Submitted:
21 July 2023
Posted:
25 July 2023
You are already at the latest version
Abstract
Brassica napus is an important vegetable and oil crop worldwide. The research is meaningful for yield and plant architecture in B. napus. In this study, one natural mutant (MT) line with determinate and capitulum-like inflorescence was chosen for further study. The paraffin sectioning detected that the inflorescence apex began to split and developed into one terminal flower, starting from five-leaf stage. Genetic analysis indicated that the segregation patterns of inflorescences in the F2 populations supported a digenic inheritance model, which was further approved by BSA-Seq technique. The BSA-Seq method detected two QTL region on C02 (14.27-18.41 Mb) and C06 (32.98-33.68 Mb) for the genetic control of determinate inflorescences in MT plants. In addition, the expression profile in MT compared with WT was analysized, and a total of 133 candidate genes for regulating the flower development (75 genes, 56.4%), shoot meristem development (29 genes, 21.8%) and inflorescence meristem development (13 genes, 9.8%) were identified. Then one joint analysis combing BSA-Seq and RNA-Seq identified two candidate gene of BncTFL1 and BncAP1 for regulating the MT phenotype. Besides, the potential utilization of the MT plants was also discussed.
Keywords:
Brassica napus
; Determinate and capitulum-like inflorescence
; terminal flower
; BncAP1
; BncTFL1
1. Introduction
Rapeseed (Brassica napus L., AACC, 2n=38) is the world’ third most important source of vegetable oil after palm and soybean [1]. In China, rapeseed accounts for about 85% acreage of oilseed Brassica, and provided the second most important vegetable oil after soybean [2]. The great importance makes it an ideal model species for theory and application research.
The precise development of inflorescences and flowers is crucial for reproductive success in flowering plants. B. napus is a simple structure typical of the Brassicaceae, and develops an indeterminate, racemetype inflorescence comprising individual lateral flowers arising immediately and sequentially from an apical inflorescence meristem (IM) [3,4]. The lateral flowers in inflorescence developed according to a well-defined program of events that gives rise to a stereotypical floral structure comprising a fixed sequence of concentric whorls with fixed numbers of floral organs (four sepals, four petals, six stamens and a central pistil) [4]. While once the flowers arise on the top of IM, naming terminal flower, the growth of inflorescence will be ceased, which is defined as determinate inflorescence. The situation of terminal flower had been found in many species, such as in Arabidopsis thaliana [5], Glycine max [6], Nicotiana tabacum [7], Brassica juncea [8], Sesamum indicum [9], Brassica napus [10].
The characteristics of inflorescences and flowers were involved in one complex genetic network, among which LEAFY (LFY), APETALA1 (AP1) and TERMINAL FLOWER1 (TFL1) played one key role. LFY is one key transcriptional regulator in the network establishing flower initiation in floral meristem (FM), and is activated by genes AGL24, SOC1, MP and ANT/AIL6 [11,12,13,14,15]. For the establishment of floral meristem identity, AP1 functions a key role and is activated by LFY, CAL and LMI2 [16]. The TFL1 gene had been drawn a widely interests for its important role in shifting indeterminate inflorescence to determinate inflorescence due to the appearance of terminal flower in the top of IM. The terminal flower mutant was firstly isolated from the recessive mutations by screening a M2 population derived from EMS mutagenized seeds of Arabidopsis thaliana ecotype Columbia [5]. Then the candidate recessive gene of tfl1 for regulating the terminal flower phenotype in Arabidopsis thaliana was mapped on the top arm of chromosome 5 [17] and cloned [18]. After that, tfl1′ homolog in Nicotiana tabacum [7] and Glycine max [6] were cloned. In Sesamum indicum, the Sidt1 gene homologous to Arabidopsis TFL1 was mapped on LG09 in a genome region of 41 kb by one ultra-dense SNP genetic map [19]. The Sdt1 homologous to Arabidopsis TFL1 gene associated with determinate feature in Brassica juncea was mapped to the linkage group B05, which was flanked by SSR markers SJ6842 and Ni4-A10 at distances of 15.9 cM and 14.0 cM, respectively [8]. One recent paper in B. napus published the discovery of one microspore culture-origin determinate mutation with terminal flower [20]. The regulator Bnsdt1, homologous to Arabidopsis TFL1, was fine mapped on one region of approximately 220 kb, between 16,627 and 16,847 kb on A10 using BC1 and BC3 populations [20]. The cooperative function of these genes could result in differential inflorescence architectures. “A unifying inflorescence model” postulated that TFL1 could increase vegetativeness (veg) and LFY could reduce veg in meristems, leading to different architectures in Arabidopsis thaliana [3]. In another model, increased TFL1 expression could lead to larger inflorescences with more and longer branches, whereas increased AP1 expression could lead to smaller inflorescences with fewer branches and flowers [21].
Except for the traditional techniques for genetic research, such as QTL mapping, many novel techniques based on sequencing were numerously emerging in recent years. RNA-Seq is a recently developed approach to transcriptome profiling using deep-sequencing technologies [22]. The obtained transcriptome included all coding mRNA and noncoding RNA sequences in the cells of one specific development stage or physiological condition. Plant transcriptome analysis is fast and considerable for providing information on highly expressed genes, differentially expressed genes, new genes for function analysis and gene screening related to studied traits [23,24,25,26]. Besides, BSA-Seq strategy was one more efficient QTL mapping method comparing with the traditional QTL mapping method. Combing the traditional BSA method and next-generation sequencing (NGS) technique, BSA-Seq technique has been widely used in QTL mapping for the precise identification of target genes [27,28,29], such as in rapeseed [30], rice [29], maize [31] and so on. Furthermore, the combined analysis of BSA-seq and RNA-seq data enabled the identification of candidate genes [32,33,34].
In the present study, we reported one natural mutant (MT) with determinate and capitulum-like inflorescence in B. napus. Then phenotypic analysis, genetic analysis, RNA-Seq and BSA-Seq methods were used to identify the candidate genes for regulating the determinate inflorescence. The results would be helpful in studying the molecular mechanism of the mutation phenotype and improving the inflorescence architecture in B. napus.
2. Results
2.1. Phenotypic characterization of the mutant (MT) plants
One mutant (MT) plant (Figure 1B/b and Figure 1D/d) was found in the self-polinated progenies of the wide-type (WT) B. napus line of GD605-2 (Figure 1A/a and Figure 1C/c) in the field. The phenotype of MT plants and the WT plants was kept the same until the plants started to bolt. Then at the apex of each inflorescence axis, the indeterminate inflorescence was terminated with a terminal flower in MT plants (Figure 1B/b and Figure 1D/d). The terminal flowers contained one pistil and six stamens, while no petals and seeds would be formed. Each inflorescence axis with buds/flowers at the apex closer to the “terminal flower” was kept to be clustered together and not prolongated, which finally developed into one capitulum-like head in MT plants (Figure 1A/a and Figure 1D/d). Each capitulum-like head had about 10.0±0.9 normal flowers/siliques in the top of each inflorescence axis. The MT plants had a plant height of about 158.8±4.3 cm, significantly (p=0.001) lower than the WT plants of 180.9±3.4 cm. Besides, no significant difference (p=0.903) in the main inflorescence was detected for the number of siliques between WT plants (58.3±8.2) and MT plants (58.0±9.9).
The development of inflorescence apex from three to seven-leaf stages of MT and WT plants was observed by paraffin sectioning technique (Figure 2). The inflorescence apex from three to four-leaf stages developed normally in all the detected MT and WT plants (Figure 2A/a and Figure 2B/b). Starting from five-leaf stage, the inflorescence apex began to split and developed into one terminal flower (Figures D-F/d-f).
2.2. Genetic analysis of the MT phenotype
Genetic determinism behind the MT phenotype was analyzed through the phenotypic characterization of F1 progenies from crosses between the MT and WT plants. All the F1 plants from 4 reciprocal crosses between MT and Ningyou7 (29 and 33 plants, respectively), Bakow (29 and 33 plants, respectively), 97009 (26 and 30 plants, respectively), 97081 (28 and 15 plants, respectively) displayed similar phenotype of indeterminate inflorescences to the WT parent plants. Since all the F1 plants displayed the normal phenotype of indeterminate inflorescences, the allele(s) responsible for the MT phenotype showed complete recessiveness over the allele(s) responsible for the normal phenotype of indeterminate inflorescences.
In the F2 population from MT×WT, 553 plants with indeterminate inflorescences and 131 plants with determinate inflorescences in 2018, fitting with a segregation ratio of 13:3 (χ2=0.073, p= 0.788). In 2020, the F2 population from MT×WT was classified into two groups: plants with indeterminate inflorescences (1,077 plants) and plants with determinate inflorescences (236 plants), fitting with a segregation ratio of 13:3 (χ2=0.519, p= 0.471). Furthermore, another F2 population from WT×MT in 2020 was also showed the same segregation ratio of 13:3 (843 plants with indeterminate inflorescences versus 172 plants with determinate inflorescences (χ2=2.169, p= 0.141). Therefore, the segregation patterns of inflorescence characterization in the F2 populations supported a digenic inheritance model in these crosss.
2.3. QTL mapping by BSA-Seq technique
BSA-Seq technique was used to rapidly map the QTL accounting for the determinate inflorescences of MT plants. The “determinate” DNA pool, “indeterminate” DNA pool and the two parental DNA pools were subjected to Illumina sequencing with an average sequencing depths of 16.25-fold. Then the clean reads were aligned to the reference genome by using BWA software, and the average proportion of mapped reads to clean reads was 97.08%. After removing low quality reads, a total of 94.04 Gbp clean data were obtained with an average Q30 rate of 80% and average of onefold coverage ratio of 92.16%. All the obtained clean data were used to develop SNP and InDel. A total of 1,500,703 SNPs and 945,276 SNPs were identified between two parents and 945,276 ones between the two DNA pools, respectively. Furthermore, a total of 400,329 InDel between two parents and 280,916 InDel between two DNA pools were identified. The ΔSNP-index and ΔInDel-index were used for QTL mapping of the candidate intervals for determinate inflorescence. As a result, two overlabbed regions were identified on C02, wihch had a size of 4.18 Mb (14.27–18.45 Mb) byΔSNP-index and 4.18 Mb (14.23–18.41 Mb) byΔInDel-index (Figure 3), respectively. On C06, two another overlabbed regions were identified on C06, wihch had a size of 0.7 Mb (32.98–33.68 Mb) byΔSNP-index and 0.72 Mb (32.97–33.69 Mb) byΔInDel-index, respectively. Finally, these two overlapped regions on C02 (14.27–18.41 Mb, 320 candidate genes) and C06 (32.98–33.68 Mb, 117 candidate genes) were chosen as the candidate regions for genetic control of determinate inflorescences in MT plants.
2.4. Gene expression profile analysis
A total of six cDNA libraries were constructed and sequenced, an average of 48,705,785 and 46,319,327 raw reads was generated from the WT and MT libraries, respectively (Table 1 and Table S1). After removing low quality reads, adapter polluted reads and higher N content (>5%) reads, an average of 47,184,448 (WT) and 44,944,151 (MT) clean reads were obtained. After blasting the reference genome of B. napus and filtering the genes that only contained one exon or encoded short peptide chains (<50 amino acid residues), a total of 86,026 genes were revealed through blasting the reference genome using DESeq2 (v1.6.3). It was found that 6,309 genes were differentially expressed, among which 4,103 ones were down-regulated and 2,209 ones were up-regulated in MT compared with WT (Table S2). Based on the FPKM values, the up-regulated and down-regulated genes between WT and MT were also revealed by a hierarchical clustering analysis in Figure S1. To verify the expression of DEGs detected by RNA-Seq, 16 candidate genes (DEGs) regulating the MT phenotype were randomly chosen for validation by qRT-PCR. The data obtained by qRT-PCR was consistent with the RNA-Seq results (Figure S2 and Table S3), suggesting the reliability of the transcriptome database.
To functionally annotate the B. napus transcriptome, the 6,309 DEGs were blasted in search of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). Finally, 4,548 DEGs were successfully annotated, among which 4,522 (71.68%) in GO and 2,127 (33.71%) in KEGG (Table S4). For the GO classification analysis of DEGs, 4,522 genes were assigned into three main GO functional categories (cellular component, biological process and molecular function) and then divided into 55 sub-categories (Figure S3 and Table S5). For the KEGG analysis of gene functions, all the annotated 6,309 DEGs were assigned into 128 pathways (Table S6) based on KEGG database. Several DEGs were assigned into more than one sub-category.
2.5. Candidate genes for regulating the determinate inflorescence
Following the annotation of GO and KEGG, the potential DEGs for regulating the flower and inflorescence mutation were further blasted with the database of Arabidopsis thaliana (https://www.arabidopsis.org/index.jsp) and NCBI (https://www.ncbi.nlm.nih.gov/) for detailed information. A total of 133 candidate genes for regulating the flower development (75 genes, 58.6%), shoot meristem development (29 genes, 22.7%) and inflorescence meristem development were identified (13 genes, 9.8%) (Figure. S4 and Table S7). Several genes were involved in more than one sub-category.
To integrate the results of BSA-Seq and RNA-sequencing, we perform an alignment analysis between the 133 candidate genes potentially related to determinate inflorescence of the MT plants and the reference genome of B. napus (https://www.genoscope.cns.fr/brassicanapus/) by the BLAST-like alignment tool [35]. Eight DEGs corresponding to seven genes (Table 2) located on C02 between 14.27 Mb and 18.41 Mb were detected, among which the widely recognized genes for regulating determinate inflorescence of BnaC02g02900D encoding TERMINAL FLOWER 1 (TFL1) (ChrC02: 1,320,657–1,321,719) was included [20,36]. In addition, three DEGs on C06 (Table 2) between 32.98 Mb and 33.68 Mb were detected, among which only one DEG of BnaC06g25500D encoding the gene of APETALA1 (AP1) (C06: 27,150,336–27,153,999) was related to the inflorescence development. Then the two candidate genes were renamed as BncTFL1 and BncAP1. The expression level of BncTFL1 and BncAP1 showed significantly increased from 5-leaf stage and started to decrease from 7-leaf stage in WT plants (Figure 4). In addition, the expression level of BncTFL1 and BncAP1 was significantly decreased from 5-leaf stage to 7-leaf stage in MT plants comparing with WT plants (Figure 4).
3. Discussion
In the present study, one natural mutation with determinate inflorescences characterizing with terminal flower and capitulum-like inflorescences in Brassiceae were reported. The mutation was characterized by paraffin sectioning, and the inflorescence apex began to split and developed into one terminal flower, starting from five-leaf stage. The segregation patterns of inflorescences in the F2 populations supported a digenic inheritance model, which was further approved by BSA-Seq technique. TERMINAL FLOWER 1 (TFL1) had been proved to be responsible for controlling the determinate inflorescences in many species [5,6,7,8,9,20]. While BncTFL1 mapped on C02 in the present study was one new allele of TFL1, differently from these genes on B05 of B. juncea [8] and on A10 of B. napus [10] in Brassiceae. In addition, AP1 had been identified as the genes controlling the development of inflorescence meristem and floral meristem [21], BncAP1 on C06 was first reported as the candidate gene of controlling determinate inflorescences. Quite different from the reported phenotype of determinate inflorescences, the capitulum-like inflorescences was also found in GD605-2. The formation of capitulum-like inflorescences had once been well exploited by two models of FL1 & LFY [3] and TFL1 & AP1 [21], in which the specific combination of gene expression levels result in the emergence of different capitulum-like inflorescences.
The special mutation with determinate and capitulum-like inflorescence could provide unique materials for the basic research of the development of inflorescence in B. napus. To broaden the utilization of the MT phenotype, we had transferred the mutation phenotype into germplasm resources with colored flowers, purple stem and leaves, muti-head inflorescence. These new materials could provide useful resources for rapeseed tourism. In the future, we would focus on the gene cloning and functional analysis of these two candidate genes of BncTFL1 and BncAP1.
4. Materials and methods
4.1. Plant materials and field experiment
In March 2014, one mutant (MT) plant with determinate and capitulum-like inflorescence was found. The MT plant was naturally mutated and found in the self-polinated progenies of one breeding line GD605-2 (F7) (wild-type, WT). In October 2017, twenty plants for each of the MT (F4) and WT (F7) lines were planted in the field of Guizhou University, Guiyang, China. In March 2018, the inflorescences from three randomly chosen plants for each of MT and WT lines were sampled, immediately frozen in liquid nitrogen, and stored at -800C for RNA extraction.
4.2. Investigation of the agronomic traits of MT and WT plants
In April 2019, twenty plants for each of MT and WT plants were used for investigation of number of flowers/siliques in the capitulum-like head, plant height, number of siliques in the main inflorescence.
4.3. Paraffin sectioning
The paraffin sectioning is mainly following the work of Zhou, et al. [37] with a minor modification. The shoot tips of MT and WT lines from three to seven-leaf stages are dissected as fast as possible after sampling. The dissected tissue samples were fixed for >24 h in FAA fixative in chemical hood. The fixed tissues were then continuously dehydrated and cleared, respectively, using ethanol and xylene in volume ratios of 3:1, 1:1, and 1:3. After the tissue samples were in melted paraffin, they were removed from the mold and embedded in 60 °C paraffin wax solution which was allowed to solidify at room temperature. Paraffin sections (10 μm) were obtained using an automatic microtome. The sections were stained with aniline blue, and observed with a microscope. Images of the anthers at different stages were captured with a camera.
4.4. BSA-Seq analysis
The BSA-Seq method was used for mapping the genes of regulating the phenotype of determinate inflorescence. The “determinate” bulk was made by mixing equal amount of DNA from 10 F2 plants with determinate inflorescence, while the “indeterminate” bulk was formed from 10 F2 plants with indeterminate inflorescence. The two bulks and the DNA samples of the parental lines of WT plants and MT plants were sequenced on an Illumina HiSeq™2000 platform (Beijing Biomarker Biotechnology Co., Beijing, China). The low-quality reads containing adaptors were filtered. The reads with more than 10% of missing bases and more than 50% of bases with Q-score lower than 10 were filtered, and the clean reads thus obtained were mapped to the Brassica napus reference genome (Genoscope v4.1, http://www.genoscope.cns.fr/brassicanapus/data/) using BWA software [29,38]. SNP-calling and SNP annotation were done using the professional softwares of ANNOVAR and SAM [38,39], MarkDuplicatePicards (http://sourceforge.net/projects/picard/), GATK [40], SnpEff [41]. The differences in allele frequency between bulked pools was used to calculate the SNP-index for identifying the candidate regions of the genome associated with determinate inflorescences [28,29,42]. In detial, Δ(SNP index) was calculated by sliding window analysis among the genome within 1 Mb width windows and 1 kb at each step [28]. Totally, all those analysis was performed with the related tools on the online open platform of BMKCloud (http://www.biocloud.com/).
4.5. RNA extraction, preparation, sequencing and data analysis
Total RNA (2 µg) was extracted from the shoots (0.2–0.5g) of three independent plants for each of WT and MT lines in 5-leaf stage using the TRIzol kit (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. The RNA purity was checked using the Kaiao K5500®Spectrophotometer (Kaiao, Beijing, China), and the RNA integrity and concentration were assessed using the RNA Nano 6000 Assay Kit for the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Then, the six RNA samples were sent to the ANOROAD GENOME company (http://www.genome.cn/) for the construction of cDNA libraries and Illumina deep sequencing according to the paper of Wang, et al. [43]. The raw RNA-sequencing data were filtered by a Perl script, following the steps of Wu, et al. [44] and our earlier study [34].
4.6. Identification and annotation of differentially expressed genes (DEGs)
DESeq2 v1.6.3 was used for differential gene expression analysis between MT and WT with three biological replicates under the theoretical basis obeys the hypothesis of negative binomial distribution for the value of count. The p-value was corrected by the BH method. Genes with q≤0.05 and |log2_ratio|≥1 were identified as differentially expressed genes (DEGs) [45]. The DEGs obtained were further annotated with Gene Ontology (GO, http://geneontology.org/) and analyzed by KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.kegg.jp/) [46,47]. The GO enrichment of DEGs was implemented by the hypergeometric test, in which the p-value is calculated and adjusted to produce the q-value, and the data background is the genes in the whole genome. GO terms with q<0.05 were considered to be significantly enriched. GO enrichment analysis was used to determine the biological functions of the DEGs. The KEGG enrichment of the DEGs was determined by the hypergeometric test, in which p-value was adjusted by multiple comparisons to produce the q-value. KEGG terms with q<0.05 were considered to be significantly enriched.
4.7. Quantitative real time-PCR (qRT-PCR) analysis
Quantitative real-time PCR (qRT-PCR) was used to verify the transcript levels of the RNA-Seq results. Total RNA was extracted using the TRIzol kit (Invitrogen), according to the manufacturer’s instructions. Then, the cDNA was synthesized by reverse transcription using PrimeScript RT reagent kits with gDNA Eraser (Takara, Dalian, China) according to the manufacturer’s instructions. Sixteen gene-specific primers for qRT-PCR were designed based on reference unigene sequences randomly chosen from the DEGs using Primer Premier 5.0. Real-time PCR was conducted using SsoAdvancedTM Universal SYBR Green Supermix (Hercules, CA) according to our earlier research [34]. The 2-ΔΔCt algorithm was used to calculate the relative level of gene expression. The β-actin gene was used as the internal control, and the WT samples served as the control. All qRT-PCR were performed with three biological replicates, and run on a Bio-Rad CFX96 Real Time System (Bio-Rad, Hercules, CA, USA).
5. Conclusions
In the present study, we reported one natural mutation with determinate and capitulum-like inflorescence in B. napus. The mutation started from 5-leaf stage in the shoots to the flowering stage in inflorescence with a phenotype of one top flower and capitulum-like inflorescence. Trough genetic analysis and BSA-Seq analysis, two QTL regions on C02 and C06 were detected. To better dig the candidate genes within the QTL regions, RNA-Seq analysis was done. Finally, one joint analysis combing BSA-Seq and RNA-Seq identified two candidate gene of BncTFL1 and BncAP1 for regulating the MT phenotype. Except for one useful material for basic research of inflorescence development, the mutation could also be used for rapeseed tourism as for its special inflorescence phenotype.
Supplementary Materials
Figure S1. Hierarchical clustering analysis of the differentially expression genes between WT and MT. The column represents individual experiment of each biological replicate of WT for WT01-03 and MT for MT01-03, and each row represents one gene. Red represents high expression, and blue represents low expression. Figure S2. qRT-PCR verification of 16 randomly chosen differentially expression genes from RNA-Seq. Relative level: log2 (fold change). Figure S3. Gene ontology (GO) function classification of differentially expressed genes (DEGs) in MT compared with WT. Figure S4. The function classification of 133 candidate genes related to the mutated phenotype of determinate and capitulum-like inflorescence. Table S1. The detailed information in transcriptome sequencing. Table S2. The list of differentially expressed genes (DEGs) by RNA-Seq. Table S3. Primers used in qRT-PCR experiment for the validation of RNA-Sequenced DEGs. Table S4. The annotated information of DEGs. Table S5. The GO classification of DEGs. Table S6. The KEGG classification of DEGs. Table S7. The genes (DEGs) related to the regulation of flower and inflorescence development.
Author Contributions
ET planned and designed the research. WW and HZ performed the experiments with the assistance of YM, ID, YB, KY, YX, WD and CD contributed to the experimental design and data interpretation. WW, HZ and ET wrote the manuscript. All authors contributed to the article and approved the submitted version. All authors have reviewed and approved the final version of the manuscript and therefore are equally responsible for the integrity and accuracy of its content.
Funding
This work was supported by the Guizhou Provincial Science and Technology Plan Project (Qian Kehe Support [2022]key026), National Natural Science Foundation of China (Grant No. 32160483), Key Laboratory of Molecular Breeding for Grain and Oil Crops in Guizhou Province (Qiankehezhongyindi (2023) 008), Key Laboratory of Functional Agriculture of Guizhou Provincial Higher Education Institutions (Qianjiaoji (2023) 007).
Acknowledgments
We are specially grateful to Department of Science and Technology of Guizhou Province and National Natural Science Foundation of China for providing enough funding for this study. We also thank the Teaching Practice Field of Guizhou University for supporting the field experiment of this study. We also want to express our gratitude to Shuchun Lin for his cooperation and great contribution in the field experiment.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Beckman, C. Vegetable oils: Competition in a changing market. Bi-weekly Bulletin Agriculture and Agri-Food Canada 2005, (18), 11.
- Fu, T. , Breeding and utilization of rapeseed hybrid. Hubei Science Technology Press (Hubei) 2000, 167–169. [Google Scholar]
- Prusinkiewicz, P.; Erasmus, Y.; Lane, B.; Harder, L. D.; Coen, E. , Evolution and development of inflorescence architectures. Science 2007, 316(5830), 1452–1456. [Google Scholar] [CrossRef]
- Plackett, A. R. G.; Powers, S. J.; Phillips, A. L.; Wilson, Z. A.; Hedden, P.; Thomas, S. G. , The early inflorescence of Arabidopsis thaliana demonstrates positional effects in floral organ growth and meristem patterning. Plant Reproduction 2018, 31(2), 171–191. [Google Scholar] [CrossRef] [PubMed]
- Shannon, S.; Meeks-Wagner, D. R. , A Mutation in the Arabidopsis TFL1 Gene Affects Inflorescence Meristem Development. The Plant Cell 1991, 3(9), 877–892. [Google Scholar]
- Bernard, R. L. , Two Genes Affecting Stem Termination in Soybeans1. Crop Science 1972, 12(2), 235–239. [Google Scholar] [CrossRef]
- Kato, H.; Honma, T.; Goto, K. CENTRORADIALIS/TERMINAL FLOWER 1 gene homolog is conserved inN. tabacum, a determinate inflorescence plant. Journal of Plant Research, 1998; 111, 2, 289–294. [Google Scholar]
- Kaur, H.; Banga, S. S. , Discovery and mapping of Brassica junceaSdt1gene associated with determinate plant growth habit. Theoretical and Applied Genetics 2015, 128(2), 235–245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Miao, H.; Li, C.; Wei, L.; Duan, Y.; Ma, Q.; Kong, J.; Xu, F.; Chang, S. , Ultra-dense SNP genetic map construction and identification of SiDt gene controlling the determinate growth habit in Sesamum indicum L. Scientific Reports 2016, 6, 31556. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tang, M.; Nelson, A.; Caligagan, H.; Zhou, X.; Clark-Wiest, C.; Ngo, R.; Brady, S. M.; Kliebenstein, D. J. , Network-Guided Discovery of Extensive Epistasis between Transcription Factors Involved in Aliphatic Glucosinolate Biosynthesis. Plant Cell 2018, 30(1), 178–195. [Google Scholar] [CrossRef]
- Parcy, F.; Nilsson, O.; Busch, M. A.; Lee, I.; Weigel, D. , A genetic framework for floral patterning. Nature 1998, 395, 561. [Google Scholar] [CrossRef] [PubMed]
- Saddic, L. A.; Huvermann, B.; Bezhani, S.; Su, Y.; Winter, C. M.; Kwon, C. S.; Collum, R. P.; Wagner, D. , The LEAFY target LMI1 is a meristem identity regulator and acts together with LEAFY to regulate expression of CAULIFLOWER. Development 2006, 133(9), 1673–1682. [Google Scholar] [CrossRef]
- Lee, J.; Oh, M.; Park, H.; Lee, I. , SOC1 translocated to the nucleus by interaction with AGL24 directly regulates LEAFY. The Plant Journal 2008, 55(5), 832–843. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Teo, Z. W. N.; Bi, Y.; Song, S.; Xi, W.; Yang, X.; Yin, Z.; Yu, H. , A conserved genetic pathway determines inflorescence architecture in Arabidopsis and rice. Developmental cell 2013, 24(6), 612–622. [Google Scholar] [CrossRef]
- Gregis, V.; Andrés, F.; Sessa, A.; Guerra, R. F.; Simonini, S.; Mateos, J. L.; Torti, S.; Zambelli, F.; Prazzoli, G. M.; Bjerkan, K. N.; Grini, P. E.; Pavesi, G.; Colombo, L.; Coupland, G.; Kater, M. M. , Identification of pathways directly regulated by SHORT VEGETATIVE PHASE during vegetative and reproductive development in Arabidopsis. Genome Biology 2013, 14(6), R56. [Google Scholar] [CrossRef]
- Wils, C. R.; Kaufmann, K. , Gene-regulatory networks controlling inflorescence and flower development in Arabidopsis thaliana. Biochim Biophys Acta Gene Regul Mech 2017, 1860(1), 95–105. [Google Scholar] [CrossRef]
- Alvarez, J.; Guli, C. L.; Yu, X.-H.; Smyth, D. R. , terminal flower: a gene affecting inflorescence development in Arabidopsis thaliana. The Plant Journal 1992, 2(1), 103–116. [Google Scholar] [CrossRef]
- Ohshima, S.; Murata, M.; Sakamoto, W.; Ogura, Y.; Motoyoshi, F. , Cloning and molecular analysis of the Arabidopsis gene Terminal Flower 1. Molecular and General Genetics MGG 1997, 254(2), 186–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, T.; Duan, M.; Song, J.; Li, X. De novo Transcriptome Analysis of Sinapis alba in Revealing the Glucosinolate and Phytochelatin Pathways. Frontiers in plant science, 2016; 7, 259. [Google Scholar] [CrossRef]
- Li, K.; Yao, Y.; Xiao, L.; Zhao, Z.; Guo, S.; Fu, Z.; Du, D. , Fine mapping of the Brassica napus Bnsdt1 gene associated with determinate growth habit. Theoretical and Applied Genetics 2018, 131(1), 193–208. [Google Scholar] [CrossRef]
- Ma, Q.; Liu, X.; Franks, R. G.; Xiang, Q.-Y. J. , Alterations of CorTFL1 and CorAP1 expression correlate with major evolutionary shifts of inflorescence architecture in Cornus (Cornaceae)—a proposed model for variation of closed inflorescence forms. The New phytologist 2017, 216(2), 519–535. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. , RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics 2009, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xu, H.; Shen, Y.; Wang, J. , Transcriptomic analysis of rice (Oryza sativa) endosperm using the RNA-Seq technique. Plant Molecular Biology 2013, 81(4), 363–378. [Google Scholar] [CrossRef]
- Jones, S. I.; Vodkin, L. O. , Using RNA-Seq to Profile Soybean Seed Development from Fertilization to Maturity. PLOS ONE 2013, 8(3), e59270. [Google Scholar] [CrossRef]
- Loraine, A. E.; McCormick, S.; Estrada, A.; Patel, K.; Qin, P. , RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant Physiol 2013, 162(2), 1092–1109. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Dong, N.; Wang, Y.; Li, G.; Wang, L.; Guo, X.; Li, J.; Wen, Z.; Wei, W. , RNA-seq transcriptome analysis of the immature seeds of two Brassica napus lines with extremely different thousand-seed weight to identify the candidate genes related to seed weight. PLOS ONE 2018, 13(1), e0191297. [Google Scholar] [CrossRef]
- Wenger, J. W.; Schwartz, K.; Sherlock, G. , Bulk segregant analysis by high-throughput sequencing reveals a novel xylose utilization gene from Saccharomyces cerevisiae. PLoS genetics 2010, 6(5), e1000942. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S. , QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. The Plant Journal 2013, 74(1), 174–183. [Google Scholar] [CrossRef]
- Guo, Z.; Cai, L.; Chen, Z.; Wang, R.; Zhang, L.; Guan, S.; Zhang, S.; Ma, W.; Liu, C.; Pan, G. , Identification of candidate genes controlling chilling tolerance of rice in the cold region at the booting stage by BSA-Seq and RNA-Seq. Royal Society Open Science 2020, 7(11), 201081. [Google Scholar] [CrossRef]
- Ye, S.; Yan, L.; Ma, X.; Chen, Y.; Wu, L.; Ma, T.; Zhao, L.; Yi, B.; Ma, C.; Tu, J. , Combined BSA-seq based mapping and RNA-seq profiling reveal candidate genes associated with plant architecture in Brassica napus. International Journal of Molecular Sciences 2022, 23(5), 2472. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.; Xiao, Y.; Conklin, P. A.; Govindarajulu, R.; Kelly, J. A.; Scanlon, M. J.; Whipple, C. J.; Bartlett, M. Bulked-segregant analysis coupled to whole genome sequencing (BSA-Seq) for rapid gene cloning in maize. G3: Genes, Genomes, Genetics 2018, 8(11), 3583–3592. [Google Scholar] [CrossRef]
- Luo, M.; Lu, B.; Shi, Y.; Zhao, Y.; Liu, J.; Zhang, C.; Wang, Y.; Liu, H.; Shi, Y.; Fan, Y. , Genetic basis of the oil biosynthesis in ultra-high-oil maize grains with an oil content exceeding 20%. Frontiers in Plant Science 2023, 14, 1168216. [Google Scholar] [CrossRef]
- Yu, K.; He, Y.; Li, Y.; Li, Z.; Zhang, J.; Wang, X.; Tian, E. , Quantitative Trait Locus Mapping Combined with RNA Sequencing Reveals the Molecular Basis of Seed Germination in Oilseed Rape. Biomolecules 2021, 11(12), 1780. [Google Scholar] [CrossRef] [PubMed]
- Khattak, A. N.; Wang, T.; Yu, K.; Yang, R.; Wan, W.; Ye, B.; Tian, E. , Exploring the basis of 2-propenyl and 3-butenyl glucosinolate synthesis by QTL mapping and RNA-sequencing in Brassica juncea. PLOS ONE 2019, 14(10), e0220597. [Google Scholar] [CrossRef]
- Kent, W. J. , BLAT—the BLAST-like alignment tool. Genome research 2002, 12(4), 656–664. [Google Scholar]
- Zhang, Y.; Wang, L.; Gao, Y.; Li, D.; Yu, J.; Zhou, R.; Zhang, X. , Genetic dissection and fine mapping of a novel dt gene associated with determinate growth habit in sesame. BMC Genetics 2018, 19(1), 38. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Z.; Ji, R.; Feng, H. Comparative transcript profiling of fertile and sterile flower buds from multiple-allele-inherited male sterility in Chinese cabbage (Brassica campestris L. ssp. pekinensis). Molecular Genetics and Genomics 2017, 292(5), 967–990. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. , Fast and accurate short read alignment with Burrows–Wheeler transform. bioinformatics 2009, 25(14), 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. , ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 2010, 38(16), e164–e164. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. , The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 2010, 20(9), 1297–1303. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L. L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S. J.; Lu, X.; Ruden, D. M. , A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. fly 2012, 6(2), 80–92. [Google Scholar] [CrossRef]
- Fekih, R.; Takagi, H.; Tamiru, M.; Abe, A.; Natsume, S.; Yaegashi, H.; Sharma, S.; Sharma, S.; Kanzaki, H.; Matsumura, H. , MutMap+: genetic mapping and mutant identification without crossing in rice. PloS one 2013, 8(7), e68529. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T. A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J. C.; Ware, D. , Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nature Communications 2016, 7, 11708. [Google Scholar] [CrossRef]
- Wu, W.; Huang, Z.; Li, Z.; Zhang, S.; Liu, X.; Gu, D. , De novo transcriptome sequencing of Cryptotermes domesticus and comparative analysis of gene expression in response to different wood species. Gene 2016, 575, (2, Part 3). 655–666. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. , DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26(1), 136–138. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B. A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M. J.; Salzberg, S. L.; Wold, B. J.; Pachter, L. , Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010, 28(5), 511–515. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D. R.; Pimentel, H.; Salzberg, S. L.; Rinn, J. L.; Pachter, L. , Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012, 7(3), 562–578. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The phenotype analysis between mutant (MT) plant and wild-type (WT) plant in flowering and pod development period. (A): one normal indeterminate inflorescence, and the top of inflorescence axis is shown in the lower-left; (B): one mutant branch with determinate inflorescence, and the top of the inflorescence axis with terminal flower is shown in the lower-left; (C): normal branches with siliques, and the top of the inflorescence axis is shown in the lower-left; (D): mutant branches with siliques, and the top of the inflorescence axis is shown in the lower-left; (E): diagram of (A); (F): diagram of (B); (G): diagram of (C); (H): diagram of (D). Arrows represent indeterminate growth.
Figure 1.
The phenotype analysis between mutant (MT) plant and wild-type (WT) plant in flowering and pod development period. (A): one normal indeterminate inflorescence, and the top of inflorescence axis is shown in the lower-left; (B): one mutant branch with determinate inflorescence, and the top of the inflorescence axis with terminal flower is shown in the lower-left; (C): normal branches with siliques, and the top of the inflorescence axis is shown in the lower-left; (D): mutant branches with siliques, and the top of the inflorescence axis is shown in the lower-left; (E): diagram of (A); (F): diagram of (B); (G): diagram of (C); (H): diagram of (D). Arrows represent indeterminate growth.

Figure 2.
The observation of inflorescence apex from three to seven-leaf stages of MT and WT plants by paraffin sectioning technique. WT: three-leaf WT (A) and MT (a) plants; four-leaf WT (B) and MT (b) plants; five-leaf WT (C) and MT (c) plants; six-leaf WT (D) and MT (d) plants; seven-leaf WT (E and F) and MT (e and f) plants.
Figure 2.
The observation of inflorescence apex from three to seven-leaf stages of MT and WT plants by paraffin sectioning technique. WT: three-leaf WT (A) and MT (a) plants; four-leaf WT (B) and MT (b) plants; five-leaf WT (C) and MT (c) plants; six-leaf WT (D) and MT (d) plants; seven-leaf WT (E and F) and MT (e and f) plants.
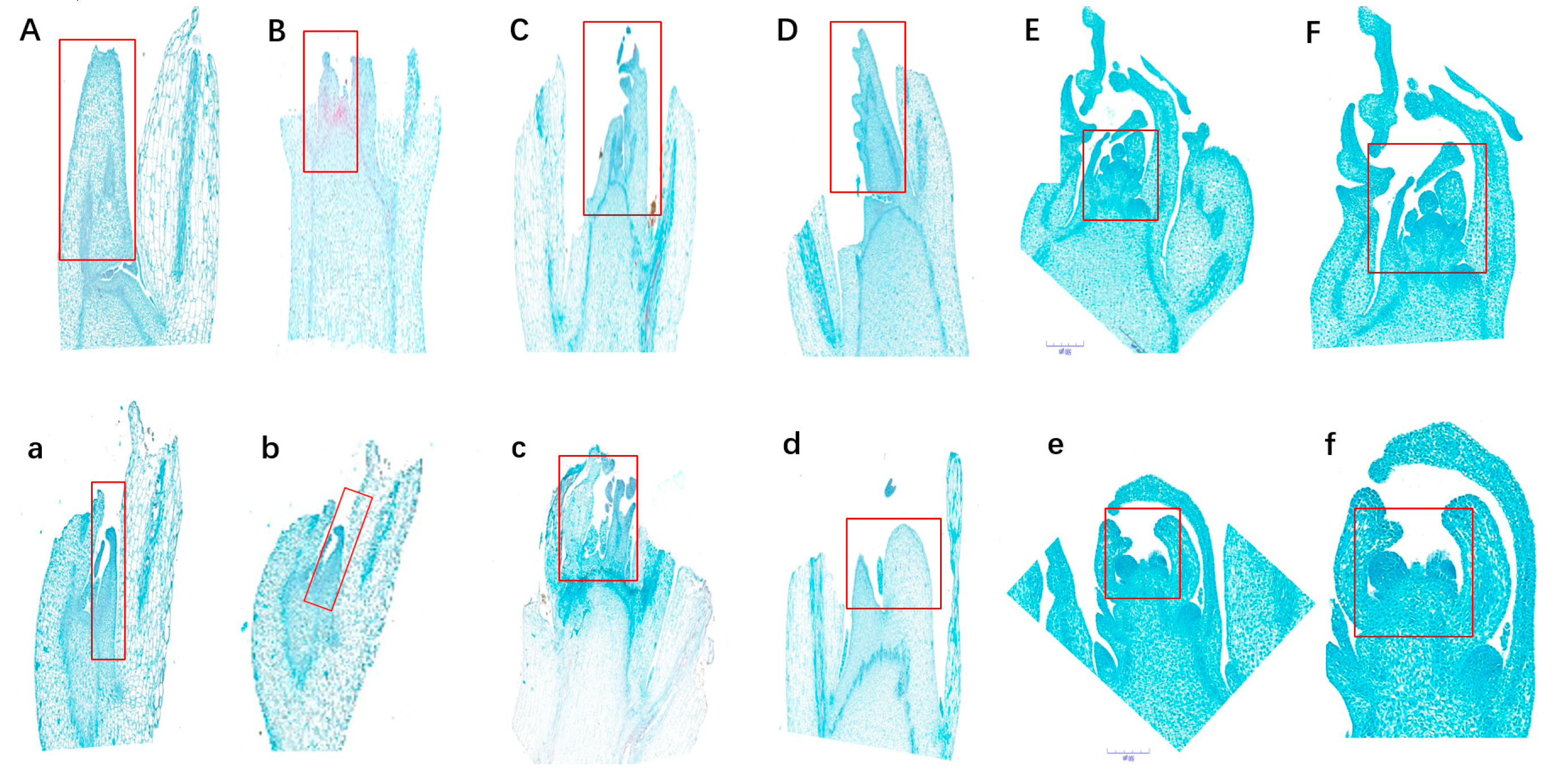
Figure 3.
QTL mapping results obtained by ΔSNP-index (A) and ΔSNP-inDel (B) method of BSA-Seq technique.
Figure 3.
QTL mapping results obtained by ΔSNP-index (A) and ΔSNP-inDel (B) method of BSA-Seq technique.
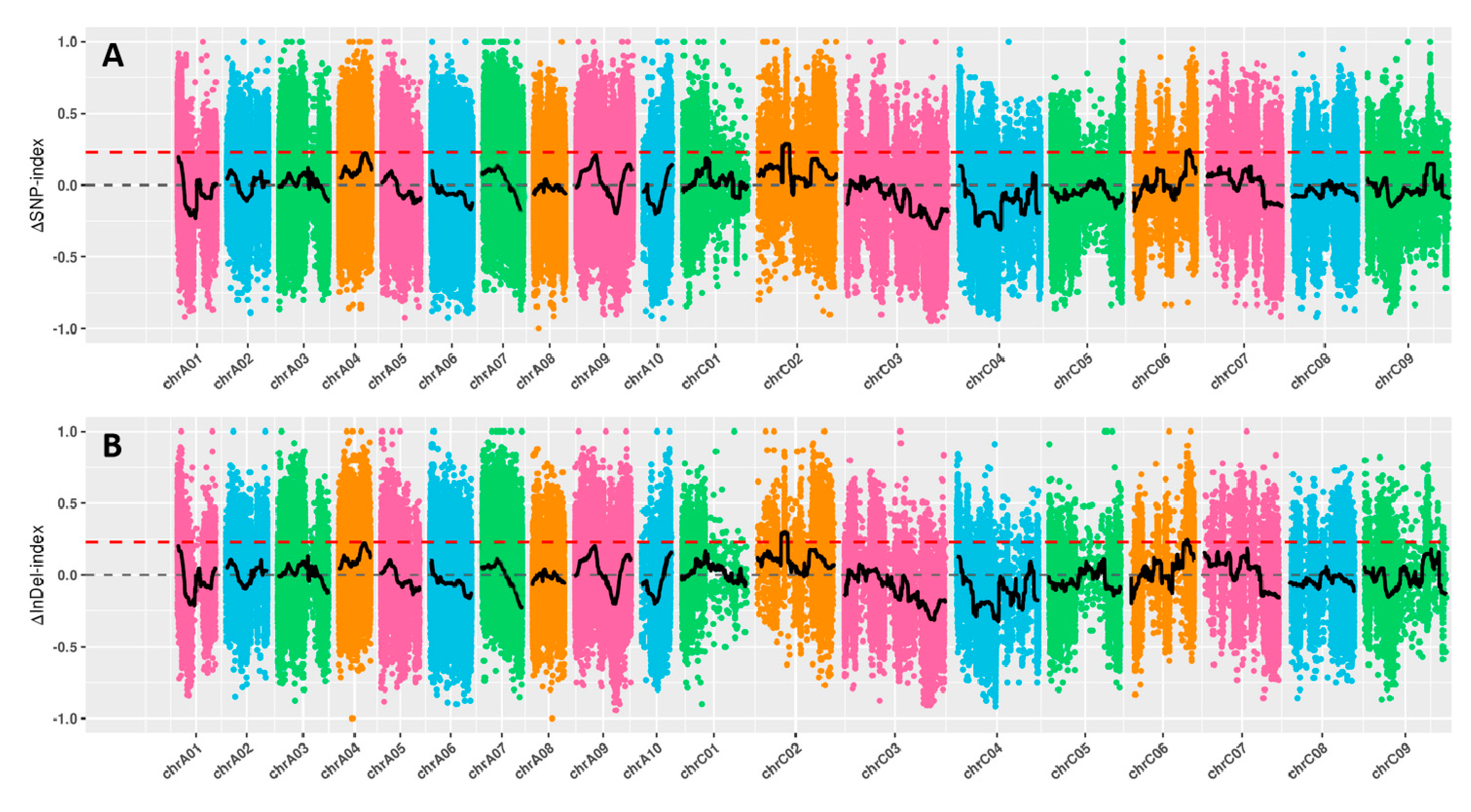
Figure 4.
Expression level of BncTFL1 (A) and BncAP1 (B) in the shoots of MT and WT lines from 2-leaf stage to 7-leaf stage. The number from 2 to 7 in X-axis indicates the sampling stages from 2-leaf stage to 7-leaf stage. ** indicates the expression level is extremely significantly difference at the level of p<0.001.
Figure 4.
Expression level of BncTFL1 (A) and BncAP1 (B) in the shoots of MT and WT lines from 2-leaf stage to 7-leaf stage. The number from 2 to 7 in X-axis indicates the sampling stages from 2-leaf stage to 7-leaf stage. ** indicates the expression level is extremely significantly difference at the level of p<0.001.
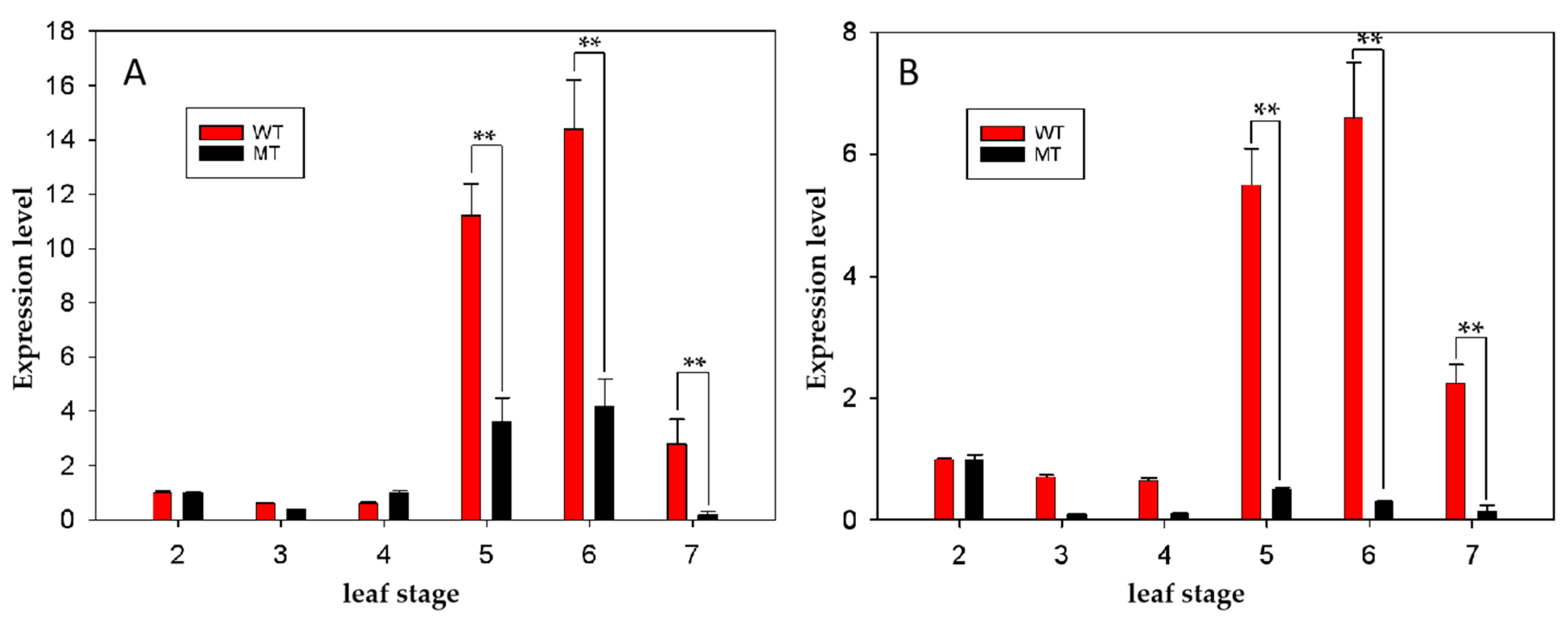

Table 1.
Summary of transcriptome sequencing data.
| Items | WT (Mean) | MT (Mean) |
|---|---|---|
| Raw Reads Number | 48,705,785 | 46,319,327 |
| Raw Bases Number | 7,305,867,800 | 6,947,899,100 |
| Clean Reads Number (%) | 47,184,448 (96.88) | 44,944,151 (96.96) |
| Clean Bases Number | 7,077,667,200 | 6,741,622,600 |
| Low-quality Reads Number (%) | 344,567 (0.71) | 360,269 (0.78) |
| Mapped Reads (%) | 41,135,862 (87.18) | 39,824,899 (88.60) |
| UnMapped Reads | 6,048,586 | 5,119,252 |
| MultiMap Reads (%) | 6,988,386 (14.81) | 6,857,621 (15.26) |
| Ns Reads Number (%) | 3,115 (0.01) | 3,694 (0.01) |
| Adapter Polluted Reads Number (%) | 1,173,656 (2.41) | 1,011,214 (2.18) |
| Raw Q30 Bases Rate (%)1 | 93.84 | 93.67 |
| Clean Q30 Bases Rate (%) | 94.14 | 94.01 |
| Exon (%) | 15,111,070 (94.36) | 14,619,066 (94.53) |
| Intron (%) | 249,023 (1.56) | 221,680 (1.43) |
| Intergenic (%) | 649,815 (4.07) | 623,039 (4.04) |
| Novel Transcripts | 160,798 | 129,446 |
Table 2.
The DEGs located on the QTL regions on C02 and C06.
| Gene ID inDarmor-bzh | AradopsisID | E-value | Gene name | Gene function |
|---|---|---|---|---|
| BnaC02g02980D | AT5G03680 | 0 | PETAL LOSS | Organ initiation and orientation |
| BnaC02g02940D | AT2G18960 | 4E-102 | OPEN STOMATA 2 | Regulation of stomatal movement |
| BnaC02g02910D | AT2G46030 | 3E-85 | UBIQUITIN-CONJUGATING ENZYME 6 | Ubiquitin-dependent protein catabolic process |
| BnaC02g02900D | AT5G03840 | 0 | TERMINAL FLOWER 1, TFL1 | Controls inflorescence meristem identity |
| BnaC02g02880D | AT5G64140 | 1E-55 | RIBOSOMAL PROTEIN S28 | Ribosomal small subunit assembly and translation |
| BnaC02g02870D | AT5G03860 | 0 | MALATE SYNTHASE | Encodes a protein with malate synthase activity |
| BnaC02g02830D | AT5G03940 | 0 | 54 CHLOROPLAST PROTEIN | Protein import into chloroplast thylakoid membrane |
| BnaC02g02820D | ||||
| BnaC06g25530D | AT1G68990 | 0 | MALE GAMETOPHYTE DEFECTIVE 3 | Transcription of mitochondrial genes |
| BnaC06g25500D | AT1G69120 | 0 | APETALA1, AP1 | Inflorescence meristem identity, specifies floral meristem and sepal identity |
| BnaC06g25460D | AT3G08900 | 2E-48 | REVERSIBLY GLYCOSYLATED POLYPEPTIDE 3 | UDP-L-arabinose metabolic process |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



