
Submitted:
25 July 2023
Posted:
26 July 2023
You are already at the latest version
Abstract
Being the major cellular component of highly dynamic tissue endometrial stromal cells (EnSC) are exposed to cycles of proliferation upon hormonal stimulation what might pose risks for mutations accumulation and malignization. However, endometrial stromal tumors are rare and unusual types of tumor. The present study aimed to uncover defense mechanisms that might underlie resistance of EnSC against oncogenic transformation. All experiments were performed in vitro using the following methods: FACS, WB, RT-PCR, IF, molecular cloning, lentiviral transduction, and CRISPR/Cas9 genome editing. We revealed that expression of the mutant HRASG12V results in senescence of EnSC. We experimentally confirmed inability of HRASG12V-expressing EnSC to bypass senescence and resume proliferation even upon oestrogen stimulation. At the molecular level, induction of oncogene-induced senescence (OIS) was accompanied by the activation of MEK/ERK, PI3K/AKT, p53/p21WAF/CIP/Rb and p38/p16INK4a/Rb pathways, however inhibiting either pathway did not prevent cell cycle arrest. PTEN loss was established as the additional feature of HRASG12V-induced senescence of EnSC. By using CRISPR-Cas9 mediated PTEN knockout, we distinguished PTEN loss-induced senescence as a reserve molecular mechanism to prevent transformation of HRASG12V-expressing EnSC. The present study highlights oncogene-induced senescence as an antitumor defense mechanism of EnSC controlled by multiple backup molecular pathways.
Keywords:
endometrial stromal cells
; senescence
; oncogenes
; cancer
; HRASG12V
1. Introduction
The endometrium is the inner lining of the uterus essential for human reproduction. During women's life this unique tissue adopts to multiple physiological states, including premenarche, menstrual cycling, pregnancy, and postmenopause [1,2]. Among these states, menstrual cycling is almost continuous during fertile period of the women with exception for pregnancy and lactation. Menstrual cycles are governed by oscillating levels of oestrogen and progesterone, which results in cyclic growth, differentiation/decidualization, shedding and further regeneration of two-thirds of the endometrium [1]. At the histological level endometrium is composed of a stromal cell layer invaginated by epithelial glands and covered by the luminal epithelium [2]. The highly dynamic nature of this tissue contributes to the accumulation of somatic mutations in cancer-associated genes, what in turn poses risks for developing cancer in adult women [3,4]. Indeed, endometrial cancer is one of the most common gynecologic malignancies worldwide [3]. Interestingly, despite the fact that both epithelial and stromal cells are affected by cyclic hormonal alterations, endometrial cancer is commonly referred to endometrial carcinoma, while endometrial stromal tumors seem to be rare and unusual types of tumor [5,6]. The latter raises the question regarding defense mechanisms that might underlie resistance of endometrial stromal cells (EnSC) against oncogenic transformation.
Senescence is well-established tumor-suppressive mechanism [7]. At the cellular level, senescence is considered as an important intrinsic stress-reaction that prevents propagation of cells bearing damages via irreversible cell cycle block [7]. Different types of senescence are commonly distinguished, including replicative and various stress-induced forms [8]. More than 20 years ago, expression of oncogenes was also shown to trigger senescence [9]. For now numerous oncogenes such as HRASG12V, NRASQ61R, BRAFV600E are proved to trigger oncogene-induced senescence (OIS) in various types of cells [reviewed in 10]. OIS shares the same basic features as the replicative and the stress-induced forms of senescence, i.e. irreversible proliferation block, enhanced expression of the inhibitors of cyclin-dependent kinases p21WAF/CIP and p16INK4a, DNA damage, increased cell size, elevated intracellular reactive oxygen species levels, senescence-associated β-galactosidase (SA-β-gal) activity [10]. A plenty of evidence provides strong arguments that OIS serves as the first barrier of defense against cancer development [11,12,13,14]. Indeed, cells with features of OIS have been detected in early neoplastic and premalignant lesions in different genetically engineered mouse models as well as in humans [11,12]. Interestingly, further progression of a subset of these lesions to more advanced cancer stages is associated with the loss of senescence features [14].
Previously, we have shown that EnSC are prone to senescence triggered by various stresses including oxidative stress, heat shock, radiation, and treatment with genotoxic agents [15,16,17]. However, the response of EnSC towards oncogenes expression remained uncovered. Within the present study, we tested the suggestion that OIS might form defense mechanism of EnSC against neoplastic transformation.
2. Results
2.1. Expression of HRASG12V results in senescence-like phenotype in EnSC
Mutations in the RAS gene occur frequently in various human cancers and are experimentally validated to be the drivers of tumor initiation and maintenance. Missense mutation G12V contributes to HRAS oncogenicity by stabilizing this protein in constitutively active GTP bound state [3,4]. To explore the outcomes of HRASG12V expression in EnSC we applied the tetracycline-controlled system (Tet-On), which allows inducible expression of the mutant oncogene HRASG12V. The system included two lentivectors: (1) the first one contained the sequence of the mutant oncogene HRASG12V, preceded by the regulatory Tet-operator (TetO) sequence; (2) the second lentivector contained coding sequences of the repressor protein TetR and of the fluorescent reporter GFP (Figure 1A). In the absence of tetracycline, TetR binds to the TetO sequence and prevents HRASG12V expression. Upon addition, tetracycline interacts with TetR, what allows unhindered expression of the oncogene. By using this system, we first assessed proliferation kinetics in HRASG12V-expressing EnSC. As shown in Figure 1B, upon tetracycline EnSC transduced with the lentivectors for HRASG12V expression demonstrated gradual decrease in proliferation rate until its complete loss on day 4 (Figure 1B). Control EnSC, which carried the same Tet-On lentiviral system but were not treated with tetracycline, preserved normal proliferation rate during the whole observation period (Figure 1B). To validate acquisition of senescent phenotype by EnSC expressing HRASG12V, we further investigated another hallmarks of senescence. As shown in microphotographs, EnSC expressing HRASG12V became flattened and vacuolated (Figure 1C). Moreover, upon expression of the oncogene EnSC gradually increased in size (Figure 1D). We next revealed SA-β-gal activity in HRASG12V-expressing EnSC (Figure 1E,F). Finally, we observed enhanced expression of the crucial components of senescence associated secretory phenotype (SASP), including IL6, MMP2, and STC1 in EnSC upon HRASG12V expression (Figure 1G). Together the set of the identified parameters provides clear evidence in favor of senescence induction in EnSC expressing HRASG12V.
3.2. EnSC expressing HRASG12V are unable to bypass senescence
The above results indicate that EnSC enter senescence upon activation of the oncogene HRASG12V. Though typically senescence is believed to be the irreversible cell state, there is also evidence indicating that some cells may overcome OIS [14,19]. It was shown that cells remained in the senescent state for prolonged periods might resume proliferation and develop features of cancer cells [14,19]. In accordance with this notion, we revealed the appearance of the small clones of proliferating cells among the senescent ones at day 13 of tetracycline treatment (Figure 2A). Moreover, around day 40 upon tetracycline small proliferating cells almost completely replaced senescent ones. To test whether these cells indeed escaped from HRASG12V-induced senescence, we modified our lentivectors and performed additional experiments. The original lentivector containing HRASG12V coding sequence also included the puromycin resistance gene sequence. The later allowed selection of HRASG12V-carrying cells on the puromycin-containing media, however such selection may be imperfect. To additionally control selection procedure, we cloned mCherry coding sequence in the same reading frame just before the HRASG12V sequence, so that both genes were under the same promoter (Figure 2B). By using our modified HRASG12V-mCherry Tet-On system we were able to visualize HRASG12V expressing EnSC by mCherry fluorescence. As expected, at day 37 of tetracycline treatment we observed large and flattened senescent EnSC, which were mCherry-positive, and colonies of small cells, which, however, did not express mCherry and thus did not carry HRASG12V (Figure 2B). This result favors the notion that mCherry-negative small cells might remain after puromycin selection. Indeed, additional round of puromycin treatment led to significant death of these small cells, which was comparable to that of the control (non-transduced) EnSC (Figure 2C). Together these data provide evidence that EnSC can not overcome HRASG12V-induced senescence and suggest tight molecular regulation of OIS stability in EnSC.
3.3. Oestrogen supplementation does not prevent proliferation loss in HRASG12V-expressing EnSC
Due to its functional role, EnSC are exposed to the cyclic influence of oestrogen that stimulates proliferation of these cells crucial for endometrial regrowth during each menstrual cycle [2]. At the same time, there are literary evidences indicating that oestogen might reduce senescence of various cell types including endothelial progenitor cells, chondrocytes and human mesenchymal stem cells [20,21,22]. Therefore, we assessed the effects of oestrogen on the main characteristics of HRASG12V-expressing EnSC. To this end, we additionally supplemented tetracycline-containing growth media with β-Estradiol. Importantly, β-Estradiol had no effect either on proliferation, or on the size and SA-β-gal activity of the HRASG12V-expressing EnSC (Figure 3). These results provide additional confirmation of the stability of senescence reaction of EnSC upon oncogene expression.
3.4. Molecular mechanisms regulating HRASG12V-induced senescence in EnSC
The main cellular outcomes of RAS activation are proliferation and survival mediated via RAS/RAF/MEK/ERK and RAS/PI3K/AKT pathways [23]. In line with that, we revealed phosphorylation of the main downstream targets of RAS – ERK1/2 and AKT – short after tetracycline addition (Figure 4A). The later resulted in a brief period of hyperproliferation of oncogene-expressing EnSC, as indicated in Figure 4B.
Commonly, hyperproliferation caused by oncogenic HRASG12V drives DNA replication stress and constitutive activation of DNA damage response (DDR) [24]. Indeed, we detected accumulation of γH2AX foci, which mark DNA damage, in HRASG12V-expressing EnSC (Figure 4C). Activation of DDR in EnSC triggered by the expression of the oncogene led to phosphorylation of the Chk2 and of the tumor suppressor protein p53 (Figure 4D). The later was expectedly followed by the enhanced expression of the inhibitor of cyclin-dependent kinases p21WAF/CIP and establishment of cell cycle arrest (Figure 4D). Another classical molecular pathway responsible for cell cycle block maintenance during senescence is p38/p16INK4a/Rb [17]. As shown in Figure 4E, stress-kinase p38 became activated in EnSC after 8 days of tetracycline supplementation, what further led to the elevated expression of p16INK4a. Thus, we detected activation of two major signaling pathways p53/p21WAF/CIP/Rb and p38/p16INK4a/Rb that mediate cell cycle block in HRASG12V-expressing EnSC. Activation of both pathways was strictly coordinated; expression p21WAF/CIP started just after tetracycline addition and remained elevated for 8 days, while expression of p16INK4a increased only at day 11 (Figure 4D,E). The revealed activation dynamics suggests that p53/p21WAF/CIP/Rb pathway is responsible for the cell cycle block initiation while p38/p16INK4a/Rb mediates its stabilization during OIS in EnSC.
Having established crucial molecular pathways that mediate initiation and progression of HRASG12V-induced senescence in EnSC, we further tested the possibility to reverse or alleviate this reaction. To this end, we applied specific inhibitors of either pathway, including (1) U0126 that inhibits the kinase activity of MEK1/2 thus preventing the activation of ERK; (2) LY294002 that blocks PI3K-dependent AKT phosphorylation and kinase activity; (3) SB203580 that inhibits catalytic activity of p38 by binding to the ATP binding pocket; (4) pifithrin-α – inhibitor of p53 activity (Figure 5A). The compounds were used at concentrations found to be efficient and non-toxic in our previous studies [15,25,26,27]. As shown in Figure 5A,B, U0126 and LY294002 prevented cell size increase and SA-β-Gal staining of HRASG12V-expressing EnSC, what demonstrates involvement of both pathways in the acquisition of senescent phenotype during OIS. At the same time, none of the compounds applied was able to prevent proliferation loss induced by the oncogene expression (Figure 5C). Of note, previously we revealed that cell cycle arrest in stress-induced senescent EnSC could be overcome by p38 inhibition [15]. These results suggest that cell cycle arrest in HRASG12V-expressing EnSC is probably controlled by several backup pathways what prevents cell cycle reentry and transformation.
3.5. PTEN loss-induced senescence is an additional backup molecular mechanism to prevent transformation of HRASG12V-expressing EnSC
Previously, it was shown that oncogenic HRAS downregulates expression of tumor suppressor PTEN that negatively regulates cell survival by opposing the activation of the PI3K/AKT/mTOR signaling network [23]. Expression of the mutant HRASG12V in EnSC resulted in decreased mRNA and protein expression of the crucial tumor-suppressor PTEN (Figure 6A,B). As shown in Figure 6A, B, both PTEN mRNA and protein levels gradually declined from day 2 reaching minimal level on day 6 under tetracycline treatment. To distinguish functional role of PTEN downregulation during the development of HRASG12V-induced senescence of EnSC, we further knocked out PTEN. As shown in Figure 6C,D, application of CRISPR/Cas9 genome editing system resulted in significant reduction of PTEN gene expression and almost completely abolished its expression at the protein level. We next assessed the main characteristics of EnSC with PTEN loss. Notably, along with complete proliferation loss EnSC with PTEN knockout demonstrated increased cell size and autofluorecesnce levels, indicating hypertrophy and lipofucine accumulation (Figure 6E–G). Moreover, we detected enhanced SA-β-gal staining of EnSC with PTEN loss (Figure 6H,I). Together, these data indicate that PTEN loss is sufficient to trigger senescence in EnSC. Thus, PTEN loss observed in HRASG12V-expressing EnSC might form additional backup mechanism to induce senescence and thus to avoid cell transformation.
3. Discussion
The present study aimed to uncover defense mechanisms of EnSC against transformation. We revealed that EnSC are prone to OIS in response to HRASG12V oncogene expression. The first evidence of HRAS-induced senescence dates back to 1997, when the authors revealed stalled mitotic activity, enlarged and flattened morphology of human diploid fibroblasts expressing mutant HRASG12V [9]. Later on this observation was significantly extended, and today OIS resulting from expression of different oncogenes is considered as the intrinsic antitumor mechanism common for various types of cells [10]. Similar to other cell types, upon HRASG12V expression EnSC acquire all features typical for senescent cells, including proliferation block, altered morphology and SA-β-Gal activity. Commonly, complete proliferation loss and development of senescent phenotype are considered as the consequences of DDR resulting from hyperreplication of genomic DNA induced by the oncogene expression [24]. In line with this notion, we revealed a brief period of hyperproliferation of HRASG12V-expressing EnSC followed by proliferation arrest and appearance of the DNA damage.
Despite the concrete mechanism of DNA damage upon oncogene expression, DDR further leads to cell cycle arrest via p53/p21WAF/CIP/Rb and/or p38/p16INK4a/Rb pathways [17]. Although the senescence as the outcome of oncogene expression may seem cell type-independent, molecular mechanisms underlying cell cycle arrest establishment during OIS largely depend on the cellular context [28]. For example, expression of p53 is crucial for HRASG12V-induced senescence development in normal human fibroblasts, since its depletion prevents proliferation arrest in oncogene-expressing cells [9,24,29]. Contrarily, normal human mammary epithelial cells as well as esophageal keratinocytes undergo HRASG12V-induced senescence via p53-independent mechanism [30,31]. The same controversy is true for p16INK4a involvement in HRASG12V-induced senescence: while progression of OIS in fibroblasts rely on p16INK4a expression, in HRASG12V-expressing normal human melanocytes depletion of p16INK4a had no effect on senescence progression [32,33]. Notably, recent study revealed that expression of oncogenic HRASG12V together with p16INK4a knockdown in EnSC induced high-grade endometrial stromal sarcoma in mice [34]. Here we detected activation of both signaling pathways p53/p21WAF/CIP/Rb and p38/p16INK4a/Rb in HRASG12V-expressing EnSC. However, neither inhibition of p53 nor downregulation of p38/p16INK4a affected OIS progression in EnSC. Moreover, inhibition of ERK and AKT – downstream targets of the major signaling pathways directly activated by RAS – reduced senescent phenotype but also did not affect cell cycle block in oncogene-expressing EnSC. Together these data suggest the existence of compensatory pathways that regulate cell cycle arrest during OIS in EnSC.
Further analysis of the molecular consequences of HRASG12V expression in EnSC uncovered gradual loss of PTEN expression. Earlier it was shown that oncogenic RAS down-regulates expression of the proapoptotic tumor suppressor PTEN in fibroblasts and epithelial cells via p53-independent pathway [23]. The authors of the above study experimentally verified that oncogenic RAS might suppress PTEN expression via the RAF/MEK/ERK/c-Jun pathway leading to cellular transformation. However, in case of HRASG12V-expressing EnSC decreased expression of PTEN did not lead to cellular transformation. By performing additional set of experiments, we uncovered that PTEN loss itself is sufficient to trigger senescence in EnSC. This result is in line with the existing literary data uncovering tumor suppressor loss-induced form of senescence triggered by reduced expression of PTEN gene [35,36]. It should be highlighted that previous studies considered both HRASG12V- and PTEN-loss-induced forms of senescence separately, while the present study provides the first evidence of the possible intersection between these forms of senescence. Together the results obtained allow speculating that PTEN loss might form backup mechanism that additionally controls proliferation arrest during HRASG12V-induced senescence of EnSC.
Despite of the tight molecular control of proliferation arrest during OIS, a growing body of evidence demonstrates that cells might escape from OIS through cell-autonomus and cell-non-autonomus mechanisms, including derepression of hTERT locus, reorganization of topologically associated domains, stemness-associated reprogramming and downregulation of histone demethylases [14,19]. For example, previous study revealed that the population of the human diploid fibroblasts could spontaneously escape from OIS induced by HRASG12V [19]. The authors demonstrated preserved proliferation and decreased expression of p16INK4a in OIS-escaped fibroblasts. Similarly to these findings, we detected the appearance of small proliferating EnSC, which later on completely replaced senescent EnSC from the population. However, we experimentally verified that these small cells originated from imperfect lentiviral transduction and further antibiotic selection. Probably, the results of the above study might also outcome from the technical batches of the transduction procedure, since OIS-escaped fibroblasts were prone to stress-induced senescence and were not able for anchorage-independent growth in soft-agar [19]. According to our data, EnSC that expressed HRASG12V remained in senescent state for prolonged periods and were not able to overcome OIS and resume proliferation.
Another possibility to escape from OIS that should be taken into account might result from the physiological oestrogen-mediated regulation of EnSC proliferation within endometrium. This possibility is reinforced by the fact that oestrogen controls telomerase activity and hTERT expression in various estrogen targeted tissues, including endometrium [37]. Indeed, oestrogen deficiency leads to telomere shortening, while, oestrogen hyperstimulation increases telomerase activity and maintains telomere length [38]. Moreover, oestrogen supplementation turned out to be effective to reduce senescence of various types of cells [20,21,22]. Notably, HRASG12V-expressing EnSC remained stably arrested and preserved all the features of senescent cells despite of oestrogen addition.
4. Materials and Methods
4.1. EnSC culture conditions
EnSC line used in the present study was obtained from the shared research facility “Vertebrate cell culture collection” of the Institute of Cytology of the Russian Academy of Sciences (supported by the Ministry of Science and Higher Education of the Russian Federation, Agreement №075-15-2021-683). Cells were cultured in DMEM/F12 (Gibco BRL, USA) at 37 ˚C in humidified incubator containing 5% CO2. Cultural medium was supplemented with 10% FBS (HyClone, USA), 1% penicillin-streptomycin (Gibco BRL, USA) and 1% glutamax (Gibco BRL, USA). Serial passaging was performed when the cells reached 80% – 90% confluence. Cells at early passages (5 – 9) were used in all experiments.
4.2. Molecular cloning
Four-steps molecular cloning was performed to insert the sequence encoding the mCherry fluorescent protein into pLenti CMV/TO RasV12 Puro (w119-1) (Addgene #22262). Firstly, mCherry sequence together with P2A sequence were amplified from pUltra-hot lentivector (Addgene #24130) using specific primers listed in Table 1. Secondly, HRASG12V sequence was amplified from pLenti CMV/TO RasV12 Puro (w119-1) using specific primers listed in Table 1. Of note, mCherry reverse and HRASG12V forward primers contained complementary sites for further overlap. The obtained sequences were then combined by amplification using two primers – mCherry forward and HRASG12V reverse (annealing temperature 67 ˚С, 2 min elongation). Final product was inserted into pLenti CMV/TO RasV12 Puro (w119-1) by restriction using XbaI (New England Biolabs, USA) and BamHI (New England Biolabs, USA) and further ligation using Quick Ligation™ Kit (New England Biolabs, USA). The obtained lentivector was named HRAS-mCherry pLenti CMV/TO RasV12 Puro.
For CRISPR-mediated PTEN knockout pCC_01—hU6-BsmBI-sgRNA(EþF)-barcode-EFS-Cas9-NLS-2A-Puro-WPRE (Addgene #139086) was used. Oligonucleotide sequences of single guide RNAs (sgRNAs) for PTEN were designed using the CCTop-CRISPR/Cas9 target online predictor and the CRISPR-ERA web applications, sequences are presented in Table 1. SgRNA coding sequences were inserted into the indicated above lentivector using BsmBI-based cloning (Thermo Fisher Scientific, USA), following the protocols described in our previous study [18]. Unmodified lentivector, containing non-targeting sgRNAs sequence, was used as non-targeting control.
All amplification procedures were performed using Encyclo Plus PCR kit (Evrogen, Russia). DNA products were cleaned up from PCR mix/agarose gels using Cleanup Standard kit (Evrogen, Russia). Plasmid DNA was extracted using Plasmid Miniprep kit (Evrogen, Russia). All procedures were performed according to manufacturer’s instructions. Plasmids were amplified using E.coli strain Stbl3.
4.3. Lentiviral transduction and tetracycline treatment
EnSC were transduced with the viruses produced with the use of the following lentivectors: (1) pLenti CMV/TO RasV12 Puro (w119-1) (Addgene #22262), (2) FgH1tUTG (Addgene #70183), (3) HRAS-mCherry pLenti CMV/TO RasV12 Puro, (4) pCC_01—hU6-BsmBI-sgRNA(EþF)-barcode-EFS-Cas9-NLS-2A-Puro-WPRE and (5) its variant for PTEN knockout. Protocols of lentiviral particles production and EnSC lentiviral transduction are described in detail in our previous article [18]. To induce HRASG12V expression EnSC transduced with the appropriate lentiviruses were cultured in medium containing 20 µM tetracycline (Sigma-Aldrich, USA). Tetracycline-containing medium was changed every two days.
4.4. Cells treatment conditions
All experimental treatments were performed in complete culture media; either inhibitor was added simultaneously with tetracycline every other day. The following inhibitors were used: 20 µM LY294002 (LY) (Sigma-Aldrich, USA), 10 µM U0126 (Sigma-Aldrich, USA), 5 µM SB203580 (SB) (Sigma-Aldrich, USA), 50 µM pifithrin-α (PFT) (Merck, Germany). For oestrogen treatment EnSC were treated with 10 nM β-Estradiol (Sigma-Aldrich, USA).
4.5. Flow cytometry
Measurements of proliferation, cell size, and autofluorescence (lipofuscin accumulation), were carried out by flow cytometry. Flow cytometry was performed using the CytoFLEX (Beckman Coulter, USA) and the obtained data were analyzed using CytExpert software version 2.0. Adherent cells were rinsed twice with PBS and harvested by trypsinization. Detached cells were pooled, resuspended in fresh medium and stained with 0.1 µg/mL 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen, USA). DAPI-negative (living) cells were then counted and analyzed for autofluorescence and forward light scattering (reflecting cell size).
4.6. SA-β-Gal staining
SA-β-gal staining was performed using senescence β-galactosidase staining kit (Cell Signaling, USA) according to manufacturer’s instructions. Quantitative analysis of images was produced with the application of MatLab package. For each experimental point not less 50 randomly selected cells were analyzed.
4.7. Western blotting
Western blotting was performed as described previously [15]. SDS-PAGE electrophoresis, transfer to nitrocellulose membrane and immunoblotting with ECL (Thermo Scientific, USA) detection were performed according to standard manufacturer’s protocols (Bio-Rad Laboratories, USA). Antibodies against the following proteins were used: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (clone 14C10) (Cell Signaling, USA), pp53 (Ser15) (clone 16G8) (Cell Signaling, USA), p21WAF/CIP (clone 12D1) (Cell Signaling, USA), p16INK4a (Affinity Biosciences, USA), pRb (Ser807/811) (Cell Signaling, USA), pChk2 (Thr68) (Cell Signaling, USA), pERK (Thr202/Tyr204) (Cell Signaling, USA), pAkt (Ser473) (Cell Signaling, USA), pp38 MAPK (Thr180/Tyr182) (clone D3F9) (Cell Signaling, USA), PTEN (clone 2F4C9) (Invitrogen, USA), horseradish peroxidase-conjugated goat anti-rabbit IgG (Cell Signaling, USA), and horseradish peroxidase-conjugated goat anti-mouse IgG (Cell Signaling, USA). Scans of uncropped blots presented in the study are available in Supplementary Table S1.
4.8. Immunofluorescence
Cells grown on coverslips were fixed with 4% formaldehyde (15 min), permeabilized with 0.1% Triton X-100 (10 min) and blocked with 1% bovine serum albumin (1 h). Cells were incubated with primary anti-γH2AX antibodies (Abcam, USA) overnight at 4 ˚C, followed by the incubation with secondary antibodies — Alexa Fluor 488 goat anti-mouse (Invitrogen, USA) for 1 h at room temperature. The slides were counterstained with 1 µg/ml DAPI (DAPI, Invitrogen, USA), mounted using 2% propyl gallate and analysed using Zeiss LSM Pascal 5 laser scanning microscope (Carl Zeiss, Germany). ZOE Fluorescent Cell Imager (BioRad, USA) was used to acquire images of living cells expressing fluorescent reporter proteins.
4.9. RNA extraction, reverse transcription and real time PCR
RNA extraction, reverse transcription and real time PCR were performed as described in our previous study [17]. Reagents for RNA extraction (ExtractRNA reagent), for reverse transcription (MMLV RT kit) and for real time PCR (HS SYBR kit) were obtained from Evrogen, Russia. Gene expression levels were assessed using the Realtime PCR BioRad CFX-96 amplifier (BioRad, USA), the following analysis of the obtained data was performed using the Bio-Rad CFX Manager software (BioRad, USA). Primer sequences and the corresponding annealing temperatures are listed in Table 2.
4.10. Statistical analysis
All quantitative data are shown as mean ± SD. To get significance in the difference between two groups Welch’s t-test was applied. For multiple comparisons between groups, one-way ANOVA with Tukey honestly significant difference (HSD) was used. Statistical analysis was performed using GraphPad Prism version 8.0.5.
5. Conclusions
To sum up, within the present study we revealed senescence as the dominant and stable reaction of EnSC towards oncogene expression. The results obtained allows suggesting that OIS is the defense mechanism that underlie resistance of EnSC against oncogenic transformation, what, at least in part, might explain the rare incidence of endometrial stromal tumors. At the same time, EnSC senescence requires further precise investigation in terms of endometrial epithelial cancer initiation and progression. A growing body of evidence highlights that senescent stromal cells within the tumor microenvironment are capable of interacting with tumor cells and via paracrine action might be involved in tumor initiation and progression [reviewed in 39]. In line with this speculation, recent study revealed significant correlation of stromal p16INK4a expression with endometrial carcinomas rather than with benign and preneoplastic lesions in a cohort including 124 cases of endometrial lesions [40]. The authors concluded that stromal p16INK4a expression is involved in the development and progression of endometrial carcinoma. Bearing in mind, that increased expression of p16INK4a is the most common marker of senescent cells in vivo, it can be logically assumed that senescent EnSC might have protumorigenic influence on epithelial component of endometrium, what however remains to be elucidated in future studies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.V.B.; methodology, A.V.B., P.I.D. and A.N.S.; software, A.L.T. and P.I.D.; validation, A.L.T.; formal analysis, A.L.T.; investigation, A.L.T., P.I.D. and A.N.S.; resources, A.V.B.; data curation, A.L.T. and P.I.D.; writing—original draft preparation, A.V.B.; writing—review and editing, A.V.B. and P.I.D.; visualization, A.L.T.; supervision, A.V.B.; project administration, A.V.B.; funding acquisition, A.V.B. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Russian Science Foundation (#19-74-10038).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analyzed during this study are included in the manuscript.
Acknowledgments
The authors are thankful to Polina Parfenova for the assistance in the PTEN knockout experiments.
Conflicts of Interest
The authors declare no conflict of interest.
List of Abbreviations:.
| EnSCs | human MSCs derived from endometrium |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DDR | DNA damage response |
| HSD | honestly significant difference |
| OIS | oncogene-induced senescence |
| SASP | senescence-associated secretory phenotype |
| SA-β-Gal | senescence-associated β-Galactosidase |
References
- Owusu-Akyaw, A.; Krishnamoorthy, K.; Goldsmith, L.T.; Morelli, S.S. The role of mesenchymal-epithelial transition in endometrial function. Hum Reprod Update. 2019, 25, 114–133. [Google Scholar] [CrossRef]
- Critchley, H.O.D.; Maybin, J.A.; Armstrong, G.M.; Williams, A.W.R. Physiology of the Endometrium and Regulation of Menstruation. Physiol Rev. 2020, 100, 1149–1179. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Leongamornlert, D.; Coorens, T.H.H.; Sanders, M.A.; Ellis, P.; et al. The mutational landscape of normal human endometrial epithelium. Nature. 2020, 580, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Nakaoka, H.; Suda, K.; Yoshihara, K.; Ishiguro, T.; et al. Spatiotemporal dynamics of clonal selection and diversification in normal endometrial epithelium. Nat Commun. 2022, 13, 943. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, D.; Sung, W.J.; Hong, J. High-Grade Endometrial Stromal Sarcoma: Molecular Alterations and Potential Immunotherapeutic Strategies. Front Immunol. 2022, 13, 837004. [Google Scholar] [CrossRef]
- Ren, X.; Liang, J.; Zhang, Y.; Jiang, N.; Xu, Y.; et al. Single-cell transcriptomic analysis highlights origin and pathological process of human endometrioid endometrial carcinoma. Nat Commun. 2022, 13, 6300. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Dimri, G.; Hara, E. Handbook of the Biology of Aging; Academic Press: New York, USA, 1996. [Google Scholar]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat Rev Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Zhu, H.; Blake, S.; Kusuma, F.K.; Pearson, R.B.; Kang, J.; Chan, K.T. Oncogene-induced senescence: From biology to therapy. Mech Ageing Dev. 2020, 187, 111229. [Google Scholar] [CrossRef]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat Rev Cancer. 2010, 10, 51–57. [Google Scholar] [CrossRef]
- Saab, R. Senescence and pre-malignancy: How do tumors progress? Semin Cancer Biol. 2011, 21, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Komseli, E.S.; Pateras, I.S.; Krejsgaard, T.; Stawiski, K.; Rizou, S.V.; et al. A prototypical non-malignant epithelial model to study genome dynamics and concurrently monitor micro-RNAs and proteins in situ during oncogene-induced senescence. BMC Genomics 2018, 19, 37, Erratum in: BMC Genomics 2022, 22, 327. [Google Scholar] [CrossRef]
- Martínez-Zamudio, R.I.; Stefa, A.; Nabuco Leva Ferreira Freitas, J.A.; Vasilopoulos, T.; Simpson, M.; et al. Escape from oncogene-induced senescence is controlled by POU2F2 and memorized by chromatin scars. Cell Genom. 2023, 3, 100293. [Google Scholar] [CrossRef] [PubMed]
- Borodkina, A.; Shatrova, A.; Abushik, P.; Nikolsky, N.; Burova, E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging. 2014, 6, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Alekseenko, L.L.; Zemelko, V.I.; Domnina, A.P.; Lyublinskaya, O.G.; Zenin, V.V.; et al. Sublethal heat shock induces premature senescence rather than apoptosis in human mesenchymal stem cells. Cell Stress Chaperones. 2014, 19, 355–366. [Google Scholar] [CrossRef]
- Deryabin, P.; Borodkina, A. Reduced Efficiency of DNA Repair and Antioxidant Defense Promotes the Accumulation of DNA Damage During Cell Senescence. Cell Tiss Biol. 2021, 15, 532–543. [Google Scholar] [CrossRef]
- Deryabin, P.; Griukova, A.; Shatrova, A.; Petukhov, A.; Nikolsky, N.; Borodkina, A. Optimization of lentiviral transduction parameters and its application for CRISPR-based secretome modification of human endometrial mesenchymal stem cells. Cell Cycle. 2019, 18, 742–758. [Google Scholar] [CrossRef]
- Kohsaka, S.; Sasai, K.; Takahashi, K.; Akagi, T.; Tanino, M.; et al. A population of BJ fibroblasts escaped from Ras-induced senescence susceptible to transformation. Biochem Biophys Res Commun. 2011, 410, 878–884. [Google Scholar] [CrossRef]
- Imanishi, T.; Hano, T.; Nishio, I. Estrogen reduces endothelial progenitor cell senescence through augmentation of telomerase activity. J Hypertens. 2005, 23, 1699–1706. [Google Scholar] [CrossRef]
- Breu, A.; Sprinzing, B.; Merkl, M.; Bechmann, V.; Kujat, R.; Jenei-Lanzl, Z.; et al. Estrogen reduces cellular aging in human mesenchymal stem cells and chondrocytes. J Orthop Res. 2011, 29, 1563–1571. [Google Scholar] [CrossRef]
- Sasaki, Y.; Ikeda, Y.; Miyauchi, T.; Uchikado, Y.; Akasaki, Y.; Ohishi, M. Estrogen-SIRT1 Axis Plays a Pivotal Role in Protecting Arteries Against Menopause-Induced Senescence and Atherosclerosis. J Atheroscler Thromb. 2020, 27, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.M.; Burikhanov, R.; Goswami, A.; Rangnekar, V.M. Suppression of PTEN expression is essential for antiapoptosis and cellular transformation by oncogenic Ras. Cancer Res. 2007, 67, 10343–10350. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Deryabin, P.I.; Borodkina, A.V.; Nikolsky, N.N.; Burova, E.B. The relationship between p53/p21/Rb and MAPK signaling pathways in human endometrium-derived stem cells under oxidative stress. Cell Tiss Biol. 2016, 10, 185–193. [Google Scholar] [CrossRef]
- Borodkina, A.V.; Shatrova, A.N.; Deryabin, P.I.; Grukova, A.A.; Nikolsky, N.N.; Burova, E.B. Tetraploidization or autophagy: The ultimate fate of senescent human endometrial stem cells under ATM or p53 inhibition. Cell Cycle. 2016, 15, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Grukova, A.A.; Shatrova, A.N.; Deryabin, P.I.; Borodkina, A.V.; Knyazev, N.A.; et al. Modulation of senescence phenotype of human endometrial stem cells under inhibition of mtor and map-kinase signaling pathways. Tsitologiia. 2017, 59, 410–420. [Google Scholar]
- Bianchi-Smiraglia, A.; Nikiforov, M.A. Controversial aspects of oncogene-induced senescence. Cell Cycle. 2012, 11, 4147–4151. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Halazonetis, T.D. Oncogene-induced senescence: The bright and dark side of the response. Curr Opin Cell Biol. 2010, 22, 816–827. [Google Scholar] [CrossRef]
- Harada, H.; Nakagawa, H.; Oyama, K.; Takaoka, M.; Andl, C.D.; et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res. 2003, 1, 729–738. [Google Scholar]
- Cipriano, R.; Kan, C.E.; Graham, J.; Danielpour, D.; Stampfer, M.; Jackson, J.W. TGF-beta signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc Natl Acad Sci USA. 2011, 108, 8668–8673. [Google Scholar] [CrossRef]
- Drayton, S.; Rowe, J.; Jones, R.; Vatcheva, R.; Cuthbert-Heavens, D.; et al. Tumor suppressor p16INK4a determines sensitivity of human cells to transformation by cooperating cellular oncogenes. Cancer Cell. 2003, 4, 301–310. [Google Scholar] [CrossRef]
- Bansal, R.; Nikiforov, M.A. Pathways of oncogene-induced senescence in human melanocytic cells. Cell Cycle. 2010, 9, 2782–2788. [Google Scholar] [CrossRef]
- Brandt, L.P.; Albers, J.; Hejhal, T.; Catalno, A.; Wild, P.J.; Frew, I.J. Oncogenic HrasG12V expression plus knockdown of Cdkn2a using ecotropic lentiviral vectors induces high-grade endometrial stromal sarcoma. PLoS ONE. 2017, 12, e0186102. [Google Scholar] [CrossRef] [PubMed]
- Parisotto, M.; Grelet, E.; El Bizri, R.; Metzger, D. Senescence controls prostatic neoplasia driven by Pten loss. Mol Cell Oncol. 2018, 6, 1511205. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Hwang, H.J.; Kang, D.; Park, H.A.; Lee, H.C.; et al. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene. 2019, 38, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Taheri, M.; Ghafouri-Fard, S.; Najafi, S.; Kallenbach, J.; Keramatfar, E.; et al. Hormonal regulation of telomerase activity and hTERT expression in steroid-regulated tissues and cancer. Cancer Cell Int. 2022, 22, 258. [Google Scholar] [CrossRef] [PubMed]
- Bayne, S.; Li, H.; Jones, M.E.; Pinto, A.R.; Van Sinderen, M.; et al. Estrogen deficiency reversibly induces telomere shortening in mouse granulosa cells and ovarian aging in vivo. Protein Cell. 2011, 2, 333–346. [Google Scholar] [CrossRef]
- Ye, M.; Huang, X.; Wu, Q.; Liu, F. Senescent Stromal Cells in the Tumor Microenvironment: Victims or Accomplices? Cancers (Basel). 2003, 15, 1927. [Google Scholar] [CrossRef]
- Yoon, G.; Koh, C.W.; Yoon, N.; Kim, J.-Y.; Kim, H.-S. Stromal p16 expression is significantly increased in endometrial carcinoma. Oncotarget. 2016, 8, 4826–4836. [Google Scholar] [CrossRef]
Figure 1.
Characteristics of EnSC expressing mutant oncogene HRASG12V. (A) Scheme of the applied two-component Tet-On system for inducible expression of HRASG12V under tetracycline (TET) stimulation. (B) Growth curves; (C) morphology, (D) cell size, (E) and (F) SA-β-Gal activity, and (G) mRNA levels of inflammatory factors STC1, MMP2, IL6 for the control (TET-) and HRASG12V-expressing (TET+) EnSC. Scale bars on the microphotographs are 100 μm. Data are presented as (B, D, G) mean ± SD, n = 3; or (F) median ± 1.5 IQR, n = 50. **p < 0.01, ***p < 0.005 by (B) two-way ANOVA with Tukey’s HSD or (D, F, G) Welch’s t-test.
Figure 1.
Characteristics of EnSC expressing mutant oncogene HRASG12V. (A) Scheme of the applied two-component Tet-On system for inducible expression of HRASG12V under tetracycline (TET) stimulation. (B) Growth curves; (C) morphology, (D) cell size, (E) and (F) SA-β-Gal activity, and (G) mRNA levels of inflammatory factors STC1, MMP2, IL6 for the control (TET-) and HRASG12V-expressing (TET+) EnSC. Scale bars on the microphotographs are 100 μm. Data are presented as (B, D, G) mean ± SD, n = 3; or (F) median ± 1.5 IQR, n = 50. **p < 0.01, ***p < 0.005 by (B) two-way ANOVA with Tukey’s HSD or (D, F, G) Welch’s t-test.
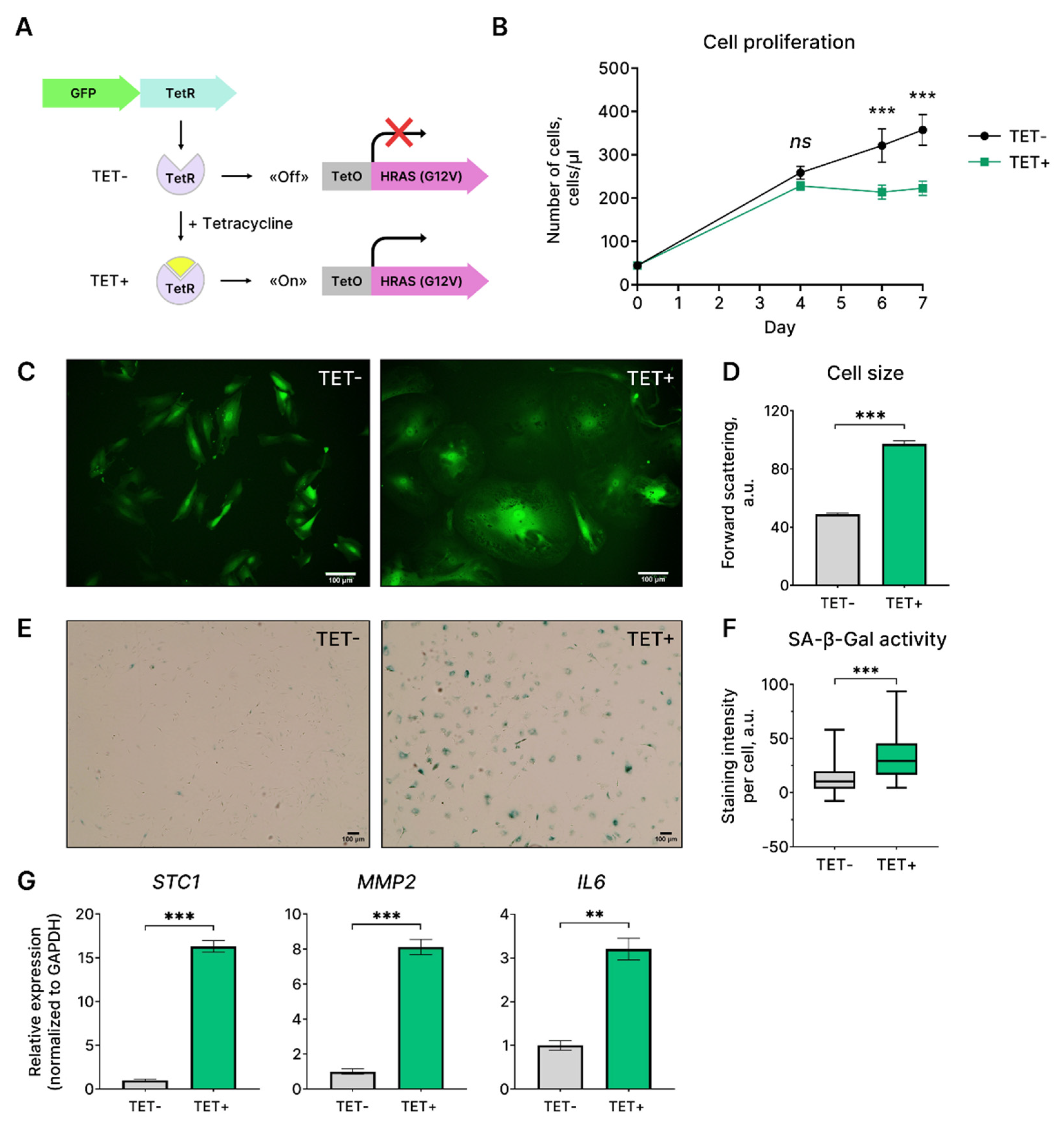
Figure 2.
Identification of colonies of small proliferating cells that appear during long-term culturing of HRASG12V-expressing EnSC as an artefact of imperfect puromycin selection. (A) and (B) Schemes of the original and modified two-component Tet-On systems for inducible expression of HRASG12V under tetracycline (TET) stimulation. Modification resulted in expression of fluorescent protein mCherry under the same promoter with HRASG12V. Microphotographs show absence of mCherry fluorescence in colonies of small cells (indicated with yellow arrows) that appear during long-term culturing of HRASG12V-expressing EnSC (indicated with white arrows). Scale bars on the microphotographs are 100 μm. (C) Relative viability of long-term clonal culture of HRASG12V-expressing (TET+) EnSC and control non-transduced EnSC under puromycin selection. Data are presented as mean ± SD, n = 3, ns p > 0.05 by two-way ANOVA.
Figure 2.
Identification of colonies of small proliferating cells that appear during long-term culturing of HRASG12V-expressing EnSC as an artefact of imperfect puromycin selection. (A) and (B) Schemes of the original and modified two-component Tet-On systems for inducible expression of HRASG12V under tetracycline (TET) stimulation. Modification resulted in expression of fluorescent protein mCherry under the same promoter with HRASG12V. Microphotographs show absence of mCherry fluorescence in colonies of small cells (indicated with yellow arrows) that appear during long-term culturing of HRASG12V-expressing EnSC (indicated with white arrows). Scale bars on the microphotographs are 100 μm. (C) Relative viability of long-term clonal culture of HRASG12V-expressing (TET+) EnSC and control non-transduced EnSC under puromycin selection. Data are presented as mean ± SD, n = 3, ns p > 0.05 by two-way ANOVA.
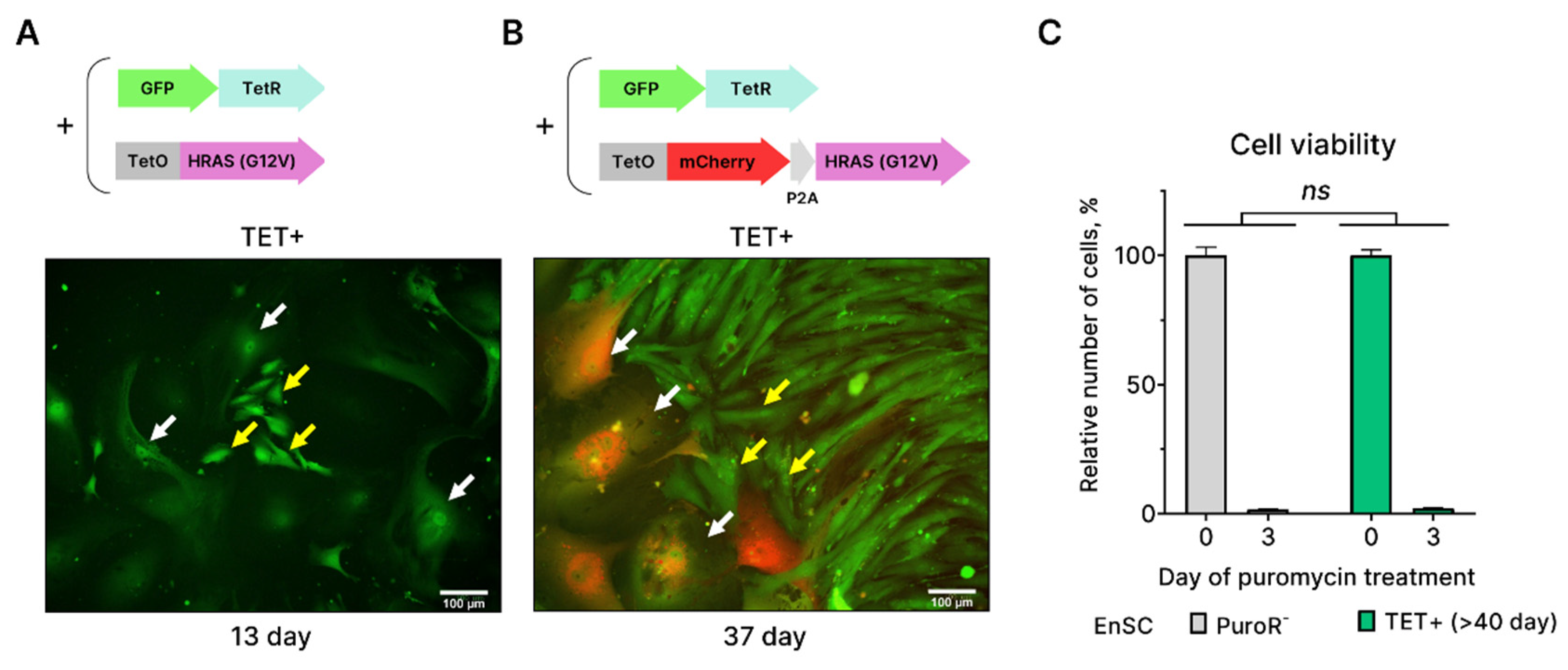
Figure 3.
Effects of oestogen stimulation of HRASG12V-expressing EnSC. (A) Cell size and (B) SA-β-Gal activity of the control (TET-), HRASG12V-expressing (TET+) EnSC, and HRASG12V-expressing (TET+ E2) EnSC additionally treated with oestrogen. (C) Growth curves. Data are presented as (A, C) mean ± SD, n = 3; or (B) median ± 1.5IQR, n = 50; ns p > 0.05 by one- or two-way ANOVA with Tukey’s HSD.
Figure 3.
Effects of oestogen stimulation of HRASG12V-expressing EnSC. (A) Cell size and (B) SA-β-Gal activity of the control (TET-), HRASG12V-expressing (TET+) EnSC, and HRASG12V-expressing (TET+ E2) EnSC additionally treated with oestrogen. (C) Growth curves. Data are presented as (A, C) mean ± SD, n = 3; or (B) median ± 1.5IQR, n = 50; ns p > 0.05 by one- or two-way ANOVA with Tukey’s HSD.
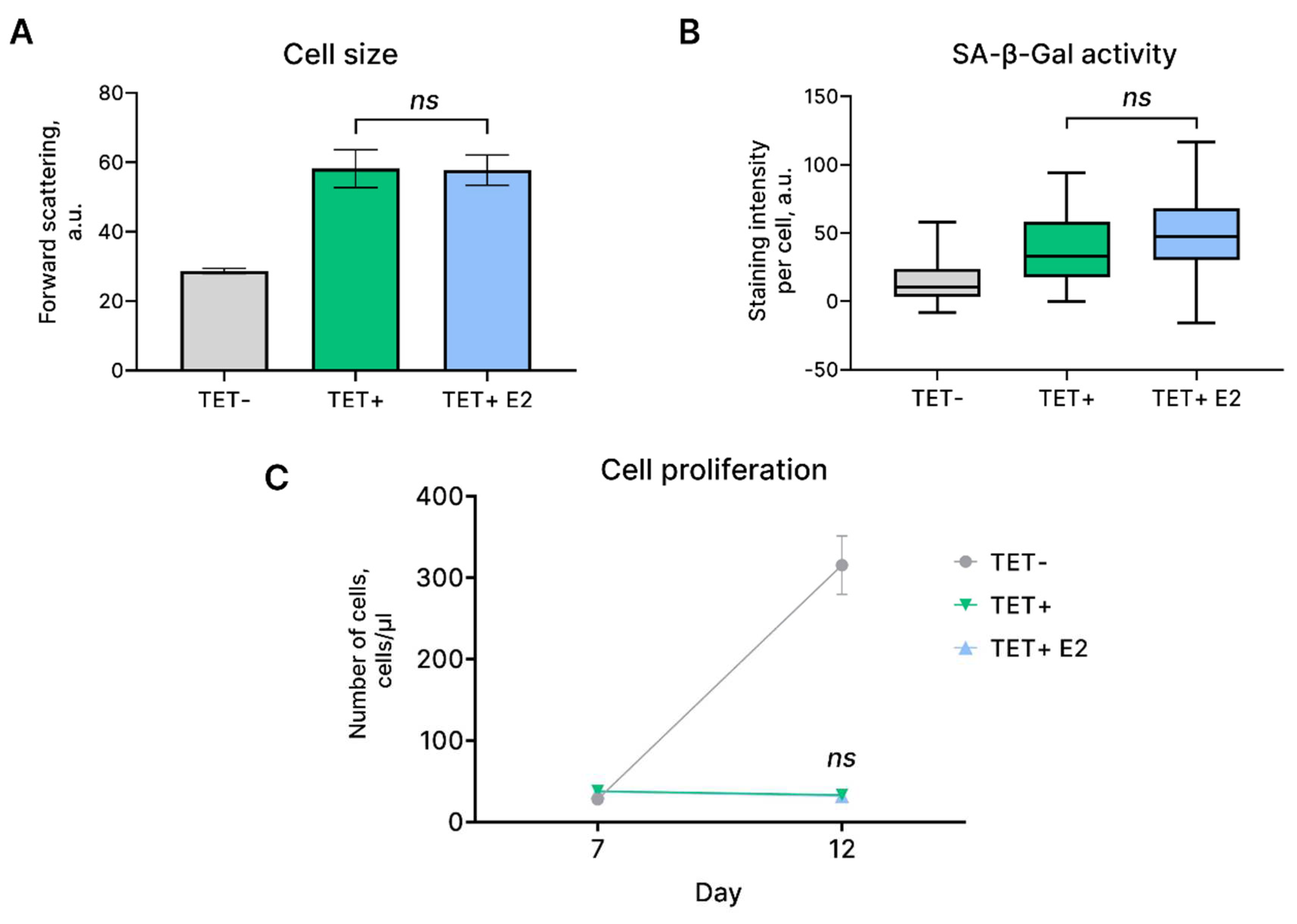
Figure 4.
Molecular features of EnSC’s OIS caused by HRASG12V expression. (A) Phosphorylation levels of AKT and ERK1/2 kinases, (B) growth curves, (C) DNA damage γH2A.X foci in the control (TET-) and HRASG12V-expressing (TET+) EnSC, and activity of (D) Chk2/p53/p21WAF/CIP/Rb and (E) p38/p16INK4a/Rb in HRASG12V-expressing (TET+) EnSC. Scale bars on the microphotographs are 50 μm or 10 μm. For blotting, GAPDH was used as loading control, representative blots are shown. Data are presented as mean ± SD; n = 3; ns p > 0.05, *p < 0.05 by two-way ANOVA with Tukey’s HSD.
Figure 4.
Molecular features of EnSC’s OIS caused by HRASG12V expression. (A) Phosphorylation levels of AKT and ERK1/2 kinases, (B) growth curves, (C) DNA damage γH2A.X foci in the control (TET-) and HRASG12V-expressing (TET+) EnSC, and activity of (D) Chk2/p53/p21WAF/CIP/Rb and (E) p38/p16INK4a/Rb in HRASG12V-expressing (TET+) EnSC. Scale bars on the microphotographs are 50 μm or 10 μm. For blotting, GAPDH was used as loading control, representative blots are shown. Data are presented as mean ± SD; n = 3; ns p > 0.05, *p < 0.05 by two-way ANOVA with Tukey’s HSD.
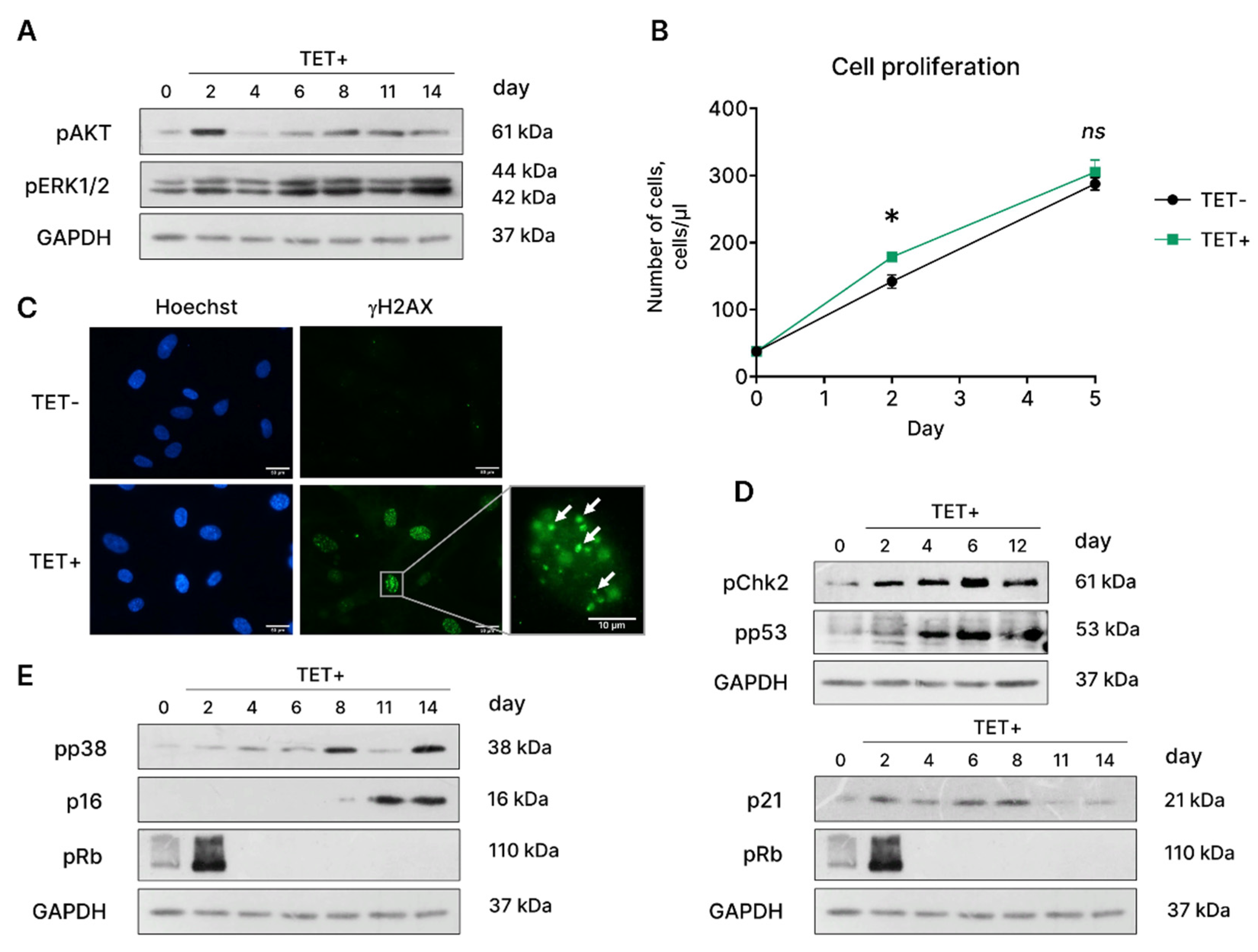
Figure 5.
Modulation of activity of the molecular pathways mediating HRASG12V-induced senescence in EnSC. (A) Сell size, (B) SA-β-Gal activity and (C) growth curves of the control (TET-), HRASG12V-expressing (TET+) EnSC, and HRASG12V-expressing EnSC treated with AKT inhibitor LY294002 (TET+ LY), p53 inhibitor pifithrin-α (TET+ PFT), p38 inhibitor SB203580 (TET+ SB), and MEK1/2 inhibitor U0126 (TET+ U). Data are presented as (A) median ± 1.5IQR, n = 50; or (B, C) mean ± SD, n = 3; ns p > 0.05, **p < 0.01, ***p < 0.005 by one- or two-way ANOVA with Tukey HSD compared to control (TET+ Ctrl) cells.
Figure 5.
Modulation of activity of the molecular pathways mediating HRASG12V-induced senescence in EnSC. (A) Сell size, (B) SA-β-Gal activity and (C) growth curves of the control (TET-), HRASG12V-expressing (TET+) EnSC, and HRASG12V-expressing EnSC treated with AKT inhibitor LY294002 (TET+ LY), p53 inhibitor pifithrin-α (TET+ PFT), p38 inhibitor SB203580 (TET+ SB), and MEK1/2 inhibitor U0126 (TET+ U). Data are presented as (A) median ± 1.5IQR, n = 50; or (B, C) mean ± SD, n = 3; ns p > 0.05, **p < 0.01, ***p < 0.005 by one- or two-way ANOVA with Tukey HSD compared to control (TET+ Ctrl) cells.
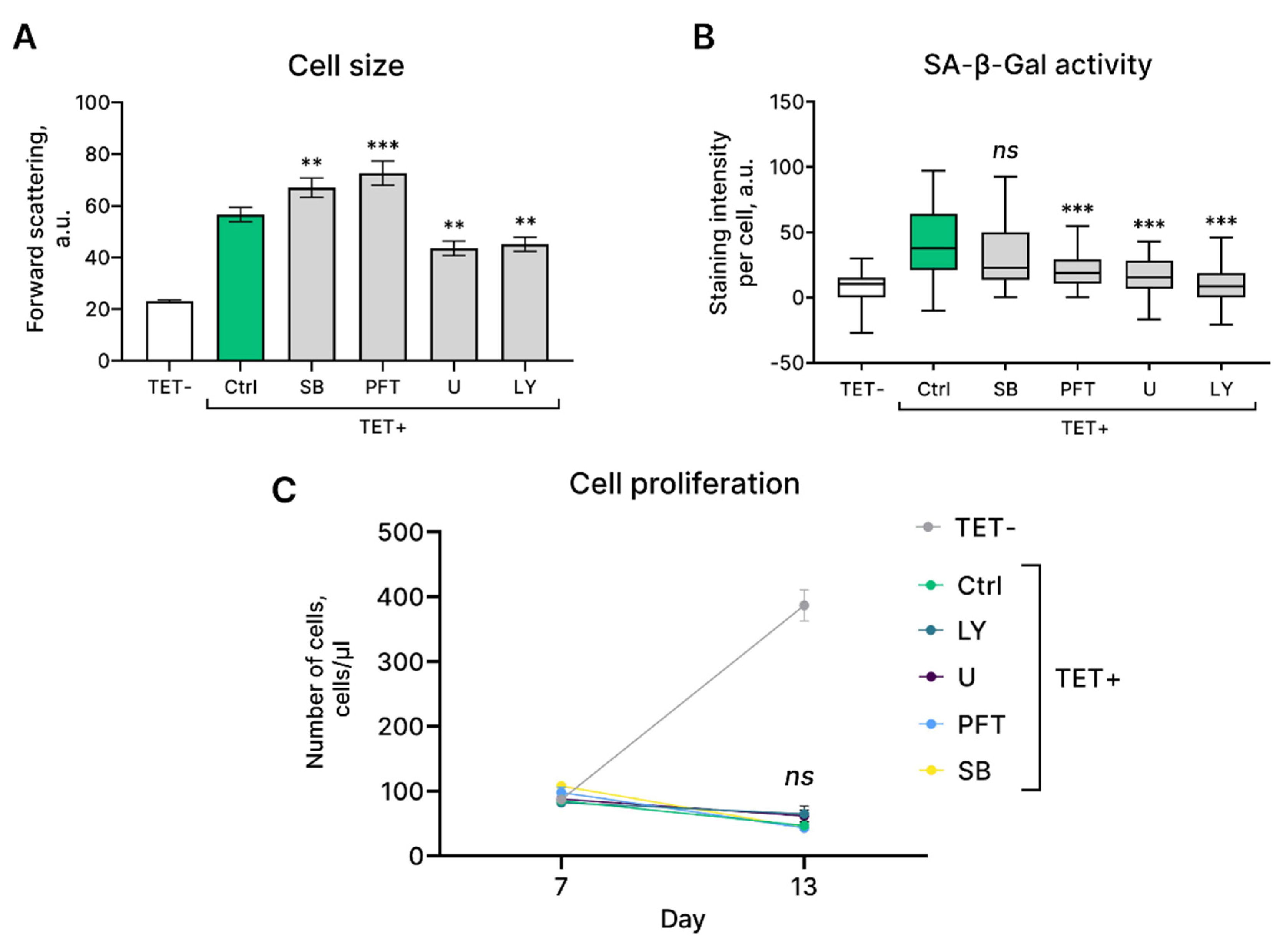
Figure 6.
Evidence of PTEN loss-induced senescence of EnSC. (A) mRNA and (B) protein levels of PTEN expression in HRASG12V-expressing (TET+) EnSC. (C) and (D) verification of CRISPR/Cas9-mediated PTEN knockout in control EnSC at mRNA and protein levels. (E) Cell size, (F) lipofuscin intracellular level, (G) growth curves, and (H) and (I) SA-β-Gal activity in control and PTEN knockout (PTEN KO) EnSC. Scale bars on the microphotographs are 100 μm. For blotting, GAPDH was used as loading control, representative blots are shown. Data are presented as (A, C, E, F, G) mean ± SD, n = 3; or (I) median ± 1.5IQR, n = 50; *p < 0.05, ***p < 0.005 by (A) one-way ANOVA with Tukey HSD compared to control cells (at day 0), (G) two-way ANOVA with Tukey HSD or (C, E, F, I) Welch’s t-test.
Figure 6.
Evidence of PTEN loss-induced senescence of EnSC. (A) mRNA and (B) protein levels of PTEN expression in HRASG12V-expressing (TET+) EnSC. (C) and (D) verification of CRISPR/Cas9-mediated PTEN knockout in control EnSC at mRNA and protein levels. (E) Cell size, (F) lipofuscin intracellular level, (G) growth curves, and (H) and (I) SA-β-Gal activity in control and PTEN knockout (PTEN KO) EnSC. Scale bars on the microphotographs are 100 μm. For blotting, GAPDH was used as loading control, representative blots are shown. Data are presented as (A, C, E, F, G) mean ± SD, n = 3; or (I) median ± 1.5IQR, n = 50; *p < 0.05, ***p < 0.005 by (A) one-way ANOVA with Tukey HSD compared to control cells (at day 0), (G) two-way ANOVA with Tukey HSD or (C, E, F, I) Welch’s t-test.
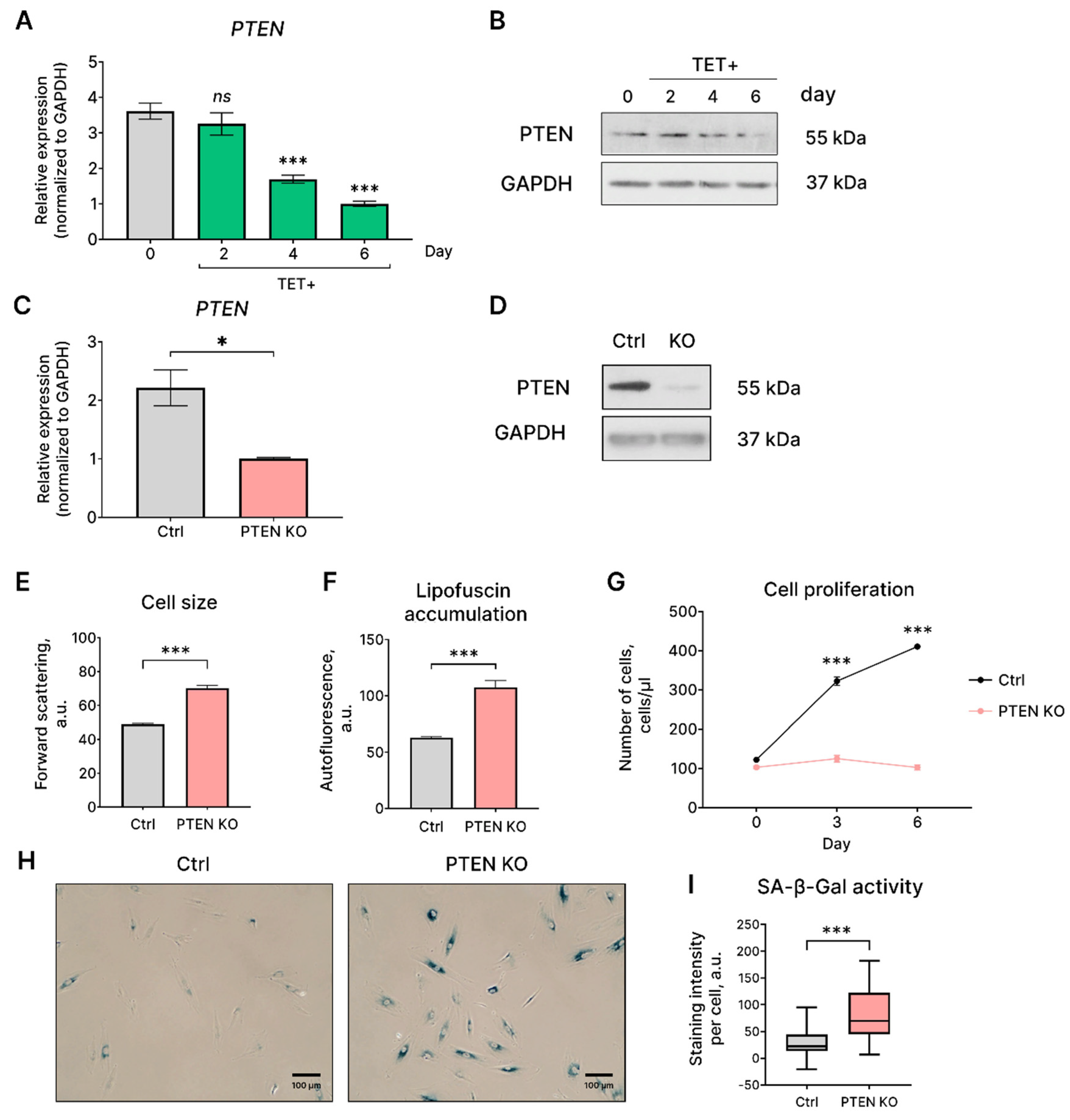

Table 1.
Oligonucleotide sequences for mCherry and sgRNAs cloning.
| # | Oligonucleotide | Sequence | Tm, °C |
|---|---|---|---|
| 1 | mCherry forward | 5’-ATATTTGGATCCGGTCCGATCCACCGGTCGC-3’ | 63.0 |
| 2 | mCherry reverse | 5’-CGTCATCGCTCCAGAAGGCCCGGGATTCTCCTCC-3’ | 63.0 |
| 3 | HRASG12V forward | 5’-GGGCCTTCTGGAGCGATGACGGAATATAAGCTGGTGG-3’ | 64.0 |
| 4 | HRASG12V reverse | 5’-GTCGAGCGGCCGCCACTGTG-3’ | 64.0 |
| 5 | PTEN sgRNA forward | 5’-CACCGAAACAAAAGGAGATATCAAG-3’ | - |
| 6 | PTEN sgRNA reverse | 5’-AAACCTTGATATCTCCTTTTGTTTC-3’ | - |
Table 2.
Primer oligonucleotide sequences.
| # | Oligonucleotide | Sequence | Tm, °C |
|---|---|---|---|
| 1 | GAPDH forward | 5’-GAGGTCAATGAAGGGGTCAT-3’ | 57.0 |
| 2 | GAPDH reverse | 5’-AGTCAACGGATTTGGTCGTA-3’ | 57.0 |
| 3 | PTEN forward | 5’-TTGAAGACCATAACCCACCA-3’ | 58.0 |
| 4 | PTEN reverse | 5’-CACATAGCGCCTCTGACTG-3’ | 58.0 |
| 5 | STC1 forward | 5’-TGAGGCGGAGCAGAATGACT-3’ | 59.5 |
| 6 | STC1 reverse | 5’-CAGGTGGAGTTTTCCAGGCAT-3’ | 59.5 |
| 7 | IL6 forward | 5’-ATGTAGCCGCCCCACACA-3’ | 58.0 |
| 8 | IL6 reverse | 5’-CCAGTGCCTCTTTGCTGCTT-3’ | 58.0 |
| 9 | MMP2 forward | 5’-AGATCTTCTTCTTCAAGGACCGGTT-3’ | 59.5 |
| 10 | MMP2 reverse | 5’-GGCTGGTCAGTGGCTTGGGGTA-3’ | 59.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



