
Submitted:
28 July 2023
Posted:
01 August 2023
You are already at the latest version
Abstract
Emerging evidence indicates that intracellular calcium (Ca2+) levels and their regulatory proteins play essential roles in normal stem cell proliferation and differentiation. Cancer stem-like cells (CSCs) are subpopulations of cancer cells that retain characteristics similar to stem cells and play an essential role in cancer progression. Recent studies have reported that the Orai3 calcium channel plays an oncogenic role in human cancer. However, its role in CSC remains underexplored. In this study, we explored the effects of Orai3 in the progression and stemness of oral/oropharyngeal squamous cell carcinoma (OSCC). Orai3 expression was increased in a stepwise manner during the progression of OSCC. In addition, Orai3 was highly enriched in CSC populations of OSCC. Ectopic Orai3 expression in non-tumorigenic immortalized oral epithelial cells increased intracellular Ca2+ levels, acquiring malignant growth and CSC properties. Conversely, silencing of endogenous Orai3 in OSCC cells suppressed CSC phenotype, indicating a pivotal role of Orai3 in CSC regulation. Moreover, Orai3 markedly increased the expression of inhibitor of DNA binding 1 (ID1), a stemness transcription factor. Orai3 and ID1 were highly expressed in CSCs compared to non-CSCs, implying the functional importance of the Orai3/ID1 axis in CSC regulation. Furthermore, suppression of ID1 abrogated CSC phenotype in the cell with ectopic Orai3 overexpression and OSCC. Our study reveals that Orai3 is a novel functional CSC regulator in OSCC and further suggests that Orai3 plays an oncogenic role in OSCC by promoting cancer stemness via ID1 upregulation.
Keywords:
Calcium
; Orai3
; OSCC
; cancer stem-like cells
; ID1
1. Introduction
Recent studies have uncovered and validated the pathophysiologic role of cancer stemness, referring to the stem cell-like phenotype of cancers, in the long-term sustenance of cancers [1]. Cancer stemness is known to be responsible for tumorigenicity, metastasis, therapy resistance, and recurrence of cancer cells, indicating its pivotal role in tumor progression and aggression [2]. The stemness phenotype has been observed in various human cancers, including OSCC [2]. OSCC, the sixth most common cancer worldwide is often preceded by clinically well-defined lesions, such as leukoplakia that is histologically classified as dysplastic or non-dysplastic leukoplakia. Dysplastic leukoplakia is defined as an oral premalignant lesion and is associated with a likely progression to cancer; however, it is not an accurate predictor of cancer risk [3,4]. Early-stage tumors can usually be managed through surgery and radiotherapy. However, successful treatment is inversely proportional to the extent of the disease at the time of treatment. A combination of chemotherapy and radiation therapy, although effective in treating 97% of early-stage tumors, was only 33% effective in treating advanced tumors [5]. Therefore, advancing our understanding of the molecular regulation of cancer stemness is crucial for developing a new generation of effective therapies for OSCC.
Intracellular Ca2+ is a universal second messenger regulating many physiological processes, and deregulation of intracellular Ca2+ is implicated in cell proliferation, metastasis, and suppression of apoptosis, indicating that altered Ca2+ homeostasis is a hallmark of cancer cells [6,7,8,9]. Emerging evidence also indicates the crucial role of Orai Ca2+ channels in carcinogenesis [10,11,12,13,14,15,16,17,18,19,20]. The Orai channel family includes three Orai homologs (Orai1, 2, and 3), and they are highly calcium-selective channels that mediate intracellular Ca2+ influx in non-excitable cells, such as cancer cells [21]. Among the three Orai homologs, Orai1, the most studied homolog was reported to play a critical role in cancer progression [22]. We demonstrated that increased Orai1 expression is necessary and sufficient for tumor progression by promoting cancer stemness in OSCC [23]. Our study further showed that the activation of Ca2+-dependent transcription factor, NFAT (nuclear factor of activated T cells) was required for Orai1-induced stemness promotion in OSCC, indicating the novel role of Orai1/NFAT axis in cancer stemness [23]. The oncogenic role of Orai2 has been demonstrated in various human cancers including OSCC [24]. Similar to Orai1, Orai2 was significantly upregulated in OSCC tissues compared to normal tissues [24]. Inhibition of Orai2 suppressed malignant growth and properties in OSCC cells [24]. These findings indicate that Orai1 and Orai2 have significant roles in OSCC; however, the role of Orai3 in OSCC remains poorly explored especially in cancer progression and stemness.
In the present study, we report for the first time that Orai3 expression is elevated in a stepwise manner in oral/oropharyngeal carcinogenesis and enriched in OSCC CSC populations. We further provided evidence that Orai3 promotes OSCC progression by enhancing cancer stemness via ID1 upregulation, suggesting a novel CSC regulatory mechanism by Orai3/ID1 axis.
2. Materials and Methods
2.1. Cell culture and reagents
Primary normal human oral keratinocytes (NHOK) were prepared from oral mucosa and cultured in Keratinocyte Growth Medium (KGM, Lonza). Non-tumorigenic immortalized oral epithelial cell lines HOK-16B were also cultured in KGM. Eleven human OSCC cell lines (BapT, FaDu, SCC4, SCC9, SCC9/TNF, SCC15, SCC105, SNU1066, UM6, UM17, and YD38) were cultured in DMEM/Ham’s F12 (Invitrogen) supplemented with 10% FBS (Gemini Bioproducts), 0.4 μg/ml hydrocortisone (Sigma-Aldrich) and 5 μg/ml Gentamycin aminoglycoside antibiotic (Invitrogen).
2.2. Laser-captured microdissection (LCM)
Following histological examination of H & E staining of oral mucosal tissues [i.e. NHOE (n = 4), precancerous oral lesions (n = 4) and OSCC tissues (n = 4)], epithelial layers from the paraffin-embedded tissue samples were excised by laser-captured microdissection (LCM) using Leica (LMD) 7000 system (Leica Microsystems Inc, Richmond, IL) at the California NanoSystems Institute at UCLA (Los Angeles, CA). LCM-derived tissue RNAs were extracted using a high pure RNA paraffin kit (Roche). RT was performed with RNA isolated from the tissue sections using a Superscript II RT kit (Invitrogen) with random hexamer primers (Promega) according to the manufacturer’s instructions. Then, qPCR was performed using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) and QuantStudio 3 qPCR System (Thermo Fisher Scientific) as described in our prior work [25]. Thermocycling conditions for all PCR reactions included an initial denaturation stage at 95°C for 10 min, followed by 50 cycles.
2.3. Quantitative real-time PCR (qPCR)
cDNA was synthesized from 2.5 μg of total RNA using SuperScript first-strand synthesis system (Invitrogen). Then, qPCR was performed using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) and QuantStudio 3 qPCR System (Thermo Fisher Scientific) as described in our prior work [23]. The primer sequences were obtained from the Universal Probe Library (Roche), and the sequences can be available upon request. Second derivative Cq value determination method was used to compare fold-differences according to the manufacturer’s instructions.
2.4. Western Blotting
Western blotting was performed as described previously (Lee, 2019). We used the following primary antibodies: anti-Orai3 (ab115558, Abcam, Cambridge, UK), anti-ID1 (SC-133104, Santa Cruz), anti-ID2 (sc-398104, Santa Cruz), anti-Bmi1 (5856, Cell Signaling), anti-EZH2 (ab186006, Abcam, Cambridge, UK), anti-Nanog (sc-293121, Santa Cruz), anti-Oct4 (sc-5279, Santa Cruz) and anti-GAPDH (sc-25778; Santa Cruz). Horseradish peroxidase (HRP)-conjugated secondary antibodies were obtained from Santa Cruz.
2.5. Tumor sphere formation assay
Three thousand cells were grown in 3 ml of serum free DMEM/F12 media supplemented with 1:50 B27 (Invitrogen), 20 ng/mL EGF, 20 ng/mL, 10μg/mL insulin, penicillin, streptomycin, and amphotericin B in Ultra-Low Attachment 6-well Plates (Corning) for 6-10 days. The number of tumor spheres formed were observed and counted under a microscope.
2.6. Migration assay
Cell migration was measured using 6.5 mm transwell chambers with 8.0 μm polycarbonate membranes (Corning: Product#3422) as described in our previous publication (Lee, 2019). Twenty thousand cells were seeded and incubated for 2 days.
2.7. Ectopic expression of Orai3
The retroviral pMSCV-CITE-eGFP-Puro vector encoding human Orai3 [26] was used to prepare viruses. The vector was transfected into GP2-293 universal packaging cells (Clonetech, Mountain View, CA, USA) along with pVSV-G envelope plasmid using lipofectamine 2000 (Life Technologies). Detailed methods of retrovirus production and infection can be found in our previous publications [23]. Infected cells were selected with 0.5μg/ ml puromycin for two weeks and used for experiments.
2.8. SOCE assay (Single-cell Ca2+ imaging)
2.9. Cell Proliferation Assay
Cell growth was determined by using the tetrazolium salt (MTT) cell proliferation assay kit (ATCC, Manassas, VA, USA) and cell counting. The cells were plated at 2 x 103 cells per well into a 96-well plate. They were then incubated in a culture medium containing various ethanol concentrations (0–300 mM) for 2 and 7 days. Absorbance at 570 nm was determined using a microplate reader. For cell counting, the cells were plated at 2 x 104 cells per well into a 6-well plate. They were then incubated for indicated days and counted.
2.10. Anchorage-independent growth
To determine colony-forming efficiency in semi-solid medium, 1 x 104 cells were plated in culture medium containing 0.3% agarose over a base layer of serum-free medium containing 0.5% agarose. Three weeks after incubation, colonies were counted. The experiment was performed in the absence of ethanol and in triplicates with 60-mm dishes.
2.11. In vivo xenograft tumor assay
Five-ten million cells were subcutaneously injected into the flank of immunocompromised mice (strain nu/ nu, Charles River Laboratories). The animal study was performed according to the protocol approved by UCLA Animal Research Committee. The kinetics of tumor growth was determined by measuring the volume in three perpendicular axes of the nodules using micro-scaled calipers.
2.12. ALDH1 assay
Using Aldehyde Dehydrogenase-Based Cell Detection Kit (STEMCELL), ALDH enzymatic activity was determined. Total of 1 x106 cells were re-suspended in the ALDEFLUOR Assay Buffer in the volume of 1ml. Fluorescent nontoxic ALDEFLUOR Reagent BODIPYTM (1.25μl) was added as a substrate to measure ALDH enzymatic activity in intact cells. Immediately after adding the substrate reagent, 0.5ml of the cell suspension was transferred into the control tube which contains specific inhibitor for ALDH, diethylaminobenzaldehyde (DEAB) for calculating background fluorescence. Then, cells were incubated at 35˚C for 30 minutes and fluorescence data acquisition was made by using a BD FACScan flow cytometer (BD Biosciences).
2.13. Small Interfering RNA (siRNA) Transfection
Orai3 siRNA (sc-76005; Santa Cruz, Dallas, TX, USA), ID1 siRNA (sc-29356; Santa Cruz) and control siRNA (sc-37007; Santa Cruz) were purchased and introduced into cells using Lipofectamine RNAiMAX (Invitrogen). Cells (2 x 105) were plated in 60-mm dishes and transfected with 10 μg siRNA. The cultures were harvested after one day post-transfection for expression and functional analyses.
2.14. Mouse models
To induce oral tumors, C57BL/6 mice (Jackson Laboratory) were exposed to 4-NQO (Sigma-Aldrich, St. Louis, MO) diluted in drinking water to the final concentration of 30 μg/ml for 16 weeks, followed by six weeks of normal drinking water, as described in our previous study (Chen W, 2018)]. At the end of experiment, tongues were harvested for histological examination and RNA isolation. All procedures involving the use of mice will be in accordance with the National Institutes of Health guidelines and are approved by the UCLA Animal Research Committee.
2.15. Statistical analysis
The statistical analyses were calculated using GraphPad Prism 5. The data were expressed as mean ± standard deviation (SD). Data between two groups were compared using parametric Student’s t test or paired t test. A value of P < 0.05 was considered as statistically significant.
3. Result
3.1. Stepwise increase of Orai3 expression during oral carcinogenesis
To investigate the role of Orai3 in oral carcinogenesis, we determined the transcript level of Orai3 in normal human oral epithelia (NHOE), dysplasia, and OSCC tissues by employing laser capture microdissection (LCM), followed by qPCR. The assay revealed a gradual increase of Orai3 expression during oral carcinogenesis (Figure 1A). The expression of Orai3 in normal human oral keratinocytes (NHOK), immortalized non-tumorigenic oral epithelial cell line (HOK-16B), and OSCC cell lines (BapT, FaDu, SCC4, SCC9, SCC9/TNF, SCC15, SCC105, SNU1066, UM6, UM17B, and YD38) were also evaluated (Figure 1B,C). Orai3 was commonly elevated in OSCC cells compared to the tested normal and immortalized cells, although with a certain degree of variability among the tested OSCC cell lines (Figure 1B). Consistent with the result from Figure 1A, a gradual increase of Orai3 expression was detected in in vitro sequential, multistep oral carcinogenesis model, i.e., NHOK→HOK-16B→BapT (Figure 1B). NHOK was immortalized by high-risk HPV-16 (HOK-16B), and HOK-16B was further transformed into oncogenic cells by the treatment of chemical carcinogen benzo(a)pyrene (BAPT) [28]. To further validate the importance of elevated Orai3 during oral carcinogenesis, we utilized a carcinogen-induced tongue cancer mouse model. As demonstrated in our prior study [29], chronic exposure of mice to 4-nitroquinoline-1-oxide (4-NQO) led to rampant oral tumor formation in the tongue, while tongue from mice exposed to DMSO (vehicle) exhibited similar histology with normal squamous epithelium. Consistent with our findings from human cancer, Orai3 was greatly elevated in the tumor-bearing tongue compared to the normal tongue, suggesting that increased Orai3 is associated with chemical-induced oral carcinogenesis (Figure 1D). Taken together, our findings indicate a stepwise increase of Orai3 during oral carcinogenesis, suggesting an important role of Orai3 in the progression of OSCC.
3.2. Orai3 is enriched in CSC populations derived from OSCC
Emerging evidence indicates that CSCs play a key role in cancer progression [30]. Thus, to explore the importance of Orai3 in CSCs, we first compared the levels of Orai3 in various CSC populations with those in their corresponding non-CSC populations derived from OSCC cells. Tumor spheres are generated from self-renewing cells and display increased CSC phenotype and stemness transcription factors (Nanog, Oct4, KLF4, Lin28, and Sox2) compared with their corresponding monolayer adherent cells, indicating that they are CSC-enriched cell population [23]. Orai3 was highly enriched in tumor spheres compared with their corresponding monolayer adherent cells derived from multiple OSCC cell lines (Figure 2A). The activity of aldehyde dehydrogenase 1 (ALDH1) is a key CSC marker for head and neck cancer, including OSCC [31]. ALDH1High cancer cells display enhanced CSC properties compared to ALDH1Low cells [31]. In our study, ALDH1High OSCC cells expressed higher levels of Orai3 transcript than ALDH1Low OSCC cells sorted from SCC4 (Figure 2B). CSC population can be enriched after treatment of chemotherapeutic drugs [31]. Thus, we also measured Orai3 expression in cisplatin-resistant SCC4 cells that were isolated from SCC4 treated with 25 μM cisplatin for 2 days [32]. Orai3 mRNA was significantly elevated in the cisplatin-resistant cells compared to their sensitive control cells (Figure 2C). Moreover, Orai3 protein was also greatly increased in the tested CSC populations compared to their corresponding non-CSC populations (Figure 2D). Our findings suggest that elevated Orai3 may be necessary to support CSCs in OSCC.
3.3. Elevated Orai3 supports CSC phenotype
Having established that increased Orai3 is necessary for CSCs, we next examined whether ectopic Orai3 expression confers CSC phenotype on oral epithelial cells. We overexpressed Orai3 in non-tumorigenic immortalized oral epithelial cells, HOK-16B, using the lentiviral vector expressing Orai3 or empty vector (EV) as a control (Figure 3A,B). The elevation of Ca2+ influx by Orai3 was also confirmed by performing Ca2+ imaging assay (Figure 3C). Then, we examined the effect of Orai3 on cell proliferation and found that Orai3 overexpression led to a robust increase in the proliferation capacity of HOK-16B in vitro (Figure 3D). Moreover, Orai3 conferred anchorage-independent growth ability to HOK-16 cells (Figure 3E). This ability has been linked to tumor cell aggressiveness in vivo, including tumorigenicity [33]. To further examine the effect of Orai3 on tumor growth in vivo, we injected the cells into nude mice and observed tumor formation (Figure 3F). No tumors developed in mice receiving HOK-16B/EV cells. However, HOK-16B/Orai3 cells began to form tumor nodules in 2nd week after injection and reached their maximum size by 4th week. The nodules then regressed, leaving only necrotic tissue in the 8th week. Our findings indicate that ectopic expression of Orai3 in the non-tumorigenic immortalized oral epithelial cells resulted in an increase in cell proliferation and the acquisition of malignant growth properties.
Next, we investigated the effect of Orai3 on CSC phenotype in the immortalized cells. Ectopic Orai3 expression significantly increased both ALDH1 mRNA by 6-fold (Figure 4A) and ALDH1High cell population by 7.8-fold (1.7% VS. 13.3%, Figure 4B) in HOK-16B, indicating that Orai3 increases the CSC population. Orai3 expression induced tumor sphere formation ability, indicating the acquisition of self-renewal capacity by Orai3 (Figure 4C). A transwell migration assay (Figure 4D) demonstrated that HOK-16B/Orai3 migrated significantly faster than HOK-16B/EV. These findings indicate that ectopic Orai3 expression is sufficient to confer CSC phenotype on the immortalized oral epithelial cells.
To further confirm the importance of elevated Orai3 expression for the maintenance of CSC phenotype, we knocked down endogenous Orai3 in OSCC cells by siRNA (Figure 5A). Knockdown of Orai3 suppressed the expression of ALDH1 mRNA by 60% (Figure 5B), self-renewal capacity by 50% (Figure 5C), and migration ability by 95% (Figure 5D) of OSCC cells. These clearly indicate that Orai3 is required for the maintenance of CSC phenotype in OSCC. Collectively, our data suggest that Orai3 promotes OSCC progression by increasing cancer stemness.
3.4. ID1 is required for Orai3-induced CSC phenotype
To understand the mechanism by which Orai3 regulates CSC phenotype, we initially examined the effect of Orai3 on the expression of 17 key CSC regulators (Figure 6A). Among the tested CSC regulators, inhibitor of DNA binding 1 (ID1), a stemness transcription factor was most upregulated by Orai3 in HOK-16B (Figure 6A,B). Conversely, silencing of Orai3 resulted in a significant decrease in ID1 expression (data not shown). Since ID1 is considered a master regulator of CSCs [34] and plays an oncogenic role in OSCC [35,36,37,38], our findings suggest that ID1 may play an important role in Orai3-mediated CSC regulation.
To evaluate the functional role of ID1 in the Orai3-induced CSC phenotype, we knocked down ID1 in HOK-16B/Orai3 by siRNA (Figure 7A). Knockdown of ID1 reduced self-renewal by 75% (Figure 7B) and migration by 60% (Figure 7C) in the cells with ectopic Orai3 overexpression. We also demonstrated that the knockdown of endogenous ID1 in OSCC (Figure 7D) suppressed self-renewal by 60% (Figure 7E) and migration ability by 50% (Figure 7F). Collectively, our data indicate that Orai3 promotes CSC phenotype via upregulation of ID1, suggesting a novel CSC regulatory mechanism by the Orai3/ID1 axis.
3.5. Orai3 and ID1 are highly expressed in CSC-enriched populations, and their expression levels are positively correlated in OSCC cells
To further examine the importance of the Orai3/ID1 axis in CSCs, we measured the levels of ID1 in various CSC populations compared with those in their corresponding non-CSC populations (Figure 8A,B). Similar to the enrichment of Orai3 in CSCs (Figure 2), ID1 was highly expressed in tumor spheres compared with their corresponding monolayer adherent cells. ALDH1High OSCC cells expressed higher ID1 than ALDH1Low OSCC cells sorted from SCC4. Moreover, the level of ID1 was much greater in cisplatin-resistant than cisplatin-sensitive SCC4 cells. These indicate that Orai3/ID1 axis is elevated in CSC-enriched OSCC populations. Furthermore, to determine whether Orai3 expression correlates with ID1 level in OSCC, we compared Orai3 and ID1 level in 13 human OSCC cell lines (Figure 8C). Our data revealed that Orai3 transcript levels are positively correlated with ID1 transcript levels in OSCC.
4. Discussion
The CSC hypothesis is a well-studied theory to explain tumor initiation and progression [31]. CSCs are a very small population within the overall heterogeneous tumor cell population and are can sustain tumor aggressiveness through their unique properties such as self-renewal capacity, migratory ability, and drug resistance [31]. CSCs have been isolated from multiple solid tumors, including OSCC, a common malignant tumor of the head and neck. We have reported various molecular determinants governing the stemness of OSCC and suggested their application for therapeutic modality [23,39,40,41]. Owing to the heterogeneity of CSC [42], unveiling novel molecular regulators for cancer stemness is of paramount importance for developing a new generation of effective therapies for OSCC. To the best of our knowledge, this is the first study to report that Orai3/ID1 signaling is a novel molecular axis for governing the stemness of OSCC. The expression of Orai3 is elevated in a stepwise manner during OSCC carcinogenesis and further enriched in CSC populations compared to that in non-CSC populations. Ectopic expression of Orai3 in immortalized oral epithelial cells induces malignant growth as well as CSC phenotype. Inhibition of Orai3 suppresses cancer stemness of OSCC. Furthermore, our data indicate that Orai3 enhances CSC phenotype via upregulation of ID1, suggesting the vital role of the Orai3/ID1 axis in OSCC CSCs.
The emerging pathophysiological role of Orai3 has been reported in various solid cancers, such as breast, prostate, lung, colorectal, and pancreatic cancer [43]. Elevated expression of Orai3 has been observed in various human cancers, including breast, prostate, and pancreatic cancer [43]. However, there is no report on the role of Orai3 in oral/oropharyngeal carcinogenesis. Our study showed that Orai3 is highly expressed in OSCC compared to precancerous and normal tissues and culture cells. Furthermore, precancerous oral epithelial cells express higher Orai3 compared to normal oral epithelial cells. These findings indicate a stepwise increase of Orai3 expression during oral/oropharyngeal carcinogenesis. Moreover, Orai3 expression was greatly elevated in a carcinogen-induced tongue cancer mouse model. Our findings indicate that Orai3 plays a vital role in the progression of OSCC in vivo.
Our study clearly demonstrated that Orai3 is required for the maintenance of malignant growth and stemness properties of OSCC. Ectopic Orai3 expression in immortalized oral epithelial cells induced not only Ca2+ influx but also malignant growth properties, including anchorage-independent growth ability and self-renewal capacity. Anchorage-independent growth ability is a well-known characteristic of cancer, which is linked to in vivo tumorigenicity [33]. Self-renewal capacity is considered as the key characteristic by which CSCs regenerate themselves, suggesting the driving force of tumorigenesis. Indeed, Orai3 increased cell proliferation capacity in vivo; however, Orai3 failed to endow the cells with full tumor-forming ability in vivo. Furthermore, Orai3 increased CSC populations and phenotype. Ectopic Orai3 expression markedly increases the ALDH1High CSC population in the non-tumorigenic oral epithelial cells. ALDH1High cancer cells displayed higher self-renewal, migration, and tumorigenic potential than ALDH1low cells [44,45,46]. Orai3 also markedly increases the motility of the non-tumorigenic cells. Conversely, inhibition of endogenous Orai3 in OSCC resulted in reduced CSC properties, such as ALDH1 activity, self-renewal capacity, and migration ability. Our finding is consistent with previous reports showing the importance of Orai3 in cell migration. Knockdown of Orai3 in invasive breast cancer cell lines decreased cell migration, whereas its overexpression promoted cellular motility [47]. However, the underlying mechanism by which Orai3 enhances oral epithelial cell migration has not been understood. Therefore, the effects of Orai3 on epithelial-to-mesenchymal transition (EMT) and metastasis-related gene expression should be warranted to investigate [48]. We conclude that Orai3 increases not only the number of CSCs but also CSC properties. Thus, we hypothesize that Orai3 promotes the malignant progression of OSCC by increasing the CSC phenotype. Collectively, our findings suggest that Orai3 could be an effective therapeutic target for OSCC.
Orai3 regulates cancer growth and migration thru multiple pathways [43]. Our study demonstrated that Orai3 enhances CSC phenotype via upregulation of ID1 (inhibitor of DNA-binding 1), a stemness transcription factor. ID proteins lack a DNA binding domain and function as dominant negative regulators of basic helix-loop-helix (bHLH) transcription factors through heterodimerization with other bHLH factors [49]. Among four ID family members (ID1-4), ID1 is the most extensively studied, and its role in cancer is generally considered a tumor promoter [50]. ID1 is overexpressed in many types of malignancies, such as breast, lung, prostate, cervical, colorectal, liver, and brain cancer. ID1 enhances tumor progression, aggressiveness, and metastasis which are responsible for mortality in cancer patients. Conversely, ID1 inhibition results in a decrease in tumor growth and metastasis [51]. ID1 elicits its tumor-promoting effects by regulating various target genes involved in cancer development [50]. For instance, ID1 promoted lung cancer growth by activating CDK4/cyclin D1 [52] and breast cancer metastasis thru S100A9 regulation [53]. Interestingly, a potential role of ID1 in the regulation of cancer stemness has been demonstrated. ID1 promoted stemness phenotype through the activation of the WNT/SHH signaling pathway in brain and colorectal cancer [54,55]. Moreover, ID1 induced breast carcinogenesis by increasing stem cell activities in transgenic mice [56]. These suggest that ID1 may mediate its tumor-promoting effects by regulating cancer stemness. Genetic inhibition of ID1 in OSCC cells suppressed cancer stemness features, including self-renewal and migration. Moreover, the ID1 inhibition in HOK-16B/Orai3 led to suppression of the CSC phenotype, indicating the functional role of increased ID1 in the Orai3-induced CSC phenotype. This suggests that Orai3/ID1 axis is a novel regulatory signaling for maintaining cancer stemness in OSCC. As secondary evidence supporting this, we found that both Orai3 and ID1 are enriched in CSC populations, and their expression is positively correlated. Thus, we hypothesize that Orai3 regulates cancer stemness via ID1; however, the underlying mechanisms by which Orai3 regulates ID1 have not been understood.
Orai channels induce Ca2+ entry, and increased intracellular Ca2+ activates downstream responses, including NFAT signaling pathway [57]. Activated NFAT signaling plays an important role in tumorigenesis by regulating various target genes involved in cancer development [58]. NFAT is a family of transcription factors composed of four members, NFATc1, NFATc2, NFATc3, and NFATc4 [59]. To test if NFAT signaling is activated by Orai3, we examined the expressions of NFAT in HOK-16B/Orai3 cells. Interestingly, among the four members, only NFATc1 expression was upregulated in the cells, suggesting that increased intracellular Ca2+ by Orai3 activates NFATc1 (data not shown), implying a novel Orai3-NFATc1-ID1 circuit. However, the role of NFATc1 on ID1 regulation has not been documented. Therefore, further investigation is necessary to examine the detailed regulatory mechanism of the Orai3-NFATc1-ID1 circuit and its role in the regulation of CSC. Moreover, the Ca2+-independent mechanism of Orai3 was reported in cancer [60], which adds an additional layer of interest for future study of Orai3.
In conclusion, the Orai3 Ca2+ channel is a novel molecular regulator of the malignant and stemness phenotype of OSCC. Orai3 promotes CSC phenotype via ID1 upregulation. The Orai3/ID1 axis is augmented in CSC populations of OSCC, suggesting that the Orai3/ID1 axis could be an important therapeutic target in OSCC. Since Orai3 and ID1 are readily inhibited by small molecular inhibitors [61,62], targeting Oria3/ID1 axis could be a selective therapeutic modality against OSCC CSCs.
Author Contributions
Conceptualization, K.-H.S. and Y.K.; methodology, K.-H.S., Y.K., and Y.G.; formal analysis, A.N., Y.S., S.H.L., C.E.M., S.S.; investigation, A.N. and A.H.K.; resources, K.-H.S. and Y.K.; data curation, K.-H.S., Y.K. Y.G., and A.N.; writing-original draft preparation, K.-H.S., and A.N.; writing-review and editing, Y.K., M.K.K., R.H.K. and N.-H.P.; funding acquisition, K.-H.S. and Y.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by UCLA School of Dentistry faculty seed grant (K.-H.S.) and TRDRP T31IP1543C (Y.K.).
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
All data are available in the text and supplementary.
Acknowledgments
The authors would like to thank all members of The Shapiro Family Laboratory of Viral Oncology and Aging Research for discussing data interpretation and sharing their knowledge.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
Ca2+ calcium; OSCC, Oral/oropharyngeal squamous cell carcinomas; CSCs, Cancer stem-like cells; ID1, inhibitor of DNA binding 1; NFAT, nuclear factor of activated T cells; NHOK, normal human oral keratinocytes; NHOE, Normal human oral epithelia; LCM, laser-captured microdissection; ALDH1, Aldehyde dehydrogenase 1; siRNA, small interfering RNA
References
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu Rev Pathol 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.-H.; Kim, R.H. An Updated Review of Oral Cancer Stem Cells and Their Stemness Regulation. 2018, 23, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S.; Reibel, J.; Bouquot, J.; Dabelsteen, E. Oral epithelial dysplasia classification systems: predictive value, utility, weaknesses and scope for improvement. J Oral Pathol Med 2008, 37, 127–133. [Google Scholar] [CrossRef]
- Brennan, M.; Migliorati, C.A.; Lockhart, P.B.; Wray, D.; Al-Hashimi, I.; Axell, T.; Bruce, A.J.; Carpenter, W.; Eisenberg, E.; Epstein, J.B.; et al. Management of oral epithelial dysplasia: a review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod, 2007; 103 Suppl, S19 e11–12. [Google Scholar] [CrossRef]
- Marcial, V.A.; Pajak, T.F.; Mohiuddin, M.; Cooper, J.S.; al Sarraf, M.; Mowry, P.A.; Curran, W.; Crissman, J.; Rodriguez, M.; Velez-Garcia, E. Concomitant cisplatin chemotherapy and radiotherapy in advanced mucosal squamous cell carcinoma of the head and neck. Long-term results of the Radiation Therapy Oncology Group study 81-17. Cancer 1990, 66, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer 2011, 11, 609–618. [Google Scholar] [CrossRef]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis 2010, 1, e75. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef]
- Liu, H.; Hughes, J.D.; Rollins, S.; Chen, B.; Perkins, E. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp Mol Pathol 2011, 91, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lkhagvadorj, S.; Lee, M.R.; Hwang, K.H.; Chung, H.C.; Jung, J.H.; Cha, S.K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem Biophys Res Commun 2014, 448, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma; 2014.
- Lee, S.H.; Rigas, N.K.; Lee, C.R.; Bang, A.; Srikanth, S.; Gwack, Y.; Kang, M.K.; Kim, R.H.; Park, N.H.; Shin, K.H. Orai1 promotes tumor progression by enhancing cancer stemness via NFAT signaling in oral/oropharyngeal squamous cell carcinoma. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hao, J.; Zhang, Y.; Yang, Z.; Cao, Y.; Lu, W.; Shu, Y.; Jiang, L.; Hu, Y.; Lv, W.; et al. Orai1 mediates tumor-promoting store-operated Ca2+ entry in human gastrointestinal stromal tumors via c-KIT and the extracellular signal-regulated kinase pathway. Tumour Biol 2017, 39, 1010428317691426. [Google Scholar] [CrossRef]
- Xia, J.; Wang, H.; Huang, H.; Sun, L.; Dong, S.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett 2016, 381, 31–40. [Google Scholar] [CrossRef]
- Deng, W.; Wang, J.; Zhang, J.; Cai, J.; Bai, Z.; Zhang, Z. Orai1, a Direct Target of microRNA-519, Promotes Progression of Colorectal Cancer via Akt/GSK3beta Signaling Pathway. Dig Dis Sci 2016, 61, 1553–1560. [Google Scholar] [CrossRef]
- Zhan, Z.Y.; Zhong, L.X.; Feng, M.; Wang, J.F.; Liu, D.B.; Xiong, J.P. Over-expression of Orai1 mediates cell proliferation and associates with poor prognosis in human non-small cell lung carcinoma. Int J Clin Exp Pathol 2015, 8, 5080–5088. [Google Scholar]
- Srikanth, S.; Gwack, Y. Chapter Eight - Molecular Regulation of the Pore Component of CRAC Channels, Orai1. In Current Topics in Membranes; Murali, P., Ed.; Academic Press, 2013; Volume 71, pp. 181–207. [Google Scholar]
- Cantonero, C.; Sanchez-Collado, J.; Gonzalez-Nunez, M.A.; Salido, G.M.; Lopez, J.J.; Jardin, I.; Rosado, J.A. Store-independent Orai1-mediated Ca(2+) entry and cancer. Cell Calcium 2019, 80, 1–7. [Google Scholar] [CrossRef]
- Lee, S.H.; Rigas, N.K.; Lee, C.R.; Bang, A.; Srikanth, S.; Gwack, Y.; Kang, M.K.; Kim, R.H.; Park, N.H.; Shin, K.H. Orai1 promotes tumor progression by enhancing cancer stemness via NFAT signaling in oral/oropharyngeal squamous cell carcinoma. Oncotarget 2016, 7, 43239–43255. [Google Scholar] [CrossRef]
- Singh, A.K.; Roy, N.K.; Bordoloi, D.; Padmavathi, G.; Banik, K.; Khwairakpam, A.D.; Kunnumakkara, A.B.; Sukumar, P. Orai-1 and Orai-2 regulate oral cancer cell migration and colonisation by suppressing Akt/mTOR/NF-kappaB signalling. Life Sci 2020, 261, 118372. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.H.; Bae, S.D.; Hong, H.S.; Kim, R.H.; Kang, M.K.; Park, N.H. miR-181a shows tumor suppressive effect against oral squamous cell carcinoma cells by downregulating K-ras. Biochemical and Biophysical Research Communications 2011, 404, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Srikanth, S.; Feske, S.; Cruz-Guilloty, F.; Oh-hora, M.; Neems, D.S.; Hogan, P.G.; Rao, A. Biochemical and functional characterization of Orai proteins. J Biol Chem 2007, 282, 16232–16243. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.D.; Srikanth, S.; Yee, M.K.; Mock, D.C.; Lawson, G.W.; Gwack, Y. ORAI1 deficiency impairs activated T cell death and enhances T cell survival. Journal of immunology 2011, 187, 3620–3630. [Google Scholar] [CrossRef]
- Park, N.H.; Gujuluva, C.N.; Baek, J.H.; Cherrick, H.M.; Shin, K.H.; Min, B.M. Combined oral carcinogenicity of HPV-16 and benzo(a)pyrene: an in vitro multistep carcinogenesis model. Oncogene 1995, 10, 2145–2153. [Google Scholar]
- Chen, W.; Kang, K.L.; Alshaikh, A.; Varma, S.; Lin, Y.-L.; Shin, K.-H.; Kim, R.; Wang, C.-Y.; Park, N.-H.; Walentin, K.; et al. Grainyhead-like 2 (GRHL2) knockout abolishes oral cancer development through reciprocal regulation of the MAP kinase and TGF-β signaling pathways. Oncogenesis 2018, 7, 38. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol 2019, 234, 8381–8395. [Google Scholar] [CrossRef]
- Shin, K.H.; Kim, R.H. An Updated Review of Oral Cancer Stem Cells and Their Stemness Regulation. Crit Rev Oncog 2018, 23, 189–200. [Google Scholar] [CrossRef]
- Martin, C.E.; Nguyen, A.; Kang, M.K.; Kim, R.H.; Park, N.H.; Shin, K.H. DYRK1A is required for maintenance of cancer stemness, contributing to tumorigenic potential in oral/oropharyngeal squamous cell carcinoma. Exp Cell Res 2021, 405. [Google Scholar] [CrossRef]
- 10. yexcr.2021.11 2656.
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Benezra, R.; Iavarone, A. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat Rev Cancer 2014, 14, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.; Goel, M.M.; Makker, A.; Bhatia, V.; Chandra, S.; Kumar, S.; Agarwal, S.P. Fibroblast Growth Factor (FGF-2) and Its Receptors FGFR-2 and FGFR-3 May Be Putative Biomarkers of Malignant Transformation of Potentially Malignant Oral Lesions into Oral Squamous Cell Carcinoma. PLoS One 2015, 10, e0138801. [Google Scholar] [CrossRef] [PubMed]
- Ozretic, L.; Wagner, S.; Huebbers, C.U.; Gattenlohner, S.; Klussmann, J.P.; Beutner, D.; Zander, T.; Buettner, R.; Quaas, A. FGFR1 amplification and co-overexpression of c-MYC in oropharyngeal squamous cell carcinoma. Oral Oncol 2016, 54, e7–e9. [Google Scholar] [CrossRef]
- Dvorak, P.; Dvorakova, D.; Hampl, A. Fibroblast growth factor signaling in embryonic and cancer stem cells. FEBS Lett 2006, 580, 2869–2874. [Google Scholar] [CrossRef]
- Gotoh, N. Control of stemness by fibroblast growth factor signaling in stem cells and cancer stem cells. Curr Stem Cell Res Ther 2009, 4, 9–15. [Google Scholar] [CrossRef]
- Lee, S.H.; Kieu, C.; Martin, C.E.; Han, J.; Chen, W.; Kim, J.S.; Kang, M.K.; Kim, R.H.; Park, N.H.; Kim, Y.; et al. NFATc3 plays an oncogenic role in oral/oropharyngeal squamous cell carcinomas by promoting cancer stemness via expression of OCT4. Oncotarget 2019, 10, 2306–2319. [Google Scholar] [CrossRef]
- Lee, C.R.; Lee, S.H.; Rigas, N.K.; Kim, R.H.; Kang, M.K.; Park, N.H.; Shin, K.H. Elevated expression of JMJD6 is associated with oral carcinogenesis and maintains cancer stemness properties. Carcinogenesis 2016, 37, 119–128. [Google Scholar] [CrossRef]
- Patel, S.S.; Shah, K.A.; Shah, M.J.; Kothari, K.C.; Rawal, R.M. Cancer stem cells and stemness markers in oral squamous cell carcinomas. Asian Pac J Cancer Prev 2014, 15, 8549–8556. [Google Scholar] [CrossRef]
- Meyer, M.J.; Fleming, J.M.; Lin, A.F.; Hussnain, S.A.; Ginsburg, E.; Vonderhaar, B.K. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res 2010, 70, 4624–4633. [Google Scholar] [CrossRef]
- Sanchez-Collado, J.; Jardin, I.; Lopez, J.J.; Ronco, V.; Salido, G.M.; Dubois, C.; Prevarskaya, N.; Rosado, J.A. Role of Orai3 in the Pathophysiology of Cancer. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Clay, M.R.; Tabor, M.; Owen, J.H.; Carey, T.E.; Bradford, C.R.; Wolf, G.T.; Wicha, M.S.; Prince, M.E. Single-Marker Identification of Head and Neck Squamous Cell Carcinoma Cancer Stem Cells with Aldehyde Dehydrogenase. Head and Neck-Journal for the Sciences and Specialties of the Head and Neck 2010, 32, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Ota, N.; Ohno, J.; Seno, K.; Taniguchi, K.; Ozeki, S. In vitro and in vivo expression of aldehyde dehydrogenase 1 in oral squamous cell carcinoma. Int J Oncol 2014, 44, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Richard, V.; Sebastian, P.; Nair, M.G.; Nair, S.N.; Malieckal, T.T.; Kumar, T.R.S.; Pillai, M.R. Multiple drug resistant, tumorigenic stem-like cells in oral cancer. Cancer Letters 2013, 338, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.F.; Miller, L.D.; Chan, X.B.; Black, M.A.; Pang, B.; Ong, C.W.; Salto-Tellez, M.; Liu, E.T.; Desai, K.V. JMJD6 is a driver of cellular proliferation and motility and a marker of poor prognosis in breast cancer. Breast Cancer Res 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Doi 10.1186/Bcr3200.
- Davis, F.M.; Peters, A.A.; Grice, D.M.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS One 2012, 7, e36923. [Google Scholar] [CrossRef]
- Perk, J.; Iavarone, A.; Benezra, R. Id family of helix-loop-helix proteins in cancer. Nat Rev Cancer 2005, 5, 603–614. [Google Scholar] [CrossRef]
- Zhao, Z.; Bo, Z.; Gong, W.; Guo, Y. Inhibitor of Differentiation 1 (Id1) in Cancer and Cancer Therapy. Int J Med Sci 2020, 17, 995–1005. [Google Scholar] [CrossRef]
- Lai, X.; Liao, J.; Lin, W.; Huang, C.; Li, J.; Lin, J.; Chen, Q.; Ye, Y. Inhibitor of DNA-binding protein 1 knockdown arrests the growth of colorectal cancer cells and suppresses hepatic metastasis in vivo. Oncol Rep 2014, 32, 79–88. [Google Scholar] [CrossRef]
- Cheng, Y.J.; Tsai, J.W.; Hsieh, K.C.; Yang, Y.C.; Chen, Y.J.; Huang, M.S.; Yuan, S.S. Id1 promotes lung cancer cell proliferation and tumor growth through Akt-related pathway. Cancer Lett 2011, 307, 191–199. [Google Scholar] [CrossRef]
- Gumireddy, K.; Li, A.P.; Kossenkov, A.V.; Cai, K.Q.; Liu, Q.; Yan, J.C.; Xu, H.; Showe, L.; Zhang, L.; Huang, Q.H. ID1 Promotes Breast Cancer Metastasis by S100A9 Regulation. Mol Cancer Res 2014, 12, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, R.; Wu, M.; Smiljanic, S.; Kaskun, O.; Ghannad-Zadeh, K.; Celebre, A.; Isaev, K.; Morrissy, A.S.; Guan, J.; Tong, J.F.; et al. ID1 Is Critical for Tumorigenesis and Regulates Chemoresistance in Glioblastoma. Cancer Research 2019, 79, 4057–4071. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lai, X.; Yu, Y.; Li, J.; Cao, L.; Lin, W.; Huang, C.; Liao, J.; Chen, W.; Li, C.; et al. Inhibitor of DNA binding 1 (Id1) mediates stemness of colorectal cancer cells through the Id1-c-Myc-PLAC8 axis via the Wnt/beta-catenin and Shh signaling pathways. Cancer Manag Res 2019, 11, 6855–6869. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Park, J.H.; Lee, J.Y.; Won, H.Y.; Jang, K.S.; Min, K.W.; Jang, S.H.; Woo, J.K.; Oh, S.H.; Kong, G. Overexpression of Id1 in transgenic mice promotes mammary basal stem cell activity and breast tumorigenesis. Oncotarget 2015, 6, 17276–17290. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol Rev 2005, 85, 757–810. [Google Scholar] [CrossRef]
- Mancini, M.; Toker, A. NFAT proteins: emerging roles in cancer progression. Nat Rev Cancer 2009, 9, 810–820. [Google Scholar] [CrossRef]
- Daniel, C.; Gerlach, K.; Väth, M.; Neurath, M.F.; Weigmann, B. Nuclear factor of activated T cells—A transcription factor family as critical regulator in lung and colon cancer. International Journal of Cancer 2014, 134, 1767–1775. [Google Scholar] [CrossRef]
- Chamlali, M.; Kouba, S.; Rodat-Despoix, L.; Todesca, L.M.; Petho, Z.; Schwab, A.; Ouadid-Ahidouch, H. Orai3 Calcium Channel Regulates Breast Cancer Cell Migration through Calcium-Dependent and -Independent Mechanisms. Cells 2021, 10. [Google Scholar] [CrossRef]
- Kim, K.D.; Srikanth, S.; Tan, Y.V.; Yee, M.K.; Jew, M.; Damoiseaux, R.; Jung, M.E.; Shimizu, S.; An, D.S.; Ribalet, B.; et al. Calcium signaling via Orai1 is essential for induction of the nuclear orphan receptor pathway to drive Th17 differentiation. J Immunol 2014, 192, 110–122. [Google Scholar] [CrossRef]
- Mistry, H.; Hsieh, G.; Buhrlage, S.; Huang, M.; Park, E.; Cuny, G.; Galinsky, I.; Stone, R.M.; Gray, N.S.; Parmar, K.; et al. Small Molecule Inhibitors of USP1 Target ID1 Degradation in Leukemic Cells and Cause Cytotoxicity. Blood 2013, 122. [Google Scholar] [CrossRef]
Figure 1.
Elevated expression of Orai3 during oral/oropharyngeal carcinogenesis. (A) Level of Orai3 mRNA was determined in normal human oral epithelia (NHOE: n=4), oral dysplasia (n=4) and OSCC (n=4) tissues by microdissection followed by qRT-PCR. The levels of Orai3 mRNA were normalized with the expression of GAPDH. (B) Level of Orai3 mRNA was determined in normal human oral keratinocyte (NHOK), non-tumorigenic immortalized oral epithelial cell line (HOK-16B) and 11 OSCC cell lines (BapT, FaDu, SCC4, SCC9, SCC9/TNF, SCC15, SCC105, SNU1066, UM6, UM17B, and YD38) by qRT-PCR. (C) Level of Orai3 protein was determined by Western blot. GAPDH was used as a loading control. (D) Level of Orai3 was determined in tongues from mice exposed to DMSO or 4-NQO by qPCR. qPCR was performed with total RNAs isolated from tongue tissues.
Figure 1.
Elevated expression of Orai3 during oral/oropharyngeal carcinogenesis. (A) Level of Orai3 mRNA was determined in normal human oral epithelia (NHOE: n=4), oral dysplasia (n=4) and OSCC (n=4) tissues by microdissection followed by qRT-PCR. The levels of Orai3 mRNA were normalized with the expression of GAPDH. (B) Level of Orai3 mRNA was determined in normal human oral keratinocyte (NHOK), non-tumorigenic immortalized oral epithelial cell line (HOK-16B) and 11 OSCC cell lines (BapT, FaDu, SCC4, SCC9, SCC9/TNF, SCC15, SCC105, SNU1066, UM6, UM17B, and YD38) by qRT-PCR. (C) Level of Orai3 protein was determined by Western blot. GAPDH was used as a loading control. (D) Level of Orai3 was determined in tongues from mice exposed to DMSO or 4-NQO by qPCR. qPCR was performed with total RNAs isolated from tongue tissues.
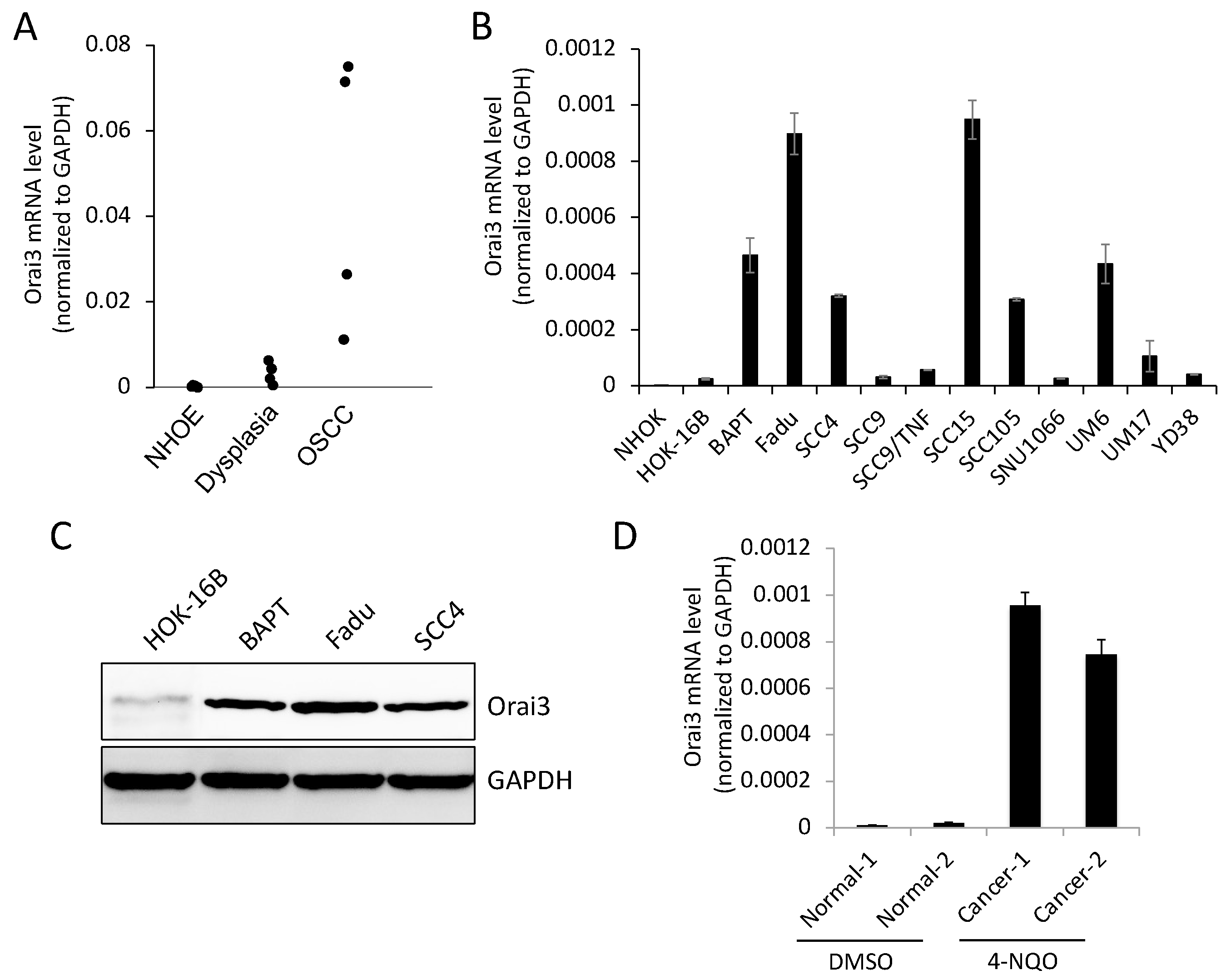
Figure 2.
Enrichment of Orai3 in CSC populations. (A) Expression of Orai3 was assessed in tumor spheres (Sph.) and their corresponding adherent monolayer cells (Mono.) derived from multiple OSCC cell lines by qPCR. (B) ALDH1HIGH (High) and ALDH1low (Low) cell populations were sorted from SCC4 cells by flow cytometry, and their Orai3 expression level were assessed by qPCR. *P < 0.05 (C) Expression of Orai3 was assessed in cisplatin-sensitive (Sens) and cisplatin-resistant (Resist) SCC4 cells by qPCR. **P < 0.01 (D) Expression of Orai3 was assessed in CSC populations and their corresponding non-CSC populations by Western blot.
Figure 2.
Enrichment of Orai3 in CSC populations. (A) Expression of Orai3 was assessed in tumor spheres (Sph.) and their corresponding adherent monolayer cells (Mono.) derived from multiple OSCC cell lines by qPCR. (B) ALDH1HIGH (High) and ALDH1low (Low) cell populations were sorted from SCC4 cells by flow cytometry, and their Orai3 expression level were assessed by qPCR. *P < 0.05 (C) Expression of Orai3 was assessed in cisplatin-sensitive (Sens) and cisplatin-resistant (Resist) SCC4 cells by qPCR. **P < 0.01 (D) Expression of Orai3 was assessed in CSC populations and their corresponding non-CSC populations by Western blot.
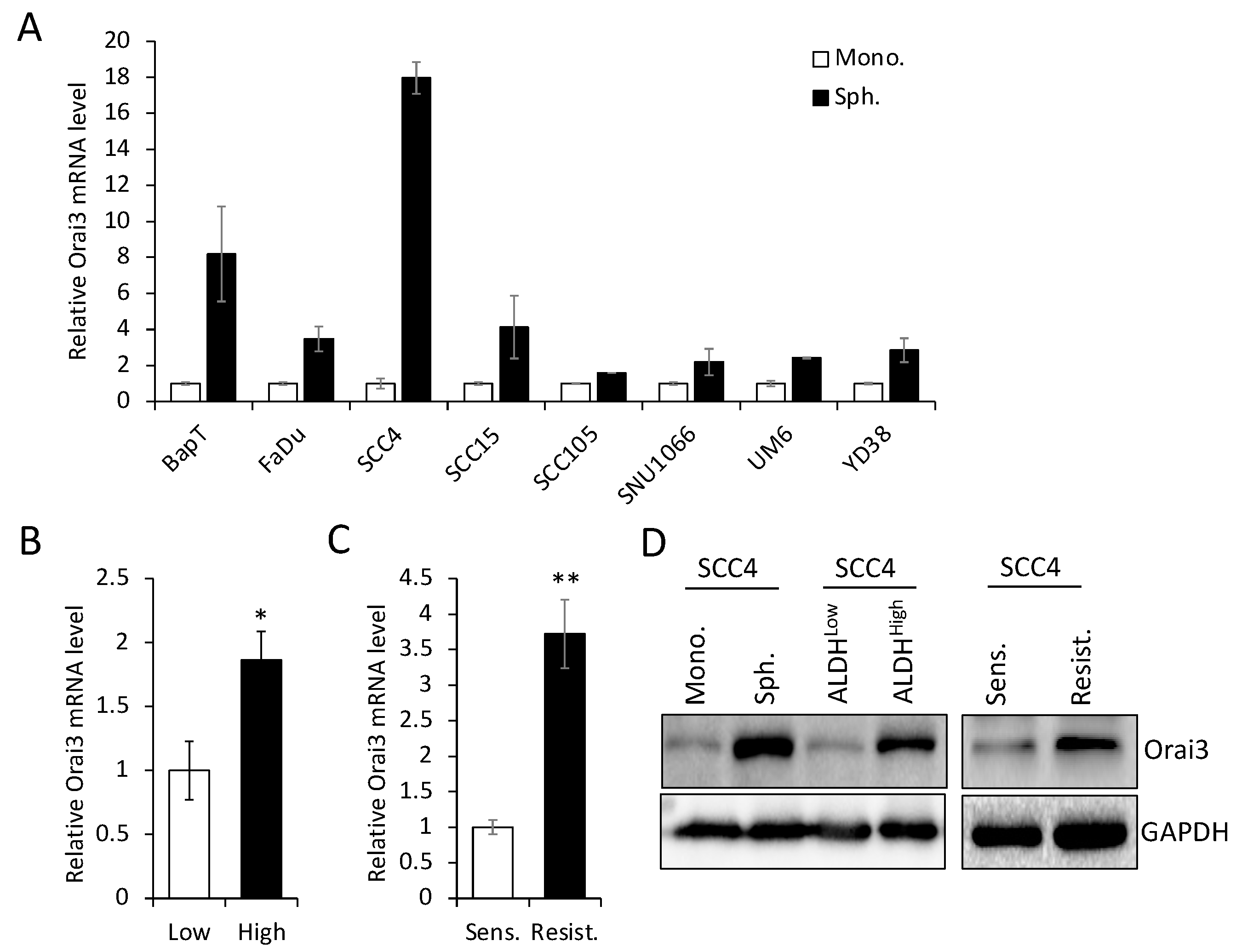
Figure 3.
Ectopic Orai13 expression induces malignant growth properties in non-tumorigenic immortalized oral epithelial cells. Orai3 was overexpressed in non-tumorigenic immortalized oral epithelial cells, HOK-16B, by infecting with retroviral vector expressing Orai3, and its overexpression was confirmed by (A) qPCR and (B) Western blot analysis. (C) Intracellular Ca2+ imaging assay was performed to confirm the activation of Orai3 function. Intracellular Ca2+ stores were depleted with 1 μM TG in the absence of extracellular Ca2+, followed by re-addition of 2 mM Ca2+. Ca2+ influx was analyzed by single-cell video imaging of Fura2-labeled, GFP+ cells. More than 30 GFP+ cells were analyzed in each experiment. (D) Effect of Orai3 on cell proliferation of HOK-16B was determined by cell counting. Data are means ± SD of triplicate experiments. *P < 0.001 (E) Effect of Orai3 on anchorage independent growth of HOK-16B was determined by soft agar assay. Data are means ± SD of triplicate experiments. *P < 0.001 by two-tailed Student’s t test. (F). Effect of Orai3 on in vivo tumorigenicity of non-tumorigenic immortalized oral epithelial cells was determined by xenograft tumor assay. HOK-16B/EV and HOK-16B/Orai3 were injected subcutaneously into 5 nude mice. Tumor sizes were measured for 8 weeks. *P < 0.01.
Figure 3.
Ectopic Orai13 expression induces malignant growth properties in non-tumorigenic immortalized oral epithelial cells. Orai3 was overexpressed in non-tumorigenic immortalized oral epithelial cells, HOK-16B, by infecting with retroviral vector expressing Orai3, and its overexpression was confirmed by (A) qPCR and (B) Western blot analysis. (C) Intracellular Ca2+ imaging assay was performed to confirm the activation of Orai3 function. Intracellular Ca2+ stores were depleted with 1 μM TG in the absence of extracellular Ca2+, followed by re-addition of 2 mM Ca2+. Ca2+ influx was analyzed by single-cell video imaging of Fura2-labeled, GFP+ cells. More than 30 GFP+ cells were analyzed in each experiment. (D) Effect of Orai3 on cell proliferation of HOK-16B was determined by cell counting. Data are means ± SD of triplicate experiments. *P < 0.001 (E) Effect of Orai3 on anchorage independent growth of HOK-16B was determined by soft agar assay. Data are means ± SD of triplicate experiments. *P < 0.001 by two-tailed Student’s t test. (F). Effect of Orai3 on in vivo tumorigenicity of non-tumorigenic immortalized oral epithelial cells was determined by xenograft tumor assay. HOK-16B/EV and HOK-16B/Orai3 were injected subcutaneously into 5 nude mice. Tumor sizes were measured for 8 weeks. *P < 0.01.
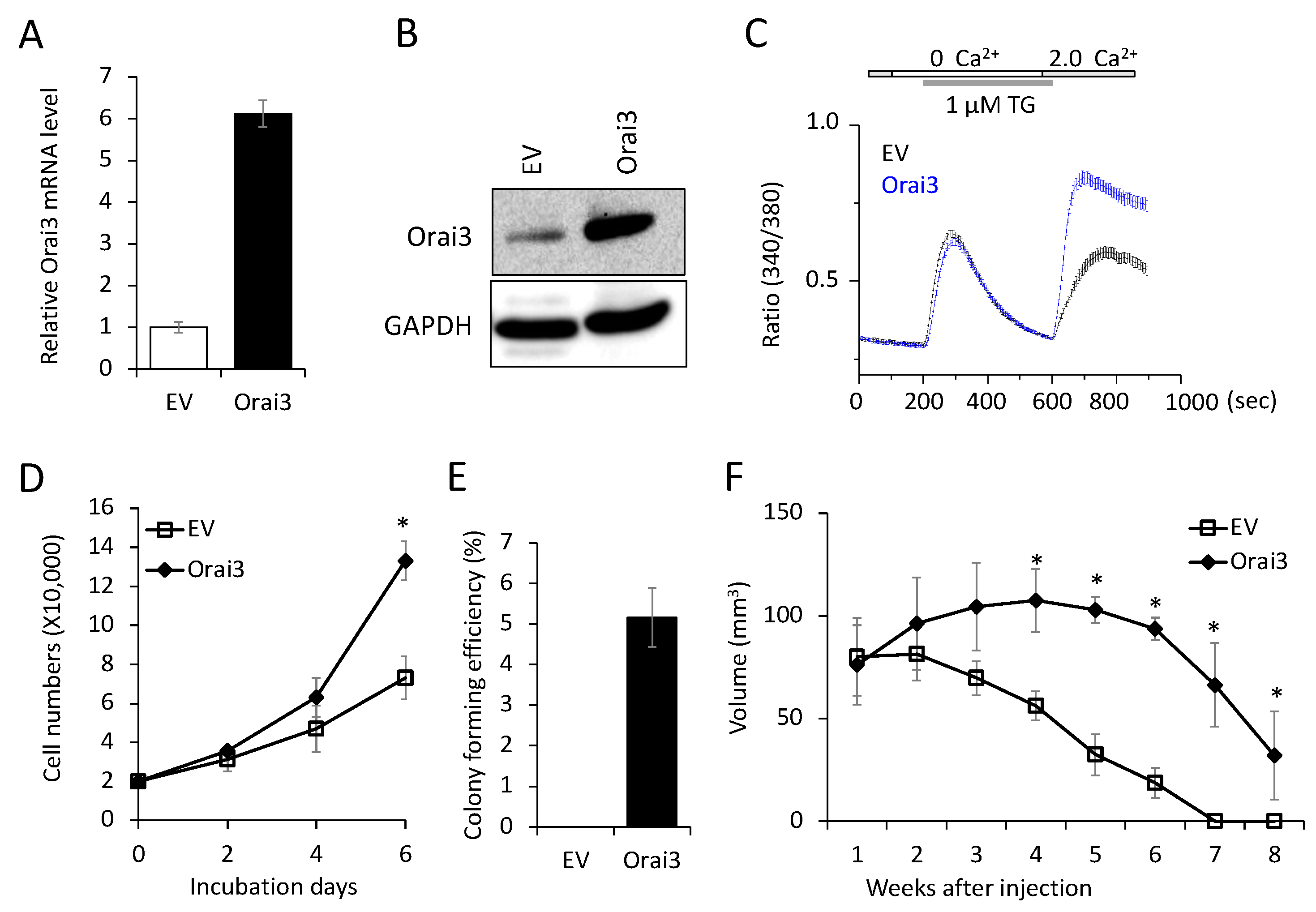
Figure 4.
Ectopic Orai3 expression enhances CSC phenotype in non-tumorigenic immortalized oral epithelial cells. (A) Effect of Orai3 on ALDH1 expression in HOK-16B was determined by qPCR. *P < 0.01 (B) Effect of Orai3 on ALDH1 activity in HOK-16B was determined by Aldefluor assay. Cells were labeled with Aldefluor combined with or without the ALDH1 inhibitor DEAB and analyzed by flow cytometry. The gate for ALDH1 + cells is determined in relation to the DEAB control (+DEAB) and shows the brightly fluorescent ALDH1 population versus the side scatter, a population that is absent/decreased in the presence of DEAB. The number shown in each panel reflects the percentage of ALDH1+ cells in each cell type. (C) Effect of Orai3 on self-renewal capacity of HOK-16B was determined by tumor sphere formation assay. Representative image of tumor spheres formed by HOK-16B/EV and HOK 16B/Orai3 are shown on the right. Bar indicates 100 μm. *P < 0.01 (D) Effect of Orai1on migration ability in HOK-16B was determined by transwell migration assay. *P < 0.01.
Figure 4.
Ectopic Orai3 expression enhances CSC phenotype in non-tumorigenic immortalized oral epithelial cells. (A) Effect of Orai3 on ALDH1 expression in HOK-16B was determined by qPCR. *P < 0.01 (B) Effect of Orai3 on ALDH1 activity in HOK-16B was determined by Aldefluor assay. Cells were labeled with Aldefluor combined with or without the ALDH1 inhibitor DEAB and analyzed by flow cytometry. The gate for ALDH1 + cells is determined in relation to the DEAB control (+DEAB) and shows the brightly fluorescent ALDH1 population versus the side scatter, a population that is absent/decreased in the presence of DEAB. The number shown in each panel reflects the percentage of ALDH1+ cells in each cell type. (C) Effect of Orai3 on self-renewal capacity of HOK-16B was determined by tumor sphere formation assay. Representative image of tumor spheres formed by HOK-16B/EV and HOK 16B/Orai3 are shown on the right. Bar indicates 100 μm. *P < 0.01 (D) Effect of Orai1on migration ability in HOK-16B was determined by transwell migration assay. *P < 0.01.
Figure 5.
Inhibition of Orai3 suppresses CSC phenotype in OSCC cells. (A) Endogenous Orai3 was knocked down in SCC4 using siRNA against Orai3 (Orai3i). The cells transfected with control siRNA (CTLi) were included for comparison. Knockdown of Orai3 was confirmed by qPCR. (B) The effect of Orai3 knockdown on ALDH1 was determined by qPCR. *P < 0.01 (C) The effect of Orai3 knockdown on self-renewal capacity was determined by tumor sphere formation assay. **P < 0.05 (D) The effect of Orai3 knockdown on migration ability was determined by transwell migration assay. *P < 0.01.
Figure 5.
Inhibition of Orai3 suppresses CSC phenotype in OSCC cells. (A) Endogenous Orai3 was knocked down in SCC4 using siRNA against Orai3 (Orai3i). The cells transfected with control siRNA (CTLi) were included for comparison. Knockdown of Orai3 was confirmed by qPCR. (B) The effect of Orai3 knockdown on ALDH1 was determined by qPCR. *P < 0.01 (C) The effect of Orai3 knockdown on self-renewal capacity was determined by tumor sphere formation assay. **P < 0.05 (D) The effect of Orai3 knockdown on migration ability was determined by transwell migration assay. *P < 0.01.
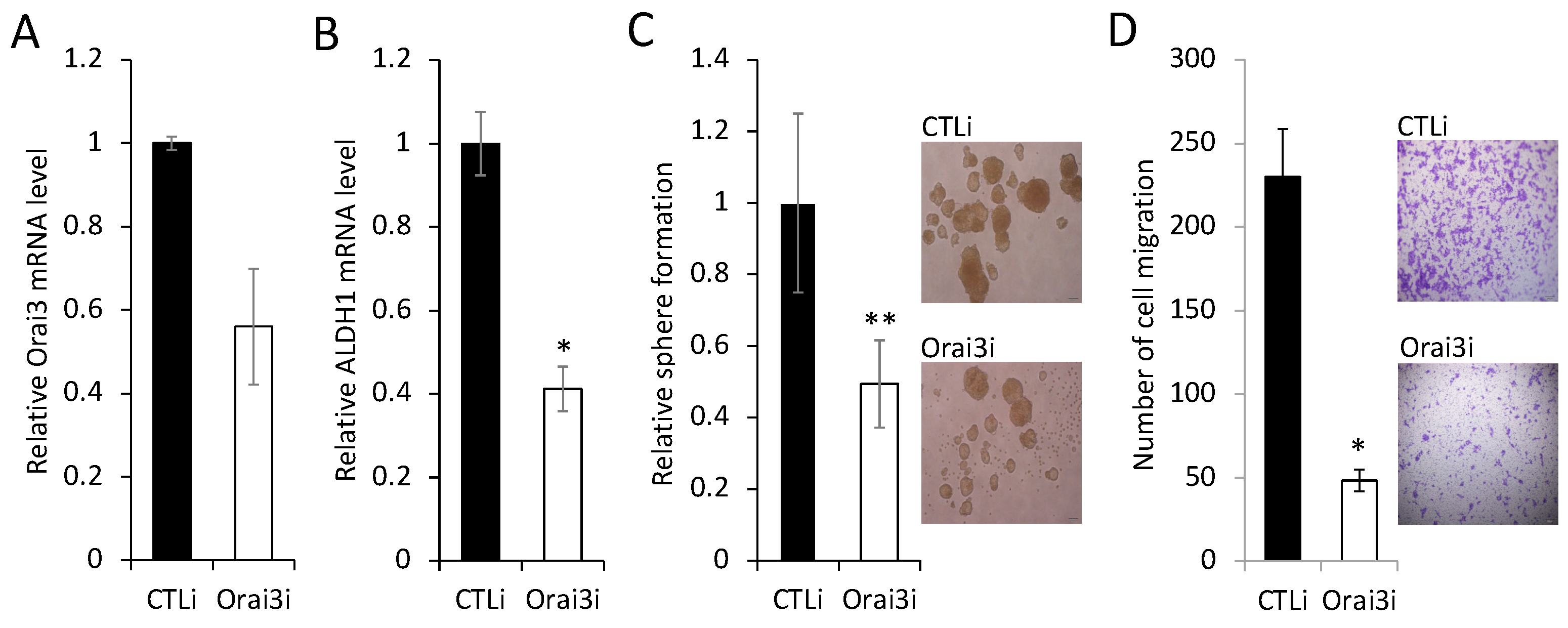
Figure 6.
Orai3 increases ID1 expression. Effect of Orai3 on CSC-related genes was determined by (A) qPCR and (B) Western bot analysis. Their levels in HOK-16B/Orai3 were plotted as fold change against those in HOK-16B/EV.
Figure 6.
Orai3 increases ID1 expression. Effect of Orai3 on CSC-related genes was determined by (A) qPCR and (B) Western bot analysis. Their levels in HOK-16B/Orai3 were plotted as fold change against those in HOK-16B/EV.
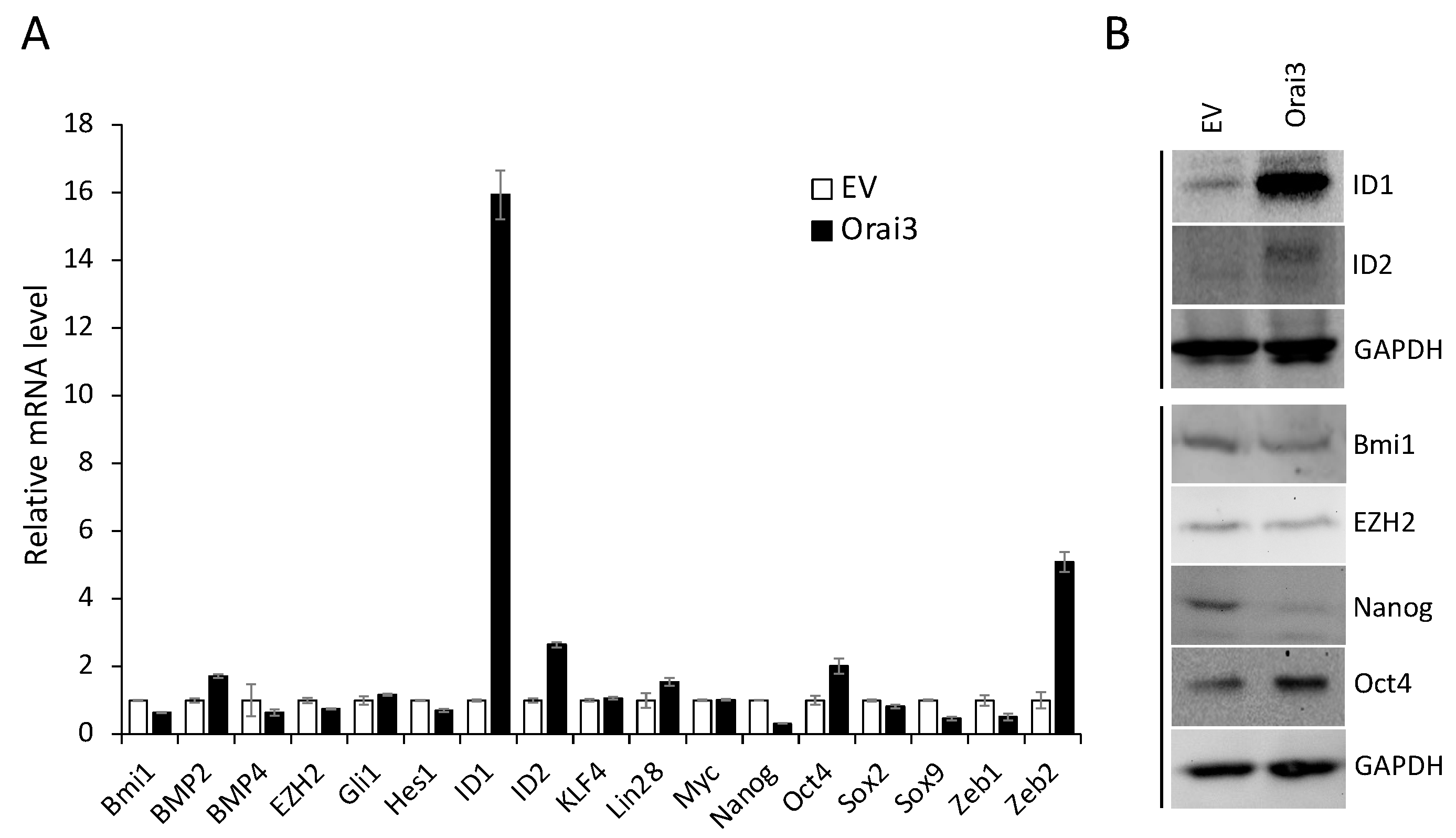
Figure 7.
ID1 is required for Orai3-induced CSC phenotype. (A) ID1 was knocked down in HOK-16B/Orai3 using siRNA against ID1 (ID1i). The cells transfected with control siRNA (CTLi) were included for comparison. Knockdown of ID1 was confirmed by qPCR. (B) The effect of ID1 knockdown on self-renewal capacity of HOK-16B/Orai3 was determined by tumor sphere formation assay. (C) The effect of ID1 knockdown on migration ability in HOK-16B/Orai3 was determined by transwell migration assay. (D) Endogenous ID1 was knocked down SCC4 using siRNA against ID1 (ID1i). The cells transfected with control siRNA (CTLi) were included for comparison. Knockdown of ID1 was confirmed by qPCR. (E) The effect of ID1 knockdown on self-renewal capacity of SCC4 was determined by tumor sphere formation assay. (F) The effect of ID1 knockdown on migration ability of SCC4 was determined by transwell migration assay. *P < 0.01, **P < 0.05.
Figure 7.
ID1 is required for Orai3-induced CSC phenotype. (A) ID1 was knocked down in HOK-16B/Orai3 using siRNA against ID1 (ID1i). The cells transfected with control siRNA (CTLi) were included for comparison. Knockdown of ID1 was confirmed by qPCR. (B) The effect of ID1 knockdown on self-renewal capacity of HOK-16B/Orai3 was determined by tumor sphere formation assay. (C) The effect of ID1 knockdown on migration ability in HOK-16B/Orai3 was determined by transwell migration assay. (D) Endogenous ID1 was knocked down SCC4 using siRNA against ID1 (ID1i). The cells transfected with control siRNA (CTLi) were included for comparison. Knockdown of ID1 was confirmed by qPCR. (E) The effect of ID1 knockdown on self-renewal capacity of SCC4 was determined by tumor sphere formation assay. (F) The effect of ID1 knockdown on migration ability of SCC4 was determined by transwell migration assay. *P < 0.01, **P < 0.05.
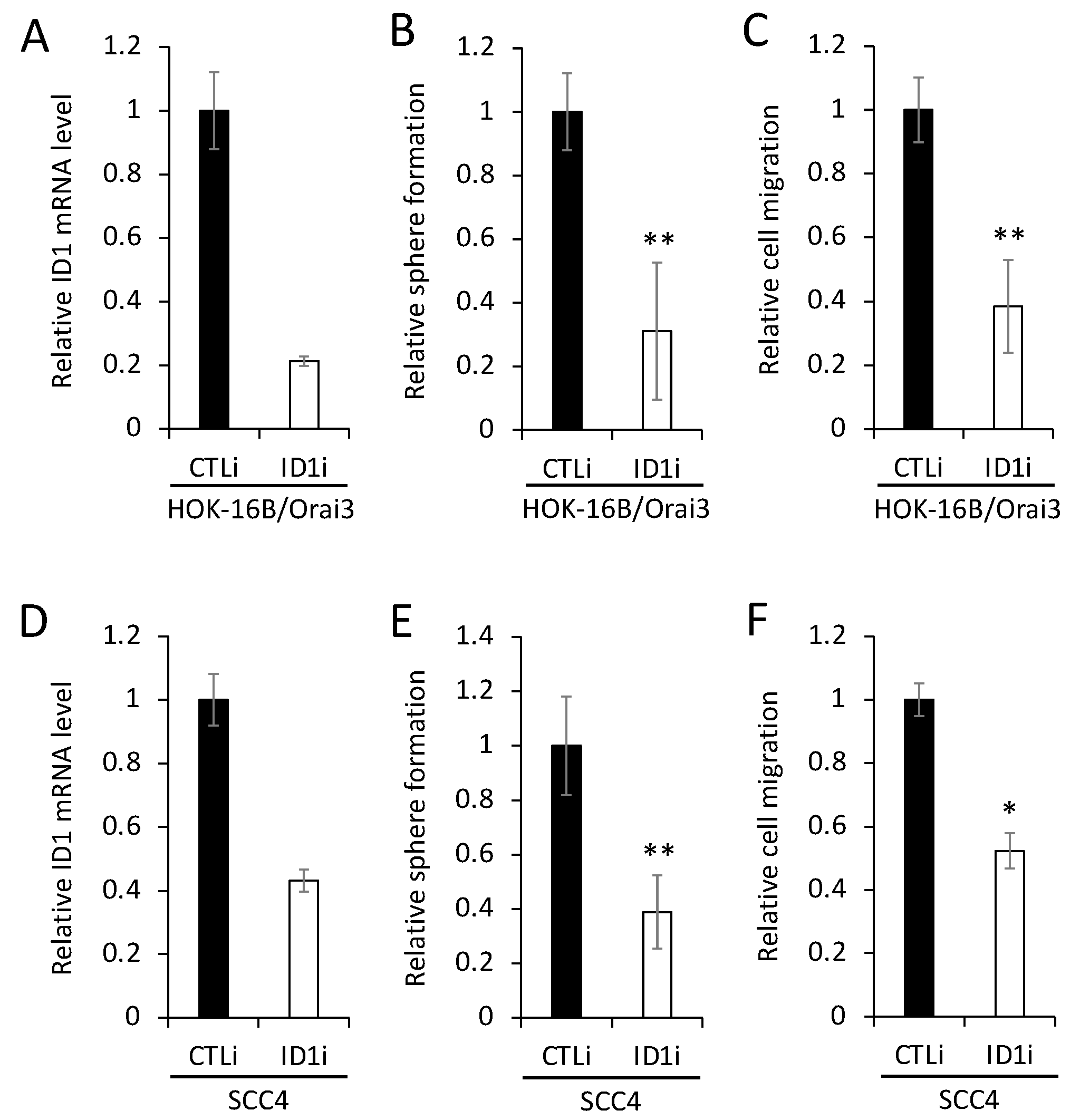
Figure 8.
Orai3/ID1 axis is enriched in CSC populations, and their mRNA levels are positively correlated in OSCC cells. Expression of ID1 is measured in CSC populations and their corresponding non-CSC populations by (A) qPCR and (B) Western blot. *P < 0.01, **P < 0.05 (C) Correlation analysis of Orai3 and ID1 mRNA was determined based on their expression levels in 13 human OSCC cell lines by qPCR.
Figure 8.
Orai3/ID1 axis is enriched in CSC populations, and their mRNA levels are positively correlated in OSCC cells. Expression of ID1 is measured in CSC populations and their corresponding non-CSC populations by (A) qPCR and (B) Western blot. *P < 0.01, **P < 0.05 (C) Correlation analysis of Orai3 and ID1 mRNA was determined based on their expression levels in 13 human OSCC cell lines by qPCR.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



