
Submitted:
25 July 2023
Posted:
26 July 2023
You are already at the latest version
Abstract
Hepatocellular carcinoma (HCC) poses a significant global health concern, with its incidence steadily increasing. The development of HCC is a multifaceted, multi-step process involving alterations in various signaling cascades. In recent years, significant progress has been made in understanding the molecular signaling pathways that play central roles in hepatocarcinogenesis. In particular, the EGFR/PI3K/Akt/mTOR signaling pathway in HCC has garnered renewed attention from both basic and clinical researchers. Preclinical studies in vitro and in vivo have shown the effectiveness of targeting the key components of this signaling pathway in human HCC cells. Thus, targeting these signaling pathways with small molecule inhibitors holds promise as a potential therapeutic option for patients with HCC. In this review, we will explore recent advancements in understanding the role of the EGFR/PI3K/Akt/mTOR signaling pathway in HCC and assess the effectiveness of targeting this signaling cascade as a potential strategy for HCC therapy based on preclinical studies
Keywords:
hepatocellular carcinoma
; EGFR/PI3K/AKT/mTOR signaling
; animal models
; Targeted Therapy
1. Introduction
Liver cancer presents a significant global health challenge due to its rising incidence and mortality rates. According to the World Health Organization, liver cancer accounted for approximately 800,000 deaths, positioning it as the fourth leading cause of cancer-related mortality[1]. Liver cancer comprises a heterogeneous group of malignant tumors with varying histologic characteristics and poor prognoses. Among these, hepatocellular carcinoma (HCC) represents the most prevalent primary liver cancer, accounting for approximately 80% of cases, followed by cholangiocarcinoma (CC), which contributes to 10-20% of primary liver cancers[2,3].
Existing treatment modalities for HCC offer limited success, with a considerably low 5-year survival rate. Surgical resection or local ablation therapy is typically favored for early-stage HCC; however, tumor recurrence occurs in approximately 70% of patients within 5 years[1,4]. For advanced HCC, systemic therapy is recommended as the standard treatment option, but overall prognosis has been unsatisfactory[1,2].
HCC is a heterogeneous tumor with diverse phenotypic and genetic characteristics, and its tumorigenesis involves a range of molecular mechanisms[5,6]. Extensive research on the molecular pathogenesis of HCC has identified several critical signaling pathways involved in tumor initiation, promotion, and metastasis. These pathways include the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), Wnt/β-catenin, Hedgehog (HH), Hippo-YAP/TAZ, and the phosphatidylinositol 3-kinase /AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathways. Recently, we have extensively reviewed the roles of MAPK/ERK, Wnt/β-catenin, HH, and Hippo-YAP/TAZ signaling in HCC development, together with preclinical and clinical studies targeting the signaling pathways in HCC[7,8].
In this review, our aim is to provide an overview of the tumor-promoting effects exerted by the PI3K/AKT/mTOR signaling pathway in hepatocellular carcinoma (HCC), which is primarily triggered by the epidermal growth factor receptor (EGFR). Additionally, we will discuss the results from recent preclinical and clinical studies that target the EGFR/PI3K/AKT/mTOR signaling cascade as a potential therapeutic approach for treating HCC.
2. Role of EGFR/PI3K/AKT/mTOR signaling pathway in HCC
2.1. Overview of EGFR/PI3K/AKT/mTOR signaling pathway
Epidermal Growth Factor Receptor (EGFR) is a cell surface receptor that belongs to the ErbB family of receptor tyrosine kinases. EGFR is activated by binding of specific ligands, such as epidermal growth factor (EGF) or transforming growth factor-alpha (TGF-α). Ligand binding induces receptor dimerization, leading to the activation of the intrinsic kinase activity of EGFR. The catalytic domain of EGFR kinases carries out transphosphorylation process by which an array of tyrosine residues at the c-terminal cytoplasmic domain of EGFR are phosphorylated by its dimerized partner. Phosphotyrosine (pY) at the cytoplasmic domain of an activated EGFR acts as a docking site for proteins containing Src homology 2 (SH2) domains, such as Phosphatidylinositol 3-kinase (PI3K). The SH2 domain of PI3K binds to phosphorylated tyrosine residues on EGFR, bringing PI3K in close proximity to the plasma membrane. Upon recruitment, PI3K converts phosphatidylinositol-4,5-disphosphate (PIP2) in the plasma membrane into phosphatidylinositol-3,4,5-trisphosphate (PIP3)[9,10]. This conversion is carried out by the lipid kinase activity of the catalytic subunit of PI3K. The conversion of PIP2 to PIP3 can be antagonized by lipid phosphatases PTEN that converts PIP3 into PIP2[11]. PIP3 serves as a docking site for proteins containing pleckstrin homology (PH) domains, such as the serine/threonine kinase AKT (also known as protein kinase B). Thus, the presence of PH domains in AKT allows the serine/threonine kinase to be recruited to the plasma membrane where AKT is phosphorylated at two critical residues: Thr308 by phosphoinositide-dependent kinase 1 (PDK1) and Ser473 by mammalian target of rapamycin complex 2 (mTORC2). These phosphorylation events lead to full activation of AKT[12]. Fully activated AKT phosphorylates and regulates numerous downstream effectors, including proteins involved in cell survival, protein synthesis, metabolism, and cell cycle progression. One of the key targets of activated AKT is the mammalian target of rapamycin (mTOR), which exists in two complexes: mTORC1 and mTORC2 (Figure 1).
The mammalian target of rapamycin(mTOR) is a serine/ threonine protein kinase and regulates tumor growth, proliferation, metabolism, cell growth and immunity[13]. mTOR has two complexes, which are mTORC1 and mTORC2. mTORC1 consists of mTOR, regulator associated protein of mTOR (Raptor), DEP domain-containing mTOR interacting protein (DEPTOR), mammalian lethal with SEC13 protein 8 (mLST8), and proline rich AKT substrate 40 (PRAS40). mTORC2 contains rapamycin insensitive companion of mTOR (Rictor), DEPTOR, mLST8, rictor-1 (Protor-1), Protor-2, and mammalian stress-activated protein kinase-interacting protein 1 (mSin1). mTORC1 is involved in cell growth and proliferation through nutritional sensing, and mTORC2 regulates the rebuilding of cytoskeletons and the cell survival[14]. To activate mTORC1, RHEB, a small GTPase, is required. However, RHEB is usually inactivated by TSC1/2, a GTPase activating protein (GAP) inhibitor[15]. The activated AKT can inhibits TSC1/2 and let RHEB activated, resulting in the activation of mTORC1 and its downstream targets. The active mTORC1 increases protein synthesis by phosphorylating the eukaryotic translation inhibition factor 4E binding protein1(4EBP1) and ribosomal proteins S6 kinase 1/2 (S6K1/2). Considering that these cellular processes are closely involved in carcinogenesis, it is no surprise that the dysregulation of mTOR pathway has been observed in multiple human solid tumors[16].
2.2. Activation of EGFR/PI3K/AKT/mTOR signaling pathway in HCC
The EGFR/PI3K/AKT/mTOR signaling pathway is a major pathway in diverse types of cancers. The pathway actively regulates various aspects of cellular processes including cell proliferation, survival, migration, and metabolism. It also plays a pivotal role in regulating tumor microenvironment via angiogenesis and recruitment of inflammatory cells[17]. As well, the EGFR/PI3K/AKT/mTOR signaling is significantly involved in therapy response and metastasis[18,19]. Considering the multifaceted roles in carcinogenesis, it is no surprise that the signaling pathway is commonly unregulated in a variety of human cancers[20].
In HCC, the EGFR/PI3K/AKT/mTOR pathway is abnormally activated in approximately 50% of cases, and this dysregulated activation is involved in various cellular processes, including cell proliferation, tumor cell differentiation, autophagy, metabolism, and epithelial-mesenchymal transition (EMT)[14]. EGFR overexpression occurs in 68% of human HCC cases and significantly correlates with metastasis, poor patient survival, and aggressive tumors[21]. Moreover, EGFR ligands are overexpressed in human liver cancer cells and tumor tissues[22]. In higher stages of HCC, this signaling pathway is associated with vascular invasion, poor differentiation, and reduced survival rates[23]. Additionally, it has been reported that 40% of HCC patients who underwent orthotopic liver transplantation exhibit elevated mTOR activity[24].
PTEN suppresses AKT activation by converting PIP3 to PIP2. Loss of the tumor suppressor PTEN and reduced expression of PTEN are primarily observed in the majority of patients with HCC in the early stages[25,26]. Somatic mutations in PTEN have been reported in HCC tissues[27,28]. Moreover, Fujiwara et al. observed 12 allelic losses of PTEN in 37 patients with HCC[29]. Loss-of-function mutations in TSC1/2 are found in approximately 20% of patients with HCC, which serve as another major suppressor of the EGFR/PI3K/AKT/mTOR signaling pathway[30].
3. In vitro studies investigating EGFR/PI3K/AKT/mTOR signaling in HCC cell lines
3.1. Tumorigenic roles of EGFR/PI3K/AKT/mTOR signaling in HCC cells
The activation of the EGFR/PI3K/AKT/mTOR pathway has been observed in various cancer types. Although ligand binding to EGFR is the natural process that induces dimerization of the receptors and subsequent phosphorylation of the cytoplasmic tails, overexpression of EGFR alone can lead to enhanced formation of its dimerization and activation of the downstream signaling pathway in the absence of its ligands. EGFR overexpression can be achieved through various mechanisms[31]. In addition to gene amplification and epigenetic upregulation of the EGFR gene, changes in positive or negative regulators of EGFR can also affect its abundance in cancer cells. For example, NT5DC2 suppresses the ubiquitination of EGFR, preventing its ubiquitin-mediated proteasome degradation, and leading to increased EGFR levels. In HCC cell lines such as MHCC97H and PLC/RLF/5, upregulated NT5DC2 induced overexpression of EGFR and activation of the downstream PI3K/AKT/mTOR signaling pathway[32]. Similarly, Song et al. reported that 14-3-3σ can interact with EGFR and stabilize the receptor, prolonging the activation of EGFR signaling in Huh7 and HepG2 cells[33]. Tropomodulin 3 (TMOD3), a member of the pointed end capping protein family, is significantly upregulated in HCCs and correlated with poor survival of patients with HCC. In various HCC cell lines, including Huh-7 and Hep3B, TMOD3 was found to facilitate the phosphorylation of the cytoplasmic tail of EGFR, triggering activation of the downstream PI3K/AKT/mTOR signaling cascade[34].
In addition to EGF/EGFR-mediated activation of the PI3K/AKT/mTOR signaling pathway, various molecules can contribute to eliciting the pathway. Recently, DEAD/DEAH box helicase 11 (DDX11) and Apelin (APLN) were identified as activators of the PI3K/AKT/mTOR signaling pathway in HCC. Overexpression and/or knockdown of these molecules substantially altered the PI3K/AKT/mTOR signaling in HCC cell lines and significantly affected the tumorigenic potentials of the HCC cells[35,36]. Alpha-fetoprotein (AFP), strongly correlated with the aggressiveness of HCC, is a serum biomarker routinely used for the diagnosis and prognosis of HCC. Recently, Wang et al. investigated the role of AFP in HCC using two HCC cell lines. They reported that AFP interacted with PTEN, a negative regulator of AKT, and activated the PI3K/AKT/mTOR signaling pathway[37]. The role of claudin-6 (CLDN6) was investigated in HepG2 cells where CLDN6 was found to activate the EGFR/PI3K/AKT/mTOR signaling pathway. Knockdown of CLDN6 led to decreases in proliferation, migration, and invasion of HepG2 cells[38]. MicroRNAs (miRNAs) can also contribute to the activation of the EGFR/PI3K/AKT/mTOR signaling pathway. For example, miR-494-3p is correlated with aggressive clinicopathological characteristics and poor prognosis in HCC patients. Lin et al. showed that miR-494-3p bound to the 3′UTR of PTEN mRNA and repressed its translation in two HCC cell lines, SMMC7721 and HCCLM3[39]. Notably, the suppression of PTEN expression by ectopic expression of miR-494-3p led to the activation of the PI3K/AKT/mTOR pathway and enhanced metastasis potentials of HCC cells[39]. Another in vitro study using the PLC/PRF/5 HCC cell line showed that Mac-2-binding protein glycan isomer (M2BPGi) activated mTOR and exerted tumor-promoting effects on HCC[40].
3.2. Anti-tumor effects of targeting EGFR/ PI3K/AKT/mTOR pathway in HCC cells
Given that the EGFR/PI3K/AKT/mTOR signaling pathway exerts strong tumor-promoting effects on HCC, it is reasonable to attempt to inhibit the pathway for the treatment of HCC. EGFR is a direct target of miRNA-133b. Overexpression of miRNA-133b significantly suppressed EGFR protein expression and led to decreased activities of PI3K, AKT, and mTOR in the HepG2 cells[41]. Of note, inhibition of EGFR/PI3K/AKT/mTOR pathway via overexpression of miRNA-133b induced activation of caspase-3/-8 and apoptotic cell deaths in HepG2 cells. Methyltransferase like 14 (METTL14) can destabilize EGFR mRNA via an N6-methyladenosine (m6A) RNA methylation. METTL14 is significantly downregulated in HCC and associated with poor prognosis of HCC patients[42]. Similarly, an m6A-binding protein, YTH-Domain Family Member 2 (YTHDF2) binds to m6A sites in 3’UTR of EGFR mRNA and promote the degradation of EGFR mRNA in HCC cells. In HCC cells such as HEP3B and SMMC7721, overexpression of YTHDF2 suppressed cell proliferation via destabilizing the EGFR mRNA and thus acted as a tumor suppressor[43]. In summary, inhibiting EGFR by overexpression of endogenous EGFR suppressors such miRNA-133b, METTL14, and YTHDF2 led to subsequent inactivation of the downstream PI3K/AKT/mTOR signaling pathway and effectively reduced the malignancy of HCC cells by inducing cell apoptosis and suppressing cell proliferation.
In line with the observations in HCC cells characterized by overexpression of endogenous suppressors of EGFR, pharmacological inhibition of EGFR in HCC cell lines elicits similar effects. Specifically, it leads to the suppression of cell proliferation and the induction of apoptosis in HCC cells through the downregulation of PI3K, p-AKT, and p-mTOR. For instance, treatment of HepG2 cells with GW2974, an EGFR inhibitor, induced the attenuation of the downstream PI3K/AKT/mTOR signaling pathway, resulting in decreased cell proliferation and increased apoptosis in HCC cells[41]. Moreover, the inhibition of EGFR using erlotinib demonstrated inhibitory effects on the migratory capabilities and cell proliferation in HCC cells[32,44]. Similarly, the administration of EGFR inhibitors AG1478 and Gefitinib led to reduced cell proliferation, decreased invasion, and enhanced apoptosis in HCC cells[38,45].
In addition to targeting EGFR, inhibition of its downstream effectors, such as PI3K, AKT, and/or mTOR exerted similar tumor-suppressing effects on HCC cells (Table 1) Treatment with LY294002 or Wortmannin, potent inhibitors of PI3K, downregulated the phosphorylated levels of AKT, and induced apoptosis in HCC cells such as MHCC97 and Huh7[46]. Treatment of HCC cells with a PI3K inhibitor 740Y-P showed similar effects[47]. These results consistently show that the inhibition of PI3K leads to the suppression of tumor growth and the induction of apoptosis. Cell cycle arrest was also observed in HCC cells when they were treated with inhibitors of PI3K. For example, treatment of Huh7 and HepG2 cells with copanlisib induced cell cycle arrest via the downregulation of CDK4/6 and cyclin D1, although the treatment had a minor effect on apoptosis[48].
MK2206 effectively interacts with the pleckstrin-homology (PH) domain of AKT and hinders its recruitment to the plasma membrane, inhibiting PDK1 binding and subsequent activation of AKT[49]. MK2206 has shown strong potency in inhibiting AKT[50]. Similar to the findings in HCC cells treated with PI3K inhibitors, treatment with MK2206 also induced growth inhibition and apoptosis in HCC cells[51,52]. AKT Inhibitor VIII, which also blocks the activity of AKT through binding to the pleckstrin homology (PH) domain in AKT, suppressed cell growth and induced apoptosis in HCC cells[46].
Recent studies indicate that mTOR plays critical roles in maintaining stemness-related functions in cancer stem cells (CSCs), and inhibition of mTOR leads to sensitization of CSCs to radiation therapy in breast cancer[53]. In line with the findings, treatment with rapamycin, the prototypic mTOR inhibitor, significantly reduced the frequency of CD133+/EpCAM+ cells in Hep3B and Huh7, which are widely considered liver cancer stem cell population[54]. RAD001, also known as everolimus, is an inhibitor of mTOR. Its binding to the FK506-binding protein12 (FKBP12) allows the RAD001-FKBP12 complex to interact with mTOR, which subsequently inhibits S6K1 and 4EBP1 phosphorylation by mTOR. Treatment with RAD001 resulted in the induction of apoptosis[51], as well as a decrease in cell proliferation in diverse HCC cell lines including Hep3B, Huh7, and HepG2[51,52].
The PI3K/mTOR dual inhibitor BEZ235 induced growth inhibition and apoptosis in HCC cells[51]. Similarly, treatment of HCC cells with Lenvatinib, which targets both AKT and mTOR, exhibited inhibitory effects on cell proliferation and migration[55]. In summary, targeting the EGFR/PI3K/AKT/mTOR signaling pathway via the inhibition of EGFR, PI3K, AKT, and/or mTOR have consistently shown anti-tumor effects on HCC cells in vitro (Table 1).
3.3. Targeting EGFR/PI3K/AKT/mTOR signaling on sorafenib-resistant HCC cells
Sorafenib is the first-line systemic therapeutic for patients with advanced HCC which inhibits both receptor tyrosine kinases (RTKs) and RAF[56]. However, the development of resistance to the drug and disease progression are nearly inevitable during the course of the treatment[57]. Sorafenib resistance appears to be associated with the activation of the PI3K/AKT/mTOR signaling[58]. Therefore, the combination of sorafenib and an inhibitor of EGFR/PI3K/AKT/mTOR signaling pathway has been proposed as an effective therapeutic approach[45,52,59].
Copanlisib, a PI3K inhibitor, down-regulates downstream targets of AKT, leading to cell cycle arrest and suppression of cell proliferation, although it has a minimal effect on apoptosis[48,60]. Copanlisib counteracted the sorafenib-induced AKT phosphorylation and synergistically enhanced anti-tumor effects on HCC when combined with sorafenib[48]. Likewise, combined treatment with sorafenib and capsaicin, an inhibitor of the PI3K/AKT/mTOR signaling pathway also showed enhanced anti-tumor effects in Hep3B and HuH7 cells[61].
Lenvatinib is another first-line treatment for HCC which also inhibits RTKs. The combination of lenvatinib and copanlisib effectively suppressed the phosphorylation of AKT, which had been induced by the treatment with lenvatinib. Copanlisib enhanced pro-apoptotic effects on HCC cell lines that were resistant to lenvatinib[45]. Moreover, sequential treatment of Huh7 cells with rapamycin, an mTOR inhibitor, following the treatment with sorafenib substantially increased the sensitivity of HCC cells to sorafenib, and decreased the frequency of liver cancer stem cell (CSC)-like cells, which are considered primary cells resistant to chemotherapy[54].
In summary, combination of sorafenib or lenvatinib with agents targeting the PI3K/AKT/mTOR signaling can enhance the anticancer activity of the RTK inhibitors and is expected to overcome the emergence of therapy-resistant cells.
4. Animal studies investigating EGFR/PI3K/AKT/mTOR signaling in HCC
4.1. Animal models for HCC induced by activated EGFR/PI3K/AKT/mTOR signaling
Studies have demonstrated the induction of hepatocarcinogenesis through modification of genes involved in the PI3K/AKT/mTOR signaling pathway (Table 2). PTEN functions as a negative regulator of PI3K/AKT/mTOR signaling because it counteracts PI3K-mediated AKT activation (Figure 1). For the inactivation of PTEN specifically in the liver, genetically engineered mice are used that carry two Pten alleles flanked by loxP sites as well as the Cre transgene under the promoter of albumin. These mice (referred to as “AlbCre; Ptenfl/fl mice”) were prone to the development of HCC as well as intrahepatic cholangiocarcinoma (ICC). Tumors from the mice exhibited significant increases in the phosphorylated level of AKT, confirming the activation of the PI3K/AKT/mTOR signaling in the Pten-deleted livers[62].
Recently, simple liver-specific transgenesis has been developed that allows liver in adult mice to be transfected with DNA. This methodology, called hydrodynamic tail vein injection or simply HTVI, employs the application of physical force through the rapid injection of a large volume of DNA solution into the lateral tail vein. This process generates increased pressure within the vena cava, propelling the DNA solution into the large hepatic vein and eventually reaching the hepatic tissue and hepatocytes[63,64]. For genome editing in the liver using the CRISPR/Cas system, plasmids containing Cas9 and sgRNA are delivered to the liver via the HTVI method[65]. Using CRISPR-based gene editing combined with the HTVI method, the Pten gene was ablated in a subset of hepatocytes in murine livers[66]. In this setting, however, tumor failed to develop in the mice (referred to as “sgPten mice”). The discrepancy in the results between the sgPten and AlbCre; Ptenfl/fl mice is thought to be partially due to the lower frequency of hepatocytes having undergone the loss of PTEN in the sgPten mice compared with that in the AlbCre; Ptenfl/fl mice. Moreover, in the case of the latter, the Pten gene is deleted during early embryogenesis, as opposed to the sgPten mice in which the deletion is induced in adult livers. Considering massive cell divisions during embryonic and fetal development, deletion of Pten in embryos is expected to create more favorable tissue environment for hepatocarcinogenesis[67]. Although tumor failed to develop in the sgPten mice, concomitant c-Met overexpression in the liver led to the formation of hepatic adenomas (HCA) as well as HCC[66].
Plasmids delivered to liver via HTVI remain as episomes in the nucleus and thus are prone to degradation. To overcome this limitation, the Sleeping Beauty transposon system is often coupled with HTVI, which allows gene of interest to be integrated into the genome. HTVI of transposons encoding an activated form of AKT (myr-AKT1) alone induced HCC after 24 weeks post HTVI[68,69]. Notably, simultaneous expression of myr-AKT1 and c-Met significantly accelerated HCC development, causing complete lethality by 8 weeks post HTVI[68]. Likewise, co-expression of myr-AKT1 and N-RasG12V led to rapid emergence of HCC[69]. The studies indicate that oncogenic AKT synergistically cooperate with another oncogene in the development of HCC.
TSC1 is as an upstream inhibitor of mTOR by suppressing an mTOR activator, RHEB (Figure 1). Liver-specific knockout of Tsc1 using a similar method as described in the AlbCre; Ptenfl/fl mice resulted in development of HCC by the age of 9-10 months old[70]. Similar experiments using the AlbCre; Tsc1fl/f mice conducted by other groups also showed HCC development with a minor presence of ICC by 40 weeks of age. Tumors from the AlbCre; Tsc1fl/f mice consistently exhibited activation of mTOR signaling[62,71]. Of note, concomitant deletion of both Tsc1 and Pten in the liver gave rise to rapid induction of HCC, usually around the age of 14 weeks, showing that simultaneous deletion of the two major negative regulators of the PI3K/AKT/mTOR signaling strongly enhanced the signaling pathway and carcinogenesis in the liver[62].
ICC; intrahepatic cholangiocarcinoma, HCA; hepatocellular adenoma, HCC, hepatocellular carcinoma.
4.2. Preclinical animal studies targeting EGFR/PI3K/AKT/mTOR signaling in HCC
Since the activation of the EGFR/PI3K/AKT/mTOR signaling pathway significantly contributes to HCC development, various inhibitors of this signaling pathway have been administered to murine models of HCC to investigate their effects on HCC in vivo (Table 3).MUC15 is a member of the high-molecular-weight glycoprotein family of Mucins. It has been shown to bind to EGFR and induce EGFR degradation, subsequently suppressing the downstream PI3K/AKT/mTOR signaling pathway[72]. In xenograft models of HCC, mice transplanted with HCCLM3 cells overexpressing MUC15 displayed fewer and smaller tumors, leading to increased survival rates compared to control mice transplanted with HCC cells overexpressing green fluorescent protein (GFP), an inert protein. Additionally, the degradation of EGFR by MUC15 resulted in fewer lung metastases in the MUC15 group compared to the control[72]. RHEB, an activator of mTOR, was also targeted in a xenograft model of HCC. The downregulation of RHEB using short hairpin RNA (shRNA) led to the suppression of tumor growth in vivo[73].
Pharmacological inhibition of PI3K, AKT, or mTOR exerted similar anti-cancer effects in xenograft models of HCC. Recently, a novel small-molecule inhibitor of PI3K, DZW-310, was developed. Administration of the drug to a Hep3B xenograft tumor model resulted in a significant decrease in tumor growth[74]. As observed in in vitro studies using HCC cells (see above in section 3.2), administration with an AKT inhibitor, MK2206 led to growth inhibition of tumors in xenograft models transplanted with Hep3B and Huh7[51]. A novel dual inhibitor was developed that simultaneously inhibits AKT and mTOR. The compound, ZJQ-24, suppressed tumor growth in HepG2 xenograft mice in a dose-dependent manner[75].
Rapamycin (sirolimus) and its derivatives, RAD001 (also known as everolimus) function as highly selective allosteric inhibitors of mTORC1. They bind tightly to its primary target, the FK506-binding protein12 (FKBP12), which allows the complex to associate with the FKBP-rapamycin binding domain (FRB domain) of mTOR, which is located proximal to the active site of mTOR kinase. The formation of the FKBP12-rapamycin-mTOR complex restricts the access of mTOR substrates, and thus inhibits phosphorylation and activation of S6K1 and 4EBP1[76,77]. In patient-derived HCC xenograft models, treatment with RAD001 led to significant reduction in tumor growth in a dose-dependent manner[78]. Molecular analysis confirmed significant decreases in the phosphorylated levels of S6K1 and 4EBP1, while the level of phosphorylated mTOR was not altered. RAD001-induced growth suppression was associated with inhibition of cell proliferation via the downregulation of Cdk-6, Cdk-2, Cdk-4, cdc-25C, cyclin B1 and c-Myc[78]. Treatment with rapamycin alone in the HepG2 xenograft model, however, showed no significant benefit to animal survival compared to control mice treated with the vehicle[79]. Instead, combination therapy with rapamycin and bevacizumab, a monoclonal antibody targeting vascular endothelial growth factor (VEGF), led to reduced tumor sizes as well as prolonged survival in the xenograft mice. Although the discrepancy in the results between RAD001 and rapamycin treatments is not clear, it is speculated that xenograft mice transplanted with HCC directly derived from patients might better represent the characteristics of human HCC compared to those transplanted with HepG2, which had been long maintained in tissue culture in vitro. Metformin inhibits mTOR via the activation of AMPK, a strong suppressor of mTOR. It is noteworthy that treatment with metformin also retarded tumor development in HCC xenograft models transplanted with Bel-7402 cells[80].
5. Clinical studies targeting EGFR/PI3K/AKT/mTOR signaling
Given the significant roles of the EGFR/PI3K/AKT/mTOR pathway in human cancer, there has been a strong emphasis on investigating its clinical potential through targeting the major components of the EGFR/PI3K/AKT/mTOR signaling pathway using specific small-molecule inhibitors. For instance, copanlisib, an inhibitor specific to PI3K, was administered to patients with cancers carrying an activating mutation in PI3K[81].
Combinatory inhibition of mTOR and VEGF using rapamycin and bevacizumab significantly reduced tumor growth in xenograft models of HCC[79](Table 3). A phase I clinical trial explored the combination of rapamycin with bevacizumab at the maximum tolerated dose in patients with HCC, demonstrating a complete response in one case and stable disease states in a majority. Phase I/II clinical trials are ongoing to further investigate efficacy of the combination therapy in HCC [82]. Erlotinib is an EGFR inhibitor that effectively suppress phosphorylation of its downstream effectors, such as AKT and mTOR. Combined inhibition of EGFR and VEGF using erlotinib and bevacizumab showed similar anti-cancer effects in patients with advanced HCC [83].
Various drugs have been employed to inhibit mTOR in clinical studies of HCC[82]. A phase IV study aims to confirm if everolimus can suppress liver tumor relapse and improve patient condition following liver transplantation (NCT02081755). HCC patients treated with the mTOR inhibitor sirolimus (rapamycin) exhibited a significantly longer overall survival (OS) compared to the control group (median survival of 12 and 8 months, respectively)[84]. A phase II clinical study demonstrated that sirolimus extended survival of patients with HCC after liver transplantation (NCT01374750). Onatasertib (CC-223), an orally administered mTOR inhibitor, has been found to induce mitochondrial dysfunction in HCC cell lines[85]. In an open-label phase II trial (NCT03591965), researchers explored the use of onatasertib in subjects with hepatitis B virus (HBV)-positive HCC who had previously undergone at least one line of therapy. Another completed phase I/II study investigated AZD8055, a novel ATP-competitive mTOR kinase inhibitor, evaluating its safety, tolerability, pharmacokinetics, and preliminary efficacy[86]. Overall, although some clinical trials have shown that mTOR inhibitors can provide a survival benefit in patients with advanced HCC, especially when they were intolerant to sorafenib (a standard first-line therapy for advanced HCC), not all trials have demonstrated a significant improvement in overall survival with the treatment of an mTOR inhibitor.
In an attempt to improve outcomes, a single-arm phase II trial combined temsirolimus and sorafenib, aiming to exploit mTOR inhibition along with sorafenib's effects on HCC [87]. However, while the combination was deemed safe, it fell short of achieving the desired target effectiveness. Despite this setback, ongoing research continues to explore drug combinations, and several clinical studies are underway[88,89]. It is noteworthy that while mTOR inhibitors as well as other therapeutics targeting the EGFR/PI3K/AKT/mTOR pathway have shown some promise in preclinical studies, their efficacy in patients with advanced HCC remains uncertain. These findings highlight the complexity of targeting the EGFR/PI3K/AKT/mTOR pathway in HCC and underscore the need for further investigation to identify more effective treatment approaches.
6. Perspectives and Conclusions
A comprehensive understanding of the molecular pathway leading to carcinogenesis is crucial for predicting patient responses to targeted therapies, significantly impacting clinical decision-making. Development of HCC is a complex process, involving various alterations in multiple signaling cascades [5,6]. Among the various oncogenic signals, the EGFR/PI3K/AKT/mTOR pathway stands out as it is activated in over 50% of HCC cases, making it a crucial target for patients with this condition. Research has emphasized the pivotal role of the EGFR/PI3K/AKT/mTOR cascade in the development of HCC. Promisingly, preclinical studies using human HCC cells have consistently demonstrated the effectiveness of targeting the key components of this signaling pathway. Such interventions have shown to suppress the proliferation of tumor cells in vitro and inhibit the growth of HCC in vivo, offering hope for potential therapeutic approaches.
While preclinical studies have shown promise in targeting the PI3K/AKT/mTOR pathway, however, clinical trials have not consistently demonstrated significant improvement in patient outcomes. The overall response rate and survival benefits observed in clinical studies targeting the EGFR/PI3K/AKT/mTOR signaling pathway have been modest or disappointing. These less-than-satisfactory outcomes can be attributed, in part, to the significant toxicities and adverse effects caused by PI3K/AKT/mTOR inhibitors, negatively impacting patients' quality of life and leading to treatment discontinuation. However, there is hope for the future as new drugs are anticipated to be developed soon, specifically targeting the EGFR/PI3K/AKT/mTOR pathway while minimizing toxicities and adverse effects.
Funding
This work was supported by a grant from Kyung Hee University in 2023 (KHU- 20230902 awarded to SWR).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat Rev Dis Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J Hepatol 2020, 72, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N Engl J Med 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Moon, H.; Ro, S.W. MAPK/ERK Signaling Pathway in Hepatocellular Carcinoma. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Park, H.; Baek, J.; Moon, H.; Ro, S.W. Target Therapy for Hepatocellular Carcinoma: Beyond Receptor Tyrosine Kinase Inhibitors and Immune Checkpoint Inhibitors. Biology (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014, 13, 140–156. [Google Scholar] [CrossRef]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Moten, A.; Lin, H.K. Akt: a new activation mechanism. Cell Res 2014, 24, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.J.; Wankell, M.; Palamuthusingam, P.; McFarlane, C.; Hebbard, L. Targeting the PI3K/Akt/mTOR Pathway in Hepatocellular Carcinoma. Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther 2021, 6, 425. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis 2020, 11, 797. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front Oncol 2022, 12, 819128. [Google Scholar] [CrossRef]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin Cancer Biol 2022, 85, 69–94. [Google Scholar] [CrossRef]
- Harada, K.; Shiota, G.; Kawasaki, H. Transforming growth factor-alpha and epidermal growth factor receptor in chronic liver disease and hepatocellular carcinoma. Liver 1999, 19, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Takeda, T.; Sakon, M.; Tsujimoto, M.; Higashiyama, S.; Noda, K.; Miyoshi, E.; Monden, M.; Matsuura, N. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br J Cancer 2001, 84, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med Oncol 2010, 27, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Sieghart, W.; Fuereder, T.; Schmid, K.; Cejka, D.; Werzowa, J.; Wrba, F.; Wang, X.; Gruber, D.; Rasoul-Rockenschaub, S.; Peck-Radosavljevic, M.; et al. Mammalian target of rapamycin pathway activity in hepatocellular carcinomas of patients undergoing liver transplantation. Transplantation 2007, 83, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Chiang, D.Y.; Newell, P.; Peix, J.; Thung, S.; Alsinet, C.; Tovar, V.; Roayaie, S.; Minguez, B.; Sole, M.; et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008, 135, 1972–1983. [Google Scholar] [CrossRef]
- Chen, D.; Li, Z.; Cheng, Q.; Wang, Y.; Qian, L.; Gao, J.; Zhu, J.Y. Genetic alterations and expression of PTEN and its relationship with cancer stem cell markers to investigate pathogenesis and to evaluate prognosis in hepatocellular carcinoma. J Clin Pathol 2019, 72, 588–596. [Google Scholar] [CrossRef]
- Kawamura, N.; Nagai, H.; Bando, K.; Koyama, M.; Matsumoto, S.; Tajiri, T.; Onda, M.; Fujimoto, J.; Ueki, T.; Konishi, N.; et al. PTEN/MMAC1 mutations in hepatocellular carcinomas: somatic inactivation of both alleles in tumors. Jpn J Cancer Res 1999, 90, 413–418. [Google Scholar] [CrossRef]
- Yao, Y.J.; Ping, X.L.; Zhang, H.; Chen, F.F.; Lee, P.K.; Ahsan, H.; Chen, C.J.; Lee, P.H.; Peacocke, M.; Santella, R.M.; et al. PTEN/MMAC1 mutations in hepatocellular carcinomas. Oncogene 1999, 18, 3181–3185. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Hoon, D.S.; Yamada, T.; Umeshita, K.; Gotoh, M.; Sakon, M.; Nishisho, I.; Monden, M. PTEN / MMAC1 mutation and frequent loss of heterozygosity identified in chromosome 10q in a subset of hepatocellular carcinomas. Jpn J Cancer Res 2000, 91, 287–292. [Google Scholar] [CrossRef]
- Ho, D.W.H.; Chan, L.K.; Chiu, Y.T.; Xu, I.M.J.; Poon, R.T.P.; Cheung, T.T.; Tang, C.N.; Tang, V.W.L.; Lo, I.L.O.; Lam, P.W.Y.; et al. TSC1/2 mutations define a molecular subset of HCC with aggressive behaviour and treatment implication. Gut 2017, 66, 1496–1506. [Google Scholar] [CrossRef]
- Wang, C.; Liao, Y.; He, W.; Zhang, H.; Zuo, D.; Liu, W.; Yang, Z.; Qiu, J.; Yuan, Y.; Li, K.; et al. Elafin promotes tumour metastasis and attenuates the anti-metastatic effects of erlotinib via binding to EGFR in hepatocellular carcinoma. J Exp Clin Cancer Res 2021, 40, 113. [Google Scholar] [CrossRef] [PubMed]
- Li, K.S.; Zhu, X.D.; Liu, H.D.; Zhang, S.Z.; Li, X.L.; Xiao, N.; Liu, X.F.; Xu, B.; Lei, M.; Zhang, Y.Y.; et al. NT5DC2 promotes tumor cell proliferation by stabilizing EGFR in hepatocellular carcinoma. Cell Death Dis 2020, 11, 335. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Liu, Y.; Liu, F.; Zhang, L.; Li, G.; Yuan, C.; Yu, C.; Lu, X.; Liu, Q.; Chen, X.; et al. The 14-3-3σ protein promotes HCC anoikis resistance by inhibiting EGFR degradation and thereby activating the EGFR-dependent ERK1/2 signaling pathway. Theranostics 2021, 11, 996–1015. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Yang, Y.; Hong, Y.G.; Wang, M.C.; Yuan, S.X.; Wang, Z.G.; Bi, F.R.; Hao, L.Q.; Yan, H.L.; Zhou, W.P. Tropomodulin 3 modulates EGFR-PI3K-AKT signaling to drive hepatocellular carcinoma metastasis. Mol Carcinog 2019, 58, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wong, C.C.; Liu, D.; Go, M.Y.Y.; Wu, B.; Peng, S.; Kuang, M.; Wong, N.; Yu, J. APLN promotes hepatocellular carcinoma through activating PI3K/Akt pathway and is a druggable target. Theranostics 2019, 9, 5246–5260. [Google Scholar] [CrossRef]
- Yu, Y.; Zhao, D.; Li, K.; Cai, Y.; Xu, P.; Li, R.; Li, J.; Chen, X.; Chen, P.; Cui, G. E2F1 mediated DDX11 transcriptional activation promotes hepatocellular carcinoma progression through PI3K/AKT/mTOR pathway. Cell Death Dis 2020, 11, 273. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, M.; Wang, Q.; Hou, Y.; Li, L.; Weng, H.; Zhao, Y.; Chen, D.; Guo, J.; Ding, H.; et al. Publisher Correction: Alpha-fetoprotein inhibits autophagy to promote malignant behaviour in hepatocellular carcinoma cells by activating PI3K/AKT/mTOR signalling. Cell Death Dis 2019, 10, 214. [Google Scholar] [CrossRef]
- Huang, L.; Zhao, C.; Sun, K.; Yang, D.; Yan, L.; Luo, D.; He, J.; Hu, X.; Wang, R.; Shen, X.; et al. Downregulation of CLDN6 inhibits cell proliferation, migration, and invasion via regulating EGFR/AKT/mTOR signalling pathway in hepatocellular carcinoma. Cell Biochem Funct 2020, 38, 541–548. [Google Scholar] [CrossRef]
- Lin, H.; Huang, Z.P.; Liu, J.; Qiu, Y.; Tao, Y.P.; Wang, M.C.; Yao, H.; Hou, K.Z.; Gu, F.M.; Xu, X.F. MiR-494-3p promotes PI3K/AKT pathway hyperactivation and human hepatocellular carcinoma progression by targeting PTEN. Sci Rep 2018, 8, 10461. [Google Scholar] [CrossRef]
- Dolgormaa, G.; Harimoto, N.; Ishii, N.; Yamanaka, T.; Hagiwara, K.; Tsukagoshi, M.; Igarashi, T.; Watanabe, A.; Kubo, N.; Araki, K.; et al. Mac-2-binding protein glycan isomer enhances the aggressiveness of hepatocellular carcinoma by activating mTOR signaling. Br J Cancer 2020, 123, 1145–1153. [Google Scholar] [CrossRef]
- Wang, X.; Zeng, J.; Wang, L.; Zhang, X.; Liu, Z.; Zhang, H.; Dong, J. Overexpression of microRNA-133b is associated with the increased survival of patients with hepatocellular carcinoma after curative hepatectomy: Involvement of the EGFR/PI3K/Akt/mTOR signaling pathway. Oncol Rep 2017, 38, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhuang, Y.; Zhang, J.; Chen, M.; Wu, S. METTL14 Inhibits Hepatocellular Carcinoma Metastasis Through Regulating EGFR/PI3K/AKT Signaling Pathway in an m6A-Dependent Manner. Cancer Manag Res 2020, 12, 13173–13184. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Liao, D.; Zhang, M.; Zeng, C.; Li, X.; Zhang, R.; Ma, H.; Kang, T. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett 2019, 442, 252–261. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, H.; Li, M.; Wu, H.; Guo, Y.; Chen, J.; Shan, J.; Chen, X.; Shen, J.; Ma, Q.; et al. KIAA1199 promotes sorafenib tolerance and the metastasis of hepatocellular carcinoma by activating the EGF/EGFR-dependent epithelial-mesenchymal transition program. Cancer Lett 2019, 454, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Liu, J.; Wang, Y.; Dong, J. Co-administration of MDR1 and BCRP or EGFR/PI3K inhibitors overcomes lenvatinib resistance in hepatocellular carcinoma. Front Oncol 2022, 12, 944537. [Google Scholar] [CrossRef] [PubMed]
- Buontempo, F.; Ersahin, T.; Missiroli, S.; Senturk, S.; Etro, D.; Ozturk, M.; Capitani, S.; Cetin-Atalay, R.; Neri, M.L. Inhibition of Akt signaling in hepatoma cells induces apoptotic cell death independent of Akt activation status. Invest New Drugs 2011, 29, 1303–1313. [Google Scholar] [CrossRef]
- Gong, C.; Ai, J.; Fan, Y.; Gao, J.; Liu, W.; Feng, Q.; Liao, W.; Wu, L. NCAPG Promotes The Proliferation Of Hepatocellular Carcinoma Through PI3K/AKT Signaling. Onco Targets Ther 2019, 12, 8537–8552. [Google Scholar] [CrossRef]
- Ye, L.; Mayerle, J.; Ziesch, A.; Reiter, F.P.; Gerbes, A.L.; De Toni, E.N. The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell Death Discov 2019, 5, 86. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, Y.; Zhang, L.; Ren, X.; Huber-Keener, K.J.; Liu, X.; Zhou, L.; Liao, J.; Keihack, H.; Yan, L.; et al. MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Mol Cancer Ther 2012, 11, 154–164. [Google Scholar] [CrossRef]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 2010, 9, 1956–1967. [Google Scholar] [CrossRef]
- Ou, D.L.; Lee, B.S.; Lin, L.I.; Liou, J.Y.; Liao, S.C.; Hsu, C.; Cheng, A.L. Vertical blockade of the IGFR- PI3K/Akt/mTOR pathway for the treatment of hepatocellular carcinoma: the role of survivin. Mol Cancer 2014, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Grabinski, N.; Ewald, F.; Hofmann, B.T.; Staufer, K.; Schumacher, U.; Nashan, B.; Jücker, M. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol Cancer 2012, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Yu, X.; Lin, X.; He, S. Inhibition of mTOR sensitizes breast cancer stem cells to radiation-induced repression of self-renewal through the regulation of MnSOD and Akt. Int J Mol Med 2016, 37, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Kahraman, D.C.; Kahraman, T.; Cetin-Atalay, R. Targeting PI3K/Akt/mTOR Pathway Identifies Differential Expression and Functional Role of IL8 in Liver Cancer Stem Cell Enrichment. Mol Cancer Ther 2019, 18, 2146–2157. [Google Scholar] [CrossRef]
- Zhao, Z.; Song, J.; Zhang, D.; Wu, F.; Tu, J.; Ji, J. Oxysophocarpine suppresses FGFR1-overexpressed hepatocellular carcinoma growth and sensitizes the therapeutic effect of lenvatinib. Life Sci 2021, 264, 118642. [Google Scholar] [CrossRef]
- Llovet, J.M.; Hernandez-Gea, V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res 2014, 20, 2072–2079. [Google Scholar] [CrossRef]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim Biophys Acta Rev Cancer 2017, 1868, 564–570. [Google Scholar] [CrossRef]
- Samarin, J.; Laketa, V.; Malz, M.; Roessler, S.; Stein, I.; Horwitz, E.; Singer, S.; Dimou, E.; Cigliano, A.; Bissinger, M.; et al. PI3K/AKT/mTOR-dependent stabilization of oncogenic far-upstream element binding proteins in hepatocellular carcinoma cells. Hepatology 2016, 63, 813–826. [Google Scholar] [CrossRef]
- Gedaly, R.; Angulo, P.; Chen, C.; Creasy, K.T.; Spear, B.T.; Hundley, J.; Daily, M.F.; Shah, M.; Evers, B.M. The role of PI3K/mTOR inhibition in combination with sorafenib in hepatocellular carcinoma treatment. Anticancer Res 2012, 32, 2531–2536. [Google Scholar]
- Schneider, P.; Schön, M.; Pletz, N.; Seitz, C.S.; Liu, N.; Ziegelbauer, K.; Zachmann, K.; Emmert, S.; Schön, M.P. The novel PI3 kinase inhibitor, BAY 80-6946, impairs melanoma growth in vivo and in vitro. Exp Dermatol 2014, 23, 579–584. [Google Scholar] [CrossRef]
- Dai, N.; Ye, R.; He, Q.; Guo, P.; Chen, H.; Zhang, Q. Capsaicin and sorafenib combination treatment exerts synergistic anti-hepatocellular carcinoma activity by suppressing EGFR and PI3K/Akt/mTOR signaling. Oncol Rep 2018, 40, 3235–3248. [Google Scholar] [CrossRef] [PubMed]
- Kenerson, H.L.; Yeh, M.M.; Kazami, M.; Jiang, X.; Riehle, K.J.; McIntyre, R.L.; Park, J.O.; Kwon, S.; Campbell, J.S.; Yeung, R.S. Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology 2013, 144, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Calvisi, D.F. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 2014, 184, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.L.; Han, K.H.; Lee, J.D.; Ro, S.W. Transgenic mouse models generated by hydrodynamic transfection for genetic studies of liver cancer and preclinical testing of anti-cancer therapy. Int J Cancer 2016, 138, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Ro, S.W.; Seo, S.H.; Jeon, Y.; Moon, H.; Kim, D.Y.; Kim, S.U. Genetically Engineered Mouse Models for Liver Cancer. Cancers (Basel) 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Hu, J.; Cao, H.; Pilo, M.G.; Cigliano, A.; Shao, Z.; Xu, M.; Ribback, S.; Dombrowski, F.; Calvisi, D.F.; et al. Loss of Pten synergizes with c-Met to promote hepatocellular carcinoma development via mTORC2 pathway. Exp Mol Med 2018, 50, e417. [Google Scholar] [CrossRef]
- Moon, H.; Park, H.; Chae, M.J.; Choi, H.J.; Kim, D.Y.; Ro, S.W. Activated TAZ induces liver cancer in collaboration with EGFR/HER2 signaling pathways. BMC Cancer 2022, 22, 423. [Google Scholar] [CrossRef]
- Hu, J.; Che, L.; Li, L.; Pilo, M.G.; Cigliano, A.; Ribback, S.; Li, X.; Latte, G.; Mela, M.; Evert, M.; et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep 2016, 6, 20484. [Google Scholar] [CrossRef]
- Ho, C.; Wang, C.; Mattu, S.; Destefanis, G.; Ladu, S.; Delogu, S.; Armbruster, J.; Fan, L.; Lee, S.A.; Jiang, L.; et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology 2012, 55, 833–845. [Google Scholar] [CrossRef]
- Menon, S.; Yecies, J.L.; Zhang, H.H.; Howell, J.J.; Nicholatos, J.; Harputlugil, E.; Bronson, R.T.; Kwiatkowski, D.J.; Manning, B.D. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci Signal 2012, 5, ra24. [Google Scholar] [CrossRef]
- Luo, Y.D.; Fang, L.; Yu, H.Q.; Zhang, J.; Lin, X.T.; Liu, X.Y.; Wu, D.; Li, G.X.; Huang, D.; Zhang, Y.J.; et al. p53 haploinsufficiency and increased mTOR signalling define a subset of aggressive hepatocellular carcinoma. J Hepatol 2021, 74, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Chen, L.; Chen, H.Y.; Hu, L.; Li, L.; Sun, H.Y.; Jiang, F.; Zhao, J.; Liu, G.M.; Tang, J.; et al. MUC15 inhibits dimerization of EGFR and PI3K-AKT signaling and is associated with aggressive hepatocellular carcinomas in patients. Gastroenterology 2013, 145, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Pan, Z.; Zhang, J.; Ni, J.; Wang, C.; Wang, Z.; Gu, F.; Dong, W.; Zhou, W.; Liu, H. Overexpression of RHEB is associated with metastasis and poor prognosis in hepatocellular carcinoma. Oncol Lett 2018, 15, 3838–3845. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, X.; Liu, M.; Qin, X.; Wu, Q.; Ding, H.; Zhao, Q. DZW-310, a novel phosphoinositide 3-kinase inhibitor, attenuates the angiogenesis and growth of hepatocellular carcinoma cells via PI3K/AKT/mTOR axis. Biochem Pharmacol 2022, 201, 115093. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Zhang, J.; Liu, D.; Bao, Y.; Chen, T.; Tang, T.; Lin, J.; Luo, Y.; Jin, Y.; et al. Indole hydrazide compound ZJQ-24 inhibits angiogenesis and induces apoptosis cell death through abrogation of AKT/mTOR pathway in hepatocellular carcinoma. Cell Death Dis 2020, 11, 926. [Google Scholar] [CrossRef]
- Hausch, F.; Kozany, C.; Theodoropoulou, M.; Fabian, A.K. FKBPs and the Akt/mTOR pathway. Cell Cycle 2013, 12, 2366–2370. [Google Scholar] [CrossRef]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell 2010, 38, 768–774. [Google Scholar] [CrossRef]
- Huynh, H.; Chow, K.H.; Soo, K.C.; Toh, H.C.; Choo, S.P.; Foo, K.F.; Poon, D.; Ngo, V.C.; Tran, E. RAD001 (everolimus) inhibits tumour growth in xenograft models of human hepatocellular carcinoma. J Cell Mol Med 2009, 13, 1371–1380. [Google Scholar] [CrossRef]
- Ong, L.C.; Song, I.C.; Jin, Y.; Kee, I.H.; Siew, E.; Yu, S.; Thng, C.H.; Huynh, H.; Chow, P.K. Effective inhibition of xenografts of hepatocellular carcinoma (HepG2) by rapamycin and bevacizumab in an intrahepatic model. Mol Imaging Biol 2009, 11, 334–342. [Google Scholar] [CrossRef]
- Ling, S.; Song, L.; Fan, N.; Feng, T.; Liu, L.; Yang, X.; Wang, M.; Li, Y.; Tian, Y.; Zhao, F.; et al. Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int J Oncol 2017, 50, 297–309. [Google Scholar] [CrossRef]
- Damodaran, S.; Zhao, F.; Deming, D.A.; Mitchell, E.P.; Wright, J.J.; Gray, R.J.; Wang, V.; McShane, L.M.; Rubinstein, L.V.; Patton, D.R.; et al. Phase II Study of Copanlisib in Patients With Tumors With PIK3CA Mutations: Results From the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol Z1F. J Clin Oncol 2022, 40, 1552–1561. [Google Scholar] [CrossRef]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol 2014, 60, 855–865. [Google Scholar] [CrossRef]
- Thomas, M.B.; Garrett-Mayer, E.; Anis, M.; Anderton, K.; Bentz, T.; Edwards, A.; Brisendine, A.; Weiss, G.; Siegel, A.B.; Bendell, J.; et al. A Randomized Phase II Open-Label Multi-Institution Study of the Combination of Bevacizumab and Erlotinib Compared to Sorafenib in the First-Line Treatment of Patients with Advanced Hepatocellular Carcinoma. Oncology 2018, 94, 329–339. [Google Scholar] [CrossRef]
- Lee, K.W.; Kim, S.H.; Yoon, K.C.; Lee, J.M.; Cho, J.H.; Hong, S.K.; Yi, N.J.; Han, S.S.; Park, S.J.; Suh, K.S. Sirolimus Prolongs Survival after Living Donor Liver Transplantation for Hepatocellular Carcinoma Beyond Milan Criteria: A Prospective, Randomised, Open-Label, Multicentre Phase 2 Trial. J Clin Med 2020, 9. [Google Scholar] [CrossRef]
- Tian, L.Y.; Smit, D.J.; Jücker, M. The Role of PI3K/AKT/mTOR Signaling in Hepatocellular Carcinoma Metabolism. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Cervello, M.; McCubrey, J.A.; Cusimano, A.; Lampiasi, N.; Azzolina, A.; Montalto, G. Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget 2012, 3, 236–260. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Joseph, N.M.; Nimeiri, H.S.; Hwang, J.; Kulik, L.M.; Ngo, Z.; Behr, S.C.; Onodera, C.; Zhang, K.; Bocobo, A.G.; et al. Phase II Trial of the Combination of Temsirolimus and Sorafenib in Advanced Hepatocellular Carcinoma with Tumor Mutation Profiling. Liver Cancer 2021, 10, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Yeo, W. Development of systemic therapy for hepatocellular carcinoma at 2013: updates and insights. World J Gastroenterol 2014, 20, 3135–3145. [Google Scholar] [CrossRef]
- Lu, X.; Paliogiannis, P.; Calvisi, D.F.; Chen, X. Role of the Mammalian Target of Rapamycin Pathway in Liver Cancer: From Molecular Genetics to Targeted Therapies. Hepatology 2021, 73 Suppl 1, 49–61. [Google Scholar] [CrossRef]
- Song, L.; Luo, Y.; Li, S.; Hong, M.; Wang, Q.; Chi, X.; Yang, C. ISL Induces Apoptosis and Autophagy in Hepatocellular Carcinoma via Downregulation of PI3K/AKT/mTOR Pathway in vivo and in vitro. Drug Des Devel Ther 2020, 14, 4363–4376. [Google Scholar] [CrossRef]
Figure 1.
Schematic illustration of EGFR/PI3K/AKT/mTOR signaling pathway. Receptor dimerization induces phosphorylation of tyrosine residues at the c-terminal cytoplasmic domain of EGFR. PI3K binds to the phosphorylated tyrosine residues on EGFR and converts phosphatidylinositol-4,5-disphosphate (PIP2) in the plasma membrane into phosphatidylinositol-3,4,5-trisphosphate (PIP3), leading to the recruitment and subsequent phosphorylation of AKT by PDK1 and mTORC2. Phosphorylated AKT in turn leads to activation of mTORC1 by regulating the activity of RHEB. The active mTOR induces phosphorylation and activvation of S6K1 and 4EBP1.
Figure 1.
Schematic illustration of EGFR/PI3K/AKT/mTOR signaling pathway. Receptor dimerization induces phosphorylation of tyrosine residues at the c-terminal cytoplasmic domain of EGFR. PI3K binds to the phosphorylated tyrosine residues on EGFR and converts phosphatidylinositol-4,5-disphosphate (PIP2) in the plasma membrane into phosphatidylinositol-3,4,5-trisphosphate (PIP3), leading to the recruitment and subsequent phosphorylation of AKT by PDK1 and mTORC2. Phosphorylated AKT in turn leads to activation of mTORC1 by regulating the activity of RHEB. The active mTOR induces phosphorylation and activvation of S6K1 and 4EBP1.
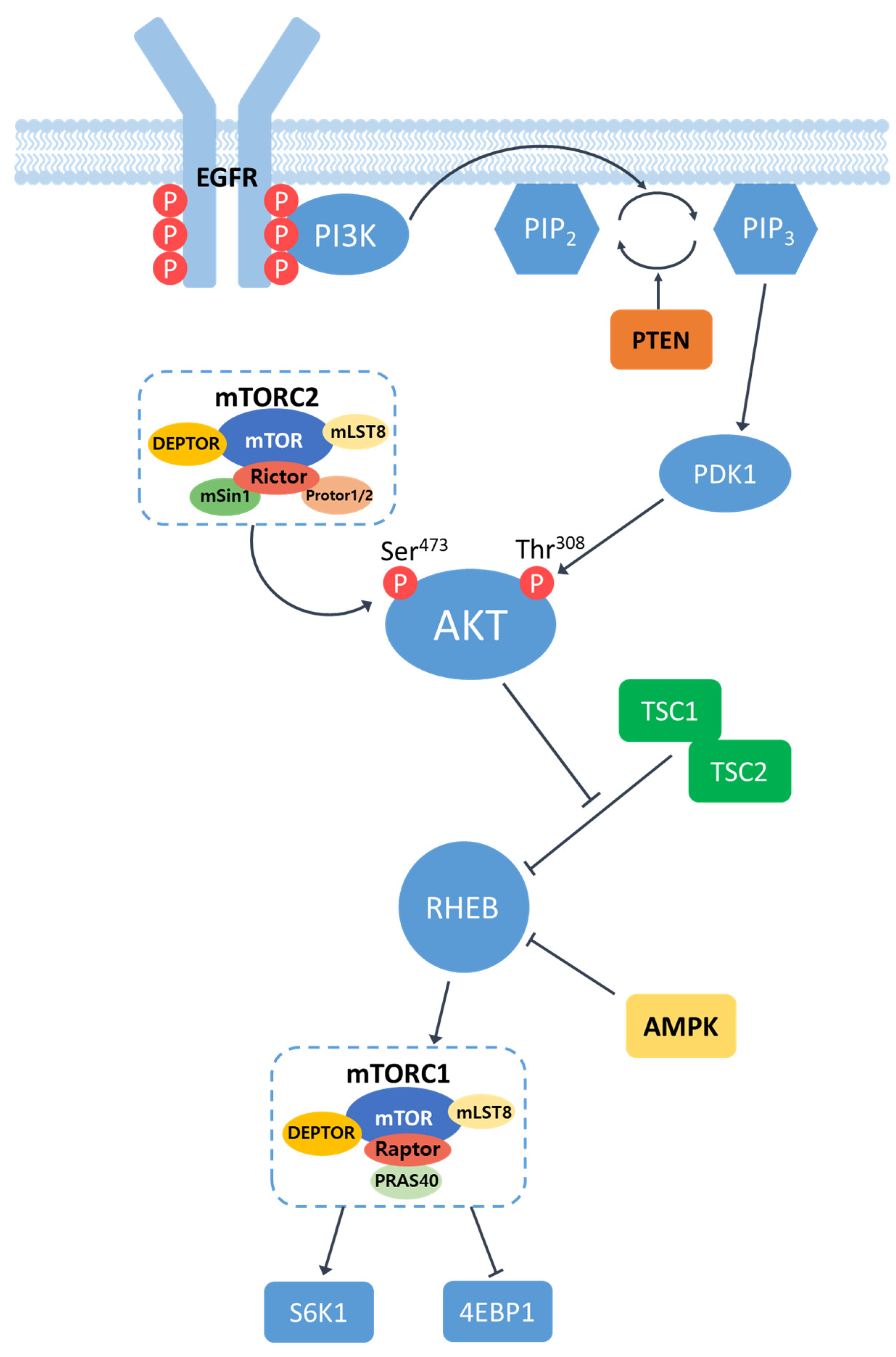

Table 1.
The inhibition of the EGFR/PI3K/AKT/mTOR pathway in HCC cell lines.
| Drug | Target | HCC Cell Line | Phenotype | Refernce |
|---|---|---|---|---|
| Erlotinib | EGFR | MHCC97H, PLC/RLF/5 | reduced cell proliferation | [32] |
| Huh7 | reduced migration | [44] | ||
| AG1478 | EGFR | HepG2 | reduced cell proliferation and invasion | [38] |
| Gefitinib | EGFR | Huh7 | Induced apoptosis | [45] |
| GW2974 | EGFR | HepG2 | reduced cell proliferation, Induced apoptosis | [41] |
| copanlisib | PI3K | Huh7, HepG2 | Induced apoptosis, inhibited cell growth, inducing cell cycle arrest | [45,48] |
| LY294002 | PI3K | MHCC97-H | Induced apoptosis | [90] |
| Huh7,Mahlavu | suppressed cell growth, Induced apoptosis | [46] | ||
| Wortmannin | PI3K | Huh7,Mahlavu | suppressed cell growth, Induced apoptosis | [46] |
| HepG2 | Induced apoptosis | [46] | ||
| 740Y-P | PI3K | SMMC-7721,MHCC-97H | reduced cell proliferation, Induced apoptosis | [47] |
| MK2206 | AKT | Hep3B, Huh7,PLC/RLF/5 | growth-inhibitory, Induced apoptosis | [51] |
| HepG2 | anti-proliferative | [52] | ||
| AKT inhibitor VIII | AKT | Huh7,Mahlavu | suppressed cell growth, Induced apoptosis | [46] |
| Rapamycin | mTOR | Hep3B | prevent enrichment of CSCs | [54] |
| RAD001 (everolimus) | mTOR | Hep3B, Huh7, PLC/RLF/5 | growth-inhibitory, Induced apoptosis | [51] |
| BEZ235 | PI3K/mTOR | Hep3B, Huh7, PLC/RLF/5 | growth-inhibitory, Induced apoptosis | [51] |
| Lenvatinib | AKT/mTOR | Hep3B, HepG2 | reduced cell proliferation, migration | [55] |
Table 2.
Mouse models for HCC induced by activated EGFR/PI3K/AKT/mTOR signaling.
| Gene | Mouse Model | Phenotype/Tumor Type | Reference |
|---|---|---|---|
| Pten | Alb-Cre; Ptenfl/fl | ballooning, steatosis / ICC, HCC | [62] |
| sgPten sgPten/c-Met |
lipid accumulation / no tumor lipid accumulation, / HCA, HCC |
[66] | |
| AKT | HA-myr-AKT1 HA-myr-AKT1/V5-c-Met |
hepatic steatosis, proliferation / HCC rapid liver tumor growth |
[68] |
| myr-AKT1 myr-AKT1/N-RasG12V |
hepatocyte proliferation/ HCA(12w), HCC(6m) spotty and pale color / nodular lesions(~4w) |
[69] | |
| Tsc1/Tsc2 | Alb-Cre; Tsc1fl/fl | dysplastic foci, nodules, hepatomas / HCC | [70] |
| Alb-Cre; Tsc1fl/fl | moderate tumor incidence rate / HCC | [71] | |
| Alb-Cre; Tsc1fl/fl Alb-Cre ; Tsc1fl/fl/Ptenfl/fl |
no steatosis / HCC, ICC great hepatomegaly(early), large tumor(14w) / HCC |
[62] |
Table 3.
HCC suppression in xenograft models by targeting the EGFR/PI3K/AKT/mTOR signaling.
| Agent | Target | HCC Cell Line Transplanted | Phenotype | References |
|---|---|---|---|---|
| MUC15 | EGFR | HCCLM3 | Delayed tumor formation, higher survival rate | [72] |
| RHEB-shRNA | RHEB | SMMC-7721 | Decrease in tumor growth | [73] |
| DZW-310 | PI3K | Hep3B | Decrease in tumor growth | [74] |
| MK2206 | AKT | Hep3B, Huh 7 | Decrease in tumor growth | [51] |
| ZJQ-24 | AKT/mTOR | HepG2 | Tumor cell death/Reduced tumor growth | [75] |
| RAD001 | mTOR | Patient-derived HCCs | Decrease in tumor growth | [78] |
| rapamycin | mTOR | HepG2 | No effects | [79] |
| rapamycin + bevacizumab | mTOR + VEGF | HepG2 | Decreased tumor size/increased survival | [79] |
| metformin | mTOR | Bel-7402 cells | Decrease in tumor growth | [80] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



