
Submitted:
01 August 2023
Posted:
02 August 2023
You are already at the latest version
Abstract
The protozoan parasite Plasmodium falciparum is the causative pathogen of the most severe form of malaria, for which novel strategies for treatment are urgently required. The primary energy supply for intraerythrocytic stages of Plasmodium is the production of ATP via glycolysis. Due to the parasite’s strong dependence on this pathway and significant structural differences of its glycolytic enzymes compared to their human counterpart, glycolysis is considered as a potential drug target. In this study, we provide the first three-dimensional protein structure of P. falciparum hexokinase (PfHK), containing novel information about the mechanisms of PfHK. We identified for the first time a Plasmodium-specific insertion which lines the active site. Moreover, we propose that this insertion plays a role upon ATP binding. Residues of the insertion further seem to affect the tetrameric interface and therefore suggest a special way of communication among the different monomers. In addition, we confirmed that PfHK is targeted and affected by oxidative posttranslational modifications (oxPTMs). Both Sglutathionylation and Snitrosation revealed an inhibitory effect on the enzymatic activity of PfHK.
Keywords:
crystal structure
; glycolysis
; hexokinase
; malaria
; posttranslational modification
; redox regulation
1. Introduction
Malaria is an infectious disease that threatens global health and life. In 2021, the World Health Organization reported 247 million cases of malaria infection worldwide, leading to 619,000 deaths [1]. These numbers and possibly imminent resistance against currently used drugs underscore the urgency to develop new strategies for antimalarial treatment [2,3]. Malaria is caused by the protozoan parasite Plasmodium. Among the five human pathogen species, infections with P. falciparum are responsible for most fatal cases and the highest mortality [1]. Symptoms occur during the infection of erythrocytes and are mainly caused by cytoadherence. The parasite’s replication within erythrocytes requires high levels of glucose as an energy supply to produce ATP via glycolysis [4,5,6]. Therefore, glycolysis is not only enhanced but indispensable for the survival of intraerythrocytic stages of Plasmodium and has attracted attention in the context of potential drug development [7,8].
PfHK is the first and simultaneously rate-limiting enzyme of glycolysis, catalyzing the phosphorylation of glucose to glucose-6-phosphate (G6P). ATP provides the phosphoryl group and the reaction requires divalent cations, usually Mg2+ [9,10,11]. The production of G6P is not only important for glycolytic flux but is also crucial and rate-limiting for the pentose phosphate pathway and the supply of NADPH as a reducing agent, which G6P dehydrogenase provides [12,13]. PfHK exists as a single copy gene on chromosome 6 without any isoenzymes [14]. Hexokinases are well conserved among Plasmodium, but the sequence identity to their human counterpart is lower than 32% [7,15]. Compared to other proteins within similar species, PfHK contains fifteen cysteines (~3%) and is therefore considered to be exceptionally cysteine rich [16]. Cysteines provide the basis for redox regulation via oxidative posttranslational modifications (oxPTM), which has already been reported to occur on various glycolytic enzymes. Previous studies suggested that S-glutathionylation and S-nitrosation target PfHK [17,18].
Within this study, we provide the first three-dimensional structure of PfHK with a 2.8 Å resolution solved via X-ray crystallography. Both glucose and citrate were identified as ligands bound to conserved residues of the active site. The structure provides novel information about the closure of the active site and the interaction of the single monomers within the tetrameric assembly of PfHK and thus allows for a better understanding of its oligomerization behavior, which is quite distinct from hexokinases of other organisms. Moreover, we confirmed that S-glutathionylation and S-nitrosation both target and inhibit the enzyme.
2. Results
2.1. Overall Structure of P. falciparum Hexokinase
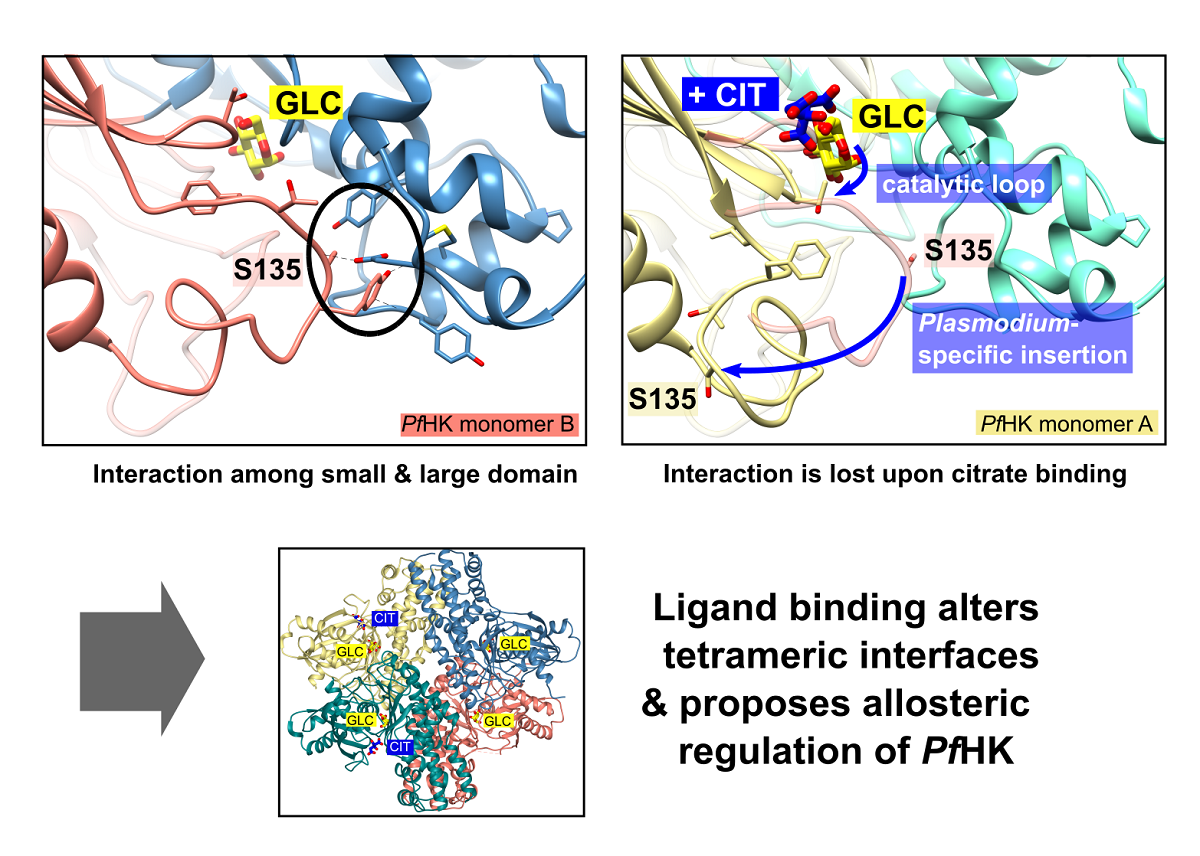
We obtained trigonal crystals of PfHK with a resolution of 2.8 Å. All structures were solved via molecular replacement. Data collection and refinement statistics are summarized in Table 1. The crystals belong to space group P3121 and contain two monomers within the asymmetric unit (Figure 1 A). The overall fold of each monomer is similar to other hexokinases and consists of two domains: a small domain comprising residues E94-L239 and a large domain comprising residues M1-Q93 and N240-P490, with the active site located between both domains. The binding sites for the substrates glucose and ATP are highly conserved among different species. The active site of both monomers contains a glucose molecule, and in one monomer a citrate molecule was additionally bound. The two monomers are essentially similar (the RMSD is 0.7 Å with 463 residues). However, the superimposition of both monomers reveals the movement of two loops. Those loops, G104-F108 (catalytic loop; C-loop) and L130-G141 (Plasmodium-specific insertion; P-insert), are located in the small domain within the cleft of the substrate-binding site and will be described in more detail in the following sections (Figure 1 B). The average B-factor of the structure is relatively high at 90 Å2, but this is to be expected since the Wilson B-factor of the data is also high (96 Å2). In monomer A, the average B-factor (103 Å2) for the P-insert region is in the same range as the average B-factor of the whole structure but is moderately increased in subunit B (124 Å2). These findings are in line with the electron density and indicate some flexibility.
2.2. Glucose and Citrate are Bound to the Active Site
Hexokinases catalyze the phosphorylation of glucose to G6P. For this purpose, ATP serves as phosphoryl donor, and after catalysis both products G6P and ADP are released. Previous X-ray crystallographic studies have demonstrated that the enzyme exhibits two conformational states: the open state, which occurs prior to glucose binding, and the closed state, where the two lobes surrounding the glucose substrate are closed upon glucose binding [19,20]. In both subunits of the present structure, glucose is coordinated by strictly conserved residues from both domains: S183, P185, T200, K201, N240, D241, I265, G269, E292, Q319, and E322 (Figure 2 A). Accordingly, comparison with the structures of the open and closed form of P. vivax hexokinase (PvHK, PDB ID: 6VYF, 6VYF) shows that this PfHK structure corresponds to the closed form of PvHK with an RMSD of 1.1 Å for 413 residues. In addition to the glucose a citrate molecule is bound in one subunit (A) of PfHK. The citrate interacts with conserved residues from both domains: G104, N107, R109, T268, and S436, which are typically involved in ADP/ATP binding (Figure 2 B).
Human HKI (PDB ID: 1DGK) is a pseudo dimer whose subdomains are connected by a helix (residues 448-474). Only in the active site of the second subdomain (475-913) a glucose and an ADP molecule are bound, which is why we used this part for the comparison. The superposition revealed an RMSD of 1.6 Å (405 residues, 33.4 % sequence identity). As described for the glucose binding site, the residues responsible for ADP/ATP binding are also highly conserved among all organisms. In PfHK, the putative ADP/ATP binding site consists of loop residues G104-N107 and β-turn residues G267-T268, both interacting with the β- and γ-phosphate of ADP/ATP. The adenine ring would be sandwiched between residues N357-S362 and L437-W443 (Figure 2 C).
Superimposition with human hexokinase structures complexed with the products, G6P (1HKB) or with ADP (1DGK), revealed that the phosphates of ADP or G6P occupied the same binding site as the citrate molecule in PfHK (Figure 2 C). Therefore, the citrate is located at the site where phosphate is normally transferred from ATP to glucose. Moreover, it interacts with residues used in homologous HK structures to bind ATP/ADP. These findings could be further confirmed with other hexokinase structures with bound G6P or ADP (PDB IDs: 1BG3, 6JJ9).
2.3. A Plasmodium-Specific Insertion (P-Insert) Lines the Active Site
Structural comparison of the two PfHK subunits and human hexokinase (PDB ID: 1DGK) reveals that the connection between a conserved β-strand and a conserved α-helix is different. In the human crystal structure, this connection consists of residues P115-S122, whereas in Plasmodium it comprises residues K131-T148 (Figure 3 A). However, the most significant differences are observed in the region of the mobile loop L130-G141 (P-insert) in PfHK, which in human involves only three residues (P115-N117). PfHK shares 89.6% sequence identity with PvHK and sequence alignment has shown that this insertion is also present in Plasmodium vivax, suggesting a Plasmodium-specific insertion [15]. In both the open and closed forms of the PvHK structures (PDB IDs: 6VYF, 6VYF), coordinates were not deposited for regions K131-K145, F167-N177, and V227-V235, nor for any ligands. This indicates that the P-insert loop was disordered in the PvHK structures.
While the P-insert is most probable a Plasmodium-specific insertion, the hexokinase from Eimeria tenella (EtHK, PDB IDs: 6KSR, 6KSJ), possesses a similar insertion, though this insertion is three amino acids shorter (Figure 3 B). E. tenella is another apicomplexan parasite with a sequence similarity of 43% compared to PfHK. Both EtHK structures are in the open state, although one (PDB ID: 6KSR) had a galactose molecule bound in the active site. There is no indication whether bound glucose instead of galactose would result in a close conformation. However, since this PfHK structure is in the closed conformation, superposition with the open conformation of EtHK (PDB ID: 6KSR, RMSD of 2.2 Å at 396 residues) shows a large shift of the small domain compared to PfHK. Consequently, the different structural arrangement of the "P-insert" is not comparable.
2.4. Does Catalysis Affect the Conformation of the C-Loop and the P-Insert?
As mentioned above, the superimposition of both PfHK monomers reveals different conformations for the C-loop (G104-F108) and the P-insert (L130-G141, Figure 4). Except for four residues in subunit B (G134-S137), the electron densities of both the P-insert and the C-loop are well defined in both monomers. Since the chain trace of regions L130-T133 and H138-G141 is very different in the two subunits, the conformation of the linking residues must also be different, thus extending over the entire P-insert.
In monomer B, P-insert residues L130-G141 adopt a completely different conformation compared to monomer A, pointing into the active site channel, and resulting in a large shift of up to 20 Å for residues G134-E139 (Supplemental information, Figure S1 A+B). In this conformation, S135 forms a hydrogen bond to loop residue D354; furthermore, S135 is now in van der Waals distance to Y329. P-insert residues T133-Y136 interact with Y312 and S353-N357 from the large domain (Figure 4). Interestingly, some of these residues are part of the interface between the two subunits, which is described in more detail in section 2.6 (Oligomerization of PfHK).
In subunit A, all these contacts have been lost, and the access to the active site is broadened by rearrangement of the conserved C-loop and the P-insert. Compared to subunit B, C-loop residue T106 is shifted by 6 Å. To be sure that these observed conformational changes are induced by citrate and not by the crystal packing, we analyzed the crystal contacts. There are no contacts between the C-loop and symmetry equivalent monomers in either subunit (Figure 1). In subunit A, P-insert residues G134-Y136 interact via a few hydrophobic interactions with a neighboring tetramer, more specifically with residues N87-P90, Y275, K287, and C260-Y261 of subunit B'. (Supplementary Information, Figure S2). In subunit B, there are no interactions of the P-insert loop with neighboring molecules, and there is enough space for the P-insert to adopt the same conformation as in subunit A. Thus, the P-insert can move freely which makes the active site accessible to the solvent and, in principal, allowing a citrate molecule to bind.
2.5. Structural Comparisons Suggest a Plasmodium-Specific Mechanism
The superposition of the two subunits (A and B) reveals that the position of the citrate molecule from subunit A would collide with the C-loop in subunit B, where the C-loop and the P-insert are oriented inwards the active site (Figure 1, Figure 3 and Figure 4). Residue S436, which was shown to form a hydrogen bond with the citrate molecule, and residue S362 are conserved residues of the ATP/ADP binding pocket (Figure 2 C). The binding of citrate requires the movement of C-loop residues (G105-N107) and subsequently a rearrangement of the P-insert. This movement of the C-loop is also seen in the structures of human glucokinase. Superimposition with human glucokinase (PDB ID: 3VEY) in complex with glucose and ATPγS, which mimics a quasi-transition state, shows that binding of ATP causes the C-loop to adopt the same conformation as in the PfHK monomer A with bound citrate (Figure 4 B). In the related structure (PDB ID: 3VEV) with solely bound glucose, the C-loop adopts the same conformation as in subunit B (no citrate) from PfHK (Figure 4 C). Since only glucose is bound, the C-loop adopts a pre-catalytic conformation. However, the same conformation is also seen in structures containing the products ADP (PDB ID: 1DGK, Figure 2 C) or G6P (PDB ID: 1HKB), so the conformation of the C-loop in these structures corresponds to a post-catalytic conformation.
As mentioned before, the citrate molecule is bound to the phosphate binding and transfer site of subunit A. Thus, it forces a rearrangement of the C-loop in this subunit followed by a rearrangement of the P-insert, resulting in a catalytically active conformation of both loops. In our structure, the bound citrate could mimic the mechanism upon ATP binding, as seen in the quasi-transition state structure of human glucokinase (PDB ID: 3VEY) in complex with glucose and ATPγS. In subunit B, the P-insert occupies the ATP-binding site via interactions with the large domain, thus inhibiting ATP binding and causing the C-loop and the P-insert to adopt a pre- or post-catalytic conformation. The conformation of the P-insert in subunit B would hinder product release, but rearrangement of the P-insert in subunit A facilitates the release of the products (Figure 5). This could be a Plasmodium-specific hexokinase mechanism that potentially affects the enzyme’s activity.
2.6. Oligomerization of P. falciparum Hexokinase
The asymmetric unit of the crystal contains a dimer (AB), forming a tetramer together with the symmetry-equivalent dimer (A'B'). The two dimers are connected by a 2-fold rotation axis (Figure 6 A). In accordance with that, size-exclusion chromatography showed that PfHK assembles as a tetramer (Figure 6 B+C). All enzymatic activities were measured with tetrameric PfHK without any reducing agent, unless described otherwise.
Due to the crystallographic symmetry relationship between the dimers, two interfaces are identical in each case so that there are only three different interfaces AB (≙A'B'), AA' (≙ BB') and AB' (≙BA'), which are shown in Figure 7 A. Analysis with the PISA server (https://www.ebi.ac.uk/pdbe/pisa/) revealed that the subunits are tightly connected via hydrogen bonds and hydrophobic interactions.
The interface AB involves mainly residues from the large domains of subunit A and B. They are strongly connected via seven different hydrogen bonds and one additional salt bridge between E28A and K348B. Moreover, interactions among the residues T302-Y312, R336-K348, and W351 from both monomers could be observed. In monomer B, residues K35-Q39 participate, whereas in monomer A, residues R32-Q39 form the interface region (Figure 7 B).
Residues from the small domains mainly form the BB’ interface (Figure 7 C). N70 from both monomers is hydrogen-bonded to I197, D198, and E214 from the other monomer. Interface residues are K67-E76, K80, T187-D198, R207-K220, and R316-R318.
In contrast to the multiple hydrogen bonds of interfaces AB and AA’, subunits A and B’ are mainly connected with hydrophobic interactions between a long helix (E49-R66) from both subunits. Further connection is mediated via a hydrogen bond between N53A and Y56B’, interactions between residues K281-Y282, D299, I423 from monomer A, and residues S45-K67 and K298-D299 from monomer B’. Interestingly, H75 from all four monomers assembles within the center of the tetramer and is part of the tetrameric interfaces (Figure 7 D). Distances between the H75 sidechains (atom NE) are ~3.6 Å of subunit B and B’ or A and A’, and 5.9 Å between subunits a (CE1), B (NE) or subunits A' and B’.
As already discussed by Srivastava and colleagues, the tetrameric assembly of Plasmodium hexokinases is unique, since most hexokinases occur in a biologically relevant manner as monomers or (pseudo-)dimers. PvHK shares 89.6% sequence identity with the present PfHK [15]. The structures (PDB IDs: 6VYG and 6VYF) of PvHK were solved via cryo-electron microscopy at a resolution of 3.3 Å and 3.5 Å, respectively and the authors suppose a bound glucose or another similar sugar molecule in its active site, but the deposited PDB file does not contain any ligands. Superimposition of the dimers resulted in 1.8 Å with 824 residues. However, in the PvHK structure regions K131-K145, F167-N177, and V227-V235 were not defined. Comparison of both Plasmodium tetramers shows differences in the interface of AA’ and AB, which involves the small domain (2.2 Å with 1,633 residues). As described above, the P-insert is rearranged in our structure upon citrate binding and probably also upon ATP binding. This movement is responsible for the loss of multiple contacts between the small and large domains in monomer A. Of particular importance appears to be the interaction between the P-insert residues L130-S135 (small domain) and region P304-Y312 (large domain), which in turn is part of the AB interface. (Figure 4; Figure 7 B). The loss of this contact alters the AB interface and affects the neighboring molecule in the tetramer. This could be an allosteric effect, possibly explaining the binding of citrate (or physiologically ATP) only to one subunit.
2.7. Oxidative Posttranslational Modifications inhibit the Enzymatic Activity of PfHK
Redox regulation via oxPTMs occurs under both physiological and pathophysiological conditions at the protein’s cysteine residues and can have a major influence on protein conformation, stability, function, and interactions with other proteins [21,22,23]. OxPTMs target hexokinases from a variety of organisms. Previous studies gained important insights into the patterns of S-glutathionylation, S-nitrosation, and S-sulfenylation within P. falciparum trophozoite stages and indicated that PfHK was a target of S-glutathionylation and S-nitrosation under both basal conditions and conditions with increased oxidative stress [17,18,24]. A prerequisite for those modifications is the presence of reactive thiols. Compared to other proteins within similar species, PfHK contains fifteen cysteines (3%) and is therefore considered to be exceptionally cysteine rich [16]. S-glutathionylation of purified, recombinant PfHK was shown via α-glutathione Western blot analysis, whereas S-nitrosation was confirmed with a biotin-switch assay followed by α-biotin Western blot analysis, described in more detail in Methods (Figure 8 A+B).
For investigating possible effects of those modifications on the enzymatic activity of PfHK, we performed kinetic analyses of modified enzyme. To ensure adequate recombinant production of PfHK prior to our analyses, we measured KM values for comparison with former studies. The KM value for ATP was 0.83 ± 0.08 mM and 0.13 ± 0.01 mM for glucose. Interestingly, the KM for glucose was almost 5-fold lower when comparing it to previously measured recombinant PfHK Michaelis-Menten constants [7].
S-Glutathionylation was induced by incubating PfHK with different concentrations of oxidized glutathione (GSSG, 0-10 mM). The enzymatic activity decreased in a dose-dependent manner, and we observed a time-dependent inhibition of PfHK activity. After 30 minutes of incubation, its enzymatic activity was reduced by more than 70% (Figure 8 C+D). To induce S-nitrosation, PfHK was incubated with varying concentrations of nitroso-cysteine (Cys-NO, 0-2 mM). Comparable to the effects observed for S-glutathionylation, S-nitrosation inhibits the enzymatic activity of PfHK in a dose- and time-dependent manner (Figure 8 E+F). The incubation with Cys-NO reduced its activity by ~40% after 30 minutes. The inhibitory effect of both modifications can be reversed by adding DTT, which was also shown to remove the signal in anti-glutathione Western blot analysis (Figure 8 G).
Theoretically, PfHK possesses 15 cysteines that could be oxidized. By using Ellman’s reagent, we identified 10-11 cysteines susceptible to oxidation of their sulfur group. This is in accordance with MS analysis of modified PfHK. From the fifteen present cysteines, not all were detected via MS, however, twelve were found to be S-nitrosated. Six out of those modified cysteines were found to be also susceptible to S-glutathionylation, whereby a specific modification pattern could not be determined (Supplemental Information, Figure S3+S4). The present three-dimensional structure further supports these findings: within the tetrameric assembly, putatively eleven cysteines are accessible for oxPTMs. According to this structure, none of the cysteines are located in the active site; therefore, modified cysteines could only indirectly affect the enzymatic activity. C186 and C273 are located adjacent to the glucose binding site (7.5 and 9 Å apart, respectively), but their accessibility for oxPTMs cannot be certainly confirmed. The modification of C77 and C85 could induce conformational changes of beta sheets, potentially resulting in decreased enzymatic activity. Due to a reductant in the crystallization buffer, residues C249 and C260 are reduced in our structure, but they could easily form a disulfide (Supplemental Information, Figure S5).
3. Discussion
In this study, we present the first PfHK structure in a closed state conformation complexed with glucose. Our structure provides novel information about the small domain involved in the unique tetrameric assembly of Plasmodium hexokinases. Among the two monomers within the asymmetric unit, we observed conformational changes in the small domain of monomer A, induced by binding a citrate molecule. The residues involved are found in a Plasmodium-specific insertion (P-insert) and give insights into a so-far unknown mechanism: the binding of citrate seems to trigger a quasi-transition state, similar to the state observed in the structure of human glucokinase in complex with glucose and ATPγS (PDB ID: 3VEY), which shows that binding of ATP causes the catalytic loop to adopt the same conformation as in monomer A with bound citrate (Figure 4 B). Additionally, PfHK is targeted by oxPTMs, more precisely by S-glutathionylation and S-nitrosation that both inhibit the enzymatic activity. Based on the structural analyses, the susceptibility of single cysteines to these modifications is discussed.
In mammalian tissues, four distinct hexokinases have been identified, referred to as type I, II, III, and IV. Mammalian hexokinase type IV is usually termed ‘glucokinase’. While types I-III have a size of ~100 kDa and are believed to have evolved from duplication of the original hexokinase gene, mammalian glucokinase (type IV) is a ~50 kDa isoenzyme [25]. For yeast, two isoenzymes were reported, both being similar to the mammalian glucokinase [20]. The domain organization observed in our structures is well conserved and was previously reported for various organisms [19,26,27]. Among different Plasmodium species, the sequence identities to P. falciparum cover a range from 85.5% (P. relictum) to 99.8% (P. reichenowi) identity (analyzed with NCBI Smart BLAST). Since PvHK (PDB IDs: 6VYG, 6VYF) and PfHK share high sequence and structure identities, many of the interface residues reported within this study confirm the data presented previously for the PvHK tetramer [15]. However, some loops in PvHK are not defined, including the P-insert (PDB IDs: 6VYG, 6VYF), so this region cannot be used for comparison with our data.
Hexokinases consist of two lobes, and upon binding glucose, the lobes move toward each other. During this process, one lobe rotates twelve degrees relative to the other, resulting in movements of the polypeptide backbone of 8 Å [19]. Binding glucose induces the closure of the cleft between the small and large domain, which envelopes the glucose molecule except for the hydroxyl group on the C6 atom, to which a phosphoryl group is subsequently transferred from ATP [11]. After catalysis, a movement of the small domain allows ADP to dissociate from the active site, followed by the release of G6P, which is the preferred order [26,28]. This is consistent with our finding that the P-insert can adopt different conformations, depending on the binding or absence of ligands at the phosphate binding site. PfHK is known to be inhibited by its products ADP and G6P up to 94% and also by high concentrations of its substrate ATP [7]. The residues participating in glucose binding are conserved among different species, involving amino acids from both domains [20,26,27,29]. In contrast to other studies, Liu and colleagues suggested an alternation between different conformational states to ensure efficient substrate binding and product release. Furthermore, they postulate that binding glucose does not inevitably lead to a closed conformation but also induces an active open state [30].
Within this study we could identify glucose and citrate as ligands within the active site of PfHK. The citrate molecule, only bound in monomer A, interacts via hydrogen bonds with residues that are typically involved in phosphate and/or ATP binding [26,31,32]. We suppose that in this study, the presence of 300 mM citrate in the crystallization buffer prevents ATP (10 mM in protein solution, further diluted by crystallization buffer) from binding, as well as the subsequent phosphate transfer to glucose, which could be the reason why we did not observe an electron density for ATP. Supplying the enzyme solution with both substrates upon crystallization certainly is an uncommon approach, since one would expect catalysis that leads to an instable enzyme and failure of crystallization. We have tried various combinations of ligands in the enzyme solutions, including ATP analogues, ADP, and G6P, without success in achieving crystal growth. Only the combination of glucose and ATP yielded crystals.
Not much is known about the regulatory impact of citrate on hexokinases: There are only scattered studies suggesting citrate activates mammalian and yeast HK, and, to the best of our knowledge, citrate is reported to inhibit noncompetitively only one hexokinase, namely from Aspergillus niger [33,34,35,36]. Nevertheless, the phosphate binding site is well conserved, which conflicts with common criteria of promising drug targets and excludes citrate to be a candidate for inhibitor design. Moreover, the high citrate concentration in the crystallization buffer supports the hypothesis of its random binding rather than having a physiological significance. We hypothesize the citrate-induced state of monomer A to mimic the hexokinase conformation upon the binding of ATP. The new position of the catalytic loop (G104-F108; Figure 4 A) in monomer A forces the P-insert (L130-G141) to rearrange. The movement of both loops seems to be ATP-specific, since in structures complexed with ADP, G6P, or glucose, the corresponding catalytic loop adopts the pre-catalytic state (Figure 4 A-C).
Hexokinases I-III are active as monomers, consisting of N- and C-terminal halves connected via a transition helix, forming a pseudo-dimer. [26,37]. For yeast hexokinases, both monomers and dimers have been reported and also Sulfulobus tokodaii hexokinase exists as a dimer, whereby the subunits are closely connected and therefore differ from the mammalian HK pseudo-dimers [20,29,38,39]. In type I and III hexokinases, the C-termini are catalytically relevant, whereas the N-terminal halves fulfill a regulatory role. Both termini are known to communicate via conformational changes. Upon ligation of G6P at the N-terminal half, for example, conformational changes lead to the inhibition of catalysis at the C-terminus via an allosteric mechanism [26,32,37,40,41]. We hypothesize that the two different conformational changes within the PfHK subunits adopt a similar way of communication among the different monomers, probably mediated via displaced interface residues. This could explain how PfHK is regulated upon catalysis, in addition to switching between the open and the closed conformation: The subunits putatively alternate substrate binding which could be why citrate is bound only in one subunit of the present dimer. In subunit B, where solely glucose is bound, residues L130-S135 (small domain), that are part of the P-insert, are in contact with region P304-Y312 (large domain), which in turn is part of the interface AB (Figure 4; Figure 7 B). In monomer A with bound citrate, this contact is lost, resulting in an altered AB interface and thus also affecting the neighboring molecule in the tetramer. This suggests an allosteric modulation, affecting the enzyme’s activity and explaining that citrate, which mimics ATP binding, is bound only in one subunit. However, this hypothesis requires further kinetic and structural analyses to show a potential regulatory mechanism.
In previous studies, PfHK was kinetically characterized and found to share various similarities with other hexokinases. The products G6P and ADP regulate the enzyme via a feedback mechanism, and ATP was shown to inhibit its activity [7]. The PfHK KM value for glucose obtained in this study was lower than those obtained in previous studies [7,12]. Interestingly, the KM for glucose measured for Cryptosporidium parvum and Toxoplasma gondii hexokinase (CpHK, TgHK) were similarly low or even lower (Table 2). A lower KM for glucose suggests low glucose concentrations within the parasite and is crucial for its survival [42,43]. However, one should note that there are minor deviations concerning the assay conditions, for example the pH values that range from 7.0 to 7.9 in the different studies, which could also lead to changed enzymatic characteristics. It has already been shown that in infected erythrocytes, hexokinase activity and glucose consumption equally are significantly increased. Consequently, this increase affects the levels of reduced glutathione (GSH) by providing NADPH for downstream redox reactions leading to a 25-fold increase of the production of GSH [12].
Interfering with the parasite’s antioxidative defense system is characteristic not only for drugs currently used in clinical practice but also for novel strategies of drug development [44,45,46,47]. Within this study, we showed for the first time that PfHK is inhibited upon both S-glutathionylation and S-nitrosation. Probably, only 10-11 cysteines are accessible within the three-dimensional structure. Cysteines near the active site (C77, C85, C186, C273) or cysteines within the tetrameric interface (C193, C346) seem likely to be modified, induce conformational changes, and thus impair proper catalysis. Interestingly, C193 is highly conserved among Plasmodium species, whereas C346 is restricted to P. falciparum and six other species infecting chimpanzees and gorillas (Supplemental Information, Figure S6). As already mentioned, C249 and C260 would allow the formation of a disulfide but were reduced in the present structure, most probable due to presence of DTT in the crystallization buffer. This disulfide would not only be more susceptible for oxPTMs and subsequent enzymatic regulation but could also be the target for redox-active proteins. Indeed, PfHK was found to be a target of thioredoxin, glutaredoxin, and the Plasmodium-specific plasmoredoxin, further indicating its importance in redox regulatory processes within the parasite [48].
Besides their important role in glycolysis, hexokinases are reported to be moonlighting proteins, which is a term for proteins performing different independent functions. These functions include the regulation of redox signaling or performing as a gene repressor, as a protein kinase, or as an immune receptor. It is known that mammalian, yeast, plant, and numerous parasitic hexokinases are posttranslationally modified as a response to environmental changes [49,50]. Many hexokinases are known to be S-nitrosated, and S-nitrosation was also predicted for PfHK [18]. Depending on the organism studied, S-nitrosation either can inhibit hexokinase activity or does not affect the enzymatic activity. Also, the incubations with HK substrates can have different effects: incubations with glucose prior to the modification prevented from inhibition, whereas incubations with ATP can either prevented, or increase an inhibition. An increase is possibly mediated by conformational changes that increase the exposure of reactive thiols to NO-donors [51,52,53].
Numerous studies provide data about S-glutathionylation of glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) and other glycolytic enzymes that this modification targets, but the effect of S-glutathionylation on hexokinases was scarcely known. S-glutathionylation patterns in P. falciparum have already been thoroughly studied and indicate redox regulation in the malaria parasite: GAPDH and pyruvate kinase were shown to be inhibited by S-glutathionylation [17], which is comparable to the present data on PfHK. The present study provides additional data that is consistent with those previous findings.
The fact that P. falciparum vitally depends on glucose as an energy supply and that PfHK is putatively massively involved in and controlled by redox regulatory processes makes PfHK a very promising drug target by combining two strategies. Moreover, the quaternary organization of Plasmodium hexokinases is unique and potentially provides unknown mechanisms of enzymatic activity regulation [15]. Due to this uniqueness, the present structure of PfHK and the novel insights into the mechanism and the potential redox regulation provide a good basis for designing new Plasmodium-specific inhibitors.
4. Materials and Methods
Heterologous Expression and Purification of Recombinant PfHK
Due to differences in codon-usage among P. falciparum and E. coli, the hexokinase gene sequence was ordered codon optimized for E. coli from General Biosystems (PlasmoDB ID: PF3D7_0624000). The sequence was cloned into pET30a-c(+) with NdeI and XhoII, leading to a C-terminal His6-tag. Heterologous expression was performed in C41(DE3) E. coli cells. Cell were grown to an OD600 of 0.6 and induced with 500 µM IPTG. Expression was performed for 18 h at 25 °C. Cells were harvested at 10,000 g and 4 °C for 15 min. After adding a protease inhibitor cocktail (4 nM cystatin, 150 nM pepstatin, 100 µM phenylmethylsulfonyl fluoride), cells were stored in 50 mM HEPES, pH 8.0, 300 mM NaCl, 20 mM imidazole, and 10% glycerol at -20 °C until lysis. Cells were lysed with 1 mg DNase, 16 mg lysozyme, 30% glycerol and 2% triton X-100 with a total volume of 50 mL per liter E. coli culture, which was adjusted with storage buffer. Lysis was conducted over night for at least 14 h at 4 °C. After three cycles of sonication (30 s, 4 °C, 70% power), cells were centrifuged (30 min, 25,000 g, 4 °C). The supernatant was applied to a Talon® metal affinity resin (Clontech, Heidelberg, Germany), using 1 ml resin per liter E. coli culture (= 1 column volume = CV). To exclude unspecifically bound proteins, the column was washed with 20 CV storage buffer before eluting hexokinase with 50, 100, and 200 mM imidazole; 4 CV respectively. Eluted fractions were checked for purity with SDS-PAGE, 12% acrylamide, and subsequently concentrated with a Vivaspin 15R centrifugal concentrator (Sartorius, MWCO 30,000 Da) to a final concentration of ~3 mg/ml or ~10 mg/ml for crystallization trials. The final concentration was measured with a Biospectrometer® basic (A280, εPfHK = 74,237.5 M-1 cm-1). Size exclusion chromatography was performed with a HiLoad 16/60 SuperdexTM 200 prepacked column connected to an ÄKTATM pure protein purification system (Cytiva, Germany) to assess the oligomerization behavior of PfHK.
Kinetic Characterization of PfHK
The activity of PfHK was measured by observing the increase of NADPH at 340 nm at 25 °C in an assay coupled to glucose-6-phosphate dehydrogenase (G6PD). The standard assay contained 15 mM D-glucose, 4 mM ATP, 1 mM NADP+, 5 U/mL G6PD (from baker’s yeast, Merck, Germany) in 100 mM triethanolamine, pH 7.5 and 10 mM MgCl2. For determining Michaelis-Menten constants, ATP was used in concentrations ranging from 0.1 to 5 mM, D-glucose was used 0.02 to 30 mM. The data was analyzed using GraphPad Prism 8 (see also ‘Data analyses and statistics’). Each value is a mean ± SD from at least three independent determinations using different biological batches of enzyme.
Crystallization
Crystals were grown by the sitting drop technique, using 20% PEG 3,350, 200 mM sodium citrate and 0.1 M sodium citrate buffer with a pH ranging from 3.4 to 4.0. The protein solution contained 5-8 mg·ml-1 PfHK, 10 mM glucose, 10 mM ATP and 5 mM DTT. As cryoprotectant, 25% ethylene glycol was used.
Data Collection and Refinement
All diffraction data was recorded at 100 K by using synchrotron radiation at the Swiss Light Source in Villigen, Switzerland (beamline X10SA, Detector: EIGER). Prior to data collection the crystals were soaked in mother liquor containing 25% ethylene glycol. The data sets were processed with XDS [54], which includes routines for space group determination. The trigonal crystals diffracted up to 2.8 Å.
The structure was solved via molecular replacement methods. The search model was generated vial homology modelling with SWISS-MODEL [55]. The open (PDB ID: 6VYF) and closed (PDB ID: 6VYG) forms of HK from P. vivax, which has 89.6% sequence identity to PfHK, were used as templates. The correct space group (P3121, with two monomers in the asymmetric unit) could be determined via molecular replacement searches using the closed form of PvHK. The initial Rfree was 47.8%, which dropped to 28% after several rounds of refinement and manual rebuilding. The final statistics are shown in Table 1. Molecular replacement and structure refinement were carried out with the PHENIX program suite [56,57], and the interactive graphics program Coot [58] was used for model building. Manually rebuilding and subsequent refinement resulted in a PfHK model, comprising residues I25 to N486, one glucose and one citrate molecule in subunit A and residues I23 to I489 and one glucose molecule in subunit B. The structure was well defined by the 2fofc electron density, except some side chains at the surface and residues G134 to S137 of subunit B.
Gel filtration Chromatography
To remove excess chemicals like DTT, GSSG, or S-nitrosocysteine (CysNO), gel filtration chromatography was conducted. For this purpose, P6 gel powder (Biorad, Feldkirchen, Germany) with a fractionation range from 1,000 to 6,000 Da was soaked for at least 1 h in PfHK storage buffer. Afterwards, 2 ml columns were loaded with the pre-soaked gel, centrifuged at 1,000 g for 2 min at 4 °C, and the flow-through was discarded. To achieve efficient desalting of protein samples, the remaining volume of gel after centrifugation should be used in a 5-fold excess over the protein volume. Protein samples were applied, and the columns were centrifuged again to obtain desalted protein in the flow-through. After each desalting step, the protein concentration was determined again.
Glutathionylation
To ensure all cysteines are reduced, PfHK was incubated with 5 mM DTT for 30 min at 4 °C. Excess DTT was removed via gel filtration chromatography as described above. Afterwards, 1 mg/ml PfHK was incubated with increasing concentrations (0 to 10 mM) of oxidized glutathione (GSSG) at 37 °C for 10 min. To stop the reaction and remove excess GSSG, the samples were immediately desalted after the respective incubation and kept at 25 °C until used for Western blot or enzymatic analysis. To investigate the time course of glutathionylation-mediated inhibition, 5 mM GSSG was used for incubations, and an aliquot was taken out to assess the enzymatic activity at different time points (0, 5, 10, 20, 30 min). To validate glutathionylation with Western blot analyses, protein samples were prepared with sample buffer and directly applied to SDS-PAGE followed by semi-dry Western blot with a glutathione antibody (Abcam, Germany; Glutathione antibody [D8] (ab19534); diluted 1:250 in TBST).
Synthesis of S-Nitrosocysteine, Nitrosation and Biotin-Switch Assay
As already described for glutathionylation, PfPK was reduced by adding DTT, which was removed via gel filtration chromatography. Nitrosation of protein samples was induced via incubations with CysNO, which was prepared separately for each experiment. All solutions were freshly solved and kept on ice and in the dark to avoid loss of concentration due to decreased stability. For synthesizing 95.24 mM CysNO, 250 mM sodium nitrite (Roth, Karlsruhe, Germany) was incubated with 250 mM L-cysteine-hydrochloride-monohydrate and 1 M HCl in a 4:4:1 ratio for at least 5 min. Before starting the nitrosation, the CysNO solution had to be neutralized with 1 M NaOH (ratio 6:1) to a final pH of 7.5. The final CysNO solution had an orange-red color. To study nitrosation, 1 mg/ml pre-reduced PfHK was incubated with different concentrations of CysNO, ranging from 0 to 5 mM or 1 mM for time course experiments. The incubations were conducted at 25 °C in the dark. To stop the reaction and investigate the enzymatic activity, samples were desalted. If the samples were analyzed via Western blot, the reaction was stopped by adding 100% ice-cold acetone followed by protein precipitation for 30 min at -20 °C. Samples were applied to a biotin-switch assay, which can indirectly detect the presence of nitroso groups within protein samples and was previously described in detail [18]. To describe the assay in brief, the samples were washed with 70% ice-cold acetone to remove excess CysNO, followed by blocking unmodified cysteines with 0.2 M iodoacetamide in blocking buffer (8 M urea, 50 mM Tris pH 8.0, 1 mM EDTA, 0.1 mM neocuproine) for 1 h at 50 °C to alkylate the thiols. Again, the incubation was stopped by adding acetone, protein precipitation at -20 °C, and three washing steps. During the next incubation, conducted in labeling buffer (4 M urea, 50 mM Tris pH 8.0, 1 mM EDTA, 0.01 neocuproine), denitrosation of thiols was achieved by adding 20 mM sodium ascorbate. Simultaneously, the denitrosated thiols were labeled with 0.2 mM iodoacetyl-PEG2-biotin for 1 h at 25 °C in the dark. Samples without sodium ascorbate served as negative controls in addition to samples without CysNO. The reaction was again stopped with acetone, and after protein precipitation and the washing steps, protein pellets were resuspended in blocking buffer and SDS-PAGE sample buffer. Finally, semi-dry Western blot analysis with a biotin antibody (Santa Cruz, USA; Biotin Antibody (33): sc-101,339; diluted 1:1000 in 5% nonfat milk in TBST) was performed to detect nitrosation.
Mass Spectrometry Analysis
To identify cysteines that are sensitive for oxPTMs, reduced PfHK was incubated with varying concentrations of GSSG and Cys-NO as described above. The MALDI-TOF MS analyses were performed at the Core Facility of Mass Spectrometry and Elemental Analysis, Philipps University Marburg (timsTOF Pro (Bruker), software: PEAKS).
Data Analyses and Statistics
To kinetically evaluate modified PfHK and to calculate Michaelis-Menten constants and Vmax, we used Microsoft Excel and GraphPad Prism 8. The elution profile of size exclusion chromatography was analyzed with UnicornTM 7.0, the control and evaluation software of the ÄKTATM system, also provided by Cytiva. Chemiluminescent imaging of Western blot analysis was conducted with ChemoStar ECL and a fluorescent imager (Intas, Göttingen, Germany).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization (KB, KFW, MD), data curation (MD, KFW, ADW, ASV); formal analysis (MD, KFW); funding acquisition (KB); investigation (MD); methodology (MD, KFW, SR); project administration (MD); resources (KB, KFW); software (MD, KFW); supervision (KB, KFW); validation (MD, ASV); visualization (MD); writing - original draft (MD); writing - review & editing (KFW, MD, ADW, SR).
Funding
This research was funded by the German Research Foundation (Grant BE1540/23-2 within the DFG Priority Program 1710 on Thiol Switches) and the LOEWE Center DRUID (Project E3) within the Hessian Excellence Program.
Data Availability Statement
Coordinates and measured reflection amplitudes have been deposited in the Worldwide Protein Data Bank RCSB PDB (http://pdb.org): code 7ZZI for PfHK.
Acknowledgments
The authors wish to thank Jude Przyborski for his support in revising the manuscript and Michaela Stumpf for her excellent technical assistance. Furthermore, they would like to thank Ilme Schlichting and Miroslaw Tarnawski for the thoughtfully arranged data collection. Diffraction data was collected at beamline X10SA, Swiss Light Source, Paul Scherrer Institute, Villigen, Switzerland, and the authors thank the beamline staff for the excellent setup.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization World Malaria Report 2022; 2022;
- Blasco, B.; Leroy, D.; Fidock, D.A. Antimalarial Drug Resistance: Linking Plasmodium Falciparum Parasite Biology to the Clinic. Nat Med 2017, 23, 917–928. [Google Scholar] [CrossRef]
- Nsanzabana, C.; Ariey, F.; Beck, H.-P.; Ding, X.C.; Kamau, E.; Krishna, S.; Legrand, E.; Lucchi, N.; Miotto, O.; Nag, S.; et al. Molecular Assays for Antimalarial Drug Resistance Surveillance: A Target Product Profile. PLoS One 2018, 13, e0204347. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K. Glucose Uptake in Plasmodium Falciparum-Infected Erythrocytes Is an Equilibrative Not an Active Process. Mol Biochem Parasitol 1996, 82, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Moxon, C.A.; Gibbins, M.P.; McGuinness, D.; Milner, D.A.; Marti, M. New Insights into Malaria Pathogenesis. Annu Rev Pathol 2020, 15, 315–343. [Google Scholar] [CrossRef]
- Roth, E.F.; Calvin, M.C.; Max-Audit, I.; Rosa, J.; Rosa, R. The Enzymes of the Glycolytic Pathway in Erythrocytes Infected with Plasmodium Falciparum Malaria Parasites. Blood 1988, 72, 1922–1925. [Google Scholar] [CrossRef]
- Harris, M.T.; Walker, D.M.; Drew, M.E.; Mitchell, W.G.; Dao, K.; Schroeder, C.E.; Flaherty, D.P.; Weiner, W.S.; Golden, J.E.; Morris, J.C. Interrogating a Hexokinase-Selected Small-Molecule Library for Inhibitors of Plasmodium Falciparum Hexokinase. Antimicrob Agents Chemother 2013, 57, 3731–3737. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Patrick, S.L.; Blanding, W.M.; Dwivedi, V.; Suryadi, J.; Golden, J.E.; Coussens, N.P.; Lee, O.W.; Shen, M.; Boxer, M.B.; et al. Identification of Novel Plasmodium Falciparum Hexokinase Inhibitors with Antiparasitic Activity. Antimicrob Agents Chemother 2016, 60, 6023–6033. [Google Scholar] [CrossRef]
- Hurley, J.H. The Sugar Kinase/Heat Shock Protein 70/Actin Superfamily: Implications of Conserved Structure for Mechanism. Annu Rev Biophys Biomol Struct 1996, 25, 137–162. [Google Scholar] [CrossRef]
- Nishimasu, H.; Fushinobu, S.; Shoun, H.; Wakagi, T. Identification and Characterization of an ATP-Dependent Hexokinase with Broad Substrate Specificity from the Hyperthermophilic Archaeon Sulfolobus Tokodaii. J Bacteriol 2006, 188, 2014–2019. [Google Scholar] [CrossRef]
- Roy, S.; Vivoli Vega, M.; Harmer, N.J. Carbohydrate Kinases: A Conserved Mechanism across Differing Folds. Catalysts 2019, 9, 29. [Google Scholar] [CrossRef]
- Roth, E.F. Malarial Parasite Hexokinase and Hexokinase-Dependent Glutathione Reduction in the Plasmodium Falciparum-Infected Human Erythrocyte. J Biol Chem 1987, 262, 15678–15682. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, K.; Berneburg, I.; Jortzik, E.; Hahn, J.; Rahbari, M.; Schulz, N.; Preuss, J.; Zapol’skii, V.A.; Bode, L.; Pinkerton, A.B.; et al. Glucose 6-Phosphate Dehydrogenase 6-Phosphogluconolactonase: Characterization of the Plasmodium Vivax Enzyme and Inhibitor Studies. Malar J 2019, 18, 22. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome Sequence of the Human Malaria Parasite Plasmodium Falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef]
- Srivastava, S.S.; Darling, J.E.; Suryadi, J.; Morris, J.C.; Drew, M.E.; Subramaniam, S. Plasmodium Vivax and Human Hexokinases Share Similar Active Sites but Display Distinct Quaternary Architectures. IUCrJ 2020, 7, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Miseta, A.; Csutora, P. Relationship between the Occurrence of Cysteine in Proteins and the Complexity of Organisms. Mol Biol Evol 2000, 17, 1232–1239. [Google Scholar] [CrossRef]
- Kehr, S.; Jortzik, E.; Delahunty, C.; Yates, J.R.; Rahlfs, S.; Becker, K. Protein S-Glutathionylation in Malaria Parasites. Antioxid Redox Signal 2011, 15, 2855–2865. [Google Scholar] [CrossRef]
- Wang, L.; Delahunty, C.; Prieto, J.H.; Rahlfs, S.; Jortzik, E.; Yates, J.R.; Becker, K. Protein S-Nitrosylation in Plasmodium Falciparum. Antioxidants & Redox Signaling 2014, 20, 2923–2935. [Google Scholar] [CrossRef]
- Bennett, W.S.; Steitz, T.A. Glucose-Induced Conformational Change in Yeast Hexokinase. Proc Natl Acad Sci U S A 1978, 75, 4848–4852. [Google Scholar] [CrossRef]
- Kuser, P.; Cupri, F.; Bleicher, L.; Polikarpov, I. Crystal Structure of Yeast Hexokinase PI in Complex with Glucose: A Classical “Induced Fit” Example Revised. Proteins 2008, 72, 731–740. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-Glutathionylation in Protein Redox Regulation. Free Radic Biol Med 2007, 43, 883–898. [Google Scholar] [CrossRef]
- Jortzik, E.; Wang, L.; Becker, K. Thiol-Based Posttranslational Modifications in Parasites. Antioxid Redox Signal 2012, 17, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Groitl, B.; Jakob, U. Thiol-Based Redox Switches. Biochim Biophys Acta 2014, 1844, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Schipper, S.; Wu, H.; Furdui, C.M.; Poole, L.B.; Delahunty, C.M.; Park, R.; Yates, J.R.; Becker, K.; Przyborski, J.M. Identification of Sulfenylation Patterns in Trophozoite Stage Plasmodium Falciparum Using a Non-Dimedone Based Probe. Mol Biochem Parasitol 2021, 242, 111362. [Google Scholar] [CrossRef]
- Wilson, J.E. Isozymes of Mammalian Hexokinase: Structure, Subcellular Localization and Metabolic Function. J Exp Biol 2003, 206, 2049–2057. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, A.E.; Zeng, C.; Bartunik, H.D.; Fromm, H.J.; Honzatko, R.B. Regulation of Hexokinase I: Crystal Structure of Recombinant Human Brain Hexokinase Complexed with Glucose and Phosphate. J Mol Biol 1998, 282, 345–357. [Google Scholar] [CrossRef]
- He, C.; Chen, J.; Wang, H.; Wan, Y.; Zhou, J.; Dan, Z.; Zeng, Y.; Xu, W.; Zhu, Y.; Huang, W.; et al. Crystal Structures of Rice Hexokinase 6 with a Series of Substrates Shed Light on Its Enzymatic Mechanism. Biochem Biophys Res Commun 2019, 515, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Kosow, D.P.; Rose, I.A. Product Inhibition of the Hexokinases. J Biol Chem 1970, 245, 198–204. [Google Scholar] [CrossRef]
- Kuettner, E.B.; Kettner, K.; Keim, A.; Svergun, D.I.; Volke, D.; Singer, D.; Hoffmann, R.; Müller, E.-C.; Otto, A.; Kriegel, T.M.; et al. Crystal Structure of Hexokinase KlHxk1 of Kluyveromyces Lactis: A Molecular Basis for Understanding the Control of Yeast Hexokinase Functions via Covalent Modification and Oligomerization. J Biol Chem 2010, 285, 41019–41033. [Google Scholar] [CrossRef]
- Liu, S.; Ammirati, M.J.; Song, X.; Knafels, J.D.; Zhang, J.; Greasley, S.E.; Pfefferkorn, J.A.; Qiu, X. Insights into Mechanism of Glucokinase Activation: Observation of Multiple Distinct Protein Conformations. J Biol Chem 2012, 287, 13598–13610. [Google Scholar] [CrossRef]
- Aleshin, A.E.; Kirby, C.; Liu, X.; Bourenkov, G.P.; Bartunik, H.D.; Fromm, H.J.; Honzatko, R.B. Crystal Structures of Mutant Monomeric Hexokinase I Reveal Multiple ADP Binding Sites and Conformational Changes Relevant to Allosteric Regulation. J Mol Biol 2000, 296, 1001–1015. [Google Scholar] [CrossRef]
- Mulichak, A.M.; Wilson, J.E.; Padmanabhan, K.; Garavito, R.M. The Structure of Mammalian Hexokinase-1. Nat Struct Biol 1998, 5, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Kosow, D.P.; Oski, F.A.; Warms, J.V.; Rose, I.A. Regulation of Mammalian Hexokinase: Regulatory Differences between Isoenzyme I and II. Arch Biochem Biophys 1973, 157, 114–124. [Google Scholar] [CrossRef]
- Colowick, S.P. The Hexokinases. In The Enzymes; Boyer, P.D., Ed.; Academic Press, 1973; Vol. 9, pp. 1–48. [CrossRef]
- Kosow, D.P.; Rose, I.A. Activators of Yeast Hexokinase. J Biol Chem 1971, 246, 2618–2625. [Google Scholar] [CrossRef]
- Steinböck, F.; Choojun, S.; Held, I.; Roehr, M.; Kubicek, C.P. Characterization and Regulatory Properties of a Single Hexokinase from the Citric Acid Accumulating Fungus Aspergillus Niger. Biochim Biophys Acta 1994, 1200, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.Y.; Alechina, O.; Aleshin, A.E.; Fromm, H.J.; Honzatko, R.B. Identification of a Phosphate Regulatory Site and a Low Affinity Binding Site for Glucose 6-Phosphate in the N-Terminal Half of Human Brain Hexokinase. J Biol Chem 1998, 273, 19548–19553. [Google Scholar] [CrossRef] [PubMed]
- Hoggett, J.G.; Kellett, G.L. Kinetics of the Monomer-Dimer Reaction of Yeast Hexokinase PI. Biochem J 1992, 287 ( Pt 2), 567–572. [Google Scholar] [CrossRef]
- Nishimasu, H.; Fushinobu, S.; Shoun, H.; Wakagi, T. Crystal Structures of an ATP-Dependent Hexokinase with Broad Substrate Specificity from the Hyperthermophilic Archaeon Sulfolobus Tokodaii. J Biol Chem 2007, 282, 9923–9931. [Google Scholar] [CrossRef]
- Hashimoto, M.; Wilson, J.E. Kinetic and Regulatory Properties of HK I(+), a Modified Form of the Type I Isozyme of Mammalian Hexokinase in Which Interactions between the N- and C-Terminal Halves Have Been Disrupted. Arch Biochem Biophys 2002, 399, 109–115. [Google Scholar] [CrossRef]
- Sebastian, S.; Wilson, J.E.; Mulichak, A.; Garavito, R.M. Allosteric Regulation of Type I Hexokinase: A Site-Directed Mutational Study Indicating Location of the Functional Glucose 6-Phosphate Binding Site in the N-Terminal Half of the Enzyme. Arch Biochem Biophys 1999, 362, 203–210. [Google Scholar] [CrossRef]
- Saito, T.; Maeda, T.; Nakazawa, M.; Takeuchi, T.; Nozaki, T.; Asai, T. Characterisation of Hexokinase in Toxoplasma Gondii Tachyzoites. Int J Parasitol 2002, 32, 961–967. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, H.; Guo, F.; Sun, M.; Zhu, G. A Unique Hexokinase in Cryptosporidium Parvum, an Apicomplexan Pathogen Lacking the Krebs Cycle and Oxidative Phosphorylation. Protist 2014, 165, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Neyaz, M.K.; Ikramul Hasan, S. Exploiting Unique Structural and Functional Properties of Malarial Glycolytic Enzymes for Antimalarial Drug Development. Malar Res Treat 2014. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Tilley, L.; Vennerstrom, J.L.; Roberts, D.; Rogerson, S.; Ginsburg, H. Oxidative Stress in Malaria Parasite-Infected Erythrocytes: Host-Parasite Interactions. Int J Parasitol 2004, 34, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.S.; Carvalho Júnior, A.R.; Lacerda, E.M. da C.B.; da Silva, L.C.N.; Marinho, C.R.F.; André, E.; Fernandes, E.S. Oxidative and Nitrosative Stresses in Cerebral Malaria: Can We Target Them to Avoid a Bad Prognosis? J Antimicrob Chemother 2020, 1363–1373. [Google Scholar] [CrossRef]
- Tiwari, S.; Sharma, N.; Sharma, G.P.; Mishra, N. Redox Interactome in Malaria Parasite Plasmodium Falciparum. Parasitol Res 2021, 120, 423–434. [Google Scholar] [CrossRef]
- Sturm, N.; Jortzik, E.; Mailu, B.M.; Koncarevic, S.; Deponte, M.; Forchhammer, K.; Rahlfs, S.; Becker, K. Identification of Proteins Targeted by the Thioredoxin Superfamily in Plasmodium Falciparum. PLOS Pathogens 2009, 5, e1000383. [Google Scholar] [CrossRef]
- Heneberg, P. Redox Regulation of Hexokinases. Antioxid Redox Signal 2019, 30, 415–442. [Google Scholar] [CrossRef]
- Rodríguez-Saavedra, C.; Morgado-Martínez, L.E.; Burgos-Palacios, A.; King-Díaz, B.; López-Coria, M.; Sánchez-Nieto, S. Moonlighting Proteins: The Case of the Hexokinases. Front Mol Biosci 2021, 8. [Google Scholar] [CrossRef]
- Miller, S.; Ross-Inta, C.; Giulivi, C. Kinetic and Proteomic Analyses of S-Nitrosoglutathione-Treated Hexokinase A: Consequences for Cancer Energy Metabolism. Amino Acids 2007, 32, 593–602. [Google Scholar] [CrossRef]
- Mongin, A.A.; Dohare, P.; Jourd’heuil, D. Selective Vulnerability of Synaptic Signaling and Metabolism to Nitrosative Stress. Antioxid Redox Signal 2012, 17, 992–1012. [Google Scholar] [CrossRef]
- Romero, J.M.; Bizzozero, O.A. Intracellular Glutathione Mediates the Denitrosylation of Protein Nitrosothiols in the Rat Spinal Cord. J Neurosci Res 2009, 87, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr D Biol Crystallogr 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr D Biol Crystallogr 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, N.W.; Tronrud, D.E.; Adams, P.D.; Karplus, P.A. A New Default Restraint Library for the Protein Backbone in Phenix: A Conformation-Dependent Geometry Goes Mainstream. Acta Cryst D 2016, 72, 176–179. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr D Biol Crystallogr 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Secondary-Structure Matching (SSM), a New Tool for Fast Protein Structure Alignment in Three Dimensions. Acta Cryst D 2004, 60, 2256–2268. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera - a Visualization System for Exploratory Research and Analysis. J Comput Chem 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
Figure 1.
PfHK overall fold. (A) The asymmetric unit of PfHK contains two monomers. Each monomer consists of two domains: a small domain (E94-L239) and a large domain (M1-Q93; N240-P490). (B) PfHK monomer A is complexed with glucose (GLC) and citrate (CIT), whereas in monomer B only GLC is bound. Superimposition of both PfHK monomers reveals a shift of two loops: the catalytic loop (C-loop: residues G104-F108) and a Plasmodium-specific insertion (P-insert: residues L130-G141), most likely evoked by the binding of a citrate within monomer A.
Figure 1.
PfHK overall fold. (A) The asymmetric unit of PfHK contains two monomers. Each monomer consists of two domains: a small domain (E94-L239) and a large domain (M1-Q93; N240-P490). (B) PfHK monomer A is complexed with glucose (GLC) and citrate (CIT), whereas in monomer B only GLC is bound. Superimposition of both PfHK monomers reveals a shift of two loops: the catalytic loop (C-loop: residues G104-F108) and a Plasmodium-specific insertion (P-insert: residues L130-G141), most likely evoked by the binding of a citrate within monomer A.
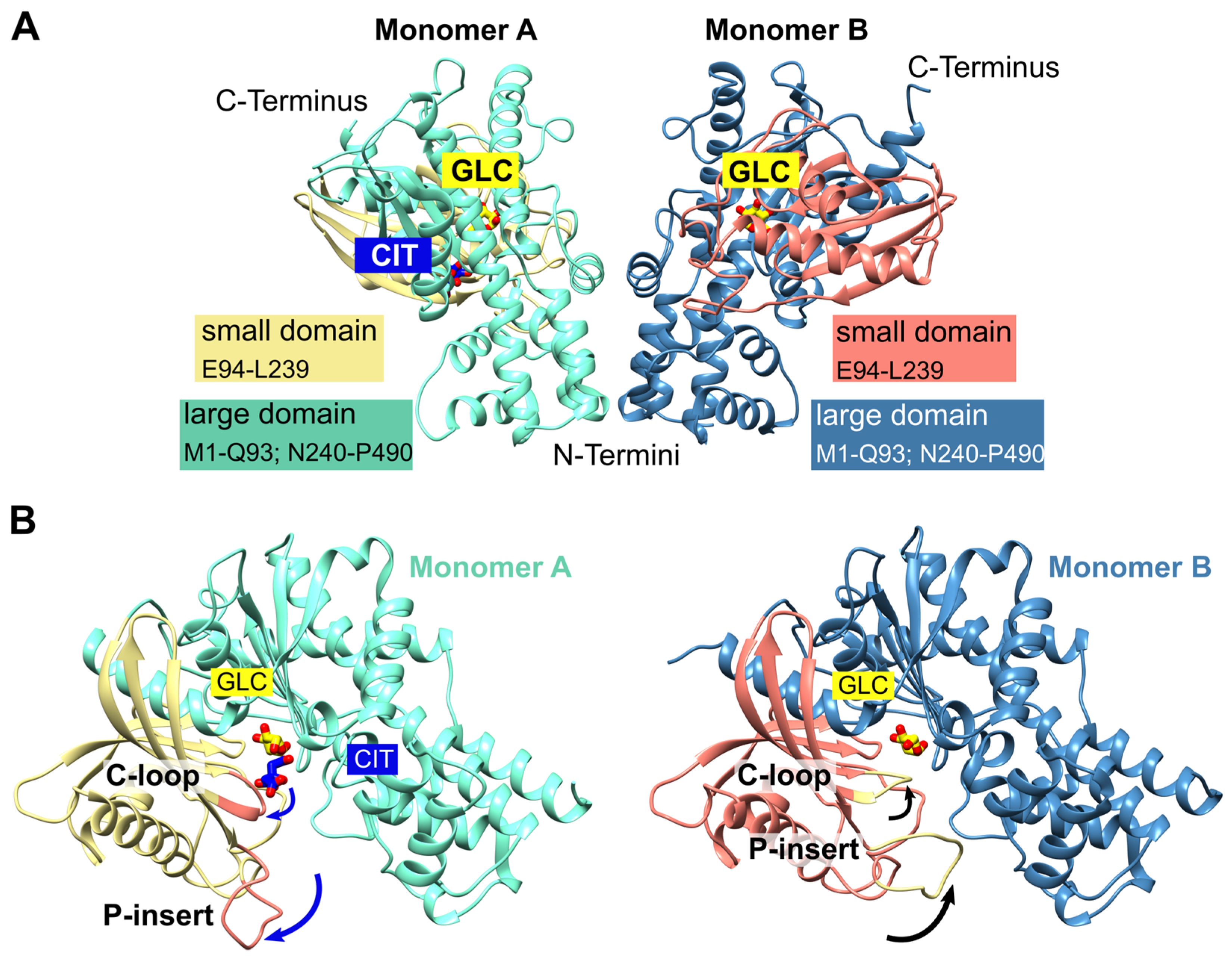
Figure 2.
PfHK monomers provide information about the highly conserved substrate binding sites. (A) Glucose (GLC) is bound within the active site of PfHK monomer B and coordinated by the highlighted residues of both domains (small domain = salmon; large domain = steel blue) that are conserved among different species. (B) Close-up into the active site of PfHK monomer A with GLC and citrate (CIT) bound. Citrate is hydrogen bonded by residues G104, N107, T268 and S436. The final 2fo-fc map, covering the substrates GLC and CIT, is contoured at 2.0 σ. (C) PfHK monomer A was superimposed with two different human hexokinase structures (PDB IDs: 1HKB, dark grey; 1DGK, light grey) to identify the corresponding glucose-6-phosphate (G6P) and ADP/ATP binding sites in PfHK. The ADP/ATP binding site is highlighted in violet. PO4, phosphate.
Figure 2.
PfHK monomers provide information about the highly conserved substrate binding sites. (A) Glucose (GLC) is bound within the active site of PfHK monomer B and coordinated by the highlighted residues of both domains (small domain = salmon; large domain = steel blue) that are conserved among different species. (B) Close-up into the active site of PfHK monomer A with GLC and citrate (CIT) bound. Citrate is hydrogen bonded by residues G104, N107, T268 and S436. The final 2fo-fc map, covering the substrates GLC and CIT, is contoured at 2.0 σ. (C) PfHK monomer A was superimposed with two different human hexokinase structures (PDB IDs: 1HKB, dark grey; 1DGK, light grey) to identify the corresponding glucose-6-phosphate (G6P) and ADP/ATP binding sites in PfHK. The ADP/ATP binding site is highlighted in violet. PO4, phosphate.
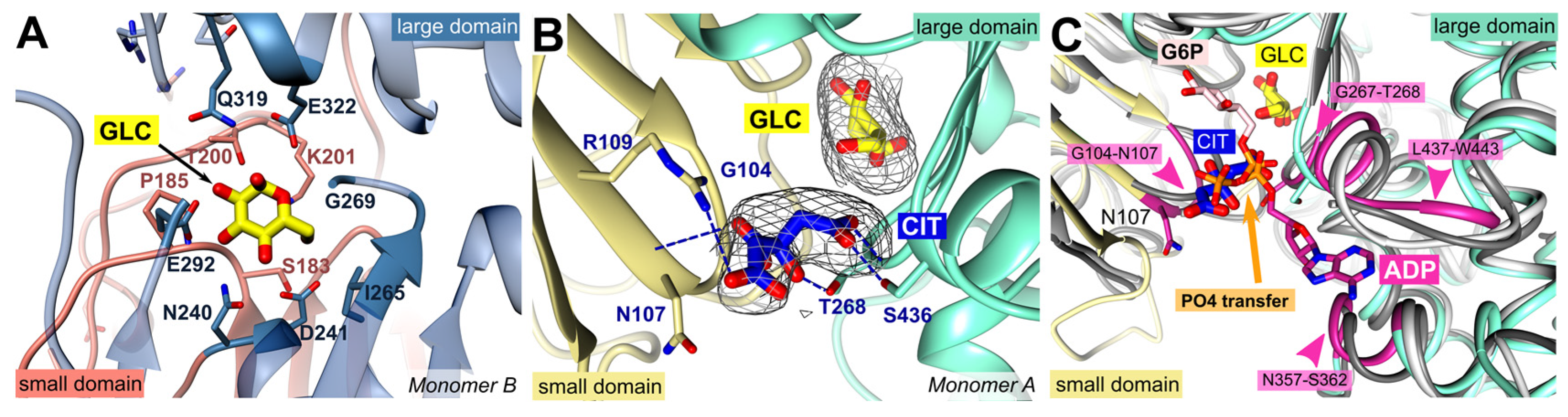
Figure 3.
Structural comparison with hexokinases from H. sapiens and E. tenella. (A) PfHK reveals Plasmodium-specific insertion (P-insert, residues K131-K141) when superimposed with human hexokinase (PDB ID: 1DGK, grey). The human structure is complexed with glucose (GLC) and phosphate (PO4). (B) PfHK monomer A and B were superimposed with two different EtHK structures (PDB IDs: 6KSR, dark grey; 6KSJ light grey) to compare the P-insert and a similar insertion from EtHK. GLA, galactose; GLC, glucose.
Figure 3.
Structural comparison with hexokinases from H. sapiens and E. tenella. (A) PfHK reveals Plasmodium-specific insertion (P-insert, residues K131-K141) when superimposed with human hexokinase (PDB ID: 1DGK, grey). The human structure is complexed with glucose (GLC) and phosphate (PO4). (B) PfHK monomer A and B were superimposed with two different EtHK structures (PDB IDs: 6KSR, dark grey; 6KSJ light grey) to compare the P-insert and a similar insertion from EtHK. GLA, galactose; GLC, glucose.
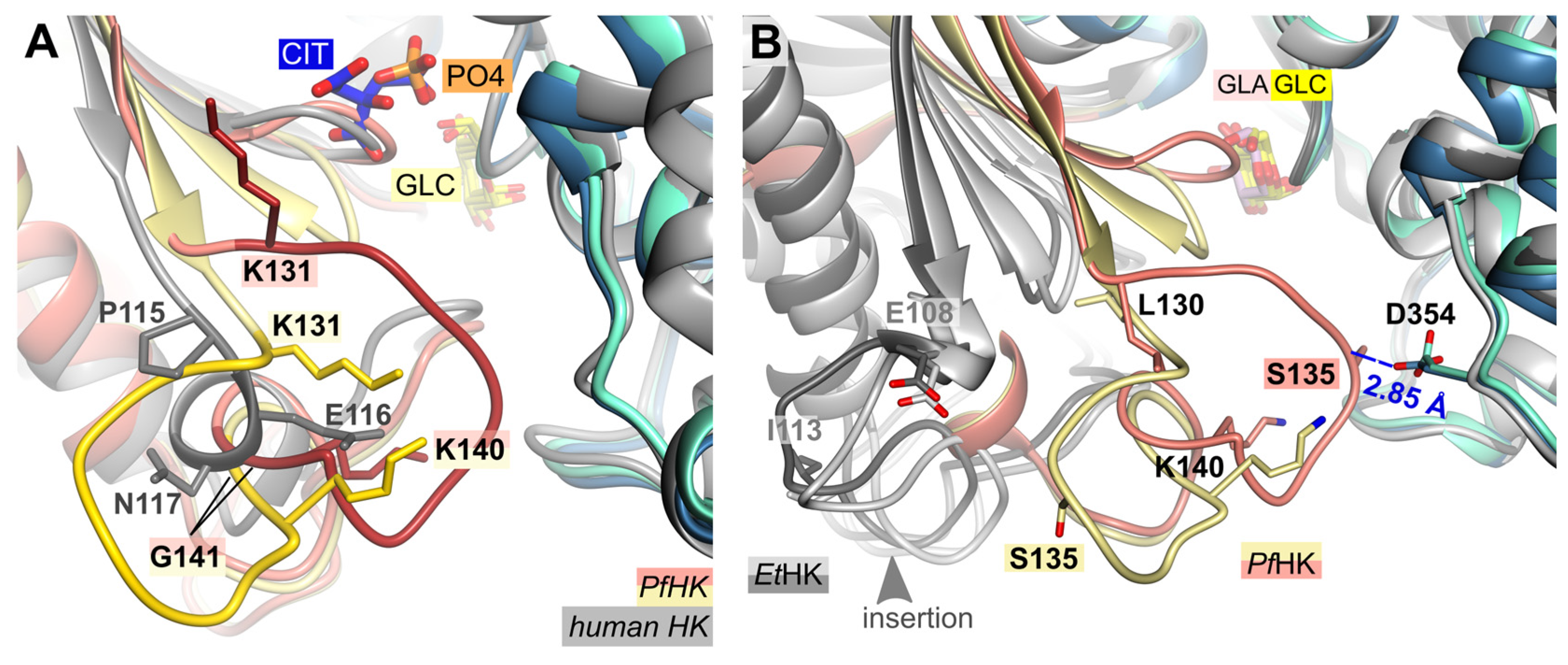
Figure 4.
Conformational changes of the small domains induced by the binding of citrate. The catalytic loop is indicated by T106. (A) Detailed view of the superimposed PfHK monomers A and B shows the 20 Å shift of residue S135 induced by binding citrate (CIT). In the tetrameric assembly, residues P304-Y312 are part of the AB interface. (B) Superimposition of PfHK monomer A with human glucokinase complexed with glucose and ATPγS (PDB ID: 3VEY). (C) Superimposition of PfHK monomer B with human glucokinase complexed with glucose (PDB ID: 3VEV).
Figure 4.
Conformational changes of the small domains induced by the binding of citrate. The catalytic loop is indicated by T106. (A) Detailed view of the superimposed PfHK monomers A and B shows the 20 Å shift of residue S135 induced by binding citrate (CIT). In the tetrameric assembly, residues P304-Y312 are part of the AB interface. (B) Superimposition of PfHK monomer A with human glucokinase complexed with glucose and ATPγS (PDB ID: 3VEY). (C) Superimposition of PfHK monomer B with human glucokinase complexed with glucose (PDB ID: 3VEV).
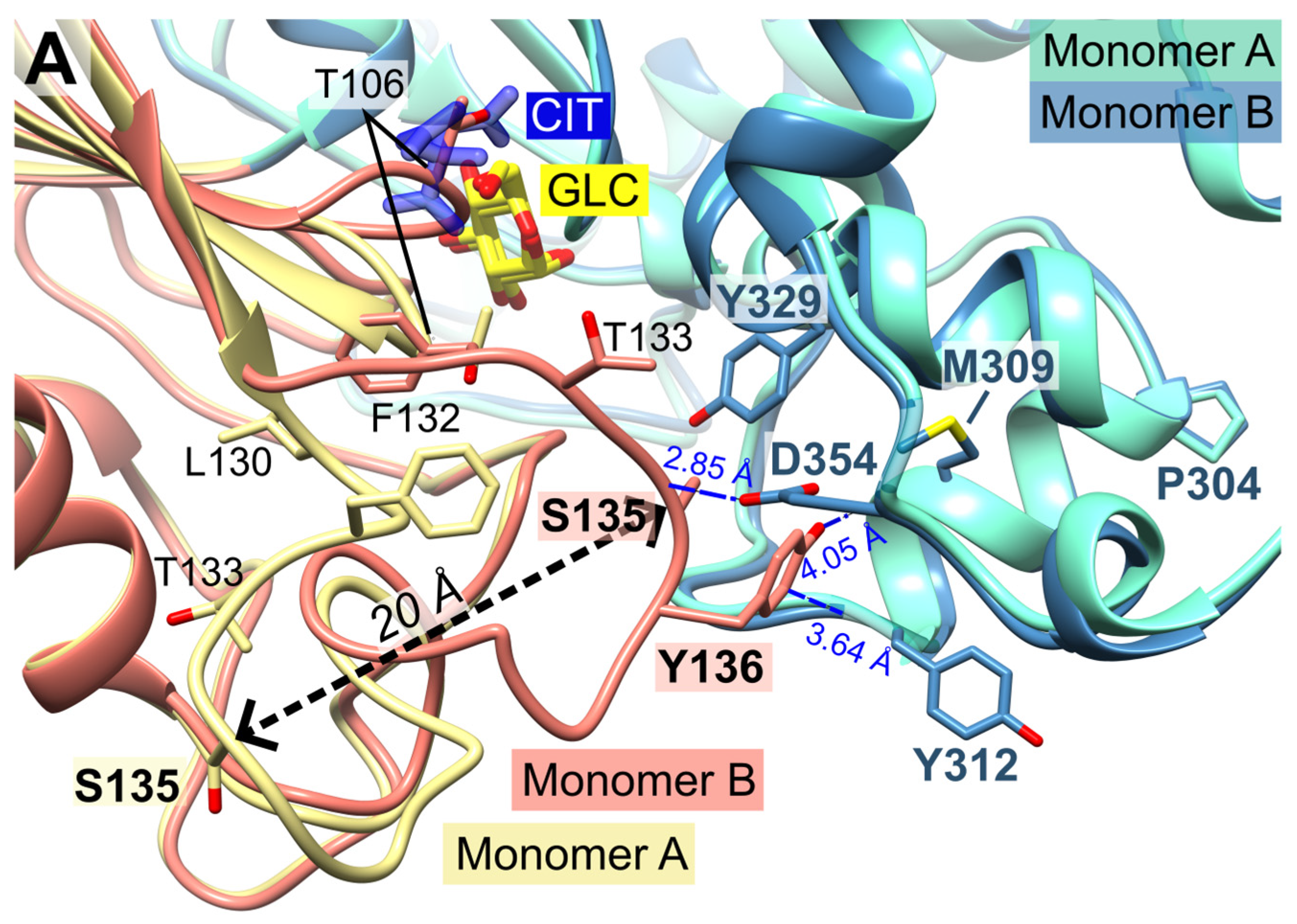
Figure 5.
Proposed mechanism upon substrate binding and product release. After the binding of glucose (GLC), the binding of ATP evokes a conformational change of resides G104-F108 and L130-G141 (shown via superimposition of human glucokinase with bound ATPγS, PDB ID: 3VEY, with PfHK monomers A and B). This broadens the active site and thereby facilitates the release of the products glucose-6-phosphate (G6P) and ADP after the transfer of phosphate.
Figure 5.
Proposed mechanism upon substrate binding and product release. After the binding of glucose (GLC), the binding of ATP evokes a conformational change of resides G104-F108 and L130-G141 (shown via superimposition of human glucokinase with bound ATPγS, PDB ID: 3VEY, with PfHK monomers A and B). This broadens the active site and thereby facilitates the release of the products glucose-6-phosphate (G6P) and ADP after the transfer of phosphate.
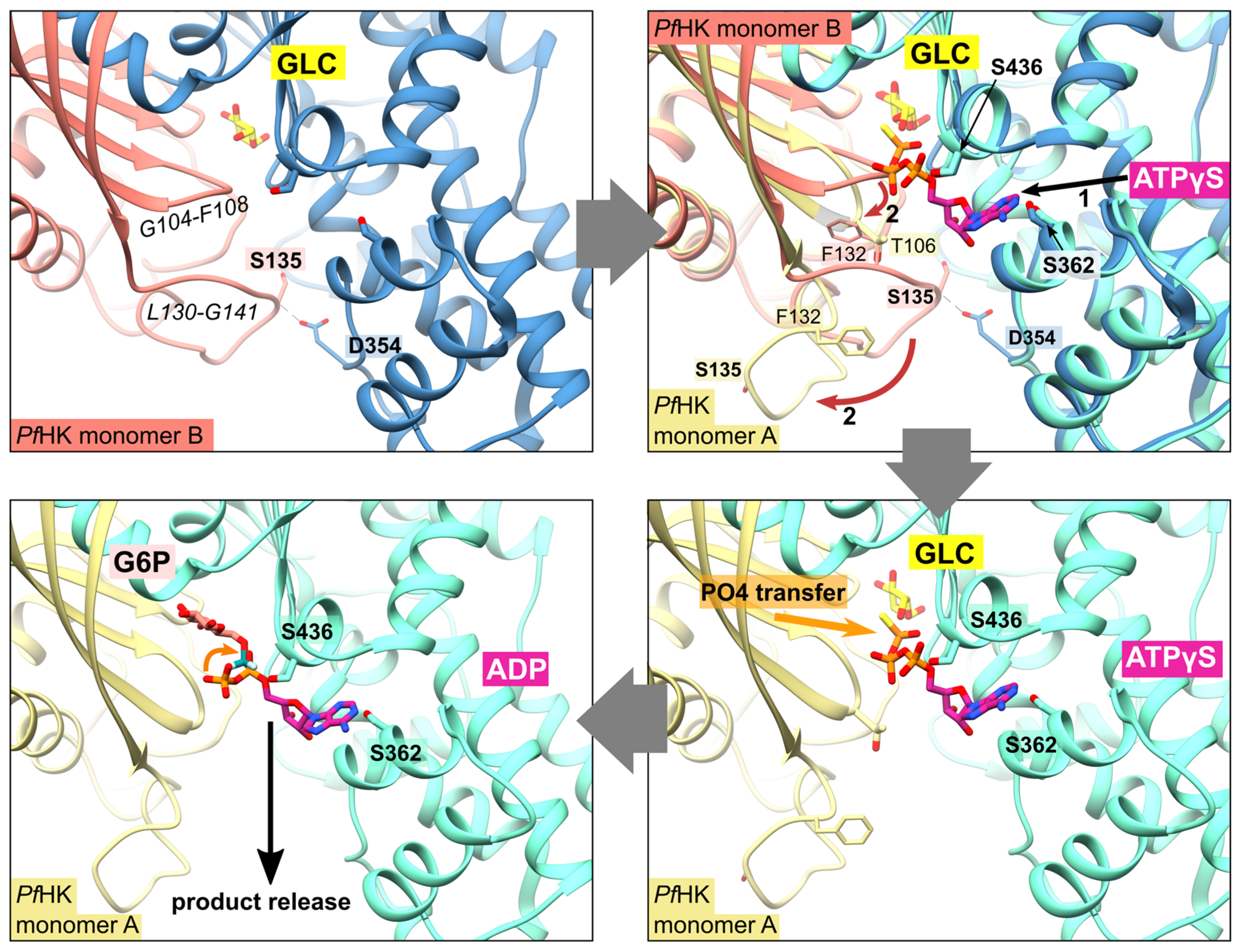
Figure 6.
Tetrameric assembly of PfHK. (A) PfHK tetramer created with a crystallographic symmetry operation. (B) PfHK was purified via Talon® metal affinity chromatography, followed by size exclusion chromatography using an ÄKTATM pure protein purification system, SuperdexTM 200 column. PfHK eluted at ~65 mL. (C) Size exclusion chromatography calibration curve for PfHK. The estimated molecular mass is indicated in the trend line as a pink square, corresponding to ~225 kDa (PfHK monomer = 56.3 kDa, expected PfHK tetramer = 225 kDa).
Figure 6.
Tetrameric assembly of PfHK. (A) PfHK tetramer created with a crystallographic symmetry operation. (B) PfHK was purified via Talon® metal affinity chromatography, followed by size exclusion chromatography using an ÄKTATM pure protein purification system, SuperdexTM 200 column. PfHK eluted at ~65 mL. (C) Size exclusion chromatography calibration curve for PfHK. The estimated molecular mass is indicated in the trend line as a pink square, corresponding to ~225 kDa (PfHK monomer = 56.3 kDa, expected PfHK tetramer = 225 kDa).
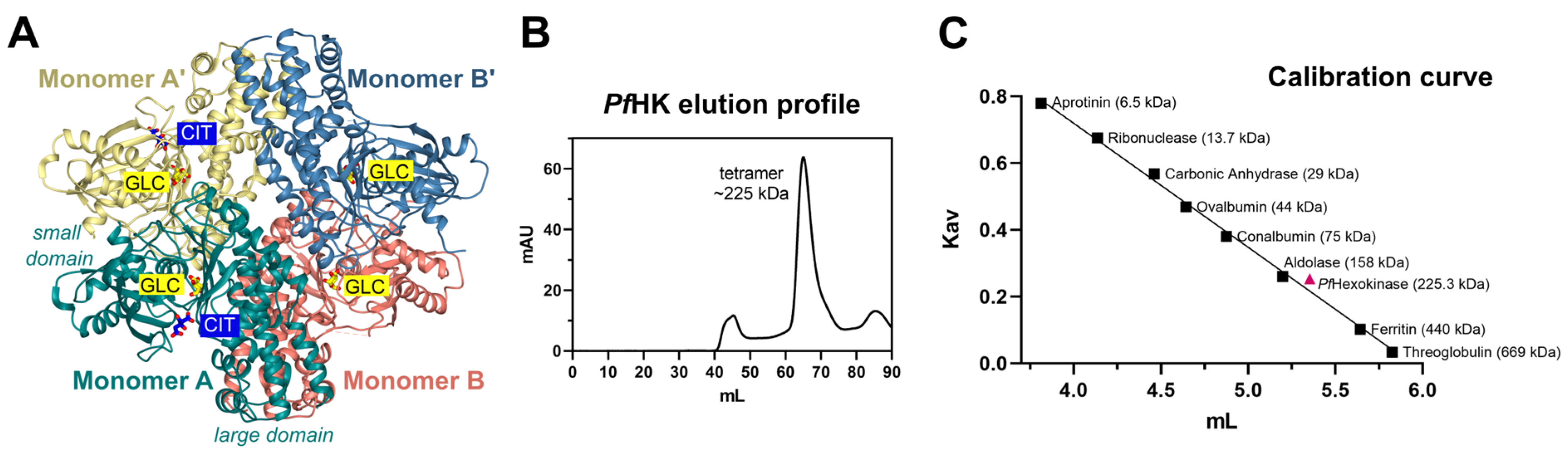
Figure 7.
PfHK unique tetrameric interfaces. (A) The PfHK tetramer has three interaction sites that are highlighted with a blue (A-B’), salmon (B-B’) and green square (A-B), respectively. (B) The A-B interface is strongly connected via seven hydrogen bonds and one additional salt bridge between E28 and K348. Besides, mainly residues from the large domains form the interface. (C) Mainly residues from the small domains connect monomers B-B’. N70 from each monomer can be hydrogen-bonded to I197, D198 and E214. (D) The A-B’ interface is formed via hydrophobic interactions between two helices and a hydrogen bond between N53 and Y56. Distances between the H75 sidechains range from 3.6 to 5.8 Å. Hydrogen bonds are indicated by black dotted lines.
Figure 7.
PfHK unique tetrameric interfaces. (A) The PfHK tetramer has three interaction sites that are highlighted with a blue (A-B’), salmon (B-B’) and green square (A-B), respectively. (B) The A-B interface is strongly connected via seven hydrogen bonds and one additional salt bridge between E28 and K348. Besides, mainly residues from the large domains form the interface. (C) Mainly residues from the small domains connect monomers B-B’. N70 from each monomer can be hydrogen-bonded to I197, D198 and E214. (D) The A-B’ interface is formed via hydrophobic interactions between two helices and a hydrogen bond between N53 and Y56. Distances between the H75 sidechains range from 3.6 to 5.8 Å. Hydrogen bonds are indicated by black dotted lines.
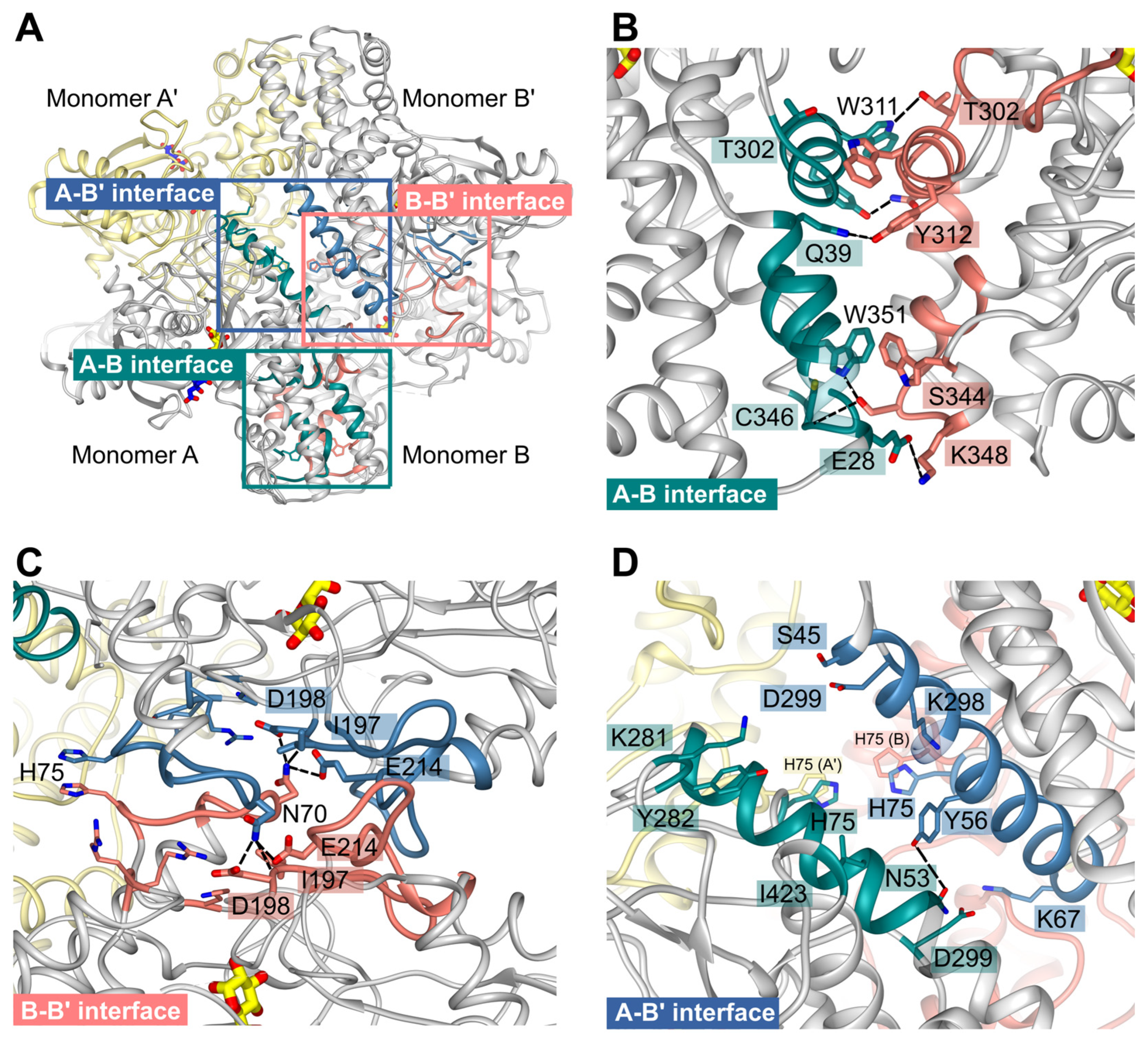
Figure 8.
PfHK is inhibited via S-glutathionylation and S-nitrosation. (A) S-glutathionylated samples from (C) were analyzed via Western blot. S-glutathionylated PfHK was detected with a glutathione antibody. SDS-PAGE loading buffer with DTT served as a chemical negative control. (B) S-nitrosated samples from (E) were analyzed via Western blot. S-nitrosated PfHK was detected with a biotin antibody. Samples without NaAsc (sodium ascorbate) served as a chemical negative control. In both (E) and (F) ~2 µg protein per lane were used and Ponceau staining served as a loading control. (C) Reduced PfHK was incubated with 0-10 mM GSSG for 10 min at 37 °C. The activity of the sample incubated with 0 mM GSSG (control) was defined as 100%. (D) Reduced PfHK was incubated with 0 mM (control) or 5 mM GSSG for 0-30 min at 37 °C. The activity of the control sample at 0 min was defined as 100%. (E) Reduced PfHK was incubated with 0-5 mM Cys-NO for 60 min at 25 °C. The activity of the sample incubated with 0 mM Cys-NO (control) was defined as 100%. (F) Reduced PfHK was incubated with 0 mM (control) or 1 mM Cys-NO for 0-120 min at 25 °C. The activity of the control sample at 0 min was defined as 100%. Values from (C)-(F) are shown as mean values ± SD. Measurements were performed in at least three biologically independent triplicates. (G) Incubation of S-glutathionylated and S-nitrosated PfHK with DTT for 30 min at 4 °C restores its enzymatic activity. Values are shown as mean values ± SD. Measurements were performed in at least three biologically independent triplicates.
Figure 8.
PfHK is inhibited via S-glutathionylation and S-nitrosation. (A) S-glutathionylated samples from (C) were analyzed via Western blot. S-glutathionylated PfHK was detected with a glutathione antibody. SDS-PAGE loading buffer with DTT served as a chemical negative control. (B) S-nitrosated samples from (E) were analyzed via Western blot. S-nitrosated PfHK was detected with a biotin antibody. Samples without NaAsc (sodium ascorbate) served as a chemical negative control. In both (E) and (F) ~2 µg protein per lane were used and Ponceau staining served as a loading control. (C) Reduced PfHK was incubated with 0-10 mM GSSG for 10 min at 37 °C. The activity of the sample incubated with 0 mM GSSG (control) was defined as 100%. (D) Reduced PfHK was incubated with 0 mM (control) or 5 mM GSSG for 0-30 min at 37 °C. The activity of the control sample at 0 min was defined as 100%. (E) Reduced PfHK was incubated with 0-5 mM Cys-NO for 60 min at 25 °C. The activity of the sample incubated with 0 mM Cys-NO (control) was defined as 100%. (F) Reduced PfHK was incubated with 0 mM (control) or 1 mM Cys-NO for 0-120 min at 25 °C. The activity of the control sample at 0 min was defined as 100%. Values from (C)-(F) are shown as mean values ± SD. Measurements were performed in at least three biologically independent triplicates. (G) Incubation of S-glutathionylated and S-nitrosated PfHK with DTT for 30 min at 4 °C restores its enzymatic activity. Values are shown as mean values ± SD. Measurements were performed in at least three biologically independent triplicates.
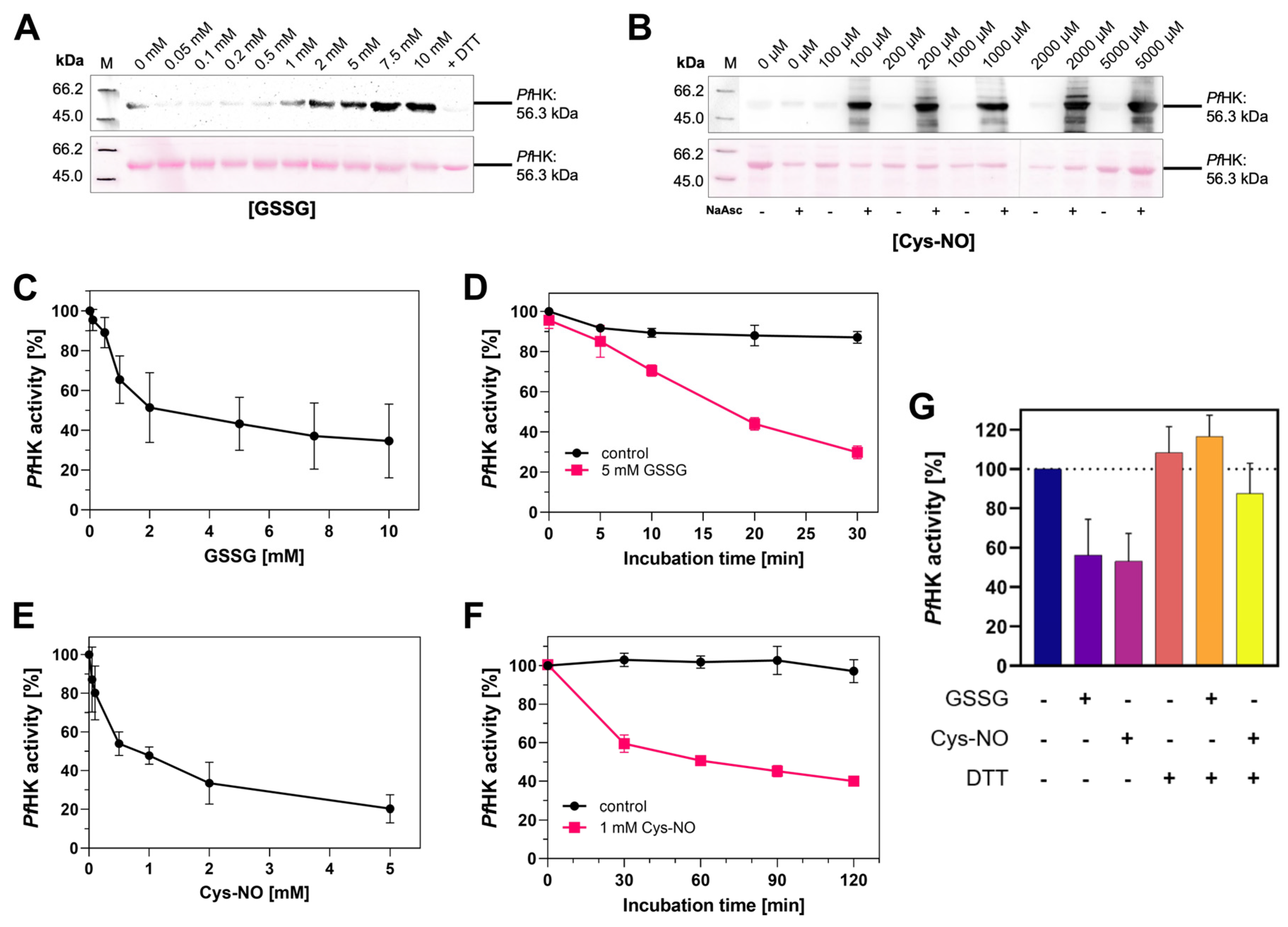

Table 1.
Data collection and refinement statistics.
| PDB accession code | 7ZZI (PfHK) |
| Space group | P3121 |
| Unit cell parameters | |
| a, b, c (Å) | 125.24 125.24 120.93 |
| α, β, γ (°) | 90 90 120 |
| Data collection | |
| Beamline | SLS beam line X10SA |
| Temperature (K) | 100 |
| Resolution range | 49.5 - 2.8 (2.9 - 2.8) |
| Wilson B-factor Å2 | 95.8 |
| Total reflections | 278108 (26474) |
| Unique reflections | 27459 (2644) |
| Multiplicity | 10.1 (10.0) |
| Completeness (%) | 99.7 (97.1) |
| Mean I/σ(I) | 13.1 (0.8) |
| Wilson B-factor | 95.8 |
| R-mergea (%) | 0.12 (3.41) |
| R-pimb (%) | 0.04 (1.12) |
| CC1/2 (%) | 0.999 (0.305) |
| Molecules per ASU | 2 |
| Refinement | |
| Reflections used in refinement | 27456 (2644) |
| Reflections used for R-free | 2746 (264) |
| R-work | 0.199 (0.428) |
| R-free | 0.268 (0.481) |
| protein residues | 930 |
| macromolecules Average B-factor Å2 | 89.9 |
| ligands Average B-factor Å2 | 82.8 |
| solvent Average B-factor Å2 | 82.2 |
| RMS deviations | |
| Bonds (Å) | 0.01 |
| Angles (°) | 1.17 |
| Ramachandran favored (%) | 96.11 |
1 Statistics for the highest-resolution shell are shown in parentheses.
Table 2.
KM values of hexokinases from different studies and species.
| KM (mM) | ||
| Glucose | ATP | |
| PfHK, present study | 0.13 ± 0.01 | 0.83 ± 0.08 |
| PfHK, Harris et al. 2013 [7] | 0.62 ± 0.06 | 0.66 ± 0.08 |
| PfHK, Roth et al. 1987 [12] | 0.431 ± 0.021 | 3.1 ± 1.4 |
| CpHK, Yu et al. 2015 [43] | 0.138 | 0.673 |
| TgHK, Saito et al. 2002 [42] | 0.0080 ± 0.0008 | 1.05 ± 0.25 |
1 rom infected erythrocytes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



