
Submitted:
31 August 2023
Posted:
31 August 2023
You are already at the latest version
Abstract
Bioactive lipids are involved in cellular signalling events with links to human disease. Many of these are involved in inflammation under normal and pathological conditions. Despite being attractive molecules from a pharmacological point of view, detection and quantification of lipids has been a major challenge. Here, we have optimised a liquid chromatography dynamic multiple reaction monitoring targeted mass spectrometry (LC-dMRM-MS) approach to profile eicosanoids and fatty acids in biological samples. In particular, by applying this analytic workflow to study a cellular model of Chronic Myeloid Leukaemia (CML), we found that intra- and extra-cellular 2-Arachidonoylglycerol (2-AG), intracellular Arachidonic Acid (AA), and extracellular Prostaglandin F2α (PGF2α), 5-Hydroxyeicosatetraenoic acid (5-HETE), Palmitic acid (PA, C16:0) and Stearic acid (SA, C18:0) were altered in response to immunomodulation by type I Interferon (IFN-I), a currently approved treatment for CML. Our observations indicate changes in eicosanoid and fatty acid metabolism with potential relevance in the context of cancer inflammation and CML.
Keywords:
Bioactive Lipids
; Eicosanoids
; Fatty Acids
; Mass Spectrometry
; Lipidomics
; Innate Immunity
; Type-I Interferon Response
; Chronic Myeloid Leukaemia
; Cancer Inflammation
; Cancer Metabolism
1. Introduction
Bioactive lipids regulate cellular functions and contribute to tissue homeostasis and pathology [1]. Four families of bioactive lipids are involved in inflammation and immune regulation; eicosanoids, specialized pro-resolving mediators, lysoglycerophospholipids/ sphingolipids and endocannabinoids, which are generated from ω-3 or ω-6 polyunsaturated fatty acids (PUFAs). Eicosanoids are key elements in inflammation, and have been linked with diseases, such as viral infections, neurodegenerative disorders [2], rheumatoid arthritis [3], atherosclerosis [4,5], acute coronary syndrome [6], cancer [7,8], systemic lupus erythematosus [9], multiple sclerosis [10], liver injury [11], celiac disease [12], diabetes [13], cystic fibrosis [14], renovascular disease [15], asthma [16] and muscle dysfunction [17]. In addition, fatty acids, the common components of complex lipids, often act as immunomodulatory factors [18,19].
In general, lipids are challenging molecules to characterize due to the limited analytical capacity to identify, quantify, and annotate them in biological samples, which has been an impediment to the advancement of discovering molecular mechanisms of disease, biomarker discovery and drug development. Mass spectrometry (MS) approaches, predominantly in combination with liquid chromatography (LC), are widely used for analysis of biomolecules [20-23], including eicosanoids [24, 25] . More recently, LC coupled with dynamic multiple-reaction monitoring (dMRM) workflows for targeted detection of specific subsets of eicosanoids [26, 27], and high-resolution (MRMHR) methods to generate a library of high-resolution fragmentation spectra [28] , have been developed. In addition, eicosanoid profiling by LC-MS has previously been performed on human plasma [29], clinical samples [30] and mouse models of pathophysiological states [31]. However, such analyses remain challenging due to limitations in the accurate assignment and detection of endogenous species [32, 33]. One reason is a lack of thoroughness in optimising truly representative sample matrix variations, instead of using “surrogate matrices” (such as BSA in sample buffer, which would be a matrix approximation), and applying more efficient extraction methodologies adapted to specific lipid subsets within specific sample types (e.g. cell and cell supernatant samples).
In this study, an intracellular and extracellular analysis (the media in which the respective cells are cultured) of eicosanoids and fatty acids was performed using a tailored extraction and dMRM method to maximize the sensitivity of detecting endogenous species. Since bioactive lipids such as eicosanoids have important roles in inflammation and cancer, we performed a proof-of-concept experiment to study the effect of the pro-inflammatory cytokine interferon α 2 (IFNα-2b; hereafter IFN-I) on HAP1 cells. HAP1 cells were derived from a chronic myeloid leukaemia (CML) patient and have been extensively used in different translational studies [34-36]. IFN-I is a cytokine secreted by most cells in response to viral infection, and with known immunostimulatory and anti-cancer properties [37]. The IFN-I subtype IFNα2 has been approved, alone and in combination with tyrosine kinase inhibitors (TKIs), for the treatment of CML [38].
Here, we found that IFN-I affected intra- and extracellular concentrations of 2-Arachidonoylglycerol (2-AG), the intracellular concentration of Arachidonic Acid (AA), and the extracellular concentrations of Prostaglandin F2α (PGF2α), 5-Hydroxyeicosatetraenoic acid (5-HETE), Palmitic acid (PA, C16:0) and Stearic acid (SA, C18:0), suggesting a role of these lipids in the inflammatory state of tumours.
2. Results
A representative group of eicosanoids and fatty acids (Figure 1a) was selected based on their inflammatory roles and potential regulation by IFN-I. We focused on improving the identification and separation of lipid standards (chromatographic separation), their ionisation (source parameters), and quantitation by developing and implementing a dMRM method. We also applied this method to biological samples, where we optimised the amount of starting material, the lipid extraction method, and determined the specific matrix effects upon each lipid in two sample types (cells and cell supernatant), in Figure 1b.
2.1. Liquid chromatography-dynamic multiple reaction monitoring-mass spectrometry (LC-dMRM-MS) method optimisation for bioactive lipid standards
Analysis of lipid standards (Table 1) was first performed in MS1 mode to detect the corresponding precursor ions. Following tandem mass spectrometry (MS/MS; MS2), an extracted ion chromatogram of the most intense fragment ion was used for lipid assignment. The MS2 spectra was then compared to either acquired or predicted MS2 spectra in the Human Metabolome Database (HMDB) for confirmation of identity, by using one to three fragment ions as a reference, Figure 2a. Once discovered, the retention time was then used to assign the retention time window in the dMRM acquisition parameters.
Implementing dMRM increases the ion of interest’s response by removal of interfering signals (noise), and acquiring data on specific product ions only when the corresponding precursor ion is present. dMRM also improves dwell times during data acquisition by applying a retention time window that restricts when data is obtained for specific transitions, resulting in superior signal-to-noise (S/N) ratios. Non-overlapping ion transmissions also optimises dwell time and therefore sensitivity. dMRM transitions of lipid standards (Table 1) were identified using the Agilent MRM optimiser software, which applies a collisional energy (CE) ramp and suggests transitions based on the abundance and the mass-to-charge ratio (m/z) values. Manual checking of these transitions was performed, as the ion implied to have the highest abundance by the software, (the quantitation ion, the “quant ion”) did not always align with the ion with the actual greatest response. By this method, we have identified up to four dMRM transitions that can be used as quant and qualifier ions (“qual ions”) (Figures 2b, 2c and Table 1). The “quant ion” is used for quantification, and the other three ions (“qual ions”) are used to corroborate identification. Analysis of lipids in the presence of ammonium acetate (which is our mobile phase modifier) can create ammonium adduct ions of lipids. In our methodology, an example of this phenomenon is the precursor ion of 5-HETE, which was an ammonium adduct with a m/z of 338.3. Structures of all lipid standards analysed and the corresponding proposed quant ion structures are shown in Figure S1.
Chromatographic separation of lipids reduces the complexity of biological samples and decreases matrix effects. The chromatogram, in Figure 2d, displays the dMRM transitions for an equimolar ratio of all the lipids profiled. On the x-axis is the elution time (counts vs. acquisition time, also known as the retention time), and on the y-axis is the response (counts, AUC = area under curve). Not all responses are the same for all the lipids, even when at the same on-column concentration, as lipids are ionised at different efficiencies. Interestingly, it was not possible to chromatographically separate the regio-isomers species PGD2 and PGE2 reliably or sufficiently enough to determine their individual concentrations, with this particular methodology. The results reported here are to be interpreted as the concentration of one, the other or both those metabolites combined. Otherwise, our method has sufficiently adequate separation, preventing overlapping MRMs, which results in longer scan times for each MRM, increasing the points per peak for each lipid. In the case of the co-eluting compounds, for example SA and PGF2α, compound specific precursor and product ions allows for their accurate identification.
We observed that the retention time for certain lipids could have a slight variation, at different column loadings, therefore the dMRM retention time window in the analysis method (used to trigger data acquisition for the respective transition at that particular time) was set wider for those lipids. In addition, peak shape can also change at different column loadings, with shoulders or tails on some peaks, therefore we have included these in our analysis. For some lipids, lower on-column concentrations appeared to result in jagged, not smooth, peaks; therefore, it was necessary to add smoothing during data processing. To further improve the ionisation efficiency of lipids, all MS parameters were adjusted to an optimal value (Table 2). For example, we found that a source gas temperature of 80 °C was optimal for identification of intact lipids by MS2, due to a lower degree of in-source fragmentation, allowing for identification of fairly intact lipids. However, for dMRM analysis, the most optimal precursor to product ion responses were obtained at a source gas temperature of 280 °C. Once the optimal MS parameters were determined, we subsequently validated the robustness and reproducibility of our optimized method by intraday variation analysis, whereby lipid standards were combined and analysed in technical triplicate at two concentrations, 0.3 and 3 pmol on-column, respectively (Figure 3). The coefficient of variation (CV) was calculated between the mean of technical replicates at both concentrations. The observed CVs, for all the lipids are lower than 20 %, and lower than 5 % for most lipids (six out of nine lipid standards), highlighting the repeatability of this method.
2.2. Bioactive lipid analysis in biological samples
To enhance the extraction of lipids from cells, three different liquid-liquid extraction (LLE) techniques, were tested (Figure 4a). To cells the bioactive lipid mix (BLM, see section 4.4. Standard solutions), containing all the lipid standards of interest, was added in the extraction buffer to yield a final concentration of 10 pmol/L on column. We established that Extraction 1 was the most optimal extraction method for PGD2, and Extraction 3 was optimal for extracting 2-AG, AA and LTC4 (Figure 4b and Table S1a). However, Extraction 2 was ideal for the majority of metabolites (5-HETE, 5-oxo-ETE, PA, PGF2α, and SA) (Figure 4b and Table S1a) and therefore it was the method of choice for lipid extraction from cells in subsequent experiments.
Metabolite extraction from the media which the cells were cultured (the cell supernatant) was performed in a similar manner to metabolite extraction from cells, with the exception that steps 1 and 2 were combined (Figure 5a). We determined that Extraction 1 was the most optimal extraction method for 2-AG and LTC4, and Extraction 2 was most optimal for extraction of 5-HETE, AA, and 5-oxo-ETE (Figure 5b and Table S1b). Extraction 3 resulted in the extraction of all lipids, with the highest extraction efficiency for four of the nine lipids analysed (PA, PGF2α, PGD2/E2 and SA), and therefore it was the method of choice for lipid extraction from cell supernatant samples in subsequent experiments
We subsequently determined the matrix effect for cell and cell supernatant samples (see section 4.8. Data analysis), to evaluate whether the complex biological mixture enhances or suppresses ionization for each lipid, by calculating a corresponding Matrix RF value (Figure 6), which ranged from 0.34 to 40.06 for cells and 0.07 to 4.81 for cell supernatants (Table S2). The high degree of variation in Matrix RF values for each lipid highlights the importance of determining a Matrix RF for every individual lipid analysed, in each specific sample type.
2.3. Analysis of the bioactive lipidome after IFN-I treatment in cancer cells
To validate our methodology, we decided to profile the nine lipids of interest in a CML-derived cell line, upon treatment with IFNα-2b; IFN-I treatment resulted in significant changes of the intracellular lipidome in cell samples (Figure 7a and 7b; Table S3c and S3d), as well as the extracellular lipidome in cell supernatant samples (Figure 7a and 7c). We observed a decrease in the intra- and extra- cellular concentrations of 2-AG upon IFN-I treatment, and a similar decrease was seen in intracellular concentrations of AA, which was in line with literature published data [39]. Although we cannot confidently say whether this was due to a decrease in the synthesis of these lipids or an increase in their degradation, their increased conversion into downstream lipid metabolites upon IFN-I treatment is another plausible mechanism. An increase in the extracellular levels of PGF2α was observed, and the intracellular concentrations of 5-HETE did not change upon IFN-I treatment. This suggests 2-AG and AA may be used to generate 5-HETE, in the presence of IFN-I. There was no significant change in LTC4 in cells or in the cell supernatant, indicating no major changes in the synthesis or usage of this lipid. Intracellular amounts of PA and SA were not affected. However, extracellular amounts of PA and SA increased significantly. This would suggest an increase in the synthesis and secretion of these fatty acids. 5-oxo-ETE, PGD2 and PGE2 were not identified in either cell or cell supernatant fractions. Nonetheless, this method can be used to confidently detect minute changes for all these lipids (even if the changes are not statistically significant).
3. Discussion
The primary objective of this article was to establish the importance of investigating the appropriate extraction technique for the specific sample type (e.g. cells or cell supernatant), in addition to considering the matrix effect of every lipid analysed within the specific sample type, and applying the methodology to unravelling the effect of interferon treatment upon the lipidome in the context of inflammation, cancer and specifically CML. We have used HAP1 cells as a model for CML, as HAP1 cells are derived from a CML patient cells and have the BCR-ABL gene, which is common in leukaemia’s, turning the myeloid cell into a chronic myeloid cell [40, 41].
Due to intrinsic molecular differences, innate variation and organic solvent preferences during extraction, there is no agreement on the best sample preparation and analysis of lipids. As a consequence, analysing different subclasses simultaneously is challenging. In this study, we have optimised extraction, chromatography and MS parameters which provide the best conditions for the identification and quantitation of a set of eicosanoids and fatty acids with well-established roles in inflammation.
Solid-phase extraction (SPE) methods, can be used to reduce the complexity, and to concentrate the samples prior to analysis. However, we found that SPE is unreliable for concentrating a variety of lipids simultaneously, resulting in the loss of some metabolites (unpublished observations). Therefore, we opted to optimise LLE methods, and concentrate samples subsequently by resuspension of the dried lipids into smaller volumes. Due to differences in natural abundance of these metabolites, and the MS response, we were able to dilute the sample for those lipids with a higher natural concentration and/or a higher ion intensity (such as intracellular 2-AG). A separate method could be implemented for the analysis of 2-AG, LTC4, and PGF2α, as even at low on-column concentrations as these lipids have a large AUC. For instance, the standards and samples could be diluted further than reported here. Pre-analytical influences may change lipid concentrations or matrices which is why we prefer to use extensive matrix-dependent normalisations, for each analyte within a “true” matrix (e.g. not a surrogate matrix), and demonstrate its application tailored to specific endogenous lipid (eicosanoids and fatty acids) detection in clinically relevant biological samples. However, to bypass normalisation calculations, to provide information on the regulatory network and to identify important metabolite conversions within the pathway, ratios between metabolites could be used instead (e.g. PGE2:PGF2α). In this analysis, there is variability in response curves for the lipids (R2 values, from 0.950 to 0.999; Table S5), and variation in the matrix effects (RF values from 0.34 to 40.06), highlighting the need to determine the responses for every lipid quantified, as the results would otherwise vary significantly from the observed amounts. Nonetheless, the reproducibility of detection for our method achieved technical CV values of less than 20% and were within 5-10% for most species (Figure 3). Our intraday variation for all lipids analysed was below 20% CV, with 5-oxo-ETE, SA, 2-AG, PA, and 5-HETE being below 5%.
Recommended options for future development of this analysis could include chemical derivatization of the lipids, exploring an analysis in negative mode electrospray ionization (ESI-), using an ultrafast LC, and a complementary proteomics analysis. Derivitisation has been implemented for enhanced lipidomic analysis by MS [42], and it may boost the MS response for some lipids. However, it would need to be considered that derivatization itself is laborious, time-consuming, different metabolites have differing derivatization efficiencies, and batch effects are common. It has also been described that lipids with carboxylic acid moieties, will benefit from being analysed in negative polarity [43, 44]. However, this may not be suitable for the simultaneous analysis of all the lipids reported here. For instance, and in line with our observations, ionisation of LTC4 has been already shown to be very efficient in positive polarity [45]. MS fast-switching between polarity modes could potentially further improve sensitivity of detection. It is important to consider that PUFAs are easily oxidized and this may lead to erroneous concentration reporting for some lipids. Using an ultrafast-LC could reduce the total analysis time, increasing throughput, reducing the cost per sample and labour time (samples would not need to be added daily). In addition, sample quality could be improved as they would not need to be stored as long in the autosampler. Integrating complementary proteomics data would lead to greater understanding and enhance confidence of the changes observed within the studied pathways. For instance, changes in the expression of some of the enzymes involved in the metabolism of eicosanoids and fatty acids have been associated with cancer development [46-49]. Pinpointing which enzymes are modified upon interferon treatment, using proteomics, may corroborate our findings and expand our knowledge of the effect of interferon treatment in CML patients.
The pro-inflammatory tumour microenvironment [50-54], and immune ‘hot’ tumours show increased levels of inflammation and anticancer immune responses, such as T cell infiltration and release of pro-inflammatory cytokines, including interferons. Defects in Interferon signaling pathways during cancer treatment have been linked to immunotherapy resistance [55]. Our results suggest that IFN-I treatment significantly reduces the intracellular concentration of the eicosanoid precursors 2-AG and AA. However, the underlying molecular mechanism is as yet still unknown. We hypothesise that this reduction may be through decreased production of their precursors (2-AG-LPA or DAG), a decreased presence or activity of the respective enzymes which produce 2-AG-LAP and DAG (for example; 2-LPAP or DAGL) or by increased metabolism to downstream eicosanoids (such as PGF2α). The latter seems to be the case as both, intracellular, and extracellular concentration of PGF2α are increasing. This may suggest that cells treated with IFN-I are secreting excess PGF2α out of the intracellular space. Increased concentrations of PGF2α have been indicated as a driver of particular cancers [56-58], although its role in CML remains unclear. Secretion of SA and PA also appear increased upon IFN-I exposure, possibly through enhanced export. These fatty acids could contribute to the tumour microenvironment, act as key metabolites in reducing tumorigenicity, and affect the interplay between tumour and immune cells. 5-oxo-ETE, PGD2 and PGE2 were not identified in the samples, indicating either their absence, that they fell below the lower limit of detection (LLOD, a signal-to-noise ratio of >3), and/or the matrix caused considerable ionisation suppression. Alternatively, perhaps there was unexpected in-source fragmentation, leading to differing parent or product ions from those that we monitored. 5-oxo-ETE could be rapidly metabolised to either 5-HETE or to Di-endoperoxidase [59, 60], and therefore with the current treatment of 36 hours, it is very likely that the changes, if happening upon IFN-I treatment, will not be observed.
We were unable to chromatographically separate standards of the isobaric species PGD2 and PGE2. However, separation is possible though, with a method specifically dedicated to the analysis of these particular lipids. Employing normal-phase chiral chromatography, and/or alternative MS approaches (employing additional separation techniques, such as ion mobility), will isolate isobaric prostaglandins based on their chirality, mobility, and/or collisional cross section value. PGD2 can undergo dehydration to form Prostaglandin J2, and other Prostaglandin metabolites, with dehydration of PGD2 being accelerated in the presence of serum albumin [61-63]. Since our tissue culture media contains serum albumin, it might be contributing to the absence of PGD2 in our analysis. Previous studies have suggested that PGE2 is reduced in CML patients [64], hence this may be why our method was unable to detect PGE2 in samples. Upon IFN-treatment, intracellular production of 5-HETE appears to be maintained, possibly through metabolism from AA, or by the reduction of 5-oxo-ETE (and NADP+) to 5-HETE (and NADPH) [60]. In addition, the extracellular quantity of 5-HETE significantly increases, which to our knowledge is novel biology and an area of CML research yet to be explored. Changes in LTC4 amount and activity, and LTC4 concentration are cell specific [65-67]. With CML researchers suggesting that there can be a steady state, an increase or a decrease in Leukotrienes in patients compared to healthy controls. Nonetheless, despite these disparities, inhibitors of Leukotriene signaling have reduced cancer growth in CML [68, 69]. When these Leukotriene inhibitors have been used in combination with traditional methods to treat CML (Tyrosine kinase inhibitors), the reduction of tumour growth was further increased [70]. Interestingly, our results, although not significant, show a slight decrease in intracellular LTC4 levels upon IFN-I stimulation. However, Leukotrienes can be readily metabolised [71], with LTC4 potentially being converted to Leukotriene D4, therefore we cannot confirm that LTC4 concentrations are affected or not by 36 hours of IFN-I treatment.
In conclusion, the present study describes an original, ultra-sensitive methodology, whereby LC and MS parameters are modified for a specific set of lipids, the extraction of these lipids from different sample types is modified accordingly and extensive matrix considerations, both in terms of the matrix effect upon each individual lipid and the matrix effect of the different sample types (cells and cell supernatant) are applied. for accurate and simultaneous analysis of a range of eicosanoids and fatty acids. Our optimization of sample preparation, technical parameters and data normalization for different sample types, plus the application of dMRM transitions maximises the sensitivity of detection, allowing detection at endogenous levels, even for very low abundance species. The observed improvements in detection and sensitivity are the hallmark of this analytical method, permitting us to resolve changes of relevant bioactive lipids in response to treatment with a pro-inflammatory cytokine in a cancer cell model. Translational application examples include the profiling of bioactive lipids in patient samples, for monitoring inflammation levels caused by pathology, or to treatment with immunodulators (i.e. immunotherapy). In closing, we believe this emphasises the benefits of using targeted MS in understanding pathophysiological states, to unravel yet unknown mechanisms of diseases.
4. Materials and Methods
4.1. Abbreviations
2-AG; 2-Arachidonoylglycerol, 2-AG-LPA; 2-Arachidonoyglycerol lysophosphatidic acid, 2LPAP; 2-Lysophosphatidic acid phosphatase, 5-HETE; 5-Hydroxyeicosatetraenoic acid, 5-HPETE; 5-hydroperoxyeicosatetraenoic acid, 5-oxo-ETE; 5-Oxo-eicosatetraenoic acid, AA; Arachidonic acid, ACN: Acetonitrile, AUC; area under the curve, BLM; bioactive lipid mix, CBR1; Carbonyl reductase 1, CE; collisional energy, CHCl3; chloroform, CML; chronic myeloid leukaemia, COX 1/2; Cytochrome C oxidase 1 or 2, CV; coefficient of variation, DAGL; Diacylglycerol lipase, dMRM; dynamic multiple-reaction monitoring, ESI; electrospray ionization, eV; electron volts, FBS; fetal bovine serum, GPX; Glutathione peroxidase, H2O; water, HMDB; Human Metabolome Database, IFNα; Interferon alpha, IMDM; Iscove's Modified Dulbecco's Medium, IPA; isopropanol, LC-MRM-QQQ; Liquid chromatography- multiple reaction monitoring- triple quadrupole, LC-MS; liquid chromatography-mass spectrometry, LLE; liquid-liquid extraction, LLOD; lower limit of detection, LLOQ; lower limit of quantification, LOX; Lysyl Oxidase, LTA4; Leukotriene A4, LTC4; Leukotriene C4, LTC4S; Leukotriene C4 synthase, LTC4S; LTC4 synthase, m/z; mass to charge ratio, MAGL; Monoacylglycerol lipase, MeOH; Methanol, MRMHR; multiple reaction monitoring high-resolution, MTBE; Methyl tert-butyl ether, NADP+ / NADPH; nicotinamide adenine dinucleotide phosphate, NT; non-treated, PA; Palmitic acid, PGD2; Prostaglandin D2, PGDS; Prostaglandin D2 synthase, PGE2; Prostaglandin E2, PGES; Prostaglandin E2 synthase, PGF2α; Prostaglandin F2α, PUFA’s; polyunsaturated fatty acids, QC; quality control, QTOF; quadrupole time of flight, RF; response factor, SD; standard deviation, S/N; signal-to-noise ratio, SA; Stearic acid, SPE; solid-phase extraction, TIMS-TOF; Trapped ion mobility – time of flight, UPLC-MS/MS; ultra-performance liquid chromatography - tandem mass spectrometry.
4.2. Figures
Figures 1a, 1b, 4a, 5a, 6 and 7d were generated in Biorender.
4.3. Reagents
Lipid standards; PGE2 (catalogue # sc-201225), PGF2α (catalogue # sc-201227), 5-HETE (catalogue # sc-205136), 5-oxo-ETE (catalogue # sc-203783) and PGD2 (catalogue # sc-201221) were purchased from Santa Cruz Biotechnology. SA (catalogue # 10011298-500 mg-CA), LTC4 (catalogue # 20210-25 ug-CAY) and PA (catalogue # 10006627-10 g-CAY) were acquired from Cayman Chemicals. AA (catalogue # ab120916) is from Abcam. 2-AG (catalogue # A8973) was purchased from Sigma. LC-MS grade water (Catalogue # 115333) and MeOH (Catalogue # 106035) was available from Merck. Amm onium acetate (Catalogue # 10365260) and IPA (Catalogue # 15686670) were from Fisher Scientific. MTBE was acquired from Acros organics (Catalogue # 3787 20010) and ACN was from Honeywell Riedel-de Haën (Catalogue # 348512.5L). IMDM was from Gibco at Thermo Fisher Scientific (reagent # 12440053) and IFN-I was from Biotechne R&D systems (catalogue # 11105-1).
4.4. Standard solutions
Stock concentrations of all lipid standards were prepared as individual aliquots at 100 µmol/L in 100% MeOH. These stocks were then pooled and serially diluted to a concentration of 100 nmol/L in 50% (v/v) MeOH to create an equimolar standard mixture, the bioactive lipid mix (BLM). The BLM was used as a spike, which was added to pooled samples (a fraction of all samples combined) to determine the matrix effects.
4.5. Tissue culture
HAP1 cells were cultured as previously described [72]. In brief, cells were cultured in IMDM media containing 10% FBS and were grown until 60-70% confluency. Cells were then treated with 1000 U/mL of IFNα-2b for 36 hours, until ~80% confluent. The surrounding media (the cell supernatant) was collected and used for analysis of extracellular lipids. Both cell supernatant and cell plates were stored at -80 °C until lipid extraction.
4.6. Lipid extraction
A simultaneous lysis and LLE was performed. For cells, the lysis was performed by scraping with 100% MeOH. The lysate was then added to 50% CHCl3 and 50% MBTE, which was then vortexed briefly and then spun at 12 rpm for 20 minutes at 4 °C. The sample was then centrifuged at 17 x G for 5 minutes at 4 °C and the resulting organic fraction was removed and stored on ice. The remaining pellet underwent another round of vortexing, spinning and centrifuging in 50% CHCl3 and 50% MTBE with the resulting organic fraction being pooled with the previous fraction. The final extraction step was performed with 50% MeOH and the sample was vortexed, spun and centrifuged again with the organic fraction being pooled with the previous fractions. Once all organic fractions were collected for each sample, a small portion of each was removed and pooled together for a pooled sample. The samples and the pooled samples were then dried in a speedvac at 30 °C until dry and then stored at -20 °C until LC-MS analysis.
To the dried lipid extracts 50% MeOH or the same volume of the BLM was added prior to analysis. The samples were briefly vortexed and then shaken at 400 rpm at 4 °C until homogenous. Cell-supernatant samples were diluted 1 in 20. Cell samples were diluted 1 in 10 for analysis of AA, PGF2α, SA and 5-HETE or diluted 1 in 100 for analysis of 2-AG, PA and LTC4. Diluted samples were then transferred to LC-MS autosampler vials.
4.7. LC-MS method
Levels of 2-AG, AA, PGD2, PGE2, PGF2α, 5-HETE, 5-oxo-ETE, LTC4, PA and SA were analysed. Metabolites were quantified using an optimised dMRM method on a triple quadrupole mass spectrometer with a JetStream ESI source (Agilent 6490) coupled to a 1290 Agilent LC system.
Lipids were separated on an ACQUITY UPLC BEH C18 column (1.7 µm, 100 x 2.1 mm i.d., Waters) with mobile phase A of 2 % IPA with 5 mM Ammonium Acetate, and mobile phase B of 100 % IPA with 5 mM Ammonium Acetate at 40 °C. The flow rate was set to 0.21 mL min-1 and sample injection volume was 1 µL, 10 µL or 15 µL (depending of the response of the metabolites). The following gradient (% mobile phase B) was used: 0 - 1.5 min at 50% B, 1.5 - 9 min 70% B, 9 - 13 min 100% B. A wash with 100% mobile phase B and a wash with 100% mobile phase A was performed to clean the column before re-equilibration to starting conditions. The autosampler was maintained at 4 °C.
The following ESI+ source parameters were used: gas temp at 280 °C, gas flow 14 L/min, nebulizer at 20 psi, sheath gas temp at 250 °C, sheath gas flow at 11 L/min, capillary voltage 3000 V, nozzle voltage 1500 V, high-pressure RF at 150 V and low-pressure RF at 60V. Transitions used in the dMRM analysis are shown in Table 1, LC and MS parameters used are shown in Table 2.
4.8. Data analysis
Initial data processing was performed using Agilent MassHunter Quantitative software. Post-processing was performed in Excel and GraphPad Prism:
The MRM AUC’s were corrected with the matrix response factor (matrix RF) (Figure 6). To determine the matrix RF, the response of the analyte as a standard is needed, as is the response in the sample (the intrinsic contribution, the matrix) and the response of the standard mixture spiked into the matrix. To calculate the matrix RF, the response of the intrinsic contribution in the sample (the matrix) is subtracted from the response of the standard spiked into the matrix, to give the effect of the matrix upon the standard (response of the standard mixture in the matrix). The response of the standard by itself divided by the response of the standard mixture in the matrix gives a matrix response factor.
To account for matrix effects in biological samples, the response of an analyte in a (biological) sample is multiplied by the corresponding matrix RF value to give the ‘real’ response of the analyte (i.e., to eliminate ionization enhancement/suppression effects on an analyte). This matrix RF corrected response can subsequently be used to calculate analyte concentration from a standard curve trendline equation (y = Mx+C). Standard curves are generated by injection of increasing concentrations of the BLM, calculating the AUC at each concentration, and plotting AUC values against the corresponding concentration
Statistical analysis of the two sample (or unpaired) t-test is used to validate difference between no treatment (NT) and treated with IFNα (+IFNα) for 36 hours. A p-value less than 0.05 (***) indicates the results have highly significant differences.
4.9. Method validation
The LLOD was calculated as a S/N ratio of >3, the lower limit of quantification (LLOQ) was a S/N ratio of >10. Intraday precision was calculated using three replicates of two concentrations over the course of one day and results are reported as CV % between replicates of one concentration. To monitor instrument performance over time and check that batches that span inter-day analysis were consistent (quality control, QC), the BLM (1 pmol/L on-column) was routinely injected. If total sample analysis time was over multiple days, the samples were briefly vortexed at the start of each day to avoid precipitation.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org. Figure S1: Chemical structures of lipids analysed and their proposed product structures. The mass-to-charge (m/z) ratios for the intact structures and product ion structures are shown next to the molecule. Figure S2: Response (calibration) curve for PGF2α. An increasing concentration of standard (from 100 zmol/L to 50 fmol/L) is injected and the response (AUC), for the quantitation ion increases. The response is plotted against the concentration (in fmol/L). Table S1a: LLE of lipids from cells using three different methods (extractions). BLM was added before extracting to yield a final concentration of 10 pmol/L on column. The mean, SD, standard error of the mean (SEM) and the CV of biological replicates (3 replicates) has been calculated for each lipid in the BLM. Table S1b: LLE of cell supernatant from cells using three different methods (extractions). BLM was added before extracting to yield a final concentration of 10 pmol/L on column. The mean, SD, standard error of the mean (SEM) and the CV of biological replicates (3 replicates) has been calculated for each lipid in the BLM. Table S2: Calculating the matrix effect: the values used in this experiment. Table S3a: This table shows the concentration in cell samples, for each biological and technical replicate. The concentration units are above each column. Table S3b: Results of cell samples. The mean concentration, SD and the number of samples (N) is used to calculate the p-value. NT samples are compared to IFNα treated samples. Table S3c: Results of cells. Biological replicates: Samples are in biological triplicate. Each biological replicate is run in technical triplicate. From this the mean, SD (std.dev) and the CV % has been calculated for biological replicates. Table S3d: Results of cells. Technical replicates: Each biological replicate is run in technical triplicate. From this the mean, SD (std.dev) and the CV % has been calculated for technical replicates. Table S4a: This table shows the concentration in cell supernatant samples, for each biological and technical replicate. The concentration units are above each column. Table S4b: Results of cell supernatant samples. The mean concentration, SD and the number of samples (N) is used to calculate the p-value. Table S4c: Results of cell supernatants. Biological replicates: Samples are in biological triplicate. Each biological replicate is run in technical triplicate. From this the mean, SD (std.dev) and the CV % has been calculated for biological replicates. Table S4d: Results of cell supernatants. Technical replicates: Each biological replicate is run in technical triplicate. From this the mean, SD (std.dev) and the CV % has been calculated for technical replicates. Table S5: Calibration curves (response curves) are generated using the response of the product ion. On the X axis is the concentration, on the Y axis is the response (AUC). The equation of the linear regression is y=Mx+C, and the accuracy of the trendline is shown by the R2.
Author Contributions
Conceptualization and writing—original draft preparation, H.C.S., B.M.K and A.P.F.; methodology, validation, formal analysis, visualization, H.C.S.; software and data curation, H.C.S. and Z.Y.; investigation, H.C.S., S.D.D. and A.P.F.; resources, supervision, project administration and funding acquisition, B.M.K. and A.P.F.; writing—review and editing, H.C.S., S.D.D., Z.Y., B.M.K. and A.P.F. All authors have read and agreed to the published version of the manuscript.
Funding
H.C.S.: S.D.D., Z.Y., B.M.K. and A.P.F. are funded by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Science (CIFMS), China (grant nr - 2018-I2M-2-002). H.C.S., S.D.D., B.M.K. and A.P.F. are also funded by Pfizer Inc.
Data Availability Statement
Data has been submitted to the public repository metabolomeXchange (Metabolights), reference MTBLS7875.
Acknowledgments
Many thanks to Alan Scott for reviewing the text prior to publication. We would also like to thank Dr. Paolo Spingardi (UCL Business Ltd) and Dr. Ashvina Segaran (Ludwig Cancer Research, University of Oxford) for their assistance in the operation and maintenance of the triple quad mass spectrometer, and for their helpful insights into method optimisation.
Conflicts of Interest
“The authors declare no conflict of interest.”
References
- Wang, C.; Yang. J.; Zhang, X. Editorial: The Role of Bioactive Lipids in Homeostasis and Pathology. Front Physiol 2021, 12, 773632. [CrossRef]
- Minhas, P.S.; Latif-Hernandez, A.; McReynolds, M.R.; Durairaj, A.S.; Wang, Q.; Rubin, A.; Joshi, A.U.; He, J.Q.; Gauba, E.; Liu, L.; Wang, C.; Linde, M.; Sugiura, Y.; Moon, P.K.; Majeti, R.; Suematsu, M.; Mochly-Rosen, D.; Weissman, I.L.; Longo, F.M.; Rabinowitz, J.D.; Andreasson, K.I. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature 2021, 590(7844), 122-128. [CrossRef]
- Lindqvist, H.M.; Winkvist, A.; Gjertsson, I.; Calder, P.C.; Armando, A.M.; Quehenberger, O.; Coras, R.; Guma, M. Influence of Dietary n-3 Long Chain Polyunsaturated Fatty Acid Intake on Oxylipins in Erythrocytes of Women with Rheumatoid Arthri-tis. Molecules 2023, 28(2), 717. [CrossRef]
- Zhou, Y.J.; Wang, J.H.; Li, L.; Yang, H.W.; Wen, D.L.; He, Q.C. Expanding expression of the 5-lipoxygenase/leukotriene B4 pathway in atherosclerotic lesions of diabetic patients promotes plaque instability. Biochem Biophys Res Commun 2007, 363(1), 30-6. [CrossRef]
- Cipollone, F.; Mezzetti, A.; Fazia, M.L.; Cuccurullo, C.; Iezzi, A.; Ucchino, S.; Spigonardo, F.; Bucci, M.; Cuccurullo, F.; Pres-cott, S.M.; Stafforini; D.M. Association between 5-lipoxygenase expression and plaque instability in humans. Arterioscler Thromb Vasc Biol 2005, 25(8),1665-70. [CrossRef]
- Zu, L.; Guo, L.; Zhou, B.; Gao, W. Relationship between metabolites of arachidonic acid and prognosis in patients with acute coronary syndrome. Thromb Res 2016, 144, 192-201. [CrossRef]
- Chang, N.; Wu, C.; Chen, D.; Yeh, C.; Lin, C. J High levels of arachidonic acid and peroxisome proliferator-activated receptor-alpha in breast cancer tissues are associated with promoting cancer cell proliferation. Nutr Biochem 2013, 24(1), 274-81. [CrossRef]
- Rolland, P.H.; Martin, P.M.; Jacquemier, J.; Rolland, A.M.; Toga, M. Prostaglandin in human breast cancer: Evidence suggesting that an elevated prostaglandin production is a marker of high metastatic potential for neoplastic cells. J Natl Cancer Inst 1980, 64(5), 1061-70. [CrossRef]
- Sipka, S.; Szántó, S.; Szucs, K.; Kovács, I.; Kiss, E.; Antal-Szamás, P.; Lakos, G.; Aleksza, M.; Illés, A.; Gergely, P.; Szegedi, G. J Decreased arachidonic acid release in peripheral blood monocytes of patients with systemic lupus erythematosus. Rheu-matol. 2001, 28(9), 2012-7.
- Prüss, H.; Rosche, B.; Sullivan, A.B.; Brommer, B.; Wengert, O.; Gronert, K.; Schwab, J.M. Proresolution lipid mediators in multiple sclerosis - differential, disease severity-dependent synthesis - a clinical pilot trial. PLoS One 2013, 8(2), e55859. [CrossRef]
- Sacerdoti, D.; Balazy, M.; Angeli, P.; Gatta, A.; McGiff, J.C. Eicosanoid excretion in hepatic cirrhosis. J Clin Invest 1997, 100(5), 1264-70. [CrossRef]
- Martín-Masot, R.; Galo-Licona, J.D.; Mota-Martorell, N.; Sol, J.; Jové, M.; Maldonado, J.; Pamplona, R.; Nestares, T. Up-Regulation of Specific Bioactive Lipids in Celiac Disease. Nutrients 2021, 13(7), 2271. [CrossRef]
- Kathir, K.; Dennis, J.M.; Croft, K.D.; Mori, T.A.; Lau, A.K.; Adams, M.R.; Stocker, R. Equivalent lipid oxidation profiles in advanced atherosclerotic lesions of carotid endarterectomy plaques obtained from symptomatic type 2 diabetic and nondiabetic subjects. Free Radic Biol Med 2010, 49(3), 481-6. [CrossRef]
- Zakrzewski, J.T.; Barnes, N.C.; Piper, P.J.; Costello, J.F. Detection of sputum eicosanoids in cystic fibrosis and in normal saliva by bioassay and radioimmunoassay. Br J Clin Pharmacol 1987, 23(1), 19-27. [CrossRef]
- Minuz, P.; Jiang, H.; Fava, C.; Turolo, L.; Tacconelli, S.; Ricci, M.; Patrignani, P.; Morganti, A.; Lechi, A.; McGiff, J.C. Altered release of cytochrome p450 metabolites of arachidonic acid in renovascular disease. Hypertension 2008, 51(5), 1379-85. [CrossRef]
- Lundström, S.L; Levänen, B.; Nording, M.; Klepczynska-Nyström, A.; Sköld, M.; Haeggström, J.Z.; Grunewald, J.; Svarten-gren, M.; Hammock, B.D.; Larsson, B.; Eklund, A.; Wheelock, A.M.; Wheelock, C.E. Asthmatics exhibit altered oxylipin profiles compared to healthy individuals after subway air exposure. PLoS One 2011, 6(8), e23864. [CrossRef]
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Bioactive Lipids and Chronic Inflammation: Managing the Fire Within. Front Immunol 2018, 9, 38. [CrossRef]
- Amine, H.; Benomar, Y.; Taouis M. Palmitic acid promotes resistin-induced insulin resistance and inflammation in SH-SY5Y human neuroblastoma. Sci Rep 2021, 11(1), 5427. [CrossRef]
- Anderson, E.K.; Hill, A.A.; Hasty, A.H.; Stearic acid accumulation in macrophages induces toll-like receptor 4/2-independent inflammation leading to endoplasmic reticulum stress-mediated apoptosis. Arterioscler Thromb Vasc Biol, 2012, 32(7), 1687-95. [CrossRef]
- Guo, B.; Chen, B.; Liu, A.; Zhu, W.; Yao, S. Liquid chromatography-mass spectrometric multiple reaction monitoring-based strategies for expanding targeted profiling towards quantitative metabolomics. Curr Drug Metab 2012, 13(9), 1226-43. [CrossRef]
- Edwards, M.E.; De Luca, T.; Ferreira, C.R.; Collins, K.S.; Eadon, M.T.; Benson, E.A.; Sobreira, T.J.P.; Cooks, R,J. Multiple reaction monitoring profiling as an analytical strategy to investigate lipids in extracellular vesicles. J Mass Spectrom 2021, 56(1), e4681. [CrossRef]
- Valli, A.; Rodriguez, M.; Moutsianas, L.; Fischer, R.; Fedele, V.; Huang, H-L.; Van Stiphout, R.; Jones, D.; Mccarthy, M.; Vin-axia, M.; Igarashi, K.; Sato, M.; Soga, T.; Buffa, F.; Mccullagh, J.; Yanes, O.; Harris, A.; Kessler, B.M. Hypoxia induces a lipogenic cancer cell phenotype via HIF1α-dependent and -independent pathways. Oncotarget 2015, 6(4), 1920-41. [CrossRef]
- Cajka, T.; Fiehn, O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. Trends Analyt Chem 2014, 61, 192-206. [CrossRef]
- Kortz, L.; Dorow, J.; Ceglarek, U. Liquid chromatography-tandem mass spectrometry for the analysis of eicosanoids and related lipids in human biological matrices: a review. J Chromatogr B Analyt Technol Biomed Life Sci 2014, 964, 1-11. [CrossRef]
- Deems, R.; M W, Buczynski, M.W.; Bowers-Gentry, R.; Harkewicz, R.; Dennis, E.A. Detection and quantitation of eicosanoids via high performance liquid chromatography-electrospray ionization-mass spectrometry. Methods Enzymol. 2007;432:59-82. [CrossRef]
- Wang, Y.; Armando, A.M.; Quehenberger, O.; Yan, C.; Dennis E.A. Comprehensive ultra-performance liquid chromato-graphic separation and mass spectrometric analysis of eicosanoid metabolites in human samples. J Chromatogr A 2014, 1359, 60-9. [CrossRef]
- Lu, L.; Mai, Z.; Zhou, H.; Guan, W.; Wu, S.; Zou, H.; Shen, M.; Zhan, Y.; Ye, F.; Qiu, M.; Shen, L.; Zhao, B.; Yang, Z. Simulta-neous profiling and quantification of 25 eicosanoids in human serum by ultrahigh-performance liquid chromatography coupled to tandem mass spectrometry. Anal Bioanal Chem 2022, 414(29-30), 8233-8244. [CrossRef]
- Sorgi, C.A.; Peti, A.P.F; Petta, T.; Meirelles, A.F.G.; Fontanari, C.; de Moraes, L.A.B.; Faccioli, L.H. Comprehensive high-resolution multiple-reaction monitoring mass spectrometry for targeted eicosanoid assays. Sci Data 2018, 21;5, 180167. [CrossRef]
- Miller, T.M.; Poloyac, S.M.; Anderson, K.B.; Waddell, B.L.; Messamore, E.; Yao, J.K. A rapid UPLC-MS/MS assay for eico-sanoids in human plasma: Application to evaluate niacin responsivity. Prostaglandins Leukot Essent Fatty Acids 2018, 136, 153-159. [CrossRef]
- Raeven, P.; Hagn, G.; Niederstaetter, L.; Brugger, J.; Bayer-Blauensteiner, S.; Domenig, C.; Hoetzenecker, K.; Posch, M.; Leit-ner, G.; Gerner, C.; Baron, D.M. Red blood cell transfusion-related eicosanoid profiles in intensive care patients-A pro-spective, observational feasibility study. Front Physiol 2023, 14, 1164926. [CrossRef]
- Wang, D.; Pan, C,; Han, J.; Zhao, Y.; Liu, S.; Li, C.; Yi, Y.; Zhang, Y.; Tang, X.; Liang, A. Involvement of p38 MAPK/cPLA2 and arachidonic acid metabolic pathway in Shengmai injection-induced pseudo-allergic reactions. Journal of Ethnopharmacol-ogy 2023, 309, 116357. [CrossRef]
- Rustam, Y.H.; Reid, G.E. Analytical Challenges and Recent Advances in Mass Spectrometry Based Lipidomics. Anal Chem 2018, 90(1), 374-397. [CrossRef]
- Murphy, R.C. Challenges in Mass Spectrometry-based Lipidomics of Neutral Lipids.Trends Analyt Chem 2018, 107, 91-98. [CrossRef]
- Heisterkamp, N.; Stam, K.; Groffen, J.; de Klein, A.; Grosveld, G. Structural organization of the bcr gene and its role in the Ph' translocation. Nature 1985, 315(6022), 758-61. [CrossRef]
- Collins, S.J.; Kubonishi, I.; Miyoshi, I.; Groudine, M.T. Altered transcription of the c-abl oncogene in K-562 and other chronic myelogenous leukemia cells. Science 1984 225(4657),72-4. [CrossRef]
- Shen, H.; McHale, C.M.; Haider, S.I.; Jung, C.; Zhang, S.; Smith, M.T.; Zhang, L. Identification of Genes That Modulate Susceptibility to Formaldehyde and Imatinib by Functional Genomic Screening in Human Haploid KBM7 Cells. Toxicol Sci 2016, 151(1), 10-22. [CrossRef]
- Aricò, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers (Basel) 2019,11(12), 1943. [CrossRef]
- Paul, F.; Pellegrini, S.; Uzé, G. IFNA2: The prototypic human alpha interferon. Gene 2015, 567(2), 132-7. [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Ni-cholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nature Protocols 2013, 8(1), 17–33. [CrossRef]
- Heisterkamp, N.; Stam, K.; Groffen, J.; de Klein, A.; Grosveld, G. Structural organization of the bcr gene and its role in the Ph' translocation. Nature. 1985, 315(6022), 758-61. [CrossRef]
- Collins, S.J.; Kubonishi, I.; Miyoshi, I.; Groudine, M.T. Altered transcription of the c-abl oncogene in K-562 and other chronic myelogenous leukemia cells. Science. 1984, 225(4657), 72-4. [CrossRef]
- Sun, D.; Meng, X.; Ren, T.; Fawcett, J.P.; Wang, H.; Gu, J. Establishment of a Charge Reversal Derivatization Strategy to Improve the Ionization Efficiency of Limaprost and Investigation of the Fragmentation Patterns of Limaprost Derivatives Via Exclusive Neutral Loss and Survival Yield Method. J Am Soc Mass Spectrom 2018, 29, 1365-1375. [CrossRef]
- Kortz, L. Dorow, J.; Becker, S.; Thiery, J.; Ceglarek, U. Fast liquid chromatography-quadrupole linear ion trap-mass spectrometry analysis of polyunsaturated fatty acids and eicosanoids in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 2013, 927, 209-13. [CrossRef]
- Song, J.; Liu, X.; Wu, J.; Meehan, M.J.; Blevitt, J.M.; Dorrestein, P.C.; Milla, M.E. A highly efficient, high-throughput lipidomics platform for the quantitative detection of eicosanoids in human whole blood. Anal Biochem 2013, 433(2), 181-8. [CrossRef]
- Sanak, M.; Gielicz, A.; Bochenek, G.; Kaszuba, M.; Niżankowska-Mogilnicka, E.; Szczeklik, A. Targeted eicosanoid lipidomics of exhaled breath condensate provide a distinct pattern in the aspirin-intolerant asthma phenotype. J Allergy Clin Immunol 2011, 127(5), 1141-7.e2. [CrossRef]
- Guzman, M. A new age for MAGL. Chem Biol 2010, 17, 4–6. [CrossRef]
- Tuo, W.; Leleu-Chavain, N.; Spencer, J.; Sansook, S.; Millet, R.; Chavatte, P. Therapeutic potential of fatty acid amide hydrolase, monoacylglycerol lipase, and N-acylethanolamine acid amidase inhibitors. J Med Chem 2017, 60, 4–46. [CrossRef]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.; Cravatt, B.F.; Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140(1), 49-61. [CrossRef]
- Giles, F.J. Kantarjian, H.M. Bekele, B.N. Cortes, J.E.; Faderl, S.; Thomas, D.A.; Manshouri, T.; Rogers, A.; Keating, M.J.; Talpaz, M.; O'Brien, S.; Albitar, M. Bone marrow cyclooxygenase-2 levels are elevated in chronic-phase chronic myeloid leukaemia and are associated with reduced survival. Br J Haematol 2002, 119(1), 38-45. [CrossRef]
- Stenke, L.; Sjolinder, M.; Miale, T.D.; Lindgren, J.A. Novel enzymatic abnormalities in AML and CML in blast crisis: elevated leucocyte leukotriene C4 synthase activity paralleled by deficient leukotriene biosynthesis from endogenous substrate. British Journal of Haematology 1998, 101, 728–736. [CrossRef]
- Cacho-Diaz, B.; García-Botello, D.R.; Wegman-Ostrosky, T.; Reyes-Soto, G.; Ortiz-Sánchez, E.; Herrera-Montalvo, L.A. Tumor microenvironment differences between primary tumor and brain metastases. J Transl Med 2020, 18, 1. [CrossRef]
- Roma-Rodrigues, C., Mendes, R., Baptista, P. V.; Fernandes, A. R. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci 2019, 20(4), 840. [CrossRef]
- Belli, C.; Trapani, D.; Viale, G.; D'Amico, P.; Duso, B.A.; Vigna, P.D.; Orsi, F.; Curigliano, G.; Targeting the microenvironment in solid tumors. Cancer Treat Rev 2018, 65, 22–32. [CrossRef]
- Aricò, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers (Basel) 2019, 11(12), 1943. [CrossRef]
- Nakamura, K.; Smyth, M.J. Targeting cancer-related inflammation in the era of immunotherapy. Immunol Cell Biol 2017, 95, 325–332. [CrossRef]
- Kim, D.; Garza, L.A. A new target for squamous cell skin cancer? Exp. Dermatol 2015, 24, 14–15. [CrossRef]
- Pabst, T.; Kortz, L.; Fiedler, G.M.; Ceglarek, U.; Idle, J.R.; Beyoğlu, D. The plasma lipidome in acute myeloid leukemia at diagnosis in relation to clinical disease features. BBA Clin 2017, 7, 105-114. [CrossRef]
- Qualtrough, D.; Kaidi, A.; Chell, S.; Jabbour, H.N.; Williams, A.N.; Paraskeva, C. Prostaglandin F(2alpha) stimulates motility and invasion in colorectal tumor cells. Int J Cancer 2007, 121(4), 734-40. [CrossRef]
- Griesser, M.; Boeglin, W.E.; Suzuki, T.; Schneider, C. Convergence of the 5-LOX and COX-2 pathways: heme-catalyzed cleavage of the 5S-HETE-derived di-endoperoxide into aldehyde fragments. J Lipid Res 2009, 50(12), 2455-62. [CrossRef]
- Powell, W.S.; Gravelle, F.; Gravel, S.; Hashefi, M. Metabolism of 5(S)-hydroxyeicosanoids by a specific dehydrogenase in human neutrophils. J Lipid Mediat 1993, 6(1-3), 361-8.
- Bie, Q.; Dong, H.; Jin, C.; Zhang, H.; Zhang, B. 15d-PGJ2 is a new hope for controlling tumor growth. Am J Transl Res 2018, 10(3), 648–658.
- Fitzpatrick, F.A.; Wynalda, M.A. Albumin-catalyzed metabolism of prostaglandin D2. Identification of products formed in vitro.. J Biol Chem. 1983 Oct 10;258(19):11713-8. [CrossRef]
- Maxey, K.M.; Hessler, E.; MacDonald, J.; Hitchingham, L. The nature and composition of 15-deoxy-Delta(12,14)PGJ(2). Prostaglandins Other Lipid Mediat. 2000 Jun;62(1):15-21. [CrossRef]
- Parise, P.; Huybrechts, E.; Grasselli, S.; Falcinelli, F.; Nenci, G.G.; Gresele, P.; Vermylen, J. Generation of arachidonic acid metabolites from stimulated whole blood in patients with chronic myeloproliferative disorders. Acta Haematol. 1991;85(2):88-92. [CrossRef]
- Tornhamre, S.; Stenke, L.; Granzelius, A.; Sjölinder, M.; Näsman-Glaser, B.; Roos, C.; Widell, S.; Lindgren, J.A. Inverse relationship between myeloid maturation and leukotriene C4 synthase expression in normal and leukemic myelopoiesis-consistent overexpression of the enzyme in myeloid cells from patients with chronic myeloid leukemia. Exp Hematol 2003, 31(2), 122-30. [CrossRef]
- Sjölinder, M.; Stenke, L.;Näsman-Glaser, B.; Widell, S.; Doucet, J.; Jakobsson, P.J.; Lindgren, J.A.; Aberrant expression of active leukotriene C(4) synthase in CD16(+) neutrophils from patients with chronic myeloid leukemia. Blood 2000, 95(4), 1456-64. [CrossRef]
- Roos, C.; Sjölinder, M.; Stenke, L.; Tornhamre, S. Abnormal LTC4 synthase RNA degradation in neutrophils from CML patients. Br J Haematol 2004, 124(6), 739-45. [CrossRef]
- Zovko, A.; Yektaei-Karin, E.; Salamon, D.; Nilsson, A.; Wallvik, J.; Stenke, L. Montelukast, a cysteinyl leukotriene receptor antagonist, inhibits the growth of chronic myeloid leukemia cells through apoptosis. Oncol Rep. 2018 Aug;40(2):902-908. [CrossRef]
- Yektaei-Kari, E; Zovko, A.; Nilsson, A.; Näsman-Glaser, B.; Kanter, L.; Rådmark, O.; Wallvik, J.; Ekblom, M.; Dolinska, M.; Qian, H.; Stenke, L. Modulation of leukotriene signaling inhibiting cell growth in chronic myeloid leukemia. Leuk Lymphoma. 2017 Aug;58(8):1903-1913. [CrossRef]
- Sveinbjörnsson, B.; Rasmuson, A.; Baryawno, N.; Wan, M.; Pettersen, I.; Ponthan, F.; Orrego, A.; Haeggström, J.Z.; Johnsen, J.I.; Kogner, P. Expression of enzymes and receptors of the leukotriene pathway in human neuroblastoma promotes tumor survival and provides a target for therapy. FASEB J. 2008 Oct;22(10):3525-36. [CrossRef]
- Dahinden, C.A.; Clancy, R.M.; Gross, M.; Chiller, J.M.; Hugli, T.E. Leukotriene C4 production by murine mast cells: evidence of a role for extracellular leukotriene A4. Proc Natl Acad Sci USA 1985, 82(19), 6632-6. [CrossRef]
- Pinto-Fernandez, A.; Salio, M.; Partridge, T.; Chen, J.; Vere, G.; Greenwood, H.; Olie, C.S.; Damianou, A.; Scott, H.C.; Pegg, H.J.; Chiarenza, A.; Díaz-Saez, L.; Smith, P.; Gonzalez-Lopez, C.; Patel, B.; Anderton, E.; Jones, N.; Hammonds, T.R.; Huber, K.; Muschel, R.; Borrow, P.; Cerundolo, V.; Kessler, B.M. Deletion of the deISGylating enzyme USP18 enhances tumour cell antigenicity and radiosensitivity. British Journal of Cancer 2021, 124, 817–830. [CrossRef]
Figure 1.
Eicosanoid pathway and methodology workflow. (a) Metabolic pathways of selected bioactive lipids. Converting enzymes are in boxes and lipids in this study are circled in grey. De novo lipogenesis leading to fatty acid synthesis, which results in the elongation of fatty acids, produces Palmitic acid and Stearic acid. Exogenous fatty acid uptake is via transmembrane transporters. (b) Optimised experimental workflow for the analysis of eicosanoids and fatty acids using ultra-high-performance liquid chromatography-dMRM-MS (UPLC-dMRM-MS). Cells were cultured in IMDM media, containing 10% fetal bovine serum (FBS), and grown until 60-70% confluent. Cells were then treated with 1000 U/ml of IFNα-2b for 36 hours, reaching ~80% confluence. The surrounding media (the cell supernatant) was collected from the plates and cells were lysed. Lipid extraction from cells and cell supernatants was performed with Methanol/ Methyl Tert-Butyl Ether/ Chloroform (MeOH/MTBE/CHCl3), and acetonitrile (ACN), respectively. Lipid extracts were subsequently dried and reconstituted in 50% (v/v) MeOH (in H2O) prior to LC-MS analysis.
Figure 1.
Eicosanoid pathway and methodology workflow. (a) Metabolic pathways of selected bioactive lipids. Converting enzymes are in boxes and lipids in this study are circled in grey. De novo lipogenesis leading to fatty acid synthesis, which results in the elongation of fatty acids, produces Palmitic acid and Stearic acid. Exogenous fatty acid uptake is via transmembrane transporters. (b) Optimised experimental workflow for the analysis of eicosanoids and fatty acids using ultra-high-performance liquid chromatography-dMRM-MS (UPLC-dMRM-MS). Cells were cultured in IMDM media, containing 10% fetal bovine serum (FBS), and grown until 60-70% confluent. Cells were then treated with 1000 U/ml of IFNα-2b for 36 hours, reaching ~80% confluence. The surrounding media (the cell supernatant) was collected from the plates and cells were lysed. Lipid extraction from cells and cell supernatants was performed with Methanol/ Methyl Tert-Butyl Ether/ Chloroform (MeOH/MTBE/CHCl3), and acetonitrile (ACN), respectively. Lipid extracts were subsequently dried and reconstituted in 50% (v/v) MeOH (in H2O) prior to LC-MS analysis.
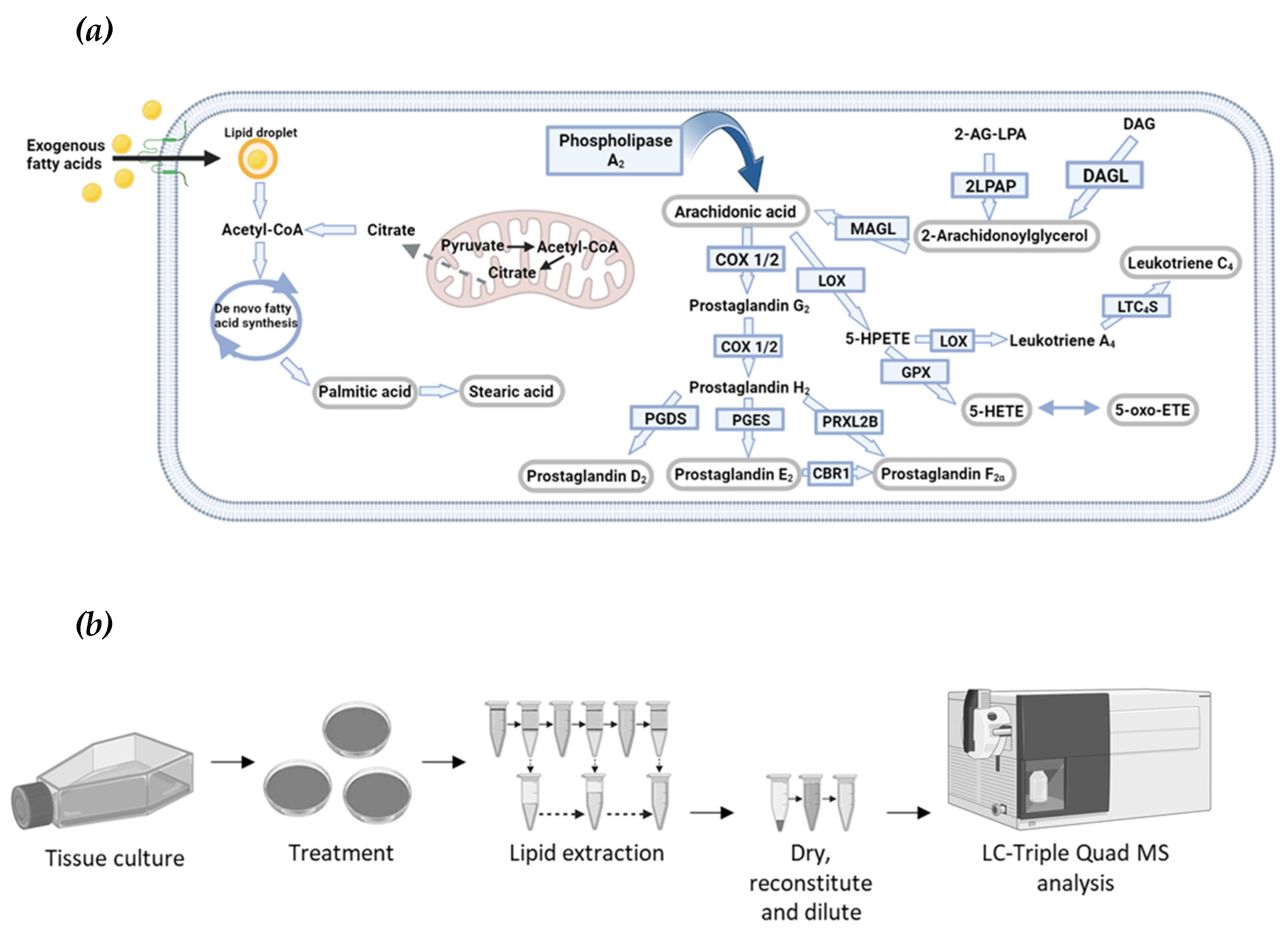
Figure 2.
Optimised eicosanoid and fatty acid profiling by targeted MS. (a) The MS2 ion spectra of PGF2α (molar mass of 354.48) using electrospray ionization in positive mode (ESI+), identifying the sodium adduct ([M+Na]+), potassium adduct ([M+K]+), dimer ([2M+H]+), and the sodiated dimer ([2M+Na]+) adduct of PGF2α. The fragments at m/z 319.2 and m/z 265.1, caused by in-source fragmentation, have formulas and structures as shown. (b) dMRM chromatogram of PGF2α. The sodium adduct (precursor ion) is exposed to a CE of 4 eV to obtain the product ion of m/z 360.3, 12 eV for product ion 342.2 m/z, 48 eV for product ion 57 m/z and a 64 eV for product ion 43.3 m/z. The product ion, 360.3 m/z is the most abundant species and is therefore used as the quant ion for determining concentration. (c) dMRM ion spectra of PGF2α. The precursor ion is indicated by the red star at m/z 377.2 The product ions are shown. (d) Product ion chromatograms of eicosanoid and fatty acid standards. Standards are in an equimolar ratio and an on-column concentration of 300 fmol/L. The chromatogram is not scaled to the largest in the chromatogram, which would make each peak have the same height (normalise the counts to the largest peak in the chromatogram). For each lipid, the most intense peak is the quant ion, the other three peaks are the respective qual ions.
Figure 2.
Optimised eicosanoid and fatty acid profiling by targeted MS. (a) The MS2 ion spectra of PGF2α (molar mass of 354.48) using electrospray ionization in positive mode (ESI+), identifying the sodium adduct ([M+Na]+), potassium adduct ([M+K]+), dimer ([2M+H]+), and the sodiated dimer ([2M+Na]+) adduct of PGF2α. The fragments at m/z 319.2 and m/z 265.1, caused by in-source fragmentation, have formulas and structures as shown. (b) dMRM chromatogram of PGF2α. The sodium adduct (precursor ion) is exposed to a CE of 4 eV to obtain the product ion of m/z 360.3, 12 eV for product ion 342.2 m/z, 48 eV for product ion 57 m/z and a 64 eV for product ion 43.3 m/z. The product ion, 360.3 m/z is the most abundant species and is therefore used as the quant ion for determining concentration. (c) dMRM ion spectra of PGF2α. The precursor ion is indicated by the red star at m/z 377.2 The product ions are shown. (d) Product ion chromatograms of eicosanoid and fatty acid standards. Standards are in an equimolar ratio and an on-column concentration of 300 fmol/L. The chromatogram is not scaled to the largest in the chromatogram, which would make each peak have the same height (normalise the counts to the largest peak in the chromatogram). For each lipid, the most intense peak is the quant ion, the other three peaks are the respective qual ions.
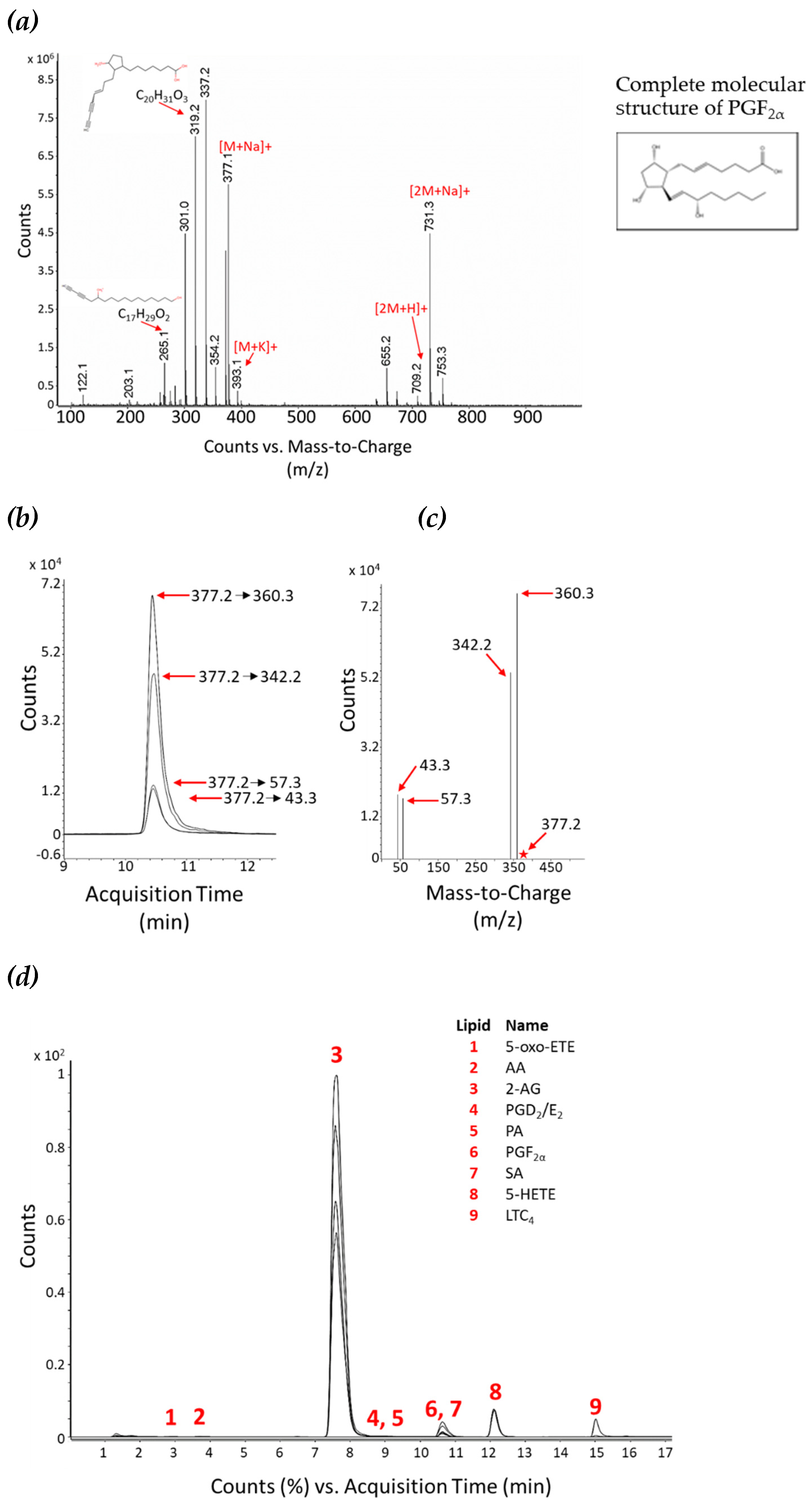
Figure 3.
Method intraday reproducibility. Lipid standards were combined and analysed in technical triplicate at two concentrations, 0.3 pmol (grey dots) and 3 pmol (black dots) on-column, respectively. The CV between technical triplicates was calculated (as %) for each standard, as a mean of the two concentrations (dashed line) or individually (dots).
Figure 3.
Method intraday reproducibility. Lipid standards were combined and analysed in technical triplicate at two concentrations, 0.3 pmol (grey dots) and 3 pmol (black dots) on-column, respectively. The CV between technical triplicates was calculated (as %) for each standard, as a mean of the two concentrations (dashed line) or individually (dots).
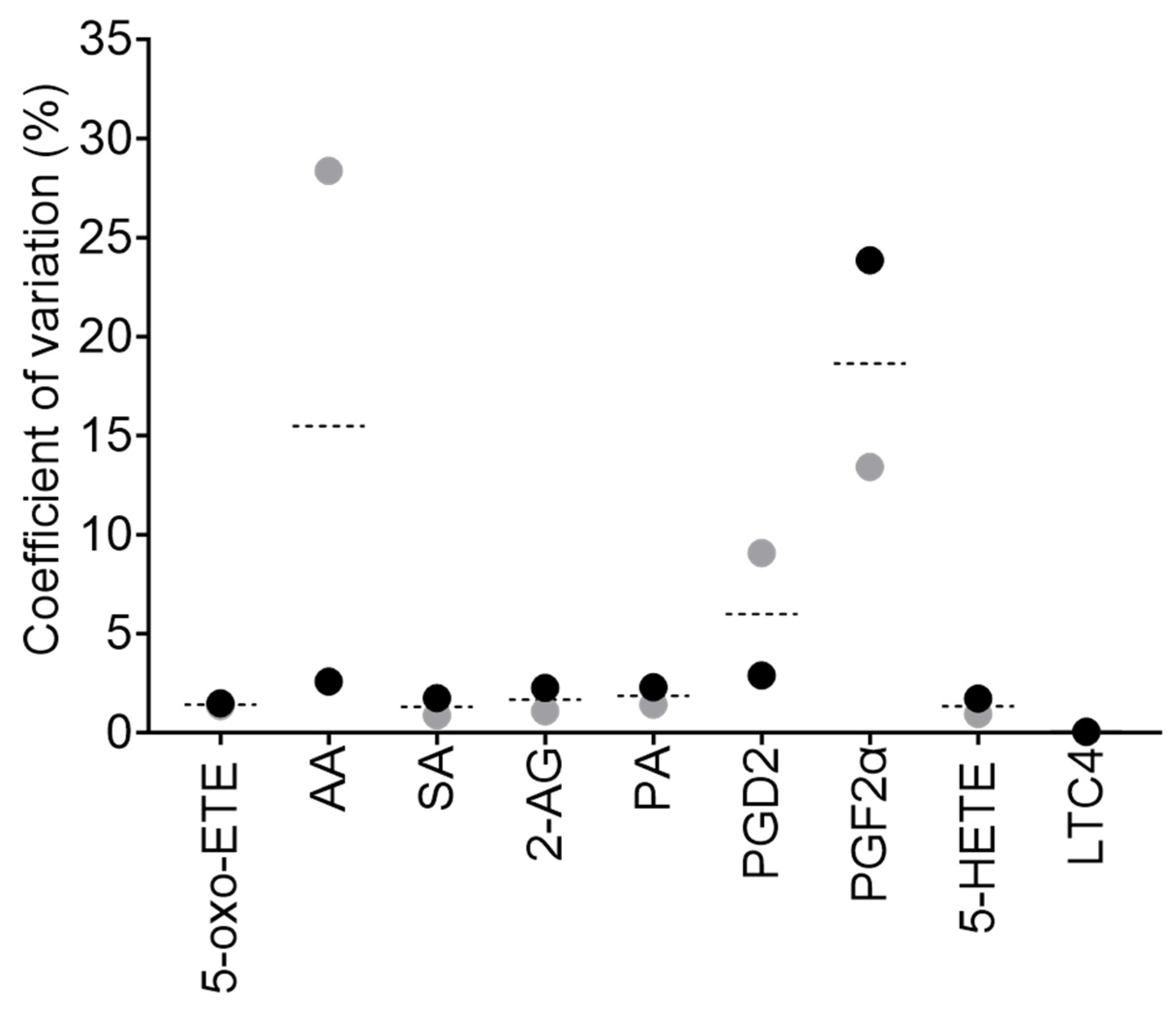
Figure 4.
Comparison of liquid-liquid extraction (LLE) methods for cellular lipids. (a) Three LLE techniques were tested. In liquid-liquid Extraction Method 1 cells are lysed with 50% (v/v) MeOH in H2O (step 1), followed by lipid extraction with MTBE (steps 2 and 3) and finally 50% (v/v) MeOH in H2O (step 4). In liquid-liquid Extraction Method 2 cells are lysed with 100% MeOH (step 1), followed by lipid extraction with MTBE:CHCl3 (1:1, v/v)(steps 2 and 3), and 50% (v/v) MeOH in H2O (step 4). In liquid-liquid Extraction Method 3 cell lysis and all lipid extraction steps are performed with 100% ACN. Three biological replicates were used for each technique. (b) The graph represents the mean of three biological replicates. Light grey bars are the results from Extraction Method 1, dark grey bars are results from Extraction Method 2, and black bars are from Extraction Method 3. Results are normalised. Normalisation parameters: 100% is defined as the largest mean in each data set and the results are given as percentages. For area under the curve (AUC) for each result (not normalised results), see Table S1a.
Figure 4.
Comparison of liquid-liquid extraction (LLE) methods for cellular lipids. (a) Three LLE techniques were tested. In liquid-liquid Extraction Method 1 cells are lysed with 50% (v/v) MeOH in H2O (step 1), followed by lipid extraction with MTBE (steps 2 and 3) and finally 50% (v/v) MeOH in H2O (step 4). In liquid-liquid Extraction Method 2 cells are lysed with 100% MeOH (step 1), followed by lipid extraction with MTBE:CHCl3 (1:1, v/v)(steps 2 and 3), and 50% (v/v) MeOH in H2O (step 4). In liquid-liquid Extraction Method 3 cell lysis and all lipid extraction steps are performed with 100% ACN. Three biological replicates were used for each technique. (b) The graph represents the mean of three biological replicates. Light grey bars are the results from Extraction Method 1, dark grey bars are results from Extraction Method 2, and black bars are from Extraction Method 3. Results are normalised. Normalisation parameters: 100% is defined as the largest mean in each data set and the results are given as percentages. For area under the curve (AUC) for each result (not normalised results), see Table S1a.
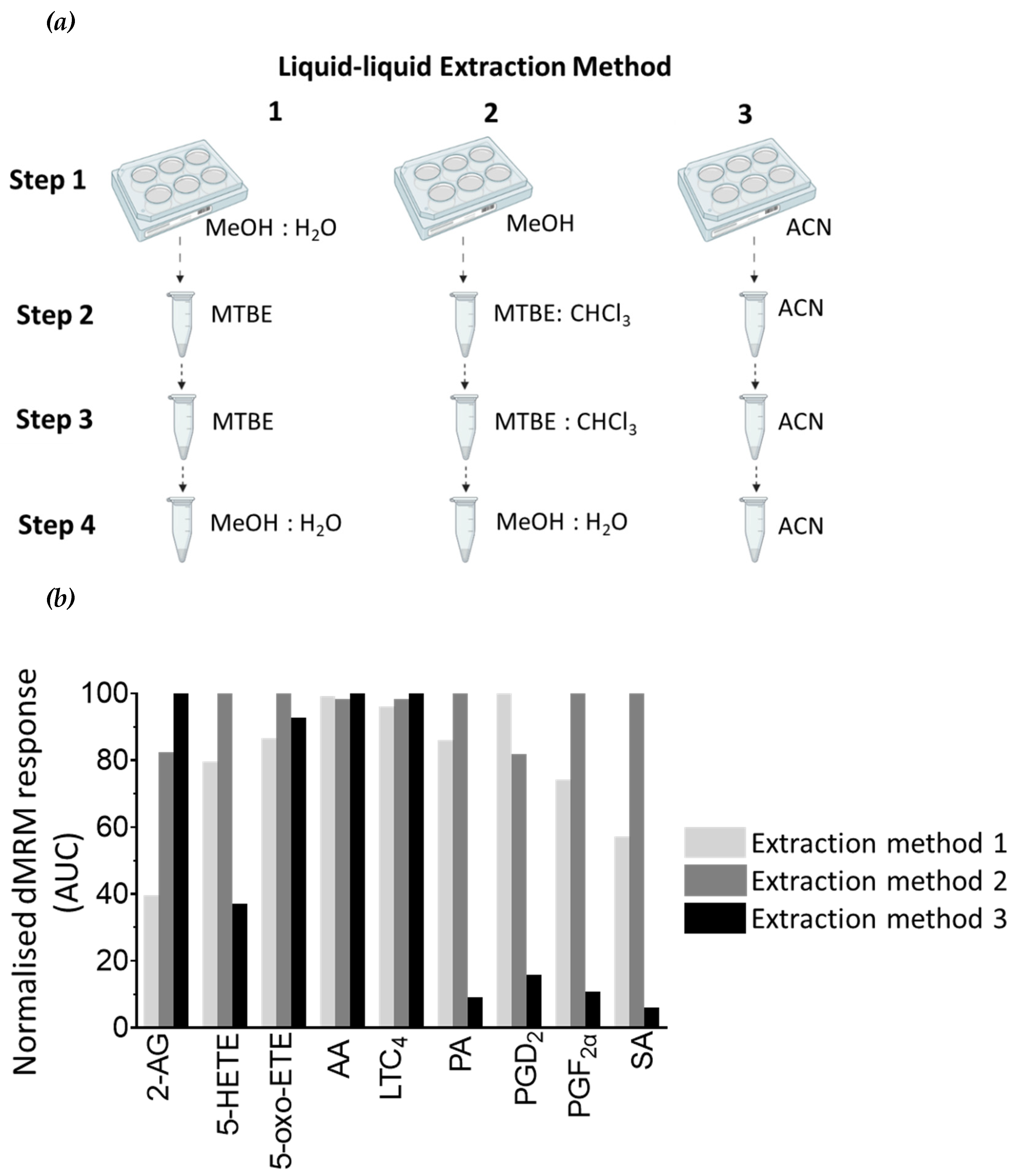
Figure 5.
Comparison of LLE techniques for secreted lipids in cell supernatant material. (a) Three LLE techniques were tested. In liquid-liquid Extraction Method 1 lipids are first extracted with MeOH:H2O:MBTE (1:1:2, v:v:v), followed by extraction with MTBE (step 2) and 50% (v/v) MeOH in H2O (step 3). In liquid-liquid Extraction Method 2 lipids are first extracted with MeOH:MTBE:CHCL3 (2:1:1, v:v:v), followed by extraction step with MTBE:CHCl3 (step 2), and 50% (v/v) MeOH in H2O (step 3). In liquid-liquid Extraction Method 3 all lipid extract steps are performed with 100% ACN. Three biological replicates were used for each technique. (b) The graph represents the mean of three biological replicates. Light grey bars are the results from Extraction Method 1, dark grey bars are results from Extraction Method 2, and black bars are from Extraction Method 3. Results are normalised. Normalisation parameters: 100% is defined as the largest mean in each data set and the results are given as percentages. For area under the curve (AUC) for each result (not normalised results), see Table S1b.
Figure 5.
Comparison of LLE techniques for secreted lipids in cell supernatant material. (a) Three LLE techniques were tested. In liquid-liquid Extraction Method 1 lipids are first extracted with MeOH:H2O:MBTE (1:1:2, v:v:v), followed by extraction with MTBE (step 2) and 50% (v/v) MeOH in H2O (step 3). In liquid-liquid Extraction Method 2 lipids are first extracted with MeOH:MTBE:CHCL3 (2:1:1, v:v:v), followed by extraction step with MTBE:CHCl3 (step 2), and 50% (v/v) MeOH in H2O (step 3). In liquid-liquid Extraction Method 3 all lipid extract steps are performed with 100% ACN. Three biological replicates were used for each technique. (b) The graph represents the mean of three biological replicates. Light grey bars are the results from Extraction Method 1, dark grey bars are results from Extraction Method 2, and black bars are from Extraction Method 3. Results are normalised. Normalisation parameters: 100% is defined as the largest mean in each data set and the results are given as percentages. For area under the curve (AUC) for each result (not normalised results), see Table S1b.
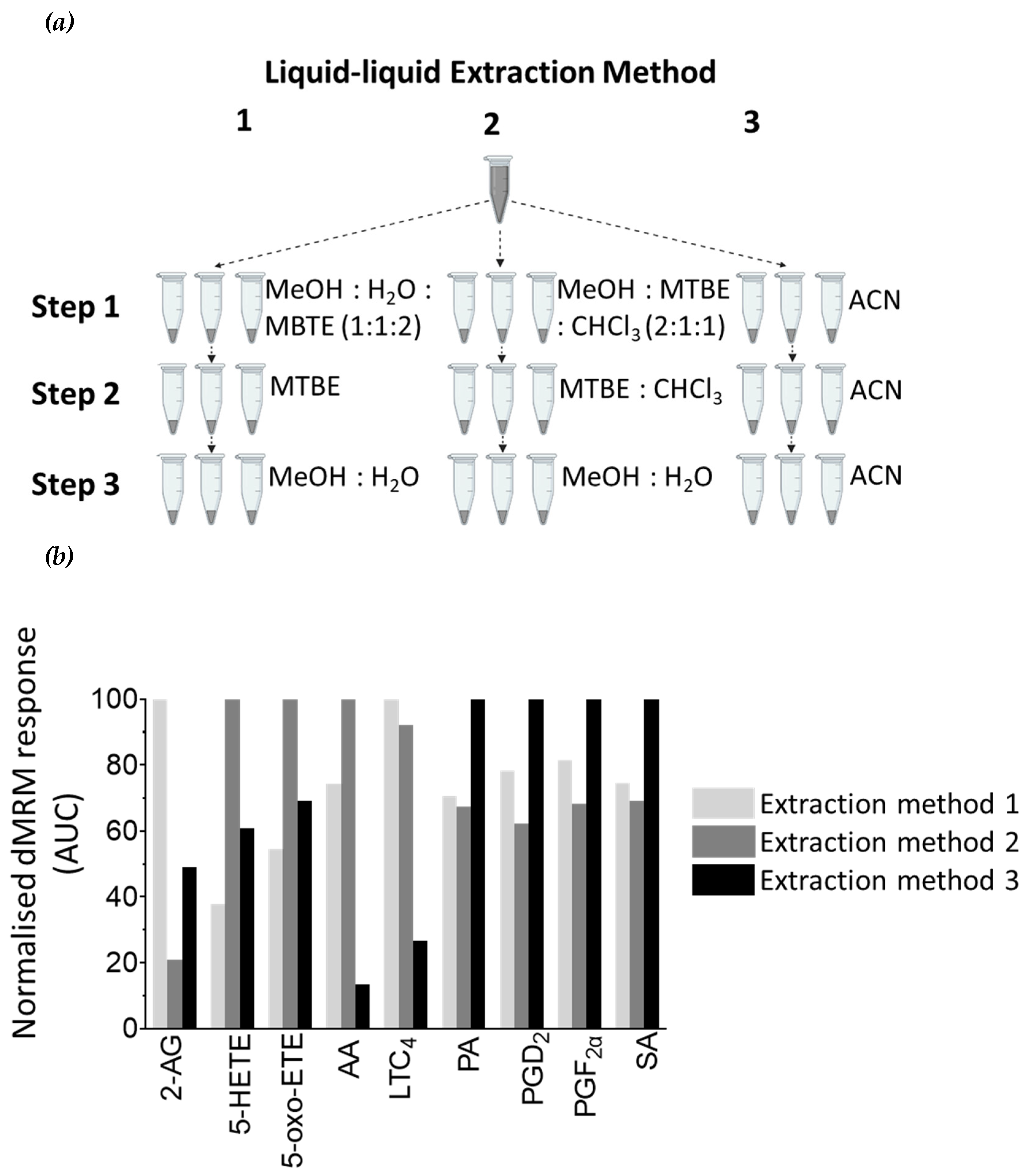
Figure 6.
Matrix effect on lipid detection. The workflow followed for determining the matrix effect for each lipid. Three types of samples must be prepared: a pool of all samples (matrix, yellow tube), a standard mixture (the BLM, red tube) and the standard mixture spiked into the matrix (grey tube). First, the intrinsic contribution of the lipid response from the matrix itself (yellow tube) is removed from the standard mixture spiked into the matrix (grey tube) by subtracting the MRM AUC for each metabolite found in the matrix. This calculation gives the BLM response when present in the matrix (orange tube). A matrix RF value is subsequently obtained by dividing the MRM AUC value for the standard mixture (red tube) by the MRM AUC value for the standard mixture when present in the matrix (orange tube). Numeric values for measured matrix RFs are provided in Table S2.
Figure 6.
Matrix effect on lipid detection. The workflow followed for determining the matrix effect for each lipid. Three types of samples must be prepared: a pool of all samples (matrix, yellow tube), a standard mixture (the BLM, red tube) and the standard mixture spiked into the matrix (grey tube). First, the intrinsic contribution of the lipid response from the matrix itself (yellow tube) is removed from the standard mixture spiked into the matrix (grey tube) by subtracting the MRM AUC for each metabolite found in the matrix. This calculation gives the BLM response when present in the matrix (orange tube). A matrix RF value is subsequently obtained by dividing the MRM AUC value for the standard mixture (red tube) by the MRM AUC value for the standard mixture when present in the matrix (orange tube). Numeric values for measured matrix RFs are provided in Table S2.
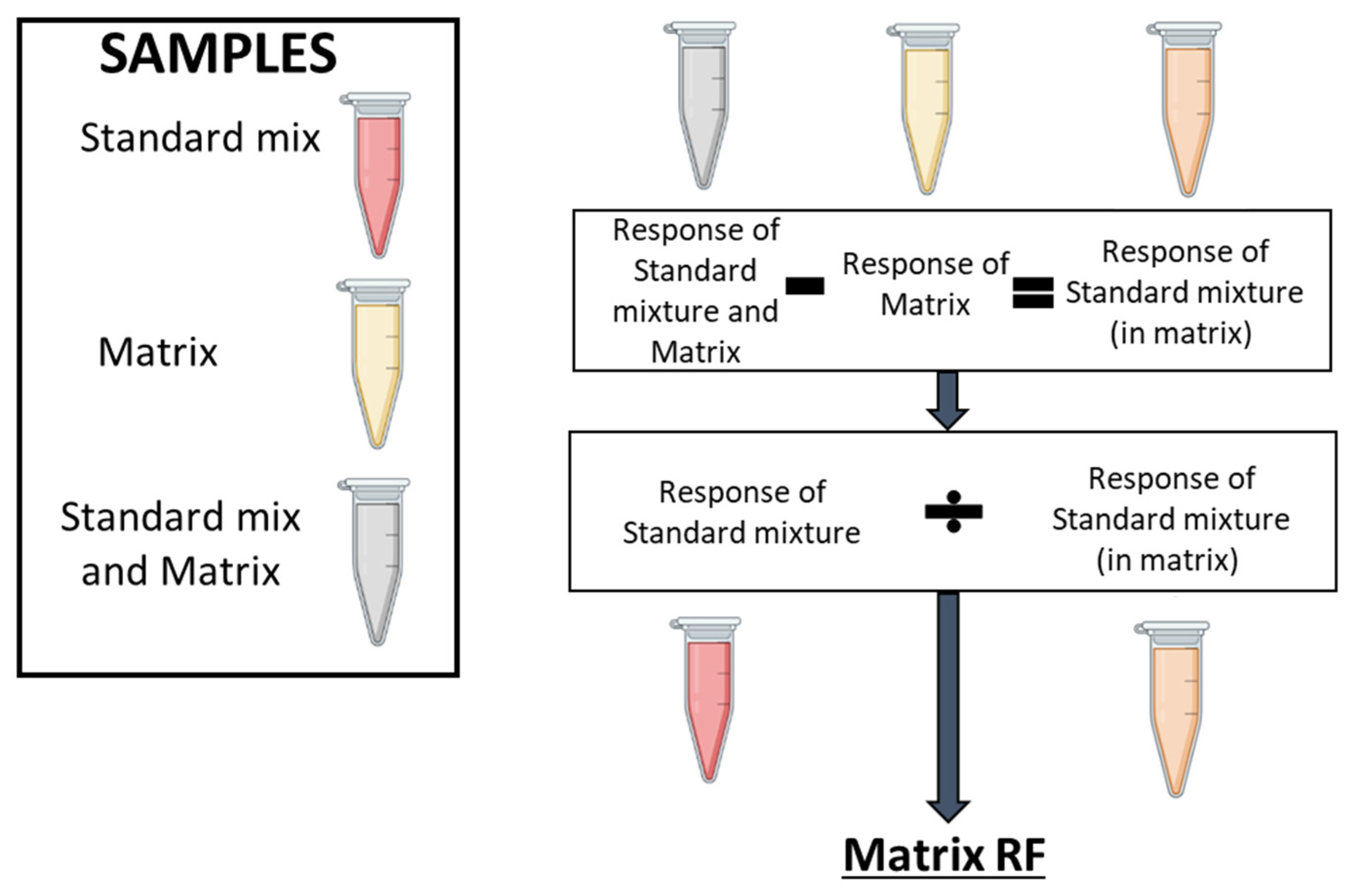
Figure 7.
Interferon alpha modulates eicosanoid and fatty acid pathways. (a) Analysis of bioactive lipids in HAP1 cells and cell supernatants after 36 hours treatment with IFN-I. (a) Graphs represent the mean with the standard error of the mean (SEM). Cell results are graphs on the left, the corresponding cell supernatant is on the right. Grey bars are for the not treated sample (NT) and grey with a white stripe pattern are results for IFN-I treated samples. The statistical analysis is the two sample (or unpaired) t-test. A p-value less than 0.05 suggests the results have highly significant differences. If there is no significance difference it is not labelled. (b) Mean percentage change of bioactive lipids in HAP1 cells, not treated (NT) and treated with IFNα for 36 hours (+ IFNα) (c) Cell supernatant from HAP1 cells, not treated (NT) and treated with IFNα for 36 hours (+ IFNα). Heat maps display normalized lipid abundances relative to the mean concentration of the nontreated samples (NT), which is represented by grey. For IFN-treatment, blue results are for a decrease in concentration, in percentage, grey is no change and red is for an increase in concentration in percentage. (d) Metabolic pathways of selected lipids after IFN-treatment. Converting enzymes are in boxes and lipids in this study are circled. Lipids circled in grey, have no change compared to non-treated cells, blue have decreased concentration compared to non-treated cells, lipids circled in red have an increased concentration compared to non-treated. Lipids discovered in the cell supernatant are outside of the cell lipid membrane. Lipids with a significant difference between non-treated and treated (a p-value of less than 0.05) are indicated by a yellow star.
Figure 7.
Interferon alpha modulates eicosanoid and fatty acid pathways. (a) Analysis of bioactive lipids in HAP1 cells and cell supernatants after 36 hours treatment with IFN-I. (a) Graphs represent the mean with the standard error of the mean (SEM). Cell results are graphs on the left, the corresponding cell supernatant is on the right. Grey bars are for the not treated sample (NT) and grey with a white stripe pattern are results for IFN-I treated samples. The statistical analysis is the two sample (or unpaired) t-test. A p-value less than 0.05 suggests the results have highly significant differences. If there is no significance difference it is not labelled. (b) Mean percentage change of bioactive lipids in HAP1 cells, not treated (NT) and treated with IFNα for 36 hours (+ IFNα) (c) Cell supernatant from HAP1 cells, not treated (NT) and treated with IFNα for 36 hours (+ IFNα). Heat maps display normalized lipid abundances relative to the mean concentration of the nontreated samples (NT), which is represented by grey. For IFN-treatment, blue results are for a decrease in concentration, in percentage, grey is no change and red is for an increase in concentration in percentage. (d) Metabolic pathways of selected lipids after IFN-treatment. Converting enzymes are in boxes and lipids in this study are circled. Lipids circled in grey, have no change compared to non-treated cells, blue have decreased concentration compared to non-treated cells, lipids circled in red have an increased concentration compared to non-treated. Lipids discovered in the cell supernatant are outside of the cell lipid membrane. Lipids with a significant difference between non-treated and treated (a p-value of less than 0.05) are indicated by a yellow star.
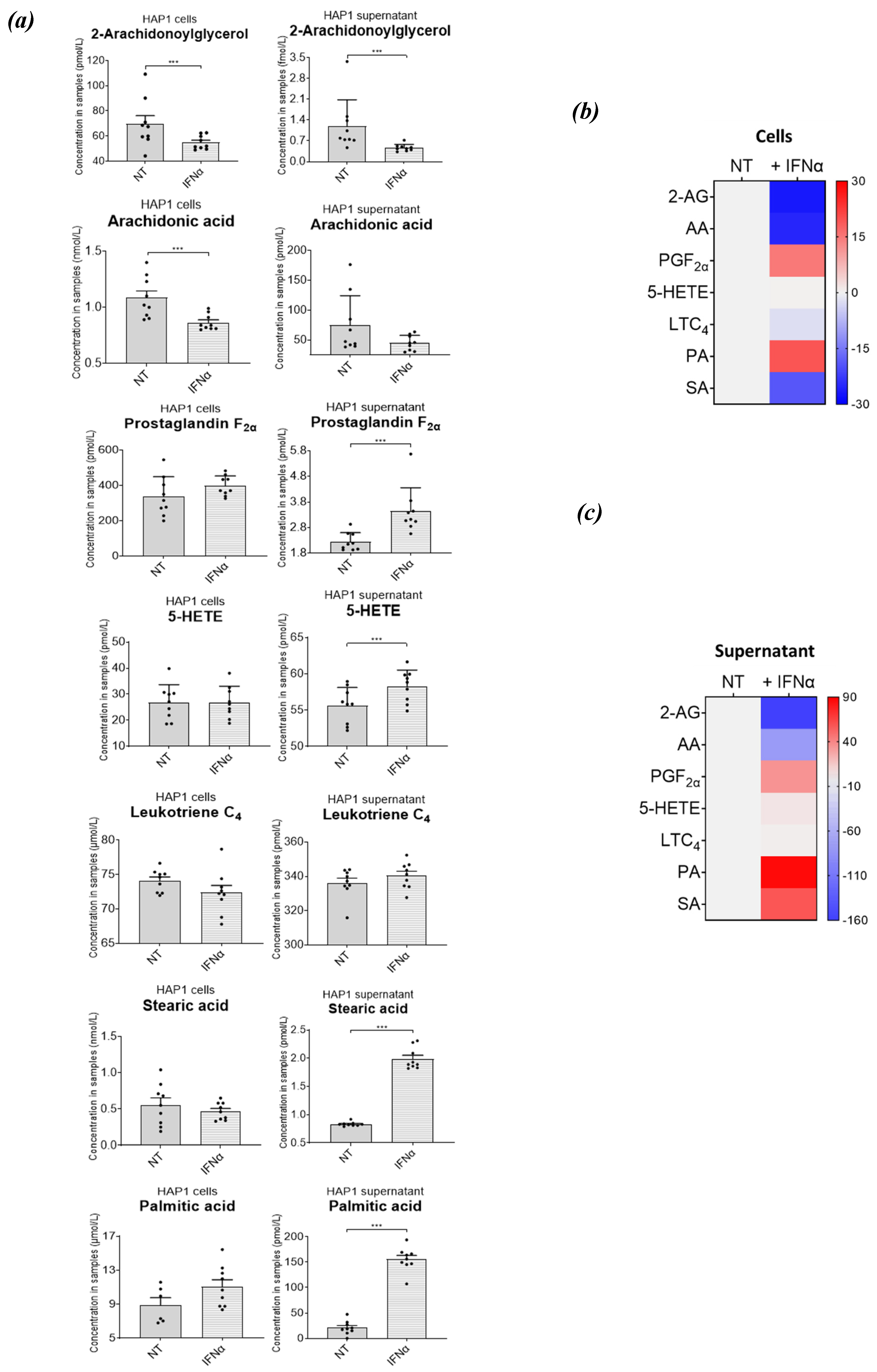
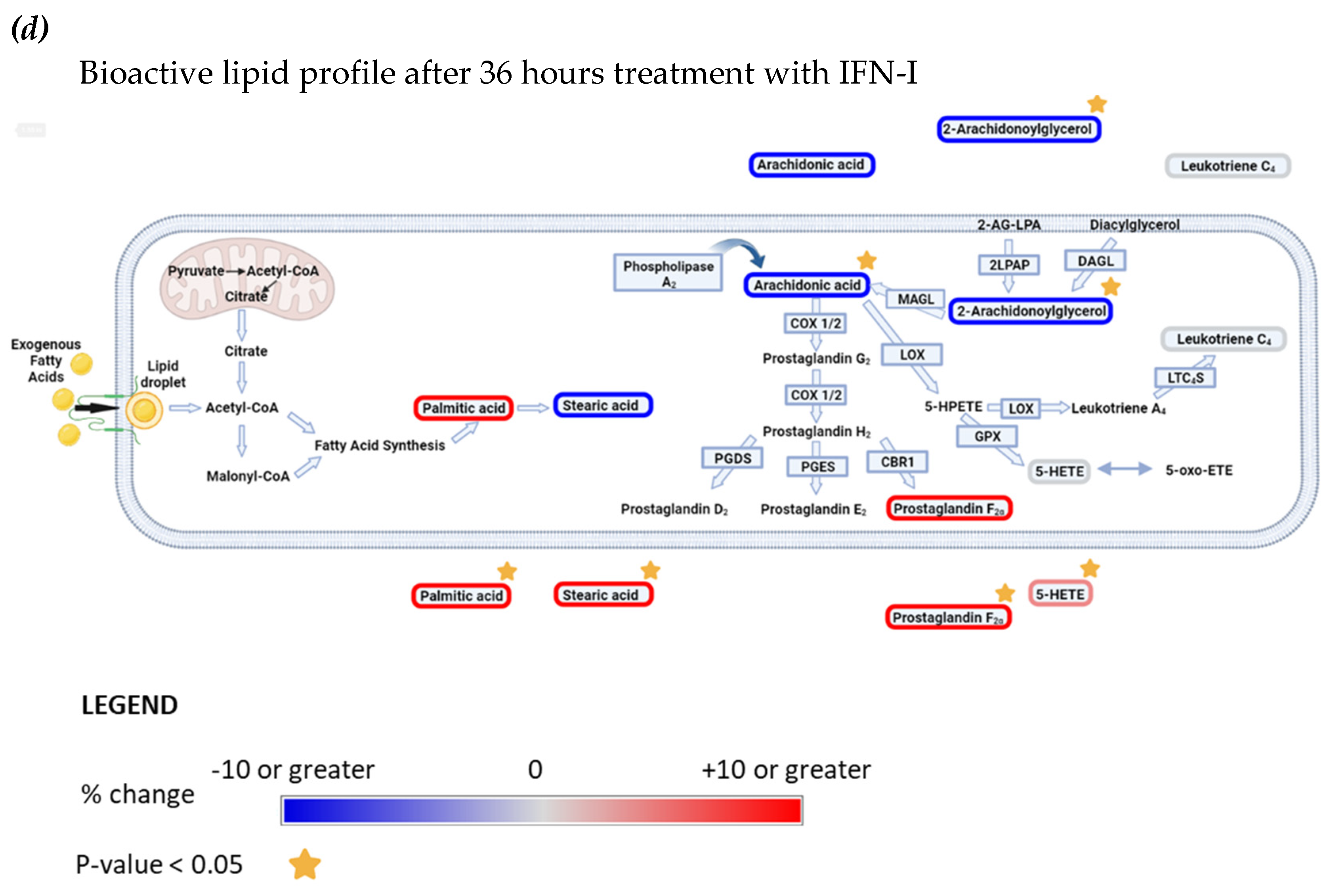

Table 1.
Optimised transitions for LC-dMRM-MS. Lipid names, dMRM parameters and lower limits of quantification (LLOQ) are listed for all eicosanoid and fatty acid standards analysed. The CE used from precursor to product ion transitions are indicated in electron volts (eV).
Table 1.
Optimised transitions for LC-dMRM-MS. Lipid names, dMRM parameters and lower limits of quantification (LLOQ) are listed for all eicosanoid and fatty acid standards analysed. The CE used from precursor to product ion transitions are indicated in electron volts (eV).
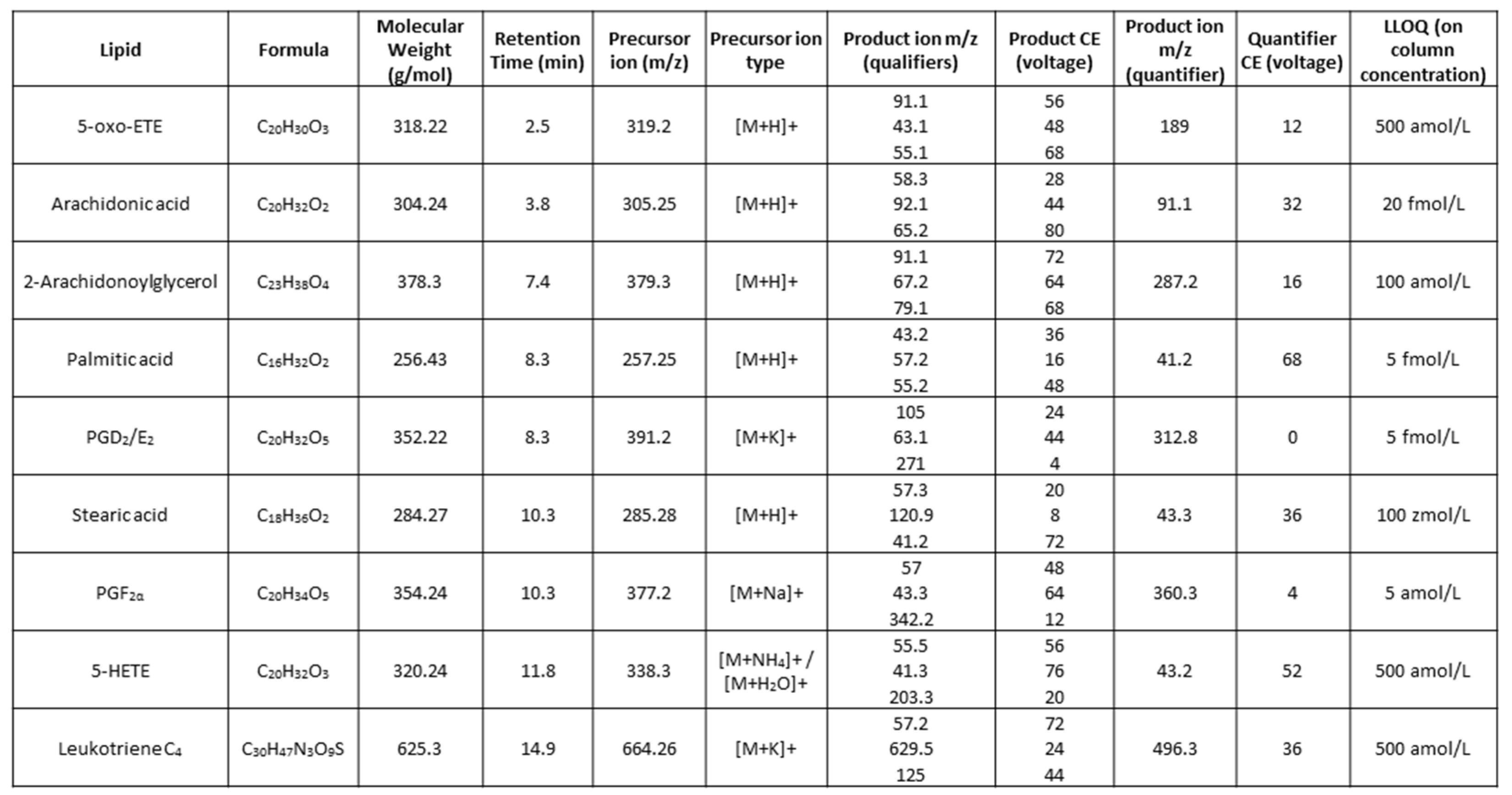 |
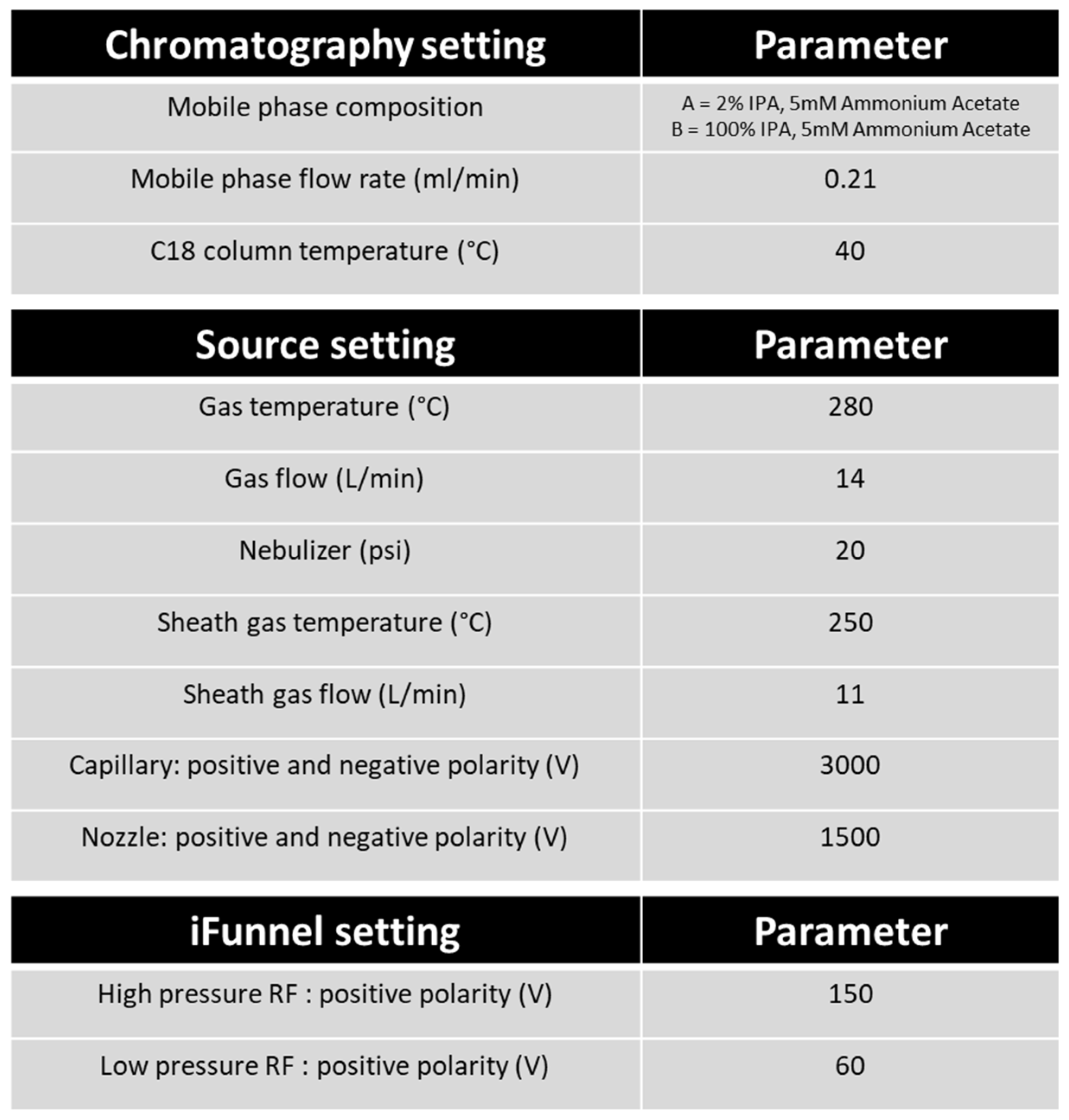
Table 2.
Chromatography and mass spectrometry parameters. This table represents the instrumentation settings used in the analysis method and parameters applied. Settings are selected for the most favourable separation and ionisation of eicosanoids and fatty acids.
Table 2.
Chromatography and mass spectrometry parameters. This table represents the instrumentation settings used in the analysis method and parameters applied. Settings are selected for the most favourable separation and ionisation of eicosanoids and fatty acids.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



