
Submitted:
19 July 2023
Posted:
21 July 2023
You are already at the latest version
Abstract
The major histocompatibility complex (MHC) genes are the most polymorphic genes in vertebrates, and their proteins play a critical role in adaptive immunity for defense against a variety of pathogens. MHC diversity was lost in many species after experiencing a decline in size. To understand the variation and evolution of MHC genes in the Siberian ibex, Capra sibirica, which has undergone a population decline, we analyzed the variation of the second exon of MHC class II DRB genes in samples collected from five geographic localities in Xinjiang, China, that belong to three diverged mitochondrial clades. Consequently, we identified a total of 26 putative functional alleles (PFAs) with 260 bp in length from 43 individuals, and found one (for 27 individuals) to three (for 5 individuals) PFAs per individual, indicating the presence of one or two DRB loci per haploid genome. The Casi-DRB1*16 was the most frequently occurring PFA, Casi-DRB1*22 came after found in only seven individuals, 14 PFAs occurred once (7 PFAs twice), implying high frequency of rare PFAs. Interestingly, more than half of the (15) PFAs were specific to clade I, only one and three PFAs were specific to clades II and III, respectively. So, we assume that the polygamy and sextual segregation nature of this species likely contributed to the allelic diversity of DRB genes. Genetic diversity indices showed that PFAs of clade II were lower in nucleotide, amino acid, and supertype diversity compared to those of the other two clades. The way of allele sharing and FST values between three clades were to some extent in agreement with pattern observed in mitochondrial DNA divergence. In addition, recombination analyses revealed no evidence for significant signatures of recombination events. Alleles shared by clades III and the other two clades diverged 6 million years ago, and systematic neighbor grids showed cross-species polymorphisms. Together with the PAML and MEME analyses, the results indicated that the DRB gene in C. sibirica evolved under balancing and positive selection. However, by comparison, it can be clearly seen that different populations were under different selective pressures. Our results are valuable in understanding the diversity and evolution of the DRB gene in a mountain living C. sibirica and in making decisions on future long-term protection strategies.
Keywords:
Mountain ungulate
; MHC DRB gene
; genetic diversity
; population fluctuation
Introduction
The way a species interacts with other species and its environment associated with its genetic diversity. A species' ability to adapt to disturbances caused by humans and the natural environment depends on variation as well. The degree of genetic diversity within a species affects that species' capacity to adapt to environmental changes [1]. The measurement of these variation, however, cannot be limited to the use of traditional neutral markers. The polymorphisms of Major Histocompatibility Complex (MHC) genes are considered to affect the functional plasticity of immune responses to diverse pathogenic stressors, making them excellent candidates to research adaptive evolutionary processes in natural populations [2,3]. This trait highlights how the immune system susceptible to environmental stress and how crucial it is for illuminating the mechanisms of adaptive genetic variation required for the long-term survival of species or populations [4,5].
The most polymorphic region of the vertebrate genome that evolved under positive and balancing selection is the MHC [5,6,7], a multigene family of the vertebrate adaptive immune system that contains highly polymorphic motifs that are strongly associated with immune response and disease resistance [8,9]. MHC genes belong to two main subfamilies, class I and class II, and encode proteins that are necessary for pathogen recognition and presentation to T cells. Functional class II proteins are heterodimers consisting of α and β chains, and DR subclasses are encoded by the DRA and DRB genes, respectively [10]. The amino acid residues that bind directly to the antigen are called antigen binding sites (ABS). ABSs were located at the α1 domain of α1 chain and the β1 domain of β chain, in which MHC polymorphisms in vertebrates mainly occur [11,12].
Alpine ungulates are a significant factor in the maintenance of the structure of vegetation and the cycling of nutrients in high-mountain ecosystems, as well as a significant source of food for predators [13,14]. However, due to its slow growth, poor rate of reproduction, vulnerability to exploitation by humans, loss of habitat, susceptibility to infectious diseases, and other reasons [15,16,17], Most of them are extremely vulnerable to extinction. Of these, a typical alpine hoofed species of the subfamily Caprinae (family Bovidae), is the Siberian ibex, Capra sibirica. This species widely habituated in the alpine regions of Central Asia, from northern India through Pakistan and Afghanistan to Russia (Siberia), and eastward to northwest China and western Mongolia [18]. According to studies, the Siberian ibex, to some extent, suffered threats from various pathogens like lethal bacteria, viruses, ticks, and mites [19,20,21]. Moreover, it shares more than 76 percent of its food with domestic animals in Chinese territory [22], indicating not only fierce food competition but also a greatly increased risk of becoming infected. Despite the importance of MHC genes for immunological fitness, an assessment of the diversity and occurrence of these genes is still lacking in the Siberian ibex, the globally ‘Near Threatened’ mammal in Central Asia, and locally urgently needs an effective conservation and management programs [18].
Because of anthropogenic impacts, Siberian ibex populations dropped globally and their range shrunk drastically in the 1970s [23,24]. In China, particularly, it had been listed as an endangered species and given Class I protection priority in 1998 [25]. The population has fortunately started to recover owing to effective conservation and management (creation of protected areas, etc.) by the Chinese government and neighboring nations in recent years, and thus its protection priority was decreased to Class II in 2021 [26]. Generally, reduced genetic diversity is associated with demographic perturbation. Natural populations of many species that underwent a reduction in size exhibited very limited MHC diversity [27,28,29]. However, both theoretical and empirical studies also showed that a longer timescale of selection maintained higher MHC diversity in a population experienced demographic fluctuations [30,31]. It is thus significant to study whether a highly genetically diverged Siberian ibex populations [32] maintained high MHC diversity during a more than half-century period of recovery.
Therefore, our objectives in this study had three facets. To begin with, we aim to comparatively evaluate the MHC diversity in different Siberian ibex populations in Xinjiang, China, and discuss our results with other species bottlenecked. In addition, we also try to ascertain if the MHC DRB1 divergence in different populations was in accordance with the results of mitochondrial genes divergence we reported previously [32]. Finally, to check if the MHC DRB1 genes in the Siberian ibex that went through population fluctuation resemble the common characteristics of MHC in other vertebrates, such as positive selection, recombination, and trans-species polymorphism, and to clarify the genetic relationships of MHC DRB1 alleles of the relic species Siberian ibex and its congenerics, including domestic goats. Our results were of importance in understanding the adaptive ability of this species and planning scientific conservation strategies to ensure long-term population development.
Materials and Methods
Samples
A total of 43 samples, including 33 feces, 5 muscle, 4 skin, and 1 liver sample, were analysed. Of these, 10 samples collected from Urumqi, 16 from Arturk, 1 from Sawan, 13 from Ulugqat, and 3 from Kagilik (Figure 1). All samples in this study were came from samples of our previous study [32]. Tissue samples either taken from individuals died of natural causes or dead individuals that were poached. Individual identity of fecal samples was established according to Abduriyim et al. [33,34]. All fecal and tissue samples were preserved in 96 % ethanol, and skin samples were directly frozen in plastic bags at -80 ℃ until use.
Experimental Procedures
The total genomic DNA of fecal samples was extracted using an Omega stool DNA extraction kit (Omega Bio-tec, Georgia, USA), and that of muscle and skin samples was extracted using a Tiangen tissue/blood DNA extraction kit (Tiangen Bio-tec, Beijing, China), following the manufacturer's instructions. After electrophoresis detection, DNA concentration and purity were measured by the Thermo Nanodrop 1000 and stored at 4 ℃ for later use.
Part of MHC class II DRB1 exon 2 (260 bp, excluding the primer sequences) was PCR amplified using primer pairs of CapDRB1.1F and CapDRB1.2R [35], because this segment is the most polymorphic region and includes all ABSs necessary for pathogen recognition [36,37]. A PCR reaction volume contained 40-150 ng of DNA, 5 pmol of each primer, 12.5 μL of Tiangen's 2 TaqPCR Master Mix, and then adjusted to a final volume of 25 μL with RNase-Free double distilled water. The PCR thermal cycling conditions were as follows: pre-denaturation at 94 ºC for 4 min, followed by 35 cycles of denaturation at 94 ºC for 30 s, annealing at 64 ºC for 30 s, extension at 72 ºC for 45 s, and final extension at 72 ºC for 5 min. The PCR products were verified by 2 % agarose gel electrophoresis and green fluorescence dye imaging under ultraviolet irradiation, and those with the expected band size were sent for Sanger sequencing bidirectionally (Qingke Biology, Xi-an, China).
PCR products assumed to contain more than one sequences were proceeded to cloning and sequencing for allele isolation. PCR products were recovered using a gel recovery kit (Tiangen, Beijing, China), connected to the PMDTM19-T plasmid vector (Takara, Tokyo, Japan), then transformed into Escherichia coli (DH5α) receptive cells. For selection of cells with positive plasmids, the bacteria were grown on LB solid medium containing ampicillin, IPTG and X-gal at 37 ºC overnight. Bacteria containing plasmids with the target PCR product were screened by blue/white selection and direct-colony PCR amplification using M13 forward and reverse primers with the same PCR condition as described earlier. At least 8 clones per sample were bidirectionally sequenced for each individual.
MHC Genotyping
All nucleotide sequences obtained were aligned using MEGA v.6.0 [38]. The unique and same sequences were screened using DnaSP v.5.10.01 [39]. The final sequences were identified as potentially genuine DRB1exon 2 sequences if they matched in the forward and reverse directions, and were detected at least twice in one individual (two independent PCR reactions for one individual) or once each from at least two individuals [40]. Single, unique sequences were omitted, as they may have been PCR chimeras or due to other PCR errors [5,37]. We verified candidate sequences with BLAST searches [41] at the National Center for Biotechnology Information (NCBI) GenBank database. Final verified sequences were named by consulting the conventions of Klein et al. [42], and Ballingall & Todd [43].
Data Analyses
The nucleotide, amino acid, super type diversity, and pairwise population fixation indices (FST) of DRB1 exon 2 for different populations were calculated by DnaSP v.5.10.01 [39], and the neutral selection was analyzed. MEGA v.6.0 were used to estimate the ratio ω (dN/dS) of non-synonymous (dN) to synonymous (dS) substitution rates [44]; this ratio provides a measure of selective pressure at the level of individual sites [45]. Values of ω > 1 indicate positive selection, while ω = 1 and ω < 1 indicate neutral evolution and purifying selection, respectively. Values of dN, dS and ω were calculated separately for presumed ABSs deduced according to Reche & Reinherz [46], non-ABSs, and all sites. HyPhy [47] implemented in MEGA was used to detect signs of positive selection. In order to examine positive selection across all sites based on maximum likelihood methods, CodeML in PAML 4.9 [48] was employed as well. The likelihood ratio tests (LRTs) were used to compare the four models: M1a, almost neutral; M2a, positive selection; M7, beta; and M8, beta and ω, and decide which model best fit our data [45,49,50]. Using LRTs, two nested models (M1a vs. M2a; M7 vs. M8) were compared. Using Bayes Empirical Bayes inference [51], positively selected locations were found. In addition, using Datamonkey v.2.0 [52], a web-based server for the HyPhy Package, a mixed-effects model of evolution, MEME [53] analysis was carried out to find codons that had been subject to positive selection.
Gene recombination analysis of DRB1 exon 2 sequences was performed in RDP4 [54]. Specific methods were first used in RDP [55], GENECONV [56], MaxChi [57] and Bootscan [58], which use default Settings to detect recombination events using Bonferroni correction for multiple comparisons. Recombination events detected by at least three of these methods were then rechecked using all RDP methods available [55]. In addition, we also use the GARD [59], provided by the Datamonkey webserver [60], to detect the signatures of recombination breakpoints. In order to avoid the impact of possible gene replication, conversion, and recombination on phylogenetic analysis, we chose Splitstree4 v.4.14.5 to construct a neighbor network of DRB1 sequences [5,61].
Result
Diversity of DRB1 Alleles
Our analytical sequences were 260 bp in length, encode 86 amino acids including 20 ABSs, accounting for 91% of the DRB1 β1 domain (Figure 2). We identified 26 presumably functional alleles (PFA) in a total of 43 individuals belong to three mtDNA clades. None of these sequences were pseudogenes. The number of PFA found in a single individual ranged from 1 to 3, indicating existence of 1 or 2 loci per haploid genome in the Siberian ibex. 26 individuals possess only one PFA, implying that these individuals were homozygous. 12 individuals with two PFAs were highly likely heterozygous individuals. Only five individuals had three PFLs (Table A1). Sum up, most of the individuals in Siberian ibex Xinjiang populations had one DRB1 locus and could be homozygous.
The most common PFA was Casi-DRB1*16, occurred in 17 individuals that belong to Clade I (12 individuals) and Clade III (5 individuals). The PFAs Casi-DRB1*22 and 19 came after, but we found only in seven and five individuals, respectively. Casi-DRB1*16 and 17 were shared by Clades I and III, Casi-DRB1*02, 03, and 05 by Clades II and III. Besides, Casi-DRB1*08 and 15 were specific to Clade Ⅱ, while Casi-DRB1*01, 18, and 24 were only found in Clade Ⅲ, and the remaining ones were exclusively occupied by Clade Ⅰ (Figure 1, Table A1).
Diversity indices showed a high level of genetic diversity at the nucleotide, amino acid and supertype sequence level for all clades. By comparison, the nucleotide diversity of individuals from Clade I and Clade Ⅲ was similar, and both were higher than that from Clade Ⅱ. In terms of amino acid and super type, Clade II had an overall lower level of diversity than Clade I and Clade III as well (Table 1). Tajima's D values were positive, except for Clade II, though none of these values were significant (Table 1). Altogether, this indicates that the MHC class II DRB1 locus in different clades was likely at the different the stage of bottleneck or selection pressures.
We calculated the Genetic differentiation by FST values, both including and excluding shared alleles between the three Clades. The FST value between clades I and Ⅲ was negative (Table 2), indicating that within-population genetic differentiation was higher than between-population genetic differentiation. After excluding the shared alleles, the value was positive but very low, implying that the shared alleles were more divergent than the unique alleles in these clades. When including shared alleles, the FST values between clades I and II, II and Ⅲ were 0.267 and 0.343, respectively (Table 2), indicating a very high level of differentiation.
Recombination and Selection on DRB1
It was hardly evident that significant recombination signatures exist in our analyses of the DRB1 exon 2 sequences of the Siberian ibex. Hence, we used all sequences in the downstream analyses. To evaluate selection pressure, we calculated the ω ratio of non-synonymous to synonymous substitution rates for positions in the presumed ABSs, non-ABS codons and all codons for three Clades. The ω ratio value for ABS codons in C. sibirica DRB1 was greater than one. Our result thus indicates that variation at the ABS codons were generated and maintained by positive selection. Comparatively, ω value for Clade II was nearly twice of that for clades I and II, indicating that the selection intensity on these clades was different (Table 3). This was in line with the results of PAML and MEME analyses that provide evidence for positive selections at the single codon level (Table 4). Likelihood ratio tests (LRT) showed that the M2a and M8 models provided significantly better fits to our data than models without selection (Table 4). The M2a and M8 models identified 13 and 14 positively selected sites in Clade I, respectively, 13 from each model in Clade III, while only five from each model in Clade II (Figure 2, Table 4), with most of the sites occurring in presumed ABS codons. Finally, the MEME analysis showed six codons under positive selection in Clade I, only one in Clade III, and none in Clade II (Figure 2).
Phylogeny of DRB1 Alleles
In the phylogenetic neighbor grid, the DRB1 exon 2 sequences of C. sibirica did not form separate groups according to the geographical population results, but merged into sequences of other species of genus Capra, forming groups of A, B, C, D, E, F, and G (Figure 3). Trans-species polymorphism (TSP) was clearly evident, with some exon 2 sequences from particular C. sibirica being more closely related to sequences from other Capra species than to those from the same species. The E and G groups are composed of four types of Capra: C. sibirica, C. aegagrus, C. hircus, C. pyrenaica DRB sequences; The remaining A, B, C, D, and F groups contain only DRB sequences of C. sibirica, C. aegagrus, and C. hircus.
Discussion
In this study, we examined the sequence diversity of the MHC Class II DRB1 exon 2 of C. sibirica from the eastern Tianshan, the middle Tianshan, and the Kunlun Mountains, with a maximum distance of about 2, 000 km and an average of 4, 500–5, 250 m-height mountain picks [62], which seriously hindered the genetic communication between populations [32]. In addition, anthropogenic factors led these populations to a drastic decline in size.
MHC DRB1 Diversity and Divergence
Indirect indicators of the immunological fitness of populations, MHC genes are adaptive genetic markers useful in wild animal populations of concern for protection [3,63]. Many species, which went through severe bottlenecks, show very low levels of genetic diversity at the MHC, for example, mountain goats, Oreamnos Americanus [64] and Galà pagos penguin, Spheniscus mendiculus [65]. Our study on MHC class II DRB1 exon 2 allowed, for the first time, a comparison of genetic variation among C. sibirica populations that genetically highly diverged and underwent population reduction in size in Xinjiang, China [18,32]. Unexpectedly, we found higher allelic diversity of MHC class II DRB1 loci in C. sibirica compared to other congenerics. Likewise, despite a rinderpest epidemic-induced bottleneck, high allelic diversity for the DRB3 gene was reported for the African buffalo, Syncerus caffer [66]. Although the 26 PFAs we detected in 43 C. sibirica individuals (Figure 1, Table A1) seem to be lower than the 22 PFAs among 25 samples reported for its domestic counterpart from six different breeds [67], only seven PFAs were found among 132 individuals of Capra pyrenaica with two subspecies, C. p. hispanica and C. p. victoria [68]. This high number of alleles is mainly attributed to this species’ sexual segregation and preference for different habitats and diets for both genders [69,70]. Though the reduction in size in C. sibirica may have an impact on the heterozygosity of the MHC DRB locus, since more than half of the studied individuals (26 out of 43 samples) possess a single PFA (Table A1).
Individuals of C. sibirica clade II had low levels of diversity at the allelic, nucleotide, amino acid, and supertype levels relative to those of Clades I and III (Table A1, Table 1), indicating that the impact of population declines and/or environmental pathogenic pressures on the different geographic populations was different [71]. We also cannot exclude the possibility that this difference is due to the low number of samples analyzed; thus, dense sampling is needed for further related studies.
Although we did not find a single allele shared by all three C. sibirica clades, but found alleles common to two clades. For instance, the alleles Casi-DRB1*16 (the most frequent one identified in individuals from east and middle Tianshan mountains, and Kunlun Mountains), Casi-DRB1*17 and Casi-DRB1*19 were shared by clades III and I, while alleles Casi-DRB1*02 and Casi-DRB1*05 were shared by clades III and II (Fig.1 and Table A1). The radiation of these clades dates back around 6.75 million years ago [32], indicating preservation of these alleles in C. sibirica for such a long evolutionary time. Retaining alleles along the evolutionary time is not specific to our study. An allele was conserved in the genus Meles for nearly 2 million years [37]; two alleles were even shared by multiple species from different genera in mustelids [72], diverged more than 11 million years ago [73]; some MHC allele surprisingly preserved among different family [5]. This is probably because C. sibirica populations in Xinjiang, China subjected to same pathogenic burdens for a long evolutionary time, as polymorphism in MHC gene was pathogen driven [74].
Meantime, we found more population- or clade-specific alleles (Figure 1, Table A1), implying high differentiation at this locus. The long radiation time (3.3–6.7 million years) of these populations or clades probably illuminates this phenomenon [32]. MHC genes also showed genetic differentiation between populations in some mammal species [71, 75-76) The FST values between Clade I and Clade II and Clade II and Clade III were greater than 0.25 (Table 2), indicating large genetic differences. What is puzzling is that the FST value between the Clade I and Clade III was negative. This is because that the differences within populations were greater than the differences between populations [77]. The negative value of FST generally interpreted as 0 [78], which means clade I and III were not differentiated. However, after excluding the shared alleles between these clades in FST estimation, it turns to be low differentiation (Table 2). Overall, this in part supports the pattern of mitochondrial DNA [32].
The number of PFAs identified for our studied individuals shows 1 or 2 loci of the DRB gene (Table A1), with a low frequency of 2 loci (5 out of 43 individuals), though. Many species in the genus Capra, including Alpine ibex (Capra ibex), Spanish ibex (Capra pyrenaica), and Himalayan tahr (Hemitragus jemlahicus), have only one locus of DRB [79]. It can be seen that the ancestor of Capra species is supposed to possess a single locus at the MHC DRB gene. Despite the small portion of individuals with two loci, they split into clades I and III, respectively. We assume that the one locus likely emerged from the other locus through gene duplication [6]. Even if no evidence supports the occurrence of recombination events due likely to the shortness of our analytical sequences, intergenic recombination or gene conversion may explain this phenomenon as well [80], and they might happen twice independently in these two clades. A population genome study on the MHC class II region will help us demonstrate this notion.
Evolution of the DRB1 Gene
Generally, MHC gene polymorphism were generated and retained by gene recombination [79,81], gene duplication [6], balancing selection [74,82], and/or positive selection [37,74,82]. In our study, we did not find any significant signature of recombination events, convincing us that gene recombination was not the reason for generation of MHC diversity. Nonetheless, we found more rare alleles than shared or high frequency alleles (Table A1). This is suggestion of balanced polymorphisms include negative-frequency-dependent selection, where rare alleles are favored. Besides, we also found a notable excess of nonsynonymous over synonymous substitutions at ABSs, in different clades (Table 3). In our phylogenetic relationship analysis, DRB1 sequences of C. sibirica were grouped with the sequences of its counterparts (Figure 3), suggesting that some alleles are phylogenetically more closely related to the alleles of other species than to those of its own, a typical trans-species polymorphism [83] which is reported for MHC genes of many species [5,37,74,82,84]. All of these were the evidence supporting the presence of long-term balancing selection in the C. sibirica, considering that the Capra species were diverged approximately 6 million years ago [32]. Moreover, the PAML CodeML and MEME analyses identified up to 12 positively selected sites, most of which coincide with the ABSs (Figure 2 and Table 4), suggested that the sequence variation of DRB1 genes was driven by positive selection due to pathogenic burdens [19,20,21]. In sum, our results together indicate that selection was the main force shaping and maintaining DRB1 gene polymorphism in C. sibirica.
It is worthy to mention that we as well as observed an exceeded nonsynonymous relative to synonymous substitutions at the none-ABSs in all clades of C. sibirica (Table 3), which is in line with the positive selection analyses that showed several positively selected sites out of ABSs (Figure 2 and Table 4). This is consistent with the results of Abduriyim et al. [37] in a species of Canidae. Considering that all MHC studies deduce the ABS locations based on human MHC structure [46], the actual location of ABSs in the MHC Class II DR β-chain of C. sibirica, radiation from humans took place as far back as 95 million years [85], may be different. This leaves an open question if ABSs of MHC molecule in all mammals were overlapped.
Conclusions
Despite the level of genetic diversity in clade II is lower than that in other clades, and thus requires close attention in future conservation plans, the overall diversity (i.e., allelic, nucleotide, amino acid and supertype diversity) of MHC class II DRB1 genes in C. sibirica Xinjiang populations after a bottleneck have not rapidly been lost. The differential preference for habitat and food of two sexes might contribute to generation of MHC diversity, too. The genetic differentiation of clades/populations was to some extent in support of the results by Wang et al. [32] on mtDNA. The diversity of MHC DRB1 genes in C. sibirica was shaped and maintained by selection, both positive and balancing selection.
Author Contributions
S.A. conceived and designed the study; D.P., W.R., and S.A. did the fieldwork and samples collection; D.P. and W.R. conducted lab experiments, D.P. and W.R. performed data analysis; D.P. and S.A. prepared the first draft, S.A. edited the manuscript. All authors approved the final version of this manuscript.
Funding
This study was funded by the National Natural Science Foundation of China (No. 32260328).
Data Availability Statement
The sequences we obtained have been deposited in the NCBI databases under accession numbers OR257668 – OR257693.
Acknowledgments
We thank Saypulla Suba in the Forest and Grassland Bureau of Hami city and Abdugini Kerim in the Forest and Grassland Bureau of Arturk county for their help in field sampling. This study was funded by the National Natural Science Foundation of China (No. 32260328).
Conflicts of Interest
The authors declare no conflict of interest.
Appendix

Table A1.
Distribution and frequency of MHC class II DRB1 alleles among 43 Capra sibirica individuals in Xinjiang, China. The clades I to III were determined based on mitochondrial sequence analyses [32].
Table A1.
Distribution and frequency of MHC class II DRB1 alleles among 43 Capra sibirica individuals in Xinjiang, China. The clades I to III were determined based on mitochondrial sequence analyses [32].
| No. | Clade | location | Individual identity | Casi-DRB1* | Number of verified alleles | ||||||||||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 | ||||||
| 1 | Ⅰ | Arturk | CSYW7 | + | 1 | ||||||||||||||||||||||||||
| 2 | Arturk | CSYWa | + | + | + | 3 | |||||||||||||||||||||||||
| 3 | Arturk | CSYWb | + | + | + | 3 | |||||||||||||||||||||||||
| 4 | Arturk | CSYWc | + | 1 | |||||||||||||||||||||||||||
| 5 | Arturk | CSYWd | + | 1 | |||||||||||||||||||||||||||
| 6 | Arturk | CSYWe | + | 1 | |||||||||||||||||||||||||||
| 7 | Arturk | CSYWi | + | 1 | |||||||||||||||||||||||||||
| 8 | Arturk | CSYWj | + | + | 2 | ||||||||||||||||||||||||||
| 9 | Arturk | CSYWk | + | 1 | |||||||||||||||||||||||||||
| 10 | Arturk | CSYWB | + | + | 2 | ||||||||||||||||||||||||||
| 11 | Arturk | CSYWD | + | + | 2 | ||||||||||||||||||||||||||
| 12 | Arturk | CSYWE | + | + | 2 | ||||||||||||||||||||||||||
| 13 | Arturk | CSYWF | + | 1 | |||||||||||||||||||||||||||
| 14 | Arturk | CSYWX1 | + | 1 | |||||||||||||||||||||||||||
| 15 | Arturk | CSYWX2 | + | 1 | |||||||||||||||||||||||||||
| 16 | Arturk | CSYWX3 | + | 1 | |||||||||||||||||||||||||||
| 17 | Sawan | SLG | + | 1 | |||||||||||||||||||||||||||
| 18 | Urumqi | CShx1 | + | + | 2 | ||||||||||||||||||||||||||
| 19 | Urumqi | CShx2 | + | + | 2 | ||||||||||||||||||||||||||
| 20 | Urumqi | CShx6 | + | 1 | |||||||||||||||||||||||||||
| 21 | Urumqi | CShx10 | + | + | 2 | ||||||||||||||||||||||||||
| 22 | Urumqi | CShx11 | + | 1 | |||||||||||||||||||||||||||
| 23 | Urumqi | CShx13 | + | 1 | |||||||||||||||||||||||||||
| 24 | Urumqi | CShx14 | + | 1 | |||||||||||||||||||||||||||
| 25 | Urumqi | CShx16 | + | 1 | |||||||||||||||||||||||||||
| 26 | Urumqi | CShx19 | + | 1 | |||||||||||||||||||||||||||
| 27 | Urumqi | CShx20 | + | + | 2 | ||||||||||||||||||||||||||
| 28 | Ⅱ | Ulugqat | CSnj1 | + | 1 | ||||||||||||||||||||||||||
| 29 | Ulugqat | CSnj3 | + | 1 | |||||||||||||||||||||||||||
| 30 | Ulugqat | CSnjP1 | + | 1 | |||||||||||||||||||||||||||
| 31 | Ulugqat | CSnjP2 | + | + | 2 | ||||||||||||||||||||||||||
| 32 | Ulugqat | CSnjP3 | + | + | 2 | ||||||||||||||||||||||||||
| 33 | Ⅲ | Ulugqat | CSnj5 | + | + | + | 3 | ||||||||||||||||||||||||
| 34 | Ulugqat | CSnj6 | + | 1 | |||||||||||||||||||||||||||
| 35 | Ulugqat | CSnj8 | + | 1 | |||||||||||||||||||||||||||
| 36 | Ulugqat | CSnj9 | + | + | + | 3 | |||||||||||||||||||||||||
| 37 | Ulugqat | CSnj10 | + | 1 | |||||||||||||||||||||||||||
| 38 | Ulugqat | CSnj11 | + | + | 2 | ||||||||||||||||||||||||||
| 39 | Ulugqat | CSnjJ1 | + | + | + | 3 | |||||||||||||||||||||||||
| 40 | Ulugqat | CSnjJ2 | + | 1 | |||||||||||||||||||||||||||
| 41 | Kagilik | CSYC1 | + | 1 | |||||||||||||||||||||||||||
| 42 | Kagilik | CSYC2 | + | 1 | |||||||||||||||||||||||||||
| 43 | Kagilik | CSYC3 | + | + | 2 | ||||||||||||||||||||||||||
| Total | 4 | 2 | 2 | 1 | 4 | 2 | 2 | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 17 | 2 | 1 | 5 | 1 | 1 | 7 | 1 | 1 | 1 | 1 | 65 | ||||
The bolded + marks represent alleles unique to that clade. The bolded and italic allele names signify identical amino acid sequences. The allele names with same color mean identical supertype sequences (ABS sequences).
References
- Ellegren, H. , Galtier, N. Determinants of genetic diversity. Nature Reviews Genetics 2016, 17, 422–433. [Google Scholar] [CrossRef]
- Kaufman, J. Unfinished business: Evolution of the MHC and the adaptive immune system of jawed vertebrates. Annual Review of Immunology 2018, 36, 383–409. [Google Scholar] [CrossRef] [PubMed]
- Sommer, S. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Frontiers in Zoology 2005, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Meyers, L.A.; Bull, J.J. Fighting change with change: Adaptive variation in an uncertain world. Trends in Ecology & Evolution 2022, 17, 551–557. [Google Scholar]
- Abduriyim, S.; Nishita, Y.; Kosintsev, P.A.; Raichev, E.; Vainola, R.; Kryukov, A.P.; Abramov, A.V.; Kaneko, Y.; Masuda, R. Evolution of MHC class I genes in Eurasian badgers, genus Meles (Carnivora, Mustelidae). Heredity 2019, 122, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Abduriyim, S.; Zou, D.H.; Zhao, H.B. Origin and evolution of the major histocompatibility complex class I region in eutherian mammals. Ecology and evolution 2019, 9, 7861–7874. [Google Scholar] [CrossRef]
- Mungall, A.J.; Palmer, S.A.; Sims, S.K.; Edwards, C.A.; Ashurst, J.L.; Wilming, L.; Jones, M.C.; Horton, R.; Hunt, S.E.; Scott, C.E.; et al. The DNA sequence and analysis of human chromosome 6. Nature 2003, 425, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Alvaro-Benito, M.; Stolzenberg, S.; Noe, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Frontiers in Immunology 2017, 8, 292–305. [Google Scholar] [CrossRef]
- Wolin, A.; Lahtela, E.L.; Anttila, V.; Petrek, M.; Grunewald, J.; van Moorsel, C.H.M.; Eklund, A.; Grutters, J.C.; Kolek, V.; Mrazek, F.; Kishore, A.; Padyukov, L.; Pietinalho, A.; Ronninger, M.; Seppanen, M.; Selroos, O.; Lokki, M.L. Snp variants in major histocompatibility complex are associate with sarcoidosis susceptibility—a joint analysis in four European populations. Frontiers in Immunology 2017, 8, 422. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular biology of the cell. 5th Ed. New York: Garland Science. 2007.
- Brown, J.H.; Jardetzky, T.S.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Strominger, J.L.; Wiley, D.C. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 1993, 364, 33–39. [Google Scholar] [CrossRef]
- Sin, Y.W.; Dugdale, H.L.; Newman, C.; Macdonald, D.W.; Burke, T. MHC class II genes in the European badger (Meles meles): characterization, patterns of variation, and transcription analysis. Immunogenetics 2012, 64, 313–327. [Google Scholar] [CrossRef]
- Karanth, K.U.; Nichols, J.D.; Kumar, N.S.; Link, W.A.; Hines, J.E. Tigers and their prey: Predicting carnivore densities from prey abundance. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 4854–4858. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, K.R.; Redpath, S.M.; Bhatnagar, Y.V.; Ramakrishnan, U.; Chaturvedi, V.; Smout, S.C.; Mishra, C. ; Impact of wild prey availability on livestock predation by snow leopards. R. Soc. Open Sci. 2017, 4, 170026. [Google Scholar] [CrossRef]
- Schaller, G.; Li, H.; Tarif; Lv, H.; Ren, J.R.; Qiu, M.J.; Wang, H.B.; Anney; Aziguli; Abrimiti. Status of large mammals in the Taxkorgan Nature Reserve, Xinjiang, China. Arid Zone Research 1987, 42, 53–71.
- Wang, X.F. Diagnosis and treatment of viral keratoconjunctivitis in Capra sibirica. Heilongjiang Animal Science and Veterinary Medicine 2007, 307, 145. [Google Scholar]
- Zhu, X.S.; Wang, M.Y.; Yang, W.K.; David, B. Ecology and biology of Capra sibirica: Current situation of studies. Chinese Journal of Ecology 2015, 34, 3553–3559. [Google Scholar]
- Reading, R.; Michel, S.; Suryawanshi, K.; Bhatnagar, Y.V. Capra sibirica. The IUCN Red List of Threatened Species 2020, e.T42398A22148720. https://dx.doi.org/10.2305/IUCN.UK.2020-2.RLTS.T42398A22148720.en. Accessed on 31 May 2023.
- Thermistor, H. Diagnosis of streptococcal disease in Capra sibirica. Chinese Journal of Animal Husbandry and Veterinary Medicine 2008, 4, 42. [Google Scholar]
- Chen, W.D.; Li, T.; Yakp. Laboratory diagnosis and prevention of death of wild Capra sibirica caused by Streptococcus pneumoniae. Xinjiang animal husbandry 2012, 7, 55–56. [Google Scholar]
- Bayendrigan; An, N. Diagnosis and control of anthrax infection in wild Capra sibirica. Journal of Animal Science and Veterinary Medicine 2015, 34, 124.
- Zhu, X.S. Studies on feeding habits and homoerotic clusters of Capra sibirica [D]. Beijing: University of Chinese Academy of Sciences, 2016.
- Yang, Q.S.; Feng, Z.J. China Red Book of Endangered Animals: Mammals. Beijing: Beijing Science Press. 1998, 314-317.
- Reading, R.; Shank, C. Capra sibirica. IUCN Red List of Threatened Species. 2009.
- Wang, S.; Xie, Y. Red List of Chinese Species. Beijing: Higher Education Press. 2004.
- National Forestry and Grassland Administration, National Park Administration, 2021, http://www.forestry.gov.cn.
- Radwan, J.; Kawako, A.; Wójcik, J.M.; Babik, W. Mhc-drb3 variation in a free-living population of the European bison, Bison bonasus. Molecular Ecology 2010, 16, 531–540. [Google Scholar] [CrossRef]
- Mainguy, J.; Worley, K.; Côté, S.D.; Coltman, D.W. Low MHC DRB class II diversity in the mountain goat: Past bottlenecks and possible role of pathogens and parasites. Conservation Genetics 2007, 8, 885–891. [Google Scholar] [CrossRef]
- Bollmer, J.L., Vargas, F.H., Parker, P.G. Low MHC variation in the endangered Galápagos penguin (Spheniscus mendiculus). Immunogenetics 2007, 59, 593–602. [CrossRef] [PubMed]
- Ejsmond, M.J.; Radwan, J. MHC diversity in bottlenecked populations: a simulation model. Conservation Genetics 2011, 12, 129–137. [Google Scholar] [CrossRef]
- Oliver, M.K.; Piertney, S.B. Selection Maintains MHC Diversity through a Natural Population Bottleneck. Mol. Biol. Evol 2012, 29, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.R.; Dong, P.P.; Hirata, D.; Abduriyim, S. Mitochondrial DNA analyses revealed distinct lineages in an alpine mammal, Siberian ibex (Capra sibirica) in Xinjiang, China. Ecology and evolution 2023. [CrossRef]
- Abduriyim, S.; Nabi, A.; Halik, M. Low genetic diversity in the goitered gazelle Gazella subgutturosa (Guldenstadt, 1780) (Artiodactyla: Bovidae) in North-western China as revealed by the mitochondrial cytochrome b gene. Acta Zoologica Bulgarrica 2018, 70, 211–218. [Google Scholar]
- Abduriyim, S.; Zibibulla, G.; Eli, S.; Ismayil, Z.; Halik, M. Phylogeny and genetic structure of the goitered gazelle (Artiodactyla, Bovidae) in north-western China indicated by the hypervariable mitochondrial control region. Systematics and Biodiversity 2018, 16, 527–537. [Google Scholar] [CrossRef]
- Amills, M.; Francino, O.; Sanchez, A. Nested PCR allows the characterization of Taql and Pstl RFLPs in the second exon of the caprine MHC class II DRB gene. Vet Immunol Immunopathol 1995, 48, 313–321. [Google Scholar] [CrossRef]
- Sin, Y.W.; Dugdale, H.L.; Newman, C.; Macdonald, D.W.; Burke, T. MHC class II genes in the European badger (Meles meles): characterization, patterns of variation, and transcription analysis. Immunogenetics 2012, 64, 313–327. [Google Scholar] [CrossRef]
- Abduriyim, S.; Nishita, Y.; Kosintsev, P.A.; Raichev, E.; Väinölä, R.; Kryukov, A.P.; Abramov, A.V.; Kaneko, Y.; Masuda, R. Diversity and evolution of MHC class II DRB gene in the Eurasian badger genus Meles (Mammalia: Mustelidae). Biological Journal of The Linnean Society 2017, 122, 258–273. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [PubMed]
- Kennedy, L.J.; Altet, L.; Angles, J.M.; Barnes, A.; Carter, S.D.; Francino, O.; Thomson, W. Nomenclature for factors of the dog major histocompatibility system (DLA), 1998: first report of the ISAG DLA Nomenclature Committee. Animal Genetics 2000, 31, 52–61. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [PubMed]
- Klein, J.; Bontrop, R.E.; Dawkins, R.L.; Erlich, H.A.; Gyllensten, U.B.; Heise, E.R.; Watkins, D.I. Nomenclature for the major histocompatibility complexes of different species: a proposal. Immunogenetics 1990, 31, 217–219. [Google Scholar]
- Ballingall, K.T.; Todd, H. An official nomenclature for the major histocompatibility complex allele sequences from the domestic goat (Capra hircus). HLA 2019, 93, 36–38. [Google Scholar] [CrossRef]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Molecular Biology and Evolution 1986, 3, 418–426. [Google Scholar]
- Yang, Z.H.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar]
- Reche, P.A.; Reinherz, E.L. Sequence variability analysis of human class I and class II MHC molecules: functional and structural correlates of amino acid polymorphisms. Journal of Molecular Biology 2003, 331, 623–641. [Google Scholar]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar]
- Yang, Z.H. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [PubMed]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [PubMed]
- Yang, Z.H.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [PubMed]
- Yang, Z.H.; Wong, W.S.W.; Nielsen, R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Pond, S.L.K. Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 2012, 8, e1002764. [Google Scholar]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrov. 2005, 21, 98–102. [Google Scholar]
- Pond, S.L.K.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: a genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar]
- Delport, W.; Poon, A.F.Y.; Frost, S.D.W.; Pond, S.L.K. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [PubMed]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies, Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar]
- Zhang, X.J.; Wang, J.; Huang, G.; Chen, Y.H.; Yang, L.M.; Li, H.J.; Li, M.; Zheng, N. Spatiotemporal Distribution of Cloud Liquid Water Volume over Three Main Mountains in Xinjiang. Arid Zone Research 2018, 35, 846–854. [Google Scholar]
- Ujvari, B.; Belov, K. Major histocompatibility complex (MHC) markers in conservation biology. Int. J. Mol. Sci. 2011, 12, 5168–5186. [Google Scholar]
- Mainguy, J.; Worley, K.; Côté, S.D.; Coltman, D.W. Low MHC DRB class II diversity in the mountain goat: Past bottlenecks and possible role of pathogens and parasites. Conservation Genetics 2007, 8, 885–891. [Google Scholar]
- Bollmer, J.L., Vargas, F.H., Parker, P.G. Low MHC variation in the endangered Galápagos penguin (Spheniscus mendiculus). Immunogenetics 2007, 59, 593–602.
- Wenink, P.W.; Groen, A.F.; Roelke-Parker, M.E.; Prins, H.H.T. African buffalo maintain high genetic diversity in the major histocompatibility complex in spite of historically known population bottlenecks. Mol. Ecol. 1998, 7, 1315–1322. [Google Scholar]
- Schwaiger, F.W.; Weyers, E.; Epplen, C.; Brün, J.; Ruff, G.; Crawford, A.; Epplen, J.T. The paradox of MHC-DRB exon/intron evolution: α-helix and β-sheet encoding regions diverge while hypervariable intronic simple repeats coevolve with β-sheet codons. J. Mol. Evol. 1993, 37, 260–272. [Google Scholar]
- Angelone, S.; Jowers, M.J.; Min, A.R.M.; Fandos, P.; Prieto, P.; Pasquetti, M.; Cano-Manuel, F.J.; Mentaberre, G.; Olvera, J.R.L.; Raez-Bravo, A.; Espinosa, J.; Perez, J.M.; Soriguer, R.C.; Rossi, L.; Granados, J.E. Hidden MHC genetic diversity in the Iberian ibex (Capra pyrenaica). BMC Genet. 2018, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Blank, D.; Wang, M.Y.; da Silva, A. A.; Yang, W.K.; Ruckstuhl, K.; Alves, J. Diet differences between males and females in sexually dimorphic ungulates: A case study on Siberian ibex. European journal of wildlife research 2020, 66, 1–10. [Google Scholar] [CrossRef]
- Han, L.; Wang, Z.; Blank, D.; Wang, M.Y.; Yang, W.K. Different environmental requirements of female and male Siberian ibex, Capra sibirica. Scientific reports 2021, 11, 6064. [Google Scholar] [CrossRef]
- Awadi, A.; Ben Slimen, H.; Smith, S.; Knauer, F.; Makni, M.; Suchentrunk, F. Positive selection and climatic effects on MHC class II gene diversity in hares (Lepus capensis.) from a steep ecological gradient. Scientific Reports 2018, 8, 11514. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Nishita, Y.; Lansink, G.M.J.; Holmala, K.; Aspi, J.; Masuda, R. Diversity of the MHC class II DRB gene in the wolverine (Carnivora: Mustelidae: Gulo gulo) in Finland. PLoS ONE 2022, 17, e0267609. [Google Scholar] [CrossRef]
- Koepfli, K.P.; Deere, K.A.; Slater, G.J.; Begg, C.; Begg, K.; Grassman, L.; Lucherini, M.; Veron, G.; Wayne, R.K. Multigene phylogeny of the Mustelidae: Resolving relationships, tempo and biogeographic history of a mammalian adaptive radiation. BMC Biol. 2008, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Nishita, Y.; Abramov, A.V.; Kosintsev, P.A.; Lin, L.K.; Watanabe, S.; Yamazaki, K.; Kaneko, Y.; Masuda, R. Genetic variation of the MHC class II DRB genes in the Japanese weasel, Mustela itatsi, endemic to Japan, compared with the Siberian weasel, Mustela sibirica. Tissue Antigens 2015, 86, 431–442. [Google Scholar]
- Amaike, Y.; Nishita, Y.; Uraguchi, K.; Masuda, R. Genetic Diversity of MHC Class II DRB1 Exon 2 in the Red Fox (Vulpes vulpes) on Hokkaido, Japan. Zoological Science 2018, 35, 402–410. [Google Scholar] [CrossRef]
- Mason, R.A.B.; Browning, T.L.; Eldridge, M.D.B. Reduced mhc class ii diversity in island compared to mainland populations of the black-footed rock-wallaby (petrogale lateralis lateralis). Conservation Genetics 2011, 12, 91–103. [Google Scholar]
- Smaragdov, M.G.; Kudinov, A.A.; Uimari, P. Assessing the genetic differentiation of Holstein cattle herds in the Leningrad region using Fst statistics. Agricultural and Food Science 2018, 27, 96–101. [Google Scholar] [CrossRef]
- Meirmans, P.G. Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 2006, 60, 2399–2402. [Google Scholar] [CrossRef] [PubMed]
- Schaschl, H.; Wandeler, P.; Suchentrunk, F.; Obexer-Ruff, G.; Goodman, S.J. Selection and recombination drive the evolution of MHC class II DRB diversity in ungulates. Heredity 2006, 97, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Reusch, T.B.; Langefors, Å. Inter- and Intralocus Recombination Drive MHC Class IIB Gene Diversification in a Teleost, the Three-Spined Stickleback Gasterosteus aculeatus. J. Mol. Evol. 2005, 61, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Bartocillo, A.M.F.; Nishita, Y.; Abramov, A.V.; Masuda, R. Molecular evolution of MHC class II DRB exon 2 in Japanese and Russian raccoon dogs, Nyctereutes procyonoides (Carnivora: Canidae). Biological Journal of The Linnean Society 2020, 129, 61–73. [Google Scholar] [CrossRef]
- Nishita, Y.; Kosintsev, P.A.; Haukisalmi, V.; Väinölä, R.; Raichev, E.G.; Murakami, T.; Abramov, A.V.; Kaneko, Y.; Masuda, R. Diversity of MHC class II DRB alleles in the Eurasian population of the least weasel, Mustela nivalis (Mustelidae: Mammalia). Biological Journal of the Linnean Society 2017, 121, 28–37. [Google Scholar] [CrossRef]
- Klein, J.; Sato, A.; Nagl, S.; O’hUigín, C. Molecular trans-species polymorphism. Annu. Rev. Immunol. 1998, 29, 1–21. [Google Scholar] [CrossRef]
- Bartocillo, A.M.F.; Nishita, Y.; Abramov, A.V.; Masuda, R. Evolution of MHC class I genes in Japanese and Russian raccoon dogs, Nyctereutes procyonoides (Carnivora: Canidae). Mamm. Res. 2021, 66, 371–383. [Google Scholar] [CrossRef]
- Eizirik, E.; Murphy, W.J.; O’Brien, S.J. Molecular Dating and Biogeography of the Early Placental Mammal Radiation. Journal of Heredity 2001, 92, 212–219. [Google Scholar] [CrossRef]
Figure 1.
Sampling locations for Capra sibirica in Xinjiang, China, in this study. Each small circle on the map indicates sampling locality and different colors signify different clades determined based on the mtDNA analyses [32]. The pie chart shows alleles frequencis (number of alleles) for geographic populations/clades, with each allele in a different color (key at the right).
Figure 1.
Sampling locations for Capra sibirica in Xinjiang, China, in this study. Each small circle on the map indicates sampling locality and different colors signify different clades determined based on the mtDNA analyses [32]. The pie chart shows alleles frequencis (number of alleles) for geographic populations/clades, with each allele in a different color (key at the right).
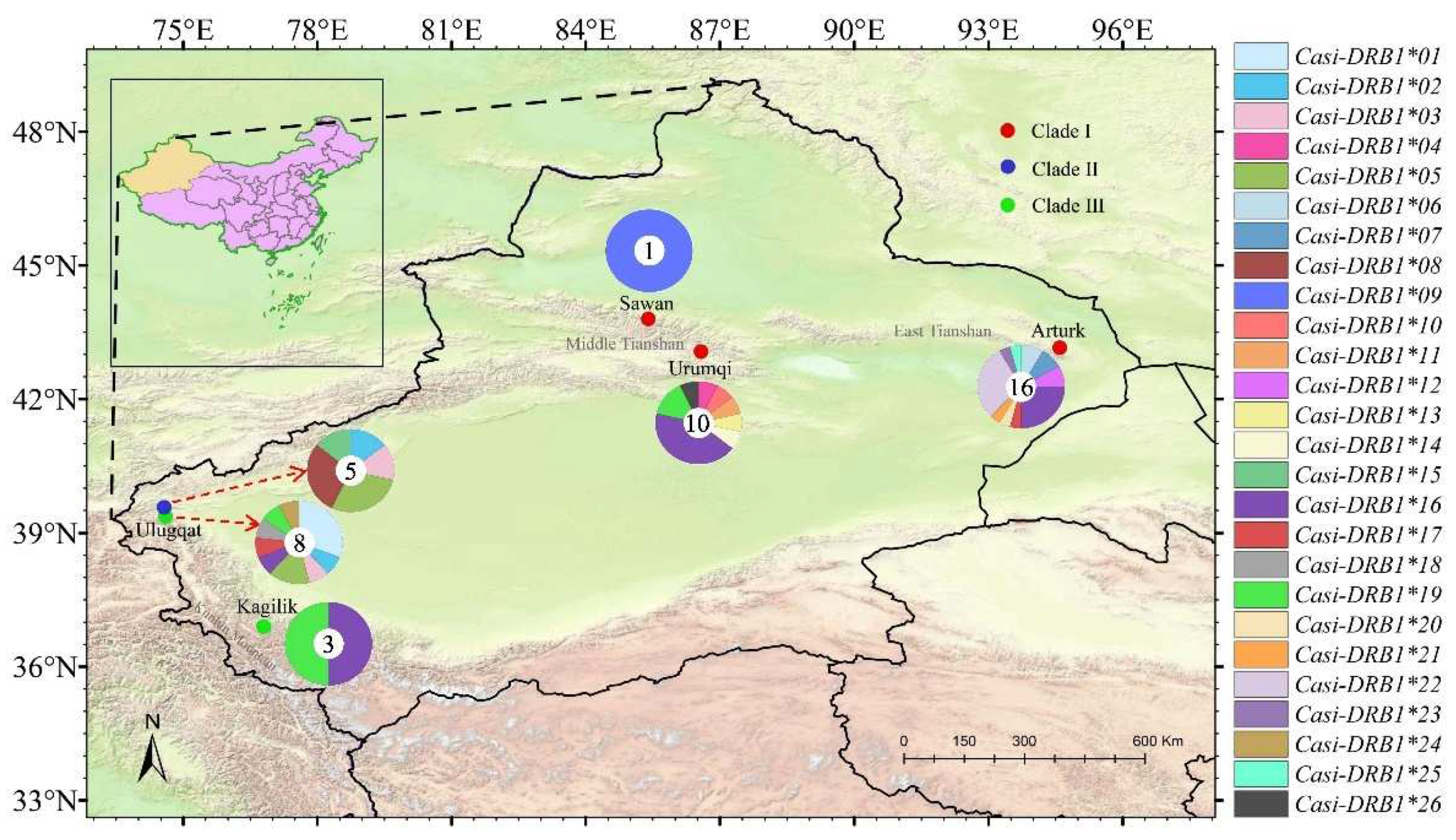
Figure 2.
Alignment of deduced amino acid sequences encoded by exon 2 of MHC class II DRB1 alleles in Capra sibirica. Numbers above the amino acid sequences indicate positions in the β1-domain of the DR protein β-chain. Dots indicate amino acids identical to those in Casi-DRB1*01. Putative ABSs as determined by Reche & Reinherz [46] are shaded. * signs at the bottom of the table indicate sites inferred to be under positive selection by MEME analysis and Bayes Empirical Bayes inference (BEB) using PAML. For the M2a and M8 models in BEB, only significant results are indicated by + (P > 95%). Clade II did not find positive selection sites by MEME analysis and is therefore not shown.
Figure 2.
Alignment of deduced amino acid sequences encoded by exon 2 of MHC class II DRB1 alleles in Capra sibirica. Numbers above the amino acid sequences indicate positions in the β1-domain of the DR protein β-chain. Dots indicate amino acids identical to those in Casi-DRB1*01. Putative ABSs as determined by Reche & Reinherz [46] are shaded. * signs at the bottom of the table indicate sites inferred to be under positive selection by MEME analysis and Bayes Empirical Bayes inference (BEB) using PAML. For the M2a and M8 models in BEB, only significant results are indicated by + (P > 95%). Clade II did not find positive selection sites by MEME analysis and is therefore not shown.

Figure 3.
Phylogenetic neighbor network of MHC II DRB1 exon 2 (233 bp) sequences from Capra species, including C. sibirica in this study and the remaining three species sequences downloaded from GenBank. The numbers represent allele names with profix Casi-DRB1*.
Figure 3.
Phylogenetic neighbor network of MHC II DRB1 exon 2 (233 bp) sequences from Capra species, including C. sibirica in this study and the remaining three species sequences downloaded from GenBank. The numbers represent allele names with profix Casi-DRB1*.
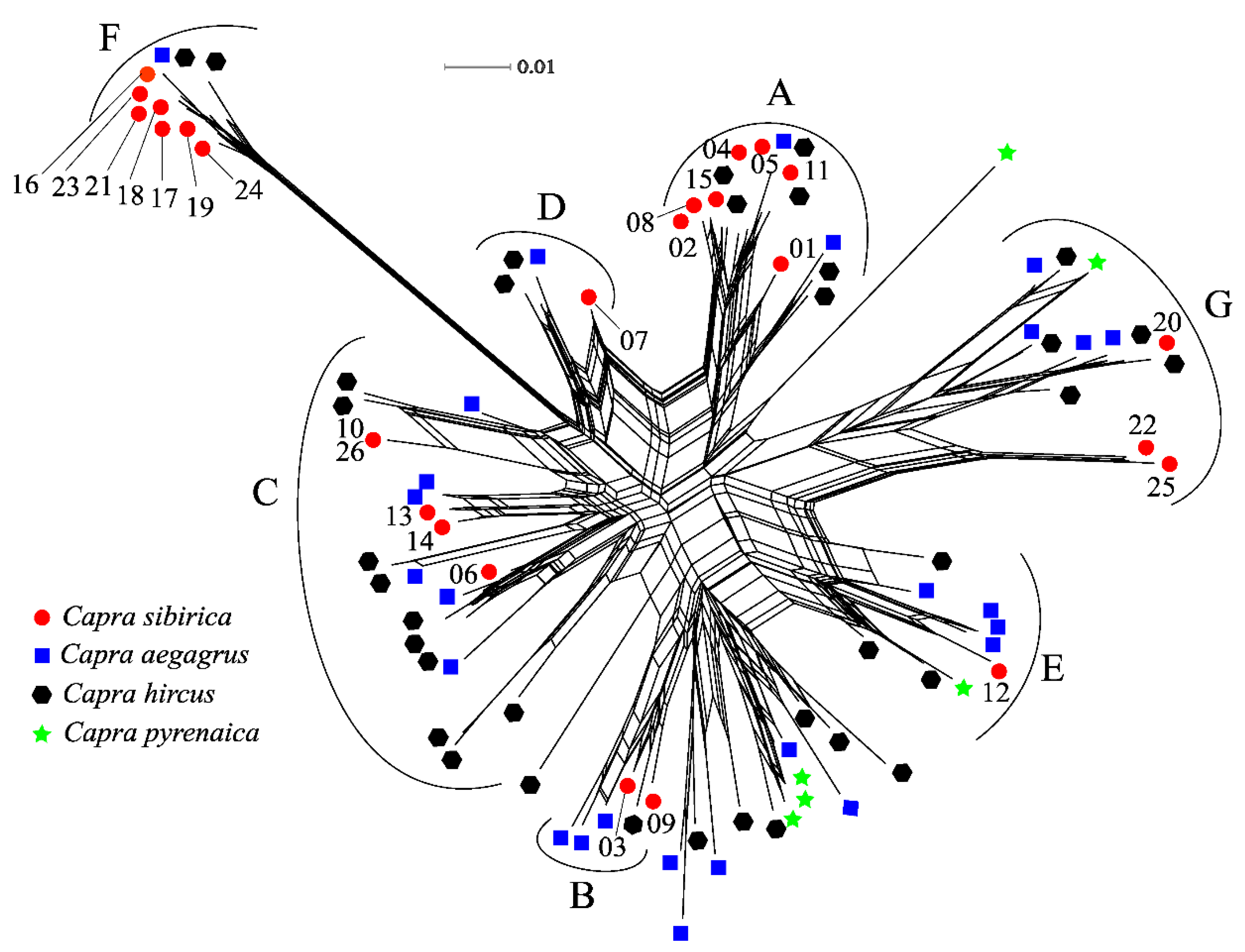
Table 1.
Genetic diversity at nucleotide, amino acid and supertype levels and neutrality test on the MHC DRB1 gene for three clades of Capra sibirica in Xinjiang, China.
Table 1.
Genetic diversity at nucleotide, amino acid and supertype levels and neutrality test on the MHC DRB1 gene for three clades of Capra sibirica in Xinjiang, China.
| Clade | Sample | Number | Diversity indices | Tajima's D | ||
| size | π | aa | Supertype | |||
| Ⅰ | 27 | 18/16/14 | 0.092 | 0.190 | 0.427 | 0.184 |
| Ⅱ | 5 | 5/5/4 | 0.035 | 0.078 | 0.228 | -0.793 |
| Ⅲ | 11 | 9/8/8 | 0.084 | 0.177 | 0.402 | 0.705 |
| Total | 43 | 26/23/20 | 0.089 | 0.179 | 0.406 | 0.277 |
Note: The Number column shows, in order, the number of alleles, the number of amino acids, and the number of supertypes.
Table 2.
Genetic differentiation (FST) of the MHC DRB1 gene among the three clades of Capra sibirica in Xinjiang, China. The FST Values were calculated including (below the diagonal) and excluding (above the diagonal) shared alleles between clades.
Table 2.
Genetic differentiation (FST) of the MHC DRB1 gene among the three clades of Capra sibirica in Xinjiang, China. The FST Values were calculated including (below the diagonal) and excluding (above the diagonal) shared alleles between clades.
| Clade | Ⅰ | Ⅱ | Ⅲ |
| Ⅰ | 0.364 | 0.030 | |
| Ⅱ | 0.267 | 0.560 | |
| Ⅲ | -0.013 | 0.343 |
Table 3.
Rates (± standard error) of non-synonymous (dN) and synonymous (dS) substitutions and their ratio (ω) for the presumed antigen binding sites (ABS), non-ABSs, and sites overall in the β1-domain of the MHC class II DRB1 genes for Capra sibirica in Xinjiang, China.
Table 3.
Rates (± standard error) of non-synonymous (dN) and synonymous (dS) substitutions and their ratio (ω) for the presumed antigen binding sites (ABS), non-ABSs, and sites overall in the β1-domain of the MHC class II DRB1 genes for Capra sibirica in Xinjiang, China.
| Substitution type | Number of codons | Clade I | Clade II | Clade III | |
| dN | ABS | 20 | 1.203 ± 0.209 | 0.244 ± 0.149 | 0.690 ± 0.186 |
| Non-ABS | 66 | 0.288 ± 0.140 | 0.051± 0.084 | 0.132 ± 0.096 | |
| Overall | 86 | 0.514 ± 0.127 | 0.101 ± 0.081 | 0.257 ± 0.100 | |
| dS | ABS | 20 | 0.658 ± 0.293 | 0.087± 0.288 | 0.392± 0.298 |
| Non-ABS | 66 | 0.159 ± 0.170 | 0.015± 0.122 | 0.068± 0.208 | |
| Overall | 86 | 0.293± 0.158 | 0.037 ± 0.144 | 0.167± 0.180 | |
| ω | ABS | 20 | 1.828 | 2.805 | 1.760 |
| Non-ABS | 66 | 1.811 | 3.400 | 1.941 | |
| Overall | 86 | 1.754 | 2.730 | 1.539 |
Table 4.
The results of codon based positive selection analyses using maximum likelihood models in CodeML for MHC DRB1 exon 2 sequences from Capra sibirica. Positively selected sites (PSS), log-likelihood (lnL), the likelihood ratio test (LRT) and probability (P) values were presented.
Table 4.
The results of codon based positive selection analyses using maximum likelihood models in CodeML for MHC DRB1 exon 2 sequences from Capra sibirica. Positively selected sites (PSS), log-likelihood (lnL), the likelihood ratio test (LRT) and probability (P) values were presented.
| Clade | Models | lnL | Parameter estimates | PSS | LRT | d.f. | P value |
| Ⅰ | M1a | -1010.99 | P0 = 0.860, P1 = 0.140, ω0 = 0.041, ω1 = 1.000 | M1a vs M2a | 2 | <0.01 | |
| M2a | -972.69 | P0 = 0.572, P1 = 0.401, P2 = 0.026, ω0 = 0.099, ω1 = 1.000, ω2 = 13.181 | 11, 13, 26, 56, 57, 66, 67, 70, 71, 73, 74, 78, 86 | ||||
| M7 | -1014.03 | P = 0.025, q = 0.155 | M7 vs M8 | 2 | <0.01 | ||
| M8 | -972.72 | P0 = 0.974, P = 0.107, q = 0.116, P1 = 0.026, ω = 13.366 | 11, 13, 26, 32, 56, 57, 66, 67, 70, 71, 73, 74, 78, 86 | ||||
| Ⅱ | M1a | -445.45 | P0 = 0.524, P1 = 0.476, ω0 = 0.000, ω1 = 1.000 | M1a vs M2a | 2 | 0.0103 | |
| M2a | -440.88 | P0 =0.858, P1 = 0.000, P2 = 0.142, ω0 = 0.000, ω1 = 1.000, ω2 = 11.496 | 13, 57, 70, 73, 74 | ||||
| M7 | -446.65 | P = 1.970, q = 0.005 | M7 vs M8 | 2 | <0.01 | ||
| M8 | -440.88 | P0 = 0.859, P = 0.005, q = 2.990, P1 = 0.142, ω = 11.496 | 13, 57, 70, 73, 74 | ||||
| Ⅲ | M1a | -640.12 | P0 = 0.737, P1 = 0.263, ω0 = 0.000, ω1= 1.000 | M1a vs M2a | 2 | <0.01 | |
| M2a | -622.57 | P0 = 0.963, P1 = 0.000, P2 = 0.037, ω0 = 0.546, ω1= 1.000, ω2 = 18.992 | 11, 13, 26, 32, 37, 56, 57, 70, 71, 73, 74, 78, 86 | ||||
| M7 | -640.26 | P = 0.005, q = 0.012 | M7 vs M8 | 2 | <0.01 | ||
| M8 | -622.47 | P0 = 0.969, P = 0.008, q = 0.005, P1 = 0.031, ω = 20.626 | 11, 13, 26, 32, 37, 56, 57, 70, 71, 73, 74, 78, 86 | ||||
| All | M1a | -1132.10 | P0 = 0.898, P1 = 0.102, ω0 = 0.039, ω1 = 1.000 | ||||
| M2a | -1081.75 | P0 = 0.978, P1 = 0.000, P2 = 0.022, ω0 = 0.468, ω1 = 1.000, ω2 = 14.736 | 11, 13, 26, 32, 57, 67, 70, 71, 73, 74, 78, 86 | M1a vs M2a | 2 | <0.01 | |
| M7 | -1134.53 | P = 0.016, q = 0.103 | |||||
| M8 | -1079.73 | P0 = 0.979, P = 0.021, q = 0.026, P1 = 0.021, ω = 14.775 | 11, 13, 26, 32, 57, 67, 70, 71, 73, 74, 78, 86 | M7 vs M8 | 2 | <0.01 |
ω equals dN to dS ratio; Pn is the proportion of amino acids in the ωn site class; P and q are parameters of the beta distribution.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



