossRef]
- Lepore Signorile, M.; Grossi, V.; Di Franco, S.; Forte, G.; Disciglio, V.; Fasano, C.; Sanese, P.; De Marco, K.; Susca, F.C.; Mangiapane, L.R.; et al. Pharmacological targeting of the novel β-catenin chromatin-associated kinase p38α in colorectal cancer stem cell tumorspheres and organoids. Cell Death Dis 2021, 12, 316. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, C.; Wang, G.; Chen, J.; Ju, S.; Huang, J.; Wang, X. SNORD1C maintains stemness and 5-FU resistance by activation of Wnt signaling pathway in colorectal cancer. Cell Death Discov 2022, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Min, H.Y.; Yong, Y.S.; Ann, J.; Nguyen, C.T.; La, M.T.; Hyun, S.Y.; Le, H.T.; Kim, H.; Kwon, H.; et al. A novel C-terminal heat shock protein 90 inhibitor that overcomes STAT3-Wnt-β-catenin signaling-mediated drug resistance and adverse effects. Theranostics 2022, 12, 105–125. [Google Scholar] [CrossRef]
- Guo, W.; Li, W.; Yuan, L.; Mei, X.; Hu, W. MicroRNA-106a-3p Induces Apatinib Resistance and Activates Janus-Activated Kinase 2 (JAK2)/Signal Transducer and Activator of Transcription 3 (STAT3) by Targeting the SOCS System in Gastric Cancer. Med Sci Monit 2019, 25, 10122–10128. [Google Scholar] [CrossRef]
- He, H.; Song, F.; Gao, Q.; Lu, Z.; Yuan, Y.; Li, X.; Chen, L.; Jia, C.; Yang, R.; Yang, J.; et al. The APEX1/miRNA-27a-5p axis plays key roles in progression, metastasis and targeted chemotherapy of gastric cancer. Int J Pharm 2021, 599, 120446. [Google Scholar] [CrossRef]
- Ma, D.; Liu, P.; Hu, C.; Zhou, Z.; Wang, P.; Wang, Y.; Zhang, Y.; Ran, Y.; Li, P.; Zhao, J.; et al. Intracellular angiopoietin-1 promotes TKI-resistance via activation of JAK/STAT5 pathway in chronic myeloid leukemia. Oncogene 2023, 42, 124–137. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Meng, J.; Guo, F.; Ren, D.; Wu, H.; Jin, X. S-palmitoylation of PCSK9 induces sorafenib resistance in liver cancer by activating the PI3K/AKT pathway. Cell Rep 2022, 40, 111194. [Google Scholar] [CrossRef]
- Chen, S.; Xia, X. Long noncoding RNA NEAT1 suppresses sorafenib sensitivity of hepatocellular carcinoma cells via regulating miR-335-c-Met. J Cell Physiol 2019, 234, 14999–15009. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Shinonaga, R.; Sakaguchi, H.; Kitagawa, Y.; Yoshida, K. NEAT1-SOD2 Axis Confers Sorafenib and Lenvatinib Resistance by Activating AKT in Liver Cancer Cell Lines. Curr Issues Mol Biol 2023, 45, 1073–1085. [Google Scholar] [CrossRef]
- Enomoto, K.; Hirayama, S.; Kumashiro, N.; Jing, X.; Kimura, T.; Tamagawa, S.; Matsuzaki, I.; Murata, S.I.; Hotomi, M. Synergistic Effects of Lenvatinib (E7080) and MEK Inhibitors against Anaplastic Thyroid Cancer in Preclinical Models. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.; Wu, C.; Xia, Q.; Xu, D. ASIC1a mediates the drug resistance of human hepatocellular carcinoma via the Ca(2+)/PI3-kinase/AKT signaling pathway. Lab Invest 2017, 97, 53–69. [Google Scholar] [CrossRef]
- Vijayakumar, G.; Swetha, U.S.; Sudhagar, S. Tamoxifen modulates mitochondrial dynamics through AMPK and MAPK during nutrition deprivation. Cell Biol Int 2022, 46, 1661–1671. [Google Scholar] [CrossRef]
- Rekha, P.; Gupta, A.; Goud, K.S.; Biswas, B.; Bhattar, S.; Vijayakumar, G.; Selvaraju, S. GPER induces mitochondrial fission through p44/42 MAPK - Drp1 pathway in breast cancer cells. Biochem Biophys Res Commun 2023, 643, 16–23. [Google Scholar] [CrossRef]
- Troiani, T.; Napolitano, S.; Vitagliano, D.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; Ciardiello, D.; Ciardiello, F.; Martinelli, E. Primary and acquired resistance of colorectal cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway activation and can be overcome by combined MEK/EGFR inhibition. Clin Cancer Res 2014, 20, 3775–3786. [Google Scholar] [CrossRef]
- Wang, Q.; Liao, C.; Tan, Z.; Li, X.; Guan, X.; Li, H.; Tian, Z.; Liu, J.; An, J. FUT6 inhibits the proliferation, migration, invasion, and EGF-induced EMT of head and neck squamous cell carcinoma (HNSCC) by regulating EGFR/ERK/STAT signaling pathway. Cancer Gene Ther 2023, 30, 182–191. [Google Scholar] [CrossRef]
- Pan, Z.; Wang, K.; Wang, X.; Jia, Z.; Yang, Y.; Duan, Y.; Huang, L.; Wu, Z.X.; Zhang, J.Y.; Ding, X. Cholesterol promotes EGFR-TKIs resistance in NSCLC by inducing EGFR/Src/Erk/SP1 signaling-mediated ERRα re-expression. Mol Cancer 2022, 21, 77. [Google Scholar] [CrossRef]
- Udagawa, S.; Ooki, A.; Shinozaki, E.; Fukuda, K.; Yamaguchi, K.; Osumi, H. Circulating Tumor DNA: The Dawn of a New Era in the Optimization of Chemotherapeutic Strategies for Metastatic Colo-Rectal Cancer Focusing on RAS Mutation. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Draškovič, T.; Zidar, N.; Hauptman, N. Circulating Tumor DNA Methylation Biomarkers for Characterization and Determination of the Cancer Origin in Malignant Liver Tumors. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu Rev Pathol 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer 2019, 18, 128. [Google Scholar] [CrossRef]
- Lin, F.; Xiong, M.; Hao, W.; Song, Y.; Liu, R.; Yang, Y.; Yuan, X.; Fan, D.; Zhang, Y.; Hao, M.; et al. A Novel Blockade CD47 Antibody With Therapeutic Potential for Cancer. Front Oncol 2020, 10, 615534. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; Toni, E.N.D.; et al. Phase 3 randomized, open-label, multicenter study of tremelimumab (T) and durvalumab (D) as first-line therapy in patients (pts) with unresectable hepatocellular carcinoma (uHCC): HIMALAYA. J Clin Oncol 2022, 40, 379–379. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Yang, Y.; Heeke, S.; Patel, S.A.; Poteete, A.; Udagawa, H.; Elamin, Y.Y.; Moran, C.A.; Kashima, Y.; Arumugam, T.; et al. CD70 is a therapeutic target upregulated in EMT-associated EGFR tyrosine kinase inhibitor resistance. Cancer Cell 2023, 41, 340–355.e346. [Google Scholar] [CrossRef]
- Marks, D.K.; Gartrell, R.D.; El Asmar, M.; Boboila, S.; Hart, T.; Lu, Y.; Pan, Q.; Yu, J.; Hibshoosh, H.; Guo, H.; et al. Akt Inhibition Is Associated With Favorable Immune Profile Changes Within the Tumor Microenvironment of Hormone Receptor Positive, HER2 Negative Breast Cancer. Front Oncol 2020, 10, 968. [Google Scholar] [CrossRef]
- Owusu-Brackett, N.; Zhao, M.; Akcakanat, A.; Evans, K.W.; Yuca, E.; Dumbrava, E.I.; Janku, F.; Meric-Bernstam, F. Targeting PI3Kβ alone and in combination with chemotherapy or immunotherapy in tumors with PTEN loss. Oncotarget 2020, 11, 969–981. [Google Scholar] [CrossRef]
- Kang, C.; Ju, S.; Kim, J.; Jung, Y. Chloroquine prevents hypoxic accumulation of HIF-1α by inhibiting ATR kinase: implication in chloroquine-mediated chemosensitization of colon carcinoma cells under hypoxia. Pharmacol Rep 2023, 75, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Cocco, S.; Leone, A.; Roca, M.S.; Lombardi, R.; Piezzo, M.; Caputo, R.; Ciardiello, C.; Costantini, S.; Bruzzese, F.; Sisalli, M.J.; et al. Inhibition of autophagy by chloroquine prevents resistance to PI3K/AKT inhibitors and potentiates their antitumor effect in combination with paclitaxel in triple negative breast cancer models. J Transl Med 2022, 20, 290. [Google Scholar] [CrossRef]
- Dong, Q.; Han, D.; Li, B.; Yang, Y.; Ren, L.; Xiao, T.; Zhang, J.; Li, Z.; Yang, H.; Liu, H. Bionic lipoprotein loaded with chloroquine-mediated blocking immune escape improves antitumor immunotherapy. Int J Biol Macromol 2023, 240, 124342. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat Rev Drug Discov 2021, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Yetisgin, A.A.; Cetinel, S.; Zuvin, M.; Kosar, A.; Kutlu, O. Therapeutic Nanoparticles and Their Targeted Delivery Applications. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Majidinia, M.; Mirza-Aghazadeh-Attari, M.; Rahimi, M.; Mihanfar, A.; Karimian, A.; Safa, A.; Yousefi, B. Overcoming multidrug resistance in cancer: Recent progress in nanotechnology and new horizons. IUBMB Life 2020, 72, 855–871. [Google Scholar] [CrossRef]
- Han, E.; Kim, D.; Cho, Y.; Lee, S.; Kim, J.; Kim, H. Development of Polymersomes Co-Delivering Doxorubicin and Melittin to Overcome Multidrug Resistance. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Zhou, S.; Ma, Y.; Xu, R.; Tang, X. Nanoparticles Loaded with GSK1059615 Combined with Sorafenib Inhibited Programmed Cell Death 1 Ligand 1 Expression by Negatively Regulating the PI3K/Akt/NF-κB Pathway, Thereby Reversing the Drug Resistance of Hepatocellular Carcinoma to Sorafenib. J Biomed Nanotechnol 2022, 18, 693–704. [Google Scholar] [CrossRef]
- Lv, W.; Wu, H.; Zhang, Y.; Li, H.; Shu, H.; Su, C.; Zhu, Y.; Wang, T.; Nie, F. cRGD-targeted gold-based nanoparticles overcome EGFR-TKI resistance of NSCLC via low-temperature photothermal therapy combined with sonodynamic therapy. Biomater Sci 2023, 11, 1677–1691. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Buhimschi, A.D.; Armstrong, H.A.; Toure, M.; Jaime-Figueroa, S.; Chen, T.L.; Lehman, A.M.; Woyach, J.A.; Johnson, A.J.; Byrd, J.C.; Crews, C.M. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton's Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57, 3564–3575. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, X.; Ding, N.; Gao, H.; Wu, Y.; Yang, Y.; Zhao, M.; Hwang, J.; Song, Y.; Liu, W.; et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Research 2018, 28, 779–781. [Google Scholar] [CrossRef]
- Desai, A.; Lovly, C.M. Strategies to overcome resistance to ALK inhibitors in non-small cell lung cancer: a narrative review. Transl Lung Cancer Res 2023, 12, 615–628. [Google Scholar] [CrossRef]
- Yang, T.; Curtis, S.; Bai, A.; Young, A.; Derosier, D.; Ripley, S.; Bai, S. CRISPR/Cas9 targeting liposomes knocked down multidrug resistance proteins in brain endothelial cells as a model to predict potential pharmacoresistance. Colloids Surf B Biointerfaces 2023, 222, 113103. [Google Scholar] [CrossRef]
- Huang, S.; Ma, Z.; Zhou, Q.; Wang, A.; Gong, Y.; Li, Z.; Wang, S.; Yan, Q.; Wang, D.; Hou, B.; et al. Genome-Wide CRISPR/Cas9 Library Screening Identified that DUSP4 Deficiency Induces Lenvatinib Resistance in Hepatocellular Carcinoma. Int J Biol Sci 2022, 18, 4357–4371. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.K.; Srivastava, S.; Hancock, A.; Shrivastava, A.; Morvant, J.; Shankar, S.; Srivastava, R.K. Inhibition of ribosome assembly factor PNO1 by CRISPR/Cas9 technique suppresses lung adenocarcinoma and Notch pathway: Clinical application. J Cell Mol Med 2023, 27, 365–378. [Google Scholar] [CrossRef]
- Qureshi, R.; Zou, B.; Alam, T.; Wu, J.; Lee, V.H.F.; Yan, H. Computational Methods for the Analysis and Prediction of EGFR-Mutated Lung Cancer Drug Resistance: Recent Advances in Drug Design, Challenges and Future Prospects. IEEE/ACM Trans Comput Biol Bioinform 2023, 20, 238–255. [Google Scholar] [CrossRef]
- Huang, Y.Q.; Sun, P.; Chen, Y.; Liu, H.X.; Hao, G.F.; Song, B.A. Bioinformatics toolbox for exploring target mutation-induced drug resistance. Brief Bioinform 2023, 24. [Google Scholar] [CrossRef]
- Fröhlich, F.; Gerosa, L.; Muhlich, J.; Sorger, P.K. Mechanistic model of MAPK signaling reveals how allostery and rewiring contribute to drug resistance. Mol Syst Biol 2023, 19, e10988. [Google Scholar] [CrossRef]
- Rosenberger, G.; Li, W.; Turunen, M.; He, J.; Subramaniam, P.S.; Pampou, S.; Griffin, A.T.; Karan, C.; Kerwin, P.; Murray, D.; et al. Network-based elucidation of colon cancer drug resistance by phosphoproteomic time-series analysis. bioRxiv 2023. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, Y.; Li, H.; Zhou, Y.; Shi, S.; Chen, Z.; He, X.; Zhang, H.; Li, F.; Yin, J.; et al. DRESIS: the first comprehensive landscape of drug resistance information. Nucleic Acids Res 2023, 51, D1263–d1275. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhang, Y.; Guo, L.; Liu, C.; Wang, P.; Ren, W. Exosomal microRNA-107 reverses chemotherapeutic drug resistance of gastric cancer cells through HMGA2/mTOR/P-gp pathway. BMC Cancer 2021, 21, 1290. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Chen, L.; Chen, R.; Xu, F.; Huang, Z.; Huang, R.; Wang, W.; Xu, Q. miR-4486 enhances cisplatin sensitivity of gastric cancer cells by restraining the JAK3/STAT3 signalling pathway. J Chemother 2022, 34, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Fang, Y.; Wang, X.; Han, Y.; Du, F.; Li, C.; Hu, H.; Liu, H.; Liu, Q.; Wang, J.; et al. miR-302a Inhibits Metastasis and Cetuximab Resistance in Colorectal Cancer by Targeting NFIB and CD44. Theranostics 2019, 9, 8409–8425. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Khan, F.; Upadhyay, T.K.; Seungjoon, M.; Park, M.N.; Kim, B. New insights about the PDGF/PDGFR signaling pathway as a promising target to develop cancer therapeutic strategies. Biomed Pharmacother 2023, 161, 114491. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Deng, Y.; Chen, X.; Zhang, J.; Chen, Y.; Li, H.; Wu, Q.; Yang, Z.; Zhang, L.; Liu, B. Inhibition of PDGFR by CP-673451 induces apoptosis and increases cisplatin cytotoxicity in NSCLC cells via inhibiting the Nrf2-mediated defense mechanism. Toxicol Lett 2018, 295, 88–98. [Google Scholar] [CrossRef]
- Yin, L.; He, J.; Xue, J.; Na, F.; Tong, R.; Wang, J.; Gao, H.; Tang, F.; Mo, X.; Deng, L.; et al. PDGFR-β inhibitor slows tumor growth but increases metastasis in combined radiotherapy and Endostar therapy. Biomed Pharmacother 2018, 99, 615–621. [Google Scholar] [CrossRef]
- Wong, J.P.; Todd, J.R.; Finetti, M.A.; McCarthy, F.; Broncel, M.; Vyse, S.; Luczynski, M.T.; Crosier, S.; Ryall, K.A.; Holmes, K.; et al. Dual Targeting of PDGFRα and FGFR1 Displays Synergistic Efficacy in Malignant Rhabdoid Tumors. Cell Rep 2016, 17, 1265–1275. [Google Scholar] [CrossRef]
- Kun, E.; Tsang, Y.T.M.; Ng, C.W.; Gershenson, D.M.; Wong, K.K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev 2021, 92, 102137. [Google Scholar] [CrossRef]
- Dudka, W.; Hoser, G.; Mondal, S.S.; Turos-Korgul, L.; Swatler, J.; Kusio-Kobialka, M.; Wołczyk, M.; Klejman, A.; Brewinska-Olchowik, M.; Kominek, A.; et al. Targeting integrated stress response with ISRIB combined with imatinib treatment attenuates RAS/RAF/MAPK and STAT5 signaling and eradicates chronic myeloid leukemia cells. BMC Cancer 2022, 22, 1254. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Carlomagno, F.; Anaganti, S.; Guida, T.; Salvatore, G.; Troncone, G.; Wilhelm, S.M.; Santoro, M. BAY 43-9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst 2006, 98, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Strumberg, D.; Clark, J.W.; Awada, A.; Moore, M.J.; Richly, H.; Hendlisz, A.; Hirte, H.W.; Eder, J.P.; Lenz, H.J.; Schwartz, B. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 2007, 12, 426–437. [Google Scholar] [CrossRef]
- Bruix, J.; Takayama, T.; Mazzaferro, V.; Chau, G.Y.; Yang, J.; Kudo, M.; Cai, J.; Poon, R.T.; Han, K.H.; Tak, W.Y.; et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol 2015, 16, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Tovar, V.; Cornella, H.; Moeini, A.; Vidal, S.; Hoshida, Y.; Sia, D.; Peix, J.; Cabellos, L.; Alsinet, C.; Torrecilla, S.; et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut 2017, 66, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Bank, T.; Malnassy, G.; Arteaga, M.; Shang, N.; Dalheim, A.; Ding, X.; Cotler, S.J.; Denning, M.F.; Nishimura, M.I.; et al. Inhibition of insulin-like growth factor 1 receptor enhances the efficacy of sorafenib in inhibiting hepatocellular carcinoma cell growth and survival. Hepatol Commun 2018, 2, 732–746. [Google Scholar] [CrossRef]
- Peng, S.W.; Ngo, M.T.; Kuo, Y.C.; Teng, M.H.; Guo, C.L.; Lai, H.C.; Chang, T.S.; Huang, Y.H. Niclosamide Revitalizes Sorafenib through Insulin-like Growth Factor 1 Receptor (IGF-1R)/Stemness and Metabolic Changes in Hepatocellular Carcinoma. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhou, X.; Xu, H.; Shi, X.; Zhao, J.; Yang, M.; Zhang, L.; Jin, X.; Hu, Y.; Li, X.; et al. Niclosamide Inhibits Cell Growth and Enhances Drug Sensitivity of Hepatocellular Carcinoma Cells via STAT3 Signaling Pathway. J Cancer 2018, 9, 4150–4155. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Armstrong, C.; Zhu, Y.; Evans, C.P.; Gao, A.C. Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. Prostate 2015, 75, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.; Chen, Y.K.; Hsu, Y.J.; Lin, B.R. Niclosamide inhibits the cell proliferation and enhances the responsiveness of esophageal cancer cells to chemotherapeutic agents. Oncol Rep 2020, 43, 549–561. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Stages of carcinogenesis. Exposure to a carcinogenic agent such as viruses, chemicals or radiation will induce DNA damage in one or a small population of healthy cells. Failure in DNA repair will lead to intrinsic or acquired mutations that will potentially affect the cell biological process such as its growth and apoptosis resulting in initiating premalignancy transformation. The transformed cells will then promote the growth of neoplastic lesion which hold so many genetic alterations. Chemopreventive agents such as natural agents derived from dietary sources (e.g., curcumin, resveratrol) or bioactive molecules (tamoxifen, raloxifene) will be used to block and suppress cell growth rates at those stages to reverse or delay the carcinogenesis process. Failure to eliminate those premalignant cells will lead to the progression stage resulting in the formation of a malignant tumour. Chemopreventive agents will be given at this stage to inhibit the cell invasion, metastasis and angiogenesis. Failure to do so will result in metastasis via bloodstream or lymphatic system. Created with BioRender.com.
Figure 1.
Stages of carcinogenesis. Exposure to a carcinogenic agent such as viruses, chemicals or radiation will induce DNA damage in one or a small population of healthy cells. Failure in DNA repair will lead to intrinsic or acquired mutations that will potentially affect the cell biological process such as its growth and apoptosis resulting in initiating premalignancy transformation. The transformed cells will then promote the growth of neoplastic lesion which hold so many genetic alterations. Chemopreventive agents such as natural agents derived from dietary sources (e.g., curcumin, resveratrol) or bioactive molecules (tamoxifen, raloxifene) will be used to block and suppress cell growth rates at those stages to reverse or delay the carcinogenesis process. Failure to eliminate those premalignant cells will lead to the progression stage resulting in the formation of a malignant tumour. Chemopreventive agents will be given at this stage to inhibit the cell invasion, metastasis and angiogenesis. Failure to do so will result in metastasis via bloodstream or lymphatic system. Created with BioRender.com.
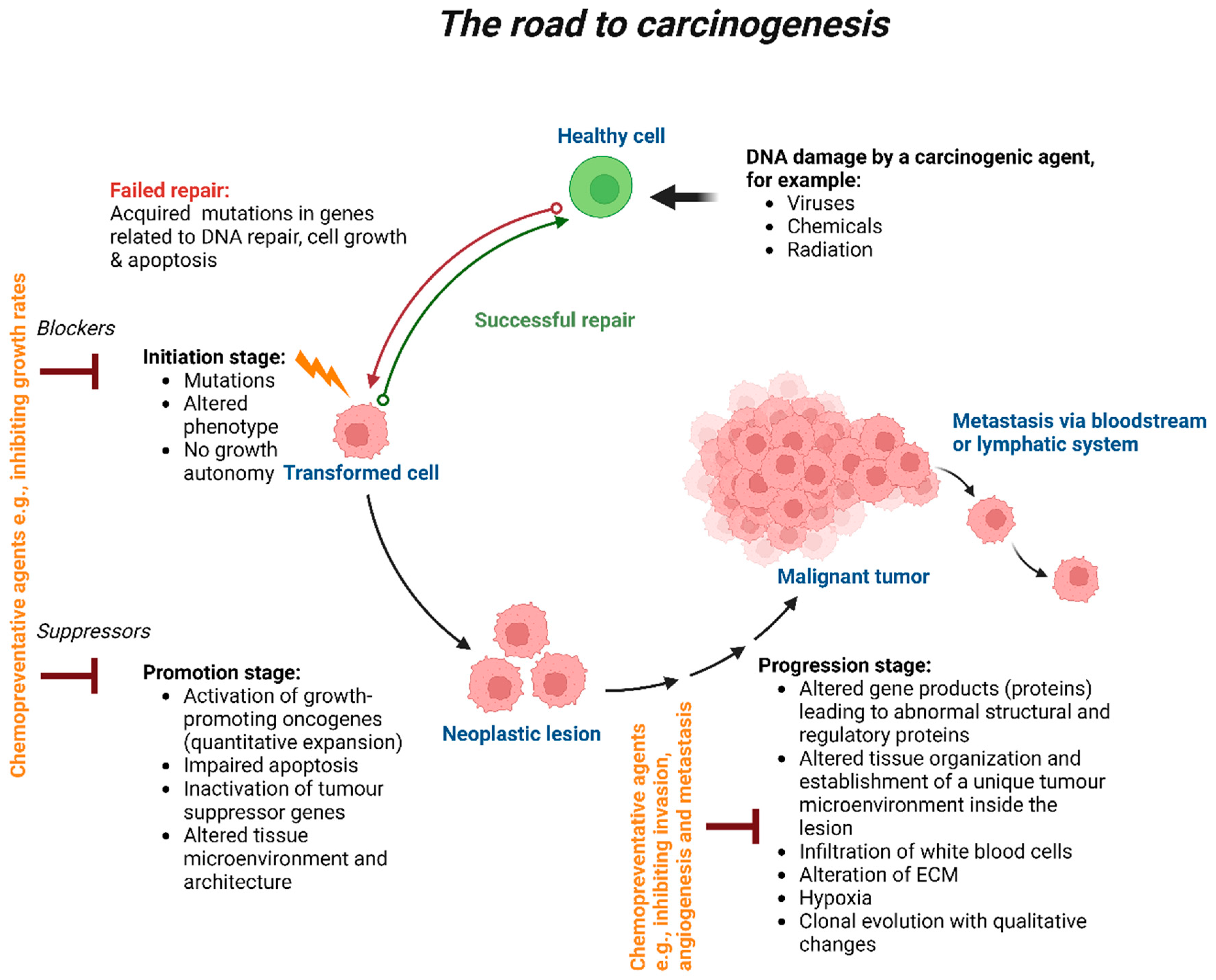
Figure 2.
Potential mechanisms in cancer drug resistance. Several mechanisms trigger drug resistance in cancer. Exposure of cancer cells to therapeutic pressure induce genomic alterations and mutations either (1) Intrinsic represented in red or (2) acquired represented in green after cycles of treatment which in both cases result in drug resistance. (3) Slow growing tumours and (4) intractable genomic drivers (e.g., MYC and TP53) play a critical role in the emergence of drug resistance. All those factors play a vital role in tumour heterogeneity leading to genetic diversity. (5) Tumour microenvironment mediate resistance by several mechanisms e.g., (a) escaping immune surveillance, (b) stimulating paracrine growth factors by tumour associated cells to promote cancer cell growth and (c) neovascularization of tumour cells by overexpressing vascular endothelial growth factor receptors (VEGFR). All those factors make a complex network that trigger drug resistance in cancer. Created with BioRender.com.
Figure 2.
Potential mechanisms in cancer drug resistance. Several mechanisms trigger drug resistance in cancer. Exposure of cancer cells to therapeutic pressure induce genomic alterations and mutations either (1) Intrinsic represented in red or (2) acquired represented in green after cycles of treatment which in both cases result in drug resistance. (3) Slow growing tumours and (4) intractable genomic drivers (e.g., MYC and TP53) play a critical role in the emergence of drug resistance. All those factors play a vital role in tumour heterogeneity leading to genetic diversity. (5) Tumour microenvironment mediate resistance by several mechanisms e.g., (a) escaping immune surveillance, (b) stimulating paracrine growth factors by tumour associated cells to promote cancer cell growth and (c) neovascularization of tumour cells by overexpressing vascular endothelial growth factor receptors (VEGFR). All those factors make a complex network that trigger drug resistance in cancer. Created with BioRender.com.
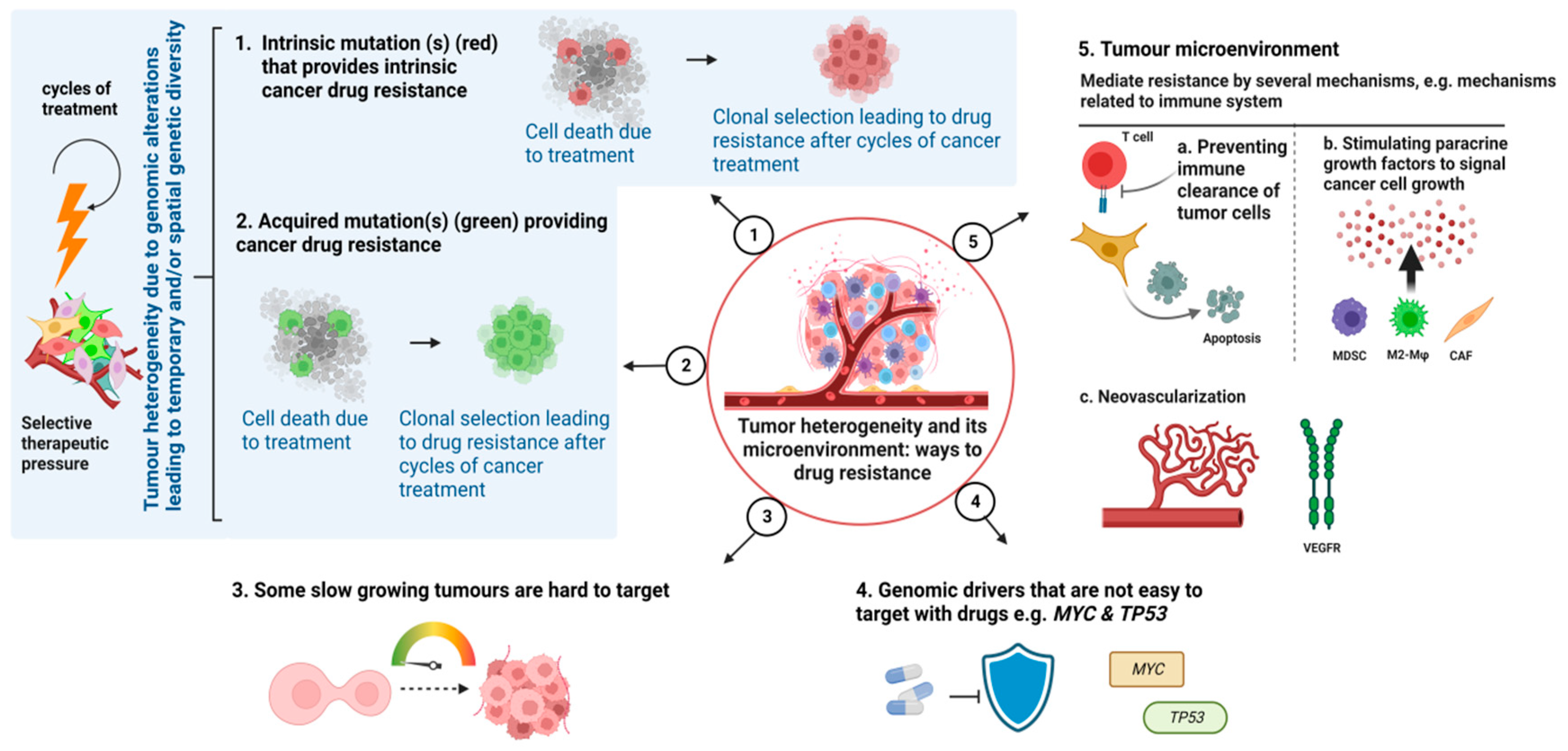
Figure 3.
Drug resistance management. Strategies used to manage and overcome drug resistance are depicted in this picture. (1) Earlier detection of actionable genomic modifications using ctDNA is a powerful tool to predict cancer recurrence influencing a more effective treatment decision-making that results in a better response to treatment. (2 & 3) Immunotherapy such as checkpoint inhibitors can be used as monotherapy or in combination to target multiple pathways simultaneously and increase treatment effectiveness. (4) Mapping cancer dependencies using DeepMap is an effective approach to predict genes responsible for drug resistance and identify new genetic targets thereby facilitating the discovery of drugs that can potentially overcome resistance. Created with BioRender.com.
Figure 3.
Drug resistance management. Strategies used to manage and overcome drug resistance are depicted in this picture. (1) Earlier detection of actionable genomic modifications using ctDNA is a powerful tool to predict cancer recurrence influencing a more effective treatment decision-making that results in a better response to treatment. (2 & 3) Immunotherapy such as checkpoint inhibitors can be used as monotherapy or in combination to target multiple pathways simultaneously and increase treatment effectiveness. (4) Mapping cancer dependencies using DeepMap is an effective approach to predict genes responsible for drug resistance and identify new genetic targets thereby facilitating the discovery of drugs that can potentially overcome resistance. Created with BioRender.com.
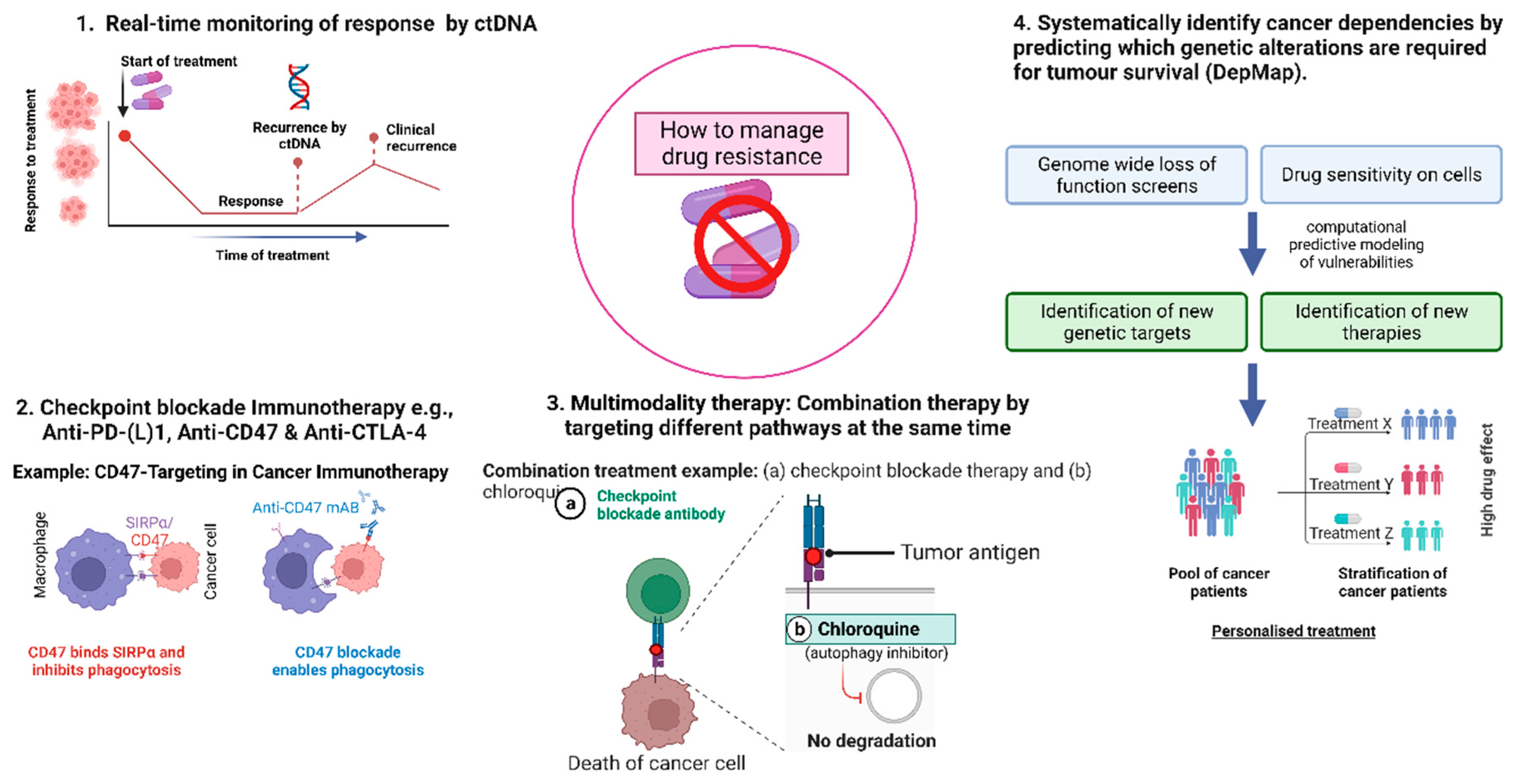
Figure 4.
Targeting multiple pathways in drug resistant cells. The impact of many therapies on signalling in drug-resistant cells is depicted in this picture. (A) When targeting a single signalling pathway such as PGFR signalling, cancer cells become resistant to PDGFR inhibitors (such as CHMFL-PDGFR-159) by activating compensatory signalling pathways of alternative RTKs (e.g., FGFR or VEGFR) leading o cell survival and migration. (B) However, resistance and signalling reactivation can be overcome by combination therapy and multi-target kinase inhibitors that target multiple signalling pathways leading to effective inhibition of cancer progression and drug resistance. Created with BioRender.com.
Figure 4.
Targeting multiple pathways in drug resistant cells. The impact of many therapies on signalling in drug-resistant cells is depicted in this picture. (A) When targeting a single signalling pathway such as PGFR signalling, cancer cells become resistant to PDGFR inhibitors (such as CHMFL-PDGFR-159) by activating compensatory signalling pathways of alternative RTKs (e.g., FGFR or VEGFR) leading o cell survival and migration. (B) However, resistance and signalling reactivation can be overcome by combination therapy and multi-target kinase inhibitors that target multiple signalling pathways leading to effective inhibition of cancer progression and drug resistance. Created with BioRender.com.
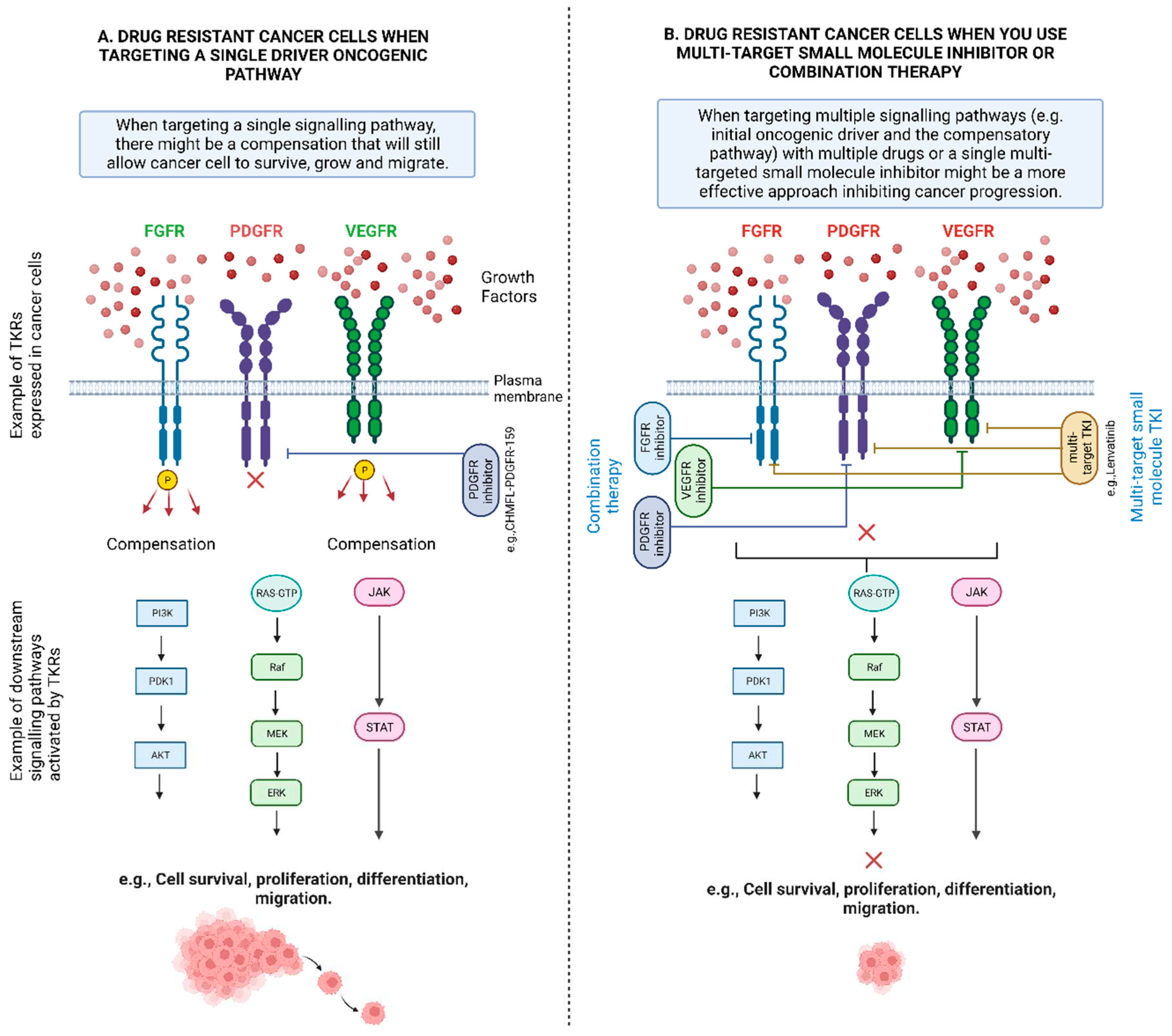

Table 1.
Recruiting phase 3 interventional clinical studies targeting receptors tyrosine kinase signalling pathways in drug resistant cancers. Interventions targeting RTK signalling are highlighted in bold.
Table 1.
Recruiting phase 3 interventional clinical studies targeting receptors tyrosine kinase signalling pathways in drug resistant cancers. Interventions targeting RTK signalling are highlighted in bold.
| Interventions | NCT Number | Type of cancer | Study Title |
|---|---|---|---|
| Targeting multiple RTKs | |||
| Famitinib (a RTKI against multiple targets, e.g., VEGFR 2/3, PDGFR, and stem cell factor receptor (c-kit)). Sunitinib (a RTKI against multiple targets, e.g., VEGFR and PDGFR) |
NCT04409223 | Gastrointestinal Stromal Tumours | Efficacy and Safety of Famitinib Versus Sunitinib in the Treatment of Advanced Gastrointestinal Stromal Tumour Patients After Failure of Imatinib |
| AL3818 (a RTKI against multiple targets, e.g., VEGFR, FGFR, PDGFR, and c-kit). Paclitaxel, Pegylated Liposomal Doxorubicin (PLD), Topotecan, Carboplatin |
NCT02584478 | Endometrial Carcinoma, Ovarian Carcinoma, Fallopian Tube Carcinoma, Primary Peritoneal Carcinoma, Cervical Carcinoma |
Phase 1/2a/3 Evaluation of Adding AL3818 to Standard Platinum-Based Chemotherapy in Subjects With Recurrent or Metastatic Endometrial, Ovarian, Fallopian, Primary Peritoneal or Cervical Carcinoma (AL3818-US-002) |
| Regorafenib (a RTKI against multiple targets, e.g., VEGF1/2/3, PDGFR, FGFR, KIT, RET, RAF-1, BRAF) Nivolumab, Docetaxel, Paclitaxel, Irinotecan, Trifluridine/Tipracil |
NCT04879368 | Gastro-Oesophageal Cancer | RegoNivo vs Standard of Care Chemotherapy in AGOC |
| Targeting EGFR | |||
| ASK120067 (Third generation EGFR TKI) Gefitinib (First generation EGFR TKI) ASK120067, Placebo Gefitinib 250 mg, Gefitinib, Placebo ASK120067 |
NCT04143607 | Locally Advanced or Metastatic NSCLC | ASK120067 Versus Gefitinib as First-line Treatment for EGFRm Locally Advanced or Metastatic NSCLC |
| Aumolertinib (Third generation EGFR TKI) Osimertinib (Second generation EGFR TKI) Pemetrexed, Cisplatin, Carboplatin. Paclitaxel, Nab paclitaxel, Gemcitabine |
NCT05493501 | NSCLC | Aumolertinib With Chemotherapy or Alone Compared With Osimertinib in Patients With Epidermal Growth Factor Receptor-Mutant Non-Small Cell Lung Cancer |
| Gefitinib (First generation EGFR TKI) Afatinib (Second generation ErbB family inhibitor Erlotinib (First generation EGFR TKI) Metformin Hydrochloride, Placebo |
NCT05445791 | NSCLC | Metformin Plus Tyrosine Kinase Inhibitors for Treatment of Patients With Non-small Cell Lung Cancer With EGFR Mutations |
| Targeting EGFR and HER-2 | |||
| Pyrotinib (EGFR and HER2 inhibitor) Trastuzumab (HER2 inhibitor) |
NCT05346861 | HER2-positive Breast Cancer, Metastatic Breast Cancer |
Pyrotinib Rechallenge in Her2-positive Metastatic Breast Cancer Pretreated With Pyrotinib and Trastuzumab |
| Targeting EGFR and VEGFR | |||
| Gefitinib (First generation EGFR TKI) Apatinib (VEGFR-2 TKIs) Placebo |
NCT02824458 | Non-Squamous NSCLC | A Study of Gefitinib With or Without Apatinib in Patients With Advanced Non-squamous Non-Small-Cell Lung Cancer Harboring EGFR Mutations |
| Osimertinib (Second generation EGFR TKI) Bevacizumab (anti-VEGF monoclonal antibody) |
NCT04181060 | Advanced Lung Non-Squamous Non-Small Cell Carcinoma, Metastatic Lung Non-Squamous Non-Small Cell Carcinoma, Recurrent Lung Non-Squamous Non-Small Cell Carcinoma, Stage IIIB Lung Cancer AJCC v8, Stage IV Lung Cancer AJCC v8 | Osimertinib With or Without Bevacizumab as Initial Treatment for Patients With EGFR-Mutant Lung Cancer |
| Targeting BCR-ABL and JAK | |||
| Dasatinib (small-molecule, BCR-ABL inhibitor) Ruxolitinib (JAK inhibitor) Prednisone, Vincristine, Daunorubicin. Pegaspargase. Erwinase®, Cyclophosphamide, Cytarabine, Mercaptopurine, Methotrexate, Blinatumomab, Bortezomib, Dexamethasone, Doxorubicin, Etoposide, Clofarabine, Vorinostat. Idarubicin. Nelarabine, Thioguanine, Asparaginase Erwinia chrysanthemi (recombinant)-rywn, Calaspargase Pegol |
NCT03117751 |
Acute Lymphoblastic Leukemia, Acute Lymphoblastic Lymphoma |
Total Therapy XVII for Newly Diagnosed Patients With Acute Lymphoblastic Leukemia and Lymphoma |
| Targeting VEGF pathway | |||
| IBI305 (anti-VEGF monoclonal antibody) Sintilimab, Pemetrexed, Cisplatin, Placebo1, Placebo2 |
NCT03802240 | Non-Squamous NSCLC | Sintilimab ± IBI305 Plus Chemotherapy (Pemetrexed + Cisplatin) for EGFRm + Locally Advanced or Metastasis Non-Squamous NSCLC Patients After EGFR-TKI Treatment Failure |
| BD0801 (anti-VEGF monoclonal antibody) Paclitaxel, Placebo, Topotecan, doxorubicin liposome |
NCT04908787 | Ovarian Cancer | A Phase III Study of BD0801 Combined With Chemotherapy in Recurrent, Platinum-resistant Epithelial Ovarian Cancer |
| Targeting mTOR | |||
| Everolimus (mTOR inhibitor) Letrozole, Bicalutamide, Itraconazole |
NCT03458221 | Recurrent Ovarian Cancer, Signal Transduction Pathway Deregulation, Therapy-Associated Cancer | Signal TrAnsduction Pathway Activity Analysis in OVarian cancER |
| Targeting HER3 | |||
| Patritumab Deruxtecan (HER3-DXd antibody attached to topoisomerase I inhibitor) Platinum-based chemotherapy |
NCT05338970 | Non-Squamous NSCLC, EGFR L858R, EGFR Exon 19 Deletion |
HERTHENA-Lung02: A Study of Patritumab Deruxtecan Versus Platinum-based Chemotherapy in Metastatic or Locally Advanced EGFRm NSCLC After Failure of EGFR TKI Therapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



