
Submitted:
31 July 2023
Posted:
02 August 2023
You are already at the latest version
Abstract
Tissue regeneration is an essential requirement for wound healing and recovery of organs’ dysfunction. It has been demonstrated that wound healing can be facilitated by activating paracrine signaling mediated by exosomes secreted from stem cells, since exosomes deliver many functional molecules including growth factors (GFs) and neurotrophic factors (NFs) effective for tissue regeneration. In this study, an exosome-rich conditioned medium (ERCM) was collected from human amniotic membrane stem cells (AMSCs) by cultivating the cells under a low oxygen tension (2% O2 and 5% CO2). The contents of GFs and NFs including keratinocyte growth factor, epidermal growth factor, fibroblast growth factor 1, transforming growth factor-β, and vascular endothelial growth factor responsible for skin regeneration were much higher (10-30 folds) in the ERCM than in normal conditioned medium. In was found that CM-DiI-labeled exosomes readily entered keratinocytes and fibroblasts, and that ERCM not only facilitated the proliferation of keratinocytes in normal condition, but also protected against H2O2 cytotoxicity in a concentration-dependent manner. In cell-migration assay, the scratch wound in keratinocyte culture dish was rapidly closed by treatment with ERCM. Such wound-healing effects of ERCM were confirmed in a rat whole skin-excision model: i.e., the wound closure was significantly accelerated by topical application of ERCM solution (4 x 109 exosome particles/100 μL) at 4-day intervals. In the wounded skin, the deposition of collagens was enhanced by treatment with ERCM, which was supported by the increased production of collagen-1 and collagen-3. The results indicate that ERCM from AMSCs containing a large amount of GFs and NFs improve wound healing through tissue regeneration not only by facilitating keratinocyte proliferation for skin repair, but also activating fibroblasts for extracellular matrix production.
Keywords:
amniotic membrane stem cell
; exosome-rich conditioned medium (ERCM)
; growth factor
; neurotrophic factor
; wound healing
; keratinocyte proliferation
; collagen synthesis
1. Introduction
When the skin is damaged, hemostasis and inflammation occur, blood vessels constrict, and fibrin and platelets aggregate, forming clots [1]. These clots release cytokines and growth factors (GFs) that initiate an inflammatory response. Neutrophils, monocytes, fibroblasts, and endothelial cells at the wound site secrete extracellular matrix (ECM), providing a scaffold in which tissue-regenerating cells can be activated and proliferate more appropriately. Inflammatory mediators accumulate, blood vessels around the wound expand, and cells infiltrate to remove invading microorganisms and cellular debris [1,2].
Fibroblasts, present during the emergence of new blood vessels, proliferate and invade the thrombus to form contractile granulation tissue [3,4]. At this time, as the wounds matures, major collagens that make up the ECM. More than 20 different types of collagen have been identified from humans, mainly types 1, 2, and 3. Collagen is one of the popular materials in protein-based scaffolds, and collagen-1 and collagen-3 play an important role in wound healing. During wound healing, the components of the ECM show certain changes: i.e., collagen-3 produced in the proliferative phase is replaced by stronger collagen-1 as the tissue matures [5,6]. Remodeling, degradation, and synthesis of the ECM will strengthen the tensile strength and restore the normal tissue structure, in which the nurturing tissue will gradually rise, the hair follicles and blood vessels will be formed, and the collagen fibers will also gradually increase [7].
It has been known that stem cells play a central role in the wound healing through paracrine effects. Notably, the paracrine effects are mediated by functional molecules including proteins and microRNAs (miRNAs) released from stem cells [8,9]. Interestingly, the functional molecules are delivered in forms of extracellular vesicles (EVs) called exosomes, by which healing efficiency is increased. Exosomes are membrane lipid vesicles with diameters of 30-250 nm and have attracted a lot of attention in the field of skin recovery and regeneration. Indeed, stem cell-derived exosomes are known to accelerate wound closure and promote wound healing [10]. As underlying mechanisms, stem cell exosomes modulate inflammatory responses, accelerate angiogenesis, increase keratinocyte and fibroblast migration and proliferation, and activate fibroblasts to synthesize collagen and elastin fibers [11,12].
Recently, we attained an exosome-rich conditioned medium (ERCM) from human amniotic membrane stem cells (AMSCs) by cultivating at a hypoxic oxygen (2% O2) tension [13]. It was confirmed that the ERCM contained tens or hundreds of functional molecules compared with a normal conditioned medium (NCM) collected at a normoxic (20% O2) culture condition. Therefore, in the present study, we assessed whether the AMSC exosomes enter keratinocytes and fibroblasts, facilitate keratinocyte proliferation and fibroblast activation, and thereby enhance wound healing in vitro and in vivo.
2. Materials and Methods
2.1. Preparation of AMSCs
AMSCs were collected under Good Manufacturing Practice conditions (Central Research Institute of Designed Cells Co., Ltd., Cheongju, Republic of Korea) as previously described [13,14]. In brief, amniotic membranes were digested with collagenase I. After removal of red blood cells, the remaining cells were suspended in Designed Cells-Exclusive Medium (DCEM; Designed Cells Co., Ltd.) supplemented with 5% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin (Invitrogen, Carlsbad, CA, USA). Cultures were maintained under 5% CO2 at 37°C in culture flask. The prepared amniotic stem cells were analyzed for their stem cell markers in a fluorescence-activated single cell sorting (FACS). The cells were confirmed to be mesenchymal stem cells (MSCs) [14].
2.2. Preparation of ERCM and Characterization
The separated AMSCs were dispersed in serum-free DCEM in Hyper flask (Nunc, Rochester, NY, USA) and cultivated under normoxic oxygen (20% O2, 5% CO2) or hypoxic oxygen (2% O2, 5% CO2) tensions at 37°C for 3 days [13]. The medium was vacuum-filtered through a PES membrane (0.22 μm) (Corning, Glendale, CA, USA). The conditioned medium was concentrated 30 times using Vivaflow-200 (Sartorius, Hannover, Germany).
Purified exosomes were adjusted to 106-107 particles/mL in PBS for nanoparticle- tracking analysis (NTA) using a Nanosight 300 equipped with v3.2.16 analytical software (Malvern Instruments. Westborough, MA, USA). The sample was photographed, and the particle concentration and size were analyzed [13].
Western blot analysis of CD9, CD63, and CD81 from isolated exosomes was performed using protein DC assay kits (Bio-Rad Laboratories, Hercules, CA, USA) [13]. An aliquot of NCM or ERCM was denatured using denaturation buffer, and then resolved via 12% SDS-PAGE. Resolved proteins were transferred onto Immobilon-P PVDF membrane and reacted with primary antibodies for CD9, CD63 or CD81 (1:1000; Abcam, Cambridge, UK) overnight at 4°C, followed by incubation with HRP-conjugated secondary anti-mouse antibody (1:2000; Abcam) at room temperature. After washing, the signal was recorded using WestFemto maximum sensitivity substrate kit under Bio-Rad ChemiDoc Imager (Bio-Rad Laboratories).
Enzyme-linked immunosorbent assay (ELISA) was conducted to analyze functional molecules, that is, GFs and neurotrophic factors (NFs), related to wound healing in NCM and ERCM [13]. ELISA kits for keratinocyte growth factor (KGF) (ab183362; Abcam), epidermal growth factor (EGF) (K0331115; Komabiotech, Seoul, Korea), fibroblast growth factor 1 (FGF1) (ab219636; Abcam), transforming growth factor-β (TGF-β) (ab100647; Abcam), and vascular endothelial growth factor (VEGF) (ab100662; Abcam) were used according to the manufacturer's instructions. Briefly, NCM or ERCM was put into the ELISA wells and incubated at room temperature. After washing 3-4 times, the primary antibodies were added, and reacted at room temperature. Following incubation with secondary antibody at room temperature, color-developing substrate was applied for 10-30 min. After treatment with a stop solution, the absorbance was measured at 450 nm.
2.3. Exosome Uptake, Cell Proliferation and Protection Assay
Exosomes in ERCM obtained from AMSCs were labeled with red CM-DiI membrane dye (C7000; Invitrogen) and prepared at a concentration of 4 × 1010 particles/mL [49–51]. HaCaT (a human keratinocyte cell line; Cell Lines Service, Heidelberg, Germany) and 3T3-L1 (a mouse fibroblast cell line; ATCC CL-173, Manassas, BA, USA) cells were cultivated in Dulbecco’s Modified Eagle’s Medium (DMEM; Biowest, Kansas City, MO, USA) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 1% L-glutamine (Gibco Life Technologies, Grand Island, NY, USA). The HaCaT and 3T3-L1 cells were seeded on 8-well chamber slides (NUNC C7182; Thermo Fisher Scientific, Waltham, USA) at 1 × 105/mL. After 24 hours, cells were damaged with 200 µM H2O2, and then incubated with labeled exosomes (50 μL/mL) for 4 hours at 37°C [13]. The cells were fixed with 4% paraformaldehyde, and treated with 0.1% Triton X-100 (Thermo Fisher Scientific). After blocking with 1% bovine serum albumin (BSA) for 1 hour, the cells were immunostained with anti-α-tubulin antibody (1:1000, ab7291; Abcam, Cambridge, UK) for 2 hours at 37°C, followed by goat anti-mouse IgG Alexa FluorTM 488 (1:500, Invitrogen) for 1 hour at room temperature. The cell nuclei were stained with DAPI (Thermo Fisher Scientific), and examined under a microscope (BX51; Olympus, Tokyo, Japan) [15,16].
HaCaT cells (1 × 104/mL) were seeded in a 96-well plate. In order to assess the cell-proliferating activity of ERCM, the cells were treated with ERCM (1-30 μL/mL). To evaluate the cytoprotective activity of ERCM against oxidative stress, the cells were exposed to 200 μM H2O2 and treated with ERCM [13]. After 24-hour culture at 37°C, the cell viability was quantified using Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan). CCK-8 assay was carried out by adding 10 µL of CCK-8 reagent into each cell culture well. After additional 2-hour incubation, the absorbance at 450 nm was measured using a microplate reader (Bio-Rad Laboratories Inc., Hercules, CA, USA).
For the in vitro wound-healing (scratched cell-migration) assay, HaCaT cells (1 × 105 cells/mL) were seeded in a 12-well plate for about 24 hours. When 90% confluence was achieved, a uniform scratch was made with a sterile pipette tip. Each well was washed with PBS, supplemented with new culture medium containing each concentration of ERCM, and then cultivated for 24 hours. Images of each scratch were observed microscopically, and captured immediately and 24 hours after scratching.
2.4. Whole-Thickness Wound Model and Wound Closure
Male Sprague-Dawley rats (7 weeks old, weighing 200-250 g) were purchased from DBL (Eumseong, Korea). The animals were housed at a standard room with a constant temperature (23 ± 2°C), relative humidity of 55 ± 10%, and 12-hour light/dark cycle, and given commercial rodent chow and purified water ad libitum.
The fur on the dorsal skin of the rats was shaved with a clipper prior to preparing the wound. Rat were lightly anesthetized with isoflurane, and then a full-thickness excision was made using a sterile 20-mm (in diameter) biopsy punch. ERCM (4 x 109 exosome particles/100 μL) or Saline solution was applied over the wound area 4 times on days 0, 4, 8, and 12. As a reference material, 3 mg Fucidin® ointment (100 μg as fusidate sodium; Donghwa Pharm, Seoul, Korea) was applied at the same schedule.
The wound areas were observed on days 0, 4, 8, 12, 16, and 19, photographed, and quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The rate of wound closure was expressed as the ratio of wound remained compared to the whole area on day 0.
2.5. Microscopic Observation and Collagen Analysis
Following tissue-processing procedures, formalin-fixed and paraffin-embedded skin tissues were stained with hematoxylin-eosin and Masson’s trichrome to observe overall structure and fibers, respectively.
In order to clearly show the collagen synthesis in fibroblasts, collagen-1 and collagen-3 were immunostained. The tissue sections were incubated with primary antibodies specific for collagen-1 (1:1,000, PA5-29569; Invitrogen) or collagen-3 (1:1,000, PA5-27828; Invitrogen) overnight at 4°C. After washing with PBS, the sections were incubated with secondary antibodies for 2 hours. The tissues were color-developed with 3,3-diaminobenzidine tetrahydrochloride (DAB; Novus Biologicals, Centennial, CO, USA) for 1-2 min. All sections were counterstained with Mayer’s hematoxylin, and examined under a high-power optical microscope (Carl Zeiss, Jena, Germany).
For western blot analysis of collagen-1 and collagen-3, the wounded skins were excised, freeze-clamped in liquid nitrogen, and stored at -80°C until use. Frozen tissue samples were homogenized in 10 volumes of RIPA buffer solution (Pierce, Rockford, IL, USA). Proteins were quantified using a BCA protein assay kit (Pierce). Proteins were denatured in 0.5 M Tris-HCl buffer (pH 6.8) containing 10% SDS and 10% ammonium persulfate, separated by 10% SDS-PAGE, and transferred to a nitrocellulose membrane in 25 mM Tris buffer containing 20% methanol, 1% SDS, and 192 mM glycine. After blocking with 3% skim milk, the membrane was incubated with a collagen-1 (1:1,000, PA5-29569; Invitrogen) or collagen-3 (1:1,000, PA5-27828; Invitrogen) overnight at 4°C, followed by a secondary goat anti-rabbit IgG conjugated with horseradish peroxidase (1:1,000, 7074; Cell Signaling Technology, Danvers, MA, USA) for 1 hour at room temperature. The membrane was then developed using an enhanced chemiluminescence solution (ATTO-TEC GmbH, Siegen, Germany). The band densities were measured using ImageJ software (National Institutes of Health) and normalized to the density of β-actin.
2.6. Statistical Analysis
Data are presented as mean ± standard deviation. Statistical analysis was performed with SPSS version 26.0 program (SPSS Inc., Chicago, IL, USA). Differences among groups were analyzed with one-way ANOVA, followed by Tukey’s HSD at a level of P < 0.05.
3. Results
3.1. Characteristics of Exosomes from AMSCs
From TEM analysis, typical exosome structures of homogeneous, spherical, and membrane-bound vesicles were observed, in which the size of the exosome particles was confirmed to be smaller than 100 nm (Figure 1A). NTA revealed the size distribution of exosomes, wherein the major peak was found to be 72 nm (Figure 1B).
The contents of exosomes in NCM and ERCM were measured by western blotting CD9, CD63, and CD81 markers. In comparison with NCM obtained in a normoxic condition (20% O2), levels of all the 3 markers in ERCM collected after a hypoxic culture (2% O2) were much higher (Figure 1C). In parallel with the exosome markers, the concentrations of GFs and NFs including KGF, EGF, FGF1, TGF-β, and VEGF were very high in ERCM, reaching 50 times those of NCM (Figure 1D).
3.2. Exosome Uptake in HaCaT and 3T3-L1 Cells
It was confirmed that the CM-DiI-labeled AMSC exosomes readily penetrate both the normal and H2O2-treated HaCaT keratinocytes and 3T3-L1 fibroblasts, although more exosomes were found in the damaged cells (Figure 2).
3.3. Proliferative and Protective Activities in HaCaT Cells
In a normal culture condition, ERCM significantly increased the HaCaT keratinocyte proliferation at concentrations of 6-30 μL/mL, reaching up to 162% for 24 hours (Figure 3A). As an oxidative stress, HaCaT cells exposed to 200 μM H2O2 for 24 hours resulted in 35% death (Figure 3B). However, simultaneous treatment with the ERCM near-fully rescued the keratinocytes from a low concentration of 1 μg/mL, and furthermore facilitated their proliferation at a high concentration (30 μg/mL).
In a model of in vitro wound-healing, ERCM facilitated the HaCaT cell migration in a concentration-dependent manner (Figure 3C,D). Although the scratch wound was closed by 53% without treatment, it was significantly facilitated by treatment with 6-30 μg/mL of ERCM, reaching 84-95%.
3.4. Wound Healing In Vivo
The skin wound applied with Saline solution was gradually closed, reaching 90% 19 days after whole-thickness excision (Figure 4A,B). The wound healing was markedly accelerated by treatment with the ERCM: i.e., the closure rate was much higher at early phase, leading to 90% on day 12, without prominent scar tissue formation. A partial effect was also achieved with fusidate sodium.
3.5. Microscopic Findings and Collagen Deposition
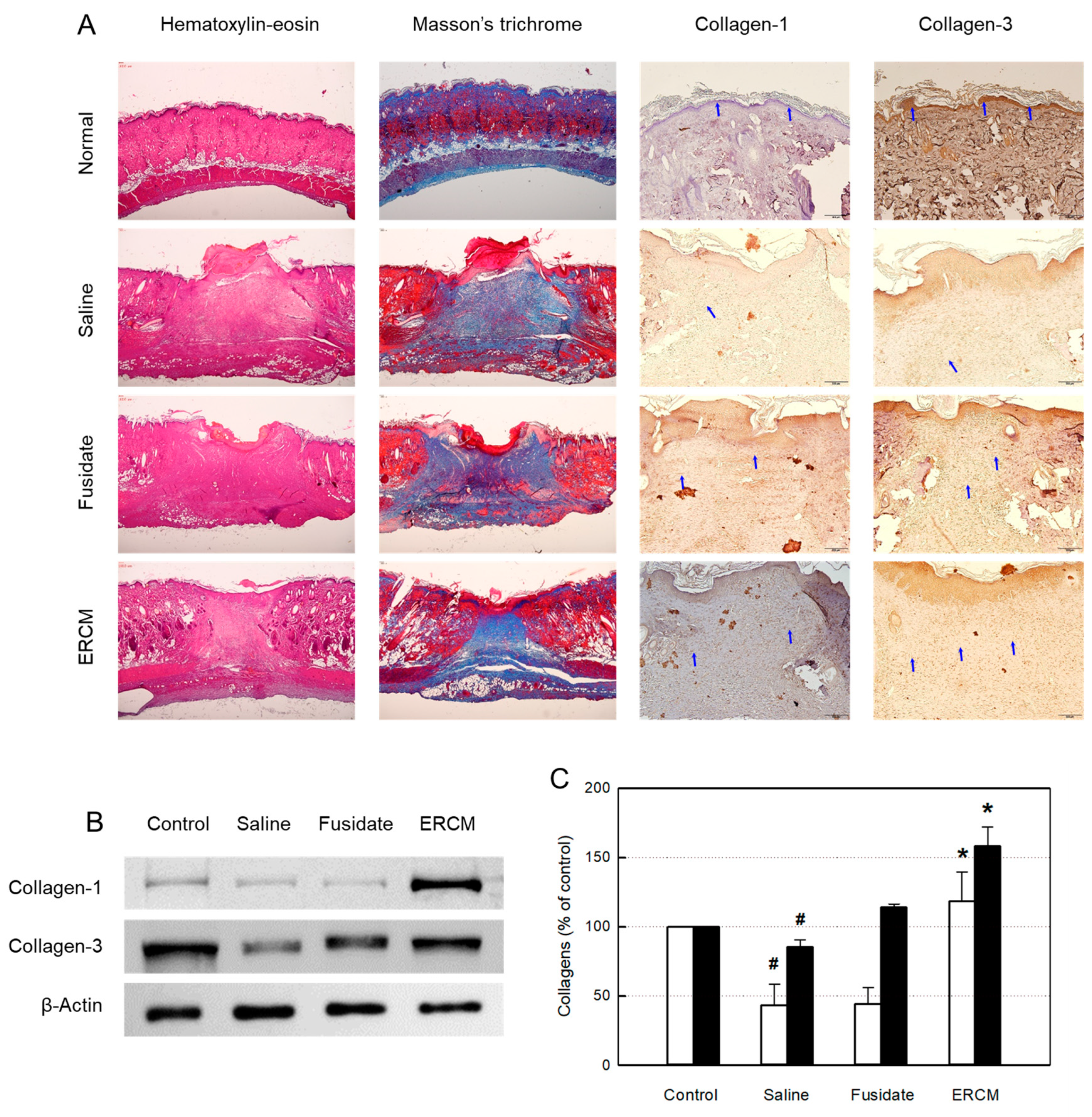
The histological structures of the regenerated dermis were analyzed on day 19 (Figure 5A). In hematoxylin-eosin stained findings, wide regenerating tissues were observed. Notably, the wound treated with ERCM was narrower than the saline-treated wound, and hair follicles were formed in the restored tissue. In the trichrome-stained findings, the facilitated wound closure with minimal scar tissues was more clearly observed when treated with ERCM, and fusidate exhibited a partial effect.
In the immunohistochemical (IHC) analysis of collagen-1 and collagen-3, the contents of the collagens were upregulated by treatment with ERCM (Figure 5A). Such increased production of collagens was confirmed by western blot analysis (Figure 5B,C). Separately, fusidate tended to increase the collagen-3 production.
4. Discussion
Wound healing proceeds through an inflammatory phase, proliferation of pre-existing cells, ECM repair by collagen deposition, and post-differentiation processes [17,18,19]. During this process, various cell types including corneocytes, fibroblasts, endothelial cells, macrophages, and platelets, are regulated, of which migration, invasion, proliferation, and differentiation are involved in inflammatory responses, new tissue formation, and ultimately wound closure. This complex process is executed and coordinated by an equally complex signaling network involving numerous GFs as well as cytokines and chemokines [17,18,19]. Hence, the topic of wound healing has attracted intensive research to understand its pathophysiology and develop new treatment strategies of a variety of diseases.
In wound healing, small EVs are considered to have the capacity influencing key biological processes for skin regeneration [20]. Interestingly, MSCs play an important role in tissue regeneration: i.e., exosomes secreted from stem cells may contribute to paracrine signaling [21]. We demonstrated that the local application of AMSC ERCM into rat whole-skin-excision wound exerted prominent repairing effects, as confirmed by more rapid closure, enhanced collagen deposition, higher rates of reepithelization and new blood vessel formation, as well as less scar tissue formation [11].
Such effects of ERCM on the enhanced wound repair could be supported by the activity on cell proliferation and migration. It is interest to note that the exosome particles readily penetrated both the normal and H2O2-exposed keratinocytes and fibroblasts, wherein more exosomes were found in the damaged cells. It is of interest to note that the particle size of AMSCs (mean 72 nm) was much smaller than exosomes from other MSCs such as adipose-derived mesenchymal stem cells (ADSCs; mean 220 nm) and umbilical cord blood-derived mesenchymal stem cells (UCBSCs; mean 120 nm) [29]. Such a small size may be advantageous for passing body membranes such as the skin and blood–brain barrier (BBB). In addition, ERCM markedly facilitated keratinocyte proliferation, protected against oxidative injury, and induced migration into a scratch wound in culture systems [12]. Interestingly, the cell-proliferative and -protective effects were observed at very low concentrations (≥3-6 μL/mL), especially displaying near-full recovery from H2O2 cytotoxicity at 1 μL/mL. The results suggest that exosomes providing GFs and NFs may directly protect keratinocytes and/or affect keratinocytes to evoke their regenerative responses, thereby accelerating proliferation and overcoming cytotoxicity. Indeed, high concentrations of KGF, EFG, and VEGF were detected in ERCM in the ELISA. Actually, it was reported that stem cells highly expressing GFs and NFs protected brain injury in a middle cerebral artery occlusion (MCAO) model [22], and that VEGF enhanced antioxidative capacity by up-regulating antioxidant enzymes [23]. Therefore, the decreased cell and tissue injury may be in part mediated by antioxidative activity of GFs/NFs released from ERCM [24,25].
Exosomes up-regulate protein expression related to angiogenesis, reepithelization, and ECM remodeling in wounded tissues [17,18,19]. Angiogenesis is known to play a central role in wound repair, wherein VEGF is the major GFs involved. The initial inflammation is a key phase in the wound healing process, which partially triggers the angiogenic response by producing high levels of VEGF [21,26].
In addition to KGF and EGF, the epithelial cell-specific GFs, very high concentrations of FGF1 and TGF-β related to fibroblast activation and proliferation were also observed in ERCM [13,27]. FGF1 is the basic GF for ECM production through collagen synthesis. Therefore, it is believed that the increased synthesis of collagen-1 and collagen-3 and their deposition in the wounded tissue may be partially induced by FGF1 from ERCM.
The up-regulation of VEGF expression increases the production of TGF-β, which is well known to play important roles in wound healing and scar prevention [28]. Actually, TGF-β was found to have important implications in all phases of wound healing, with recent dramatic findings implicating treatment options for early wound control. From the effects of TGF-β on monocytes, fibroblasts, endothelial cells, and keratinocytes, it was confirmed that this growth factor has pivotal roles in each phase of wound healing [28]. In general, TGF-β is released from degranulating platelets and secreted by all of the major cell types participating in the repair process, including lymphocytes, macrophages, endothelial cells, smooth muscle cells, epithelial cells, and fibroblasts [29,30]. Importantly, ERCM contained high levels of TGF-β governed the early phase of wound repair, leading to facilitated wound closure as well as decreased inflammatory complications.
In late proliferative phase, ECM production, angiogenesis, and reepithelization are major phenomena. In final maturation phase, formation of myofibroblasts for wound contraction is commonly observed [31]. In addition to early role, TGF-β is also involved in the inflammation, angiogenesis, reepithelization, and connective tissue regeneration. That is, TGF-β regulates angiogenic VEGF expression, collagen production (particularly collagen-1 and collagen-3) composing ECM, and remodeling of wounds affecting scar tissue formation [32,33]. Thus, the beneficial effects, increased wound repair and decreased scar tissue formation, might came in part from TGF-β in ERCM.
In microscopic IHC findings, increased collagen deposition was observed in the deep wound treated with ERCM. Such findings were confirmed by the western blot analysis of collagen-1 and collagen-3, indicative of the accelerated wound healing showing earlier closure of the open injury [34,35]. The histological analysis on day 19 confirmed that the wound healing entered the remodeling stage, in which gradual loss of cells and blood vessels, with changes in collagen types in ECM [36]. It was demonstrated that exosomes from ADSCs inhibited trans-differentiation of fibroblasts-to-myofibroblasts, and hypertrophic scar formation via miR-192-5p–IL-17RA–Smad axis [37]. In the control wound applied with saline, the surface tissue crust showed inverted contraction. Interestingly, however, the crust formation was minimal in the wound treated with ERCM. So, it is expected that the mild late-stage skin reaction following treatment with ERCM should make more smooth surface healing remaining minimal scar tissue.
Collectively, our research findings demonstrate that ERCM from AMSCs can promote skin wound healing and reduce scar tissue formation by facilitating proliferation of keratinocytes and optimizing the properties of fibroblasts [38]. Although additional follow-up studies to clarify underlying mechanisms, it is suggested that ERCM containing a large amount of GFs and NFs could be a good candidate for clinical trials to manage diverse acute and chronic skin lesions.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Figure S1.
Author Contributions
Conceptualization, T.M.K., E.-K.C. and Y.-B.K.; methodology, T.M.K.; validation, D.G. and S.-C.H.; investigation, C.H.N., S.P., H.R.S. and K-E.T; resources, S.-C.H.; data curation, A.-y.L.; writing—original draft preparation, T.M.K.; writing—review and editing, Y.-B.K.; supervision, Y.-B.K.; project administration, Y.-B.K.; funding acquisition, S.-C.H. and Y.-B.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI22C1650).
Institutional Review Board Statement
Human amniotic membrane tissues were obtained through caesarean section from a healthy female donor. All sample collection procedures from human beings were approved by IRB of Korea University Anam Hospital (Approval No. 2020AN0305). All procedures for animal experiments were approved and carried out in accordance with the Institutional Animal Care and Use Committee of Laboratory Animal Research Center at Chungbuk National University, Republic of Korea (Approval No. CBNUA-1483-21-01).
Informed Consent Statement
Written informed consent on the collection of amniotic membrane tissues has been obtained from the donors.
Data Availability Statement
Data are contained within the article.
Acknowledgments
This work was conducted during the research year of Chungbuk National University in 2022.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, GC. Wound healing: A cellular perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef] [PubMed]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, AJ.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Davison-Kotler, E.; Marshall, W.S.; García-Gareta, E. Sources of collagen for biomaterials in skin wound healing. Bioengineering 2019, 6, 56. [Google Scholar] [CrossRef]
- Hochstein, A.O.; Bhatia, A. Collagen: Its role in wound healing. Wound Manag. 2014, 4, 104–109. [Google Scholar]
- Diller, R.B.; Tabor, A.J. The role of the extracellular matrix (ECM) in wound healing. A review. Biomimetics 2022, 7, 87. [Google Scholar] [CrossRef]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Sun, BK.; Siprashvili, Z.; Khavari, P.A. Advances in skin grafting and treatment of cutaneous wounds. Science 2014, 346, 941–945. [Google Scholar] [CrossRef]
- Record, M.; Carayon, K.; Poirot, M.; Silvente-Poirot, S. Exosomes as new vesicular lipid transporters involved in cell–cell communication and various pathophysiologies. Biochim. Biophy. Acta (BBA)—Mol. Cell Biol. Lipids 2014, 1841, 108–120. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Ann. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Baglio, S.R.; Pegtel, D.M.; Baldini, N. Mesenchymal stem cell secreted vesicles provide novel opportunities in (stem) cell-free therapy. Front. Physiol. 2012, 3, 359. [Google Scholar] [CrossRef]
- Hu, L.I.; Wang, J.; Zhou, X.; Xiong, Z.; Zhao, J.; Yu, R.; Chen, L. Exosomes derived from human adipose mensenchymal stem cells accelerates cutaneous wound healing via optimizing the characteristics of fibroblasts. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Hu, Y.; Rao, S.S.; Wang, Z.X.; Ca, J.; Tan, Y.J.; Luo, J.; Xie, H. Exosomes from human umbilical cord blood accelerate cutaneous wound healing through miR-21-3p-mediated promotion of angiogenesis and fibroblast function. Theranostics 2018, 8, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Seong, H.-R.; Noh, C.H.; Park, S.; Cho, S.; Hong, S.J.; Lee, A.Y.; Geum, D.; Hong, S.-C.; Park, D.; Kim, T.M.; et al. Intraocular pressure-lowering and retina-protective effects of exosome-rich conditioned media from human amniotic membrane stem cells in a rat model of glaucoma. Int. J. Mol. Sci. 2023, 24, 8073. [Google Scholar] [CrossRef]
- Kang, D.; Kang, M.J.; Kong, D.; Lee, J.E.; Lee, A.Y.; Geum, D.H.; Kim, B.S.; Kim, Y.S.; Hong, S.C. Effect of human amniotic epithelial stem cell transplantation on preterm premature rupture of fetal membrane using the amniotic pore culture technique in vitro. Gynecol. Obstet. Investig. 2022, 87, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Andrade, W.; Seabrook, T.J.; Johnston, M.G.; Hay, J.B. The use of the lipophilic fluorochrome CM-DiI for tracking the migration of lymphocytes. J. Immunol. Methods 1996, 194, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Santelices, J.; Ou, M.; Hui, W.W.; Maegawa, G.H.; Edelmann, M.J. Fluorescent labeling of small extracellular vesicles (EVs) isolated from conditioned media. Bio Protoc. 2022, 12, e4447. [Google Scholar] [CrossRef]
- Guillamat-Prats, R. The role of MSC in wound healing, scarring and regeneration. Cells 2021, 10, 1729. [Google Scholar] [CrossRef]
- Gonzalez, A.C.D.O.; Costa, T.F.; Andrade, Z.D.A.; Medrado, A.R.A.P. Wound healing-A literature review. An. Bras. Dermatol. 2016, 91, 614–620. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Wei, F.; Wang, A.; Wang, Q.; Han, W.; Rong, R.; Wang, L.; Li, Y. Plasma endothelial cells-derived extracellular vesicles promote wound healing in diabetes through YAP and the PI3K/Akt/mTOR pathway. Aging 2020, 12, 12002. [Google Scholar] [CrossRef]
- Tonnesen, M.G.; Feng, X.; Clark, RA. Angiogenesis in wound healing. J. Investig. Dermatol. Symp Proc. 2000, 5, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Shin, K.; Cha, Y.; Ban, Y.H.; Park, S.K.; Jeong, H.S.; Park, D.; Choi, E.K.; Kim, Y.B. Neuroprotective effects of human neural stem cells over-expressing choline acetyltransferase in a middle cerebral artery occlusion model. J. Chem. Neuroanat. 2020, 103, 101730. [Google Scholar] [CrossRef]
- Abid, M.R.; Schoots, I.G.; Spokes, K.C.; Wu, S.Q.; Mawhinney, C.; Aird, W.C. Vascular endothelial growth factor-mediated induction of manganese superoxide dismutase occurs through redox-dependent regulation of forkhead and IκB/NF-κB. J. Biol. Chem. 2004, 279, 44030–44038. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Molecular and biological properties of vascular endothelial growth factor. J. Mol. Med. 1999, 77, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Sadat, S.; Gehmert, S.; Song, Y.H.; Yen, Y.; Bai, X.; Gaiser, S.; Klein, H.; Alt, E. The cardioprotective effect of mesenchymal stem cells is mediated by IGF-I and VEGF. Biochem. Biophys. Res. Commun. 2007, 363, 674–679. [Google Scholar] [CrossRef]
- Galiano, R.D.; Tepper, O.M.; Pelo, C.R.; Bhatt, K.A.; Callaghan, M.; Bastidas, N.; Gurtner, G.C. Topical vascular endothelial growth factor accelerates diabetic wound healing through increased angiogenesis and by mobilizing and recruiting bone marrow-derived cells. Am. J. Pathol. 2004, 164, 1935–1947. [Google Scholar] [CrossRef]
- Zhang, B.; Tian, X.; Hao, J.; Xu, G.; Zhang, W. Mesenchymal stem cell-derived extracellular vesicles in tissue regeneration. Cell Transplant. 2020, 29, 1–14. [Google Scholar] [CrossRef]
- Riedel, K.; Riedel, F.; Goessler, U.R.; Germann, G.; Sauerbier, M. TGF-β antisense therapy increases angiogenic potential in human keratinocytes in vitro. Arch. Med. Res. 2007, 38, 45–51. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Kane, C.J.; Hebda, P.A.; Mansbridge, J.N.; Hanawalt, P.C. Direct evidence for spatial and temporal regulation of transforming growth factor β1 expression during cutaneous wound healing. J. Cell. Physiol. 1991, 148, 157–173. [Google Scholar] [CrossRef]
- Xue, M.; Jackson, C.J. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef]
- Ramirez, H.; Patel, S.B.; Pastar, I. The role of TGFβ signaling in wound epithelialization. Adv. Wound Care 2014, 3, 482–491. [Google Scholar] [CrossRef] [PubMed]
- White, L.A.; Mitchell, T.I.; Brinckerhoff, C.E. Transforming growth factor β inhibitory element in the rabbit matrix metalloproteinase-1 (collagenase-1) gene functions as a repressor of constitutive transcription. Biochim. Biophys. Acta – Gene Struct. Express. 2000, 1490, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Tutuianu, R.; Rosca, A.M.; Iacomi, D.M.; Simionescu, M.; Titorencu, I. Human mesenchymal stromal cell-derived exosomes promote in vitro wound healing by modulating the biological properties of skin keratinocytes and fibroblasts and stimulating angiogenesis. Int. J. Mol. Sci. 2021, 22, 6239. [Google Scholar] [CrossRef]
- Zhang, J.; Guan, J.; Niu, X.; Hu, G.; Guo, S.; Li, Q.; Wang, Y. Exosomes released from human induced pluripotent stem cells-derived MSCs facilitate cutaneous wound healing by promoting collagen synthesis and angiogenesis. J. Transl. Med. 2015, 13, 1–14. [Google Scholar] [CrossRef]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Shi, J.; Liu, K.; Wang, X.; Jia, Y.; He, T.; Shen, K.; Wang, Y.; Liu, J.; et al. Exosomes derived from human adipose mesenchymal stem cells attenuate hypertrophic scar fibrosis by miR-192-5p/IL-17RA/Smad axis. Stem Cell Res. Ther. 2021, 12, 221. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, B.; Shi, H.; Qian, H.; Xu, W. MSC-exosome: A novel cell-free therapy for cutaneous regeneration. Cytotherapy 2018, 20, 291–301. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Isolation and characterization of exosomes from amniotic membrane stem cells (AMSCs). (A) Representative transmission electron microscopic findings of exosomes. (B) Particle size distribution of AMSC exosomes analyzed by a Nanoparticle-Tracking Analysis system. (C) Western blot analysis of CD9-, CD63-, and CD81-positive exosomes in normal conditioned medium (NCM) and exosome-rich conditioned medium (ERCM). (D) Concentrations of growth factors (GFs) and neurotrophic factors (NFs) in NCM (white bars) and ERCM (black bars). KGF: keratinocyte growth factor, EGF: epidermal growth factor, FGF1: fibroblast growth factor 1, TGF-β: transforming growth factor-β, VEGF: vascular endothelial growth factor. *Significantly different from NCM (P<0.05).
Figure 1.
Isolation and characterization of exosomes from amniotic membrane stem cells (AMSCs). (A) Representative transmission electron microscopic findings of exosomes. (B) Particle size distribution of AMSC exosomes analyzed by a Nanoparticle-Tracking Analysis system. (C) Western blot analysis of CD9-, CD63-, and CD81-positive exosomes in normal conditioned medium (NCM) and exosome-rich conditioned medium (ERCM). (D) Concentrations of growth factors (GFs) and neurotrophic factors (NFs) in NCM (white bars) and ERCM (black bars). KGF: keratinocyte growth factor, EGF: epidermal growth factor, FGF1: fibroblast growth factor 1, TGF-β: transforming growth factor-β, VEGF: vascular endothelial growth factor. *Significantly different from NCM (P<0.05).
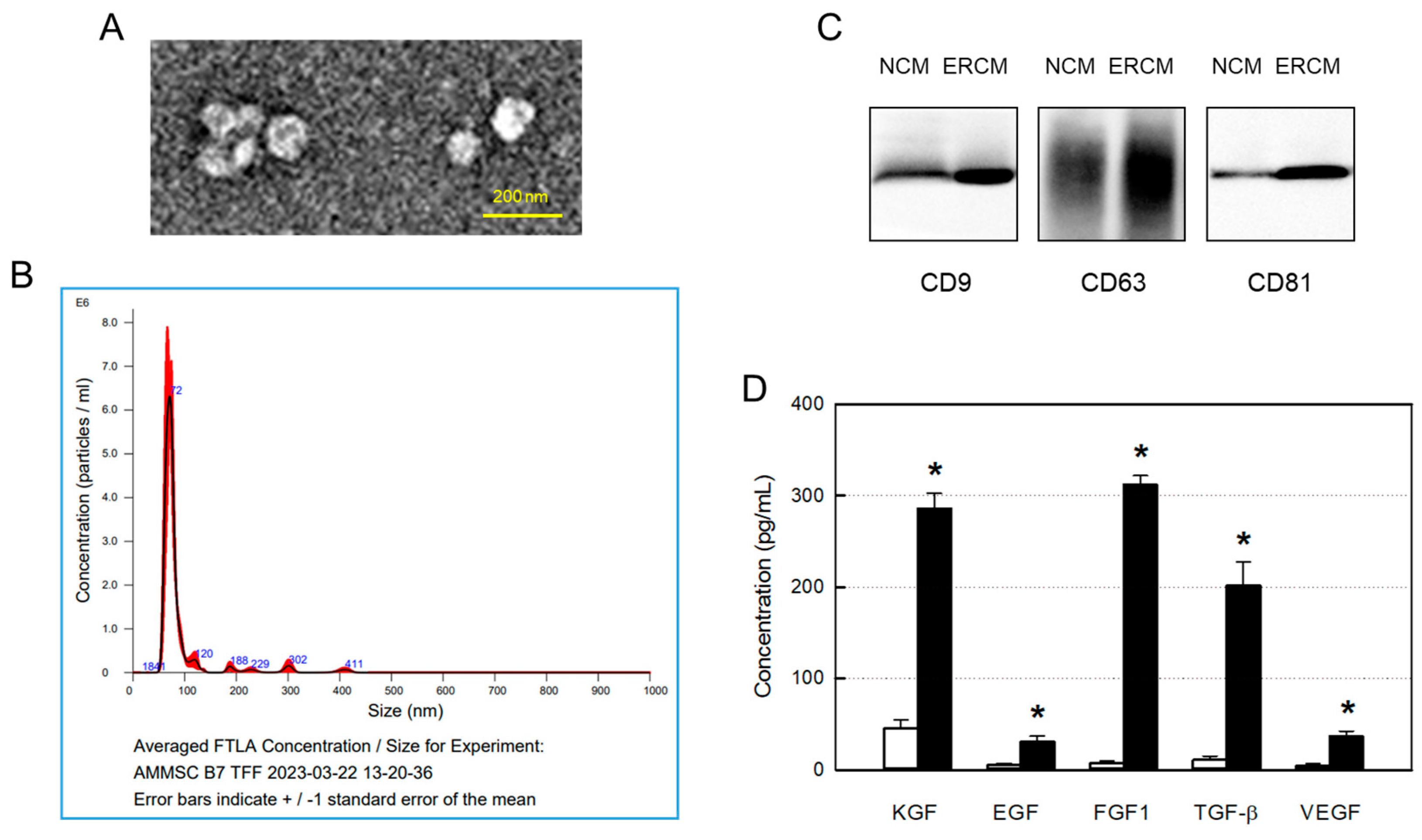
Figure 2.
Penetration of CM-DiI-labeled AMSC exosomes into HaCaT keratinocytes (A) and 3T3-L1 fibroblasts exposed to 200 μM H2O2 or its vehicle (Normal).
Figure 2.
Penetration of CM-DiI-labeled AMSC exosomes into HaCaT keratinocytes (A) and 3T3-L1 fibroblasts exposed to 200 μM H2O2 or its vehicle (Normal).
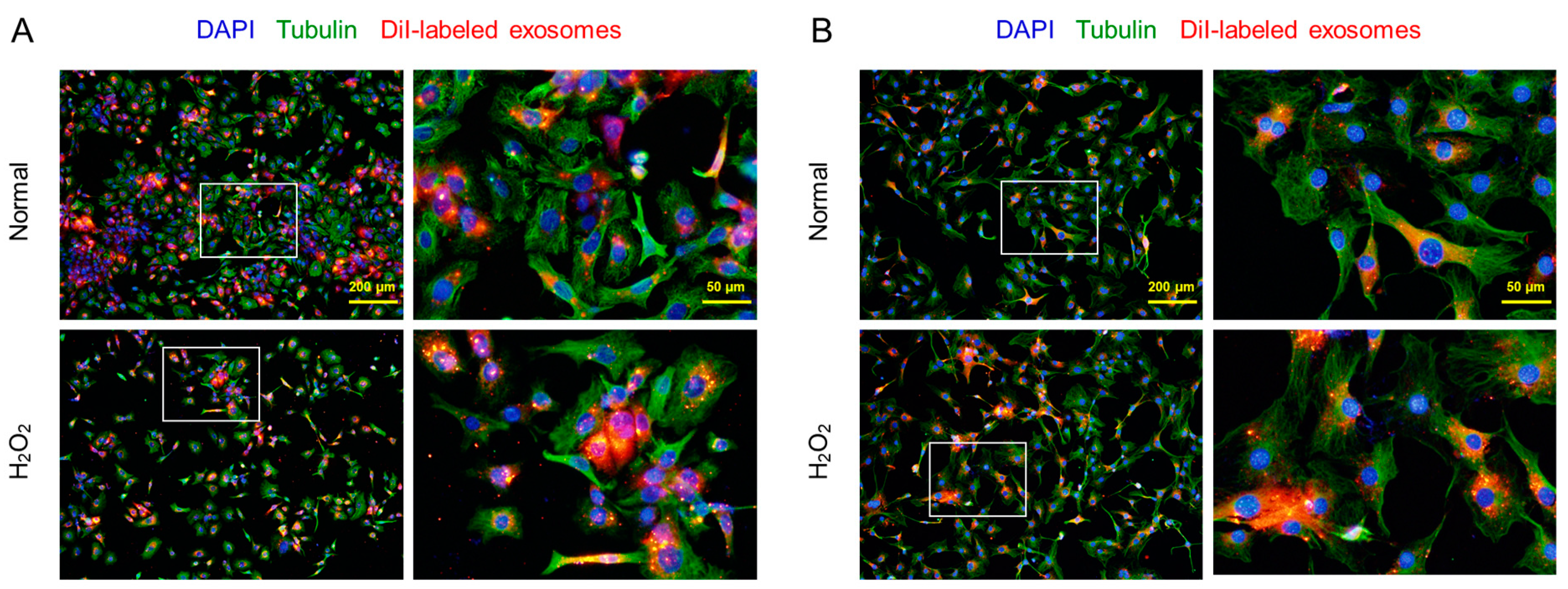
Figure 3.
HaCaT keratinocyte-proliferative and -protective activities of exosome-rich conditioned medium (ERCM). (A) HaCaT cell proliferation by ERCM. (B) HaCaT cell protection by ERCM against oxidative stress (200 μM H2O2). (C & D) Facilitation of HaCaT cell migration (scratch-healing) by ERCM. #Significantly different from Normal control (P<0.05). *Significantly different from Scratch alone (P<0.05).
Figure 3.
HaCaT keratinocyte-proliferative and -protective activities of exosome-rich conditioned medium (ERCM). (A) HaCaT cell proliferation by ERCM. (B) HaCaT cell protection by ERCM against oxidative stress (200 μM H2O2). (C & D) Facilitation of HaCaT cell migration (scratch-healing) by ERCM. #Significantly different from Normal control (P<0.05). *Significantly different from Scratch alone (P<0.05).
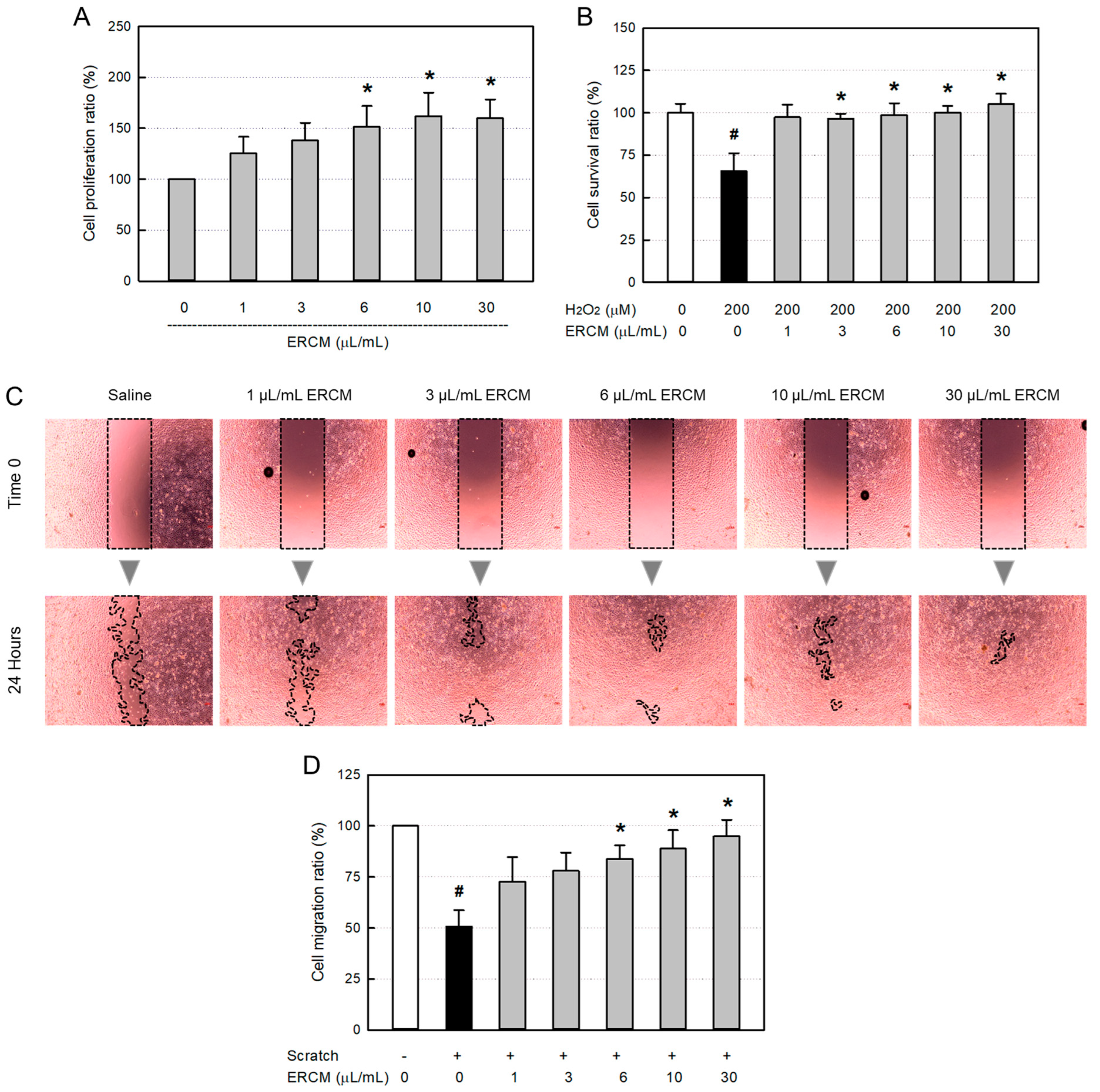
Figure 4.
Gross findings of wound healing in rats dermally applied with fusidate sodium or exosome-rich conditioned medium (ERCM). (A) Representative findings. (B) Quantitative analysis of wound closure. ○: Normal, ●: Fusidate sodium, ■: ERCM. *Significantly different from Normal control (P < 0.05).
Figure 4.
Gross findings of wound healing in rats dermally applied with fusidate sodium or exosome-rich conditioned medium (ERCM). (A) Representative findings. (B) Quantitative analysis of wound closure. ○: Normal, ●: Fusidate sodium, ■: ERCM. *Significantly different from Normal control (P < 0.05).
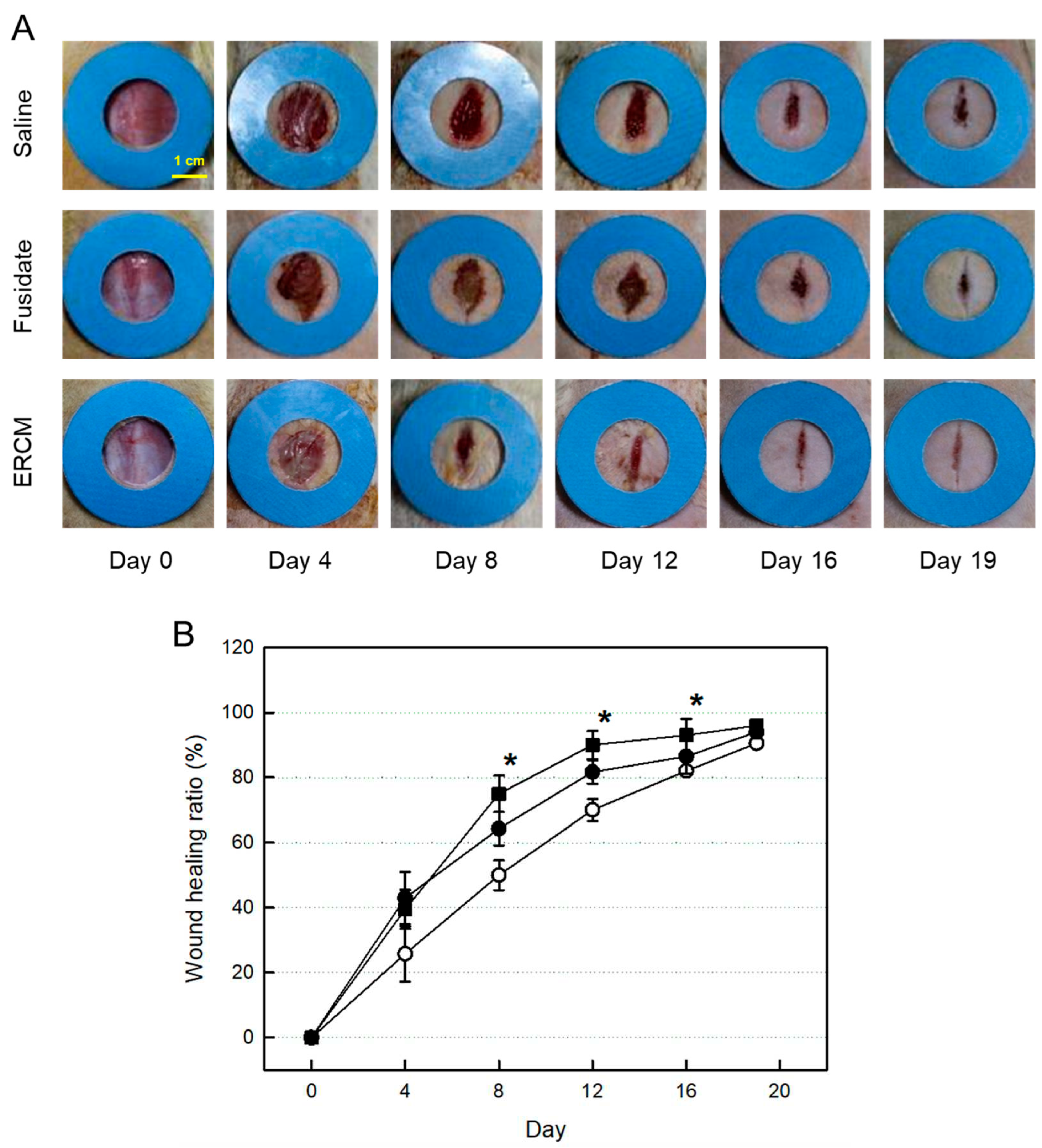
Figure 5.
Microscopic findings of wound healing and collagen synthesis in rats dermally applied with fusidate sodium or exosome-rich conditioned medium (ERCM). (A) Representative findings stained with hematoxylin-eosin, Masson’s trichrome or immunostained with antibodies specific for collagen-1 and collagen-3. (B & C) Western blot analysis of collagen-1 (white cars) and collagen-3 (black bars). #Significantly different from Normal control (P<0.05). *Significantly different from Saline (Wound alone) (P<0.05).
Figure 5.
Microscopic findings of wound healing and collagen synthesis in rats dermally applied with fusidate sodium or exosome-rich conditioned medium (ERCM). (A) Representative findings stained with hematoxylin-eosin, Masson’s trichrome or immunostained with antibodies specific for collagen-1 and collagen-3. (B & C) Western blot analysis of collagen-1 (white cars) and collagen-3 (black bars). #Significantly different from Normal control (P<0.05). *Significantly different from Saline (Wound alone) (P<0.05).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



