
Submitted:
25 July 2023
Posted:
27 July 2023
You are already at the latest version
Abstract
ABSTRACT: Recurrent glioblastoma (rGBM) is a highly aggressive form of brain cancer that poses a significant challenge for treatment in neurooncology, and the survival status of patients after relapse usually means rapid deterioration, and also the leading cause of death among patients. In recent years, immunotherapy has emerged as a promising strategy for the treatment of recurrent glioblastoma by stimulate the body's immune system to recognize and attack cancer cells , which could be used as a in combination with other treatments such as surgery, radiation, and chemotherapy to improve outcomes for patients with recurrent glioblastoma, This therapy combines several key methods such as the use of monoclonal antibodies, chimeric antigen receptor T cell (CAR-T) therapy, checkpoint inhibitors, oncolytic viral therapy cancer vaccines, and combination strategies. In this review, we mainly document the latest immunotherapies for the treatment of glioblastoma and focus on the rGBM especially.
Keywords:
Keywords: recurrent glioblastoma
; immunotherapy
; CAR-T therapy
; immune checkpoint inhibitor
; cancer vaccine
; oncolytic viral therapy
1. Introduction
Gliomas have traditionally been classified by the World Health Organization (WHO) as grades I and II (low-grade gliomas), III and IV (high-grade gliomas) according to their malignancy and histopathological features[1]. Glioblastoma (GBM) is the most common and most aggressive brain tumor (WHO grade IV), accounting for approximately 14.2% of all brain tumors[2]. The current standard-of-care for GBM consists of maximum safe surgical resection followed by radiotherapy and temozolomide (TMZ) chemotherapy[3,4]. Unfortunately, patient outcomes remain almost universally fatal with a median overall survival (OS) of 14.6 to 20.5 months, and the prognosis is worse in older patients, with an average survival of less than 8.5 months after diagnosis[5,6]. Since most patients experience recurrence, recurrent glioblastoma (rGBM) is a condition with bleak outlook as treatment options are very limited with no universally held standard of care[7,8]. At present, bevacizumab is the only drug which is approved by the Food and Drug Administration (FDA) to treat recurrent rGBM[9]. The randomized clinical trial (RCT) results with bevacizumab as first-line therapy for rGBM[10,11] in recurrence[12] have been consistent: decrease in the intensity and volume of contrast enhancement, decrease in peritumoral edema, decrease in corticosteroid use, statistically significant prolongation in PFS, but no improvement in OS[10,11,12]. Resection is not widely adopted nor regarded as effective since most patients (70~75%) are not candidates for repeat gross total resection at recurrence, resulting in a large unmet need for this patient population[13,14,15]. Thus, new therapeutic strategies are urgently needed for rGBM.
Therapeutic failure, in part, is due to extensive intratumoral heterogeneity at the cellular, genetic, and functional levels. This heterogeneity may be explained by a distinct subset of cells called GBM stem cells (GSCs), which are capable of self-renewal, differentiation, and plasticity[16]. It is believed that this subpopulation of GSCs, after undergoing selective pressures from primary GBM (pGBM) therapy, become chemotherapy and radiotherapy resistant, and seed formation of the therapy-resistant recurrent tumors[17,18,19]. Multiple studies have identified and isolated GSCs with tumor-initiating properties[20,21]. Tumor cells adjacent to GSCs will inhibit GSCs through paracrine and cell contact, so that GSCs enter a dormant state. Under certain circumstances, when non-functional GSCs are separated from surrounding tumor cells, their proliferative capacity will be reactivated leading to tumor recurrence[22,23]. Other feature of GBM contributing to poor prognosis include the existence of the blood-brain barrier (BBB). BBB plays a protective role in the normal brain with two lines of defense, physical barrier and chemical barrier, which can prevent macromolecular substances and unnecessary cells from entering the brain. However, it also prevents effective therapeutic drugs including small molecules and antibodies from reaching tumor cells in GBM. In addition, the central nervous system (CNS) has long been considered as an immune-privileged site with restricted access that profoundly affects the capacity of T cells to exert their functions[24]. This special microenvironment prevented T-cell priming and re-stimulation, and ultimately impaired anti-tumor immune response[25]. Moreover, GBM cells can exert local immunosuppressive effects in many ways. On the one hand, GBM cells themselves can secrete various protumor cytokines and/or chemokines, which can influence macrophage polarization, promote regulatory T cell (Treg) recruitment, and inhibit dendritic cell (DC) maturation and natural killer (NK) cell function. On the other hand, GBM cells can express immunosuppressive molecules, such as programmed cell death protein 1 ligand (PD-L1), which can prevent T cell proliferation and activation[26]. The immune microenvironment in the pGBM and rGBM displays similar suppressive changes. One study confirmed that glioma-associated microglia/macrophages (GAMs), as the dominant infiltrating immunocytes, present great inter- and intra-tumoral heterogeneity and that GAMs increased exhausted T cells, infiltrating Tregs, and nonfunctional NK cells contribute to local immune suppressive characteristics[27]. Thus, opportunities and challenges remain in finding more efficient treatments against rGBM.
Fortunately, advances in decades of investment in molecular pathogenesis of glioblastoma are rapidly translated into innovative clinical trials, utilizing improved genomic, epigenetic, transcriptomic and proteomic characterization of glioblastoma as well as the brain microenvironment and immune system interactions[28]. Researchers have also achieved certain results in CNS drug delivery methods, increased the survival of patients[29]. Immunotherapy, which harnesses the body’s immune system to against cancer, has led to important clinical advances over the past few years[30,31,32]. On the basis of therapeutic gains made in immune checkpoint blockade and chimeric antigen receptor-modified T (CAR-T) cells, Science journal awarded cancer immunotherapy its ‘Breakthrough of the Year’ in 2013[30]. Subsequently, The Nobel Prize in Physiology or Medicine 2018 awarded discovery of cancer therapy by inhibition of negative immune regulation. These excellent findings laid the foundation for the clinical development of immunotherapy, which have dramatically improved outcomes for many people with cancer. In recent years, lots of immunotherapy drugs, from monoclonal antibody against cytotoxic-T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1) and PD-L1, to CAR T cell therapy, are approved by U.S. FDA for cancer treatment[30,31,33]. In spite of the consideration of the CNS’s immune-privilege, immunotherapy for GBM still obtains considerable achievements[34,35,36,37]. Ongoing studies are using combinatorial therapies[38,39]. Thus, immunotherapy holds great promise in rGBM treatment. In this review, we present an overview of the current immunotherapy for rGBM, including vaccines, CAR-T, checkpoint inhibitors, and oncolytic virotherapy, and discuss the challenges and future directions of them, provide an reference for the immunotherapy of rGBM patients and improve the overall survival.
2. Immunotherapy for the Treatment of Recurrent Glioblastoma
2.1. CAR-T therapy
2.1.1. The background of CAR-T therapy
T cells engineered to express chimeric antigen receptors to identify and attack specific markers expressed on the surface of malignant tumor cells have shown remarkable success in many tumors, particularly in hematological malignancies. [40,41] Tumor-specific CAR-T cells can be activated without antigen-presenting procedure and MHC molecules, which makes them can be modified to accurately target most antigens in the human body.[42,43] In the recent ten years, CAR-T therapies have transformed the management of many cancers with its high efficiency and fewer adverse events. Due to the protection of the BBB, the CNS used to be regarded as an immune-privileged environment in humans.[44,45] However, this strict mechanism was also found to be changed in some special situations, and then peripheral immune cells could enter the CNS from areas through high blood vessels regions like the choroid plexus and subarachnoid space. Particularly, when some pathogens invade or pathogenic damage happens, such as some latent infectiones, the peripheral immune cells will cross the BBB and assist in keeping the homeostasis of CNS.[46] Previous studies have found the existence of various T cells in the tumor microenvironment (TME) of rGBM, while some tumor-related cells and factors in rGBM could inhibit the proliferation and protection function of these T cells .[47,48] Meanwhile, rGBM has some very special target substances, which are different from normal neurons and glial cells, making it suitable for CAR-T to identify and design. These characteristics of rGBM provide us with a theoretical possibility of CAR-T to effectively control the development of rGBM tumor cells with a slight neurotoxicity. Certainly, there are also many challenges in this process, just as the clinical practices tell.
2.1.2. The latest development of CAR-T therapy
To date, there are many modified CAR-T therapies with different targets in vitro experiments, animal experiments, and clinical trials. The therapeutic targets of CAR-T that have completed clinical trials include EGFRvIII, IL13Ra2, HER2, etc. which are the most focused targets for rGBM.
The first preclinical study of CAR-T therapy on glioblastoma was conducted by Kahlon et al in 2004 targeting the interleukin 13 receptor α2 (IL13Rα2).[49] ILRα2 is highly expressed in glioblastoma but has low expression in the normal brain and most normal tissues[50]. Therefore, IL13Rα2 became one of the most common targets for rGBM, and the first target treated in the clinical human body by CAR-T therapy. In 2015, Brown et al. conducted the first human trial in three rGBM patients targeting IL13Rα2 to explore the safety and effect of CAR-T therapy in rGBM.[51] This treatment was found to be well tolerated with a transient anti-tumor activity in two of the three patients. Although this phase 1 study finally failed to increase patients’ overall survival rate (OS) significantly, these findings provide the first promising human clinical experience for the treatment of rGBM with intracranial administration of IL13Rα2-directed CAR-T. The feasibility and safety of CAR-T for rGBM proved by Brown et al. successfully set the foundation for future improvement of CAR-T therapy. Most recently, they also reported their phase 1 trial results of the off-the-shelf, allogeneic IL13Rα2-directed CAR-T product for the treatment of rGBM.[52] This allogeneic product was proven to have the feasibility, safety, and therapeutic potential for rGBM patients, which would dramatically reduce the costs of CAR-T therapies and increase their accessibility in the clinic.
Besides IL13Rα2, EGFRvIII is another very interesting and well-known target for rGBM. EGRFvIII is a deletion mutation of the epidermal growth factor receptor (EGFR) and is often expressed in most tumors.[53] In GBM, about 40% of newly diagnosed patients have EGFR gene amplification, and about 50% of patients with EGFR-amplified GBM show constitutive activation of the oncogenic variant, EGFRvIII.[54] The first preclinical research of CAR-T targeting EGFRvIII in glioblastoma was reported in 2009, which showed that the modified T cells have effective and specific cytotoxic activity against glioblastoma tumor cells expressing EGRFvIII in vivo.[55] In the experiment by Donald et al. in 2017, clinical results of the EGFRvIII-directed CAR-T therapy were first observed. In their work, the EGFRvIII-directed CAR-T was intravenously injected into 10 treated patients with rGBM. [56] All patients involved had a transient proliferation of CAR-T-EGFRvIII in peripheral blood. For 7 patients who underwent further procedural intervention, it was found that CAR-T-EGFRvIII was successfully transported to the rGBM region, and antigen reduction occurred in 5 of these 7 patients. Besides, in Donald’s research, they found no cross-reactivity of wild-type EGFR when patients used the CAR-T therapy. This further proved that CAR-T was a feasible and safe therapy for rGBM. And in May 2021, a successfully prolonged survival case following EGFRvIII- CAR-T treatment for rGBM was reported by Joseph et al..[57] A 59-year-old patient, who received a single peripheral infusion of CAR-T-EGFRvIII, survived 36 months after GBM recurrence, far exceeding the expected survival for rGBM. And the EGFRvIII-directed T cells persisted in her peripheral circulation for 29 months of follow-up, which is the longest persistence reported in rGBM CAR-T trials to date.
Another commonly studied target for rGBM is Human Epidermal Growth Factor Receptor 2 (HER2). HER2 is found to be overexpressed in many kinds of cancers and approximately 80% of GBM, however, it is also expressed to some extent in most normal tissues. The first preclinical research targeting HER2 by CAR-T therapy was published in 2010 by Ahmed et al.[58] But until 2017, the results of the first clinical trials were firstly reported and showed that infusion of autologous HER2-directed CAR-T to 17 patients was well tolerated without dose-limiting toxicity in rGBM.[59] This clinical report also showed some clinical benefits of CAR-T therapy for rGBM patients involved through transient tumor reduction and/or tumor necrosis effects. Despite of this encouraging result, , considering the expression of HER2 in some important organs, the safety of HER2-directed drugs still needs more strict experiments in the future before it is widely used in clinic.
In addition to the above well-known targets, the B7-Homolog3 (B7-H3, also known as CD276), extracellular matrix metalloproteinase inducer (EMMPIRIN, also known as CD147), dissialoganglioside (GD2), matrix metalloprotease 2 (MMP2), CD133, CD70, etc. are other interesting targets in recent years for rGBM and have gradually entered different experimental stages.[47] For instance, in May 2022, a study reported that the use of CAR-T therapy targeting CD133 in mice with human GBM was considered successful because it reduced more than 80% of tumor burden in these mice and successfully improved their survival rates.[60] Overall, although these different targets have performed well in vitro or animal experiments, there is still a distance for them to be truly clinically used and improve the OS of patients with rGBM. Main clinic researches of CAR-T on the rGBM are listed in the Table 1 below, which contains the completed and undergoing trials but not terminated ones.
2.1.3. The limitation of CAR-T therapy
Despite the advantages and feasibility of CAR-T therapy, there are plenty of reasons that hinder the application of CAR-T therapy in rGBM.
(1) Whereas researhers have made numerous efforts in the molecular characteristics research of rGBM, only a few molecules remain suitable for further experiments. This is mainly due to the strong heterogeneity of rGBM. There is currently no target that can be ubiquitously present in all tumor cells and significantly distinguished from normal tissues. Therefore, we still need a more comprehensive and in-depth understanding of rGBM.
(2) The infiltration rate of T cells in rGBM remains inherently low due to the specificity of CNS and the protection of BBB. The peripheral immune cells thus are difficult to enter the CNS, including modified T cells by intravenous injection. Meanwhile, TME of rGBM also has strong immunosuppressive effects on T cells, so how to enhance the chemotaxis and function of T cells is also one of the challenges.[48]
(3) At present, the toxic and side effects of CAR-T are relatively small compared with other mainstream therapies, but some types of it still have dose toxicity when they are intravenously injected. For example, in a 2019 incremental dose experiment targeting EGFRvIII, there was a death case reported.[61] Besides, due to the specificity limitation of some targets, there are some problems such as off-target toxicity. For example, a common target HER2 is also expressed in some normal tissues of important organs. One of the main concerns of HER2-CAR-T therapy is the risk of attacking normal tissue.[62] Even so, the side effects of CAR-T treatment for rGBM are still acceptable. Most patients only suffer from transient discomfort. The systemic cytokine release syndrome (CRS) which is a common risk in CAR-T therapy has not yet been reported.
(4) In addition, CAR-T currently also has a certain drug resistance. Taking CAR-T therapy targeting EGFRvII as an example, although the continuous existence of CAR-T can be seen in peripheral blood and the tumor will be controlled in a short term, after the administration of EGFRvIII-specific CAR-T, the loss/down-regulation of tumor EGFRvIII occurs. Then new relapsed tumors thus lack the specific target, which will result in the failure of CAR T.[56] Similarly, the same situation occurred after the administration of IL13Rα2-CAR-T.[63] In sum, although CAR-T therapy is currently being developed at various clinical stages, it has not yet resulted in a significant improvement and change of OS in patients with rGBM. However, we cannot judge the superiority of a treatment method merely by the OS increase. At the same level of OS, it would also be valuable if the CAR-T therapy could have less torturous adverse events or alleviate patients’ suffering compared with other treatments.
2.1.4. The prospective of CAR-T therapy
CAR-T therapy has great improvement possibilities in the future from the treatment methods and modes for rGBM. The optimization in multiple aspects could be carried out.
(1) The development of better targets is needed to improve the specificity and efficiency of all targeted drugs including CAR-T. Given the high degree of heterogeneity in most solid tumors, a single effective target now is much insufficient.
(2) Try to better recruit peripheral immune cells to glioblastoma and increase the infiltration rate of CAR-T in the CNS. Now some physical ways such as non-invasive micro bubble-enhanced focused ultrasound (MBF), or some biological ways like the up-expression of chemokines are proven the ability to increase the permeability of BBB. And some chemotactic enhancement methods for CAR-T are also stable and feasible in preclinical experiments.[64] Moreover, to avoid consumption in the peripheral blood, direct local injection or intracavitary injection can be useful. And its safety and advantages have been shown in clinical experiments targeting multiple targets such as IL13Rα2.[65]
(3) In addition to the use of a single target, multiple rGBM targets can be used in combination. The combination of multiple tumor-specific targets, the combination of tumor-specific targets and anti-targets for normal tissues, the combination of tumor targets and targets for inhibitory cells, etc., can improve the targeting ability of CAR-T and reduce its off-target toxicity. For instance, Bryan et al. developed a bicistronic construct to drive the expression of a CAR specific for EGFRvIII, and a bispecific T-cell engager (BiTE) against normal EGFR.[66] The treatment with this CAR-T secreting BiTEs circumvented antigen escape without detectable toxicity and resulted in the nearly complete disappearance of glioblastoma in mice. Niaz et al. developed a novel CAR-T targeting IL13Rα2 and EphA2 for enhanced glioblastoma therapy and proved its tumor control effect better than any single targeting CAR-T.[67]
(4) The combination of CAR-T therapy and other therapies can be adopted. For example, CAR-T is combined with chemotherapy or radiotherapy. Radiotherapy will release pro-inflammatory cytokines to increase the infiltration of immune cells into the tumor, and the radiation itself can also change the permeability of BBB.[68,69] There has been found that the combination with radiotherapy can improve the efficacy of CAR-T therapy in rGBM and some other solid tumor models.[70,71] Besides, the combination of CAR-T with some small molecule cancer inhibitors has also shown synergistic effects, such as the tyrosine kinase inhibitor (TKI).
(5) Other ways to improve the function of CAR-T, like the structure optimization of CAR-T or the control of dysfunction effect in the rGBM TME. There are still a lot of possibilities for CAR-T therapy for rGBM in the future.
In conclusion, although current clinical CAR-T therapy, like many other treatments for rGBM, has not yet significantly increased the OS in patients with rGBM, its higher specificity, limited adverse events, and broad optimization space make it a new hope for rGBM. Still, there are many new-generation CAR-T therapy trials that have demonstrated high efficiency in the control of rGBM development in the preclinical phase and we believe these all efforts of humans to conquer cancer will eventually pay back in the future.
2.2. Immune checkpoint inhibitor
2.2.1. CTLA-4 inhibitors
In the tumor immune response, antigen-presenting cells activate T cells via MHC molecules and costimulatory signals, with the CD28/B7 pathway being an important costimulatory pathway. CTLA-4 (CD152) is expressed in both activated T cells and Tregs, and it functions as a potent competitive inhibitor of CD28/B7 as its affinity for B7 (CD80/86) is 10- to 20-fold higher than that of CD28.[72] So generally, CTLA-4 acts as one of the immune checkpoints that inhibits T cell activation, and thus effectively inhibiting anti-tumor immune response in TME. Studies have found that GBM patients with lower CTLA-4 expression on T lymphocytes tend to have a better prognosis,[73] indicating CTLA-4's value as a prognostic factor in GBM.[74] One of the earliest studies evaluating the effect of CTLA-4 blocker in glioma was conducted in 2007. The result showed that in glioma model mice, CTLA-4 blocking was linked to increased tumor-infiltrating T cells.[75] Subsequent studies in 2016 and 2019 demonstrated that CTLA-4 inhibitors, which disrupt the formation of the CTLA-4/CD80 complex within the tumor, improved the survival of GBM-bearing model mice.[76,77] Afterwards, several clinical trials have proven its efficacy and safety in tumor immunotherapy and more are ongoing.[74,78,79,80,81]
Although pre-clinical trials have shown potential, and some antibody-mediated immune checkpoint blockades (ICB) of CTLA-4 have shown positive effects in patients with glioma,[82] available clinical data on the use of CTLA-4 inhibitors as monotherapy in GBM haven’t been convincing to date. Currently, ipilimumab is the only CTLA-4 blocking antibody that has been approved by FDA, but its efficacy in GBM has not been demonstrated yet.
As for combination therapies involving CTLA-4 inhibitors, one clinical trialfound that compared to using Nivolumab alone, combination therapy of Nivolumab with ipilimumab showed lower tolerability and no obvious improvement of PFS/OS in patients with recurrent GBM.[83] In a case series study in 2016, 20 patients with rGBM were treated with ipilimumab and bevacizumab, and about 31% showed a partial response.[84] Some studies showed that ipilimumab may be particularly efficacious in patients with recurrent hypermutant GBM when applied in combination with other immunotherapy modalities in adjuvant and neoadjuvant settings.[85] Other clinical trials investigating the safety, tolerability, and efficacy of ipilimumab combination therapies in rGBM include (NCT03233152, NCT04403649, NCT03707457 and, NCT03430791). Another phase II trial (NCT02794883) evaluated tremelimumab (also an anti-CTLA-4 monoclonal antibody) and anti-PD-L1 antibody as monotherapies and combination therapy in patients with rGBM. About 41.7% patients treated with tremelimumab alone showed grade 5 disease progression, and only 18.2% in combination strategy. The median overall survival for tremelimumab group was 7.2 months.
Immunotherapy targeting CTLA-4 still face some challenges, like adverse effects of CTLA-4 inhibitors and unsatisfactory therapeutic results in most GBM patients. CTLA-4 blockade monotherapy is not as effective in GBM as in other cancers owing to the GBM’s unique characteristics. In the future, combination therapies could be a potential way out as T cells typically have multiple checkpoints. Moreover, further investigation of CTLA-4 expression profile is needed to determine drug concentration in clinical trials, and predictive biomarkers are also required to increase efficacy of trial and therapies.[86]
2.2.2. PD1/PDL1
The PD-1/PD-L1 pair is one of the most representative ICB that can reactivate T-cell function and promote anti-tumor activity upon inhibition. Due to encouraging outcomes observed in other malignancies [87,88], there is substantial interest in investigating the efficacy of PD-1/PD-L1 blockade in GBM including rGBM. In general, PD-1 blockade is mainly evaluated in CNS malignancies, including rGBM, as it does not necessitate crossing the BBB to locally inhibit the pathway.
Despite a study by Berghoff et al. indicating that PD-L1 is expressed in 72.2% rGBM [89], PD-L1 inhibitor failed to meet the activity threshold when combine with VEGFR inhibitor axitinib [90]. In an open-label, randomized, multicenter phase III trial CheckMate-143 (NCT02017717), the use of PD-1 blockade as monotherapy did not demonstrate survival benefits compared to bevacizumab in patients with rGBM [91]. The median OS for nivolumab group was 9.8 months while that of the bevacizumab group was 10 months. Similarly, in a phase II trial (NCT02337491), another agent, pembrolizumab, showed ineffectiveness as monotherapy or in combination with bevacizumab in treating rGBM [92]. However, a subgroup analysis revealed that patients with MGMT-methylated tumors and no baseline corticosteroid treatment had a median OS of 17 months, compared to 10.1 months for similar tumors treated with bevacizumab [91].
Some patients with hypermutated rGBM who have biallelic mismatch repair deficiency may benefit from PD-1 blockade [93], which is consistent with many other cancers [94,95]. However, Touat et al. suggested that the hypermutational burden induced by chemotherapy may not enhance the response to PD-1 blockade [96]. Additional trials are underway to further evaluate the responsiveness of hypermutated rGBM - NCT02658279, NCT04145115. While there is still controversy surrounding how to utilize mutation burdens to predict anti-PD-1 response, approaches that promote intratumorally lymphocyte infiltration are necessary for most patients.
Neoadjuvant administration of PD-1 blockade is one approach that has been proposed to enhance intratumorally lymphocyte infiltration in patients with rGBM. This approach may prime an effective systemic immunity, potentially facilitating local lymphocyte infiltration while the tumor is surgically removed [97]. Two trials have been conducted based on this hypothesis. One study (NCT02852655) with 35 rGBM patients found that neoadjuvant pembrolizumab led to a median OS of 13.9 months compared to 7.6 months for adjuvant pembrolizumab only [98]. In the other single-arm study (NCT02550249), 30 patients (27 rGBM and 3 ndGBM) were treated with nivolumab pre- and post-operatively, but the median OS for these patients was 7.3 months which was not superior to the existing strategy [99]. Difference between the two studies could lead by different drugs utilized, small numbers of participants, and/or selection bias while only certain patients with rGBM are eligible for additional surgeries. In a serial study, scientists noticed a population with enriched BRAF/PTPN11 mutations in 30% rGBM that responded to PD-1 blockade [100]. Further investigation revealed that ERK1/2 activation in rGBM is favorable to PD-1 blockade and promotes tumor-infiltrating myeloid cells and microglia expressing more MHC class II and associated genes[101]. Another ongoing study (NCT02337686) is devoted to evaluating immune effector function in this neoadjuvant setting. Though extra caution is needed before drawing any conclusions, both studies demonstrated similar intratumorally and systemic immune changes, suggesting that combinations with other immune and non-immune agents may be worth exploring.
To date, the majority of studies have focused on combining anti-PD-1/PD-L1 with other treatment modalities. Some groups have focused on combining with conventional methods like radiotherapy, chemotherapy, and anti-VEGF therapies, but with adjusted strategies. Novel procedures like stereotactic radiation (NCT04977375, NCT02866747, NCT02829931), laser interstitial thermotherapy (NCT02311582, NCT03277638, NCT03341806), and tumor treating fields (NCT03430791) are tested in multiple ongoing trials, with the hope of generating enough local immune reaction. Upregulation of multiple alternative immune checkpoints on T cells and/or tumor cells has been observed in other solid tumors as resistance to ICB [102]. Clinical trials targeting IDO1 (NCT03532295), CTLA-4 (NCT02794883), LAG-3 (NCT03493932), CD137 (NCT02658981) along with PD-1, are underway in rGBM. Combined with other immunotherapies like tumor vaccine and oncolytic virus also harbored lots of interest with multiple ongoing trials (Table 2).
2.2.3. Negative immune regulation
T cell exhaustion plays a significant role in the local immunosuppression and immune dysfunction observed in GBM. Worenieck et al. have unveiled T cell exhaustion signature in various tumors and highlighted LAG-3 as one of the T cell immune checkpoints upregulated in GBM that lead to severe T cell exhaustion.[103] LAG-3 can inhibit the function of CD8+ T cells and enhance the immunosuppressive activity of regulatory T cells (Tregs).[104] Another study by Shen et al. showed that patients with LAG-3 expression on peripheral blood CD8+cells exhibited poorer responses to ICB antibodies. Therefore, LAG-3 could serve as an independent biomarker to guide treatment as well as an actionable target for standard-ICB-resistant patients, showing significantly correlations with response, survival, and progression-free survival in various cancer types.[105] Clinical trials have already shown the anti-tumor activity of anti-LAG-3 agents, although modest.[106] Currently, a phase I clinical trial (NCT02658981) is evaluating the efficacy and safety of anti-LAG-3 agents in rGBM. Moreover, the previous study by Worenieck showed that compared to T cells with only one checkpoint, T cells expressing multiple immune checkpoints were more dysfunctional.[103] So this phase I trial is also evaluating the combination of anti-LAG-3 and anti-PD-1, whose anti-tumor activity has already been demonstrated in a clinical trial involving unselected patients with cancer.[107] Initial data from another phase II/III trial (NCT03470922) has also shown that compared to using anti-PD-1 alone, melanoma patients receiving combination therapy showed improved PFS. In conclusion, LAG-3 is a unique, non-redundant checkpoint that limits the efficacy of standard ICB therapies such as PD-1 and CTLA-4 inhibitors. It holds the potential as a biomarker that guides treatment, a candidate for novel agents or combinations, and a promising immune target for standard ICB-resistant patients.
T cell immunoglobulin and mucin-domain containing-3 (TIM-3) is another immune checkpoint involved in negative immune regulation. It was demonstrated to induce CD8+T cell apoptosis and exhaustion, as well as inhibit T cell response in glioma.[108] This has led to disappointing outcomes in patients receiving anti-PD-1 therapy and lower survival rate of GBM patients.[109] Currently, several phase I studies are underway to evaluate the potential of TIM-3 as a therapeutic target. One of these studies (NCT02817633) has reported tolerability and promising efficacy of TSR-022, an anti-TIM-3 monoclonal antibody, in patients with advanced solid tumors (AMBER). It may help patients who showed no response to standard ICB therapy.[110,111]
Other immune checkpoints for negative immune regulation include T-cell immunoglobulin and ITIM (Immunoreceptor Tyrosine-based Inhibitory Motif) domain (TIGIT), VISTA, and B7-H3 (CD276).[112] They’re all potential ICB targets under research, but currently, no trials are evaluating their efficacy in GBM.
2.2.4. positive immune regulation
Inducible co-stimulator (ICOS) is a novel immune checkpoint and an independent prognostic factor for glioma. It is expressed on the surface of activated T cells and enhances the secretion of multiple immune cytokines.[114] ICOS participates in positive immune regulation as ICOS/ICOSL pathway was shown to promote T cell differentiation, proliferation, and activation.[113] [114] But on the other hand, it also induces Tregs activation, especially in GBM, in which its negative effects outweigh its positive effects.[115,116] So, ICOS played a negative role in the immune microenvironment of glioma and GBM through promoting tumor formation, development, and drug-resistance.[117] Wang et al. discovered a positive correlation between ICOS expression and glioma malignancy. In general, higher ICOS often indicates shorter life expectancy.[116] Therapeutic strategies targeting ICOS for glioma hold promise as it has already exhibited anti-tumor effect in some malignancies.[118,119] Wang’s work also revealed synergistic interactions between ICOS and other important immune checkpoints, suggesting the possibility for combination therapy. To date, several clinical trials has been testing combination therapy of anti-ICOS and anti-CTLA-4/anti-PD/PD-1.[112] [118,119]Further studies and experiments are required to evaluate the efficacy and safety of anti-ICOS therapy in treating GBM.
Glucocorticoid-induced TNFR-related gene (GITR) and OX40 belong to the tumor tumor necrosis factor (TNF) superfamily, and they also play positive roles in immune regulation. They reduce T cell apoptosis, boost T cell proliferation and increase T cell activity. Until now, several agonist antibodies for GITR and OX40 are under investigation (NCT02598960, NCT02628574, NCT01862900).[112]In general, many patients who receive ICB therapy targeting PD-1/PD-L1 and/or CTLA-4 haven’t shown promising responses thus far. But novel immune checkpoints listed above show promise in improving the situation. They may offer potential benefits for patients who have exhibited unsatisfactory responses to standard therapy. They also got their own advantages over standard ICB. For example, the intracellular tail of TIM-3 has no ITIM or immunoreceptor tyrosine-based switch motifs (ITSM).[109] Moreover, many of them are directly implicated in the progression of GBM and are involved in immune response recruitment and activation. Although currently there are not many studies investigating their therapeutic efficacy in GBM, hopefully novel immune checkpoints may be of greater importance and become the focus of future research.
2.2.5. Challenges and future directions of ICB in rGBM
Unlike other immunotherapies, ICB is extremely dependent on the intact immune system, from antigen presentation to effector lymphocytes activation. This is the major challenge in achieving positive results in rGBM as monotherapy given the local and systemically suppressed immune environment created by GBM [120]. Specifically, T cell dysfunction has been considered a hallmark of GBM, which would not be an easy fix by ICB [48]. Furthermore, immunosuppressive therapies such as chemotherapy or steroids that rGBM patients may go through could further limit the benefits of ICB [121]. Additional constraints unique to CNS tumors include the restricted access of drugs to the CNS. Many trials have attempted to circumvent this issue by applying ICB directly within the tumor. Duerinck et al. tested the idea by injecting ipilimumab and nivolumab intracerebrally in 27 patients (NCT03233152)[122]. The treatment appears to be safe and feasible, with a median OS of 9.5 months. Further studies are needed to determine whether local administration within tumors is required for optimal efficacy.
A simple modification to treatment regimens may help the situation, neoadjuvant treatment appears to be an attractive strategy for rGBM. Despite the lack of responses or partial responses in OS, pro-inflammatory changes in the tumor microenvironment are encouraging. It is possible that other checkpoints may be more predominant in rGBM, and thus PD-1 blockades may only improve lymphocyte activation without reversing the effects controlled by other checkpoints. Thus, novel checkpoints such as VISTA [123], Siglec-15 [124], and HHLA2 [125] may be worth testing once their role in rGBM is confirmed. Nevertheless, ICB seems to be a good addition for lots of current immunotherapies relying on cytotoxic T cell functions with highly expressed intertumoral immune checkpoints in rGBM. In turn, other immunotherapies may compensate for the limitations of ICB by presenting antigens, creating a local immune response, or overcoming the immunosuppressive tumor microenvironment. The search for biomarkers to identify patients who are more responsive to ICB is also a promising avenue for further exploration.
2.3. Cancer vaccination therapy for rGBM
2.3.1. The background of cancer vaccination therapy for rGBM
The use of anti-tumor vaccines, another form of immunotherapy, has also garnered significant interest in the treatment of rGBM due to its demonstrated potential and promise in both preventive and therapeutic effects. [126,127]This therapeutic approach typically targets tumor antigens to induce adaptive immune responses against tumors. Given the realatively low tumor mutational burden (TMB) observed in rGBM, the antigen targets selected are most often tumor-associated antigens, with only a minority of mutations serving as tumor-specific antigens (TSA)[82]. According to the immune subtypes of GBM classified by Han Lin et al., immune subtype 3 (IS3) exhibits the poorest prognosis but derives the greastest benifit from vaccination therapy.[128] Overall, numerous vaccination approaches are currently under investigation[130], with the majority still in early stages of clinical development and clinical trials.
Generally, GBM vaccines can be categorized into several groups, including peptide vaccines, immune cell-based vaccines (DC cell-based, B cell-based), and nucleic acid vaccines. Table 3 presents the primary vaccines that have been studied or tested in rGBM. Next, we will provide a detailed discussion of each type of them.
2.3.2. Peptide vaccines
Peptide vaccines are typically 8-30 AA (amino acids) in length. They function by encompassing TSA or TAA (tumor-associated antigens).
Among the TSA peptide vaccines, Rindopepimut (CDX-110) has garnered significant interest, which is characterized by low off-target toxicity. However, its patient eligibility is limited as it specifically targets EGFRvIII, a mutant form of EGFR only heterogeneously expressed in 25-30% of GBM, with 82% of tumors not expressing it upon recurrence. Several clinical trials have been conducted to evaluate its efficacy.
Early studies include three uncontrolled phase II trials reported in 2010,2011,2015. In these trials, GBM patients who underwent gross total resection and chemoradiotherapy received rindopepimut vaccination. The results showed a median overall survival of 24 months, represeting a modest improvement over historical controls. [135,136,137] In a phase II trial in 2015 by Reardon et al., the combination of rindopepimut and bevacizumab was shown to have promising therapeutic activity and tolerability in patients with rGBM[138]. In 2017, Weller M et al. reported that patients with minimal residual disease who received rindopepimut with TMZ didn’t show an improvement in OS compared with patients receiving TMZ alone in a double-blind, placebo-controlled, multicenter phase III trial ACTIV, but the data demonstrate decent humoral immune response.[139] In 2020, Reardon DA et al. reported favorable outcomes when exploring the efficacy of rindopepimut plus bevacizumab in a smaller cohort of EGFRvlll-positive GBM.[140] Overall, these studies suggest that rindopepimut may have some activity in carefully selected patient cohorts, but further investigation is required to determine the optimal treatment regimen. Those contradictory and inconsistent results questioned the effect of the single antigen-targeted vaccine and lend support to combination strategies and multi-epitope vaccines.
Isocitrate dehydrogenase-1 (IDH1) mutations, on the other hand, create TSA as a potential target for vaccination therapy. The frequency of IDH mutations was found to be less than 10% in pGBM whereas it exceeds 70% in rGBM, thus indicating a broader application regimen for rGBM compared to vaccinations targeting EGFRvIII. Preclinical studies have already shown that peptide vaccines spanning the IDH1 mutation may elicit antitumor T cell responses. In 2021, a phase I trial reported that approximately 90% of patients with glioma exhibited an immune response following treatment with an IDH1-R132H+-specific vaccine.[132] Combination therapy involving PD-L1 checkpoint inhibition has also been proposed.[141]
Wilm’ tumor 1 is another notable antigen in GBM, with a particularly high presence reaching 94%.[142]. Unlike other single antigen-targeted vaccines, the risk of immune escape is relatively low for WT1 vaccine, as the loss of WT1 expression was shown to halt tumor proliferation and induce cancer cell death. In 2020, Rudnick, J. D et al. reported the clinical responses to WT1 vaccination in patients with rGBM who were positeve for human leukocyte antigen HLA-A24 in a phase I/II study, the results were limited with a 9.5% overall response rate and 20 weeks of progression-free survival (PFS) time.[133]
The vaccination approaches discussed above are all single antigen-targeted, but multiple-epitope peptide vaccines are believed to hold greater potency and efficiency due to their ability to induce more robust and comprehensive immune responses.
IMA950 is a novel therapeutic peptide vaccine that includes 11 synthetic TAA. It enables the stimulation of specific cytotoxic T lymphocytes (CTL) to eliminate malignant tumor cells. A a phase I/II trial completed in 2019 evaluated IMA950 in combination with poly-ICLC and TMZ. The overall cohort of patients showed a median OS of 21 months from the date of surgery, compared with 19 months in the GBM-only cohort. PFS of patients in the overall cohort were 93% and 56% at 6 and 9 months, respectively.[143] However, IMA950 has not shown any benefit in rGBM patients so far.[130] It is worth mentioning that the peptide set selected from the IMA950 may have potential applications in immunotherapy of grades II and III gliomas, which is different from other peptide vaccines.[144]
TAS0313 is a multi-epitope long peptide vaccine targeting multiple TAAs in rGBM. In 2022, it was demonstrated to have promising efficacy and acceptable safety in rGBM patients [145].
Heat-shock protein peptide complex-96 (HSPPC-96) is another vaccine approach that targets multiple tumor antigens. In 2014, Bloch et al. investigated the efficacy and safety of HSPPC-96 vaccination in rGBM patients in a phase II trial and reported a median OS time of 42.6 weeks.[146] Another phase I study showed a 2.3 folds increase in tumor-specific immune response of ndGBM patients after they were treated with HSPPC-96 vaccine.[147] Many researchers are exploring the potential of the HSPPC-96 vaccine when combined with radiotherapy and chemotherapy (NCT00905060) for primary GBM and with bevacizumab (NCT01814813) in the treatment of recurrent GBM at present.[148]
The lack of high expression of GBM-specific antigen due to low TMB and extensive heterogeneity of GBM between individual patients has been posing challenges to GBM therapy, which means there is not a one-fit-all vaccination approach. Recent advances in next-generation sequencing and novel bioinformatics tools, however, enable us to systemically discover tumor-specific neoantigens as suitable targets, which have the potential to solve the problem and thus garnering significant attention. Through whole exome sequencing of patient tumor cells and peripheral blood, we can explore expressed mutations in tumors and then rank candidate targets for synthesizing to generate vaccines.[150]. Those personalized, neoantigen-based vaccines have shown robust tumor-specific immunogenicity and preliminary evidence of anti-tumor activity in patients with melanoma and other cancers.[149] Moreover, it elicits much lower toxicity compared to TAA- targeted vaccines. Based on these promising findings, two phase I/Ib studies of multi-epitope, personalized antigen vaccines were carried out and reported in 2019, in which Keskin et al. demonstrated the generation of circulating polyfunctional neoantigen-specific CD4+, CD8+ T cells that were enriched in a memory phenotype and found an increase of tumor-infiltrating T cells (TILs). It suggests that neoantigen vaccines have the potential to transform a “cold” tumor environment into a “hot” one. The study also provided evidence that neoantigen-specific T cells can migrate into an intracranial GBM. But disappointingly, all patients in the trial still experienced tumor recurrence and ultimately died.[151]In another similar phase I trial conducted by Hilf et al., comparable findings were reported, showing acceptable safety profile and sustained T cell response.[152] In 2019, a phase III trial of personalized peptide vaccination for rGBM was conducted in HLA-A24 positive patients too, but neither the primary endpoint (OS) nor the secondary endpoint was reached.[134] Other trials have also investigated the safety of combination therapy with radiation therapy (RT) or immune checkpoint inhibitors (ICIs).[150,153] In conclusion, this strategy requires further exploration of its efficacy and more improvement to overcome challenges like tumor-intrinsic defects and immunosuppressive factors in the microenvironment. Combination therapies may offer a potential solution to address these obstacles. It is also noteworthy that the process of neoantigen identification and vaccination development is time-consuming (about 3 months) [154], which poses another limitation to its application. Detecting recurrent and shared neoantigens holds promise in addressing this issue. Subunit vaccines have acceptable safety profiles, significant efficacy and are considered logistically feasible [155]. In comparison to whole protein or pathogen vaccines, these domain-based vaccines offer notable advantages. In 2021, Mahmoud Gharbavi et al. reported that they designed and synthesized a multi-domain recombinant vaccine for glioblastoma multiform. The process involved the selection of the most potent domains of TAAs using immune-informatics analysis and their combination to elicit an immune response in the host the potency of this novel multi-domain subunit vaccine was demostrated through physicochemical analysis, And its antigenicity was estimated at 0.78. The multi-domain vaccine could potentially provide both prophylactic and therapeutic benefits.[156]
The Mannan-BAM, TLR Ligands, Anti-CD40 Antibody (MBTA) vaccine represents another personalized vaccination approach that targets multiple TSA. This vaccine offers distinct advantages because it circumvents the long process of silico tumor-neoantigen enrichment required for personalized neoantigen peptide vaccination by enabling tumor-specific neoantigen to be processed in vivo through endogenous pathways that activate the innate immune system.[157] In this way, it allows the innate immune system to select antigenic targets through natural processing mechanisms. Furthermore, the MBTA vaccine has shown potential to overcome the challenges associated with immunosuppression and intratumoral heterogeneity.[158]
2.3.3. Cell-based vaccines
Currently, about half of the ongoing phase II/III trials on GBM involve cell-based vaccines, the majority of which use a dendritic cell (DC) carrier. Other cell-based vaccines include B cell-based vaccines have also gained much attention due to their high mobility and convenience to be manufactured ex vivo.[160] The subsequent section provides comprehensive descriptions of these vaccine types.
Dendritic cell (DC) vaccines
DC vaccines are of great interest due to the critical role played by DCs in immune regulation and antigen presentation. They can target tumor antigens or serve as immune-boosting adjuvants in vaccination therapy.[161] DC vaccination targeting tumor peptides has demonstrated auspicious results in rGBM patient treatment. Adjuvant DC immunotherapy in rGBM patients was also shown to induce long-term survival. Typically, DC vaccination was generated ex vivo from DCs harvested from patients and subsequentlly stimulated by either tumor antigens, cell lysates, recombinant proteins, or nucleic acids before administration. The commonly utilized DC types include Mo-DCs and leukemia-derived DCs (DCleu).
Several studies have revealed the clinical efficacy of DC-based vaccines, but there’ve also been conflicting results. In 2020, researchers reported a phase II clinical trial of alpha-type-1 polarized DC-based vaccination in newly diagnosed high-grade glioma, which showed a significant survival-prolonging effect in treated patients.[162] Vaccination using DCs loaded with TAAs and mRNA of neoantigens extended patients’ mOS to 19 months.[163] In 2022, a meta-analysis encompassing 15 clinical trials (comprising 452 cases and 629 controls) assessing the efficacy of DCV in newly-diagnosed GBM (ndGBM) patients revealed that DCV had no impact on 6-month PFS or 6-month OS, but led to significantly longer 1-year OS and longer 2-year OS. Its delayed effect suggests the necessity for additional therapies to facilitate earlier action of DCV.[164]
However, two meta-analyses in 2021 concluding that DCV has no obvious impact on the prognosis of ndGBM. But these two analyses had relatively small sample sizes, which may have influenced the conclusions drawn.[165,166]
Besides, the observed heterogeneity in the results of DCV studies may also be attributed to variations in methods employed and differences in patient populations recruited. Studies have indicated that patients with low B7-H4 expression treated who received DCV treatment experienced significantly prolonged OS. Furthermore, methylated MGMT promoter, wild-type IDH, and mutation-type TERT are also linked to better response to DCV.[167] A short life expectancy for GBM may mask the effect of DCV too as it typically requires a minimum period of 6-month to become evident. Based on these studies, stratification of GBM patients based on molecular biomarkers to identify more sensitive groups may be necessary prior to DCV therapy.
Among the single targeted DC vaccine candidates, Wilms' tumor 1 (WT1)-pulsed autologous DCs and cytomegalovirus phosphoprotein 65 RNA (CMV pp65)-pulsed DCs have shown promise. The efficacy and safety of the WT1 DC vaccine in rGBM patients were already demonstrated in a phase I trial.[161] Researchers have also found that compared to WT1 peptide vaccination therapy, DC-based vaccination induces and activates more tumor antigen-specific cytotoxic T cells in rGBM, which may lead to prolonged survival in rGBM patients. Several phase I trials of CMV pp65 DC vaccine have shown promising results as well,[168] and currently a randomized phase II trial is recruiting newly diagnosed GBM patients (NCT02465268).
However, as rGBM is highly heterogenous, several studies have revealed that vaccines targeting a single tumor antigen have difficulty achieving optimal clinical effects unless the antigen is widely expressed in tumor cells. Therefore, there is a growing focus on the development of vaccines that target multiple antigens.
ICT107 is a DC vaccine pulsed with six synthetic peptides. It is specifically designed for GBM and is produced through the ex vivo incubation of patient-derived DCs with six GBM TAAs. Its safety and therapeutic activity in HLA-A2 positive patients have already been demonstrated in some early phase clinical trials, which led to a phase III trial (NCT02546102) carried out in HLA-A2 positive patients with ndGBM. However, this phase III trial was suspended in 2017 due to inadequate funding, halting further progress in its evaluation.
The autologous tumor cell lysate-pulsed dendritic cell (DC) vaccine can target multiple antigens too. This personalized vaccination therapy also addresses the heterogeneity of glioblastoma (GBM) by utilizing patient-derived autologous antigens rather than standardized antigens. DCVax-L, for instance, employs autologous whole tumor lysate to pulse patient-derived DCs, targeting the full repertoire of antigens and minimizing immune escape. Theoretically, this kind of vaccine should be more efficient but carry a higher risk of autoimmune response. As promising results have been observed in preclinical models and early-stage clinical trials, a phase III prospective externally controlled cohort trial (NCT00045968) was conducted in ndGBM. By 2018, this phase III trial showed that the overall intent-to-treat (ITT) population had a median OS of 23.1 months from surgery and a low incidence of grade 3 or 4 adverse events(2.1%), superior to the median OS of 15–17 months reported in past studies and clinical trials.[169] In 2023, the same trial reported that the median OS for ndGBM patients treated with DCVax-L was 19.3 months compared to 16.5 months in the control group. The 48-month survival rate from randomization was 15.7% compared to 9.9%. For rGBM patients, DCVax-L also showed advantages compared to the control group. Moreover, a better response was observed in patients with methylated MGMT [131]. This study demonstrated that adding DCVax-L to SOC resulted in a clinically meaningful and statistically significant extension of survival in both ndGBM and rGBM patients compared to external controls who received SOC alone. Overall, the addition of DCVax-L to standard therapy has shown feasibility, safety, and the potential to extend survival in GBM patients. Another randomized phase II trial (NCCT03014804) on Vax-L is currently underway.
AV-GBM-1 is an autologous tumor-initiating cell pulsed DC vaccine, which is different from DCVax-L (utilize fresh whole tumors). A multicenter phase II trial was designed to evaluate AV-GBM-1 and reported that the treatment was well-tolerated with a prolonged median PFS, though no median OS improvement was observed.[170] Another phase III trial for AV-GBM-1 has been approved by the FDA and is underway (NCT05100641).
Similar to the advantages of neoantigen-targeted peptide vaccines over TAA peptide vaccines, personalized neoantigen-pulsed DC vaccines have also been considered more effective than TAA-pulsed DC vaccines [171]. Numerous trials utilizing personalized neoantigen-pulsed DC vaccines are currently ongoing.[172]
Combinatorial therapy of DC vaccines with chemotherapy and checkpoint inhibitors is also under active research, as it has been demonstrated that the efficacy of DC vaccines enhanced through this approach.[173]
Although the administration of inactivated tumor cells or patient-derived tumor cell lysates have exhibited superiority, their efficacy is hampered by their inability to kill tumor cells before inducing immune responses, which can be fatal as GBM progresses rapidly. In 2023, Chen et al. developed a bifunctional cancer cell-based vaccine (Therapeutic tumor cells) that drives direct tumor killing and antitumor immunity simultaneously. It represents a promising cell-based immunotherapy as it has shown therapeutic efficacy in a recurrent GBM mice model.[159]
Finally, DC vaccine immunotherapy still faces several challenges, including the presence of an immunosuppressive TME, and intrinsic drawbacks like high costs as well as time-consuming processes, which limit its widespread application [174] However, despite all of these challenges, DCV still represents a promising new strategy for GBM and other malignancies with validated safety and feasibility.
B cell vaccines
B cell vaccine is another emerging cell-based vaccine for glioblastomathat harbors great potential. Lee-Chang et al. developed BVax, which was shown to migrate into secondary lymphoid organs to activate T cells for the removal of GBM cells. In a trial conducted on GBM model mice, the combination of PD-L1, BVax, and radiation therapy led to 80% tumor eradication and sustained potent immunological memory, effectively preventing tumor re-growth.[160]
2.3.4. Nucleic acid vaccines
Nucleic acid vaccines, including mRNA vaccines and DNA vaccines, offer several advantages over peptide vaccines. For instance, they can encode entire tumor antigens and are not restricted by the patient’s HLA type compared to conventional vaccination.[175] Additionally, they have the capability to deliver multiple antigens and exhibit greater resistance to drug resistance.[176] Moreover, the production of nucleic acid vaccine can be more rapid and cost-effective compared to peptide vaccines.
In 2022, Amit S et al. employed the UNITE platform to develop a multi-antigen targeted DNA vaccine (ITI-1001) encoding human cytomegalovirus (HCMV) proteins that are expressed in GBM cells. The vaccine elicited robust humoral and cellular immune responses and led to improved survival in GBM-bearing mice.[177] This therapy is partiularly suitable for certain patients whose medical conditions do not allow leukapheresis and autologous DC immunity. In the same year, a combination therapy involving the DNA vaccine pTOP and immune checkpoint blockades in orthotopic unresectable GBM model mice was shown to improve effector T/Treg ratios and infiltration of CD8 T cells in tumor, opening a new prospective for GBM treatment.[178]
Compared to DNA vaccines discussed above, mRNA vaccines have higher expression efficacy and are easier to design and modify, making them well-suited for individualized treatment approaches. Moreover, mRNA vaccines offer enhanced safety as they do not require integration into the patient’s genome. The efficacy of mRNA vaccines has been evaluated in various types of tumors, yielding promising results. In 2022, Han Lin et al. reported using gene expression profiling interactive analysis (GEPIA) to evaluate the expression profile of GBM antigens as well as their clinical influence. They selected six TAA and TSA that were highly correlated with GBM prognosis to be potential targets for developing mRNA vaccine, and found that GBMs of the immune-cold subtype I3 were more likely to benefit from vaccination. So screening mRNA-sensitive patients (for example, IS3) before treatment is important.[128] Also in 2022, another similar research selected nine antigen candidates, adding to the previous research.[129] 2.3.5 The limitations existed and strategies to enhance cancer vaccines for rGBM
As for vaccination therapy for rGBM, there are still many challenges waiting to be addressed, including: (I) systemic and local immunosuppression in the tumor microenvironment (II) high tumor heterogeneity and deficiency of specific tumor antigens (due to low TMB) within GBM;[179] (III)BBB which prevents peripheral immune cells from entering CNS;(IIII) severe adverse effects of some vaccines. Efforts have been made to overcome these challenges and we compile some possible ways below. For example, to overcome the local immunosuppressive environment, studies have demonstrated that certain agonists targeting tumor-associated macrophages (TAM), such as poly-ICLC, resquimod, and imiquimod, can be used as vaccine adjuvants to enhance the efficacy of vaccine therapy. It can prolong the median PFS of GBM patients to 21 months post-diagnosis.[180] The mechanism underlying is that these agonists can repolarize TAM, which make up 80% of immune cells in the tumor microenvironment. M2 phenotype TAMs, in particular, contribute to tumor progression and invasion through several mechanisms.[181] Another strategy to make TME “hotter” is to utilize personalized neoantigen-targeted vaccination therapy.[151] Besides, accumulating evidence showed that the gut microbiota can regulate immunity and metabolism in the GBM microenvironment making it a potential therapeutic target to modulate the immunosuppressive TME of GBM too.[148] To find TSA and overcome intertumoral heterogeneity, personalized neoantigen-targeted vaccine hold promise with effectively reduced off-target toxicity.[172] To avoid immune escape and solve the problem of individual heterogeneity, we may utilize tumor cell-pulsed DCV or add other therapeutic modalities such as molecular targeted therapy to immunotherapy. To disrupt BBB and enable the access of immune cells, combination therapy with MRI-guided laser ablation (MLA) may be beneficial.[182] However, despite these encouraging results of preclinical and phase I/II clinical trials, and even success in a few case reports, the phase II/III transition remains particularly challenging. To date, no successful phase III clinical trials with large patient cohorts for GBM immunotherapy have been reported.[38]
In conclusion, vaccination therapy has been considered one of the most promising approaches to improve the outcomes of rGBM patients. From trials that have been conducted so far, it is evident that single-agent immunotherapy has limited efficacy for rGBM, so rational combinatorial treatment strategies worth more attention. In the future, we also need to better understand the mechanisms of GBM immunosuppression. Tumor-specific antigenic profiles that are more effective are urgently needed too. Finally, although several vaccines have already shown efficacy and safety in phase I and II trials, overall results of phase III clinical trials are still disappointing, without significant improvement in the prognosis of rGBM. Accordingly, more phase III trials are needed.
2.5. Oncolytic viral therapy in recurrent GBM (rGBM).
In recent years, oncolytic virus (OV) therapy has demonstrated great potential in prolonging survival, improving patients' quality of life, and less adverse effects. Contrast to OV, following traditional therapy, such as surgery, radio- or chemotherapy, the median survival of patients suffering from pGBM is approximately 14.6 months[126].
The clinical trials and animal experiments evidence showed in Table 4.
The mechanism of the Oncolytic virus is still unclear and the oncolytic procedure is multi-related and multi-staged. Nevertheless, there are two dominant perspectives: one is that OVs directly destroy GB cells, and the other is that OVs induce tumor cell lysis by virus-specific infection of tumor cells and the release of viral progeny to induce tumor cell lysis[127,128,129,130,131,132,133].

Table 4.
Oncolytic viral therapy trial in recurrent GBM (rGBM).
| Agents | Year | Study design | Subjects | Experiment time | Registration Number |
|---|---|---|---|---|---|
| Herpes simplex virus (HSV-1716) |
2000 | Phrase Ⅰ trial | Patients had biopsy proven high grade glioma | 24 months | PMID10845724[134] |
| G207 | 2009 | Phrase Ⅰ b trial | Patients had an initial histologically confirmed diagnosis of glioblastoma multiforme | 19 months | F05041106[135] |
| G207 | 2014 | Phrase Ⅰ trial | Patients had pathologically confirmed residual/recurrent glioblastoma multiforme, gliosarcoma, or astrocytoma | 11-51 months | NCT00157703[136] |
| G207 | 2015 | Case report | A 52-year-old Caucasian female had a GBM with an infltrative glial tumor | More than 5.5 years | NCT00028158[137] |
| G207 | 2022 | Cross-sectional study (a Gene Expression Analyses) | Patients are from the phase Ib G207 clinical trial (NCT00028158) | / | /[138] |
| G47Δ | 2022 | Phrase Ⅱ trial | Patients who had a pathologically confirmed diagnosis of glioblastoma with a persistent or recurrent tumor | 2-5 years | UMIN000015995[139] |
| Herpes simplex virus Expressing Interleukin-12 (M002) | 2012 | Animal experiment | Specific-pathogen-free female SCID and B6D2F1 mice | More than 80 days | /[140] |
| Herpes simplex virus type 1 thymidine kinase suicide gene therapy (HSV1-tk) | 1998 | Phrase Ⅰ/Ⅱ trial | Patients had a recurrence of primary glioblastoma | 830 days | /[141] |
| Herpes simplex thymidine kinase gene (HSV-tk) | 1999 | Phrase Ⅱ trial | Patients with relapsed GBM | More than 15 months | /[142] |
| Adenovirus mediated HSV-tk gene therapy (AdvHSV-tk) | 2004 | RCT | All patients with operable primary or recurrent highgrade glioma | More than 200 weeks | /[143] |
| Delta-24-RGD | 2018 | Phrase Ⅰ trial | Patients with recurrent malignant glioma | More than 3 years | NCT00805376[144] |
| Delta-24-RGD | 2022 | Animal experiment | 95 mice | More than 100 days | /[145] |
| Reovirus | 2008 | Phrase Ⅰ trial | Patients had a diagnosis of GBM | More than 234 weeks | /[146] |
| Reovirus | 2014 | Phrase Ⅰ trial | Patients had either first, second, or third occurrence of a supratentorial tumor with a histologic diagnosis consistent with glioblastoma multiforme | More than 989 days | /[147] |
| NDV-HUJ Oncolytic Virus | 2005 | Phase I/II Trial | Patients had been diagnosed with GBM based on histology and gadolinium-enhanced (Gd+) MRI, and all had a recurrence of GBM | More than 66 weeks | /[148] |
| G207& ganciclovir | 2000 | Animal experiment | Six-week-old female A/J mice | More than 30 days | /[149] |
| Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex | 2003 | Phase I Trial | Patients had histologically confirmed malignant glioma, defined as GBM | More than 248 weeks | /[150] |
There are four prominent OV families tested in human or animal trials, which are Herpes simplex virus-1 based (HSV-1-based), AdenovirusBased, ReovirusBased, and Newcastle Disease VirusBased.
2.5.1. Herpes simplex virus-1 based (HSV-1-based)
HSV ⁃1 is a large double-stranded DNA virus, a common human pathogen with a long-term latent and lifelong potential for infection in humans[151]. It is a neurotropic virus, and the genes involved in tumor lysis differ from neurotoxic genes, allowing tumor cells to replicate and manipulate tumor lysis genes[152] conditionally.
Currently, three HSV ⁃1 lysosomal strain (including HSV1716, G207, and G47Δ) have completed phase I clinical trials in glioma patients and clinical trials to evaluate the efficacy and safety of two other HSV ⁃1 lysosomal strain (M032 and QNestin34.5) are ongoing[153].
HSV1716
HSV1716 is a double-copy neurotoxic gene γ134.5-deficient generation lysogenic HSV that selectively replicates in actively dividing cells[154]. In 2000, Rampling, R et al. reported the first evidence in support of the safety of HSV1716 in rGBM treatment in humans[134]. In that study, nine patients, who had previous surgery and radiotherapy, three each received 103, 104, and 105 pfu of HSV 1716 by stereotactic injection directly into the tumor. Five of nine died after the injection from 8 weeks to 9 months during the follow-up. Three underwent further surgery; one died of tumor progression at nine months, and two were alive and well at least 17 months. The other two patients remained well at 14 and 24 months, respectively. They concluded the feasibility of using replication competent HSV as part of a combination therapy regimen in rGBM,
G207
G207 is a double-copy γ134.5 gene deletion and insertion of the exogenous gene lacZ into the UL39 gene [138], thus inactivating ICP6, which supports conditional replication of the virus in actively dividing cells. The effectiveness was demonstrated in mouse and non-human primate experiments[149,155,156,157]. James M Markert et al. showed the safety of inoculation G207 in the brain surrounding a glioma resection cavity[135]. The maximum dose in this 1b trial (registration number: F05041106) is 1×109 pfu. Three of the six subjects improved Karnofsky's performance following the G207 injection. The median survival was 6.6 months (range: 2–20.75 months) from G207 inoculation. No patients did further chemotherapy, which indicated G207 administration in any decrease in tumor progression. None of the deaths or complications could be attributed to G207 administration in the tumor or brain tissue next to the resection cavity. Five years later, this research group conducted a phase 1 trial (registration number: NCT00157703) to show the safety and potential clinical response of single-dose stereotactic intratumoral administration by G207 in rGBM patients[136]. Nine people received one dose of G207 and then were treated focally with 5 Gy radiation. Six patients had stable conditions or partial response for at least one point. The progression-free survival time was approximately 2.5 months (95% confidence interval: 1–5.75), and the estimated median survival time was 7.5 months (95% confidence interval: 3.0–12.7) from G207 injection. One year later, A US team reported that a 52-year-old Caucasian female extended a tumor progression-free interval of 6 years with G207 oncolytic therapy and brief exposure to further treatments after the first treatment doing aggressive tumor resection, radiotherapy, and chemotherapy[137]. Recent gene research has revealed that the immune activity differences in post-G207 and pre-G207 samples are associated with survival duration in patients with rGBM. The tremendous change following the G207 injection is the increasing proportion of CD4 and CD8, CD8+ T-cell to exhausted CD8+ T-cell ratio, and the NK CD56 dim to total tumor-infiltrating lymphocytes ratio. The survival data showed that four of six survived longer than the median survival of GBM recurrence, four months.
G47Δ
G47Δ was constructed by deleting the α47 gene in G207 viral mutant[158]. Tomoki Todo et al. have published their newest results of a phase 2 trial (registration number: UMIN000015995) for applying G47Δ in residual or recurrent glioblastoma treatment in 2022[139]. The research showed the median overall survival time was 20.2 (95% confidence interval: 16.8–23.6) months after G47∆ initiation and 28.8 (95% confidence interval: 20.1–37.5) months after the initial surgery. 17 of 19 patients suffered from fever as the most common adverse event. The only serious side effects (grade 2) occurred in one patient (5.3%), leading to a prolongation of hospitalization. G47Δ therapy indicated good efficacy and safety in rGBM treatment, which approved it as the first oncolytic virus product from the Japanese Pharmaceuticals and Medical Devices Agency.
Genetically Engineered Herpes Simplex Virus Expressing Interleukin-12 (M002)
James M. Markert et al. compared M002 with R3659, R8306, and G207 and found that: M002 indicated superior antitumor activity, no significant imaging or clinical evidence of toxicity in mice right frontal lobes of A. nancymae, and stimulating mice producing IL-12 which activates A. nancymae lymphocytes in vitro[140]. This evidence supports M002 to be trailed in a phase 1 study for patients with rGBM.
2.5.2. Adenovirus-Based
Adenovirus is a double-stranded, envelope-free DNA virus that is a common human pathogen that usually causes mild upper respiratory tract infections[151].
Delta-24-RGD (DNX⁃2401)
Delta ⁃24⁃RGD adenovirus (DNX⁃2401) is modified from Human adenovirus 5 (HAd5), which is deleted 24 base pairs in the E1A gene, and RGD⁃motif is inserted into the H⁃loop region of the adenovirus, thus enhancing the selective replication of the virus[162,163].
DNX-2401 conducted in rGBM treatment has only happened in the last few years. In 2018, the first report of a phase 1 study was published[144]. Thirty-seven patients were assigned to A (n=25) and B groups (n=12). On day 0, both groups executed stereotactic tumor injection of DNX-2401 (1 x 107 – 3 x 1010 vp). Then group A followed up and assessed the toxicity and response, while group B did En bloc tumor resection along with catheter and intramural injection of DNX-2401 (1 x 107 – 3 x 108 vp) at day 14, biological studies, and toxicity and survival studies. In group A, 72% (n=18) of patients showed tumor reductions with 9.5 months median survival duration. Besides, five people survived longer than three years from the surgery, and three of five demonstrated a dramatic reduction (≥ 95%) in tumor size. Because of resection on day 14, group B can only provide survival information, two of the twelve had more than two years of survival, and the overall median survival was 13 months. Furthermore, DNX-2401 replicates and spreads within the tumor in group B; a histopathologic check showed CD8+, and T-bet+ cells infiltrated the tumor, indicating direct virus-induced oncolysis. It proved that DNC-2401 therapy caused direct oncolytic effects and anti-glioma response, which led to immune responses and long-term survival in patients with rGBM. In addition, a Japanese team found that patient-derived bone marrow human mesenchymal stem cells (PD-BM-hMSCs) loaded with Delta-24-RGD (PD-BM-MSC-D24) can either eradicate gliomas tissue in vitro or improve the survival of mice harboring U87MG gliomas in vivo[145]. It provides evidence for using PD-BM-hMSCs to deliver DNX⁃2401 to treat brain tumors.
2.5.3. Reovirus-Based
Reovirus (respiratory enteric orphan virus) is a naturally occurring double-stranded RNA virus that can be isolated from humans' respiratory and gastrointestinal tracts. A phase 1 study indicated that after injecting reovirus at 1×107, 1×108, or 1×109 tissue culture infectious dose 50 in a volume of 0.9 ml[146]. Karnofsky's Performance scores of seven patients increased without showing grade III or IV adverse events (AEs). Ten patients had tumor progression; the other two either remained stable or were not evaluable. The overall median survival was 21 weeks (range: 6-234 weeks), with one of them alive at the discontinued point. Generally, a maximum dose did not reach, and the results demonstrated good tolerance to using these doses and schedules in patients with rGBM. A fellow dose escalation study was conducted in 2014[147]. Fifteen adult patients were injected with 1×108 to 1×109 tissue culture infectious dose 50; two people had stable disease as their best performance at the follow-up endpoint, one had a partial response, and 12 patients had tumor progression. For survival issues, 13 patients survived approximately two years, and the rest two were alive in the following 3 and 5 years, respectively.
2.5.4. Newcastle Disease Virus Based
NDV-HUJ Oncolytic Virus
NDV is a single-stranded RNA virus whose natural host is poultry, and NDV-HUJ is the oncolytic HUJ strain of the Newcastle disease virus[148]. A phrase 1/2 study determined NDV-HUJ safety and tumor response. Initially, 14 patients were enrolled and completed an accelerated intrapatient dose-escalation protocol, from 0.1 to 11 billion infectious units (BIU) of NDV-HUJ (1 BIU = 1×109 EID50 50% egg infectious dose). Then they received the highest preclinical tested dosage (55 BIU) for three cycles. Secondly, the patients received two to three cycles of 11 BIU depending on their tumor progression. Grade I/II constitutional fever was the most common adverse effect, possibly related to treatment, among the patients. The maximum tolerated dose was not observed. These findings encouraged the continued evaluation of NDV-HUJ in rGBM.
2.5.5. The future directions of Oncolytic viral therapy in (rGBM)
There are still many issues to be explored in treating rGBM with oncolytic viruses, including mechanism of action, safety and maximum dose, and mode of inoculation. The use of oncolytic viruses in combination with standard conventional therapeutic regimens and other agents, such as immune checkpoint inhibitors, will also be the focus of further research. In addition, OVs can also serve as innate adjuvants to enhance antitumor immune response and combine with other immunotherapies to improve the immunosuppressive microenvironment. In the future, OVs and related combination therapeutic strategies to improve the outcome of glioma treatment are promising.
2.6. Combination Strategies for GBM
2.6.1. Chemotherapy and radiotherapy
In 1970, there was clinical evidence that patients with GBM with lomustine plus radiotherapy achieved median survival of 11.5 months, which was longer than that of patients receiving radiotherapy alone[164]. Subsequently, it was found that TMZ was treated concurrently with radiotherapy, and maintenance chemotherapy for 6 weeks improved the survival of GBM patients to 14.6 months[165]. A large number of clinical trials have been conducted in people under 60 to 70 years of age, so most clinicians consider TMZ plus radiotherapy to be the standard of care for GBM patients under 65 years of age. In recent years, there have been many experimental data from elderly patients that have also demonstrated better results during TMZ added to radiotherapy[166,167,168]. At the same time, there are results that support TMZ therapy for longer survival in patients with MGMT promoter methylation tumors[169]. This suggests that the status of MGMT can be used to select patients who benefit more from treatment, avoiding toxic and expensive treatment for patients with poor prognosis. Especially in older patients, individualized treatment should be based on performance status, degree of resection of the lesion, and MGMT status, including radiation dose and whether or not to combine chemotherapy[167,170]. However, TMZ treatment has limitations. Combination chemotherapy and radiation therapy can lead to comorbidities, including bone marrow suppression and infection. Common side effects are neutropenia and thrombocytopenia[171]. There is no evidence that changing the dose of the TMZ regimen or extending its administration beyond 6 months improves survival. Furthermore, the effect of TMZ is correlated with MGMT promoter methylation. Chemical resistance to alkylating agents in GBM patients leads to research to explore more targeted treatments, such as exploring new drugs O6-benzylguanine (O6-BG) and O6-(4-bromothenyl) guanine (O6-BTG), RNAi, and viral proteins targeting MGMT to improve the anti-tumor effects of TMZ[172].
2.6.2. Molecularly targeted drugs
Bevacizumab (BVZ) was approved in 2009 in countries such as the United States and Switzerland for the treatment of rGBM, but data from two large phase III European Organization for Research and Treatment of Cancer (EORTC) trials did not show that it extended overall survival (OS) in patients with GBM[173]. However, it has significantly improved progression-free survival (PFS) rates and reduced demand for steroids, which can improve quality of life[174]. Much of the current research is looking for a combination of BVZ and immunomodulators or other drugs.
2.6.3. Tumor Treatment Fields (TTFields)
The Phase III registration trial demonstrated that TTFields has the same efficacy as chemotherapy and bevacizumab, and the Food and Drug Administration (FDA) approved the TTFields for the treatment of rGBM[175]. Since then, multiple clinical trials have shown that TTFields have better results in combination with surgery and chemoradiotherapy. The trial of Felix Bokstein et al. confirmed that TTFields in combination with chemotherapy and radiation therapy has a good effect, and does not increase the toxicity of chemotherapy or radiotherapy. In addition to the appearance of adverse effects of scalp irritation, this combination therapy is safe and feasible. They are preparing to conduct a phase II study to further test the protocol[176]. Experiments on newly diagnosed GBM patients have shown that TTFields combined with TMZ and CCNU is safe and feasible, and has potentially beneficial therapeutic effects[177]. Clark et al. found in in vitro cell experiments that the antitumor efficacy of TTFields was not affected by the MGMT status of cells[178]. The most common adverse effect of this therapy is localized skin disease, but it causes much less haematological toxicity and gastrointestinal irritation than radiotherapy and chemotherapy. The use of dexamethasone may reduce the therapeutic effect of TFields and radiotherapy. Gregory's research illustrates that placement of TTFields arrays does not significantly affect target volume coverage[179]. The modeling results of Eric et al. show that the therapeutic effect of TTFields is limited by the location of the tumor in the brain, and larger tumors may require longer treatment times[175].
Table 5.
Non-immunotherapy combination therapy for GBM .
| Clinical Trails | Phase | Interventions | Arms | Combined Therapy |
|---|---|---|---|---|
| NCT00684567 | Ⅱ | Drug: TMZ Radiation: RT |
Single arm:TMZ + RT | Chemotherapy and radiotherapy |
| NCT01730950 | Ⅱ |
Biological: BVZ Radiation: RT |
Arm 1: BVZ Arm 2: BVZ + RT |
Radiation therapy with bevacizumab for the rGBM |
| NCT01894061 | Ⅱ | Biological: BVZ Device: NovoTTF-l00A Other: Quality of Life Assessment |
Arm 1: BVZ + NovoTTF-100A | NovoTTF-100A With Bevacizumab (Avastin) for the rGBM |
| NCT01849146 | Ⅰ | Drug: Adavosertib, TMZ Radiation: RT |
Arm 1: Adavosertib + TMZ + RT Arm 2: adavosertib + TMZ |
Adavosertib, RT, and TMZfor the Newly Diagnosed GBM or rGBM |
| NCT00650923 | Ⅰ |
Drug: Ziv-aflibercept, TMZ, Procedure: RT, pharmacological study, laboratory biomarker analysis |
Arm 1: ziv-aflibercept + RT + TMZ |
Aflibercept, RT, and TMZ for the Newly Diagnosed GBM or rGBM |
2.6.4. Combination Strategies of immunotherapy
Immunotherapy has made advances in the treatment of rGBM patients, however, there are several causes, that make single immunotherapy treatments less successful.
Due to the intricately regulated immune system in rGBM, inhibiting the PD-1/PD-L1 pathway alone in rGBM is insufficient to activate sufficient effector T cells to destroy tumor cells in rGBM[180,181,182,183,184,185,186]. Additionally, adaptive resistance to anti-PD-1 therapy, such as the exhaustion of cytotoxic T cells brought on by coinhibitory molecules, results in unfavorable therapies[187,188]. In addition, despite the fact that anti-PD-1 therapy can kill certain tumor cells, many subclonal tumor cells are able to survive and grow continuously as a result of the complex and varied biological characteristics of rGBM[180,189].
CAR-T cell therapy has been developed as an effector for lymphocytes to increase immune response in GB because the blood-brain barrier makes it challenging for immune cells and medications to enter tumor tissues in rGBM[190]. CAR-TR cells, on the other hand, have limited infiltration and a brief lifetime, which results in a low cytotoxic impact on curing rGBM[191,192,193]. The heterogeneity of tumor cells is also blamed for contributing to the recurrence of rGBM[180,189].
Although numerous tumor vaccines have been proposed to treat rGBM, there are a number of obstacles that hinder vaccinations from working[194,195,196,197,198]. For instance, GB is characterized as lacking efficient treatment targets due to its poor immunogenicity and tumo[199]r mutational burden[200]. High rGBM heterogeneity and difficult activated cytotoxic cell transition through blood-brain barriers are also attributed to vaccine treatment failure[180,189].
As a result, combined immunotherapy is used as a treatment option for rGBM more successfully. In comparison to DC vaccination alone, it has been found that anti-PD-1/PD-L1 medication dramatically enhances the immunological response of rGBM patients following vaccination. The mechanism may be that the DC vaccine increases PD-1 expression, and that anti-PD-1 therapy administered after the DC vaccine increases its efficacy and promote tumor cell cytolysis[201]. Besides, EGFRvIII-specific CAR-T cell therapy was found to be beneficial in the treatment of rGBM patients when combined with anti-PD-1/PD-L1 therapy[202]. Also, there is the experimental proof that using anti-PD-1 and CD19 CAR-T cells together dramatically increased therapeutic success in refractory diffuse large B-cell lymphoma[203]. Based on these, when paired with anti-PD-1/PD-L1 therapy, CAR-T cell therapy targeting additional peptides, such as IL-13R2, EphA2, or HER2, may benefit to treating rGBM as well.
Besides PD-1, other marker genes downregulating T cell function on the surface of T lymphocytes may serve as inhibitory receptors, including CTLA-4, TIM-3, and LAG-3[204,205,206,207]. LAG-3, a T cell exhaustion marker that is abundantly expressed in GB tumors, is one example. Anti-LAG-3 antibodies may thus be used in combination with other immune checkpoint inhibitors to treat rGBM. A study has discovered that compared with the control group in mice, the mice in the anti-LAG-3 combined with anti-PD-1 therapy achieved a significant improvement regarding survival benefits[208]. Additionally, several clinical studies and experiments are being conducted to investigate the effectiveness of combination treatments that simultaneously target CTLA-4, LAG-3, TIM-3, and PD-1/PD-L1[209,210]. Costimulatory molecules are highly expressed in T lymphocytes, including 4-1BB and OX40, and they can also be utilized to combine anti-PD-1 antibodies to effectively treat rGBM [211,212,213,214,215].
There are also several studies investigating combination therapy to increase the positive effects of vaccinations, taking into account how chemotherapy and DC vaccines might complement one another. For instance, it has been shown that chemotherapy given after vaccination considerably increased the survival duration of rGBM when compared to chemotherapy or vaccine given alone[216].
Depending on whether the rGBM is positioned in a resectable anatomical site, surgical resection and reradiation are also effective treatment options for the condition[217]. More importantly, it has been discovered that radiation and surgical resection dramatically improve rGBM when used in conjunction with other treatments like anti-PD-1 therapy. There is evidence that treating rGBM with neoadjuvant anti-PD-1 therapy plus surgical resection, and subsequently, adjuvant anti-PD-1 therapy is effective strategy: Neoadjuvant anti-PD-1 therapy helps to stop the progression of the cell cycle and proliferation by triggering the IFN-γ response; resection was performed to reduce the tumor burden and maintain tumor-specific T cell function; adjuvant anti-PD-1 therapy helps to further kill any remaining tumor cells in the rGBM[218]. The combination of radiotherapy and anti-PD-1 therapy, known as neoadjuvant anti-PD-1 plus radiotherapy, followed by adjuvant anti-PD-1 therapy, has been shown to have a synergistic effect on tumors. This is due to radiotherapy can accelerate the clinical effect of anti-PD-1 on tumors via activating immunogenic cell death and TCR diversity with increased IFN-γ release[219,220].
2.6.5. Virus-based Combination Strategies
Though each treatment received positive responses in some patients, some still suffered AEs or died, attributed to tumor progression. Thus, in 2003, Isabelle M. Germano et al. combined Adenovirus, Herpes simplex-thymidine kinase, and ganciclovir[150]. At the time of recurrence, researchers performed volumetric resection of the tumor and injected ADV/HSV-tk complex in the tumor bed, then administered GCV (10 mg/kg/day) within 24 h after surgery for seven days. 11 patients were assigned to 3 sub-cohorts, who received 2.5×1011, 3.0×1011, and 9.0×1011 VP ADV/HSV-tk complex, respectively. 3 months later, 8/10 patients’ Karnofsky score was maintained ≥70 and 5/9 in 6 months. Ten of eleven patients survived longer than 52 weeks; the survival is 112.3 weeks. One was still alive 248 weeks after diagnosis. It indicated that the used doses of the complex were safe and that the whole treatment schedule was tolerable.
2.6.6. The current situation and prospect of combination strategies for GBM
Currently, the FDA has approved five drugs and one device to treat GBM: TMZ, lomustine, intravenous carmustine, carmustine wafer implants, BVZ, and TTFields. The radiotherapy and TMZ chemotherapy consider to be the standard of care for GBM. TTFields is the only treatment that has been shown to improve OS (20.5 vs. 15.6 months) and PFS6 (56% vs. 37%) in comparison to the current standard of care (SOC)[221], but has not been universally accepted yet as part of SOC. The bevacizumab is the only FDA approved drug for recurrent GBM. More researches should be conducted to find the real SOC for recurrent GBM.
There are still numerous issues to be resolved even though combination immunotherapy for rGBM has shown promising outcomes. To maximize treatment effectiveness, the best combination immunotherapy sequence should first be confirmed. For instance, it was discovered that administering an anti-PD-1 antibody after an agonist anti-OX40 antibody could increase its effectiveness in preventing tumor growth, but administering both antibodies at the same time could counteract the antitumor effects of an agonist anti-OX40 alone in the rGBM model[214,215]. Second, the timing of immunotherapy is crucial and should be confirmed when used in conjunction with other forms of treatment. Take LAG-3 as an example, as an early marker of T cell exhaustion in glioblastoma specimens, the mice in the combined anti-LAG-3 administered on the 10th day after tumor and anti-PD-1 therapy achieved an unideal survival benefit compared to the mice in the combined anti-LAG-3 administered on the 7th day after tumor and anti-PD-1 therapy, suggesting anti-LAG-3 are more effective in the early stage of the tumor when combined with other immunotherapy[208]. Third, compared to immunotherapy alone, there are many more combination tactics available, and validating each potential immunotherapy combination approach requires an inordinate amount of time and money. Therefore, massive parallel combination immunotherapy arrays or computational immunotherapy combination methods are needed urgently in the community to decrease costs significantly in discovering promising combination immunotherapy. Fourth, finding efficient therapeutic biomarkers is necessary to direct the development of efficient combination immunotherapy. Due to the great heterogeneity and low immunogenicity of rGBM, it will be advantageous to find reliable and attractive molecular targets for possible combination immunotherapy. With the development of technology, there are more and more good practices to identify and evaluate potential disease-associated therapeutic molecular targets and develop prediction methods to predict the efficacy of combination therapy in common diseases[222,223,224,225,226]. Last but not least, developing strategies to maximize CAR-T cell longevity, increase cell infiltration, and circumvent blood-brain barrier issues are effective directions to increase the survival of rGBM patients after receiving combination immunotherapy.
3. Conclusion and future perspective of immunotherapy to recurrent GBM
Immunotherapy has emerged as a promising strategy for the treatment of GBM, as it seeks to harness the power of the immune system to recognize and attack cancer cells. Several types of immunotherapies have been studied for GBM, including CAR-T, checkpoint inhibitors, cancer vaccines, and oncolytic viruses, and combination strategies. (Figure 1).
Checkpoint inhibitors target proteins that regulate the immune system, such as PD-1 and CTLA-4, and can enhance the ability of T cells to attack cancer cells. Cancer vaccines can prime the immune system to recognize and attack cancer cells, and even can be developed using a patient's own tumor cells to generate a personalized vaccine. CAR-T therapy can specifically target cancer cells by isolating and multiplying T cells, while oncolytic viruses infect and destroy cancer cells.
To improve the efficacy of immunotherapy for GBM, several approaches are being explored. As the future direction of immunotherapy for recurrent GBM, combination therapies will likely involve a combination of different approaches, which aim to target multiple pathways involved in cancer growth and immune evasion, have shown promise. Despite the promise of immunotherapy for GBM, clinical trials have had mixed results. Some studies have shown modest improvements in survival and quality of life, while others have not shown significant benefits over traditional therapies. The heterogeneity of GBM, as well as the immunosuppressive tumor microenvironment, may play a role in the variable response to immunotherapy.
While there is still much to learn about the optimal use of immunotherapy for recurrent GBM, the field holds great promise for improving outcomes and quality of life for patients with this devastating disease. Continued research is needed to address the challenges and identify the most effective combination of immunotherapy approaches, as well as to develop new biomarkers and delivery methods to improve outcomes for patients with recurrent GBM.
Author Contributions
X.L., W.D., Z.Z., D.C., X.P., K.L., Q.S., L.F and Y.D. were involved in the drafting of the manuscript. X.L. and Z.Z. were involved in the editing of the manuscript. W.D. helped with designing figures for the manuscript. S.W. was involved in the drafting and editing of the manuscript. X.L. was involved in planning, detailing the content, and editing the manuscript. All authors have read and agreed to the published version of the manuscript.Z.Z., W.D. contributed equally and could be considered co-first authors.
Conflicts of Interest
All authors have no conflict of interest to declare.
References
- Board, W.C.o.T.E. Central Nervous System Tumours.
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015-2019. Neuro-Oncology 2022, 24, v1-v95. [Google Scholar] [CrossRef] [PubMed]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacology & Therapeutics 2015, 152, 63-82. [Google Scholar] [CrossRef]
- R, S.; Wp, M.; Mj, v.d.B.; M, W.; B, F.; Mj, T.; K, B.; Aa, B.; C, M.; U, B.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine 2005, 352. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A. Neuro-oncology in 2015: Progress in glioma diagnosis, classification and treatment. Nature Reviews. Neurology 2016, 12, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. The New England Journal of Medicine 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- van Linde, M.E.; Brahm, C.G.; de Witt Hamer, P.C.; Reijneveld, J.C.; Bruynzeel, A.M.E.; Vandertop, W.P.; van de Ven, P.M.; Wagemakers, M.; van der Weide, H.L.; Enting, R.H.; et al. Treatment outcome of patients with recurrent glioblastoma multiforme: a retrospective multicenter analysis. Journal of Neuro-Oncology 2017, 135, 183-192. [Google Scholar] [CrossRef]
- Weller, M.; Le Rhun, E.; Preusser, M.; Tonn, J.-C.; Roth, P. How we treat glioblastoma. ESMO Open 2019, 4, e000520. [Google Scholar] [CrossRef]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Bevacizumab (Avastin®) as Treatment of Recurrent Glioblastoma Multiforme. The Oncologist 2012, 17, 1482. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. New England Journal of Medicine 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. New England Journal of Medicine 2014, 370, 709–722. [Google Scholar] [CrossRef]
- EORTC 26101 phase III trial exploring the combination of bevacizumab and lomustine in patients with first progression of a glioblastoma. Journal of Clinical Oncology.
- Mooney, J.; Bernstock, J.D.; Ilyas, A.; Ibrahim, A.; Yamashita, D.; Markert, J.M.; Nakano, I. Current Approaches and Challenges in the Molecular Therapeutic Targeting of Glioblastoma. World Neurosurgery 2019, 129, 90–100. [Google Scholar] [CrossRef]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma--are we there yet? Neuro-Oncology 2013, 15, 4-27. [Google Scholar] [CrossRef]
- Sampson, J.H.; Singh Achrol, A.; Aghi, M.K.; Bankiewiecz, K.; Bexon, M.; Brem, S.; Brenner, A.; Chandhasin, C.; Chowdhary, S.; Coello, M.; et al. Targeting the IL4 Receptor with MDNA55 in Patients with Recurrent Glioblastoma: Results of a Phase 2b Trial. Neuro-Oncology 2023, noac285. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, G.; Amiji, M.M. Cancer stem cell-targeted therapeutics and delivery strategies. Expert Opinion on Drug Delivery 2017, 14, 997–1008. [Google Scholar] [CrossRef]
- Magrath, J.W.; Kim, Y. Salinomycin's potential to eliminate glioblastoma stem cells and treat glioblastoma multiforme (Review). International Journal of Oncology 2017, 51, 753–759. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Molecular Cancer 2006, 5, 67. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bar-Lev, L.; Sharife, H.; Grunewald, M.; Mogilevsky, M.; Licht, T.; Goveia, J.; Taverna, F.; Paldor, I.; Carmeliet, P.; et al. Identification of vascular cues contributing to cancer cell stemness and function. Angiogenesis 2022, 25, 355–371. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Research 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Bischof, J.; Westhoff, M.-A.; Wagner, J.E.; Halatsch, M.-E.; Trentmann, S.; Knippschild, U.; Wirtz, C.R.; Burster, T. Cancer stem cells: The potential role of autophagy, proteolysis, and cathepsins in glioblastoma stem cells. Tumour Biology: The Journal of the International Society for Oncodevelopmental Biology and Medicine 2017, 39, 1010428317692227. [Google Scholar] [CrossRef] [PubMed]
- Campos, B.; Gal, Z.; Baader, A.; Schneider, T.; Sliwinski, C.; Gassel, K.; Bageritz, J.; Grabe, N.; von Deimling, A.; Beckhove, P.; et al. Aberrant self-renewal and quiescence contribute to the aggressiveness of glioblastoma. The Journal of Pathology 2014, 234, 23–33. [Google Scholar] [CrossRef]
- Jackson, C.M.; Lim, M.; Drake, C.G. Immunotherapy for brain cancer: recent progress and future promise. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 2014, 20, 3651-3659. [Google Scholar] [CrossRef] [PubMed]
- Biollaz, G.; Bernasconi, L.; Cretton, C.; Püntener, U.; Frei, K.; Fontana, A.; Suter, T. Site-specific anti-tumor immunity: differences in DC function, TGF-beta production and numbers of intratumoral Foxp3+ Treg. European Journal of Immunology 2009, 39, 1323–1333. [Google Scholar] [CrossRef]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro-Oncology 2015, 7 Suppl 7, vii9-vii14. [Google Scholar] [CrossRef]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-Cell Atlas Reveals Complexity of the Immunosuppressive Microenvironment of Initial and Recurrent Glioblastoma. Frontiers in Immunology 2020, 11, 835. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Xing, W.-k.; Shao, C.; Qi, Z.-y.; Yang, C.; Wang, Z. The role of Gliadel wafers in the treatment of newly diagnosed GBM: a meta-analysis. Drug Design, Development and Therapy 2015, 9, 3341-3348. [Google Scholar] [CrossRef]
- McNutt, M. Cancer immunotherapy. Science (New York, N.Y. Science (New York, N.Y.) 2013, 342, 1417. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Topalian, S.L.; Weiner, G.J.; Pardoll, D.M. Cancer immunotherapy comes of age. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 2011, 29, 4828-4836. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Seyfrid, M.; Maich, W.T.; Shaikh, M.V.; Tatari, N.; Upreti, D.; Piyasena, D.; Subapanditha, M.; Savage, N.; McKenna, D.; Mikolajewicz, N.; et al. CD70 as an actionable immunotherapeutic target in recurrent glioblastoma and its microenvironment. Journal for ImmunoTherapy of Cancer 2022, 10, e003289. [Google Scholar] [CrossRef] [PubMed]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832-844. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Brennan, C.W.; DeAngelis, L.M.; Omuro, A.M. Emerging therapies for glioblastoma. JAMA neurology 2014, 71, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.-T.; Song, H.; et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Human Gene Therapy 2012, 23, 1043–1053. [Google Scholar] [CrossRef]
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: current state and future directions. Journal of Experimental & Clinical Cancer Research 2022, 41, 142. [Google Scholar] [CrossRef]
- News, N. Searching for a Cure for Deadly Brain Tumors. Neuroscience News 2022. [Google Scholar]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat Biomed Eng 2018, 2, 377–391. [Google Scholar] [CrossRef]
- Galea, I.; Bechmann, I.; Perry, V.H. What is immune privilege (not)? Trends Immunol 2007, 28, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Salinas, S.; Schiavo, G.; Kremer, E.J. A hitchhiker's guide to the nervous system: the complex journey of viruses and toxins. Nat Rev Microbiol 2010, 8, 645–655. [Google Scholar] [CrossRef]
- Arima, Y.; Harada, M.; Kamimura, D.; Park, J.H.; Kawano, F.; Yull, F.E.; Kawamoto, T.; Iwakura, Y.; Betz, U.A.; Marquez, G.; et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell 2012, 148, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front Neurosci 2021, 15, 662064. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin Cancer Res 2018, 24, 3792–3802. [Google Scholar] [CrossRef]
- Kahlon, K.S.; Brown, C.; Cooper, L.J.; Raubitschek, A.; Forman, S.J.; Jensen, M.C. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res 2004, 64, 9160–9166. [Google Scholar] [CrossRef]
- Brown, C.E.; Warden, C.D.; Starr, R.; Deng, X.; Badie, B.; Yuan, Y.C.; Forman, S.J.; Barish, M.E. Glioma IL13Ralpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS One 2013, 8, e77769. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin Cancer Res 2015, 21, 4062–4072. [Google Scholar] [CrossRef]
- Brown, C.E.; Rodriguez, A.; Palmer, J.; Ostberg, J.R.; Naranjo, A.; Wagner, J.R.; Aguilar, B.; Starr, R.; Weng, L.; Synold, T.W.; et al. Off-the-shelf, steroid-resistant, IL13Ralpha2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol 2022, 24, 1318–1330. [Google Scholar] [CrossRef]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J 2013, 280, 5350–5370. [Google Scholar] [CrossRef]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A 1992, 89, 4309–4313. [Google Scholar] [CrossRef]
- Bullain, S.S.; Sahin, A.; Szentirmai, O.; Sanchez, C.; Lin, N.; Baratta, E.; Waterman, P.; Weissleder, R.; Mulligan, R.C.; Carter, B.S. Genetically engineered T cells to target EGFRvIII expressing glioblastoma. J Neurooncol 2009, 94, 373–382. [Google Scholar] [CrossRef]
- O'Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017, 9. [Google Scholar] [CrossRef]
- Durgin, J.S.; Henderson, F., Jr.; Nasrallah, M.P.; Mohan, S.; Wang, S.; Lacey, S.F.; Melenhorst, J.J.; Desai, A.S.; Lee, J.Y.K.; Maus, M.V.; et al. Case Report: Prolonged Survival Following EGFRvIII CAR T Cell Treatment for Recurrent Glioblastoma. Front Oncol 2021, 11, 669071. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res 2010, 16, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832-844 e836. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J Immunother 2019, 42, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Ralpha2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol Res 2017, 5, 571–581. [Google Scholar] [CrossRef]
- Lechpammer, M.; Rao, R.; Shah, S.; Mirheydari, M.; Bhattacharya, D.; Koehler, A.; Toukam, D.K.; Haworth, K.J.; Pomeranz Krummel, D.; Sengupta, S. Advances in Immunotherapy for the Treatment of Adult Glioblastoma: Overcoming Chemical and Physical Barriers. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol Ther 2018, 26, 31–44. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol 2019, 37, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Wang, R.; Li, W.; Zhang, Z.; Chang, Y.; Hu, Y.; Zhao, J.; Zheng, X.; Mao, Q.; Xia, H. A novel TanCAR targeting IL13Ralpha2 and EphA2 for enhanced glioblastoma therapy. Mol Ther Oncolytics 2022, 24, 729–741. [Google Scholar] [CrossRef]
- Lugade, A.A.; Sorensen, E.W.; Gerber, S.A.; Moran, J.P.; Frelinger, J.G.; Lord, E.M. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol 2008, 180, 3132–3139. [Google Scholar] [CrossRef]
- Portella, L.; Scala, S. Ionizing radiation effects on the tumor microenvironment. Semin Oncol 2019, 46, 254–260. [Google Scholar] [CrossRef]
- Jin, L.; Ge, H.; Long, Y.; Yang, C.; Chang, Y.E.; Mu, L.; Sayour, E.J.; De Leon, G.; Wang, Q.J.; Yang, J.C.; et al. CD70, a novel target of CAR T-cell therapy for gliomas. Neuro Oncol 2018, 20, 55–65. [Google Scholar] [CrossRef]
- DeSelm, C.; Palomba, M.L.; Yahalom, J.; Hamieh, M.; Eyquem, J.; Rajasekhar, V.K.; Sadelain, M. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol Ther 2018, 26, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zeng, G. Cancer and innate immune system interactions: translational potentials for cancer immunotherapy. J Immunother 2012, 35, 299–308. [Google Scholar] [CrossRef]
- Fong, B.; Jin, R.; Wang, X.; Safaee, M.; Lisiero, D.N.; Yang, I.; Li, G.; Liau, L.M.; Prins, R.M. Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PLoS One 2012, 7, e32614. [Google Scholar] [CrossRef]
- Liu, F.; Huang, J.; Liu, X.; Cheng, Q.; Luo, C.; Liu, Z. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int 2020, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Ochiai, H.; Mitchell, D.A.; Grossi, P.M.; Sweeney, A.E.; Archer, G.E.; Cummings, T.; Allison, J.P.; Bigner, D.D.; Sampson, J.H. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res 2007, 13, 2158–2167. [Google Scholar] [CrossRef] [PubMed]
- Galstyan, A.; Markman, J.L.; Shatalova, E.S.; Chiechi, A.; Korman, A.J.; Patil, R.; Klymyshyn, D.; Tourtellotte, W.G.; Israel, L.L.; Braubach, O.; et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat Commun 2019, 10, 3850. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol Res 2016, 4, 124–135. [Google Scholar] [CrossRef]
- Xu, S.; Tang, L.; Li, X.; Fan, F.; Liu, Z. Immunotherapy for glioma: Current management and future application. Cancer Lett 2020, 476, 1–12. [Google Scholar] [CrossRef]
- Ghouzlani, A.; Kandoussi, S.; Tall, M.; Reddy, K.P.; Rafii, S.; Badou, A. Immune Checkpoint Inhibitors in Human Glioma Microenvironment. Front Immunol 2021, 12, 679425. [Google Scholar] [CrossRef]
- O'Malley, D.M.; Randall, L.M.; Jackson, C.G.; Coleman, R.L.; Hays, J.L.; Moore, K.N.; Naumann, R.W.; Rocconi, R.P.; Slomovitz, B.M.; Tewari, K.S.; et al. RaPiDS (GOG-3028): randomized Phase II study of balstilimab alone or in combination with zalifrelimab in cervical cancer. Future Oncol 2021, 17, 3433–3443. [Google Scholar] [CrossRef]
- Perets, R.; Bar, J.; Rasco, D.W.; Ahn, M.J.; Yoh, K.; Kim, D.W.; Nagrial, A.; Satouchi, M.; Lee, D.H.; Spigel, D.R.; et al. Safety and efficacy of quavonlimab, a novel anti-CTLA-4 antibody (MK-1308), in combination with pembrolizumab in first-line advanced non-small-cell lung cancer. Ann Oncol 2021, 32, 395–403. [Google Scholar] [CrossRef]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol 2018, 20, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Carter, T.; Shaw, H.; Cohn-Brown, D.; Chester, K.; Mulholland, P. Ipilimumab and Bevacizumab in Glioblastoma. Clin Oncol (R Coll Radiol) 2016, 28, 622-626. [Google Scholar] [CrossRef]
- Youssef, G.; Dietrich, J. Ipilimumab: an investigational immunotherapy for glioblastoma. Expert Opin Investig Drugs 2020, 29, 1187–1193. [Google Scholar] [CrossRef]
- Fathi, M.; Razavi, S.M.; Sojoodi, M.; Ahmadi, A.; Ebrahimi, F.; Namdar, A.; Hojjat-Farsangi, M.; Gholamin, S.; Jadidi-Niaragh, F. Targeting the CTLA-4/B7 axes in glioblastoma: preclinical evidence and clinical interventions. Expert Opin Ther Targets 2022, 26, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wohrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol 2015, 17, 1064–1075. [Google Scholar] [CrossRef]
- Awada, G.; Ben Salama, L.; De Cremer, J.; Schwarze, J.K.; Fischbuch, L.; Seynaeve, L.; Du Four, S.; Vanbinst, A.M.; Michotte, A.; Everaert, H.; et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: a stratified, open-label, single-center phase 2 clinical trial (GliAvAx). J Immunother Cancer 2020, 8. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; de Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin Cancer Res 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124-128. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Blake, S.J.; Yong, M.C.; Harjunpaa, H.; Ngiow, S.F.; Takeda, K.; Young, A.; O'Donnell, J.S.; Allen, S.; Smyth, M.J.; et al. Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov 2016, 6, 1382–1399. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inoges, S.; de Andrea, C.; Lopez-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Author Correction: Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 2019, 25, 1022. [Google Scholar] [CrossRef]
- Arrieta, V.A.; Chen, A.X.; Kane, J.R.; Kang, S.J.; Kassab, C.; Dmello, C.; Zhao, J.; Burdett, K.B.; Upadhyayula, P.S.; Lee-Chang, C.; et al. ERK1/2 phosphorylation predicts survival following anti-PD-1 immunotherapy in recurrent glioblastoma. Nat Cancer 2021, 2, 1372–1386. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Andrews, L.P.; Workman, C.J.; Vignali, D.A.A. Intratumoral regulatory T cells: markers, subsets and their impact on anti-tumor immunity. Immunology 2019, 157, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Postow, M.A.; Adamow, M.; Arora, A.; Hannum, M.; Maher, C.; Wong, P.; Curran, M.A.; Hollmann, T.J.; Jia, L.; et al. LAG-3 expression on peripheral blood cells identifies patients with poorer outcomes after immune checkpoint blockade. Sci Transl Med 2021, 13. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Melero, I.; Bhatia, S.; Bono, P.; Sanborn, R.E.; Lipson, E.J.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S.; et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti-LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti-PD-1/PD-L1 therapy. Journal of Clinical Oncology 2017, 35. [Google Scholar] [CrossRef]
- Kraman, M.; Faroudi, M.; Allen, N.L.; Kmiecik, K.; Gliddon, D.; Seal, C.; Koers, A.; Wydro, M.M.; Batey, S.; Winnewisser, J.; et al. FS118, a Bispecific Antibody Targeting LAG-3 and PD-L1, Enhances T-Cell Activation Resulting in Potent Antitumor Activity. Clin Cancer Res 2020, 26, 3333–3344. [Google Scholar] [CrossRef]
- Anderson, A.C. Tim-3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol Res 2014, 2, 393–398. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, X.; Yu, Z.; Zhou, Y.; Zhu, S.; Zhang, Y.H.; Lin, X.P.; Mou, Y.; Zhang, J. Correlation of Tim-3 expression with chemokine levels for predicting the prognosis of patients with glioblastoma. J Neuroimmunol 2021, 355, 577575. [Google Scholar] [CrossRef]
- Mohsenzadegan, M.; Nowroozi, M.R.; Fotovvat, A.; Bavandpour Baghshahi, P.; Bokaie, S.; Inanloo, S.H.; Sharifi, L. The prospect of targeting T cell immunoglobulin and mucin-domain containing-3 in renal cell carcinoma immunotherapy. Scand J Immunol 2022, 96, e13197. [Google Scholar] [CrossRef]
- Mohsenzadegan, M.; Bavandpour, P.; Nowroozi, M.R.; Amini, E.; Kourosh-Arami, M.; Momeni, S.A.; Bokaie, S.; Sharifi, L. The Potential of T Cell Immunoglobulin and Mucin-Domain Containing-3 (Tim-3) in Designing Novel Immunotherapy for Bladder Cancer. Endocr Metab Immune Disord Drug Targets 2021, 21, 2131–2146. [Google Scholar] [CrossRef]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef]
- Stone, E.L.; Pepper, M.; Katayama, C.D.; Kerdiles, Y.M.; Lai, C.Y.; Emslie, E.; Lin, Y.C.; Yang, E.; Goldrath, A.W.; Li, M.O.; et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity 2015, 42, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Wikenheiser, D.J.; Stumhofer, J.S. ICOS Co-Stimulation: Friend or Foe? Front Immunol 2016, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.; Zurgil, N.; Langevitz, P.; Ehrenfeld, M.; Deutsch, M. The immunosuppressive effect of methotrexate in active rheumatoid arthritis patients vs. its stimulatory effect in nonactive patients, as indicated by cytometric measurements of CD4+T cell subpopulations. Immunological Investigations 2004, 33, 351-362. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shi, F.; Shan, A. Transcriptome profile and clinical characterization of ICOS expression in gliomas. Front Oncol 2022, 12, 946967. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, W.; Fan, W.; Ren, C.; Xu, J.; Zeng, F.; Bao, Z.; Jiang, T.; Zhao, Z. Comprehensive transcriptomic characterization reveals core genes and module associated with immunological changes via 1619 samples of brain glioma. Cell Death Dis 2021, 12, 1140. [Google Scholar] [CrossRef]
- Le, K.S.; Thibult, M.L.; Just-Landi, S.; Pastor, S.; Gondois-Rey, F.; Granjeaud, S.; Broussais, F.; Bouabdallah, R.; Colisson, R.; Caux, C.; et al. Follicular B Lymphomas Generate Regulatory T Cells via the ICOS/ICOSL Pathway and Are Susceptible to Treatment by Anti-ICOS/ICOSL Therapy. Cancer Res 2016, 76, 4648–4660. [Google Scholar] [CrossRef]
- Mo, L.J.; Chen, Q.M.; Zhang, X.J.; Shi, X.J.; Wei, L.L.; Zheng, D.P.; Li, H.W.; Gao, J.M.; Li, J.L.; Hu, Z.M. Depletion of regulatory T cells by anti-ICOS antibody enhances anti-tumor immunity of tumor cell vaccine in prostate cancer. Vaccine 2017, 35, 5932–5938. [Google Scholar] [CrossRef]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Kostine, M.; Mauric, E.; Tison, A.; Barnetche, T.; Barre, A.; Nikolski, M.; Rouxel, L.; Dutriaux, C.; Dousset, L.; Prey, S.; et al. Baseline co-medications may alter the anti-tumoural effect of checkpoint inhibitors as well as the risk of immune-related adverse events. Eur J Cancer 2021, 157, 474–484. [Google Scholar] [CrossRef]
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: a phase I clinical trial. J Immunother Cancer 2021, 9. [Google Scholar] [CrossRef]
- Villarroel-Espindola, F.; Yu, X.; Datar, I.; Mani, N.; Sanmamed, M.; Velcheti, V.; Syrigos, K.; Toki, M.; Zhao, H.; Chen, L.; et al. Spatially Resolved and Quantitative Analysis of VISTA/PD-1H as a Novel Immunotherapy Target in Human Non-Small Cell Lung Cancer. Clin Cancer Res 2018, 24, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, J.; Liu, L.N.; Flies, D.B.; Nie, X.; Toki, M.; Zhang, J.; Song, C.; Zarr, M.; Zhou, X.; et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat Med 2019, 25, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Ren, X.; Galbo, P.M., Jr.; Moerdler, S.; Wang, H.; Sica, R.A.; Etemad-Gilbertson, B.; Shi, L.; Zhu, L.; Tang, X.; et al. KIR3DL3-HHLA2 is a human immunosuppressive pathway and a therapeutic target. Sci Immunol 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat Rev Immunol 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Barber, G.N. Cytoplasmic DNA innate immune pathways. Immunol Rev 2011, 243, 99–108. [Google Scholar] [CrossRef]
- Hengel, H.; Koszinowski, U.H.; Conzelmann, K.K. Viruses know it all: new insights into IFN networks. Trends Immunol 2005, 26, 396–401. [Google Scholar] [CrossRef]
- O'Neill, L.A.; Bowie, A.G. Sensing and signaling in antiviral innate immunity. Curr Biol 2010, 20, R328–333. [Google Scholar] [CrossRef]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Rampling, R.; Cruickshank, G.; Papanastassiou, V.; Nicoll, J.; Hadley, D.; Brennan, D.; Petty, R.; MacLean, A.; Harland, J.; McKie, E.; et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34. 5 null mutant 1716) in patients with recurrent malignant glioma. Gene therapy 2000, 7, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Markert, J.M.; Liechty, P.G.; Wang, W.; Gaston, S.; Braz, E.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Lakeman, A.D.; Palmer, C.A.; et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Molecular therapy : the journal of the American Society of Gene Therapy 2009, 17, 199-207. [Google Scholar] [CrossRef]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Molecular therapy : the journal of the American Society of Gene Therapy 2014, 22, 1048-1055. [Google Scholar] [CrossRef] [PubMed]
- Whisenhunt, T.R., Jr.; Rajneesh, K.F.; Hackney, J.R.; Markert, J.M. Extended disease-free interval of 6 years in a recurrent glioblastoma multiforme patient treated with G207 oncolytic viral therapy. Oncolytic virotherapy 2015, 4, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.E.; Cassady, K.A.; Roth, J.C.; Clements, J.; Schieffer, K.M.; Leraas, K.; Miller, A.R.; Prasad, N.; Leavenworth, J.W.; Aban, I.B.; et al. Immune Activity and Response Differences of Oncolytic Viral Therapy in Recurrent Glioblastoma: Gene Expression Analyses of a Phase IB Study. Clinical cancer research : an official journal of the American Association for Cancer Research 2022, 28, 498-506. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: a phase 2 trial. Nature medicine 2022, 28, 1630–1639. [Google Scholar] [CrossRef]
- Markert, J.M.; Cody, J.J.; Parker, J.N.; Coleman, J.M.; Price, K.H.; Kern, E.R.; Quenelle, D.C.; Lakeman, A.D.; Schoeb, T.R.; Palmer, C.A.; et al. Preclinical evaluation of a genetically engineered herpes simplex virus expressing interleukin-12. Journal of virology 2012, 86, 5304–5313. [Google Scholar] [CrossRef]
- Klatzmann, D.; Valery, C.A.; Bensimon, G.; Marro, B.; Boyer, O.; Mokhtari, K.; Diquet, B.; Salzmann, J.L.; Philippon, J. A phase I/II study of herpes simplex virus type 1 thymidine kinase "suicide" gene therapy for recurrent glioblastoma. Study Group on Gene Therapy for Glioblastoma. Human gene therapy 1998, 9, 2595–2604. [Google Scholar] [CrossRef]
- Shand, N.; Weber, F.; Mariani, L.; Bernstein, M.; Gianella-Borradori, A.; Long, Z.; Sorensen, A.G.; Barbier, N. A phase 1-2 clinical trial of gene therapy for recurrent glioblastoma multiforme by tumor transduction with the herpes simplex thymidine kinase gene followed by ganciclovir. GLI328 European-Canadian Study Group. Human gene therapy 1999, 10, 2325-2335. [Google Scholar] [CrossRef] [PubMed]
- Immonen, A.; Vapalahti, M.; Tyynela, K.; Hurskainen, H.; Sandmair, A.; Vanninen, R.; Langford, G.; Murray, N.; Yla-Herttuala, S. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Molecular therapy : the journal of the American Society of Gene Therapy 2004, 10, 967-972. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018, 36, 1419-1427. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Gumin, J.; Gao, F.; Hossain, A.; Shpall, E.J.; Kondo, A.; Parker Kerrigan, B.C.; Yang, J.; Ledbetter, D.; Fueyo, J.; et al. Characterization of patient-derived bone marrow human mesenchymal stem cells as oncolytic virus carriers for the treatment of glioblastoma. Journal of neurosurgery 2022, 136, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, P.; Roldan, G.; George, D.; Wallace, C.; Palmer, C.A.; Morris, D.; Cairncross, G.; Matthews, M.V.; Markert, J.; Gillespie, Y.; et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Molecular therapy : the journal of the American Society of Gene Therapy 2008, 16, 627-632. [Google Scholar] [CrossRef] [PubMed]
- Kicielinski, K.P.; Chiocca, E.A.; Yu, J.S.; Gill, G.M.; Coffey, M.; Markert, J.M. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Molecular therapy : the journal of the American Society of Gene Therapy 2014, 22, 1056-1062. [Google Scholar] [CrossRef]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Molecular therapy : the journal of the American Society of Gene Therapy 2006, 13, 221-228. [Google Scholar] [CrossRef]
- Todo, T.; Rabkin, S.D.; Martuza, R.L. Evaluation of ganciclovir-mediated enhancement of the antitumoral effect in oncolytic, multimutated herpes simplex virus type 1 (G207) therapy of brain tumors. Cancer gene therapy 2000, 7, 939–946. [Google Scholar] [CrossRef]
- Germano, I.M.; Fable, J.; Gultekin, S.H.; Silvers, A. Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex: preliminary results of a phase I trial in patients with recurrent malignant gliomas. Journal of neuro-oncology 2003, 65, 279-289. [Google Scholar] [CrossRef]
- Wollmann, G.; Ozduman, K.; van den Pol, A.N. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J 2012, 18, 69–81. [Google Scholar] [CrossRef]
- Parker, J.N.; Bauer, D.F.; Cody, J.J.; Markert, J.M. Oncolytic viral therapy of malignant glioma. Neurotherapeutics 2009, 6, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum Gene Ther Clin Dev 2016, 27, 69–78. [Google Scholar] [CrossRef] [PubMed]
- McKie, E.A.; MacLean, A.R.; Lewis, A.D.; Cruickshank, G.; Rampling, R.; Barnett, S.C.; Kennedy, P.G.; Brown, S.M. Selective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours--evaluation of a potentially effective clinical therapy. Br J Cancer 1996, 74, 745-752. [Google Scholar] [CrossRef] [PubMed]
- Hunter, W.D.; Martuza, R.L.; Feigenbaum, F.; Todo, T.; Mineta, T.; Yazaki, T.; Toda, M.; Newsome, J.T.; Platenberg, R.C.; Manz, H.J.; et al. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates. Journal of virology 1999, 73, 6319–6326. [Google Scholar] [CrossRef]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nature medicine 1995, 1, 938–943. [Google Scholar] [CrossRef]
- Sundaresan, P.; Hunter, W.D.; Martuza, R.L.; Rabkin, S.D. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation in mice. Journal of virology 2000, 74, 3832–3841. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Tufaro, F.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.; Prabhu, S.; Fuller, G.; Aldape, K.; et al. First-in-Human Phase I Clinical Trial of Oncolytic Delta-24-Rgd (Dnx-2401) with Biological Endpoints: Implications for Viro- Immunotherapy. Neuro Oncol. 2014, 16 (Suppl 3), iii39. [Google Scholar] [CrossRef]
- Klatzmann, D.; Valéry, C.A.; Bensimon, G.; Marro, B.; Boyer, O.; Mokhtari, K.; Diquet, B.; Salzmann, J.L.; Philippon, J. A phase I/II study of herpes simplex virus type 1 thymidine kinase "suicide" gene therapy for recurrent glioblastoma. Study Group on Gene Therapy for Glioblastoma. Human gene therapy 1998, 9, 2595–2604. [Google Scholar] [CrossRef]
- Smitt, P.S.; Driesse, M.; Wolbers, J.; Kros, M.; Avezaat, C. Treatment of relapsed malignant glioma with an adenoviral vector containing the herpes simplex thymidine kinase gene followed by ganciclovir. Molecular therapy : the journal of the American Society of Gene Therapy 2003, 7, 851-858. [Google Scholar] [CrossRef]
- Immonen, A.; Vapalahti, M.; Tyynelä, K.; Hurskainen, H.; Sandmair, A.; Vanninen, R.; Langford, G.; Murray, N.; Ylä-Herttuala, S. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Molecular therapy : the journal of the American Society of Gene Therapy 2004, 10, 967-972. [Google Scholar] [CrossRef]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.X.; Levin, V.A.; Yung, W.K.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef]
- Whyte, P.; Williamson, N.M.; Harlow, E. Cellular targets for transformation by the adenovirus E1A proteins. Cell 1989, 56, 67–75. [Google Scholar] [CrossRef]
- Hochberg, F.H.; Linggood, R.; Wolfson, L.; Baker, W.H.; Kornblith, P. Quality and Duration of Survival in Glioblastoma Multiforme: Combined Surgical, Radiation, and Lomustine Therapy. JAMA 1979, 241, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New England Journal of Medicine 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.; Laperriere, N.; O'Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. New England Journal of Medicine 2017, 376, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Malmström, A.; Grønberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. The Lancet Oncology 2012, 13, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Gzell, C.; Wheeler, H.; Guo, L.; Kastelan, M.; Back, M. Elderly patients aged 65–75 years with glioblastoma multiforme may benefit from long course radiation therapy with temozolomide. Journal of Neuro-Oncology 2014, 119, 187-196. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Godard, S.; Dietrich, P.-Y.; Regli, L.; Ostermann, S.; Otten, P.; Van Melle, G.; de Tribolet, N.; Stupp, R. Clinical Trial Substantiates the Predictive Value of O-6-Methylguanine-DNA Methyltransferase Promoter Methylation in Glioblastoma Patients Treated with Temozolomide. Clinical Cancer Research 2004, 10, 1871–1874. [Google Scholar] [CrossRef]
- Mir, T.; Pond, G.; Greenspoon, J.N. Outcomes in Elderly Patients with Glioblastoma Multiforme Treated with Short-Course Radiation Alone Compared to Short-Course Radiation and Concurrent and Adjuvant Temozolomide Based on Performance Status and Extent of Resection. Current Oncology 2021, 28, 2399–2408. [Google Scholar] [CrossRef]
- Stupp, R.; Dietrich, P.-Y.; Kraljevic, S.O.; Pica, A.; Maillard, I.; Maeder, P.; Meuli, R.; Janzer, R.; Pizzolato, G.; Miralbell, R.; et al. Promising Survival for Patients With Newly Diagnosed Glioblastoma Multiforme Treated With Concomitant Radiation Plus Temozolomide Followed by Adjuvant Temozolomide. Journal of Clinical Oncology 2002, 20, 1375–1382. [Google Scholar] [CrossRef]
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O6-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Frontiers in Oncology 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.M.; Umemura, Y.; Leung, D. Bevacizumab and Glioblastoma: Past, Present, and Future Directions. The Cancer Journal 2018, 24. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Roth, P.; Rushing, E.J.; Weller, J.; Andratschke, N.; Hofer, S.; Korol, D.; Regli, L.; Pangalu, A.; Pless, M.; et al. Bevacizumab may improve quality of life, but not overall survival in glioblastoma: an epidemiological study. Annals of Oncology 2018, 29, 1431–1436. [Google Scholar] [CrossRef]
- Swanson, K.D.; Lok, E.; Wong, E.T. An Overview of Alternating Electric Fields Therapy (NovoTTF Therapy) for the Treatment of Malignant Glioma. Current Neurology and Neuroscience Reports 2016, 16, 8. [Google Scholar] [CrossRef]
- Bokstein, F.; Blumenthal, D.; Limon, D.; Harosh, C.B.; Ram, Z.; Grossman, R. Concurrent Tumor Treating Fields (TTFields) and Radiation Therapy for Newly Diagnosed Glioblastoma: A Prospective Safety and Feasibility Study. Frontiers in Oncology 2020, 10. [Google Scholar] [CrossRef]
- Lazaridis, L.; Schäfer, N.; Teuber-Hanselmann, S.; Blau, T.; Schmidt, T.; Oster, C.; Weller, J.; Tzaridis, T.; Pierscianek, D.; Keyvani, K.; et al. Tumour Treating Fields (TTFields) in combination with lomustine and temozolomide in patients with newly diagnosed glioblastoma. Journal of Cancer Research and Clinical Oncology 2020, 146, 787–792. [Google Scholar] [CrossRef]
- Clark, P.A.; Gaal, J.T.; Strebe, J.K.; Pasch, C.A.; Deming, D.A.; Kuo, J.S.; Robins, H.I. The effects of tumor treating fields and temozolomide in MGMT expressing and non-expressing patient-derived glioblastoma cells. J Clin Neurosci 2017, 36, 120–124. [Google Scholar] [CrossRef]
- Stachelek, G.C.; Grimm, J.; Moore, J.; Huang, E.; Spoleti, N.; Redmond, K.J.; Lim, M.; Bettegowda, C.; Kleinberg, L. Tumor-Treating Field Arrays Do Not Reduce Target Volume Coverage for Glioblastoma Radiation Therapy. Adv Radiat Oncol 2020, 5, 62–69. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. International Journal of Radiation Oncology* Biology* Physics 2013, 86, 343-349. [Google Scholar] [CrossRef]
- Huang, B.Y.; Zhan, Y.P.; Zong, W.J.; Yu, C.J.; Li, J.F.; Qu, Y.M.; Han, S. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PloS one 2015, 10, e0134715. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer cell 2017, 32, 253-267. e255. [Google Scholar] [CrossRef]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget 2017, 8, 91779. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nature medicine 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. Journal of Experimental Medicine 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nature communications 2016, 7, 10501. [Google Scholar] [CrossRef]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in GlioblastomaT-Cell Exhaustion Signatures in Glioblastoma. Clinical Cancer Research 2018, 24, 4175–4186. [Google Scholar] [CrossRef]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges . Neuro-oncology 2018, 20, 1429-1438. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N engl j Med 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. New England Journal of Medicine 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. New England Journal of Medicine 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- !!! INVALID CITATION !!! [15,16].
- Saikali, S.; Avril, T.; Collet, B.; Hamlat, A.; Bansard, J.-Y.; Drenou, B.; Guegan, Y.; Quillien, V. Expression of nine tumour antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL-13Rα2, gp100 and TRP-2 for immunotherapy. Journal of neuro-oncology 2007, 81, 139-148. [Google Scholar] [CrossRef]
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Bigner, D.D. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. In Proceedings of the Seminars in immunology; 2008; pp. 267–275. [Google Scholar]
- Zhu, X.; Prasad, S.; Gaedicke, S.; Hettich, M.; Firat, E.; Niedermann, G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget 2015, 6, 171. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Sun, W.; Hussain, S.F.; Dey, M.; Crutcher, L.; Aldape, K.; Gilbert, M.; Hassenbusch, S.J.; Sawaya, R.; Schmittling, B. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: case study. Neuro-oncology 2008, 10, 98-103. [Google Scholar] [CrossRef]
- Bhowmik, A.; Khan, R.; Ghosh, M.K. Blood brain barrier: a challenge for effectual therapy of brain tumors. BioMed research international 2015. [Google Scholar] [CrossRef]
- Okada, H.; Kalinski, P.; Ueda, R.; Hoji, A.; Kohanbash, G.; Donegan, T.E.; Mintz, A.H.; Engh, J.A.; Bartlett, D.L.; Brown, C.K. Induction of CD8+ T-cell responses against novel glioma–associated antigen peptides and clinical activity by vaccinations with α-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. Journal of clinical oncology 2011, 29, 330. [Google Scholar]
- Yang, T.; Kong, Z.; Ma, W. PD-1/PD-L1 immune checkpoint inhibitors in glioblastoma: Clinical studies, challenges and potential. Human Vaccines & Immunotherapeutics 2021, 17, 546-553. [Google Scholar]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Science translational medicine 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: refueling the CAR. Blood, The Journal of the American Society of Hematology 2017, 129, 1039-1041. [Google Scholar] [CrossRef]
- Maes, W.; Van Gool, S.W. Experimental immunotherapy for malignant glioma: lessons from two decades of research in the GL261 model. Cancer immunology, immunotherapy 2011, 60, 153-160. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Feng, S.; Xu, L.; Shi, W.; Wang, X.; Wang, H.; Yu, C.; Dong, T.; Xu, M.; Liang, G. Tim-3 on peripheral CD4+ and CD8+ T cells is involved in the development of glioma. DNA and cell biology 2014, 33, 245–250. [Google Scholar] [CrossRef]
- Li, G.; Wang, Z.; Zhang, C.; Liu, X.; Cai, J.; Wang, Z.; Hu, H.; Wu, F.; Bao, Z.; Liu, Y. Molecular and clinical characterization of TIM-3 in glioma through 1,024 samples. Oncoimmunology 2017, 6, e1328339. [Google Scholar] [CrossRef] [PubMed]
- Granier, C.; De Guillebon, E.; Blanc, C.; Roussel, H.; Badoual, C.; Colin, E.; Saldmann, A.; Gey, A.; Oudard, S.; Tartour, E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO open 2017, 2, e000213. [Google Scholar] [CrossRef] [PubMed]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. International journal of cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L. Tumor-Infiltrating Lymphocytes in Glioblastoma Are Associated with Specific Genomic Alterations and Related to Transcriptional ClassTumor-Infiltrating Lymphocytes in Glioblastoma. Clinical Cancer Research 2013, 19, 4951–4960. [Google Scholar] [CrossRef]
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. 2019, 38, 1-13. Journal of Experimental & Clinical Cancer Research 2019, 38, 1–13. [Google Scholar]
- Shindo, Y.; Yoshimura, K.; Kuramasu, A.; Watanabe, Y.; Ito, H.; Kondo, T.; Oga, A.; Ito, H.; Yoshino, S.; Hazama, S. Combination immunotherapy with 4-1BB activation and PD-1 blockade enhances antitumor efficacy in a mouse model of subcutaneous tumor. Anticancer research 2015, 35, 129–136. [Google Scholar]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunological reviews 2009, 229, 173–191. [Google Scholar] [CrossRef]
- Munks, M.W.; Mourich, D.V.; Mittler, R.S.; Weinberg, A.D.; Hill, A.B. 4-1BB and OX40 stimulation enhance CD8 and CD4 T-cell responses to a DNA prime, poxvirus boost vaccine. Immunology 2004, 112, 559–566. [Google Scholar] [CrossRef]
- Messenheimer, D.J.; Jensen, S.M.; Afentoulis, M.E.; Wegmann, K.W.; Feng, Z.; Friedman, D.J.; Gough, M.J.; Urba, W.J.; Fox, B.A. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40Timing Is Critical for OX40 plus PD-1 Combination. Clinical Cancer Research 2017, 23, 6165–6177. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Ahmad, S.; Verma, V.; Zeng, P.; Ananth, S.; Gaur, P.; Gittelman, R.M.; Yusko, E.; Sanders, C.; Robins, H. Concurrent PD-1 blockade negates the effects of OX40 agonist antibody in combination immunotherapy through inducing T-cell apoptosis. Cancer immunology research 2017, 5, 755–766. [Google Scholar] [CrossRef]
- Wheeler, C.J.; Das, A.; Liu, G.; Yu, J.S.; Black, K.L. Clinical responsiveness of glioblastoma multiforme to chemotherapy after vaccination. Clinical Cancer Research 2004, 10, 5316–5326. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Portnow, J.; Ammirati, M.; Baehring, J.; Brem, H.; Butowski, N.; Fenstermaker, R.A.; Forsyth, P.; Hattangadi-Gluth, J.; Holdhoff, M. NCCN guidelines insights: central nervous system cancers, version 1. 2017. Journal of the National Comprehensive Cancer Network 2017, 15, 1331–1345. [Google Scholar] [CrossRef]
- Faghfuri, E.; Faramarzi, M.A.; Nikfar, S.; Abdollahi, M. Nivolumab and pembrolizumab as immune-modulating monoclonal antibodies targeting the PD-1 receptor to treat melanoma. Expert review of anticancer therapy 2015, 15, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. JNCI: Journal of the National Cancer Institute 2013, 105, 256-265. [Google Scholar] [CrossRef]
- Teng, F.; Kong, L.; Meng, X.; Yang, J.; Yu, J. Radiotherapy combined with immune checkpoint blockade immunotherapy: achievements and challenges. Cancer letters 2015, 365, 23–29. [Google Scholar] [CrossRef]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9. [Google Scholar] [CrossRef]
- Pan, X.; Burgman, B.; Wu, E.; Huang, J.H.; Sahni, N.; Yi, S.S. i-Modern: Integrated multi-omics network model identifies potential therapeutic targets in glioma by deep learning with interpretability. Computational and Structural Biotechnology Journal 2022, 20, 3511–3521. [Google Scholar] [CrossRef]
- Pan, X.; Burgman, B.; Sahni, N.; Yi, S.S. Deep learning based on multi-omics integration identifies potential therapeutic targets in breast cancer. bioRxiv 2022. [Google Scholar]
- Liu, B.; Liu, Y.; Pan, X.; Li, M.; Yang, S.; Li, S.C. DNA methylation markers for pan-cancer prediction by deep learning. Genes 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Kovács, I.A.; Barabási, A.-L. Network-based prediction of drug combinations. Nature communications 2019, 10, 1197. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Gautam, P.; Kononov, A.; Potdar, S.; Saarela, J.; Wennerberg, K.; Aittokallio, T. Prediction of drug combination effects with a minimal set of experiments. Nature machine intelligence 2019, 1, 568–577. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The overview of the immunotherapy for the treatment of recurrent glioblastoma. A: CAR-T therapy can target antioens that are highly expressed on the GBM cell suraces, including EGFRvIII, IL13Ra2, HER2, B7-H3, EMMPIRIN, GD2, MMP2, CD133, CD70, CD276, CSPG4, NKG2D, CAIX, EphA2, TROP2. B: lmmune checkpoint inhibitors are monoclonal antibodies that target immune checkpoints to block immune cell inhibition, such as IDO1, CTLA-4/B7, LAG-3, CD137, VISTA, Siglec-15, HHLA2, and LAG-3/MHC, TIM3/CEACAM-1, VISTAL/VISTA, TIGIT/CD155, CD112 for negative immune regulation, ICOS/ICOSL, OX40/CD252, GITR/GITRL, CTLA-4/CD80 or CD86 for positive immune regulation. C: Vaccine therapy depends on dendritic cells, which present antigens or peptides to cytotoxic T cells via MHC class I-TCR interaction leading to T cell activation. Then, the cytotoxic T cells eradicate GBM cells via MHC class -TCR interaction, the lasted vaccine therapy for recurrent GBM including DCVax-L plus SOC, Allogeneic Tumor Lysate-Pulsed Autologous Dendritic Cell Vaccination, VXM01 (DNA plasmid vaccine for VEGFR-2) and avelumab (anti-PD-L1), Pembrolizumab With Autologous Tumor Lysate-Pulsed Dendritic Cell Vaccination, IDH1-R132H peptide vaccine, TAS0313, Neoadjuvant PD-1 Antibody Alone or Combined With Autologous Glioblastoma Stem-like Cell Antigens-primed DC Vaccines, IDH1-R132H+-specific vaccine, PEPIDH1M vaccines, HSPPC-96 vaccination, HSPPC-96 vaccine with bevacizumab, Rindopepimut and bevacizumab, WT1 vaccination, Personalized peptide vaccination, EGFR(V)-EDV-Dox. D: Oncolytic viral therapy utilizes genetic engineered viruses, which could selectively infect and replicate in GBM cells, resulting in cell lysis and release of tumor antigens. This can further trigger an adaptive antitumor immune response by stimulating antigen presenting cells, which including HSV-1716, G207& ganciclovir, G47Δ, M002, HSV-tk, Delta-24-RGD, Reovirus, NDV-HUJ Oncolytic Virus, Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex. E: As the future direction of immunotherapy for recurrent GBM, combination strategies could be the future direction of immunotherapy for recurrent GBM, which involve a combination of different therapies, have shown more promising outcome than single therapy.
Figure 1.
The overview of the immunotherapy for the treatment of recurrent glioblastoma. A: CAR-T therapy can target antioens that are highly expressed on the GBM cell suraces, including EGFRvIII, IL13Ra2, HER2, B7-H3, EMMPIRIN, GD2, MMP2, CD133, CD70, CD276, CSPG4, NKG2D, CAIX, EphA2, TROP2. B: lmmune checkpoint inhibitors are monoclonal antibodies that target immune checkpoints to block immune cell inhibition, such as IDO1, CTLA-4/B7, LAG-3, CD137, VISTA, Siglec-15, HHLA2, and LAG-3/MHC, TIM3/CEACAM-1, VISTAL/VISTA, TIGIT/CD155, CD112 for negative immune regulation, ICOS/ICOSL, OX40/CD252, GITR/GITRL, CTLA-4/CD80 or CD86 for positive immune regulation. C: Vaccine therapy depends on dendritic cells, which present antigens or peptides to cytotoxic T cells via MHC class I-TCR interaction leading to T cell activation. Then, the cytotoxic T cells eradicate GBM cells via MHC class -TCR interaction, the lasted vaccine therapy for recurrent GBM including DCVax-L plus SOC, Allogeneic Tumor Lysate-Pulsed Autologous Dendritic Cell Vaccination, VXM01 (DNA plasmid vaccine for VEGFR-2) and avelumab (anti-PD-L1), Pembrolizumab With Autologous Tumor Lysate-Pulsed Dendritic Cell Vaccination, IDH1-R132H peptide vaccine, TAS0313, Neoadjuvant PD-1 Antibody Alone or Combined With Autologous Glioblastoma Stem-like Cell Antigens-primed DC Vaccines, IDH1-R132H+-specific vaccine, PEPIDH1M vaccines, HSPPC-96 vaccination, HSPPC-96 vaccine with bevacizumab, Rindopepimut and bevacizumab, WT1 vaccination, Personalized peptide vaccination, EGFR(V)-EDV-Dox. D: Oncolytic viral therapy utilizes genetic engineered viruses, which could selectively infect and replicate in GBM cells, resulting in cell lysis and release of tumor antigens. This can further trigger an adaptive antitumor immune response by stimulating antigen presenting cells, which including HSV-1716, G207& ganciclovir, G47Δ, M002, HSV-tk, Delta-24-RGD, Reovirus, NDV-HUJ Oncolytic Virus, Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex. E: As the future direction of immunotherapy for recurrent GBM, combination strategies could be the future direction of immunotherapy for recurrent GBM, which involve a combination of different therapies, have shown more promising outcome than single therapy.
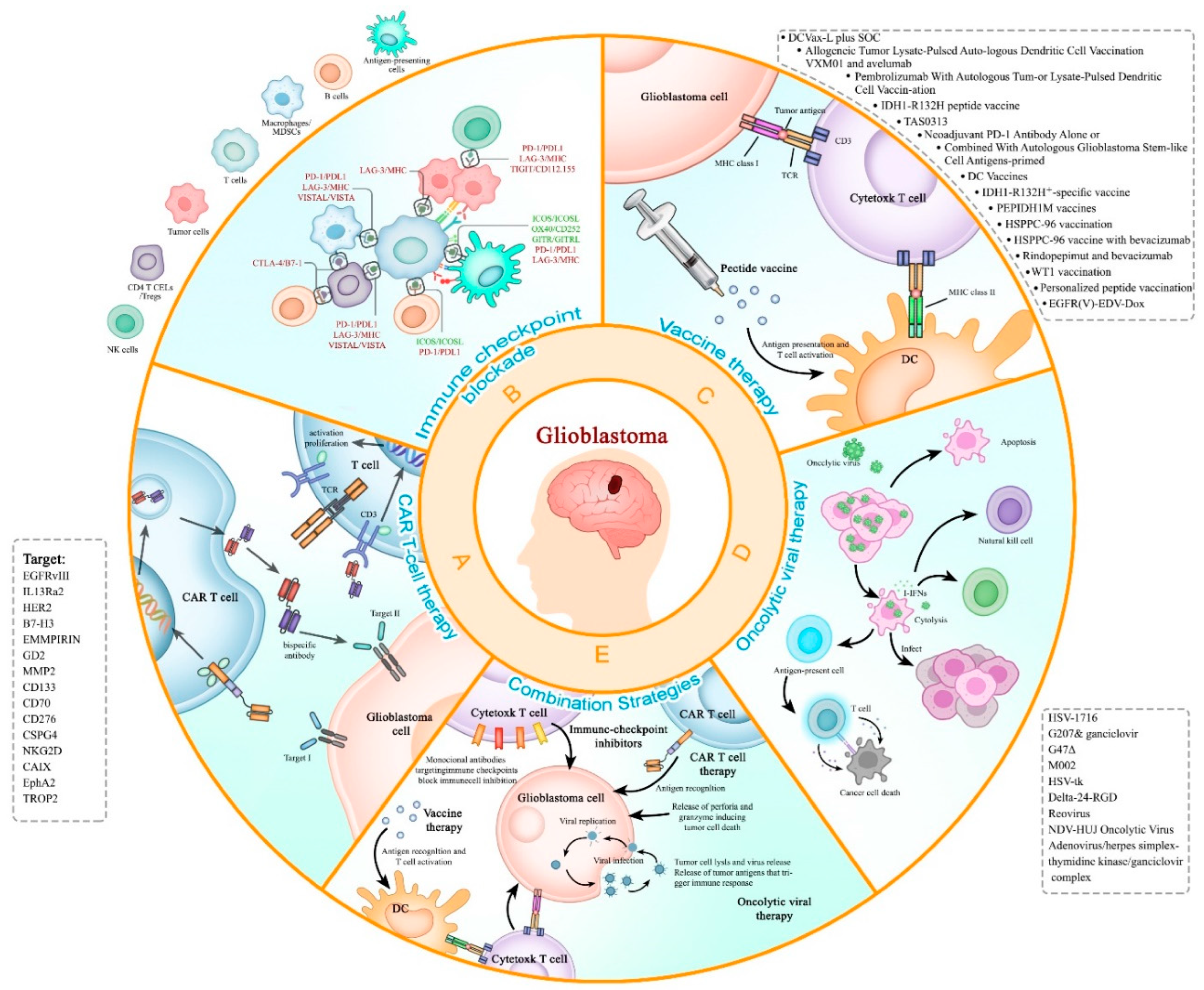
Table 1.
The clinical trials of CAR-T therapy on rGBM in ClinicalTrials.gov.
| Project name | Target | Clinic Phase |
Start Date | Estimated or Actual Completion Date |
Estimated or actual Enrollment |
Status |
|---|---|---|---|---|---|---|
| NCT00730613 | IL-13Rα2 | Phase 1 | Feb-02, 2002 | Aug-11, 2011 | 3 participants | done |
| NCT01082926 | IL-13Rα2 | Phase 1 | May-10, 2010 | Sep-1, 2013 | 6 participants | done |
| NCT02208362 | IL-13Rα2 | Phase 1 | May-15, 2015 | Jun-18, 2023 | 82 participants | going |
| NCT04003649 | IL-13Rα2 | Phase 1 | Dec-19, 2019 | Dec-31, 2023 | 60 participants | going |
| NCT02209376 | EGFRvIII | Phase 1 | Nov-14, 2014 | Apr-1, 2018 | 11 participants | done |
| NCT01454596 | EGFRvIII | Phase 1 | May-12, 2012 | May-1, 2012 | 18 participants | done |
| NCT03726515 | EGFRvIII | Phase 1 | Mar-11, 2019 | Feb-27, 2021 | 7 participants | done |
| NCT05024175 | EGFRvIII and EGFR | Phase 1 | Dec-1, 2021 | Aug-1, 2039 | 18 participants | going |
| NCT05168423 | EGFR and IL13Rα2 | Phase 1 | Mar-19, 2023 | Dec-19, 2029 | 18 participants | going |
| NCT01109095 | HER2 | Phase 1 | Oct-1, 2010 | Mar-1, 2018 | 16 participants | done |
| NCT03389230 | HER2 | Phase 1 | Aug-14, 2018 | Dec-15, 2023 | 42 participants | going |
| NCT03383978 | HER2 | Phase 1 | Dec-1, 2017 | Dec-31, 2023 | 42 participants | going |
| NCT04045847 | CD147 | Phase 1 | Oct-30, 2020 | May-30, 2022 | 31 participants | Unknown |
| NCT05627323 | MMP2 | Phase 1 | Feb-1, 2023 | Jan-1, 2041 | 42 participants | going |
| NCT04214392 | MMP2 | Phase 1 | Feb-26, 2020 | Feb-26, 2020 | 36 participants | going |
| NCT04385173 | B7-H3 | Phase 1 | Dec-1, 2022 | May-1, 2024 | 12 participants | going |
| NCT05241392 | B7-H3 | Phase 1 | Jan-27, 2022 | Dec-31, 2024 | 30 participants | going |
| NCT04077866 | B7-H3 | Phase 1/2 | Jun-1, 2023 | Aug-1, 2025 | 40 participants | going |
| NCT05366179 | B7-H3 | Phase 1 | Sep-2, 2022 | May, 2030 | 36 participants | going |
| NCT05474378 | B7-H3 | Phase 1 | Jul-12, 2022 | Aug-1, 2025 | 39 participants | |
| NCT05353530 | CD70 | Phase 1 | Oct-1, 2022 | Dec, 2040 | 18 participants | going |
| NCT04717999 | NKG2D | Unknown | Sep-1, 2021 | Dec-21, 2023 | 20 participants | going |
Table 2.
Ongoing rGBM trails combined with PD-1/PD-L1 blockades.
| Clinical Trail | Phase | Interventions | Arms | Combined Therapy |
|---|---|---|---|---|
| NCT05700955 | I | Drug: pembrolizumab and TMZ | Single arm: neoadjuvant pembrolizumab + TMZ | Neoadjuvant chemotherapy |
| NCT03661723 | II | Drug: pembrolizumab, bevacizumab Radiation: re-RT |
Arm 1: pembrolizumab + RT (lead-in) Arm 2: pembrolizumab + bevacizumab + RT (lead-in) Arm 3: pembrolizumab + RT Arm 4: pembrolizumab + bevacizumab + RT |
Adjusted RT, VEGFA inhibitor |
| NCT03743662 | II | Drug: pembrolizumab, bevacizumab Radiation: re-RT Procedure: re-resection |
Arm 1: re-RT + bevacizumab + Nivolumab Arm 2: re-RT + bevacizumab + Nivolumab + re-resection |
re-RT, bevacizumab, re-resection |
| NCT04977375 | I/II | Drug: pembrolizumab radiation: stereotactic RT |
Single arm: pembrolizumab + stereotactic RT + surgical resection | Stereotactic RT |
| NCT02866747 | I/II | Drug: durvalumab Radiation: HFSRT |
Arm 1: RT alone Arm 2: RT + durvalumab |
HFSRT |
| NCT02829931 | I | Radiation: HFSRT Drug: nivolumab, bevacizumab, ipilimumab |
Single arm: HFSRT + ipilimumab + nivolumab + bevacizumab | VEGFA, CTLA-4 inhibitors, HFSRT |
| NCT03722342 | I | Drug: TTAC-0001, pembrolizumab | Arm 1: TTAC-0001 12 mg/kg on D1, D8 and D15 + pembrolizumab 200 mg on D1 Arm 2: TTAC-0001 16 mg/kg on D1, D8 and D15 + pembrolizumab 200 mg on D1 Arm 3: TTAC-0001 8 mg/kg on D1, D8 and D15 + pembrolizumab 200 mg on D1 |
VEGFR2 inhibitor |
| NCT02311582 | I/II | Drug: pembrolizumab Procedure: LITT |
Arm 1: pembrolizumab + LITT Arm 2: pembrolizumab only |
Thermotherapy |
| NCT03277638 | I/II | Drug: pembrolizumab Procedure: LITT |
Single arm: pembrolizumab + LITT | Thermotherapy |
| NCT03341806 | I | Drug: avelumab Procedure: LITT |
Arm 1: avelumab Arm 2: avelumab + LITT |
Thermotherapy |
| NCT03430791 | I/II | Drug: nivolumab, ipilimumab Device: TTF |
Arm 1: nivolumab + TTF Arm 2: nivolumab + ipilimumab +TTF |
CTLA-4 inhibitor, tumor treating fields |
| NCT03532295 | II | Drug: epacadostat, retifanlimab, bevacizumab Radiation: RT |
Arm 1: retifanlimab + RT + bevacizumab Arm 2: retifanlimab + RT + bevacizumab + epacadostat |
RT, VEGFA, and IDO1 inhibitor |
| NCT02794883 | II | Drug: durvalumab, tremelimumab | Arm 1: durvalumab Arm 2: durvalumab + tremelimumab Arm 3: tremelimumab |
CTLA-4 inhibitor |
| NCT03493932 | I | Drug: BMS-986016, nivolumab | Single arm: BMS-986016 + nivolumab | LAG-3 inhibitor |
| NCT02658981 | I | Drug: BMS-986016, urelumab, nivolumab | Arm 1: BMS-986016 Arm 2: BMS-986016 + nivolumab Arm 3: urelumab + nivolumab |
LAG-3, CD137 inhibitors |
| NCT05465954 | II | Drug: efineptakin alfa, pembrolizumab | Single arm: efineptakin alfa + pembrolizumab, before and after surgery | Neoadjuvant IL7 |
| NCT04201873 | I | Biological: DC tumor cell lysate vaccine Drug: pembrolizumab, poly ICLC |
Arm 1: pembrolizumab + ATL-DC + poly ICLC Arm 2: placebo + ATL-DC + poly ICLC |
DC vaccine |
| NCT04013672 | II | Drug: pembrolizumab, surVaxM, sargramostim, montanide ISA 51 | Arm 1: have not received immunotherapy Arm 2: have failed prior anti-PD1 therapy |
Peptide-based vaccine |
| NCT03665545 | I/II | Drug: IMA950/Poly-ICLC and pembrolizumab | Arm 1: IMA950/Poly-ICLC Arm 2: IMA950/Poly-ICLC + pembrolizumab |
Peptide-based vaccine |
| NCT05084430 | I/II | Drug: M032, pembrolizumab | Single arm: pembrolizumab + M032 | Oncolytic herpes simplex virus |
| NCT04479241 | II | Drug: lerapolturev, pembrolizumab | Single arm: lerapolturev + pembrolizumab | Oncolytic poliovirus |
| NCT02798406 | II | Biological: DNX-2401 Drug: pembrolizumab |
Single arm: DNX-2401 + pembrolizumab | Oncolytic adenovirus |
| NCT05463848 | II | Drug: pembrolizumab, olaparib, TMZ | Arm 1: pembrolizumab + olaparib + TMZ Arm 2: pembrolizumab monotherapy |
PARP inhibitor, chemotherapy |
| NCT02430363 | I/II | Drug: pembrolizumab Biological: suppressor of the PI3K/Akt pathways |
Single arm: pembrolizumab + suppressors of the PI3K/Akt pathways | PI3K/Akt suppressors |
| NCT05053880 | I/II | Drug: ACT001, pembrolizumab | Arm 1: pembrolizumab Arm 2: pembrolizumab+ACT001 |
PAI-1 inhibitor |
TMZ: temozolomide, RT: radiation therapy, HFSRT: hypofractionated stereotactic irradiation, LITT: laser interstitial thermotherapy, TTF: Tumor Treating Fields, DC: dendritic cell.
Table 3.
The latest clinical trials on vaccination therapies for rGBM.
| type | Last reported | Therapy | phase | Registration number |
|---|---|---|---|---|
| DC vaccines | 2023 | Allogeneic Tumor Lysate-Pulsed Autologous Dendritic Cell Vaccination | Early Phase I | NCT03360708 |
| Peptide vaccines | 2023 | Allogeneic tumor lysate vaccine | Phase I | NCT04642937 |
| Nucleic acid vaccines | 2022 | VXM01 (DNA plasmid vaccine for VEGFR-2) and avelumab (anti-PD-L1) | Phase I/II | NCT03750071 |
| DC vaccines | 2022 | DCVax-L plus SOC | Phase III | NCT00045968 |
| DC vaccines | 2022 | Pembrolizumab With Autologous Tumor Lysate-Pulsed Dendritic Cell Vaccination | Phase I | NCT04201873 |
| DC vaccines | 2022 | mRNA tumor antigen-pulsed autologous DCs | Phase I | NCT02808364 |
| Peptide vaccines | 2022 | TAS0313 | Phase II | JapicCTI-183824 |
| Peptide vaccines | 2022 | VBI-1901 (targeting CMV antigen gB and pp65) | Phase I/II | NCT03382977 |
| DC vaccines | 2021 | Neoadjuvant PD-1 Antibody Alone or Combined With Autologous Glioblastoma Stem-like Cell Antigens-primed DC Vaccines | Phase II | NCT04888611 |
| DC vaccines | 2021 | allogeneic glioblastoma stem-like cell line-pulsed DC cell | Phase I | NCT02010606 |
| Peptide vaccines | 2021 | PEPIDH1M vaccines | Phase I | NCT02193347 |
| Peptide vaccines | 2021 | HSPPC-96 vaccine | Phase II | NCT00293423 |
| Peptide vaccines | 2021 | HSPPC-96 vaccine with bevacizumab | Phase II | NCT01814813 |
| Peptide vaccines | 2020 | Rindopepimut and bevacizumab | Phase II | NCT01498328 |
| Peptide vaccines | 2020 | HSPPC-96 vaccine | Phase I | NCT02722512 |
| DC vaccines | 2020 | Autologous tumor cell-pulsed DCs (ADCTA) | Phase III | NCT04277221 |
| Peptide vaccines | 2019 | Personalized peptide vaccination | Phase III | AMED number: 16ck0106086h0003 |
| Nucleic acid vaccines | 2019 | EGFR(V)-EDV-Dox | Phase I | NCT02766699 |
| DC vaccines | 2019 | Autologous tumor lysate-loaded DCs | Phase I | NCT04002804 |
| DC vaccines | 2019 | Tumor lysate-pulsed DCs | Phase II | NCT00576537 |
| DC vaccines | 2019 | GSC (Glioma Stem Cells) -Loaded Dendritic Cells | Phase I | NCT02820584 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



