
Submitted:
18 July 2023
Posted:
20 July 2023
You are already at the latest version
Abstract
The DnaA protein has long been considered to play the key role in the initiation of chromosome replication in modern bacteria. Many questions about this role, however, remain unanswered. Here, we raise these questions within a framework based on the dynamics of hyperstructures alias large assemblies of molecules and macromolecules that perform a function. In this dynamics, hyperstructures can (1) emit and receive signals or (2) fuse and separate from one another. We ask whether the DnaA-based initiation hyperstructure acts as a logic gate receiving information from the membrane, the chromosome and metabolism to trigger replication; we try to phrase some of these questions in terms of DNA supercoiling, strand opening, glycolytic enzymes, SeqA, ribonucleotide reductase, the Macromolecular Synthesis operon, post-translational modifications and metabolic pools. Finally, we ask whether, underpinning the regulation of the cell cycle, there is a physico-chemical clock inherited from the first protocells, and whether this clock emits a single signal that triggers both chromosome replication and cell division.
Keywords:
Charles E. Helmstetter Prize
; E. coli
; ribonucleotide reductase
; sequestration
; oriC
; macromolecular crowding
; differentiation
; macromolecular synthesis operon
; integrative suppression
; L-form
1. Introduction
The introduction should briefly place the study in a broad context and highlight why it is important. It should define the purpose of the work and its significance. The current state of the research field should be carefully reviewed and key publications cited. Please highlight controversial and diverging hypotheses when necessary. Finally, briefly mention the main aim of the work and highlight the principal conclusions. As far as possible, please keep the introduction comprehensible to scientists outside your particular field of research. References should be numbered in order of appearance and indicated by a numeral or numerals in square brackets—e.g., [1] or [2,3], or [4,5,6]. See the end of the document for further details on references. The coordination of cell growth and chromosome replication is achieved by mechanisms that are still being uncovered. One approach to investigating this coordination is genetic and, over the last half century, this has led to the isolation of conditional lethal mutants of cell division or DNA synthesis. As part of these investigations, in early 1960, MK started to isolate mutants of Escherichia coli K12 that fail to grow at high temperature. With his collaborators, he found that nearly 1% of colonies obtained at 30°C from a mutagenized culture failed to grow at 42°C but, on examining each clone for DNA or protein syntheses and morphological changes after transfer to this temperature, he found only a few mutants affected in DNA synthesis with the majority being those defective in protein synthesis or in cell division [1]. Isolation of temperature-sensitive (ts) mutants continued at the Pasteur Institute and its collection has been beneficial to studies on the cell cycle such as the discovery of FtsZ ring, which is essential for division [2], and to studies on metabolism such as those on the ribonucleotide reductase [3].
The first priority was the elucidation of the regulatory mechanism of chromosome replication as hypothesized in the Replicon Theory [4] according to which DNA replication starts from the genetically defined point (oriC) by the action of an initiator. MK therefore sought mutants that failed to initiate replication at high temperature and found two [5]. These mutations were mapped to the same locus and the gene was called dnaA [6].
Further characterization of these dnaA mutants demonstrated a close connection between initiation of replication and cell cycle control: at non-permissive temperature, a dnaA mutant temporarily stops dividing and forms filamentous cells [1]; division later resumes towards one end of the filament to produce normal-sized cells that lack DNA [7]. The fact that the size of these anucleate cells is relatively constant (but see [8]) is consistent with the idea that DnaA is involved, directly or indirectly, in the positioning of the division site.
In fact, the possibility that DnaA protein acts as a regulator of gene expression was raised by Hansen a few years after the isolation of the first mutant [9] and DnaA was subsequently shown to regulate many operons [10]. These observations therefore help make DnaA a candidate for the role of coordinator of the cell cycle.
To explore this proposal, it is essential to characterise the biochemical properties of the DnaA protein. A large part of this was done by Kornberg and his collaborators using genetic engineering only 20 years after the first isolation of a dnaA mutant [11]. They found that the DnaA protein is an ATPase possessing a high affinity for the replication origin (oriC) via DnaA boxes constituted of nine bases. The consequence of this interaction is the opening of oriC, which allows the insertion of DNA helicase into oriC in order to start DNA synthesis after the loading of DNA polymerase III. This interaction between DnaA and DnaA-boxes seems to be important in most of processes in which DnaA is involved [12,13].
Here, we raise questions that need to be addressed to clarify the roles of DnaA and related proteins in the cell cycle. We do this in the theoretical framework of hyperstructure dynamics and in the context of an origins-of-life scenario in which the early cells or protocells had a cell cycle regulated by a cellular clock or timer based on physical chemistry. We consider the possibility that DnaA—and the hyperstructures with which it is associated—are the heirs to the original clock and that they integrate different sorts of information in order to coordinate the cell cycle with the environment.
2. The theoretical framework for the questions
We propose that the precursors of cells, alias protocells, had a physico-chemical ‘clock’ that emitted a single signal to trigger simultaneously the processes of both DNA replication and cell division. If so, it is conceivable that this clock still functions in modern cells but that this functioning now involves sophisticated macromolecules and the complex structures (hyperstructures) into which these macromolecules assemble in order to function. In this framework, we ask how the different hyperstructures involving DnaA and related proteins might be central to the operation of this clock. Along with its partners, DnaA-based hyperstructures integrate environmental information via the structures of the membrane and chromosome, and via metabolites and ions (Figure 1). The output of this integration is the signal for the cell to make the transition from the non-replicating to the replicating state; the latter state ends with the division that produces daughter cells with possibly different phenotypes as modulated by DnaA. This integration has characteristics with actions typical of both AND and OR logic gates insofar as some but not all the information integrated by the DnaA-based initiation hyperstructure is required for it to trigger replication.
3. Crowding, phase separation and the cell cycle
Biomolecular condensates are a class of hyperstructures that play major roles in prokaryotic and eukaryotic physiology (for a review see [14]). They form because of phase separation, which is due to transient, low-affinity, cohesive interactions between their constituent polymers whilst stereo-specific, macromolecular interactions only play a secondary role [15]. These interactions are determined by molecular and macromolecular crowding [16]. Different crowding agents can have different effects as illustrated by the compaction and DNA-binding of HU [17]. This paves the way for hyperstructures to respond in different ways in transducing information from inside and outside the cell. In the case of the initiation of chromosome replication, a low membrane occupancy is needed for the activation or rejuvenation of DnaA [18,19] whilst macromolecular crowding is required for the replication of oriC in vitro [20]; in the case of chromosome segregation, phase separation is proposed to underpin the segregation of newly replicated chromosomes [21]; in the case of cell division, macromolecular crowding is required for the formation of FtsZ droplets in vitro [22]. Fundamental questions here include: are crowding and phase separation the primary determinants of hyperstructure dynamics? Do they determine the assembly and disassembly of hyperstructures? Do they control the fusion and fission of hyperstructures? In short, are they the essence of the clock that controls the cell cycle?
4. Is there a chromosomal DnaA hyperstructure?
The chromosomal datA site is a 1 kb region that contains binding sites for DnaA and that helps in its inactivation [23]. An excess of datA sites results in a delay to the initiation of replication whilst the lack of datA results in extra initiations and it was originally suggested that the binding of DnaA to newly duplicated datA during oriC sequestration could help prevent premature reinitiation when oriC is desequestered [23]. When datA is negatively supercoiled, DnaA –ATP oligomers are stabilised, datA-IHF interactions and DnaA-ATP hydrolysis are promoted [24]. These results are consistent with a datA-based chromosomal hyperstructure helping regulate initiation. The two chromosomal intergenic regions, DnaA-reactivating sequence 1 (DARS1) and DnaA-reactivating sequence 2 (DARS2), each contain a cluster of DnaA binding sites; these sites promote regeneration of DnaA-ATP from DnaA-ADP by nucleotide exchange, and thereby help to promote the initiation of replication. [25]. (note that DnaA-ADP is mainly monomeric and unable to go from high-affinity binding sites to nucleate polymerisation at the low-affinity binding sites in the origin of replication). It is thought that DARS1 is mainly involved in maintaining the origin concentration whereas DARS2 is also involved in maintaining single cell synchrony [26].
After initiation, the ATPase activity of DnaA is stimulated by the regulatory inactivation of DnaA (RIDA) complex composed of the Hda protein interacting with DNA-loaded β-clamp [27]. Recently, it has been found that only the disruption of RIDA has a major effect on initiation (since DARS and datA can compensate for one another) [28]. All of this raises the question of whether the inactivation of DnaA takes place within a chromosomal DnaA hyperstructure.
DnaA is also a sequence-specific transcriptional regulator. Such regulators bind to sites that are distributed on the chromosome with a periodicity consistent with a solenoidal-type organization [29]; this organization would bring together a regulator and its sites into a hyperstructure. If this hyperstructure does indeed exist, what is its relationship with the datA and the DARS sites? Do the above constitute separate hyperstructures and, if so, how do they interact? Or are they all part of an initiation hyperstructure?
5. Is there a membrane DnaA hyperstructure?
The involvement of the membrane in the initiation of replication has long been known [30,31,32], and it is tempting to speculate that the initiation hyperstructure may also contain acidic phospholipids such as cardiolipin and indeed that the hyperstructure is physically associated with the bilayer itself, possibly in a fluid state. Indeed, around 10% of the DnaA in the cell is associated with the membrane [33]. In this association both domain II and the hydrophobic domain III of DnaA are important [34]. DnaA association with the membrane may dissociate ADP from DnaA depending on the degree of protein crowding on the membrane [18,19]. Insertion into or association with membrane is fundamental to transertion (the coupled transcription, translation and insertion of proteins into membrane) and to the existence of transertion hyperstructures [35,36,37]; this raises the question of whether a DnaA hyperstructure based on transertion might exist for part of the cell cycle and, moreover, whether such transertion might be important for the existence and/or operation of a chromosomal DnaA or other hyperstructure.
6. Does the initiation hyperstructure contain glycolytic enzymes?
Metabolism is coupled to DNA synthesis to nutrient richness and growth rate in a variety of ways. One way this occurs is via (p)ppGpp [38] with the transcription of dnaA in E. coli [39] and the level of DnaA protein in C. crescentus [40] being lowered by (p)ppGpp. Another way is via central carbon metabolism that, in E. coli, can: (1) promote DnaA to its active DnaA-ATP form and its binding to oriC by cAMP (a regulator of this part of metabolism) [41]; (2) suppress the defects of the dnaA46 mutant by changes in pyruvate and acetate metabolism [42]; (3) inhibit DnaA conversion to DnaA-ATP and its binding to oriC (via acetylation of DnaA, with acetyl-CoA and acetyl-phosphate as donors) (for references see [43]). Evidence for the involvement of the central carbon metabolism in DNA replication in B. subtilis includes: (1) subunits of pyruvate dehydrogenase (PdhC) and related enzymes bind the origin of replication region, DnaC and DnaG inhibit the initiation of replication; (2) mutations in the genes of central carbon metabolism suppress initiation and elongation defects in dnaC, dnaG, and dnaE mutants; (3) mutations in gapA (which encodes glyceraldehyde 3-phosphate dehydrogenase) perturb the metabolic control of replication; (4) pyruvate kinase (PykA) can both inhibit initiation and stimulate elongation via proposed interactions with DnaC, DnaG, and DnaE as modulated perhaps by phosphorylation [43,44].
Given that metabolic enzymes can exist as hyperstructures in their own right, a question that arises here is whether metabolic enzymes are also part of an initiation hyperstructure. And a related question is whether the metabolites themselves are directly part of initiation and/or replication hyperstructures not just by binding to the constituent proteins (as in the case of DnaA and cAMP [41]) or by being used to modify the protein (as in the case of DnaA and acetyl-CoA) but also by binding directly to RNA or DNA. If metabolites were indeed to bind the origin region, this would make the connection with the Ring World, an origins-of-life scenario in which small, double-stranded DNA rings were selected firstly because they catalysed the reactions of central carbon metabolism [45].
7. Does the DnaA-initiation hyperstructure contain SeqA?
E. coli avoids multiple re-initiations of chromosome replication by a sequestration mechanism that depends on the SeqA protein binding preferentially to newly replicated, hemi-methylated GATC sites, many of which are clustered in oriC. This sequestration, which involves the membrane, occurs only when oriC is hemi-methylated [46] and when SeqA, which has an affinity for membrane, is present [47,48]; the result is an inhibition of initiation [49]. SeqA forms multimers and SeqA-DNA complexes can cover 100 kb of DNA and are close or integral to the replication hyperstructure(s) (and have a bidirectional movement that differs from that of the origins (which go to the poles) [50,51]. GATC sites are clustered not only in the oriC region but also in many genes involved in the replication and repair of DNA (such as dnaA, dnaC, dnaE, gyrA, topA, hepA, lhr, parE, mukB, recB, recD, and uvrA) as well as genes involved in the synthesis of the precursors of DNA (such as nrdA, purA, purF, purL, pyrD and pyrI) consistent perhaps with the presence of these genes and their products in a replication hyperstructure [52]. One related question is whether the DnaA-initiation hyperstructure in its earliest form contains SeqA and, reciprocally, a second question is whether the replication hyperstructure contains DnaA?
8. What is the relationship between strand opening and DnaA binding?
Katayama’s group analysed the opening of M13-oriC DNA in vitro using various types of mutated oriC, DnaA and the Nucleoid-Associated Protein, IHF; the results obtained were consistent with those obtained in vivo [53]. We should, however, be cautious in attributing the results of P1 nuclease cutting at DUE (DNA unwinding element) to oriC opening; this caution is needed because the interpretation is based solely on measuring nuclease cutting that, moreover, only affected half of the substrate added [53].
Recently, Strick’s group performed a single molecule analysis on DnaA–oriC(2kb) interaction using an optical magnetic tweezer to follow the rapid kinetics of double-stranded DNA opening. They observed formation of stable complexes between supposedly DnaA-ATP oligomers and oriC with different degrees of positive supercoiling. The formation of these complexes occurred using an oriC that lacked the DUE, raising the question of whether they were studying a non-canonical reaction. Other questions include why the kinetics of the complex formation was not studied, why the formation of the complex did not occur constantly [54] and whether DnaA was actually present in the complex. It should be pointed out that the use of optical magnetic tweezers is technically demanding: it requires the attachment of oriC DNA to a magnetic bead followed by the selection of intact oriC-containing beads (which are easily damaged and consequently in a minority).
Techniques based on minicircles of DNA facilitate the detection of fine-scale modifications to the DNA structure. Using an oriC minicircle of 641bp with 3 negative supercoils, Landoulsi and Kohiyama found that around 80% of this substrate was positively twisted 3 times during incubation with DnaA and that the efficiency of unwinding was affected by the degree of negative superhelicity of the minicircle (3 negative turns proved more effective in causing unwinding than 2 or 4 negative turns). Unwinding of this oriC minicircle by DnaA was verified by Bal31 sensitivity (rather than by P1 nuclease sensitivity) whilst the presence of DnaA on the unwound minicircle was confirmed by an anti-DnaA antiserum. The problem raised by this work is the unwinding did not require ATP [55] despite the demonstration by Kornberg’s group that the unwinding of oriC plasmid DNA by DnaA does require ATP [56]. It should also be noted that the above work on oriC minicircles depends on a sophisticated technique that requires the formation of circles from a linear 641bp oriC fragment that can only be achieved in a glass capillary after overnight incubation in the presence of DNA ligase and ethidium bromide, which introduces superhelicity; modification of the superhelicity of minicircles resulting from DnaA action is scored after Topo I treatment and is not directly measured.
This work raised the question of whether or not the ATP-dependent opening of oriC by DnaA (as demonstrated by P1 nuclease sensitivity) is the unique pathway for the initiation of replication. The fact that the mutant isolated first, dnaA46, which has lost the ATP binding site, can grow normally at low temperature indicates the existence of an alternative pathway whereby oriC can fire without ATP. Consistent with this, the growth of dnaA46 is more sensitive than the wild type to gyrase inhibitors [57] whilst the opening of oriC minicircles by DnaA is sensitive to negative supercoiling densities [55] (see above). Unfortunately, no data are presently available from X-ray crystallography of the whole molecule of DnaA or from cryoEM analysis of DnaA-oriC.
9. Does DnaA participate in differentiation?
In the strand segregation hypothesis, a coherent phenotypic diversity is generated by the segregation of certain hyperstructures with only one of the parental DNA strands [58]; candidate hyperstructures for such asymmetric segregation include those containing the Nucleoid-Associated Proteins (NAPs) and the topoisomerases. An asymmetric segregation of a chromosomal DnaA hyperstructure is another seductive possibility: could DnaA play a particular role in generating phenotypic diversity (e.g., in preparing a population to confront stresses via its role in modulating gene expression) or in connecting different phenotypes with different patterns of the cell cycle—or indeed both?
10. What modifications does DnaA undergo and what are their roles?
It has been proposed that a hyperstructure might be assembled if enzymes (such as protein kinases) and their NAP substrates were to associate with one another in a positive feedback loop in which, for example, the modification of a NAP by its cognate kinase increases the probability of colocation of both the NAPs and the kinase [58]. In line with this, the acetylation of a lysine residue (K178) prevents DnaA from binding to ATP and inhibits initiation whilst the acetylation of another lysine residue (K243) also inhibits initiation but does not affect the ATP/ADP binding affinity of DnaA or the ability of DnaA to bind to the dnaA promoter region and to DARS1 [59].
DnaA binds cAMP with a Kd of a similar order to that with which it binds to ATP; indeed, the affinity of DnaA for cAMP is such that most of the cell’s DnaA should be bound to cAMP when the latter is present at the physiological concentration of 1 µM [41]. cAMP bound to DnaA is chased by ATP but not by ADP (note that there is only one cAMP binding site on the protein [41]). in vitro, cAMP stimulates DnaA binding to oriC and to DnaA sites elsewhere in the chromosome [41]; in vivo, the addition of cAMP to a cya mutant (which encodes the adenylate cyclase that catalyses the production of cAMP) increased the level of DnaA [60]. Significantly, despite DnaA’s stability in vivo, it has recently been shown to be degraded in vivo in ATP-depletion conditions [61] and one possibility is that cAMP helps both protect DnaA from degradation and regenerate DnaA-ATP from DnaA-ADP (by causing the release of the bound ADP). This raises the question of whether the state of the environment as reflected in a cAMP signal is transduced by the level of DnaA and by the DnaA-ATP: DnaA-ADP ratio into the expression of DnaA-regulated genes and cell cycle timing.
In the case of Caulobacter crescentus, the phosphorylation status of CtrA is central to cell cycle progress [62]. The many possible post-translational modifications to DnaA and to other proteins in the initiation and replication hyperstructures therefore include phosphorylation, and several other modifications such as succinylation, methylation, proprionylation, malonylation, deamidation of asparagines, and glycosylation (for references see [58]). Another post-translational modification—and one that is largely ignored—is the covalent addition of poly-(R)-3-hydroxybutyrate (PHB) to proteins [63]; one proposed function of such addition to NAPs would be to regulate their interaction with nucleic acids [64]. An important question is therefore whether DnaA undergoes modifications like the addition of PHB and, if so, does such modification help the type of hyperstructure into which DnaA assembles?
11. Is DnaA a controller of chromosomal copy numbers rather than a timer?
Fralick found that the timing of initiation and the number of replicating chromosomes per cell (and the DNA /mass ratio) could be varied independently of one another in a temperature-sensitive dnaA(ts) mutant grown at different temperatures. These results were interpreted as DnaA being an essential component of the ‘replication apparatus’ but not itself being the signal that triggers initiation [65,66]. This interpretation would be consistent with the finding that the time of initiation is not advanced by a 50% increase in the concentration of DnaA-ATP, with the authors concluding that although DnaA protein is required for initiation of synchronous and well-timed replication cycles, the accumulation of DnaA-ATP does not control the time of initiation [67]. It should be noted that stopping the transcription of dnaA only led to a small increase in cell size as DnaA was diluted by growth whilst only disrupting RIDA had a major effect on initiation [28]. Finally, a mathematical model has recently been proposed that combines the titration and activation of DnaA strategies to explain how initiation might be timed at fast and slow growth rates to give both a precise volume per origin and the addition of a constant volume between initiations [68].
12. Does the MMS operon play an important role in initiation?
The macromolecular synthesis (MMS) operon is highly conserved [69]; in E. coli, it comprises three genes: rpsU, which encodes the S21 ribosomal protein, dnaG, which encodes the DNA primase involved in the initiation of chromosome replication, and rpoD, which encodes the principal sigma subunit of RNA polymerase. This operon is subject to a complex pattern of internal and external regulation in which it is possible to regulate each of its three genes independently of the others. In a series of investigations of heterogeneous responses to environmental stresses in Listeria monocytogenes, it was found that acid and other stresses primarily selected for rpsU variants, in some of which 116 genes were upregulated, mainly those controlled by the alternative stress sigma factor SigB; this leads to the hypothesis (1) that single amino acid substitutions in RpsU enable L. monocytogenes to switch between high fitness-low stress resistance and low fitness-high stress resistance and (2) that RpsU interacts with the stressosome, a stress-related hyperstructure responsible for integrating information about multiple environmental stresses and transmitting this as signals [70,71]. How might this relate to hyperstructure dynamics? If the MMS operon exists as a hyperstructure based on coupled transcription-translation, speculative hypotheses that might be entertained include an MMS hyperstructure being physically associated with an initiation/replication hyperstructure or a ribosomal hyperstructure; such association could then supply newly synthesized proteins directly to the appropriate hyperstructure (in the case of the primase, for lagging strand synthesis). Alternatively, the association between the MMS hyperstructure and another hyperstructure could result in the sequestering of newly synthesized proteins leading in E. coli, for example, to a reduction in the level of sigma70 (the ‘house-keeping’ sigma) thereby favouring the other sigma factors and the emergence of a stress-adapted phenotype.
13. Does DnaA or a DnaA-based initiation hyperstructure also trigger division?
DNA replication is clearly coupled to cell division insofar as signalling systems exist to prevent division when DNA has been damaged [72]. These include the SOS system that, when induced by DNA damage, produces the SulA/SfiA protein (along with forty other proteins) to interfere with the action of the key protein in cell division, FtsZ [73]. DNA replication is also coupled to cell division insofar as several ts mutants affected in the initiation or elongation steps of DNA replication stop dividing normally at non-permissive temperature and form filamentous cells that resemble those formed by a thy mutant during thymine starvation [74]. This cessation of division does not, however, necessarily mean that some aspect of the replication of the chromosome (including termination of replication) is responsible for the initiation of cell division. Indeed, after further cultivation of ts replication mutants at the non-permissive temperature, division resumes towards the ends of the filamentous cells to produce cells that lack chromosomal DNA; these anucleate cells are of almost normal size. This production of anucleate cells occurs with dnaA, dnaC, dnaG (parB) and dnaB ts mutants and requires the absence of the inhibitor of division, SulA/SfiA, and a mutation in ftsZ (sfiB). How might this production occur?
It may be significant that the above anucleate cell production also requires cAMP (via either the activity of the wild type cya gene or an exogenous supply of cAMP) along with the cAMP receptor protein, CAP [74]. CAP regulates the transcription of over a 100 genes in E. coli, including those in the lac operon. It is therefore conceivable that (1) there are major differences in the structure of the membrane in the presence and absence of cAMP and CAP and (2) these differences could affect transertion (e.g., via Lac permease) and hence the membrane domain dynamics that are proposed to time and position division [75,76]. If membrane dynamics do indeed underpin the regulation of the cell cycle at a fundamental level, it would make sense for proteins such as DnaA to respond to this fundamental system too, given that these sophisticated proteins presumably evolved some time after protocells had achieved some mastery over replication and division.
It could also be argued that would have made sense for the earliest ‘protocells’ to have had the same signalling mechanism leading to both DNA replication and cell division. This is because the RNA and/or DNA in these protocells was probably short and, in one scenario, in the form of a population of rings [45,77]; hence, the fundamental problem that protocells had to solve was not how to divide after replicating a long stretch of DNA but rather how to proceed successfully through a complete cell cycle, which is a single decision. VN has proposed that making this decision requires both intensity-sensing (does a cellular constituent risk becoming limiting for growth) and quantity-sensing (is there enough material to make viable daughter cells?) [78]. Once a signalling mechanism had been adopted, it would be understandable if modern cells had been constrained to have retained the essence of this mechanism (even if overlain by the complex web of modern macromolecules). It turns out that there is some evidence based on the relationship between the physical properties of the membrane and the distribution of the nucleoids consistent with the idea that the initiation of replication and the initiation of division might indeed be triggered at the same time and, if so, logically by the same process (like transertion) [79]. Phospholipids are not just associated with the initiation hyperstructure but also with the division hyperstructure, and in B. subtilis, for example, most of the phospholipid synthases are located in the membrane part of the hyperstructure, which is enriched in cardiolipin and phosphatidylethanolamine. Another finding consistent with a close relationship between the DNA replication and cell division is that an excess of DnaA (or an effective excess due to a deletion of datA) resulted in cell division in the absence of replication to generate anucleate cells [80]. In terms of modern hyperstructures, one explanation for the dependence of anucleate cell production on cAMP by dna(ts) mutants is that the membrane dynamics driving both replication and division hyperstructures is affected by DnaA binding to cAMP.
14. Do variations in the speed of the elongation step of DNA replication matter?
Oscillations in the speed of the replisome along the E. coli chromosome have been reported and possibly explained as due to the initiation of new replisomes slowing the progress of existing ones [81]. Temporal oscillations in the speed of the replisome have also been found by others in E. coli, Vibrio cholerae and B. subtilis with short pauses at ribosomal genes [82]. These oscillations also showed a time-dependent or bilateral symmetry about the origin, consistent with global variations in the availability of an element essential for replication (see below). Significant variations in the level of ATP between individual E. coli cells have been reported [83] whilst complex oscillatory variations in this level occur in individual cells during the cell cycle with an average maximum of 2.4 mM and minimum of 1.2 mM [84]. Variations in the speed of replication in different places in the chromosome have been proposed to help determine the phenotype [85]. Could such variations be studied at the level of single cells? One technique that might be used is the CIS technique (for Combing and Imaging by Secondary Ion Mass Spectrometry), which can detect individual DNA fragments labelled in vivo with stable isotopes on the scale of a few hundred base pairs [86,87].
15. Does an initiation hyperstructure sense DNA supercoiling?
The supercoiling state of chromosomal DNA varies according to the growth phase and to extracellular stresses such as osmotic shock, heat, pH, and antibiotics [88,89,90]. It can also vary along the chromosome and can form a spatiotemporal gradient running from replication origin to terminus on both arms of the E. coli chromosome [91]. DNA gyrase has been proposed to act as a negative regulator of DnaA-dependent replication initiation from oriC in B. subtilis since gyrase activity decreases DnaA association with oriC and inhibits replication initiation [92]. A deficiency of Topoisomerase I increases negative supercoiling, which results in the formation of transcription-associated RNA-DNA hybrids (R-loops), and DnaA- and oriC-independent constitutive stable DNA replication [93]. In other words, the initiation hyperstructure can take more than one form in response to different inputs such as supercoiling and the state of DnaA, thereby acting as an OR gate.
16. Is ribonucleotide reductase an essential constituent of the initiation hyperstructure?
The initiation and elongation steps of chromosome replication are tightly coordinated and mutually dependent in all organisms: the inhibition of initiation results in an increase in elongation rates and vice versa [94,95,96,97,98]. The observation of a negative correlation between initiation and elongation suggests that either directly or indirectly, initiation of DNA replication and elongation of DNA synthesis are interdependent. In eukaryotes, under physiological conditions, a clear negative correlation has been observed between replicon size (length of DNA replicated bidirectionally) and DNA replication fork rates, while inhibiting elongation at replication forks induces the activation of additional replication origins termed “dormant origins” [99]. Could this interrelationship between DNA replication initiation and elongation involve ribonucleoside diphosphate reductase (RNR), which supplies the deoxyribonucleotides (dNTPs) that are essential for replication? Could there be a relationship between the oscillations in ATP during cell growth [84], the oscillations in the speed of the replisome with its pauses at ribosomal genes [82] and the activity of RNR—indeed could a need to divert ribonucleotides into dNTPs be one explanation why there is no transcription during S-phase in eukaryotes?
The pool of available dNTPs is critical for successful replication since, with a defective supply, the DNA is likely to be damaged. Decreases in the size of the dNTP pool result in increases in the C period and vice versa, consistent with a major role for this pool in replication speed [100,101,102,103]. It would make no apparent sense then for initiation of replication to occur without the availability of this pool—and without the ability of RNR to supply dNTPs continuously at the right rate. Localisation of RNR is consistent with this enzyme being part of a replication hyperstructure [104,105] and, importantly, only a very small pool of dNTPs accumulates in the cell, which would allow for no more than one half minute of replication [106,107]. This would suggest the need for ribonucleotide reductase to be present and active at or near the replication forks both at the time of initiation and during elongation.
The question then is whether RNR must be functioning for initiation to occur—and, possibly, functioning in the right place? Put differently, could RNR act as a sensor—or allow the initiation hyperstructure to act as a sensor—in order to couple metabolism and cell growth with replication? If so, could the very activity of RNR determine its presence in the initiation hyperstructure and the ability of this hyperstructure to trigger replication, as proposed for functioning-dependent structures [108]?
The rate of replication fork movement in all organisms depends on the activity of the enzyme RNR, which controls the level and balance of dNTP pool sizes during DNA synthesis (the S-phase in eukaryotes and the C-period in bacteria). The rate of replication fork movement also depends on a number of other factors including the lagging strand DNA polymerase DnaE in B. subtilis [109] and the replication elongation factor DnaX in E. coli [110]. Consistent with an initiation-elongation regulatory circuit, it has been found in E. coli that DnaA regulates the nrdAB gene, which encodes RNR [111,112,113,114]. Low levels/concentrations of DnaA-ATP stimulate nrdAB expression (presumably prior to initiation) while high levels inhibit nrdAB expression (presumably at the time of initiation). Various studies have shown that DnaA-ATP modulates the level of nrdAB transcription and RNR activity, such that the active DnaA-ATP form of the protein correlates with both the number of replication forks and dNTP levels [113,114]. Genetic evidence for the existence of such a regulatory circuit has been established with the identification of suppressors of elongation mutants (dnaX2016) in E. coli that usually map to the dnaA gene in both E. coli and B. subtilis [110,115], while suppressors of the mutant hda gene, which overinitiates DNA replication, map to the nrdAB gene [116,117]. Suppressors of the dnaAcos mutant, which also overinitiates DNA replication, likewise map to the nrdAB locus [118,119].
As noted above, initiation in E. coli is a complex process involving the formation of a multi-component replication hyperstructure (composed of RNR, DnaA and possibly SeqA and other factors) that activates initiation at a specific cell mass called the “initiation mass” [104]. The initiation mass is independent, it appears, of cell growth rate. How the cell “senses” the initiation mass, and therefore “knows” when to initiate chromosome duplication has remained a mystery since the initiation mass concept was first introduced over fifty years ago. It is known, however, that neither DnaA, which controls the frequency of initiation, nor RNR, which controls the rate of DNA synthesis, appears to be the primary determinant of the timing of initiation or of the setting the initiation mass (Flatten 2015, [114] (see above ‘Is DnaA a controller of chromosomal copy numbers rather than a timer?’).
Cells with reduced dNTP levels, however, initiate DNA replication earlier in the cell cycle (immediately after cell division) compared to wild type cells, but at a relatively larger cell size and hence at the same initiation mass [102]. This observation might suggest a link between the rate of elongation and the initiation mass itself, since DNA replication is coupled to cell growth albeit by an unknown mechanism [109]. This raises an interesting question: does the rate of elongation, which is coupled to cell growth in the mother cell, set the initiation mass in the daughter cells, instead of the initiation mass setting the time of initiation in the daughter cell cycles? If so, then the rate of elongation rather than the initiation mass itself might be the decisive parameter that determines the major events driving the bacterial cell cycle. Rephrasing the question more precisely: is the initiation mass a passive consequence of the coupling between replication and growth or is it, as commonly believed, an active cause of initiation and its timing in the cell cycle?
The independence of the initiation mass from the cell growth rate strongly suggests a metabolic link to the signalling of replication initiation, a link that remains poorly elucidated to date. In eukaryotes, DNA synthesis takes place during the reductive (biosynthetic) phase of the cell cycle and coincides with an abrupt rise in reactive oxygen species (ROS) at the G1 oxidative phase/ S-phase transition, which suggests that the cellular metabolic state plays a role in signalling the timing of initiation at a critical cell physiology/mass, at least in eukaryotes [120,121,122]. As mentioned above (see Does the initiation hyperstructure contain glycolytic enzymes?), in B. subtilis, a number of enzymes involved in metabolism have been shown to be associated with both replication initiation and elongation (specifically the DnaC helicase, DnaG primase and DnaE lagging strand polymerase) whilst in E. coli, carbon metabolism plays an important role in DNA replication fidelity and correlates with DNA synthesis [123,124,125].
It is tempting to speculate that levels and balances in metabolite pool sizes play a significant regulatory role in signalling and controlling important cell cycle processes such as the accumulation of the initiation mass, the timing of the initiation of DNA replication, the rate of chromosome elongation and cell division. Clearly, these events are tightly coordinated and co-regulated. One way is via post-translational modifications such as the acetylation of DnaA (see above What modifications does DnaA undergo and what are their roles?) and it may be significant that the acetylation of RNR in human cells results in the reduction of the dNTP pool and DNA replication fork stalling [126]. That said, questions concerning the role of metabolism in coordinating and regulating critical cell cycle functions have yet to be fully answered. It would be interesting, for example, to investigate how NADP(H):NAD+, ATP:ADP and NTP:dNTP pool sizes co-vary during the cell cycle and whether or not they might play a role in determining the initiation mass, and thus prove informative in revealing the mysterious mechanism(s) by which the initiation mass appears to coordinate and control the major events of the cell cycle—if in fact it does.
17. Miscellaneous questions
Eberle and collaborators performed a series of experiments that largely entailed shifting a growing culture of a dnaA(ts) strain (and sometimes a dnaC(ts) strain) to the non-permissive temperature for a hour and then returning that culture to the permissive temperature in the presence or absence of chloramphenicol; in the former case, this resulted in 4 to 5 initiation events as opposed to just one in the latter case [127]. Only ten minutes of inhibition of protein synthesis were needed to produce these extra initiations [128]. Could seeing initiation in terms of hyperstructure dynamics help explain these results? For example, is it possible that the inhibition of protein synthesis, which would disrupt a MMS hyperstructure (or perturb a hyperstructure to which the MMS operon would normally contribute), would therefore inhibit an initiation hyperstructure? A complementary possibility is that the drop in temperature resulted in the decondensation of ions from oriC and associated proteins within a hyperstructure leading to the opening of the strands and initiation [78].
As mentioned above (Does an initiation hyperstructure sense DNA supercoiling?), E. coli can grow despite inactivation of oriC and dnaA provided cells lack enzymes such as RNase H, which removes RNA-DNA hybrids in the form of R-loops [129]. This is because replication can be initiated at multiple ectopic oriK sites (for which a consensus sequence has yet to be defined) elsewhere on the chromosome [130,131]. It has been proposed that this ‘constitutive stable replication’ may be a relic of the replication used by early cells [132]. Assuming that increasing transcription at an oriK increases the probability of replication, it is tempting to speculate that such coupling could provide an intensity-sensing mechanism to allow replication of DNA before it becomes limiting for growth [133]. That said, it is difficult to square this simple mechanism with the apparently normal timing of minichromosome replication in conditions in which chromosome replication itself is random (see below).
Eliasson and Nordstrom used an integratively suppressed strain to investigate minichromosome replication [134]. In this strain, the chromosomal oriC is inactive and replication occurs at random from a plasmid origin (P1); despite this, the rounds of replication of minichromosomes as seen using density-shifts occurred at cell cycle intervals, consistent with an signal for initiation still being generated at the normal time [134]. The authors argued against an artefact due to a lengthening of the eclipse period—a period during which a newly replicated origin is refractory to a second initiation event [135]—but rather proposed that the minichromosome replication they observed was not being triggered by the process of chromosome replication; in other words, the system that normally triggers chromosome replication continued working even when chromosome replication was random [134]. This result therefore appears to call into question models based on the chromosome being an integral part of the cell cycle clock. Is it possible to explain the result by invoking the operation of a ‘primitive’ physico-chemical clock based, for example, on hyperstructure dynamics? In particular, could the result be explained by the cyclically changing states of metabolic, non-equilibrium hyperstructures [78]? Could it even be based on some sort of long-term cellular memory based on hyperstructures, analogous to the memory of exposure to inducer conferred by the existence of a Lac hyperstructure that, once created by a level of inducer, maintains the capacity to metabolise lactose in the subsequent absence of this high level [136,137,138]? If such a memory depended on the segregation of hyperstructures with the DNA strands over the generations, it could give a distribution of growth rates and corresponding cell cycle periods in the population [139].
L-forms are bacteria that manage to grow in the absence of a peptidoglycan layer. They can be obtained by different methods and can have different styles of growth [140]. Division still occurs in E. coli L-forms even though FtsZ levels are five-fold lower than in the cells from which the L-forms are derived [141]; indeed, division can occur in a B. subtilis L-form in the absence of FtsZ [142], which gives a possible insight into the mechanism of division in early cells [143]. What then of DNA replication and its relationship to cell division? The high ratio in a B. subtilis L-form of the number of genomes (as detected by hybridisation) to the number of colony-forming units was attributed to a weaker coupling between chromosome replication and cell division [144]. A similarly high ratio was found in an L-form of Listeria monocytogenes though it was noted that a third of the L-forms could not form colonies [145]; it was also found in this study that, for large L-form cells, those with a high concentration of DNA divided more frequently than those with a low concentration, which was interpreted as the high density of DNA in the former case contributing to the initiation of membrane perturbations and shape changes [145]. That said, it is important to note that the volume of L-form cells can be much greater than that of their walled parents, in which case the L-forms might contain less DNA per volume unit than parental cells. An E. coli L-form revealed a dependence on calcium concentrations in the growth medium with optimum growth at 32 and 37ºC, in 0.1 or 1.0 mM Ca2+, respectively [146]; this is an intriguing result given the relationship between temperature and ion condensation [147] and the putative role for ion condensation in hyperstructure dynamics and cell cycle regulation [78]. Open questions include whether initiation in L-forms depends on DnaA and oriC and where DnaA is located.
18. Discussion
The principles of Molecular Biology have been to isolate and characterise gene products. The success of this reductionist approach has laid the foundations for the complementary, integrative approach based on physics and physical chemistry that shows how these products interact. This is the approach to the bacterial cell cycle that we have adopted here. Jun and collaborators recently proposed a variant of the initiation-titration model [148] that fully exploits the existence of two forms of DnaA, their interconversion and the distribution of two types of binding sites on the chromosome that they validated using a physics-based approach [149]. Kleckner and collaborators proposed that, following the completion of ‘chromosomal and divisome-related events’, what they term a ‘progression control complex’—in other words, a type of hyperstructure—would form [150]; in combination with mass increase, the changes in this complex would trigger cell division and the release of the terminus regions (for generation 1) along with licensing the subsequent triggering of replication via DnaA etc. (for generation 2). In this hypothesis, the two events of cell division and nucleoid transition (which leads to initiation of replication) are independent of one another and could be the separate results of a common upstream event. Boye and Nordstrom argued for chromosome replication and cell division having their own, independent, cycles that are coupled by checkpoints to ensure the correct the order of events with replication and division cycles for E. coli and replication and mitotic cycles for Schizosaccharomyces pombe [151]. The independence of these cycles in bacteria is evidenced when the checkpoints fail: blocking cell division with penicillin does not block chromosome replication whilst blocking replication in the absence of the SOS system does not (ultimately) block division. In eukaryotic cells, S. pombe cells can go through mitosis without a preceding S phase, whilst in meiosis, cells can go through two consecutive reductive cell divisions without intervening DNA replication. They and others propose that these cycles operate in parallel and that they may even be initiated around the same time (for references see [151]). We subscribe to this view but adopt a different approach.
Our approach has been to ask what problems confront systems in general when they must adapt to an environment so as to profit from opportunities for growth and yet survive stresses: this is ‘life on the scales’ [78]. Successful adaptation requires the selection of regulatory criteria that include: sensing when their components risk limiting their growth, sensing when they have enough material for reproducing, sensing when they are becoming too big, avoiding having networks that interfere with one another, and anticipating environmental changes by differentiating. In bacteria, these requirements are met via the cell cycle. To take the case of differentiation, for example, the two daughter cells that result from the cell cycle naturally have different phenotypes unless the species has been selected to prevent this from occurring (here, one might ask whether the cell cycle corresponds to a spandrel [152]).
Once a regulatory system with many interactions between essential components has been constructed it is well-nigh impossible to replace it completely. With this, and with the above criteria in mind, we have tried to formulate hypotheses for the regulation of the cell cycle that can be grounded in a plausible origins-of-life scenario [45,153,154]. Since this system evolved before the emergence of sophisticated macromolecules, it probably depended on the physical chemistry of the interactions of a host of simple molecules in the form of ‘composomes’ [155], the putative ancestors of hyperstructures. This physical chemistry probably included phase separation, molecular crowding, membrane domain formation, ion condensation and, in general, the mechanisms responsible for hyperstructure dynamics. In accord with Occam’s Razor, we speculate that the regulation of the ‘cell cycle’ of the early cells was a single triggering event.
In the context of a physico-chemical approach based on hyperstructures, we have tried here to frame questions about the actors in the regulation of the cell cycle of modern bacteria. The principal actor in the initiation of chromosome replication is DnaA. The sorts of questions that therefore need answering include whether there are chromosomal hyperstructures that depend on DnaA binding to its different sites in the origin, in DARS, in datA and elsewhere, whether there is a membrane hyperstructure that depends on DnaA interacting with lipids, and whether these proposed hyperstructures can form part of a single larger hyperstructure that has a trajectory based on membrane dynamics, DNA supercoiling, crowding, phase separation etc. (see for example [21]). In this trajectory, the DnaA hyperstructure would initiate not only chromosome replication but also, perhaps, cell division. This hyperstructure might act as a logic gate and take into account: the state of transcription, translation and replication as interpreted via the putative hyperstructure created by the MMS operon; the state of metabolism as interpreted via the presence of ribonucleotide reductase or via the binding of metabolites to hyperstructure constituents (like that of cAMP to DnaA) or via the putative hyperstructure created by glycolytic enzymes; the state of the chromosome as interpreted via hyperstructures created by supercoiling. The sorts of questions that need answering include ‘does the DnaA-initiation hyperstructure contain SeqA?’, ‘what is the relationship between strand opening and DnaA binding?’ and ‘what modifications does DnaA undergo and what are their roles?’. At a deeper level, questions also include ‘does DnaA participate in differentiation?’ and ‘does DnaA or a DnaA-based initiation hyperstructure also trigger division?’.
At a still deeper level, the fundamental question is whether the initiation of cell cycle events involves a dialogue between separate hyperstructures (e.g., via the exchange of molecules and macromolecules) or whether this initiation involves a single hyperstructure undergoing changes in structure and composition (or both …). Answering this question may require the development of new techniques, for example by using electro-optic fluorescence microscopy [156], by combining Secondary Ion Mass Spectrometry [157] and toponomics [58,158], and by revisiting often forgotten papers [127,129,134].
Author Contributions
All authors contributed to the conceptualization and writing of the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
MK thanks the Internal Medicine group of Bichat Hospital for providing facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kohiyama, M.; Cousin, D.; Ryter, A.; Jacob, F. [thermosensitive mutants of escherichia coli k 12. I. Isolation and rapid characterization]. Annales de l’Institut Pasteur 1966, 110, 465–486. [Google Scholar] [PubMed]
- Bi, E.F.; Lutkenhaus, J. Ftsz ring structure associated with division in escherichia coli. Nature 1991, 354, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A.; Reichard, P.; Thelander, L. Enzymatic synthesis of deoxyribonucleotides, 8. The effects of atp and datp in the cdp reductase system from e. Coli. Proceedings of the National Academy of Sciences of the United States of America 1965, 54, 830–836. [Google Scholar] [CrossRef]
- Jacob, F.; Brenner, S. [on the regulation of DNA synthesis in bacteria: The hypothesis of the replicon]. Comptes rendus hebdomadaires des seances de l’Academie des sciences 1963, 256, 298–300. [Google Scholar] [PubMed]
- Kohiyama, M.; Lanfrom, H.; Brenner, S.; Jacob, F. [modifications of indispensable functions in thermosensitive eschcerichia coli mutants. On a mutation preventing replication of the bacterial chromosome]. Comptes rendus hebdomadaires des seances de l’Academie des sciences 1963, 257, 1979–1981. [Google Scholar] [PubMed]
- Hirota, Y.; Ryter, A.; Jacob, F. Thermosensitive mutants of e. Coli affected in the processes of DNA synthesis and cellular division. Cold Spring Harbor symposia on quantitative biology 1968, 33, 677–693. [Google Scholar] [CrossRef]
- Hirota, Y.; Jacob, F. [production of bacteria without DNA]. C R Acad Hebd Seances Acad Sci D 1966, 263, 1619–1621. [Google Scholar]
- Mulder, E.; Woldringh, C.L. Actively replicating nucleoids influence positioning of division sites in escherichia coli filaments forming cells lacking DNA. Journal of bacteriology 1989, 171, 4303–4314. [Google Scholar] [CrossRef]
- Hansen, F.G.; Rasmussen, K.V. Regulation of the dnaa product in escherichia coli. Mol Gen Genet 1977, 155, 219–225. [Google Scholar] [CrossRef]
- Hansen, F.G.; Atlung, T. The dnaa tale. Frontiers in microbiology 2018, 9, 319. [Google Scholar] [CrossRef]
- Fuller, R.S.; Kornberg, A. Purified dnaa protein in initiation of replication at the escherichia coli chromosomal origin of replication. Proceedings of the National Academy of Sciences of the United States of America 1983, 80, 5817–5821. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, J.E.; Leonard, A.C. Blocking, bending, and binding: Regulation of initiation of chromosome replication during the escherichia coli cell cycle by transcriptional modulators that interact with origin DNA. Frontiers in microbiology 2021, 12, 732270. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Ozaki, S.; Kawakami, H.; Katayama, T. Single-stranded DNA recruitment mechanism in replication origin unwinding by dnaa initiator protein and hu, an evolutionary ubiquitous nucleoid protein. Nucleic acids research 2023. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, W.; Chang, R.; Zhang, S.; Yang, G.; Zhao, G. Liquid-liquid phase separation: Unraveling the enigma of biomolecular condensates in microbial cells. Frontiers in microbiology 2021, 12, 751880. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, A. On the role of phase separation in the biogenesis of membraneless compartments. The EMBO journal 2022, 41, e109952. [Google Scholar] [CrossRef]
- Azaldegui, C.A.; Vecchiarelli, A.G.; Biteen, J.S. The emergence of phase separation as an organizing principle in bacteria. Biophys J 2021, 120, 1123–1138. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.N.; Wuite, G.J.L.; Dame, R.T. Effect of different crowding agents on the architectural properties of the bacterial nucleoid-associated protein hu. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Aranovich, A.; Gdalevsky, G.Y.; Cohen-Luria, R.; Fishov, I.; Parola, A.H. Membrane-catalyzed nucleotide exchange on dnaa. Effect of surface molecular crowding. The Journal of biological chemistry 2006, 281, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Aranovich, A.; Braier-Marcovitz, S.; Ansbacher, E.; Granek, R.; Parola, A.H.; Fishov, I. N-terminal-mediated oligomerization of dnaa drives the occupancy-dependent rejuvenation of the protein on the membrane. Bioscience reports 2015, 35. [Google Scholar] [CrossRef]
- Fuller, R.S.; Kaguni, J.M.; Kornberg, A. Enzymatic replication of the origin of the escherichia coli chromosome. Proceedings of the National Academy of Sciences of the United States of America 1981, 78, 7370–7374. [Google Scholar] [CrossRef]
- Woldringh, C.L. The bacterial nucleoid: From electron microscopy to polymer physics-a personal recollection. Life (Basel) 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Monterroso, B.; Zorrilla, S.; Sobrinos-Sanguino, M.; Robles-Ramos, M.A.; Lopez-Alvarez, M.; Margolin, W.; Keating, C.D.; Rivas, G. Bacterial ftsz protein forms phase-separated condensates with its nucleoid-associated inhibitor slma. EMBO reports 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, R.; Ozaki, T.; Moriya, S.; Ogawa, T. Negative control of replication initiation by a novel chromosomal locus exhibiting exceptional affinity for escherichia coli dnaa protein. Genes & development 1998, 12, 3032–3043. [Google Scholar]
- Kasho, K.; Tanaka, H.; Sakai, R.; Katayama, T. Cooperative dnaa binding to the negatively supercoiled data locus stimulates dnaa-atp hydrolysis. The Journal of biological chemistry 2017, 292, 1251–1266. [Google Scholar] [CrossRef]
- Fujimitsu, K.; Senriuchi, T.; Katayama, T. Specific genomic sequences of e. Coli promote replicational initiation by directly reactivating adp-dnaa. Genes & development 2009, 23, 1221–1233. [Google Scholar]
- Frimodt-Moller, J.; Charbon, G.; Krogfelt, K.A.; Lobner-Olesen, A. DNA replication control is linked to genomic positioning of control regions in escherichia coli. PLoS Genet 2016, 12, e1006286. [Google Scholar] [CrossRef]
- Kato, J.; Katayama, T. Hda, a novel dnaa-related protein, regulates the replication cycle in escherichia coli. The EMBO journal 2001, 20, 4253–4262. [Google Scholar] [CrossRef]
- Knoppel, A.; Brostrom, O.; Gras, K.; Elf, J.; Fange, D. Regulatory elements coordinating initiation of chromosome replication to the escherichia coli cell cycle. Proceedings of the National Academy of Sciences of the United States of America 2023, 120, e2213795120. [Google Scholar] [CrossRef]
- Kepes, F. Periodic transcriptional organization of the e.Coli genome. J Mol Biol 2004, 340, 957–964. [Google Scholar] [CrossRef]
- Yung, B.Y.; Kornberg, A. Membrane attachment activates dnaa protein, the initiation protein of chromosome replication in escherichia coli. Proceedings of the National Academy of Sciences of the United States of America 1988, 85, 7202–7205. [Google Scholar] [CrossRef]
- Castuma, C.E.; Crooke, E.; Kornberg, A. Fluid membranes with acidic domains activate dnaa, the initiator protein of replication in escherichia coli. The Journal of biological chemistry 1993, 268, 24665–24668. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Dowhan, W. In vivo evidence for the involvement of anionic phospholipids in initiation of DNA replication in escherichia coli. Proceedings of the National Academy of Sciences of the United States of America 1995, 92, 783–787. [Google Scholar] [CrossRef]
- Regev, T.; Myers, N.; Zarivach, R.; Fishov, I. Association of the chromosome replication initiator dnaa with the escherichia coli inner membrane in vivo: Quantity and mode of binding. PLoS One 2012, 7, e36441. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Kumar, P.; Aggarwal, M.; Sarkari, F.; Wolcott, K.M.; Chattoraj, D.K.; Crooke, E.; Saxena, R. The linker domain of the initiator dnaa contributes to its atp binding and membrane association in e. Coli chromosomal replication. Sci Adv 2022, 8, eabq6657. [Google Scholar] [CrossRef] [PubMed]
- Norris, V.; Madsen, M.S. Autocatalytic gene expression occurs via transertion and membrane domain formation and underlies differentiation in bacteria: A model. J Mol Biol 1995, 253, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Woldringh, C.L. The role of co-transcriptional translation and protein translocation (transertion) in bacterial chromosome segregation. Molecular microbiology 2002, 45, 17–29. [Google Scholar] [CrossRef]
- Fishov, I.; Namboodiri, S. A nonstop thrill ride from genes to the assembly of the t3ss injectisome. Nature communications 2023, 14, 1973. [Google Scholar] [CrossRef]
- Fernandez-Coll, L.; Maciag-Dorszynska, M.; Tailor, K.; Vadia, S.; Levin, P.A.; Szalewska-Palasz, A.; Cashel, M. The absence of (p)ppgpp renders initiation of escherichia coli chromosomal DNA synthesis independent of growth rates. mBio 2020, 11. [Google Scholar] [CrossRef]
- Chiaramello, A.E.; Zyskind, J.W. Coupling of DNA replication to growth rate in escherichia coli: A possible role for guanosine tetraphosphate. Journal of bacteriology 1990, 172, 2013–2019. [Google Scholar] [CrossRef]
- Gonzalez, D.; Collier, J. Effects of (p)ppgpp on the progression of the cell cycle of caulobacter crescentus. Journal of bacteriology 2014, 196, 2514–2525. [Google Scholar] [CrossRef]
- Hughes, P.; Landoulsi, A.; Kohiyama, M. A novel role for camp in the control of the activity of the e. Coli chromosome replication initiator protein, dnaa. Cell 1988, 55, 343–350. [Google Scholar] [CrossRef]
- Tymecka-Mulik, J.; Boss, L.; Maciag-Dorszynska, M.; Matias Rodrigues, J.F.; Gaffke, L.; Wosinski, A.; Cech, G.M.; Szalewska-Palasz, A.; Wegrzyn, G.; Glinkowska, M. Suppression of the escherichia coli dnaa46 mutation by changes in the activities of the pyruvate-acetate node links DNA replication regulation to central carbon metabolism. PLoS One 2017, 12, e0176050. [Google Scholar] [CrossRef] [PubMed]
- Horemans, S.; Pitoulias, M.; Holland, A.; Pateau, E.; Lechaplais, C.; Ekaterina, D.; Perret, A.; Soultanas, P.; Janniere, L. Pyruvate kinase, a metabolic sensor powering glycolysis, drives the metabolic control of DNA replication. BMC Biol 2022, 20, 87. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Pitoulias, M.; Soultanas, P.; Janniere, L. The replicative dnae polymerase of bacillus subtilis recruits the glycolytic pyruvate kinase (pyka) when bound to primed DNA templates. Life (Basel) 2023, 13. [Google Scholar] [CrossRef]
- Norris, V.; Demongeot, J. The ring world: Eversion of small double-stranded polynucleotide circlets at the origin of DNA double helix, rna polymerization, triplet code, twenty amino acids, and strand asymmetry. International journal of molecular sciences 2022, 23. [Google Scholar] [CrossRef]
- Ogden, G.B.; Pratt, M.J.; Schaechter, M. The replicative origin of the e. Coli chromosome binds to cell membranes only when hemimethylated. Cell 1988, 54, 127–135. [Google Scholar] [CrossRef]
- Shakibai, N.; Ishidate, K.; Reshetnyak, E.; Gunji, S.; Kohiyama, M.; Rothfield, L. High-affinity binding of hemimethylated oric by escherichia coli membranes is mediated by a multiprotein system that includes seqa and a newly identified factor, seqb. Proceedings of the National Academy of Sciences of the United States of America 1998, 95, 11117–11121. [Google Scholar] [CrossRef]
- d’Alencon, E.; Taghbalout, A.; Kern, R.; Kohiyama, M. Replication cycle dependent association of seqa to the outer membrane fraction of e. Coli. Biochimie 1999, 81, 841–846. [Google Scholar] [CrossRef]
- Landoulsi, A.; Malki, A.; Kern, R.; Kohiyama, M.; Hughes, P. The e. Coli cell surface specifically prevents the initiation of DNA replication at oric on hemimethylated DNA templates. Cell 1990, 63, 1053–1060. [Google Scholar] [CrossRef]
- Hiraga, S.; Ichinose, C.; Niki, H.; Yamazoe, M. Cell cycle-dependent duplication and bidirectional migration of seqa-associated DNA-protein complexes in e. Coli. Molecular cell 1998, 1, 381–387. [Google Scholar] [CrossRef]
- Helgesen, E.; Saetre, F.; Skarstad, K. Topoisomerase iv tracks behind the replication fork and the seqa complex during DNA replication in escherichia coli. Scientific reports 2021, 11, 474. [Google Scholar] [CrossRef] [PubMed]
- Norris, V.; Fralick, J.; Danchin, A. A seqa hyperstructure and its interactions direct the replication and sequestration of DNA. Molecular microbiology 2000, 37, 696–702. [Google Scholar] [CrossRef]
- Sakiyama, Y.; Kasho, K.; Noguchi, Y.; Kawakami, H.; Katayama, T. Regulatory dynamics in the ternary dnaa complex for initiation of chromosomal replication in escherichia coli. Nucleic acids research 2017, 45, 12354–12373. [Google Scholar] [CrossRef]
- Zorman, S.; Seitz, H.; Sclavi, B.; Strick, T.R. Topological characterization of the dnaa-oric complex using single-molecule nanomanipuation. Nucleic acids research 2012, 40, 7375–7383. [Google Scholar] [CrossRef]
- Landoulsi, A.; Kohiyama, M. Dnaa protein dependent denaturation of negative supercoiled oric DNA minicircles. Biochimie 2001, 83, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Sekimizu, K.; Bramhill, D.; Kornberg, A. Atp activates dnaa protein in initiating replication of plasmids bearing the origin of the e. Coli chromosome. Cell 1987, 50, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Filutowicz, M. Requirement of DNA gyrase for the initiation of chromosome replication in escherichia coli k-12. Mol Gen Genet 1980, 177, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Norris, V.; Kayser, C.; Muskhelishvili, G.; Konto-Ghiorghi, Y. The roles of nucleoid-associated proteins and topoisomerases in chromosome structure, strand segregation and the generation of phenotypic heterogeneity in bacteria. FEMS Microbiol Rev 2022. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Q.; Xu, Z.; Yao, Y.F. Acetylation of lysine 243 inhibits the oric binding ability of dnaa in escherichia coli. Frontiers in microbiology 2017, 8, 699. [Google Scholar] [CrossRef] [PubMed]
- Landoulsi, A.; Kohiyama, M. Initiation of DNA replication in delta cya mutants of escherichia coli k12. Biochimie 1999, 81, 827–834. [Google Scholar] [CrossRef]
- Charbon, G.; Mendoza-Chamizo, B.; Campion, C.; Li, X.; Jensen, P.R.; Frimodt-Moller, J.; Lobner-Olesen, A. Energy starvation induces a cell cycle arrest in escherichia coli by triggering degradation of the dnaa initiator protein. Frontiers in molecular biosciences 2021, 8, 629953. [Google Scholar] [CrossRef] [PubMed]
- Coppine, J.; Kaczmarczyk, A.; Petit, K.; Brochier, T.; Jenal, U.; Hallez, R. Regulation of bacterial cell cycle progression by redundant phosphatases. Journal of bacteriology 2020, 202. [Google Scholar] [CrossRef]
- Seebach, D. No life on this planet without phb. Helvetica Chimica Acta 2023, 106, e202200205. [Google Scholar] [CrossRef]
- Reusch, R.N.; Shabalin, O.; Crumbaugh, A.; Wagner, R.; Schroder, O.; Wurm, R. Posttranslational modification of e. Coli histone-like protein h-ns and bovine histones by short-chain poly-(r)-3-hydroxybutyrate (cphb). FEBS letters 2002, 527, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Fralick, J.A. Studies on the regulation of initiation of chromosome replication in escherichia coli. J Mol Biol 1978, 122, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Fralick, J.A. Is dnaa the ‘pace-maker’ of chromosome replication? An old paper revisited. Molecular microbiology 1999, 31, 1011–1012. [Google Scholar] [CrossRef]
- Flatten, I.; Fossum-Raunehaug, S.; Taipale, R.; Martinsen, S.; Skarstad, K. The dnaa protein is not the limiting factor for initiation of replication in escherichia coli. PLoS Genet 2015, 11, e1005276. [Google Scholar] [CrossRef]
- Berger, M.; Wolde, P.R.T. Robust replication initiation from coupled homeostatic mechanisms. Nature communications 2022, 13, 6556. [Google Scholar] [CrossRef]
- Versalovic, J.; Koeuth, T.; Britton, R.; Geszvain, K.; Lupski, J.R. Conservation and evolution of the rpsu-dnag-rpod macromolecular synthesis operon in bacteria. Molecular microbiology 1993, 8, 343–355. [Google Scholar] [CrossRef]
- Metselaar, K.I.; den Besten, H.M.; Boekhorst, J.; van Hijum, S.A.; Zwietering, M.H.; Abee, T. Diversity of acid stress resistant variants of listeria monocytogenes and the potential role of ribosomal protein s21 encoded by rpsu. Frontiers in microbiology 2015, 6, 422. [Google Scholar] [CrossRef]
- Koomen, J.; Huijboom, L.; Ma, X.; Tempelaars, M.H.; Boeren, S.; Zwietering, M.H.; den Besten, H.M.W.; Abee, T. Amino acid substitutions in ribosomal protein rpsu enable switching between high fitness and multiple-stress resistance in listeria monocytogenes. International journal of food microbiology 2021, 351, 109269. [Google Scholar] [CrossRef]
- Huisman, O.; D’Ari, R.; George, J. Inducible sfi dependent division inhibition in escherichia coli. Mol Gen Genet 1980, 177, 629–636. [Google Scholar] [CrossRef]
- Jones, C.; Holland, I.B. Role of the sulb (ftsz) protein in division inhibition during the sos response in escherichia coli: Ftsz stabilizes the inhibitor sula in maxicells. Proceedings of the National Academy of Sciences of the United States of America 1985, 82, 6045–6049. [Google Scholar] [CrossRef]
- Jaffe, A.; D’Ari, R.; Norris, V. Sos-independent coupling between DNA replication and cell division in escherichia coli. Journal of bacteriology 1986, 165, 66–71. [Google Scholar] [CrossRef]
- Norris, V.; Woldringh, C.; Mileykovskaya, E. A hypothesis to explain division site selection in escherichia coli by combining nucleoid occlusion and min. FEBS letters 2004, 561, 3–10. [Google Scholar] [CrossRef]
- Matsumoto, K.; Hara, H.; Fishov, I.; Mileykovskaya, E.; Norris, V. The membrane: Transertion as an organizing principle in membrane heterogeneity. Frontiers in microbiology 2015, 6, 572. [Google Scholar] [CrossRef] [PubMed]
- Demongeot, J.; Moreira, A. A possible circular rna at the origin of life. J Theor Biol 2007, 249, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Norris, V.; Amar, P. Chromosome replication in escherichia coli: Life on the scales. Life (Basel) 2012, 2, 286–312. [Google Scholar] [CrossRef]
- Fishov, I.; Woldringh, C.L. Visualization of membrane domains in escherichia coli. Molecular microbiology 1999, 32, 1166–1172. [Google Scholar] [CrossRef]
- Morigen, M.; Flatten, I.; Skarstad, K. The escherichia coli data site promotes proper regulation of cell division. Microbiology (Reading, England) 2014, 160, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Bhat, D.; Hauf, S.; Plessy, C.; Yokobayashi, Y.; Pigolotti, S. Speed variations of bacterial replisomes. eLife 2022, 11. [Google Scholar] [CrossRef]
- Huang, D.; Johnson, A.E.; Sim, B.S.; Lo, T.W.; Merrikh, H.; Wiggins, P.A. The in vivo measurement of replication fork velocity and pausing by lag-time analysis. Nature communications 2023, 14, 1762. [Google Scholar] [CrossRef] [PubMed]
- Yaginuma, H.; Kawai, S.; Tabata, K.V.; Tomiyama, K.; Kakizuka, A.; Komatsuzaki, T.; Noji, H.; Imamura, H. Diversity in atp concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Scientific reports 2014, 4, 6522. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.H.; Jacobs-Wagner, C. Connecting single-cell atp dynamics to overflow metabolism, cell growth, and the cell cycle in escherichia coli. Curr Biol 2022, 32, 3911–3924 e3914. [Google Scholar] [CrossRef] [PubMed]
- Norris, V.; Koch, I.; Amar, P.; Kepes, F.; Janniere, L. Hypothesis: Local variations in the speed of individual DNA replication forks determine the phenotype of daughter cells. Medical Research Archives 2017, 5. [Google Scholar]
- Cabin-Flaman, A.; Monnier, A.F.; Coffinier, Y.; Audinot, J.N.; Gibouin, D.; Wirtz, T.; Boukherroub, R.; Migeon, H.N.; Bensimon, A.; Janniere, L. , et al. Combed single DNA molecules imaged by secondary ion mass spectrometry. Anal Chem 2011, 83, 6940–6947. [Google Scholar] [CrossRef]
- Cabin-Flaman, A.; Monnier, A.F.; Coffinier, Y.; Audinot, J.N.; Gibouin, D.; Wirtz, T.; Boukherroub, R.; Migeon, H.N.; Bensimon, A.; Janniere, L. , et al. Combining combing and secondary ion mass spectrometry to study DNA on chips using (13)c and (15)n labeling. F1000Research 2016, 5, 1437. [Google Scholar] [CrossRef]
- Dorman, C.J. Flexible response: DNA supercoiling, transcription and bacterial adaptation to environmental stress. Trends Microbiol 1996, 4, 214–216. [Google Scholar] [CrossRef]
- Blot, N.; Mavathur, R.; Geertz, M.; Travers, A.; Muskhelishvili, G. Homeostatic regulation of supercoiling sensitivity coordinates transcription of the bacterial genome. EMBO reports 2006, 7, 710–715. [Google Scholar] [CrossRef]
- Lal, A.; Dhar, A.; Trostel, A.; Kouzine, F.; Seshasayee, A.S.; Adhya, S. Genome scale patterns of supercoiling in a bacterial chromosome. Nature communications 2016, 7, 11055. [Google Scholar] [CrossRef]
- Muskhelishvili, G.; Forquet, R.; Reverchon, S.; Meyer, S.; Nasser, W. Coherent domains of transcription coordinate gene expression during bacterial growth and adaptation. Microorganisms 2019, 7. [Google Scholar] [CrossRef]
- Samadpour, A.N.; Merrikh, H. DNA gyrase activity regulates dnaa-dependent replication initiation in bacillus subtilis. Molecular microbiology 2018, 108, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Leela, J.K.; Raghunathan, N.; Gowrishankar, J. Topoisomerase i essentiality, dnaa-independent chromosomal replication, and transcription-replication conflict in escherichia coli. Journal of bacteriology 2021, 203, e0019521. [Google Scholar] [CrossRef]
- Hand, R. Regulation of DNA replication on subchromosomal units of mammalian cells. The Journal of cell biology 1975, 64, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Conti, C.; Sacca, B.; Herrick, J.; Lalou, C.; Pommier, Y.; Bensimon, A. Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol Biol Cell 2007, 18, 3059–3067. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 promotes replication fork progression by controlling replication initiation. Proceedings of the National Academy of Sciences of the United States of America 2010, 107, 16090–16095. [Google Scholar] [CrossRef]
- Guzmán, E.; Salguero, I.; Mata Martín, C.; López-Acedo, E.; Guarino Almeida, E.; Sánchez-Romero, M.; Norris, V.; Jiménez-Sánchez, A. Relationship between fork progression and initiation of chromosome replication in e. Coli. In DNA replication, Seligmann, H., Ed. InTechOpen: Rijeka, Croatia, 2011; pp 203-220.
- Menolfi, D.; Lee, B.J.; Zhang, H.; Jiang, W.; Bowen, N.E.; Wang, Y.; Zhao, J.; Holmes, A.; Gershik, S.; Rabadan, R. , et al. Atr kinase supports normal proliferation in the early s phase by preventing replication resource exhaustion. Nature communications 2023, 14, 3618. [Google Scholar] [CrossRef]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How dormant origins promote complete genome replication. Trends in biochemical sciences 2011, 36, 405–414. [Google Scholar] [CrossRef]
- Zaritsky, A.; Pritchard, R.H. Changes in cell size and shape associated with changes in the replication time of the chromosome of escherichia coli. Journal of bacteriology 1973, 114, 824–837. [Google Scholar] [CrossRef]
- Churchward, G.; Bremer, H. Determination of deoxyribonucleic acid replication time in exponentially growing escherichia coli b/r. Journal of bacteriology 1977, 130, 1206–1213. [Google Scholar] [CrossRef]
- Odsbu, I.; Morigen; Skarstad, K. A reduction in ribonucleotide reductase activity slows down the chromosome replication fork but does not change its localization. PLoS One 2009, 4, e7617. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Dai, X.; Guo, W.; Ge, Z.; Yang, M.; Wang, H.; Wang, Y.P. Manipulating the bacterial cell cycle and cell size by titrating the expression of ribonucleotide reductase. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Guzman, E.C.; Caballero, J.L.; Jimenez-Sanchez, A. Ribonucleoside diphosphate reductase is a component of the replication hyperstructure in escherichia coli. Molecular microbiology 2002, 43, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Romero, M.A.; Molina, F.; Jimenez-Sanchez, A. Organization of ribonucleoside diphosphate reductase during multifork chromosome replication in escherichia coli. Microbiology 2011, 157, 2220–2225. [Google Scholar] [CrossRef]
- Werner, R. Nature of DNA precursors. Nature: New biology 1971, 233, 99–103. [Google Scholar] [CrossRef]
- Pato, M.L. Alterations of deoxyribonucleoside triphosphate pools in escherichia coli: Effects on deoxyribonucleic acid replication and evidence for compartmentation. Journal of bacteriology 1979, 140, 518–524. [Google Scholar] [CrossRef]
- Thellier, M.; Legent, G.; Amar, P.; Norris, V.; Ripoll, C. Steady-state kinetic behaviour of functioning-dependent structures. The FEBS journal 2006, 273, 4287–4299. [Google Scholar] [CrossRef]
- Nouri, H.; Monnier, A.F.; Fossum-Raunehaug, S.; Maciag-Dorszynska, M.; Cabin-Flaman, A.; Kepes, F.; Wegrzyn, G.; Szalewska-Palasz, A.; Norris, V.; Skarstad, K. , et al. Multiple links connect central carbon metabolism to DNA replication initiation and elongation in bacillus subtilis. DNA Res 2018, 25, 641–653. [Google Scholar] [CrossRef]
- Skovgaard, O.; Lobner-Olesen, A. Reduced initiation frequency from oric restores viability of a temperature-sensitive escherichia coli replisome mutant. Microbiology (Reading, England) 2005, 151, 963–973. [Google Scholar] [CrossRef]
- Fuchs, J.A.; Karlstrom, H.O. Mapping of nrda and nrdb in escherichia coli k-12. Journal of bacteriology 1976, 128, 810–814. [Google Scholar] [CrossRef]
- Augustin, L.B.; Jacobson, B.A.; Fuchs, J.A. Escherichia coli fis and dnaa proteins bind specifically to the nrd promoter region and affect expression of an nrd-lac fusion. Journal of bacteriology 1994, 176, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Gon, S.; Camara, J.E.; Klungsoyr, H.K.; Crooke, E.; Skarstad, K.; Beckwith, J. A novel regulatory mechanism couples deoxyribonucleotide synthesis and DNA replication in escherichia coli. The EMBO journal 2006, 25, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Olliver, A.; Saggioro, C.; Herrick, J.; Sclavi, B. Dnaa-atp acts as a molecular switch to control levels of ribonucleotide reductase expression in escherichia coli. Molecular microbiology 2010, 76, 1555–1571. [Google Scholar] [CrossRef]
- Janniere, L.; Canceill, D.; Suski, C.; Kanga, S.; Dalmais, B.; Lestini, R.; Monnier, A.F.; Chapuis, J.; Bolotin, A.; Titok, M. , et al. Genetic evidence for a link between glycolysis and DNA replication. PLoS One 2007, 2, e447. [Google Scholar] [CrossRef] [PubMed]
- Fujimitsu, K.; Su’etsugu, M.; Yamaguchi, Y.; Mazda, K.; Fu, N.; Kawakami, H.; Katayama, T. Modes of overinitiation, dnaa gene expression, and inhibition of cell division in a novel cold-sensitive hda mutant of escherichia coli. Journal of bacteriology 2008, 190, 5368–5381. [Google Scholar] [CrossRef] [PubMed]
- Babu, V.M.P.; Itsko, M.; Baxter, J.C.; Schaaper, R.M.; Sutton, M.D. Insufficient levels of the nrdab-encoded ribonucleotide reductase underlie the severe growth defect of the deltahda e. Coli strain. Molecular microbiology 2017, 104, 377–399. [Google Scholar] [CrossRef]
- Charbon, G.; Campion, C.; Chan, S.H.; Bjorn, L.; Weimann, A.; da Silva, L.C.; Jensen, P.R.; Lobner-Olesen, A. Re-wiring of energy metabolism promotes viability during hyperreplication stress in e. Coli. PLoS Genet 2017, 13, e1006590. [Google Scholar] [CrossRef] [PubMed]
- Charbon, G.; Riber, L.; Lobner-Olesen, A. Countermeasures to survive excessive chromosome replication in escherichia coli. Curr Genet 2018, 64, 71–79. [Google Scholar] [CrossRef]
- Havens, C.G.; Ho, A.; Yoshioka, N.; Dowdy, S.F. Regulation of late g1/s phase transition and apc cdh1 by reactive oxygen species. Molecular and cellular biology 2006, 26, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Burhans, W.C.; Heintz, N.H. The cell cycle is a redox cycle: Linking phase-specific targets to cell fate. Free radical biology & medicine 2009, 47, 1282–1293. [Google Scholar]
- Kirova, D.G.; Judasova, K.; Vorhauser, J.; Zerjatke, T.; Leung, J.K.; Glauche, I.; Mansfeld, J. A ros-dependent mechanism promotes cdk2 phosphorylation to drive progression through s phase. Developmental cell 2022, 57, 1712–1727 e1719. [Google Scholar] [CrossRef] [PubMed]
- Maciag, M.; Nowicki, D.; Janniere, L.; Szalewska-Palasz, A.; Wegrzyn, G. Genetic response to metabolic fluctuations: Correlation between central carbon metabolism and DNA replication in escherichia coli. Microb Cell Fact 2011, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Maciag, M.; Nowicki, D.; Szalewska-Palasz, A.; Wegrzyn, G. Central carbon metabolism influences fidelity of DNA replication in escherichia coli. Mutation research 2012, 731, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.; Maciag-Dorszynska, M.; Wosinski, A.; Gaffke, L.; Morcinek-Orlowska, J.; Rintz, E.; Bielanska, P.; Szalewska-Palasz, A.; Muskhelishvili, G.; Wegrzyn, G. The role of metabolites in the link between DNA replication and central carbon metabolism in escherichia coli. Genes (Basel) 2020, 11. [Google Scholar] [CrossRef]
- Chen, G.; Luo, Y.; Warncke, K.; Sun, Y.; Yu, D.S.; Fu, H.; Behera, M.; Ramalingam, S.S.; Doetsch, P.W.; Duong, D.M. , et al. Acetylation regulates ribonucleotide reductase activity and cancer cell growth. Nature communications 2019, 10, 3213. [Google Scholar] [CrossRef]
- Eberle, H.; Forrest, N.; Hrynyszyn, J.; Van Knapp, J. Regulation of DNA synthesis and capacity for initiation in DNA temperature sensitive mutants of escherichia coli i. Reinitiation and chain elongation. Mol Gen Genet 1982, 186, 57–65. [Google Scholar] [CrossRef]
- Eberle, H.; Forrest, N. Regulation of DNA synthesis and capacity for initiation in DNA temperature sensitive mutants of escherichia coli. Ii. Requirements for acquisition and expression of initiation capacity. Mol Gen Genet 1982, 186, 66–70. [Google Scholar] [CrossRef]
- Kogoma, T.; Subia, N.L.; von Meyenburg, K. Function of ribonuclease h in initiation of DNA replication in escherichia coli k-12. Mol Gen Genet 1985, 200, 103–109. [Google Scholar] [CrossRef]
- Maduike, N.Z.; Tehranchi, A.K.; Wang, J.D.; Kreuzer, K.N. Replication of the escherichia coli chromosome in rnase hi-deficient cells: Multiple initiation regions and fork dynamics. Molecular microbiology 2014, 91, 39–56. [Google Scholar] [CrossRef]
- Veetil, R.T.; Malhotra, N.; Dubey, A.; Seshasayee, A.S.N. Laboratory evolution experiments help identify a predominant region of constitutive stable DNA replication initiation. mSphere 2020, 5. [Google Scholar] [CrossRef]
- von Meyenburg, K.; Boye, E.; Skarstad, K.; Koppes, L.; Kogoma, T. Mode of initiation of constitutive stable DNA replication in rnase h-defective mutants of escherichia coli k-12. Journal of bacteriology 1987, 169, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Norris, V. Hypothesis: Transcriptional sensing and membrane-domain formation initiate chromosome replication in escherichia coli. Molecular microbiology 1995, 15, 985–987. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, A.; Nordstrom, K. Replication of minichromosomes in a host in which chromosome replication is random. Molecular microbiology 1997, 23, 1215–1220. [Google Scholar] [CrossRef]
- Zaritsky, A.; Rabinovitch, A.; Liu, C.; Woldringh, C.L. Does the eclipse limit bacterial nucleoid complexity and cell width? Synthetic and systems biotechnology 2017, 2, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Novick, A.; Weiner, M. Enzyme induction as an all-or-none phenomenon. Proceedings of the National Academy of Sciences of the United States of America 1957, 43, 553–566. [Google Scholar] [CrossRef]
- Cohn, M.; Horibata, K. Inhibition by glucose of the induced synthesis of the beta-galactoside-enzyme system of escherichia coli. Analysis of maintenance. Journal of bacteriology 1959, 78, 601–612. [Google Scholar] [CrossRef]
- Laurent, M.; Charvin, G.; Guespin-Michel, J. Bistability and hysteresis in epigenetic regulation of the lactose operon. Since delbruck, a long series of ignored models. Cellular and molecular biology (Noisy-le-Grand, France) 2005, 51, 583–594. [Google Scholar]
- Norris, V.; Ripoll, C. Generation of bacterial diversity by segregation of DNA strands. Frontiers in microbiology 2021, 12. [Google Scholar] [CrossRef]
- Glover, W.A.; Yang, Y.; Zhang, Y. Insights into the molecular basis of l-form formation and survival in escherichia coli. PLoS One 2009, 4, e7316. [Google Scholar] [CrossRef]
- Onoda, T.; Enokizono, J.; Kaya, H.; Oshima, A.; Freestone, P.; Norris, V. Effects of calcium and calcium chelators on growth and morphology of escherichia coli l-form nc-7. Journal of bacteriology 2000, 182, 1419–1422. [Google Scholar] [CrossRef]
- Leaver, M.; Dominguez-Cuevas, P.; Coxhead, J.M.; Daniel, R.A.; Errington, J. Life without a wall or division machine in bacillus subtilis. Nature 2009, 457, 849–853. [Google Scholar] [CrossRef]
- Mercier, R.; Kawai, Y.; Errington, J. Excess membrane synthesis drives a primitive mode of cell proliferation. Cell 2013, 152, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.N.; Allan, E.J.; Amijee, F.; Undril, V.J.; Glover, L.A. An investigation of enumeration and DNA partitioning in bacillus subtilis l-form bacteria. Journal of Applied Bacteriology 1994, 77, 497–503. [Google Scholar] [CrossRef]
- Studer, P.; Staubli, T.; Wieser, N.; Wolf, P.; Schuppler, M.; Loessner, M.J. Proliferation of listeria monocytogenes l-form cells by formation of internal and external vesicles. Nature communications 2016, 7, 13631. [Google Scholar] [CrossRef] [PubMed]
- Onoda, T.; Oshima, A. Effects of ca2+ and a protonophore on growth of an escherichia coli l-form. J Gen Microbiol 1988, 134, 3071–3077. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, C.; Norris, V.; Thellier, M. Ion condensation and signal transduction. BioEssays 2004, 26, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hansen, F.G.; Christensen, B.B.; Atlung, T. The initiator titration model: Computer simulation of chromosome and minichromosome control. Res Microbiol 1991, 142, 161–167. [Google Scholar] [CrossRef]
- Fu, H.; Xiao, F.; Jun, S. Replication initiation in bacteria: Precision control based on protein counting. bioRxiv, 2005. [Google Scholar]
- Kleckner, N.E.; Chatzi, K.; White, M.A.; Fisher, J.K.; Stouf, M. Coordination of growth, chromosome replication/segregation, and cell division in e. Coli. Frontiers in microbiology 2018, 9, 1469. [Google Scholar] [CrossRef]
- Boye, E.; Nordstrom, K. Coupling the cell cycle to cell growth. EMBO reports 2003, 4, 757–760. [Google Scholar] [CrossRef]
- Amir, A. Is cell size a spandrel? eLife 2017, 6. [Google Scholar] [CrossRef]
- Hunding, A.; Kepes, F.; Lancet, D.; Minsky, A.; Norris, V.; Raine, D.; Sriram, K.; Root-Bernstein, R. Compositional complementarity and prebiotic ecology in the origin of life. Bioessays 2006, 28, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Hassenkam, T.; Deamer, D. Visualizing rna polymers produced by hot wet-dry cycling. Scientific reports 2022, 12, 10098. [Google Scholar] [CrossRef] [PubMed]
- Segre, D.; Ben-Eli, D.; Lancet, D. Compositional genomes: Prebiotic information transfer in mutually catalytic noncovalent assemblies. Proceedings of the National Academy of Sciences of the United States of America 2000, 97, 4112–4117. [Google Scholar] [CrossRef]
- Bowman, A.J.; Huang, C.; Schnitzer, M.J.; Kasevich, M.A. Wide-field fluorescence lifetime imaging of neuron spiking and subthreshold activity in vivo. Science (New York, N.Y 2023, 380, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- Lechene, C.; Hillion, F.; McMahon, G.; Benson, D.; Kleinfeld, A.M.; Kampf, J.P.; Distel, D.; Luyten, Y.; Bonventre, J.; Hentschel, D. , et al. High-resolution quantitative imaging of mammalian and bacterial cells using stable isotope mass spectrometry. J Biol 2006, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Schubert, W.; Gieseler, A.; Krusche, A.; Serocka, P.; Hillert, R. Next-generation biomarkers based on 100-parameter functional super-resolution microscopy tis. N Biotechnol 2012, 29, 599–610. [Google Scholar] [CrossRef]
Figure 1.
Control of the bacterial cell cycle by hyperstructures and DnaA. The boxes represent hyperstructures. A clock or timer based on physical chemical processes provides the intensity and quantity (IQ) sensing needed for a successful cell cycle. This clock sends a signal to initiate both chromosome replication (via a DnaA-based initiation hyperstructure) and cell division. The initiation and replication hyperstructures can inhibit the cell division process (orange arrows).
Figure 1.
Control of the bacterial cell cycle by hyperstructures and DnaA. The boxes represent hyperstructures. A clock or timer based on physical chemical processes provides the intensity and quantity (IQ) sensing needed for a successful cell cycle. This clock sends a signal to initiate both chromosome replication (via a DnaA-based initiation hyperstructure) and cell division. The initiation and replication hyperstructures can inhibit the cell division process (orange arrows).
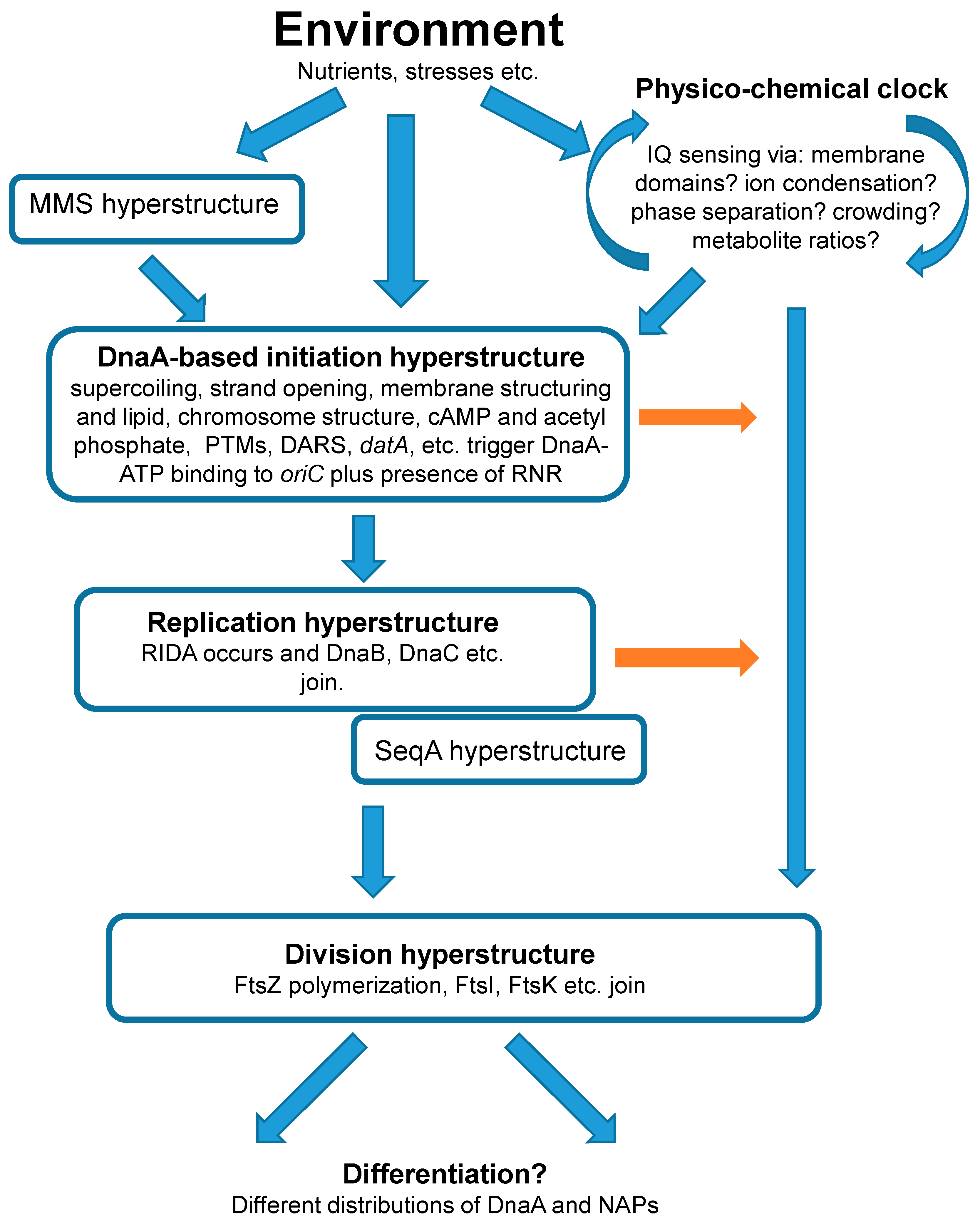
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



