
You are currently viewing a beta version of our website. If you spot anything unusual, kindly let us know.
Preprint
Article
Enhancing Uranium Extraction Efficiency Using Protonated Amines and Quaternary Ammoniums-Based Ionic Liquids: Mechanistic Insights and Non-Linearities Analysis
Altmetrics
Downloads
115
Views
57
Comments
0
A peer-reviewed article of this preprint also exists.



This version is not peer-reviewed
Abstract
This study investigates uranium solvent extraction under AMEX process conditions. The use of pure extractants without diluents or phase modifiers allows not only to reduce the use of volatile organic compounds, but also to provide higher extraction yields without third phase formation. Pure extractants are protonated amines or quaternary ammoniums with suitable counter ion, which act at the interface between ion pairs and protic ionic liquids. The mixture of sulfates anion (SO42-) and bis(trifluoromethanesulfonyl)imide anion (NTf2-) revealed unexpected nonlinear extraction behaviors, which appear highly important to rationalize for optimized application. Spectroscopic analysis (NMR, UV-vis, FT-IR, and EXAFS) showed that uranium extraction occurs via protonated amine and three sulphates. Nonlinear extraction could further be interpreted by considering water and acid transfer between the two phases: at lower sulphate ratios, the release of acid from the organic phase into the aqueous phase was shown to influence the number of protonated amines in the organic phase, affecting uranium extraction before its enhancement. Furthermore, the extraction loss at higher sulphate ratios, was assigned to the destabilization of bidentate uranium-sulphate complexes due to a competition between water and sulphates.
Keywords:
Subject: Chemistry and Materials Science - Analytical Chemistry
1. Introduction
Industrial plants use large volumes of organic diluents (usually kerosene) for solvent extraction in mixer-settler. AMine EXtraction (AMEX) process is based on mixtures of alkyl amines in aliphatic diluent with fatty alcohol [1]. Despites its extensive application for uranium purification in the front end of the nuclear fuel cycle, this process is still facing several problems. Non-ideal uranium separation, requirement of a phase modifier (fatty alcohol) to prevent third phase formation and possible extractant degradation due to the presence of vanadium and phase modifier [2,3,4].
In particular, the Somaïr uranium extraction and purification plant in Niger has a stock of around 900 m3 of solvent, 75 % of which is replaced each year due to formulation degradation (crud formation), loss through entrainment of the organic phase in the aqueous phase, or evaporation.
Numerous alternatives are currently being studied to avoid the use of volatile organic compounds for metal separation. For example, solid-liquid extraction [5] supercritical extraction [6] and flotation [7] have been proposed. However, they remain difficult to apply to uranium extraction, as they require a complete rethink of the industrial process and facilities. Interesting alternatives also involve a simple replacement of organic solvents with non-volatile solvents such as ionic liquids (ILs) or deep eutectic solvents [8,9].
Under AMEX process conditions [1,10] it has been demonstrated and patented that the use of pure extractant (trialkylamines), without diluent and phase modifier enables higher extraction yields without third phase formation [11]. Pure extractants are quaternary ammoniums or trialkylamines previously protonated, and associated with a counter anion. These pure amines and quaternary ammoniums actually lie at the interface between amine (ion pair) and protic ionic liquid (quaternary ammonium) chemistry and have already been used as ionic liquids for liquid-liquid extraction [12,13,14]. Such ionic liquids are often used as diluents to increase the extraction capacity of a ligand, but they are more than a plain diluent. They modify the extraction mechanism through the participation of their anion or cation in the metal complex, or by inducing a different organization of the organic phase [15,16,17]. Many studies have been dedicated to the mechanisms of solvent extraction in ILs. Billard et al. [18] proposed two equilibria (cationic and anionic exchanges) to summarize the possible ion exchanges mechanisms when ILs are used as diluents. It was found out that ion exchange is the dominant mechanism for uranium and actinides extraction.
A wide range of ILs have also been used as extractants in aliphatic diluents to which phase modifiers are sometimes added [19,20,21,22]. Depending on the anions and cations they contain, ILs can be highly hydrophobic and present a sufficiently low-viscosity at room temperature to be used both as an extraction agent and as a hydrophobic phase. In this case, dilution in an aliphatic diluent is not necessary. Extraction performance and selectivity is usually improved compared to conventional solvents [23,24,25,26,27].
Mixtures of ILs may also be applied as they generally combine the properties of two different ILs. For instance, a "diluting" IL, with advantageous transport properties (low viscosity and high conductivity), can be mixed with a "complexing" IL whose transport properties are prohibitive for use at low temperatures [28,29,30].
In the present study, diluent free quaternary ammoniums or trialkylamines and their mixtures were assessed in the condition on the AMEX process. Topological formula of involved anion and cation are given in Figure 1 where TOAH+ stands for trioctylammonium, MTOA+ for methyltrioctylammonium and where the commercial Aliquat 336+ is composed of a mixture of MTOA+ and methyltridecylammonium (MTDA+). Sulphate (SO42−), chloride (Cl−) and bis(trifluoromethanesulfonyl)imide (NTf2−) were chosen to evaluate the anion effect on uranium extraction.
The various tested mixtures provided high extraction efficiency with nonlinear properties. A mechanistic studies is proposed to elucidate the uranyl extraction mechanisms of the [TOAH]2[SO4] + [TOAH]2[NTf2] mixture. As it might be very interesting to design mixtures with optimized extraction properties, origin of these non-linearities has been investigated in details in this study. Coordination of uranyl was evaluated by NMR, UV-vis and EXAFS spectroscopy. Considering water and acid transfer between the two phase, FT-IR spectroscopy was further exploited to elucidate the various regimes of extraction of this mixture.
2. Materials and Methods
2.1. Chemicals and reagents
Trioctylamine (TOA) 98 %, sulphuric acid 96 % and ammonium sulphate were purchased from Sigma-Aldrich. Aliquat 336 and lithium bis(trifluoromethanesulfonyl)imide (LiNTf2) were supplied by Alfa Aesar and ABCR respectively. Methyltrioctylammonium chloride ([MTOA][Cl]) and methyltrioctylammonium bis(trifluoromethanesulfonyl)imide ([MTOA][NTf2]) were purchased from ABCR. All chemicals were used as recieved, without any further purification.
2.2. Instrumentation and analysis
Metal concentration was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES Spectro Arcos - AMETEK Materials Analysis, ThermoFisher Scientific). Prior to concentration measurements, aliquots of uranium and iron stock and working solutions were diluted with a v:v 0.99% nitric acid solution in order to bring the concentration in the 1-15 ppm range. Metal quantification was performed at several wavelengths. Uranium was detected at 393.203 nm, 393.203 nm, 385.466 nm and 385.958 nm; iron was detected at 239.562 nm, 240.488 nm and 259.940 nm. Calibration samples were prepared from 1000 mg·L−1 uranium and iron, standards (SCP Science PlasmaCal). Uncertainties in metal concentrations were determined statistically by repeated measurements.
The amount of extracted acid was determined by acid-base titration using a Metrohm 905 titrando automatic potentiometer. A sample of aqueous phase was taken and dosed with a 0.01 mol·L−1 NaOH solution. The concentration of sulfuric acid in the organic phases was given by subtracting the acidity of the aqueous phase measured at equilibrium from that of the initial aqueous phase. The water present in the organic phases was determined by the Karl Fischer method, using a Metrohm coulometer.
The speciation of uranium complexes in ionic liquid media was achieved by ultra violet-visible and Fourier transform infrared spectroscopies (UV-vis and FT-IR). UV-vis spectra were recorded between 200 and 800 nm on a dual-beam Varian Cary 5000 spectrophotometer in plastic cells with a 2 mm optical path. Absorbances of contacted, pre-contacted and non-contacted samples were measured using an empty cell in the second beam as a reference. Subtractions were calculated in post-processing. At a given wavelength λ, the absorbance A of a mixture of n absorbing species is the sum of the individual absorbance. The signal from species formed by water and acid can therefore be subtracted to highlight uranium-related signals. Infrared spectra of ionic liquid phases were recorded on a Bruker Vertex 70 FT-IR spectrometer equipped with a diamond ATR. A blank was run on air before each measurement. The Fourier transform was calculated using Perkin Elmer’s Spectrum software, then the background noise was subtracted. The measurement range extends from 4000 to 615 cm−1. Many absorption bands were found in the printed part of the spectrum of interest in this study. The pre-contacted sample signal was subtracted to highlight the uranium-related absorption bands. A Voigt function was used to best fit the signal and calculate the area under the curves of the highlighted spectra.
X-ray absorption spectroscopy (Extended X-Ray Absorption Fine Structure-EXAFS) was carried out on the MARS (Multi-Analyses on Radioactive Samples) light line at the SOLEIL synchrotron (Saint-Aubin, France) (proposal ID: 20210696) [31,32]. Spectra were recorded at the uranium LIII threshold (17167 eV). Calibrations were carried out at the yttrium K threshold (17039 eV).
In practice, samples were placed in double-confinement EXAFS cells, enabling active samples to be measured. The cells can hold a maximum of 12 samples. The volume of each sample is 0.25 mL.
Data were processed using the Athena program. EXAFS oscillations are extracted in several steps. The ionization energy threshold E0 is defined by taking the maximum of the derivative at the threshold jump. After signal normalization using linear and cubic functions, the signal can be converted from E (eV) energy space to k (Å−1) wavenumber space. To increase the amplitude of oscillations observed at high wavenumbers, a k² or k3 factor can be applied. Once the oscillations have been extracted, conversion to real distance space R (Å) is performed by applying a Fourier transform to the EXAFS k3 signal, χ(k) over a defined wavenumber range (in the remainder of this study, this range is 3.75 < k < 14). Finally, the pseudo-radial distribution function is obtained as a function of R. This pseudo-radial distribution function schematically represents the probability of atoms being present around the absorber atom as a function of their distance increased by a phase shift φ. It is not the actual distance R from the absorber atom. Fits were then made in the Artemis software using model structure compounds (references found in the literature where available + FEFF software). The discrepancy between the fitted data and the experimentally found data can be expressed by calculating the R factor based on the method of least squares.
Fluorine-19 nuclear magnetic resonance spectroscopy (19F-NMR) was also used to study the impact of metal extraction on the chemical shift of the fluorine function of NTf2. Double-walled tubes were used so as not to alter the chemistry of the organic phase, and so that a reference solvent (deuterated DMSO) could be applied externally.
2.3. Extraction experiments
Several organic extractants based on protonated tertiary amines or quaternary ammoniums and their mixtures were evaluated in this work for the extraction of uranium in sulfuric media. Their synthesis is described in ESI 1.
Organic phases were prepared by mixing protonated tertiary amines or quaternary ammoniums (defined as Am 1 and Am 2) in different proportions. Samples were classified according to their molar ratio of Am 1, xAm 1, defined as follows:
The aqueous phases used in this study were prepared by dissolving the uranium sulphate, UO2SO4⋅6.38H2O, M = 479.43 g·mol−1 and iron sulphate, Fe2(SO4)3⋅8.3H2O, M = 549.46 g·mol−1.
The acidity of the solution was controlled by adding sulfuric acid (v:v H2SO4 96%, M = 98.08 g·mol−1) and the sulphate concentration was increased by adding ammonium sulphate ((NH4)2SO4, M = 132.14 g·mol−1).
The aqueous and organic phases are brought into contact in eppendorf tubes of appropriate volume and stirred (500 rpm) for 1 hour at 20 °C in a thermostatically-controlled cell (Infor-ht® ecotron). The phases are then separated after a centrifugation step (Rotina 380R) lasting between 5 and 20 min. at a rotation speed of 8000 rpm. The phases are then separated in 2 different flasks before sampling for analysis.
where stands for the sum of the chemical activities of a solute M in all its forms in the organic phase and , for the sum of the chemical activities of the same solute in all its forms in the aqueous phase. In dilute media, activity is equal to molar concentration
3. Results and discussion
Several commercial quaternary ammonium ligands and their mixtures were evaluated for uranium extraction from sulphuric media. Their performance were compared with the classical trioctylamine extractant (taken as reference for the AMEX process) solubilised in dodecane in presence of 1-octanol as phase modifier. The evaluated cations and anions are illustrated in Figure 1. Distribution ratios of uranium obtained with these ammoniums salts and mixtures are displayed in Figure 2.
Results show that uranium extraction is not only driven by the nature of cations and anions but also by the ratio between them. When similar cation are considered, systems with SO42− extract better than with Cl− (see Figure 2b). Whatever the cation, no extraction occurred when NTf2− was used as anion (see left side of Figure 2a–d and green line in Figure 2d). Uranium extraction increases with sulphates concentrations, which is consistent with the recognized extraction mechanisms for such systems: as demonstrated by Sukhbaatar et al., [33] uranium extraction from sulphuric media requires the formation of three sulphates uranyl complexes.
Looking at the effect of the cations, Figure 2 shows moreover that TOAH+ extracts more uranium than MTOA+ and than the Aliquat 336 when the counter anion is a sulphate (right side of Figure 2a–c).
Interestingly, uranium extraction is not linear with the molar ratio of one quaternary ammonium to another, but shows non-linearities with a synergistic peak when mixtures of anions are involved (see Table 1 for position of the synergistic peaks).
Overall, nonlinear extraction is observed when the cations are identical and when the anion is modified. In contrast, when the anion is identical and the cation is modified, linear extraction is obtained. We can therefore conclude that the non-linearities observed are due to anion mixing and not cation mixing.
As it might be very interesting to design mixtures with optimized extraction properties, origin of these non-linearities has been investigated in more details for the mixture [TOAH]2[SO4] and [TOAH][NTf2]. This peculiar system is interesting as it is the most efficient one toward uranium extraction, but also because its extraction non-linearities show three distinct regimes (Figure 2a): extraction is initially antagonistic (lower than the linear mixture), then synergistically increased, and finally decreased for the higher sulphate ratios. Extraction mechanisms of the [TOAH]2[SO4] + [TOAH]2[NTf2] mixture were therefore investigated with a spectroscopic study combined with a detailed analysis of the sulphate effect on uranium, acid and water transfer between the organic and aqueous phases.
3.1. Effect of the amount of sulphates in the aqueous phase
Like sulphates in the organic phase, the amount of sulphates in the aqueous phase may affect uranium extraction. In order to evaluate this effect, the sulphate concentration in the aqueous phase was varied by changing the concentration of ammonium sulphate. Effects on uranium extraction, acid and water transfer are presented in Figure 3 for sulphate concentrations of [(NH4)2SO4] = 0.1 and 1 mol·L−1 in the aqueous phase.
Figure 3a shows that increasing sulphate concentration in the aqueous phase decreases uranium extraction for all x[TOAH]2[SO4] ratios. It is also interesting to note that the decrease in DU between 75 and 100 % sulphate is most marked for [(NH4)2SO4] = 1 mol·L−1. Water concentration (Figure 3b) is unaffected by the amount of ammonium sulphate, while acid transfer between the two phases is little affected (Figure 3c). The acid transfer is actually unexpected. Instead of being extracted in the organic phase with the metal (as it is usually the case in solvent extraction), acid is released from the organic toward the aqueous phase. This acid release is assigned to the deprotonation of the TOAH+ originally present in the organic phase. It appears to be poorly affected by the (NH4)2SO4 concentration for the low x[TOAH]2[SO4] ratios, while it is slightly increased above x[TOAH]2[SO4] = 50 %.
This experiment shows therefore that uranium extraction is both dependent on the amount of sulphate present in the aqueous and in the organic phase. Most importantly, it shows also that the decrease in uranium extraction at high x[TOAH]2[SO4] ratios is concomitant with an increase in the release of H+ protons in the aqueous phase (expected to decrease the concentration of protonated TOA in the organic phase), as well as with a strong increase in water extraction in the organic phase.
To further understand the nonlinear uranium extraction with the x[TOAH]2[SO4] ratios, the remainder of this paper is devoted to characterizing the uranyl complexes extracted in the organic phase. It is indeed important to evaluate if uranium is chelated as in the conventional medium (TOA in dodecane) or if the NTf2 anion is involved to form mixed complexes for some x[TOAH]2[SO4] ratios.
3.2. Characterizing the extracted species
3.1.1. NTf2 participation in the uranium complex
To obtain information about the chemical environment of NTf2− in the binary mixtures before and after uranium extraction, 19F-NMR and FT-IR measurements were carried out. Note that NTf2− gives only one resonance band in the 19F-NMR spectra. Its evolution provides information on the environmental change of NTf2−.19F-NMR spectra of [TOAH]2[SO4] + [TOAH][NTf2] mixtures are plotted in Figure 4a for x[TOAH]2[SO4] = 50%. It shows that the chemical shift for NTf2− is not modified before and after uranium extraction, which indicates that NTf2− does not directly coordinate uranyl.
This conclusion is reinforced by a Fourier transform infra-red (FT-IR) analysis shown in Figure 4b. It can be observed that the characteristic bands of NTf2 [34] are not affected by the presence of uranium. NTf2 therefore does not participate directly in the complex with uranium, and the extraction non-linearities of the mixture cannot be explained by the formation of a synergistic or antagonistic mixed complex of uranium with sulphates and NTf2.
3.1.2. UV-vis spectroscopy and EXAFS, study of first neighbors
To determine whether the origin of the nonlinear extraction is due to uranium speciation, UV-vis uranium spectroscopy and X-ray absorption spectroscopy (EXAFS) measurements were performed for various x[TOAH]2[SO4] ratios.
UV-vis spectra recorded after contact with a uranium solution (Figure 5a), show the same type of fingerprint signal for 25 to 100 % ratios as for the conventional medium (TOA + dodecane + octanol) shown in dotted orange line. The different trend observed for the ratio 0% is due to the absence of uranium in this sample. As demonstrated by Servaes et al. who studied the speciation of uranyl nitrate complexes in acetonitrile and ionic liquid [35], this fingerprint is characteristic of a trigonal symmetric uranium complex. In their study, spectra of UO2(NO3)3− complexes in acetonitrile or in [C4mim][NTf2] show signals similar to those observed for UO2(CO3)34− and UO2(CH3COO3)3 complexes [36]. By comparing the experimental spectra with those reported in the literature, it can therefore be concluded that for all x[TOAH]2[SO4] ratios, the uranyl sulphate complex exhibits D3h trigonal symmetry, suggesting that uranyl is complexed with 3 sulphates in an ionic liquid medium. EXAFS spectroscopy measurements were carried out to confirm this hypothesis.
Figure 5b displays EXAFS spectra measured at the L3 threshold of uranium and the corresponding Fourier transforms. No EXAFS spectra could be measured for x[TOAH]2[SO4] ratios lower than 25 % as the uranium concentrations was too small in these samples. For the higher x[TOAH]2[SO4] ratios, it shows that experimental spectra (solid lines) obtained with the [TOAH]2[SO4] + [TOAH][NTf2] mixtures are very similar to the one measured in conventional media (orange spectrum, TOA + dodecane). It can also be noted that in addition to the first U-Oyl contribution of linear transdioxo bonds (R + φ = 1.2 Å) and that of the equatorial U-Oeq oxygen coordination layer (R + φ = 1.8 to 2.4 Å), the spectra show intense contributions in the second coordination sphere (R + φ > 2.5 Å) due to sulfur atoms of sulphate anions coordinated with UO22+ [33,37]
In a previous study carried out in a conventional medium (TOA + dodecane), coupling a classical fit with molecular dynamics simulations enabled to unravel the speciation of uranium in the organic phase: the complex consists of three sulphate anions coordinated to uranyl via U-O bonds in the first coordination sphere [33]. The study also showed that uranyl trisulphate complexes shift from a 3 bidentate sulphate configuration to a 2 bidentate and 1 monodentate configuration, with the most favorable configuration remaining the 3 bidentate sulphate configuration.
In order to probe more precisely the local structure of uranyl in [TOAH]2[SO4] + [TOAH][NTf2] mixtures, a fit of the EXAFS spectra was performed by applying a structural model of three bidentate sulphates in the first uranium sphere. The fitted parameters are presented in Table 2 and the corresponding fitted spectra are plotted as dotted lines in Figure 5b.
Good fits are obtained for all the x[TOAH]2[SO4] ratios, confirming the structure with three bidentate sulphates. As expected from the qualitative study of the data, U-Oyl, U-Oeq and U-S bond lengths do not vary. However, although this does not lead to outliers, the quality of fit deteriorates for the higher x[TOAH]2[SO4] ratios: the R factor increases from 3.9 % to 6.0 % (see Figure 6a). Furthermore, as shown in Figure 6b, the Debye-Waller factor increases suggesting more disorder in the complexes. The evolution of these two parameters indicates that the U-3 sulphate complex is destabilized, which could explain a loss of uranium extraction for the high x[TOAH]2[SO4] ratios.
In conclusion, 19F-NMR, UV-vis and EXAFS data indicate that there is no mixed complexes formed with sulphates and NTf2 in the first coordination sphere. However, destabilization of the bidentate sulphate complex is observed when the x[TOAH]2[SO4] ratio reaches 100 %, which may be related to the decrease of uranium extraction observed for these high ratios.
As extracted water is very important at these ratios, it would have been interesting to evaluate its contribution to the complexes. However, it was not possible to include it distinctly in the fits, as the signatures of the monodentate U-SO4 and U-H2O bonds are too similar (same distance, same properties). EXAFS therefore does not allow us to conclude if water competes or not with uranium sulphate.
3.1.3. FT-IR spectroscopy, looking at the second coordination sphere and beyond
Fourier transform infrared (FT-IR) spectra of protonated tertiary amines mixtures are presented in Figure 7 and ESI 2 and 3. Three main bands can be distinguished in the region 800-1000 cm−1. Identification of resonance bands has been performed according to results proposed in the literature based on a IR study dedicated to the structure of tridecylammonium sulphate in presence of uranium [38]. The three bands were therefore assigned to monodentate water-bound sulphates ν (S-OH) at 855 cm−1, to the uranyl motif ν (UO22+) at 914 cm−1 and to bidentate uranium-bound sulphates ν (S-(O...U)2) at 967 cm−1.
A linear shift of the ν(S-OH) absorption band towards the lower frequencies can be observed with the molar ratio of sulphate. It indicates that sulphate becomes increasingly free (see ESI 4). The area under these ν(S-OH) band curve was plotted in Figure 8a. It shows an increase after the ratio x[TOAH]2[SO4] = 30% that follows the trend of the water extraction profile (see Figure 3b). This first observation suggests that the sulphate are not only involved in the uranyl complexes but also more and more connected with water which concentration is significantly increasing for the high x[TOAH]2[SO4] ratios.
To highlight the bands related to uranium, it was necessary to subtract the signal of the pre-contacted samples. After subtraction, each band of interest was fitted with a Voigt function. The area under the curve (AUC) is plotted in Figure 8b,c as a function of x[TOAH]2[SO4] for the ν (UO22+) and ν (S-(O...U)2) bands.
It is interesting to note that the area under the bands attributed to ν (UO22+) and ν (S-(O...U)2) bonds follows a similar trend to that of the uranium extraction profile (plotted in Figure 2a): it is initially constant up to x[TOAH]2[SO4] = 12.5%, then intensifies up to x[TOAH]2[SO4] = 75 %, and decreases thereafter.
This infrared study indicates therefore that for x[TOAH]2[SO4] ratios above 75 %, the uranium-sulphate bond is less present, to the benefit of the sulphate-water bond, suggesting a competition between the sulphate-uranium and sulphate-water complexes.
To interpret the overall uranium extraction profile of the [TOAH]2[SO4] + [TOAH][NTf2] mixture (Figure 2a), it is necessary to take into account all the data obtained by the spectroscopic methods, as well as the water extraction data and the acid release from the organic to the aqueous phase (Figure 3b,c). Spectroscopic data indicates that the NTf2 anion is not involved in the extraction of uranium. As in the conventional medium (TOA + dodecane), uranium is extracted by TOAH+ protonated amines and three bidendate sulphates in the first coordination sphere.
For low x[TOAH]2[SO4] ratios of 0-25 %, uranium extraction does not increase, while the amount of sulphates introduced into the organic phase does. This effect can be explained by the increased release of H+ protons from the organic phase into the aqueous phase. This release reduces the number of protonated amines available, inhibiting uranium extraction.
From x[TOAH]2[SO4] = 25 %, proton release stabilizes and then decreases, leading to an increase in the number of TOAH+ protonated amines available. This increase in TOAH+ accompanied by an increase in sulphates with x[TOAH]2[SO4] is therefore consistent with the very sharp increase in uranium extraction between x[TOAH]2[SO4] = 25 and 75 %.
Beyond this ratio, the very sharp increase in extracted water leads, as previously suggested, to a competition between the sulphate-uranium and sulphate-water complexes, which destabilizes the uranyl tri-sulphate complexes and leads to saturation, then to a decrease in uranium extraction.
This study shows that the main elements responsible for uranium extraction are sulphates and protonated amines, and that the availability of the latter is regulated by the concentration of extracted water, as well as by the release of acidic protons from the organic phase to the aqueous phase.
A deeper understanding of these phenomena, would require to interprete the nonlinear behavior of water extraction and the origin of H+ proton release. The increased extraction of water can be partially explained by the difference in hydrophilicity between NTf2− and SO42− anions, but its nonlinear extraction might be related to the activity of the aqueous phase and to the chemical potential equilibria between the species present in the aqueous and organic phases. The chemical potential equilibrium may also be responsible for the release of protons from the organic phase to the aqueous phase. These equilibria are however very difficult to write down and predict.
4. Conclusions
Several organic extractant based on protonated tertiary amines or quaternary ammoniums and their mixtures were evaluated for the extraction of uranium in the AMEX process conditions. Irrespective of the leachate and the cation used, non-linearities in uranium extraction were observed as a function of the sulphate ratio in the organic phase. This effect, which allows obtaining very high extraction efficiency for some mixtures, can be advantageously considered for optimized uranium extraction or when physico-chemical properties (as viscosity or density) of the final solvent need to be adjusted. Origin of these non-linearities were therefore investigated with a mechanistic study.
An extensive analysis of the amounts of extracted water, acid and uranium combined with detailed spectroscopic studies (NMR, UV-vis, FT-IR and EXAFS) of contacted organic phases was carried out on the [TOAH]2[SO4] + [TOAH][NTf2] system to understand the three regimes of extraction observed along with the sulphate ratios. Whatever the x[TOAH]2[SO4] ratio, the NTf2− anion was shown to be absent of the first coordination sphere. It was shown that uranium extraction takes place via four protonated amines and three bidendate sulphates, as in the conventional AMEX process. Analysis of EXAFS data showed moreover that the bidentate uranium-sulphate complex is destabilized for the high x[TOAH]2[SO4] ratios. Infrared spectroscopy revealed along with this destabilization, that the number of sulphate-uranium bonds decreases in favor of sulphate-water bonds. It is assumed that a competition between sulphates-water and sulphates uranyl complexes governs the extraction loss observed at high x[TOAH]2[SO4] ratios.
To understand the observed non-linearities over the whole range of x[TOAH]2[SO4] ratios, the release of acid from the organic phase into the aqueous phase as well as the nonlinear water extraction were further considered. The nonlinear extraction of uranium was finally related to the concentration of protonated amines and sulphates: (i) by influencing the number of available protonated amines in the organic phase, acid release negatively affects uranium extraction at low x[TOAH]2[SO4] ratios; (ii) in the medium x[TOAH]2[SO4] ratios, the saturation of acid release induces a strong increase of the available protonated amines leading to a strong increase of uranium extraction; (iii) at higher x[TOAH]2[SO4] ratios, the strong increase of water extraction destabilizes the uranyl-sulphate complexes because of a competition between sulphates-water and sulphates uranyl complexes.
Water extraction is therefore one of the main parameters ruling the extraction mechanisms of such systems. As it was already observed on different ionic liquid systems, it is also likely to govern physicochemical properties as density and viscosity. Finding an optimized mixture of ionic liquids for solvent extraction requires therefore to elucidate the water extraction properties of the systems.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Guerinoni E.: Investigation, data curation, writing—original draft. Dourdain S.: Writing, review. Lu Z.: Investigation, data curation. Dumas T.: EXAFS spectroscopy measurements and analysis. Arrachart G.: NMR spectroscopy measurements, review. Giusti F.: review. Solari P.L.: local contact for EXAFS spectroscopy. Pellet-Rostaing S.: Project administration, Resources, Supervision, Funding acquisition.
Funding
We acknowledge the NEEDS program as well as CEA for their funding.
Data Availability Statement
Data will be made available on request.
Acknowledgments
We acknowledge SOLEIL and beamline MARS for provision of synchrotron radiation facilities and assistance. We also thank Beatrice Baus-Lagarde for her help with ICP-OES measurements, David Lemire for his participation to the EXAFS experiment.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Crouse, D.J.; Brown, K.B. The Amex Process for Extracting Thorium Ores with Alkyl Amines. Ind. Eng. Chem. 1959, 51, 1461–1464. [Google Scholar] [CrossRef]
- Sato, T.; Watanabe, H.; Suzuki, H. Liquid-Liquid Extraction of Molybdenum(VI) from Aqueous Acid Solutions by High-Molecular Weight Amines. Solvent Extraction and Ion Exchange 1986, 4, 987–998. [Google Scholar] [CrossRef]
- Sato, T.; Watanabe, H. The Extraction of Zirconium(IV) from Sulfuric Acid Solutions by Long-Chain Alkyl Quaternary Ammonium Compound. Separation Science and Technology 1982, 17, 625–634. [Google Scholar] [CrossRef]
- Chagnes, A.; Fosse, C.; Courtaud, B.; Thiry, J.; Cote, G. Chemical degradation of trioctylamine and 1-tridecanol phase modifier in acidic sulfate media in the presence of vanadium (V). Hydrometallurgy 2011, 105, 328–333. [Google Scholar] [CrossRef]
- Solgy, M.; Taghizadeh, M.; Ghoddocynejad, D. Adsorption of uranium(VI) from sulphate solutions using Amberlite IRA-402 resin: Equilibrium, kinetics and thermodynamics study. Annals of Nuclear Energy 2015, 75, 132–138. [Google Scholar] [CrossRef]
- Kumar, P.; Pal, A.; Saxena, M.K.; Ramakumar, K.L. Supercritical fluid extraction of uranium and thorium from solid matrices. Desalination 2008, 232, 71–79. [Google Scholar] [CrossRef]
- Smolinski, T.; Wawszczak, D.; Deptula, A.; Lada, W.; Olczak, T.; Rogowski, M.; Pyszynska, M.; Chmielewski, A.G. Solvent extraction of Cu, Mo, V, and U from leach solutions of copper ore and flotation tailings. J. Radioanal. Nucl. Chem. 2017, 314, 69–75. [Google Scholar] [CrossRef]
- Quijada-Maldonado, E.; Olea, F.; Sepúlveda, R.; Castillo, J.; Cabezas, R.; Merlet, G.; Romero, J. Possibilities and challenges for ionic liquids in hydrometallurgy. Separation and Purification Technology 2020, 251, 117289. [Google Scholar] [CrossRef]
- Yan, Q.; Cai, Y.; Wang, Z.; Dong, X.; Yuan, L.; Feng, W.; Chen, J.; Xu, C. Separation of americium from lanthanide by a Task-Specific ionic liquid decorated with 2,6-Bis-Triazolyl-Pyridine moiety. Separation and Purification Technology 2022, 299, 121752. [Google Scholar] [CrossRef]
- Brown, K.B.; Coleman, C.F.; Crouse, D.J.; Denis, J.O.; Moore, J.G. THE USE OF AMINES AS EXTRACTANTS FOR URANIUM FROM ACIDIC SULFATE LIQUORS. A Preliminary Report. 1954. [Google Scholar] [CrossRef]
- Lu, Z.; Dourdain, S.; Pellet-Rostaing, S.; Arrachart, G.; Giusti, F. Mélanges de Sels d’ammonium Quaternaire Pour l’extraction de l’uranium(VI) de Solutions Aqueuses d’acide Sulfurique. 3 June 2022. [Google Scholar]
- Ghandi, K. A Review of Ionic Liquids, Their Limits and Applications. GSC 2014, 04, 44–53. [Google Scholar] [CrossRef]
- Greaves, T.L.; Drummond, C.J. Protic Ionic Liquids: Properties and Applications. Chem. Rev. 2008, 108, 206–237. [Google Scholar] [CrossRef]
- Keshapolla, D.; Srinivasarao, K.; Gardas, R.L. Influence of temperature and alkyl chain length on physicochemical properties of trihexyl- and trioctylammonium based protic ionic liquids. The Journal of Chemical Thermodynamics 2019, 133, 170–180. [Google Scholar] [CrossRef]
- Billard, I.; Ouadi, A.; Gaillard, C. Is a universal model to describe liquid–liquid extraction of cations by use of ionic liquids in reach? Dalton Transactions 2013, 42, 6203–6212. [Google Scholar] [CrossRef] [PubMed]
- Dukov, I.L.; Atanassova, M. Effect of the diluents on the synergistic solvent extraction of some lanthanides with thenoyltrifluoroacetone and quaternary ammonium salt. Hydrometallurgy 2003, 68, 89–96. [Google Scholar] [CrossRef]
- Dietz, M.L.; Dzielawa, J.A.; Laszak, I.; Young, B.A.; Jensen, M.P. Influence of solvent structural variations on the mechanism of facilitated ion transfer into room-temperature ionic liquids. Green Chemistry 2003, 5, 682–685. [Google Scholar] [CrossRef]
- Billard, I.; Ouadi, A.; Jobin, E.; Champion, J.; Gaillard, C.; Georg, S. Understanding the Extraction Mechanism in Ionic Liquids: UO22+/HNO3/TBP/C4-mimTf2N as a Case Study. Solvent Extraction and Ion Exchange 2011, 29, 577–601. [Google Scholar] [CrossRef]
- Wionczyk, B.; Apostoluk, W. Solvent extraction of chromium(III) from alkaline media with quaternary ammonium compounds. Part I. Hydrometallurgy 2004, 72, 185–193. [Google Scholar] [CrossRef]
- Rout, A.; Venkatesan, K.A.; Srinivasan, T.G.; Vasudeva Rao, P.R. Ionic liquid extractants in molecular diluents: Extraction behavior of europium (III) in quarternary ammonium-based ionic liquids. Separation and Purification Technology 2012, 95, 26–31. [Google Scholar] [CrossRef]
- Jaree, A.; Khunphakdee, N. Separation of concentrated platinum(IV) and rhodium(III) in acidic chloride solution via liquid–liquid extraction using tri-octylamine. Journal of Industrial and Engineering Chemistry 2011, 17, 243–247. [Google Scholar] [CrossRef]
- Biswas, S.; Basu, S. Extraction of zirconium(IV) from hydrochloric acid solutions by tri-octylamine and neutral donors. Journal of Radioanalytical and Nuclear Chemistry 1999, 242, 253–258. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Yang, A.; Lv, P.; Zhang, L.; Li, Y.; Yang, Y. Extraction and separation on Au(III) and Pt(IV) from HCl media using novel piperazine-based ionic liquid as an ionic exchanger. Journal of Molecular Liquids 2022, 353, 118846. [Google Scholar] [CrossRef]
- Xue, W.; Liu, R.; Liu, X.; Wang, Y.; Lv, P.; Yang, Y. Selective extraction of Nd(III) by novel carboxylic acid based ionic liquids without diluent from waste NdFeB magnets. Journal of Molecular Liquids 2022, 364, 119919. [Google Scholar] [CrossRef]
- Micheau, C.; Lejeune, M.; Arrachart, G.; Draye, M.; Turgis, R.; Michel, S.; Legeai, S.; Pellet-Rostaing, S. Recovery of tantalum from synthetic sulfuric leach solutions by solvent extraction with phosphonate functionalized ionic liquids. Hydrometallurgy 2019, 189, 105107. [Google Scholar] [CrossRef]
- Hu, Q.; Zhao, J.; Wang, F.; Huo, F.; Liu, H. Selective extraction of vanadium from chromium by pure [C8mim][PF6]: An anion exchange process. Separation and Purification Technology 2014, 131, 94–101. [Google Scholar] [CrossRef]
- Zuo, Y.; Liu, Y.; Chen, J.; Li, D.Q. The Separation of Cerium(IV) from Nitric Acid Solutions Containing Thorium(IV) and Lanthanides(III) Using Pure [C8mim]PF6 as Extracting Phase. Ind. Eng. Chem. Res. 2008, 47, 2349–2355. [Google Scholar] [CrossRef]
- Villemejeanne, B.; Legeai, S.; Meux, E.; Dourdain, S.; Mendil-Jakani, H.; Billy, E. Halide based ionic liquid mixture for a sustainable electrochemical recovery of precious metals. Journal of Environmental Chemical Engineering 2021, 10, 107063. [Google Scholar] [CrossRef]
- Ouadi, A.; Klimchuk, O.; Gaillard, C.; Billard, I. Solvent extraction of U(vi) by task specific ionic liquids bearing phosphoryl groups. Green Chem. 2007, 9, 1160–1162. [Google Scholar] [CrossRef]
- Rout, A.; Binnemans, K. Solvent Extraction of Neodymium(III) by Functionalized Ionic Liquid Trioctylmethylammonium Dioctyl Diglycolamate in Fluorine-free Ionic Liquid Diluent. Ind. Eng. Chem. Res. 2014, 53, 6500–6508. [Google Scholar] [CrossRef]
- Llorens, I.; Solari, P.L.; Sitaud, B.; Bes, R.; Cammelli, S.; Hermange, H.; Othmane, G.; Safi, S.; Moisy, P.; Wahu, S.; et al. X-ray absorption spectroscopy investigations on radioactive matter using MARS beamline at SOLEIL synchrotron. Radiochimica Acta 2014, 102, 957–972. [Google Scholar] [CrossRef]
- Sitaud, B.; Solari, P.L.; Schlutig, S.; Llorens, I.; Hermange, H. Characterization of radioactive materials using the MARS beamline at the synchrotron SOLEIL. Journal of Nuclear Materials 2012, 425, 238–243. [Google Scholar] [CrossRef]
- Sukhbaatar, T.; Duvail, M.; Dumas, T.; Dourdain, S.; Arrachart, G.; Solari, P.L.; Guilbaud, P.; Pellet-Rostaing, S. Probing the existence of uranyl trisulfate structures in the AMEX solvent extraction process. Chem. Commun. 2019, 55, 7583–7586. [Google Scholar] [CrossRef] [PubMed]
- Hanke, K.; Kaufmann, M.; Schwaab, G.; Havenith, M.; Wolke, C.T.; Gorlova, O.; Johnson, M.A.; Kar, B.P.; Sander, W.; Sanchez-Garcia, E. Understanding the ionic liquid [NC4111][NTf2] from individual building blocks: an IR-spectroscopic study. Phys. Chem. Chem. Phys. 2015, 17, 8518–8529. [Google Scholar] [CrossRef] [PubMed]
- Servaes, K.; Hennig, C.; Billard, I.; Gaillard, C.; Binnemans, K.; Görller-Walrand, C.; Van Deun, R. Speciation of Uranyl Nitrato Complexes in Acetonitrile and in the Ionic Liquid 1-Butyl-3-methylimidazolium Bis(trifluoromethylsulfonyl)imide. European Journal of Inorganic Chemistry 2007, 2007, 5120–5126. [Google Scholar] [CrossRef]
- Görller-Walrand, C.; De Jaegere, S. Étude comparative des spectres d’absorption de complexes d’uranyle en solution et a l’état solide. Complexes de symétrie Cs, D2h (6) ET D3h (6). J. Chim. Phys. 1972, 69, 726–736. [Google Scholar] [CrossRef]
- Hennig, C.; Kraus, W.; Emmerling, F.; Ikeda, A.; Scheinost, A.C. Coordination of a Uranium(IV) Sulfate Monomer in an Aqueous Solution and in the Solid State. Inorg. Chem. 2008, 47, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- Lipovskii, A.A.; Kuzina, M.G. The Infrared Absorption Spectra and Structure of Tridecylammonium Sulphate, Tridecylammonium Hydrogensulphate, and Tridecylammonium Dioxotrisulphatouranate (VI). 1965, 10, 740–745. [Google Scholar]
Figure 1.
Structure of cations investigated (top): trioctylammonium ([TOAH]+), methyltrioctylammonium ([MTOA]+) and Aliquat 336. Structure of anions tested (bottom): sulphate (SO42−), chloride (Cl−) and bis(trifluoromethanesulfonyl)imide (NTf2−).
Figure 1.
Structure of cations investigated (top): trioctylammonium ([TOAH]+), methyltrioctylammonium ([MTOA]+) and Aliquat 336. Structure of anions tested (bottom): sulphate (SO42−), chloride (Cl−) and bis(trifluoromethanesulfonyl)imide (NTf2−).
Figure 2.
(a) Distribution coefficients of U of quaternary ammoniums mixtures carrying same cation and different anions (a–c) or different cations (d). The initial aqueous solution contains 0.1 mol·L−1 sulfuric acid (pH = 1), 0.1 mol·L−1 ammonium sulphate, 250 ppm U (VI) and 250 ppm Fe (III). A/O = 2. Distribution coefficients of U measured in the same conditions for the reference system TOA-dodecane are presented in dashed orange line on all graphs. Composition of Aliquat 336 is given in supplementary materials. Molar ratios higher than 50 % are not presented in Figure 2b because the mixtures were not liquid/soluble.
Figure 2.
(a) Distribution coefficients of U of quaternary ammoniums mixtures carrying same cation and different anions (a–c) or different cations (d). The initial aqueous solution contains 0.1 mol·L−1 sulfuric acid (pH = 1), 0.1 mol·L−1 ammonium sulphate, 250 ppm U (VI) and 250 ppm Fe (III). A/O = 2. Distribution coefficients of U measured in the same conditions for the reference system TOA-dodecane are presented in dashed orange line on all graphs. Composition of Aliquat 336 is given in supplementary materials. Molar ratios higher than 50 % are not presented in Figure 2b because the mixtures were not liquid/soluble.
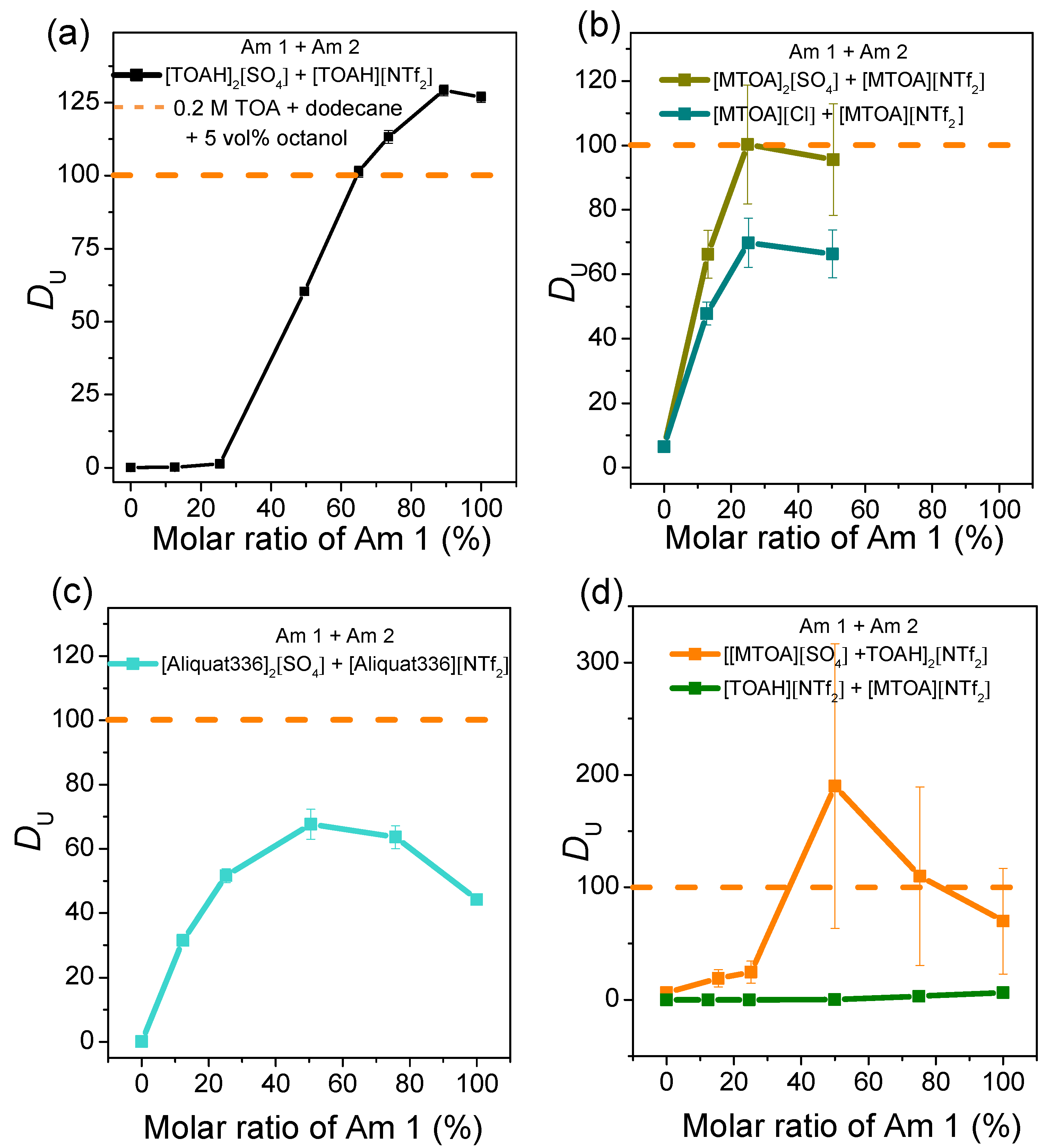
Figure 3.
(a) Uranium distribution coefficient, (b) concentration of water in the organic phase after contact and (c) concentration of acid in the aqueous phase after contact, measured for each mixture [TOAH]2[SO4] + [TOAH][NTf2] for 2 concentrations [(NH4)2SO4] = 0.1 and 1 mol·L−1 in the aqueous phase. A/O = 2. Composition of aqueous phase: 250 ppm U (VI), 250 ppm Fe (III), 0.1 mol·L−1 H2SO4.
Figure 3.
(a) Uranium distribution coefficient, (b) concentration of water in the organic phase after contact and (c) concentration of acid in the aqueous phase after contact, measured for each mixture [TOAH]2[SO4] + [TOAH][NTf2] for 2 concentrations [(NH4)2SO4] = 0.1 and 1 mol·L−1 in the aqueous phase. A/O = 2. Composition of aqueous phase: 250 ppm U (VI), 250 ppm Fe (III), 0.1 mol·L−1 H2SO4.
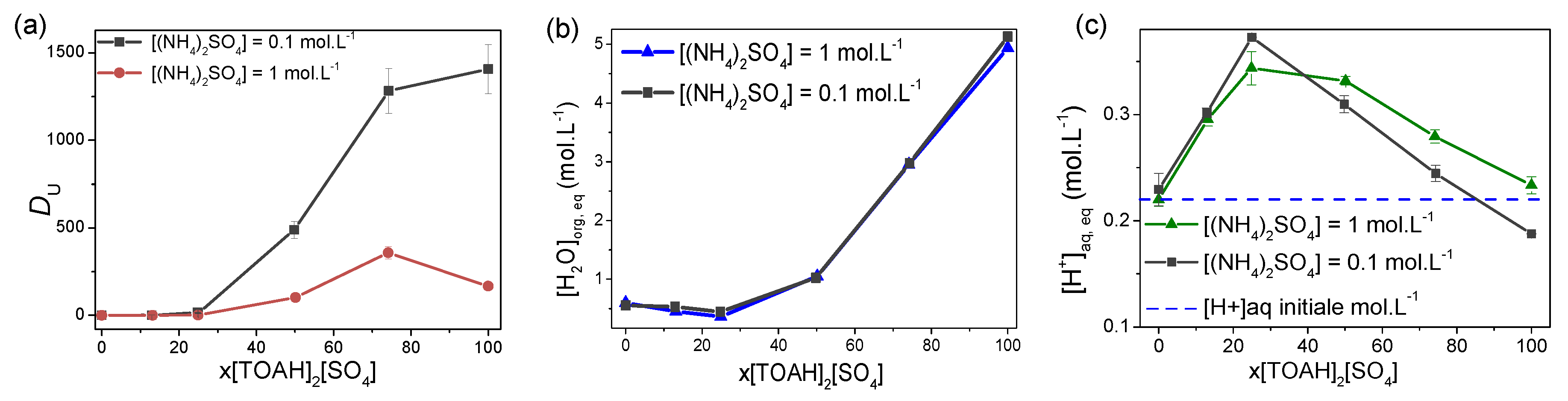
Figure 4.
(a) 19F-NMR spectrum and (b) FT-IR spectra of the mixture x[TOAH]2[SO4] = 50 %. Black before and red after contact with an aqueous phase containing 2500 ppm U(VI), 0.1 mol·L−1 sulfuric acid and 1 mol·L−1 ammonium sulphates. A/O = 4.
Figure 4.
(a) 19F-NMR spectrum and (b) FT-IR spectra of the mixture x[TOAH]2[SO4] = 50 %. Black before and red after contact with an aqueous phase containing 2500 ppm U(VI), 0.1 mol·L−1 sulfuric acid and 1 mol·L−1 ammonium sulphates. A/O = 4.
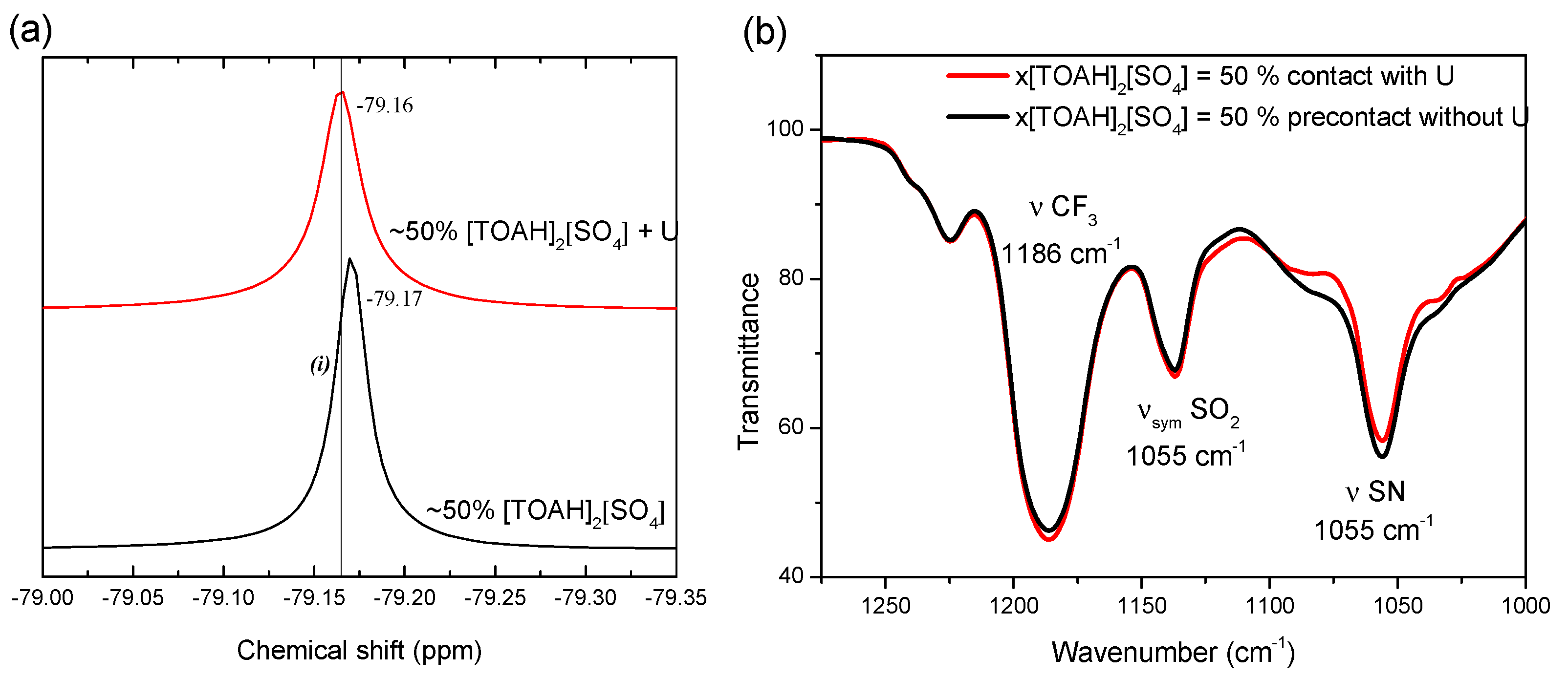
Figure 5.
(a) Raw UV-vis data for uranyl in the mixtures [TOAH]2[SO4] + [TOAH][NTf2] (x[TOAH]2[SO4]= 25 to 100%) and in the reference medium TOA + dodecane. (b) Experimental (solid lines) and fitted (dotted lines) EXAFS spectra: k3-weighted Fourier transforms of the 3-sulphate bidentate model around uranyl. The orange line represents the reference system, the blue, green and red lines the compositions x[TOAH]2[SO4] = 25, 50 and 100 % respectively. (b) Structure of uranium sulphate, the different contributions to the EXAFS spectra are indicated by red dotted lines.
Figure 5.
(a) Raw UV-vis data for uranyl in the mixtures [TOAH]2[SO4] + [TOAH][NTf2] (x[TOAH]2[SO4]= 25 to 100%) and in the reference medium TOA + dodecane. (b) Experimental (solid lines) and fitted (dotted lines) EXAFS spectra: k3-weighted Fourier transforms of the 3-sulphate bidentate model around uranyl. The orange line represents the reference system, the blue, green and red lines the compositions x[TOAH]2[SO4] = 25, 50 and 100 % respectively. (b) Structure of uranium sulphate, the different contributions to the EXAFS spectra are indicated by red dotted lines.
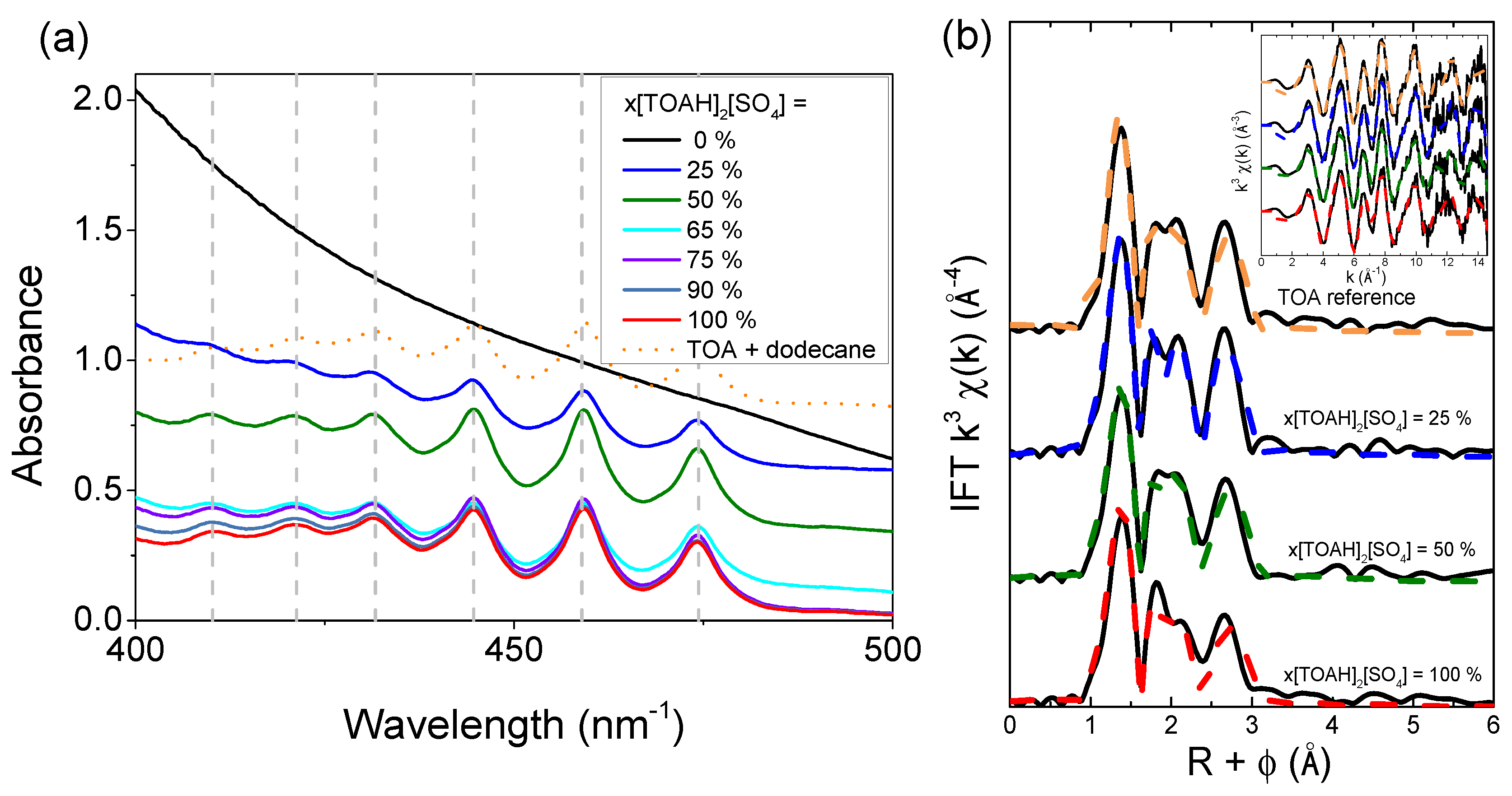
Figure 6.
Evolution of (a) R-factor and (b) Debye-Waller-factor accounting for disorder.
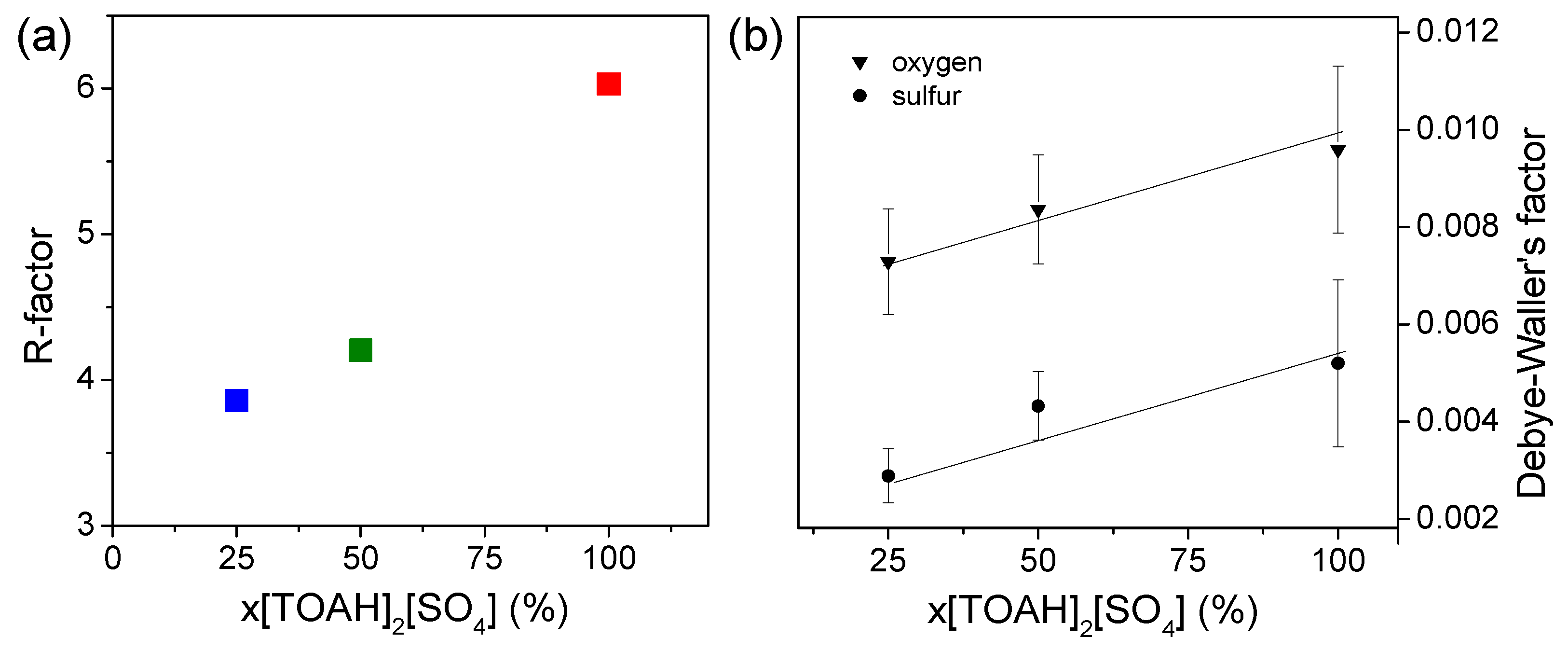
Figure 7.
FT-IR spectra between 800 and 1000 cm−1 of the mixtures [TOAH]2[SO4] + [TOAH][NTf2] after contact with an aqueous phase containing 2500 ppm U(VI), 0.1 mol·L−1 sulfuric acid and 1 mol·L−1 ammonium sulphate. A/O = 4.
Figure 7.
FT-IR spectra between 800 and 1000 cm−1 of the mixtures [TOAH]2[SO4] + [TOAH][NTf2] after contact with an aqueous phase containing 2500 ppm U(VI), 0.1 mol·L−1 sulfuric acid and 1 mol·L−1 ammonium sulphate. A/O = 4.
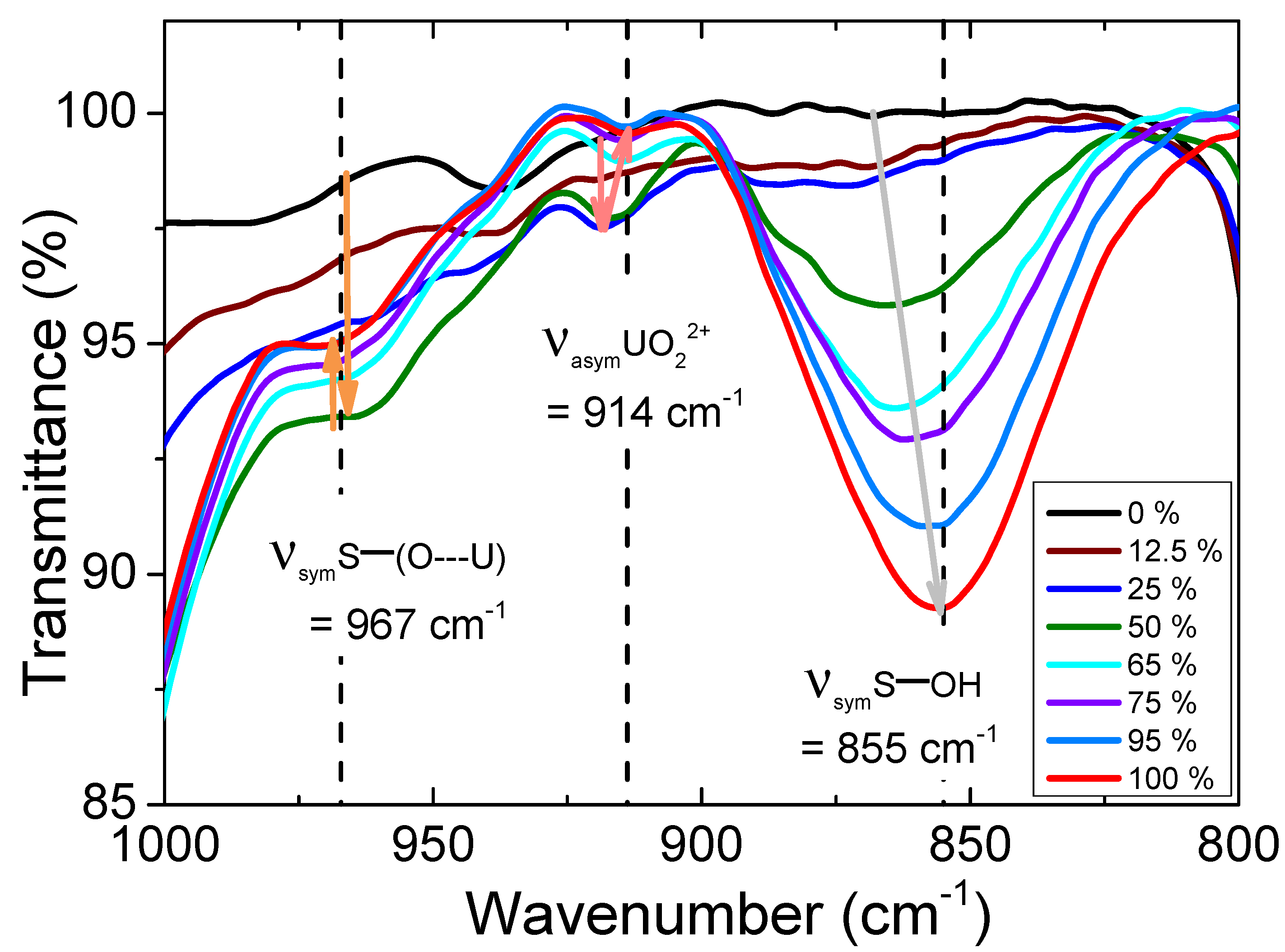
Figure 8.
Integration of the characteristic bands of (a) ν (S-OH), (b) ν (UO22+) and (c) ν (S-(O...U)2) as a function of x[TOAH]2[SO4].
Figure 8.
Integration of the characteristic bands of (a) ν (S-OH), (b) ν (UO22+) and (c) ν (S-(O...U)2) as a function of x[TOAH]2[SO4].
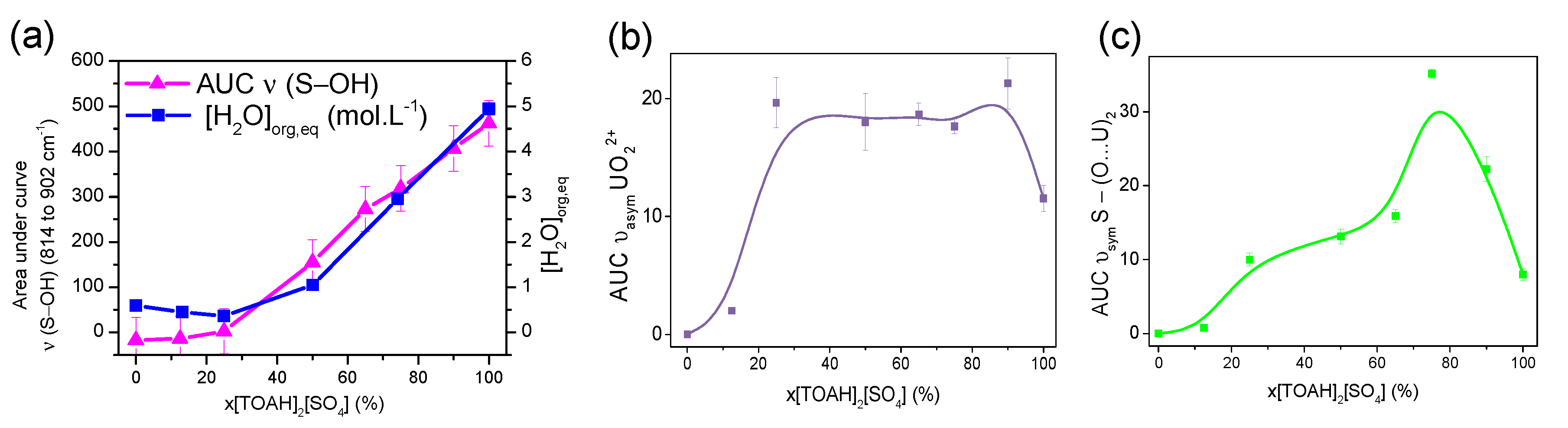

Table 1.
Synergistic peak position and distribution coefficient of uranium (data extracted from Figure 2).
Table 1.
Synergistic peak position and distribution coefficient of uranium (data extracted from Figure 2).
| [MTOA]2[SO4] + [MTOA][NTf2] | [MTOA][Cl] + [MTOA][NTf2] | [Aliquat 336]2[SO4] + [Aliquat 336][NTf2] | [MTOA][NTf2] + [TOAH]2[SO4] | [TOAH]2[SO4] + [TOAH][NTf2] | |
|---|---|---|---|---|---|
| Ratio (%) | 25 | 25 | 50 | 50 | 90 |
| DU | 100 | 70 | 78 | 196 | 128 |
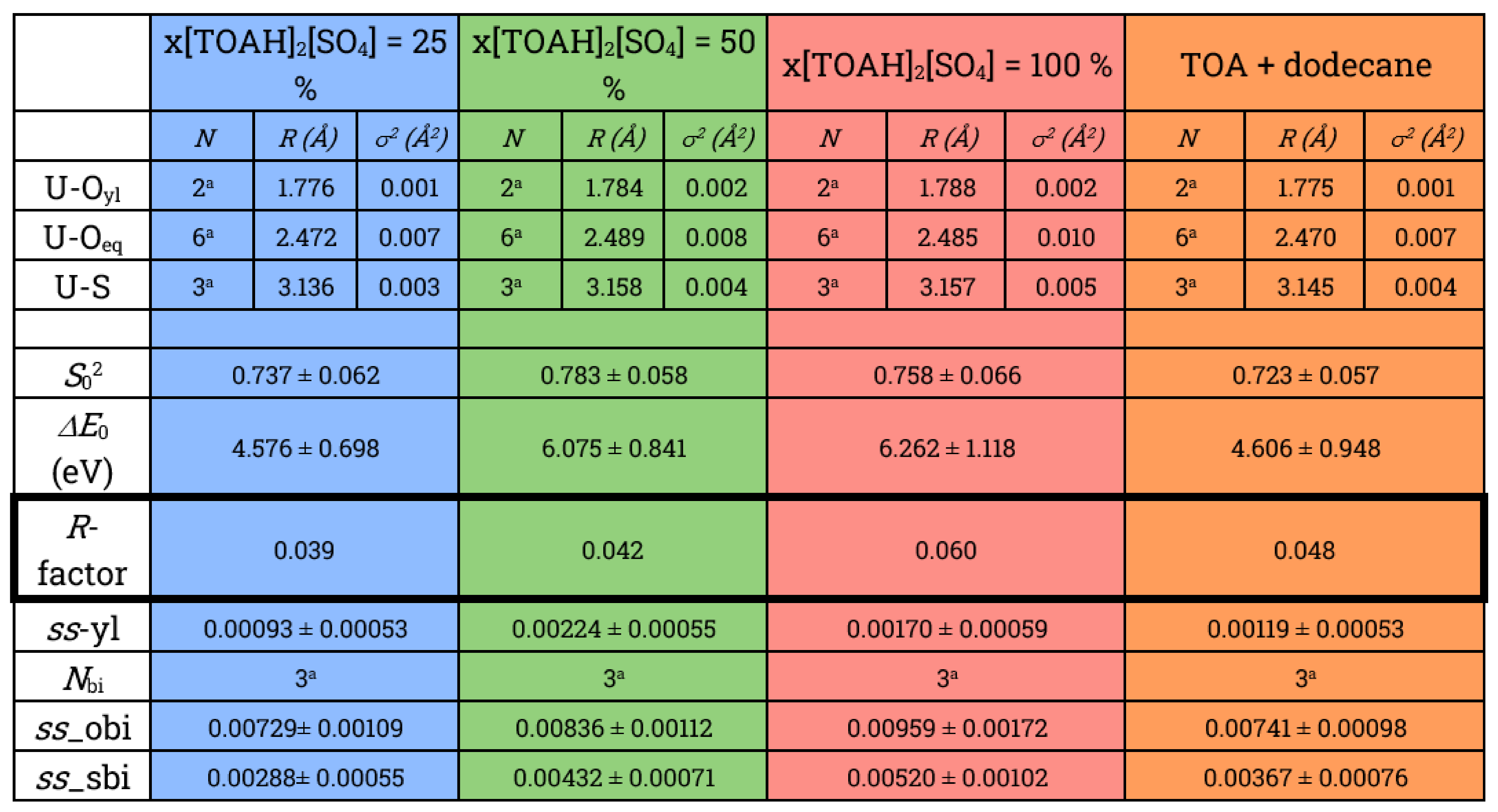
Table 2.
Parameters of the best fit obtained for EXAFS spectra of [UO2(SO4)3]4− complexes. (σ2 is the Debye-Waller factor accounting for disorder, ΔE0 is the energy fit, S02 is the amplitude factor, R-factor is the tuning factor.
Table 2.
Parameters of the best fit obtained for EXAFS spectra of [UO2(SO4)3]4− complexes. (σ2 is the Debye-Waller factor accounting for disorder, ΔE0 is the energy fit, S02 is the amplitude factor, R-factor is the tuning factor.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
supplementary.docx (543.09KB )
Submitted:
31 July 2023
Posted:
02 August 2023
You are already at the latest version
Alerts
A peer-reviewed article of this preprint also exists.
supplementary.docx (543.09KB )
This version is not peer-reviewed
Submitted:
31 July 2023
Posted:
02 August 2023
You are already at the latest version
Alerts
Abstract
This study investigates uranium solvent extraction under AMEX process conditions. The use of pure extractants without diluents or phase modifiers allows not only to reduce the use of volatile organic compounds, but also to provide higher extraction yields without third phase formation. Pure extractants are protonated amines or quaternary ammoniums with suitable counter ion, which act at the interface between ion pairs and protic ionic liquids. The mixture of sulfates anion (SO42-) and bis(trifluoromethanesulfonyl)imide anion (NTf2-) revealed unexpected nonlinear extraction behaviors, which appear highly important to rationalize for optimized application. Spectroscopic analysis (NMR, UV-vis, FT-IR, and EXAFS) showed that uranium extraction occurs via protonated amine and three sulphates. Nonlinear extraction could further be interpreted by considering water and acid transfer between the two phases: at lower sulphate ratios, the release of acid from the organic phase into the aqueous phase was shown to influence the number of protonated amines in the organic phase, affecting uranium extraction before its enhancement. Furthermore, the extraction loss at higher sulphate ratios, was assigned to the destabilization of bidentate uranium-sulphate complexes due to a competition between water and sulphates.
Keywords:
Subject: Chemistry and Materials Science - Analytical Chemistry
1. Introduction
Industrial plants use large volumes of organic diluents (usually kerosene) for solvent extraction in mixer-settler. AMine EXtraction (AMEX) process is based on mixtures of alkyl amines in aliphatic diluent with fatty alcohol [1]. Despites its extensive application for uranium purification in the front end of the nuclear fuel cycle, this process is still facing several problems. Non-ideal uranium separation, requirement of a phase modifier (fatty alcohol) to prevent third phase formation and possible extractant degradation due to the presence of vanadium and phase modifier [2,3,4].
In particular, the Somaïr uranium extraction and purification plant in Niger has a stock of around 900 m3 of solvent, 75 % of which is replaced each year due to formulation degradation (crud formation), loss through entrainment of the organic phase in the aqueous phase, or evaporation.
Numerous alternatives are currently being studied to avoid the use of volatile organic compounds for metal separation. For example, solid-liquid extraction [5] supercritical extraction [6] and flotation [7] have been proposed. However, they remain difficult to apply to uranium extraction, as they require a complete rethink of the industrial process and facilities. Interesting alternatives also involve a simple replacement of organic solvents with non-volatile solvents such as ionic liquids (ILs) or deep eutectic solvents [8,9].
Under AMEX process conditions [1,10] it has been demonstrated and patented that the use of pure extractant (trialkylamines), without diluent and phase modifier enables higher extraction yields without third phase formation [11]. Pure extractants are quaternary ammoniums or trialkylamines previously protonated, and associated with a counter anion. These pure amines and quaternary ammoniums actually lie at the interface between amine (ion pair) and protic ionic liquid (quaternary ammonium) chemistry and have already been used as ionic liquids for liquid-liquid extraction [12,13,14]. Such ionic liquids are often used as diluents to increase the extraction capacity of a ligand, but they are more than a plain diluent. They modify the extraction mechanism through the participation of their anion or cation in the metal complex, or by inducing a different organization of the organic phase [15,16,17]. Many studies have been dedicated to the mechanisms of solvent extraction in ILs. Billard et al. [18] proposed two equilibria (cationic and anionic exchanges) to summarize the possible ion exchanges mechanisms when ILs are used as diluents. It was found out that ion exchange is the dominant mechanism for uranium and actinides extraction.
A wide range of ILs have also been used as extractants in aliphatic diluents to which phase modifiers are sometimes added [19,20,21,22]. Depending on the anions and cations they contain, ILs can be highly hydrophobic and present a sufficiently low-viscosity at room temperature to be used both as an extraction agent and as a hydrophobic phase. In this case, dilution in an aliphatic diluent is not necessary. Extraction performance and selectivity is usually improved compared to conventional solvents [23,24,25,26,27].
Mixtures of ILs may also be applied as they generally combine the properties of two different ILs. For instance, a "diluting" IL, with advantageous transport properties (low viscosity and high conductivity), can be mixed with a "complexing" IL whose transport properties are prohibitive for use at low temperatures [28,29,30].
In the present study, diluent free quaternary ammoniums or trialkylamines and their mixtures were assessed in the condition on the AMEX process. Topological formula of involved anion and cation are given in Figure 1 where TOAH+ stands for trioctylammonium, MTOA+ for methyltrioctylammonium and where the commercial Aliquat 336+ is composed of a mixture of MTOA+ and methyltridecylammonium (MTDA+). Sulphate (SO42−), chloride (Cl−) and bis(trifluoromethanesulfonyl)imide (NTf2−) were chosen to evaluate the anion effect on uranium extraction.
The various tested mixtures provided high extraction efficiency with nonlinear properties. A mechanistic studies is proposed to elucidate the uranyl extraction mechanisms of the [TOAH]2[SO4] + [TOAH]2[NTf2] mixture. As it might be very interesting to design mixtures with optimized extraction properties, origin of these non-linearities has been investigated in details in this study. Coordination of uranyl was evaluated by NMR, UV-vis and EXAFS spectroscopy. Considering water and acid transfer between the two phase, FT-IR spectroscopy was further exploited to elucidate the various regimes of extraction of this mixture.
2. Materials and Methods
2.1. Chemicals and reagents
Trioctylamine (TOA) 98 %, sulphuric acid 96 % and ammonium sulphate were purchased from Sigma-Aldrich. Aliquat 336 and lithium bis(trifluoromethanesulfonyl)imide (LiNTf2) were supplied by Alfa Aesar and ABCR respectively. Methyltrioctylammonium chloride ([MTOA][Cl]) and methyltrioctylammonium bis(trifluoromethanesulfonyl)imide ([MTOA][NTf2]) were purchased from ABCR. All chemicals were used as recieved, without any further purification.
2.2. Instrumentation and analysis
Metal concentration was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES Spectro Arcos - AMETEK Materials Analysis, ThermoFisher Scientific). Prior to concentration measurements, aliquots of uranium and iron stock and working solutions were diluted with a v:v 0.99% nitric acid solution in order to bring the concentration in the 1-15 ppm range. Metal quantification was performed at several wavelengths. Uranium was detected at 393.203 nm, 393.203 nm, 385.466 nm and 385.958 nm; iron was detected at 239.562 nm, 240.488 nm and 259.940 nm. Calibration samples were prepared from 1000 mg·L−1 uranium and iron, standards (SCP Science PlasmaCal). Uncertainties in metal concentrations were determined statistically by repeated measurements.
The amount of extracted acid was determined by acid-base titration using a Metrohm 905 titrando automatic potentiometer. A sample of aqueous phase was taken and dosed with a 0.01 mol·L−1 NaOH solution. The concentration of sulfuric acid in the organic phases was given by subtracting the acidity of the aqueous phase measured at equilibrium from that of the initial aqueous phase. The water present in the organic phases was determined by the Karl Fischer method, using a Metrohm coulometer.
The speciation of uranium complexes in ionic liquid media was achieved by ultra violet-visible and Fourier transform infrared spectroscopies (UV-vis and FT-IR). UV-vis spectra were recorded between 200 and 800 nm on a dual-beam Varian Cary 5000 spectrophotometer in plastic cells with a 2 mm optical path. Absorbances of contacted, pre-contacted and non-contacted samples were measured using an empty cell in the second beam as a reference. Subtractions were calculated in post-processing. At a given wavelength λ, the absorbance A of a mixture of n absorbing species is the sum of the individual absorbance. The signal from species formed by water and acid can therefore be subtracted to highlight uranium-related signals. Infrared spectra of ionic liquid phases were recorded on a Bruker Vertex 70 FT-IR spectrometer equipped with a diamond ATR. A blank was run on air before each measurement. The Fourier transform was calculated using Perkin Elmer’s Spectrum software, then the background noise was subtracted. The measurement range extends from 4000 to 615 cm−1. Many absorption bands were found in the printed part of the spectrum of interest in this study. The pre-contacted sample signal was subtracted to highlight the uranium-related absorption bands. A Voigt function was used to best fit the signal and calculate the area under the curves of the highlighted spectra.
X-ray absorption spectroscopy (Extended X-Ray Absorption Fine Structure-EXAFS) was carried out on the MARS (Multi-Analyses on Radioactive Samples) light line at the SOLEIL synchrotron (Saint-Aubin, France) (proposal ID: 20210696) [31,32]. Spectra were recorded at the uranium LIII threshold (17167 eV). Calibrations were carried out at the yttrium K threshold (17039 eV).
In practice, samples were placed in double-confinement EXAFS cells, enabling active samples to be measured. The cells can hold a maximum of 12 samples. The volume of each sample is 0.25 mL.
Data were processed using the Athena program. EXAFS oscillations are extracted in several steps. The ionization energy threshold E0 is defined by taking the maximum of the derivative at the threshold jump. After signal normalization using linear and cubic functions, the signal can be converted from E (eV) energy space to k (Å−1) wavenumber space. To increase the amplitude of oscillations observed at high wavenumbers, a k² or k3 factor can be applied. Once the oscillations have been extracted, conversion to real distance space R (Å) is performed by applying a Fourier transform to the EXAFS k3 signal, χ(k) over a defined wavenumber range (in the remainder of this study, this range is 3.75 < k < 14). Finally, the pseudo-radial distribution function is obtained as a function of R. This pseudo-radial distribution function schematically represents the probability of atoms being present around the absorber atom as a function of their distance increased by a phase shift φ. It is not the actual distance R from the absorber atom. Fits were then made in the Artemis software using model structure compounds (references found in the literature where available + FEFF software). The discrepancy between the fitted data and the experimentally found data can be expressed by calculating the R factor based on the method of least squares.
Fluorine-19 nuclear magnetic resonance spectroscopy (19F-NMR) was also used to study the impact of metal extraction on the chemical shift of the fluorine function of NTf2. Double-walled tubes were used so as not to alter the chemistry of the organic phase, and so that a reference solvent (deuterated DMSO) could be applied externally.
2.3. Extraction experiments
Several organic extractants based on protonated tertiary amines or quaternary ammoniums and their mixtures were evaluated in this work for the extraction of uranium in sulfuric media. Their synthesis is described in ESI 1.
Organic phases were prepared by mixing protonated tertiary amines or quaternary ammoniums (defined as Am 1 and Am 2) in different proportions. Samples were classified according to their molar ratio of Am 1, xAm 1, defined as follows:
The aqueous phases used in this study were prepared by dissolving the uranium sulphate, UO2SO4⋅6.38H2O, M = 479.43 g·mol−1 and iron sulphate, Fe2(SO4)3⋅8.3H2O, M = 549.46 g·mol−1.
The acidity of the solution was controlled by adding sulfuric acid (v:v H2SO4 96%, M = 98.08 g·mol−1) and the sulphate concentration was increased by adding ammonium sulphate ((NH4)2SO4, M = 132.14 g·mol−1).
The aqueous and organic phases are brought into contact in eppendorf tubes of appropriate volume and stirred (500 rpm) for 1 hour at 20 °C in a thermostatically-controlled cell (Infor-ht® ecotron). The phases are then separated after a centrifugation step (Rotina 380R) lasting between 5 and 20 min. at a rotation speed of 8000 rpm. The phases are then separated in 2 different flasks before sampling for analysis.
where stands for the sum of the chemical activities of a solute M in all its forms in the organic phase and , for the sum of the chemical activities of the same solute in all its forms in the aqueous phase. In dilute media, activity is equal to molar concentration
3. Results and discussion
Several commercial quaternary ammonium ligands and their mixtures were evaluated for uranium extraction from sulphuric media. Their performance were compared with the classical trioctylamine extractant (taken as reference for the AMEX process) solubilised in dodecane in presence of 1-octanol as phase modifier. The evaluated cations and anions are illustrated in Figure 1. Distribution ratios of uranium obtained with these ammoniums salts and mixtures are displayed in Figure 2.
Results show that uranium extraction is not only driven by the nature of cations and anions but also by the ratio between them. When similar cation are considered, systems with SO42− extract better than with Cl− (see Figure 2b). Whatever the cation, no extraction occurred when NTf2− was used as anion (see left side of Figure 2a–d and green line in Figure 2d). Uranium extraction increases with sulphates concentrations, which is consistent with the recognized extraction mechanisms for such systems: as demonstrated by Sukhbaatar et al., [33] uranium extraction from sulphuric media requires the formation of three sulphates uranyl complexes.
Looking at the effect of the cations, Figure 2 shows moreover that TOAH+ extracts more uranium than MTOA+ and than the Aliquat 336 when the counter anion is a sulphate (right side of Figure 2a–c).
Interestingly, uranium extraction is not linear with the molar ratio of one quaternary ammonium to another, but shows non-linearities with a synergistic peak when mixtures of anions are involved (see Table 1 for position of the synergistic peaks).
Overall, nonlinear extraction is observed when the cations are identical and when the anion is modified. In contrast, when the anion is identical and the cation is modified, linear extraction is obtained. We can therefore conclude that the non-linearities observed are due to anion mixing and not cation mixing.
As it might be very interesting to design mixtures with optimized extraction properties, origin of these non-linearities has been investigated in more details for the mixture [TOAH]2[SO4] and [TOAH][NTf2]. This peculiar system is interesting as it is the most efficient one toward uranium extraction, but also because its extraction non-linearities show three distinct regimes (Figure 2a): extraction is initially antagonistic (lower than the linear mixture), then synergistically increased, and finally decreased for the higher sulphate ratios. Extraction mechanisms of the [TOAH]2[SO4] + [TOAH]2[NTf2] mixture were therefore investigated with a spectroscopic study combined with a detailed analysis of the sulphate effect on uranium, acid and water transfer between the organic and aqueous phases.
3.1. Effect of the amount of sulphates in the aqueous phase
Like sulphates in the organic phase, the amount of sulphates in the aqueous phase may affect uranium extraction. In order to evaluate this effect, the sulphate concentration in the aqueous phase was varied by changing the concentration of ammonium sulphate. Effects on uranium extraction, acid and water transfer are presented in Figure 3 for sulphate concentrations of [(NH4)2SO4] = 0.1 and 1 mol·L−1 in the aqueous phase.
Figure 3a shows that increasing sulphate concentration in the aqueous phase decreases uranium extraction for all x[TOAH]2[SO4] ratios. It is also interesting to note that the decrease in DU between 75 and 100 % sulphate is most marked for [(NH4)2SO4] = 1 mol·L−1. Water concentration (Figure 3b) is unaffected by the amount of ammonium sulphate, while acid transfer between the two phases is little affected (Figure 3c). The acid transfer is actually unexpected. Instead of being extracted in the organic phase with the metal (as it is usually the case in solvent extraction), acid is released from the organic toward the aqueous phase. This acid release is assigned to the deprotonation of the TOAH+ originally present in the organic phase. It appears to be poorly affected by the (NH4)2SO4 concentration for the low x[TOAH]2[SO4] ratios, while it is slightly increased above x[TOAH]2[SO4] = 50 %.
This experiment shows therefore that uranium extraction is both dependent on the amount of sulphate present in the aqueous and in the organic phase. Most importantly, it shows also that the decrease in uranium extraction at high x[TOAH]2[SO4] ratios is concomitant with an increase in the release of H+ protons in the aqueous phase (expected to decrease the concentration of protonated TOA in the organic phase), as well as with a strong increase in water extraction in the organic phase.
To further understand the nonlinear uranium extraction with the x[TOAH]2[SO4] ratios, the remainder of this paper is devoted to characterizing the uranyl complexes extracted in the organic phase. It is indeed important to evaluate if uranium is chelated as in the conventional medium (TOA in dodecane) or if the NTf2 anion is involved to form mixed complexes for some x[TOAH]2[SO4] ratios.
3.2. Characterizing the extracted species
3.1.1. NTf2 participation in the uranium complex
To obtain information about the chemical environment of NTf2− in the binary mixtures before and after uranium extraction, 19F-NMR and FT-IR measurements were carried out. Note that NTf2− gives only one resonance band in the 19F-NMR spectra. Its evolution provides information on the environmental change of NTf2−.19F-NMR spectra of [TOAH]2[SO4] + [TOAH][NTf2] mixtures are plotted in Figure 4a for x[TOAH]2[SO4] = 50%. It shows that the chemical shift for NTf2− is not modified before and after uranium extraction, which indicates that NTf2− does not directly coordinate uranyl.
This conclusion is reinforced by a Fourier transform infra-red (FT-IR) analysis shown in Figure 4b. It can be observed that the characteristic bands of NTf2 [34] are not affected by the presence of uranium. NTf2 therefore does not participate directly in the complex with uranium, and the extraction non-linearities of the mixture cannot be explained by the formation of a synergistic or antagonistic mixed complex of uranium with sulphates and NTf2.
3.1.2. UV-vis spectroscopy and EXAFS, study of first neighbors
To determine whether the origin of the nonlinear extraction is due to uranium speciation, UV-vis uranium spectroscopy and X-ray absorption spectroscopy (EXAFS) measurements were performed for various x[TOAH]2[SO4] ratios.
UV-vis spectra recorded after contact with a uranium solution (Figure 5a), show the same type of fingerprint signal for 25 to 100 % ratios as for the conventional medium (TOA + dodecane + octanol) shown in dotted orange line. The different trend observed for the ratio 0% is due to the absence of uranium in this sample. As demonstrated by Servaes et al. who studied the speciation of uranyl nitrate complexes in acetonitrile and ionic liquid [35], this fingerprint is characteristic of a trigonal symmetric uranium complex. In their study, spectra of UO2(NO3)3− complexes in acetonitrile or in [C4mim][NTf2] show signals similar to those observed for UO2(CO3)34− and UO2(CH3COO3)3 complexes [36]. By comparing the experimental spectra with those reported in the literature, it can therefore be concluded that for all x[TOAH]2[SO4] ratios, the uranyl sulphate complex exhibits D3h trigonal symmetry, suggesting that uranyl is complexed with 3 sulphates in an ionic liquid medium. EXAFS spectroscopy measurements were carried out to confirm this hypothesis.
Figure 5b displays EXAFS spectra measured at the L3 threshold of uranium and the corresponding Fourier transforms. No EXAFS spectra could be measured for x[TOAH]2[SO4] ratios lower than 25 % as the uranium concentrations was too small in these samples. For the higher x[TOAH]2[SO4] ratios, it shows that experimental spectra (solid lines) obtained with the [TOAH]2[SO4] + [TOAH][NTf2] mixtures are very similar to the one measured in conventional media (orange spectrum, TOA + dodecane). It can also be noted that in addition to the first U-Oyl contribution of linear transdioxo bonds (R + φ = 1.2 Å) and that of the equatorial U-Oeq oxygen coordination layer (R + φ = 1.8 to 2.4 Å), the spectra show intense contributions in the second coordination sphere (R + φ > 2.5 Å) due to sulfur atoms of sulphate anions coordinated with UO22+ [33,37]
In a previous study carried out in a conventional medium (TOA + dodecane), coupling a classical fit with molecular dynamics simulations enabled to unravel the speciation of uranium in the organic phase: the complex consists of three sulphate anions coordinated to uranyl via U-O bonds in the first coordination sphere [33]. The study also showed that uranyl trisulphate complexes shift from a 3 bidentate sulphate configuration to a 2 bidentate and 1 monodentate configuration, with the most favorable configuration remaining the 3 bidentate sulphate configuration.
In order to probe more precisely the local structure of uranyl in [TOAH]2[SO4] + [TOAH][NTf2] mixtures, a fit of the EXAFS spectra was performed by applying a structural model of three bidentate sulphates in the first uranium sphere. The fitted parameters are presented in Table 2 and the corresponding fitted spectra are plotted as dotted lines in Figure 5b.
Good fits are obtained for all the x[TOAH]2[SO4] ratios, confirming the structure with three bidentate sulphates. As expected from the qualitative study of the data, U-Oyl, U-Oeq and U-S bond lengths do not vary. However, although this does not lead to outliers, the quality of fit deteriorates for the higher x[TOAH]2[SO4] ratios: the R factor increases from 3.9 % to 6.0 % (see Figure 6a). Furthermore, as shown in Figure 6b, the Debye-Waller factor increases suggesting more disorder in the complexes. The evolution of these two parameters indicates that the U-3 sulphate complex is destabilized, which could explain a loss of uranium extraction for the high x[TOAH]2[SO4] ratios.
In conclusion, 19F-NMR, UV-vis and EXAFS data indicate that there is no mixed complexes formed with sulphates and NTf2 in the first coordination sphere. However, destabilization of the bidentate sulphate complex is observed when the x[TOAH]2[SO4] ratio reaches 100 %, which may be related to the decrease of uranium extraction observed for these high ratios.
As extracted water is very important at these ratios, it would have been interesting to evaluate its contribution to the complexes. However, it was not possible to include it distinctly in the fits, as the signatures of the monodentate U-SO4 and U-H2O bonds are too similar (same distance, same properties). EXAFS therefore does not allow us to conclude if water competes or not with uranium sulphate.
3.1.3. FT-IR spectroscopy, looking at the second coordination sphere and beyond
Fourier transform infrared (FT-IR) spectra of protonated tertiary amines mixtures are presented in Figure 7 and ESI 2 and 3. Three main bands can be distinguished in the region 800-1000 cm−1. Identification of resonance bands has been performed according to results proposed in the literature based on a IR study dedicated to the structure of tridecylammonium sulphate in presence of uranium [38]. The three bands were therefore assigned to monodentate water-bound sulphates ν (S-OH) at 855 cm−1, to the uranyl motif ν (UO22+) at 914 cm−1 and to bidentate uranium-bound sulphates ν (S-(O...U)2) at 967 cm−1.
A linear shift of the ν(S-OH) absorption band towards the lower frequencies can be observed with the molar ratio of sulphate. It indicates that sulphate becomes increasingly free (see ESI 4). The area under these ν(S-OH) band curve was plotted in Figure 8a. It shows an increase after the ratio x[TOAH]2[SO4] = 30% that follows the trend of the water extraction profile (see Figure 3b). This first observation suggests that the sulphate are not only involved in the uranyl complexes but also more and more connected with water which concentration is significantly increasing for the high x[TOAH]2[SO4] ratios.
To highlight the bands related to uranium, it was necessary to subtract the signal of the pre-contacted samples. After subtraction, each band of interest was fitted with a Voigt function. The area under the curve (AUC) is plotted in Figure 8b,c as a function of x[TOAH]2[SO4] for the ν (UO22+) and ν (S-(O...U)2) bands.
It is interesting to note that the area under the bands attributed to ν (UO22+) and ν (S-(O...U)2) bonds follows a similar trend to that of the uranium extraction profile (plotted in Figure 2a): it is initially constant up to x[TOAH]2[SO4] = 12.5%, then intensifies up to x[TOAH]2[SO4] = 75 %, and decreases thereafter.
This infrared study indicates therefore that for x[TOAH]2[SO4] ratios above 75 %, the uranium-sulphate bond is less present, to the benefit of the sulphate-water bond, suggesting a competition between the sulphate-uranium and sulphate-water complexes.
To interpret the overall uranium extraction profile of the [TOAH]2[SO4] + [TOAH][NTf2] mixture (Figure 2a), it is necessary to take into account all the data obtained by the spectroscopic methods, as well as the water extraction data and the acid release from the organic to the aqueous phase (Figure 3b,c). Spectroscopic data indicates that the NTf2 anion is not involved in the extraction of uranium. As in the conventional medium (TOA + dodecane), uranium is extracted by TOAH+ protonated amines and three bidendate sulphates in the first coordination sphere.
For low x[TOAH]2[SO4] ratios of 0-25 %, uranium extraction does not increase, while the amount of sulphates introduced into the organic phase does. This effect can be explained by the increased release of H+ protons from the organic phase into the aqueous phase. This release reduces the number of protonated amines available, inhibiting uranium extraction.
From x[TOAH]2[SO4] = 25 %, proton release stabilizes and then decreases, leading to an increase in the number of TOAH+ protonated amines available. This increase in TOAH+ accompanied by an increase in sulphates with x[TOAH]2[SO4] is therefore consistent with the very sharp increase in uranium extraction between x[TOAH]2[SO4] = 25 and 75 %.
Beyond this ratio, the very sharp increase in extracted water leads, as previously suggested, to a competition between the sulphate-uranium and sulphate-water complexes, which destabilizes the uranyl tri-sulphate complexes and leads to saturation, then to a decrease in uranium extraction.
This study shows that the main elements responsible for uranium extraction are sulphates and protonated amines, and that the availability of the latter is regulated by the concentration of extracted water, as well as by the release of acidic protons from the organic phase to the aqueous phase.
A deeper understanding of these phenomena, would require to interprete the nonlinear behavior of water extraction and the origin of H+ proton release. The increased extraction of water can be partially explained by the difference in hydrophilicity between NTf2− and SO42− anions, but its nonlinear extraction might be related to the activity of the aqueous phase and to the chemical potential equilibria between the species present in the aqueous and organic phases. The chemical potential equilibrium may also be responsible for the release of protons from the organic phase to the aqueous phase. These equilibria are however very difficult to write down and predict.
4. Conclusions
Several organic extractant based on protonated tertiary amines or quaternary ammoniums and their mixtures were evaluated for the extraction of uranium in the AMEX process conditions. Irrespective of the leachate and the cation used, non-linearities in uranium extraction were observed as a function of the sulphate ratio in the organic phase. This effect, which allows obtaining very high extraction efficiency for some mixtures, can be advantageously considered for optimized uranium extraction or when physico-chemical properties (as viscosity or density) of the final solvent need to be adjusted. Origin of these non-linearities were therefore investigated with a mechanistic study.
An extensive analysis of the amounts of extracted water, acid and uranium combined with detailed spectroscopic studies (NMR, UV-vis, FT-IR and EXAFS) of contacted organic phases was carried out on the [TOAH]2[SO4] + [TOAH][NTf2] system to understand the three regimes of extraction observed along with the sulphate ratios. Whatever the x[TOAH]2[SO4] ratio, the NTf2− anion was shown to be absent of the first coordination sphere. It was shown that uranium extraction takes place via four protonated amines and three bidendate sulphates, as in the conventional AMEX process. Analysis of EXAFS data showed moreover that the bidentate uranium-sulphate complex is destabilized for the high x[TOAH]2[SO4] ratios. Infrared spectroscopy revealed along with this destabilization, that the number of sulphate-uranium bonds decreases in favor of sulphate-water bonds. It is assumed that a competition between sulphates-water and sulphates uranyl complexes governs the extraction loss observed at high x[TOAH]2[SO4] ratios.
To understand the observed non-linearities over the whole range of x[TOAH]2[SO4] ratios, the release of acid from the organic phase into the aqueous phase as well as the nonlinear water extraction were further considered. The nonlinear extraction of uranium was finally related to the concentration of protonated amines and sulphates: (i) by influencing the number of available protonated amines in the organic phase, acid release negatively affects uranium extraction at low x[TOAH]2[SO4] ratios; (ii) in the medium x[TOAH]2[SO4] ratios, the saturation of acid release induces a strong increase of the available protonated amines leading to a strong increase of uranium extraction; (iii) at higher x[TOAH]2[SO4] ratios, the strong increase of water extraction destabilizes the uranyl-sulphate complexes because of a competition between sulphates-water and sulphates uranyl complexes.
Water extraction is therefore one of the main parameters ruling the extraction mechanisms of such systems. As it was already observed on different ionic liquid systems, it is also likely to govern physicochemical properties as density and viscosity. Finding an optimized mixture of ionic liquids for solvent extraction requires therefore to elucidate the water extraction properties of the systems.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Guerinoni E.: Investigation, data curation, writing—original draft. Dourdain S.: Writing, review. Lu Z.: Investigation, data curation. Dumas T.: EXAFS spectroscopy measurements and analysis. Arrachart G.: NMR spectroscopy measurements, review. Giusti F.: review. Solari P.L.: local contact for EXAFS spectroscopy. Pellet-Rostaing S.: Project administration, Resources, Supervision, Funding acquisition.
Funding
We acknowledge the NEEDS program as well as CEA for their funding.
Data Availability Statement
Data will be made available on request.
Acknowledgments
We acknowledge SOLEIL and beamline MARS for provision of synchrotron radiation facilities and assistance. We also thank Beatrice Baus-Lagarde for her help with ICP-OES measurements, David Lemire for his participation to the EXAFS experiment.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Crouse, D.J.; Brown, K.B. The Amex Process for Extracting Thorium Ores with Alkyl Amines. Ind. Eng. Chem. 1959, 51, 1461–1464. [Google Scholar] [CrossRef]
- Sato, T.; Watanabe, H.; Suzuki, H. Liquid-Liquid Extraction of Molybdenum(VI) from Aqueous Acid Solutions by High-Molecular Weight Amines. Solvent Extraction and Ion Exchange 1986, 4, 987–998. [Google Scholar] [CrossRef]
- Sato, T.; Watanabe, H. The Extraction of Zirconium(IV) from Sulfuric Acid Solutions by Long-Chain Alkyl Quaternary Ammonium Compound. Separation Science and Technology 1982, 17, 625–634. [Google Scholar] [CrossRef]
- Chagnes, A.; Fosse, C.; Courtaud, B.; Thiry, J.; Cote, G. Chemical degradation of trioctylamine and 1-tridecanol phase modifier in acidic sulfate media in the presence of vanadium (V). Hydrometallurgy 2011, 105, 328–333. [Google Scholar] [CrossRef]
- Solgy, M.; Taghizadeh, M.; Ghoddocynejad, D. Adsorption of uranium(VI) from sulphate solutions using Amberlite IRA-402 resin: Equilibrium, kinetics and thermodynamics study. Annals of Nuclear Energy 2015, 75, 132–138. [Google Scholar] [CrossRef]
- Kumar, P.; Pal, A.; Saxena, M.K.; Ramakumar, K.L. Supercritical fluid extraction of uranium and thorium from solid matrices. Desalination 2008, 232, 71–79. [Google Scholar] [CrossRef]
- Smolinski, T.; Wawszczak, D.; Deptula, A.; Lada, W.; Olczak, T.; Rogowski, M.; Pyszynska, M.; Chmielewski, A.G. Solvent extraction of Cu, Mo, V, and U from leach solutions of copper ore and flotation tailings. J. Radioanal. Nucl. Chem. 2017, 314, 69–75. [Google Scholar] [CrossRef]
- Quijada-Maldonado, E.; Olea, F.; Sepúlveda, R.; Castillo, J.; Cabezas, R.; Merlet, G.; Romero, J. Possibilities and challenges for ionic liquids in hydrometallurgy. Separation and Purification Technology 2020, 251, 117289. [Google Scholar] [CrossRef]
- Yan, Q.; Cai, Y.; Wang, Z.; Dong, X.; Yuan, L.; Feng, W.; Chen, J.; Xu, C. Separation of americium from lanthanide by a Task-Specific ionic liquid decorated with 2,6-Bis-Triazolyl-Pyridine moiety. Separation and Purification Technology 2022, 299, 121752. [Google Scholar] [CrossRef]
- Brown, K.B.; Coleman, C.F.; Crouse, D.J.; Denis, J.O.; Moore, J.G. THE USE OF AMINES AS EXTRACTANTS FOR URANIUM FROM ACIDIC SULFATE LIQUORS. A Preliminary Report. 1954. [Google Scholar] [CrossRef]
- Lu, Z.; Dourdain, S.; Pellet-Rostaing, S.; Arrachart, G.; Giusti, F. Mélanges de Sels d’ammonium Quaternaire Pour l’extraction de l’uranium(VI) de Solutions Aqueuses d’acide Sulfurique. 3 June 2022. [Google Scholar]
- Ghandi, K. A Review of Ionic Liquids, Their Limits and Applications. GSC 2014, 04, 44–53. [Google Scholar] [CrossRef]
- Greaves, T.L.; Drummond, C.J. Protic Ionic Liquids: Properties and Applications. Chem. Rev. 2008, 108, 206–237. [Google Scholar] [CrossRef]
- Keshapolla, D.; Srinivasarao, K.; Gardas, R.L. Influence of temperature and alkyl chain length on physicochemical properties of trihexyl- and trioctylammonium based protic ionic liquids. The Journal of Chemical Thermodynamics 2019, 133, 170–180. [Google Scholar] [CrossRef]
- Billard, I.; Ouadi, A.; Gaillard, C. Is a universal model to describe liquid–liquid extraction of cations by use of ionic liquids in reach? Dalton Transactions 2013, 42, 6203–6212. [Google Scholar] [CrossRef] [PubMed]
- Dukov, I.L.; Atanassova, M. Effect of the diluents on the synergistic solvent extraction of some lanthanides with thenoyltrifluoroacetone and quaternary ammonium salt. Hydrometallurgy 2003, 68, 89–96. [Google Scholar] [CrossRef]
- Dietz, M.L.; Dzielawa, J.A.; Laszak, I.; Young, B.A.; Jensen, M.P. Influence of solvent structural variations on the mechanism of facilitated ion transfer into room-temperature ionic liquids. Green Chemistry 2003, 5, 682–685. [Google Scholar] [CrossRef]
- Billard, I.; Ouadi, A.; Jobin, E.; Champion, J.; Gaillard, C.; Georg, S. Understanding the Extraction Mechanism in Ionic Liquids: UO22+/HNO3/TBP/C4-mimTf2N as a Case Study. Solvent Extraction and Ion Exchange 2011, 29, 577–601. [Google Scholar] [CrossRef]
- Wionczyk, B.; Apostoluk, W. Solvent extraction of chromium(III) from alkaline media with quaternary ammonium compounds. Part I. Hydrometallurgy 2004, 72, 185–193. [Google Scholar] [CrossRef]
- Rout, A.; Venkatesan, K.A.; Srinivasan, T.G.; Vasudeva Rao, P.R. Ionic liquid extractants in molecular diluents: Extraction behavior of europium (III) in quarternary ammonium-based ionic liquids. Separation and Purification Technology 2012, 95, 26–31. [Google Scholar] [CrossRef]
- Jaree, A.; Khunphakdee, N. Separation of concentrated platinum(IV) and rhodium(III) in acidic chloride solution via liquid–liquid extraction using tri-octylamine. Journal of Industrial and Engineering Chemistry 2011, 17, 243–247. [Google Scholar] [CrossRef]
- Biswas, S.; Basu, S. Extraction of zirconium(IV) from hydrochloric acid solutions by tri-octylamine and neutral donors. Journal of Radioanalytical and Nuclear Chemistry 1999, 242, 253–258. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Yang, A.; Lv, P.; Zhang, L.; Li, Y.; Yang, Y. Extraction and separation on Au(III) and Pt(IV) from HCl media using novel piperazine-based ionic liquid as an ionic exchanger. Journal of Molecular Liquids 2022, 353, 118846. [Google Scholar] [CrossRef]
- Xue, W.; Liu, R.; Liu, X.; Wang, Y.; Lv, P.; Yang, Y. Selective extraction of Nd(III) by novel carboxylic acid based ionic liquids without diluent from waste NdFeB magnets. Journal of Molecular Liquids 2022, 364, 119919. [Google Scholar] [CrossRef]
- Micheau, C.; Lejeune, M.; Arrachart, G.; Draye, M.; Turgis, R.; Michel, S.; Legeai, S.; Pellet-Rostaing, S. Recovery of tantalum from synthetic sulfuric leach solutions by solvent extraction with phosphonate functionalized ionic liquids. Hydrometallurgy 2019, 189, 105107. [Google Scholar] [CrossRef]
- Hu, Q.; Zhao, J.; Wang, F.; Huo, F.; Liu, H. Selective extraction of vanadium from chromium by pure [C8mim][PF6]: An anion exchange process. Separation and Purification Technology 2014, 131, 94–101. [Google Scholar] [CrossRef]
- Zuo, Y.; Liu, Y.; Chen, J.; Li, D.Q. The Separation of Cerium(IV) from Nitric Acid Solutions Containing Thorium(IV) and Lanthanides(III) Using Pure [C8mim]PF6 as Extracting Phase. Ind. Eng. Chem. Res. 2008, 47, 2349–2355. [Google Scholar] [CrossRef]
- Villemejeanne, B.; Legeai, S.; Meux, E.; Dourdain, S.; Mendil-Jakani, H.; Billy, E. Halide based ionic liquid mixture for a sustainable electrochemical recovery of precious metals. Journal of Environmental Chemical Engineering 2021, 10, 107063. [Google Scholar] [CrossRef]
- Ouadi, A.; Klimchuk, O.; Gaillard, C.; Billard, I. Solvent extraction of U(vi) by task specific ionic liquids bearing phosphoryl groups. Green Chem. 2007, 9, 1160–1162. [Google Scholar] [CrossRef]
- Rout, A.; Binnemans, K. Solvent Extraction of Neodymium(III) by Functionalized Ionic Liquid Trioctylmethylammonium Dioctyl Diglycolamate in Fluorine-free Ionic Liquid Diluent. Ind. Eng. Chem. Res. 2014, 53, 6500–6508. [Google Scholar] [CrossRef]
- Llorens, I.; Solari, P.L.; Sitaud, B.; Bes, R.; Cammelli, S.; Hermange, H.; Othmane, G.; Safi, S.; Moisy, P.; Wahu, S.; et al. X-ray absorption spectroscopy investigations on radioactive matter using MARS beamline at SOLEIL synchrotron. Radiochimica Acta 2014, 102, 957–972. [Google Scholar] [CrossRef]
- Sitaud, B.; Solari, P.L.; Schlutig, S.; Llorens, I.; Hermange, H. Characterization of radioactive materials using the MARS beamline at the synchrotron SOLEIL. Journal of Nuclear Materials 2012, 425, 238–243. [Google Scholar] [CrossRef]
- Sukhbaatar, T.; Duvail, M.; Dumas, T.; Dourdain, S.; Arrachart, G.; Solari, P.L.; Guilbaud, P.; Pellet-Rostaing, S. Probing the existence of uranyl trisulfate structures in the AMEX solvent extraction process. Chem. Commun. 2019, 55, 7583–7586. [Google Scholar] [CrossRef] [PubMed]
- Hanke, K.; Kaufmann, M.; Schwaab, G.; Havenith, M.; Wolke, C.T.; Gorlova, O.; Johnson, M.A.; Kar, B.P.; Sander, W.; Sanchez-Garcia, E. Understanding the ionic liquid [NC4111][NTf2] from individual building blocks: an IR-spectroscopic study. Phys. Chem. Chem. Phys. 2015, 17, 8518–8529. [Google Scholar] [CrossRef] [PubMed]
- Servaes, K.; Hennig, C.; Billard, I.; Gaillard, C.; Binnemans, K.; Görller-Walrand, C.; Van Deun, R. Speciation of Uranyl Nitrato Complexes in Acetonitrile and in the Ionic Liquid 1-Butyl-3-methylimidazolium Bis(trifluoromethylsulfonyl)imide. European Journal of Inorganic Chemistry 2007, 2007, 5120–5126. [Google Scholar] [CrossRef]
- Görller-Walrand, C.; De Jaegere, S. Étude comparative des spectres d’absorption de complexes d’uranyle en solution et a l’état solide. Complexes de symétrie Cs, D2h (6) ET D3h (6). J. Chim. Phys. 1972, 69, 726–736. [Google Scholar] [CrossRef]
- Hennig, C.; Kraus, W.; Emmerling, F.; Ikeda, A.; Scheinost, A.C. Coordination of a Uranium(IV) Sulfate Monomer in an Aqueous Solution and in the Solid State. Inorg. Chem. 2008, 47, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- Lipovskii, A.A.; Kuzina, M.G. The Infrared Absorption Spectra and Structure of Tridecylammonium Sulphate, Tridecylammonium Hydrogensulphate, and Tridecylammonium Dioxotrisulphatouranate (VI). 1965, 10, 740–745. [Google Scholar]
Figure 1.
Structure of cations investigated (top): trioctylammonium ([TOAH]+), methyltrioctylammonium ([MTOA]+) and Aliquat 336. Structure of anions tested (bottom): sulphate (SO42−), chloride (Cl−) and bis(trifluoromethanesulfonyl)imide (NTf2−).
Figure 1.
Structure of cations investigated (top): trioctylammonium ([TOAH]+), methyltrioctylammonium ([MTOA]+) and Aliquat 336. Structure of anions tested (bottom): sulphate (SO42−), chloride (Cl−) and bis(trifluoromethanesulfonyl)imide (NTf2−).
Figure 2.
(a) Distribution coefficients of U of quaternary ammoniums mixtures carrying same cation and different anions (a–c) or different cations (d). The initial aqueous solution contains 0.1 mol·L−1 sulfuric acid (pH = 1), 0.1 mol·L−1 ammonium sulphate, 250 ppm U (VI) and 250 ppm Fe (III). A/O = 2. Distribution coefficients of U measured in the same conditions for the reference system TOA-dodecane are presented in dashed orange line on all graphs. Composition of Aliquat 336 is given in supplementary materials. Molar ratios higher than 50 % are not presented in Figure 2b because the mixtures were not liquid/soluble.
Figure 2.
(a) Distribution coefficients of U of quaternary ammoniums mixtures carrying same cation and different anions (a–c) or different cations (d). The initial aqueous solution contains 0.1 mol·L−1 sulfuric acid (pH = 1), 0.1 mol·L−1 ammonium sulphate, 250 ppm U (VI) and 250 ppm Fe (III). A/O = 2. Distribution coefficients of U measured in the same conditions for the reference system TOA-dodecane are presented in dashed orange line on all graphs. Composition of Aliquat 336 is given in supplementary materials. Molar ratios higher than 50 % are not presented in Figure 2b because the mixtures were not liquid/soluble.
Figure 3.
(a) Uranium distribution coefficient, (b) concentration of water in the organic phase after contact and (c) concentration of acid in the aqueous phase after contact, measured for each mixture [TOAH]2[SO4] + [TOAH][NTf2] for 2 concentrations [(NH4)2SO4] = 0.1 and 1 mol·L−1 in the aqueous phase. A/O = 2. Composition of aqueous phase: 250 ppm U (VI), 250 ppm Fe (III), 0.1 mol·L−1 H2SO4.
Figure 3.
(a) Uranium distribution coefficient, (b) concentration of water in the organic phase after contact and (c) concentration of acid in the aqueous phase after contact, measured for each mixture [TOAH]2[SO4] + [TOAH][NTf2] for 2 concentrations [(NH4)2SO4] = 0.1 and 1 mol·L−1 in the aqueous phase. A/O = 2. Composition of aqueous phase: 250 ppm U (VI), 250 ppm Fe (III), 0.1 mol·L−1 H2SO4.
Figure 4.
(a) 19F-NMR spectrum and (b) FT-IR spectra of the mixture x[TOAH]2[SO4] = 50 %. Black before and red after contact with an aqueous phase containing 2500 ppm U(VI), 0.1 mol·L−1 sulfuric acid and 1 mol·L−1 ammonium sulphates. A/O = 4.
Figure 4.
(a) 19F-NMR spectrum and (b) FT-IR spectra of the mixture x[TOAH]2[SO4] = 50 %. Black before and red after contact with an aqueous phase containing 2500 ppm U(VI), 0.1 mol·L−1 sulfuric acid and 1 mol·L−1 ammonium sulphates. A/O = 4.
Figure 5.
(a) Raw UV-vis data for uranyl in the mixtures [TOAH]2[SO4] + [TOAH][NTf2] (x[TOAH]2[SO4]= 25 to 100%) and in the reference medium TOA + dodecane. (b) Experimental (solid lines) and fitted (dotted lines) EXAFS spectra: k3-weighted Fourier transforms of the 3-sulphate bidentate model around uranyl. The orange line represents the reference system, the blue, green and red lines the compositions x[TOAH]2[SO4] = 25, 50 and 100 % respectively. (b) Structure of uranium sulphate, the different contributions to the EXAFS spectra are indicated by red dotted lines.
Figure 5.
(a) Raw UV-vis data for uranyl in the mixtures [TOAH]2[SO4] + [TOAH][NTf2] (x[TOAH]2[SO4]= 25 to 100%) and in the reference medium TOA + dodecane. (b) Experimental (solid lines) and fitted (dotted lines) EXAFS spectra: k3-weighted Fourier transforms of the 3-sulphate bidentate model around uranyl. The orange line represents the reference system, the blue, green and red lines the compositions x[TOAH]2[SO4] = 25, 50 and 100 % respectively. (b) Structure of uranium sulphate, the different contributions to the EXAFS spectra are indicated by red dotted lines.
Figure 6.
Evolution of (a) R-factor and (b) Debye-Waller-factor accounting for disorder.
Figure 7.
FT-IR spectra between 800 and 1000 cm−1 of the mixtures [TOAH]2[SO4] + [TOAH][NTf2] after contact with an aqueous phase containing 2500 ppm U(VI), 0.1 mol·L−1 sulfuric acid and 1 mol·L−1 ammonium sulphate. A/O = 4.
Figure 7.
FT-IR spectra between 800 and 1000 cm−1 of the mixtures [TOAH]2[SO4] + [TOAH][NTf2] after contact with an aqueous phase containing 2500 ppm U(VI), 0.1 mol·L−1 sulfuric acid and 1 mol·L−1 ammonium sulphate. A/O = 4.
Figure 8.
Integration of the characteristic bands of (a) ν (S-OH), (b) ν (UO22+) and (c) ν (S-(O...U)2) as a function of x[TOAH]2[SO4].
Figure 8.
Integration of the characteristic bands of (a) ν (S-OH), (b) ν (UO22+) and (c) ν (S-(O...U)2) as a function of x[TOAH]2[SO4].
Table 1.
Synergistic peak position and distribution coefficient of uranium (data extracted from Figure 2).
Table 1.
Synergistic peak position and distribution coefficient of uranium (data extracted from Figure 2).
| [MTOA]2[SO4] + [MTOA][NTf2] | [MTOA][Cl] + [MTOA][NTf2] | [Aliquat 336]2[SO4] + [Aliquat 336][NTf2] | [MTOA][NTf2] + [TOAH]2[SO4] | [TOAH]2[SO4] + [TOAH][NTf2] | |
|---|---|---|---|---|---|
| Ratio (%) | 25 | 25 | 50 | 50 | 90 |
| DU | 100 | 70 | 78 | 196 | 128 |
Table 2.
Parameters of the best fit obtained for EXAFS spectra of [UO2(SO4)3]4− complexes. (σ2 is the Debye-Waller factor accounting for disorder, ΔE0 is the energy fit, S02 is the amplitude factor, R-factor is the tuning factor.
Table 2.
Parameters of the best fit obtained for EXAFS spectra of [UO2(SO4)3]4− complexes. (σ2 is the Debye-Waller factor accounting for disorder, ΔE0 is the energy fit, S02 is the amplitude factor, R-factor is the tuning factor.
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
N,N’-Bis(salicylidene)ethylenediamine (Salen) as an Active Compound for the Recovery of Ni(II), Cu(II), and Zn(II) Ions from Aqueous Solutions
Katarzyna Witt
et al.
Membranes,
2020
Comparative Extraction of Aluminum Group Metals Using Acetic Acid Derivatives with Three Different-Sized Frameworks for Coordination
Keisuke Ohto
et al.
Separations,
2021



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated