Submitted:
28 July 2023
Posted:
31 July 2023
You are already at the latest version
Abstract
Calcium (Ca2+) ions act as second messenger, regulating several cell functions. Mitochondria are critical organelles for the regulation of intracellular Ca2+. Mitochondrial calcium (mtCa2+) uptake is ensured by the presence in the inner mitochondrial membrane (IMM) of the mitochondrial calcium uniporter (MCU) complex, a macromolecular structure composed of pore-forming and regulatory subunits. MtCa2+ uptake plays a crucial role in the regulation of oxidative metabolism and cell death. Many evidences demonstrate that dysregulation of mtCa2+ homeostasis can have serious pathological outcomes. In this review, we briefly discuss the molecular structure and the function of the MCU complex and then we focus our attention on human diseases in which a dysfunction in mtCa2+ has been showed.
Keywords:
MCU
; mitochondrial Ca2+ signaling
; cancer
; cardiovascular diseases
; metabolic diseases
; skeletal muscle diseases
; neurodegenerative disorders
1. MCU Complex Structure and Function
The MCU complex is composed of pore forming subunits, i.e. MCU and its dominant-negative isoform, MCUb, the essential MCU regulator (EMRE), which allows interaction with the MICU mitochondrial calcium uptake regulatory subunits (MICU1, MICU2 and MICU3), and possibly other modulators, such as the mitochondrial calcium uniporter regulator 1 (MCUR1) (Figure 1).
The molecular identity of MCU was discovered in 2011 by two different studies. MCU is a 40 KDa protein, highly conserved and ubiquitously expressed, located in the IMM. These reports show that the downregulation of MCU strongly reduces mtCa2+ uptake in cells after Ca2+ release from the ER after treatment with IP3-generating agonist. Notably, these changes occur without impinging on mitochondrial morphology and membrane potential. Coherently, MCU overexpression strongly enhances mtCa2+ uptake after agonist-induced stimulation [1,2]. Structure analyses revealed that both the N- and C-termini of the protein face the mitochondrial matrix and that MCU contains two transmembrane domains linked by a highly conserved loop located in the intermembrane space, containing a “DIME” motif with negatively charged amino acids critical for Ca2+ permeation [3].
Raffaello et al., in 2013 described an alternative isoform of MCU, MCUb, a 33 KDa protein which shares high structural similarity with MCU. However, critical amino acids substitutions in the DIME motif explains why it exerts a dominant-negative effect, reducing the [Ca2+]mt rise evoked by agonist stimulation. The opposite effect on mtCa2+ uptake was obtained by MCUb silencing, confirming its role as a negative regulator of the channel [4]. The MCU/MCUb ratio varies greatly between different mammalian tissues, and this impinges on the intrinsic capacity of mitochondria of a tissue to rapidly accumulate Ca2+. Not surprisingly, the heart, that undergoes repetitive Ca2+ spiking, at the risk of mitochondrial Ca2+ overload, displays a high MCUb/MCU ratio, while skeletal muscle ensures maximal and sustained metabolic upregulation in phasic responses though a very low expression of MCUb. In general, the great variability in expression and stoichiometry of MCU and MCUb accounts, at least in part, for the wide differences in the MCU currents measured in different mammalian tissues [5].
The Essential MCU Regulator (EMRE) is a 10 KDa metazoan-specific protein, identified by quantitative mass spectrometry of affinity-purified MCU complex components [6]. It is inserted in the IMM by a single transmembrane domain and is required for the interaction between MCU and MICU1, acting as a bridge between the pore-forming and the regulatory subunits of the complex. Experiments performed on EMRE knockout cells clearly demonstrate that mtCa2+ uptake is impaired, phenocopying the effect of MCU silencing. This evidence enlightens the critical role of EMRE as an essential component of the MCU complex, required for its activity [6]. Coherently, reconstitution of the human MCU protein alone in yeast cells is not sufficient for uniporter activity, because the MCU channel is active only if MCU and EMRE are co-expressed [7]. It is not surprising that the proteolytic regulation of EMRE is a finely-tuned process, essential for the formation of a functional MCU complex [8].
MICU1 was the first member of the MCU complex to be identified in 2010. It is a 54 KDa protein located in the IMS where it regulates the activity of the MCU channel in Ca2+-dependent way [9]. Indeed, it contains two EF-hand Ca2+ binding domains at its N-terminal sequence. This regulation ensured by MICU1 explains the sigmoidal response to cytCa2+ levels of mtCa2+ uptake. On one hand, at low cytCa2+ concentrations, MICU1 keeps the channels closed and mtCa2+ uptake is negligible, thus avoiding a constant entry of Ca2+ into the mitochondrial matrix. On the other hand, when cytCa2+ concentration increases, MICU1 acts as cooperative activator of MCU, leading to an exponential increase of mtCa2+ uptake [10]. An alternative splicing isoform of MICU1, MICU1.1, characterized by the addiction of a short exon (4 amino acids), was shown to be expressed predominantly in skeletal muscle, and at a lower level in the brain. Similar to MICU1, MICU1.1 act as a positive modulator of the channel, leading to even higher increases of mtCa2+ uptake after stimulation. Interestingly, MICU1.1 when co-expressed with MICU2 does not show any reduction of [Ca2+] peaks, implying that the MICU1.1/MICU2 heterodimer is less sensitive to negative regulation of MICU2 (see below) [11].
Two paralogs of MICU1 residing in the IMS contributing to the regulation of mtCa2+ handling: MICU2 and MICU3 [12]. MICU2 has an expression pattern similar to MICU1, it contains two EF-hand domains and interacts with MICU1 and MCU, forming obligate heterodimers with MICU1 [13]. Interestingly, MICU2 protein stability seems to be dependent on the presence of MICU1. Indeed, MICU1 silencing leads to the loss also of MICU2 [12]. Patron et al., proposed an inhibitory effect of MICU2 on MCU activity at low cytCa2+ concentration [13], while Kamer et al., proposed that both MICU1 and MICU2 act as gatekeeping of the channel [14]. Notably, another study indicates that MICU2 can regulate the Ca2+ threshold of MCU activation [15].
MICU3 is another MICU1 paralog mainly expressed in the nervous system, and to a lower extent in skeletal muscle. Similarly to MICU1 and MICU2, also MICU3 contains two EF-hand domains. Patron et al. demonstrated that MICU3 forms heterodimers exclusively with MICU1, and not with MICU2. Notably, MICU3 has a reduced gatekeeping activity compared to MICU1, mediating more rapid responses to elevated cytCa2+ levels. Thus, MICU3 is proposed to be a positive modulator of MCU channel ensuring mtCa2+ uptake in response to fast cytCa2+ rises, typical of neuronal stimulation [16].
MCUR1 is a 35KDa protein located in the IMM that interacts with MCU [17]. Its downregulation leads to a decrease in mtCa2+ uptake and ATP production [18]. In contrast to this view, Paupe et al., showed that MCUR1 is not a regulator of the MCU complex, but it is a cytochrome c oxidase assembly factor [19]. Thus, the precise role of MCUR1 related to mtCa2+ homeostasis still remains to be fully elucidated.
2. Cardiovascular Diseases
The dysregulation of mitochondrial calcium (mtCa2+) homeostasis occurring in acute myocardial ischaemia reperfusion (I/R) injury points to a critical role of MCU in cardiovascular diseases. Upon reperfusion, mtCa2+ overload leads to excessive reactive oxygen species (ROS) production, promoting the opening of the mitochondrial permeability transition pore (PTP), which triggers cardiomyocyte death [20]. Thus, MCU was conceptually expected to be involved in ischemia-dependent sensitization of cardiac muscle to reperfusion damage.
The analysis of hearts from MCU knockout mice showed not only unaffected basal cardiac parameters, but, surprisingly, were not protected from ischemia/reperfusion damage, as expected of a role of mitochondrial Ca2+ loading upstream of permeability transition pore (PTP) opening. In addition, the hearts of MCU-/- mice were not protected by treatment with the pore desensitizer cyclosporin A (CsA) [21]. The same lack of protection was also observed in a transgenic mouse model in which MCU downregulation was achieved by the overexpression of a dominant-negative form of MCU (DN-MCU) [22]. Overall, these results suggest that constitutive ablation of MCU (and hence loss of mtCa2+ signals) from the embryonic phase could lead to mitochondrial compensatory mechanisms (e.g. in sensitivity to Ca2+ or modulators of PTP or prevalence of other cell death pathways).
To overcome this problem, a mouse model with acute deletion of MCU in adult cardiomyocytes was generated [23,24]. In contrast to constitutive MCU deletion, the conditional knockout model showed protection from I/R injury and cell death, thus confirming the view of MCU-dependent dysregulated Ca2+ signals upstream of PTP opening and myocardial cell death.
Interestingly, MCUb and mNCLX gene expression increases after ischemic damage to the heart, and their overexpression, that leads to a decrease in mtCa2+ by limiting the uptake or enhancing the efflux respectively, is protective against I/R injury [25,26].
In this contest the role of MICU1 is still controversial. Indeed, while on one hand it is protective in early stages after reperfusion since its knockdown worsens the I/R damage, on the other hand MICU1 is also found to be upregulated during the late stages after reperfusion. However, the mechanism behind this increase is still not clear [25,27].
Together, these findings highlight the relevance of MCU modulation as potential therapeutic approach in the treatment of cardiovascular diseases. However, further studies are needed to translate these findings in a clinical approach.
3. Metabolic Diseases
Normal blood glucose concentrations are ensured by glucose-induced insulin secretion from pancreatic β-cells [28]. In this process mitochondrial oxidative metabolism plays a key role. Indeed, an increase in cytosolic ATP level [29] results in the closure of ATP-sensitive K+ channels (KATP) [30], leading to plasma membrane depolarization. This in turn promotes the opening of voltage-gated Ca2+ channels, allowing Ca2+ entry into the cell, ultimately leading to insulin release [31].
Considering the critical role of mitochondrial oxidative metabolism in glucose-induced insulin secretion, a properly functional MCU complex is required in pancreatic β-cells for the correct functioning of this process [32]. ATP rise upon glucose stimulation is impaired when MCU is downregulated [33], resulting in a decrease in insulin secretion [34]. Interestingly, not only the pore-forming subunit MCU, but also the regulatory subunit MICU1 is important for insulin secretion. Similar to MCU silencing, MICU1 downregulation leads to a decrease in ATP levels and glucose-induced insulin secretion in pancreatic β-cells [34]. Surprisingly, the strongest reduction in insulin secretion in β-cells is observed when the regulatory subunit MICU2 is downregulated [35]. Although the precise mechanism by which different subunits of the MCU complex influence insulin release still needs to be fully elucidated, the critical role of the MCU complex in the secretory function of pancreatic β-cells is undoubted.
MCU complex components are upregulated in insulin-resistant adipocytes and in mouse and human visceral adipose tissue (VAT) in conditions of obesity and diabetes. Interestingly, normal levels of MCU expression are restored in VAT of patients after bariatric surgery-induced weight loss. As for insulin secretion in pancreatic β-cells, also in this scenario not all the MCU complex components behave similarly, but a critical role emerges for MICU1, the only MCU complex component strongly upregulated during the transition from obesity to diabetes [36]. These data suggest a key role of mitochondrial Ca2+ dysregulation in obesity and diabetes, highlighting the relevance of MCU as a putative therapeutic target for the treatment of these metabolic diseases.
4. Cancer
A large number of studies have been published in the last decade associating MCU with development and progression of different tumors. This is not surprising, given the multifarious role of mtCa2+ on key aspects of carcinogenesis. On the one hand, oncogenes of the Bcl-2 family have been shown to reduce mtCa2+ loading by reducing ER Ca2+ levels and/or desensitizing the inositol 1,4,5 phosphate receptor (IP3R), thereby impairing apoptosis [37,38,39,40]. On the other hand, metastatic tumors have been shown to upregulate MCU, and the consequent increase of matrix Ca2+ favors motility and invasiveness, via a ROS/HIF-1α mitochondria-to-nucleus signaling pathway [41,42] (Figure 2). In the large number of reports highlighting a role for MCU is cancer, heterogeneity is thus the key word, not only between different cancer types, but also between different cell lines of the same cancer.
4.1. Breast Cancer
The majority of studies about MCU and cancer are related to breast cancer. The picture that emerges from these studies, however, is quite complex, with marked differences in the various cell models [43]. One of the most aggressive breast tumor subtypes is the triple-negative breast cancer (TNBC). Tosatto et al., demonstrated that in TNBC the expression of MCU correlates with tumor size and lymph node infiltration, suggesting a potential role of MCU in tumor growth and metastatic potential. Accordingly, the authors showed that in xenografts models of TNBC, MCU downregulation reduced tumor size and cell motility as well as metastatic infiltrations. Finally, a positive correlation of MCU levels in TNBC with mROS and HIF-1α signaling was revealed [41]. Recently, a novel mechanism was proposed to explain the link between MCU and TNBC progression. Filadi et al., found that spontaneous and sustained inositol 1,4,5-trisphosphate (IP3)-dependent Ca2+ oscillations occurs in TNBC cells and they are associated with the regulation of fatty acid (FA) metabolism. The modulation of mitochondrial FA metabolism mediated by Ca2+ concentration has a deeply effect on TNBC cell migration, although further studies will be needed to get full mechanistic insight [44]. However, the critical role of MCU regulation of cell metabolism in supporting tumor growth and proliferation was recently further confirmed by Fernandez Garcia et al. [45].
The role of MCU in breast cancer was also studied in another widely used breast carcinoma cell line, MDA-MB-231. Curry at al., showed that MCU downregulation in this model did not cause changes in proliferation or viability. Interestingly, distinct effects of MCU downregulation were observed on cell death pathways. The authors showed that MCU silencing increased the cytotoxicity in MDA-MB-231 cells only in a caspase-independent way. Indeed, cell death was increased only when MCU silencing was accompanied by a treatment with ionomycin, while no effects were observed in combination with the Bcl-2 inhibitor ABT-263, initiator of caspase-dependent cell death [46]. In this case, similarly to data obtained by the same authors in other cancers, the authors postulate that impaired mtCa2+ uptake reduces Ca2+ buffering, and thus favors Ca2+-dependent apoptotic pathways.
A way to induce cell-death through MCU modulation was also investigated by De Mario et al., in a study that identified positive and negative MCU modulators. Among the negative modulators, benzethonium emerged as an effective compound. In MDA-MB-231 cells, benzethonium, by negatively modulating MCU, delays tumor cell growth and migration [47]. Conversely, another recent study showed that MCU upregulation can be critical for MDA-MB-231 cells survival. Xue et al. showed that MCU is required for the pro-apoptotic effect of RY10-4, a protoapigenone analog, on MDA-MB-231 cells. Treatment with this compound causes a strong MCU upregulation which results in mtCa2+ overload and finally apoptosis [48].
Interestingly, similarly to TNBC cells, also in MDA-MB-231 cells MCU levels correlate with metastasis and invasion and this was correlated with an effect on SOCE-dependent breast cancer cell migration, since MCU silencing abolished thapsigargin-induced SOCE activity [49]. Hall et al., further confirmed that poorer disease outcome was associated with MCU overexpression. Interestingly, the opposite effect was observed with MICU1, since its downregulation was associated with a negative disease outcome. Surprisingly, contrary to what was observe in TNBC cells, downregulation of MCU did not affect ROS production and that MCU was dispensable for clonogenic cell survival of MDA-MB-231 cells [50]. Finally, Yu et al., demonstrated that MCU overexpression in MC7F, a human breast carcinoma line, increase the invasiveness and metastatic potential of these cells [51].
4.2. Pancreatic Cancer
In hepatocellular carcinoma cells (HCC) cells, HINT2 was shown to promote cell death via apoptosis, however the molecular mechanism remained elusive [52]. In pancreatic cancer, HINT2 also promotes cell death, and Chen et al., showed that treatment with ruthenium red, an inhibitor of MCU, reduced HINT2-dependent induced apoptosis. Interestingly, overexpression of HINT2 increases the expression of EMRE and decreases the expression of MICU1 and MICU2. The authors hypothesized a possible mechanism in which a constitutively active channel, lacking the regulation of the MICUs subunits, could account for mtCa2+ overload and consequent cell death [53]. In line with this study, Xie et al., showed that overexpression of EMRE positively correlates with pancreatic ductal adenocarcinoma (PDAC) prognosis [54]. Recently, elevated mtCa2+ uptake was associated to metastasis formation in PDAC. The effect of MCU in PDCA metastasis is through the activation of the KEap-Nrf2 antioxidant program [55]. Overall, these studies suggest that MCU plays a role also in the pathogenesis of pancreatic cancer.
4.3. Colon Cancer
Marchi et al., identified miRNA-25, a cancer-related MCU-targeting microRNA. Experiments performed on HeLa cells, showed that miRNA-25 overexpression decreases mtCa2+ uptake, without affecting cytosolic Ca2+ levels. The expression of this miRNA is upregulated in human colon cancers, where MCU expression is accordingly downregulated, and this correlates with apoptotic death resistance [56]. However, recently Yu et al., proposed that the lncRNA CERS6 antisense RNA 1 (CERS6-AS1) promotes colon cancer progression via upregulation of MCU [57]. Recently, another miRNA was found to target MCU in colorectal cancer (CRC), miR-138-5p. In CRC, miR-138-5p is downregulated, increasing MCU expression [58]. A further study by Zeng et al., showed that mtCa2+ uptake promotes colorectal cancer progression, through the interaction between RIPK1, a signaling molecule essential for cell survival, and MCU [59]. Although further studies are needed to clarify the role of mtCa2+ in colon cancer progression, these studies underly the importance of miRNA and lncRNA, instead of MCU directly, as possible target to modulate MCU activity in pathological contests.
4.4. Hepatocellular Carcinoma
Ren et al., demonstrated that in HCC cells the expression of MCU is enhanced, MICU1 is downregulated, while MICU2, MCUb and EMRE expression level is unaffected. Moreover, MCU upregulation is associated with poor survival and metastasis in HCC patients. The authors showed that the strong increase in mtCa2+ uptake promotes ROS production by downregulating NAD+, sirtuin3 (SIRT3) and superoxide dismutase 2 (SOD2) activity. High ROS levels, in turn, stimulates metalloproteinase-2 activity increasing cell motility [60]. In a second publication by the same group it was showed that also the regulator of MCU complex, MCUR1, was enhanced in HCC cells. The consequent increase in mtCa2+ uptake lead to increase ROS production and ROS-dependent p53 degradation, promoting cancer cell survival [61]. Another study investigating the expression profile of long noncoding RNA in sub-lethal heat-treated HCC cells, showed a downregulation of MCU. This model characterizes a transition zone of radiofrequency ablation (RFA), a treatment insufficient to kill all tumor cells, leading to residual cancer occurrence [62]. Further studies are needed to understand the contribution of MCU in this model.
4.5. Other Cancer Types
An emerging role of MCU in many other cancer types is emerging in the recent years.
A recent work by Stejerean-Todoran et al., enlightened the role of the MCU complex in melanoma, showing that silencing of MCU suppresses melanoma cell growth, but it promotes cell migration and invasion reducing the sensitivity to immunotherapies [42].
High levels of MCU expression were also found in ovarian cancer, and its silencing reduces ovarian cancer cells proliferation and migration. This was correlated to a decreased ROS production [63]. Similarly, reduction of MCU expression in renal cell carcinoma, through overexpression of EFHD1, a negative regulator of MCU activity, leads to reduction in cell migration and metastatic potential [64].
5. Skeletal Muscle Diseases
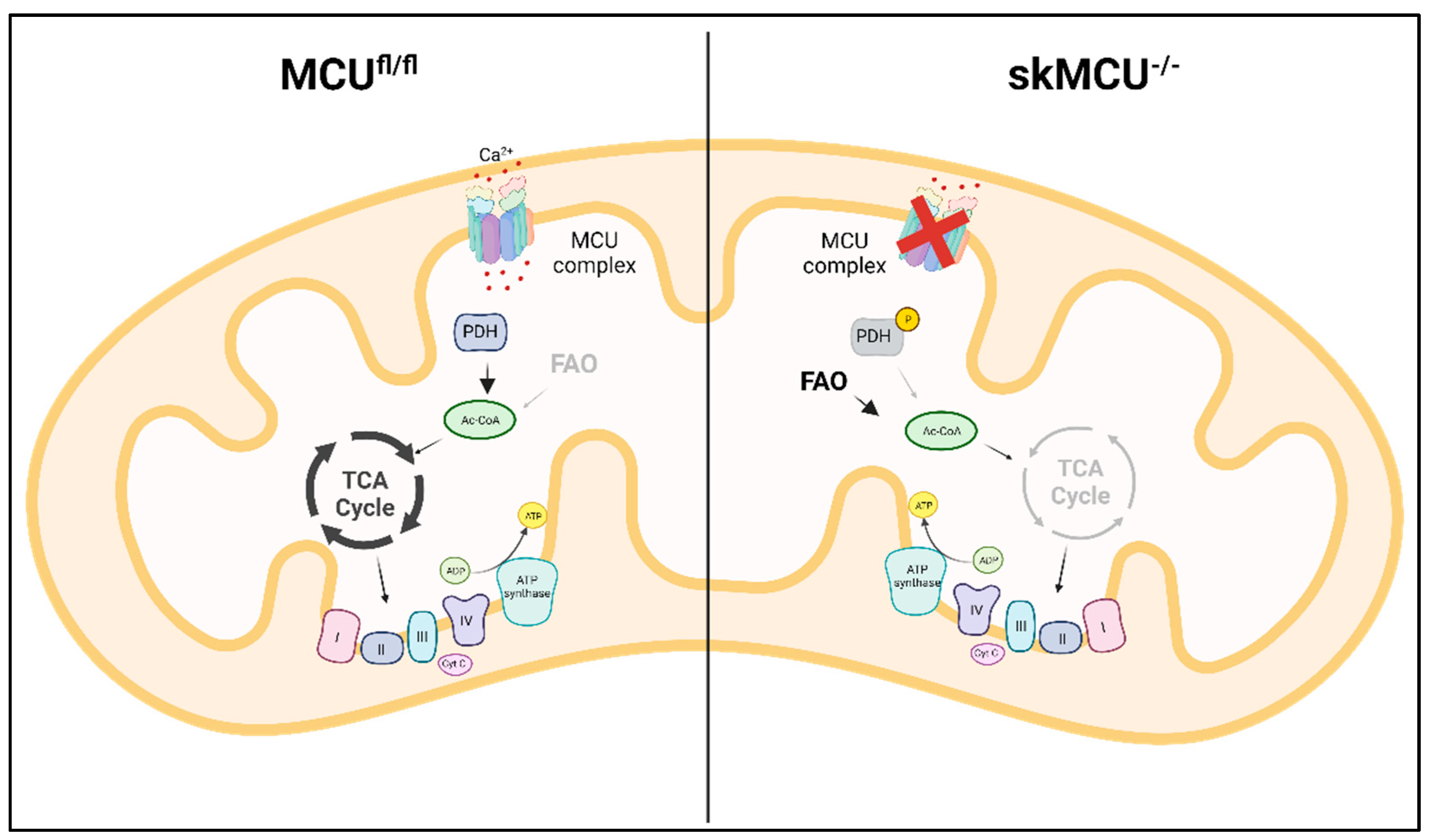
The molecular identification of MCU was followed by intensive studies on skeletal muscle aimed at characterizing the role of mtCa2+ homeostasis in this tissue characterized with a specific physiology and Ca2+ signaling repertoire [65]. The study of the first global MCU knockout mouse model exhibited the most prominent alterations in the skeletal muscle. As expected, in this model both resting mtCa2+ concentrations and stimulated mtCa2+ uptake were reduced. These alterations caused an impairment in mitochondrial oxidative metabolism with an increase in the phosphorylation level of pyruvate dehydrogenase, leading to a reduction in TCA cycle activity. The defective mitochondrial energetic control is responsible for the reduction in exercise performance and muscle force [21]. Mammucari et al., studied the role of MCU in adult skeletal muscle, avoiding compensatory effects that can be present in the global knockout model. MCU expression was modulated through silencing and overexpression in vivo: overexpression of MCU caused muscle hypertrophy, while the silencing of MCU led to muscle atrophy. Interestingly, the control of muscle size and trophism by MCU, observed in both developing and adult muscles, did not depend on the effect on aerobic metabolism, but on the regulation of two major pathways of skeletal muscle hypertrophy, IGF1-Akt and PGC-1α4 [66]. To further characterize the role of MCU in skeletal muscle physiology, Gherardi et al., generated a skeletal muscle-specific MCU knockout, characterized by myofiber-specific impairment of mtCa2+ uptake. This triggered a decrease in muscle exercise performance and force, and a fiber-type switch, from slow to fast MHC expression. Notably, loss of MCU rewired skeletal muscle metabolism toward fatty acid oxidation [67] (Figure 3).
Interestingly, another MCU complex component, MCUb, the dominant-negative form of MCU, plays a key role during skeletal muscle regeneration, by modulating macrophage-driven stimulation and differentiation of satellite cells after muscle damage. In particular, MCUb was shown to drive macrophage polarization from the pro-inflammatory phenotype to the anti-inflammatory phenotype, with secretion of cytokines that promote satellite cells differentiation and fusion [68].
The role of the MCU complex in skeletal muscle physiology is critical, and mutations in MICU1 gene were reported in human patients with a disease phenotype characterized by learning difficulties, a progressive extrapyramidal movement disorder and learning difficulties. Clinically, the disease was characterized by early onset proximal muscle weakness, intellectual impairment and elevated levels of serum creatine kinases. At the genetic level, different loss-of-function mutations were found, resulting in the loss of MICU1 protein. This leads to in increased mtCa2+ load, increasing sensitivity to cell death stimuli but also resulting in lower cytoplasmic Ca2+ level, potentially impinging on muscle contraction and synaptic transmission [69]. To further understand the mechanisms behind this neuromuscular disease, Debattisti et al., characterized patient cells and skeletal muscle-specific MICU1 knockout mice. Lack of MICU1 was associated with a low threshold for MCU-mediated Ca2+ uptake. Notably, MICU1 loss causes muscle atrophy and a decrease in force. The alterations in mtCa2+ uptake during sarcolemma injury, leads to an ineffective muscle repair [70]. Recently, the potential other side of the coin, the neural pathogenesis, was characterized by Singh et al. They generated a neuron-specific MICU1-KO mouse model showing progressive motor and cognitive degeneration. MICU1-KO neurons are more susceptible to mtCa2+ overload and cell death, and this is reverted by the inhibition of the mPTP [71].
MICU1 was shown to be critical also in another skeletal muscle disorder, the Barth syndrome, characterized by cardiolipin deficiency. Ghosh et al., utilized several Barth syndrome models including yeast, mouse model, and patient cells, and showed that cardiolipin is required for the stability of MICU1, which is reduced in Barth syndrome patient-derived cells, together with MCU and MICU2. The reduction in mtCa2+ uptake results in reduced mitochondrial respiration [72].
Finally, mtCa2+ uptake was found to be critical in embryonal rhabdomyosarcoma (ERMS). Indeed, MCU expression is upregulated in ERMS and its downregulation causes mROS level decrease, and an increased propensity to differentiate, inhibiting the oncogenic phenotype [73].
6. Neurodegenerative Diseases
Neurodegenerative diseases comprise a large group of heterogeneous disorders, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis, that are all characterized by selective cell death of neuronal subtypes. A common feature of these disorders is mtCa2+ overload, that can trigger the opening of mPTP, leading to cell death [74].
6.1. Alzheimer’s Disease
The pathogenic mechanisms of Alzheimer’s disease (AD) are still poorly characterized, but a hallmark is the aberrant processing of the amyloid precursor protein (APP) mediated by γ-secretase. The catalytic components of γ-secretase, presenilin-1 and -2 are enriched in the mitochondria-associate ER membranes (MAMs) in cell models of AD [75]. Zampese et al., showed that overexpression and the silencing of presenilin-2 modulate the transfer of Ca2+ between ER and mitochondria. In particular, overexpression of presenilin-2 mutants found in familial AD increased the physical interaction between ER and mitochondria, increasing mtCa2+ entry [76]. Cheung et al., showed that familial AD presenilin-1 and -2 mutants interact with 1,4,5-trisphosphate receptor (InsP3R) to promote Ca2+ release from ER [77]. Importantly, this leads to mtCa2+ overload and an increase in the open probability of mPTP [78]. Notably, the pathogenesis of familial AD was ameliorated in mouse model with the suppression of InsP3R-mediated Ca2+ signaling [79]. Interestingly, amyloid-beta and prion peptides can also induce the release of Ca2+ from the ER [80]. Reduction of NCLX activity, another way to increase the susceptibility to Ca2+-induced cell death, was found in AD neurons, supporting the notion of mtCa2+ overload as crucial in AD progression [81].
6.2. Parkinson’s Disease
Parkinson’s disease (PD) is a neurological disorder associated with the loss of dopaminergic neurons in the substantia nigra, characterized by typical motor symptoms including tremors and muscle rigidity [82]. Models of PD caused by alpha-synuclein overexpression show excessive mtCa2+ uptake, enhancing ROS production and impairing oxidative metabolism [83]. Mutations in PINK1 are also causative of PD, and Gandhi et al., investigated the alterations in mtCa2+ homeostasis in PINK1-deficient neurons. PINK1 deficiency causes mtCa2+ overload, increasing ROS production and impairing respiration, resulting in neuronal cell death [84]. Genetic and pharmacological inactivation of MCU in a pink1-mutant zebrafish prevents dopaminergic neuronal cell death via rescue of mitochondrial respiration [85]. Interestingly, also mutations in the E3 ubiquitin-ligase Parkin cause familial PD, and Matteucci et al., showed that Parkin is required for the proteasome-mediated degradation of MICU1. Probably in indirect way, also MICU2 stability was affected upon Parkin overexpression. This study suggests that Parkin loss could contribute to the impairment in mtCa2+ handling through its regulation of MICUs proteins [86]. Finally, mutations in leucin-rich repeat kinase 2 (LRRK2), a common genetic cause of PD, increased the expression of MCU and MICU2, enhancing mtCa2+ uptake in cortical neurons and familial PD cells [87].
6.3. Huntington’s Disease
Huntington’s disease (HD) is a progressive neurodegenerative disorder in which the huntingtin gene is expanded by CAG triplet repeat leading to a N-terminal polyglutamine strand of variable length [88]. Panov et al., showed a reduction in mtCa2+ uptake in HD cells from patients together with a reduction in membrane potential [89]. However, the susceptibility to mPTP opening is increased in mitochondria isolated from HD cells [90]. Another study conducted on striatal neurons of HD showed that mitochondria were unable to handle large Ca2+ loads, maybe for the increasing sensitivity to mPTP, which reduces membrane potential leading to Ca2+ release [91]. Further studies are needed to elucidate the contribution of mtCa2+ in the generation and progression of this disease.
6.4. Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal motoneuron disease characterized by muscle weakness, atrophy and eventually paralysis that leads to death [92]. The role of mtCa2+ in ALS changes during the progression of the disease. Indeed, in cultured embryonic motor neurons from ALS mouse model, MCU is upregulated and its pharmacological inhibition is protective against excitotoxicity. Instead, MCU expression is reduced in motor neurons from symptomatic ALS mouse model [93]. However, another study showed that in motor neurons of ALS end stage, MCU and MICU1 were upregulated [94]. Thus, a different MCU modulation can be protective depending on the distinct phase of the disease.
7. Conclusions and Future Perspectives
In summary, since the molecular identification of MCU, research on this essential signaling component of mitochondria has literally boomed, with several hundreds of papers providing a deeper insight into the association with other proteins, the regulation of its activity and its role in the physiology of a broad variety of tissues. It has become clear that the essential constituent of the pore region, MCU itself, is part of a larger complex, and that the molecular composition of the latter (i.e. the presence of the dominant-negative MCUb pore subunit, or the identity of the associated regulatory elements) provides wide flexibility to the molecular machinery of a specific cell type, in agreement with the physiological properties of the cell. It is also becoming increasing clear that alterations of this finely tuned process have effects on events as diverse as metabolism, cell death and inflammation. In this review, we have summarized the increasing evidence associating dysregulation of MCU with the pathogenesis of diseases of high prevalence and social impact. While mechanistic insight in most cases still needs to be fully elucidated, these data highlight the mitochondrial calcium signaling machinery as a promising pharmacological target and suggest that the clarification of the role of the individual molecular components may lead to new drugs of high precision in these diseases.
Author Contributions
D.D.A. and R.R. designed the manuscript; D.D.A. and R.R. drafted the text; D.D.A. prepared the figures; D.D.A. and R.R. critically discussed the content and R.R. revised the manuscript; R.R. is the funding recipient. All authors have read and agreed to the published version of the manuscript.
Funding
The research in the authors’ laboratory is funded by grants by the Italian Ministry of University and Research (PRIN, PNRR MUR Mission 4, Component 2) and of Health (Ricerca Finalizzata), the Cariparo Foundation, the Italian Association for Cancer Research (IG18633) and Telethon-Italy (GGP16029).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The images of this review were created with BioRender, which we acknowledge.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011 Jun 19;476(7360):341-5. PMID: 21685886; PMCID: PMC3486726. [CrossRef]
- De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011 Jun 19;476(7360):336-40. PMID: 21685888; PMCID: PMC4141877. [CrossRef]
- Baradaran R, Wang C, Siliciano AF, Long SB. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nature. 2018 Jul;559(7715):580-584. Epub 2018 Jul 11. PMID: 29995857; PMCID: PMC6336196. [CrossRef]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013 Aug 28;32(17):2362-76. Epub 2013 Jul 30. PMID: 23900286; PMCID: PMC3771344. [CrossRef]
- Fieni F, Lee SB, Jan YN, Kirichok Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun. 2012;3:1317. PMID: 23271651; PMCID: PMC3818247. [CrossRef]
- Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013 Dec 13;342(6164):1379-82. Epub 2013 Nov 14. PMID: 24231807; PMCID: PMC4091629. [CrossRef]
- Yamamoto T, Yamagoshi R, Harada K, Kawano M, Minami N, Ido Y, Kuwahara K, Fujita A, Ozono M, Watanabe A, Yamada A, Terada H, Shinohara Y. Analysis of the structure and function of EMRE in a yeast expression system. Biochim Biophys Acta. 2016 Jun;1857(6):831-9. Epub 2016 Mar 18. [CrossRef] [PubMed]
- König T, Tröder SE, Bakka K, Korwitz A, Richter-Dennerlein R, Lampe PA, Patron M, Mühlmeister M, Guerrero-Castillo S, Brandt U, Decker T, Lauria I, Paggio A, Rizzuto R, Rugarli EI, De Stefani D, Langer T. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol Cell. 2016 Oct 6;64(1):148-162. Epub 2016 Sep 15. [CrossRef] [PubMed]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010 Sep 16;467(7313):291-6. Epub 2010 Aug 8. PMID: 20693986; PMCID: PMC2977980. [CrossRef]
- Csordás G, Golenár T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, Perez SF, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnóczky G. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca²⁺ uniporter. Cell Metab. 2013 Jun 4;17(6):976-987. PMID: 23747253; PMCID: PMC3722067. [CrossRef]
- Vecellio Reane D, Vallese F, Checchetto V, Acquasaliente L, Butera G, De Filippis V, Szabò I, Zanotti G, Rizzuto R, Raffaello A. A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca2+ Uptake Machinery of Skeletal Muscle. Mol Cell. 2016 Nov 17;64(4):760-773. Epub 2016 Nov 3. [CrossRef] [PubMed]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One. 2013;8(2):e55785. Epub 2013 Feb 7. PMID: 23409044; PMCID: PMC3567112. [CrossRef]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De Stefani D, Rizzuto R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell. 2014 Mar 6;53(5):726-37. Epub 2014 Feb 20. PMID: 24560927; PMCID: PMC3988891. [CrossRef]
- Kamer KJ, Grabarek Z, Mootha VK. High-affinity cooperative Ca2+ binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep. 2017 Aug;18(8):1397-1411. Epub 2017 Jun 14. PMID: 28615291; PMCID: PMC5538426. [CrossRef]
- Payne R, Hoff H, Roskowski A, Foskett JK. MICU2 Restricts Spatial Crosstalk between InsP3R and MCU Channels by Regulating Threshold and Gain of MICU1-Mediated Inhibition and Activation of MCU. Cell Rep. 2017 Dec 12;21(11):3141-3154. PMID: 29241542; PMCID: PMC5734103. [CrossRef]
- Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019 Jan;26(1):179-195. Epub 2018 May 3. PMID: 29725115; PMCID: PMC6124646. [CrossRef]
- Adlakha J, Karamichali I, Sangwallek J, Deiss S, Bär K, Coles M, Hartmann MD, Lupas AN, Hernandez Alvarez B. Characterization of MCU-Binding Proteins MCUR1 and CCDC90B - Representatives of a Protein Family Conserved in Prokaryotes and Eukaryotic Organelles. Structure. 2019 Mar 5;27(3):464-475.e6. Epub 2019 Jan 3. [CrossRef] [PubMed]
- Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, Csordás G, Madireddi P, Yang J, Müller M, Miller R, Kolesar JE, Molgó J, Kaufman B, Hajnóczky G, Foskett JK, Madesh M. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012 Dec;14(12):1336-43. Epub 2012 Nov 25. Erratum in: Nat Cell Biol. 2013 Jan;15(1):123. Erratum in: Nat Cell Biol. 2015 Jul;17(7):953. PMID: 23178883; PMCID: PMC3511605. [CrossRef]
- Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab. 2015 Jan 6;21(1):109-16. [CrossRef] [PubMed]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009 Nov;1787(11):1395-401. Epub 2009 Jul 1. PMID: 19576166; PMCID: PMC2730424. [CrossRef]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013 Dec;15(12):1464-72. Epub 2013 Nov 10. PMID: 24212091; PMCID: PMC3852190. [CrossRef]
- Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, Luczak ED, Wang Q, Rokita AG, Wehrens XH, Song LS, Anderson ME. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015 Jan 20;6:6081. Erratum in: Nat Commun. 2015;6:7241. PMID: 25603276; PMCID: PMC4398998. [CrossRef]
- Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015 Jul 7;12(1):23-34. Epub 2015 Jun 25. PMID: 26119731; PMCID: PMC4517182. [CrossRef]
- Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015 Jul 7;12(1):15-22. Epub 2015 Jun 25. PMID: 26119742; PMCID: PMC4497842. [CrossRef]
- Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, Tsai EJ, Molkentin JD, Chen X, Madesh M, Houser SR, Elrod JW. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature. 2017 May 4;545(7652):93-97. Epub 2017 Apr 26. PMID: 28445457; PMCID: PMC5731245. [CrossRef]
- Lambert JP, Luongo TS, Tomar D, Jadiya P, Gao E, Zhang X, Lucchese AM, Kolmetzky DW, Shah NS, Elrod JW. MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation. 2019 Nov 19;140(21):1720-1733. Epub 2019 Sep 19. PMID: 31533452; PMCID: PMC6996560. [CrossRef]
- Xue Q, Pei H, Liu Q, Zhao M, Sun J, Gao E, Ma X, Tao L. MICU1 protects against myocardial ischemia/reperfusion injury and its control by the importer receptor Tom70. Cell Death Dis. 2017 Jul 13;8(7):e2923. PMID: 28703803; PMCID: PMC5550843. [CrossRef]
- Rutter, GA. Visualising insulin secretion. The Minkowski Lecture 2004. Diabetologia. 2004 Nov;47(11):1861-72. Epub 2004 Nov 17. [CrossRef] [PubMed]
- Kennedy HJ, Pouli AE, Ainscow EK, Jouaville LS, Rizzuto R, Rutter GA. Glucose generates sub-plasma membrane ATP microdomains in single islet beta-cells. Potential role for strategically located mitochondria. J Biol Chem. 1999 May 7;274(19):13281-91. [CrossRef] [PubMed]
- Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984 Nov 29-Dec 5;312(5993):446-8. [CrossRef] [PubMed]
- Wollheim CB, Sharp GW. Regulation of insulin release by calcium. Physiol Rev. 1981 Oct;61(4):914-73. [CrossRef] [PubMed]
- Georgiadou E, Haythorne E, Dickerson MT, Lopez-Noriega L, Pullen TJ, da Silva Xavier G, Davis SPX, Martinez-Sanchez A, Semplici F, Rizzuto R, McGinty JA, French PM, Cane MC, Jacobson DA, Leclerc I, Rutter GA. The pore-forming subunit MCU of the mitochondrial Ca2+ uniporter is required for normal glucose-stimulated insulin secretion in vitro and in vivo in mice. Diabetologia. 2020 Jul;63(7):1368-1381. Epub 2020 Apr 29. PMID: 32350566; PMCID: PMC7286857. [CrossRef]
- Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R, Rutter GA. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLoS One. 2012;7(7):e39722. Epub 2012 Jul 19. PMID: 22829870; PMCID: PMC3400633. [CrossRef]
- Alam MR, Groschner LN, Parichatikanond W, Kuo L, Bondarenko AI, Rost R, Waldeck-Weiermair M, Malli R, Graier WF. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic β-cells. J Biol Chem. 2012 Oct 5;287(41):34445-54. Epub 2012 Aug 17. PMID: 22904319; PMCID: PMC3464549. [CrossRef]
- Vishnu N, Hamilton A, Bagge A, Wernersson A, Cowan E, Barnard H, Sancak Y, Kamer KJ, Spégel P, Fex M, Tengholm A, Mootha VK, Nicholls DG, Mulder H. Mitochondrial clearance of calcium facilitated by MICU2 controls insulin secretion. Mol Metab. 2021 Sep;51:101239. Epub 2021 Apr 28. PMID: 33932586; PMCID: PMC8163986. [CrossRef]
- Wright LE, Vecellio Reane D, Milan G, Terrin A, Di Bello G, Belligoli A, Sanna M, Foletto M, Favaretto F, Raffaello A, Mammucari C, Nitti D, Vettor R, Rizzuto R. Increased mitochondrial calcium uniporter in adipocytes underlies mitochondrial alterations associated with insulin resistance. Am J Physiol Endocrinol Metab. 2017 Dec 1;313(6):E641-E650. Epub 2017 Aug 8. PMID: 28790027; PMCID: PMC6109647. [CrossRef]
- Pinton P, Ferrari D, Magalhães P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol. 2000 Mar 6;148(5):857-62. PMID: 10704437; PMCID: PMC2174537. [CrossRef]
- Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001 Jun 1;20(11):2690-701. PMID: 11387204; PMCID: PMC125256. [CrossRef]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003 Apr 4;300(5616):135-9. Epub 2003 Mar 6. [CrossRef] [PubMed]
- Rosa N, Ivanova H, Wagner LE 2nd, Kale J, La Rovere R, Welkenhuyzen K, Louros N, Karamanou S, Shabardina V, Lemmens I, Vandermarliere E, Hamada K, Ando H, Rousseau F, Schymkowitz J, Tavernier J, Mikoshiba K, Economou A, Andrews DW, Parys JB, Yule DI, Bultynck G. Bcl-xL acts as an inhibitor of IP3R channels, thereby antagonizing Ca2+-driven apoptosis. Cell Death Differ. 2022 Apr;29(4):788-805. Epub 2021 Nov 8. PMID: 34750538; PMCID: PMC8990011. [CrossRef]
- Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, Duchen MR, Rosato A, Bogeski I, Szabadkai G, Rizzuto R, Mammucari C. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol Med. 2016 May 2;8(5):569-85. PMID: 27138568; PMCID: PMC4864890. [CrossRef]
- Stejerean-Todoran I, Zimmermann K, Gibhardt CS, Vultur A, Ickes C, Shannan B, Bonilla Del Rio Z, Wölling A, Cappello S, Sung HM, Shumanska M, Zhang X, Nanadikar M, Latif MU, Wittek A, Lange F, Waters A, Brafford P, Wilting J, Urlaub H, Katschinski DM, Rehling P, Lenz C, Jakobs S, Ellenrieder V, Roesch A, Schön MP, Herlyn M, Stanisz H, Bogeski I. MCU controls melanoma progression through a redox-controlled phenotype switch. EMBO Rep. 2022 Nov 7;23(11):e54746. Epub 2022 Sep 26. PMID: 36156348; PMCID: PMC9638851. [CrossRef]
- Vultur A, Gibhardt CS, Stanisz H, Bogeski I. The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflugers Arch. 2018 Aug;470(8):1149-1163. Epub 2018 Jun 21. [CrossRef] [PubMed]
- Filadi R, De Mario A, Audano M, Romani P, Pedretti S, Cardenas C, Dupont S, Mammucari C, Mitro N, Pizzo P. Sustained IP3-linked Ca2+ signaling promotes progression of triple negative breast cancer cells by regulating fatty acid metabolism. Front Cell Dev Biol. 2023 Mar 13;11:1071037. PMID: 36994106; PMCID: PMC10040683. [CrossRef]
- Fernandez Garcia E, Paudel U, Noji MC, Bowman CE, Rustgi AK, Pitarresi JR, Wellen KE, Arany Z, Weissenrieder JS, Foskett JK. The mitochondrial Ca2+ channel MCU is critical for tumor growth by supporting cell cycle progression and proliferation. Front Cell Dev Biol. 2023 Jun 8;11:1082213. PMID: 37363724; PMCID: PMC10285664. [CrossRef]
- Curry MC, Peters AA, Kenny PA, Roberts-Thomson SJ, Monteith GR. Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun. 2013 May 10;434(3):695-700. Epub 2013 Apr 18. [CrossRef] [PubMed]
- De Mario A, Tosatto A, Hill JM, Kriston-Vizi J, Ketteler R, Vecellio Reane D, Cortopassi G, Szabadkai G, Rizzuto R, Mammucari C. Identification and functional validation of FDA-approved positive and negative modulators of the mitochondrial calcium uniporter. Cell Rep. 2021 Jun 22;35(12):109275. PMID: 34161774; PMCID: PMC8242467. [CrossRef]
- Xue P, Chen Q, Ren X, Liu D, Yang X. A novel protoapigenone analog RY10-4 induces apoptosis of breast cancer cells by exacerbating mitochondrial Ca2+ influx through mitochondrial calcium uniporter. Toxicol Appl Pharmacol. 2021 Dec 15;433:115776. Epub 2021 Oct 28. [CrossRef] [PubMed]
- Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, Cai G, Meng X, Zou F. Mitochondrial Ca²⁺ uniporter is critical for store-operated Ca²⁺ entry-dependent breast cancer cell migration. Biochem Biophys Res Commun. 2015 Feb 27;458(1):186-93. Epub 2015 Jan 29. [CrossRef] [PubMed]
- Hall DD, Wu Y, Domann FE, Spitz DR, Anderson ME. Mitochondrial calcium uniporter activity is dispensable for MDA-MB-231 breast carcinoma cell survival. PLoS One. 2014 May 6;9(5):e96866. PMID: 24802861; PMCID: PMC4011874. [CrossRef]
- Yu C, Wang Y, Peng J, Shen Q, Chen M, Tang W, Li X, Cai C, Wang B, Cai S, Meng X, Zou F. Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget. 2017 Jul 31;8(48):83831-83844. PMID: 29137386; PMCID: PMC5663558. [CrossRef]
- Martin J, Maurhofer O, Bellance N, Benard G, Graber F, Hahn D, Galinier A, Hora C, Gupta A, Ferrand G, Hoppeler H, Rossignol R, Dufour JF, St-Pierre MV. Disruption of the histidine triad nucleotide-binding hint2 gene in mice affects glycemic control and mitochondrial function. Hepatology. 2013 May;57(5):2037-48. [CrossRef] [PubMed]
- Chen L, Sun Q, Zhou D, Song W, Yang Q, Ju B, Zhang L, Xie H, Zhou L, Hu Z, Yao H, Zheng S, Wang W. HINT2 triggers mitochondrial Ca2+ influx by regulating the mitochondrial Ca2+ uniporter (MCU) complex and enhances gemcitabine apoptotic effect in pancreatic cancer. Cancer Lett. 2017 Dec 28;411:106-116. Epub 2017 Sep 23. [CrossRef] [PubMed]
- Xie KF, Guo DD, Luo XJ. SMDT1-driven change in mitochondrial dynamics mediate cell apoptosis in PDAC. Biochem Biophys Res Commun. 2019 Apr 2;511(2):323-329. Epub 2019 Feb 16. [CrossRef] [PubMed]
- Wang X, Li Y, Li Z, Lin S, Wang H, Sun J, Lan C, Wu L, Sun D, Huang C, Singh PK, Hempel N, Trebak M, DeNicola GM, Hao J, Yang S. Mitochondrial Calcium Uniporter Drives Metastasis and Confers a Targetable Cystine Dependency in Pancreatic Cancer. Cancer Res. 2022 Jun 15;82(12):2254-2268. PMID: 35413105; PMCID: PMC9203979. [CrossRef]
- Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corrà F, Giorgi C, De Marchi E, Poletti F, Gafà R, Lanza G, Negrini M, Rizzuto R, Pinton P. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol. 2013 Jan 7;23(1):58-63. Epub 2012 Dec 13. PMID: 23246404; PMCID: PMC3540261. [CrossRef]
- Yu J, Chen X, Li J, Wang F. CERS6 antisense RNA 1 promotes colon cancer via upregulating mitochondrial calcium uniporter. Eur J Clin Invest. 2023 May;53(5):e13951. Epub 2023 Feb 23. [CrossRef] [PubMed]
- Zhu J, Zhang C, Wang Z, Shi L, Li L, Wu H, Liu M. miR-138-5p targets MCU to inhibit mitochondrial biogenesis and colorectal cancer growth. J Cell Mol Med. 2023 Jun 1. Epub ahead of print. [CrossRef] [PubMed]
- Zeng F, Chen X, Cui W, Wen W, Lu F, Sun X, Ma D, Yuan Y, Li Z, Hou N, Zhao H, Bi X, Zhao J, Zhou J, Zhang Y, Xiao RP, Cai J, Zhang X. RIPK1 Binds MCU to Mediate Induction of Mitochondrial Ca2+ Uptake and Promotes Colorectal Oncogenesis. Cancer Res. 2018 Jun 1;78(11):2876-2885. Epub 2018 Mar 12. [CrossRef] [PubMed]
- Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, Guo X, Ji L, Huang Q, Zhang H, Yang H, Xing J. MCU-dependent mitochondrial Ca2+ inhibits NAD+/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene. 2017 Oct 19;36(42):5897-5909. Epub 2017 Jun 26. [CrossRef] [PubMed]
- Ren T, Wang J, Zhang H, Yuan P, Zhu J, Wu Y, Huang Q, Guo X, Zhang J, Ji L, Li J, Zhang H, Yang H, Xing J. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxid Redox Signal. 2018 Apr 20;28(12):1120-1136. Epub 2017 Nov 1. [CrossRef] [PubMed]
- Deng Q, Chen S, Fu C, Jiang J, Zou M, Tan Y, Wang X, Xia F, Feng K, Ma K, Bie P. Long noncoding RNA expression profiles in sub-lethal heat-treated hepatoma carcinoma cells. World J Surg Oncol. 2017 Jul 21;15(1):136. PMID: 28732507; PMCID: PMC5521104. [CrossRef]
- Zhao L, Jiang M, Tian T, Wang G, Mei Y, Fu G, Zhou N. Effects of MCU mediated Ca2+ homeostasis on ovarian cancer cell SKOV3 proliferation, migration and transforming. Curr Mol Med. 2022 Jun 17. Epub ahead of print. [CrossRef] [PubMed]
- Meng K, Hu Y, Wang D, Li Y, Shi F, Lu J, Wang Y, Cao Y, Zhang CZ, He QY. EFHD1, a novel mitochondrial regulator of tumor metastasis in clear cell renal cell carcinoma. Cancer Sci. 2023 May;114(5):2029-2040. Epub 2023 Feb 26. PMID: 36747492; PMCID: PMC10154798. [CrossRef]
- Gherardi G, De Mario A, Mammucari C. The mitochondrial calcium homeostasis orchestra plays its symphony: Skeletal muscle is the guest of honor. Int Rev Cell Mol Biol. 2021;362:209-259. Epub 2021 May 20. [CrossRef] [PubMed]
- Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, Cagnin S, Braga A, Zanin S, Pallafacchina G, Zentilin L, Sandri M, De Stefani D, Protasi F, Lanfranchi G, Rizzuto R. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 2015 Mar 3;10(8):1269-79. Epub 2015 Feb 26. PMID: 25732818; PMCID: PMC4351162. [CrossRef]
- Gherardi G, Nogara L, Ciciliot S, Fadini GP, Blaauw B, Braghetta P, Bonaldo P, De Stefani D, Rizzuto R, Mammucari C. Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ. 2019 Jan;26(2):362-381. Epub 2018 Sep 19. PMID: 30232375; PMCID: PMC6329801. [CrossRef]
- Feno S, Munari F, Reane DV, Gissi R, Hoang DH, Castegna A, Chazaud B, Viola A, Rizzuto R, Raffaello A. The dominant-negative mitochondrial calcium uniporter subunit MCUb drives macrophage polarization during skeletal muscle regeneration. Sci Signal. 2021 Nov 2;14(707):eabf3838. Epub 2021 Nov 2. [CrossRef] [PubMed]
- Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, Bonthron DT, Pysden KA, Whyte T, Munteanu I, Foley AR, Wheway G, Szymanska K, Natarajan S, Abdelhamed ZA, Morgan JE, Roper H, Santen GW, Niks EH, van der Pol WL, Lindhout D, Raffaello A, De Stefani D, den Dunnen JT, Sun Y, Ginjaar I, Sewry CA, Hurles M, Rizzuto R; UK10K Consortium; Duchen MR, Muntoni F, Sheridan E. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat Genet. 2014 Feb;46(2):188-93. Epub 2013 Dec 15. [CrossRef] [PubMed]
- Debattisti V, Horn A, Singh R, Seifert EL, Hogarth MW, Mazala DA, Huang KT, Horvath R, Jaiswal JK, Hajnóczky G. Dysregulation of Mitochondrial Ca2+ Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell Rep. 2019 Oct 29;29(5):1274-1286.e6. PMID: 31665639; PMCID: PMC7007691. [CrossRef]
- Singh R, Bartok A, Paillard M, Tyburski A, Elliott M, Hajnóczky G. Uncontrolled mitochondrial calcium uptake underlies the pathogenesis of neurodegeneration in MICU1-deficient mice and patients. Sci Adv. 2022 Mar 18;8(11):eabj4716. Epub 2022 Mar 18. PMID: 35302860; PMCID: PMC8932652. [CrossRef]
- Ghosh S, Zulkifli M, Joshi A, Venkatesan M, Cristel A, Vishnu N, Madesh M, Gohil VM. MCU-complex-mediated mitochondrial calcium signaling is impaired in Barth syndrome. Hum Mol Genet. 2022 Feb 3;31(3):376-385. PMID: 34494107; PMCID: PMC8825335. [CrossRef]
- Chiu HY, Loh AHP, Taneja R. Mitochondrial calcium uptake regulates tumour progression in embryonal rhabdomyosarcoma. Cell Death Dis. 2022 Apr 30;13(4):419. PMID: 35490194; PMCID: PMC9056521. [CrossRef]
- Britti E, Delaspre F, Tamarit J, Ros J. Mitochondrial calcium signalling and neurodegenerative diseases. Neuronal Signal. 2018 Nov 16;2(4):NS20180061. PMID: 32714593; PMCID: PMC7373239. [CrossRef]
- Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL, Schon EA. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012 Nov 5;31(21):4106-23. Epub 2012 Aug 14. PMID: 22892566; PMCID: PMC3492725. [CrossRef]
- Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci U S A. 2011 Feb 15;108(7):2777-82. Epub 2011 Feb 1. PMID: 21285369; PMCID: PMC3041131. [CrossRef]
- Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008 Jun 26;58(6):871-83. PMID: 18579078; PMCID: PMC2495086. [CrossRef]
- Toglia P, Ullah G. The gain-of-function enhancement of IP3-receptor channel gating by familial Alzheimer's disease-linked presenilin mutants increases the open probability of mitochondrial permeability transition pore. Cell Calcium. 2016 Jul;60(1):13-24. Epub 2016 May 7. [CrossRef] [PubMed]
- Shilling D, Müller M, Takano H, Mak DO, Abel T, Coulter DA, Foskett JK. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer's disease pathogenesis. J Neurosci. 2014 May 14;34(20):6910-23. PMID: 24828645; PMCID: PMC4019804. [CrossRef]
- Ferreiro E, Oliveira CR, Pereira CMF. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol Dis. 2008 Jun;30(3):331-342. Epub 2008 Feb 20. [CrossRef] [PubMed]
- Jadiya P, Kolmetzky DW, Tomar D, Di Meco A, Lombardi AA, Lambert JP, Luongo TS, Ludtmann MH, Praticò D, Elrod JW. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer's disease. Nat Commun. 2019 Aug 29;10(1):3885. PMID: 31467276; PMCID: PMC6715724. [CrossRef]
- Kalia LV, Lang AE. Parkinson's disease. Lancet. 2015 Aug 29;386(9996):896-912. Epub 2015 Apr 19. [CrossRef] [PubMed]
- Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int J Biochem Cell Biol. 2009 Oct;41(10):2015-24. Epub 2009 May 19. [CrossRef] [PubMed]
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, Abramov AY. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009 Mar 13;33(5):627-38. PMID: 19285945; PMCID: PMC2724101. [CrossRef]
- Soman S, Keatinge M, Moein M, Da Costa M, Mortiboys H, Skupin A, Sugunan S, Bazala M, Kuznicki J, Bandmann O. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1-/- zebrafish. Eur J Neurosci. 2017 Feb;45(4):528-535. Epub 2016 Dec 28. PMID: 27859782; PMCID: PMC5324670. [CrossRef]
- Matteucci A, Patron M, Vecellio Reane D, Gastaldello S, Amoroso S, Rizzuto R, Brini M, Raffaello A, Calì T. Parkin-dependent regulation of the MCU complex component MICU1. Sci Rep. 2018 Sep 21;8(1):14199. Erratum in: Sci Rep. 2019 Mar 12;9(1):4665. PMID: 30242232; PMCID: PMC6155109. [CrossRef]
- Verma M, Callio J, Otero PA, Sekler I, Wills ZP, Chu CT. Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD-Associated LRRK2 Mutants. J Neurosci. 2017 Nov 15;37(46):11151-11165. Epub 2017 Oct 16. PMID: 29038245; PMCID: PMC5688524. [CrossRef]
- Walker, FO. Huntington's disease. Lancet. 2007 Jan 20;369(9557):218-28. [CrossRef] [PubMed]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002 Aug;5(8):731-6. [CrossRef] [PubMed]
- Panov AV, Lund S, Greenamyre JT. Ca2+-induced permeability transition in human lymphoblastoid cell mitochondria from normal and Huntington's disease individuals. Mol Cell Biochem. 2005 Jan;269(1-2):143-52. [CrossRef] [PubMed]
- Lim D, Fedrizzi L, Tartari M, Zuccato C, Cattaneo E, Brini M, Carafoli E. Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J Biol Chem. 2008 Feb 29;283(9):5780-9. Epub 2007 Dec 21. [CrossRef] [PubMed]
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001 May 31;344(22):1688-700. [CrossRef] [PubMed]
- Tadić V, Adam A, Goldhammer N, Lautenschlaeger J, Oberstadt M, Malci A, Le TT, Sengupta S, Stubendorff B, Keiner S, Witte OW, Grosskreutz J. Investigation of mitochondrial calcium uniporter role in embryonic and adult motor neurons from G93AhSOD1 mice. Neurobiol Aging. 2019 Mar;75:209-222. Epub 2018 Nov 23. [CrossRef] [PubMed]
- Mühling T, Duda J, Weishaupt JH, Ludolph AC, Liss B. Elevated mRNA-levels of distinct mitochondrial and plasma membrane Ca(2+) transporters in individual hypoglossal motor neurons of endstage SOD1 transgenic mice. Front Cell Neurosci. 2014 Nov 14;8:353. PMID: 25452714; PMCID: PMC4231948. [CrossRef]
Figure 1.
MCU complex composition and activity. The MCU complex localize in the IMM and is composed of pore-forming subunits, MCU and MCUb, regulatory subunits, MICUs family, and the structural protein EMRE. The MICUs proteins sense increases in [Ca2+]i via EF-hand domains, undergoing conformational changes and allowing Ca2+ entry into the mitochondrial matrix.
Figure 1.
MCU complex composition and activity. The MCU complex localize in the IMM and is composed of pore-forming subunits, MCU and MCUb, regulatory subunits, MICUs family, and the structural protein EMRE. The MICUs proteins sense increases in [Ca2+]i via EF-hand domains, undergoing conformational changes and allowing Ca2+ entry into the mitochondrial matrix.
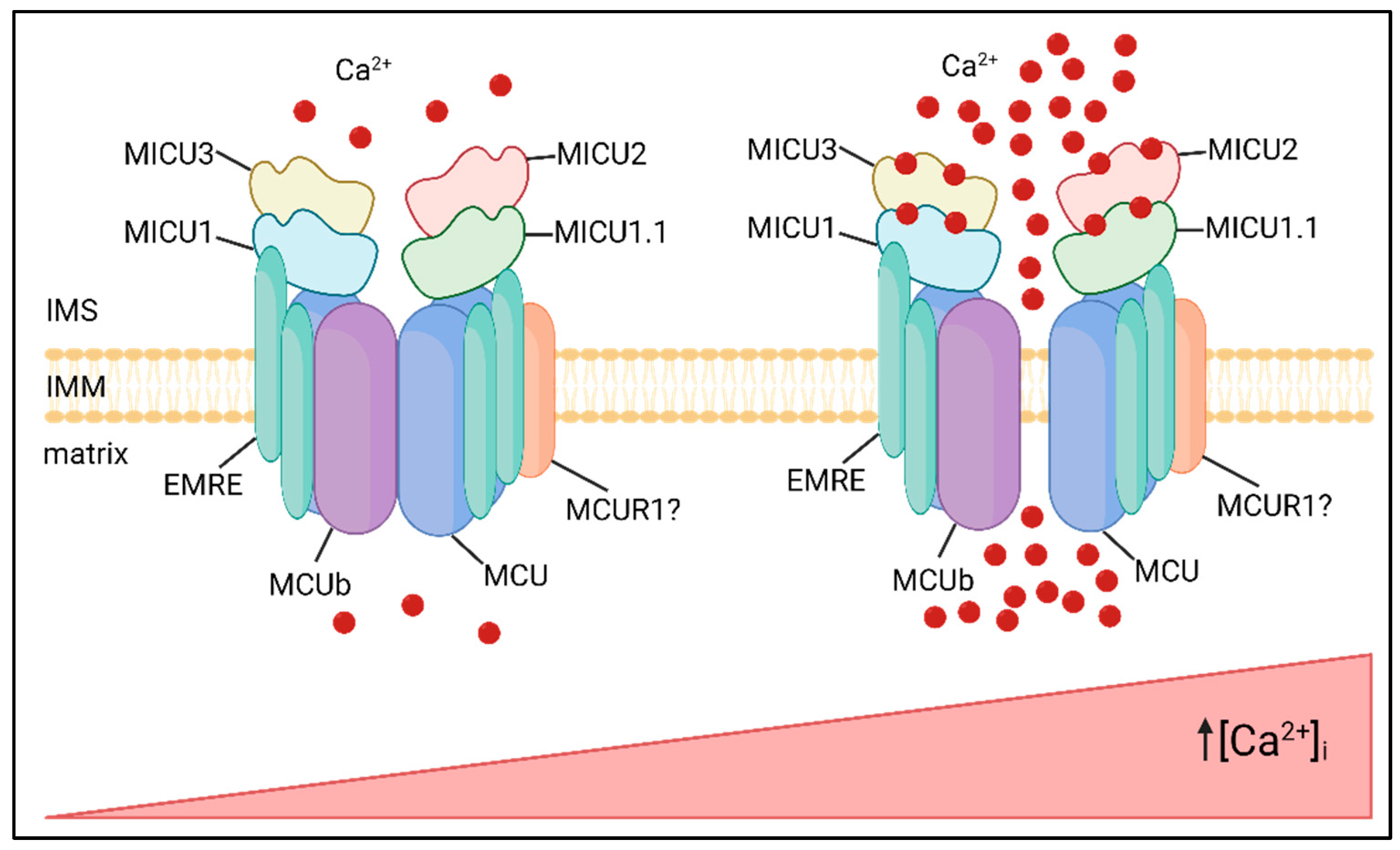
Figure 2.
MtCa2+ dysfunctions in cancer. Left panel: anti-apoptotic Bcl-2 proteins reduce mtCa2+ loading, by reducing ER Ca2+ loading and/or desensitizing IP3R, impairing apoptosis. Right panel: MCU is upregulated in metastatic tumors, and the consequent increase in mtCa2+ uptake promotes tumor progression via ROS/HIF-1α mitochondria-to-nucleus signaling pathway.
Figure 2.
MtCa2+ dysfunctions in cancer. Left panel: anti-apoptotic Bcl-2 proteins reduce mtCa2+ loading, by reducing ER Ca2+ loading and/or desensitizing IP3R, impairing apoptosis. Right panel: MCU is upregulated in metastatic tumors, and the consequent increase in mtCa2+ uptake promotes tumor progression via ROS/HIF-1α mitochondria-to-nucleus signaling pathway.
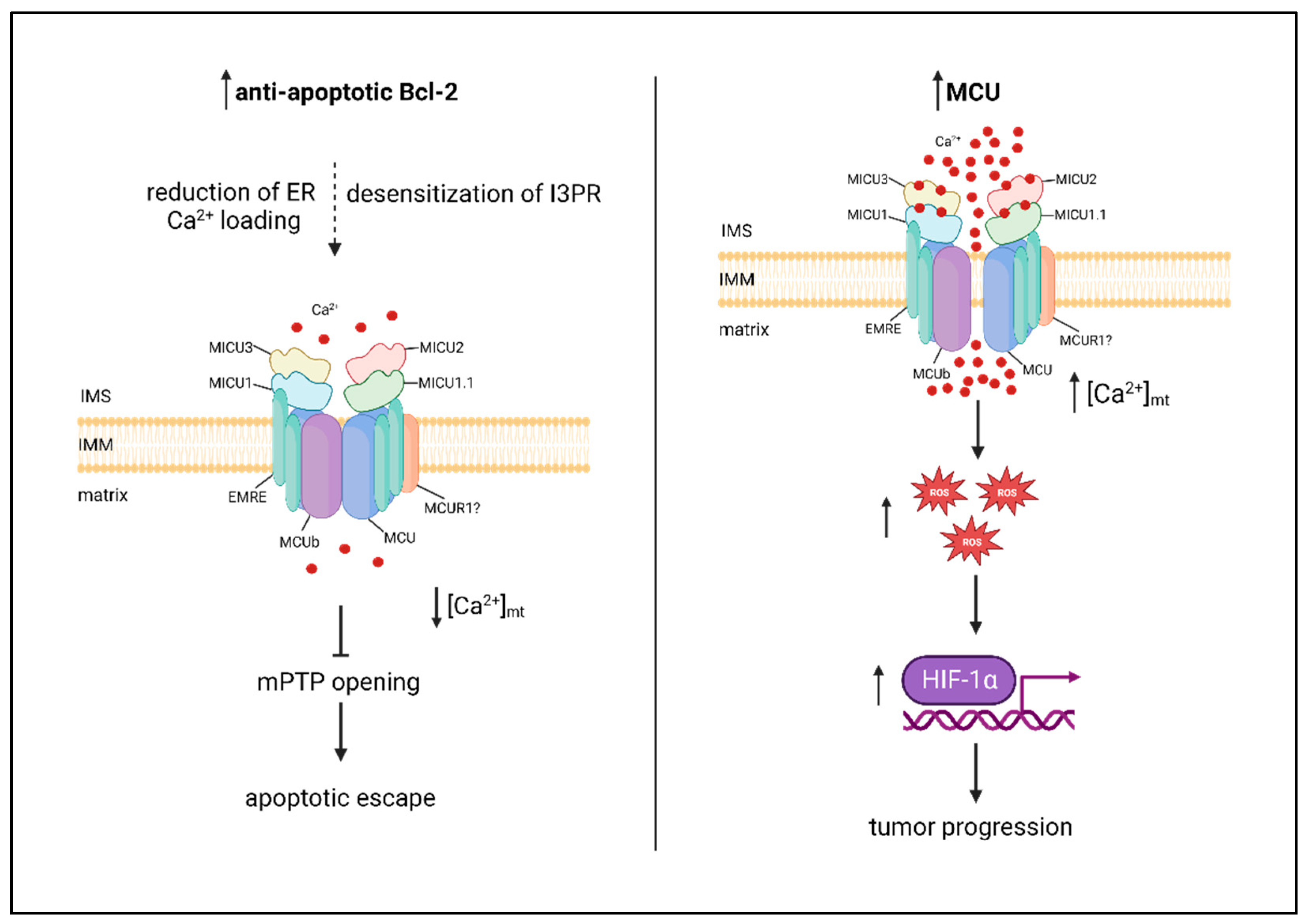
Figure 3.
MCU deletion in skeletal muscle rewires metabolism towards FA utilization. In MCU-/- muscles the loss of mtCa2+ uptake leads to PDH inactivity, impairment in pyruvate oxidation and decrease in TCA cycle activity. The impairment in glucose oxidation is partially compensated by an increase in FA oxidation.
Figure 3.
MCU deletion in skeletal muscle rewires metabolism towards FA utilization. In MCU-/- muscles the loss of mtCa2+ uptake leads to PDH inactivity, impairment in pyruvate oxidation and decrease in TCA cycle activity. The impairment in glucose oxidation is partially compensated by an increase in FA oxidation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



