
Submitted:
28 July 2023
Posted:
31 July 2023
You are already at the latest version
Abstract
Hepatic macrophages act as the liver’s first line of defense against injury. Their differentiation into pro-inflammatory or anti-inflammatory subpopulations is a critical event that maintains a delicate balance between liver injury and repair. In our investigation, we explored the influence of the small heterodimer partner (SHP), a nuclear receptor primarily associated with metabolism, on macrophage differentiation during the innate immune response. During macrophage differentiation, we observed significant alterations in Shp mRNA expression. Deletion of SHP promoted M1 differentiation while interfering with M2 polarization. Conversely, overexpression of SHP resulted in increased expression of peroxisome proliferator activated receptor gamma (Pparg), a master regulator of anti-inflammatory macrophage differentiation, thereby inhibiting M1 differentiation. Upon lipopolysaccharide (LPS) injection, there was a notable increase in the pro-inflammatory M1-like macrophages, accompanied by exacerbated infiltration of monocyte-derived macrophages (MDMs) into the livers of Shp myeloid cell specific knockout (Shp-MKO). Concurrently, we observed significant induction of tumor necrosis factor alpha (Tnfa) and chemokine (C-C motif) ligand 2 (Ccl2) expression in LPS-treated Shp-MKO livers. Additionally, the mitogen-activated protein kinase (MAPK) and nuclear factor kappa B (NF-kB) pathways were activated in LPS-treated Shp-MKO livers. Consistently, both pathways were hindered in SHP overexpression macrophages. Finally, we demonstrated that SHP interacts with p65, thereby influencing macrophage immune repones. In summary, our study uncovered a previously unrecognized role of SHP in promoting anti-inflammatory macrophage differentiation during the innate immune response. This was achieved by SHP acting as a regulator for the Pparg, MAPK and NF-kB pathways.
Keywords:
nuclear receptor
; small heterodimer partner (SHP)
; knockout
; macrophage
; differentiation
1. Introduction
The liver serves as a primary target for the innate immune response due to its continuous exposure to microorganisms and products originating from the gut [1,2]. Within the liver, monocytes/macrophages, along with granulocytes and dendritic cells, act as key effector cells of the innate immune system. Hepatic macrophages are primarily composed of two distinct types: resident macrophages known as Kupffer cells (KCs), which originate from erythromyeloid progenitors derived from the yolk sac, and monocyte-derived macrophages (MDM). In the context of inflammation, monocytes migrate from the peripheral circulation to the liver, where they differentiate into tissue macrophages. These macrophages play crucial roles in functions such as phagocytosis of foreign particles and cellular debris, antigen presentation to lymphocytes, secretion of cytokines, modulation of immune responses, and the restoration of a normal tissue environment [3,4].
Macrophages possess remarkable plasticity, allowing them to adapt their phenotypes and functions in response to environmental cues. In vitro studies have revealed that macrophages can be broadly categorized into two major populations based on their distinct phenotypes: classically activated proinflammatory M1 macrophages and alternatively activated anti-inflammatory M2 macrophages [5,6]. However, it is worth noting that certain macrophages, such as tumor-associated macrophages, may exhibit characteristics that overlap between these two groups [7]. The prototypical signals triggering M1 proinflammatory activation include interferon gamma (IFN-gamma), lipopolysaccharide (LPS), and tumor necrosis factor alpha (TNFα), while interleukin 4 (IL4) serves as a key signal for anti-inflammatory M2 activation. Although the M1/M2 classification may oversimplify the intricate biological response of macrophages in vivo, numerous studies have substantiated that macrophage differentiation into distinct pro-inflammatory or anti-inflammatory phenotypes significantly influences host defense and the pathogenesis of various liver diseases [8,9,10]. Therefore, the identification of heterogeneous macrophage populations and a comprehensive understanding of the molecular mechanisms governing macrophage heterogeneity are crucial for assessing disease progression, evaluating treatment outcomes, and developing targeted therapeutics that specifically modulate macrophage function [11,12,13,14,15].
Our study aimed to investigate the involvement of small heterodimer partner (Nr0b2, Homo sapiens SHP; Mus musculus Shp) in macrophage differentiation during the innate immune response. SHP is a nuclear receptor lacking DNA binding domain and known endogenous ligands [16]. It functions as a negative regulator of gene transcription and plays a crucial role in the regulation of bile acid, glucose, and energy metabolism through its interactions with other nuclear receptors and transcription factors [16,17,18,19,20]. Recent studies have shed light on a novel function of SHP in inflammation, where it acts as a negative regulator of immune response [21]. Mice lacking SHP are more susceptible to endotoxin-induced sepsis and concanavalin A-induced hepatitis [16,22,23,24] while inducing SHP expression has been found to ameliorate systemic inflammatory responses [25]. Moreover, we have recently discovered an anti-inflammatory role of SHP during the development of non-alcoholic steatohepatitis (NASH), where the loss of SHP in hepatocytes triggers nuclear factor kappa B (NF-κB) activation and the release of chemokine (C-C motif) ligand 2 (CCL2), exacerbating liver inflammation and fibrosis [26,27].
While the role of SHP in repressing innate immune activation has been well-documented, its involvement in macrophage differentiation during the innate immune response remains unclear. To address this gap in knowledge, we utilized a cell type-specific knockout mouse model to examine the role of SHP in macrophage differentiation. We found that Shp mRNA was down-regulated in pro-inflammatory M1 macrophages, but up-regulated in anti-inflammatory M2 macrophages. Deletion of Shp promoted M1 macrophage differentiation while interfering with M2 macrophage polarization. Conversely, overexpression of SHP resulted in increased expression of peroxisome proliferator activated receptor gamma (Pparg) with decreased M1 differentiation. Consistently, in Shp myeloid cell specific knockout (Shp-MKO) mouse model, we observed increased hepatic infiltration of monocytes and M1 macrophage differentiation following LPS challenge, accompanied by augmented activation of the mitogen-activated protein kinase (MAPK) and NF-κB pathways resulting from the loss of macrophage SHP in Shp-MKO. In summary, our study sheds light on the crucial role of SHP in modulating hepatic macrophage differentiation, contributing to the regulation of the inflammatory response and immune balance in the liver.
2. Materials and Methods
2.1. Cell lines, chemicals, plasmids, and antibodies
Mouse macrophage RAW 264.7 cells (ATCC TIB-71) were cultured in Dulbecco's Modified Eagle's Medium supplemented with 100 units/ml penicillin G-streptomycin sulfate and 10% heat-inactivated fetal bovine serum. To achieve overexpression of FLAG-SHP, a lentiviral vector pMSCV-puro was employed, and stable overexpression cells were selected using puromycin (Fisher, A1113802). The cells were treated with 100 ng/ml lipopolysaccharide (LPS, Sigma, L2654) for various time intervals (0, 5, 10, 30, and 60 minutes) for subsequent western blot analysis. For western blotting, immunohistochemistry staining, and immunoprecipitation, the following antibodies were utilized: β-actin (Sigma, A-1978), phospho-JNK (Thr-183/Tyr-185) (Cell Signaling Technology, 4668), JNK (Cell Signaling Technology, 9252), phospho-c-Jun (Ser-63) (Cell Signaling Technology, 2361), α-tubulin (Sigma, T6074), histone H3 (Cell Signaling Technology, 14269), phosphor-TAK1 (Ser-412) (Cell Signaling Technology, 9339), TAK1 (Cell Signaling Technology, 4505), phospho-SEK1/MKK4 (Ser257) (Cell Signaling Technology, 4514), SEK1/MKK4 (Cell Signaling Technology, 9152), phospho-IKKα (Ser176)/IKKβ (Ser177) (Cell Signaling Technology, 2078), IKKβ (Cell Signaling Technology, 8943), phospho-IκBα (Ser32/36) (Cell Signaling Technology, 9246), IκBα (Cell Signaling Technology, 4814), NF-κB p65 (Cell Signaling Technology, 8242), and F4/80 (Cell Signaling Technology, 70076).
2.2. Animal studies
C57BL/6J mice (stock no. 000664) were procured from the Jackson Laboratory. Shpflox/flox mice, generously provided by Drs. Johan Auwerx and Kristina Schoonjans at the Ecole Polytechnique de Lausanne were backcrossed into the C57BL/6J background for 10 generations. Shpflox/flox mice were crossed with LysM-Cre mice (Jackson Laboratory, Stock No: 004781) to generate heterozygous mice. Subsequently, the heterozygous mice were bred to obtain Shp myeloid cell specific knockout (Shp-MKO represents Shp flox/flox; LysMcre positive) and their littermate wild-type controls (WT represents Shp flox/flox; LysMcre negative). Mice were housed in a virus-free facility with a 12-h light/dark cycle (lights on from 6 a.m. to 6 p.m.) and maintained at a temperature of 25 °C, with ad libitum access to food and water. Male mice aged 8-10 weeks were used for the experiments, unless otherwise stated (n = 5/group). In the LPS injection experiment, both WT and Shp-MKO mice received intraperitoneal injection of LPS at 1 mg/kg body weight. Samples were collected at 0-, 3-, and 7-hour post-injection. For the bone marrow-derived macrophage polarization experiment, a published protocol was followed [28]. In brief, the femur and tibia were collected from the mice, and bone marrow cells were differentiated into macrophages using mouse macrophage colony-stimulating factor (M-CSF, R&D Systems™, 416ML010) at 10 ng/ml for 7 days. On the 7th day, the differentiated macrophages were cultured with IFN-gamma (100 ng/mL) or IL4 (50 ng/ml) for 24 hours to induce M1 or M2 macrophage polarization, respectively. All experiments were conducted in compliance with relevant guidelines and regulations approved by the Institutional Animal Care and Use Committee (ICAUC) at the University of Kansas Medical Center.
2.3. Hepatic cell isolation and flow cytometry analysis
Hepatic cell isolation and purification were conducted at the Kansas University Medical Center Cell Isolation Core, following a previously described method [29] with slight modifications. In brief, mouse livers were perfused with 25 ml of solution I (9.5 g/liter Hanks’ balanced salt solution, 0.5 mmol/liter EGTA, pH 7.2), followed by 50 ml of solution II (9.5 g/liter Hanks’ balanced salt solution, 0.14 g/liter collagenase IV, and 40 mg/liter trypsin inhibitor, pH 7.5). After digestion, a single-cell suspension was obtained and filtered through a 100-μm Falcon cell strainer (Fisher Scientific, 08-771-19). The cells were centrifuged at 50 × g for 5 min at 4 °C to pellet hepatocytes. The supernatant containing nonparenchymal cells (NPCs) was then centrifuged at 300 × g for 10 min at 4 °C to enrich NPCs. In hepatic macrophage polarization experiment, macrophages were captured from NPCs by CD11b MicroBeads (Miltenyi Biotec Inc. 130-049-601) and differentiated into M1 or M2 macrophages using DMEM media supplemented with IFN-gamma (100 ng/mL) or IL4 (50 ng/ml) for 24 hours, respectively. In flow cytometry experiment, approximately 1 × 106 NPCs were incubated with anti-mouse CD16/CD32 (TruStain FcX, BioLegend, USA, cat. 101319) diluted in FACS buffer (2 mM EDTA, 10% FBS in PBS) for 15 minutes on ice to block non-specific antibody binding. Subsequently, the cells were incubated with the Brilliant Violet 605™ CD45 (BioLegend, USA, 103139), Brilliant Violet 421™ CD11b (BioLegend, USA, 101235), PE/Cyanine7 Ly-6C (BioLegend, USA, 128017) anti-mouse antibodies, and the fixable viability dye (Zombie Aqua, BioLegend, 423101) for 30 minutes on ice. After centrifugation (300 × g) for 5 minutes, the cells were washed twice with 1 ml of PBS for 5 minutes and finally resuspended in 300 µl of FACS buffer. The cells were then analyzed using a FACS Calibur instrument (BD, Franklin Lakes, New Jersey). FlowJo-V10 software was used for data analysis.
2.4. Liver histology and immunohistochemistry
Fresh liver tissues were fixed with 10% formalin (Fisher, SF100) to preserve their structural integrity. Paraffin sections of 5 μm thickness were prepared and subjected to staining with hematoxylin and eosin (H&E) for general tissue examination, as well as immunohistochemical staining. For the immunohistochemistry staining of F4/80, the paraffin sections were rehydrated and treated with 0.3% hydrogen peroxide in PBS for 15 minutes to block endogenous peroxidase activity. Antigen retrieval was achieved by boiling the sections in sodium citrate buffer (pH 6.0) for 5 minutes using a pressure cooker. Subsequently, the slides were treated with 5% normal serum for 30 minutes to block non-specific binding, followed by overnight incubation with rabbit anti-mouse F4/80 antibody at 4 °C. For the final detection, an ImmPRESS peroxidase polymer detection kit (Vector Laboratories, MP-7444) and ImmPACT 3,3'-diaminobenzidine peroxidase substrate (Vector Laboratories, SK-4105) were utilized. After thorough washing, the sections were counterstained with hematoxylin, dehydrated, cleared, and mounted. Microscopic images were captured using a BX60 microscope, and the area of positive staining for DAB (3,3'-diaminobenzidine) was quantified using ImageJ software.
2.5. Real-time quantitative PCR
The real-time quantitative PCR (qPCR) analysis was performed using the SYBR Green PCR master mix (Applied Biosystems), following the previously described protocols [26,27,30]. The specific primer sequences utilized for the qPCR are provided in Table S1. The hsa-miR-34a-5p LNA™ PCR primer set (Exiqon, 204486) has been used to measure the expression level of miR-34a. The abundance of PCR products was quantified using threshold cycle (Ct) values, and the relative ratio of specific genes to the housekeeping gene actin was determined. The resulting values were then presented as the fold change in the tested group compared to the control group.
2.6. Western blotting and immunoprecipitation
Mouse liver tissues were prepared for protein analysis using the following procedures. First, the tissues were homogenized using a PowerGen 700 homogenizer (Fisher Scientific) in lysis buffer containing protease inhibitors (Fisher Scientific, protease inhibitor mixture PI78410). The lysis buffer consisted of 50 mM Tris (pH 7.5), 1% Nonidet P-40, 150 mM NaCl, 0.5% sodium deoxycholate, and 0.1% SDS, ensuring efficient extraction of whole protein lysates. For the extraction of nuclear and cytoplasmic proteins, a commercial kit (Fisher, PI78833) was utilized according to the manufacturer's instructions. Next, protein lysates (60 μg) were separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were then blocked and incubated with primary antibodies specific to the target proteins. Subsequently, horseradish peroxidase-conjugated secondary antibodies were applied, allowing for the detection of antibody binding. The visualization of antibody-bound proteins was achieved using either SuperSignal West Pico Plus Chemiluminescent Substrate (Fisher, PI34580) or SuperSignal West Femto Chemiluminescent Substrate (Fisher, PI34094). Images were captured using a LI-COR imaging system. To ensure equal protein loading, loading controls such as β-actin, α-tubulin, and histone H3 were included and verified. Quantitative analysis of band intensity was performed using Image Studio Lite software, and the relative expression levels were normalized to the loading controls. For the immunoprecipitation experiment, 1000 μg of whole protein lysates from control PMSCV cells and PMSCV-SHP cells overexpressing FLAG-SHP were incubated with 2 μg of anti-FLAG M2 magnetic beads (Sigma, M8823). The immune complexes were captured using a magnetic stand, and subsequent elution was performed using 2× SDS loading buffer. The pulldown of p65 and FLAG-SHP was detected by Western blotting. A TrueBlot® anti-rabbit IgG HRP (Rockland, RL18-8816-33) was used as a secondary antibody as this antibody does not interfere with the immunoprecipitation of immunoglobulin heavy and light chains, ensuring accurate detection.
2.7. Statistical analysis
GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA) was used for data analysis. The quantitative data are presented as the mean ± SEM. Statistical analysis was performed using Student's t-test to determine the significant difference between two groups. For comparisons among multiple groups, one-way analysis of variance (ANOVA) was conducted, followed by Duncan's test. Statistical significance was considered at a 95% confidence level.
3. Results
3.1. Macrophage differentiation alters Shp mRNA expression
To investigate the impact of macrophage differentiation on the expression of Shp, we conducted experiments using hepatic macrophages isolated from C57BL/6J liver. The macrophages were differentiated into either pro-inflammatory M1 or anti-inflammatory M2 macrophages by treating them with IFN-gamma (100 ng/ml) or IL4 (50 ng/ml), respectively. Following a twenty-four-hour incubation, successful differentiation into M1 or M2 macrophages was confirmed by observing significant upregulation of genes encoding pro-inflammatory markers Tnfa and nitric oxide synthase 2 (Nos2) in M1 macrophages, as well as the anti-inflammatory markers arginase 1 (Arg1) and CD163 antigen (Cd163) in M2 macrophages (Figure 1). Interestingly, we observed that M1 differentiation led to the inhibition of Shp mRNA expression, while M2 differentiation resulted in its increased expression (Figure 1). These findings strongly suggest that macrophage differentiation alters Shp mRNA expression.
3.2. Shp deletion in macrophages enhances M1 polarization but impairs M2 differentiation
To assess the role of SHP in regulating macrophage differentiation, we crossed Shpflox/flox with LysMcre mice and generated Shp-MKO and WT controls. Confirmation of Shp deletion from myeloid cells was achieved through qPCR analysis of peritoneal macrophages isolated from both the Shp-MKO and littermate WT controls (Figure 2A). Subsequently, we performed macrophage differentiation experiments using bone marrow cells obtained from the WT and Shp-MKO mice, which were cultured with macrophage colony-stimulating factor (M-CSF) for 7 days to generate bone marrow-derived macrophages (BMDMs). On the seventh day, the differentiated BMDMs were treated with either IFN-gamma or IL4. Remarkably, IFN-gamma treatment significantly increased the mRNA expression of proinflammatory M1 markers, Tnfa and Nos2, in WT macrophages (Figure 2B). Strikingly, this effect was augmented in the Shp knockout macrophages (Figure 2B), indicating that the absence of Shp enhanced M1 macrophage polarization. Conversely, M2 anti-inflammatory macrophage differentiation was impaired in the Shp knockout macrophages, resulting in a reduced induction of M2 marker mannose receptor C type 1 (Mrc1 or Cd206) mRNA compared to the WT after IL4 treatment (Figure 2B). These findings clearly demonstrate that knocking out Shp in macrophages enhanced M1 polarization while impairing M2 polarization.
3.3. SHP overexpression increases the expression of Pparg and inhibits M1 differentiation
To investigate whether SHP overexpression in macrophages could reverse the observed effects in Shp-MKO BMDMs, we utilized lentiviral transduction to introduce a Flag-SHP fusion protein into the murine macrophage cell line RAW 264.7. Stable SHP overexpression cells (PMSCV-SHP) were then selected using puromycin, while cells infected with the lentiviral vector PMSCV served as a control. The successful overexpression of SHP in PMSCV-SHP cells was confirmed through Western blot analysis (Figure 2C). Considering that Pparg acts as a master regulator of anti-inflammatory macrophage differentiation [31,32], and our previous study revealed a close relationship between Shp and Pparg, with Shp deletion decreasing Pparg mRNA expression [27], we hypothesized that SHP overexpression would increase Pparg expression. Given that Shp inhibits the expression of miR-34a [33] and miR-34a can target the 3’-untranslated region (3’UTR) of Pparg mRNA [34] to decrease Pparg mRNA expression [35], we further speculated that overexpression of SHP would result in decreased miR-34a expression and increased Pparg expression. To test this hypothesis, we examined the mRNA expression of miR-34a and Pparg in PMSCV-SHP RAW cells and vector control cells. As expected, PMSCV-SHP cells exhibited a downregulation of miR-34a and upregulation of Pparg compared to vector control cells (Figure 2C). This indicates that SHP overexpression indeed increases the expression of Pparg, which should theoretically inhibit M1 macrophage differentiation. Indeed, the anticipated effect of SHP overexpression was observed, as it significantly inhibited the expression of Nos2, a marker of M1 proinflammatory macrophages, both in the control condition and after IFN-gamma stimulation (Figure 2D). Furthermore, we noticed a decrease in Pparg mRNA after IFN-gamma treatment in both PMSCV-SHP RAW cells and vector controls; However, the PMSCV-SHP RAW cells exhibited persistently higher expression of Pparg (Figure 2D). These results demonstrate that SHP plays a critical role as a regulator of macrophage polarization, with Pparg likely involved in the underlying mechanism. Overall, our findings emphasize the importance of SHP in modulating macrophage differentiation in vitro, as its absence promotes M1 polarization and impairs M2 polarization. Conversely, SHP overexpression increases Pparg and inhibits M1 macrophage differentiation, highlighting its potential as an important regulator of macrophage differentiation during the innate immune response.
3.4. Shp knockout leads to a persistent hepatic infiltration of proinflammatory monocytes and M1 macrophage differentiation following LPS challenge
To assess the impact of abnormal macrophage polarization resulting from Shp deletion on the immune response to endotoxin challenge, we conducted an intraperitoneal injection of a low dose of LPS (1 mg/kg body weight) in both WT and Shp-MKO mice. Samples were collected at 0-, 3-, and 7-hour intervals after injection to determine the extent of the immune response (Figure 3A). Notably, the low dose of LPS did not cause significant changes in mouse body weights, liver weights, or liver-to-body weight ratios between the WT and Shp-MKO mice (Figure 3B). Histological examination of liver sections using H&E staining did not reveal any evident differences between the two groups after LPS injection (Figure 3C). However, immunohistochemical staining for adhesion G protein-coupled receptor E1 (Emr1 or F4/80), a surface marker of macrophages, indicated a significant increase in macrophage numbers in both WT and Shp-MKO livers after 3 hours of LPS injection (Figure 3D). Subsequently, macrophage numbers in WT livers returned to basal levels after 7 hours of LPS challenge. In contrast, the Shp-MKO livers maintained elevated macrophage numbers at the 7-hour timepoint following LPS injection (Figure 3D). These findings suggest that myeloid Shp knockout mice sustain a pro-inflammatory signal after LPS challenge and lack an anti-inflammatory mechanism to halt macrophage accumulation during the resolution phase of inflammation.
To further investigate the inflammatory phenotype and origin of hepatic macrophages, flow cytometry analysis was performed. Our hypothesis centered on the idea that the lack of Shp in myeloid cells would result in enhanced differentiation of M1 pro-inflammatory macrophages within the liver. This effect was likely attributable to the enhanced infiltration of pro-inflammatory monocytes into the hepatic tissue due to loss of Shp, particularly following the LPS challenge. To test this hypothesis, LPS (1 mg/kg body weight) was administered intraperitoneally to both WT and Shp-MKO mice. After 3 hours, liver perfusion was performed, followed by the isolation of liver non-parenchymal cells (NPCs). Those NPCs were then subjected to staining with specific cell markers, including hematopoietic cell marker protein tyrosine phosphatase receptor type C (PTPRC or CD45), macrophage marker F4/80, monocyte marker integrin alpha M (Itgam or CD11b), and M1 proinflammatory cell marker Ly6-C antigen (Ly6C). Subsequently, the leukocyte population was isolated based on forward scatter (FSC) vs side scatter (SSC) and then gated for CD45 expression. As anticipated, the population of pro-inflammatory M1-like macrophages, identified as F4/80+Ly6CHigh by flow cytometry, displayed a significant increase in Shp-MKO livers compared to WT controls, both under basal and LPS challenge conditions (Figure 4A). Similarly, the population of pro-inflammatory CD11b+Ly6CHigh monocytes was significantly higher in Shp-MKO livers compared to WT controls (Figure 4B). Additionally, under basal conditions, the population of monocyte derived macrophages (MDMs) within the liver, identified as CD11bHighF4/80Intermediate, was significantly elevated in Shp-MKO livers compared to WT livers (Figure 4C). Furthermore, the LPS challenge induced a substantial increase in MDM infiltration into the liver, which was nearly tripled in Shp-MKO livers (Figure 4C). These findings strongly suggest that the targeted deletion of Shp in myeloid cells fosters the infiltration of pro-inflammatory monocytes into the liver and enhances pro-inflammatory M1 macrophage differentiation in response to endotoxin.
3.5. Shp deletion in myeloid cells leads to increased cytokine production in response to LPS challenge
Our previous study has shown that LPS treatment decreased Shp mRNA expression in hepatocytes [26]. Consistent with our previous findings, we observed a sharp reduction in hepatic Shp mRNA levels at both 3-hour and 7-hour time points following LPS challenge in WT mice (Figure 5A). Notably, there was a partial recovery of Shp mRNA expression in WT livers at the 7-hour time point. In contrast, Shp mRNA was nearly undetectable in Shp-MKO liver after LPS challenge at both time points (Figure 5A). LPS challenge significantly increased the mRNA levels of pro-inflammatory genes Ccl2, Cd11b, and Ly6C in WT livers, and this response was further augmented in Shp-MKO livers (Figure 5A). Importantly, we observed a sustained elevation in Cd11b and Ly6C mRNA levels in the Shp-MKO liver after 7 hours of LPS challenge (Figure 5A). This finding aligns with the increased presence of pro-inflammatory MDMs and M1 macrophages in the Shp-MKO liver after LPS challenge, as detected through flow cytometry analysis (Figure 4). Consistent with the changes in Ccl2 mRNA levels observed in WT livers, the serum concentration of CCL2 increased after 3 hours of LPS challenge and returned to basal levels after 7 hours (Figure 5B). A similar pattern was observed for serum TNFα levels in WT mice. Strikingly, Shp-MKO mice exhibited approximately 2-3 times higher induction of serum CCL2 and TNFα compared to WT controls at all time points after LPS challenge (Figure 5B). These results collectively suggest that the loss of macrophage Shp leads to the absence of an anti-inflammatory mechanism in Shp-MKO mice, highlighting the important regulatory role of myeloid Shp in controlling inflammatory responses.
3.6. Shp deletion results in hyperactivation of both MAPK and NF-κB signaling pathways
Both the MAPK and NF-κB signaling pathways are well-established as promoters of pro-inflammatory M1 macrophage differentiation [36,37,38]. To investigate whether the abnormal macrophage polarization observed in Shp-MKO mice could be attributed to the hyperactivation of MAPK and NF-κB signaling, we examined the activation status of several intermediate and effector proteins in these pathways in the livers of WT and Shp-MKO mice following LPS injection. After 3 hours of LPS challenge, we observed significantly higher levels of phosphorylated proteins associated with MAPK signaling, including p-TAK1 Ser412, p-JNK Thr183/Tyr185, and p-c-Jun Ser63, in the Shp-MKO livers compared to WT controls (Figure 6A). Furthermore, after 7 hours of LPS challenge, we observed continuous high levels of p-MKK Ser257, p-JNK Thr183/Tyr185, and p-c-Jun Ser63 in the Shp-MKO liver (Figure 6A). These findings indicate that Shp-MKO mice exhibit augmented MAPK activation following LPS challenge, sustaining an overall higher amplification of the immune response in Shp-MKO mice.
Additionally, we found that the phosphorylated proteins associated with NF-κB signaling, including p-IKKα/β Ser176/177 and p-IκBα Ser32/36, were significantly higher in the Shp-MKO livers compared to WT livers after both the 3-hour and 7-hour LPS challenges (Figure 6A). This suggests a hyperactivation of the NF-κB signaling pathway in the Shp-MKO liver compared to WT liver following LPS injection. To further confirm this observation, we assessed the nuclear translocation of p65, a marker of NF-κB pathway activation, and found significantly higher inductions at both the 3-hour and 7-hour timepoints after LPS challenge in the Shp-MKO liver compared to WT controls (Figure 6B). Collectively, these observations indicate that myeloid SHP negatively regulates the activation of both the MAPK and NF-κB pathways following LPS challenge.
To gain deeper insights into the role of SHP in regulating these pathways, we conducted experiments using RAW cells overexpressing SHP (PMSCV-SHP) and vector control PMSCV RAW cells. These cells were treated with LPS (100 ng/ml) for varying durations (0, 5, 10, 30, and 60 minutes), and we examined the expression of proteins involved in the MAPK and NF-κB pathways. Our results demonstrated that LPS treatment led to time-dependent changes in the expression of p-MKK4 Ser257, p-JNK Thr183/Tyr185, p-c-Jun Ser63, p-IKKα/β Ser176/177, and p-IκBα Ser32/36 (Figure 7A). Remarkably, the overexpression of SHP in macrophages resulted in a reduction of these phosphorylated proteins after LPS treatment (Figure 7A). Prior studies have highlighted SHP's ability to negatively regulate the function of various transcription factors and nuclear receptors through direct protein-protein interactions [16]. Building on this knowledge, we delved further into the potential interactions between SHP and proteins in the MAPK and NF-κB pathways. Employing co-immunoprecipitation (Co-IP) and western blot analyses, we successfully identified proteins interaction between SHP and p65 (Figure 7B). These findings conclusively establish that SHP plays a pivotal role in regulating immune response by inhibiting both MAPK and NF-κB signaling pathways.
4. Discussion
Hepatic macrophages are key players in innate immunity and vital components of the liver. These macrophages display remarkable heterogeneity and plasticity, allowing them to respond to diverse stimuli in different physiological and pathological conditions [39,40,41,42]. Traditionally, macrophages have been classified into two extreme groups: M1 classically activated proinflammatory macrophages and M2 alternatively activated anti-inflammatory macrophages. However, emerging research employing single-cell RNA sequencing has unveiled the intricate nature of macrophage differentiation, showcasing a multitude of activation states that surpass the conventional M1/M2 classification. Nevertheless, despite this newfound complexity, the M1/M2 classification continues to serve as a valuable framework for understanding macrophage function and gene roles, offering a useful overview in the study of macrophage biology. SHP is an atypical nuclear receptor that plays a critical role in various pathophysiological processes, including inflammation, metabolism, and energy homeostasis [17,19,23]. Our previous research has highlighted the anti-inflammatory role of hepatic SHP in a mouse model of nonalcoholic steatohepatitis [26,27]. Building upon these findings, our current study aimed to investigate the role of myeloid SHP in macrophage polarization during acute innate immune response. We discovered that SHP regulates macrophage polarization, and its influence on M1 and M2 differentiation is an important mechanism through which SHP inhibits inflammation. Mechanistically, we made the novel discovery that myeloid SHP modulates macrophage differentiation by regulating Pparg, MAPK, and NF-κB pathways.
An intriguing finding of our study is the alteration of SHP expression during macrophage polarization. Specifically, anti-inflammatory M2 macrophage differentiation led to an increase in Shp mRNA expression, while pro-inflammatory M1 macrophage differentiation resulted in its decrease. Although the precise mechanisms underlying this regulation are unknown and beyond the scope of our current study, previous studies have demonstrated that several nuclear receptors and transcription factors bind to the Shp gene promoter and influence its expression [16]. For instance, PPARg can bind to the PPAR response element on the Shp gene promoter and induce Shp mRNA expression [43]. Macrophage-stimulating factor (MSP) increases Shp mRNA expression through the activation of AMP-activated protein kinase (AMPK) pathway [44,45]. Considering that PPARg and AMPK pathways are upregulated during M2 macrophage differentiation [46,47], it is tempting to speculate that the increase in Shp mRNA in M2 macrophages may be attributed to the activation of these pathways. Conversely, JNK activation suppresses Shp transcription in hepatocytes [26] and JNK activation is required for M1 macrophage polarization [48]. Hence, it is possible that JNK activation in M1 macrophages leads to the decreased Shp mRNA expression. Further investigations are warranted to explore whether manipulating PPARg, AMPK, or JNK in macrophages can alter SHP expression during macrophage differentiation.
Motivated by the differential SHP expression observed in M1 and M2 macrophages, we sought to determine whether SHP plays a functional role in macrophage polarization. To this end, we generated a genetic mouse model lacking Shp specifically in myeloid cells using LysM-Cre-mediated knockout. We isolated BMDMs from both Shp-MKO and WT controls and found that Shp loss inhibited the polarization of BMDMs toward an M2 phenotype while promoting polarization toward an M1 state. Moreover, overexpression of SHP in the macrophage cell line RAW cells increased Pparg mRNA expression and inhibited macrophage polarization toward a M1 phenotype. Encouraged by these in vitro results, we conducted in vivo studies by injecting a low dose of LPS into Shp-MKO and WT controls. We observed a sustained increase in liver macrophage numbers in Shp-MKO mice following LPS challenge. Notably, flow cytometry revealed a higher population of CD11bHigh F4/80Intermediate monocyte-derived macrophages in Shp-MKO livers compared to WT controls after LPS challenge, suggesting that the loss of SHP in myeloid cells enhances monocyte infiltration into the liver, replenishing hepatic macrophage populations. Monocyte recruitment to the liver is finely regulated by chemokines, among which CCL2 plays a crucial role. Inhibition of CCL2 or genetic knockout of Ccl2 specifically in myeloid cells has been shown to reduce monocyte infiltration into the liver during both acute and chronic hepatic injury [49,50,51]. In our previous study, we found that the loss of Shp in hepatocytes triggers the production of CCL2, leading to the initiation of monocyte recruitment [26]. Hence, we postulated that the increased monocyte infiltration observed in Shp-MKO livers after LPS challenge might be due to elevated CCL2 levels following SHP loss in myeloid cells. Our observations confirmed this speculation, as we noted a significant increase in hepatic Ccl2 mRNA expression and elevated serum CCL2 levels in Shp-MKO mice. These results suggest that SHP universally regulates CCL2 expression in various cell types, contributing to the enhanced monocyte infiltration observed in Shp-MKO livers during the response to LPS challenge.
Our flow cytometry analysis yielded a significant discovery, revealing increased populations of F4/80+Ly6CHigh and CD11b+Ly6CHigh cells in Shp-MKO livers compared to WT controls. Ly6C, a member of the lymphocyte antigen-6 (Ly6)/urokinase-type plasminogen activator receptor protein superfamily, is closely associated with infiltrating monocytes and is involved in the production of pro-inflammatory cytokines and chemokines, such as interleukin 1 (IL-1), interleukin 18 (IL-18), and Ccl2 [52]. Ly6CHigh monocytes are recognized as a pro-inflammatory subset contributing to tissue inflammation and T-cell activation [53]. Notably, Ly6CHigh monocytes infiltration and their subsequent differentiation into pro-inflammatory M1 macrophages are considered crucial early steps in liver inflammation [51]. In our study, flow cytometry analysis demonstrated a notable increase in pro-inflammatory CD11b+Ly6CHigh monocytes and F4/80+Ly6CHigh M1-like macrophages in Shp-MKO livers compared to WT controls, both under basal condition and after LPS challenge. These findings suggest that the loss of Shp in myeloid cells promotes the infiltration of pro-inflammatory monocytes into the liver and enhances their differentiation into pro-inflammatory M1 macrophages. The precise mechanisms by which SHP regulates the expression of Ly6C remain unclear. Additional studies are needed to unravel the molecular pathways and signaling events through which SHP modulates Ly6C expression in myeloid cells.
Pparg, MAPK, and NF-κB are critical players in the regulation of macrophage activation and differentiation [37,54]. In our study, we observed that mice lacking myeloid Shp exhibited a pronounced activation of the MAPK and NF-κB pathways in the liver following LPS challenge. In contrast, when SHP was overexpressed in macrophages, it resulted in increased Pparg expression and inhibited the activation of MAPK and NF-κB signaling. Additionally, we made an interesting discovery of a protein-protein interaction between SHP and p65. While previous research has established that SHP interacts with p65 and TRAF6, leading to the inhibition of NF-κB signaling [23], our study provides the first evidence of SHP’s regulatory role in modulating Pparg expression and impeding MKK4 and JNK activation. These findings further underscore the significance of SHP in shaping macrophage differentiation during the innate immune response.
One limitation of our study is the use of the LyzCre system, which achieves high level gene knockout in myeloid cells, including monocytes and macrophages [55]. However, the LyzCre knock-out strategy also affects a subset of neutrophils [56]. Therefore, some of the pro-inflammatory phenotypes observed in the Shp-MKO mice, such as increased cytokine and chemokine production, may also be attributed to Shp loss in neutrophils. Future studies employing alternative conditional macrophage-specific gene knockout models will be necessary to confirm our results obtained from the LyzCre system.
5. Conclusions
Our study has unveiled that the absence of Shp in myeloid cells results in an augmented infiltration of pro-inflammatory monocytes and their subsequent differentiation into pro-inflammatory M1 macrophages upon LPS challenge. This sustained accumulation of pro-inflammatory macrophages in the liver highlights the crucial role of SHP in regulating macrophage polarization and its significant impact on the immune response during LPS-induced inflammation. These effects can be attributed to the dysregulation of Pparg, MAPK, and NF-κB signaling pathways due to the loss of Shp in macrophages, further contributing to the persistent accumulation of pro-inflammatory M1 macrophages. Overall, our findings shed light on the intricate mechanisms through which SHP influences macrophage behavior, thereby significantly contributing to our understanding of the complex interplay between SHP, macrophage polarization, and the innate immune response. This research enhances our comprehension of the underlying processes involved in immune regulation and may have implications in the development of novel therapeutic approaches for inflammatory conditions.
Funding
This work was supported by National Institutes of Health grants R01DK119131 and K22CA184146. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of Interest
The authors declare that they have no conflicts of interest with contents of this article.
Abbreviations
SHP: small heterodimer partner; Pparg, peroxisome proliferator activated receptor gamma; LPS, lipopolysaccharide; MDM, monocyte-derived macrophages; BMDM, bone marrow-derived macrophages; Shp-MKO, Shp myeloid cell specific knockout; Tnfa, tumor necrosis factor alpha; Ccl2, chemokine (C-C motif) ligand 2; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; KC, Kupffer cells; IFN-gamma, interferon gamma; IL4, interleukin 4; NASH, non-alcoholic steatohepatitis; Nos2, nitric oxide synthase 2; Arg1, arginase 1; Cd163, CD163 antigen; Mrc1, mannose receptor C type 1; 3’UTR, 3’-untranslated region; Emr1 or F4/80, adhesion G protein-coupled receptor E1; NPCs, non-parenchymal cells; PTPRC or CD45, protein tyrosine phosphatase receptor type C; Itgam or CD11b, monocyte marker integrin alpha M; Ly6C, Ly6-C antigen; FSC, forward scatter; SSC, side scatter; Co-IP, co-immunoprecipitation; MSP, Macrophage-stimulating factor; AMPK, AMP-activated protein kinase; IL-1, interleukin 1; IL-18, interleukin 18.
References
- Gao, B.; Jeong, W.I.; Tian, Z. Liver: An organ with predominant innate immunity. Hepatology 2008, 47, 729-736. [CrossRef]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res 2001, 7, 167-202.
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat Rev Immunol 2011, 11, 762-774. [CrossRef]
- Silva, M.T. When two is better than one: macrophages and neutrophils work in concert in innate immunity as complementary and cooperative partners of a myeloid phagocyte system. J Leukoc Biol 2010, 87, 93-106. [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014, 41, 14-20. [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 2004, 25, 677-686. [CrossRef]
- Cheng, K.; Cai, N.; Zhu, J.; Yang, X.; Liang, H.; Zhang, W. Tumor-associated macrophages in liver cancer: From mechanisms to therapy. Cancer Commun (Lond) 2022, 42, 1112-1140. [CrossRef]
- Lichtnekert, J.; Kawakami, T.; Parks, W.C.; Duffield, J.S. Changes in macrophage phenotype as the immune response evolves. Curr Opin Pharmacol 2013, 13, 555-564. [CrossRef]
- Luo, W.; Xu, Q.; Wang, Q.; Wu, H.; Hua, J. Effect of modulation of PPAR-gamma activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep 2017, 7, 44612. [CrossRef]
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol 2021, 18, 45-56. [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat Rev Immunol 2003, 3, 23-35. [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008, 8, 958-969. [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front Biosci 2008, 13, 453-461. [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J Hepatol 2017, 66, 1300-1312. [CrossRef]
- Peiseler, M.; Schwabe, R.; Hampe, J.; Kubes, P.; Heikenwalder, M.; Tacke, F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease - novel insights into cellular communication circuits. J Hepatol 2022, 77, 1136-1160. [CrossRef]
- Zhang, Y.; Hagedorn, C.H.; Wang, L. Role of nuclear receptor SHP in metabolism and cancer. Biochim Biophys Acta 2011, 1812, 893-908. [CrossRef]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 2000, 6, 517-526. [CrossRef]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 2000, 6, 507-515. [CrossRef]
- Wang, L.; Liu, J.; Saha, P.; Huang, J.; Chan, L.; Spiegelman, B.; Moore, D.D. The orphan nuclear receptor SHP regulates PGC-1alpha expression and energy production in brown adipocytes. Cell Metab 2005, 2, 227-238. [CrossRef]
- Suh, Y.H.; Kim, S.Y.; Lee, H.Y.; Jang, B.C.; Bae, J.H.; Sohn, J.N.; Bae, J.H.; Suh, S.I.; Park, J.W.; Lee, K.U.; et al. Overexpression of short heterodimer partner recovers impaired glucose-stimulated insulin secretion of pancreatic beta-cells overexpressing UCP2. J Endocrinol 2004, 183, 133-144. [CrossRef]
- Yuk, J.M.; Jin, H.S.; Jo, E.K. Small Heterodimer Partner and Innate Immune Regulation. Endocrinol Metab (Seoul) 2016, 31, 17-24. [CrossRef]
- Johansson, L.; Bavner, A.; Thomsen, J.S.; Farnegardh, M.; Gustafsson, J.A.; Treuter, E. The orphan nuclear receptor SHP utilizes conserved LXXLL-related motifs for interactions with ligand-activated estrogen receptors. Mol Cell Biol 2000, 20, 1124-1133. [CrossRef]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Kim, J.J.; Kim, S.W.; Jin, H.S.; Yang, C.S.; Park, K.A.; Chanda, D.; Kim, D.K.; et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat Immunol 2011, 12, 742-751. [CrossRef]
- Noh, J.R.; Kim, Y.H.; Kim, D.K.; Hwang, J.H.; Kim, K.S.; Choi, D.H.; Lee, S.J.; Lee, H.G.; Lee, T.G.; Weng, H.L.; et al. Small heterodimer partner negatively regulates C-X-C motif chemokine ligand 2 in hepatocytes during liver inflammation. Sci Rep 2018, 8, 15222. [CrossRef]
- Yang, C.S.; Yuk, J.M.; Kim, J.J.; Hwang, J.H.; Lee, C.H.; Kim, J.M.; Oh, G.T.; Choi, H.S.; Jo, E.K. Small heterodimer partner-targeting therapy inhibits systemic inflammatory responses through mitochondrial uncoupling protein 2. PLoS One 2013, 8, e63435. [CrossRef]
- Zou, A.; Magee, N.; Deng, F.; Lehn, S.; Zhong, C.; Zhang, Y. Hepatocyte nuclear receptor SHP suppresses inflammation and fibrosis in a mouse model of nonalcoholic steatohepatitis. J Biol Chem 2018, 293, 8656-8671. [CrossRef]
- Magee, N.; Zou, A.; Ghosh, P.; Ahamed, F.; Delker, D.; Zhang, Y. Disruption of hepatic small heterodimer partner induces dissociation of steatosis and inflammation in experimental nonalcoholic steatohepatitis. J Biol Chem 2020, 295, 994-1008. [CrossRef]
- Toda, G.; Yamauchi, T.; Kadowaki, T.; Ueki, K. Preparation and culture of bone marrow-derived macrophages from mice for functional analysis. STAR Protoc 2021, 2, 100246. [CrossRef]
- Vrochides, D.; Papanikolaou, V.; Pertoft, H.; Antoniades, A.A.; Heldin, P. Biosynthesis and degradation of hyaluronan by nonparenchymal liver cells during liver regeneration. Hepatology 1996, 23, 1650-1655. [CrossRef]
- Magee, N.; Ahamed, F.; Eppler, N.; Jones, E.; Ghosh, P.; He, L.; Zhang, Y. Hepatic transcriptome profiling reveals early signatures associated with disease transition from non-alcoholic steatosis to steatohepatitis. Liver Res 2022, 6, 238-250. [CrossRef]
- Heming, M.; Gran, S.; Jauch, S.L.; Fischer-Riepe, L.; Russo, A.; Klotz, L.; Hermann, S.; Schafers, M.; Roth, J.; Barczyk-Kahlert, K. Peroxisome Proliferator-Activated Receptor-gamma Modulates the Response of Macrophages to Lipopolysaccharide and Glucocorticoids. Front Immunol 2018, 9, 893. [CrossRef]
- Gautier, E.L.; Chow, A.; Spanbroek, R.; Marcelin, G.; Greter, M.; Jakubzick, C.; Bogunovic, M.; Leboeuf, M.; van Rooijen, N.; Habenicht, A.J.; et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J Immunol 2012, 189, 2614-2624. [CrossRef]
- Lee, J.; Padhye, A.; Sharma, A.; Song, G.; Miao, J.; Mo, Y.Y.; Wang, L.; Kemper, J.K. A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via microRNA-34a inhibition. J Biol Chem 2010, 285, 12604-12611. [CrossRef]
- Li, X.; Chen, Y.; Wu, S.; He, J.; Lou, L.; Ye, W.; Wang, J. microRNA-34a and microRNA-34c promote the activation of human hepatic stellate cells by targeting peroxisome proliferator-activated receptor gamma. Mol Med Rep 2015, 11, 1017-1024. [CrossRef]
- Zarkesh, M.; Tabaei, K.; Akbarzadeh, M.; Daneshafrooz, A.; Zadeh-Vakili, A. Association of miR-34a and miR-143 levels with PPARgamma gene expression in adipose tissues of non-diabetic adults. J Physiol Anthropol 2022, 41, 13. [CrossRef]
- Wu, X.; Wang, Z.; Shi, J.; Yu, X.; Li, C.; Liu, J.; Zhang, F.; Chen, H.; Zheng, W. Macrophage polarization toward M1 phenotype through NF-kappaB signaling in patients with Behcet's disease. Arthritis Res Ther 2022, 24, 249. [CrossRef]
- Kerneur, C.; Cano, C.E.; Olive, D. Major pathways involved in macrophage polarization in cancer. Front Immunol 2022, 13, 1026954. [CrossRef]
- Tian, L.; Li, W.; Yang, L.; Chang, N.; Fan, X.; Ji, X.; Xie, J.; Yang, L.; Li, L. Cannabinoid Receptor 1 Participates in Liver Inflammation by Promoting M1 Macrophage Polarization via RhoA/NF-kappaB p65 and ERK1/2 Pathways, Respectively, in Mouse Liver Fibrogenesis. Front Immunol 2017, 8, 1214. [CrossRef]
- Krenkel, O.; Hundertmark, J.; Abdallah, A.T.; Kohlhepp, M.; Puengel, T.; Roth, T.; Branco, D.P.P.; Mossanen, J.C.; Luedde, T.; Trautwein, C.; et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 2020, 69, 551-563. [CrossRef]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thone, T.; Browaeys, R.; et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022, 185, 379-396 e338. [CrossRef]
- Vonderlin, J.; Chavakis, T.; Sieweke, M.; Tacke, F. The Multifaceted Roles of Macrophages in NAFLD Pathogenesis. Cell Mol Gastroenterol Hepatol 2023, 15, 1311-1324. [CrossRef]
- Li, W.; Chang, N.; Li, L. Heterogeneity and Function of Kupffer Cells in Liver Injury. Front Immunol 2022, 13, 940867. [CrossRef]
- Kim, H.I.; Koh, Y.K.; Kim, T.H.; Kwon, S.K.; Im, S.S.; Choi, H.S.; Kim, K.S.; Ahn, Y.H. Transcriptional activation of SHP by PPAR-gamma in liver. Biochem Biophys Res Commun 2007, 360, 301-306. [CrossRef]
- Du, J.; Xiang, X.; Xu, D.; Cui, K.; Pang, Y.; Xu, W.; Mai, K.; Ai, Q. LPS Stimulation Induces Small Heterodimer Partner Expression Through the AMPK-NRF2 Pathway in Large Yellow Croaker (Larimichthys crocea). Front Immunol 2021, 12, 753681. [CrossRef]
- Chung, H.T. SHP gains citizenship of the AMPK kingdom. Cell Mol Immunol 2011, 8, 450-452. [CrossRef]
- Weng, S.Y.; Schuppan, D. AMPK regulates macrophage polarization in adipose tissue inflammation and NASH. J Hepatol 2013, 58, 619-621. [CrossRef]
- Yao, Q.; Liu, J.; Zhang, Z.; Li, F.; Zhang, C.; Lai, B.; Xiao, L.; Wang, N. Peroxisome proliferator-activated receptor gamma (PPARgamma) induces the gene expression of integrin alpha(V)beta(5) to promote macrophage M2 polarization. J Biol Chem 2018, 293, 16572-16582. [CrossRef]
- Chen, J.Y.; Song, C.X.; Lei, S.Y.; Li, J.; Zuo, A.J.; Xu, D.; Guo, Y. CTRP9 induces macrophages polarization into M1 phenotype through activating JNK pathway and enhances VSMCs apoptosis in macrophages and VSMCs co-culture system. Exp Cell Res 2020, 395, 112194. [CrossRef]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416-426. [CrossRef]
- Ehling, J.; Bartneck, M.; Wei, X.; Gremse, F.; Fech, V.; Mockel, D.; Baeck, C.; Hittatiya, K.; Eulberg, D.; Luedde, T.; et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut 2014, 63, 1960-1971. [CrossRef]
- Song, P.; Zhang, J.; Zhang, Y.; Shu, Z.; Xu, P.; He, L.; Yang, C.; Zhang, J.; Wang, H.; Li, Y.; et al. Hepatic recruitment of CD11b+Ly6C+ inflammatory monocytes promotes hepatic ischemia/reperfusion injury. Int J Mol Med 2018, 41, 935-945. [CrossRef]
- Brempelis, K.J.; Crispe, I.N. Infiltrating monocytes in liver injury and repair. Clin Transl Immunology 2016, 5, e113. [CrossRef]
- Yang, P.; Liu, L.; Sun, L.; Fang, P.; Snyder, N.; Saredy, J.; Ji, Y.; Shen, W.; Qin, X.; Wu, Q.; et al. Immunological Feature and Transcriptional Signaling of Ly6C Monocyte Subsets From Transcriptome Analysis in Control and Hyperhomocysteinemic Mice. Front Immunol 2021, 12, 632333. [CrossRef]
- Mussbacher, M.; Derler, M.; Basilio, J.; Schmid, J.A. NF-kappaB in monocytes and macrophages - an inflammatory master regulator in multitalented immune cells. Front Immunol 2023, 14, 1134661. [CrossRef]
- Abram, C.L.; Roberge, G.L.; Hu, Y.; Lowell, C.A. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods 2014, 408, 89-100. [CrossRef]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Forster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 1999, 8, 265-277. [CrossRef]
Figure 1.
Macrophage differentiation alters Shp mRNA expression. The livers of C57BL/6 mice were perfused and digested to harvest nonparenchymal cells (NPCs). Hepatic macrophages were then isolated from NPCs by CD11b MicroBeads and differentiated into M1 or M2 macrophages using DMEM media supplemented with IFN-gamma (100 ng/mL) or IL4 (50 ng/ml) for 24 hours, respectively. The relative mRNA levels of M1 markers (Tnfa and Nos2), M2 markers (Arg1 and Cd163), and Shp were determined using quantitative PCR (qPCR). Data are presented as mean ± SEM for 3 samples/group. *p < 0.05 and **p < 0.01 between the indicated groups.
Figure 1.
Macrophage differentiation alters Shp mRNA expression. The livers of C57BL/6 mice were perfused and digested to harvest nonparenchymal cells (NPCs). Hepatic macrophages were then isolated from NPCs by CD11b MicroBeads and differentiated into M1 or M2 macrophages using DMEM media supplemented with IFN-gamma (100 ng/mL) or IL4 (50 ng/ml) for 24 hours, respectively. The relative mRNA levels of M1 markers (Tnfa and Nos2), M2 markers (Arg1 and Cd163), and Shp were determined using quantitative PCR (qPCR). Data are presented as mean ± SEM for 3 samples/group. *p < 0.05 and **p < 0.01 between the indicated groups.
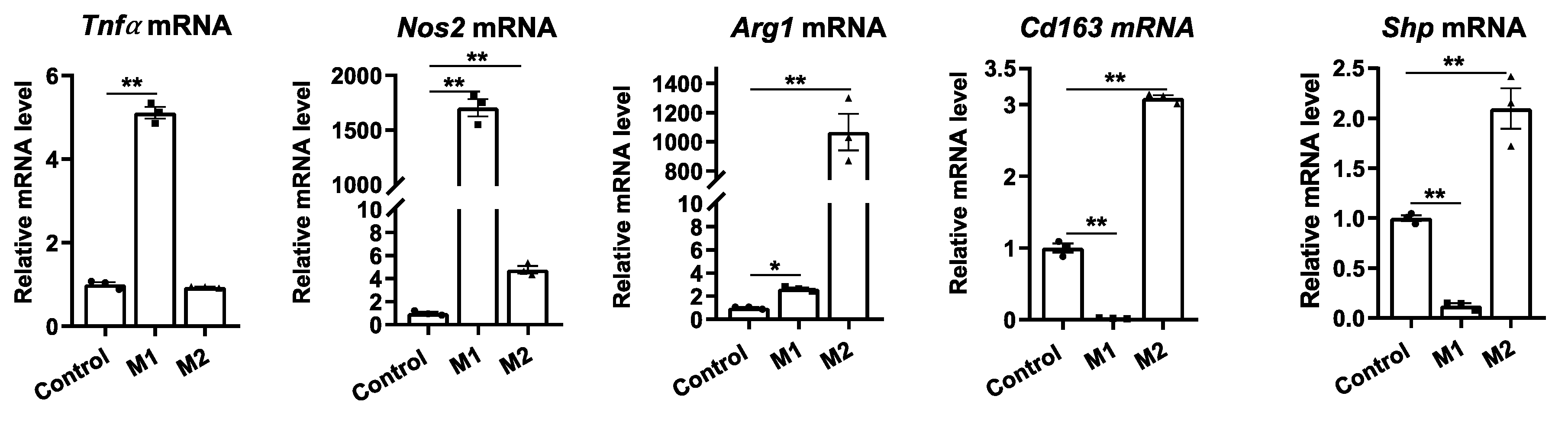
Figure 2.
Shp deletion in macrophages enhances M1 polarization but impairs M2 differentiation. (A) Peritoneal macrophages were isolated from WT and Shp-MKO mice, and the relative mRNA levels of Shp were determined by qPCR. (B) Bone marrow cells were isolated from WT and Shp-MKO mice and differentiated into macrophages using M-CSF (10 ng/ml) for 7 days. On the 7th day, the differentiated macrophages were cultured with IFN-gamma (100 ng/mL) or IL4 (50 ng/ml) for 24 hours to induce M1 or M2 macrophage polarization, respectively. The mRNA expression of Tnfa, Nos2, and Cd206 was determined using qPCR. (C) Left, western blot confirmed the overexpression of Flag-SHP in mouse macrophage RAW cells. Right, the expression of miR-34a and Pparg was determined by qPCR. (D) RAW cells with or without SHP overexpression were treated with IFN-gamma (100 ng/mL) for 24 hours. qPCR was employed to measure mRNA levels of Nos2 and Pparg. The data are presented as mean ± SEM for 3 samples/group. * p < 0.05 and **p < 0.01 between the indicated groups.
Figure 2.
Shp deletion in macrophages enhances M1 polarization but impairs M2 differentiation. (A) Peritoneal macrophages were isolated from WT and Shp-MKO mice, and the relative mRNA levels of Shp were determined by qPCR. (B) Bone marrow cells were isolated from WT and Shp-MKO mice and differentiated into macrophages using M-CSF (10 ng/ml) for 7 days. On the 7th day, the differentiated macrophages were cultured with IFN-gamma (100 ng/mL) or IL4 (50 ng/ml) for 24 hours to induce M1 or M2 macrophage polarization, respectively. The mRNA expression of Tnfa, Nos2, and Cd206 was determined using qPCR. (C) Left, western blot confirmed the overexpression of Flag-SHP in mouse macrophage RAW cells. Right, the expression of miR-34a and Pparg was determined by qPCR. (D) RAW cells with or without SHP overexpression were treated with IFN-gamma (100 ng/mL) for 24 hours. qPCR was employed to measure mRNA levels of Nos2 and Pparg. The data are presented as mean ± SEM for 3 samples/group. * p < 0.05 and **p < 0.01 between the indicated groups.
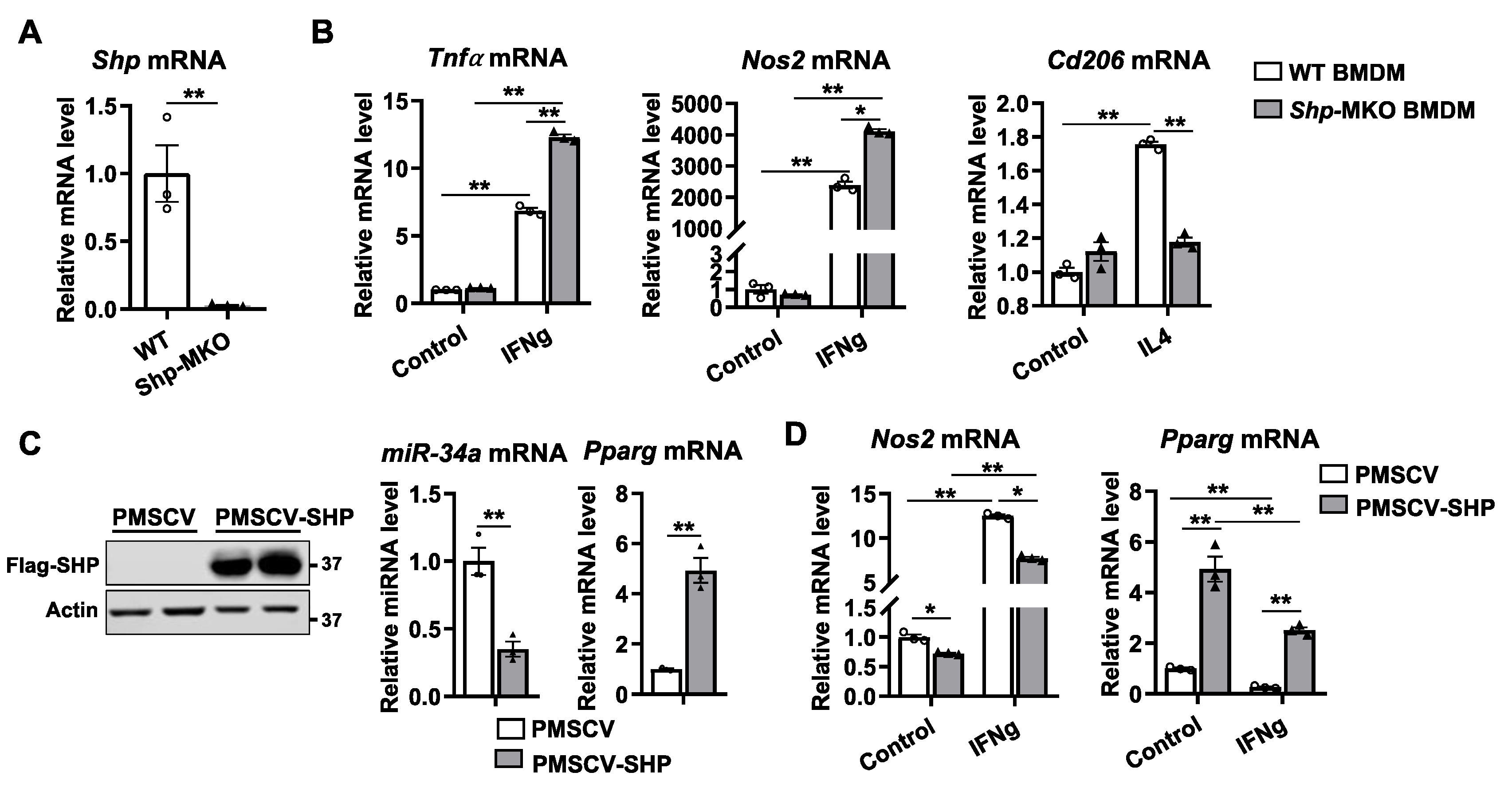
Figure 3.
Shp knockout results in a persistent hepatic accumulation of macrophages following LPS challenge. (A) Shp myeloid cell specific knockout (Shp-MKO) was generated by breeding Shpflox/flox with LysM-Cre mice. Both WT and Shp-MKO mice were subjected to intraperitoneal LPS (1 mg/kg body weight) injection, and samples were collected at 0-, 3-, and 7-hour post injection. (B) Mouse body weight, liver weight, and liver-to-body weight ratio. (C) Liver sections were stained with hematoxylin and eosin (H&E) to examine the histological changes in the liver. (D) Left, representative images of liver sections stained with macrophage marker F4/80. Original magnification, X40. Right, quantification of the DAB-positive staining area. n=3/group. The data are presented as mean ± SEM for 3 samples/group. * p < 0.05 between the indicated groups.
Figure 3.
Shp knockout results in a persistent hepatic accumulation of macrophages following LPS challenge. (A) Shp myeloid cell specific knockout (Shp-MKO) was generated by breeding Shpflox/flox with LysM-Cre mice. Both WT and Shp-MKO mice were subjected to intraperitoneal LPS (1 mg/kg body weight) injection, and samples were collected at 0-, 3-, and 7-hour post injection. (B) Mouse body weight, liver weight, and liver-to-body weight ratio. (C) Liver sections were stained with hematoxylin and eosin (H&E) to examine the histological changes in the liver. (D) Left, representative images of liver sections stained with macrophage marker F4/80. Original magnification, X40. Right, quantification of the DAB-positive staining area. n=3/group. The data are presented as mean ± SEM for 3 samples/group. * p < 0.05 between the indicated groups.
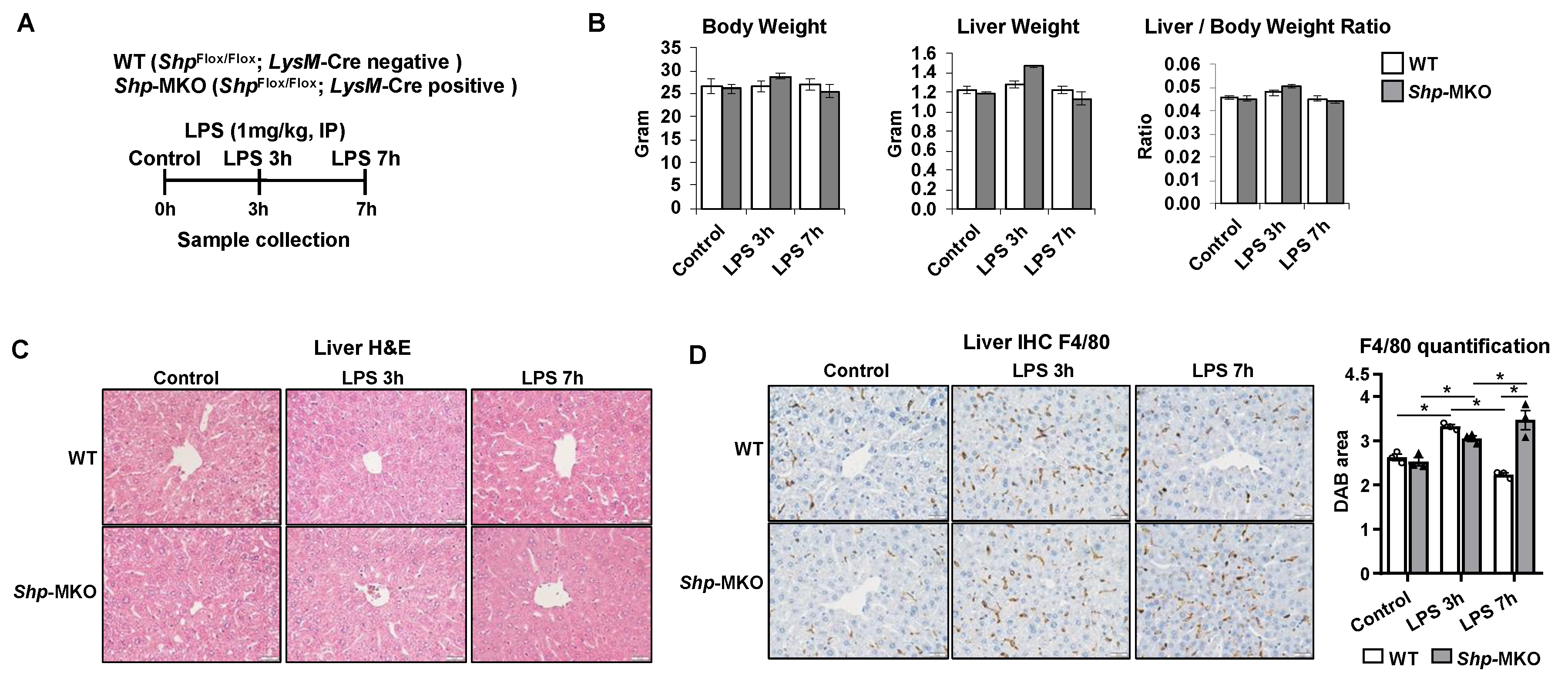
Figure 4.
Flow cytometry analysis of composition of hepatic macrophages and monocytes following LPS challenge. WT and Shp-MKO mice were intraperitoneally injected with or without LPS (1 mg/kg body weight). After 3 hours, mouse livers were perfused and digested to isolate nonparenchymal cells (NPCs). Approximately 1 × 106 NPCs were labeled with specific antibodies and prepared for flow cytometry analysis. Single cells were gated based on FSC-A and FSC-H to exclude doublets. Dead cells stained with Zombie Aqua were excluded from the analysis. Live cells positive for CD45 expression were gated, and the populations of interest were calculated, including F4/80+Ly6CHigh pro-inflammatory M1 macrophages (A), CD11b+Ly6CHigh pro-inflammatory monocytes (B), and CD11bHighF4/80Intermediate monocyte-derived macrophages (C). The data are presented as mean ± SEM for 3 samples/group. * p < 0.05 and **p < 0.01 between the indicated groups.
Figure 4.
Flow cytometry analysis of composition of hepatic macrophages and monocytes following LPS challenge. WT and Shp-MKO mice were intraperitoneally injected with or without LPS (1 mg/kg body weight). After 3 hours, mouse livers were perfused and digested to isolate nonparenchymal cells (NPCs). Approximately 1 × 106 NPCs were labeled with specific antibodies and prepared for flow cytometry analysis. Single cells were gated based on FSC-A and FSC-H to exclude doublets. Dead cells stained with Zombie Aqua were excluded from the analysis. Live cells positive for CD45 expression were gated, and the populations of interest were calculated, including F4/80+Ly6CHigh pro-inflammatory M1 macrophages (A), CD11b+Ly6CHigh pro-inflammatory monocytes (B), and CD11bHighF4/80Intermediate monocyte-derived macrophages (C). The data are presented as mean ± SEM for 3 samples/group. * p < 0.05 and **p < 0.01 between the indicated groups.
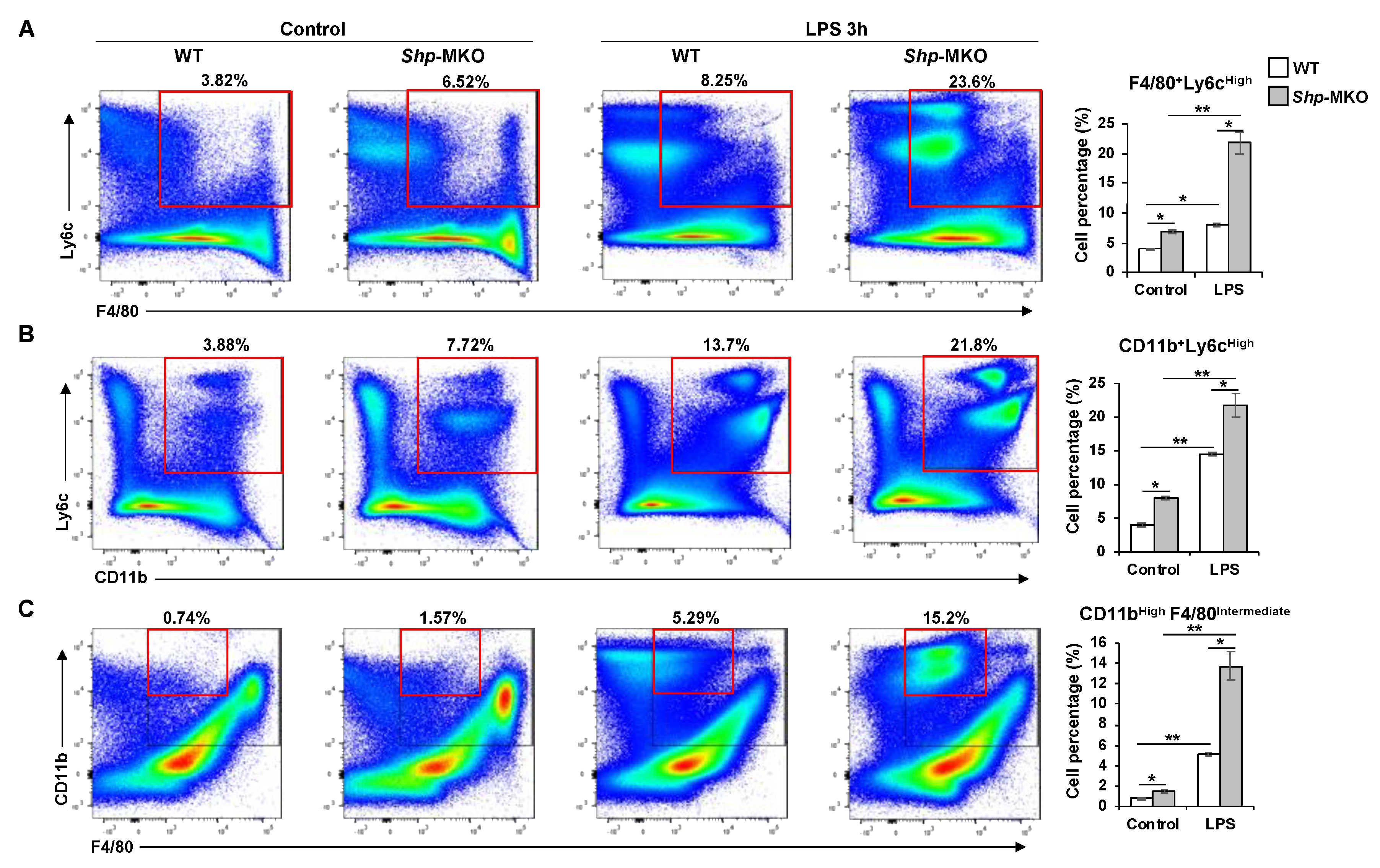
Figure 5.
Myeloid cell specific deletion of Shp leads to enhanced chemokines production in response to LPS challenge. WT and Shp-MKO mice were intraperitoneally injected with LPS (1 mg/kg body weight), and samples were collected at 0-, 3-, and 7-hour post-injection. (A) The mRNA levels of Shp, Ccl2, Cd11b, and Ly6c in liver tissues were quantified using qPCR. (B) The serum levels of CCL2 and TNFα were measured using ELISA to evaluate the circulating levels of these chemokines. The data are presented as mean ± SEM for 3 samples/group. * p < 0.05 and **p < 0.01 between the indicated groups.
Figure 5.
Myeloid cell specific deletion of Shp leads to enhanced chemokines production in response to LPS challenge. WT and Shp-MKO mice were intraperitoneally injected with LPS (1 mg/kg body weight), and samples were collected at 0-, 3-, and 7-hour post-injection. (A) The mRNA levels of Shp, Ccl2, Cd11b, and Ly6c in liver tissues were quantified using qPCR. (B) The serum levels of CCL2 and TNFα were measured using ELISA to evaluate the circulating levels of these chemokines. The data are presented as mean ± SEM for 3 samples/group. * p < 0.05 and **p < 0.01 between the indicated groups.
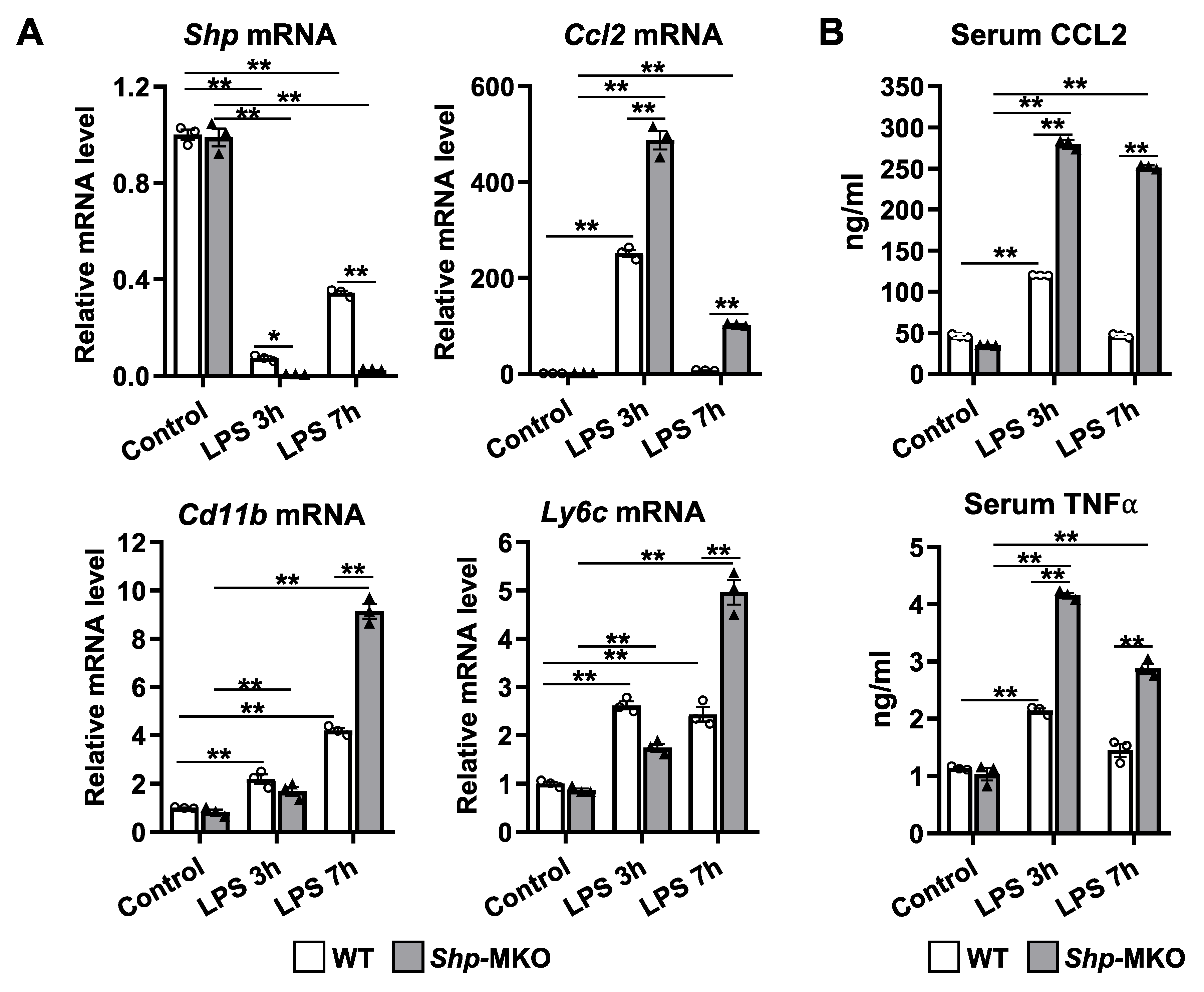
Figure 6.
Myeloid cell specific deletion of Shp results in hyperactivation of MAPK and NF-κB pathways in response to LPS challenge. WT and Shp-MKO mice were intraperitoneally injected with LPS (1 mg/kg body weight), and samples were collected at 0-, 3-, and 7-hour post-injection. (A) Left, Western blot analysis of whole protein lysates from liver tissues revealed the expression and phosphorylation levels of proteins involved in MAPK and NF-κB signaling pathways. Right, the protein band density was quantified using Image Studio software, and the relative expression levels were normalized to the loading control β-actin. (B) Left, Western blot analysis of cytoplasmic and nuclear fractions demonstrated the expression and phosphorylation levels of proteins involved in NF-κB signaling. Right, the protein band density was quantified using Image Studio software, and the relative levels of proteins were normalized to the nuclear loading control Histone H3 and the cytoplasmic loading control α-tubulin, respectively. The data are presented as mean ± SEM. Western blots were repeated for 3 times and one represented image was included in the figure. * p < 0.05 and **p < 0.01 between the indicated groups.
Figure 6.
Myeloid cell specific deletion of Shp results in hyperactivation of MAPK and NF-κB pathways in response to LPS challenge. WT and Shp-MKO mice were intraperitoneally injected with LPS (1 mg/kg body weight), and samples were collected at 0-, 3-, and 7-hour post-injection. (A) Left, Western blot analysis of whole protein lysates from liver tissues revealed the expression and phosphorylation levels of proteins involved in MAPK and NF-κB signaling pathways. Right, the protein band density was quantified using Image Studio software, and the relative expression levels were normalized to the loading control β-actin. (B) Left, Western blot analysis of cytoplasmic and nuclear fractions demonstrated the expression and phosphorylation levels of proteins involved in NF-κB signaling. Right, the protein band density was quantified using Image Studio software, and the relative levels of proteins were normalized to the nuclear loading control Histone H3 and the cytoplasmic loading control α-tubulin, respectively. The data are presented as mean ± SEM. Western blots were repeated for 3 times and one represented image was included in the figure. * p < 0.05 and **p < 0.01 between the indicated groups.
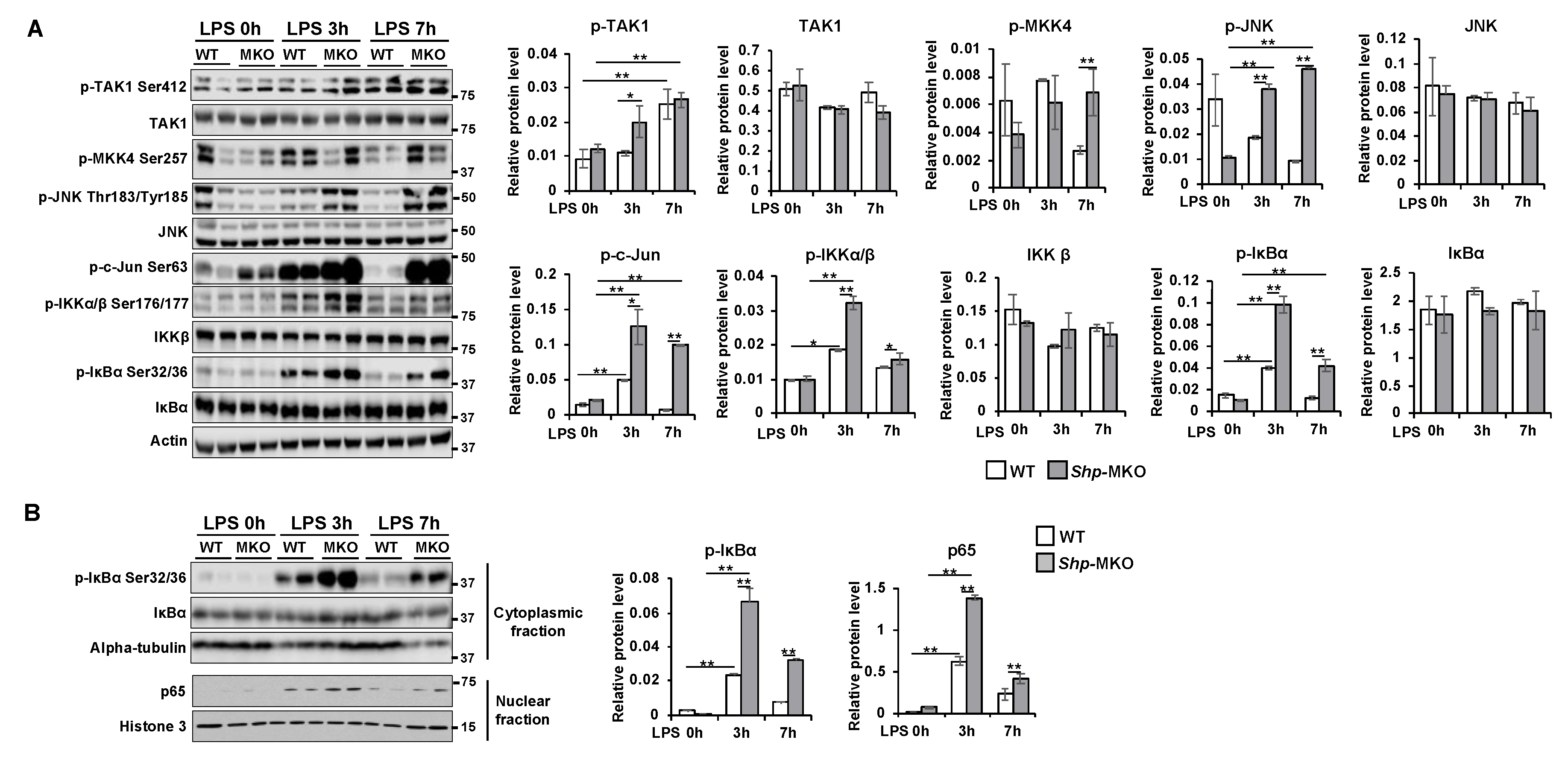
Figure 7.
SHP overexpression hinders the activation of MAPK and NF-κB pathways in response to LPS challenge. (A) Left, mouse macrophage RAW cells with or without SHP overexpression were treated with 100 ng/ml LPS for different durations (0, 5, 10, 30, and 60 minutes). Whole cell lysates were collected and subjected to western blot analysis to assess the expression and phosphorylation levels of proteins involved in MAPK and NF-κB signaling pathways. Right, the protein band density was quantified using Image Studio software, and the relative expression levels were normalized to the loading control β-actin. (B) Co-immunoprecipitation experiments were conducted using whole protein lysates from RAW cells with or without FLAG-SHP overexpression. The protein-protein interaction of SHP with p65 was detected by western blot analysis. The data are presented as mean ± SEM. Western blots were repeated for 3 times and one represented image was included in the figure. * p < 0.05 and **p < 0.01 between the indicated groups.
Figure 7.
SHP overexpression hinders the activation of MAPK and NF-κB pathways in response to LPS challenge. (A) Left, mouse macrophage RAW cells with or without SHP overexpression were treated with 100 ng/ml LPS for different durations (0, 5, 10, 30, and 60 minutes). Whole cell lysates were collected and subjected to western blot analysis to assess the expression and phosphorylation levels of proteins involved in MAPK and NF-κB signaling pathways. Right, the protein band density was quantified using Image Studio software, and the relative expression levels were normalized to the loading control β-actin. (B) Co-immunoprecipitation experiments were conducted using whole protein lysates from RAW cells with or without FLAG-SHP overexpression. The protein-protein interaction of SHP with p65 was detected by western blot analysis. The data are presented as mean ± SEM. Western blots were repeated for 3 times and one represented image was included in the figure. * p < 0.05 and **p < 0.01 between the indicated groups.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



