
Submitted:
19 July 2023
Posted:
20 July 2023
You are already at the latest version
Abstract
Leptospirosis is a zoonotic disease of global importance. The current killed vaccine provides serovar-specific protection without sterilizing immunity. Several surface antigens, including Leptospira immunoglobulin-like proteins (LigA and LigB), have been identified as subunit vaccine candidates; however, they require potent adjuvants. Bacterial Lipopolysaccharide (LPS), including lipid A, is a well-known immunostimulatory agent, and the formulation of Monophosphoryl Lipid A (MPLA) in Alum is a clinical adjuvant. Being less endotoxic, we tested the adjuvant activity of lipid A purified from L. interrogans serovar Pomona (PLA) for its ability to activate innate and enhance antigen-specific adaptive immune response. PLA-induced activation of macrophages similar to levels induced by MPLA, albeit at a much higher dose, indicating that it is less stimulatory than MPLA. Mice immunized with a Variable portion of LigA (LAV) formulated in Alum and PLA (LAV-Alum-PLA) induced significantly higher levels of LAV-specific humoral and cellular immune response than Alum but similar to levels induced by Alum-MPLA. The adjuvant activity of PLA seems to be quite similar to MPLA and primarily mediated through enhanced recruitment, activation, and uptake of antigens by innate immune cells. Moreover, like MPLA, the PLA formulation was able to generate a long-term memory response. Most importantly, PLA demonstrated better potency than MPLA formulation and generated sterilizing immunity against challenge in a hamster model of leptospirosis. Altogether, our study has provided important insight into the adjuvant activity of Leptospira lipid A and has opened avenues for the development of LPS-based vaccines against this dreadful zoonosis.
Keywords:
Lig A
; Lipid A
; Adjuvant
; Vaccine
; MPLA
; Alum
; Leptospirosis
1. Introduction
Leptospirosis, a zoonotic disease, continues to be a significant global public health concern, posing a threat as its prevalence escalates due to the effects of climate change and global warming. Each year, approximately one million cases of human leptospirosis are reported, resulting in an estimated fatality rate of 60,000 [1]. Despite the availability of a wide range of antibiotics, their effectiveness diminishes when the bacteria infiltrate vital organs and causes considerable damage, often due to delayed diagnosis. To combat this disease effectively, vaccination offers a cost-effective and secure preventive measure. However, the current killed vaccine provides only short-term, serovar-specific immunity and fails to prevent bacterial shedding through urine. Hence, it is imperative to develop potent vaccines capable of inducing sterilizing immunity against various serovars.
Subunit vaccines are advanced next-generation candidates that prioritize safety by utilizing acellular microbial outer membrane components such as lipopolysaccharide, surface proteins, glycoproteins, and toxoids [2,3]. Various outer membrane and surface proteins of Leptospira have been examined as potential subunit vaccines and have demonstrated varying degrees of protection in animal models [4]. Among these proteins, Leptospira immunoglobulin-like protein A (LigA), specifically its C-terminal or variable region (LAV), has emerged as a highly promising candidate, supported by the findings of several investigators [5,6]. Adjuvants are critical in enhancing vaccine efficacy by augmenting the immune response without directly contributing to antigen-specific protection [7]. Several surface proteins of Leptospira have been tested alongside potent adjuvants like Freund's, liposomes, xanthan gum, PLGA-microparticles, and emulsions such as AddaVax and Emulsigen-D. These combinations have imparted varying degree of protection (ranging from 50% to 70%) however, failed to provide sterilizing immunity [5,8,9,10,11]. It is important to note that, except Alum, all these are preclinical adjuvants. Fortunately, new generation of clinical adjuvants, including MF59, Montanide, and Adjuvant Systems (AS03, AS04), have become available [12]. AS04, for instance, combines the TLR4 agonist Monophosphoryl lipid A (MPLA) with an aluminum salt, and it has been tested in vaccines against HPV and HBV [13]. Our recent study has demonstrated that LAV formulated in AS04 exhibited superior immune response and protective efficacy in a hamster model of leptospirosis [14]. These findings highlight the potential AS04 or similar formulation in development of potent vaccine against Leptospirosis.
Lipopolysaccharide (LPS) is a major antigen of a gram-negative bacteria [15]. It acts as a TLR4 agonist, stimulating the activation and maturation of antigen-presenting cells (APCs). However, LPS toxicity has prevented its use as an adjuvant in human vaccines [16]. Removal of phosphate from lipid A derived from LPS of Salmonella Minnesota has reduced its toxicity and led to the development of a less toxic derivative, Monosphosphoryl lipid A (MPLA) [17]. Although both LPS and MPLA are recognized by TLR4, MPLA signals through a less inflammatory pathway involving a TRIF adaptor [18]. MPLA, when formulated in Alum (AS04) can enhance the antigen specific immune response and is also capable of modulating towards a mixed TH1/TH2 or a polarized Th1 cell response. In contrast, Alum predominantly elicits a biased Th2 response [19,20]. Much of the adjuvant activity of AS04 is attributed to the immunostimulatory activity of MPLA, although alum helps in prolonging this stimulation [20]. Leptospira LPS is a major antigen that induces protective immunity in animal models [21]. It is known to be naturally less endotoxic and atypical, signalling through TLR4 and TLR2 [22]. Given the significant roles of TLR2 and TLR4 in protecting against leptospirosis and considering the potent adjuvanticity of MPLA, it was of interest to evaluate the adjuvant activity of naturally less endotoxic Leptospira LPS (specifically lipid A) against leptospirosis [14,23].
In the present study, we isolated lipid A from Leptospira interrogans serovar Pomona (PLA) and conducted experiments to examine its immunostimulatory activity on mouse macrophages. Subsequently, we assessed the adjuvant activity of PLA in mice in terms of enhancing the immune response first against model antigen Ovalbumin (OVA) and then against Leptospira surface antigen (LAV). The immune response generated was compared with antigens formulated with Alum alone or Alum with MPLA. To gain insight into the adjuvant mechanism of PLA, we tested its ability to induce a local inflammatory response and generation of long-term memory. Finally, we tested the immune response and protective efficacy of PLA-based formulation in a hamster model of leptospirosis.
2. Material and Methods
2.1. Study Design
In this study, we tested the immunostimulatory and adjuvant activity of Leptospira LipidA, specifically Lpid A isolated from pathogenic Leptospira interrogans serovar Pomona (PLA). The activity was compared with Monophosphoryl Lipid A (MPLA), a well-known stimulatory agent and a clinical adjuvant. The stimulatory activity of PLA was tested in mouse macrophages in terms of their ability to induce pro-inflammatory cytokines and maturation markers. To test the ability of PLA to enhance antigen-specific humoral and cellular immune response, it was formulated first with model antigen Ovalbumin (OVA) in 2% Allohydrogel to determine the optimum PLA dose and then with variable portion of Leptospira immunoglobulin-like protein A (LAV) in 2% Allohydrogel. The immune response was analyzed in mice immunized with these formulations following standard procedures as described in the methodology. To identify key innate immune cell mediators and gain insight into their mechanism of action, we injected PLA or MPLA-based formulation containing fluorescent antigen (LAV) in mice and then evaluated the recruitment, antigen uptake, and activation status of immune cells in Draining lymph nodes using Flow cytometry and RT-PCR. To evaluate the long-term memory response induced by PLA-based formulation, we analyzed the LAV-specific antibody levels till 24 weeks and then analyzed both B and T cell memory after in vitro stimulation with recall antigen (LAV). Finally, we tested the efficacy of PLA-based vaccine formulation against the challenge with virulent Leptospira in a hamster model. The efficacy was assessed in terms of survivors, bacterial load, and assessing the lesions in vital organs by histopathology.
2.2. Chemicals and Reagents
Unless stated otherwise, most of the chemical and cell culture reagents utilized in the study were obtained from Sigma-Aldrich. ELISA kits were procured from R&D Biosystems, and flow cytometry antibodies were procured from BD Biosciences. Monophosphoryl Lipid-A (MPLA) and 2% Alhydrogel® (Alum) were purchased from InvivoGen.
2.3. Animals and Housing Conditions
The animal experiments were approved by the Institutional Animal Ethics Committee (IAEC) as per the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India. Female C57BL/6J mice, aged 5 to 6 weeks, were initially procured from the Jackson Laboratory in the USA. These mice were inbred and housed at Animal Resource and Experimental Facility at NIAB. Male Golden Syrian hamsters, aged 4 to 5 weeks, were procured from the Charles River and maintained at Jeeva Life Sciences Pvt. Ltd. in Hyderabad, where the experiments were conducted. The animals were kept in a controlled environment with a diurnal lighting cycle (12 hours light:12 hours dark) at 23-24°C temperature and 45% humidity, following standard pathogen-free conditions. They had access to food and water ad libitum at the facility. Throughout the study, the mice and hamsters were visually monitored twice daily for vital signs and signs of illness. At the end of the study, the mice or hamsters were euthanized using 5% isoflurane as per the CPCSEA's Euthanasia Guidelines. Confirmation of death was ensured through cervical dislocation performed two minutes after the cessation of breathing.
2.4. Antigen Preparation
OVA was reconstituted in endotoxin-free water (Millipore) at 1 mg/mL and stored at -20°C. The LAV was purified using a previously described method [14]. Briefly, E. coli BL21(DE3) harboring the expression plasmid pET28a-LAV were cultured in LB broth with 40 μg/mL kanamycin, and protein expression was induced by adding 1 mM isopropyl β-D-1-thiogalactoside (IPTG). The cells were harvested by centrifugation at 8,000 rpm for 5 minutes and lysed with 100 mM Tris-HCl and 150 mM NaCl at pH 8.0, followed by sonication. Debris was removed by centrifugation, and the supernatant was subjected to affinity chromatography using a Ni-NTA beads column from GE Healthcare. The eluted protein was dialyzed against 1×PBS and passed through Detox-Gel from Pierce (USA) to remove E. coli LPS. The protein was checked further for LPS contamination by the Limulus amoebocyte lysate (LAL) assay kit (Thermo Fischer), indicating that the endotoxin level was below 0.011 EU/mL. The protein purity and size was analyzed through SDS-PAGE, and its concentration was determined using the Bradford reagent (Sigma-Aldrich).
2.5. Leptospira Culture
The virulent strain of L. interrogans serovar Pomona was grown at 28°C under aerobic conditions in liquid Ellinghausen-McCullough-Johnson-Harris (EMJH) medium (Difco, BD, USA) supplemented with 10% (vol/vol) EMJH enrichment medium (BD, USA). To make it virulent, the strain underwent a series of passages in golden Syrian hamsters, followed by its isolation from kidneys. The virulent Leptopires were maintained in the semi-solid culture.
2.6. Lipid A Isolation and TLC
The isolation of Lipid A from Leptospira was carried out following the published method with minor modifications [24]. In brief, log phase cultures of Leptospira were pelleted, washed twice with DPBS, and then the cell pellet was suspended in a single-phase Bligh-Dyer mixture of chloroform, methanol, and water (1:2:0.8 v/v) followed by agitation for 20 minutes at room temperature. The cell fragments were separated by centrifugation at 2000 rpm for 20 minutes, and lipid A from LPS was separated by boiling the sample in hydrolysis buffer (50 mM sodium acetate, pH 4.5, 1% sodium dodecyl sulfate, pH 4.5) for 1 hour. After cooling to room temperature, the samples were re-extracted using two-phase Bligh-Dyer mixtures (chloroform, methanol, and water in 2:2:1.8, v/v/v ration). The samples were centrifuged at 2000 rpm for 20 minutes to separate the phases, and the lower phase containing lipid A was transferred and dried in a rota evaporator. The quality of lipid A was assessed by silica thin-layer chromatography (TLC) using the previously described method [24]. Briefly, a 10 µL lipid A dissolved in chloroform and methanol (4:1, v/v) was spotted on a silica plate using a microcapillary glass pipette and air-dried for 15 minutes. The plate was placed in a pre-equilibrated tank containing the mobile phase (chloroform: pyridine: 88% formic acid: water; 50:50:16:5 v/v). Once the solvent front reached the top, the plate was removed, air-dried, and immersed in a 10% sulfuric acid-ethanol mixture. Afterward, the plate was air-dried in a fume hood and visualized by placing it on a 250°C ceramic hot plate until the lipid A charred and became visible.
2.7. Cell Stimulation Assay
Isolated Lipid A was weighed and dissolved in endotoxin-free water (Millipore) at 1 mg/mL concentration by keeping it in a water bath Sonicator (Cole-Parmer) for 10min at 37°C. To determine the optimal lipid A dose, RAW264.7 cells were seeded onto the 24well plates, followed by stimulation with various concentrations of PLA (0.1/0.5/1/2/5/10/20/50 µg/mL) or MPLA (0.5/1/2 µg/mL) for 24h at 37 °C, and cytokine (IL-6) response was measured in the culture supernatant by Sandwich ELISA kit (R&D systems) following the manufacturer instructions.
2.8. Vaccine Formulation
The test/control formulations were prepared fresh on the day of dosing by the method provided below. The optimum dose (20 or 40 µg/animal) for in vivo adjuvant activity of PLA was determined with Ovalbumin. The different vaccine formulations were prepared by mixing 100 µg 2% Alhydrogel (Alum) with MPLA (5 µg) or PLA (20 or 40 µg) with an equal volume of DPBS containing antigen OVA or LAV (10 µg 1st immunization, 5 µg as booster) in 1:1 ratio. In contrast, formulations for hamster immunization were made with LAV (50 µg first immunization or 25 µg for booster) in Alum with MPLA (20 µg) or PLA (80 µg). To prepare HKL, log phase culture of Pomona was counted using the Petroff-Hausser Counter. The cells were washed twice with DPBS followed by heat-inactivation at 56 °C for 30 min and then suspended in DPBS to a concentration of 2 × 109 cells/mL combined with 100 µg alhydrogel in a ratio of 1:1 v/v.
2.9. Animal Immunizations and Sample Collection
Male C57BL/6 mice (6 animals/group) were immunized subcutaneously (100 μl/animal total) with various vaccine formulations and then boosted, as detailed above. The animals were bled before immunization (pre-bled) and before and after one week of a booster. On the 28th day, animals were euthanized, and blood, spleen, and lymph nodes were collected for further analysis. To evaluate the long-term immune response and generation of immunological memory, the animals after booster (3rd week) were bled every alternative week till 24 weeks (~6 months) and finally euthanized with or without giving additional booster to collect blood, spleen, and lymph nodes to analyze the memory response. The tissues from the injection site of mice were collected after 4 and 24 hours and preserved in RNAprotect (Qiagen) solution. Male golden Syrian hamsters, aged 4 to 5 weeks, were immunized subcutaneously using different formulations: PBS or Heat killed bacterin (109) or LAV (50 μg/animal) in Alum, or LAV -Alum-MPLA, or LAV-Alum-PLA, in a total volume of 200 μl and then boosted on day 21 with 25 µg of antigen. Before immunization, the hamsters were anesthetized using an intraperitoneal injection of ketamine (10 mg/mL) and xylazine (1 mg/mL) at 100µL per 130 g of body weight. The animals were bled at day 0, 21 and 35 and finally euthanized at day 35 and spleens were collected to determine antigen specific immune response.
2.9. ELISA for Serum Antibody Levels
The antibody levels in serum obtained from mice and hamsters were determined by ELISA following the standard protocol as published elsewhere [25]. In brief, 96-well microtiter plates (Nunc, Denmark) were coated with either OVA or LAV (100 ng/well for mice and 200 ng/well for hamster experiments) in 0.1 M bicarbonate buffer and incubated overnight at 4°C. The next day, the plates were washed thrice with 1 × PBS containing 0.05% Tween 20 (PBST, blocked with 1% BSA for 1 hour at room temperature. Plates were washed and added 100 µl/well of diluted serum (1:100 to 1:100,000 in PBST) and incubated for 2 hours at 37°C in a humid chamber. After washing, 100 µl/well HRP-conjugated (dilution of 1:6,000) goat anti-mouse secondary antibodies (IgG, or IgG1, or IgG2c, or IgA) or mouse anti-hamster antibodies (anti-hamster total IgG, IgG1, or IgG2/3) were added and incubated for 1 hour at room temperature. The plates were washed five times, and TMB substrate (100 µl/well) was added and placed at room temperature without exposure to direct light for 20 minutes. The enzymatic reaction was stopped by adding 50 µL 2N H2SO4, and the optical density was measured at 450 and 540 nm using an ELISA reader (Perkin Elmer).
2.10. Cell Proliferation and Cytokine Estimation
The proliferation of Lymphocytes obtained from various groups of immunized mice or hamsters was determined by stimulating the cells with recall antigen (OVA or LAV) and counting them after 48-72 hrs as described previously[14]. Briefly, Splenocytes (1×105 cells/well) were seeded in a 24-well plate and stimulated with varying concentrations (1, 2.5, and 5 µg/mL) of OVA or LAV for 48 to 72 hours. Cells were collected by gentle pipetting to determine the proliferation and counted using the trypan blue size exclusion method. The levels of IL-4 and IFN-γ released in the culture supernatant after 48hrs were determined by sandwich ELISA kits (R&D Systems) according to the manufacturer's instructions. The proliferation of lymphocytes isolated from hamsters was determined by stimulating them with varying LAV concentrations (1, 2, and 10 µg/mL) for 48 to 72 hours. The cells were collected and stored in RNA later to determine the expression of IL-4 and IFN-γ by RT-PCR.
2.11. Flow Cytometry
To study the role of PLA treatment on macrophage activation, RAW264.7 cells were seeded in 12-well plates (0.1 × 106/well) and treated with either MPLA (1 µg/mL) or lipid A (2 or 5 µg/mL) for 24 hours. The cells were rinsed twice with prechilled PBS, harvested, and blocked with Rat anti-Mouse CD16/CD32 (FC antibody; 553141, BD Biosciences) dissolved in PBS with 0.5% (w/v) BSA and 2% (v/v) FBS for 30 minutes. The cells were washed thrice and incubated on ice with antibodies PerCp Cy5.5 Rat anti-Mouse I-A/I-E (MHC-II; 562363), APC Hamster anti-Mouse CD80; 560016, PE Rat anti-Mouse CD86; 560601, and BV421 Rat anti-Mouse CD40; 562846 obtained from BD Biosciences. The cells were washed thrice to remove the unbound antibody and fixed with 1% paraformaldehyde. To study adjuvant recruitment, antigen uptake and activation status of APCs, LAV labelled with Alexa Fluor™ 488 (Invitrogen-A10235) formulated in different adjuvant was injected and DLNs were isolated at 4- and 24-hours post-injection. Cells in DLNs were washed, and subjected to Fc receptor blocking at 4°C for 30 minutes. These cells were then stained with BV-421 Hamster Anti-mouse CD11c; 562782 and PerCp Cy5.5 Rat anti-Mouse I-A/I-E (MHC-II; 562363) (for dendritic cells), BV510 Rat anti-Mouse CD11b; 562950/ PE-CF 594 Rat anti-Mouse F4/80; 565613 (for monocytes/macrophages), BV510 Rat anti-Mouse CD11b; 562950/ BV605 Rat anti-Mouse Ly6C; 563011 (for monocytes), BV510 Rat anti-Mouse CD11b; 562950/ PE-Cy7 Rat anti-Mouse Ly6G; 560601 (for neutrophils), APC Hamster anti-Mouse CD80; 560016, PE Rat anti-Mouse CD86; 560601, and PerCp Cy5.5 Rat anti-Mouse I-A/I-E (MHC-II; 562363) to identify specific cell types and determine their activation status. In another set of experiments, the splenocytes obtained from immunized mice (28th day or 24w, or 25w) or hamsters (35th day) from different groups were stained with BD Biosciences Rat anti-Mouse FITC-CD3; 555274/ BV421-CD4; 562891, PE-CD8a; 567630/ APC-Cy 7-CD44; 560568/ BV650-CD62L; 564108 (for mice) or eBiosciences APC Rat anti-Mouse CD4; 17-0041-82/ PE Mouse anti-Rat CD8b; 12-0080-82 (for hamsters) to determine the T cell population. The stained cells were washed and fixed with 1% paraformaldehyde. The BD LSRFortessa™ Cell Analyzer was used to acquire data (100,000 events/sample) and was analyzed utilizing FlowJo software (Tree Star Inc.).
2.12. Generation of Bone Marrow-Derived DCs.
The bone marrow-derived dendritic cells were prepared using a previously described method [14]. Briefly, the bone marrow cells obtained from mice were cultured in a 100 mm dish (1x107 cells) containing complete DMEM medium supplemented with GM-CSF (20 ng/mL; Preprotech) and IL-4 (5 ng/mL; Preprotech). On days 2 and 7, half of the medium in each well was replaced with fresh complete DMEM media supplemented with GM-CSF (40 ng/mL) and IL-4 (10 ng/mL). On day 9, the non-adherent floating cells were discarded, and adherent cells were scrapped. Cell count and viability were determined and further utilized for specific assays.
2.13. CTL Assay
The Cytotoxic T cells generated on the 28th day against OVA or LAV in mice immunized with different formulations were determined by LDH-based cytotoxicity assay as previously described [14]. Briefly, 3×107 spleenocytes were suspended in 10mL of complete RPMI supplemented with 0.2 ng/mL IL-2 and cultured in 25-cm2 tissue culture flasks. These flasks were maintained upright for 5 days, after which the non-adherent cells were collected and used as the effector (E) cells. The dendritic cells (DCs; 5×105) were seeded into 12 well plates and treated with 2 µg/mL OVA or LAV or BSA (Bovine serum albumin) for 16 hours at 37°C, 5% CO2 and used as target cells (T). The DCs were treated with mytomycin C (50 μg/mL) for 45 minutes. The E-cells were enumerated and incubated with DCs (T) at different E: T ratios (10:1, 25:1, and 50:1) and further incubated for 5 hrs at 37C, 5% CO2. The DCs pulsed with OVA or LAV served as specific target cells, whereas DCs pulsed with BSA represented the non-specific target cells. The cytotoxicity was determined using the LDH assay kit (Cytotox 96, Promega) for the specific target cell lysis by CTLs, as described previously [26]. Maximum release was achieved by lysis of DCs with Triton X-100 at a final concentration of 1% (vol/vol). The LDH released by untreated cells was designated the spontaneous release. Cytotoxicity was calculated as follows:
2.14. Lymph Node Sectioning and Immunofluorescent Staining
The inguinal draining lymph nodes were collected from different immunized animals on the 28th day and processed as previously described 1. Briefly, the lymph nodes were fixed in a 4% paraformaldehyde (PFA) solution for an hour and then gradually dehydrated in sucrose solutions of 10%, 20%, and 30% at 4°C. Subsequently, the lymph nodes were frozen in a cryomold using liquid nitrogen and stored at -80°C until further processing. The Leica Cryostat (Leica Geosystems, Switzerland) was used to make the sections with a thickness of 12 μm. The sections were rehydrated in Phosphate Buffered Saline (PBS; pH 7.4) for three hours and then blocked (5% BSA and 0.05% Tween 20 in PBS) for an hour. They were stained with Rat anti-Mouse FITC-conjugated CD3 (555274, BD Biosciences, 1:300), Rat anti-Mouse Per-CP-conjugated CD45R (B220; 130-102-815, Miltenyi Biotec, 1:100), Rat anti-Mouse PE-conjugated T- and B-Cell Activation Antigen (GL-7; 561530, BD Biosciences, 1:200), and Rabbit anti-Mouse CXCR5 (ab254415, Abcam, 1:500) overnight in a moist chamber at 4°C, washed five times with PBS and incubated with the secondary antibody, Goat Anti-Rabbit Alexa Fluor® 405-conjugated IgG H&L (ab175652, Abcam, 1:300), for two hours at room temperature. The antibody-stained slides were mounted using VECTASHIELD® Antifade Mounting Medium (Vector Laboratories and imaging was done using a Carl Zeiss Axio Scope VII fluorescent microscope with a 20× Plan Apochromat 0.45 NA objective and an EXFO X-Cite metal halide light source. The images were captured (Hamamatsu ORCA-ER CCD) in tile scanning mode and processed (stitched) using Zen Blue.
2.15. qRT-PCR
The expression of various cytokine and chemokines genes was determined by the standard protocol described previously [14]. Briefly, 800 µl of TRIzol (Invitrogen, Carlsbad, CA) and 200 µl of chloroform were added to the tissues from mice or cell pellets from hamsters, lysed, and centrifuged at 12,000 rpm for 5 minutes at 4°C. The resulting aqueous phase was processed through RNA easy mini columns (MN) following the manufacturer's protocol to purify the RNA. The quality of the RNA was assessed by running it on a formaldehyde gel to detect the presence of 16s and 18s RNA bands, as well as on a Bioanalyzer. UV spectroscopy (Invitrogen) was used to determine the RNA quantity and purity at a 260/280 ratio. The PrimeScript 1st strand cDNA Synthesis Kit (Takara) was used according to the manufacturer's instructions to synthesize the first-strand cDNA. qRT-PCR was conducted in 96-well microtiter plates on a Bio-Rad system. The two-step amplification involved a 10-µL reaction volume containing 50 ng of cDNA, 10 pM of each primer (Supplementary Table 3), and SYBR green (Bio-Rad). The samples were run in triplicate, and the data were analyzed using the fold change (2-ΔΔCt). The gene fold changes of treatment at different time points were determined relative to the control. The mRNA levels of the analyzed genes were normalized with the respective sample housekeeping gene (β-actin or GAPDH). All the primers used in the study were synthesized by IDT, and their sequences are given in Supplementary Table 3.
2.16. Infection Experiments
2.16.1. Challenge and Organ Collection
The hamsters were challenged intraperitoneally at day 35 ( two weeks after booster) with 100 times the effective dose (ED50) of virulent L. interrogans serovar Pomona. The ED50 was determined as previously described [27]. The following clinical signs were considered as endpoint criteria: hematuria, loss of appetite, gait or breathing difficulty, ruffled fur, hunched posture, prostration, or weight loss exceeding 20%. These clinical signs were monitored three times daily for four weeks. Hamsters displaying severe clinical signs (moribund) were euthanized after blood collection and recorded as deceased. The hamsters that survived the challenge were bled and sacrificed at the end of the observation period. Aseptic collection of the kidneys, liver, and lungs was conducted to assess bacterial load and histopathology.
2.17. Determination of Bacterial Load
We determined the bacterial load in the organs of all infected animals that met the specified endpoint criteria or survived until the end of the experiment and then euthanized. We employed the quantitative RT-PCR method to determine the bacterial load as previously described [28]. The kidneys, liver, and lungs tissues were sliced into small pieces and stored in RNAprotect (Qiagen) until the process, and total DNA was isolated using a standard protocol. The quantification was conducted in Bio-Rad Real-Time PCR System using 2 × SYBR Green PCR Master Mix (Bio-Rad) and specific primers for leptospiral 16s rRNA and LipL32. To create a leptospiral DNA standard curve, we utilized 10-fold serially diluted DNA of L. interrogans serovar Pomona, equivalent to a range of 2 × 101 to 2 × 109 cells/mL.
2.18. Histopathology
The structural integrity of hamster tissues was maintained by collecting, fixing, and preserving in a 10% neutral buffered formalin solution. These tissues were then sectioned (5μm) using a microtome, stained (with hematoxylin and eosin), and observed using light microscopy. A board-certified veterinary pathologist evaluated Leptospira-induced lesions in the infected organs in the blindfold samples. The severity of tubulointerstitial nephritis was determined using a grading system ranging from 0 (indicating normal) to 3 (representing severe). This grading system was based on previously established criteria [28]. Similarly, lung and liver pathology was assessed by counting the average number of inflammatory foci in 10× fields. The grading scale for lung and liver pathology ranged from 0 (indicating normal) to 3 (representing more than 7 inflammatory foci).
2.19. Statistical Analysis
In most of the experiments conducted, the results were analyzed using the one-way analysis of variance (ANOVA) with the Dunnett hypothesis test unless otherwise specified. The data were expressed as the mean of triplicates with the standard error of the mean (SEM). Statistical significance was determined with a threshold of p < 0.05.
3. Results
3.1. Leptospira Lipid A Is a Potent Adjuvant Capable of Inducing Strong Innate and Antigen-Specific Adaptive Immune Responses
We purified lipid A from Leptospira interrogans serovars Pomona (PLA) using a standard protocol and analyzed it by thin layer chromatography (Figure 1A). To test its immunostimulatory activity, we stimulated mouse macrophages with varying dose of PLA and then analyzed the production of pro-inflammatory cytokines and assessed upregulation of costimulatory molecules (CD80, CD86, CD40), and maturation marker (MHCII). Our result shows that similar to MPLA, PLA-induced activation of mouse macrophages as evident from the significant levels of IL-6 and upregulation of CD80, CD86, CD40, and MHCII; however, it was achieved at much higher doses indicating that it is less stimulatory than MPLA (Figure 1B,C). Since MPLA is known to enhance the antigen-specific immune response, we investigated the similar effect of PLA when formulated with OVA and Alum. The results demonstrated that OVA-Alum-PLA induced significantly higher levels of antibodies compared to OVA-Alum alone, but similar to the levels induced by OVA-Alum-MPLA (Figure 1D). Notably, OVA-Alum-PLA showed higher levels of both IgG1 and IgG2c, but failed to induce mucosal antibody IgA (Figure 1D). Further to analyze the T cell response, splenocytes were isolated from different groups on day 28 and stimulated with the recall antigen OVA. The results showed that splenocytes isolated from OVA-Alum-PLA exhibited significantly higher levels of proliferation compared to the control group (LAV-Alum) but similar to the levels induced by splenocytes from OVA-Alum-MPLA (Figure 1E). Analysis of cytokines in the culture supernatant showed that Alum predominantly induced a Th2 response with mainly IL-4 and low levels of IFN-γ, both PLA and MPLA induced a mixed Th1/Th2 response with significantly higher levels of both IL-4 and IFN-γ (Figure 1F). Additionally, we investigated whether the OVA-Alum-PLA formulation could induce cytotoxic T cells (CTLs) by assessing the lysis of target cells (DCs stimulated with OVA) using the LDH assay. The results indicated that CTLs obtained from animals immunized with either OVA-Alum-PLA or OVA-Alum-MPLA demonstrated a higher level of target cell lysis (70-80%) compared to OVA-Alum (20-30%) at the highest E: T ratio (Figure 1G). It is important to note that effectors obtained from both OVA-Alum-PLA and OVA-Alum-MPLA did not exhibit lysis of non-specific targets (DCs stimulated with unrelated antigen BSA), thus confirming the specificity of CTLs (Data not shown). Collectively, our results revealed that similar to MPLA, PLA acts as a potent adjuvant capable of enhancing antigen-specific humoral and cellular immune responses.
3.2. PLA Enhances the Adaptive Immune Response against Leptospira Surface Antigen
To evaluate the potential role of PLA in boosting the immune response to bacterial antigens, we focused on Leptospira immunoglobulin-like protein A (LigA). This surface protein is expressed during infection and is a promising vaccine candidate. We employed a previously described method to clone, express, and purify the variable portion of Leptospira immunoglobulin-like protein A (LAV) in soluble form [29]. Mice were then immunized with LAV in combination with Alum, or Alum-PLA, or Alum-MPLA, and we measured the resulting humoral and cellular immune responses specific to LAV. Our results indicate that similar to OVA, PLA enhanced the production of LAV-specific antibodies (IgG and various isotypes) as well as the T cell response (Figure 2A,B). Specifically, LAV-Alum-PLA stimulated a balanced Th1/Th2 response, leading to the production of both IgG2a/IFN-γ and IgG1/IL-4 (Figure 2C). Additionally, both PLA and MPLA-containing formulations enhanced the generation of LAV-specific CD4+ and CD8+ T cells (Figure 2D). Further, both PLA and MPLA formulation generated LAV specific cytotoxic T cells (CTLs). Notably, these CTLs exhibited a higher rate of target cell lysis (50-70%) as compared to CTLs obtained from LAV-Alum, which induced 20-30% lysis at the highest E: T ratio (Figure 2E). Furthermore, effector cells derived from PLA or MPLA-based formulations did not show lysis of non-specific targets (DCs stimulated with an unrelated antigen, OVA), thereby confirming the specificity of the CTL response (Data not shown). Collectively, our results demonstrate that PLA can enhance the adaptive immune response against the Leptospira surface antigen, LAV.
3.3. PLA Mediates Its Adjuvant Effect by Enhanced Recruitment and Activation of Innate Immune Cells at Draining Lymph Nodes
To comprehend the adjuvant mechanism of PLA, we conducted an analysis of the local inflammatory response and activation of innate cells at the site of vaccine administration and the draining lymph nodes (DLNs). This examination was carried out at two different time points, namely, 4 hours and 24 hours post-injection. Pooled cells from DLNs were analyzed by flow cytometry to assess their numbers, types, and activation status (Figure 3A). Our findings revealed that although PLA prompted significant recruitment of neutrophils (CD11b+Ly6G+) at 4 hours, the recruitment of DCs (CD11c+MCHII+) and granulocytes/monocytes (CD11b+Ly6C+) in PLA was considerably higher than in MPLA and Alum at 24 hours post-injection (Figure 3B). On the other hand, MPLA-alum induced early recruitment of neutrophils (CD11b+Ly6G+) and granulocytes/monocytes (CD11b+Ly6C+) (Figure 2B). Our tSNE analysis demonstrated that both PLA and MPLA significantly enhanced the expression of costimulatory molecules (CD80, CD86) and a maturation marker (MHCII) in the recruited immune cells (Figure 3C). Once we established that the PLA formulation generally led to robust cell migration and activation, we proceeded to analyze the uptake of the antigen (LAV labelled with Alexa FluorTM 488) by these cells. Flow cytometry analysis indicated that at 4 hours, there was rapid detection and a significantly higher number of LAV+ cells in both LAV-Alum-PLA and LAV-Alum-MPLA groups compared to LAV-Alum (Figure 3D). Neutrophils and DCs predominantly took up the antigens in PLA formulation, whereas macrophages did so in MPLA-immunized animals (Figure 3D). As adjuvants induce the expression of various cytokines and chemokines at the injection site to create a pro-inflammatory environment, we examined the expression of cytokines, chemokines, and their receptors induced by PLA using qRT-PCR. Our gene expression analysis at 4 hours and 24 hours demonstrated that PLA modulated the expression of several cytokines/chemokines or their receptors (Figure 3E). While MPLA enhanced the expression of (ccl2, ccl5, ccl10, il-1b, mip1a, and cxcl10) at an early time point (4 hours), PLA enhanced the expression of these cytokines/chemokines at both 4 hours and 24 hours. Mock injection with PBS induced a basal expression of these cytokines/chemokines due to needle-induced injury (Figure 3E).
3.4. PLA-Formulated Vaccine Induced Long-Term Immune Response and Generation of Immunological Memory
Since we observed that the PLA-formulated vaccine stimulated a robust innate and subsequent adaptive immune response, we were interested in examining its ability to generate a long-lasting memory response, which is a crucial criterion for an effective vaccine. We analyzed antibody levels in different groups up to 24 weeks (approximately 6 months) after immunization to assess the long-term memory response. Both the LAV-Alum-PLA and LAV-Alum-MPLA formulations displayed a sustained humoral response, as evidenced by detectable antibody levels even at 24 weeks post-immunization (Figure 4A). However, the LAV-Alum group exhibited significantly lower antibody levels with a subsequent decline in titers at 24 weeks. Both LAV-Alum-PLA and LAV-Alum-MPLA induced significantly higher levels of IgG and IgG1, while only LAV-Alum-PLA triggered a significant increase in IgG2c levels (Figure 4A). To evaluate the induction of immunological memory by these adjuvants, we administered an antigen booster without adjuvant and observed an enhanced antibody response in both the LAV-Alum-PLA and LAV-Alum-MPLA groups (Figure 4A). To assess the generation of memory T cells, we isolated splenocytes and stimulated them with a recall antigen (LAV). Our results showed significantly higher lymphocyte proliferation and cytokine production (IL-4, IFN-g) in both the LAV-Alum-PLA and LAV-Alum-MPLA groups (Figure 4B). Likewise, lymphocytes obtained after the booster displayed significantly increased proliferation and cytokine induction (Figure 4B). We then examined the memory phenotype of CD4 and CD8 T cells, characterized as TCM (CD44high and CD62Lhigh; central memory), and TEM (CD44high and CD62Llow; effector memory). Animals immunized with LAV-Alum-PLA or LAV-Alum-MPLA showed significantly higher levels of both central and effector memory CD4 and CD8 T cells compared to LAV-Alum (Figure 4C). The formation of Germinal Centers (GCs) is crucial for the generation of long-lived plasma cells that secrete high-affinity antibodies and the development of immunological memory. To determine if the higher and long-lasting antibody response induced by LAV-Alum-PLA is correlated with its modulation of GCs, we isolated inguinal lymph nodes from euthanized animals 28 days post immunization and stained sections with fluorescent antibodies against B220 (B-cell marker), CD3 (T-cell marker), GL7 (B cell GC marker), and CXCR5 (GC resident T follicular helper cells). Immunofluorescence analysis revealed a greater frequency of B220+GL7+ and CD3+ CXCR5+ cells in mice immunized with either LAV-Alum-PLA or LAV-Alum-MPLA, indicating an increased number of lymph node GCs compared to LAV-Alum (Figure 5).
3.5. PLA Formulated Vaccine-Induced Sterile Immunity against the Disease in a Hamster Model
Next we tested the effectiveness of the PLA-formulated vaccine in generating a strong immune response and its correlation with disease protection in a hamster model of leptospirosis. We compared the efficacy of this formulation with the standard Killed vaccine (HKL). Analysis of the antibody response on day 35 (2 weeks after booster) revealed significantly higher levels of IgG in both LAV-Alum-PLA and LAV-Alum-MPLA formulations compared to LAV-Alum, with a predominance of both IgG1 and IgG2/3 (Figure 5A). Furthermore, lymphocytes isolated from the LAV-Alum-PLA or LAV-Alum-MPLA immunized group exhibited significantly higher levels of proliferation and enhanced expression of IL-4 and IFN-γ transcripts compared to LAV-Alum (Figure 6B). The HKL-immunized animals did not show significant levels of humoral and cell-mediated response specific to the LAV antigen (Figure 6A,B). T-cell analysis using flow cytometry showed a significant enhancement of CD4+ cells in all the immunized groups except the PBS group. However, CD8 T cells were only enhanced by the PLA and MPLA-based formulations (Figure 6C). To determine if the immune response corresponds to protection, we subjected the animals to a challenge with virulent Leptospira on the 35th day and evaluated protective efficacy based on progressive weight loss, survival, histopathology, and bacterial load. As previously reported in similar studies, we considered ≥20% weight loss as the endpoint criterion to prevent spontaneous death. The control group (PBS) showed typical signs of acute leptospirosis with necrosis and small foci of gross and microscopic pulmonary hemorrhage (Figure 6D). While the LAV-Alum and LAV-Alum-MPLA groups exhibited alleviated features, the LAV-Alum-PLA and HKL groups appeared close to normal (Figure 6D). The PBS group experienced progressive loss in body weight, with animals unable to survive beyond 12 days post-challenge (Figure 6E,F). The LAV-Alum and LAV-Alum-MPLA groups showed progressive weight loss initially but regained weight after 18 days post-challenge, with survival rates of 50% and 67%, respectively. In contrast, the LAV-Alum-PLA and HKL groups did not experience a decrease in body weight, and 100% animals survived in at least three independent experiments (Figure 6E,F & Sup. Table.1). We measured the bacterial load in the infected organs in terms of DNA copy number per milligram of tissue using qRT-PCR. The results demonstrated that the bacterial load in the liver, lung, and kidney of the PBS group was significantly higher (p > 0.0001) than in the other vaccinated groups (Figure 6G). Animals immunized with LAV-Alum or LAV-Alum-MPLA had a significantly lower bacterial load than the control group, but they exhibited a significantly higher bacterial load in the lungs (p > 0.01) and liver (p > 0.05), with no significant difference in the kidney when compared to HKL-immunized animals. LAV-Alum-PLA showed a similar or lower bacterial load than HKL, and in the organs of some animals, it was below the detection limit (Figure 6G). The histopathological analysis is a sensitive method for detecting sublethal leptospiral infection and is an important parameter for assessing vaccine efficacy. Examination of the liver, lung, and kidney revealed varying degrees of lesions, necrosis, and cell infiltration (Figure 6H). The PBS control animals exhibited severe lesions in the kidneys characterized by marked chronic tubulointerstitial nephritis, severe atrophy, fibrosis, and lymphocyte infiltration. The liver showed centrilobular necrosis with numerous inflammatory foci, while the lungs displayed oedema and foci of intense haemorrhages (Figure 6H). Pathological scoring of the organs from these vaccine groups indicated that while 80% of animals were normal, 20% had mild lesions in the HKL group, whereas 85-90% of animals were normal, and 10-15% had mild lesions in LAV-Alum-PLA group. The LAV-Alum-MPLA group showed 40% normal animals, while 35% had moderate and 25% had mild lesions. In contrast, in the LAV-Alum group, all the animals showed lesions where 20% had mild, 45% had moderate, and around 35% had severe lesions (Figure 6H & Sup. Table.2).
4. Discussion
Currently, the primary means of treating and preventing leptospirosis during an outbreak is through empirical broad-spectrum antibiotics used as therapeutics or prophylaxis [30]. However, using antibiotics to protect all susceptible humans and animals from infection and spread after a leptospirosis outbreak is impractical and may lead to resistance [31]. Vaccination is the most effective and low-cost intervention; however, the current killed vaccine induces short-term and serovar-specific protection and is unable to provide sterilizing immunity [21]. It is used mainly in animals, and due to toxicity, its use is precluded in humans [32]. To overcome the limitations of traditional whole cell-vaccine, several protective surface antigens have been identified for the development of new-era subunit vaccines [4,33]. However, these vaccines require potent adjuvants [34]. Bacterial LPS, including lipid A, has been widely tested as immunostimulatory agents, and various studies have focused on the structure-activity relationship of these TLR4 ligands and their role in actively developing as adjuvants [35]. Since the clinical approval of MPLA as an adjuvant, mainly in the AS0X series (GSK), its use is already expanding as an important component in anti-cancer, anti-protozoan, and anti-malarial vaccines [36,37,38]. The development of synthetic Glucopyranosyl lipid A (GLA) further highlights the importance of bacterial lipid A as an adjuvant [39]. Our recent study has also demonstrated that MPLA can enhance the antigen-specific immune response and protective efficacy in a hamster model of leptospirosis [14]. Since LPS is a major Leptospira surface antigen, protective immunity generated against whole-cell vaccines is believed to be primarily mediated by anti-LPS IgMs [40]. In fact, several studies have demonstrated the role of LPS in mediating protection against challenges in animal models [41]. This is also supported by the fact that the TLR4 receptor, of which LPS is a ligand, plays an important role in protection against the disease [22]. Leptospira LPS is naturally less endotoxic and atypical as it recognizes both TLR2 and TLR4 to induce inflammatory response [23,42]. Further, it is not necessary to structurally modify LPS or lipid A to minimize toxicity while maintaining the adjuvant effect. Keeping these reports in mind, it was tempting to evaluate the adjuvant potential of Leptospira lipid A.
We isolated LPS and Lipid A from three important pathogenic Leptospira interrogans serovars (Icterhaemorrhaigie, Hardjo, Pomona) infecting various hosts and tested its innate activity on mouse macrophages. Irrespective of the serovar, it was purified, all lipid A’s induced TLR4 MyD88 dependent pro-inflammatory response and activation of macrophages, however, at a dose much higher than MPLA (unpublished data). Interestingly, LPS isolated from serovar Pomona induced macrophage activation via signaling through a less inflammatory TRIF pathway, which is similar to MPLA (unpublished data) [18]. Based on these results, it was interesting to test PLA's potency in enhancing the antigen-specific immune response. Our in vitro result shows that PLA seems to be less stimulatory than MPLA as it induces pro-inflammatory cytokines and expression of activation and maturation markers at a much higher dose than MPLA (Figure 1B). This difference in activity might be due to difference in source as PLA is lab-purified and MPLA is a highly pure commercial product. This difference in immunostimulatory activity between MPLA and PLA may also be attributed to some of the structural differences between the two lipid A’s. Although Lipid A structure within serovars seems to be conserved as we could not find any structural differences in lipid A isolated from three pathogenic serovars (unpublished data), however, there is structural diversity in lipid A across the genus of Leptospira as has been reported recently [43].
Our in vivo data show that Alum-PLA induced significantly higher levels of OVA and LAV-specific humoral and cellular immune response than induced by Alum alone but similar to levels induced by Alum-MPLA; however, this was achieved at a much higher dose (20 µg) as compared to MPLA (5 µg) further confirming the overall less stimulatory activity of PLA than MPLA (Figure 1 and Figure 2). The mechanism by which PLA enhances the antigen-specific humoral and cellular immune response seems be similar to MPLA and primarily mediated by enhanced recruitment, activation, maturation, and uptake of antigen by APCs resulting in subsequent activation of B and T cell responses [20]. This is supported by our result showing the rapid appearance of Ag-loaded and activated APCs in the draining lymph nodes in both PLA and MPLA-formulated vaccines (Figure 6B,D). The cytokines induced at the injection site by PLA formulated vaccine correlate to the cellular events detected in the draining lymph nodes (Figure 6E) [44]. Chemokines such as CCL2 and CCL3, enhanced by both PLA and MPLA formulations, are known to promote the recruitment of monocytes and immature DCs [45]. Differentiated APCs are key mediators for the induction of adaptive responses, as they can take up antigens and migrate to the draining lymph nodes [45]. Our result shows that formulation of PLA with Alum has led to an optimal APC recruitment and activation in the draining lymph node, which could be partly contributed by Alum in prolonging the cytokine response to PLA. The direct activation of APCs like DCs by PLA or MPLA may also be crucial for a sustained antibody response. In fact, it is believed that direct activation of DCs by TLR agonists rather than cytokine alone may enhance their ability to promote Ag-specific immune response [46]. The immunostimulatory activity of PLA or MPLA may indirectly play an important role in enhancing antibody response as pro-inflammatory cytokines are known to stimulate T helper cells specialized in providing help to B cells [47]. Although we have not tested the direct effect of PLA on T cells, it is possible that it could directly activate these cells through TLR4 and enhance the expression of activation markers (CD69) and costimulatory ligands (CD40L) [48].
The strong and similar levels of innate and antigen-specific adaptive immune response generated by both MPLA and PLA may be attributed to some of the structural similarities and differences between the two immunostimulatory molecules. Both lipid A’s have amide linkages (although PLA has a higher number) which may induce more stable and stronger interaction with TLR4 leading to strong innate and subsequent adaptive immune response [49,50]. This has been observed previously during N. meningitidis infection where generation of robust immune response is due to strong interaction of its lipid A (having amide-linked fatty acids) with TLR4 [51]. Our result has shown that both MPLA and PLA-based vaccine formulations have induced long-term and persistent responses leading to the generation of immunological memory, which may also be attributed to the amide linkage of their lipid A. Amide linkages contribute to stability and providing resistance to hydrolysis by acylase which allows this lipid A to persist longer and sustain the immune response. Another structural difference between PLA and MPLA is that the former has two unsaturated fatty acyl chains. Unsaturated fatty acids in the lipid A structure have been shown to enhance TLR recognition and influence host immune response [52]. The unsaturated fatty acids can cause structural modifications in Lipid A, such as the formation of kinks which allow them to better fit into the hydrophobic pocket of MD-2, an accessory protein required for TLR4 signaling. This enhanced binding affinity leads to increased activation of TLR4-mediated immune responses. Modification of lipid A with unsaturated fatty acids in Salmonella enterica serovar Minnesota has been associated with increased cytokine production and inflammation in the host cells [53]. Molecular docking studies have also confirmed that the kinks in lipid A from E. coli, caused by unsaturated fatty acids, lead to more extensive interactions with TLR4 and MD-2 compared to lipid A with saturated fatty acids [54].
To assess if the strong humoral and cell-mediated immune response generated by LAV-Alum-PLA is able to induce protection against disease, we tested the efficacy of the formulation in a hamster model of leptospirosis. Our result shows that hamsters immunized with LAV-Alum-PLA induced significantly higher levels of antigen-specific antibodies and enhanced levels of both IL-4 and IFN-gamma than those immunized with LAV-Alum (Figure 6A,B). The immune response correlated to a higher level of protection, as evident from enhanced survival, reduced bacterial load, and lesions in vital organs (Figure 6D–F). Although LAV-Alum-PLA and LAV-Alum-MPLA induced similar immune responses, the former induced much better protection (similar to HKL) in terms of survivors and reducing the bacterial burden in organs (Figure 6G). The significantly higher level of immune response and protection induced by LAV-Alum-MPLA than LAV-Alum is consistent with previous studies, including ours, where MPLA-based formulation imparted significantly higher level of protection (ranging from 60-67%) than Alum [10,11,14]. Interestingly, PLA induced better protection than MPLA-based formulation and even sterilizing immunity in some animals in spite of the similar levels of immune response they generated. We speculate that anti-PLA antibodies may contribute in enhanced efficacy of this formulation since anti-LPS antibodies play an important role in protection against leptospirosis [55]. It might also be attributed to the activation of additional innate pathways, particularly those involving signalling through TLR2 [56]. However, these hypotheses need to be tested and validated. It is important to note that the protection imparted by HKL is through the generation of the immune response against a repertoire of surface antigens, including LPS; however, it was not contributed by LAV-specific antibodies and T cells as LigA protein is not expressed in bacteria cultured in vitro [57]. Since O-antigen varies among different serovars and Lipid A is conserved among pathogenic species, it is expected that PLA-formulated vaccine may induce cross-protection against heterologous challenge with different serovars; however, this is an active area of investigation, and experiments are ongoing.
In conclusion, our study has demonstrated Leptospira lipid A as a potent adjuvant capable of activating innate and enhancing antigen-specific long-term protective immune response. PLA-based vaccine demonstrated superior efficacy than the MPLA-formulated vaccine but was equivalent to the standard killed vaccine (HKL). Since the killed vaccine is associated with toxicity and cannot be used in humans, the PLA-formulated vaccine could be an alternative as it is naturally less endotoxic, and we have not observed any local or systemic toxicity in mice and hamsters. Lipid A is an important protective antigen; however, without T cell help, it induces a short-term immune response and fails to generate memory. Since physically mixing PLA with protein antigens (OVA or LAV) could enhance antigen-specific immune response, hence conjugating PLA with LAV would be a better strategy as antigen co-delivery and co-activation of APCs could lead to the generation of a robust immune response simultaneously against both LAV and PLA. Hence, future effort efforts should be directed towards conjugating LAV with synthetic PLA to develop a conjugate vaccine against this important zoonosis.
Supplementary Files
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. The supplementary data contains, Table. 1: The survival data of immunized animals following infection with a virulent strain of Leptospira for 28 days. Table. 2: Histopathological scores of different organs in hamsters following infection with virulent Leptospira. Table. 3: The primers used for qRT-PCR analysis in the study.
Author’s Contribution
SMF conceived the idea and designed the experiments. VPV, MK, and SK performed the experiments. VPV, MK and SMF analysed the data. VPV and SMF wrote the initial draft, and SMF edited the manuscript. All authors approved the final version of the manuscript.
Funding and Acknowledgments
This work was supported by the DST-SERB-funded project- EMR/2017/002468 (SP046) on developing Leptospira vaccines. The project was funded to SMF by the Department of Science and Technology under the Ministry of Science and Technology, Government of India. Financial support from the NIAB core fund is duly acknowledged. The authors would like to thank the Director, NIAB, for providing the necessary infrastructural facility and support for the execution of the above study. We thank Dr. Jayant Hole, In-charge Small Animal Facility, for assistance in the animal experiments. VPV was supported by a CSIR fellowship and registered for the Ph.D. program at Manipal University, Manipal. MK was supported by a UGC fellowship and registered for the Ph.D. program at RCB, Faridabad.
Competing Interest
The authors declare no competing financial interests.
References
- Costa, F.; Hagan, J.E.; Calcagno, J.; Kane, M.; Torgerson, P.; Martinez-Silveira, M.S.; Stein, C.; Abela-Ridder, B.; Ko, A.I. Global Morbidity and Mortality of Leptospirosis: A Systematic Review. PLoS Negl Trop Dis 2015, 9, e0003898. [Google Scholar] [CrossRef]
- Greenwood, B. The contribution of vaccination to global health: past, present and future. Philos Trans R Soc Lond B Biol Sci 2014, 369, 20130433. [Google Scholar] [CrossRef]
- Delany, I.; Rappuoli, R.; De Gregorio, E. Vaccines for the 21st century. EMBO Mol Med 2014, 6, 708–720. [Google Scholar] [CrossRef]
- Barazzone, G.C.; Teixeira, A.F.; Azevedo, B.O.P.; Damiano, D.K.; Oliveira, M.P.; Nascimento, A.; Lopes, A.P.Y. Revisiting the Development of Vaccines Against Pathogenic Leptospira: Innovative Approaches, Present Challenges, and Future Perspectives. Front Immunol 2021, 12, 760291. [Google Scholar] [CrossRef]
- Faisal, S.M.; Yan, W.; McDonough, S.P.; Chang, Y.F. Leptospira immunoglobulin-like protein A variable region (LigAvar) incorporated in liposomes and PLGA microspheres produces a robust immune response correlating to protective immunity. Vaccine 2009, 27, 378–387. [Google Scholar] [CrossRef]
- Ko, A.I.; Goarant, C.; Picardeau, M. Leptospira: the dawn of the molecular genetics era for an emerging zoonotic pathogen. Nat Rev Microbiol 2009, 7, 736–747. [Google Scholar] [CrossRef]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine adjuvants: putting innate immunity to work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef]
- Evangelista, K.V.; Lourdault, K.; Matsunaga, J.; Haake, D.A. Immunoprotective properties of recombinant LigA and LigB in a hamster model of acute leptospirosis. PLoS One 2017, 12, e0180004. [Google Scholar] [CrossRef]
- Teixeira, A.F.; Fernandes, L.G.V.; Cavenague, M.F.; Takahashi, M.B.; Santos, J.C.; Passalia, F.J.; Daroz, B.B.; Kochi, L.T.; Vieira, M.L.; Nascimento, A. Adjuvanted leptospiral vaccines: Challenges and future development of new leptospirosis vaccines. Vaccine 2019, 37, 3961–3973. [Google Scholar] [CrossRef]
- Techawiwattanaboon, T.; Barnier-Quer, C.; Palaga, T.; Jacquet, A.; Collin, N.; Sangjun, N.; Komanee, P.; Patarakul, K. A Comparison of Intramuscular and Subcutaneous Administration of LigA Subunit Vaccine Adjuvanted with Neutral Liposomal Formulation Containing Monophosphoryl Lipid A and QS21. Vaccines (Basel) 2020, 8. [Google Scholar] [CrossRef]
- Techawiwattanaboon, T.; Courant, T.; Brunner, L.; Sathean-Anan-Kun, S.; Krangvichian, P.; Iadsee, N.; Nakornpakdee, Y.; Sangjun, N.; Komanee, P.; Collin, N.; et al. Designing Adjuvant Formulations to Promote Immunogenicity and Protective Efficacy of Leptospira Immunoglobulin-Like Protein A Subunit Vaccine. Front Cell Infect Microbiol 2022, 12, 918629. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, I.; Garg, R.; van Drunen Littel-van den Hurk, S. Selection of adjuvants for vaccines targeting specific pathogens. Expert Rev Vaccines 2019, 18, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Bazin-Lee, H.; Evans, J.T.; Casella, C.R.; Mitchell, T.C. MPL Adjuvant Contains Competitive Antagonists of Human TLR4. Front Immunol 2020, 11, 577823. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.P.; Kadivella, M.; Kumar, A.; Kavela, S.; Faisal, S.M. LigA formulated in AS04 or Montanide ISA720VG induced superior immune response compared to alum, which correlated to protective efficacy in a hamster model of leptospirosis. Front Immunol 2022, 13, 985802. [Google Scholar] [CrossRef]
- Avila-Calderon, E.D.; Ruiz-Palma, M.D.S.; Aguilera-Arreola, M.G.; Velazquez-Guadarrama, N.; Ruiz, E.A.; Gomez-Lunar, Z.; Witonsky, S.; Contreras-Rodriguez, A. Outer Membrane Vesicles of Gram-Negative Bacteria: An Outlook on Biogenesis. Front Microbiol 2021, 12, 557902. [Google Scholar] [CrossRef]
- Verma, S.K.; Mahajan, P.; Singh, N.K.; Gupta, A.; Aggarwal, R.; Rappuoli, R.; Johri, A.K. New-age vaccine adjuvants, their development, and future perspective. Front Immunol 2023, 14, 1043109. [Google Scholar] [CrossRef]
- Bentala, H.; Verweij, W.R.; Huizinga-Van der Vlag, A.; van Loenen-Weemaes, A.M.; Meijer, D.K.; Poelstra, K. Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock 2002, 18, 561–566. [Google Scholar] [CrossRef]
- Mata-Haro, V.; Cekic, C.; Martin, M.; Chilton, P.M.; Casella, C.R.; Mitchell, T.C. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 2007, 316, 1628–1632. [Google Scholar] [CrossRef]
- Casella, C.R.; Mitchell, T.C. Putting endotoxin to work for us: monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol Life Sci 2008, 65, 3231–3240. [Google Scholar] [CrossRef]
- Didierlaurent, A.M.; Morel, S.; Lockman, L.; Giannini, S.L.; Bisteau, M.; Carlsen, H.; Kielland, A.; Vosters, O.; Vanderheyde, N.; Schiavetti, F.; et al. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J Immunol 2009, 183, 6186–6197. [Google Scholar] [CrossRef]
- Srikram, A.; Zhang, K.; Bartpho, T.; Lo, M.; Hoke, D.E.; Sermswan, R.W.; Adler, B.; Murray, G.L. Cross-protective immunity against leptospirosis elicited by a live, attenuated lipopolysaccharide mutant. J Infect Dis 2011, 203, 870–879. [Google Scholar] [CrossRef]
- Chassin, C.; Picardeau, M.; Goujon, J.M.; Bourhy, P.; Quellard, N.; Darche, S.; Badell, E.; d'Andon, M.F.; Winter, N.; Lacroix-Lamande, S.; et al. TLR4- and TLR2-mediated B cell responses control the clearance of the bacterial pathogen, Leptospira interrogans. J Immunol 2009, 183, 2669–2677. [Google Scholar] [CrossRef]
- Nahori, M.A.; Fournie-Amazouz, E.; Que-Gewirth, N.S.; Balloy, V.; Chignard, M.; Raetz, C.R.; Saint Girons, I.; Werts, C. Differential TLR recognition of leptospiral lipid A and lipopolysaccharide in murine and human cells. J Immunol 2005, 175, 6022–6031. [Google Scholar] [CrossRef]
- Henderson, J.C.; O'Brien, J.P.; Brodbelt, J.S.; Trent, M.S. Isolation and chemical characterization of lipid A from gram-negative bacteria. J Vis Exp 2013, e50623. [Google Scholar] [CrossRef]
- Quach, H.Q.; Ovsyannikova, I.G.; Poland, G.A.; Kennedy, R.B. Detection of SARS-CoV-2 peptide-specific antibodies in Syrian hamster serum by ELISA. J Immunol Methods 2022, 505, 113275. [Google Scholar] [CrossRef]
- Hayashi, T.; Hideshima, T.; Akiyama, M.; Raje, N.; Richardson, P.; Chauhan, D.; Anderson, K.C. Ex vivo induction of multiple myeloma-specific cytotoxic T lymphocytes. Blood 2003, 102, 1435–1442. [Google Scholar] [CrossRef]
- REED, L.J.; MUENCH, H. A SIMPLE METHOD OF ESTIMATING FIFTY PER CENT ENDPOINTS12. American Journal of Epidemiology 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Wunder, E.A., Jr.; Figueira, C.P.; Santos, G.R.; Lourdault, K.; Matthias, M.A.; Vinetz, J.M.; Ramos, E.; Haake, D.A.; Picardeau, M.; Dos Reis, M.G.; et al. Real-Time PCR Reveals Rapid Dissemination of Leptospira interrogans after Intraperitoneal and Conjunctival Inoculation of Hamsters. Infect Immun 2016, 84, 2105–2115. [Google Scholar] [CrossRef]
- Kumar, A.; Varma, V.P.; Sridhar, K.; Abdullah, M.; Vyas, P.; Ashiq Thalappil, M.; Chang, Y.F.; Faisal, S.M. Deciphering the Role of Leptospira Surface Protein LigA in Modulating the Host Innate Immune Response. Front Immunol 2021, 12, 807775. [Google Scholar] [CrossRef]
- Ricaldi, J.N.; Vinetz, J.M. Leptospirosis in the tropics and in travelers. Curr Infect Dis Rep 2006, 8, 51–58. [Google Scholar] [CrossRef]
- Liegeon, G.; Delory, T.; Picardeau, M. Antibiotic susceptibilities of livestock isolates of leptospira. Int J Antimicrob Agents 2018, 51, 693–699. [Google Scholar] [CrossRef]
- Wunder, E.A.; Adhikarla, H.; Hamond, C.; Owers Bonner, K.A.; Liang, L.; Rodrigues, C.B.; Bisht, V.; Nally, J.E.; Alt, D.P.; Reis, M.G.; et al. A live attenuated-vaccine model confers cross-protective immunity against different species of the Leptospira genus. Elife 2021, 10. [Google Scholar] [CrossRef]
- Felix, S.R.; Hartwig, D.D.; Argondizzo, A.P.; Silva, E.F.; Seixas, F.K.; Neto, A.C.; Medeiros, M.A.; Lilenbaum, W.; Dellagostin, O.A. Subunit approach to evaluation of the immune protective potential of leptospiral antigens. Clin Vaccine Immunol 2011, 18, 2026–2030. [Google Scholar] [CrossRef]
- Pulendran, B.; P, S.A.; O'Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat Rev Drug Discov 2021, 20, 454–475. [Google Scholar] [CrossRef]
- Duthie, M.S.; Windish, H.P.; Fox, C.B.; Reed, S.G. Use of defined TLR ligands as adjuvants within human vaccines. Immunol Rev 2011, 239, 178–196. [Google Scholar] [CrossRef]
- Cluff, C.W. Monophosphoryl lipid A (MPL) as an adjuvant for anti-cancer vaccines: clinical results. Adv Exp Med Biol 2010, 667, 111–123. [Google Scholar] [CrossRef]
- Song, P.; He, S.; Zhou, A.; Lv, G.; Guo, J.; Zhou, J.; Han, Y.; Zhou, H.; Hao, Z.; Cong, H. Vaccination with toxofilin DNA in combination with an alum-monophosphoryl lipid A mixed adjuvant induces significant protective immunity against Toxoplasma gondii. BMC Infect Dis 2017, 17, 19. [Google Scholar] [CrossRef]
- Fries, L.F.; Gordon, D.M.; Richards, R.L.; Egan, J.E.; Hollingdale, M.R.; Gross, M.; Silverman, C.; Alving, C.R. Liposomal malaria vaccine in humans: a safe and potent adjuvant strategy. Proc Natl Acad Sci U S A 1992, 89, 358–362. [Google Scholar] [CrossRef]
- Arias, M.A.; Van Roey, G.A.; Tregoning, J.S.; Moutaftsi, M.; Coler, R.N.; Windish, H.P.; Reed, S.G.; Carter, D.; Shattock, R.J. Glucopyranosyl Lipid Adjuvant (GLA), a Synthetic TLR4 agonist, promotes potent systemic and mucosal responses to intranasal immunization with HIVgp140. PLoS One 2012, 7, e41144. [Google Scholar] [CrossRef]
- Fraga, T.R.; Barbosa, A.S.; Isaac, L. Leptospirosis: aspects of innate immunity, immunopathogenesis and immune evasion from the complement system. Scand J Immunol 2011, 73, 408–419. [Google Scholar] [CrossRef]
- Jost, B.H.; Adler, B.; Vinh, T.; Faine, S. A monoclonal antibody reacting with a determinant on leptospiral lipopolysaccharide protects guinea pigs against leptospirosis. J Med Microbiol 1986, 22, 269–275. [Google Scholar] [CrossRef]
- Werts, C.; Tapping, R.I.; Mathison, J.C.; Chuang, T.H.; Kravchenko, V.; Saint Girons, I.; Haake, D.A.; Godowski, P.J.; Hayashi, F.; Ozinsky, A.; et al. Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nat Immunol 2001, 2, 346–352. [Google Scholar] [CrossRef]
- Petrosova, H.; Mikhael, A.; Culos, S.; Giraud-Gatineau, A.; Gomez, A.M.; Sherman, M.E.; Ernst, R.K.; Cameron, C.E.; Picardeau, M.; Goodlett, D.R. Lipid A structural diversity among members of the genus Leptospira. Front Microbiol 2023, 14, 1181034. [Google Scholar] [CrossRef]
- Le, Y.; Zhou, Y.; Iribarren, P.; Wang, J. Chemokines and chemokine receptors: their manifold roles in homeostasis and disease. Cell Mol Immunol 2004, 1, 95–104. [Google Scholar]
- Joffre, O.; Nolte, M.A.; Sporri, R.; Reis e Sousa, C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev 2009, 227, 234–247. [Google Scholar] [CrossRef]
- Guo, H.; Zhang, Q.Y.; Shi, Y.; Wang, D.H. Evaluation of the International Vehicle Emission (IVE) model with on-road remote sensing measurements. J Environ Sci (China) 2007, 19, 818–826. [Google Scholar] [CrossRef]
- Eddahri, F.; Denanglaire, S.; Bureau, F.; Spolski, R.; Leonard, W.J.; Leo, O.; Andris, F. Interleukin-6/STAT3 signaling regulates the ability of naive T cells to acquire B-cell help capacities. Blood 2009, 113, 2426–2433. [Google Scholar] [CrossRef]
- Ismaili, J.; Rennesson, J.; Aksoy, E.; Vekemans, J.; Vincart, B.; Amraoui, Z.; Van Laethem, F.; Goldman, M.; Dubois, P.M. Monophosphoryl lipid A activates both human dendritic cells and T cells. J Immunol 2002, 168, 926–932. [Google Scholar] [CrossRef]
- Peri, F.; Calabrese, V. Toll-like receptor 4 (TLR4) modulation by synthetic and natural compounds: an update. J Med Chem 2014, 57, 3612–3622. [Google Scholar] [CrossRef]
- Stephenson, H.N.; John, C.M.; Naz, N.; Gundogdu, O.; Dorrell, N.; Wren, B.W.; Jarvis, G.A.; Bajaj-Elliott, M. Campylobacter jejuni lipooligosaccharide sialylation, phosphorylation, and amide/ester linkage modifications fine-tune human Toll-like receptor 4 activation. J Biol Chem 2013, 288, 19661–19672. [Google Scholar] [CrossRef]
- Coureuil, M.; Jamet, A.; Bille, E.; Lecuyer, H.; Bourdoulous, S.; Nassif, X. Molecular interactions between Neisseria meningitidis and its human host. Cell Microbiol 2019, 21, e13063. [Google Scholar] [CrossRef]
- Hwang, D.H.; Kim, J.A.; Lee, J.Y. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur J Pharmacol 2016, 785, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Cian, M.B.; Giordano, N.P.; Masilamani, R.; Minor, K.E.; Dalebroux, Z.D. Salmonella enterica Serovar Typhimurium Uses PbgA/YejM To Regulate Lipopolysaccharide Assembly during Bacteremia. Infect Immun 2019, 88. [Google Scholar] [CrossRef]
- Scott, A.J.; Oyler, B.L.; Goodlett, D.R.; Ernst, R.K. Lipid A structural modifications in extreme conditions and identification of unique modifying enzymes to define the Toll-like receptor 4 structure-activity relationship. Biochim Biophys Acta Mol Cell Biol Lipids 2017, 1862, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, X.; Wu, D.; Zhang, S.; Zhang, W.; Cao, Y. The pre-activated immune response induced by LPS protects host from leptospirosis. PLoS One 2020, 15, e0242742. [Google Scholar] [CrossRef]
- Novak, A.; Pupo, E.; Van't Veld, E.; Rutten, V.; Broere, F.; Sloots, A. Activation of Canine, Mouse and Human TLR2 and TLR4 by Inactivated Leptospira Vaccine Strains. Front Immunol 2022, 13, 823058. [Google Scholar] [CrossRef] [PubMed]
- Figueira, C.P.; Croda, J.; Choy, H.A.; Haake, D.A.; Reis, M.G.; Ko, A.I.; Picardeau, M. Heterologous expression of pathogen-specific genes ligA and ligB in the saprophyte Leptospira biflexa confers enhanced adhesion to cultured cells and fibronectin. BMC Microbiol 2011, 11, 129. [Google Scholar] [CrossRef]
Figure 1.
Immunostimulatory and adjuvant activity of Leptospira Lipid A (PLA). (A) Purification of PLA. PLA was purified, and bands were resolved and visualized by Thin Layer Chromatography (TLC) as described in the materials and methods. (B) Estimation of PLA-induced pro-inflammatory cytokine. RAW264.7 cell lines were stimulated with MPLA (0.5 - 2 µg/mL) or PLA (0.1 - 50 μg/ml) for 24 h, and culture supernatant was used to determine the IL-6 release by sandwich ELISA. (C) Analysis of PLA-induced expression of surface markers on mouse macrophages. The RAW264.7 cells were treated with MPLA (1 µg/mL) or PLA (2 or 5 μg/mL) for 24 hours, and the cells were harvested and stained with fluorochrome-conjugated antibodies and analyzed by flow cytometry. The control group indicates the uninduced samples. (D-G) Adjuvant activity of PLA in vivo. Mice were immunized with OVA formulated in Alum with or without PLA or MPLA, and antigen-specific humoral and cellular immune response was determined as described in materials and methods. (D) Humoral immune response. The OVA-specific antibodies (IgG, IgG1, IgG2c, IgA) in the serum obtained at day 28 were determined ELISA as detailed in the materials and methods section. (E) Lymphocyte proliferation. Spleenocytes isolated from various groups were stimulated with OVA (1 or 2.5, or 5 µg/mL), and cell proliferation was assessed after 72 hours using the Trypan blue size exclusion assay. (F) Cytokine analysis. The levels of IL-4 and IFN-γ in the culture supernatant of the stimulated spleenocytes were analyzed by ELISA. (G) Analysis of Cytotoxic T cells (CTLs). The activity of CTLs obtained from various groups was determined by the LDH cytotoxicity kit as specified in the materials and methods section. The one-way ANOVA was employed to calculate significant variations, with "***" denoting P ≤ 0.001 and "ns" indicating non-significance.
Figure 1.
Immunostimulatory and adjuvant activity of Leptospira Lipid A (PLA). (A) Purification of PLA. PLA was purified, and bands were resolved and visualized by Thin Layer Chromatography (TLC) as described in the materials and methods. (B) Estimation of PLA-induced pro-inflammatory cytokine. RAW264.7 cell lines were stimulated with MPLA (0.5 - 2 µg/mL) or PLA (0.1 - 50 μg/ml) for 24 h, and culture supernatant was used to determine the IL-6 release by sandwich ELISA. (C) Analysis of PLA-induced expression of surface markers on mouse macrophages. The RAW264.7 cells were treated with MPLA (1 µg/mL) or PLA (2 or 5 μg/mL) for 24 hours, and the cells were harvested and stained with fluorochrome-conjugated antibodies and analyzed by flow cytometry. The control group indicates the uninduced samples. (D-G) Adjuvant activity of PLA in vivo. Mice were immunized with OVA formulated in Alum with or without PLA or MPLA, and antigen-specific humoral and cellular immune response was determined as described in materials and methods. (D) Humoral immune response. The OVA-specific antibodies (IgG, IgG1, IgG2c, IgA) in the serum obtained at day 28 were determined ELISA as detailed in the materials and methods section. (E) Lymphocyte proliferation. Spleenocytes isolated from various groups were stimulated with OVA (1 or 2.5, or 5 µg/mL), and cell proliferation was assessed after 72 hours using the Trypan blue size exclusion assay. (F) Cytokine analysis. The levels of IL-4 and IFN-γ in the culture supernatant of the stimulated spleenocytes were analyzed by ELISA. (G) Analysis of Cytotoxic T cells (CTLs). The activity of CTLs obtained from various groups was determined by the LDH cytotoxicity kit as specified in the materials and methods section. The one-way ANOVA was employed to calculate significant variations, with "***" denoting P ≤ 0.001 and "ns" indicating non-significance.
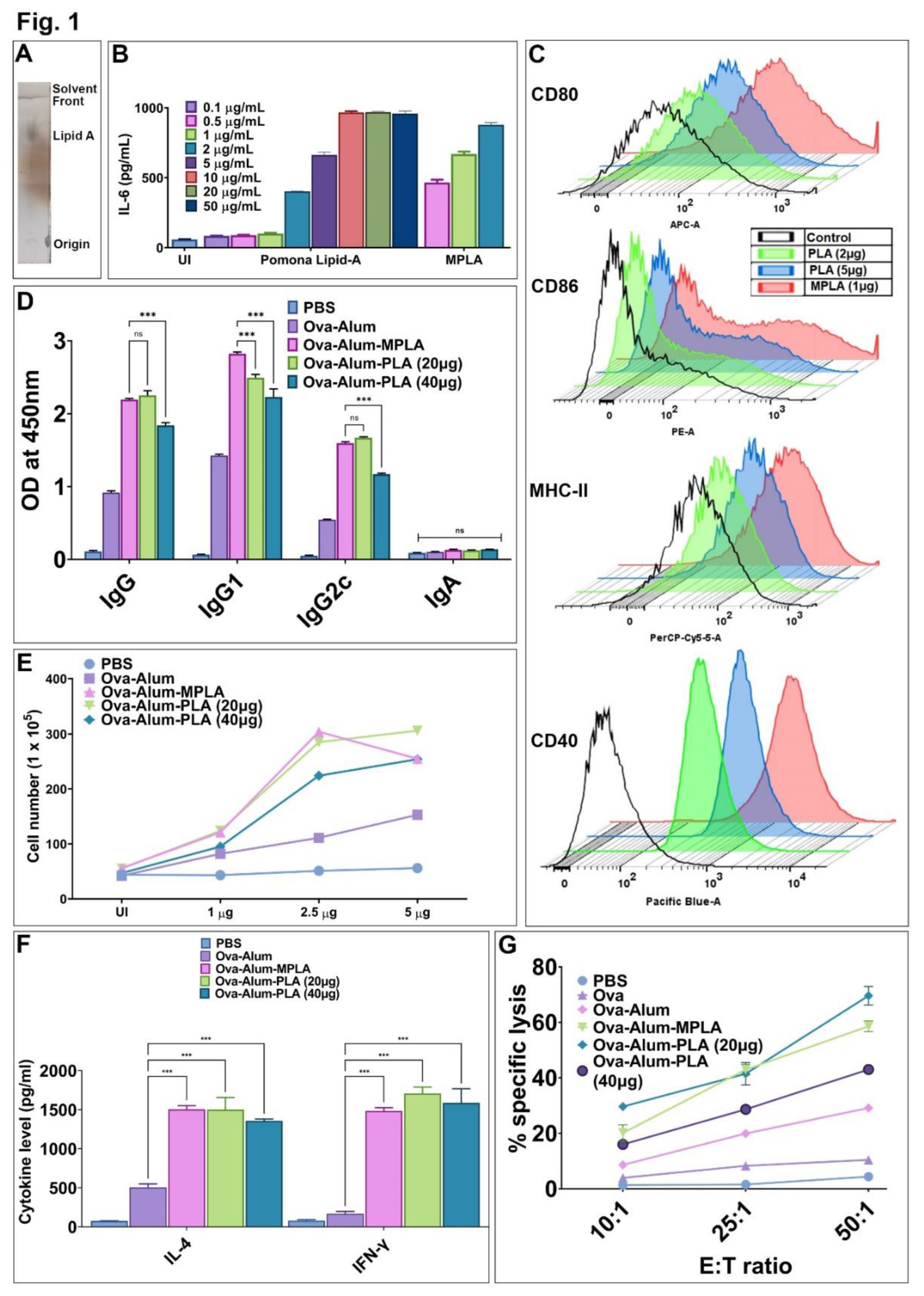
Figure 2.
PLA is a potent adjuvant that can enhance the immune response against Leptospira surface antigen. Mice were immunized with LAV formulated in Alum with or without PLA or MPLA, and LAV-specific humoral and cellular immune response was determined as described in materials and methods. A) Evaluation of LAV-specific antibody response. The level of LAV-specific antibodies (IgG, IgG1, and IgG2c) induced after immunization was analyzed by ELISA, as detailed in the method section. B) Assessment of LAV-specific lymphocyte proliferation. On day 28, splenocytes from the different immunized groups were isolated and stimulated with (LAV) for 72 hours. Cell proliferation was evaluated using the trypan blue size exclusion assay. C) Analysis of cytokine production. Splenocytes were stimulated with LAV (5 µg/mL) for 48 hours, and the release of IL-4 and IFN-γ cytokines in the culture supernatant was quantified using a sandwich ELISA kit following the manufacturer's instructions. D) Examination of T-cell phenotype. Spleenocytes stimulated with LAV (2.5 µg/mL) for 24 h were stained with anti-mouse CD3, CD4, and CD8 and then analyzed using flow cytometry. E) CTL assay. The activity of LAV-specific CTLs obtained from various groups was determined by the LDH cytotoxicity kit as specified in the materials and methods section. Significant differences were determined using one-way ANOVA. Statistical significance was denoted by "***" for P ≤ 0.001, and non-significant results were represented as "ns.".
Figure 2.
PLA is a potent adjuvant that can enhance the immune response against Leptospira surface antigen. Mice were immunized with LAV formulated in Alum with or without PLA or MPLA, and LAV-specific humoral and cellular immune response was determined as described in materials and methods. A) Evaluation of LAV-specific antibody response. The level of LAV-specific antibodies (IgG, IgG1, and IgG2c) induced after immunization was analyzed by ELISA, as detailed in the method section. B) Assessment of LAV-specific lymphocyte proliferation. On day 28, splenocytes from the different immunized groups were isolated and stimulated with (LAV) for 72 hours. Cell proliferation was evaluated using the trypan blue size exclusion assay. C) Analysis of cytokine production. Splenocytes were stimulated with LAV (5 µg/mL) for 48 hours, and the release of IL-4 and IFN-γ cytokines in the culture supernatant was quantified using a sandwich ELISA kit following the manufacturer's instructions. D) Examination of T-cell phenotype. Spleenocytes stimulated with LAV (2.5 µg/mL) for 24 h were stained with anti-mouse CD3, CD4, and CD8 and then analyzed using flow cytometry. E) CTL assay. The activity of LAV-specific CTLs obtained from various groups was determined by the LDH cytotoxicity kit as specified in the materials and methods section. Significant differences were determined using one-way ANOVA. Statistical significance was denoted by "***" for P ≤ 0.001, and non-significant results were represented as "ns.".

Figure 3.
Evaluation of the long-term memory response induced by PLA-based vaccine formulation. The animals immunized with PBS or LAV-Alum or LAV-Alum-MPLA, or LAV-Alum-PLA and boosted on the 21st day were bled every alternative week till 24 week. The animals were euthanized or given additional antigen booster (without adjuvant) and spleens were collected after one week to determine memory T cells. A) The humoral (antibody) memory response. The levels of LAV-specific IgG at various time points or isotypes (IgG1 or IgG2c) in the serum were analysed by ELISA as described in the materials and methods. B) The T-cell memory response. Spleenocytes isolated from mice euthanized on 24w and 25w were stimulated with LAV for 72 h cell proliferation was assessed by Trypan Blue size exclusion assay. C) Estimation of cytokine levels. Forty-eight hours after splenocytes stimulation with antigen (LAV; 5 µg/mL), using ELISA, the IL-4 and IFN-γ cytokines in culture supernatant as per the manufacturer's instructions. D) Analysis of memory phenotype. The spleenocytes were stained with fluorescent-labeled antibodies specific against CD3, CD4, CD8, CD62L, and CD44, and then performed flow cytometry analysis. Representative flow cytometry plots demonstrated the gating strategy for analyzing central (CD44high and CD62Lhigh) and effector (CD44high, CD62Llow) memory response in CD4 and CD8 T cell populations. The number of positive cells expressing CD44 and CD62L over the total of CD4+/CD8+ T cells were plotted as histograms. The significant differences were calculated using one-way or two-way ANOVA, with the symbols ****, ***, **, *, and ns indicating P ≤ 0.0001, P ≤ 0.001, P ≤ 0.001, P ≤ 0.05, and non-significant, respectively.
Figure 3.
Evaluation of the long-term memory response induced by PLA-based vaccine formulation. The animals immunized with PBS or LAV-Alum or LAV-Alum-MPLA, or LAV-Alum-PLA and boosted on the 21st day were bled every alternative week till 24 week. The animals were euthanized or given additional antigen booster (without adjuvant) and spleens were collected after one week to determine memory T cells. A) The humoral (antibody) memory response. The levels of LAV-specific IgG at various time points or isotypes (IgG1 or IgG2c) in the serum were analysed by ELISA as described in the materials and methods. B) The T-cell memory response. Spleenocytes isolated from mice euthanized on 24w and 25w were stimulated with LAV for 72 h cell proliferation was assessed by Trypan Blue size exclusion assay. C) Estimation of cytokine levels. Forty-eight hours after splenocytes stimulation with antigen (LAV; 5 µg/mL), using ELISA, the IL-4 and IFN-γ cytokines in culture supernatant as per the manufacturer's instructions. D) Analysis of memory phenotype. The spleenocytes were stained with fluorescent-labeled antibodies specific against CD3, CD4, CD8, CD62L, and CD44, and then performed flow cytometry analysis. Representative flow cytometry plots demonstrated the gating strategy for analyzing central (CD44high and CD62Lhigh) and effector (CD44high, CD62Llow) memory response in CD4 and CD8 T cell populations. The number of positive cells expressing CD44 and CD62L over the total of CD4+/CD8+ T cells were plotted as histograms. The significant differences were calculated using one-way or two-way ANOVA, with the symbols ****, ***, **, *, and ns indicating P ≤ 0.0001, P ≤ 0.001, P ≤ 0.001, P ≤ 0.05, and non-significant, respectively.
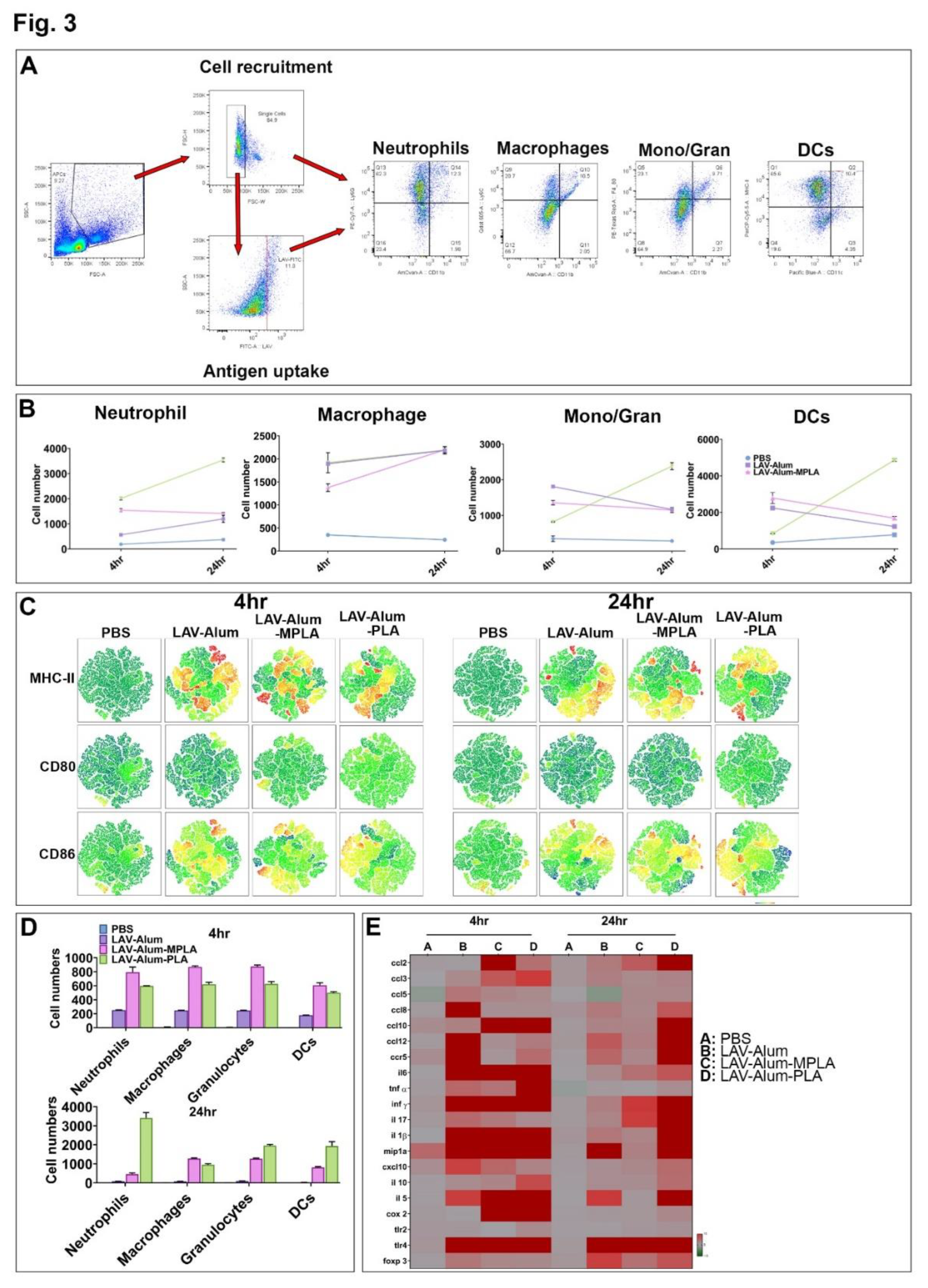
Figure 4.
Formation of Germinal Centres (GCs) in the draining lymph nodes. The formation of GCs in DLNs of various groups was assessed on the 28th-day post-immunization. DLNs were processed as frozen tissue sections and later stained with fluorochrome-conjugated anti-mouse CD3/CD45R/B220/GL-7/CXCR5 antibodies as described in the methodology. The sections were mounted and examined under a fluorescent microscope using tile scanning and then processed using the ZEN software (stitching tool) as described in the methodology section. Data represent three different experiments. Significant differences were calculated using the one-way ANOVA (****, ***, **, *, and ns indicate P ≤ 0.0001, P ≤ 0.001, < 0.01, P ≤ 0.05, and non-significant, respectively).
Figure 4.
Formation of Germinal Centres (GCs) in the draining lymph nodes. The formation of GCs in DLNs of various groups was assessed on the 28th-day post-immunization. DLNs were processed as frozen tissue sections and later stained with fluorochrome-conjugated anti-mouse CD3/CD45R/B220/GL-7/CXCR5 antibodies as described in the methodology. The sections were mounted and examined under a fluorescent microscope using tile scanning and then processed using the ZEN software (stitching tool) as described in the methodology section. Data represent three different experiments. Significant differences were calculated using the one-way ANOVA (****, ***, **, *, and ns indicate P ≤ 0.0001, P ≤ 0.001, < 0.01, P ≤ 0.05, and non-significant, respectively).
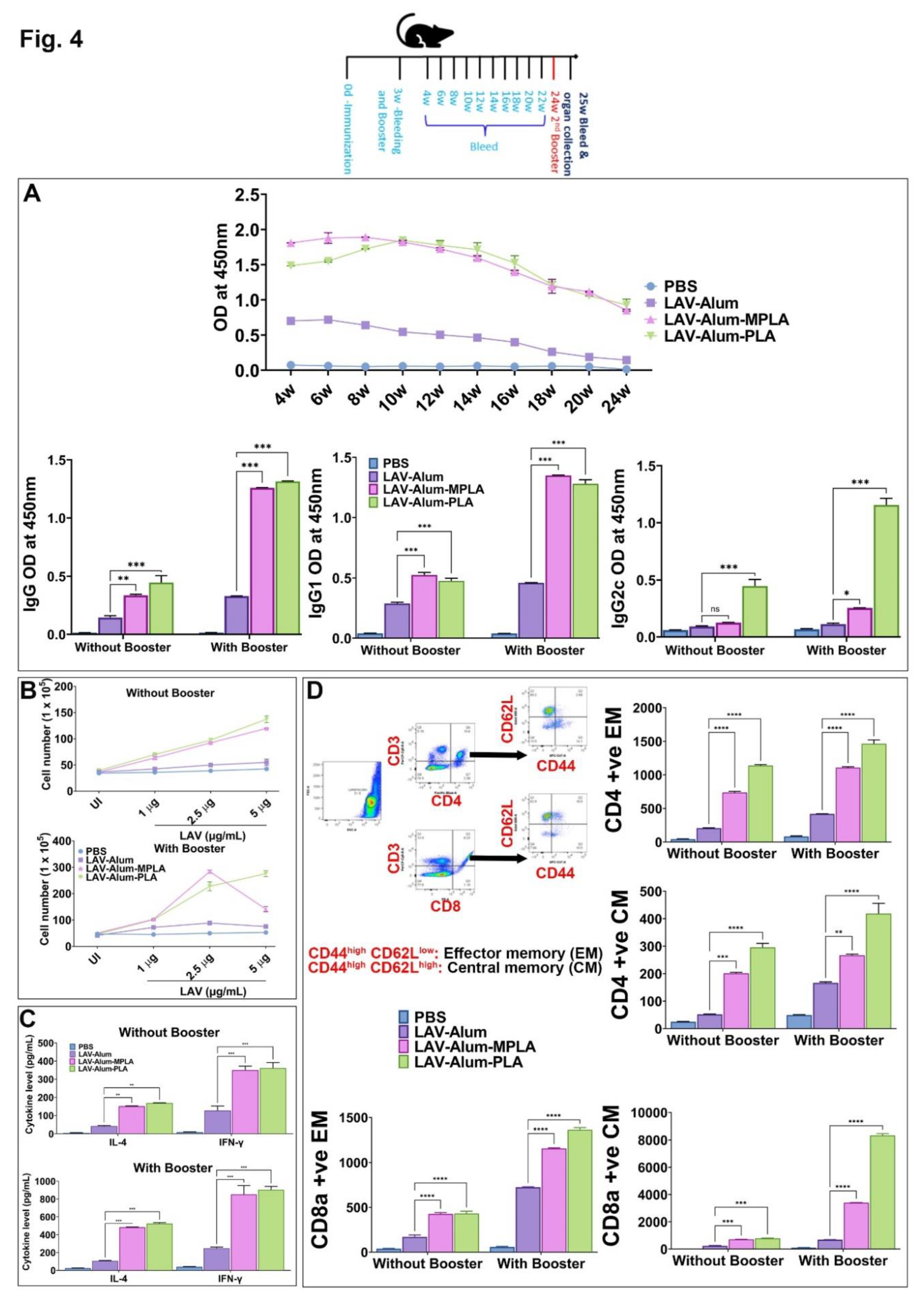
Figure 5.
PLA enhances the recruitment, activation, and antigen uptake by innate cells at the site of vaccine administration. To evaluate PLA-induced recruitment, antigen uptake and activation of innate immune cells tissue from the injection site and DLNs were isolated at 4 h and 24 h for further analysis. A) Gating strategy. The cells obtained from DLNs were stained with marker-specific fluorescent labelled antibodies as described in the methodology and then gated to identify the appropriate cell population. B) Analysis of cells recruited in DLNs. The specific cell type recruited by each vaccine formulation at different time points was analyzed as detailed in the methodology. C) Activation status of cells recruited in DLNs. The expression of surface markers (CD80, CD86, CD40, and MHC-II) was analyzed by flow cytometry and visualized by a tSNE plot. The heatmaps were generated to describe the intensity of specific markers; Dark blue regions correspond to the lowest intensity of fluorescent antibody staining, while red indicates the highest intensity, with heatmap ranging from −1622 to 262856. D) Analysis of antigen uptake by APC. The uptake of Alexa Fluor 488 labeled LAV by a different type of recruited APCs at 4 h and 24 h were analyzed by Flow cytometry. F) Inflammatory responses at the injection site. qRT-PCR analysis of cytokines, chemokines, and receptors at the injection site 4 and 24 h post-injection.
Figure 5.
PLA enhances the recruitment, activation, and antigen uptake by innate cells at the site of vaccine administration. To evaluate PLA-induced recruitment, antigen uptake and activation of innate immune cells tissue from the injection site and DLNs were isolated at 4 h and 24 h for further analysis. A) Gating strategy. The cells obtained from DLNs were stained with marker-specific fluorescent labelled antibodies as described in the methodology and then gated to identify the appropriate cell population. B) Analysis of cells recruited in DLNs. The specific cell type recruited by each vaccine formulation at different time points was analyzed as detailed in the methodology. C) Activation status of cells recruited in DLNs. The expression of surface markers (CD80, CD86, CD40, and MHC-II) was analyzed by flow cytometry and visualized by a tSNE plot. The heatmaps were generated to describe the intensity of specific markers; Dark blue regions correspond to the lowest intensity of fluorescent antibody staining, while red indicates the highest intensity, with heatmap ranging from −1622 to 262856. D) Analysis of antigen uptake by APC. The uptake of Alexa Fluor 488 labeled LAV by a different type of recruited APCs at 4 h and 24 h were analyzed by Flow cytometry. F) Inflammatory responses at the injection site. qRT-PCR analysis of cytokines, chemokines, and receptors at the injection site 4 and 24 h post-injection.
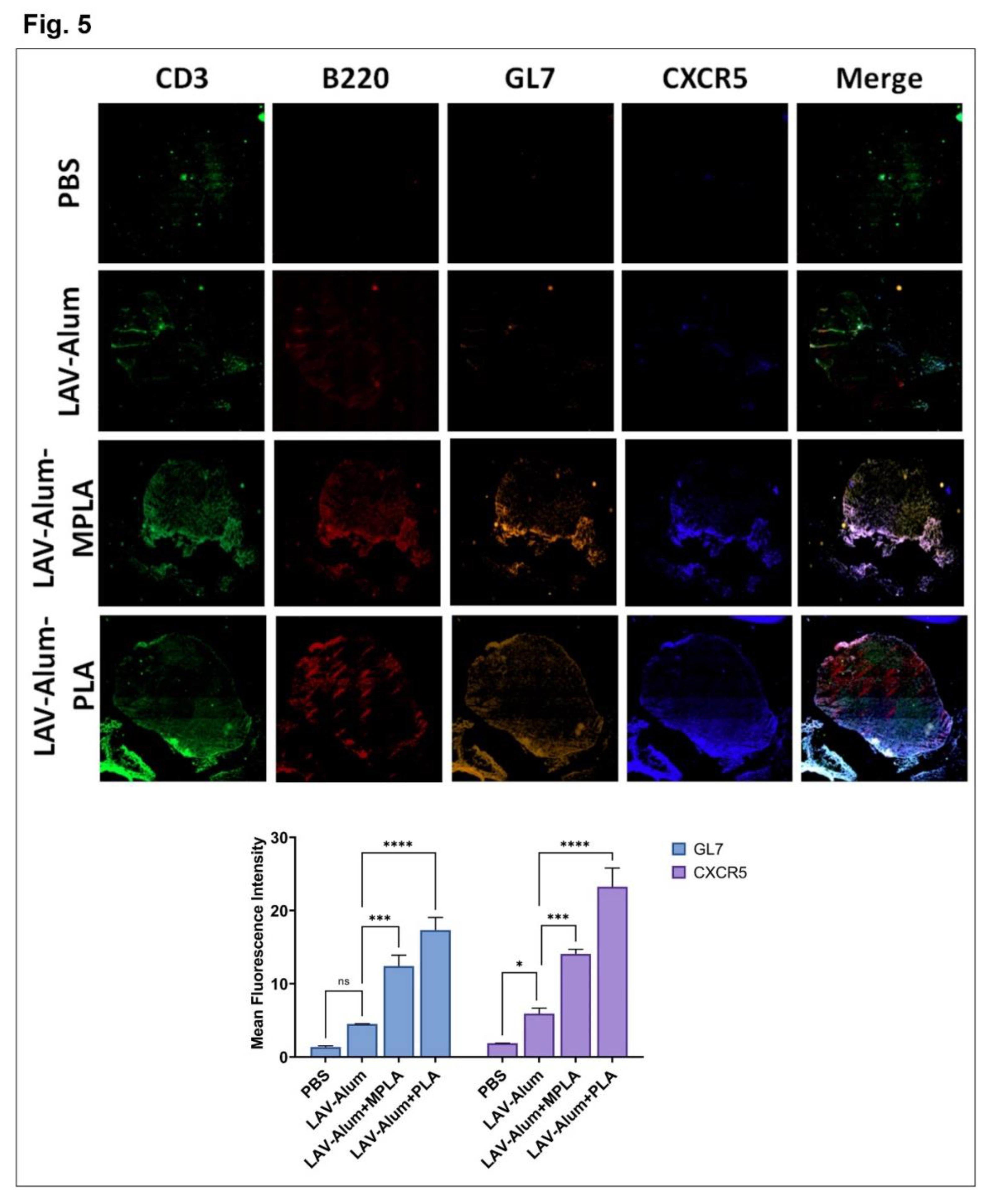
Figure 6.
PLA enhances the immune response and protective efficacy in the hamster Leptospirosis model. Immune response and protective efficacy of various vaccine formulations were assessed in the hamster model as detailed in the methodology. A) Serum Antibody. LAV-specific total IgG and subtype (IgG1 and IgG2/3) antibody levels were measured in immunized animal serum on the 35th day using ELISA. B) Cell proliferation and cytokine response. The splenocytes obtained from various groups were stimulated with LAV, and cell proliferation was analyzed using Trypan blue size exclusion after 48-72 h. Stimulated cells were harvested at 24h, and IL-4 and IFN-γ cytokine expression was analyzed using qRT-PCR. C) T-cell analysis. LAV-specific T cells phenotype were identified by gating on a selection of CD4+ CD8b+ T cells using Flowcytometry. D) Clinical manifestations. The challenged animals were observed by examining the gross appearance of the organs of the animals that either died or were euthanized according to the predetermined endpoint criteria. E) Bodyweight. The animals challenged on day 35 were monitored for any changes in body weight till day 63. Individual body weights are shown as a percentage compared with the pre-challenge weight. F) Survival analysis. The survival rate of the animals was monitored till 28 days post-challenge and was analyzed using Kaplan-Meier's plot based on criteria described in the methodology. G) Leptospira colonization in the organs. Bacterial load per gram of tissue (lung, liver, and kidney) was determined by RT-PCR quantifying the LipL32/16s gene. H) Histopathological analysis. Representative images of Hematoxylin and Eosin-stained tissues (lung, liver, and kidney) were obtained from various groups of animals which died or survived after the challenge (scale bars, 10 μm). Significant differences were determined using either one-way or two-way ANOVA. The symbols ****, ***, **, *, and ns represent P-values of ≤0.0001, ≤0.001, ≤0.001, ≤0.05, and non-significant, respectively.
Figure 6.
PLA enhances the immune response and protective efficacy in the hamster Leptospirosis model. Immune response and protective efficacy of various vaccine formulations were assessed in the hamster model as detailed in the methodology. A) Serum Antibody. LAV-specific total IgG and subtype (IgG1 and IgG2/3) antibody levels were measured in immunized animal serum on the 35th day using ELISA. B) Cell proliferation and cytokine response. The splenocytes obtained from various groups were stimulated with LAV, and cell proliferation was analyzed using Trypan blue size exclusion after 48-72 h. Stimulated cells were harvested at 24h, and IL-4 and IFN-γ cytokine expression was analyzed using qRT-PCR. C) T-cell analysis. LAV-specific T cells phenotype were identified by gating on a selection of CD4+ CD8b+ T cells using Flowcytometry. D) Clinical manifestations. The challenged animals were observed by examining the gross appearance of the organs of the animals that either died or were euthanized according to the predetermined endpoint criteria. E) Bodyweight. The animals challenged on day 35 were monitored for any changes in body weight till day 63. Individual body weights are shown as a percentage compared with the pre-challenge weight. F) Survival analysis. The survival rate of the animals was monitored till 28 days post-challenge and was analyzed using Kaplan-Meier's plot based on criteria described in the methodology. G) Leptospira colonization in the organs. Bacterial load per gram of tissue (lung, liver, and kidney) was determined by RT-PCR quantifying the LipL32/16s gene. H) Histopathological analysis. Representative images of Hematoxylin and Eosin-stained tissues (lung, liver, and kidney) were obtained from various groups of animals which died or survived after the challenge (scale bars, 10 μm). Significant differences were determined using either one-way or two-way ANOVA. The symbols ****, ***, **, *, and ns represent P-values of ≤0.0001, ≤0.001, ≤0.001, ≤0.05, and non-significant, respectively.
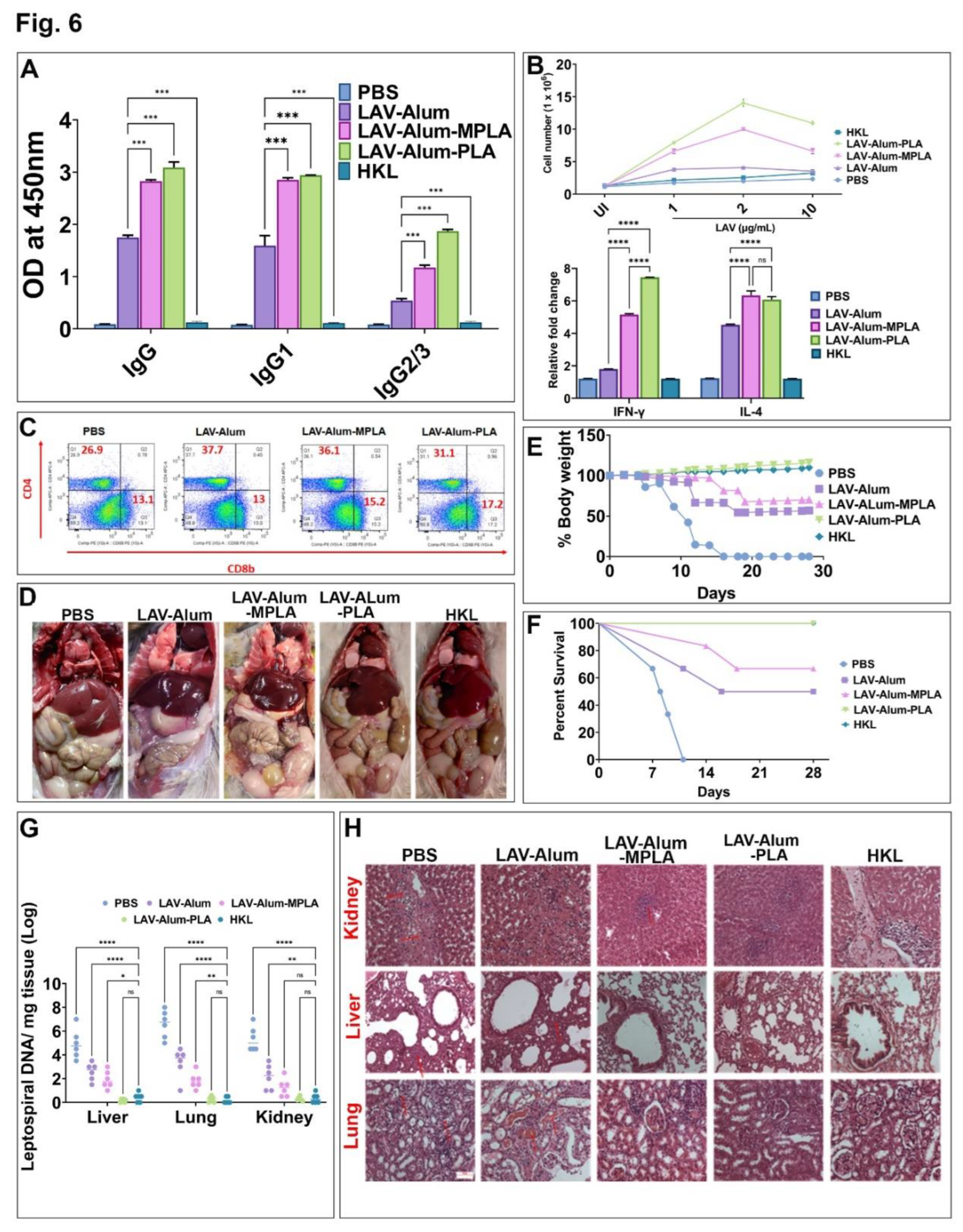
Figure 7.
Schematic representation depicting the difference in immune response and protective efficacy induced by PLA or MPLA-based vaccine formulation. (A) Immune response in mice. Both the formulation increased recruitment and activation of innate immune cells at sight of injection and Draining Lymph node. While PLA based formulation increased recruitment and activation of dendritic cells and neutrophils, MPLA promoted the activation of monocytes. Both the formulations induced mixed Th1/Th2 response with generation of both IgG1/IL-4 and IgG2c/ IFN-γ. While both formulations generated long term memory response, PLA induced significantly higher levels of GC reaction correlating to higher levels of Th1 associated antibodies. It also induced higher levels of central and effector memory T cells.(B) Immune response and protective efficacy in a Hamster model. Both the formulation induced strong antibody response with significant levels of IgG1 but PLA induced significantly higher levels of IgG2/3. PLA induced a greater number of CD4 and CD8T cells with significantly higher levels of IFN-g. The immune response correlated to significantly higher level of protective efficacy as PLA based formulation had more number of survivors having reduced or undetectable bacterial load with mild or no lesions in vital organs.
Figure 7.
Schematic representation depicting the difference in immune response and protective efficacy induced by PLA or MPLA-based vaccine formulation. (A) Immune response in mice. Both the formulation increased recruitment and activation of innate immune cells at sight of injection and Draining Lymph node. While PLA based formulation increased recruitment and activation of dendritic cells and neutrophils, MPLA promoted the activation of monocytes. Both the formulations induced mixed Th1/Th2 response with generation of both IgG1/IL-4 and IgG2c/ IFN-γ. While both formulations generated long term memory response, PLA induced significantly higher levels of GC reaction correlating to higher levels of Th1 associated antibodies. It also induced higher levels of central and effector memory T cells.(B) Immune response and protective efficacy in a Hamster model. Both the formulation induced strong antibody response with significant levels of IgG1 but PLA induced significantly higher levels of IgG2/3. PLA induced a greater number of CD4 and CD8T cells with significantly higher levels of IFN-g. The immune response correlated to significantly higher level of protective efficacy as PLA based formulation had more number of survivors having reduced or undetectable bacterial load with mild or no lesions in vital organs.
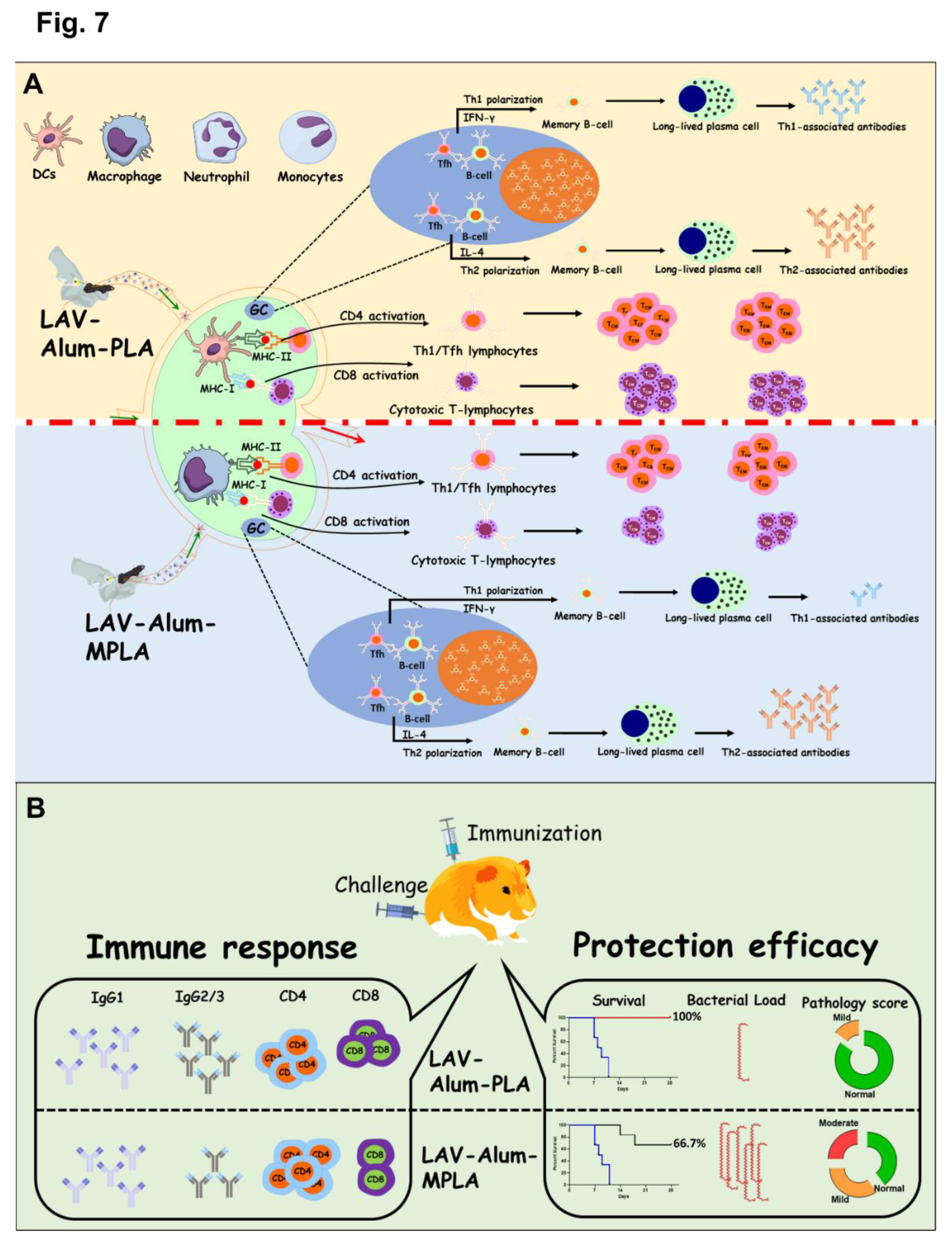
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



