
Submitted:
23 July 2023
Posted:
24 July 2023
You are already at the latest version
Abstract
Cellular senescence is characterized by permanent proliferation and migration arrest, senescence-associated secretory phenotype (SASP), and oxidative stress. Senescent vascular smooth muscle cells (VSMCs) contribute to cardiovascular diseases and atherosclerotic plaque instability. To establish and characterize a model of replicative senescence (RS), human aortic VSMCs were serially passaged to represent different stages of RS such as young proliferating cells and old/senescent non-proliferating cells. More than 50% of old cells stained positive for the senescence-associated β-galactosidase compared to 20% of young cells. Old cells have a slower proliferation rate, a migratory activity reduced by 50%, but increased levels of TP53 and of cell cycle inhibitors p21/p16 expression, and accumulate in the G1 phase. Old cells showed a flattened appearance and enlarged and regular nuclei, and downregulation of the expression of LMNB1 and HMGB1. Old cells showed also an increased expression of SASP molecules (IL1β, IL6, IL8, and MMP3). Moreover, among a set of 12 manually selected long non-coding RNAs (lncRNAs), we detected significant upregulation of PURPL and NEAT1. We observed also increased levels of RRAD mRNA. The detection of novel molecular markers of senescence, such as RRAD, PURPL, and NEAT1, could be helpful for future studies on potential anti-aging factors.
Keywords:
aging
; biomarkers
; lncRNA
; NEAT1
; PURPL
; RRAD
; senescence
; smooth muscle cells
1. Introduction
The proportion of the world’s population aged 60 years, or more, is expected to double in the next four decades, and the WHO estimates that 1 in 6 people, or 2.1 billion, will be over age 60 by 2030 [1]. Ageing plays a previously unrecognized significant role in the pathophysiology of human body [2]: it is associated with a progressive degeneration of the tissues negatively impacting on the structure and function of vital organs and it is among the most important known risk factors for chronic diseases (such as atherosclerosis or neurodegeneration). Aging includes several diverse mechanisms such as oxidative stress, genomic instability, progenitor cell exhaustion or dysfunction, telomeric and epigenetic changes, altered nutrient sensing, mitochondrial dysfunction, chronic low-grade inflammation, altered protein homeostasis, fibrosis, microbiome dysregulation and cellular senescence [3]. Senescence of various cell types, including endothelial cells, vascular smooth muscle cells (VSMCs), macrophages and T cells, has been implicated in the pathogenesis of degenerative diseases such as atherosclerosis. VSMCs, as basic component of the vascular wall and the sole cell type in the arterial medial layer, play critical roles in vascular physiological functions [4,5], are the major source of atherosclerotic plaque cells and contribute to plaque development and progression [6].
At the cellular level, senescent cells may form throughout the entire lifespan accumulating and secreting different factors that may cause deleterious effects on surrounding tissues [1]. Cellular senescence is an irreversible loss of proliferation potential [7]. Growth arrest results from silencing of proliferation-promoting genes upon activation of the tumor suppressor gene p53 and the cyclin-dependent kinase inhibitor (cdki) p21 and it is then stabilized by activation of the cdki p16 and hypophosphorylation of the retinoblastoma protein [7]. Cellular senescence can also be induced by various stimuli such as oncogene activation (oncogene-induced senescence) or DNA damaging agents and oxidative stress, called ‘stress-induced premature senescence’ [8].
Besides permanent growth arrest, senescent cells display morphological and functional changes, including flattened and hypertrophic morphology, nuclear enlargement and expression of the ‘senescence-associated β-galactosidase’ (SA-β-gal), a pH-sensitive enzyme whose activity reflects an increased lysosomal mass [5]. Senescent cells stimulate reactive oxygen species (ROS) production thus enforcing the inhibition of cell proliferation [9]. Moreover, they also show a ‘senescence-associated secretory phenotype’ (SASP), with the production proapoptotic and pro-fibrotic factors [10] and the secretion of pro-inflammatory cytokines (e.g., IL6 and CXCL8 (IL8)), chemokines (e.g., CCL2) and proteases (e.g., matrix metalloproteases (MMPs)) thus contributing to tissue inflammation [5,11].
Cellular senescence program initiation and sustainment are based on transcriptional and post-transcriptional, as well as epigenetic, changes involving not only proteins but also different types of RNAs, including long non-coding RNAs (lncRNAs) [12,13,14,15]. LncRNAs are a wide and heterogeneous class of RNA molecules longer than 200 nucleotides with a fundamental role in the control of gene expression through different mechanisms [14,15] and known to be involved in key biological processes [16,17]. More specifically concerning aging, several lncRNAs have been shown to be involved in different hallmarks of senescence, among which cell cycle arrest, apoptosis, telomere stability and inflammation [18]. Despite mounting evidence linking lncRNAs to senescence, only a few of them have been associated to the formation of senescent VSMCs so far [19,20].
Currently, there are no unanimously agreed senescence markers in human VSMCs. Our aim was to characterize a cellular model of replicative senescence in human VSMCs by means of multi-biomarkers approaches, performing an in-deep cellular morphological analysis and evaluating the expression of manually selected senescence-associated genes and lncRNAs. Our investigation led to the discovery of newly associated senescence biomarkers such as PURPL, NEAT1 and RRAD.
2. Materials and Methods
Cell cultures
Human aortic vascular smooth muscle cells (VSMCs) (PCS-100-012, ATCC, Manassas, USA) were cultured in ATCC Vascular Cell Basal Medium (PCS-100-030, ATCC; 500 ml added with 500 µl ascorbic acid, 500 µl rh EGF, 500 µl rh insulin and rh FGF-b, 25 ml glutamine), 5% fetal bovine serum (FBS, ATCC Vascular Smooth Muscle Growth Kit), and 5 ml Penicillin-Streptomycin 100X (Euroclone, Milan, Italy). In our experiments, VSMCs were used at the 5-7th passage as young (proliferating cells) and at 15-17th as old (non-proliferating cells) to represent different stages of replicative senescence. Cultures were maintained at 37°C in a 5% CO2 incubator.
Senescence-Associated β-Galactosidase Staining
Young and old VSMCs were plated in 24-well plates at a density of 2×104 cells/well. After 3 days the activity of senescence-associated-β-galactosidase (SA-β-gal) was evaluated by staining VSMCs with the Senescence Cells Histochemical Staining Kit (CS0030, Sigma-Aldrich), following manufacturer’s instructions [11].
Cell proliferation
VSMCs were seeded in 24-well plates at a density of 3 ×104 cells/well. After 24 h, medium was removed, and cells were incubated for 72 h with medium containing 0.4% FBS to synchronize cells at G0 phase of the cell cycle. After 72 h, control dishes were counted with a Coulter Counter (Beckman Coulter, Life Scientific, Milan, Italy) and this was considered the “basal” number of cells at T0. Subsequently, medium was removed and replaced with 10% of FBS for 24, 48, and 96 h. Cells number was measured and compared to the zero time-point. Results were also used to calculate the doubling time [21].
Cell migration
VSMCs were seeded in 24-well plates and, after reaching 80-90% confluence, were detached and resuspended in 0,4% serum medium and placed on a membrane with 8 μm pores for 6 h. The number of migrated cells was counted in five randomly chosen areas of the membranes at 10X magnification [22].
Cell cycle measurement
The cell cycle was determined after seeding VSMCs in 12-well plates and reaching 80-90% confluence. Then cells were washed with PBS, trypsinized, and fixed with ice-cold ethanol 66%. After fixation, cells were subsequently collected and stained with a propidium iodide flow cytometry kit (Abcam, ab139418). The cell-cycle phases were analyzed with a NovoCyte 3000 Flow Cytometer using the NovoExpress software v.1.3.3.
Immufluorescence analysis and Nuclear/Cell Size Measurement
Young and senescent VSMCs were seeded on glass-coverslips in a 24-well plate. After 3 days in culture, cells were fixed with 4% paraformaldehyde in 10 mM PBS for 30 minutes at room temperature and then washed three times with 10 mM PBS. Cells were permeabilized at 4°C in PBS containing 0.1% triton X-100 for 3 min and blocked with 5% BSA in PBS for 15 min. Then, cells were incubated with the primary antibody anti-RRAD (Thermo-Fisher) in 0.2% BSA in PBS, overnight (4°C) and then were incubated with Alexa Fluor 488-conjugated secondary antibody (Invitrogen) for 1h and washed with PBS. To evaluate nuclear and cell size changes, cells were incubated with fluorescent phalloidin (Alexa Fluor 488 phalloidin, ThermoFisher Scientific, Waltham, USA) for 1 hour at room temperature and then washed with PBS. DNA was stained with DAPI solution (Invitrogen, 1:1000 in PBS) for 10 min and slides were mounted with Fluoromount Acquous Mounting Medium (Sigma-Aldrich). The immunofluorescence (IF) signals of RRAD were acquired for each experimental group with an epi-fluorescence microscope by Nikon (Nikon Eclipse Ti) using a 100X objective, for nuclear and cell size changes images were acquired using a 20X fluorescence objective (AXIOVERT 200 Fluorescent, Carl Zeiss). Nuclear or cell size measurements and nuclear morphometric analysis were performed with the Image J software v.2.1.0 [23].
RNA isolation and retrotranscription
Total RNA was extracted from VSMCs using the Direct-zolTM RNA MiniPrep Plus kit (Zymo Research, Irvine, USA). The concentration and purity of RNA were measured using the Nanodrop 1000 spectrophotometer (ThermoFisher Scientific). All RNA samples had an A260/280 value of 1.8–2.1. The quality of RNA was also evaluated using the Tape Station 2200 instrument (Agilent). All the samples had a RIN value ≥ 9. One μg of total RNA was treated with the RQ1 RNase-Free Dnase (M6101, Promega) and then cDNA was synthetized in 20 μl reactions using the High-Capacity cDNA Reverse Transcription Kit (4368814, Applied Biosystems), according to manufacturer’s instructions. Alternatively, the RNA samples were retrotranscribed using the iScript gDNA Clear cDNA Synthesis kit (1725035, BIO RAD, Berkley, USA).
Selection of genes and lncRNAs associated with senescence
A panel of 24 transcripts, including protein-coding genes and lncRNAs, were manually selected from literature according to their association with senescence and by querying specific online resources such as The Human Ageing Genomic Resources (HAGR) [https://genomics.senescence.info/CellAge (v3 (23/04/2023)], Reactome [https://reactome.org/ (v.84,29/03/2023)] and Human Protein Atlas [HPAhttps://www.proteinatlas.org/, v.23]. A set of 12 protein–coding genes was selected as involved in different functions such as cell cycle, DNA damage and SASP, as well as novel biomarkers (Suppl Table 1). For lncRNAs, we focused on a list of 12 lncRNAs (Table 1) involved in several pathways and in the expression and secretion of SASP components.
Quantitative RT-PCR (qRT-PCR)
Quantitative RT-PCR was performed with the QuantStudio 5 thermocycler (Applied Biosystems) in 384-wells plates using the GoTaq qPCR Master Mix (A6002, Promega). 10 μl PCR reactions were prepared containing 2 μl of reverse transcriptase product and 0.2 μl of each primer (10 µM) for specific genes (Suppl Table 2). The PCR mixtures were initially denatured at 95 °C for 2 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 1 m. The results were analyzed with the QuantStudio Design & Analysis Software v1.5.2 (Applied Biosystems). The melting curve showed a single product peak, indicating good product specificity. The calculation of gene expression levels was based on the n ΔΔCt method using the geometric mean of the expression values of three normalizer genes (CYC1, EIF4A2 and RPSA). Fold changes were calculated using 2-(ΔΔCt) and comparing senescent versus young/proliferating cells.
Protein isolation, quantification, SDS page and Western Blot
For the preparation of total cell lysates, cells were washed with ice-cold PBS and lysed with lysis buffer (NaCl 150 mM, TRIS 50 mM pH 7.6, NONIDET P-40 0.5% and protease inhibitors (Merck, Milan, Italy)). Protein concentration was determined using a Pierce BCA Protein Assay Kit (Pierce, Rockford, IL, USA) and 15 μg of samples was run on SDS-PAGE. The different proteins (PCNA, LMNB1, p21, P53, RRAD, IL1b, IL6, MMP3) were detected using specific primary and secondary antibodies as needed (Suppl Table 3, which contains also antibody dilutions). Quantification of western blot bands was performed by densitometric analysis using the Image Studio Lite software v 3.1from Li-Cor Bioscience (Lincoln, NE, USA).
Statistical analysis and data visualization
Data are presented as the mean ± SD of 3 experiments performed in triplicates and were analyzed with GraphPad Prism 9 software. The analysis was performed with the unpaired Student’s t-test with Welch correction or 2way ANOVA, followed by Šidák’s multiple comparisons test. Statistical significance was set at p < 0.05. The heatmap of fold change (FC) qRT-PCR data was created by Morpheus online tool [https://software.broadinstitute.org/morpheus/].
3. Results
To establish our replicative senescence model, human VSMCs have been serially passaged. Then, we analyzed two different groups of cells: VSMCs that have been serially passaged 5-7 times (representing the “young” proliferating cells), and VSMCs that have been passaged 15-17 times (representing the “old/senescent” non-proliferating cells). Then, the two groups of cells have been deeply characterized as described below.
3.1. Senescent VSMCs show increased SA-β-gal activity
First, we evaluated the activity of SA-β-gal, which is a lysosomal hydrolytic enzyme up-regulated in senescent cells [32]. As shown in Figure 1A and B, at least 50% of old cells are positive for SA-β-gal staining (indicated by the blue color) compared to about 20% of young cells, indicating an increased SA-β-gal activity in senescent VSMCs. We also evaluated the expression of the β-gal encoding gene GLB1 by qRT-PCR. GLB1 gene expression level in old cells is 50% higher compared to young cells (Figure 1C).
3.2. Senescent VSMCs display altered cell and nuclei morphology
Senescence is normally accompanied by significant morphological changes at cell as well as at nuclear levels [33,34]. As shown in Figure 2A, old/senescent cells become flat, enlarged and vacuolized compared to young ones.
Nuclear changes have been well documented in senescent cells, which show prominent and at times multiple nuclei, with severe chromatin condensation detected as large and fluorescent after staining with DAPI [35]. To evaluate nuclear area and shape, cells have been stained with DAPI and F-actin. As shown in Figure 2B and C, old cells show a statistically significant increase of cell and nuclear area compared to young cells.
A nuclear morphometric analysis (NMA) was also performed using a FIJI plug-in [11,23]. This approach allowed us to divide cells into various groups based on their nuclear morphometry. These groups included: normal, irregular, small regular (apoptotic), small (mitotic), small irregular, large regular (senescent), large irregular nuclei [11,23]. We observed a reduced percentage of normal nuclei in old compared to young cells (88% vs 66%), as well as an increased percentage of large and regular (senescent; 29% vs 8%) and irregular nuclei (5% vs 3%) (Figure 2D).
3.3. Senescent VSMCs display reduced proliferation and cell cycle arrest
One feature of senescent cells is the reduced proliferative rate [7]. Therefore, we evaluated the expression of the aging biomarker proliferating cell nuclear antigen (PCNA), which is involved in DNA repair and whose expression is decreased in aged cells [36]. As shown in Figure 3A, old VSMCs have a significantly reduced PCNA expression at both mRNA (60% reduction of mRNA levels, p<0.001 vs young) and protein (65% reduction, p<0.05 vs young) levels. This led to a reduced cell proliferation (Figure 3B) and an increased doubling time for old cells (42 vs 25 hours in young cells). As expected, when we evaluated the cell cycle, we observed an accumulation of old VSMCs in G1 phase (Figure 3C). In addition, old VSMCs have a reduced migratory potential (70% reduction, p<0.01 vs young cells, Figure 3D).
3.4. Senescent VSMCs express well-known senescence biomarkers
Next, we evaluated in young and old cells the expression of lamin B1 (LMNB1), a protein component on nuclear lamina whose downregulation causes a detachment of chromatin domains normally attached to the nuclear lamina leading to the redistribution of hetero-chromatin from the nuclear periphery into the interior. This may trigger senescence, as reported before [37,38]. Figure 4A demonstrates that old VSMCs have a significantly reduced LMNB1 expression at both mRNA (65% reduction, p<0.01 vs young) and protein (60% reduction, p<0.05 vs young) levels, indicating an altered nuclear membrane in old cells.
During senescence, SASP-related chromatin folding and RNA homeostasis are coordinated by the extracellular senescence factor high mobility group box 1 (HMGB1) [39]. As shown in Figure 4B, HMGB1 mRNA levels are significantly reduced in old VSMCs. We observed also an upregulation of the expression of p53, whose activation regulates the expression of a large set of genes involved in cell cycle arrest [40]. In fact, we observed a doubling of the expression of p53 protein, although the mRNA levels were slightly reduced (Figure 4C, D). In line with this data, old-senescent cells showed an increased expression of p16 and p21, two tumor suppressors that, by preventing retinoblastoma phosphorylation, regulate cell cycle leading to cell growth arrest (Figures 4C, D).
The Ras Related glycolysis inhibitor and calcium channel regulator (RRAD) has been recently shown to be a negative regulator of cellular senescence [41], and therefore it was included in our list of protein-coding genes (Suppl Table 1). In our model, we observed a consistent and statistically significant upregulation of RRAD mRNA levels (fold change>6 in old cells vs. young cells) in senescent VSMCs (Figure 5A), although the analysis at the protein level did not show a similar direction (Figure 5B). As reported in the Human Protein database (HPA), the screening of RRAD gene expression levels in 44 human normal tissues (tissue atlas), as well as in 15 different cell types (single cell atlas), indicates a specific expression in tissues and cells belonging to the musculoskeletal and cardiovascular systems [https://www.proteinatlas.org/ENSG00000166592-RRAD]. Although RRAD expression was reported in SMCs in the HPA database, its subcellular localization is not publicly available. By means of immunofluorescence, we showed RRAD localization in cellular areas of the plasma membrane, nucleoplasm, and Golgi apparatus (Figure S1) both in young cells and in old/senescent VSMC cells. Of note, the signal of RRAD was more dispersed and diffuse in the old cells, possibly due to their altered cellular morphology.
3.5. Senescent VSMCs exhibit a change in inflammatory biomarkers
Senescent cells exhibit significant changes in their secretome including the expression of a variety of inflammatory proteins such as cytokines and interleukins [42], a phenomenon known as SASP [43]. To have a more complete characterization of our RS model, we evaluated the gene expression of different inflammatory markers that are typical of the SASP. The mRNA levels of IL1β, IL6, IL8 and MMP3 are significantly increased up to nine-fold in old cells (Figure 6A). A significant increase of IL1β, IL6 and MMP3 protein levels is also displayed by senescent VSMCs (Figure 6B).
3.6. LncRNAs PURPL and NEAT1 are markers of senescent VMSCs
To further expand the characterization of senescent VSMCs we also evaluated the expression of a panel of lncRNAs. LncRNAs have recently drawn attention as important regulators of pathways associated to senescence also displaying altered expression levels in various models of cellular senescence [18]. Based on literature, we selected 12 lncRNAs involved in senescence (see Table 1 and Materials and methods) and we measured their expression levels by qRT-PCR in old and young VMSCs. Our quantitative analysis revealed a significant strong up-regulation of two lncRNAs, namely P53-Upregulated Regulator of P53 Levels (PURPL; Fold Changes ranging from 3.2 to 22.5, mean average = 15.21) and Nuclear Paraspeckle Assembly Transcript 1 (NEAT1; FC ranging from 1.7 to 7.9, mean average = 4.18), and a slight down-regulation of MEG3 and up-regulation of SBLC, although not statistically significant, in senescent cells (Figure 7). For the remaining lncRNAs we observed a more heterogeneous behavior in senescent VSMCs due also to their low expression levels.
4. Discussion
Our current study provides new insights into the molecular mechanism(s) that regulate aging in human aortic VSMCs. We created a cellular model of replicative senescence by serially passing human VSMCs. In our experimental conditions, the old/senescent VSMCs express many of the typical senescence-associated markers already described in the literature. In this cellular model of VSMC senescence, we also demonstrated the expression of novel senescence-associated markers such as RRAD and the lncRNAs PURPL and NEAT1.
Old VSMCs display an increased percentage of SA-β-gal expressing cells and a different morphology, showing a flat and enlarged cell body and presence of vacuolization, a reduced percentage of normal nuclei and a parallel increase of irregular, enlarged and senescent nuclei characterized by chromatin condensation. We also observed a significant reduction of the expression of LMNB1, a dramatically reduced proliferation and growth rate, a longer doubling time and an accumulation in the G1 phase of cell cycle. Moreover, the expression of the aging biomarker PCNA, which is involved in cell proliferation and DNA repair, is decreased by up to 65% at both mRNA and protein levels. In addition, we observed an increased expression of p53, a transcription factor that can both upregulate or downregulate the expression of specific target genes by binding their promoter region [40]. In addition, the cell cycle inhibitors p16 and p21 are stimulated by more than 200% and 400%, respectively, compared to young VSMCs.
Senescent cells exhibit significant changes in their secretome including the expression of a variety of proteins such as cytokines, interleukins, chemokines, proteases, growth factors, degradative enzymes like MMPs and insoluble proteins or extracellular matrix components [42]. During senescence, SASP-related chromatin folding, and RNA homeostasis is coordinated by the extracellular senescence factor HMGB1 [39], a transcription factor or transcription inducer that is secreted or released by stressed cells and serves as an alarmin, cytokine or growth factor to activate the immune response. Suppression of HMGB1 induces cell cycle arrest and senescence in association with p21 upregulation [44]. In accordance with these observations, in our old VSMCs, HMGB1 expression is significantly reduced by 40%, and in parallel we observed strikingly increased levels of both mRNAs and proteins of inflammatory markers such as IL1β, IL6, IL8 and MMP3.
Interestingly, we observed a significant upregulation of RRAD mRNA levels. To our knowledge, this is the first report showing an increased expression of RRAD in senescent human VSMCs. RRAD is predicted to be involved in small GTPase mediated signal transduction, it has been implicated in some types of cancer [45] and is considered a biomarker of congestive heart failure [46,47]. RRAD expression appears to be stimulated by oxidative stress [48], and it has been recently associated with cellular senescence in human skin fibroblasts as a negative regulator [41]. It has been proposed that increased levels of RRAD may serve as a negative feedback mechanism in the effort to reduce the level of ROS thus countering cellular senescence [41]. In our replicative senescence model of VSMCs, although the RRAD gene expression level was upregulated in old cells, western blot analysis showed lower protein expression. The correlation between mRNAs and protein expression is affected by multiple layers of regulation and therefore it is not surprising to observe contrasting results [49]. We hypothesized that the increased gene expression of RRAD in old VMSCs is the response to the oxidative stress induced in cellular senescence. To determine if the fate of the RRAD protein in old cells is altered, the study of protein stability is needed. Intriguingly, the investigation of the subcellular localization of RRAD in young and senescent VSMC cells by IF indicates a more dispersed and diffuse distribution in old cells. A deep biochemical analysis of RRAD in our model of senescence will be the objective of further investigations.
Among the set of 12 lncRNAs manually selected for being involved in senescence, we detected by qRT-PCR a significant up-regulation of PURPL and NEAT1 in old (non-proliferating) cells compared to the young (proliferative) ones. PURPL is known to be a p53 target, as its promoter contains p53-response elements. In colorectal cancer cells, PURPL is transcriptionally activated by p53 and, in return, it can decrease the levels of p53 and of its targets, such as p21. Mechanistically, PURPL can negatively regulate p53 stability by inhibiting its interaction with the MYBBP1A protein, that can bind and stabilize p53 [28]. This negative regulation of p53 by PURPL was also observed in melanoma cells, where PURPL was shown to repress autophagic cell death by associating with mTOR and modulating ULK1 phosphorylation [50]. On the contrary, in liver cancer cells, PURPL expression is still activated by p53, but its negative effect on p53 levels is lost. Instead, upon p53 activation, loss of PURPL leads to downregulation of genes involved in mitosis, indicating a role of PURPL in cell cycle progression and mitosis [51]. Up-regulation of PURPL was consistently observed in several cellular models of replicative and induced senescence, including a model of ionizing radiation-induced senescence of human aortic endothelial cells, which is the other major cell type present in the aortic wall [29]. Our data confirm PURPL as a robust marker of senescence condition also in human VMSCs.
Moreover, the lncRNA NEAT1 was found significantly up-regulated in senescent VMSCs. NEAT1 is also a p53 target and it is a core structural component of paraspeckles, nuclear bodies with a key role in gene expression regulation through several mechanisms, including regulation of transcription, regulation of translation and modulation of miRNA processing [52,53,54,55]. NEAT1 has been also associated with diseases such as cancer, immune inflammation, and neurodegeneration [56]. Several lines of evidence show that NEAT1 expression, as well as paraspeckle formation, is induced by p53, and that NEAT1 is part of an autoregulatory negative feedback loop that attenuates p53 activity [57,58,59]. NEAT1 is also able to inhibit p21 expression by guiding the epigenetic repressor Enhancer of Zeste Homolog 2 (EZH2) to p21 promoter [60]. On the other hand, it was shown to activate pro-inflammatory cytokine IL8 transcription [54]. Importantly, NEAT1 has a key role in VSMCs switching from a contractile to a proliferative phenotype by epigenetically repressing the expression of smooth muscle-specific genes [61]. In our work, increased levels of p53 and p21 were observed in senescent VMSCs, as expected in a state of cell cycle arrest. In this context, we hypothesize that PURPL and NEAT1 up-regulation may be part of a negative feedback response to buffer excessive p53 and downstream targets levels and activity, and partly trying to restore proliferative capacity. At the same time, NEAT1 may promote the SASP by increasing IL8 levels.
Senescence may exert both beneficial and negative effects after tissue injury depending on the context. Although senescent cells may maintain a healthy physiology, senescence of various cell types has been implicated in the pathogenesis of atherosclerosis including endothelial cells, VSMCs, macrophages and T cells. The accumulation of senescent VSMCs contributes to aging as well as age-related diseases of the cardiovascular system [62], and VSMCs were reported to be one of the key pro-inflammatory senescent cell populations, and were found in unstable, rupture-prone atherosclerotic plaques [63,64]. Like other aging cells, senescent VSMCs have a low ability of mitotic division and show changes in cell signaling pathways and senescent markers, such as SA-β-gal activity, levels of p16, p38, p53, p21, and express the SASP [8]. Due to the loss of proliferative potential of senescent cells, senescence can trigger plaque instability directly by reducing the VSMC content of the fibrous cap and compromising its repair after rupture [5]. However, more recent evidence suggests a more active role of VSMC senescence in promoting plaque destabilization by driving plaque inflammation and matrix degradation and defective autophagy [5,65]. In this scenario, the involvement of lncRNAs in atherosclerosis is clearly emerging, by regulating several key processes such as cholesterol homeostasis, vascular inflammation, VSMC phenotypic switch and cell death, among others [66,67]. As discussed above, upregulation of NEAT1 in senescent VSMCs may be a fundamental contributor to plaque inflammation and destabilization by setting a ‘macrophage-like’ state with increased SASP levels.
5. Conclusions
Cell senescence could be targeted to prevent or delay the aging process and tissue dysfunction extending lifespan [68,69]. Some biochemical agents with anti-senescence potential called ‘senolytics’, compounds that selectively target and eliminate senescent cells by inducing apoptosis, have been tested in the prevention/treatment of diseases [1]. However, we need better senescence-associated targets, that may also include lncRNAs, to ameliorate our therapeutic approach. In our experimental settings, we characterized the senescence state of human VSMC by means of multi-levels approaches including fine morphological evaluations, together with the expression analysis of essential and promising cell specific molecular markers of vascular senescence. It is a basic knowledge that cellular senescence is the result of complex interplay of various biological and metabolic cell- and tissue-specific processes, so our experimental characterization is still incomplete. The definition of the functional role of PURPL, NEAT1, and RRAD in senescent human VSMCs, was beyond the scope of the present investigation but future investigations are planned. However, we think that these newly discovered senescent markers are promising molecular tools to be used in combination with landmark senescence markers to better discriminate senescent VSMCs implicated in atherogenesis and atherosclerosis complications and may help in designing new strategies for promoting healthy vascular aging.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Immunofluorescence images of RRAD localization in young and old vascular smooth muscle cells; Suppl Table 1: Bioformatic annotations of selected protein coding genes; Suppl Table 2: Primers used for q-RT-PCR experiments (protein-coding genes and lncRNAs); Suppl Table 3: Antibodies used for Western blot experiments .
Author Contributions
Conceptualization, C.B., S.B., C.R. and M.V.; formal analysis, C.R., M.V. and J.G.; investigation, C.R., J.G. and A.F.; data curation, C.B.; writing—original draft preparation, S.B.; writing—review and editing, C.B., M.V. and A.C.; supervision, S.B. and C.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by ‘Piano di sostegno alla Ricerca 2020′ Grant (Università degli Studi di Milano, award no. PSR2020_BATTAGLIA_LINEA_B to CB and MV) and ‘Piano di sostegno alla Ricerca 2021′ Grant (Università degli Studi di Milano, award no. PSR2021_BATTAGLIA_LINEA_C to CB, and award no. PSR2021_VENTURIN_LINEA_C to MV).
Institutional Review Board Statement
The study did not require ethical approval.
Acknowledgments
CR is recipient of a scholarship of Ph.D. in Pharmacological Biomolecular Sciences, Experimental and Clinical, Università degli Studi di Milano.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular Senescence and Senolytics: The Path to the Clinic. Nat Med 2022, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.A.; Krizhanovsky, V. Physiological and Pathological Consequences of Cellular Senescence. Cellular and Molecular Life Sciences 2014, 71, 4373–4386. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Bennett, M.R. Vascular Smooth Muscle Cells in Atherosclerosis: Time for a Re-Assessment. Cardiovasc Res 2021, 117, 2326–2339. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; de Meyer, G.R.Y. Vascular Smooth Muscle Cell Death, Autophagy and Senescence in Atherosclerosis. Cardiovasc Res 2018, 114, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular Smooth Muscle Cells in Atherosclerosis. Nat Rev Cardiol 2019, 16, 727–744. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat Rev Mol Cell Biol 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Chi, C.; Li, D.-J.; Jiang, Y.-J.; Tong, J.; Fu, H.; Wu, Y.-H.; Shen, F.-M. Vascular Smooth Muscle Cell Senescence and Age-Related Diseases: State of the Art. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2019, 1865, 1810–1821. [Google Scholar] [CrossRef]
- Ngoi, N.YL.; Liew, A.QX.; Chong, S.J.F.; Davids, M.S.; Clement, M.-V.; Pervaiz, S. The Redox-Senescence Axis and Its Therapeutic Targeting. Redox Biol 2021, 45, 102032. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLoS Biol 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Stojanović, S.D.; Fuchs, M.; Kunz, M.; Xiao, K.; Just, A.; Pich, A.; Bauersachs, J.; Fiedler, J.; Sedding, D.; Thum, T. Inflammatory Drivers of Cardiovascular Disease: Molecular Characterization of Senescent Coronary Vascular Smooth Muscle Cells. Front Physiol 2020, 11. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Lanigan, F.; Geraghty, J.G.; Bracken, A.P. Transcriptional Regulation of Cellular Senescence. Oncogene 2011, 30, 2901–2911. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long Non-Coding RNAs: Insights into Functions. Nat Rev Genet 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Yao, R.-W.; Wang, Y.; Chen, L.-L. Cellular Functions of Long Noncoding RNAs. Nat Cell Biol 2019, 21, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Fatica, A.; Bozzoni, I. Long Non-Coding RNAs: New Players in Cell Differentiation and Development. Nat Rev Genet 2014, 15, 7–21. [Google Scholar] [CrossRef]
- Perry, R.B.-T.; Ulitsky, I. The Functions of Long Noncoding RNAs in Development and Stem Cells. Development 2016, 143, 3882–3894. [Google Scholar] [CrossRef] [PubMed]
- Puvvula, P.K. LncRNAs Regulatory Networks in Cellular Senescence. Int J Mol Sci 2019, 20, 2615–2634. [Google Scholar] [CrossRef]
- Tan, P.; Guo, Y.-H.; Zhan, J.-K.; Long, L.-M.; Xu, M.-L.; Ye, L.; Ma, X.-Y.; Cui, X.-J.; Wang, H.-Q. LncRNA-ANRIL Inhibits Cell Senescence of Vascular Smooth Muscle Cells by Regulating MiR-181a/Sirt1. Biochemistry and Cell Biology 2019, 97, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Wu, J.; Wang, X.; Song, C. Noncoding RNAs in Vascular Aging. Oxid Med Cell Longev 2020, 2020, 1–14. [Google Scholar] [CrossRef]
- Damiani, I.; Castiglioni, S.; Sochaj-Gregorczyk, A.; Bonacina, F.; Colombo, I.; Rusconi, V.; Otlewski, J.; Corsini, A.; Bellosta, S. Purification and In Vitro Evaluation of an Anti-HER2 Affibody-Monomethyl Auristatin E Conjugate in HER2-Positive Cancer Cells. Biology (Basel) 2021, 10, 758. [Google Scholar] [CrossRef]
- Piegari, E.; De Angelis, A.; Cappetta, D.; Russo, R.; Esposito, G.; Costantino, S.; Graiani, G.; Frati, C.; Prezioso, L.; Berrino, L.; et al. Doxorubicin Induces Senescence and Impairs Function of Human Cardiac Progenitor Cells. Basic Res Cardiol 2013, 108. [Google Scholar] [CrossRef] [PubMed]
- Filippi-Chiela, E.C.; Oliveira, M.M.; Jurkovski, B.; Callegari-Jacques, S.M.; Silva, V.D. da; Lenz, G. Nuclear Morphometric Analysis (NMA): Screening of Senescence, Apoptosis and Nuclear Irregularities. PLoS One 2012, 7, e42522. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Shen, Z.; Chakraborty, A.; Giri, S.; Freier, S.M.; Wu, X.; Zhang, Y.; Gorospe, M.; Prasanth, S.G.; Lal, A.; et al. Long Noncoding RNA MALAT1 Controls Cell Cycle Progression by Regulating the Expression of Oncogenic Transcription Factor B-MYB. PLoS Genet 2013, 9, e1003368. [Google Scholar] [CrossRef] [PubMed]
- Grammatikakis, I.; Panda, A.C.; Abdelmohsen, K.; Gorospe, M. Long Noncoding RNAs (LncRNAs) and the Molecular Hallmarks of Aging. Aging 2014, 6, 992–1009. [Google Scholar] [CrossRef]
- Montes, M.; Lubas, M.; Arendrup, F.S.; Mentz, B.; Rohatgi, N.; Tumas, S.; Harder, L.M.; Skanderup, A.J.; Andersen, J.S.; Lund, A.H. The Long Non-Coding RNA MIR31HG Regulates the Senescence Associated Secretory Phenotype. Nat Commun 2021, 12, 2459. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Tu, C.; Liu, Y. Role of LncRNAs in Aging and Age-Related Diseases. Aging Medicine 2018, 1, 158–175. [Google Scholar] [CrossRef]
- Li, X.L.; Subramanian, M.; Jones, M.F.; Chaudhary, R.; Singh, D.K.; Zong, X.; Gryder, B.; Sindri, S.; Mo, M.; Schetter, A.; et al. Long Noncoding RNA PURPL Suppresses Basal P53 Levels and Promotes Tumorigenicity in Colorectal Cancer. Cell Rep 2017, 20, 2408–2423. [Google Scholar] [CrossRef]
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome Signature of Cellular Senescence. Nucleic Acids Res 2019, 47, 7294–7305. [Google Scholar] [CrossRef]
- Xu, C.L.; Sang, B.; Liu, G.Z.; Li, J.M.; Zhang, X.D.; Liu, L.X.; Thorne, R.F.; Wu, M. SENEBLOC, a Long Non-Coding RNA Suppresses Senescence via P53-Dependent and Independent Mechanisms. Nucleic Acids Res 2020, 48, 3089–3102. [Google Scholar] [CrossRef]
- Haemmig, S.; Yang, D.; Sun, X.; Das, D.; Ghaffari, S.; Molinaro, R.; Chen, L.; Deng, Y.; Freeman, D.; Moullan, N.; et al. Long Noncoding RNA SNHG12 Integrates a DNA-PK–Mediated DNA Damage Response and Vascular Senescence. Sci Transl Med 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Valieva, Y.; Ivanova, E.; Fayzullin, A.; Kurkov, A.; Igrunkova, A. Senescence-Associated β-Galactosidase Detection in Pathology. Diagnostics 2022, 12, 2309. [Google Scholar] [CrossRef]
- Son, H.N.; Chi, H.N.Q.; Chung, D.C.; Long, L.T. Morphological Changes during Replicative Senescence in Bovine Ovarian Granulosa Cells. Cell Cycle 2019, 18, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Heckenbach, I.; Mkrtchyan, G. v.; Ezra, M. ben; Bakula, D.; Madsen, J.S.; Nielsen, M.H.; Oró, D.; Osborne, B.; Covarrubias, A.J.; Idda, M.L.; et al. Nuclear Morphology Is a Deep Learning Biomarker of Cellular Senescence. Nat Aging 2022, 2, 742–755. [Google Scholar] [CrossRef]
- Machado-Oliveira, G.; Ramos, C.; Marques, A.R.A.; Vieira, O. v. Cell Senescence, Multiple Organelle Dysfunction and Atherosclerosis. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Moriwaki, S.; Sugiyama, Y.; Endo, Y.; Yamazaki, K.; Mori, T.; Takigawa, M.; Inoue, S. Decreased Gene Expression Responsible for Post-Ultraviolet DNA Repair Synthesis in Aging: A Possible Mechanism of Age-Related Reduction in DNA Repair Capacity. Journal of Investigative Dermatology 2005, 124, 435–442. [Google Scholar] [CrossRef]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Lukášová, E.; Kovařík, A.; Kozubek, S. Consequences of Lamin B1 and Lamin B Receptor Downregulation in Senescence. Cells 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Sofiadis, K.; Josipovic, N.; Nikolic, M.; Kargapolova, Y.; Übelmesser, N.; Varamogianni-Mamatsi, V.; Zirkel, A.; Papadionysiou, I.; Loughran, G.; Keane, J.; et al. HMGB1 Coordinates SASP-related Chromatin Folding and RNA Homeostasis on the Path to Senescence. Mol Syst Biol 2021, 17. [Google Scholar] [CrossRef] [PubMed]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of P53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Guo, H.; Qin, J.; Lu, S.; Liu, Q.; Zhang, X.; Zou, Y.; Gong, Y.; Shao, C. Pan-Senescence Transcriptome Analysis Identified RRAD as a Marker and Negative Regulator of Cellular Senescence. Free Radic Biol Med 2019, 130, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes Dev 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat Cell Biol 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, J.; Wen, T.; Zeng, W.; Peng, C.; Yan, S.; Tan, J.; Yang, K.; Liu, S.; Guo, A.; et al. Overexpression of HMGB1 in Melanoma Predicts Patient Survival and Suppression of HMGB1 Induces Cell Cycle Arrest and Senescence in Association with P21 (Waf1/Cip1) up-Regulation via a P53-Independent, Sp1-Dependent Pathway. Oncotarget 2014, 5, 6387–6403. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Li, Y.; Tan, X.; Liu, W.; He, X.; Pan, D.; Li, E.; Xu, L.; Long, L. Friend or Foe: Regulation, Downstream Effectors of RRAD in Cancer. Biomolecules 2023, 13, 477. [Google Scholar] [CrossRef]
- Kim, H.K.; Lee, I.; Kim, S.T.; Lee, J.; Kim, K.-M.; Park, J.O.; Kang, W.K. RRAD Expression in Gastric and Colorectal Cancer with Peritoneal Carcinomatosis. Sci Rep 2019, 9, 19439. [Google Scholar] [CrossRef]
- Belbachir, N.; Portero, V.; al Sayed, Z.R.; Gourraud, J.-B.; Dilasser, F.; Jesel, L.; Guo, H.; Wu, H.; Gaborit, N.; Guilluy, C.; et al. RRAD Mutation Causes Electrical and Cytoskeletal Defects in Cardiomyocytes Derived from a Familial Case of Brugada Syndrome. Eur Heart J 2019, 40, 3081–3094. [Google Scholar] [CrossRef]
- Halter, B.; Gonzalez de Aguilar, J.L.; Rene, F.; Petri, S.; Fricker, B.; Echaniz-Laguna, A.; Dupuis, L.; Larmet, Y.; Loeffler, J.P. Oxidative Stress in Skeletal Muscle Stimulates Early Expression of Rad in a Mouse Model of Amyotrophic Lateral Sclerosis. Free Radic Biol Med 2010, 48, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Buccitelli, C.; Selbach, M. MRNAs, Proteins and the Emerging Principles of Gene Expression Control. Nat Rev Genet 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Han, S.; Li, X.; Wang, K.; Zhu, D.; Meng, B.; Liu, J.; Liang, X.; Jin, Y.; Liu, X.; Wen, Q.; et al. PURPL Represses Autophagic Cell Death to Promote Cutaneous Melanoma by Modulating ULK1 Phosphorylation. Cell Death Dis 2021, 12, 1070. [Google Scholar] [CrossRef]
- Hartford, C.C.R.; Shrestha, R.L.; Pongor, L.; Zhao, Y.; Chen, X.; Fromont, C.; Chaudhary, R.; Li, X.L.; Pasterczyk, K.R.; Kumar, R.; et al. Context-Dependent Function of Long Noncoding RNA PURPL in Transcriptome Regulation during P53 Activation. Mol Cell Biol 2022, 42, e0028922. [Google Scholar] [CrossRef]
- Jiang, L.; Shao, C.; Wu, Q.-J.; Chen, G.; Zhou, J.; Yang, B.; Li, H.; Gou, L.-T.; Zhang, Y.; Wang, Y.; et al. NEAT1 Scaffolds RNA-Binding Proteins and the Microprocessor to Globally Enhance Pri-MiRNA Processing. Nat Struct Mol Biol 2017, 24, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, K. V.; Prasanth, S.G.; Xuan, Z.; Hearn, S.; Freier, S.M.; Bennett, C.F.; Zhang, M.Q.; Spector, D.L. Regulating Gene Expression through RNA Nuclear Retention. Cell 2005, 123, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Imamachi, N.; Akizuki, G.; Kumakura, M.; Kawaguchi, A.; Nagata, K.; Kato, A.; Kawaguchi, Y.; Sato, H.; Yoneda, M.; et al. Long Noncoding RNA NEAT1-Dependent SFPQ Relocation from Promoter Region to Paraspeckle Mediates IL8 Expression upon Immune Stimuli. Mol Cell 2014, 53, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Virnicchi, G.; Tanigawa, A.; Naganuma, T.; Li, R.; Kimura, H.; Yokoi, T.; Nakagawa, S.; Bénard, M.; Fox, A.H.; et al. NEAT1 Long Noncoding RNA Regulates Transcription via Protein Sequestration within Subnuclear Bodies. Mol Biol Cell 2014, 25, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Chen, S.; Wang, X.; Yang, X.; Chen, L.; Huang, T.; Zheng, Y.; Zheng, X.; Wu, X.; Sun, Y.; et al. Exercise Mitigates Endothelial Pyroptosis and Atherosclerosis by Downregulating NEAT1 Through N6-Methyladenosine Modifications. Arterioscler Thromb Vasc Biol 2023, 43, 910–926. [Google Scholar] [CrossRef]
- Idogawa, M.; Ohashi, T.; Sasaki, Y.; Nakase, H.; Tokino, T. Long Non-coding RNA NEAT1 Is a Transcriptional Target of P53 and Modulates P53-induced Transactivation and Tumor-suppressor Function. Int J Cancer 2017, 140, 2785–2791. [Google Scholar] [CrossRef]
- Adriaens, C.; Standaert, L.; Barra, J.; Latil, M.; Verfaillie, A.; Kalev, P.; Boeckx, B.; Wijnhoven, P.W.G.; Radaelli, E.; Vermi, W.; et al. P53 Induces Formation of NEAT1 LncRNA-Containing Paraspeckles That Modulate Replication Stress Response and Chemosensitivity. Nat Med 2016, 22, 861–868. [Google Scholar] [CrossRef]
- Mello, S.S.; Sinow, C.; Raj, N.; Mazur, P.K.; Bieging-Rolett, K.; Broz, D.K.; Imam, J.F.C.; Vogel, H.; Wood, L.D.; Sage, J.; et al. Neat1 Is a P53-Inducible LincRNA Essential for Transformation Suppression. Genes Dev 2017, 31, 1095–1108. [Google Scholar] [CrossRef]
- Wang, S.; Zuo, H.; Jin, J.; Lv, W.; Xu, Z.; Fan, Y.; Zhang, J.; Zuo, B. Long Noncoding RNA Neat1 Modulates Myogenesis by Recruiting Ezh2. Cell Death Dis 2019, 10, 505. [Google Scholar] [CrossRef]
- Ahmed, A.S.I.; Dong, K.; Liu, J.; Wen, T.; Yu, L.; Xu, F.; Kang, X.; Osman, I.; Hu, G.; Bunting, K.M.; et al. Long Noncoding RNA NEAT1 (Nuclear Paraspeckle Assembly Transcript 1) Is Critical for Phenotypic Switching of Vascular Smooth Muscle Cells. Proceedings of the National Academy of Sciences 2018, 115, E8660–E8667. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Wnuk, M.; Przybylska, D.; Grabowska, W.; Lewinska, A.; Alster, O.; Korwek, Z.; Cmoch, A.; Myszka, A.; Pikula, S.; et al. A Comparison of Replicative Senescence and Doxorubicin-Induced Premature Senescence of Vascular Smooth Muscle Cells Isolated from Human Aorta. Biogerontology 2014, 15, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Minamino, T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front Cardiovasc Med 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Uryga, A.K.; Bennett, M.R. Ageing Induced Vascular Smooth Muscle Cell Senescence in Atherosclerosis. J Physiol 2016, 594, 2115–2124. [Google Scholar] [CrossRef]
- Sweeney, M.; Cook, S.A.; Gil, J. Therapeutic Opportunities for Senolysis in Cardiovascular Disease. FEBS J 2023, 290, 1235–1255. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, J.; Deng, L.; Ishimwe, N.; Pauli, J.; Wu, W.; Shan, S.; Kempf, W.; Ballantyne, M.D.; Kim, D.; et al. INKILN Is a Novel Long Noncoding RNA Promoting Vascular Smooth Muscle Inflammation via Scaffolding MKL1 and USP10. Circulation 2023, 148, 47–67. [Google Scholar] [CrossRef]
- Josefs, T.; Boon, R.A. The Long Non-Coding Road to Atherosclerosis. Curr Atheroscler Rep 2020, 22, 55. [Google Scholar] [CrossRef]
- Cohn, R.L.; Gasek, N.S.; Kuchel, G.A.; Xu, M. The Heterogeneity of Cellular Senescence: Insights at the Single-Cell Level. Trends Cell Biol 2023, 33, 9–17. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of P16Ink4a-Positive Senescent Cells Delays Ageing-Associated Disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
Figure 1.
Senescence-associated β-gal activity and GLB1 expression in young and old human VSMCs. Cells have been plated and the activity of SA-β-gal and GLB1 expression have been evaluated. (A) Representative images of SA-β-gal staining in young and senescent VSMCs, at two different magnifications (5x and 10x). The histograms indicate the percentage of (B) SA-β-gal positive cells and (C) GLB1 mRNA levels in young (black or grey) and old (blue or light blue) cells. Unpaired t test with Welch’s correction: * p < 0.05 vs YOUNG.
Figure 1.
Senescence-associated β-gal activity and GLB1 expression in young and old human VSMCs. Cells have been plated and the activity of SA-β-gal and GLB1 expression have been evaluated. (A) Representative images of SA-β-gal staining in young and senescent VSMCs, at two different magnifications (5x and 10x). The histograms indicate the percentage of (B) SA-β-gal positive cells and (C) GLB1 mRNA levels in young (black or grey) and old (blue or light blue) cells. Unpaired t test with Welch’s correction: * p < 0.05 vs YOUNG.
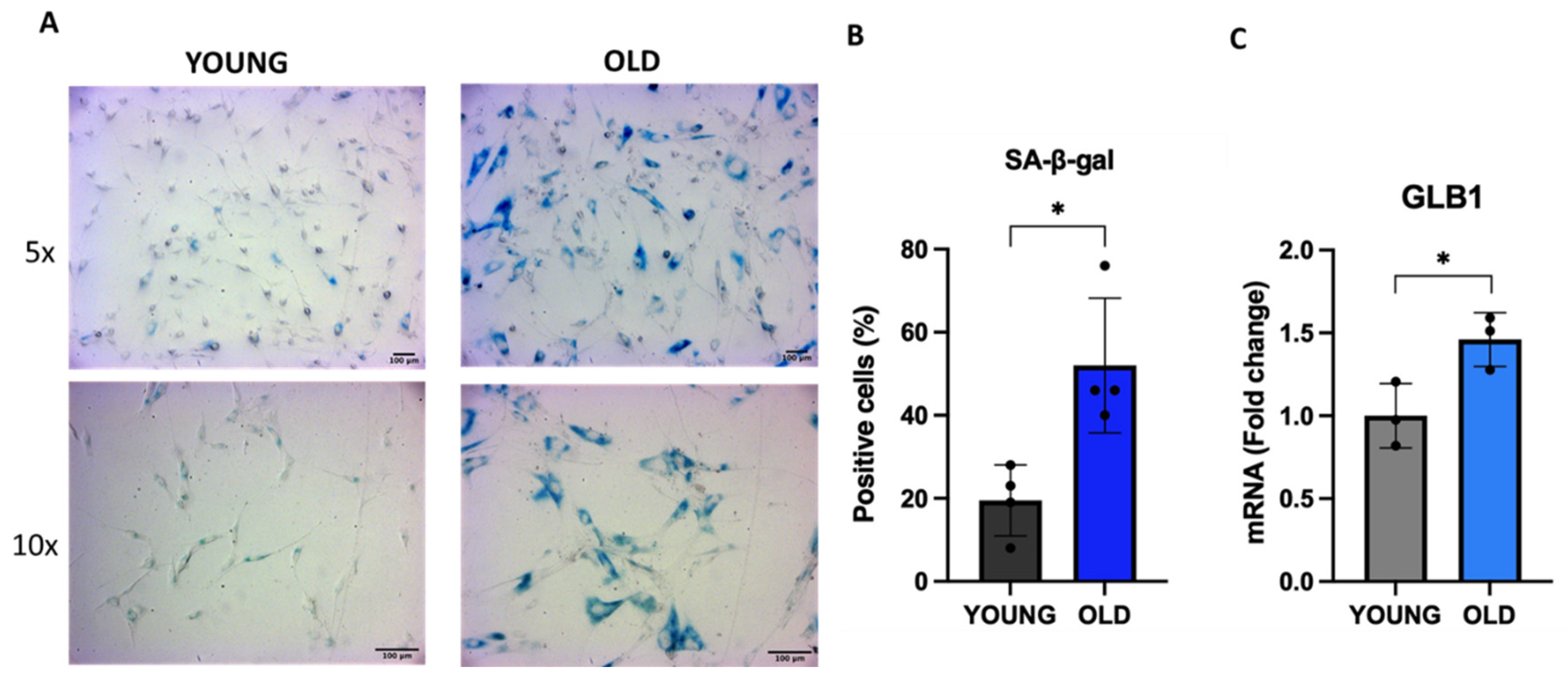
Figure 2.
Morphological and nuclear changes in old VSMCs compared to young cells. (A) Representative pictures of cell morphology, and (B) F-Actin staining. (C) The graphs show the mean ± SD of cell and nuclear area. Unpaired t test with Welch’s correction. ***p<0.001 vs YOUNG. (D) Nuclear morphometric analysis of young and old VSMCs. 2way ANOVA followed by Šidák’s multiple comparisons test; *** p < 0.001 vs YOUNG.
Figure 2.
Morphological and nuclear changes in old VSMCs compared to young cells. (A) Representative pictures of cell morphology, and (B) F-Actin staining. (C) The graphs show the mean ± SD of cell and nuclear area. Unpaired t test with Welch’s correction. ***p<0.001 vs YOUNG. (D) Nuclear morphometric analysis of young and old VSMCs. 2way ANOVA followed by Šidák’s multiple comparisons test; *** p < 0.001 vs YOUNG.
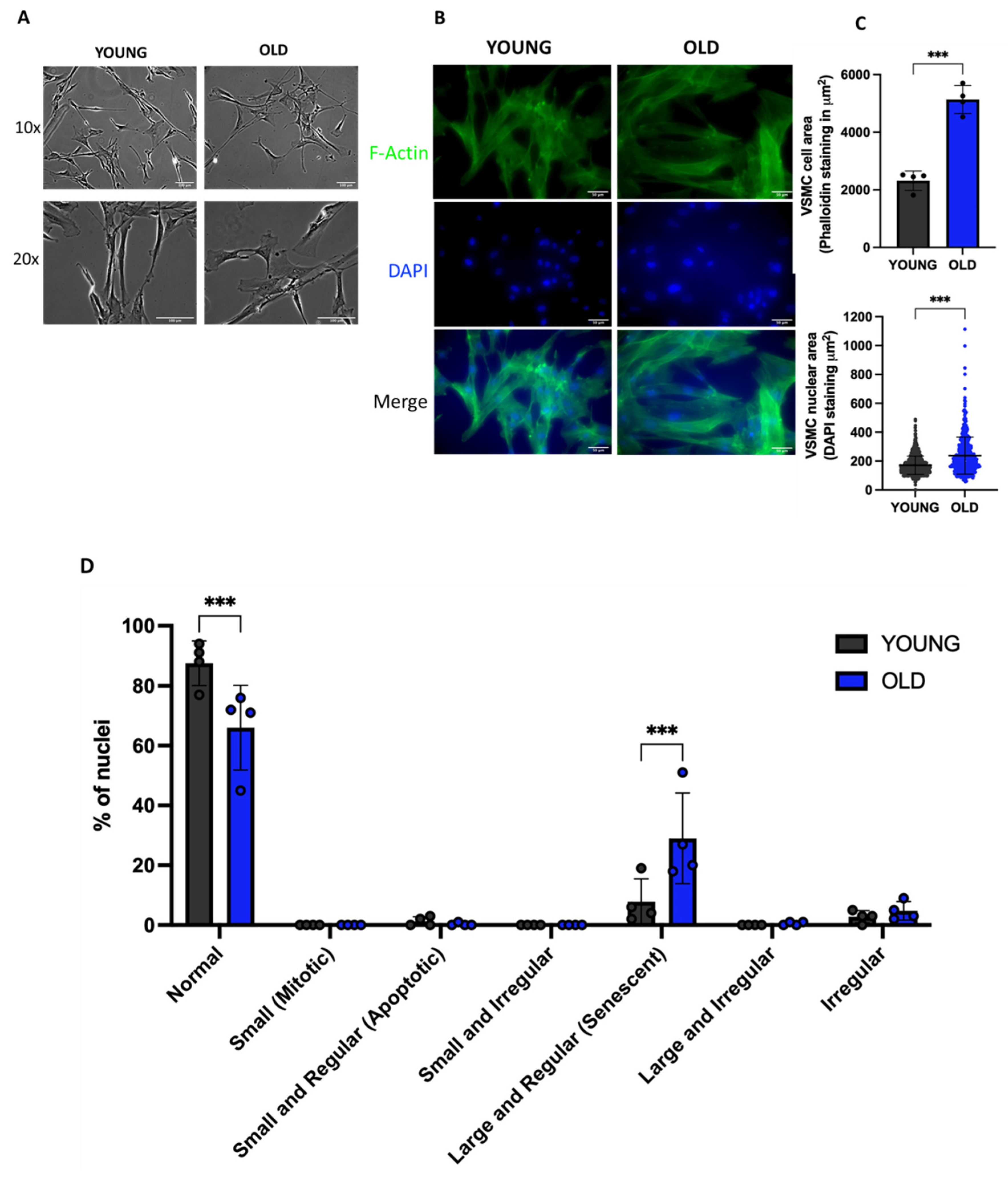
Figure 3.
Proliferative state of young and old VSMCs. (A) Expression levels of the proliferative marker PCNA have been measured by q-RT-PCR and western blot. Unpaired t test with Welch’s correction. Cell proliferation (B) and cell cycle (C) have been evaluated. 2way ANOVA. Cell migration (D) has been assessed by Boyden chamber. Unpaired t test with Welch’s correction. * p<0.05; ** p<0.01; *** p < 0.001 vs YOUNG.
Figure 3.
Proliferative state of young and old VSMCs. (A) Expression levels of the proliferative marker PCNA have been measured by q-RT-PCR and western blot. Unpaired t test with Welch’s correction. Cell proliferation (B) and cell cycle (C) have been evaluated. 2way ANOVA. Cell migration (D) has been assessed by Boyden chamber. Unpaired t test with Welch’s correction. * p<0.05; ** p<0.01; *** p < 0.001 vs YOUNG.
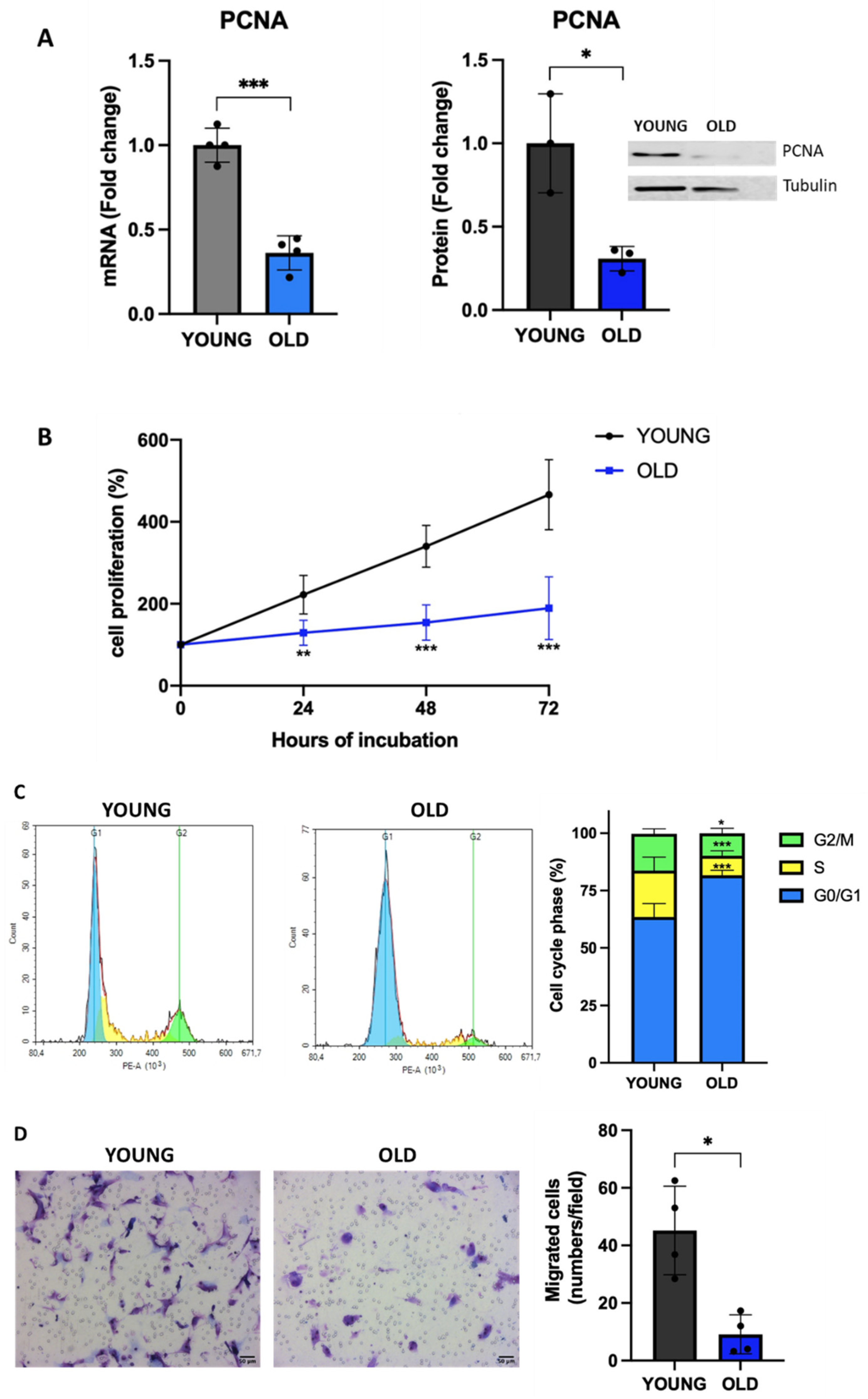
Figure 4.
Expression of known senescence-associated biomarkers. The expression levels of LMNB1 (A), HMGB1 (B), and the cell cycle inhibitors p16, p21 and p53 (C, D) have been analysed in young and old VSMCs. Unpaired t test with Welch’s correction: * p<0.05; ** p<0.01; *** p < 0.001 vs YOUNG.
Figure 4.
Expression of known senescence-associated biomarkers. The expression levels of LMNB1 (A), HMGB1 (B), and the cell cycle inhibitors p16, p21 and p53 (C, D) have been analysed in young and old VSMCs. Unpaired t test with Welch’s correction: * p<0.05; ** p<0.01; *** p < 0.001 vs YOUNG.
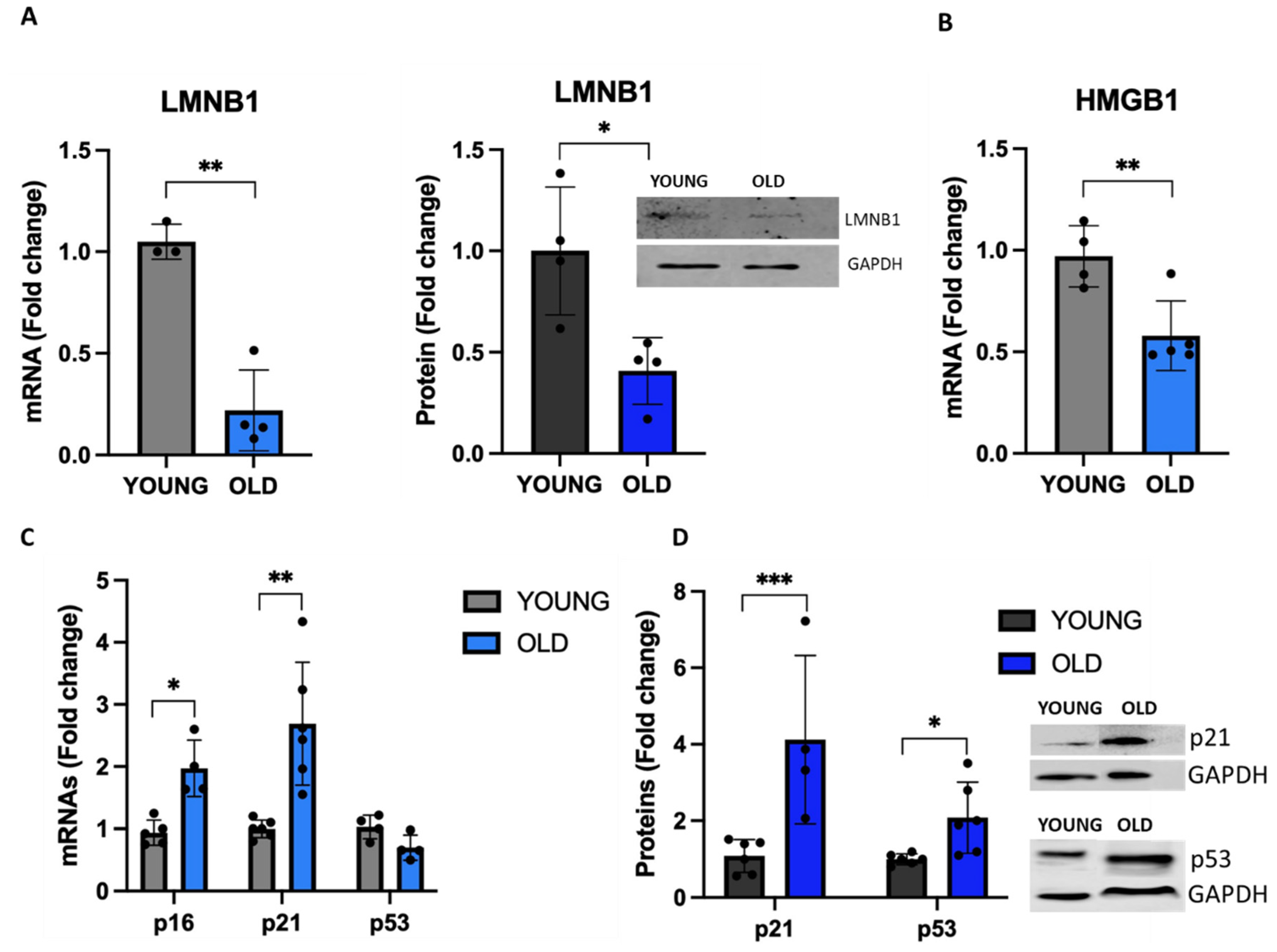
Figure 5.
Expression of the new senescence-associated biomarker RRAD. The expression of RRAD was analysed by qRT-PCR and western blot. Unpaired t test with Welch’s correction: * p<0.05; ** p < 0.01 vs YOUNG.
Figure 5.
Expression of the new senescence-associated biomarker RRAD. The expression of RRAD was analysed by qRT-PCR and western blot. Unpaired t test with Welch’s correction: * p<0.05; ** p < 0.01 vs YOUNG.
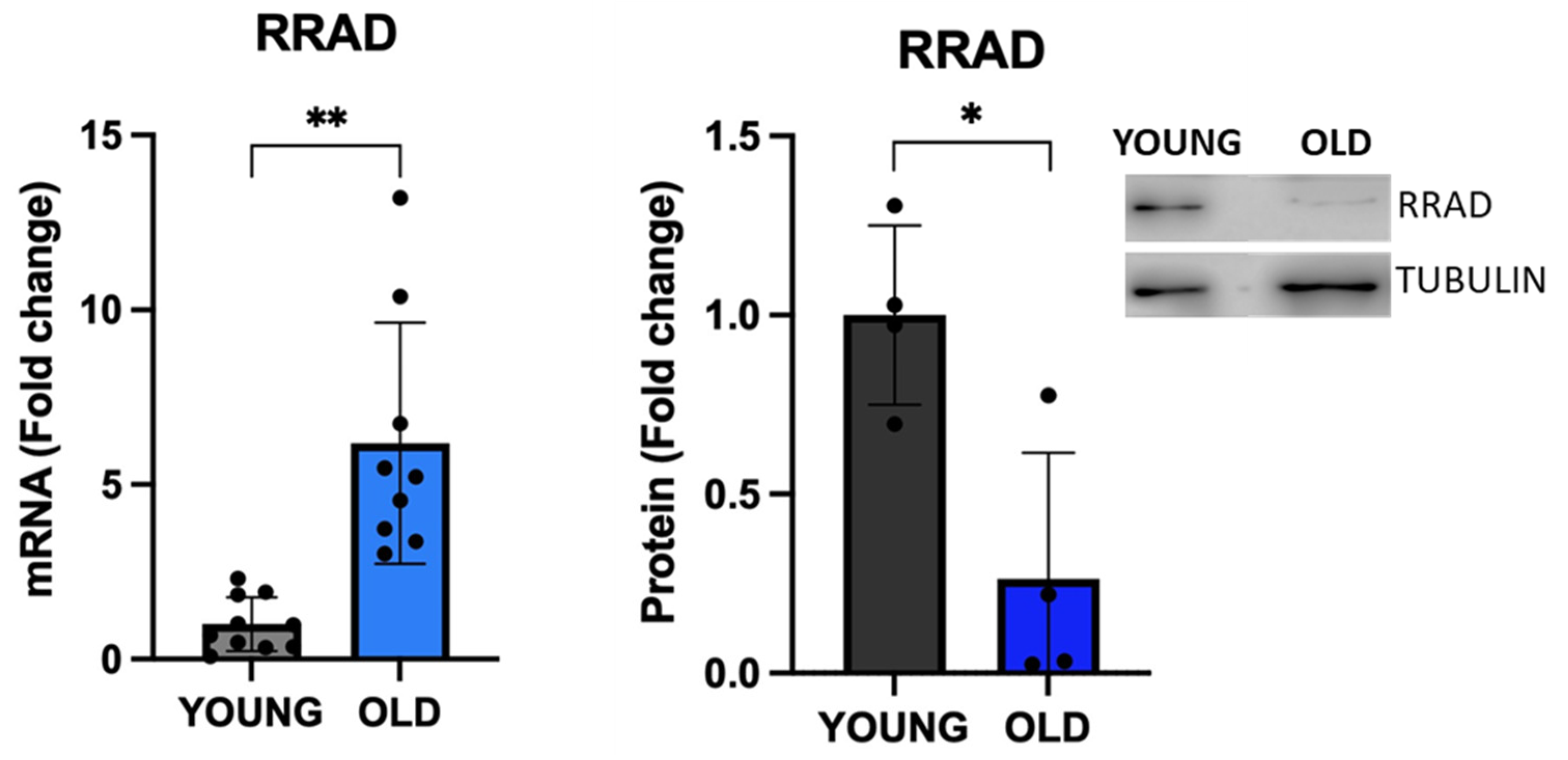
Figure 6.
Expression of SASP markers in young and old VSMCs. Expression of inflammatory markers analysed at both mRNA (A) and protein (B) levels. Unpaired t test with Welch’s correction: * p<0.05; ** p<0.01; *** p < 0.001 vs YOUNG.
Figure 6.
Expression of SASP markers in young and old VSMCs. Expression of inflammatory markers analysed at both mRNA (A) and protein (B) levels. Unpaired t test with Welch’s correction: * p<0.05; ** p<0.01; *** p < 0.001 vs YOUNG.
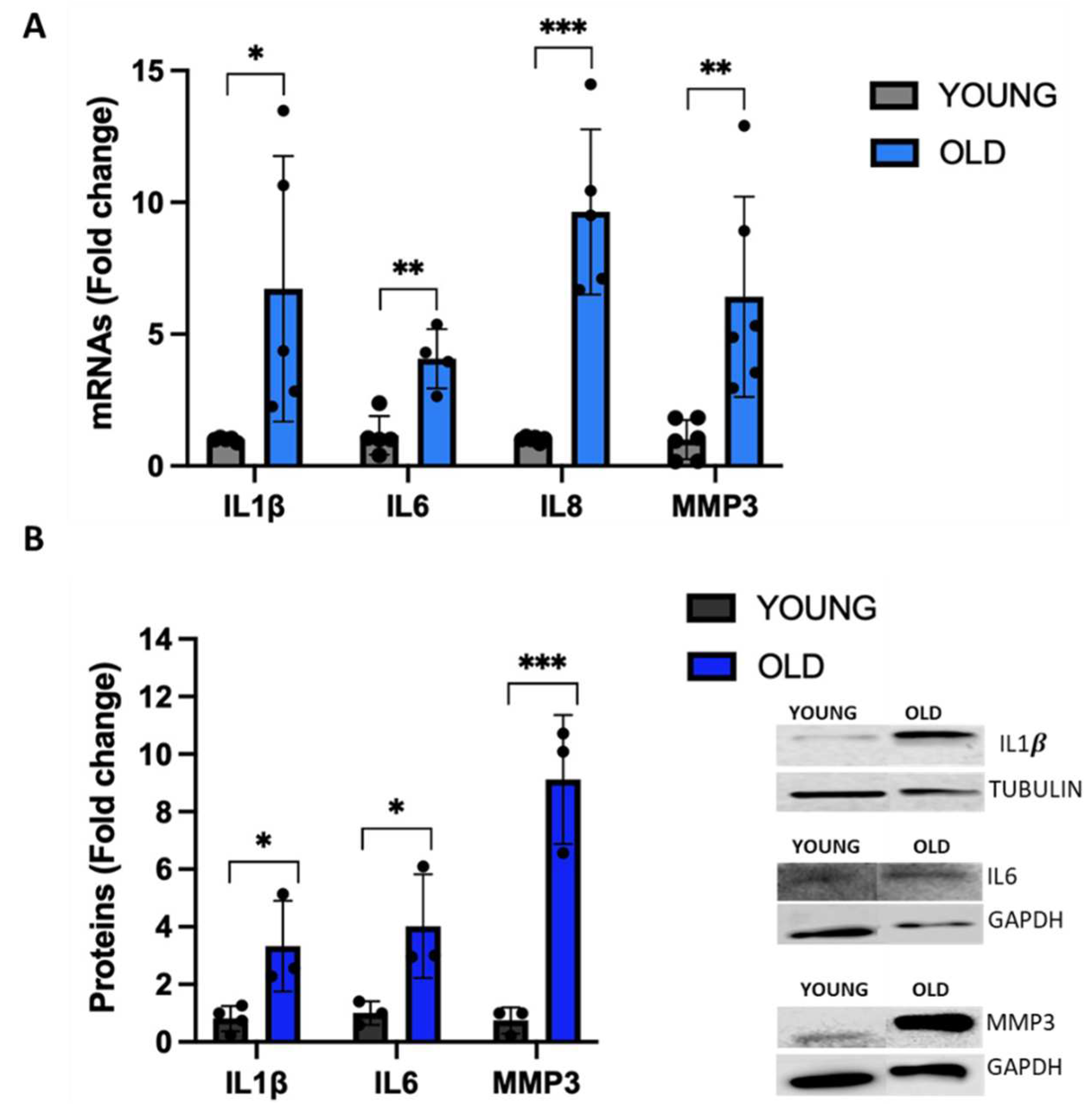
Figure 7.
Heatmap of gene expression levels of selected 12 lncRNAs. The fold change values of young cells and old cells are indicated in each dot of the heatmap (6 replicates for condition). Fold changes were calculated using 2-(ΔΔCt) and comparing senescent versus young cells. Blue and red color grade dots indicate levels of down-regulation and upregulation. Unpaired t-test with Welch’s correction: * p<0.05; vs YOUNG. The heatmap was created with the Morpheus online tool [https://software.broadinstitute.org/morpheus/].
Figure 7.
Heatmap of gene expression levels of selected 12 lncRNAs. The fold change values of young cells and old cells are indicated in each dot of the heatmap (6 replicates for condition). Fold changes were calculated using 2-(ΔΔCt) and comparing senescent versus young cells. Blue and red color grade dots indicate levels of down-regulation and upregulation. Unpaired t-test with Welch’s correction: * p<0.05; vs YOUNG. The heatmap was created with the Morpheus online tool [https://software.broadinstitute.org/morpheus/].
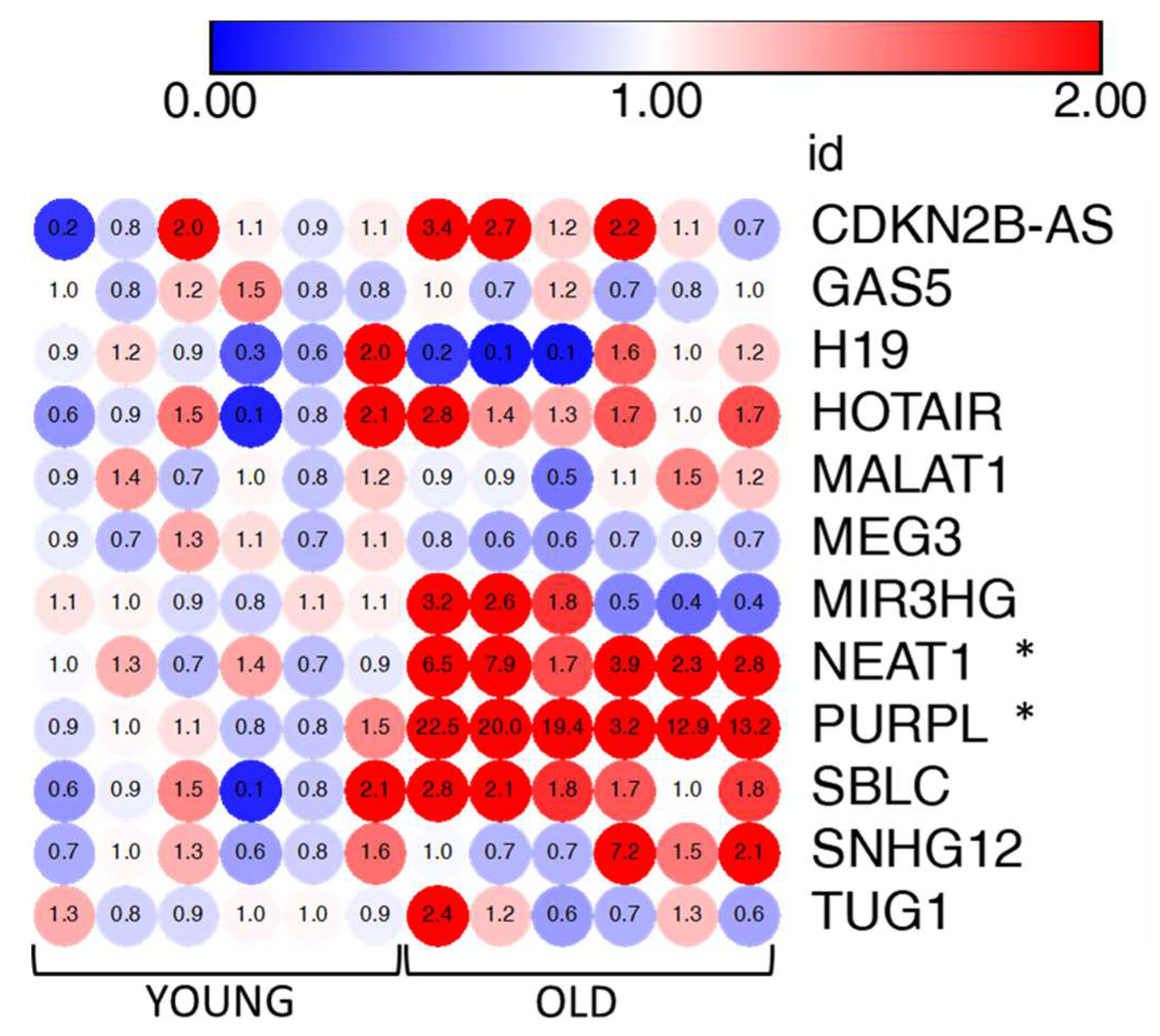

Table 1.
List of lncRNAs selected for this study and their published role in senescence.
| lncRNA symbol | lncRNA name | Expression in cellular senescence | Function/Mechanism of action | References |
|---|---|---|---|---|
| CDKN2B-AS1 | CDKN2B antisense RNA 1 | Up/Down | Suppresses the expression of CDKN2A/p16 and CDKN2B/p15 by recruiting the repressive Polycomb complexes | [18] |
| H19 | H19 imprinted maternally expressed transcript | Up/Down | Inhibits p53, CDKN1C, IL6/STAT3/p21 pathway (anti-senescence function). Derepresses β-catenin (pro-senescence function). | [18] |
| HOTAIR | HOX transcript antisense RNA | Up | Activates NF-κB/IL6 and p53/p21 pathways through DNA damage response. | [18] |
| GAS5 | growth arrest specific 5 | - | Sponges miR-223 which inhibits the anti-senescence NAMPT enzyme. | [20] |
| MALAT1 | metastasis associated lung adenocarcinoma transcript 1 | Down | Controls cell cycle progression by regulating p53 activity and the expression of B-MYB transcription factor. | [24] |
| MEG3 | maternally expressed 3 | Up | Enhances p53 transcription and reduces p53 degradation. Promotes p53 binding to target promoters. | [18,25] |
| MIR31HG | MIR31 host gene | Up | Activates the expression and secretion of SASP components. | [26] |
| NEAT1 | nuclear paraspeckle assembly transcript 1 | - | Facilitates the expression of IL8. | [27] |
| PURPL | p53 upregulated regulator of p53 levels | Up | Negatively controls p53 levels by interfering with p53-MYBBP1A complex. | [28,29] |
| SBLC | Senebloc | Down | Promotes p53 degradation. Mediates epigenetic silencing of p21 through regulation HDAC5. | [30] |
| SNHG12 | small nucleolar RNA host gene 12 | - | Inhibits p16, p21 and γH2AX expression. Regulates DNA damage response via interaction with DNA-PK kinase. | [31] |
| TUG1 | taurine up regulated 1 | Up | Growth arrestor induced by p53 upon DNA damage. Inhibits the pro-proliferation HOXB7 transcription factor | [27] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



