
Submitted:
24 July 2023
Posted:
25 July 2023
You are already at the latest version
Abstract
Papain-like lysosomal cysteine proteases include 11 human cysteine cathepsins which act as endopeptidases and/or exopeptidases. They are involved in numerous physiological and pathological processes. Among them, only cathepsins B, H, C, and X/Z exhibit exopeptidase activity. Their activities are tightly regulated in various ways to prevent potentially hazardous effects on the cell. In this review, we focus on the structural and functional aspects of these four cysteine cathepsins and their role in neurodegeneration and cancer.
Neurodegenerative disorders of aging share an endolysosomal dysfunction and accumulation and spread of oligomeric forms of neurotoxic proteins. The accumulation of various protein aggregates activates the microglia, thus inducing the activation and release of cysteine cathepsins and proinflammatory cytokines, leading to neurodegeneration.
In cancer, cysteine cathepsins participate in tumor progression and metastasis. Tumor–stromal crosstalk leads to activation of the stroma and overexpression and secretion of proteolytic enzymes, triggering extracellular matrix degradation and the release of soluble factors. Activated stromal cells (i.e., macrophages, fibroblasts, mast cells) secrete additional growth factors, cytokines, and chemokines that are responsible for the regulation of processes leading to tumor progression and metastasis. Therefore, a better understanding of the role of cysteine cathepsins in neurodegeneration and cancer could lead to novel targeted therapeutic approaches.
Keywords:
cysteine cathepsins
; exopeptidases
; neurodegeneration
; cancer
; neurodegenerative disorders
1. Cysteine Cathepsins
Almost 80 years ago Christian de Duve discovered lysosomes, termed ''suicide bags'', the single membrane-bound cytoplasmic organelles containing five hydrolases including cathepsin D [1]. The consequence of the release of enzymes from an injured lysosome was the destruction and death of their own cell [2,3]. This discovery was crucial for the beginning of understanding intracellular degradation of proteins and other macromolecules. Further studies show that lysosomes are present in almost all eukaryotic cells and contain over 50 different acid hydrolases. It is now becoming evident that the main function of lysosomes with their proteolytic/degradation potential is not to kill the cell but to be responsible for cellular homeostasis and recycling of cellular components, which has been confirmed in numerous physiological processes (reviewed in [4,5,6,7,8]. This classical view has changed more recently, after reports that cysteine cathepsins were also found in the nucleus, mitochondria, cytoplasm and the extracellular space [8,9,10,11,12]. Recent developments in quantitative proteomics and in vivo imaging have led to a better understanding of protease specificity profiling and identification of physiological substrates [13], thus resulting in a change in the concept of proteases, including cysteine cathepsins, as degrading enzymes to proteases as key signaling molecules [14,15]. A typical example of signaling is the activation of the proapoptotic protein Bid, a member of Bcl-2 family, thus initiating apoptosis [16,17,18].
Lysosomal dysfunction includes changes in expression and activity of lysosomal enzymes, changes in lysosomal size and their number, pH, and cellular positioning and changes in lysosomal membrane properties [19]. When released from the lysosomes, cathepsins are potentially hazardous and are frequently associated with various human pathologies including cancer [20,21,22], cardiovascular diseases [23,24], neurodegeneration [25,26], bone disorders, inflammatory diseases [27,28,29], and coronavirus disease SARS-CoV-2 [30,31]. Although they are less investigated, there are also diseases caused by a genetic deficiency of cysteine cathepsins F, K, C, and H, and lysosomal storage diseases [32].
Proteases catalyze irreversible hydrolytic reactions and therefore their proteolytic activity must be strictly regulated. This can be achieved at multiple levels by various mechanisms such as gene expression, post-translational modification, activation of their inactive zymogens autocatalytically or by other proteases, targeting to specific compartments, proteolysis, degradation, oxidants and endogenous protein inhibitors or exogenous inhibitors [15,33,34]. Once activated, the mature enzymes are proteolytically active and must be under physiological and pathological states regulated by the inhibitors. The inhibitors can be divided into two groups: the emergency and the regulatory inhibitors. The difference between these two groups is in their localization. The emergency inhibitors are normally localized in different cellular compartments from the enzyme, a typical example of which are cystatins, while the regulatory inhibitors are often colocalized with their target [15,35,36]. The most studied are the cystatins (family I25), which consist of three subfamilies, namely, stefins (I25A), cystatins (I25B), and kininogens (I25C). They are competitive, reversible, tight-binding inhibitors able to discriminate between endo- and exopeptidases (more in reviews by [37,38,39]). It has to be noted that the determination of the crystal structure of human stefin B in complex with papain resulted in the discovery of a new mechanism of interaction between cystatins and papain-like enzymes [40]. More recently, the thyropins, new protein inhibitors structurally different from cystatins, were discovered. They belong to the family I31 of the clan IX [41]. The most physiologically important inhibitor of this family is the p41 fragment of the invariant chain, which inhibits several cathepsins and is involved in the regulation of MHC-II antigen presentation [42,43]. There are numerous synthetic inhibitors, among them epoxysuccinate derivatives such as the first inhibitors of cysteine cathepsins [44]. One of them, CA030 (ethyl ester of epoxysuccinyl-Ile-Pro-OH), is used in the crystal structure in a complex with cathepsin B. This structure revealed for the first time a substrate-like interaction in the S1’ and S2’ residues [45].
Among lysosomal hydrolases, proteases (also termed peptidases), play a very important role. There are 15 cathepsins in humans, classified according to their catalytic types: serine proteases (cathepsins A and G), aspartic proteases (cathepsins D and E), and cysteine proteases cathepsins B, C, F, H, K, L, O, S, V, W, and X/Z. For consistency throughout the manuscript, the name cathepsin X is used.
These 11 lysosomal cysteine cathepsins are members of the papain family (C1A) of the cysteine peptidases clan (CA). They are predominantly endopeptidases, although cathepsins C and X are strictly exopeptidases. In addition, cathepsin B is also a carboxydipeptidase and cathepsin H is an aminopeptidase. All their amino acid sequences have been determined and confirmed by a bioinformatic analysis of the draft sequence of the human genome [46]. While the majority of them are ubiquitously expressed, the other four cathepsins, K, S, V, and W (also named lymphopain), show a more restricted cell- or tissue-specific distribution, suggesting their specific cellular functions [47]. Cysteine cathepsins are optimally active at acidic pH (pH 3.5-6.0) and in a reducing environment, and are mostly inactivated and unstable at neutral pH, with the exception of cathepsin S which is stable and active at neutral or slightly alkaline pH [48]. It was earlier reported that heparin-like glycosaminoglycans can potentiate the endopeptidase activity of cathepsin B at alkaline pH after the interaction of heparin and heparane sulfate with occluding loop of the enzyme [49]. Very recently, it was confirmed that cathepsin B displays dual activities, namely, dipeptidylcarboxypeptidase and endopeptidase activities, at both acidic and neutral pH conditions [50]. These findings may be important for cathepsin B functioning under different cellular pH conditions.
Lysosomal cathepsins are synthesized as preproenzymes. After removal of the N-terminal signal peptide in the endoplasmic reticulum, the resulting inactive proenzymes are transported to late endosomes or lysosomes where the prodomain (propeptide) is removed by limited proteolytic processing in order to obtain active mature enzymes. This activation process occurs autocatalytically at acidic pH as a combination of unimolecular and bimolecular processes [51]. Later, we proposed the model for autocatalytic activation of cysteine cathepsins, involving low catalytic activity of procathepsin B in dissociation of the propeptide from the active-site cleft as the first unimolecular step during zymogen activation. The second step is bimolecular proteolytic removal of the propeptide [52]. The activation process can be facilitated by glycosaminoglycans [53,54]. In contrast, procathepsin C can be activated to its mature form by cathepsin L and S, and not by autocatalytic processing [55]. Similarly, procathepsin X is incapable of autocatalytic processing but can be processed in vitro under reducing conditions by cathepsin L [56].
From the crystal structures of human procathepsin B [57], human procathepsin L [58] and some other procathepsins, it is evident that the propeptides show remarkable similarity in their folds despite their different amino acid sequences and lengths. Most propeptides contain about 100 amino acid residues; the shortest propeptide of cathepsin X contains only 38 residues [56,59], while the longest are cathepsin C, with 206 amino acid residues [60], and cathepsin F, which has 251 residues and contains a cystatin-like domain, unique among cysteine cathepsin zymogens [61]. Propeptides fold on the surface of the enzymes, covering the catalytic site and acting as inhibitors, suggesting that this mode of inhibition is common to all enzymes from the papain superfamily [58,62]. The propeptides unfold at acidic pH, thus opening the active site of the enzyme, suggesting the mechanism for acidic zymogen activation [63].
The mature forms of all cysteine cathepsins share similar sequences and a typical papain-like fold which consists of two domains forming a ''V'' active site cleft with a catalytic dyad Cys25 and His159 on the opposite sides of the domains, forming a thiolate–imidazolium ion pair responsible for the enzyme activity [64]. They are all monomers with an MW of about 30 kDa, with the exception of the tetrameric cathepsin C, which is about 200 kDa [65], and the active homodimer of cathepsin X, which has a MW of about 55 kDa [66]. From the determined crystal structures of exopeptidases cathepsin B [67], cathepsin H [68], cathepsin C [69] and cathepsin X [70], it is evident that their exopeptidase activities are a result of additional structural elements such as loops (cathepsins B and X) and propeptide regions (cathepsins C and H) (Figure 1). Later, it was reported that cathepsin X is a carboxymonopeptidase [71]. For more details, see the original structural papers and reviews [47,64].
2. Cathepsins B, H, C and X/Z in Neurodegenerative and Neuropsychiatric Disorders
Neurodegenerative disorders of aging, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal dementia (FTD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS) and, more recently, multiple sclerosis (MS), are often called proteinopathies due to the presence of misfolded and aggregated proteins that lose their physiological roles and acquire neurotoxic properties [72,73]. Notably, most neurodegenerative disorders share an endolysosomal dysfunction due to the accumulation and spread of oligomeric forms of neurotoxic proteins [72,74], in which cathepsins have been found to play an important role [25,75,76,77,78,79,80,81,82]. Moreover, many proteins associated with neurodegenerative diseases have been identified as cathepsin substrates [83]. Recently, cysteine cathepsins were also found to be involved in neuroinflammation [84,85,86,87,88,89,90,91,92,93,94,95], a process that was also recently found to be tightly linked to synaptic dysfunction and neurodegeneration [96,97,98]. In addition to neurodegenerative disorders, neuroinflammatory processes are also a critical contributor to the pathology of neuropsychiatric disorders [99]. Therefore, the role of cathepsins in neuropsychiatric disorders is of emerging interest [100,101].
Activated microglia release lysosomal cysteine proteases, such as cathepsin B [102,103,104,105] and cathepsin X [90,91], which have been proposed to induce neuronal damage under various pathological conditions [103,104,106]. α-Synuclein (α-Syn) aggregation clinically detected in the inclusion bodies of the postmortem brain tissues of PD patients [107] has been suggested to activate microglia [108,109,110]. Taken together, neuroinflammation in activated microglia appears to be neurotoxic to neurons [90,102,110,111,112].
In Figure 2, we present a schematic model, thus highlighting the involvement of cathepsins in neurodegenerative disorders. The accumulation of protein aggregates such as amyloid-β (Aβ) in AD, α-Syn in PD, and mutated huntingtin in HD, among others, activates microglia, inducing the activation and release of cysteine cathepsins (i.e., B, H, C, X) and proinflammatory cytokines, including interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), which further enhance this self-propelling neurotoxicity leading to neurodegeneration. Since the specific cathepsin(s) involved is context dependent, it will be addressed accordingly in the following text.
2.1. Roles of Cathepsins B and X in AD Pathology
AD is a progressive neurodegenerative disease most often associated with memory deficits and cognitive decline [113,114]. The neuropathological hallmarks of AD include extracellular Aβ and amyloid precursor protein (APP) deposits, intracellular neurofibrillary tangles (NFTs), dystrophic neuritis and amyloid angiopathy [115]. In the 1990s, Hardy and Higgins proposed the “amyloid cascade hypothesis”, indicating that the deposition of Aβ, as the main component of plaques, is the causative agent of Alzheimer’s pathology and that neurofibrillary tangles, cell loss, vascular damage, and dementia follow as a direct result of this deposition [116]. Although this concept has influenced and guided much of the academic and pharmaceutical research, Aβ appears to be necessary, but not sufficient, to cause AD [117]. On the other hand, Cataldo and Nixon proposed that APP within senile plaques is processed by lysosomal proteases principally derived from degenerating neurons. Therefore, the release of cathepsins from the stringently regulated intracellular milieu provides a basis for an abnormal sequence of proteolytic cleavages of accumulating amyloid precursor protein [118]. Ten years after this proposal, Nixon proposed a pathway to AD termed the "protease activation cascade" that is relevant to sporadic AD pathogenesis and involves the early and progressive activation of proteolytic systems, including, but not limited to, the calpain-calpastatin and endosomal–lysosomal systems [77]. Very recently, Lambeth and Julian studied the proteolysis of Aβ by cathepsins B, H, L and D using thioflavin T fluorescence and liquid chromatography combined with mass spectrometry, thus showing that all Aβ fibril morphologies are resistant to cathepsin digestion [119]. Notably, the authors found that the acid-grown fibrils prevented digestion primarily in the C-terminal portion of the sequence, whereas the neutral-grown fibrils were proteolytically resistant throughout the sequence [119], thus highlighting the pH dependence of the process, as well as the involvement of different cathepsins based on different pH stabilities and specificities.
Although many efforts have been made to develop therapeutic agents for AD based on the amyloid cascade hypothesis, there is currently no effective therapeutic agent. To date, much attention has also been paid to the “amyloid cascade-inflammatory hypothesis”. McGeer and McGeer [120] proposed that AD may result from the inflammatory response induced by extracellular Aβ deposits, which later become enhanced by tau aggregates. The inflammatory response, which is driven by activated microglia, increases over time as the disease progresses. Importantly, cathepsins support the roles of activated microglia in chronic neuroinflammation [94,105].
Notably, cathepsins and other lysosomal hydrolases accumulate within senile plaques in the AD brain, whereas cathepsins were found to be involved in the initiation and mediation of apoptosis and other forms of cell death, suggesting that dysfunction within the lysosomal system is a potential pathogenic mechanism in AD-related neurodegeneration [77,103,121]. Cathepsin X was also found to be associated with plaques in transgenic mouse models [122,123] and in AD patients [122].
Moreover, Sun and colleagues proposed the ''cystatin C-cathepsin B axis'' and showed that cystatin C regulates soluble Aβ and Aβ-associated neuronal deficits by inhibiting cathepsin B-induced Aβ degradation [124]. Bernstein and Keilhoff recently reviewed the putative roles of cathepsin B in AD pathology and highlighted that, on the one hand, they show a neuroprotective effect by lowering Aβ levels and improving neuronal dysfunction, while on the other hand possibly contributing to AD pathology by acting as a β-secretase and generating pyroglutamate Aβ [125]. Recently, Nixon provided growing evidence that implicates AD gene–driven endosomal–lysosomal network disruptions as not only the antecedent pathobiology that underlies β-amyloidogenesis but also as the essential partner with APP and its metabolites that drive the development of AD, including tauopathy, synaptic dysfunction and neurodegeneration [126].
Bai and colleagues showed that oxidative stress activates the NLRP3 inflammasome through upregulation of cathepsin B activity, thus supporting cathepsin B’s role in neuroinflammation and as a potential target in AD therapy [127]. Recently, Nakanishi reviewed the roles of microglial cathepsin B in inflammatory brain diseases and brain aging through both intracellular and extracellular proteolytic mechanisms [105]. Nuclear factor-κB (NF-κB) is activated by the proteolytic degradation of the inhibitor of κBα (IκBα), an endogenous inhibitor of NF-κB, and the subsequent nuclear translocation of NF-κB. The ubiquitin–proteasome system is generally involved in signal-induced IκBα degradation [128]. However, there is accumulating evidence demonstrating the involvement of autophagy machinery in IκBα degradation [129]. In activated microglia, cathepsin B induces autophagic degradation of IκBα, leading to chronic neuroinflammation [130]. Various studies have shown the involvement of microglial cathepsin B in cell death and Aβ clearance. Therefore, it is likely that the phagocytic clearance of Aβ by microglia may play a significant role in promoting the resolution of chronic neuroinflammation in AD [105]. Recently, Ni and Wu reviewed the molecular mechanisms that govern the crosstalk between systemic inflammation and neuroinflammation. The authors suggested that inflammation spreading indicates a negative spiral between systemic diseases and AD and proposed that inhibition of cathepsins B or S may delay the onset of and act as early intervention for AD [130]. A systematic review of human postmortem immunohistochemical studies and bioinformatics analyses unveiled the complexity of AD reactive astrogliosis, thus involving cathepsins [131].
In addition, Thygesen and colleagues showed the involvement of cathepsin X expressed in central nervous system (CNS) myeloid cells in AD [132].
Hwang and colleagues showed that lysosomal perturbation contributes to synaptic and cognitive decay, whereas safely enhancing protein clearance through modulated cathepsin B ameliorates compromised synapses and cognition, thus supporting the view that early cathepsin B upregulation is a disease-modifying therapy that may also slow the development of the mild cognitive impairment to dementia continuum [133].
Extensive work from Hook's lab indicates the role of cathepsin B in the behavioral and memory deficits and neuropathology of AD, traumatic brain injury (TBI), and related brain disorders [82,134,135]. Lysosomal leakage occurs in AD and TBI and is related to neurodegeneration, thus suggesting that cathepsin B is redistributed from the lysosome to the cytosol, where it initiates cell death and inflammation processes associated with neurodegeneration [82]. Therefore, cathepsin B was proposed as a possible target of AD preventive/therapeutic strategies [136,137,138,139].
On the one hand, Dunlop and Carney reported that L-serine selectively induced the activity of the autophagic-lysosomal enzymes cathepsins B and L, but not any of the proteasome-hydrolyzing activities, thus contributing to its neuroprotective effect [139]. Moreover, Cecarini and colleagues showed that metabolites such as phenyl-γ-valerolactones exert neuroprotective activity by regulating intracellular proteolysis and confirmed the role of cathepsin B in autophagy [137].
On the other hand, the repertoire of potent small molecules that act as potential cathepsin B inhibitors is expanding, thus involving E64d [136] and pyridine, acetamide, and benzohydrazide compounds [138], and various natural and synthetic heterocyclic scaffolds [140], among others.
More recently, Liu and colleagues proposed “cascaded pocket” nanosystems with spatiotemporal release in response to metal ions and cathepsin B as a novel therapeutic strategy for the treatment of AD and other brain diseases [141].
2.2. Roles of Cathepsins B and X in PD Pathology
PD is the second most common age-associated neurodegenerative disorder and is characterized by the loss of dopaminergic neurons and the presence of α-Syn-containing aggregates in the substantia nigra pars compacta. One of the hallmarks of PD pathophysiology is chronic neuroinflammation [142], where microglial cathepsin B was proposed as a key driver of inflammatory brain diseases and brain aging [105].
To investigate the mechanisms for astrocyte ATP13A2-regulated lysosomal function and neuroinflammation following 1-methyl-4-phenylpyridinium (MPP+) treatment, Qiao and colleagues used a PD model of cultured primary neurons and astrocytes from the mouse midbrain [143]. The authors showed that the lack of ATP13A2 increased lysosomal membrane permeabilization and cathepsin B release, which in turn exacerbated activation of the NLRP3 inflammasome to produce excess IL-1β from astrocytes, thus suggesting a direct link between astrocyte lysosomes and neuroinflammation [143].
Codolo and colleagues demonstrated that although the monomeric and fibrillar α-Syn forms were able to promote the expression of pro-IL-1β, following the engagement of Toll-like receptor (TLR) 2, the secretion of the mature cytokine was a peculiarity of the fibrillated protein, a process that involves NLRP3 inflammasome activation [144]. The latter relies on the phagocytosis of fibrillar α-Syn, followed by increased production of reactive oxygen species (ROS) and cathepsin B release into the cytosol [144]. In addition, Freeman and colleagues reported that α-Syn aggregates can induce the rupture of lysosomes following their endocytosis in neuronal cell lines by a mechanism that induces a cathepsin B-dependent increase in ROS in target cells [145]. They also observed that α-Syn aggregates can induce inflammasome activation in THP-1 cells [145]. NLRP3 inflammasome activation by α-Syn upon microglial endocytosis and subsequent lysosomal cathepsin B release was also confirmed in the midbrain of PD model mice and the serum of PD patients [146]. All of the above suggests that fibrillar α-Syn, released upon neuronal degeneration, acts as an endogenous trigger of the cathepsin B-mediated inflammatory response in PD, which likely precedes neurodegeneration [144,145,146].
On the other hand, cysteine cathepsin activity was found to be essential in the lysosomal degradation of α-Syn [147] and C-terminal α-Syn truncations in PD [148]. Hu and colleagues showed that α-Syn was mainly degraded in lysosomes, whereas the LRRK2 G2019S mutation, the most common genetic cause of PD, inhibited the degradation of α-Syn and promoted its aggregation. The authors also reported that LRRK2 G2019S decreased the activities of lysosomal enzymes, including cathepsins B and L, indicating that the inhibitory effect of LRRK2 G2019S on α-Syn degradation could underlie the pathogenesis of aberrant α-Syn aggregation in PD with LRRK2 mutation [149].
In addition, α-Syn fibril-induced intracellular aggregate formation requires lysosomal function, which was confirmed to be dependent on cathepsin B and not aspartic cathepsin D [150]. Recently, Blauwendraat and colleagues showed a decrease in active cathepsin B protein levels in iPSC-derived neurons from GBA variant carriers compared to noncarriers, suggesting a further reduction in lysosomal protease function in these cases. Moreover, the authors showed that the α-Syn levels remained unchanged in forebrain neurons carrying the GBA variant, thus suggesting that the overall reduction in lysosomal proteases allows for a faster accumulation of α-Syn aggregates as neurons age [151]. On the other hand, Nelson and colleagues showed that, while cathepsin D activity was significantly decreased in the late-stage PD temporal cortex, neither cathepsin B nor glucocerebrosidase (GCase) activity were. Moreover, the authors found a significant correlation between a decrease in GCase activity and an increase in p129S-α-Syn, whereas there was no significant correlation between either cathepsin D or cathepsin B and α-Gal A activity or levels, suggesting that a causal relationship between cathepsin activity and enzymes/lipids of the glycosphingolipid metabolism pathway and their relative contributions to the pathological accumulation of α-Syn species in PD would require further confirmation [152].
Recently, Pišlar and colleagues reported an upregulation of cathepsin X in the 6-hydroxydopamine (6-OHDA) model of PD, which was thus restricted to activated glial cells [80]. Dopamine neuron cell death upon 6-OHDA treatment induces loss of tyrosine hydroxylase, caspase-3 activation, intracellular ROS generation and mitochondrial dysfunction, including the release of cytochrome c and an imbalanced Bax/Bcl-2 ratio [81]. This process was prevented by the cathepsin X inhibitor AMS36, which interfered with NF-κB activation by blocking the degradation of IκBα and preventing NF-κB nuclear translocation [81].
In addition, Lee and colleagues showed that PC12 cells exposed to 6-OHDA resulted in lysosomal dysregulation, caspase activation and cell death, an effect that was attenuated using the inhibitors pepstatin A and DEVD-Cho, respectively, whereas the cathepsin B inhibitor CA-074Me failed to protect cells [153]. On the other hand, Wu and colleagues reported that the autophagy/lysosomal pathway is involved in the 6-OHDA-induced death process of PC12 cells. The authors demonstrated overactive autophagy due to mitochondrial disability, increased cathepsin B expression, and diminished Bcl-2 expression, whereas necrostatin-1 exerted a protective effect against injury on dopaminergic neurons [154].
2.3. Roles of Cathepsins B, H and X in HD
HD is a progressive, fatal, autosomal dominant neurodegenerative disorder characterized by uncontrolled excessive motor movements and cognitive and emotional deficits [157,158,159,160].
Early studies by Mantle and colleagues reported a significant increase in protease activities, especially of cathepsins H and D, in brain tissue from HD patients [76]. Nagata and colleagues then provided direct evidence for abnormalities in HD tissues outside the brain under basal conditions by examining patient lymphoblasts. The authors reported a pronounced vacuole formation containing huntingtin remnants and cathepsin B staining, thus suggesting autophagy [161]. Later, Zhang and colleagues used an HD mouse model to demonstrate the involvement of the p53 pathway in signaling both autophagy and apoptosis, a process that involved active cathepsins B and D [162].
Moreover, Kegel and colleagues reported that the endosomal–lysosomal pathway is the main pathway for the removal of excess huntingtin and that lysosomal activity may regulate the cleavage of N-terminal fragments that later aggregate in nuclear and cytoplasmic inclusions of HD neurons [163]. In this regard, several proteases, including cathepsins B, L, X, and D, caspases, calpain, metalloproteases and proteasomes, were reported to contribute to the N-terminal proteolysis of mutant huntingtin [79,163,164,165,166,167]. Interestingly, using cathepsin-deficient cells and pharmacological inhibitors, cathepsins L and X were found to be responsible for degrading polyQ proteins and peptides but no other aggregation-prone proteins, suggesting that they may have a crucial role in host defense against the toxic accumulation of polyQ proteins [168]. Lai and colleagues reported that scyllo-inositol promotes robust degradation of mutant huntingtin protein mediated by the lysosome and by the proteasome but not autophagosomes. The rescue of degradation pathways was due to a reduction in mutant polyQ-huntingtin protein levels and was not a direct result of the action of the compound on the lysosome or proteasome [169].
2.4. Roles of Cathepsins B, H and X in ALS Pathology
ALS is a motor neuron degenerative disease of complex etiology involving protein misfolding. This is a common feature shared with other neurodegenerative diseases, although there is a distinct common thread among ALS genes that connects them to the cascade of autophagy [170]. To clarify the possible association of ALS neurodegeneration with the endolysosomal system, Kikuchi and colleagues examined the pathological expression of cysteine cathepsins B, H, and L and aspartic cathepsin D in the anterior horns of 15 ALS cases and five controls [78]. The authors found that among the cathepsins examined, only the expression of cathepsin B was increased, thus suggesting that cathepsin B may play an important role in the motor neuron degeneration in ALS [78]. More recently, Mori and colleagues showed that autophagy is a common degradation pathway for Bunina Bodies and TDP-43 inclusions, which may explain the frequent coexistence of these inclusions in anterior horn cells in sporadic ALS [171].
Lee and colleagues demonstrated that proteasome inhibitors, but not cathepsin B inhibitors, indeed increased SOD1 aggregate formation but did not increase cell death, indicating that there is no association between SOD1 aggregates and cell death in familial ALS [172].
A cDNA microarray analysis on postmortem spinal cord specimens of four sporadic ALS patients compared to four age-matched non-neurological controls revealed major changes in the mRNA expression of 60 genes, including an increase in cathepsins B and D, apolipoprotein E, epidermal growth factor receptor, ferritin, and lysosomal trafficking regulator [173]. Since the results from sporadic ALS patients and the SOD1 transgenic mouse model were in good agreement, the authors suggested that the examined genes may play a specific role in the pathogenesis of ALS [173]. In addition, Boutahar and colleagues evaluated the effect of oxidative or excitotoxic stress on the transcriptional profile of ALS-linked mutant SOD1 cultured neurons and observed that both ubiquitin–proteasome and endosome–lysosome systems were upregulated in transgenic neuron culture [174]. Moreover, a meta-analysis of gene expression profiling in ALS confirmed that the differential expression of cathepsins B and D, GFAP and SERPINA3 was repeatedly found to be significant in both the mouse model and ALS patients [175]. In addition, Fukada and colleagues analyzed the gene expression in the spinal cord of SOD1(L126delTT) TgM using a cDNA microarray and identified four genes (Crym, Hspb1/HSP27, CtsH, and Paip1) that may be related to the pathogenesis of familial ALS, including the progression of reactive astrocytes and the inflammatory response of microglial cells. In particular, the authors detected the presence of cathepsin H in reactive astrocytes and microglial cells, suggesting that its overexpression might be associated with the reaction against misfolded protein due to failure of the ubiquitin–proteasome system [176].
Gene profiling of skeletal muscle in an ALS mouse model showed that before the onset of overt clinical symptoms and motor neuron death, early changes affected genes involved in detoxification, regeneration, tissue degradation and cell death. Notably, cathepsin X, metallothionein-1 and -2, ATF3, and galectin-3 genes appeared, among others, to be commonly regulated in both the skeletal muscle and spinal motor neurons of paralyzed ALS mice [177]. In addition, Wendt and colleagues showed that cathepsin X is an important player in degenerative processes during normal aging and in pathological conditions, as it was found to be upregulated in numerous glial cells in degenerating brain regions in a transgenic mouse model of ALS [122]. Moreover, a neurodegeneration-specific gene expression signature of acutely isolated microglia from an ALS mouse model revealed coregulated genes in the lysosome pathway, which include a large group of cathepsins (A, B, D, L, S, X and E), a host of lysosome enzymes (i.e., HexA), membrane markers (i.e., Cd68, Cd63, Lamp1) and components of the lysosomal ATPase (i.e., Atp6v0d1) [178]. Based on the above, the authors hypothesized that cathepsins may be involved in the removal of mutant SOD1 aggregates and neuronal debris in ALS mice [178]. On the other hand, Ulbrich and colleagues reported evidence for a reciprocal influence of SOD1 and stefin B/cystatin B at the gene expression level and for a direct interaction of the two proteins [179]. In addition, Watanabe and colleagues demonstrated that cystatin C, a main component of Bunina bodies in ALS, is an endogenous neuroprotective factor that acts through coordinated activation of two distinct neuroprotective pathways, namely, induction of autophagy and inhibition of aberrant cathepsin B activity [180].
2.5. Roles of Cathepsins B, H, C and X in MS Pathology
MS is a disease that affects the CNS and is characterized by inflammation, demyelination and neurodegeneration [181,182]. Earlier studies have shown an increase in cathepsin B levels in monocytes and macrophages, cells known to be activated in the peripheral blood of MS patients and implicated as effectors of demyelination [183]. On the other hand, even though the mean activity in MS tissue containing demyelinating lesions was higher than that in normal-appearing white matter, which was higher than that in normal control specimens, the differences were not statistically significant [184]. The authors suggested that the increased cathepsin B activity in the MS brain could be due to monocytes, macrophages and reactive astrocytes [184]. Since proteasomal dysfunction was observed in the brain white matter and gray matter of MS patients, an increase in cathepsin B activity may represent a compensatory mechanism for intracellular protein degradation [185].
In an attempt to identify the proteases involved in MS pathogenesis, a cDNA microarray analysis was carried out in the brains of proteolipid protein transgenic (plptg/-) mice, an animal model that closely mimics the failure of remyelination in MS [186]. Cathepsins B, H and L were found to be upregulated in microglia/macrophages of the brain white matter, whereas elevated expression of cystatin C was found in astrocytes, thus suggesting that the imbalance between cathepsins and their inhibitor may be cytotoxic for neurons (axons) and oligodendrocytes [186]. Allan and Yates utilized cathepsin gene knockout mice to show that cathepsin L deficiency attenuates myelin oligodendrocyte glycoprotein (MOG) antigen presentation and the development of experimental autoimmune encephalomyelitis (EAE). On the other hand, neither cathepsin B nor cathepsin S deficiency had any effect, whereas their double-mutant mice attenuate MOG led to antigen presentation and the development of EAE, an animal model of MS [187]. Moreover, Okada and colleagues showed that cathepsin H deficiency impaired TLR3-mediated activation of IRF3 and secretion of interferon-β (IFN-β) from dendritic cells, leading to an enhancement of Th1 cell differentiation, thus resulting in early-onset EAE, an animal model for MS [188]. Therefore, the existence of functional redundancy between cathepsins B, L and S in EAE suggests that the inhibition of multiple cysteine cathepsins may improve autoimmune disorders such as MS. In contrast, inhibition of cathepsin H may have an adverse effect on MS.
Recently, Liang and colleagues demonstrated that the absence of the cystatin F gene and the resulting disinhibition of cathepsin C aggravate demyelination. The authors suggested that this finding may be related to the increased expression of the glia-derived chemokine CXCL2, which may attract inflammatory cells to sites of myelin sheath damage, an effect that was reversed by knockdown of the cathepsin C gene [87]. Moreover, Shimizu and colleagues showed that the balance between cathepsin C and cystatin F controls remyelination in the brains of Plp1-overexpressing mice, a chronic demyelinating disease model [189]. From the same group, Durose and colleagues confirmed that cathepsin C and cystatin F are strongly associated with inflammatory demyelination. The authors showed that the severity of EAE was reduced in the absence of cathepsin C, whereas increased microglial cathepsin C expression enhanced clinical severity, suggesting that the interaction of cathepsin C-cystatin F plays an essential role in the inflammatory demyelination pathogenesis of EAE [190].
In addition, cathepsin X was found to be involved in the propagation of IL-1β-driven neuroinflammation, thus providing mechanistic support for an epigenetic risk factor in MS [88]. Haves-Zburof and colleagues evaluated whether the expression levels of cathepsins B and S and their inhibitors cystatins B and C are affected by the MS disease state and therapies (IFN-β and methylprednisolone) and whether they are associated with the IFN-β response phenotype. The authors showed that cathepsin S expression levels were aberrantly elevated in patients with MS in contrast to cathepsin B and suggested that further validation studies are required to assess the value of cathepsin S and cystatin C as predictive biomarkers for disease type and response to therapy and as a possible basis for the development of new targeted therapies for immune-mediated disorders such as MS [191].
2.5. Roles of cathepsins B and C in neuropsychiatric disorders
Transcriptome analysis of inbred mouse lines, selecting for low or high anxiety-related behavior with depression-like behavior, showed that cathepsin B is responsible for low anxiety in female mice [192]. The assessment of anxiety-related and depression-like behaviors of cathepsin B-deficient mice revealed an increase in depression-like behavior in females. In contrast, cathepsin C aggravates neuroinflammation involved in disturbances of behavior and neurochemistry in acute and chronic stress-induced murine models of depression [193]. On the other hand, cathepsin C knockdown partially prevented inflammation, which may help alleviate the symptoms of depression in mice.
The “monoamine hypothesis of depression” postulates that the underlying pathophysiologic basis of depression is decreased levels of 5-hydroxytriotamin, noradrenalin and/or dopamine in the CNS. More recently, the “neuroplasticity hypothesis of depression” proposed that dysfunction of neural plasticity is the pathophysiologic basis of depression [194]. The role of cathepsin C in the promotion of anxiety- and depressive-like behaviors may be due to the involvement of cathepsin C in neuroinflammation caused by activated microglia [85,93], because depression-like behavior induced by cathepsin C overexpression was associated with increased neuroinflammation and the resultant decreased 5-hydroxytryptamine levels [193]. On the other hand, cathepsin B is also involved in the induction of neuroinflammation by activated microglia [94,105], whereas cathepsin B protects against anxiety- and depressive-like behaviors. Therefore, the mechanism underlying the protective effect of cathepsin B against these disorders may stem from the role of cathepsin B in activity-dependent neuronal plasticity through activation of matrix metalloprotease-9 [195,196]. In any case, the specific pathophysiological roles of cathepsins in neuropsychiatric disorders should be elucidated in future studies.
3. Cathepsin B, H, C and X in Cancer
Accumulating evidence supports the prominent role of cysteine proteases in multiple molecular pathways involved in tumor progression and metastasis [20]. Cathepsin B, the most abundant and ubiquitously expressed exopeptidase of the papain family, is associated with tumor progression in numerous cancer types, including colorectal, breast, lung, pancreatic and gastric cancer [197,198,199,200,201,202,203]. Expression of cathepsin B was shown to correlate with increased malignancy and poor prognosis, and was thus proposed as a predictive biomarker for oral squamous cell carcinoma [204], cervical cancer [205], endometrial cancer [206] and colorectal cancer [202]. Notably, another carboxypeptidase of the clan CA/C1 cysteine protease family, cathepsin X, was shown to be primarily implicated in the development of gastrointestinal cancers that include colorectal [207,208], gastric [209], liver [210] and pancreatic cancer [211]. Moreover, a study by Wang and colleagues demonstrated the involvement of cathepsin X in the regulation of epithelial-to-mesenchymal transition (EMT) and invasion in hepatocellular carcinoma [210]. Aminopeptidases H and C have also been reported to be highly expressed in various cancers and involved in malignant transformation [212,213,214,215]. As such, it was shown that cathepsin H can regulate the processing of talin, a large focal adhesion protein, thus promoting PC3 prostate cancer cell progression through modulation of integrin activation and adhesion strength [216].
Although lysosomal cysteine cathepsins have predominantly intracellular residency, their secretion to the extracellular space has been demonstrated for multiple physiological and pathological conditions [9,20]. Identified cellular mechanisms involved in the secretion of cysteine cathepsins are often accompanied by acidification of the extracellular milieu [217], which is a characteristic feature of the tumor microenvironment (TME). Notably, the slightly acidic pH of tumors provides a favorable environment for extracellular cathepsin activity and thereby promotes the execution of their functions. In addition to tumor cells secreting substantial levels of cathepsins, tumor stromal cells, such as endothelial cells, mast cells, tumor-associated macrophages and fibroblasts, are important contributors to increased levels of cysteine cathepsins in the TME [10,218] (Figure 3).
Tumor–stromal crosstalk leads to activation of the stroma and overexpression and secretion of proteolytic enzymes, including cathepsins (i.e., B, H, C, X), triggering ECM degradation and the release of soluble factors. Activated stromal cells (i.e., macrophages, fibroblasts, mast cells) secrete additional growth factors, cytokines, and chemokines responsible for the regulation of numerous interrelated events leading to tumor progression and metastasis.
As such, it has been shown that the bulk of cathepsin B and X activity in several types of cancer emanates from immune cells of the myeloid lineage, such as peritumoral macrophages [219,220,221,222] and myeloid-derived suppressor cells [223]. Once secreted, cysteine cathepsins can participate in extracellular matrix (ECM) protein degradation, such as E-cadherin [224], collagen IV [225,226] or Tenascin-C [227]. Nevertheless, more specific roles of cysteine cathepsins have recently been discovered in modulation of extra and intracellular signal transduction pathways, which can also be executed through shedding of receptors and adhesion molecules or processing of respective cytokines and growth factors [10]. It was recently demonstrated that cathepsin C can promote proliferation and metastasis in hepatocellular carcinoma through activation of the TNF-α/MAPK (p38) signaling pathway [228]. Moreover, cathepsin B expression has been implicated in the regulation of TGF-β1 signaling [229] and MAP kinase and PI3 kinase pathways in malignant meningiomas [230].
Genetically engineered mouse models in combination with genetic ablation or overexpression of specific proteases appeared to be a valuable research tool for the characterization of multiple roles of cathepsins in the tumorigenesis and progression of cancer. A critical role of cathepsins B and X in the carcinogenesis and progression of breast cancer metastasis was discovered using a transgenic MMTV-PymT model of metastasizing breast cancer [219,222,231]. Furthermore, the impact of cathepsin B on tumor formation or progression was confirmed in multiple models, including the pancreatic cancer RIP1-Tag2 model [224] and renal cell carcinoma xenograft model [232]. Although no data are available on the role of cathepsin H in MMTV-PymT breast cancer progression, the depletion of cathepsin H significantly impaired the establishment and maintenance of the tumor vasculature and reduced tumor burden in the RIP1-Tag2 model of pancreatic islet carcinogenesis [233]. Notably, a cathepsin C tumor-promoting effect has been demonstrated in a squamous cell carcinoma K14-HPV16 model [234] but not in RIP1-Tag2 [224] or MMTV-PymT [234] transgenic mouse models. Taken together, these results indicate that the function of individual proteases may be hardwired into the specific tissue paradigm and thus depend on the type of cancer and the biology of the primary and metastatic lesion host tissue.
Cathepsin activity exists within a larger integrated network of protease activity known as the protease web [235]. Through interactions with other proteases and their inhibitors, cathepsins can alter general proteolytic activity within the tumor microenvironment. In addition to direct regulation of multiple processes involved in tumor progression and metastasis, it is now evident that many proteases can have an indirect impact through activation of multiple cascades of enzymatic activities [236,237]. It can be illustrated by the processing of urokinase-type plasminogen activator (pro-uPA) pro-form by cathepsin B, leading to conversion of plasminogen into plasmin [238], which may activate zymogens of matrix metalloproteinases, and thus together with precursor proteases of this proteolytic activation cascade execute their numerous functions associated with tumor progression and metastasis [239]. Notably, these proteolytic webs or networks can interact with other important signaling pathways in tumor biology, involving cytokines, chemokines, and kinases [240]. Taken together, as the essential elements of the proteolytic network balance, cysteine cathepsins B, X, C and H were found to be involved in multiple steps of cancer development and progression. Therefore, a better understanding of their role in tumor biology and the regulation of relevant signaling pathways can be utilized in novel targeted approaches with anticancer therapeutics.
4. Conclusions
In conclusion, from the discovery of lysosomes and lysosomal cysteine cathepsins until recently, these proteases were primarily considered responsible for terminal protein degradation. In the last thirty years, we have witnessed rapid advances resulting in the determination of the cathepsin’s structures and their mechanisms of action and interaction with endogenous and synthetic inhibitors. Further studies have revealed that lysosomal cathepsins were found in the nucleus, mitochondria, cytoplasm, and in the extracellular space, which was the beginning of the understanding of their role in numerous pathologies. The finding that cathepsins act as signaling molecules necessitates the understanding of their signaling pathways. The identification of physiological substrates is another unexplored area, which is expected to be solved by mass spectroscopy. All of these as well as other approaches are oriented towards the development of better diagnostics, new drugs, and their application in clinical trials. This review discusses the current status of cysteine cathepsins B, H, C, and X in neurodegenerative diseases and cancer in detail. Due to rapid progress in the understanding of various pathologies, new therapeutic approaches are expected in the near future.
Author Contributions
VS and VT participated in the conception of the manuscript. VS and OV prepared the figures. All authors participated in writing the manuscript. All authors have read and approved its final version.
Funding
VS and VT were supported by grants J1-2473 and P1-0140 from the Slovenian Research Agency.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgements
The authors are grateful to Dr. Iztok Dolenc for assistance on reference formatting.
Conflicts of Interest
The authors declare no conflict of interests. The funders had no role in the design of the study, collection, analysis, or interpretation of data, writing of the manuscript or the decision to publish the results.
References
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed]
- de Duve, C. Lysosomes revisited. Eur. J. Biochem. 1983, 137, 391–397. [Google Scholar] [CrossRef] [PubMed]
- de Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Brix, K.; Dunkhorst, A.; Mayer, K.; Jordans, S. Cysteine cathepsins: cellular roadmap to different functions. Biochimie 2008, 90, 194–207. [Google Scholar] [CrossRef]
- Turk, B.; Turk, V. Lysosomes as "suicide bags" in cell death: myth or reality? J. Biol. Chem. 2009, 284, 21783–21787. [Google Scholar] [CrossRef]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 2012, 1824, 22–33. [Google Scholar] [CrossRef]
- Brix, K.; McInnes, J.; Al-Hashimi, A.; Rehders, M.; Tamhane, T.; Haugen, M.H. Proteolysis mediated by cysteine cathepsins and legumain-recent advances and cell biological challenges. Protoplasma 2015, 252, 755–774. [Google Scholar] [CrossRef]
- Biasizzo, M.; Javoršek, U.; Vidak, E.; Zarić, M.; Turk, B. Cysteine cathepsins: A long and winding road towards clinics. Mol. Aspects Med. 2022, 88, 101150. [Google Scholar] [CrossRef]
- Vizovišek, M.; Fonović, M.; Turk, B. Cysteine cathepsins in extracellular matrix remodeling: Extracellular matrix degradation and beyond. Matrix Biol. 2019, 75-76, 141–159. [Google Scholar] [CrossRef]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine Cathepsins and their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9. [Google Scholar] [CrossRef]
- Wang, H.; Inoue, A.; Lei, Y.; Wu, H.; Hong, L.; Cheng, X.W. Cathepsins in the extracellular space: Focusing on non-lysosomal proteolytic functions with clinical implications. Cell. Signal. 2023, 103, 110531. [Google Scholar] [CrossRef]
- Vizovišek, M.; Vidmar, R.; Drag, M.; Fonović, M.; Salvesen, G.S.; Turk, B. Protease Specificity: Towards In Vivo Imaging Applications and Biomarker Discovery. Trends Biochem. Sci. 2018, 43, 829–844. [Google Scholar] [CrossRef]
- Turk, B.; Stoka, V. Protease signalling in cell death: caspases versus cysteine cathepsins. FEBS Lett. 2007, 581, 2761–2767. [Google Scholar] [CrossRef]
- Turk, B.; Turk, D.; Turk, V. Protease signalling: the cutting edge. EMBO J. 2012, 31, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Stoka, V.; Turk, B.; Schendel, S.L.; Kim, T.H.; Cirman, T.; Snipas, S.J.; Ellerby, L.M.; Bredesen, D.; Freeze, H.; Abrahamson, M.; Bromme, D.; Krajewski, S.; Reed, J.C.; Yin, X.M.; Turk, V.; Salvesen, G.S. Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro-caspases, is the most likely route. J. Biol. Chem. 2001, 276, 3149–3157. [Google Scholar] [CrossRef] [PubMed]
- Stoka, V.; Turk, B.; Turk, V. Lysosomal cysteine proteases: structural features and their role in apoptosis. IUBMB Life 2005, 57, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef]
- Wang, F.; Gómez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef]
- Vasiljeva, O.; Sevenich, L.; Reinheckel, T. Analyzing the Role of Proteases in Breast Cancer Progression and Metastasis Using Primary Cells from Transgenic Oncomice. Methods Mol. Biol. 2021, 2294, 275–293. [Google Scholar]
- Liu, C.L.; Guo, J.; Zhang, X.; Sukhova, G.K.; Libby, P.; Shi, G.P. Cysteine protease cathepsins in cardiovascular disease: from basic research to clinical trials. Nat. Rev. Cardiol. 2018, 15, 351–370. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, S.; Wang, M.; Shi, G.P. Cysteinyl cathepsins in cardiovascular diseases. Biochim Biophys Acta Proteins Proteom 2020, 1868, 140360. [Google Scholar] [CrossRef]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev 2016, 32, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim Biophys Acta Proteins Proteom 2020, 1868, 140443. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.; Reinheckel, T.; Peters, C.; Turk, D.; Turk, V.; Turk, B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr. Pharm. Des. 2007, 13, 387–403. [Google Scholar] [CrossRef]
- Hamon, Y.; Legowska, M.; Hervé, V.; Dallet-Choisy, S.; Marchand-Adam, S.; Vanderlynden, L.; Demonte, M.; Williams, R.; Scott, C.J.; Si-Tahar, M.; Heuzé-Vourc'h, N.; Lalmanach, G.; Jenne, D.E.; Lesner, A.; Gauthier, F.; Korkmaz, B. Neutrophilic Cathepsin C Is Maturated by a Multistep Proteolytic Process and Secreted by Activated Cells during Inflammatory Lung Diseases. J. Biol. Chem. 2016, 291, 8486–8499. [Google Scholar] [CrossRef]
- Vizovišek, M.; Vidak, E.; Javoršek, U.; Mikhaylov, G.; Bratovš, A.; Turk, B. Cysteine cathepsins as therapeutic targets in inflammatory diseases. Expert Opin. Ther. Targets 2020, 24, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Yang, W.L.; Yang, F.Y.; Zhang, L.; Huang, W.J.; Hou, W.; Fan, C.F.; Jin, R.H.; Feng, Y.M.; Wang, Y.C.; Yang, J.K. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct Target Ther 2021, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, S.; Gomez-Auli, A.; Hillebrand, L.E.; Petrera, A.; Ketscher, A.; Reinheckel, T. Inherited diseases caused by mutations in cathepsin protease genes. Febs j 2017, 284, 1437–1454. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Bond, J.S. Proteases: multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed]
- Lalmanach, G.; Saidi, A.; Bigot, P.; Chazeirat, T.; Lecaille, F.; Wartenberg, M. Regulation of the Proteolytic Activity of Cysteine Cathepsins by Oxidants. Int. J. Mol. Sci. 2020, 21, 1944. [Google Scholar] [CrossRef]
- Turk, B.; Turk, D.; Salvesen, G.S. Regulating cysteine protease activity: essential role of protease inhibitors as guardians and regulators. Curr. Pharm. Des. 2002, 8, 1623–1637. [Google Scholar] [CrossRef]
- Tušar, L.; Usenik, A.; Turk, B.; Turk, D. Mechanisms Applied by Protein Inhibitors to Inhibit Cysteine Proteases. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Turk, V.; Bode, W. The cystatins: protein inhibitors of cysteine proteinases. FEBS Lett. 1991, 285, 213–219. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef]
- Kordis, D.; Turk, V. Phylogenomic analysis of the cystatin superfamily in eukaryotes and prokaryotes. BMC Evol. Biol. 2009, 9, 266. [Google Scholar] [CrossRef]
- Stubbs, M.T.; Laber, B.; Bode, W.; Huber, R.; Jerala, R.; Lenarcic, B.; Turk, V. The refined 2.4 A X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: a novel type of proteinase inhibitor interaction. EMBO J. 1990, 9, 1939–1947. [Google Scholar] [CrossRef]
- Rawlings, N.D. Peptidase inhibitors in the MEROPS database. Biochimie 2010, 92, 1463–1483. [Google Scholar] [CrossRef] [PubMed]
- Unanue, E.R.; Turk, V.; Neefjes, J. Variations in MHC Class II Antigen Processing and Presentation in Health and Disease. Annu. Rev. Immunol. 2016, 34, 265–297. [Google Scholar] [CrossRef] [PubMed]
- Mihelic, M.; Turk, D. Two decades of thyroglobulin type-1 domain research. Biol. Chem. 2007, 388, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Katunuma, N. Structure-based development of specific inhibitors for individual cathepsins and their medical applications. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 29–39. [Google Scholar] [CrossRef]
- Turk, D.; Podobnik, M.; Popovic, T.; Katunuma, N.; Bode, W.; Huber, R.; Turk, V. Crystal structure of cathepsin B inhibited with CA030 at 2.0-A resolution: A basis for the design of specific epoxysuccinyl inhibitors. Biochemistry 1995, 34, 4791–4797. [Google Scholar] [CrossRef]
- Rossi, A.; Deveraux, Q.; Turk, B.; Sali, A. Comprehensive search for cysteine cathepsins in the human genome. Biol. Chem. 2004, 385, 363–372. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef]
- Kirschke, H.; Wiederanders, B.; Brömme, D.; Rinne, A. Cathepsin S from bovine spleen. Purification, distribution, intracellular localization and action on proteins. Biochem. J. 1989, 264, 467–473. [Google Scholar] [CrossRef]
- Almeida, P.C.; Nantes, I.L.; Chagas, J.R.; Rizzi, C.C.; Faljoni-Alario, A.; Carmona, E.; Juliano, L.; Nader, H.B.; Tersariol, I.L. Cathepsin B activity regulation. Heparin-like glycosaminogylcans protect human cathepsin B from alkaline pH-induced inactivation. J. Biol. Chem. 2001, 276, 944–951. [Google Scholar] [CrossRef]
- Yoon, M.C.; Hook, V.; O'Donoghue, A.J. Cathepsin B Dipeptidyl Carboxypeptidase and Endopeptidase Activities Demonstrated across a Broad pH Range. Biochemistry 2022, 61, 1904–1914. [Google Scholar] [CrossRef]
- Rozman, J.; Stojan, J.; Kuhelj, R.; Turk, V.; Turk, B. Autocatalytic processing of recombinant human procathepsin B is a bimolecular process. FEBS Lett. 1999, 459, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Pungercar, J.R.; Caglic, D.; Sajid, M.; Dolinar, M.; Vasiljeva, O.; Pozgan, U.; Turk, D.; Bogyo, M.; Turk, V.; Turk, B. Autocatalytic processing of procathepsin B is triggered by proenzyme activity. Febs j 2009, 276, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.; Dolinar, M.; Pungercar, J.R.; Turk, V.; Turk, B. Recombinant human procathepsin S is capable of autocatalytic processing at neutral pH in the presence of glycosaminoglycans. FEBS Lett. 2005, 579, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Caglic, D.; Pungercar, J.R.; Pejler, G.; Turk, V.; Turk, B. Glycosaminoglycans facilitate procathepsin B activation through disruption of propeptide-mature enzyme interactions. J. Biol. Chem. 2007, 282, 33076–33085. [Google Scholar] [CrossRef]
- Dahl, S.W.; Halkier, T.; Lauritzen, C.; Dolenc, I.; Pedersen, J.; Turk, V.; Turk, B. Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry 2001, 40, 1671–1678. [Google Scholar] [CrossRef]
- Sivaraman, J.; Nägler, D.K.; Zhang, R.; Ménard, R.; Cygler, M. Crystal structure of human procathepsin X: a cysteine protease with the proregion covalently linked to the active site cysteine. J. Mol. Biol. 2000, 295, 939–951. [Google Scholar] [CrossRef]
- Turk, D.; Podobnik, M.; Kuhelj, R.; Dolinar, M.; Turk, V. Crystal structures of human procathepsin B at 3.2 and 3.3 Angstroms resolution reveal an interaction motif between a papain-like cysteine protease and its propeptide. FEBS Lett. 1996, 384, 211–214. [Google Scholar] [CrossRef]
- Coulombe, R.; Grochulski, P.; Sivaraman, J.; Ménard, R.; Mort, J.S.; Cygler, M. Structure of human procathepsin L reveals the molecular basis of inhibition by the prosegment. EMBO J. 1996, 15, 5492–5503. [Google Scholar] [CrossRef]
- Nägler, D.K.; Ménard, R. Human cathepsin X: a novel cysteine protease of the papain family with a very short proregion and unique insertions. FEBS Lett. 1998, 434, 135–139. [Google Scholar] [CrossRef]
- Paris, A.; Strukelj, B.; Pungercar, J.; Renko, M.; Dolenc, I.; Turk, V. Molecular cloning and sequence analysis of human preprocathepsin C. FEBS Lett. 1995, 369, 326–330. [Google Scholar] [CrossRef]
- Nägler, D.K.; Sulea, T.; Ménard, R. Full-length cDNA of human cathepsin F predicts the presence of a cystatin domain at the N-terminus of the cysteine protease zymogen. Biochem. Biophys. Res. Commun. 1999, 257, 313–318. [Google Scholar] [CrossRef]
- Fox, T.; de Miguel, E.; Mort, J.S.; Storer, A.C. Potent slow-binding inhibition of cathepsin B by its propeptide. Biochemistry 1992, 31, 12571–12576. [Google Scholar] [CrossRef]
- Jerala, R.; Zerovnik, E.; Kidric, J.; Turk, V. pH-induced conformational transitions of the propeptide of human cathepsin L. A role for a molten globule state in zymogen activation. J. Biol. Chem. 1998, 273, 11498–11504. [Google Scholar] [CrossRef]
- Turk, B.; Turk, D.; Turk, V. Lysosomal cysteine proteases: more than scavengers. Biochim. Biophys. Acta 2000, 1477, 98–111. [Google Scholar] [CrossRef]
- Dolenc, I.; Turk, B.; Pungercic, G.; Ritonja, A.; Turk, V. Oligomeric structure and substrate induced inhibition of human cathepsin C. J. Biol. Chem. 1995, 270, 21626–21631. [Google Scholar] [CrossRef]
- Dolenc, I.; Štefe, I.; Turk, D.; Taler-Verčič, A.; Turk, B.; Turk, V.; Stoka, V. Human cathepsin X/Z is a biologically active homodimer. Biochim Biophys Acta Proteins Proteom 2021, 1869, 140567. [Google Scholar] [CrossRef]
- Musil, D.; Zucic, D.; Turk, D.; Engh, R.A.; Mayr, I.; Huber, R.; Popovic, T.; Turk, V.; Towatari, T.; Katunuma, N.; et al. The refined 2.15 A X-ray crystal structure of human liver cathepsin B: the structural basis for its specificity. EMBO J. 1991, 10, 2321–2330. [Google Scholar] [CrossRef]
- Guncar, G.; Podobnik, M.; Pungercar, J.; Strukelj, B.; Turk, V.; Turk, D. Crystal structure of porcine cathepsin H determined at 2.1 A resolution: location of the mini-chain C-terminal carboxyl group defines cathepsin H aminopeptidase function. Structure 1998, 6, 51–61. [Google Scholar] [CrossRef]
- Turk, D.; Janjić, V.; Stern, I.; Podobnik, M.; Lamba, D.; Dahl, S.W.; Lauritzen, C.; Pedersen, J.; Turk, V.; Turk, B. Structure of human dipeptidyl peptidase I (cathepsin C): exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 2001, 20, 6570–6582. [Google Scholar] [CrossRef]
- Guncar, G.; Klemencic, I.; Turk, B.; Turk, V.; Karaoglanovic-Carmona, A.; Juliano, L.; Turk, D. Crystal structure of cathepsin X: a flip-flop of the ring of His23 allows carboxy-monopeptidase and carboxy-dipeptidase activity of the protease. Structure 2000, 8, 305–313. [Google Scholar] [CrossRef]
- Puzer, L.; Cotrin, S.S.; Cezari, M.H.; Hirata, I.Y.; Juliano, M.A.; Stefe, I.; Turk, D.; Turk, B.; Juliano, L.; Carmona, A.K. Recombinant human cathepsin X is a carboxymonopeptidase only: a comparison with cathepsins B and L. Biol. Chem. 2005, 386, 1191–1195. [Google Scholar] [CrossRef]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; Mallucci, G.R.; Kroemer, G.; Levine, B.; Eskelinen, E.L.; Mochel, F.; Spedding, M.; Louis, C.; Martin, O.R.; Millan, M.J. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov 2018, 17, 660–688. [Google Scholar] [CrossRef]
- Schattling, B.; Engler, J.B.; Volkmann, C.; Rothammer, N.; Woo, M.S.; Petersen, M.; Winkler, I.; Kaufmann, M.; Rosenkranz, S.C.; Fejtova, A.; Thomas, U.; Bose, A.; Bauer, S.; Träger, S.; Miller, K.K.; Brück, W.; Duncan, K.E.; Salinas, G.; Soba, P.; Gundelfinger, E.D.; Merkler, D.; Friese, M.A. Bassoon proteinopathy drives neurodegeneration in multiple sclerosis. Nat. Neurosci. 2019, 22, 887–896. [Google Scholar] [CrossRef]
- Wang, C.; Telpoukhovskaia, M.A.; Bahr, B.A.; Chen, X.; Gan, L. Endo-lysosomal dysfunction: a converging mechanism in neurodegenerative diseases. Curr. Opin. Neurobiol. 2018, 48, 52–58. [Google Scholar] [CrossRef]
- Ii, K.; Ito, H.; Kominami, E.; Hirano, A. Abnormal distribution of cathepsin proteinases and endogenous inhibitors (cystatins) in the hippocampus of patients with Alzheimer's disease, parkinsonism-dementia complex on Guam, and senile dementia and in the aged. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 423, 185–194. [Google Scholar] [CrossRef]
- Mantle, D.; Falkous, G.; Ishiura, S.; Perry, R.H.; Perry, E.K. Comparison of cathepsin protease activities in brain tissue from normal cases and cases with Alzheimer's disease, Lewy body dementia, Parkinson's disease and Huntington's disease. J. Neurol. Sci. 1995, 131, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. A "protease activation cascade" in the pathogenesis of Alzheimer's disease. Ann. N. Y. Acad. Sci. 2000, 924, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Yamada, T.; Furuya, H.; Doh-ura, K.; Ohyagi, Y.; Iwaki, T.; Kira, J. Involvement of cathepsin B in the motor neuron degeneration of amyotrophic lateral sclerosis. Acta Neuropathol. 2003, 105, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, T.; Chighladze, E.; Waldron, E.; Hirschhorn, R.R.; Ross, C.A. Cysteine proteases bleomycin hydrolase and cathepsin Z mediate N-terminal proteolysis and toxicity of mutant huntingtin. J. Biol. Chem. 2011, 286, 12578–12589. [Google Scholar] [CrossRef]
- Pišlar, A.; Tratnjek, L.; Glavan, G.; Živin, M.; Kos, J. Upregulation of Cysteine Protease Cathepsin X in the 6-Hydroxydopamine Model of Parkinson's Disease. Front. Mol. Neurosci. 2018, 11, 412. [Google Scholar] [CrossRef] [PubMed]
- Pišlar, A.H.; Zidar, N.; Kikelj, D.; Kos, J. Cathepsin X promotes 6-hydroxydopamine-induced apoptosis of PC12 and SH-SY5Y cells. Neuropharmacology 2014, 82, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Hook, V.; Yoon, M.; Mosier, C.; Ito, G.; Podvin, S.; Head, B.P.; Rissman, R.; O'Donoghue, A.J.; Hook, G. Cathepsin B in neurodegeneration of Alzheimer's disease, traumatic brain injury, and related brain disorders. Biochim Biophys Acta Proteins Proteom 2020, 1868, 140428. [Google Scholar] [CrossRef] [PubMed]
- Drobny, A.; Prieto Huarcaya, S.; Dobert, J.; Kluge, A.; Bunk, J.; Schlothauer, T.; Zunke, F. The role of lysosomal cathepsins in neurodegeneration: Mechanistic insights, diagnostic potential and therapeutic approaches. Biochim Biophys Acta Mol Cell Res 2022, 1869, 119243. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Li, D.; Zhang, Y.; Han, C.; Liang, J.; Hou, C.; Xiao, H.; Ikenaka, K.; Ma, J. The induction of neuronal death by up-regulated microglial cathepsin H in LPS-induced neuroinflammation. J. Neuroinflammation 2015, 12, 54. [Google Scholar] [CrossRef]
- Fan, K.; Wu, X.; Fan, B.; Li, N.; Lin, Y.; Yao, Y.; Ma, J. Up-regulation of microglial cathepsin C expression and activity in lipopolysaccharide-induced neuroinflammation. J. Neuroinflammation 2012, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef]
- Liang, J.; Li, N.; Zhang, Y.; Hou, C.; Yang, X.; Shimizu, T.; Wang, X.; Ikenaka, K.; Fan, K.; Ma, J. Disinhibition of Cathepsin C Caused by Cystatin F Deficiency Aggravates the Demyelination in a Cuprizone Model. Front. Mol. Neurosci. 2016, 9, 152. [Google Scholar] [CrossRef]
- Allan, E.R.O.; Campden, R.I.; Ewanchuk, B.W.; Tailor, P.; Balce, D.R.; McKenna, N.T.; Greene, C.J.; Warren, A.L.; Reinheckel, T.; Yates, R.M. A role for cathepsin Z in neuroinflammation provides mechanistic support for an epigenetic risk factor in multiple sclerosis. J. Neuroinflammation 2017, 14, 103. [Google Scholar] [CrossRef]
- Pišlar, A.; Božić, B.; Zidar, N.; Kos, J. Inhibition of cathepsin X reduces the strength of microglial-mediated neuroinflammation. Neuropharmacology 2017, 114, 88–100. [Google Scholar] [CrossRef]
- Pišlar, A.; Tratnjek, L.; Glavan, G.; Zidar, N.; Živin, M.; Kos, J. Neuroinflammation-Induced Upregulation of Glial Cathepsin X Expression and Activity in vivo. Front. Mol. Neurosci. 2020, 13, 575453. [Google Scholar] [CrossRef]
- Pišlar, A.; Bolčina, L.; Kos, J. New Insights into the Role of Cysteine Cathepsins in Neuroinflammation. Biomolecules 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Campden, R.I.; Zhang, Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2019, 670, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, Y.; Liu, S.; Liu, Y.; Yang, X.; Liu, G.; Shimizu, T.; Ikenaka, K.; Fan, K.; Ma, J. Cathepsin C promotes microglia M1 polarization and aggravates neuroinflammation via activation of Ca(2+)-dependent PKC/p38MAPK/NF-κB pathway. J. Neuroinflammation 2019, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H. Cathepsin regulation on microglial function. Biochim Biophys Acta Proteins Proteom 2020, 1868, 140465. [Google Scholar] [CrossRef]
- Kos, J.; Mitrović, A.; Perišić Nanut, M.; Pišlar, A. Lysosomal peptidases-intriguing roles in cancer progression and neurodegeneration. FEBS Open Bio 2022. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Friedman, B.A.; Dejanovic, B.; Sheng, M. Microglia in Brain Development, Homeostasis, and Neurodegeneration. Annu. Rev. Genet. 2019, 53, 263–288. [Google Scholar] [CrossRef]
- Xu, Y.; Jin, M.Z.; Yang, Z.Y.; Jin, W.L. Microglia in neurodegenerative diseases. Neural Regen Res 2021, 16, 270–280. [Google Scholar]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; Camus, V. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef]
- Réus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.C.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef]
- Niemeyer, C.; Matosin, N.; Kaul, D.; Philipsen, A.; Gassen, N.C. The Role of Cathepsins in Memory Functions and the Pathophysiology of Psychiatric Disorders. Front Psychiatry 2020, 11, 718. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Vilalta, A. How microglia kill neurons. Brain Res. 2015, (Pt B), 288–297. [Google Scholar] [CrossRef]
- Kingham, P.J.; Pocock, J.M. Microglial secreted cathepsin B induces neuronal apoptosis. J. Neurochem. 2001, 76, 1475–1484. [Google Scholar] [CrossRef]
- Nakanishi, H. Neuronal and microglial cathepsins in aging and age-related diseases. Ageing Res Rev 2003, 2, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H. Microglial cathepsin B as a key driver of inflammatory brain diseases and brain aging. Neural Regen Res 2020, 15, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Hook, V.; Funkelstein, L.; Wegrzyn, J.; Bark, S.; Kindy, M.; Hook, G. Cysteine Cathepsins in the secretory vesicle produce active peptides: Cathepsin L generates peptide neurotransmitters and cathepsin B produces beta-amyloid of Alzheimer's disease. Biochim. Biophys. Acta 2012, 1824, 89–104. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 7737–7741. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; Lee, S.J. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 2013, 4, 1562. [Google Scholar] [CrossRef]
- Roodveldt, C.; Christodoulou, J.; Dobson, C.M. Immunological features of alpha-synuclein in Parkinson's disease. J. Cell. Mol. Med. 2008, (5b), 1820–1829. [Google Scholar] [CrossRef]
- Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. The relation between α-synuclein and microglia in Parkinson's disease: Recent developments. Neuroscience 2015, 302, 47–58. [Google Scholar] [CrossRef]
- Gallegos, S.; Pacheco, C.; Peters, C.; Opazo, C.M.; Aguayo, L.G. Features of alpha-synuclein that could explain the progression and irreversibility of Parkinson's disease. Front. Neurosci. 2015, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Ghio, S.; Kamp, F.; Cauchi, R.; Giese, A.; Vassallo, N. Interaction of α-synuclein with biomembranes in Parkinson's disease--role of cardiolipin. Prog. Lipid Res. 2016, 61, 73–82. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Lansbury, P.J. Alzheimer’s disease is the most common neurodegenerative disorder. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6 ed.; Siegel, J.G., Agranoff, B.W., Albers, R.W., Fisher, S.K., Eds.; Uhler, M. Eds. Lippincott-Raven: Philadelphia, USA, 1999; pp. 101–102. [Google Scholar]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer's disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer's disease. Indian J. Psychiatry 2009, 51, 55–61. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: time, space and 'wingmen'. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Nixon, R.A. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 3861–3865. [Google Scholar] [CrossRef]
- Lambeth, T.R.; Julian, R.R. Proteolysis of Amyloid β by Lysosomal Enzymes as a Function of Fibril Morphology. ACS Omega 2021, 6, 31520–31527. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef]
- Nakamura, Y.; Takeda, M.; Suzuki, H.; Hattori, H.; Tada, K.; Hariguchi, S.; Hashimoto, S.; Nishimura, T. Abnormal distribution of cathepsins in the brain of patients with Alzheimer's disease. Neurosci. Lett. 1991, 130, 195–198. [Google Scholar] [CrossRef]
- Wendt, W.; Zhu, X.R.; Lübbert, H.; Stichel, C.C. Differential expression of cathepsin X in aging and pathological central nervous system of mice. Exp. Neurol. 2007, 204, 525–540. [Google Scholar] [CrossRef]
- Hafner, A.; Glavan, G.; Obermajer, N.; Živin, M.; Schliebs, R.; Kos, J. Neuroprotective role of γ-enolase in microglia in a mouse model of Alzheimer's disease is regulated by cathepsin X. Aging Cell 2013, 12, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhou, Y.; Halabisky, B.; Lo, I.; Cho, S.H.; Mueller-Steiner, S.; Devidze, N.; Wang, X.; Grubb, A.; Gan, L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron 2008, 60, 247–257. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Keilhoff, G. Putative roles of cathepsin B in Alzheimer's disease pathology: The good, the bad, and the ugly in one? Neural Regen Res 2018, 13, 2100–2101. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar] [CrossRef]
- Bai, H.; Yang, B.; Yu, W.; Xiao, Y.; Yu, D.; Zhang, Q. Cathepsin B links oxidative stress to the activation of NLRP3 inflammasome. Exp. Cell Res. 2018, 362, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Colleran, A.; Ryan, A.; O'Gorman, A.; Mureau, C.; Liptrot, C.; Dockery, P.; Fearnhead, H.; Egan, L.J. Autophagosomal IkappaB alpha degradation plays a role in the long term control of tumor necrosis factor-alpha-induced nuclear factor-kappaB (NF-kappaB) activity. J. Biol. Chem. 2011, 286, 22886–22893. [Google Scholar] [CrossRef]
- Ni, J.; Wu, Z. Inflammation Spreading: Negative Spiral Linking Systemic Inflammatory Disorders and Alzheimer's Disease. Front. Cell. Neurosci. 2021, 15, 638686. [Google Scholar] [CrossRef]
- Viejo, L.; Noori, A.; Merrill, E.; Das, S.; Hyman, B.T.; Serrano-Pozo, A. Systematic review of human post-mortem immunohistochemical studies and bioinformatics analyses unveil the complexity of astrocyte reaction in Alzheimer's disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12753. [Google Scholar] [CrossRef]
- Thygesen, C.; Ilkjær, L.; Kempf, S.J.; Hemdrup, A.L.; von Linstow, C.U.; Babcock, A.A.; Darvesh, S.; Larsen, M.R.; Finsen, B. Diverse Protein Profiles in CNS Myeloid Cells and CNS Tissue From Lipopolysaccharide- and Vehicle-Injected APP(SWE)/PS1(ΔE9) Transgenic Mice Implicate Cathepsin Z in Alzheimer's Disease. Front. Cell. Neurosci. 2018, 12, 397. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Estick, C.M.; Ikonne, U.S.; Butler, D.; Pait, M.C.; Elliott, L.H.; Ruiz, S.; Smith, K.; Rentschler, K.M.; Mundell, C.; Almeida, M.F.; Stumbling Bear, N.; Locklear, J.P.; Abumohsen, Y.; Ivey, C.M.; Farizatto, K.L.G.; Bahr, B.A. The Role of Lysosomes in a Broad Disease-Modifying Approach Evaluated across Transgenic Mouse Models of Alzheimer's Disease and Parkinson's Disease and Models of Mild Cognitive Impairment. Int. J. Mol. Sci. 2019, 20, 4432. [Google Scholar] [CrossRef]
- Hook, G.; Reinheckel, T.; Ni, J.; Wu, Z.; Kindy, M.; Peters, C.; Hook, V. Cathepsin B Gene Knockout Improves Behavioral Deficits and Reduces Pathology in Models of Neurologic Disorders. Pharmacol. Rev. 2022, 74, 600–629. [Google Scholar] [CrossRef]
- Hook, G.; Kindy, M.; Hook, V. Cathepsin B Deficiency Improves Memory Deficits and Reduces Amyloid-β in hAβPP Mouse Models Representing the Major Sporadic Alzheimer's Disease Condition. J. Alzheimers Dis. 2023, 93, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Hook, G.; Hook, V.; Kindy, M. The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer's disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J. Alzheimers Dis. 2011, 26, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Cecarini, V.; Cuccioloni, M.; Zheng, Y.; Bonfili, L.; Gong, C.; Angeletti, M.; Mena, P.; Del Rio, D.; Eleuteri, A.M. Flavan-3-ol Microbial Metabolites Modulate Proteolysis in Neuronal Cells Reducing Amyloid-beta (1-42) Levels. Mol. Nutr. Food Res. 2021, 65, e2100380. [Google Scholar] [CrossRef]
- Chitranshi, N.; Kumar, A.; Sheriff, S.; Gupta, V.; Godinez, A.; Saks, D.; Sarkar, S.; Shen, T.; Mirzaei, M.; Basavarajappa, D.; Abyadeh, M.; Singh, S.K.; Dua, K.; Zhang, K.Y.J.; Graham, S.L.; Gupta, V. Identification of Novel Cathepsin B Inhibitors with Implications in Alzheimer's Disease: Computational Refining and Biochemical Evaluation. Cells 2021, 10. [Google Scholar] [CrossRef]
- Dunlop, R.A.; Carney, J.M. Mechanisms of L-Serine-Mediated Neuroprotection Include Selective Activation of Lysosomal Cathepsins B and L. Neurotox. Res. 2021, 39, 17–26. [Google Scholar] [CrossRef]
- Saroha, B.; Kumar, G.; Kumari, M.; Kaur, R.; Raghav, N.; Sharma, P.K.; Kumar, N.; Kumar, S. A decennary update on diverse heterocycles and their intermediates as privileged scaffolds for cathepsin B inhibition. Int. J. Biol. Macromol. 2022, (Pt B), 2270–2308. [Google Scholar] [CrossRef]
- Liu, R.; Yang, J.; Qiu, X.; Ji, W.; Shen, J.; Li, Y.; Lu, Z.; Wu, Y.; Wang, W.; Wang, J.; Hao, J.; Zhang, X. "Cascaded Rocket" Nanosystems with Spatiotemporal Separation for Triple-Synergistic Therapy of Alzheimer's Disease. Adv Healthc Mater 2022, 11, e2101748. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson's disease and its potential as therapeutic target. Transl Neurodegener 2015, 4, 19. [Google Scholar] [CrossRef]
- Qiao, C.; Yin, N.; Gu, H.Y.; Zhu, J.L.; Ding, J.H.; Lu, M.; Hu, G. Atp13a2 Deficiency Aggravates Astrocyte-Mediated Neuroinflammation via NLRP3 Inflammasome Activation. CNS Neurosci. Ther. 2016, 22, 451–460. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS One 2013, 8, e55375. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O'Connor, C.; Wiethoff, C.M.; Campbell, E.M. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 2013, 8, e62143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson's disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Lee, J.C. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 9322–9327. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Lacy, S.M.; Huffer, K.E.; Tayebi, N.; Sidransky, E.; Lee, J.C. C-terminal α-synuclein truncations are linked to cysteine cathepsin activity in Parkinson's disease. J. Biol. Chem. 2019, 294, 9973–9984. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Niu, J.Y.; Xiong, J.; Nie, S.K.; Zeng, F.; Zhang, Z.H. LRRK2 G2019S Mutation Inhibits Degradation of α-Synuclein in an In Vitro Model of Parkinson's Disease. Curr Med Sci 2018, 38, 1012–1017. [Google Scholar] [CrossRef]
- Tsujimura, A.; Taguchi, K.; Watanabe, Y.; Tatebe, H.; Tokuda, T.; Mizuno, T.; Tanaka, M. Lysosomal enzyme cathepsin B enhances the aggregate forming activity of exogenous α-synuclein fibrils. Neurobiol. Dis. 2015, 73, 244–253. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; Bonet-Ponce, L.; Alcalay, R.N.; Hassin-Baer, S.; Greenbaum, L.; Iwaki, H.; Leonard, H.L.; Grenn, F.P.; Ruskey, J.A.; Sabir, M.; Ahmed, S.; Makarious, M.B.; Pihlstrøm, L.; Toft, M.; van Hilten, J.J.; Marinus, J.; Schulte, C.; Brockmann, K.; Sharma, M.; Siitonen, A.; Majamaa, K.; Eerola-Rautio, J.; Tienari, P.J.; Pantelyat, A.; Hillis, A.E.; Dawson, T.M.; Rosenthal, L.S.; Albert, M.S.; Resnick, S.M.; Ferrucci, L.; Morris, C.M.; Pletnikova, O.; Troncoso, J.; Grosset, D.; Lesage, S.; Corvol, J.C.; Brice, A.; Noyce, A.J.; Masliah, E.; Wood, N.; Hardy, J.; Shulman, L.M.; Jankovic, J.; Shulman, J.M.; Heutink, P.; Gasser, T.; Cannon, P.; Scholz, S.W.; Morris, H.; Cookson, M.R.; Nalls, M.A.; Gan-Or, Z.; Singleton, A.B. Genetic modifiers of risk and age at onset in GBA associated Parkinson's disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef]
- Nelson, M.P.; Boutin, M.; Tse, T.E.; Lu, H.; Haley, E.D.; Ouyang, X.; Zhang, J.; Auray-Blais, C.; Shacka, J.J. The lysosomal enzyme alpha-Galactosidase A is deficient in Parkinson's disease brain in association with the pathologic accumulation of alpha-synuclein. Neurobiol. Dis. 2018, 110, 68–81. [Google Scholar] [CrossRef]
- Lee, D.C.; Close, F.T.; Goodman, C.B.; Jackson, I.M.; Wight-Mason, C.; Wells, L.M.; Womble, T.A.; Palm, D.E. Enhanced cystatin C and lysosomal protease expression following 6-hydroxydopamine exposure. Neurotoxicology 2006, 27, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Nakanishi, H. Lessons from Microglia Aging for the Link between Inflammatory Bone Disorders and Alzheimer's Disease. J Immunol Res 2015, 2015, 471342. [Google Scholar] [CrossRef] [PubMed]
- Milanowski, L.M.; Hou, X.; Bredenberg, J.M.; Fiesel, F.C.; Cocker, L.T.; Soto-Beasley, A.I.; Walton, R.L.; Strongosky, A.J.; Faroqi, A.H.; Barcikowska, M.; Boczarska-Jedynak, M.; Dulski, J.; Fedoryshyn, L.; Janik, P.; Potulska-Chromik, A.; Karpinsky, K.; Krygowska-Wajs, A.; Lynch, T.; Olszewska, D.A.; Opala, G.; Pulyk, A.; Rektorova, I.; Sanotsky, Y.; Siuda, J.; Widlak, M.; Slawek, J.; Rudzinska-Bar, M.; Uitti, R.; Figura, M.; Szlufik, S.; Rzonca-Niewczas, S.; Podgorska, E.; McLean, P.J.; Koziorowski, D.; Ross, O.A.; Hoffman-Zacharska, D.; Springer, W.; Wszolek, Z.K. Cathepsin B p.Gly284Val Variant in Parkinson's Disease Pathogenesis. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Lim, K.L. Genetic insights into sporadic Parkinson's disease pathogenesis. Curr Genomics 2013, 14, 486–501. [Google Scholar] [CrossRef]
- Finkbeiner, S. Huntington's Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington's Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef]
- Pandey, M.; Rajamma, U. Huntington's disease: the coming of age. J Genet 2018, 97, 649–664. [Google Scholar] [CrossRef]
- Nagata, E.; Sawa, A.; Ross, C.A.; Snyder, S.H. Autophagosome-like vacuole formation in Huntington's disease lymphoblasts. Neuroreport 2004, 15, 1325–1328. [Google Scholar] [CrossRef]
- Zhang, X.D.; Wang, Y.; Wang, Y.; Zhang, X.; Han, R.; Wu, J.C.; Liang, Z.Q.; Gu, Z.L.; Han, F.; Fukunaga, K.; Qin, Z.H. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy 2009, 5, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Kegel, K.B.; Kim, M.; Sapp, E.; McIntyre, C.; Castaño, J.G.; Aronin, N.; DiFiglia, M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 2000, 20, 7268–7278. [Google Scholar] [CrossRef]
- Qin, Z.H.; Gu, Z.L. Huntingtin processing in pathogenesis of Huntington disease. Acta Pharmacol. Sin. 2004, 25, 1243–1249. [Google Scholar]
- Kim, Y.J.; Sapp, E.; Cuiffo, B.G.; Sobin, L.; Yoder, J.; Kegel, K.B.; Qin, Z.H.; Detloff, P.; Aronin, N.; DiFiglia, M. Lysosomal proteases are involved in generation of N-terminal huntingtin fragments. Neurobiol. Dis. 2006, 22, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.P.; Holcomb, J.; Al-Ramahi, I.; de Haro, M.; Gafni, J.; Zhang, N.; Kim, E.; Sanhueza, M.; Torcassi, C.; Kwak, S.; Botas, J.; Hughes, R.E.; Ellerby, L.M. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington's disease. Neuron 2010, 67, 199–212. [Google Scholar] [CrossRef]
- Liang, Q.; Ouyang, X.; Schneider, L.; Zhang, J. Reduction of mutant huntingtin accumulation and toxicity by lysosomal cathepsins D and B in neurons. Mol. Neurodegener. 2011, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Piccirillo, R.; Hourez, R.; Venkatraman, P.; Goldberg, A.L. Cathepsins L and Z are critical in degrading polyglutamine-containing proteins within lysosomes. J. Biol. Chem. 2012, 287, 17471–17482. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Lan, C.P.; Hasan, S.; Brown, M.E.; McLaurin, J. scyllo-Inositol promotes robust mutant Huntingtin protein degradation. J. Biol. Chem. 2014, 289, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.K.H.; Thombre, R.; Wang, J. Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 697, 34–48. [Google Scholar] [CrossRef]
- Mori, F.; Miki, Y.; Kon, T.; Tanji, K.; Wakabayashi, K. Autophagy Is a Common Degradation Pathway for Bunina Bodies and TDP-43 Inclusions in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2019, 78, 910–921. [Google Scholar] [CrossRef]
- Lee, J.P.; Gerin, C.; Bindokas, V.P.; Miller, R.; Ghadge, G.; Roos, R.P. No correlation between aggregates of Cu/Zn superoxide dismutase and cell death in familial amyotrophic lateral sclerosis. J. Neurochem. 2002, 82, 1229–1238. [Google Scholar] [CrossRef]
- Offen, D.; Barhum, Y.; Melamed, E.; Embacher, N.; Schindler, C.; Ransmayr, G. Spinal cord mRNA profile in patients with ALS: comparison with transgenic mice expressing the human SOD-1 mutant. J. Mol. Neurosci. 2009, 38, 85–93. [Google Scholar] [CrossRef]
- Boutahar, N.; Wierinckx, A.; Camdessanche, J.P.; Antoine, J.C.; Reynaud, E.; Lassabliere, F.; Lachuer, J.; Borg, J. Differential effect of oxidative or excitotoxic stress on the transcriptional profile of amyotrophic lateral sclerosis-linked mutant SOD1 cultured neurons. J. Neurosci. Res. 2011, 89, 1439–1450. [Google Scholar] [CrossRef]
- Saris, C.G.; Groen, E.J.; Koekkoek, J.A.; Veldink, J.H.; van den Berg, L.H. Meta-analysis of gene expression profiling in amyotrophic lateral sclerosis: a comparison between transgenic mouse models and human patients. Amyotroph Lateral Scler Frontotemporal Degener 2013, 14, 177–189. [Google Scholar] [CrossRef]
- Fukada, Y.; Yasui, K.; Kitayama, M.; Doi, K.; Nakano, T.; Watanabe, Y.; Nakashima, K. Gene expression analysis of the murine model of amyotrophic lateral sclerosis: studies of the Leu126delTT mutation in SOD1. Brain Res. 2007, 1160, 1–10. [Google Scholar] [CrossRef]
- Gonzalez de Aguilar, J.L.; Niederhauser-Wiederkehr, C.; Halter, B.; De Tapia, M.; Di Scala, F.; Demougin, P.; Dupuis, L.; Primig, M.; Meininger, V.; Loeffler, J.P. Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model. Physiol. Genomics 2008, 32, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Morimoto, E.T.; Goodarzi, H.; Liao, J.T.; O'Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; Myers, R.M.; Maniatis, T. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef]
- Ulbrich, L.; Cozzolino, M.; Marini, E.S.; Amori, I.; De Jaco, A.; Carrì, M.T.; Augusti-Tocco, G. Cystatin B and SOD1: protein–protein interaction and possible relation to neurodegeneration. Cell. Mol. Neurobiol. 2014, 34, 205–213. [Google Scholar] [CrossRef]
- Watanabe, S.; Hayakawa, T.; Wakasugi, K.; Yamanaka, K. Cystatin C protects neuronal cells against mutant copper-zinc superoxide dismutase-mediated toxicity. Cell Death Dis. 2014, 5, e1497. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Wegner, C.; Brück, W. Inflammation, demyelination, and degeneration - recent insights from MS pathology. Biochim. Biophys. Acta 2011, 1812, 275–282. [Google Scholar] [CrossRef]
- Bever, C.T. Jr.; Panitch, H.S.; Johnson, K.P. Increased cathepsin B activity in peripheral blood mononuclear cells of multiple sclerosis patients. Neurology 1994, 44, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Bever, C.T. Jr.; Garver, D.W. Increased cathepsin B activity in multiple sclerosis brain. J. Neurol. Sci. 1995, 131, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Bizzozero, O.A. Decreased activity of the 20S proteasome in the brain white matter and gray matter of patients with multiple sclerosis. J. Neurochem. 2011, 117, 143–153. [Google Scholar] [CrossRef]
- Ma, J.; Tanaka, K.F.; Yamada, G.; Ikenaka, K. Induced expression of cathepsins and cystatin C in a murine model of demyelination. Neurochem. Res. 2007, 32, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Allan, E.R.; Yates, R.M. Redundancy between Cysteine Cathepsins in Murine Experimental Autoimmune Encephalomyelitis. PLoS One 2015, 10, e0128945. [Google Scholar] [CrossRef] [PubMed]
- Okada, R.; Zhang, X.; Harada, Y.; Wu, Z.; Nakanishi, H. Cathepsin H deficiency in mice induces excess Th1 cell activation and early-onset of EAE though impairment of toll-like receptor 3 cascade. Inflamm. Res. 2018, 67, 371–374. [Google Scholar] [CrossRef]
- Shimizu, T.; Wisessmith, W.; Li, J.; Abe, M.; Sakimura, K.; Chetsawang, B.; Sahara, Y.; Tohyama, K.; Tanaka, K.F.; Ikenaka, K. The balance between cathepsin C and cystatin F controls remyelination in the brain of Plp1-overexpressing mouse, a chronic demyelinating disease model. Glia 2017, 65, 917–930. [Google Scholar] [CrossRef]
- Durose, W.W.; Shimizu, T.; Li, J.; Abe, M.; Sakimura, K.; Chetsawang, B.; Tanaka, K.F.; Suzumura, A.; Tohyama, K.; Ikenaka, K. Cathepsin C modulates myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J. Neurochem. 2019, 148, 413–425. [Google Scholar] [CrossRef]
- Haves-Zburof, D.; Paperna, T.; Gour-Lavie, A.; Mandel, I.; Glass-Marmor, L.; Miller, A. Cathepsins and their endogenous inhibitors cystatins: expression and modulation in multiple sclerosis. J. Cell. Mol. Med. 2011, 15, 2421–2429. [Google Scholar] [CrossRef]
- Czibere, L.; Baur, L.A.; Wittmann, A.; Gemmeke, K.; Steiner, A.; Weber, P.; Pütz, B.; Ahmad, N.; Bunck, M.; Graf, C.; Widner, R.; Kühne, C.; Panhuysen, M.; Hambsch, B.; Rieder, G.; Reinheckel, T.; Peters, C.; Holsboer, F.; Landgraf, R.; Deussing, J.M. Profiling trait anxiety: transcriptome analysis reveals cathepsin B (Ctsb) as a novel candidate gene for emotionality in mice. PLoS One 2011, 6, e23604. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, K.; Liu, Y.; Liu, G.; Yang, X.; Ma, J. Cathepsin C Aggravates Neuroinflammation Involved in Disturbances of Behaviour and Neurochemistry in Acute and Chronic Stress-Induced Murine Model of Depression. Neurochem. Res. 2018, 43, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Höger, N.; Feige, B.; Blechert, J.; Normann, C.; Nissen, C. Fear extinction as a model for synaptic plasticity in major depressive disorder. PLoS One 2014, 9, e115280. [Google Scholar] [CrossRef] [PubMed]
- Padamsey, Z.; McGuinness, L.; Bardo, S.J.; Reinhart, M.; Tong, R.; Hedegaard, A.; Hart, M.L.; Emptage, N.J. Activity-Dependent Exocytosis of Lysosomes Regulates the Structural Plasticity of Dendritic Spines. Neuron 2017, 93, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.P.; Silver, J. Cathepsins in neuronal plasticity. Neural Regen Res 2021, 16, 26–35. [Google Scholar]
- Talieri, M.; Papadopoulou, S.; Scorilas, A.; Xynopoulos, D.; Arnogianaki, N.; Plataniotis, G.; Yotis, J.; Agnanti, N. Cathepsin B and cathepsin D expression in the progression of colorectal adenoma to carcinoma. Cancer Lett. 2004, 205, 97–106. [Google Scholar] [CrossRef]
- Ebert, M.P.; Krüger, S.; Fogeron, M.L.; Lamer, S.; Chen, J.; Pross, M.; Schulz, H.U.; Lage, H.; Heim, S.; Roessner, A.; Malfertheiner, P.; Röcken, C. Overexpression of cathepsin B in gastric cancer identified by proteome analysis. Proteomics 2005, 5, 1693–1704. [Google Scholar] [CrossRef]
- Hwang, J.H.; Lee, S.H.; Lee, K.H.; Lee, K.Y.; Kim, H.; Ryu, J.K.; Yoon, Y.B.; Kim, Y.T. Cathepsin B is a target of Hedgehog signaling in pancreatic cancer. Cancer Lett. 2009, 273, 266–272. [Google Scholar] [CrossRef]
- Nouh, M.A.; Mohamed, M.M.; El-Shinawi, M.; Shaalan, M.A.; Cavallo-Medved, D.; Khaled, H.M.; Sloane, B.F. Cathepsin B: a potential prognostic marker for inflammatory breast cancer. J. Transl. Med. 2011, 9, 1. [Google Scholar] [CrossRef]
- Gong, F.; Peng, X.; Luo, C.; Shen, G.; Zhao, C.; Zou, L.; Li, L.; Sang, Y.; Zhao, Y.; Zhao, X. Cathepsin B as a potential prognostic and therapeutic marker for human lung squamous cell carcinoma. Mol. Cancer 2013, 12, 125. [Google Scholar] [CrossRef]
- Abdulla, M.H.; Valli-Mohammed, M.A.; Al-Khayal, K.; Al Shkieh, A.; Zubaidi, A.; Ahmad, R.; Al-Saleh, K.; Al-Obeed, O.; McKerrow, J. Cathepsin B expression in colorectal cancer in a Middle East population: Potential value as a tumor biomarker for late disease stages. Oncol. Rep. 2017, 37, 3175–3180. [Google Scholar] [CrossRef]
- Sun, T.; Jiang, D.; Zhang, L.; Su, Q.; Mao, W.; Jiang, C. Expression profile of cathepsins indicates the potential of cathepsins B and D as prognostic factors in breast cancer patients. Oncol. Lett. 2016, 11, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.E.; Ho, C.C.; Yang, S.F.; Lin, S.H.; Yeh, K.T.; Lin, C.W.; Chen, M.K. Cathepsin B Expression and the Correlation with Clinical Aspects of Oral Squamous Cell Carcinoma. PLoS One 2016, 11, e0152165. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, H.; Li, Z.; Wang, L.; Zheng, F.; Jiang, J.; Gao, Y.; Zhong, H.; Huang, Y.; Suo, Z. Cathepsin B may be a potential biomarker in cervical cancer. Histol. Histopathol. 2012, 27, 79–87. [Google Scholar]
- Devetzi, M.; Scorilas, A.; Tsiambas, E.; Sameni, M.; Fotiou, S.; Sloane, B.F.; Talieri, M. Cathepsin B protein levels in endometrial cancer: Potential value as a tumour biomarker. Gynecol. Oncol. 2009, 112, 531–536. [Google Scholar] [CrossRef]
- Jechorek, D.; Votapek, J.; Meyer, F.; Kandulski, A.; Roessner, A.; Franke, S. Characterization of cathepsin X in colorectal cancer development and progression. Pathol. Res. Pract. 2014, 210, 822–829. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, D.; Hu, T.; Zhao, H.; Zhao, X.; Lou, Z.; He, Y.; Qin, W.; Xia, J.; Zhang, X.; Ye, L.C. KMT2A histone methyltransferase contributes to colorectal cancer development by promoting cathepsin Z transcriptional activation. Cancer Med 2019, 8, 3544–3552. [Google Scholar] [CrossRef] [PubMed]
- Krueger, S.; Kalinski, T.; Hundertmark, T.; Wex, T.; Küster, D.; Peitz, U.; Ebert, M.; Nägler, D.K.; Kellner, U.; Malfertheiner, P.; Naumann, M.; Röcken, C.; Roessner, A. Up-regulation of cathepsin X in Helicobacter pylori gastritis and gastric cancer. J. Pathol. 2005, 207, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, L.; Li, Y.; Guan, X.Y. Overexpression of cathepsin Z contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. PLoS One 2011, 6, e24967. [Google Scholar] [CrossRef]
- Lines, K.E.; Chelala, C.; Dmitrovic, B.; Wijesuriya, N.; Kocher, H.M.; Marshall, J.F.; Crnogorac-Jurcevic, T. S100P-binding protein, S100PBP, mediates adhesion through regulation of cathepsin Z in pancreatic cancer cells. Am. J. Pathol. 2012, 180, 1485–1494. [Google Scholar] [CrossRef]
- del Re, E.C.; Shuja, S.; Cai, J.; Murnane, M.J. Alterations in cathepsin H activity and protein patterns in human colorectal carcinomas. Br. J. Cancer 2000, 82, 1317–1326. [Google Scholar]
- Staack, A.; Tolic, D.; Kristiansen, G.; Schnorr, D.; Loening, S.A.; Jung, K. Expression of cathepsins B, H, and L and their inhibitors as markers of transitional cell carcinoma of the bladder. Urology 2004, 63, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.M.; Huang, Y.H.; Yeh, C.T.; Tsai, M.M.; Liao, C.H.; Cheng, W.L.; Chen, W.J.; Lin, K.H. Cathepsin H regulated by the thyroid hormone receptors associate with tumor invasion in human hepatoma cells. Oncogene 2011, 30, 2057–2069. [Google Scholar] [CrossRef]
- Khaket, T.P.; Singh, M.P.; Khan, I.; Bhardwaj, M.; Kang, S.C. Targeting of cathepsin C induces autophagic dysregulation that directs ER stress mediated cellular cytotoxicity in colorectal cancer cells. Cell. Signal. 2018, 46, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Jevnikar, Z.; Rojnik, M.; Jamnik, P.; Doljak, B.; Fonovic, U.P.; Kos, J. Cathepsin H mediates the processing of talin and regulates migration of prostate cancer cells. J. Biol. Chem. 2013, 288, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Fang, Y.Q.; Zhang, T.Y.; Ge, B.; Tang, R.J.; Huang, J.F.; Jiang, L.M.; Tan, N. Acidic extracellular microenvironment promotes the invasion and cathepsin B secretion of PC-3 cells. Int. J. Clin. Exp. Med. 2015, 8, 7367–7373. [Google Scholar]
- Jakoš, T.; Pišlar, A.; Jewett, A.; Kos, J. Cysteine Cathepsins in Tumor-Associated Immune Cells. Front. Immunol. 2019, 10, 2037. [Google Scholar] [CrossRef]
- Vasiljeva, O.; Papazoglou, A.; Krüger, A.; Brodoefel, H.; Korovin, M.; Deussing, J.; Augustin, N.; Nielsen, B.S.; Almholt, K.; Bogyo, M.; Peters, C.; Reinheckel, T. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006, 66, 5242–5250. [Google Scholar] [CrossRef]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Akkari, L.; Gocheva, V.; Kester, J.C.; Hunter, K.E.; Quick, M.L.; Sevenich, L.; Wang, H.W.; Peters, C.; Tang, L.H.; Klimstra, D.S.; Reinheckel, T.; Joyce, J.A. Distinct functions of macrophage-derived and cancer cell-derived cathepsin Z combine to promote tumor malignancy via interactions with the extracellular matrix. Genes Dev. 2014, 28, 2134–2150. [Google Scholar] [CrossRef]
- Vasiljeva, O.; Korovin, M.; Gajda, M.; Brodoefel, H.; Bojic, L.; Krüger, A.; Schurigt, U.; Sevenich, L.; Turk, B.; Peters, C.; Reinheckel, T. Reduced tumour cell proliferation and delayed development of high-grade mammary carcinomas in cathepsin B-deficient mice. Oncogene 2008, 27, 4191–4199. [Google Scholar] [CrossRef]
- Edgington-Mitchell, L.E.; Rautela, J.; Duivenvoorden, H.M.; Jayatilleke, K.M.; van der Linden, W.A.; Verdoes, M.; Bogyo, M.; Parker, B.S. Cysteine cathepsin activity suppresses osteoclastogenesis of myeloid-derived suppressor cells in breast cancer. Oncotarget 2015, 6, 27008–27022. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006, 20, 543–556. [Google Scholar] [CrossRef]
- Victor, B.C.; Anbalagan, A.; Mohamed, M.M.; Sloane, B.F.; Cavallo-Medved, D. Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Res. 2011, 13, R115. [Google Scholar] [CrossRef]
- Mitrović, A.; Mirković, B.; Sosič, I.; Gobec, S.; Kos, J. Inhibition of endopeptidase and exopeptidase activity of cathepsin B impairs extracellular matrix degradation and tumour invasion. Biol. Chem. 2016, 397, 165–174. [Google Scholar] [CrossRef]
- Mai, J.; Sameni, M.; Mikkelsen, T.; Sloane, B.F. Degradation of extracellular matrix protein tenascin-C by cathepsin B: an interaction involved in the progression of gliomas. Biol. Chem. 2002, 383, 1407–1413. [Google Scholar] [CrossRef]
- Zhang, G.P.; Yue, X.; Li, S.Q. Cathepsin C Interacts with TNF-α/p38 MAPK Signaling Pathway to Promote Proliferation and Metastasis in Hepatocellular Carcinoma. Cancer Res. Treat. 2020, 52, 10–23. [Google Scholar] [CrossRef]
- Gogineni, V.R.; Gupta, R.; Nalla, A.K.; Velpula, K.K.; Rao, J.S. uPAR and cathepsin B shRNA impedes TGF-β1-driven proliferation and invasion of meningioma cells in a XIAP-dependent pathway. Cell Death Dis. 2012, 3, e439. [Google Scholar] [CrossRef] [PubMed]
- Tummalapalli, P.; Spomar, D.; Gondi, C.S.; Olivero, W.C.; Gujrati, M.; Dinh, D.H.; Rao, J.S. RNAi-mediated abrogation of cathepsin B and MMP-9 gene expression in a malignant meningioma cell line leads to decreased tumor growth, invasion and angiogenesis. Int. J. Oncol. 2007, 31, 1039–1050. [Google Scholar] [PubMed]
- Sevenich, L.; Schurigt, U.; Sachse, K.; Gajda, M.; Werner, F.; Müller, S.; Vasiljeva, O.; Schwinde, A.; Klemm, N.; Deussing, J.; Peters, C.; Reinheckel, T. Synergistic antitumor effects of combined cathepsin B and cathepsin Z deficiencies on breast cancer progression and metastasis in mice. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 2497–2502. [Google Scholar] [CrossRef]
- Chen, C.H.; Bhasin, S.; Khanna, P.; Joshi, M.; Joslin, P.M.; Saxena, R.; Amin, S.; Liu, S.; Sindhu, S.; Walker, S.R.; Catalano, P.; Frank, D.A.; Alper, S.L.; Bhasin, M.; Bhatt, R.S. Study of Cathepsin B inhibition in VEGFR TKI treated human renal cell carcinoma xenografts. Oncogenesis 2019, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Chen, X.; Peters, C.; Reinheckel, T.; Joyce, J.A. Deletion of cathepsin H perturbs angiogenic switching, vascularization and growth of tumors in a mouse model of pancreatic islet cell cancer. Biol. Chem. 2010, 391, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Cottone, L.; Junankar, S.; Johansson, M.; DeNardo, D.G.; Korets, L.; Reinheckel, T.; Sloane, B.F.; Bogyo, M.; Coussens, L.M. Cathepsin C is a tissue-specific regulator of squamous carcinogenesis. Genes Dev. 2013, 27, 2086–2098. [Google Scholar] [CrossRef]
- Overall, C.M.; Dean, R.A. Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metastasis Rev. 2006, 25, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.; Turk, B. Dual contrasting roles of cysteine cathepsins in cancer progression: apoptosis versus tumour invasion. Biochimie 2008, 90, 380–386. [Google Scholar] [CrossRef]
- Affara, N.I.; Andreu, P.; Coussens, L.M. Delineating protease functions during cancer development. Methods Mol. Biol. 2009, 539, 1–32. [Google Scholar]
- Skrzydlewska, E.; Sulkowska, M.; Koda, M.; Sulkowski, S. Proteolytic-antiproteolytic balance and its regulation in carcinogenesis. World J. Gastroenterol. 2005, 11, 1251–1266. [Google Scholar] [CrossRef]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Mason, S.D.; Joyce, J.A. Proteolytic networks in cancer. Trends Cell Biol. 2011, 21, 228–237. [Google Scholar] [CrossRef]
- Wang, J.; Youkharibache, P.; Zhang, D.; Lanczycki, C.J.; Geer, R.C.; Madej, T.; Phan, L.; Ward, M.; Lu, S.; Marchler, G.H.; Wang, Y.; Bryant, S.H.; Geer, L.Y.; Marchler-Bauer, A. iCn3D, a web-based 3D viewer for sharing 1D/2D/3D representations of biomolecular structures. Bioinformatics. 2020, 36, 131–135. [Google Scholar] [CrossRef]
Figure 1.
Structural elements of cysteine cathepsins exhibiting exopeptidase activity. Ribbon representation of the 3D structure of cathepsins B (blue), H (red), C (exclusion domain – maroon, heavy chain – light green, light chain – orange), and X (dark green) superimposed on the endopeptidase papain, the representative member of this family (magenta). The figure was made using iCn3D [241].
Figure 1.
Structural elements of cysteine cathepsins exhibiting exopeptidase activity. Ribbon representation of the 3D structure of cathepsins B (blue), H (red), C (exclusion domain – maroon, heavy chain – light green, light chain – orange), and X (dark green) superimposed on the endopeptidase papain, the representative member of this family (magenta). The figure was made using iCn3D [241].
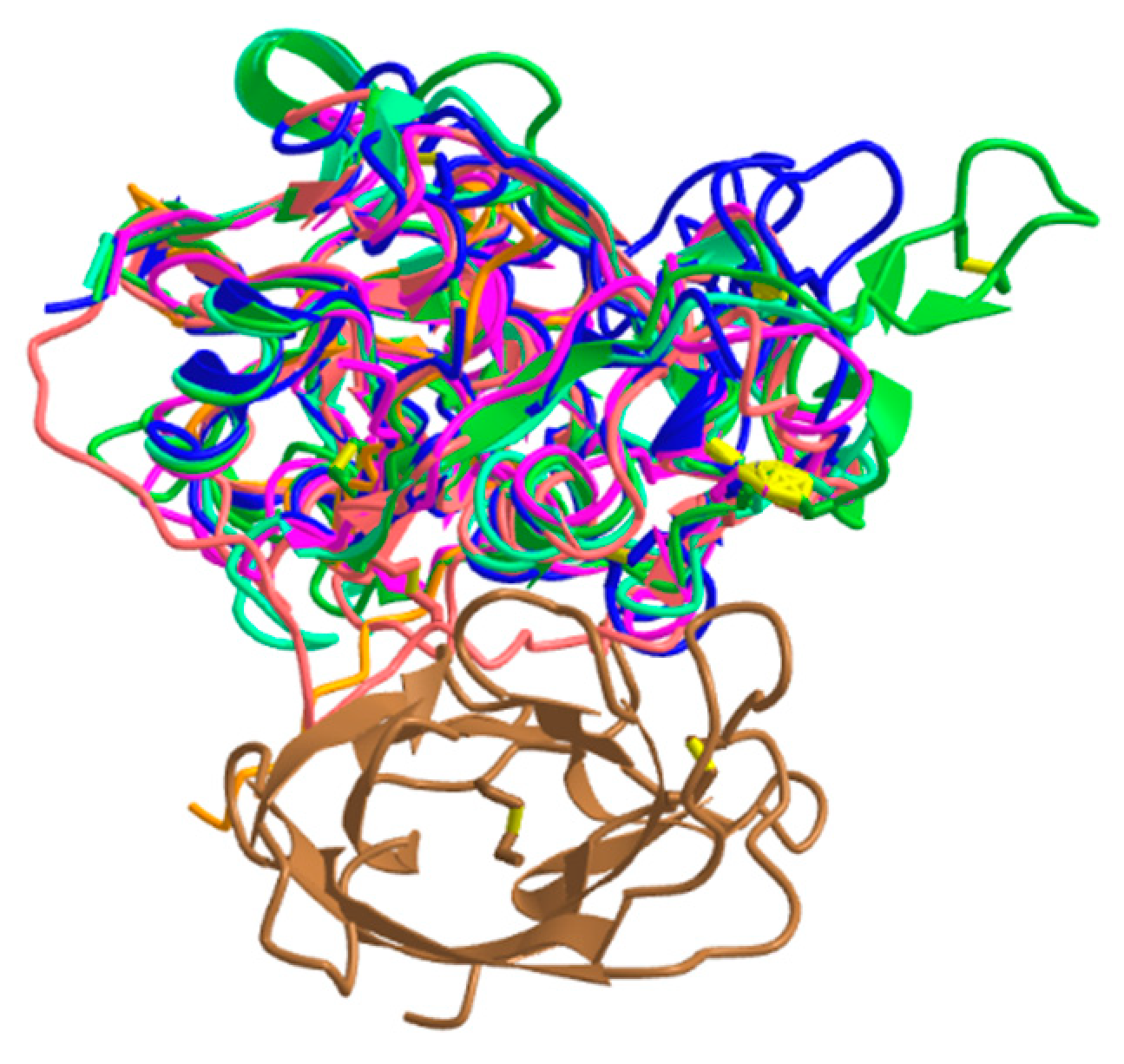
Figure 2.
Schematic model of neuronal–microglial crosstalk with emphasis on the role of cathepsins in neurodegenerative diseases.
Figure 2.
Schematic model of neuronal–microglial crosstalk with emphasis on the role of cathepsins in neurodegenerative diseases.
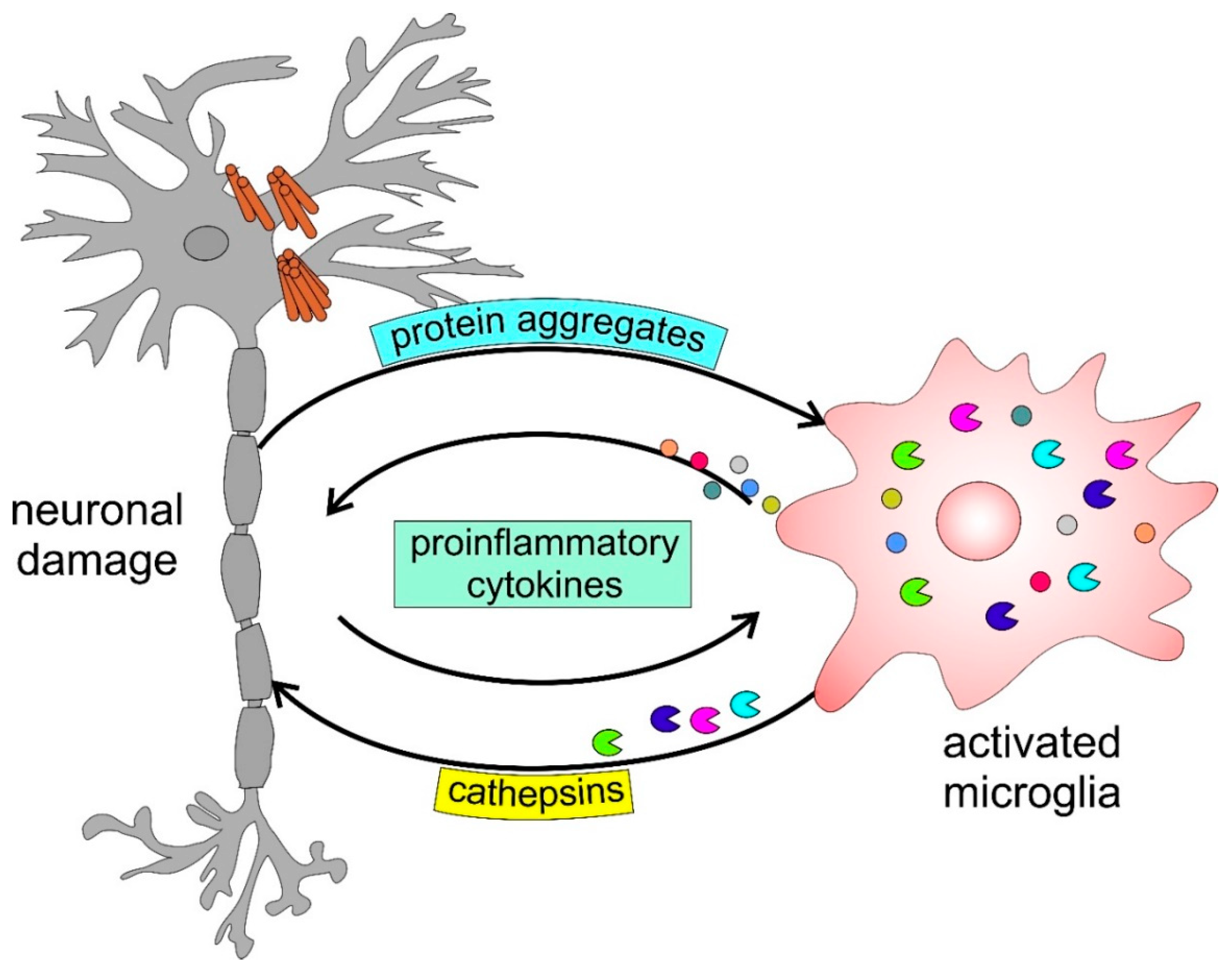
Figure 3.
Schematic model of the role of reciprocal interactions between tumor and stromal cells in promoting tumor progression.
Figure 3.
Schematic model of the role of reciprocal interactions between tumor and stromal cells in promoting tumor progression.
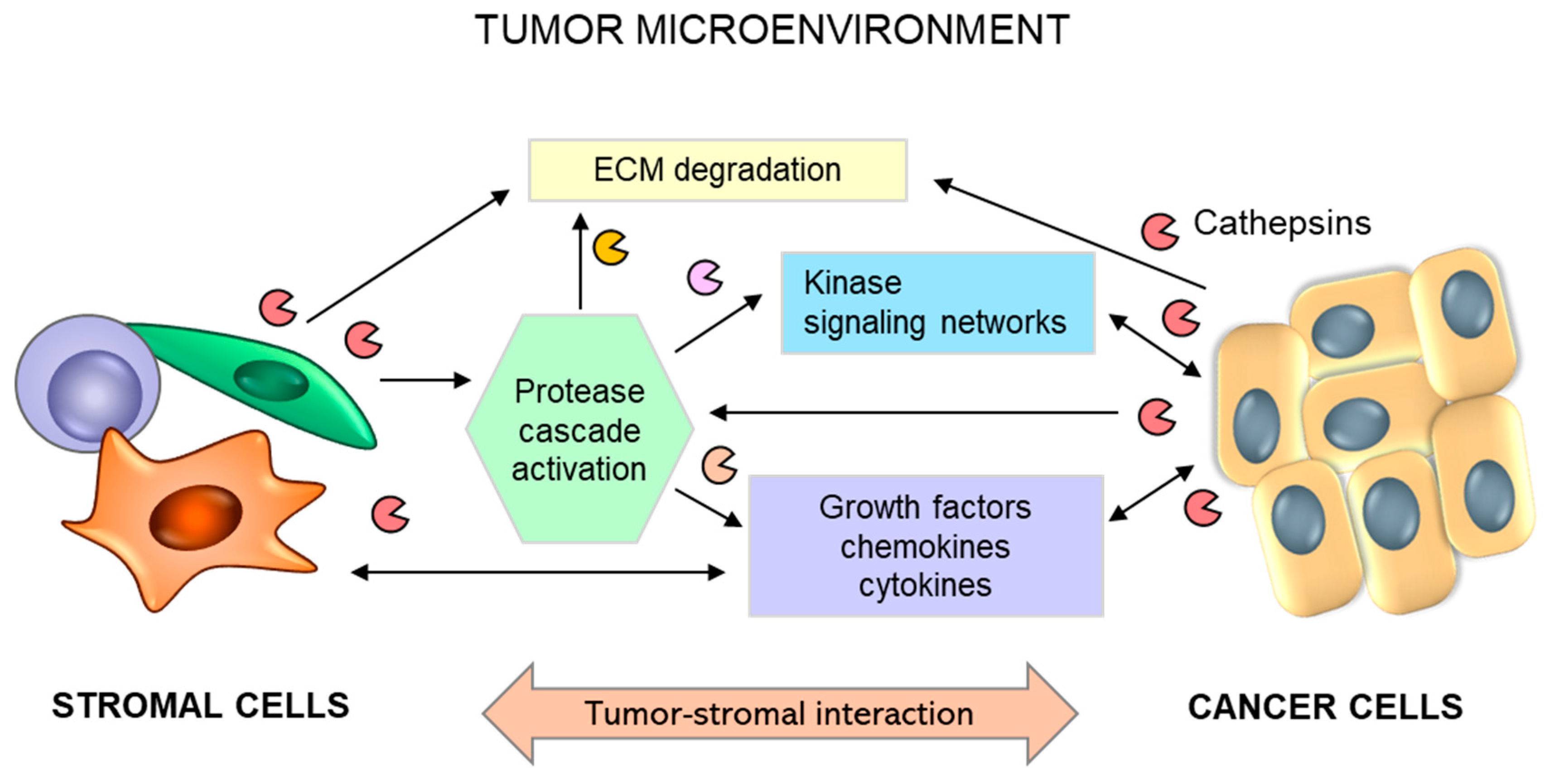
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



