
Submitted:
28 July 2023
Posted:
31 July 2023
You are already at the latest version
Abstract
Endometrial cancer (EC) is the most frequent gynaecological malignancy. The ESGO/ESTRO/ESP 2020 guidelines identify prognostic groups based on morpho-molecular characteristics. This study aims to evaluate the clinical applicability of NGS analysis to define an appropriate risk class and application for a better diagnostic and prognostic stratification of ECs. Cases of serous carcinoma (OHEC), high (HGEC) and low (LGEC) grade endometrioid carcinoma diagnosed with the morphological and immunohistochemical (IHC) protocols were considered. After a standardized pre-analytical phase, the tumor DNA was semi-automatically extracted and analyzed by NGS with a panel of 14 genes. A total of 63 cases were considered. NGS analysis was successful in 60 cases; all of these were classified according to the new diagnostic algorithm. The molecular risk classification showed a good correlation with the morphological (k=0.8). The study showed that the protocols of the pre-analytical and analytical phases used are robust and can lead to molecular results that fall within the standards required for use in clinical practice for a more precise diagnostic-therapeutic management of patients. The implementation of the classification is particularly relevant for better prognostic stratification of HGECs. In addition, the identification of a suspicious VUS in POLE questions the classification of truncating variants.
Keywords:
Endometrial Cancer (EC)
; Multigene-NGS panel
; POLE
; TP53
1. Introduction
Endometrial cancer (EC) is the most common gynaecological malignancy affecting women in developed countries. According to 2020 GLOBOCAN estimates, more than 417,000 new cases were diagnosed and nearly 97,000 women died worldwide from the disease. In Italy, EC is the third most common cancer in women aged 50-69; it is expected to grow in the next few years to be the sixth most frequent cancer overall by 2030 [1,2,3].
Historically, EC were classified into Type I and Type II carcinomas [4]. Type I included low grade endometrioid carcinoma (LGEC), usually diagnosed at an early stage with a good prognosis. Conversely, type II was high-grade tumors (HGEC), usually diagnosed in an advanced stage and characterized by a worse prognosis. This group mainly consisted of serous EC and are characterized by early TP53 mutation [5]. However, this dualistic model has shown shortcomings in describing the complexity and heterogeneity of EC. In particular, it was noted that the high-grade endometrioid histotype showed intermediate immunomorphological features between the two groups [6].
In 2013 The Cancer Genome Atlas (TCGA) proposed a new stratification based on genomic data identifying four subgroups of EC with distinct genetic profiles: DNA polymerase ε (POLE, ultramutated), microsatellite instability (MSI, hypermutated), Copy Number Low, and Copy Number High [7]. Even though clinical outcomes are correlated to these molecular subgroups, the TCGA molecular classification was not feasible in routine diagnostic procedures. In order to create a more affordable diagnostic algorithm based on feasible techniques [8,9,10], surrogate markers (POLE exonuclease domain mutation, loss of mismatch repair proteins expression, abnormal p53 expression and absence of the other markers) have been elaborated from other research groups. This led to the definition of four molecular prognostic groups: i) “POLE-mutated” (POLEmut) group, characterized by the most favorable prognosis; ii) “mismatch repair-deficient” (MMRd) group; iii) “no specific molecular profile” (NSMP) group; iv) “TP53- mutant” (TP53mut) group, characterized by poor prognosis [10,11,12,13].
Because of this, the ESGO-ESTRO-ESP 2020 guidelines have revised the stratification risk by introducing the morpho-molecular data [14, 15]. Even though the importance of molecular data has therefore been recognized, to date only few centers use these guidelines to classify EC, probably due to the lack of standardized methods to sequence POLE.
In the last few years, several Next Generation Sequencing (NGS) protocols have been developed to analyze tumor DNA. A multigene panel testing is a common approach to analyze cancer susceptibility genes, with timing and cost efficiency useful for EC molecular classification. In this scenario, we aimed to design and validate an NGS-multigene panel for EC; the EC most frequently mutated genes will be analyzed, and the obtained results will be discussed in order to classify the lesions according to the revised TCGA classification criteria defining an appropriate risk class. The combination of molecular data with established clinicopathologic risk factors could be useful for tailoring adjuvant therapy, especially in the high-intermediate-risk group, for which clinical trials are currently under evaluation [16,17,18].
2. Materials and Methods
2.1. Study Cohort
Samples from all the patients with a diagnosis of high grade endometrioid EC (HGEC) and other high-grade endometrial carcinoma (OHEC) who underwent bilateral hystero-adnexectomy in the period 2018-2020 were collected. In the latter group, only serous EC (the prototype of type II EC) were collected.
Instead, the bigger low grade endometrioid EC (LGEC) cohort, was consecutively chosen since 2018 matching HGEC and OHEC cohort. All cases were reviewed by pathologists’ expert in gynaecological pathology (VGV and PM) and selected according to the following criteria: optimal fixation/storage, high representativeness of the entire neoplasia (higher than 30%), high tumor cellularity, low percentage of stroma cell, fibrosis and necrosis. Specimens were prepared according to standardized pre-analytical procedures [19]. The most Formalin-Fixed Paraffin-Embedded (FFPE) blocks representative of the entire neoplasm was selected and manual macrodissection was performed.
2.2. DNA Extraction and NGS Sequencing
DNA was extracted from FFPE sections using automatic procedures (GeneRead DNA FFPE Treatment Kit on QIASymphony, Qiagen). For sample with low starting material, QIAamp DNA FFPE Tissue Kit was preferred. The DNA concentration was assessed by the Qubit 3.0 Fluorometer (ThermoFisher Scientific) and quality were assessed by the Agilent 4200 Tapestation with High Sensitivity D1000 ScreenTape Kit (Agilent Technologies). Samples with concentration <2.5 ng/µl along with DNA Integrity Number (DIN) <2 were excluded from sequencing. Molecular analysis was performed by NGS technology on Ion Torrent S5 platform in combination with Oncomine On Demand Tumor Specific custom panel including 14 genes (BRIP1, CTNNB1, KRAS, MLH1, MLH3, MSH2, MSH6, PALB2, PMS2, POLE, PTEN, TP53, RAD51C and RAD51D). Genes were retrieved from literature and selected on the basis of EC association [7, 20, 21]. Among these, few genes associated with ovarian cancer (BRIP1, PALB2, RAD51C, RAD51D) have been included in the panel for research purpose.
2.3. IHC Methods
IHC assays were performed on FFPE tissue sections using the automated ultraView Universal DAB procedure on the BenchMark ULTRA IHC/ISH Staining Module, Ventana.
Two patterns of p53 expression (clone DO7, prediluted, Ventana) were considered: aberrant expression (diffuse strong nuclear positivity involving at least 80% of the tumor cells or complete absence of p53 expression with internal positive control) and wild type expression (variable proportion of tumor cell nuclei staining with variable intensity). MMR proteins expression was evaluated with the following antibodies: MLH1 (Clone M1, Ventana), MSH2 (Clone G219, Ventana), MSH6 (Clone SP93, Ventana), and PMS2 (Clone A16-4, Ventana). A complete lack of tumor nuclear staining for one or more MMRPs (with internal positive control) was categorized as MMRP-deficient while a positive nuclear staining for all four MMRPs indicated a MMRP-retained status.
2.4. Variant Analysis and Classification
Parameters for analysis excluded variants with: variant allele frequency (VAF) <5%, coverage <500X, quality score (PHRED) <30, strand bias >0.65, minor allele frequency (MAF) >1% and genomic position > 20bp. Variants were classified as pathogenic/likely pathogenic (collectively termed pathogenic) according to American College of Medical Genetics and Genomics (ACMG) recommendations [22] and to Cancer Variant Interpretation Group UK (CanVIG-UK) Gene-Specific Guidance for MMR and TP53 genes [23]. Variants of Uncertain Significance (VUS) along with benign/likely benign variants were discarded. Copy number variations (CNV) have not been evaluated. All filtered variants were verified via visual inspection of .bam alignment files on Alamut Visual Plus v.1.6. Variants with ambiguous allele frequencies were analyzed by Sanger sequencing and traces were visualized on MinorVariantFinder software (ThermoFisher Scientific).
2.5. Statistical Analysis
Associations of clinicopathological parameters with molecular subtypes were compared using Two-way Chi-squared test. For the concordance of EC risk profile on a histo-morphological and molecular basis, kappa value was calculated. The histopathological parameters of the patients across the TP53 mutation spectrum were compared using and Easy Fisher Exact Test Calculator. p < 0.05 was considered statistically significant.
3. Results
3.1. Clinicopathological Characteristics
A total of 63 samples were selected for molecular analysis. The clinical and pathological characteristics of the 63 EC specimens were summarized in Table 1.
The median age was 72.1; considering age as a categorical variable related to the seniority threshold of 65 years, 43 patients (68.2%) were older and 20 (31.7%) were younger than 65.
Of the 63 cases, 47 (74.6%) were endometrioid and 16 (25.4%) were serous. Endometrioid cohort comprises 31 (65.9%) LGEC and 16 (34.1%) HGEC; among LGEC, 12 (38.7%) were G1 whereas 19 (61.3%) were G2. The OHEC cohort consisted of 16 cases, all of high-grade serous histotype.
Regarding the stage of disease, 48 cases were I-II (76.2%) and 15 were III-IV (23.8%) stage according to the FIGO classification. Lymphovascular invasion (LVSI) was also evaluated, resulting positive in 23 (36.5%) and negative in 40 (63.5%) cases.
The evaluation of MSI highlighted 49 cases (77.8%) with preserved expression and 12 (19.0%) cases with instability, of which 11 with loss of expression of MLH1/PMS2 (17.5%) and 1 (1.6%) with loss of MSH2/MSH6; in two cases (3.2%) it was not possible to determine the status of the microsatellites. The p53 expression was aberrant in 18 cases (28.6%), including 4 HGEC and 14 serous OHEC; the remaining 45 wild-type cases (71.4%) were LGEC (n=31), HGEC (n=12) and OHEC (n=2).
3.2. Multigene-NGS Panel
All samples showed good quality parameters with DIN values ranged from 2.0 to 4.8 and DNA concentration from 2.66 to 81.2 ng/µl, hence were suitable to sequencing (Supplementary Table S1). Four separate sequencing runs were performed, with a mean depth of 2893 (range 1015-4980) and 93.5% of target base coverage at 500X. Three samples were not compliant to the quality parameters showing a mean depth <1000 and target base coverage at 500X <60% and were excluded from the subsequent analysis. Sequencing metrics along with alignment quality are summarized in Supplementary Table S2.
The most frequently affected genes in our series were PTEN (55.0%) and TP53 (33.3%) followed by KRAS (18.3%), MMR (15.0%), POLE (13.3%) and CTNNB1 (11.7%); a pathogenic variant was also found in RAD51C (1.7%) and BRIP1 (1.7%), both in association with other genes (Figure 1). All pathogenic variants are enlisted in Supplementary Table S3. To evaluate whether type of mutation and type of tumor are correlate, a comparative analysis was performed and no connection was observed. The Catalogue Of Somatic Mutation In Cancer (COSMIC) was interrogated to identify a correlation between recurrent variants (Supplementary Table S4) and histological classification, and no mutational signatures were identified [24].
3.3. Molecular Typing and Risk Classification
Based on the new integrated morpho-molecular classification [15], the three histogroups were categorized as POLEmut (8/60), MMRd (4/60), NSMP (30/60), TP53mut (18/60). All cases were previously stratified for risk according to histopathological and morphological features; advanced-metastatic, high-intermediate, intermediate, high and low risk classes were assigned (Supplementary Table S5). Molecular results allowed to redefine a risk profile in 8 cases (Supplementary Table S6); a statistically significant correlation (k=0.818) was observed (Table 2).
3.4. POLE and TP53 Profiles
In out of 60 cases analyzed, 8 (15%) harbored a pathogenic variant in exonuclease domain of POLE, according to the literature [25-28]. The majority of these are endometrioid (4 HGEC, 3 LGEC), while one was OHEC. To the best of our knowledge, no POLE pathogenic variants were described in EC of serous histology [20]; therefore, a second histological evaluation was performed, which confirmed the histotype. A POLE pathogenic variant (c.857C>G; p.(Prp286Arg)) was also identified in a LGEC with allele frequency below the threshold of 5%. In order to confirm or exclude this finding, a more accurate selection of tumor area was performed; the Sanger sequencing traces were analyzed with Minor Variant Finder to call variants as low as 5%, with negative results.
Pathogenic variants in TP53 were observed in 20 out of 60 (33.3%) cases, of which 6 endometrioid (3 LGEC, 3 HGEC) and 14 OHEC. Among LGEC cases, two were carriers of additional clearly pathogenic variants, a POLE missense variant (EC-61) and two MMR variants (splice and nonsense variants, respectively; EC-33), so the risk profile was evaluated related to these findings. Of note, OHEC cohort presented a mutation profile with TP53 alone whereas endometrioid cohort presented TP53 combined to other pathogenic variants (Table 3). There was a discordant case (EC-06) histologically classified as OHEC but carrying double pathogenic variants; a review of the IHC slides was required, and a new HGEC phenotype was assigned. Consequently, the risk class also changed from “high” to “intermediate”. A statistically significant relationship was found between TP53 mutational status and histological definition (p < 0.05) [29].
Of the 20 cases harboring a mutation in TP53, two were not congruent with the IHC analysis (Supplementary Table S7). In particular, sample EC-50 showed a vegetating neoplasm that occupies anterior, posterior and fundus walls for a longitudinal extension of 4.5 cm. The neoplasm affected the uterine cavity circumferentially and infiltrated the myometrium within the inner half; for this sample, p53 expression was positive in 90% of the neoplastic elements (aberrant expression). The first NGS analysis resulted wild-type, strongly disagree with the high reliability of the IHC results. For this reason, an area with higher neoplastic density was selected on different sampling and DNA extraction with a scrape from glass slide was performed, identifying the c.796G>A; p.(Gly266Arg) pathogenic variant (VAF 18.25%). Conversely, in EC-58 the neoplasm was found on the basis of a polypoid endometrium with complex hyperplasia associated to atypia and p53 expression <1%. The NGS analysis identified the c.734G>A; p.(Gly245Asp) pathogenic variant (VAF 29.15%), also confirmed by Sanger sequencing. This missense change is in the DNA binding domain and experimental studies have shown that affects TP53 function [30,31,32,33,34,35,36,37]. This variant was reviewed by expert panel (Accession VCV000012356.51) and classified as pathogenic. As already reported [38], IHC is not reliable as the molecular analysis is, stating the importance of the proper clinical interpretation of negative results.
4. Discussion
The high heterogeneity of EC represents an important challenge in diagnostic setting and in the definition of the risk classification. Therefore, literature [39, 40] has highlighted the importance of combined diagnosis on a morphological and molecular basis in order to precisely focus the lesion for the definition of an appropriate risk class and to improve clinical management. Our study aimed to type DNA from FFPE samples by NGS sequencing in order to compare the molecular characteristics of the LGEC, HGEC and OHEC groups and to redefine the risk class of the patients.
Our study population consists of 60 EC, the majority of which were LGEC (n=31) followed by OHEC (n=16) and HGEC (n=13), according to published guidance statements for the validation of NGS-based oncology panels [19, 41].
The 14-genes NGS panel identified pathogenic variants in 57 out of 60 cases, with a mutation detection rate similar to the rates reported in the literature [20, 29, 42–45]. These results allowed us to classify our cohort with the new diagnostic algorithm for the integrated morpho-molecular classification of EC [15]. In particular, there were 20 (33.3%) tumors harboring somatic pathogenic variant of TP53 identified by NGS, of which 18 were classified as TP53mut. Despite several studies have shown a correlation between p53 IHC and the TP53 mutation [46,47,48], we found an inconsistency in 2 out of 20 cases, highlighting how an appropriate molecular analysis carried out by personnel properly trained in oncological genetics is decisive in the diagnosis of a heterogeneous pathology such as EC. Regarding MMRd, we classified 4 (6.7%) tumors with pathogenic variants in MMR genes, only one concordant with IHC results. Interestingly, all discordant cases (3/4) carried a truncating variant in MSH6 with an allele frequency between 6 and 15%. The low allele frequency could justify the conservation of the protein in the tissue. The POLE-mut tumors (8 out of 60, 15%) harbors pathogenic variants in exonuclease domain of POLE with allele frequencies ranging from 12 to 35%. All variants were reported in the literature [42] and designated as ‘hotspot’ POLE mutations. Castillo and collegues reports them frequently mutated in endometrial tissue, as already confirmed in COSMIC entries. Although they are reported uncertain on dbSNP, it should be specified that in the somatic state they could be considered likely pathogenic. However, since there is a functional test that demonstrates a reduction in activity compared to the wild type and also considering their localization, they can be classified as pathogenic [27]. We also identified a conspicuous fraction (28%) of VUS in POLE, the majority outside of the exonuclease domain (data not shown). Nevertheless, there were two variants (c.901G>A; c.907C>T) in exon 9, both close to the splice junction and with relatively low allele frequency (5.5% and 7.45% respectively). The c.901G>A p.(Asp301Asn) variant was recorded but not classified in the ClinVar database (Variation ID: 405876) and considered as a VUS via the ACMG Standards (https://varsome.com/) with 1 points applied to the PM2/PP3/BP1 supporting criteria. This alteration is predicted to be tolerated by in silico analysis, is not present in population databases and has not been reported in the literature in individuals affected with POLE-related conditions. Conversely, the c.907C>T p.(Gln303*) variant was reported as likely pathogenic in the Varsome database but recorded with conflicting interpretations of pathogenicity in ClinVar (Variation ID: 473841). This alteration is expected to result in loss of function by premature protein truncation or nonsense-mediated mRNA decay. However, loss of function via haploinsufficiency in POLE has not yet been clearly established as a mechanism of disease. For these reasons, clinical significance of POLE truncating variants still remains unclear and functional studies to characterize their pathogenicity are needed. The higher percentage of cases were NSMP, with PTEN pathogenic variations significantly associated with endometrioid carcinomas, particularly LGEC. This finding strengthens the theory that PTEN mutations arise in an early stage of the carcinogenesis of Type I carcinomas [45].
The NGS data confirmed the extreme heterogeneity and the different prognosis of high-grade endometrioid carcinomas. In fact, HGECs have been molecularly classified into three distinct classes: 4 POLEmut cases, 6 NSMP cases and 3 TP53mut cases. They can exhibit a sluggish biological behavior if associated with a POLE pathogenic variant, on the contrary, if associated with a TP53 pathogenic variant they can be extremely aggressive, even overcoming serous carcinomas. On the other hand, the OHECs confirmed their biological aggressiveness as they are almost all TP53mut, except one which was found to be POLEmut (EC-49) and for which follow-up is mandatory. Accordingly, we collected clinical data for the all 8 POLEmut cases; as reported in Table 4, the serous histotype is predominantly on the poor prognosis, regardless of POLE mutational status. Of the eight cases included in the cohort, only five (62.5%) had the indication for POLE genetic testing; for all of these, no recurrences were reported, confirming the favorable prognosis associated to POLEmut carcinoma, with the only exception of the OHEC case. A good prognosis was observed also in a LGEC (EC-61), for which the indication for POLE genetic testing was absent considering the low risk and the stage of the disease. Of the remaining two EC cases with no indication for testing, one was lost at the follow up; despite the low-grade malignancy, multi-infarct leukoencephalopathy was recorded for this patient and therefore the suspicion is for a poor prognosis.
The results we obtained allowed us to determine the risk classification on a molecular basis [15]. Comparison with the histological classification revealed that there is an excellent agreement (k = 0.818) between the two classifications. This applies to LGEC and OHEC, in which in almost all cases the risk class that had been assigned exclusively on a histo-morphological basis was confirmed. This points out how morphology is still important, accompanied by molecular analysis which is confirmed to be crucial for the definition of the prognosis and follow-up. The risk classification has changed in five HGEC; among these, three were POLEmut and two TP53mut. These data confirm again how HGECs are a highly heterogeneous group that needs molecular analysis in order to be stratified in the best possible way since there are forms that fall within group with good or poor prognosis.
There are some limitations of our study. The NGS panel we used does not cover some of the variations known to be associated with EC, such as ARID1A [49]. In addition, we were unable to evaluate somatic copy number changes and the state of methylation of the MLH1 promoter was not performed. Finally, the implementation of multi-omics data as well as functional assay to evaluate pathogenicity of POLE variant might provide more useful subtyping.
5. Conclusion
The NGS technology for the identification of pathogenic variants on FFPE tissue gave good results and confirmed the feasibility of their clinical application.
The correct risk stratification, based on morphological, molecular and clinical parameters, associated with the early diagnosis of EC, allows a better clinical-pathological management of the patient by better defining the risk class and relative treatment. This is especially true for the high-grade endometrioid carcinomas, which comprise a heterogeneous group of neoplasms with markedly different prognoses. NGS analysis confirmed that HGECs are a heterogeneous group of neoplasms that exhibit intermediate morpho-molecular characteristics between LGECs and OHECs. Therefore, the molecular analysis on tumor tissue is particularly important to improve the diagnostic and prognostic definition of EC, especially if diagnosed in the early stage of the disease.
Furthermore, the molecular risk classification had excellent agreement (k=0.818) with the histological classification for LGEC and OEHC. This confirms the importance of the morphological data for a correct classification of the lesions, accompanied by a thorough molecular analysis especially in genes associated with a favorable prognosis such as POLE.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1 - Quality parameters of the 63 EC specimen; Table S2 - Sequencing metrics and alignment quality of the NGS runs; Table S3 - List of pathogenic variants detected in EC cases; Table S4 - Recurrent pathogenic variants identified in the 60 EC cases analyzed by NGS; Table S5 - Risk profiles according to histopathological and morphological features of EC; Table S6 - Risk profiles according to molecular classification of EC; Table S7 - Concordance between TP53 molecular findings and IHC analysis.
Author Contributions
RD performed molecular analysis, data interpretation and manuscript writing; PM performed histological case evaluation, selection and revision together with VGV and IM; AG, VF and CG performed molecular analysis, results validation and literature review; TL, BG, MS, BF and FS achieved clinical diagnosis and management; PA and RP provided technical support; CG and GV performed critical reading of the manuscript; VGV and GV developed the study concept and design. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from Italian Ministry of Health (Ricerca Corrente) to MS (no grant number) and from Lega Italiana per la Lotta contro I Tumori (LILT) to FS (no grant number).
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki and the Finnish legislation for the use of archived tissue specimens and associated clinical information. The study has been approved by the Ligurian Ethical Committee (46/2020 – DB id 10320).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data sets generated during this study as well as digital images of all cases stained with H&E are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- R. L. Siegel, K. D. Miller, and A. Jemal, “Cancer statistics, 2019,” CA Cancer J Clin, vol. 69, no. 1, pp. 7–34, Jan. 2019. [CrossRef]
- H. Sung et al., “Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries,” CA Cancer J Clin, vol. 71, no. 3, pp. 209–249. [CrossRef]
- AIOM Associazione Italiana di Oncologia Medica, “I NUMERI DEL CANCRO IN ITALIA 2022,” Intermedia EDITORE, Dec. 2022, Accessed: Apr. 06, 2023. [Online]. Available: https://www.aiom.it/wp-content/uploads/2022/12/2022_AIOM_NDC-web.pdf.
- J. V. Bokhman, “Two pathogenetic types of endometrial carcinoma,” Gynecol Oncol, vol. 15, no. 1, pp. 10–17, Feb. 1983. [CrossRef]
- M. E. Urick and D. W. Bell, “Clinical actionability of molecular targets in endometrial cancer,” Nat Rev Cancer, vol. 19, no. 9, pp. 510–521, Sep. 2019. [CrossRef]
- G. F. Zannoni et al., “Does high-grade endometrioid carcinoma (grade 3 FIGO) belong to type I or type II endometrial cancer? A clinical-pathological and immunohistochemical study,” Virchows Arch, vol. 457, no. 1, pp. 27–34, Jul. 2010. [CrossRef]
- Cancer Genome Atlas Research Network et al., “Integrated genomic characterization of endometrial carcinoma,” Nature, vol. 497, no. 7447, pp. 67–73, May 2013. 20 May. [CrossRef]
- E. Stelloo et al., “Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative,” Mod Pathol, vol. 28, no. 6, pp. 836–844, Jun. 2015. [CrossRef]
- Talhouk and J., N. McAlpine, “New classification of endometrial cancers: the development and potential applications of genomic-based classification in research and clinical care,” Gynecol Oncol Res Pract, vol. 3, p. 14, 2016. [CrossRef]
- Talhouk et al., “Confirmation of ProMisE: A simple, genomics-based clinical classifier for endometrial cancer,” Cancer, vol. 123, no. 5, pp. 802–813, Mar. 2017. [CrossRef]
- Talhouk et al., “A clinically applicable molecular-based classification for endometrial cancers,” Br J Cancer, vol. 113, no. 2, pp. 299–310, Jul. 2015. [CrossRef]
- S. Kommoss et al., “Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series,” Ann Oncol, vol. 29, no. 5, pp. 1180–1188. [CrossRef]
- R. Sahu and S. P. Pattanayak, “Strategic Developments & Future Perspective on Gene Therapy for Breast Cancer: Role of mTOR and Brk/ PTK6 as Molecular Targets,” Curr Gene Ther, vol. 20, no. 4, pp. 237–258, 2020. [CrossRef]
- N. Concin et al., “ESGO/ESTRO/ESP guidelines for the management of patients with endometrial carcinoma,” Int J Gynecol Cancer, vol. 31, no. 1, pp. 12–39, Jan. 2021. [CrossRef]
- N. Colombo et al., “ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: diagnosis, treatment and follow-up,” Ann Oncol, vol. 27, no. 1, pp. 16–41, Jan. 2016. [CrossRef]
- M. Alexa, A. Hasenburg, and M. J. Battista, “The TCGA Molecular Classification of Endometrial Cancer and Its Possible Impact on Adjuvant Treatment Decisions,” Cancers (Basel), vol. 13, no. 6, p. 1478, Mar. 2021. [CrossRef]
- S. V. M. van den Heerik et al., “PORTEC-4a: international randomized trial of molecular profile-based adjuvant treatment for women with high-intermediate risk endometrial cancer,” Int J Gynecol Cancer, vol. 30, no. 12, pp. 2002–2007, Dec. 2020. [CrossRef]
- L. Vermij, V. Smit, R. Nout, and T. Bosse, “Incorporation of molecular characteristics into endometrial cancer management,” Histopathology, vol. 76, no. 1, pp. 52–63, Jan. 2020. [CrossRef]
- D. Rivera et al., “Implementing NGS-based BRCA tumour tissue testing in FFPE ovarian carcinoma specimens: hints from a real-life experience within the framework of expert recommendations,” J Clin Pathol, vol. 74, no. 9, pp. 596–603, Sep. 2021. [CrossRef]
- D. W. Bell and L. H. Ellenson, “Molecular Genetics of Endometrial Carcinoma,” Annu Rev Pathol, vol. 14, pp. 339–367, Jan. 2019. [CrossRef]
- B. Spurdle, M. A. Bowman, J. Shamsani, and J. Kirk, “Endometrial cancer gene panels: clinical diagnostic vs research germline DNA testing,” Mod Pathol, vol. 30, no. 8, pp. 1048–1068, Aug. 2017. [CrossRef]
- S. Richards et al., “Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology,” Genet Med, vol. 17, no. 5, pp. 405–424, 15. 20 May. [CrossRef]
- Garrett et al., “Cancer Variant Interpretation Group UK (CanVIG-UK): an exemplar national subspecialty multidisciplinary network,” J Med Genet, vol. 57, no. 12, pp. 829–834, Dec. 2020. [CrossRef]
- L. B. Alexandrov et al., “Signatures of mutational processes in human cancer,” Nature, vol. 500, no. 7463, pp. 415–421, Aug. 2013. [CrossRef]
- D. N. Church et al., “DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer,” Hum Mol Genet, vol. 22, no. 14, pp. 2820–2828, Jul. 2013. [CrossRef]
- S. Jumaah, M. M. Salim, H. S. Al-Haddad, K. A. McAllister, and A. A. Yasseen, “The frequency of POLE-mutation in endometrial carcinoma and prognostic implications: a systemic review and meta-analysis,” J Pathol Transl Med, vol. 54, no. 6, pp. 471–479, Nov. 2020.
- E. Shinbrot et al., “Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication,” Genome Res, vol. 24, no. 11, pp. 1740–1750, Nov. 2014. [CrossRef]
- S. Briggs and I. Tomlinson, “Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers,” J Pathol, vol. 230, no. 2, pp. 148–153, Jun. 2013. [CrossRef]
- M. Schultheis et al., “TP53 Mutational Spectrum in Endometrioid and Serous Endometrial Cancers,” Int J Gynecol Pathol, vol. 35, no. 4, pp. 289–300, Jul. 2016. [CrossRef]
- M. E. Lomax, D. M. Barnes, T. R. Hupp, S. M. Picksley, and R. S. Camplejohn, “Characterization of p53 oligomerization domain mutations isolated from Li-Fraumeni and Li-Fraumeni like family members,” Oncogene, vol. 17, no. 5, pp. 643–649, Aug. 1998. [CrossRef]
- S. Kato et al., “Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis,” Proc Natl Acad Sci U S A, vol. 100, no. 14, pp. 8424–8429, Jul. 2003. [CrossRef]
- O. Giacomelli et al., “Mutational processes shape the landscape of TP53 mutations in human cancer,” Nat Genet, vol. 50, no. 10, pp. 1381–1387, Oct. 2018. [CrossRef]
- E. Kotler et al., “A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation,” Mol Cell, vol. 71, no. 1, pp. 178-190.e8, Jul. 2018. [CrossRef]
- P. Monti et al., “Transcriptional functionality of germ line p53 mutants influences cancer phenotype,” Clin Cancer Res, vol. 13, no. 13, pp. 3789–3795, Jul. 2007. [CrossRef]
- W. Hanel, N. Marchenko, S. Xu, S. X. Yu, W. Weng, and U. Moll, “Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis,” Cell Death Differ, vol. 20, no. 7, pp. 898–909, Jul. 2013. [CrossRef]
- Y. Zerdoumi et al., “Germline TP53 mutations result into a constitutive defect of p53 DNA binding and transcriptional response to DNA damage,” Hum Mol Genet, vol. 26, no. 14, pp. 2591–2602, Jul. 2017. [CrossRef]
- P. Monti et al., “Dominant-negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes,” Mol Cancer Res, vol. 9, no. 3, pp. 271–279, Mar. 2011. [CrossRef]
- D. Kandioler et al., “TP53 genotype but not p53 immunohistochemical result predicts response to preoperative short-term radiotherapy in rectal cancer,” Ann Surg, vol. 235, no. 4, pp. 493–498, Apr. 2002. [CrossRef]
- E. Howitt et al., “Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1,” JAMA Oncol, vol. 1, no. 9, pp. 1319–1323, Dec. 2015. [CrossRef]
- F. A. Eggink et al., “Immunological profiling of molecularly classified high-risk endometrial cancers identifies POLE-mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition,” Oncoimmunology, vol. 6, no. 2, p. e1264565, 2017. [CrossRef]
- L. J. Jennings et al., “Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists,” J Mol Diagn, vol. 19, no. 3, pp. 341–365. = =. [CrossRef]
- León-Castillo et al., “Interpretation of somatic POLE mutations in endometrial carcinoma,” J Pathol, vol. 250, no. 3, pp. 323–335, Mar. 2020. [CrossRef]
- N. Bansal, V. Yendluri, and R. M. Wenham, “The molecular biology of endometrial cancers and the implications for pathogenesis, classification, and targeted therapies,” Cancer Control, vol. 16, no. 1, pp. 8–13, Jan. 2009. [CrossRef]
- L. M. Peterson et al., “Molecular characterization of endometrial cancer: a correlative study assessing microsatellite instability, MLH1 hypermethylation, DNA mismatch repair protein expression, and PTEN, PIK3CA, KRAS, and BRAF mutation analysis,” Int J Gynecol Pathol, vol. 31, no. 3, pp. 195–205. [CrossRef]
- Bilbao et al., “The relationship between microsatellite instability and PTEN gene mutations in endometrial cancer,” Int J Cancer, vol. 119, no. 3, pp. 563–570, Aug. 2006. [CrossRef]
- S. Timmerman et al., “Analysis of 108 patients with endometrial carcinoma using the PROMISE classification and additional genetic analyses for MMR-D,” Gynecol Oncol, vol. 157, no. 1, pp. 245–251, Apr. 2020. [CrossRef]
- M. Köbel, B. M. Ronnett, N. Singh, R. A. Soslow, C. B. Gilks, and W. G. McCluggage, “Interpretation of P53 Immunohistochemistry in Endometrial Carcinomas: Toward Increased Reproducibility,” Int J Gynecol Pathol, vol. 38 Suppl 1, no. Iss 1 Suppl 1, pp. S123–S131, Jan. 2019. [CrossRef]
- Y. Kobayashi et al., “Molecular Evaluation of Endometrial Dedifferentiated Carcinoma, Endometrioid Carcinoma, Carcinosarcoma, and Serous Carcinoma Using a Custom-Made Small Cancer Panel,” Pathol Oncol Res, vol. 27, p. 1610013, 2021. [CrossRef]
- Toumpeki et al., “The Role of ARID1A in Endometrial Cancer and the Molecular Pathways Associated With Pathogenesis and Cancer Progression,” In Vivo, vol. 33, no. 3, pp. 659–667, 2019. [CrossRef]
Figure 1.
Pathogenic variants detected in EC cases.
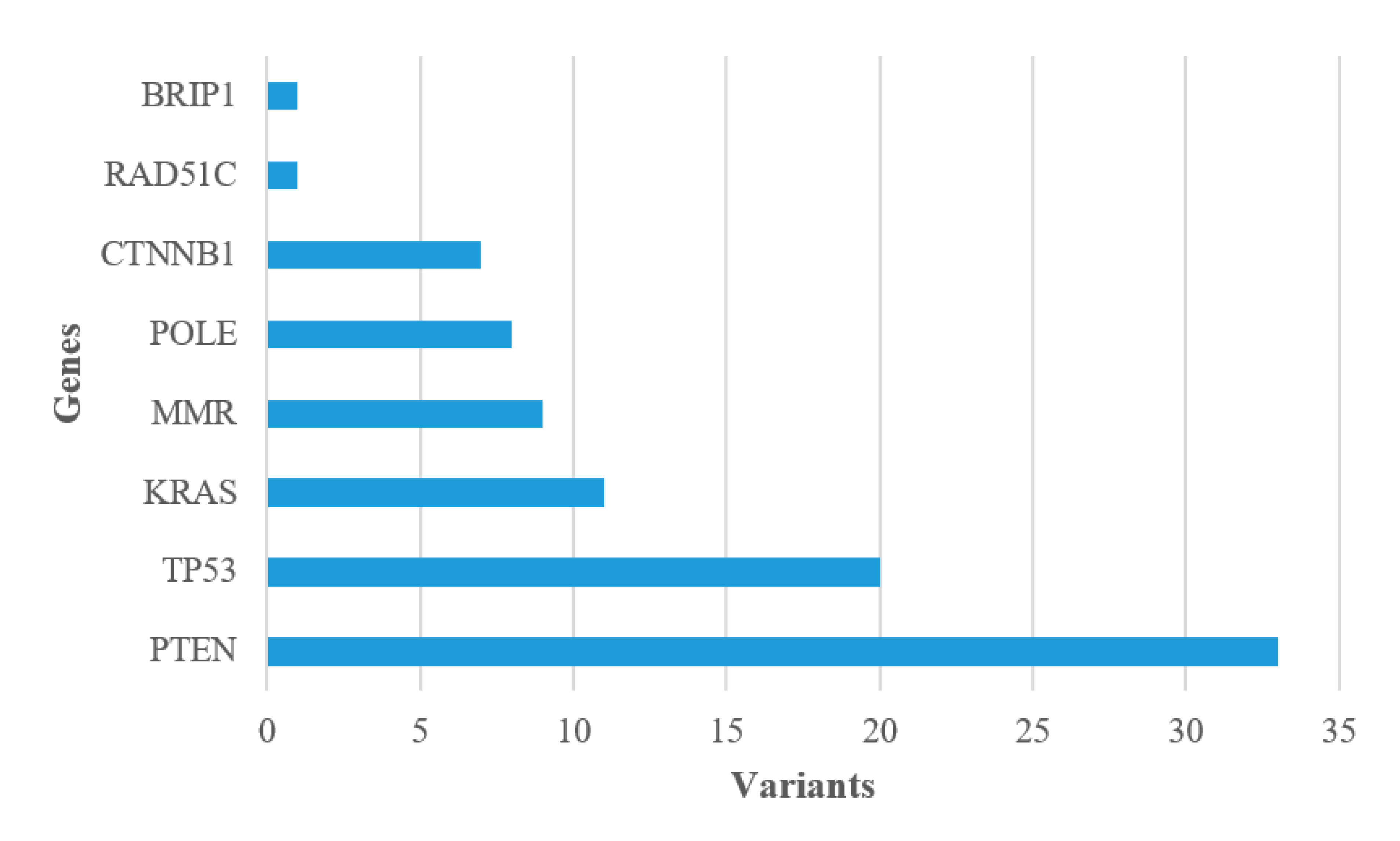

Table 1.
Clinicopathological features of the 63 EC analyzed in this study.
| Characteristics | N | % |
|---|---|---|
| Age, years | ||
| >65 | 43 | 68.2 |
| <65 | 20 | 31.7 |
| Histology | ||
| Low-grade endometrioid EC (LGEC) | 31 | 49.2 |
| High-grade endometrioid EC (HGEC) | 16 | 25.4 |
| High-grade serous EC (OHEC) | 16 | 25.4 |
| FIGO stage | ||
| I-II | 48 | 76.2 |
| III-IV | 15 | 23.8 |
| LVSI | ||
| Negative | 40 | 63.5 |
| Positive | 23 | 36.5 |
| p53 expression | ||
| Wild-type | 45 | 71.4 |
| Aberrant | 18 | 28.6 |
| Microsatellite | ||
| Conserved | 49 | 77.8 |
| Lost | 12 | 19.0 |
| Nda | 2 | 3.2 |
a Not determined.
Table 2.
Concordance of molecular and histo-morphological evaluation of EC.

Table 3.
Univariable associations of TP53 mutation profile with histological classification.
| TP53 alone | TP53 combined | p-value | |
|---|---|---|---|
| Serous | 13 | 1 | 0.0022 |
| Endometriod | 1 | 5 |
Table 4.
Follow up of the 8 POLEmut cases.
| Sample no. | Age (years) | Follow up | Adjuvant therapy | Staging | POLE indication |
|---|---|---|---|---|---|
| EC-15-B | 65 | DWDa | - | pT3a | No |
| EC-16-A | 74 | NEDb | - | pT1a/G3/N0(sn) | Yes |
| EC-36-A | 60 | NED | Radiotherapy | pT2/G1/N0(sn) | Yes |
| EC-38-A | 73 | LTFUc | - | pT1a/G2/Nx | No |
| EC-42-A | 54 | NED | Radiotherapy | pT2G3pNx | Yes |
| EC-43-A | 59 | NED | Brachytherapy | pT1bG3pN0 LVSI+ | Yes |
| EC-49-A | 84 | DWD | - | pT1bG3 serous | Yes |
| EC-61-A | 51 | NED | - | pT1A/G2/N0(sn) | No |
a Died With Disease. b No Evidence of Disease. c Lost To Follow Up.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



