
Submitted:
20 July 2023
Posted:
20 July 2023
You are already at the latest version
Abstract
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy’s disease, is a debilitating neuromuscular disease characterized by progressive muscular weakness and neuronal degeneration, affecting 1-2 individuals per 100,000 globally. While SBMA is relatively rare, recent studies have shown a significantly higher prevalence of the disease within the indigenous population of Western Canada compared to the general population. The disease is caused by a pathogenic expansion of polyglutamine residues in the androgen receptor protein, which acts as a key transcriptional regulator for numerous genes. SBMA has no cure, and current treatments are primarily supportive and focused on symptom management. Recently, a form of precision medicine known as antisense therapy has gained traction as a promising therapeutic option for numerous neuromuscular diseases. Antisense therapy uses small synthetic oligonucleotides to confer therapeutic benefit by acting on pathogenic mRNA molecules, serving to either degrade pathogenic mRNA transcripts or helping to modulate splicing. Recent studies have explored the suitability of antisense therapy for the treatment of SBMA, primarily focused on antisense-mediated mRNA knockdown approaches. Advancements in understanding the pathogenesis of SBMA and the development of targeted therapies offer hope for improved quality of life for individuals affected by this debilitating condition. Continued research is essential to optimize these genetic approaches, ensuring their safety and efficacy.
Keywords:
spinal and bulbar muscular atrophy
; antisense therapy
; oligonucleotide
; splice switching
; mRNA knockdown
; androgen receptor
; AR45
1. Introduction
1.1 Clinical Features and Prevalence in the Indigenous Population of Western Canada
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy’s disease, is an X-linked recessive neuromuscular disease with slow progression, caused by mutations in the androgen receptor gene [1,2]. The disease typically only affects males, but females can be carriers and sometimes experience muscle cramps. It typically has an onset at around 30-40 years of age in patients, with symptoms such as hand tremors, muscle cramps, and back pain arising first [1,3]. As the disease progresses, patients begin to experience muscle weakness, usually starting from the lower limbs. Within a few years after the onset of muscle weakness, many patients will require a handrail to climb the stairs [3]. Fasciculations also appear early in the disease, commonly appearing in the lower areas of the face. As the bulbar muscles start to weaken, SBMA patients notice difficulties with speech and swallowing [2]. Large natural history studies have identified that these bulbar symptoms begin to appear at an average age of 50 [3]. Near the end of the disease, patients begin needing assistance from canes or wheelchairs. A common cause of death in SBMA patients is aspiration pneumonia due to bulbar weakness [3].
Beyond the neuromuscular symptoms, SBMA also has an effect on endocrine function. Changes in the functionality of the androgen receptor result in partial androgen insensitivity in many patients, which often appears prior to any muscular symptoms [4,5,6]. Gynecomastia is the most common manifestation of androgen insensitivity in SBMA, with over 70% of patients experiencing it, according to multiple studies [4,5,6]. Other common symptoms include erectile dysfunction, testicular atrophy, and fertility problems. SBMA also has an effect on metabolism, and many patients display above-average levels of cholesterol and triglycerides, as well as elevated rates of diabetes, insulin resistance, and non-alcoholic fatty liver disease [6,7]. No cure currently exists for SBMA [8]. Typical management strategies include occupational and speech therapies, screening for respiratory problems, and pharmacological therapies to manage symptoms [9]. However, these approaches fail to address the underlying disease.
SBMA has a prevalence of 1-2 individuals per 100,000 people [2,10]. The mutation causing SBMA appears to have occurred independently in various populations worldwide, with different founder haplotypes of the disease found in the Scandinavian countries, Japan, Germany, Italy, Australia, and Canada [11]. A higher prevalence of the disease in certain areas of the world is associated with founder effects. For example, in the region of Vaasa, Finland, the prevalence of SBMA is estimated to be around 7.65 per 100,000 [12,13]. This high prevalence has been attributed to a founder effect resulting from an ancient mutation in Western Finland [13]. A similar founder effect was also observed in a group of Japanese SBMA patients through genetic analysis [14].
The highest prevalence of SBMA in a population found thus far is in the Indigenous population of Saskatchewan, Canada, where the estimated prevalence is 14.7 per 100,000 people [12]. The authors of the study, Leckie et al., also noticed a high representation of patients with Saulteaux background (either from the clinic they were studying or reported relatives), and predicted an even higher prevalence of 184.24 per 100,000 people in the Saulteaux community of Saskatchewan [12]. Notably, two participants in the study were recruited from the neighboring province of Alberta, indicating that this high prevalence may extend beyond Saskatchewan [12]. Despite already demonstrating a much higher SBMA prevalence than any other population, the authors of this paper believe that these numbers could still be underestimated, as many individuals living with SBMA in the Indigenous communities do not currently go to the neuromuscular clinic [12]. [12]Their findings suggest that the prevalence in Indigenous populations in Saskatchewan may represent the highest carrier rate for SBMA worldwide.
The high prevalence in the Indigenous communities of Saskatchewan has been associated with a founder effect, with the majority of patients in Leckie et al.’s study sharing the same haplotype [12]. Their findings suggest the founder effect likely originated approximately 250 years ago in this population. A second, distinct haplotype found in the study may also point to further founder effects in Métis and other Indigenous populations [12]. However, the authors mention that further research incorporating relatedness analyses, comprehensive genetic investigations, and larger ethnically matched control groups is necessary to strengthen these findings and gain more insights into the origin of the mutations.
Leckie et al. also explored whether Indigenous individuals with SBMA have different phenotypes compared to those documented previously in other parts of the world [12]. Like previous findings, their study indicates a slow progression of the disease, with the accumulation of weakness over time, as well as an inverse correlation between age at onset of SBMA and the size of the expanded androgen receptor (AR) repeat [12]. However, future studies with a larger sample size and more extended observation of the patients over time are needed to confirm potential phenotype differences in Indigenous populations.
The study also highlights the significant health disparities that Indigenous individuals still face in Canada, stemming from Canada’s colonial history and the wide array of social, economic, and political barriers present for Indigenous Canadians [12,15]. Indigenous populations often face barriers to accessing specialists for diagnosis and treatment in Canada, and are often not included in genetic research [16]. Recently, more care has been taken to involve Indigenous populations in genetic research whilst respecting their concerns, allowing better representation and the potential for improved future treatment of genetic conditions in Indigenous communities [12,17]. In terms of SBMA, Leckie et al suggest that a Canadian disease registry for SBMA may help further research into this disease and guide clinical efforts towards critical areas [12].
1.2 Mechanism and genetics
Spinal and bulbar muscular atrophy results from mutations in the AR gene encoding for the androgen receptor. The androgen receptor protein is one of many steroid hormone receptors belonging to the nuclear receptor superfamily and works to facilitate the effect of androgens on the body [18,19]. First discovered in humans in 1988, the AR gene is located on the long arm of the X chromosome at Xq11-12 and encodes for a 110 kDa protein product [20,21,22,23,24]. The AR gene is comprised of 8 exons that encode the three major domains of AR protein known as the transactivation domain, the DNA-binding domain, and the ligand-binding domain [18,25]. Interestingly, these three functional domains demonstrate differing degrees of conservation as the transactivation domain, coded by exon 1, is highly variable and known to have repeated glutamine, proline, and glycine residues [20]. In contrast, the DNA-binding domain encoded by exons 2 and 3 of the AR gene is conserved in all members of the steroid receptor superfamily [20,26]. The remaining exons 4-8 are less conserved and code for the ligand-binding domain of the AR gene [20,27].
SBMA belongs to a class of diseases known as trinucleotide repeat expansion disorders and results from a pathologic polyglutamine expansion in the first exon of the androgen receptor gene, characterized by glutamine residues encoded at a higher-than-normal range near the transcriptional activation domain of the AR protein [28]. In a healthy individual there are typically between 9-36 CAG repeats in this region, and SBMA begins to manifest with more than 38 repeats present [29]. Although it is not the most well-known of the group, SBMA holds the title as the first identified repeat expansion disease. In addition to SBMA, 8 other diseases stem from CAG-polyglutamine repeat expansions, all of which are characterized by their progressive neurodegenerative effects on humans [30]. These include Huntington’s disease, dentatorubral-pallidoluysian atrophy (DRPLA), and Spinocerebellar ataxia types 1, 2, 3, 6,7, and 17 [30].
The androgen receptor functions as a ligand-dependent transcription factor that regulates, in coordination with other co-regulatory proteins, the transcription of the androgen-responsive genes via binding to their regulatory sites when in its androgen-bound form [18]. In its inactive form, AR is localized to the cytoplasm where it, like other members of the steroid hormone receptors family, forms a complex with heat shock proteins [31]. Upon androgen (testosterone or dihydrotestosterone) binding, AR undergoes a conformational change and a subsequent detachment of heat shock proteins, preparing AR for translocation into the nucleus [28,31,32,33,34,35,36,37]. Following androgen binding, AR undergoes dimerization and is targeted to the nucleus where it binds to androgen-responsive elements found on androgen-regulated genes, regulating their expression [30].
SBMA is a disease characterized by both gain- and loss-of-function mechanisms [38] (Figure 1). The neurotoxic effects seen across the majority of polyglutamine expansion diseases indicate a toxic gain-of-function as the cause of neurodegeneration, consistent with the dominant pattern of inheritance in almost all these diseases [39,40]. This suggests that the encoded polyQ-AR protein is structurally modified in a way that enables it to introduce a new function that is pathogenic to neurons [39]. Interestingly, a secondary loss-of-function component is also believed to contribute to the pathogenesis of polyglutamine diseases, which manifests as partial androgen insensitivity [38].
Figure 1.
The effects of androgens binding on Wild-Type (WT) and polyglutamine (PolyQ) androgen receptors (AR) and their function in cells. Androgen binding to AR in the cytoplasm induces a conformational change in the AR before its subsequent translocation into the nucleus, where it functions as a transcription factor. However, in the case of polyQ AR, a distinct formation of aggregates upon Androgen binding is evident. These PolyQ AR aggregates are characterized by their gain and loss of function mechanisms. Some of the gain of function mechanisms resulting from polyQ AR aggregates include neuronal degeneration, altered protein-protein interactions, sequestration of cellular components, and inhibition of cellular transport. On the other hand, a Loss of function mechanism that results from polyQ AR aggregates is the loss of AR trophic support via the downregulation of the transcription of growth factors like VEGF.
Figure 1.
The effects of androgens binding on Wild-Type (WT) and polyglutamine (PolyQ) androgen receptors (AR) and their function in cells. Androgen binding to AR in the cytoplasm induces a conformational change in the AR before its subsequent translocation into the nucleus, where it functions as a transcription factor. However, in the case of polyQ AR, a distinct formation of aggregates upon Androgen binding is evident. These PolyQ AR aggregates are characterized by their gain and loss of function mechanisms. Some of the gain of function mechanisms resulting from polyQ AR aggregates include neuronal degeneration, altered protein-protein interactions, sequestration of cellular components, and inhibition of cellular transport. On the other hand, a Loss of function mechanism that results from polyQ AR aggregates is the loss of AR trophic support via the downregulation of the transcription of growth factors like VEGF.
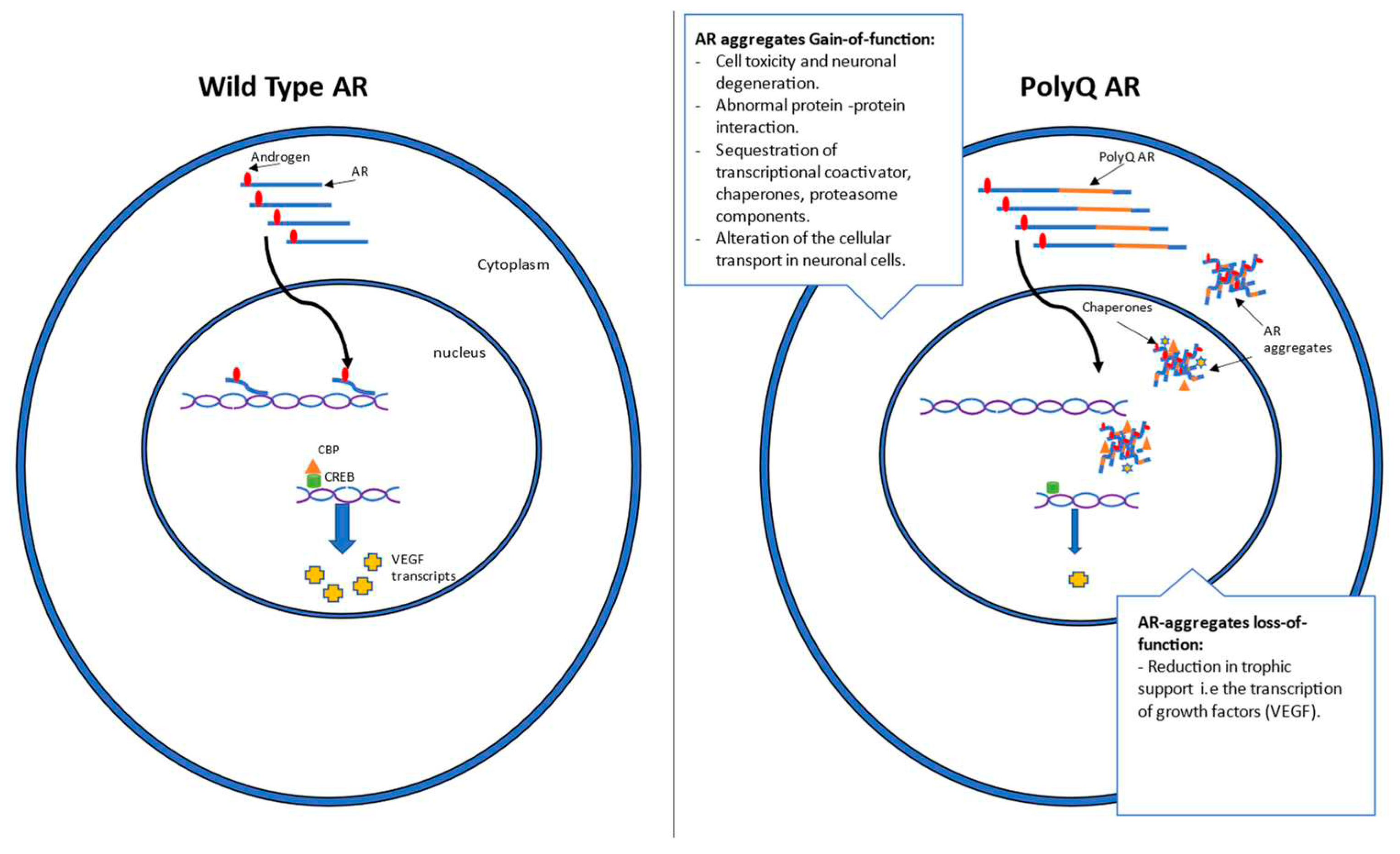
The mechanism underlying the neurodegenerative gain-of-function symptoms present in SBMA is not yet fully understood [41]. A leading hypothesis is that the well-documented accumulation and aggregation of polyQ-AR in the nucleus could be contributing to disease and motor neuron degeneration. The formation of nuclear aggregates of mutant AR is evident in both motor neurons of the spinal cord and the brain stem as well as some non-neural tissue [42]. The expansion of the polyglutamine tract in AR affects the folding of the final AR product and is associated with an increase in α-helical structures [43,44,45]. Further, it increases the stability of α-helices via the unconventional hydrogen binding of the glutamine side chain and main chain carbonyl group [43,45]. It has also been reported that this abnormal hydrogen bonding plays a role in the formation of antiparallel β-strands of polyglutamine repeats into sheets or barrels [46]. These structural changes may result in abnormal protein-protein interaction and/or the subsequent degradation of mutant proteins [39]. Others have argued that aggregation arises as the expanded polyglutamine tract could serve as a substrate for the catalysis of cross-linked protein products via transglutaminase activity, leading to the formation of aggregates and their possible breakdown [39,40,47]. While it remains unclear exactly how these protein aggregates cause disease, they are thought to be closely related to the impaired axonal transport and ultimate neuronal degeneration observed in SBMA [48,49,50].
The loss-of-function mechanism has been better characterized and is a combination of androgen insensitivity and reduced trophic support. Androgen insensitivity arises due to a reduction in the appropriate function of polyQ-AR protein, corresponding with a decrease in expression of genes typically activated by testosterone and dihydrotestosterone [27]. Additionally, polyQ-AR has been shown to display altered binding to CREB binding protein (CBP), leading to reduced transcriptional activity for genes modulated by this complex, most notably vascular endothelial growth factor (VEGF) [51]. VEGF has previously been reported to be downregulated in SBMA mice, and it is thought that this loss of trophic support could also contribute to the motor neuron pathogenesis of SBMA [38,51].
2. Current antisense approaches for SBMA
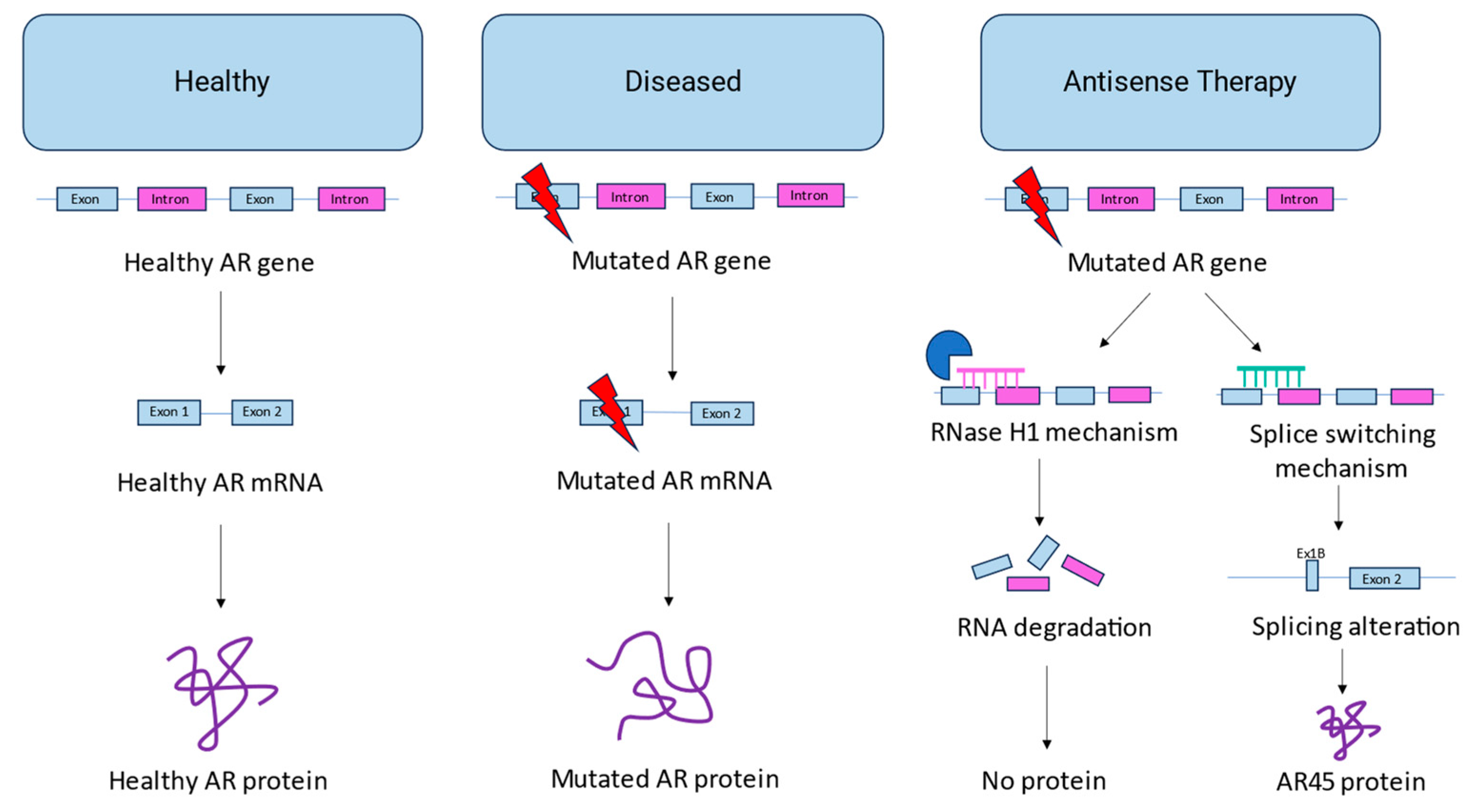
Antisense oligonucleotide therapy is a promising new area of drug development, and multiple drugs using this technology have received FDA approval in recent years [52]. This form of therapy uses oligonucleotides – often referred to as AONs or ASOs - to bind via Watson-Crick base pairing to a target sequence of RNA, where it interferes with gene expression [52,53]. One way this interference can occur is through the RNase H1 enzyme, which cleaves the targeted RNA, resulting in RNA degradation [54,55]. Another mechanism commonly used is splicing alteration, in which the oligonucleotide interferes with the normal splicing patterns of the target RNA, resulting in changes in exon skipping or inclusion [56] (Figure 2). Here, we aim to provide an overview of current antisense-based approaches to treat SBMA.
Figure 2.
Overview of Antisense Therapy for SBMA. In healthy patients, the AR gene encodes a healthy AR mRNA, which produces a functional androgen receptor protein. In patients with SBMA, a mutation in exon 1 of the AR gene results in a mutated mRNA and a mutated protein. Antisense therapy developed for SBMA employs two major mechanisms: the RNase H1 mechanism (which degrades the mutated RNA) and the splice switching mechanism (which alters splicing to replace exon 1 with exon 1B to form an AR45 isoform instead).
Figure 2.
Overview of Antisense Therapy for SBMA. In healthy patients, the AR gene encodes a healthy AR mRNA, which produces a functional androgen receptor protein. In patients with SBMA, a mutation in exon 1 of the AR gene results in a mutated mRNA and a mutated protein. Antisense therapy developed for SBMA employs two major mechanisms: the RNase H1 mechanism (which degrades the mutated RNA) and the splice switching mechanism (which alters splicing to replace exon 1 with exon 1B to form an AR45 isoform instead).
2.1. AON-mediated androgen receptor knockdown
The purpose of antisense oligonucleotide (AON)-mediated AR knockdown is to degrade mutant AR transcripts which are known to accumulate in the nucleus [50] (Figure 2). By targeting AONs containing a ribonuclease-inducing core to the AR gene, the pathogenic AR mRNA and protein levels can be reduced, leading to an improved phenotype.
Lieberman et al. aimed to explore the effect of peripheral polyQ-AR suppression in transgenic SBMA mouse models [57]. Specifically, they focused on whether AON-mediated knockdown of polyQ-AR could improve the peripheral muscular pathology of SBMA in these mice. The authors first identified 2 different 2′,4′-constrained ethyl (cEt) gapmers which caused a dose-dependent reduction in the AR mRNA of HUVEC cells, denoted ASO1 and ASO2. ASO1 targets a region in the AR transcript conserved between human and murine transcripts, whereas ASO2 targets a region specific to humans [57]. ASO1 and ASO2 were further validated in vivo in the AR113Q or humanized BAC fxAR121 SBMA mouse models, respectively. Consistent with their in vitro findings, the authors identified that subcutaneous ASO treatment led to significant knockdown of AR mRNA and a nearly complete reduction (~90% for ASO1, 95% for ASO2) in AR protein levels in the mouse quadriceps muscle. This was accompanied by a clear improvement in certain aspects of the disease phenotype, with significant amelioration of grip strength, lean body mass, muscle fiber size, and lifespan in mice treated with either ASO1 or ASO2. Notably, AR expression was unaffected in the spinal cord following subcutaneous treatment, as gapmers with this chemistry are unable to permeate the blood-brain barrier [57].
In a following study, Sahashi et al. explored AON-mediated knockdown of AR transcripts in SBMA mice but focused on neuronal polyQ-AR expression rather than peripheral [58]. The SBMA mouse model AR-97Q, which expresses both murine and transgenic human AR protein, was treated with intracerebroventricular (ICV) injection of either ASO-AR1 or ASO-2, both of which are 2′-MOE gapmers. ASO-AR1 used the same sequence as Liberman et al.’s ASO1 targeting both human and murine AR, while ASO-AR2 was a distinct sequence specific to mice [57,58]. Treatment with either ASO led to a ~50% decrease in mutant AR mRNA and protein in the spinal cord and brain, and ASO-AR1 showed an additional ~90% reduction in murine AR mRNA. AR levels in peripheral muscle were unaffected. Mice treated with either ASO also showed marked improvement in clinical phenotype, demonstrating significantly improved survival, grip strength, and rotarod performance compared to controls. Immunohistochemical analysis of ASO-AR1 treated mice also demonstrated numerous markers of improvement, such as reduced motor neuron shrinkage, reduced neuronal degeneration, and improved neuromuscular junction endplate maturation. Furthermore, despite negligible uptake of ASO into the skeletal muscle following ICV injection, the muscles of ASO-AR1 treated mice showed restored fiber size and reduced atrophy compared to controls [58].
Finally, Evers et al. aimed to identify a generic AON candidate which could knock down transcripts from all diseases arising from CAG repeats, such as Huntington’s disease, SBMA, and the spinocerebellar ataxias [59]. Despite showing some benefit for Huntington’s, their ASO candidate failed to induce notable knockdown of AR mRNA when tested in vitro and is therefore unlikely to be applicable for SBMA [59]. Beyond the scope of SBMA, several groups have also explored antisense-mediated knockdown of AR to treat prostate cancer, with dozens of AONs screened and several successful candidates identified [60,61]. While these AONs have not been explored in the context of SBMA, they further confirm the ability of AONs to knock down AR transcripts both in vitro and in vivo.
Taken together, these studies demonstrate that antisense-mediated knockdown of AR is highly effective. Furthermore, the findings of Lieberman et al. and Sahashi et al. demonstrate that AR knockdown in both peripheral tissues and the CNS is associated with an improved clinical phenotype for SBMA, suggesting that AONs may have therapeutic potential for this indication [57,58]. Of note, both groups were limited by the inability of their gapmers to cross the blood-brain barrier, requiring mutually exclusive treatment of either the CNS or peripheral muscles based on injection type. Interestingly, recent studies have identified numerous nanocarriers and apoptotic bodies which can successfully facilitate oligonucleotide penetration of the blood-brain barrier [62,63,64]. While not yet explored for SBMA, these carriers could potentially enable the treatment of both CNS and peripheral tissues with a single injection. Given that SBMA is a disease with both CNS and peripheral muscle involvement, this could be a major next step in improving the therapeutic efficacy and applicability of antisense-mediated knockdown for SBMA. Overall, AON-mediated knockdown appears to be a promising strategy for the treatment of SBMA, and recent antisense-based innovations could help to boost efficacy even further.
2.2. AON-mediated androgen receptor splice switching
As an alternative to AR knockdown, AONs can also be leveraged to induce therapeutic splice switching. Rather than recruiting ribonuclease, splice-switching AONs modulate splicing to favor the therapeutic inclusion or exclusion of a target exon, leading to the production of a functional protein product [56,65,66]. In recent years, splice-switching AONs have demonstrated great promise for the treatment of other neuromuscular disorders, and several AONs have already received FDA approval for the treatment of Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA) [67,68,69]. In the context of SBMA, designing AONs to induce skipping of exon 1, which contains the pathogenic polyQ repeat motif, could help to treat the disease (Figure 2).
Numerous isoforms of the AR protein are known to arise due to both internal translation initiation sites and alternative splicing, and isoform production has been shown to be linked to certain AR pathologies such as androgen insensitivity, SBMA, and prostate cancer [70,71,72]. One such example is AR-V7, a truncated AR isoform containing only exons 1-3 which is active even in the absence of androgens, and which is used as a biomarker for resistance to androgen-targeted therapy (ATT) in patients with prostate cancer [73,74,75]. In the healthy population, the most salient isoform is known as AR45, also referred to as AR isoform 2 [70]. AR45 arises from alternative splicing of a small exon called “1B” located within intron 1 approximately 22 kb downstream of the usual exon 1 [76]. The replacement of exon 1 with exon 1B leads to the production of a 45 kDa AR isoform which notably does not contain the polyglutamine motif which is expanded in cases of SBMA.
Recently, Lim et al. found that AR45 can regulate the transcriptional activity of full-length AR by reducing the binding of AR to its transcriptional cofactor BRD4, downregulating AR-mediated transcription [76]. In the context of SBMA where mutant AR-mediated gene expression contributes to disease pathogenesis, this downregulation was theorized to have a potential therapeutic effect. To assess this, Lim et al. overexpressed AR45 using AAV9 vectors in AR100Q mice [76]. Compared to untreated controls, AR45-overexpressing mice displayed significantly increased lifespan, delayed onset of pathological weight loss, improved rotarod performance, and improved grip strength, suggesting a notable improvement in clinical phenotype following AR45 overexpression.
To the best of the authors’ knowledge, no published work has explored splice-switching AONs for the purpose of treating SBMA. This is likely due to the fact that promoting an alternative start site using ASOs is a complex and challenging approach, with less precedent than exon inclusion or exclusion [56,77].However, given the therapeutic potential of overexpressing the AR45 isoform as identified by Lim et al. and the established efficacy of AONs as splice switching tools, this seems to be a potential direction for future research [56,76]. By designing AONs which induce the alternative splicing of exon 1B, AR45 expression could be upregulated from the patient’s own gene, simultaneously increasing the amount of AR45 and decreasing the amount of pathogenic polyQ-AR.One of the major advantages of splice switching versus knockdown approaches is that splice switching still preserves the expression of some full-length AR protein, which is important to avoid exacerbating symptoms that arise due to the absence of functional AR [76]. By limiting the pathogenic transcriptional activity of polyQ-AR without fully eliminating full length AR production, splice switching has the potential to be an effective and highly tolerable therapeutic option that should be explored in future studies.
3. Summary
Overall, both AON-mediated AR knockdown and AON-mediated AR splice switching have the potential to be valuable therapeutic approaches for the treatment of SBMA. Comparing the results from Lim et al. and Lieberman et al., it appears that AR knockdown results in a more robust alleviation of the SBMA phenotype than AR45 overexpression [57,76]. However, this comparison is heavily limited by different treatment methods (AAV versus AON), different dosages, and different SBMA mouse models, and thus should be used as a loose benchmark at best. At this time, it is uncertain how the therapeutic efficacy of AON-mediated AR splice switching would compare with AON-mediated AR knockdown. Assuming that both knockdown and AR45 overexpression can similarly alleviate the SBMA phenotype caused by polyQ-AR transcriptional activation, splice switching may be the more attractive option given that it preserves AR expression and therefore will not inhibit other important functions of the AR protein. Future studies exploring AON-based therapies for SBMA would do well to explore both splice switching and knockdown approaches to properly compare efficacy and validate this claim. Due to their rising use to treat DMD, splice-switching AONs have been an extremely popular field of research in recent years, which has led to numerous advantageous innovations [78,79]. As previously mentioned, the use of nanocarriers to allow blood-brain barrier penetration of AONs has opened the door to improved treatment of diseases which affect the CNS, such as SBMA [62,63,64]. Other studies have identified protein-AON conjugates which promote specific tissue uptake and endosomal escape of AONs, improving their bioavailability and efficacy [78,80]. As AON-based innovations continue to be discovered, their therapeutic potential steadily rises, and so does their potential ability to effectively treat SBMA.
Author Contributions
Conceptualization, H.W.C, A.A. and J.Y.; methodology, H.W.C, A.A. and J.Y..; data curation, H.W.C, A.A. and J.Y.; writing—original draft preparation, H.W.C, A.A. and J.Y.; writing—review and editing, H.W.C, A.A., J.Y., and T.Y.; supervision, T.Y.; funding acquisition, T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are grateful for funding provided by the H Jean McDiarmaid Scholarship (HWC) and the Women and Children’s Health Research Institute (WCHRI) (A.A.). T.Y is grateful for the support provided by the Women and Children’s Health Research Institute (WCHRI), Canadian Institute of Health Research (CIHR), Heart and Stroke Foundation Canada, Defeat Duchenne Canada, and the United States Department of Defense.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable.
Acknowledgments
The authors are grateful for the support provided by Muscular Dystrophy Canada, the Friends of Garrett Cumming Research Fund, the HM Toupin Neurological Science Research Fund, Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, and the Women and Children’s Health Research Institute (WCHRI).
Conflicts of Interest
H.W.C, A.A, and J.Y declare no conflicts of interest. T.Y. is a founder and shareholder of OligomicsTx, which aims to commercialize antisense oligonucleotide technology.
References
- Kennedy, W.R.; Alter, M.; Sung, J.H. Progressive Proximal Spinal and Bulbar Muscular Atrophy of Late Onset. Neurology 1968, 18, 671–671. [Google Scholar] [CrossRef] [PubMed]
- Banno, H.; Katsuno, M.; Suzuki, K.; Tanaka, F.; Sobue, G. Pathogenesis and Molecular Targeted Therapy of Spinal and Bulbar Muscular Atrophy (SBMA). Cell Tissue Res 2012, 349, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Atsuta, N.; Watanabe, H.; Ito, M.; Banno, H.; Suzuki, K.; Katsuno, M.; Tanaka, F.; Tamakoshi, A.; Sobue, G. Natural History of Spinal and Bulbar Muscular Atrophy (SBMA): A Study of 223 Japanese Patients. Brain 2006, 129, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Dejager, S.; Bry-Gauillard, H.; Bruckert, E.; Eymard, B.; Salachas, F.; LeGuern, E.; Tardieu, S.; Chadarevian, R.; Giral, P.; Turpin, G. A Comprehensive Endocrine Description of Kennedy’s Disease Revealing Androgen Insensitivity Linked to CAG Repeat Length. J Clin Endocrinol Metab 2002, 87, 3893–3901. [Google Scholar] [CrossRef] [PubMed]
- Rosenbohm, A.; Hirsch, S.; Volk, A.E.; Grehl, T.; Grosskreutz, J.; Hanisch, F.; Herrmann, A.; Kollewe, K.; Kress, W.; Meyer, T.; et al. The Metabolic and Endocrine Characteristics in Spinal and Bulbar Muscular Atrophy. J Neurol 2018, 265, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Querin, G.; Bertolin, C.; Da Re, E.; Volpe, M.; Zara, G.; Pegoraro, E.; Caretta, N.; Foresta, C.; Silvano, M.; Corrado, D.; et al. Non-Neural Phenotype of Spinal and Bulbar Muscular Atrophy: Results from a Large Cohort of Italian Patients. J Neurol Neurosurg Psychiatry 2016, 87, 810–816. [Google Scholar] [CrossRef]
- Francini-Pesenti, F.; Querin, G.; Martini, C.; Mareso, S.; Sacerdoti, D. Prevalence of Metabolic Syndrome and Non-Alcoholic Fatty Liver Disease in a Cohort of Italian Patients with Spinal-Bulbar Muscular Atrophy. Acta Myologica 2018, 37, 204. [Google Scholar]
- Tanaka, F.; Katsuno, M.; Banno, H.; Suzuki, K.; Adachi, H.; Sobue, G. Current Status of Treatment of Spinal and Bulbar Muscular Atrophy. Neural Plast 2012, 2012. [Google Scholar] [CrossRef]
- Pradat, P.F.; Bernard, E.; Corcia, P.; Couratier, P.; Jublanc, C.; Querin, G.; Morélot Panzini, C.; Salachas, F.; Vial, C.; Wahbi, K.; et al. The French National Protocol for Kennedy’s Disease (SBMA): Consensus Diagnostic and Management Recommendations. Orphanet Journal of Rare Diseases 2020 15:1 2020, 15, 1–21. [Google Scholar] [CrossRef]
- Guidetti, D.; Sabadini, R.; Ferlini, A.; Torrente, I. Epidemiological Survey of X-Linked Bulbar and Spinal Muscular Atrophy, or Kennedy Disease, in the Province of Reggio Emilia, Italy. Eur J Epidemiol 2001, 17, 587–591. [Google Scholar] [CrossRef]
- Lund, A.; Udd, B.; Juvonen, V.; Andersen, P.M.; Cederquist, K.; Davis, M.; Gellera, C.; Kölmel, C.; Ronnevi, L.O.; Sperfeld, A.D.; et al. Multiple Founder Effects in Spinal and Bulbar Muscular Atrophy (SBMA, Kennedy Disease) around the World. European Journal of Human Genetics 2001 9:6 2001, 9, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Leckie, J.N.; Joel, M.M.; Martens, K.; King, A.; King, M.; Korngut, L.W.; Koning, A.P.J. de; Pfeffer, G.; Schellenberg, K.L. Highly Elevated Prevalence of Spinobulbar Muscular Atrophy in Indigenous Communities in Canada Due to a Founder Effect. Neurol Genet 2021, 7, e607. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.; Udd, B.; Juvonen, V.; Andersen, P.M.; Cederquist, K.; Ronnevi, L.O.; Sistonen, P.; Sörensen, S.A.; Tranebjærg, L.; Wallgren-Pettersson, C.; et al. Founder Effect in Spinal and Bulbar Muscular Atrophy (SBMA) in Scandinavia. European Journal of Human Genetics 2000 8:8 2000, 8, 631–636. [Google Scholar] [CrossRef]
- Tanaka, F.; Doyu, M.; Ito, Y.; Matsumoto, M.; Mitsuma, T.; Abe, K.; Aoki, M.; Itoyama, Y.; Fischbeck, K.H.; Sobue, G. Founder Effect in Spinal and Bulbar Muscular Atrophy (SBMA). Hum Mol Genet 1996, 5, 1253–1257. [Google Scholar] [CrossRef]
- King, M.; Smith, A.; Gracey, M. Indigenous Health Part 2: The Underlying Causes of the Health Gap. The Lancet 2009, 374, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Miller, A.P.; Quesnel-Vallée, A.; Caron, N.R.; Vissandjée, B.; Marchildon, G.P. Canada’s Universal Health-Care System: Achieving Its Potential. The Lancet 2018, 391, 1718–1735. [Google Scholar] [CrossRef]
- Caron, N.R.; Chongo, M.; Hudson, M.; Arbour, L.; Wasserman, W.W.; Robertson, S.; Correard, S.; Wilcox, P. Indigenous Genomic Databases: Pragmatic Considerations and Cultural Contexts. Front Public Health 2020, 8, 529095. [Google Scholar] [CrossRef]
- Yong, E.L.; Loy, C.J.; Sim, K.S. Androgen Receptor Gene and Male Infertility. Hum Reprod Update 2003, 9, 1–7. [Google Scholar] [CrossRef]
- Lu, N.Z.; Wardell, S.E.; Burnstein, K.L.; Defranco, D.; Fuller, P.J.; Giguere, V.; Hochberg, R.B.; Mckay, L.; Renoir, J.M.; Weigel, N.L.; et al. International Union of Pharmacology. LXV. The Pharmacology and Classification of the Nuclear Receptor Superfamily: Glucocorticoid, Mineralocorticoid, Progesterone, and Androgen Receptors. Pharmacol Rev 2006, 58, 782–797. [Google Scholar] [CrossRef]
- Palazzolo, I.; Gliozzi, A.; Rusmini, P.; Sau, D.; Crippa, V.; Simonini, F.; Onesto, E.; Bolzoni, E.; Poletti, A. The Role of the Polyglutamine Tract in Androgen Receptor. J Steroid Biochem Mol Biol 2008, 108, 245–253. [Google Scholar] [CrossRef]
- Brown, C.J.; Goss, S.J.; Lubahn, D.B.; Joseph, D.R.; Wilson, E.M.; French, F.S.; Willard, H.F. Androgen Receptor Locus on the Human X Chromosome: Regional Localization to Xq11-12 and Description of a DNA Polymorphism. Am J Hum Genet 1989, 44, 264. [Google Scholar] [PubMed]
- Lubahn, D.B.; Joseph, D.R.; Sullivan, P.M.; Willard, H.F.; French, F.S.; Wilson, E.M. Cloning of Human Androgen Receptor Complementary DNA and Localization to the X Chromosome. Science (1979) 1988, 240, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Kokontis, J.; Liao, S. Molecular Cloning of Human and Rat Complementary DNA Encoding Androgen Receptors. Science (1979) 1988, 240, 324–326. [Google Scholar] [CrossRef] [PubMed]
- van Laar, J.H.; Vries, J.B. de; Voorhorst-Ogink, M.M.; Brinkmann, A.O. The Human Androgen Receptor Is a 110 KDa Protein. Mol Cell Endocrinol 1989, 63, 39–44. [Google Scholar] [CrossRef]
- Kuiper, G.G.J.M.; Faber, P.W.; Van Rooij, H.C.J.; Van der Korput, J.A.G.M.; Ris-Stalpers, C.; Klaassen, P.; Trapman, J.; Brinkmann, A.O. STRUCTURAL ORGANIZATION OF THE HUMAN ANDROGEN RECEPTOR GENE. J Mol Endocrinol 1989, 2, R1–R4. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.J.; O’Malley, B.W. Molecular Mechanisms of Action of Steroid/Thyroid Receptor Superfamily Members. Annu Rev Biochem 1994, 63, 451–486. [Google Scholar] [CrossRef]
- Brinkmann, A.O. Molecular Basis of Androgen Insensitivity. Mol Cell Endocrinol 2001, 179, 105–109. [Google Scholar] [CrossRef]
- Fischbeck, K.H.; Lieberman, A.; Bailey, C.K.; Abel, A.; Merry, D.E. Androgen Receptor Mutation in Kennedy’s Disease. Philosophical Transactions of the Royal Society B: Biological Sciences 1999, 354, 1075. [Google Scholar] [CrossRef]
- Fratta, P.; Collins, T.; Pemble, S.; Nethisinghe, S.; Devoy, A.; Giunti, P.; Sweeney, M.G.; Hanna, M.G.; Fisher, E.M.C. Sequencing Analysis of the Spinal Bulbar Muscular Atrophy CAG Expansion Reveals Absence of Repeat Interruptions. Neurobiol Aging 2014, 35, 443.e1. [Google Scholar] [CrossRef]
- Stoyas, C.A.; La Spada, A.R. The CAG–Polyglutamine Repeat Diseases: A Clinical, Molecular, Genetic, and Pathophysiologic Nosology. Handb Clin Neurol 2018, 147, 143–170. [Google Scholar] [CrossRef]
- Gelmann, E.P. Molecular Biology of the Androgen Receptor. J Clin Oncol 2002, 20, 3001–3015. [Google Scholar] [CrossRef] [PubMed]
- Ing, N.H.; Beekman, J.M.; Tsai, S.Y.; Tsai, M.-J.; O’malleyl, B.W.; Sagami, I.; Tsai, S.Y.; Wang, H.; Tsii, M.-J.; Malley, O. ’; et al. THE JOURNAL OF BIOLOGICAL CHEMISTRY Members of the Steroid Hormone Receptor Superfamily Interact with TFIIB (S300-11)" The S300-I1 Factor Was Discovered as an Activator of Ovalbumin Gene Transcription with the Chicken Oval-Bumin Upstream Promoter-Transcription Factor (COUP-T%’ Although S300-I1 Does Not Bind DNA Selec-Tively, It Stabilizes the Binding of COUP-TF to Its Cis-Element. Journal of Biological Chemistry 1992, 267, 17617–17623. [Google Scholar] [CrossRef]
- Baniahmad, A.; Ha, I.; Reinberg, D.; Tsai, S.; Tsai, M.J.; O’Malley, B.W. Interaction of Human Thyroid Hormone Receptor Beta with Transcription Factor TFIIB May Mediate Target Gene Derepression and Activation by Thyroid Hormone. Proceedings of the National Academy of Sciences 1993, 90, 8832–8836. [Google Scholar] [CrossRef] [PubMed]
- Brou, C.; Chaudhary, S.; Davidson, I.; Lutz, Y.; Wu, J.; Egly, J.M.; Tora, L.; Chambon, P. Distinct TFIID Complexes Mediate the Effect of Different Transcriptional Activators. EMBO J 1993, 12, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Brou, C.; Wu, J.; Ali, S.; Scheer, E.; Lang, C.; Davidson, I.; Chambon, P.; Tora, L. Different TBP-Associated Factors Are Required for Mediating the Stimulation of Transcription in Vitro by the Acidic Transactivator GAL-VP16 and the Two Nonacidic Activation Functions of the Estrogen Receptor. Nucleic Acids Res 1993, 21, 5. [Google Scholar] [CrossRef] [PubMed]
- Mcewan, I.J.; Gustafsson, J.Å. Interaction of the Human Androgen Receptor Transactivation Function with the General Transcription Factor TFIIF. Proc Natl Acad Sci U S A 1997, 94, 8485. [Google Scholar] [CrossRef]
- Schulman, I.G.; Chakravarti, D.; Juguilon, H.; Romo, A.; Evans, R.M. Interactions between the Retinoid X Receptor and a Conserved Region of the TATA-Binding Protein Mediate Hormone-Dependent Transactivation. Proc Natl Acad Sci U S A 1995, 92, 8288. [Google Scholar] [CrossRef]
- Thomas, P.S.; Fraley, G.S.; Damien, V.; Woodke, L.B.; Zapata, F.; Sopher, B.L.; Plymate, S.R.; La Spada, A.R. Loss of Endogenous Androgen Receptor Protein Accelerates Motor Neuron Degeneration and Accentuates Androgen Insensitivity in a Mouse Model of X-Linked Spinal and Bulbar Muscular Atrophy. Hum Mol Genet 2006, 15, 2225–2238. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Orr, H.T. Glutamine Repeats and Neurodegeneration. Annu Rev Neurosci 2000, 23, 217–247. [Google Scholar] [CrossRef]
- Ross, C.A. When More Is Less: Pathogenesis of Glutamine Repeat Neurodegenerative Diseases. Neuron 1995, 15, 493–496. [Google Scholar] [CrossRef]
- Craig, T.J.; Henley, J.M. Fighting Polyglutamine Disease by Wrestling with SUMO. J Clin Invest 2015, 125, 498–500. [Google Scholar] [CrossRef]
- Li, M.; Nakagomi, Y.; Kobayashi, Y.; Merry, D.E.; Tanaka, F.; Doyu, M.; Mitsuma, T.; Hashizume, Y.; Fischbeck, K.H.; Sobue, G. Nonneural Nuclear Inclusions of Androgen Receptor Protein in Spinal and Bulbar Muscular Atrophy. Am J Pathol 1998, 153, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.J.; Merry, D.E. Molecular Mechanisms and Therapeutics for SBMA/Kennedy’s Disease. Neurotherapeutics 2019 16:4 2019, 16, 928–947. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Watt, K.; Kelly, S.M.; Clark, C.; Price, N.C.; McEwan, I.J. Consequences of Poly-Glutamine Repeat Length for the Conformation and Folding of the Androgen Receptor Amino-Terminal Domain. J Mol Endocrinol 2008, 41, 301–314. [Google Scholar] [CrossRef]
- Escobedo, A.; Topal, B.; Kunze, M.B.A.; Aranda, J.; Chiesa, G.; Mungianu, D.; Bernardo-Seisdedos, G.; Eftekharzadeh, B.; Gairí, M.; Pierattelli, R.; et al. Side Chain to Main Chain Hydrogen Bonds Stabilize a Polyglutamine Helix in a Transcription Factor. Nature Communications 2019 10:1 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Perutz, M.F.; Johnson, T.; Suzuki, M.; Finch, J.T. Glutamine Repeats as Polar Zippers: Their Possible Role in Inherited Neurodegenerative Diseases. Proc Natl Acad Sci U S A 1994, 91, 5355. [Google Scholar] [CrossRef] [PubMed]
- Green, H. Human Genetic Diseases Due to Codon Reiteration: Relationship to an Evolutionary Mechanism. Cell 1993, 74, 955–956. [Google Scholar] [CrossRef]
- Morfini, G.; Pigino, G.; Szebenyi, G.; You, Y.; Pollema, S.; Brady, S.T. JNK Mediates Pathogenic Effects of Polyglutamine-Expanded Androgen Receptor on Fast Axonal Transport. Nature Neuroscience 2006 9:7 2006, 9, 907–916. [Google Scholar] [CrossRef]
- Rgyi Szebenyi, G.; Morfini, G.A.; Babcock, A.; Gould, M.; Selkoe, K.; Stenoien, D.L.; Young, M.; Faber, P.W.; Macdonald, M.E.; Mcphaul, M.J.; et al. Neuropathogenic Forms of Huntingtin and Androgen Receptor Inhibit Fast Axonal Transport Lengths of 20-25 Glutamines to Pathological Expansions of 40. Neurological Symptoms Typically Appear in Mid-Life, Although Longer PolyQ Repeats Are Associated with Earlier Onset of Symptoms. Aside from the PolyQ Tracts, Gene Products Associated with PolyQ Diseases Exhibit Minimal Sequence Homology. Knockouts of Genes En-Coding PolyQ-Containing Proteins Result in Different Phe-1. Neuron 2003, 40, 41–52. [Google Scholar]
- Piccioni, F.; Pinton, P.; Simeoni, S.; Pozzi, P.; Fascio, U.; Vismara, G.; Martini, L.; Rizzuto, R.; Poletti, A. Androgen Receptor with Elongated Polyglutamine Tract Forms Aggregates That Alter Axonal Trafficking and Mitochondrial Distribution in Motor Neuronal Processes. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2002, 16, 1418–1420. [Google Scholar] [CrossRef]
- Sopher, B.L.; Thomas, P.S.; Lafevre-Bernt, M.A.; Holm, I.E.; Wilke, S.A.; Ware, C.B.; Jin, L.W.; Libby, R.T.; Ellerby, L.M.; La Spada, A.R. Androgen Receptor YAC Transgenic Mice Recapitulate SBMA Motor Neuronopathy and Implicate VEGF164 in the Motor Neuron Degeneration. Neuron 2004, 41, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X. hai Antisense Technology: An Overview and Prospectus. Nature Reviews Drug Discovery 2021 20:6 2021, 20, 427–453. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous Sarcoma Virus Replication and Cell Transformation by a Specific Oligodeoxynucleotide. Proc Natl Acad Sci U S A 1978, 75, 280. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. Journal of Biological Chemistry 2004, 279, 17181–17189. [Google Scholar] [CrossRef]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-Switching Antisense Oligonucleotides as Therapeutic Drugs. Nucleic Acids Res 2016, 44, 6549. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Yu, Z.; Murray, S.; Peralta, R.; Low, A.; Guo, S.; Yu, X.X.; Cortes, C.J.; Bennett, C.F.; Monia, B.P.; et al. Peripheral Androgen Receptor Gene Suppression Rescues Disease in Mouse Models of Spinal and Bulbar Muscular Atrophy. Cell Rep 2014, 7, 774–784. [Google Scholar] [CrossRef]
- Sahashi, K.; Katsuno, M.; Hung, G.; Adachi, H.; Kondo, N.; Nakatsuji, H.; Tohnai, G.; Iida, M.; Bennett, F.F.; Sobue, G. Silencing Neuronal Mutant Androgen Receptor in a Mouse Model of Spinal and Bulbar Muscular Atrophy. Hum Mol Genet 2015, 24, 5985–5994. [Google Scholar] [CrossRef]
- Evers, M.M.; Pepers, B.A.; van Deutekom, J.C.T.; Mulders, S.A.M.; den Dunnen, J.T.; Aartsma-Rus, A.; van Ommen, G.J.B.; van Roon-Mom, W.M.C. Targeting Several CAG Expansion Diseases by a Single Antisense Oligonucleotide. PLoS One 2011, 6, e24308. [Google Scholar] [CrossRef]
- Hamy, F.; Brondani, V.; Spoerri, R.; Rigo, S.; Stamm, C.; Klimkait, T. Specific Block of Androgen Receptor Activity by Antisense Oligonucleotides. Prostate Cancer Prostatic Dis 2003, 6, 27–33. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Loriot, Y.; Beraldi, E.; Zhang, F.; Wyatt, A.W.; Nakouzi, N. Al; Mo, F.; Zhou, T.; Kim, Y.; Monia, B.P.; et al. Generation 2.5 Antisense Oligonucleotides Targeting the Androgen Receptor and Its Splice Variants Suppress Enzalutamide-Resistant Prostate Cancer Cell Growth. Clin Cancer Res 2015, 21, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pang, J.; Wang, Q.; Yan, L.; Wang, L.; Xing, Z.; Wang, C.; Zhang, J.; Dong, L.; Wang, Y.; et al. Delivering Antisense Oligonucleotides across the Blood-Brain Barrier by Tumor Cell-Derived Small Apoptotic Bodies. Advanced Science 2021, 8, 2004929. [Google Scholar] [CrossRef]
- Sun, Y.; Kong, J.; Ge, X.; Mao, M.; Yu, H.; Wang, Y. An Antisense Oligonucleotide-Loaded Blood-Brain Barrier Penetrable Nanoparticle Mediating Recruitment of Endogenous Neural Stem Cells for the Treatment of Parkinson’s Disease. ACS Nano 2023, 17, 4414–4432. [Google Scholar] [CrossRef] [PubMed]
- Min, H.S.; Kim, H.J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B.S.; Fukushima, S.; Anraku, Y.; Miyata, K.; et al. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood–Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angewandte Chemie International Edition 2020, 59, 8173–8180. [Google Scholar] [CrossRef] [PubMed]
- Echevarría, L.; Aupy, P.; Goyenvalle, A. Exon-Skipping Advances for Duchenne Muscular Dystrophy. Hum Mol Genet 2018, 27, R163–R172. [Google Scholar] [CrossRef]
- Bauman, J.; Jearawiriyapaisarn, N.; Kole, R. Therapeutic Potential of Splice-Switching Oligonucleotides. Oligonucleotides 2009, 19, 1. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Dzierlega, K.; Yokota, T. Optimization of Antisense-Mediated Exon Skipping for Duchenne Muscular Dystrophy. Gene Ther 2020, 27, 407–416. [Google Scholar] [CrossRef]
- Wurster, C.D.; Ludolph, A.C. Nusinersen for Spinal Muscular Atrophy. Ther Adv Neurol Disord 2018, 11. [Google Scholar] [CrossRef]
- Ahrens-Fath, I.; Politz, O.; Geserick, C.; Haendler, B. Androgen Receptor Function Is Modulated by the Tissue-Specific AR45 Variant. FEBS J 2005, 272, 74–84. [Google Scholar] [CrossRef]
- Wilson, C.M.; Mcphaul, M.J. A and B Forms of the Androgen Receptor Are Present in human Genital Skin Fibroblasts. Proc Natl Acad Sci U S A 1994, 91, 1234. [Google Scholar] [CrossRef]
- Dehm, S.M.; Tindall, D.J. Alternatively Spliced Androgen Receptor Variants. Endocr Relat Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen Receptor Splice Variant-7 Expression Emerges with Castration Resistance in Prostate Cancer. J Clin Invest 2018, 129, 192. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N Engl J Med 2014, 371, 1028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Karsh, L.I.; Nissenblatt, M.J.; Canfield, S.E. Androgen Receptor Splice Variant, AR-V7, as a Biomarker of Resistance to Androgen Axis-Targeted Therapies in Advanced Prostate Cancer. Clin Genitourin Cancer 2020, 18, 1–10. [Google Scholar] [CrossRef]
- Lim, W.F.; Forouhan, M.; Roberts, T.C.; Dabney, J.; Ellerington, R.; Speciale, A.A.; Manzano, R.; Lieto, M.; Sangha, G.; Banerjee, S.; et al. Gene Therapy with AR Isoform 2 Rescues Spinal and Bulbar Muscular Atrophy Phenotype by Modulating AR Transcriptional Activity. Sci Adv 2021, 7. [Google Scholar] [CrossRef]
- Ham, K.A.; Keegan, N.P.; McIntosh, C.S.; Aung-Htut, M.T.; Zaw, K.; Greer, K.; Fletcher, S.; Wilton, S.D. Induction of Cryptic Pre-MRNA Splice-Switching by Antisense Oligonucleotides. Scientific Reports 2021 11:1 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Woo, S.; Melo, D.; Huang, Y.; Dzierlega, K.; Shah, M.N.A.; Aslesh, T.; Roshmi, R.R.; Echigoya, Y.; Maruyama, R.; et al. Development of DG9 Peptide-Conjugated Single- and Multi-Exon Skipping Therapies for the Treatment of Duchenne Muscular Dystrophy. Proc Natl Acad Sci U S A 2022, 119. [Google Scholar] [CrossRef]
- Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.A.; Aoki, Y. Peptide-Conjugate Antisense Based Splice-Correction for Duchenne Muscular Dystrophy and Other Neuromuscular Diseases. EBioMedicine 2019, 45, 630. [Google Scholar] [CrossRef]
- Moulton, H.M.; Moulton, J.D. Morpholinos and Their Peptide Conjugates: Therapeutic Promise and Challenge for Duchenne Muscular Dystrophy. Biochimica et Biophysica Acta (BBA) - Biomembranes 2010, 1798, 2296–2303. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



