
Submitted:
27 July 2023
Posted:
28 July 2023
You are already at the latest version
Abstract
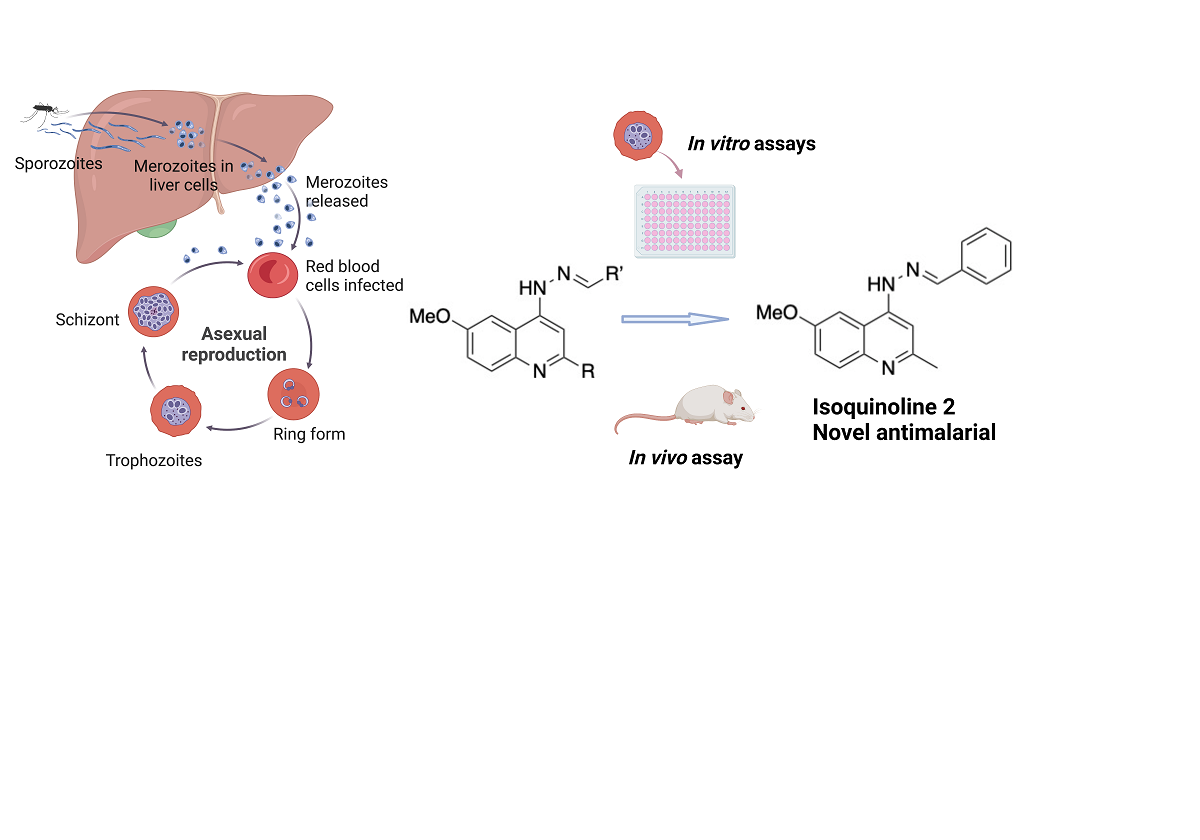
The emergence of resistance to first-line antimalarial drugs calls for development of new therapies for drug-resistant malaria. The efficacy of quinoline-based antimalarial drugs has prompted the development of novel quinolines. A panel of 4-aminoquinoline hydrazone analogues were tested on Plasmodium falciparum strains: IC50 values after a 48-hour cycle ranged from 0.60 - 49 µM, while the 72-hour cycle ranged from 26-219 nM on the multi-drug resistant K1 strain. Time-course assays were carried out to define the activity of the lead compounds which inhibited over 50 % growth in 24 hours and 90% growth in 72 hours. Cytotoxicity assays with HepG2 cells showed IC50 values of 0.87-11.1 M, whereas in MDBK cells IC50 values ranged from 1.66-11.7 M. High selectivity indices were observed for the lead compounds screened at 72 hours on P. falciparum. Analyses of stage-specificity revealed that the ring stage of the parasite life cycle were most affected. Based on antimalarial efficacy and in vitro safety profiles, lead compound 4-(2-benzylidenehydrazinyl)-6-methoxy-2-methylquinoline 2 was progressed to drug combination studies for the detection of synergism, with a combinatory index of 0.599 at IC90 for the combination of with artemether, indicating a synergistic antimalarial activity. Compound 2 was screened on different strains of P. falciparum (3D7, Dd2) which maintained similar activity to K1, suggesting no cross-resistance between multi-drug resistance and sensitive parasite strains. In vivo analysis with 2 showed suppression of parasitaemia with P. yoelii NL treated mice (20 mg/kg and 5 mg/kg).
Keywords:
4-aminoquinoline
; hydrazone
; antimalarial
; antimalarial drug interaction
; drug-resistant malaria
1. Introduction
Malaria remains a major public health threat, with 247 million malaria cases and 619 000 estimated deaths in 2021. Furthermore, an additional 63 000 estimated deaths were recorded due to service disruptions during the COVID-19 pandemic [1]. Although there has been a decline in global malaria deaths, the disease continues to cause significant mortality and morbidity burdens with severe consequences especially for pregnant women and children in Africa [2]. Malaria treatment and prevention currently relies on chemotherapy, vector control and more recently, approved vaccines used in selected regions and recommended for children living in areas with moderate to high transmission [3][4]. Chemotherapy, specifically artemisinin-based combination therapy (ACT), is the first-line treatment option against uncomplicated P. falciparum malaria [5]. Although this treatment regime is very effective, there is a growing body of evidence that artemisinin-tolerant P. falciparum parasite strains are emerging and consequently threatening the therapeutic utility of ACT regimen [6]–[11]. Furthermore, current artemisinin treatment is not broadly accessible in some developing countries because of the costs associated with supplying these drugs [12]. Therefore, effective, and affordable drugs are urgently needed.
The quinoline scaffold in synthetic and natural compounds has been shown to have a range of pharmacological activities [13], including anti-malarial. The significant impact on malaria morbidity and mortality of the quinoline compound chloroquine in the 1940s fuelled a belief that malaria might be completely eradicated from the world [14]. Following chloroquine resistance, a series of quinoline drugs including amodiaquine, piperaquine, primaquine and mefloquine were developed and found to be very active against malaria [13][15]. The relevance of novel analogues of quinoline compounds has been unequivocally established, particularly in the context of providing leads with improved pharmacological and toxicological profiles for overcoming chloroquine resistance [16].
We have previously synthesised and evaluated a series of 4-aminoquinoline hydrazone compounds as NQO2 inhibitors, a potential therapeutic target in cancer chemotherapy [17]. P. falciparum possesses a type II NADH:quinone oxidoreductase known as PfNDH2, that is structurally dissimilar to NQO2 (due to a divergent evolutionary event) but catalyses the same reaction, the oxidation of NADH to NAD+ and back to the oxidised form using ubiquinone [18]. Although PfNDH2 and NQO2 show high structural divergence as they evolved independently into enzymes with similar function, there is a possibility that their cofactor pockets have similarities in their binding and constitute the rationale for the screening of the 4-aminoquinoline hydrazone compounds against P. falciparum.
2. Results
2.1. Determination of IC50 values for the activity of 4-aminoquinoline hydrazones against K1 strain of P. falciparum
Sixteen 4-amino-6-methoxy-quinoline hydrazone compounds (see Figure S1 for full structures), with methyl and phenyl substituents at the 2-position (R = Me, Ph) and various substituents at R’ (Table 1), were tested for their antimalarial activities. A 1% parasitized culture at the trophozoite stage was treated with the compounds for 48 hours at concentrations of 3.05 nM – 200 µM (4-fold serial dilution) against the P. falciparum K1 strain and the growth was measured using the SYBR Green-based plate reader assay. The IC50 values of the quinolones ranged from 0.6 - 49 µM (Figure S2 for graphs), with the majority of the methyl-substituted analogues being more active than the phenyl-substituted analogues, supporting a smaller hydrophobic substituent at the 2-position. Compound 9, with a benzoic acid substituent on the hydrazone group was inactive, suggesting that a carboxylate anion impedes antimalarial activity. Chloroquine showed an IC50 in line with the published data (0.255 ± 0.049 µM) [19].
2.2. Time-course assay
A panel of compounds were selected based on activity and progressed in the time-course assay to define the activity timelines of the compounds. The assay was performed on 1% unsynchronized P. falciparum K1 cultures treated with previously determined IC50 (Table 1) doses of the lead compounds for 24, 48 and 72 hours. Following 24 hours incubation, the compounds exhibited activity with more than 50% reduction in parasitaemia levels, suggesting early onset antimalarial activity (Figure 1). Compounds 3 and 13 showed delayed activity, although they had shown better activity on the dose-response assay in comparison to some of the compounds that showed early onset activity. It was also observed that the activity of the compounds became more pronounced as the time progressed up until 72 hours of exposure.
2.3. Dose-response of lead compounds after 72 hours incubation period
The 72 hours IC50 assay was carried out based on the observation from the time-course assay, which showed that the hydrazone compounds were more active as time progressed. We set up drug efficacy assays for the 6 lead compounds (selected based on the best antimalarial activities) (Table 1), with 72 hours incubation on parasitised culture. A dose response experiment was initiated on 1% synchronous culture at ring stages of P. falciparum K1 strain. The selected compounds were tested at 3-fold serial dilution from 6.86 - 5000 nM. The IC50 of most of the compounds after 72 hours exposure time were observed in the low nano-molar range ((Table 2), Figure S3 for data), in comparison to the 48 hours assay (Table 1). The selectivity indices (IC50 human cells/IC50 parasites) [4] were calculated based on the 72 hours exposure studies and are presented in Table 2. Compounds 1 and 2 were found to be 10-fold more active with high selectivity indexes on the MDBK cells. Compounds 3, 4 and 14 on the other hand showed narrow selectivity indexes, although the anti-Plasmodium activity increased significantly. No direct correlation could explain these differences, however when compounds 5 (R = methyl, R’ = 2-hydroxy-3-methoxyphenyl) and 12 (R = phenyl, R’ = 2-hydroxy-3-methoxyphenyl) were compared, it was observed that the substituents on the aromatic ring (R) have a noticeable effect on cytotoxicity of the compounds, although not much difference was observed on the inhibitory activity of P. falciparum parasites.
2.4. MTT cytotoxicity assay
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay was performed to investigate the toxicity of the 4-aminoquinoline hydrazone compounds. HepG2 and MDBK cells were incubated with 2- fold serial dilution at dose ranges from 0.39 -25 µM for 5 days. Cisplatin was used as a control drug to validate the MTT assay. The cell viability data is provided in Table 1. Considering that these compounds were initially designed as anticancer drugs and have shown toxicity on SKOV-3 ovarian cancer cell line [17], we anticipated that the compounds might be more toxic on the HepG2 cancer cell line. Hence, the compounds were screened on MDBK cell line, which showed slightly less toxicity. Compound 4 showed similar toxicity on both the HepG2 and MDBK with IC50 values 1.59 μM and 1.66 μM, respectively, while the rest of the compounds showed lower toxicity on MDBK cells (see Figures S4 & S5). Compounds 5 and 11 were particularly toxic on the HepG2 cells, whereas they were not toxic on the MDBK cells. No obvious correlation was observed between the toxicity of the compounds with respect to their structures.
2.5. Stage–specificity analysis
The stage-specificity was evaluated by setting drug susceptibility experiments on synchronous cultures either at early rings or schizonts stages (as detailed in the experimental section). Each synchronous culture was treated with 1.6 - 100 times the respective IC50 (Table 1) concentrations of the eight lead compounds (1-5, 12-14) along with chloroquine as a control. Cultures were exposed to drug treatment for 24 hours, after which compounds were washed out and incubated in complete RPMI media for a further 24 hours. It was observed that all compounds primarily affected the ring stage of the parasite (Figure 2). An effect was observed on the schizont stage for compounds 5 and 12, inhibiting more than 25 % parasitaemia growth at 50 – 100 times IC50. Compound 13 was observed to have a similar effect on both the schizont stage and ring stage.
2.6. In vitro IC50 values against 3D7 and Dd2 P. falciparum strains
The lead compounds 1 and 2 were further evaluated against chloroquine-resistant P. falciparum Dd2 strain and chloroquine-sensitive P. falciparum 3D7. The drug activity assay was initiated on 1% infected blood at ring stages of the parasites at 4-fold serial dilution from 3.05 nM – 200 µM. The growth was measured after 72 hours using SYBR green-based plate reader assay. Both compounds exhibited anti-plasmodial activity at lower micro-molar ranges against both strains (Table 3, Figure S6). The results showed that the inhibitory potencies observed for compound 1 and 2 on both the K1 and Dd2 multidrug-resistant strains are similar to the sensitive strain 3D7, suggesting no cross-resistance with any of the multi-drug resistant strains tested.
2.7. In vivo assay on P. yoelii mouse model
We next assessed the anti-malarial activity of compound 2 in vivo within a P. yoelii murine model of blood-stage malaria. The experiment was conducted in a blind mode, where the researcher analysing the parasitemia was not aware of which group of mice have received the compound or the vehicle. Two dosages of the drug were tested, 5 mg/kg and 20 mg/kg, while another group of mice was administered with the drug vehicle alone (Figure 3). The lead compound 2 were injected once a day for 4 days, starting at day 11 when the infection level reached ~ 5% parasitaemia. Mice were monitored for a total of 20 days in order to detect signs of infection rebound or eventual toxicity. Comparing drug-treated mice to the placebo-vehicle-control, the parasitaemia of all mice treated with drug always remained detectable, although, at very low levels, less than 40 %, even a few days after drug treatment was suspended. Parasitaemia in the control steadily increased up to >80%. There were no detectable signs of significant toxicity or infection relapses as physical or behavioural distress was not detectable in treated mice for over three weeks. 20 mg/kg doses produced almost complete suppression of parasitaemia and cured the mice, while the 5 mg/kg doses showed delayed activity.
2.8. CalcuSyn assay for combination therapy assay
The CalcuSyn method has been previously validated in our laboratory for antimalarial drug interaction analysis [20][21]. The atovaquone-proguanil combination was used to establish the robustness of CalcuSyn drug interactivity software. The compounds were combined in accordance to the previously determined ED50 values. Briefly, a 2-fold serial dilution was carried out based on ED50 values. The mid-point equated to the previously determined ED50 values to each drug. 1% ring stage parasites were treated with either atovaquone or proguanil solely or in combination (Table 4).
The samples were analysed after 72 hours using SYBR Green plate reader method and further analysed with CalcuSyn software. The dose-effect curve, median-effect plot and isobologram (see Figure S7 for data) showed improved potency for the combination of atovaquone and proguanil in comparison to the individual drugs for validation of methodology. The combination index (CI) at ED50, ED75 and ED90 was used to determine synergism where CI > 1 was classified as antagonism, CI = 1 additive and CI < 1 synergism [22]. CI values of 0.215, 0.388 and 0.698 at IC50, IC75 and IC90 proved the strong synergistic interaction between atovaquone and proguanil.
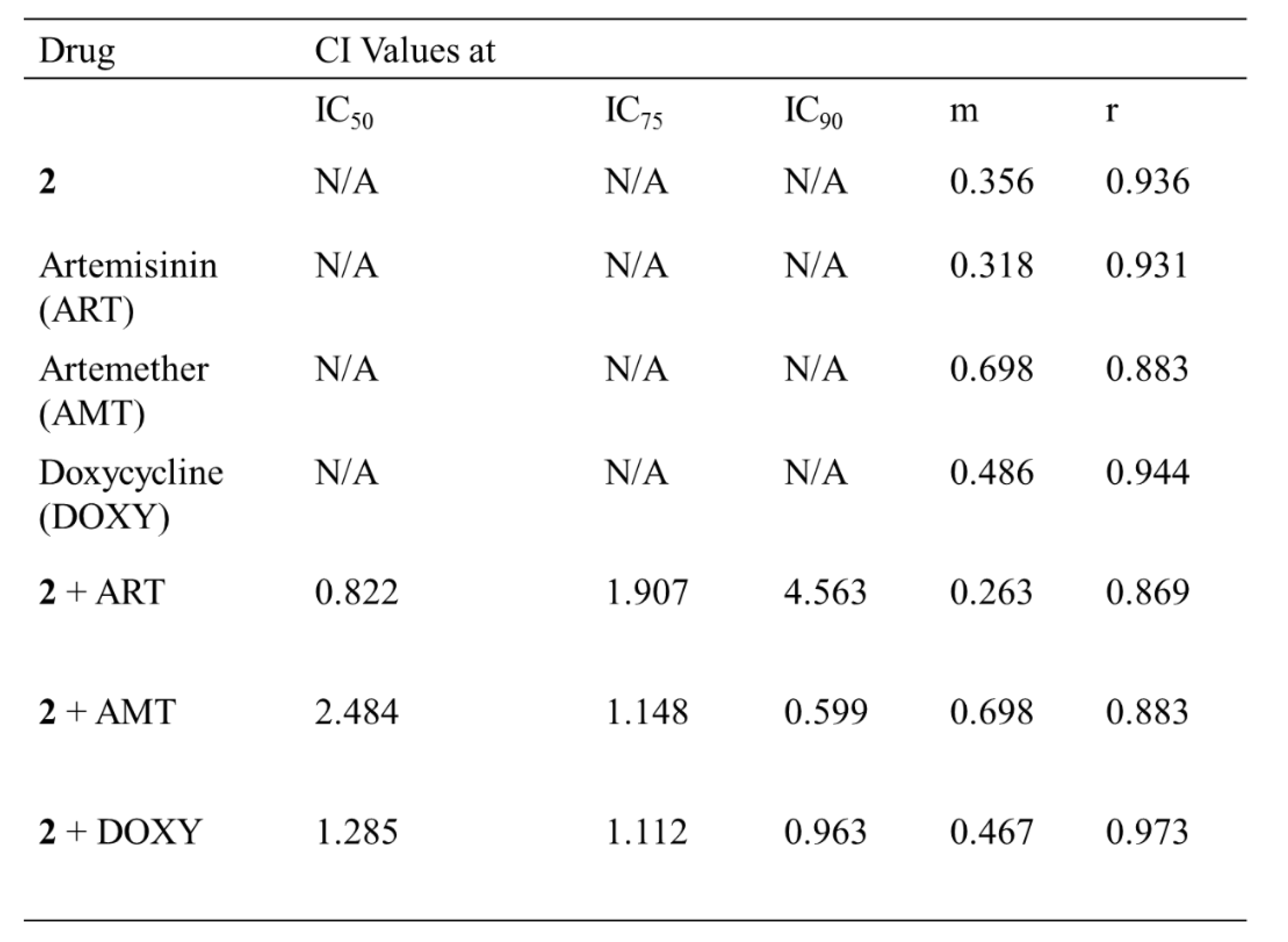
Upon validation of the atovaquone-proguanil combination, the interaction between the lead compound 2 and artemisinin, artemether and doxycycline was studied. The doses were selected based on the respective predetermined IC50 values (from 72-hours assay) of 2. The combination of compound 2 and artemisinin was classified as antagonism at IC90 and IC75, while slight synergism was observed at IC50. The combination of compound 2 and doxycycline was classified as nearly additive at IC90 and observed to be antagonism at IC50 and IC75. Synergism was observed between compound 2 and artemether at IC90 and nearly additive at IC50 and IC75 (Table 5) and dose-response data (Figure S8).
3. Discussion
The 4-aminoquinoline hydrazone compounds were initially developed as anticancer drugs. In this study these compounds have shown to have antimalarial activity with IC50 values ranging from 0.60 - 49 μM against the multi-drug resistant K1 strain of P. falciparum after 48 hours of drug exposure. The time-course experiment indicated that the activity of the compounds became more pronounced as time progressed. We then assessed the activity of the lead compounds (chosen based on their activity at 48 hours assay and structural differences) at 72 hours. We observed that the compounds were now active at low nano-molar IC50 values with the most active compounds 1 and 2 having IC50 values of 25 nM and 32nM, respectively. This suggested that the compounds are potent but are either acting more effectively with some delay or are stage specific. The latter was indeed confirmed, as most of our compounds showed predominant sensitivity for the ring stage.
One of the key fundamentals when developing anti-malaria treatments is that the drug candidate must be well tolerated and safe for use, especially for pregnant women and children. We investigated the toxicity of the 4-aminoquinoline hydrazones using the in vitro MTT assay in HepG2 and MDBK cell lines. The compounds displayed toxicity with low IC50 values ranging from 0.87 - 11.1 μM for HepG2 and 1.66 - 11.7 μM for MDBK. Interestingly, when the incubation time was increased on the Plasmodium dose response assay to 72 hours, it was observed that all of the compounds except for compound 4 (Figure S7) were safe, with therapeutic selectivity indices of >30 for the MDBK cell line. Although the assessed toxicity effect suggested that the compounds might be safe, appropriate toxicity investigation to support these results can only be concluded with the use of in vivo models. The lead compound 2 was evaluated with the P. yoelii NL mouse model and shown to be efficacious, with a daily dose of 20 mg/kg clearing the parasitaemia by day 1. The mice were fully cured from malaria infection, indicating that the 20 mg/kg dose is still higher than the minimum effective concentration and more experiments need to be performed to assess at even lower dosage between 5-20mg/kg for the treatment.
The use of combination regimes has shown to have a therapeutic advantage in comparison to monotherapy. The WHO recommends the use of artemisinin-based combination to address the failures of monotherapy. The combination regimes can kill the majority of parasites over several days by one mechanism and the partner drug can prevent recrudescence. Three existing antimalarial drugs: artemether, artemisinin and doxycycline were investigated for their interaction with compound 2. The artemisinin-2 combination was classified as antagonistic suggesting that the mode of action of the compounds overlap. Compound 2 was found to exhibit mild synergistic activity (additive) with doxycycline. The synergistic activity of compound 2 with artemether presents an exciting option for antimalarial therapy as the WHO recommends artemisinin and derivatives-based combinations to increase antimalarial potency and prolong the shelf-lives of the frontline antimalarials.
4. Materials and Methods
Drug preparation and synthesis
Compounds 1 - 16 were synthesised using the experimental procedures described previously [17]. The purity of the compounds was checked by 1H and 13C NMR spectroscopy and LC-MS, and were found to be >95% pure prior to biological experiments. NMR spectra were recorded on a Bruker 300 MHz spectrometer. Chemical shift is quoted in parts per million (ppm) and referenced to solvent peak. LC-MS was carried out using the Acquity UPLC H-class system. The mass spectrometry data was acquired in the positive [ES +] and negative [ES -] modes scanned from 100 - 1000 m/z. The LC data was obtained for Waters Acquity UPLC PDA detector scanning from 210 - 400 nm. Drug stock solutions were prepared in DMSO at 50 mM. The stock solutions were stored at 4 °C. Serial dilution in complete media was prepared from stock solutions immediately before use.
Plasmodium falciparum parasite cultivation
The K1, 3D7 (from MR4 and in lab maintained and isolated clone A10) and Dd2 strain of Plasmodium falciparum were cultured with RPMI 1640 media containing 25 mM HEPES and 0.3 g/L L-glutamine (Gibco, Life Technologies, UK) [27]. The media was supplemented with 2.5 g AlbuMax (Sigma, UK) and sterile solutions of 2.5 ml hypoxanthine (Sigma, UK), 2.5 ml 40% glucose (Dextrose Anhydrous, Fisher Scientific, UK) and 0.5 ml Gentamycin (Sigma, UK). Human erythrocytes served as host cells. Cultures were maintained at 37 °C under gas mixture of 5% CO2, 5% O2 in N2 (BOC limited, UK).
Synchronization of K1 Plasmodium falciparum
K1 strain of P. falciparum maintained at 5% haematocrit was used to establish synchrony. The culture was cultivated to ~ 8% parasitaemia, with prevalence of ring stages. The culture was centrifuged at 3500 rpm for 5 min. Supernatant was removed and parasitized RBCs were re-suspended in 5% sorbitol, incubated at room temperature for 5 min and then centrifuged at 3500 rpm for 5 min. The supernatant was removed and the pellet was washed 3 times with complete medium before setting up a new culture.
Determination of IC50 in Plasmodium falciparum
1% infected RBCs at ring or trophozoites stage was exposed to 9 dilutions (4-fold serial dilution) of compounds from 50 mM stock solutions in DMSO. 100 µl of culture volume was added in 96-well plates with 100 µl drug dilutions. Plates were either incubated for 48 hours or 72 hours at 37 °C, 5% CO2, 5% O2 in N2. Following the incubation, 150 µl of medium was carefully removed and 150 µl of 5X SYBR Green solution (prepared by adding 2 µl of 10 000 X SYBR Green in 4 ml of PBS) was added to each well. Plates were kept in the dark for 45 minutes. Fluorescence intensity was measured using a micro plate reader (Genius Tecan) with an excitation of 485 nm and emission of 535 nm.
Time–course assay
The time course assay was used to define the activity time-lines of the 4-aminoquinoline hydrazone compounds. Parasite growth of K1 strain P. falciparum in the presence of 4-aminoquinoline hydrazone compounds was assessed on asynchronous culture. The growth impairment of K1 culture was initiated at 1% parasitaemia for 24, 48 and 72 hours. The assay was analysed by flow cytometry as described below.
Flow Cytometry
Following drug efficacy experiments, 100 µl from the control and drug-treated on a 96-well plate were transferred to a micro-centrifuge tube containing 1ml of PBS. The samples were centrifuged at 1200 rpm for 2 min. The supernatant was removed and the samples were re-suspended in 1 ml of 5X SYBR Green solution and incubated in the dark for 20 min at room temperature. After staining, the samples were centrifuged for 2 min at 1200 rpm and the supernatant was discarded. The samples were fixed with 0.4% formaldehyde solution (250 µl). The samples were placed in the fridge and incubated at 4 °C for 10 - 15 min and subsequently washed 3 times with PBS. The pellet was re-suspended in PBS (1 ml) and parasitaemia was determined by SYBR Green fluorescence using the FITC channel of the BD FACs Verse flow cytometer system (Blue laser, excitation laser line 488 nM Exmax 494/ EMmax 520 nM) and cell size (forward scatter, FSC-A). Fluorescent events in drug-treated samples were compared with infected and uninfected blood counterparts and gated accordingly to obtain the percentage parasitaemia.
MTT assay for testing cell cytotoxicity
Human hepatoma (HepG2) and the Madin-Darby Bovine Kidney (MDBK) cell lines were purchased from American Type Culture Collection (ATCC), USA. The MDBK cell line was cultured with Dulbecco’s Modified Eagle’s Medium (DMEM) containing 4.5 g/l (+)-D-glucose and L-glutamine while the HepG2 cell line was cultured with RPMI 1640 containing 2mM L-glutamine and HEPES. The culture medium was supplemented with 10% Fetal Bovine Serum (FBS) and 1% penicillin. Cells were maintained in the presence of 5% CO2 atmosphere at 37 °C. The assay was carried out on a 96-well plate, with each well containing 4 x 103 cells. Plates were firstly incubated for 24 hours at 37 °C to ensure cell adherence. The cells were then treated with 100 µl of drug solution prepared at various concentrations and further incubated for 5 days. After the incubation period, 50 µl of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma, UK) solution was added to each well and incubated for a further 3 hours. The MTT solution and media was aspirated, 150 µl of DMSO was added to each well and results were read on the Ascent plate reader.
Stage-specificity assay
Plasmodium falciparum K1 was synchronised twice with 5% sorbitol at 0 hours and 31 hours to obtain early rings (up to 3 hours old) [28]. In order to obtain late trophozoites to early schizonts, the culture was synchronised twice at 0 hours and 6 hours. Each synchronous stage was diluted to 1% parasitaemia. On a 96-well plate, the two synchronous stages were incubated for 24 hours at 37 °C with a two-fold serial dilution of the 6 most active 4-aminoquinoline hydrazone compounds. The dilution ranged from 1.6 - 100X fold of the previously determined IC50 of each compound in a standard 48-hours assay. After 24 hours incubation period, plates were washed 4 times resulting in about 100-fold dilution of free compound. The plates were further incubated for an additional 24 hours at 37 °C and analysed using SYBR Green staining assays as previously described [29].
Derivation of the dose-response curves and IC50 values
IC50 values were determined using Graph Pad Prism 5.0. Values were calculated using non-linear regression by using log-transformed drug concentration plotted against dose response. Parasitaemia was calculated and normalised relative to response on the controls (cultures without drug).
Drug interaction assay for 4-aminoquinoline hydrazones
The drug combination was determined using CalcuSyn method. This method relies on predetermined IC50, which was conducted as previously explained. 0.12 - 8 times the ED50 of each drug was combined together and 2-fold serial dilution was carried out. Ring stage parasites were treated with drug-dilution and incubated for 72 hours in a 96-well plate format. Drug susceptibility was determined by the SYBR Green plate reader method while the median effect was determined using CalcuSyn method. The drug interaction was determined according to the median-effect principle by Chou Ting – Chao [30][22]. The CalcuSyn software is able to generate the isobologram plots, dose-effect curves, median-effect curves and Combination Indexes (CI) to assess eventual drug to drug interaction. The combination index which represents the pharmacological interaction, taking into account both the potency and the shape of the dose response curve has been extensively studied and can be interpreted according to CI < 1 (synergy), CI = 1 (additive) and CI > 1 (antagonist) [22].
P. yoelii NL mice screening with compound 2
C57BL/6 mice were purchased from Charles River, UK, and were maintained at the University of Manchester in individually ventilated cages. Cryopreserved P. yoelii NL parasites were passed once through mice before infection experiment. Male/female mice 6-10 weeks were infected with 1x104 parasitised red blood cells (pRBCs) intravenously. Parasitaemia growth was monitored via Giemsa-stained blood smears. The group of mice (placebo, drug dose of 5mg/kg or 20 mg/kg) were used for this experiment. Compound 2 was injected once a day for 4 days starting at the day the infection level reached ~ 5% parasitaemia. After the injection, mice were monitored for 20 days to detect eventual signs of both infection rebound and toxicity.
All animal work was approved following local ethical review by the Manchester Animal Procedures and ethics committee and was performed in strict accordance with the United Kingdom Home Office Animals (Scientific Procedure) Act 1986 (P8829D3B4).
5. Conclusions
The 4-aminoquinoline hydrazone compounds have shown good activities and low cytotoxicity, suggesting that they can be used as lead compounds as novel antimalarials. Moreover, they have shown rapid onset of action, which is very important to relieve patient symptoms early and minimise parasite resistance [23][24]. The activity of these compounds warrants further investigation as potential drugs in chemotherapy. The World Health Organisation (WHO) recommends artemisinin-based combination therapy (ACT) to confront drug-resistant P. falciparum malaria [5]. Moreover, ACT can improve antimalarial efficacy, favourable synergistic interaction and decreased toxicity [25][26]. Therefore, it is of interest to further evaluate the 4-aminoquinoline hydrazone compounds as partner drugs for combination therapy.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: Structures of the 4-aminoquinoline hydrazone compounds that were tested for their antimalarial activities; Figure S2: The effect of the 4-aminoquinoline hydrazone compounds along with chloroquine tested on the P. falciparum K1 strain (trophozoite stage). The 4-aminoquinoline hydrazones were tested in 4-fold dilution of dose range from 3.05 nM - 200 µM, while Chloroquine was tested in a dose of 9.3 - 600 nM. The compounds were incubated for 48 hours and read using SYBER green-based plate reader assay. The experiments were performed three times in triplicates. The data was analysed using GraphPad prism, Figure S3: The effect dose of the 4-aminoquinoline hydrazone compounds tested on the P. falciparum K1 strain (ring stage). The compounds were tested in 4-fold dilution of dose range from 3.05nM - 200 µM, incubated for 72 hours and read using SYBER green-based plate reader assay. The experiments were performed three times in triplicates. The data was analysed using GraphPad prism; Figure S4: MTT assay was assessed on Hep G2 cells. The cells were seeded at 4000 cells per well. The HepG2 cells were incubated with 2- fold serial dilution of the compounds at dose range of 0.39 -25 µM for 5 days. Cell viability was determined using the standard MTT assay. Data was analysed using GraphPad prism. The experiments were performed three times; each concentration was repeated three times per experiment; Figure S5: MTT assay was assessed on MDBK cells. The cells were seeded at 4000 cells per well. The MDBK cells were incubated with 2- fold serial dilution of compounds at dose range of 0.39 -25 µM for 5 days. Cell viability was determined using the standard MTT assay. Data was analysed using GraphPad prism. The experiments were performed three times; each concentration was repeated three times per experiment; Figure S6: The effect dose of the lead 4-aminoquinoline hydrazone compounds 1 & 2 tested on the P. falciparum 3D7 ig A10 (A) and Dd2 (B) in 4-fold dilution of dose range from 3.05 nM - 200 µM. The compounds were incubated for 72 hours and read using SYBER green-based plate reader assay. The experiments were performed three times in triplicates. The data was analysed using GraphPad prim; Figure S7: Dose effect analysis of atovaquone and proguanil combination on P. falciparum K1 strain. The dose-response data was analysed with CalcuSyn software to give a dose-effect curve and isobologram graph; Figure S8: Dose effect analysis of atovaquone and proguanil combination on P. falciparum K1 strain. The dose-response data was analysed with CalcuSyn software to give dose-effect curve and isobologram graph.
Author Contributions
Conceptualization, Rachael Magwaza and Sally Freeman; Data curation, Rachael Magwaza; Formal analysis, Rachael Magwaza; Funding acquisition, Sally Freeman; Investigation, Rachael Magwaza, Muna Abubaker, Michael Haley and Niroshini Nirmalan; Methodology, Rachael Magwaza and Niroshini Nirmalan; Project administration, Niroshini Nirmalan; Resources, Buthaina Hussain; Supervision, Kevin Couper, Sally Freeman and Niroshini Nirmalan; Writing – original draft, Rachael Magwaza and Sally Freeman; Writing – review & editing, Rachael Magwaza, Kevin Couper, Sally Freeman and Niroshini Nirmalan.
Funding
This research was funded by Aspen Pharmacare (SA) and National Research Foundation of South Africa (grant no: 107881).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the main text or the supplementary material.
Acknowledgments
RM would like to thank Dr Manikandan Kadirvel for technical support.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- World Health Organization, “World Malaria Report 2022.”.
- C. J. Sutherland, N. Tanomsing, D. Nolder, M. Oguike, C. Jennison, S. Pukrittayakamee, C. Dolecek, T. T. Hien, V. E. do Rosário, A. P. Arez, J. Pinto, P. Michon, A. A. Escalante, F. Nosten, M. Burke, R. Lee, M. Blaze, T. D. Otto, J. W. Barnwell, A. Pain, J. Williams, N. J. White, N. P. J. Day, G. Snounou, P. J. Lockhart, P. L. Chiodini, M. Imwong, and S. D. Polley, “ Two Nonrecombining Sympatric Forms of the Human Malaria Parasite Plasmodium ovale Occur Globally ,” J. Infect. Dis., vol. 201, no. 10, pp. 1544–1550, 2010. [CrossRef]
- K. Karunamoorthi, “Vector control: A cornerstone in the malaria elimination campaign,” Clin. Microbiol. Infect., vol. 17, no. 11, pp. 1608–1616, 2011. [CrossRef]
- M. Kaiser, P. Mäser, L. P. Tadoori, J. R. Ioset, R. Brun, and D. J. Sullivan, “Antiprotozoal activity profiling of approved drugs: A starting point toward drug repositioning,” PLoS One, vol. 10, no. 8, pp. 1–10, 2015.
- World Health Organization, World Malaria Report 2019. Geneva. 2019. [CrossRef]
- E. B. Magbity, N. T. Marbiah, G. Maude, C. F. Curtis, D. J. Bradley, B. M. Greenwood, E. Petersen, and J. D. Lines, “Effects of community-wide use of lambdacyhalothrin-impregnated bednets on malaria vectors in rural Sierra Leone,” Med. Vet. Entomol., vol. 11, no. 1, pp. 79–86, 1997. [CrossRef]
- A. Ouattara and M. B. Laurens, “Vaccines against malaria,” Clin. Infect. Dis., vol. 60, no. 6, pp. 930–936, 2015.
- J. Okombo and K. Chibale, “Recent updates in the discovery and development of novel antimalarial drug candidates,” Medchemcomm, vol. 9, no. 3, pp. 437–453, 2018.
- S. R. Meshnick, T. E. Taylor, and S. Kamchonwongpaisan, “Artemisinin and the antimalarial endoperoxides: from herbal remedy to targeted chemotherapy.,” Microbiol. Rev., vol. 60, no. 2, pp. 301–315, 1996.
- H. Noedl, Y. Se, K. Schaecher, B. L. Smith, D. Socheat, and M. M. Fukuda, “Evidence of artemisinin-resistant malaria in Western Cambodia,” N. Engl. J. Med., vol. 359, no. 24, pp. 2619–2620, 2008. [CrossRef]
- A. M. Dondorp, F. Nosten, P. Yi, D. Das, A. P. Phyo, J. Tarning, D. Ph, K. M. Lwin, F. Ariey, W. Hanpithakpong, S. J. Lee, P. Ringwald, K. Silamut, T. Herdman, S. S. An, S. Yeung, D. Socheat, and N. J. White, “Artemisinin Resistance in,” Drug Ther. (NY)., vol. 361, no. 5, pp. 455–467, 2009.
- A. B. Panosian, “Economic Access to Effective Drugs for Falciparum Malaria,” Clin. Infect. Dis., vol. 40, no. 5, pp. 713–717, 2005. [CrossRef]
- R. Musiol, T. Magdziarz, and A. Kurczyk, “Quinoline scaffold as a privileged substructure in antimicrobial drugs,” Sci. against Microb. Pathog. Commun. Curr. Res. Technol. Adv., no. January, pp. 72–83, 2011.
- F. Loeb, W. M. Clark, G. R. Coatney, L. T. Coggeshall, F. R. Dieuaide, A. R. Dochez, E. G. Hakansson, E. K. Marshall Jr., C. S. Marvel, O. R. McCoy, J. J. Sapero, W. H. Sebrell, J. A. Shannon, and G. A. Carden Jr., “Activity of a new antimalarial agent, chloroquine (SN 7618): Statement approved by the board for coordination of malarial studies,” J. Am. Med. Assoc., vol. 130, no. 16, pp. 1069–1070, 1946.
- P. A. Winstanley, S. A. Ward, and R. W. Snow, “Clinical status and implications of antimalarial drug resistance,” Microbes Infect., vol. 4, no. 2, pp. 157–164, 2002. [CrossRef]
- K. Kaur, M. Jain, R. P. Reddy, and R. Jain, “Quinolines and structurally related heterocycles as antimalarials,” Eur. J. Med. Chem., vol. 45, no. 8, pp. 3245–3264, 2010. [CrossRef]
- B. Hussein, B. Ikhmais, M. Kadirvel, R. N. Magwaza, G. Halbert, R. A. Bryce, I. J. Stratford, and S. Freeman, “Discovery of potent 4-aminoquinoline hydrazone inhibitors of NRH:quinoneoxidoreductase-2 (NQO2),” Eur. J. Med. Chem., vol. 182, p. 111649, 2019. [CrossRef]
- N. Fisher, P. G. Bray, S. A. Ward, and G. A. Biagini, “The malaria parasite type II NADH : quinone oxidoreductase : an alternative enzyme for an alternative lifestyle,” TRENDS Parasitol., vol. 23, no. 7, 2007. [CrossRef]
- S. Kondaparla, P. Agarwal, K. Srivastava, S. K. Puri, and S. B. Katti, “Design, synthesis and in vitro antiplasmodial activity of some bisquinolines against chloroquine-resistant strain,” Chem. Biol. Drug Des., vol. 89, no. 6, pp. 901–906, 2017. [CrossRef]
- H. Matthews, J. Deakin, M. Rajab, M. Idris-Usman, and N. J. Nirmalan, “Investigating antimalarial drug interactions of emetine dihydrochloride hydrate using CalcuSyn-based interactivity calculations,” PLoS One, vol. 12, no. 3, pp. 1–19, 2017.
- P. Panwar, K. K. Burusco, M. Abubaker, H. Matthews, A. Gutnov, E. Fernández-Álvaro, R. A. Bryce, J. Wilkinson, and N. Nirmalan, “Lead optimization of dehydroemetine for repositioned use in malaria,” Antimicrob. Agents Chemother., vol. 64, no. 4, pp. 1–22, 2020.
- T. C. Chou, “Drug combination studies and their synergy quantification using the chou-talalay method,” Cancer Res., vol. 70, no. 2, pp. 440–446, 2010. [CrossRef]
- M. A. Biamonte, J. Wanner, and K. G. Le Roch, “Recent advances in malaria drug discovery,” Bioorganic Med. Chem. Lett., vol. 23, no. 10, pp. 2829–2843, 2013. [CrossRef]
- J. N. Burrows, R. Hooft Van Huijsduijnen, J. J. Möhrle, C. Oeuvray, and T. N. Wells, “Designing the next generation of medicines for malaria control and eradication,” Malar. J., vol. 12, no. 1, pp. 1–20, 2013.
- D. A. Fidock, P. J. Rosenthal, S. L. Croft, R. Brun, and S. Nwaka, “Antimalarial drug discovery: Efficacy models for compound screening,” Nat. Rev. Drug Discov., vol. 3, no. 6, pp. 509–520, 2004. [CrossRef]
- R. T. Eastman and D. A. Fidock, “Artemisinin-based combination therapies: A vital tool in efforts to eliminate malaria,” Nat. Rev. Microbiol., vol. 7, no. 12, pp. 864–874, 2009. [CrossRef]
- H. A. Poindexter, “Human Malaria Parasites in Continuous Culture,” J. Natl. Med. Assoc., vol. 68, no. 6, pp. 530–533, 1976.
- C. Le Manach, C. Scheurer, S. Sax, S. Schleiferböck, D. G. Cabrera, Y. Younis, T. Paquet, L. Street, P. Smith, X. C. Ding, D. Waterson, M. J. Witty, D. Leroy, K. Chibale, and S. Wittlin, “Fast in vitro methods to determine the speed of action and the stage-specificity of anti-malarials in Plasmodium falciparum,” Malar. J., vol. 12, no. 1, pp. 1–7, 2013. [CrossRef]
- H. Matthews, M. Usman-idris, F. Khan, M. Read, and N. Nirmalan, “Drug repositioning as a route to anti-malarial drug discovery : preliminary investigation of the in vitro anti-malarial efficacy of emetine dihydrochloride hydrate,” Malar. J., vol. 12, no. 395, pp. 1–11, 2013. [CrossRef]
- T. C. Chou, “Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies,” Pharmacol. Rev., vol. 58, no. 3, pp. 621–681, 2006. [CrossRef]
Figure 1.
Time–course analysis of 4-aminoquinoline hydrazone compounds. Asynchronous cultures of P. falciparum were treated with predetermined IC50 doses (Table 1) for 72 hours and analysed at 24 hours intervals. Drug susceptibility was analysed using the SYBR Green flow cytometer method. Error bars present the standard deviation of two biological replicates. The significant difference between the control vs each compound is indicated by ***p <0.001 while the significant difference between each compound at different time intervals is indicated by ns for p > 0.05 and * for p < 0.05. The significant difference was determined by two-way ANOVA.
Figure 1.
Time–course analysis of 4-aminoquinoline hydrazone compounds. Asynchronous cultures of P. falciparum were treated with predetermined IC50 doses (Table 1) for 72 hours and analysed at 24 hours intervals. Drug susceptibility was analysed using the SYBR Green flow cytometer method. Error bars present the standard deviation of two biological replicates. The significant difference between the control vs each compound is indicated by ***p <0.001 while the significant difference between each compound at different time intervals is indicated by ns for p > 0.05 and * for p < 0.05. The significant difference was determined by two-way ANOVA.
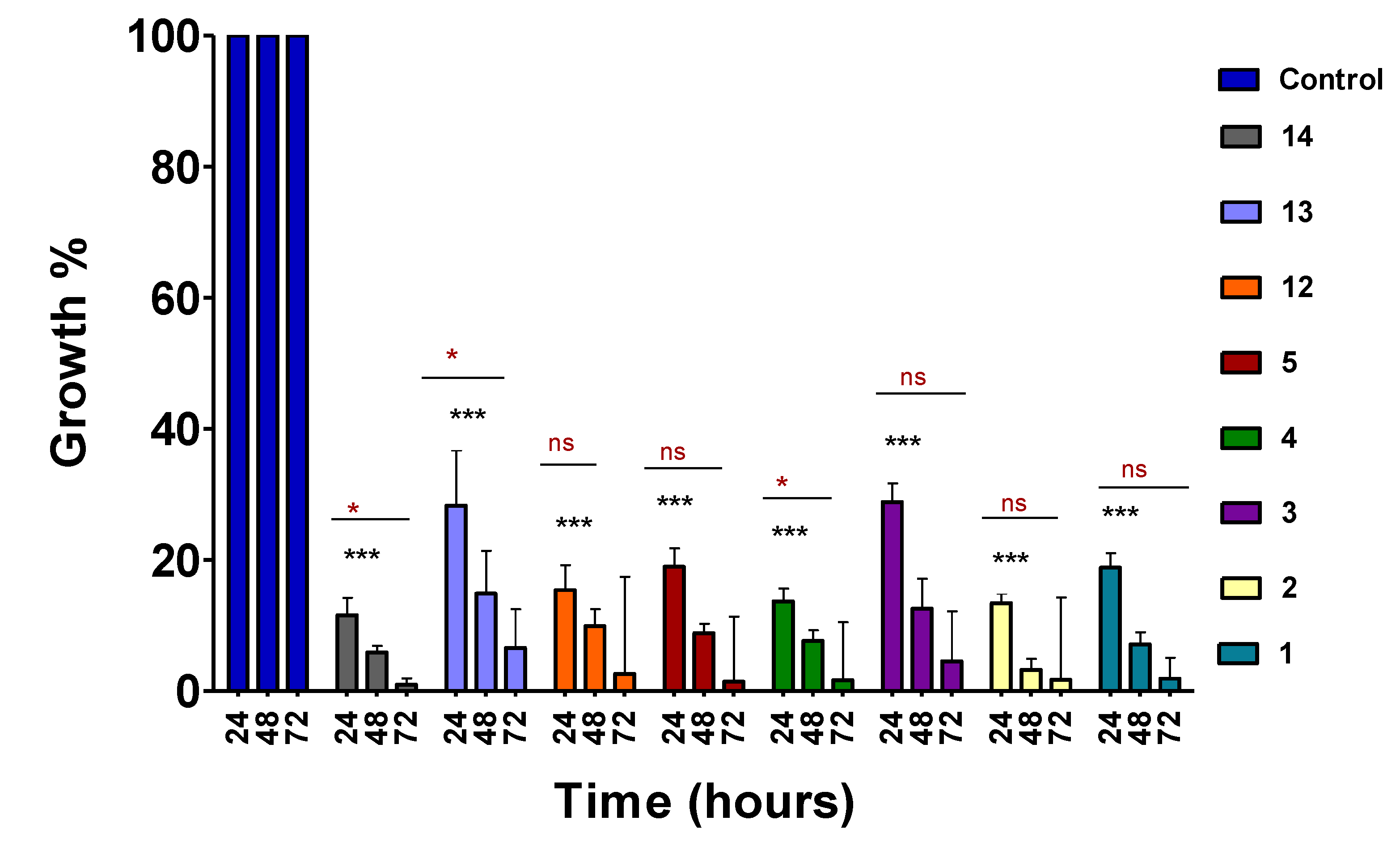
Figure 2.
Stage–specific effect of 4-aminoquinoline hydrazone compounds (1-5, 12-14) on synchronous cultures of P. falciparum K1 strain. The drug effects are expressed as a percentage of growth of the respective development stage relative to untreated control. Error bars represent the standard errors of the results from experiments performed three times with each concentration. The significant difference between the ring and schizont stage for each compound at various concentrations is indicated by ns for p > 0.05 and * for p < 0.05, ** for p < 0.01 and ***p <0.001. The significant difference was determined by two-way ANOVA.
Figure 2.
Stage–specific effect of 4-aminoquinoline hydrazone compounds (1-5, 12-14) on synchronous cultures of P. falciparum K1 strain. The drug effects are expressed as a percentage of growth of the respective development stage relative to untreated control. Error bars represent the standard errors of the results from experiments performed three times with each concentration. The significant difference between the ring and schizont stage for each compound at various concentrations is indicated by ns for p > 0.05 and * for p < 0.05, ** for p < 0.01 and ***p <0.001. The significant difference was determined by two-way ANOVA.
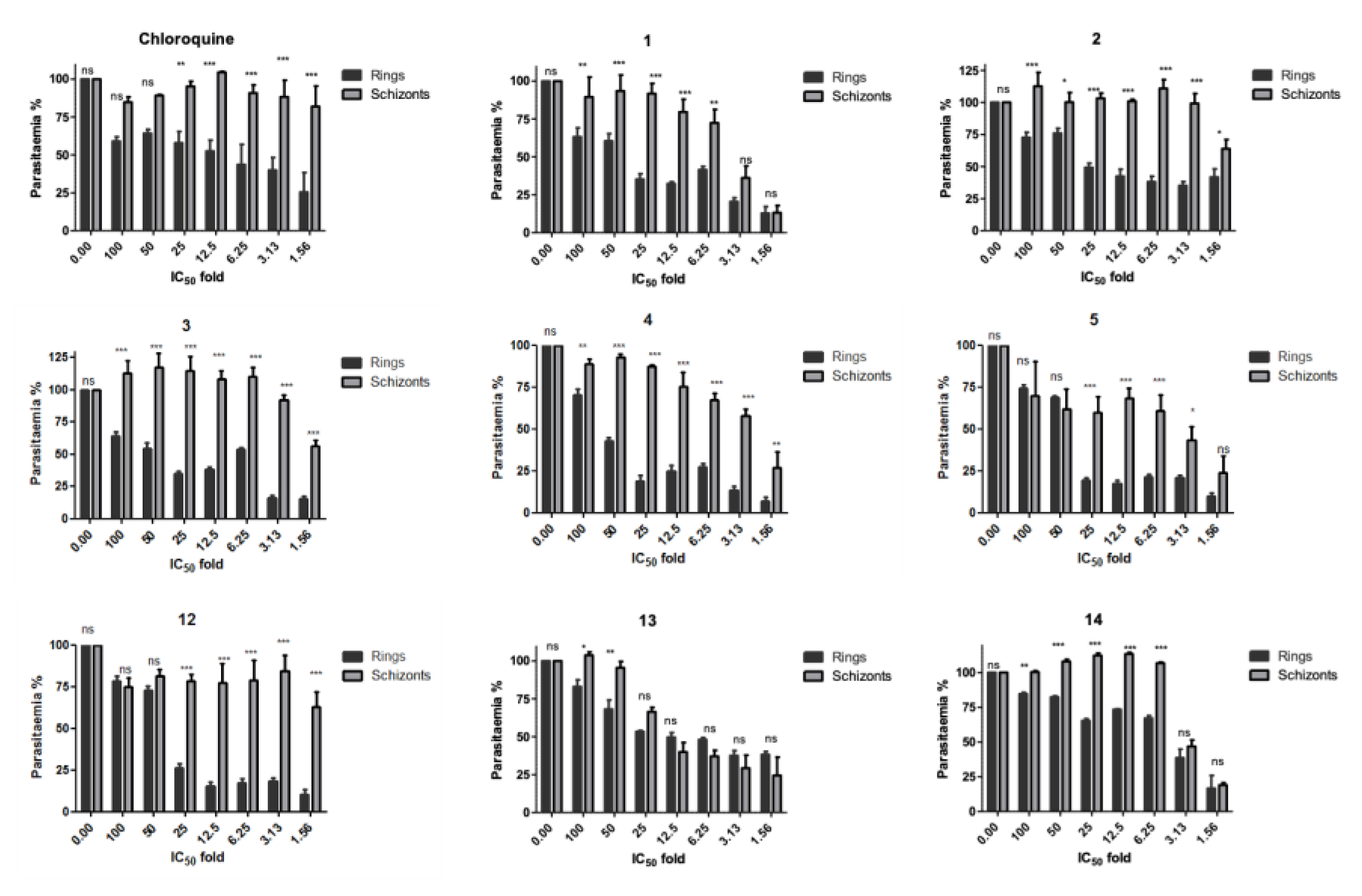
Figure 3.
Parasitaemia in infected (P. yoelii) mice during and after treatment with compound 2 at 5 mg/kg or 20 mg/kg doses and placebo for 4 days.
Figure 3.
Parasitaemia in infected (P. yoelii) mice during and after treatment with compound 2 at 5 mg/kg or 20 mg/kg doses and placebo for 4 days.
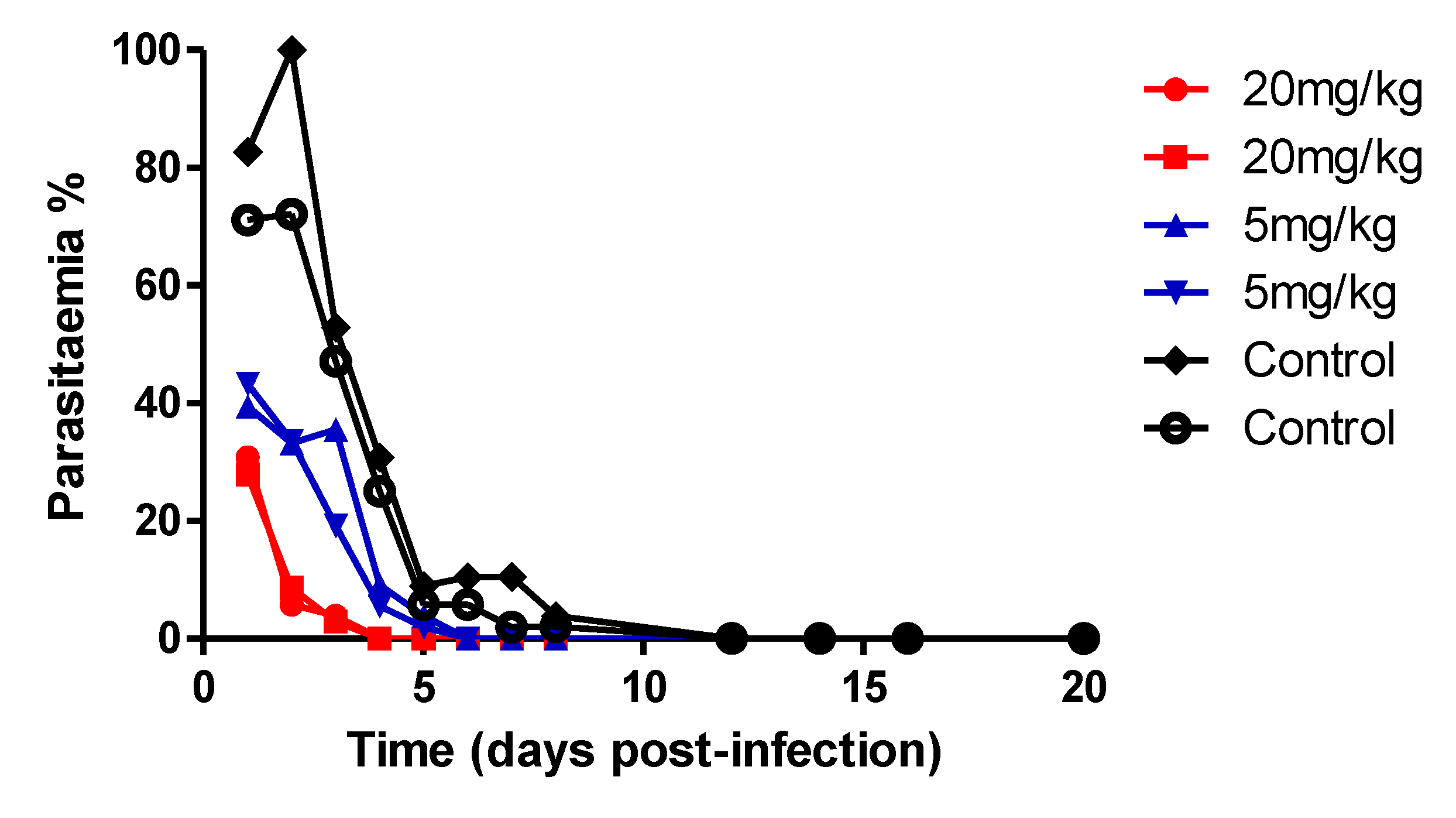

Table 1.
Inhibition of P. falciparum asexual growth by the 4-aminoquinoline hydrazones (1-16) was measured after an incubation period of 48 hours using the SYBR Green-based plate reader assay to give IC50 values (µM). Compound toxicity was measured in HepG2 and MDBK cells using the MTT assay. Both experiments were performed in triplicates to give standard error bars. Data was analysed using Graph Pad Prism. (Wide: standard error could not be calculated).
Table 1.
Inhibition of P. falciparum asexual growth by the 4-aminoquinoline hydrazones (1-16) was measured after an incubation period of 48 hours using the SYBR Green-based plate reader assay to give IC50 values (µM). Compound toxicity was measured in HepG2 and MDBK cells using the MTT assay. Both experiments were performed in triplicates to give standard error bars. Data was analysed using Graph Pad Prism. (Wide: standard error could not be calculated).
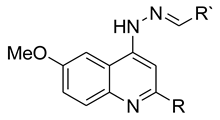 | |||||
| Plasmodium IC50(µM) ± SE | Cell toxicity IC50(µM) ± SE | Cell toxicity IC50(µM) ± SE | |||
| Compound | R | R’ | HepG2 | MDBK | |
| 1 | -Me | 4-Fluorophenyl | 0.612 ± 0.35 | 0.872 ± 0.79 | 3.77 ± 1.5 |
| 2 | -Me | Phenyl | 2.26 ± 2.2 | 1.90 ± 0.11 | 3.44 ± 2.7 |
| 3 | -Me | 3-Pyridinyl | 3.29 ± 1.6 | 3.00 ± 0.27 | 7.34 ± 7.6 |
| 4 | -Me | 2-Nitrofuranyl | 0.600 ± 0.84 | 1.59 ± 0.18 | 1.66 ± 0.21 |
| 5 | -Me | 2-Hydroxy-3-methoxyphenyl | 3.82 (wide) | 6.73 ± 0.73 | >25 |
| 6 | -Me | 4-Imidazoyl | 48.8 (wide) | 7.26 ± 1.00 | 11.7 ± 2.5 |
| 7 | -Me | 4-Nitrophenyl | 20.1 (wide) | 1.69 (wide) | 6.68 ± 1.7 |
| 8 | -Me | 4-Hydroxyphenyl | 22.3 (wide) | 1.97 ± 0.42 | 3.73 ± 1.2 |
| 9 | -Me | 4-Benzoic acid | >200 | >25 | >25 |
| 10 | -Me | 3-Hydroxyphenyl | 18.9 (wide) | 2.64 ± 0.20 | 9.71 ± 3.5 |
| 11 | -Me | 3,5-Dihydroxylphenyl | 35.9 ± 43 | 11.1 ± 2.9 | >25 |
| 12 | -Ph | 2-Hydroxy-3-methoxyphenyl | 5.67 ± 13 | 1.80 ± 0.78 | 6.60 |
| 13 | -Ph | Benzyl | 3.33 ± 2.4 | >25 | >25 |
| 14 | -Ph | 4-Nitrophenyl | 2.20 ± 1.5 | 0.89 ± 0.13 | 4.24 ± 1.0 |
| 15 | -Ph | 4-Imidazoyl | 28.3 ± 29 | 2.21 ± 0.15 | 9.31 ± 3.5 |
| 16 | -Ph | 4-N,N-dimethylaniline | 8.06 ± 4.5 | 4.15 ± 2.4 | 6.24 (wide) |
| Chloroquine | 0.199 ± 0.04 | ||||
| Cisplatin | 3.92 ± 0.64 | 9.15 ± 3.5 | |||
Table 2.
Inhibition of P. falciparum asexual growth in 72 hours. IC50 values (µM) of 4-aminoquinoline hydrazone compounds measured after an incubation period of 72 hours using SYBR Green-based plate reader assay. Experiments were performed three times, giving the Standard Error (SE). The selectivity indices calculated from cell viability IC50 determined in Table 1. Data were analysed using Graph Pad Prism. a Not Available (cytotoxicity IC50 > 50 μM)
Table 2.
Inhibition of P. falciparum asexual growth in 72 hours. IC50 values (µM) of 4-aminoquinoline hydrazone compounds measured after an incubation period of 72 hours using SYBR Green-based plate reader assay. Experiments were performed three times, giving the Standard Error (SE). The selectivity indices calculated from cell viability IC50 determined in Table 1. Data were analysed using Graph Pad Prism. a Not Available (cytotoxicity IC50 > 50 μM)
| Compound |
Plasmodium IC50 (µM) ± SE |
Selectivity indices (MDBK) |
Selectivity indices (HepG2) |
| 1 | 0.0257 ± 0.14 | 147 | 34 |
| 2 | 0.0329 ± 0.007 | 101 | 58 |
| 3 | 0.175 ± 0.04 | 42 | 17 |
| 4 | 0.219 ± 0.02 | 7 | 7 |
| 5 | 0.174 ± 0.03 | N/Aa | 39 |
| 12 | 0.133 ± 0.06 | 50 | 14 |
| 13 | 1.61 (wide) | N/Aa | N/Aa |
| 14 | 0.176 ± 0.06 | 31 | 5 |
Table 3.
The effective dose of compounds 1 and 2 tested in 4-fold serial dilutions from 3.05 nM – 200 µM on P. falciparum 3D7 and Dd2 strains. The parasites were treated with the compounds for 72 hours. The growth was read using the SYBR green-based plate reader assay. The experiment is compared to the K1 assay (also in Table 2). The experiments were performed at each concentration 3 times in triplicate. Data was analysed with GraphPad prism.
Table 3.
The effective dose of compounds 1 and 2 tested in 4-fold serial dilutions from 3.05 nM – 200 µM on P. falciparum 3D7 and Dd2 strains. The parasites were treated with the compounds for 72 hours. The growth was read using the SYBR green-based plate reader assay. The experiment is compared to the K1 assay (also in Table 2). The experiments were performed at each concentration 3 times in triplicate. Data was analysed with GraphPad prism.
| Compounds |
3D7 |
Dd2 IC50 ± SE (µM) |
K1 |
| 1 | 0.183 ± 1.04 | 0.133 ± 1.18 | 0.0257 ± 0.14 |
| 2 | 0.0554 ± 1.04 | 0.0244 ± 1.13 | 0.0329 ± 0.007 |
Table 4.
CalcuSyn outputs for the atovaquone-proguanil combination. The combinatory index values (CI) are shown for atovaquone and proguanil at the IC50, IC75 and IC90 levels of inhibition. The m and r values are also reported for all sets of data. The m value refers to the kinetic order and shape of the curve m=1, > 1 and < 1 indicates hyperbolic, sigmoidal and negative sigmoidal shape, respectively. The r value represents the linear correlation coefficient for the median effect plot and indicates conformity to the mass action law.
Table 4.
CalcuSyn outputs for the atovaquone-proguanil combination. The combinatory index values (CI) are shown for atovaquone and proguanil at the IC50, IC75 and IC90 levels of inhibition. The m and r values are also reported for all sets of data. The m value refers to the kinetic order and shape of the curve m=1, > 1 and < 1 indicates hyperbolic, sigmoidal and negative sigmoidal shape, respectively. The r value represents the linear correlation coefficient for the median effect plot and indicates conformity to the mass action law.
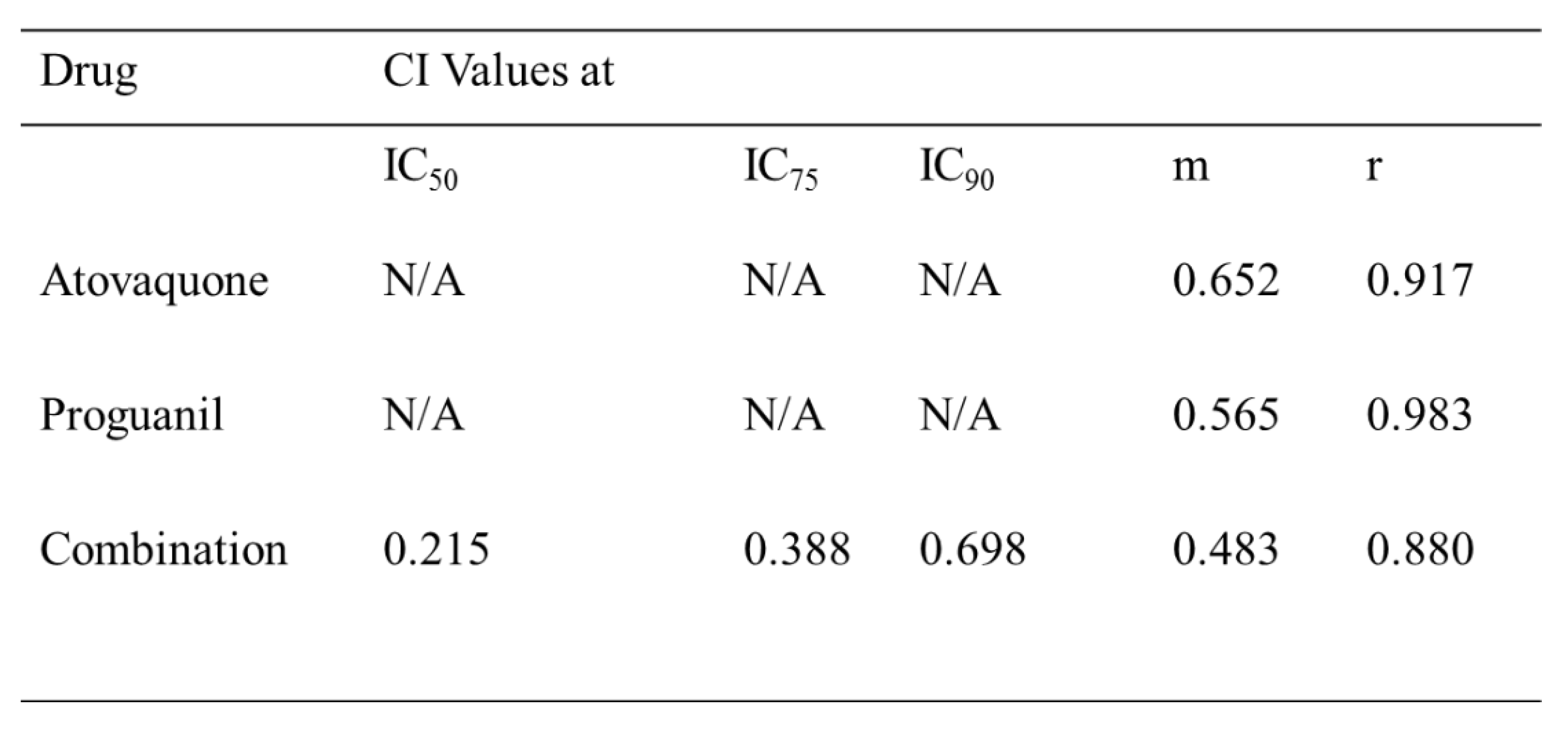
Table 5.
CalcuSyn outputs for the combination of known antimalarial drugs and compound 2. The combinatory index values (CI) are shown at the IC50, IC75 and IC90 levels of inhibition. The m and r values are also reported for all sets of data. The m values refer to the kinetic order and shape of the curve m=1, > 1 and < 1 indicates hyperbolic, sigmoidal and negative sigmoidal shape, respectively. The r value represents the linear correlation coefficient for the median effect plot and indicates conformity to the mass action law.
Table 5.
CalcuSyn outputs for the combination of known antimalarial drugs and compound 2. The combinatory index values (CI) are shown at the IC50, IC75 and IC90 levels of inhibition. The m and r values are also reported for all sets of data. The m values refer to the kinetic order and shape of the curve m=1, > 1 and < 1 indicates hyperbolic, sigmoidal and negative sigmoidal shape, respectively. The r value represents the linear correlation coefficient for the median effect plot and indicates conformity to the mass action law.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



