
Submitted:
01 August 2023
Posted:
02 August 2023
You are already at the latest version
Abstract
Transition metal alloys have emerged as promising electrocatalysts due to their ability to modulate key parameters such as d-band electron filling, Fermi level energy, and interatomic spacing, thereby influencing their affinity towards reaction intermediates. However, the structural stability of alloy electrocatalysts during alkaline hydrogen evolution reaction (HER) remains a subject of debate. In this study, we systematically investigate the structural evolution and catalytic activity of the c-Co/Co3Mo electrocatalyst under alkaline HER conditions. We reveal that the instability of the Co3Mo alloy and H0.9MoO3 during alkaline HER leads to the destruction of the crystal structure, resulting in the formation of hexagonal cobalt (h-Co) from hexagonal Co(OH)2 as an intermediate species. Furthermore, we explore the re-adsorption and surface coordination of the Mo element, which enhances the catalytic activity for alkaline HER. This work provides valuable insights into the dynamic behavior of alloy-based electrocatalysts, shedding light on their structural stability and catalytic activity during electrochemical reduction processes
Keywords:
transition metal alloys
; electrocatalysts
; structural stability
; alkaline hydrogen evolution reaction (HER)
; c-Co/Co3Mo electrocatalyst
1. Introduction
Reducing CO2 emissions has emerged as a global development goal, driving the search for clean and low-carbon renewable energy sources [1-4]. Among these, hydrogen energy has garnered significant attention as a clean and zero-carbon emission alternative [5-6]. Electrochemical water splitting, powered by electricity from renewable sources, presents a promising technology for producing "green hydrogen" [7-9]. However, the widespread adoption of this technology is hindered by the low efficiency of hydrolysis, particularly in alkaline water electrolysis cells, due to the higher operating overpotentials of the hydrogen evolution reaction (HER) [10]. Unlike the direct formation of hydrogen intermediates (H*) from proton reduction (H+) in acidic media, the alkaline HER process involves the splitting of water molecules to generate hydrogen intermediates, resulting in increased energy consumption and reduced efficiency [11-12]. Therefore, the development and design of highly efficient and stable electrocatalysts for alkaline HER is crucial for enhancing the efficiency and reducing the cost of alkaline water electrolysis cells.
To date, noble metal-based materials have been considered as the benchmark electrocatalysts for the hydrogen evolution reaction (HER). However, their high cost limits their widespread application [11,13-14]. In order to replace noble metal-based electrocatalysts, cost-effective alloy electrocatalysts composed of transition metals have been developed for alkaline HER. The alloying of transition metals allows for the tuning of d-band electron filling, Fermi level energy, and interatomic spacing, all of which can impact the affinity of the alloy electrocatalysts towards reaction intermediates [15-16]. However, the structural stability of alloy electrocatalysts, particularly those containing Mo elements, during the alkaline HER process has been a subject of controversy. For instance, in Ni4Mo alloy, Mo was found to dissolve and form MoO42- during alkaline HER, with the surface-adsorbed Mo2O72- promoting the HER activity of metal Ni [17]. This can be attributed to the high affinity between Mo and oxygen, facilitating the bonding of Mo atoms with hydroxyl groups in the alkaline electrolyte [18]. Similarly, in IrMo alloy, it has been confirmed that the surface of IrMo exhibits easier adsorption of OH compared to pure Ir [18]. Additionally, for Co-based electrocatalysts, in-situ cobalt metal formed by electrochemical reduction has been identified as the true catalytically active species for HER [19-20]. Therefore, it is necessary to explore the structural stability of Co3Mo alloys, despite their widely reported and studied catalytic activity as excellent electrocatalysts. In this study, we systematically investigate the formation of hexagonal cobalt (h-Co) from hexagonal Co(OH)2 as an intermediate species, along with the re-adsorption and surface coordination of Mo elements, leading to enhanced catalytic activity for alkaline HER.
2. Results and Discussion
The c-Co/Co3Mo electrocatalyst on carbon cloth was synthesized by annealing the Co(OH)2/CoMoO4 precursor, which was obtained through hydrothermal treatment of α-Co(OH)2 nanosheet arrays in a NaMoO4 solution (refer to the Experimental section in the Supporting Information). Carbon cloth, chosen for its high conductivity, three-dimensional (3D) microstructure, and excellent chemical/mechanical stability, served as a binder-free substrate for supporting the catalyst [21]. Furthermore, the crystal structure and composition of the c-Co/Co3Mo electrocatalysts can be optimized by adjusting the annealing and hydrothermal conditions of the precursors [22]. XRD patterns (Figure S1a) revealed that at a reduced annealing temperature of 400 °C, the annealing products mainly consisted of CoO, with no formation of c-Co and H0.9MoO3. However, when the annealing temperature was increased to 600 °C, the content of H0.9MoO3 decreased, and the primary crystal phase became Co3Mo alloy, indicating that higher temperatures favored the formation of Co3Mo alloy and promoted thermal reduction reactions [23]. Similarly, adjusting the annealing time (Figure S1b) showed that even after annealing at 500 °C for 1 hour, c-Co could be formed. Moreover, the annealing time could regulate the proportion of phase components, with the H0.9MoO3 phase initially increasing and then decreasing as the annealing time was increased from 1 hour to 3 hours. However, Co3Mo alloy was not observed even after extending the annealing time to 3 hours, highlighting the critical role of annealing temperature. Additionally, the crystal structure and composition of the Co(OH)2/CoMoO4 precursor before annealing were controlled by adjusting the hydrothermal temperature, time, and concentration (details in the supporting information, S2-S4). Experimental results demonstrated that increasing the temperature, concentration, and time of the hydrothermal reaction reduced the content of Co(OH)2 and increased the content of CoMoO4 in the Co(OH)2/CoMoO4 precursor. Subsequently, after annealing at 500 °C for 2 hours, a series of electrocatalysts with different component ratios were obtained, indicating that the structure and composition of the Co(OH)2/CoMoO4 precursor could be adjusted to achieve the optimization of target electrocatalysts. Moreover, SEM images (Figure S5-8) showed that the nanosheet arrays of the target electrocatalysts were preserved after adjusting the temperature and time of the hydrothermal reaction and annealing process, respectively. Subsequently, the HER performance of the target electrocatalysts in an alkaline electrolyte was analyzed through electrochemical tests to select the optimal electrocatalyst samples. As depicted in Figure S9-11, the target electrocatalyst (c-Co/Co3Mo) obtained by annealing sample-120℃-8h-0.5M at 500 °C for 2 hours in a mixed hydrogen-argon flow exhibited the lowest overpotential at the same current density compared to the other control samples. The optimized samples were further characterized and analyzed in detail in the subsequent discussion.
The X-ray diffraction (XRD) pattern exhibits two strong diffraction peaks at 12.2° and 44.2°, which can be attributed to the (200) facet of monoclinic H0.9MoO3 (PDF#53-1024) and the (111) facet of cubic Co (c-Co, PDF#15-0806), respectively, confirming the formation of c-Co and H0.9MoO3. The scanning electron microscopy (SEM) image in Figure 1b shows that the c-Co/Co3Mo nanosheet arrays uniformly cover the surface of the carbon cloth. Upon further magnification in SEM image (Figure 1c), it is observed that the porous nanosheet is composed of smaller-sized nanoparticles. To gain further insight into the crystal structure and nanomorphology of the catalyst, transmission electron microscopy (TEM) was employed. The TEM image in Figure 1d reveals that the nanoparticles have sizes ranging from 20 to 30 nm. The high-resolution TEM (HRTEM) image in Figure 1e clearly shows a lattice d-spacing of 0.206 nm, corresponding to the (111) facet of the c-Co phase. In Figure 1g, the lattice d-spacings of 0.245 nm and 0.195 nm can be assigned to the (021) facet of CoMoO4 and the (201) facet of Co3Mo, respectively. Furthermore, the lattice d-spacing of 0.193 nm corresponds to the (004) facet of H0.9MoO3 (Figure 1h), and the larger layer spacing of approximately 0.72 nm, attributed to the (200) facet of H0.9MoO3, is observed due to its two-dimensional layered structure. These HRTEM images indicate that during the thermal reduction process, Co(OH)2 and only a portion of CoMoO4 in the Co(OH)2/CoMoO4 precursor are transformed into c-Co and Co3Mo alloy, respectively. Additionally, a portion of CoMoO4 in the precursor is reduced to H0.9MoO3. These results demonstrate that the synthesized electrocatalyst consists of multiple crystalline phases, including c-Co, Co3Mo, H0.9MoO3, and CoMoO4. Moreover, the scanning TEM (STEM) image and corresponding energy dispersive X-ray spectroscopy (EDX) elemental mapping images (Figure 1j) confirm that Co, Mo, and O elements are uniformly distributed in the porous nanosheets.
To assess the catalytic activity and stability of the c-Co/Co3Mo electrocatalyst, a series of electrochemical tests were conducted in alkaline electrolytes. For comparison, the hydrogen evolution reaction (HER) performance of c-Co and Co3O4/CoMoO4 catalysts was also evaluated under the same conditions. The linear sweep voltammetry (LSV) curves in Figure 2a demonstrate that c-Co/Co3Mo exhibits significantly enhanced HER performance, requiring only an overpotential of 28 mV to achieve a current density of 10 mA cm-2. This overpotential is lower than that of c-Co (280 mV) and Co3O4/CoMoO4 (171 mV). Moreover, the Tafel slope of c-Co/Co3Mo is 28 mV dec-1, which is considerably lower than that of c-Co (331 mV dec-1) and Co3O4/CoMoO4 (179 mV dec-1). This suggests that the c-Co/Co3Mo electrocatalyst exhibits faster kinetics, attributed to the Volmer-Tafel mechanism, which is different from the Volmer-Heyrovsky mechanism observed in c-Co and Co3O4/CoMoO4 [24]. Electrochemical impedance spectroscopy (EIS) was employed to investigate the charge-transfer behavior at the interface between the electrocatalyst and the electrolyte [25]. The Nyquist plots in Figure 2c reveal that the c-Co/Co3Mo electrocatalyst exhibits the lowest charge transfer resistance (Rct) compared to the control samples of c-Co and Co3O4/CoMoO4. This indicates that the c-Co/Co3Mo catalyst promotes electron transfer at the interface, facilitating the alkaline HER process [26]. Furthermore, the electrochemical surface area (ECSA) values were calculated to explore the intrinsic activity of the electrocatalysts. The ECSA values were obtained from the corresponding electrochemical double layer capacitance (Cdl) derived from cyclic voltammetry (CV) curves at different scan rates in the non-faradaic region [27]. As shown in Figure 2d, the c-Co/Co3Mo electrocatalyst exhibits a larger Cdl value of 23.2 mF cm-2 compared to c-Co (14.9 mF cm-2) and Co3O4/CoMoO4 (9.5 mF cm-2). The larger ECSA suggests that the c-Co/Co3Mo catalyst exposes more catalytically active sites during the alkaline HER process [28]. The ECSA-normalized LSV curves in Figure 2e further confirm that the c-Co/Co3Mo electrocatalyst exhibits the highest intrinsic activity among the control samples [29]. In addition to catalytic activity, stability is also crucial for practical applications [10]. The stability of the c-Co/Co3Mo electrocatalyst was evaluated using chronopotentiometry (CP). As depicted in Figure 2f, the applied potential shows almost no increase after continuous operation for over 40 hours, indicating excellent stability during the alkaline HER process.
Furthermore, a series of cyclic voltammetry (CV) curves were recorded for 10 cycles in 1 M KOH at a scan rate of 50 mV s-1 between 0.124 and -0.676 V to observe the electrochemical behavior of the electrocatalysts before obtaining stable LSV curves. Interestingly, as shown in Figure 3a, the evolutive CV curves indicate that the c-Co/Co3Mo electrocatalyst undergoes rapid electrochemical activation from the 1st to the 5th CV in 1 M KOH. As the number of CV cycles increases up to 10, the CV curves become almost perfectly coincident, suggesting stable electrochemical performance. In the inset of Figure 3a, a distinct electrochemical redox peak is observed from 0.124 to -0.1 V, which may be attributed to the electrochemical transition between cobalt hydroxide and cobalt metal [30]. However, similar electrochemical activation and redox peaks were not observed in the control samples of c-Co and Co3O4/CoMoO4. For the control samples, both c-Co and Co3O4/CoMoO4 exhibited significant electrochemical instability in the 1st CV curve and then became stable in the 2nd CV curve (Figure 3b, c). The difference is that c-Co showed a slight decrease in electrochemical performance in the 2nd CV curve, while Co3O4/CoMoO4 showed an enhanced electrochemical performance in the 2nd CV curve.
To investigate the electrochemical activation, we further characterized the crystal and electronic structures of the electrocatalysts. XRD patterns (Figure S13) reveal that the diffraction peaks corresponding to phase H0.9MoO3 and c-Co in the c-Co/Co3Mo electrocatalyst significantly decrease after the HER test, indicating a clear evolution in the crystal structure due to electrochemical activation. In contrast, no significant changes in crystal structure were observed in the control samples of c-Co and Co3O4/CoMoO4 (Figure S14). Additionally, Raman spectroscopy was employed to analyze the variation in lattice vibration of the electrocatalyst before and after the HER test. As shown in Figure 4a, four peaks located at 211, 307, 385, and 906 cm-1 can be attributed to the bending vibration of O-Mo-O and the stretching vibration of Co-O-Mo, respectively [23,31-32]. After the HER test, the intensity of the vibration modes related to O-Mo-O and Co-O-Mo slightly decreases [33], while a new peak at 673 cm-1, corresponding to the stretching vibration of Co-OH, emerges. This suggests that H0.9MoO3 and a small amount of CoMoO4 in the c-Co/Co3Mo electrocatalyst exhibit poor stability during the alkaline HER process, leading to the formation of cobalt hydroxide.
The electronic structures of the c-Co/Co3Mo electrocatalyst were further investigated using X-ray photoelectron spectroscopy (XPS). In the Co 2p spectra before the HER test (Figure 4b), two peaks at 778.8 eV and 793.9 eV, corresponding to Co 2p3/2 and Co 2p1/2 of metallic Co, were observed, along with two peaks at 780.6 eV and 796.3 eV for Co-O [34-35], accompanied by satellite peaks at 786.3 eV and 802.0 eV [36-37]. After the HER test, the intensity of the metallic Co peaks increased, and the two Co-O peaks slightly shifted to higher energies, indicating the presence of Co-OH and confirming the formation of more metallic Co and new cobalt hydroxide [36]. In the Mo 3d region before the HER test (Figure 4c), two peaks at 232.4 eV and 235.6 eV were observed, corresponding to Mo5+ in H0.9MoO3, while the other two peaks at 230.1 eV and 233.5 eV were assigned to Mo4+ [38-39]. After the HER test, the two peaks related to Mo5+ slightly shifted to higher energies, indicating the presence of Mo6+ from MoO42- on the surface. This suggests that the Mo element in the electrocatalyst first dissolves in the alkaline electrolyte, undergoes electrooxidation into MoO42- ions, and then re-adsorbs onto the electrode surface [17,19]. In the O 1s region (Figure 4d), three peaks at 530.5 eV, 531.6 eV, and 533.2 eV were observed, corresponding to the Co-O/Mo-O bond, oxygen vacancy, and adsorbed water, respectively [40]. After the HER test, the peak associated with the Co-O/Mo-O bond shifted to higher binding energy, indicating the formation of cobalt hydroxide. These results suggest that during the electrochemical activation, Mo elements are leached from the electrocatalyst into the electrolyte and then re-adsorbed onto the electrode surface, accompanied by the formation of cobalt hydroxide. Combined with the previous observation of redox peaks (Figure 3a), it is speculated that cobalt hydroxide may undergo further reduction to form metallic cobalt during the alkaline HER process.
To further investigate the structural evolution of the c-Co/Co3Mo electrocatalyst, electron microscopy techniques were employed to observe the nanomorphology and microscopic crystal structure. The SEM image in Figure 5a shows that the nanomorphology of the c-Co/Co3Mo electrocatalyst remains unchanged after the HER test. However, TEM images reveal the presence of coated nanoparticles, unlike the individually dispersed nanoparticles observed before the HER test, indicating surface reconstruction in the outer layer of the nanoparticles (Figure 5b). Furthermore, HRTEM images provide insight into the microscopic crystal structure of the c-Co/Co3Mo electrocatalyst after the HER test. In Figure 5c, the lattice d-spacing of 0.207 nm corresponds to the (111) facet of c-Co, while the lattice d-spacings of 0.193 nm and 0.202 nm can be attributed to the (101) and (002) facets of hexagonal Co (h-Co). In the outer region of the nanoparticles (Figure 5d), the observed lattice d-spacing of 0.239 nm is assigned to the (101) facets of hexagonal Co(OH)2. Additionally, the (022) facet of CoMoO4 and the (101) and (002) facets of h-Co are also observed in Figure 5e, confirming the formation of h-Co. Notably, HRTEM images do not show lattice fringes corresponding to H0.9MoO3 and Co3Mo in the c-Co/Co3Mo electrocatalyst after the HER test, indicating their instability during alkaline HER. STEM images and corresponding EDX elemental mapping images reveal the homogeneous and overlapping distribution of Co, Mo, and O throughout the nanosheets (Figure S17).
To investigate the stability of the Co3Mo alloy, we prepared an electrocatalyst consisting of highly crystalline Co3Mo alloys by annealing the sample at 120℃ for 8 hours in a 0.5 M solution, followed by heating at 600°C for 2 hours. Characterization of the electrocatalyst was performed. The cyclic voltammetry (CV) curves in Figure S15 reveal an electrochemical activation of the Co3Mo alloys from the 1st to the 10th CV in a 1 M KOH solution, accompanied by a distinct electrochemical redox peak. X-ray diffraction (XRD) patterns indicate a significant reduction in the intensity of the diffraction peak of the Co3Mo alloy, suggesting its instability during the alkaline HER process. Scanning electron microscopy (SEM) images exhibit the characteristic hexagonal nanoplates of Co(OH)2, confirming its formation. These findings suggest that the Co3Mo alloy undergoes dissolution in the electrolyte during the alkaline HER process, with the dissolved cobalt ions subsequently redepositing on the electrode surface to form Co(OH)2. Consistent with previous reports, the dissolved Mo ions also readhere to the electrode surface, forming coordination polyhedra that regulate the electronic structure of the catalytic site and enhance the catalytic activity during alkaline HER. Therefore, it is reasonable to speculate that both the Co3Mo alloy and H0.9MoO3 are destroyed during the alkaline HER process, resulting in the formation of soluble Mo and Co ions. The Mo ions are then re-adsorbed on the surface to form coordination polyhedral ions, while the Co ions are re-deposited to form Co(OH)2. Finally, under a negative electric field, Co(OH)2 is further reduced to h-Co.
3. Conclusions
In summary, this study comprehensively investigated the structural evolution and electrochemical activation of the c-Co/Co3Mo electrocatalyst during alkaline HER. The instability of the Co3Mo alloy and H0.9MoO3 in the alkaline HER process results in the disruption of the crystal structure, leading to the adsorption of the Mo element and the redeposition and reduction of the Co element. This process gives rise to the formation of coordinated MoO42- ions and h-Co, with Co(OH)2 serving as an intermediate species. Importantly, these structural changes are accompanied by a significant enhancement in the catalytic activity for alkaline HER. These findings provide valuable insights into the investigation of the structural stability and dynamic catalytic activity of alloy-based electrocatalysts, thus facilitating the rational design of highly efficient catalysts for energy conversion applications.
4. Materials and Methods
4.1. Chemicals
Co(NO3)2·6H2O, hexamethylenetetramine (HMT), NaMoO4·2H2O, and KOH were bought from Sinopharm Chemical Reagent Co., Ltd, Shanghai, China. All the chemicals were directly used as received without further purification.
4.2. Synthesis of Co(OH)2/CoMoO4 Precursor on Carbon Cloth
In accordance with previous reports, α-Co(OH)2 nanosheet arrays were loaded onto a carbon cloth substrate. To prepare the Co(OH)2/CoMoO4 precursor (sample-120℃-8h-0.5 M), 2.42 g of NaMoO4·2H2O and 20 mL of deionized water were mixed thoroughly in a 45 mL Teflon autoclave to form a 0.5 M NaMoO4 solution. A piece of carbon cloth (2×2.5 cm) with α-Co(OH)2 was then added to the same autoclave. The autoclave was subsequently sealed and subjected to a hydrothermal treatment at 120 ℃ for 8 hours. After the reaction had naturally cooled down, the carbon cloth was removed, washed several times with water, and dried at 60 ℃ under vacuum conditions. Control samples were prepared by adjusting the temperature (90 ℃, 150 ℃) of the hydrothermal reaction (sample-90℃-8h-0.5M, sample-150℃-8h-0.5M), as well as by varying the reaction time (4 h, 12 h) (sample-120℃-4h-0.5M, sample-120℃-12h-0.5M). Control samples with different NaMoO4 solution concentrations (0.1 M, 2 M) were also obtained by adjusting the concentration (sample-120℃-8h-0.1M, sample-120℃-8h-2M).
4.3. Synthesis of c-Co/Co3Mo on Carbon Cloth
The Co(OH)2/CoMoO4 precursor (sample-120℃-8h-0.5M) was typically subjected to annealing at 500 °C for 2 hours in the central position of a tube furnace. The annealing process was carried out under a flow of Ar/H2 gas (100 sccm, 10% H2), with a heating rate of 5 °C/min. After the annealing process, the sample was allowed to cool down naturally, resulting in the formation of c-Co/Co3Mo on the carbon cloth substrate. To investigate the effect of annealing conditions, the temperature (300 °C, 500 °C) and duration (1 hour, 3 hours) of the annealing process were adjusted accordingly.
4.4. Synthesis of c-Co on Carbon Cloth
To synthesize c-Co on carbon cloth, the same annealing conditions as c-Co/Co3Mo were employed, with the only difference being the replacement of Co(OH)2/CoMoO4 with α-Co(OH)2 as the precursor. The α-Co(OH)2 precursor was annealed at the same temperature (500 °C) for the same duration (2 hours) in the central position of a tube furnace under an Ar/H2 flow (100 sccm, 10% H2) with a heating rate of 5 °C/min.
4.5. Synthesis of Co3O4/CoMoO4 on Carbon Cloth
For the synthesis of Co3O4/CoMoO4 on carbon cloth, the as-obtained Co(OH)2/CoMoO4 precursor (sample-120℃-8h-0.5M) was annealed at 500 °C for 2 hours in the central position of a tube furnace under an Ar flow. The heating rate during the annealing process was maintained at 5 °C/min.
4.6. Characterizations
The structures and morphologies of the samples were characterized using various techniques:
4.6.1. Scanning Electron Microscopy (SEM)
The samples were examined using a Hitachi S-4800 field emission scanning electron microscope, which provides high-resolution images of the surface morphology.
4.6.2. Transmission Electron Microscopy (TEM) with Energy Dispersive X-ray Spectroscopy (EDX)
The JEOL JEM-2100 TEM operating at 200 kV was utilized to investigate the internal structures of the samples. EDX analysis was performed simultaneously to obtain elemental composition information.
4.6.3. Powder X-ray Diffraction (XRD)
XRD patterns were obtained using a Bruker D8 ADVANCE instrument equipped with a Cu Kα radiation source (λ = 0.154178 nm). This technique provides information about the crystal structure and phase composition of the samples.
4.6.4. X-ray Photoelectron Spectroscopy (XPS)
Analysis was carried out using a Thermo Fisher ESCALAB XI+ XPS instrument. This technique allows for the characterization of the elemental composition and chemical states of the samples.
4.6.5. Raman Spectroscopy
Raman spectra were recorded using a microscopic Raman spectrometer (HORIBA Lab RAM HR Evolution) with a laser wavelength of 532 nm. Raman spectroscopy provides information about the vibrational modes and molecular structure of the samples.
These characterization techniques collectively provide valuable insights into the structures, morphologies, elemental composition, crystal phases, chemical states, and vibrational properties of the synthesized samples.
4.7. Electrochemical Measurements
The electrochemical measurements were conducted using the following setup and procedures: Three-Electrode System: A standard three-electrode system was used, consisting of the prepared samples on carbon cloth as the working electrode, Hg/HgO (1 M KOH) as the reference electrode, and a graphite rod as the counter electrode. Electrolyte: 1 M KOH solution was used as the electrolyte for all measurements. Electrochemical Activation: The electrochemical activation of the working electrode was performed by cyclic voltammetry (CV) in the potential range of 0.124 V to -0.676 V (vs RHE) at a scan rate of 50 mV s-1 in 1 M KOH solution. Working Electrode Surface Area: The working surface area of the electrode was controlled to 1×0.5 cm for all experiments. Temperature: All tests were conducted at ambient temperature. Potential Conversion: All measured potentials were converted to a reversible hydrogen electrode (RHE) using the equation: E(RHE) = E(Hg/HgO) + 0.059 × pH + 0.098 V. Potential Calibration: All measured potentials were calibrated with iR compensation. Electrochemical Double Layer Capacitance (Cdl): To determine the electrochemical double layer capacitance (Cdl), cyclic voltammetry (CV) was performed in the potential range of 0.024 V to 0.074 V (vs RHE) at various scan rates (20, 40, 60, 80, and 100 mV s-1). This potential range was chosen as no apparent faradaic reactions were observed in this range for all the electrodes. Electrochemical Impedance Spectroscopy (EIS): EIS measurements were carried out at -0.1 V (vs RHE) over a frequency range of 100 kHz to 0.01 Hz with an AC amplitude of 5 mV. Long-Term Stability Tests: Chronopotentiometry (CP) curves were recorded at a current density of 10 mA cm-2 to assess the long-term stability of the electrodes.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. The following supporting information can be found in the online version.
Author Contributions
Data curation, Long Chen and Liwen Jiang; Formal analysis, Long Chen; Investigation, Long Chen; Writing – original draft, Liwen Jiang; Writing – review & editing, Jianjun Wang. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (Grant No. 2279075), Shandong Provincial Natural Science Foundation (Grant No. ZR2020YQ09).
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
References
- Yang, L.; Shi, L.; Chen, H.; Liang, X.; Tian, B.; Zhang, K.; Zou, Y.; Zou, X. A Highly Active, Long-Lived Oxygen Evolution Electrocatalyst Derived from Open-Framework Iridates. Adv. Mater. 2023, 35, 2208539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, Y.; Zhu, S.S.; Liu, Y.Y.; Zhang, X.; Wang, J.J.; Braun, A. Covalent S-O Bonding Enables Enhanced Photoelectrochemical Performance of Cu2S/Fe2O3 Heterojunction for Water Splitting. Small, 2021, 17, 2100320. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.N.; Shi, R.; Li, Z.H.; Zhao, J.Q.; Su, C.L.; Zhang, T.R. Triphase Photocatalytic CO2 Reduction over Silver-Decorated Titanium Oxide at a Gas–Water Boundary. Angew. Chem. Int. Ed. 2022, 61, e202200802. [Google Scholar] [CrossRef]
- Miao, Z.Y.; Huang, Y.; Xin, J.P.; Su, X.W.; Sang, Y.H.; Liu, H.; Wang, J.J. High Performance Symmetric Supercapacitor Constructed Using Carbon Cloth Boosted by Engineering Oxygen-containing Functional Groups, ACS Appl. Mater. Interfaces, 2019, 11, 18044–18050. [Google Scholar] [CrossRef]
- Yi, P.; Song, Y.Y.; Li, C.Y.; Liu, R.Z.; Sun, J.K. Heterostructured Mn-doped NiSx/NiO/Ni3N Nanoplate Arrays as Bifunctional Electrocatalysts for Energy-Saving Hydrogen Production and Urea Degradation. Appl. Surf. Sci. 2023, 619, 156789. [Google Scholar] [CrossRef]
- Jiao, S.; Fu, X.; Wang, S.; Zhao, Y. Perfecting the Electrocatalysts via Imperfections: Towards Large-Scale Deployment of Water Electrolysis Technology. Energy Environ. Sci. 2021, 14, 1722–1770. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, L.W.; Liu, X.L.; Tan, T.; Liu, H.; Wang, J.J. Precisely engineering the electronic structure of active sites boosts the activity of iron-nickel selenide on nickel foam for highly efficient and stable overall water splitting. Appl. Catal. B 2021, 299, 120678. [Google Scholar] [CrossRef]
- Yao, Z.C.; Tang, T.; Jiang, Z.; Wang, L.; Hu, J.S.; Wan, L.J. Electrocatalytic Hydrogen Oxidation in Alkaline Media: From Mechanistic Insights to Catalyst Design. ACS Nano 2022, 16, 5153–5183. [Google Scholar] [CrossRef]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 0003. [Google Scholar] [CrossRef]
- Zhai, W.; Ma, Y.; Chen, D.; Ho, J.C.; Dai, Z.; Qu, Y. Recent progress on the long-term stability of hydrogen evolution reaction electrocatalysts. InfoMat 2022, 4, e12357. [Google Scholar] [CrossRef]
- Zhu, J.; Hu, L.; Zhao, P.; Lee, L.Y.S.; Wong, K.Y. Recent Advances in Electrocatalytic Hydrogen Evolution Using Nanoparticles. Chem. Rev. 2020, 120, 851–918. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhao, P.; Sheng, W. Hydrogen Evolution and Oxidation: Mechanistic Studies and Material Advances. Adv. Mater. 2019, 31, 1808066. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.Y.; Duan, Y.; Feng, X.Y.; Yu, X.; Gao, M.R.; Yu, S.H. Clean and Affordable Hydrogen Fuel from Alkaline Water Splitting: Past, Recent Progress, and Future Prospects. Adv. Mater. 2021, 33, 2007100. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhang, Y. Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 2015, 44, 5148–5180. [Google Scholar] [CrossRef]
- Faber, M.S.; Jin, S. Earth-abundant inorganic electrocatalysts and their nanostructures for energy conversion applications. Energy Environ. Sci. 2014, 7, 3519–3542. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Kong, H.; Kim, J.; Choi, S.; Ciucci, F.; Hao, Y.; Yang, S.; Shao, Z.; Lim, J. Non-precious-metal catalysts for alkaline water electrolysis: operando characterizations, theoretical calculations, and recent advances. Chem. Soc. Rev. 2020, 49, 9154–9196. [Google Scholar] [CrossRef]
- Du, W.; Shi, Y.; Zhou, W.; Yu, Y.; Zhang, B. Unveiling the In Situ Dissolution and Polymerization of Mo in Ni4Mo Alloy for Promoting Hydrogen Evolution Reaction. Angew. Chem. Int. Ed. 2021, 60, 7051–7055. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, L.; Liu, J.; Zhong, C.; Tu, Y.; Li, P.; Du, L.; Chen, S.; Cui, Z. OH spectator at IrMo intermetallic narrowing activity gap between alkaline and acidic hydrogen evolution reaction. Nat. Commun. 2022, 13, 5497. [Google Scholar] [CrossRef]
- Jiang, L.W.; Huang, Y.; Zou, Y.; Meng, C.; Xiao, Y.; Liu, H.; Wang, J.J. Boosting the Stability of Oxygen Vacancies in α-Co(OH)2 Nanosheets with Coordination Polyhedrons as Rivets for High-Performance Alkaline Hydrogen Evolution Electrocatalyst. Adv. Energy Mater. 2022, 12, 2202351. [Google Scholar] [CrossRef]
- Jin, Q.; Ren, B.; Li, D.; Cui, H.; Wang, C. In situ promoting water dissociation kinetic of Co based electrocatalyst for unprecedentedly enhanced hydrogen evolution reaction in alkaline media. Nano Energy 2018, 49, 14–22. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Norskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, 4998. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xing, Y.; Xu, S.; Lu, Y.; Sun, S.; Jiang, D. Interfacing Co3Mo with CoMoOx for synergistically boosting electrocatalytic hydrogen and oxygen evolution reactions. Chem. Eng. J. 2022, 431, 133240. [Google Scholar] [CrossRef]
- Chen, J.; Ge, Y.; Feng, Q.; Zhuang, P.; Chu, H.; Cao, Y.; Smith, W.R.; Dong, P.; Ye, M.; Shen, J. Nesting Co3Mo Binary Alloy Nanoparticles onto Molybdenum Oxide Nanosheet Arrays for Superior Hydrogen Evolution Reaction. ACS Appl. Mater. Interfaces 2019, 11, 9002–9010. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, V.R.; Strmcnik, D.; Lopes, P.P.; Markovic, N.M. Energy and fuels from electrochemical interfaces. Nat. Mater. 2016, 16, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lv, H.; Sun, L.; Jia, F.; Liu, B. Ordered Mesoporous Intermetallic Trimetals for Efficient and pH-Universal Hydrogen Evolution Electrocatalysis. Adv. Energy Mater. 2022, 12, 2201478. [Google Scholar] [CrossRef]
- Shah, A.H.; Zhang, Z.; Huang, Z.; Wang, S.; Zhong, G.; Wan, C.; Alexandrova, A.N.; Huang, Y.; Duan, X. The role of alkali metal cations and platinum-surface hydroxyl in the alkaline hydrogen evolution reaction. Nat. Catal. 2022, 5, 923–933. [Google Scholar] [CrossRef]
- Wang, J.; Xin, S.; Xiao, Y.; Zhang, Z.; Li, Z.; Zhang, W.; Li, C.; Bao, R.; Peng, J.; Yi, J.; Chou, S. Manipulating the Water Dissociation Electrocatalytic Sites of Bimetallic Nickel-Based Alloys for Highly Efficient Alkaline Hydrogen Evolution. Angew. Chem. Int. Ed. 2022, 61, e202202518. [Google Scholar] [CrossRef]
- Joo, J.; Kim, T.; Lee, J.; Choi, S.I.; Lee, K. Morphology-Controlled Metal Sulfides and Phosphides for Electrochemical Water Splitting. Adv. Mater. 2019, 31, e1806682. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, W.; Shi, Z.; Yang, L.; Tang, Y. Structural Design and Electronic Modulation of Transition-Metal-Carbide Electrocatalysts toward Efficient Hydrogen Evolution. Adv. Mater. 2019, 31, 1802880. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, J.; Niu, X.; Zhang, K.; Wang, Z.; Cui, Y.; Wang, R. Chemical Interactions and Mechanisms of Different pH Regulators on Copper and Cobalt Removal Rate of Copper Film CMP for GLSI. ECS J. Solid State Sci. Technol. 2019, 8, P99–P105. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Y.; Huang, Z.; Ren, L.; Qi, X.; Wei, X.; Zhong, J. Facile hydrothermal synthesis of NiMoO4@CoMoO4 hierarchical nanospheres for supercapacitor applications. Phys. Chem. Chem. Phys. 2015, 17, 20795–20804. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Chen, J.; Yang, R.; Mao, S.; Qin, M.; Wang, Y. Multi-hierarchical cobalt-based electrocatalyst towards high rate H2 production. Appl. Catal. B 2022, 316, 121666. [Google Scholar] [CrossRef]
- Rivas-Murias, B.; Salgueiriño, V. Thermodynamic CoO–Co3O4 crossover using Raman spectroscopy in magnetic octahedron-shaped nanocrystals. J. Raman Spectrosc. 2017, 48, 837–841. [Google Scholar] [CrossRef]
- Huang, Y.; Jiang, L.W.; Liu, H.; Wang, J.J. Electronic structure regulation and polysulfide bonding of Co-doped (Ni, Fe)1+xS enable highly efficient and stable electrocatalytic overall water splitting. Chem. Eng. J. 2022, 441, 136121. [Google Scholar] [CrossRef]
- Men, Y.N.; Jia, S.F.; Li, P.; Tan, Y.; Wang, J.B.; Zhao, P.P.; Cheng, G.Z.; Chen, S.L.; Luo, W. Boosting alkaline hydrogen evolution electrocatalysis through electronic communicating vessels on Co2P/Co4N heterostructure catalyst. Chem. Eng. J. 2022, 433, 133831. [Google Scholar] [CrossRef]
- Jiang, L.W.; Huang, Y.; Chen, B.B.; Zhou, J.Q.; Liu, H.; Wang, J.J. Electrochemically induced in-situ generated Co(OH)2 nanoplates to promote the Volmer process toward efficient alkaline hydrogen evolution reaction. Int. J. Hydrogen Energy 2021, 46, 8497–8506. [Google Scholar] [CrossRef]
- Yuan, L.; Liu, S.; Xu, S.; Yang, X.; Bian, J.; Lv, C.; Yu, Z.; He, T.; Huang, Z.; Boukhvalov, D.W.; Cheng, C.; Huang, Y.; Zhang, C. Modulation of Volmer step for efficient alkaline water splitting implemented by titanium oxide promoting surface reconstruction of cobalt carbonate hydroxide. Nano Energy 2021, 82, 105732. [Google Scholar] [CrossRef]
- Yin, Z.H.; Huang, Y.; Jiang, L.W.; Meng, C.; Wu, Y.Z.; Liu, H.; Wang, J.J. Revealing the In Situ Evolution of Tetrahedral NiMoO4 Micropillar Array for Energy-Efficient Alkaline Hydrogen Production Assisted by Urea Electrolysis. Small Struct. 2023, 2300028. [Google Scholar] [CrossRef]
- Gao, W.Q.; Zou, Y.; Zang, Y.M.; Zhao, X.L.; Zhou, W.J.; Dai, Y.; Liu, H.; Wang, J.J.; Ma, Y.D.; Sang, Y.H. Magnetic-field-regulated Ni-Fe-Mo ternary alloy electrocatalysts with enduring spin polarization enhanced oxygen evolution reaction, Chem. Eng. J. 2023, 455, 140821. [Google Scholar]
- Yang, C.; Zhong, W.; Shen, K.; Zhang, Q.; Zhao, R.; Xiang, H.; Wu, J.; Li, X.; Yang, N. Electrochemically Reconstructed Cu-FeOOH/Fe3O4 Catalyst for Efficient Hydrogen Evolution in Alkaline Media. Adv. Energy Mater. 2022, 12, 2200077. [Google Scholar] [CrossRef]
Figure 1.
(a) XRD pattern, (b, c) SEM images, (d) TEM image, (e-i) HRTEM images and (j) the corresponding EDX elemental mapping images of c-Co/Co3Mo. Scale bars, 3 nm (g), 3 nm (h), 200 nm (j).
Figure 1.
(a) XRD pattern, (b, c) SEM images, (d) TEM image, (e-i) HRTEM images and (j) the corresponding EDX elemental mapping images of c-Co/Co3Mo. Scale bars, 3 nm (g), 3 nm (h), 200 nm (j).

Figure 2.
(a) LSV curves, (b) Tafel slopes, (c) Nyquist plots, (d) Cdl values, and (e) ECSA-normalized LSV curves of c-Co, c-Co/Co3Mo, Co3O4/CoMoO4. (f) Chronopotentiometry curve of c-Co/Co3Mo at a constant current density of 10 mA cm-2.
Figure 2.
(a) LSV curves, (b) Tafel slopes, (c) Nyquist plots, (d) Cdl values, and (e) ECSA-normalized LSV curves of c-Co, c-Co/Co3Mo, Co3O4/CoMoO4. (f) Chronopotentiometry curve of c-Co/Co3Mo at a constant current density of 10 mA cm-2.
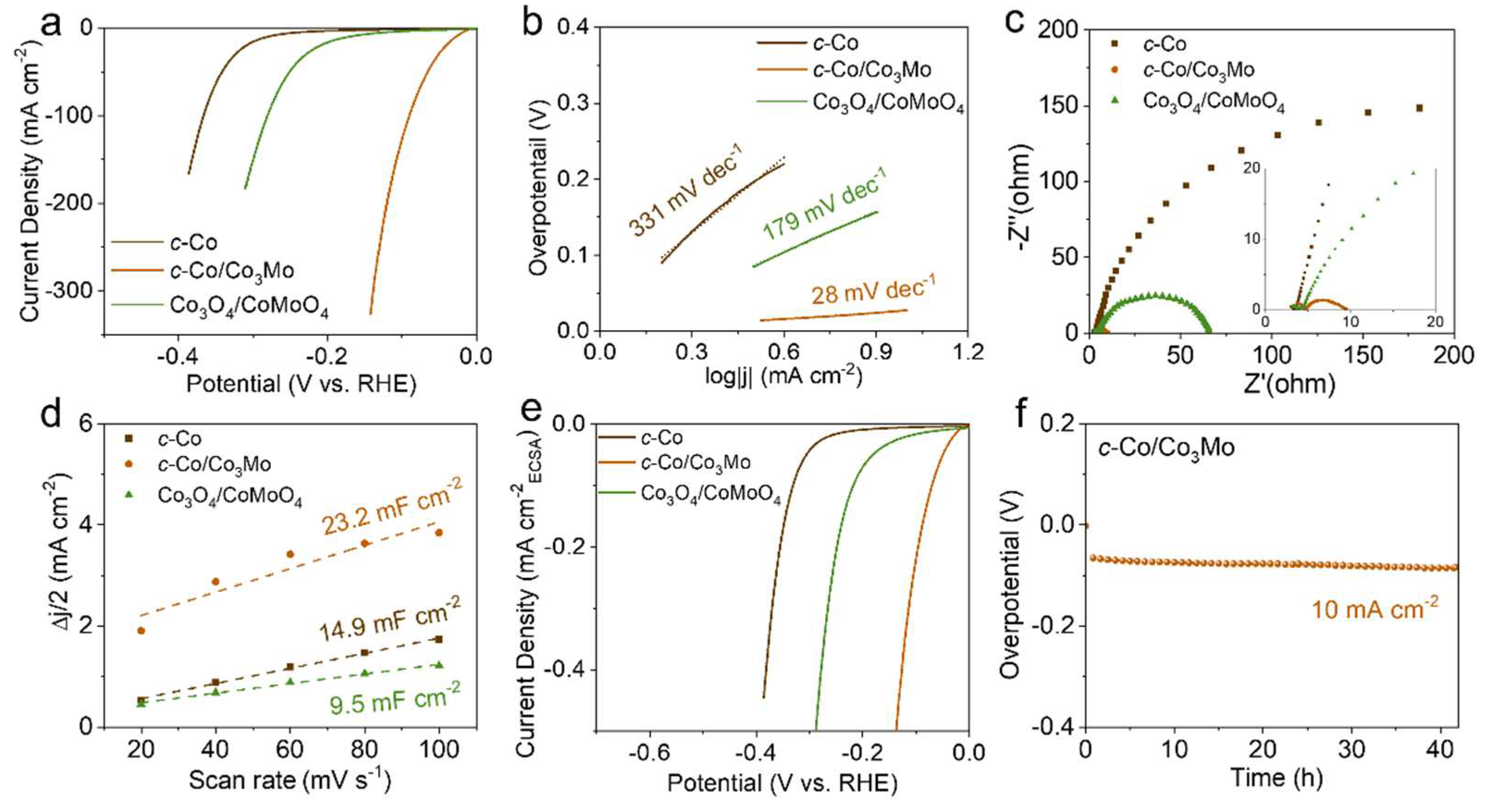
Figure 3.
Evolutive CV curves for (a) c-Co/Co3Mo, (b) c-Co and Co3O4/CoMoO4 from the 1st to the 10th CV in 1 M KOH at 50 mV s-1 between 0.124 and -0.676 V (vs RHE). The inset shows the redox peaks from 0.124 to -0.1 V (vs RHE).
Figure 3.
Evolutive CV curves for (a) c-Co/Co3Mo, (b) c-Co and Co3O4/CoMoO4 from the 1st to the 10th CV in 1 M KOH at 50 mV s-1 between 0.124 and -0.676 V (vs RHE). The inset shows the redox peaks from 0.124 to -0.1 V (vs RHE).
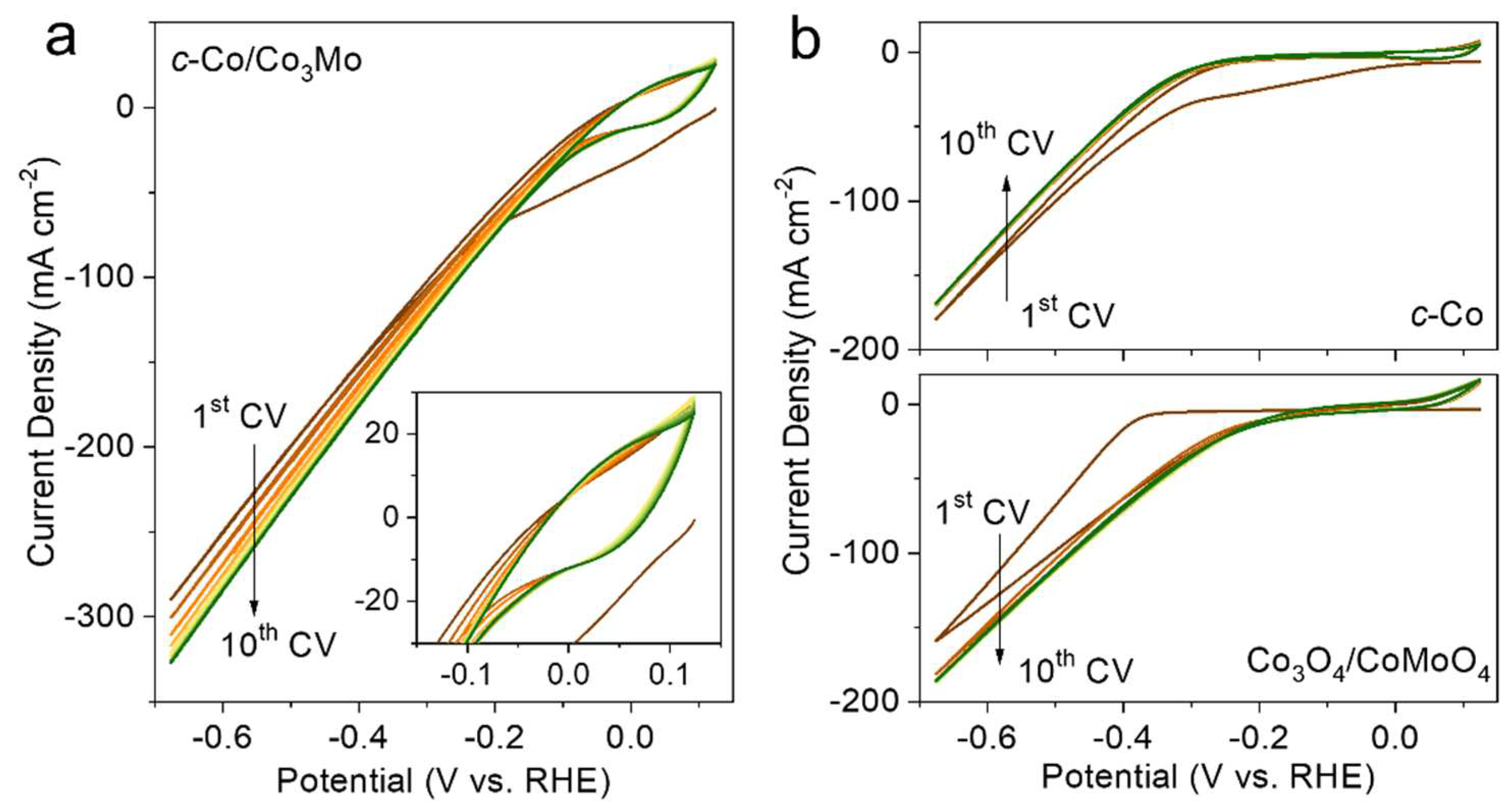
Figure 4.
(a) Raman spectra, High-resolution XPS spectra of (b) Co 2p, (c) Mo 3d, and (d) O 1s of c-Co/Co3Mo before and after HER test.
Figure 4.
(a) Raman spectra, High-resolution XPS spectra of (b) Co 2p, (c) Mo 3d, and (d) O 1s of c-Co/Co3Mo before and after HER test.
Figure 5.
(a) SEM image, (d) TEM image, and (c-e) HRTEM images of c-Co/Co3Mo after HER test.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



