Submitted:
24 July 2023
Posted:
25 July 2023
You are already at the latest version
Abstract
Proteins in biological systems function at the interface of single molecule and bulk chemistry and thus provide novel insights into the basic physical chemistry of scaling and emergent phenomena. For example, a binary mechanical model based on the chemistry of muscle contraction unifies molecular mechanics and thermodynamics and provides an explicit solution to the Gibbs paradox. Using the same model system, here I show that chemical activities of molecular states have no effect on chemical kinetics or energetics. Specifically, while the concentration or number of molecules in a molecular state is widely thought to contribute to reaction free energies, here I show that it is not the physical presence of molecules that pushes a reaction forward but the number of microstates, , accessible in a chemical state that pulls a reaction toward equilibrium with an entropic force down an entropic funnel. With the derivation of an entropic contribution to chemical kinetics, I develop a novel chemical kinetic formulation that fully describes the chemical thermodynamics of both equilibrium and non-equilibrium reactions in terms of an a priori system reaction energy landscape.
Keywords:
Entropy
; Chemical Thermodynamics
; Free Energy
; Reaction Energy Landscape
; Microstates
; Micropathways
; Non-Equilibrium
; Kinetics
; Entropic Funnel
Introduction
Technological advances over the past 70 years have fueled a molecular revolution in the biological sciences, providing remarkably detailed descriptions of the structure and function of a myriad of biomolecules. The question remains, however, to what extent is biological function the sum of these molecular parts versus an emergent property of systems of molecules? While this is fundamentally a physical chemical question, insights from small molecular ensembles in biological systems are informing new perspectives on this important chemical thermodynamic problem. For example, a model system based on the chemistry of muscle contraction offers a new perspective on entropic forces in enzyme-catalyzed reactions [1,2]. Using this model system, the mechanisms required to contain entropic forces have been clearly characterized, providing an explicit solution to the Gibbs paradox [3]. Here I show this entropic force, not some physical force exerted by molecules, balances chemical reactions.
Specifically, emergent properties of a system are broadly described by chemical activities, and the chemical activity, ai, of a molecular state, i, in a reaction is typically described by the number, Ni, or concentration of molecules, [i], in that molecular state. However, while N molecules have chemical activity, here I show that this chemical activity does not in general change with a change in the molecular state of those N molecules. What is referred to as a concentration of a molecular state in fact describes the number of microstates accessible to a system within a given chemical state. It follows that the “concentration” of a molecular state describes an entropic reaction free energy, not a chemical potential, and the “concentration”-dependence of reaction rates are physically determined by the number of micropathways between microstates, not by the activity or active mass of a molecular state.
Reaction Free Energy
Chemical systems have properties that are described both by the state of the system (T, P, V) and by the state of molecules within the system (a system containing yellow molecules appears yellow). In a given system state, the standard free energy, ΔG°, for a reaction is a molecular property. For example, under standard conditions, ΔG° for the hydrolysis of ATP to ADP and Pi can be physically defined for one molecule as the change in free energy associated with breaking a single phosphoanhydride bond. The reaction free energy, ΔrG, on the other hand, is a system property because it includes free energy changes that do not exist within individual molecules or molecular bonds.
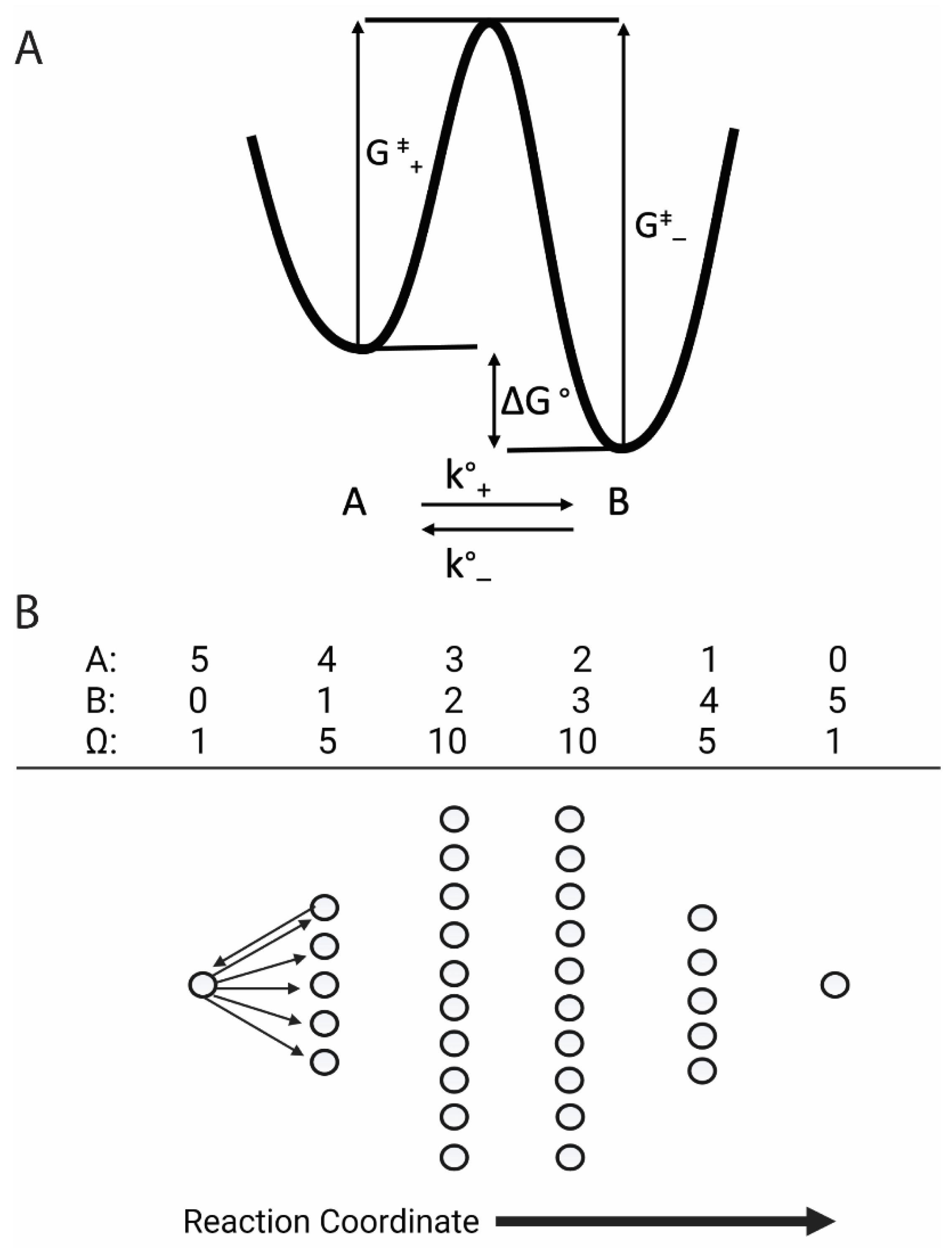
Figure 1A describes the physical chemistry of a single molecule in a solution held at constant T,P, and V that reversibly switches between molecular states A and B with forward and reverse rate constants, k°+ and k°–, and a free energy difference between A and B of ΔG° = ΔH° – TΔS°, where ΔH° and ΔS° are the molecular enthalpic and entropic contributions to ΔG° [4]. The free energy landscape of the molecule (Figure 1A) describes changes in free energy along the reaction (or conformational) coordinate of that molecule [5]. The inverse times to diffuse in both forward, k°+, and reverse, k°–, directions over an activation energy barrier separating A and B (Figure 1A) are related to ΔG° as k°+/k°- = exp(–ΔG°/kBT) [6]. For an isolated single molecule, the time-averaged occupancy of molecular states A and B is then
NB/NA = exp(-ΔGo/kT).
For an ensemble of N such molecules contained in a solution held at constant T, P, and V, the reaction occurs along a system reaction coordinate that describes changes in free energy of the system, ΔrG, with changes in the chemical (not molecular) state of the system [7,8]. In other words, in addition to ΔGo, ΔrG accounts for non-ΔGo changes in the free energy of the system with a chemical step from{NA,NB} to {NA–1,NB+1}. These non-ΔGo free energy changes include PΔV work performed with changes in solution volume, ΔV; non-PΔV work (F·d-work) performed with molecular displacements, d, against system forces, F [9]; and changes in system entropy, ΔSsys [7]. In general [7],
The terms on the right hand side of Equation (2) are the chemical forces that (when ΔrG < 0) energetically drive a reaction from {NA,NB} to {NA–1,NB+1}, or that (when ΔrG > 0) energetically drive a reaction from {NA–1,NB+1} to {NA,NB}, or that (when ΔrG = 0) balance the reaction.
ΔrG = ΔG° + PΔV + F·d – TΔSsys.
In 1864, Waage and Guldberg [10] proposed that the forces that balance chemical reactions are Newtonian, arguing that an active mass, NA, of molecules in state A pushes against an equal and opposite reactive mass, NB, of molecules in state B, such that NA·k°+ = NB·k°–. However, no term in Equation (2) is consistent with the concept of an active mass. It was subsequently shown that a change in the number of molecules, N, (the chemical activity) in a system affects the volume of a system, which through Equation (1) (PΔV) accounts for the partial pressure of a gas. In the early 1900’s, this approach was modified in an attempt to account for the forces that balance chemical reactions. Specifically, it was proposed that the chemical activity of N molecules fundamentally changes when the molecular state of those N molecules change. However, the state of a molecule is a property of that molecule and is in general fully defined by the molecular reaction energy landscape (Figure 1A) independent of system chemical activities. This is evident in the nonsensical description below that results from defining a molecular property as a chemical activity.
In most chemistry textbooks today, a term kBT·ln(NB/NA) [or the molar equivalent RT·ln([B]/[A])] is derived from the envisaged chemical activities of molecular states A and B, and Equation (1) takes the form
which at equilibrium (ΔrG = 0 and forces balance the chemical reaction) becomes
ΔrG = ΔG° + kBT·ln(NB/NA),
(NB/NA)eq = exp(–ΔGo/kBT).
Equation (3) is equivalent to Equation (1), implying the following circular argument: if chemical activities assigned to molecular states, NA and NB, are defined by ΔG° (Equation (3)), then NA and NB are molecular (ΔG°) properties (Equation (1)), not non-ΔG° chemical activities. Chemical activities describe interactions between an individual molecule and the system of N molecules within which it is contained, which means chemical activities contribute to a system reaction energy landscape with reaction coordinates {NA,NB} not a molecular energy landscape with reaction coordinates A and B (Figure 1A). Equation (2) nonsensically describes N molecules diffusing within the reaction free energy landscape of a single molecule. Perhaps the intent is that each of N molecular energy landscapes has one molecule diffusing within it, in which case there is no chemical activity – the whole is the sum of its molecular ΔG° parts with no emergent non-ΔG° system behaviors – and if there is no chemical activity, the question remains what are the forces that balance chemical reactions?
Here, I develop a formulation that describes chemical entropy, TΔSsys, as the force that balances chemical reactions. I show that while active masses and chemical activities purportedly push reactions forward, chemical entropy kinetically and energetically pulls chemical reactions down an entropic funnel.
System Entropy
For a system of N molecules (Figure 1A) contained in a solution held at constant T,P, and V, Equation (1) is
ΔrG = ΔG° – TΔSsys.
According to Boltzmann, Ssys = kB·ln(ΩNA,NB), where ΩNA,NB is the number of microstates available to the system in chemical state {NA,NB}. Specifically, ΩNA,NB = N!/(NA!NB!). The change in entropy with a single chemical step from {NA,NB} to {NA–1,NB+1} is then
Equation (4) resembles Equation (3), only here NB and NA are reaction coordinates {NA,NB} in a system energy landscape that are defined independent from the physical occupancy of states A and B.
ΔSsys = –kB·ln[((NA – 1)!(NB + 1)!)/(NA!NB!)] = –kB·ln[(NB + 1))/(NA)], and so
ΔrG = ΔG° + kBT·ln[(NB + 1))/(NA)].
The change in system entropy associated with δN chemical steps from.
Here again the gradient established by δN is not a physical gradient that energetically drives the reaction. The reaction is driven by the average change in entropy per chemical step, kBT·ln[(NB + δN/2)/(NA – δN/2)], summed over δN chemical steps, where the average change in entropy is defined by the system reaction coordinate {NA,NB} independent from the physical occupancy of states A and B.
{NA,NB} to {NA– δN,NB+δN} is
ΔSsys = –kB·ln[((NA – δN)!(NB + δN)!)/(NA!NB!)] ≈ –δN·kB·ln[(NB + δN/2))/(NA – δN/2)], and so
ΔrG = δN·ΔG° + δN·kBT·ln[(NB + δN/2)/(NA – δN/2)].
A change in ΔSsys relative to Ssys = 0 occurs from {N/2,N/2} to {N/2–δN, N/2+δN}, or
where δN/N is a fractional change in the extent of the reaction relative to unity. Assuming δN = 1 mol, Equation (6) can be written
ΔSsys = –δN·kB·ln[(N/2 + δN/2))/(NA – δN/2), or
ΔSsys = –δN·kB·ln[(1 + δN/N))/(1 – δN/N),
ΔSsys = –R·ln([B]/[A]), and
ΔrG = ΔG° + RT·ln([B]/[A])
While this demonstrates consistency with the results of a chemical activity analysis, it does not demonstrate that physical concentration gradients drive chemical reactions. Again, here [B]/[A] is not a physical concentration gradient. At equilibrium [B]/[A] equals exp(–ΔG°/RT) not because [A] + [B] molecules physically equilibrate within the energy landscape of a single molecule, but because ΔG° a priori determines the approximate point along the system reaction coordinate, {NA,NB}, at which the system equilibrates. This is illustrated in Figure 1B and Figure 2 for small numbers of molecules.
System Reaction Coordinate
Figure 1B illustrates entropic changes along the reaction coordinate for a system of N = 5 molecules each occupying either molecular state A or B. When the system is in state {5,0}, Ω5,0 = 1. Unlike in chemical activity models where kB·ln(0/5) is undefined, here the system entropy, kB·ln(1), is zero. After one net forward step, the system enters state {4,1}, increasing the number of microstates from 1 to 5 and increasing the system entropy from 0 to kB·ln(5). This increase in the number of microstates physically pulls the reaction forward since there are five-times more micropathways in the forward direction than in the reverse direction. This contrasts with the chemical forces of molecules that purported push a reaction forward. When ΔGo = 0, the reaction continues until a maximum system entropy is reached along the reaction coordinate, equilibrating in system state {3,2} when (NB + 1)/NA = exp(0) (Equation (3)).
Figure 2A is a plot of Equation (3), illustrating both the change in entropic energy, kBT·ln[NB+1,NA], and the change in molecular free energy, ΔGo, along the system reaction coordinate {NA,NB} for a system containing N = 10 molecules. Figure 2A illustrates that the change in system entropy increases logarithmically with each step whereas the change in molecular free energy, ΔGo, is the same for each step (the effect of ΔGo on ΔrG is a constant offset). When ΔGo decreases from 0 to –1.5 kBT (Figure 2A, down arrow) the equilibrium state of the system changes from {5.5,4.5} to {2,8} (Figure 2, right arrow) corresponding to a change from (NB + 1)/NA = exp(0) to (NB + 1)/NA = exp(1.5).
Figure 2.
Change in Free energy along a reaction coordinate. (A) For N = 10, Equation (3) is plotted for a ΔG° of 0 (green line) and –1.5 kBT (blue line). For ΔG° = 0, the reaction equilibrates (ΔrG = 0) at {5.5,4.5}. When ΔG° is decreased from 0 to –1.5 kBT (down arrow) the system re-equilibrates (right arrow) at {2,8}. (B) The system free energy, G, (the integral of ΔrG over the reaction coordinate) is calculated and plotted for ΔG° of 0 (green line) and –1.5 kBT (blue line) values in panel A. The same trajectory in panel A describing re-equilibration following a decrease in ΔG° is illustrated in panel B (arrows).
Figure 2.
Change in Free energy along a reaction coordinate. (A) For N = 10, Equation (3) is plotted for a ΔG° of 0 (green line) and –1.5 kBT (blue line). For ΔG° = 0, the reaction equilibrates (ΔrG = 0) at {5.5,4.5}. When ΔG° is decreased from 0 to –1.5 kBT (down arrow) the system re-equilibrates (right arrow) at {2,8}. (B) The system free energy, G, (the integral of ΔrG over the reaction coordinate) is calculated and plotted for ΔG° of 0 (green line) and –1.5 kBT (blue line) values in panel A. The same trajectory in panel A describing re-equilibration following a decrease in ΔG° is illustrated in panel B (arrows).
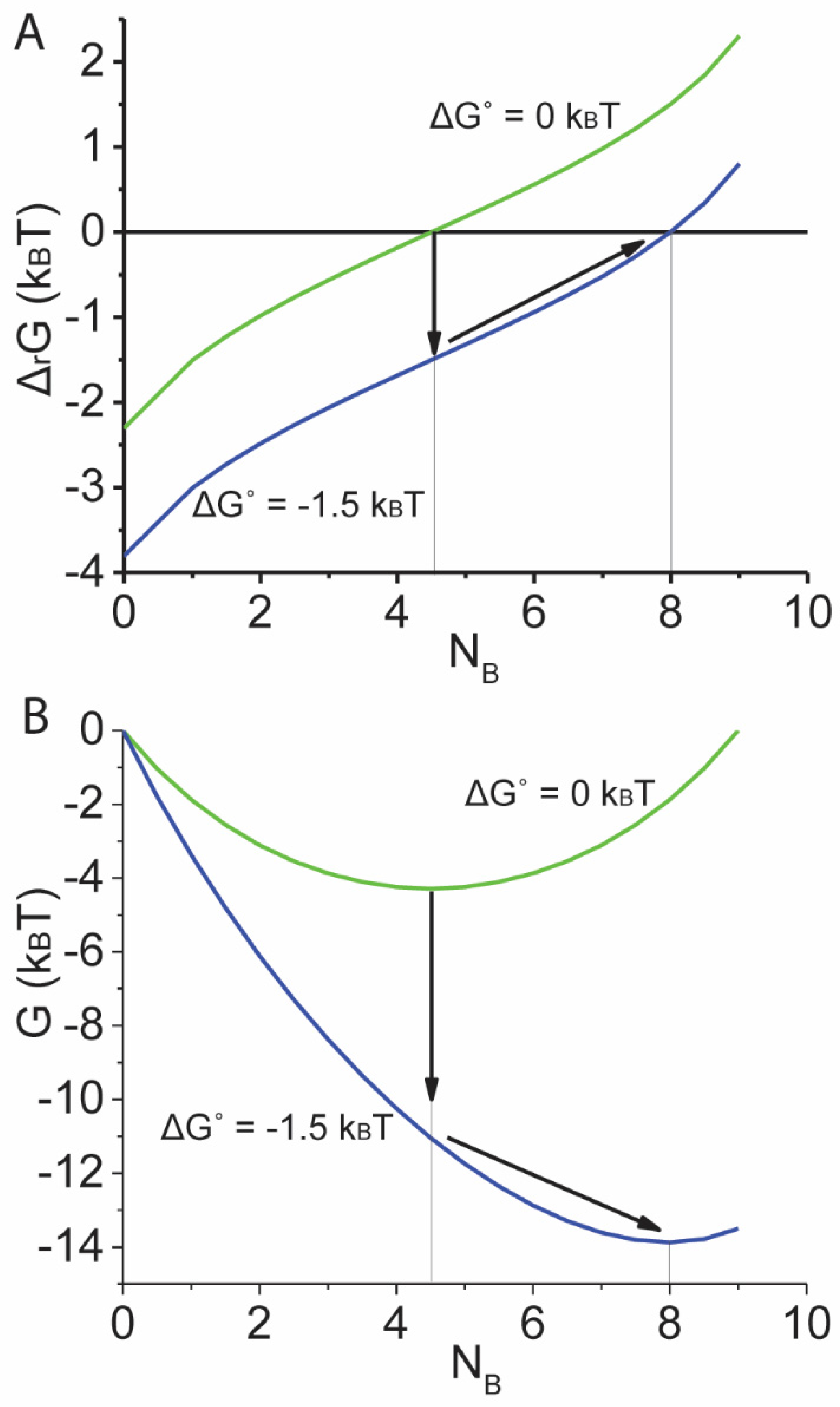
Because ΔrG is the change in free energy, G, of the system with a chemical step, the integral of Equation (3) over the reaction coordinate is the free energy, G, of the system. The two graphs in Figure 2A are integrated and replotted in Figure 2B as free energy, G. These are the system free energy landscapes described by Gibbs within which the reaction equilibrates at a point along the reaction coordinate{NA,NB} where G is a minimum. According to Gibbs, the walls of the energy landscape represent the energy that balances a reaction at equilibrium, and here the walls of the energy landscape are TSsys (the integral of TΔSsys). In other words, entropic forces, TΔSsys, (Figure 2A) drive a reaction toward equilibrium down an entropic funnel, TSsys (Figure 2B).
Figure 2B illustrates how changes in ΔG° affect the energy landscape. Changes in non-ΔG° chemical activities have similar effects [2]. However, changes in NA and NB have no effect on the energy landscape because they are reaction coordinates, not chemical activities, and changes in reaction coordinates describe movement along the landscape, not changes to the landscape. Because reaction kinetics and energetics are defined by reaction energy landscapes, NA and NB have no effect on the kinetics or energetics of a reaction.
Equilibrium Energetics
At equilibrium (ΔrG = 0), the probability of finding the system in state {NA+1,NB–1} relative to {NA,NB} is exp[–ΔG°/kBT + ln(ΩNA-1,NB+1/ΩNA,NB)], where ln(ΩNA-1,NB+1/ΩNA,NB) = TΔSsys/kBT. Thus, when ΔG° = 0, ΩNA,NB is the probability density function, PNA,NB. For comparison, according to chemical activity models the probability of finding a molecule in state B relative to state A is exp[–ΔG°/kBT], and when ΔG° = 0, PNA,NB is a binomial distribution. The equilibrium constant, K = (ΩNA,NB/ΩNA-1,NB+1)eq = exp[–ΔG°/kBT] is K = (NB(eq) + 1)/NA(eq), and describes the reaction coordinate {NA,NB}eq at equilibrium (Figure 2). For comparison, in chemical activity models the equilibrium constant K = NB(eq)/NA(eq) describes the physical distribution of N molecules within the energy landscape of a single molecule (Figure 1A).
Non-Equilibrium Energetics
When the system is perturbed from equilibrium by a change in the internal energy of the system, δE, [i.e., a change in ΔG° or kBT·ln[(ΩNA-1,NB+1/ΩNA,NB) ], the free energy equation becomes
where δE is a non-equilibrium (ne) perturbation to any of the energy term on the right-hand side of Equation (1). Here,
ΔrG = [ΔG° – kBT·ln(ΩNA-1,NB+1/ΩNA,NB)] eq + δE = δE,
δE = [ΔG° – kBT·ln(ΩNA-1,NB+1/ΩNA,NB)]ne.
As illustrated in Figure 2, if the system perturbation is a change in ΔG°, the system relaxes to a new equilibrium state, {NA,NB}eq, as kBT·ln(ΩNA-1,NB+1/ΩNA,NB) approaches the new ΔG°. Through this process, ΔrG returns to zero and δE remains in the system in the form of a change in system entropy. If the system perturbation is a change in kBT·ln(ΩNA-1,NB+1/ΩNA,NB) [an irreversible transfer of molecules between molecular states] the system relaxes back to the original equilibrium state, {NA,NB}eq, as kBT·ln(ΩNA-1,NB+1/ΩNA,NB)ne approaches ΔG°. Through this process the entropic δE is lost from the system as heat. This analysis becomes more complex when non-PΔV work is performed, in which case upon a chemical relaxation, in addition to exchanges between kBT·ln(ΩNA-1,NB+1/ΩNA,NB) and ΔG°, δE can be exchanged with internal mechanical potentials. And, in addition to being lost from the system as heat or stored in the system as entropy, δE can be lost from the system as work performed on the surroundings [2].
Equilibrium Kinetics
Chemical kinetics, like energetics, have both molecular and system components. The net rate at which the system transitions from chemical state {NA,NB} to chemical state {NA–1,NB+1} is
where k+ and k– are the forward and reverse transition rates between these states. At equilibrium, the probability of finding the system in state {NA+1,NB–1} relative to state {NA,NB} is
Here, k°+ and k°– are molecular contributions to k+ and k– and exp[–TΔSsys/kBT] is the system contribution to k+ and k–. The influence of –ΔSsys/kB on kinetics can be understood energetically as a tilt of the system landscape that adds to the tilt of the molecular landscape, ΔG°. It can also be understood kinetically as
where the factor ΩNA-1,NB+1/ΩNA,NB describes the number of micropathways available for the forward reaction relative to the reverse reaction. For example, in Figure 1B there are five micropathways from {5,0} to {4,1} and one micropathway back from {4,1} to {5,0}, which means that the forward reaction is entropically five-fold more likely than the reverse reaction (ΩNA-1,NB+1/ΩNA,NB = 5). This implies forward and reverse rate constants of
To demonstrate consistency with the chemical activity approach,
where here again NA(eq) and NB(eq) are not physical, active numbers of molecules, they are the system reaction coordinates {NA,NB}eq at equilibrium.
d{NA,NB}/dt = k+ – k–,
k+(eq)/k–(eq) = exp[(–ΔG° + kBT·ln[ΩNA-1,NB+1/ΩNA,NB]eq)/kBT] or
k+(eq)/k–(eq) = (k°+/k°–)·exp[kBT·ln(ΩNA-1,NB+1/ΩNA,NB)eq/kBT]
k+(eq)/k–(eq) = (k°+/k°–)·(ΩNA-1,NB+1/ΩNA,NB)eq
k+(eq) = k°+·ΩNA-1,NB+1(eq)
k–(eq) = k°–·ΩNA,NB(eq).
At equilibrium, k+(eq) = k–(eq), or
k°+·ΩNA-1,NB+1(eq) = k°–·ΩNA,NB(eq).
k+(eq) = k°+·NA(eq)
k–(eq) = k°–·(NB(eq) + 1)
Non-Equilibrium Kinetics
Chemical kinetics is fully determined from the molecular and system tilt of the system energy landscape. It follows that non-equilibrium kinetics is fully determined by the non-equilibrium tilt, δE, of these landscapes, where a non-equilibrium perturbation to ΔG° affects the molecular rates k°+/k°–, and an irreversible transfer of molecules between molecular states affects ΩNA,NB/ΩNA-1,NB+1. In general,
k+(t)/k–(t) = (k°+/k°–)(ΩNA-1,NB+1(t)/ΩNA,NB(t)) = exp[–δE(t)/kBT].
According to Equation (7), for a negative δE(t), k+(t) > k–(t) which is consistent with the energetic requirement that a negative δE(t) drives the reaction forward. This forward reaction increases TΔSsys (Figure 2) until δE(t) = 0, at which point k+(t) = k–(t). For a positive δE(t), k+(t) > k–(t) which is consistent with the energetic requirement that a positive δE(t) drives the reaction backward. This reaction decreases TΔSsys (Figure 2) until δE(t) = 0, at which point k+(t) = k–(t). Not only do kinetics fully account for thermodynamics, they also describe the constraints placed on mechanisms through which δE is exchanged among ΔG°, TΔSsys, and other energetic components (right side of Equation (2)) in minimizing the free energy of the system.
The non-equilibrium rate equation is the same as the equilibrium rate equation
For comparison with a chemical activity analysis, from above
where one last time, NA(t) and NB(t) describe the time dependence of steps along the system reaction coordinate {NA,NB}, not the time-dependence of the physical numbers of molecules in different states. Changes in NB(t) + 1 are equal and opposite changes in NA(t) along the system reaction coordinate, and so
which means that NA(t) decreases exponentially with time as
with a relaxation rate, k°+ + k°–, where NAo is the starting point on the reaction coordinate {NA,NB}o.
d{NA,NB}/dt = k°+·ΩNA-1,NB+1 – k°–·ΩNA,NB(t).
dNA/dt = k°+·NA(t) – k°–·(NB(t) + 1)
dNA/dt = –NA(t)·[k°– + k°+]
NA(t) = NAo·exp(–(k°+ + k°–)t),
Conclusion
In summary, a molecular energy landscape (Figure 1A) a priori defines ΔG° and rate constants, k°+ and k°–, for an isolated molecule (Figure 1A) independent of the physical existence of that molecule [5]. Similarly, an entropic energy landscape a priori defines system energetics and rates for N molecules (Figure 1B and Figure 2) independent of the existence of those molecules. Together ΔG° and entropy create a system energy landscape within which chemical entropy contains ΔG° and entropic forces balance a reaction against ΔG° by driving a reaction down an entropic funnel. The entropic contribution to chemical kinetics results in a new kinetic formalism that fully accounts for the chemical thermodynamics of a system both under equilibrium and non-equilibrium conditions.
Funding
JEB was funded by a grant from the National Institutes of Health 1R01HL090938.
Data and materials availability
All data are available in the main text.
Acknowledgments
I thank LEB, JWG, Julie, my students, colleagues, and mentors who have over many years inspired and guided this work. This was funded by a grant from the National Institutes of Health 1R01HL090938-01.
Competing interests
Author declares that they have no competing interest.
References
- J.E. Baker and D.D. Thomas, A Thermodynamic Muscle Model and a Chemical Basis for A.V. Hill’s Muscle Equation. J Muscle Res Cell Motil. 2000, 21, 335. [CrossRef] [PubMed]
- J.E. Baker, Thermodynamics and Kinetics of a Binary Mechanical System: Mechanisms of Muscle Contraction. Langmuir 2022, 38, 15905. [CrossRef] [PubMed]
- J.E. Baker. Cells Solved the Gibbs Paradox by Learning How to Contain Entropic Forces. arxiv 2023.
- L. Styer. Biochemistry; W.H. Freeman and Company: New York, NY, USA, 1995. [Google Scholar]
- H. Frauenfelder, S.G. Sligar, P.G. Wolynes, H. Frauenfelder, S.G. Sligar, and P. G. Wolynes. The Energy Landscapes and Motions of Proteins. 1991, 254, 1598.
- J. Howard, Mechanics of Motor Proteins and the Cytoskeleton, 1st ed. (Sinauer Associates, 2001).
- C. Kittel and H. Kroemer, Thermal Physics (1980).
- J. Gibbs, Elementary Principles in Statistical Mechanics Developed with Especial Reference to the Rational Foundation of Thermodynamics (1902).
- J.E. Baker, L.E.W. LaConte, I. Brust-Mascher, and D. D. Thomas, Mechanochemical Coupling in Spin-Labeled, Active, Isometric Muscle. Biophys. J. 1999, 77, 2657. [CrossRef] [PubMed]
- J. Quílez. On the Early Thermodynamic and Kinetic Deductions of the Equilibrium Constant Equation. Found. Chem. 2021, 23, 85. [CrossRef]
Figure 1.
The molecular and system contributions to the reaction free energy. (A) The molecular contribution to the reaction free energy is described by a molecular energy landscape. The free energy difference between the two metastable states, A and B, is ΔG°. The rate, k°+, at which a molecule transitions from molecular state A to B varies exponentially with –G‡+, where G‡+ is the height of the activation barrier in the forward direction. The rate, k°–, at which a molecule transitions from B to A varies exponentially with –G‡–, where G‡– is the height of the activation energy barrier in the reverse direction. (B) The system contribution to the reaction free energy is entropic. For N = 5, there are 6 system states, {NA,NB}, along the reaction coordinate each with ΩNA,NB = N!/(NA!NB!) microstates, illustrated with circles, and entropy kB·ln(ΩNA,NB). There are five micropathways (right arrows) by which a molecule in state {5,0} can transition to state {4,1}, and one micropathway (left arrow) by which a molecule in state {4,1} can transition to state {5,0}.
Figure 1.
The molecular and system contributions to the reaction free energy. (A) The molecular contribution to the reaction free energy is described by a molecular energy landscape. The free energy difference between the two metastable states, A and B, is ΔG°. The rate, k°+, at which a molecule transitions from molecular state A to B varies exponentially with –G‡+, where G‡+ is the height of the activation barrier in the forward direction. The rate, k°–, at which a molecule transitions from B to A varies exponentially with –G‡–, where G‡– is the height of the activation energy barrier in the reverse direction. (B) The system contribution to the reaction free energy is entropic. For N = 5, there are 6 system states, {NA,NB}, along the reaction coordinate each with ΩNA,NB = N!/(NA!NB!) microstates, illustrated with circles, and entropy kB·ln(ΩNA,NB). There are five micropathways (right arrows) by which a molecule in state {5,0} can transition to state {4,1}, and one micropathway (left arrow) by which a molecule in state {4,1} can transition to state {5,0}.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



