
Submitted:
02 August 2023
Posted:
03 August 2023
You are already at the latest version
Abstract
Atherosclerotic cardiovascular disease (ASCVD) remains the leading cause of death worldwide and the risk of a major cardiovascular event is highest among those with established disease. Ongoing management of these patients relies on the accurate assessment of their response to any prescribed therapy, and their residual risk, in order to optimize treatment. Recent international guidelines and position statements concur that the plasma concentration of apolipoprotein B (apoB) is the most accurate measure of lipoprotein associated ASCVD risk. This is especially true for the growing number of individuals with diabetes, obesity or the metabolic syndrome, and those on statin therapy. Most guidelines, however, continue to promote LDL-C as the primary risk marker due to uncertainty as to whether the greater accuracy of apoB is sufficient to warrant a paradigm shift. Recommendations regarding apoB measurement vary, and the information provided on how to interpret apoB results is sometimes insufficient, particularly for the non-lipid specialist. Misinformation regarding the reliability of the assays is also frequently repeated despite its equivalent or better standardization than many other diagnostic assays. Thus, demand for apoB testing is relatively low, which means there is little incentive to increase its availability or reduce its cost. In this review, we examine the results of recent clinical outcomes studies and meta-analyses on the relative values of apoB, LDL-C and non-HDL-C as markers of ASCVD risk. Although there is seemingly minimal difference among these markers when only population-based metrics are considered, it is evident from our analysis that, from a personalized or precision medicine standpoint, a great many individuals would benefit, at a negligible total cost, if apoB measurement were better integrated into the diagnosis and treatment of ASCVD.
Keywords:
lipoproteins
; atherosclerosis
; dyslipidaemia
; apolipoprotein B
; residual risk
; triglyceride-rich lipoprotein
; remnant lipoprotein
1. Introduction
Despite recent treatment advances, atherosclerotic cardiovascular disease (ASCVD) remains the leading cause of death worldwide [1]. Secondary prevention is aimed at reducing the risk of a major adverse cardiovascular event (MACE) in patients with established ASCVD. It typically involves the use of high-intensity statins, often in conjunction with relatively expensive add-on therapies, such as proprotein convertase subtilisin/kexin type 9 inhibitors (PCSK9i) and bempedoic acid. Given the high stakes of over- and under-treatment, it is of great importance that the correct therapeutic decisions are made, which relies on using the most accurate markers of ASCVD risk.
It is well-established that the trapping of apolipoprotein (apo) B-containing lipoproteins, or β-lipoproteins, and retention of their cholesterol in the arterial wall is an early step in atherosclerotic plaque formation. Hence, the measurement of cholesterol in the blood emerged early on as a key ASCVD risk marker. Initially, total plasma cholesterol (TC) concentration was used, but in the 1950s investigators like Gofman et al [2] and Olson [3] found that only the apoB-containing lipoproteins are positively associated with coronary artery disease. The seminal discovery of the role of the low-density lipoprotein (LDL) receptor (LDLR) in lipoprotein metabolism and atherosclerosis by Brown and Goldstein in 1974 [4] further solidified the importance of LDL in the pathogenesis of ASCVD. Consequently, the National Cholesterol Education Program (NCEP) in 1988 promoted LDL cholesterol (LDL-C) as the primary target for therapy [5], continuing the tradition of emphasizing lipoprotein cholesterol content in risk assessment. This coincided with the development of the first statin, lovastatin, which was the first effective and well-tolerated therapy for achieving substantial LDL-C reductions.
The attributes of β-lipoproteins that are frequently considered as being potentially relevant for understanding their atherogenicity are the type of β-lipoprotein, their cholesterol and triglyceride content, particle size and particle number. Numerous studies have recently established that the particle number of atherogenic lipoproteins (apoB-containing lipoproteins), and not their cholesterol content nor their type, is the most important attribute for determining ASCVD risk [6,7,8,9,10]. Given that all apoB-containing lipoproteins are atherogenic to varying degrees, and that apoB exists as a single copy on all of these lipoproteins, apoB is a convenient way to measure atherogenic particle number.
In 2009, the AACC Lipoprotein and Vascular Diseases Division Working Group on Best Practices published a position statement describing why apoB is the best risk marker for clinical practice [6]. In 2013, the same group supported the adoption of apoB measurement in ASCVD risk assessment and favoured treatment guidelines that utilize apoB [11]. An even stronger rationale exists today for leveraging the benefits of apoB for ASCVD risk assessment, given the growing number of patients with obesity, type II diabetes mellitus (DM2) or the metabolic syndrome (MetS). These patients are known to have abnormal lipoprotein profiles, with high triglycerides (TG), low high-density lipoprotein cholesterol (HDL-C) and elevated small, dense LDL (sdLDL) particle number, but normal or only slightly elevated LDL-C. This profile often leads to discordance between LDL particle number (LDL-P), for which apoB is a close proxy, and LDL-C, and may lead to erroneous LDL-C-based therapeutic decisions [12,13]. The discordance between apoB and LDL-C is also of particular relevance in statin-treated patients, whose LDL-C and non-HDL-C are reduced to a greater extent than their LDL-P (apoB) [14,15,16].
The Canadian Cardiovascular Society has provided percentile equivalent apoB cut-points and treatment targets alongside those for LDL-C and non-HDL-C in their guidelines since 2003, and apoB is an insured test in all but one province in Canada [17]. The most recent European Society of Cardiology and European Atherosclerosis Society (ESC/EAS) guideline also provides both secondary non-HDL-C and apoB targets and states that apoB may be the preferred test in patients with hypertriglyceridaemia [18]. The 2018 US Multisociety guideline for lipid management, however, only recommends apoB as a risk enhancer test in those patients with an intermediate 10-year risk score, or as an optional secondary target for high-risk patients. They state that a TG of ≥2.3 mmol/L may be a relative indication to measure apoB [19].
In this review, we examine the evidence from the past 15 years on the relative value of apoB versus LDL-C and non-HDL-C as ASCVD risk markers. We focus on the more recent clinical outcomes trials that show how treating to apoB targets would improve clinical outcomes for a substantial number of individuals compared to LDL-C or non-HDL-C targets.
2. ApoB Biochemistry and Lipoprotein Metabolism
ApoB is a large hydrophobic protein that is present as a single copy on LDL [20] and triglyceride-rich lipoproteins (TRLs), including chylomicrons (CMs) [21], very low-density lipoproteins (VLDLs) [22], and intermediate-density lipoproteins (IDLs) (remnant lipoproteins) [22]. There are two isoforms of apoB in circulation, apoB100 and apoB48, both of which are detected in apoB assays [20]. ApoB100 comprises 4563 amino acids and is synthesized in the liver [23]. After lipidation by microsomal triglyceride transfer protein (MTP), apoB100 is secreted as the main structural protein on VLDL [20,24]. It also contains a positively charged ligand binding domain for uptake of LDL by the LDLR [23]. ApoB48 is synthesized in the small intestine and is secreted as the main structural protein on CMs which carry dietary lipids to the lymph [21]. It is about 48% the length of apoB100 owing to a stop codon introduced during mRNA editing [25]. ApoB48 lacks the C-terminal, LDLR binding domain of apoB100, and CM remnants are instead cleared via apoE-binding receptors.
Metabolism of TRLs (Figure 1A) results in compositional changes and remodelling to other types of β-lipoproteins [26]. Both CMs and VLDLs are large lipoprotein particles (CM >670 nm, VLDL 27-70 nm) that contain mostly TG in their core. Through lipolysis to fatty acids, this TG is delivered to peripheral tissues for energy production or storage [20]. Alternatively, it may be transferred to other lipoproteins by cholesteryl ester transfer protein (CETP) [20]. As TG is depleted from their core, phospholipids are removed from their shell, and TRLs shrink to form remnant particles. As they shrink, the increasing surface tension [27] causes some of the exchangeable apolipoproteins, such as apoC-II and apoE, that modulate lipoprotein metabolism and cellular uptake, to dissociate from the TRL remnants [28]. CM remnants are removed rapidly by the liver and have a half-life of approximately 10 minutes in circulation [29,30].
During VLDL metabolism, remnant particles become enriched in cholesteryl esters, which are transferred to them from HDL particles by CETP [20,24,31]. VLDL is converted to IDL and much of this is subsequently converted to LDL, which contains approximately 6 to 7 times more cholesterol than TG [20,26]. LDL is removed by the liver, via the LDLR, and it has a half-life in plasma of approximately 3 days [20,31]. Thus, there is approximately 10 times more apoB100 than apoB48 in a fasting plasma sample and about 90% of the apoB in circulation is on LDL.
Owing to the small diameter of LDL particles, averaging about 20 nm, LDL can readily enter the vessel wall and be trapped by the extracellular matrix in the intima. Within the arterial intima, modified LDL is taken up by macrophages and other cells to induce foam cell formation, which eventually leads to atherosclerosis by a complex process that is amplified by inflammation [20,24,31]. Given that it roughly estimates of the concentration of LDL in circulation, LDL-C is associated with ASCVD events [32]. Unlike cholesterol, apoB does not exchange between lipoproteins, and there is thus a fixed amount of apoB per LDL particle. Therefore, it provides a more accurate measure of atherogenic lipoprotein particle number and is strongly associated with ASCVD [33,34,35]. The other apoB-containing lipoprotein particles are also atherogenic once they undergo sufficient lipolysis and become small enough to enter the vessel wall.
The size of a lipoprotein particle is also a determinant of its cholesterol-carrying capacity [20]. For example, larger LDL particles (20-22 nm) carry more cholesterol than do small LDL particles (19-20 nm) [36], and typically account for about 60-70% of total LDL-C. As depicted in Figure 1B, the transfer of TG from large VLDL particles to HDL by CETP may be reduced during hypertriglyceridemia, resulting in TG-enriched IDL and LDL particles [26]. The combined action of lipoprotein lipase and hepatic lipase on these particles results in the generation of small, cholesterol-poor LDL particles [26]. This explains the classic type B phenotype commonly seen in hypertriglyceridemia, in which LDL-C is normal or only slightly elevated, whereas apoB is almost always elevated [13,34]. As will be discussed in more detail, many studies have shown that when apoB and LDL-C are discordant, apoB is a better ASCVD marker than LDL-C [9,37]. Non-HDL-C, which is cholesterol on all apoB-containing lipoproteins, is less affected by this issue but, in most studies, it was also found inferior to apoB as an ASCVD biomarker [38].
It is hypothesized that there may be 4 separate pathways for TRL metabolism [26], which depends on their size and apolipoprotein cargo. Here, two pathways are depicted. Arrow weight indicates flux through each pathway relative to “normal”. Translucence in color of enzymes or receptors indicates decreased activity or affinity. (a) The hypothesized pathway for average sized VLDL and LDL. The weights of the arrows are almost all the same, to indicate “average” flux, but CETP transfers CE more readily than TG. Intermediate-sized LDL is the preferred ligand for the LDLR. LDL also delivers cholesterol to steroid-producing tissues, which endocytose the particle via the LDLR. (b) The hypothesized pathway that predominates in hypertriglyceridemia. In hypertriglyceridemic patients, it seems that larger VLDL-1 is produced, that CETP may be rate-limiting for the transfer of TG from VLDL-1 to HDL, and that LPL is less active. This results in larger, more TG-rich IDL species, which are not bound as readily by their hepatic receptors. Instead, they are processed by HL, which may have increased activity, resulting in small, cholesterol-poor LDL. This means more FA is delivered to the liver and less to peripheral tissues. The LDLR also binds sdLDL less readily, whereas a change in apoB conformation and possibly a loss of sialic acid, means sdLDL binds readily to endothelial cell-surface proteoglycans. They are also more readily modified by oxidative processes. These damaged particles are removed by macrophages via scavenger receptors. Created with BioRender.com
3. Clinical Utility of ApoB in Primary Prevention
Statins, the main lipid-lowering therapy, inhibit hepatic cholesterol synthesis and upregulate the LDLR, leading to lower LDL-C levels [39]. Clear evidence supporting the currently recommended LDL-C treatment targets and percentage reduction strategies is somewhat lacking [40], however, resulting in discrepant recommendations by the various guidelines [17,18,19]. In addition, despite reaching low LDL-C treatment goals, a large proportion of patients still experience atherosclerosis progression or ASCVD events [41]. This phenomenon of residual risk suggests that a singular focus on LDL-C measurement with fixed population-based treatment goals is not optimal for many patients [42].
Several guidelines propose non-HDL-C or apoB as secondary treatment targets, as intensifying lipid-lowering therapy to achieve these secondary targets mitigates the residual risk [43]. Treatment goals for non-HDL-C and apoB were originally derived from the LDL-C targets (Table 1) [17,18]. The American College of Cardiology and American Heart Association (ACC/AHA) Multisociety guideline does not include these parameters in its primary ASCVD prevention strategy but incorporates non-HDL-C in its secondary prevention algorithm for managing high-risk patients [19]. The non-HDL-C cut-offs are arbitrarily set to 0.8 mmol/L above the LDL-C cut-offs. It is important to emphasize that this value is based on the Friedewald equation, which incorrectly assumes that the ratio of TG to VLDL-C is fixed at 2.2 [44]. In contrast, the ESC/EAS recommend either apoB or non-HDL-C for risk assessment and provide targets for both parameters for primary prevention. They further suggest that apoB may be the preferred marker in patients with hypertriglyceridemia, obesity, or DM2 and promote it as an alternative to LDL-C for assessing ASCVD risk [18]. The Canadian Cardiovascular Society strongly recommends using non-HDL-C or apoB instead of LDL-C as the primary risk marker for ASCVD. They provide targets for both in their graphical algorithm, increasing the likelihood that clinicians will understand and use these measures [17]. Both the ESC/EAS and Canadian guidelines acknowledge the superiority of apoB over non-HDL-C, but neither unequivocally recommends apoB to be prioritized as the therapeutic target.
From a pathophysiological perspective, apoB is likely to be superior to both LDL-C and non-HDL-C as a biomarker as it represents the total atherogenic particle concentration rather than simply the cholesterol content of these particles. This is important because cholesterol content varies widely within and between particle types. For example, in 50% of individuals either smaller, cholesterol-depleted LDL particles or larger, cholesterol-enriched particles predominate [45]. In these individuals LDL-C would underestimate or overestimate the LDL-P, respectively. The importance of this issue was validated clinically in numerous discordance analyses, including the Coronary Artery Risk Development in Young Adults (CARDIA) Study [46], the Women's Health Study [47], the INTERHEART study [48], the Framingham Heart Study [49], and the Copenhagen general population study [50], all of which support the concept that, when discordant, the risk of ASCVD is more closely related to the concentration of atherogenic lipoprotein particles than to the amount of cholesterol they carry. Such discordance is common in hypertriglyceridemia [51], obesity [52], MetS and DM2 [53], all of which are becoming more common throughout the world. These high-risk individuals have predominant small, dense cholesterol-poor LDL particles, which explains why they have relatively normal LDL-C that underestimates the ASCVD risk that is evidenced by their higher LDL-P or apoB.
Another important issue is that statins increase the proportion of small dense sdLDL compared to large buoyant LDL (lbLDL) [14]. This is because the LDLR has greater affinity for lbLDL [15]. Thus, although statins reduce the concentration of all LDL particles, they have a disproportionately larger effect in reducing LDL-C, leading to an underestimation of risk, particularly in patients with high levels of sdLDL with a higher LDL-P [8,10,11]. This phenomenon is evident in the discordant responses of LDL-C and apoB to statin treatment, where statins induced an LDL-C decrease of around 34% with a concomitant decrease of only 24% in apoB [54].
To our knowledge there are no trials that specifically examined apoB as a therapeutic target. Instead, it was studied in meta-analyses comparing non-HDL-C and apoB, with some contradictory findings. The 2007 emerging risk factors collaboration (ERFC) meta-analysis showed that the hazard ratios (HRs) for ASCVD of non-HDL-C and apoB were similar through the quintiles [55]. There were, however, several limitations in this meta-analysis, such as inclusion of several studies that used non-standardized methods to measure apoB. In addition, it included several studies that were not published and therefore could not be fully evaluated. In 2012 Boekholdt et al performed a meta-analysis of 8 randomized studies evaluating the evidence for LDL-C, non-HDL-C and apoB in patients treated with statins. The HRs for cardiovascular events were virtually identical and clinically indistinguishable for residual risk (HR 1.13, 1.16 and 1.14 for LDL-C, non-HDL-C and apoB, respectively) [56]. The power of this study to determine superior precision amongst these markers was called into question by Thanassoulis et al, however, who performed a meta-analysis in 2014 of the 7 largest and most important placebo-controlled statin trials. They concluded that apoB reduction confers a better ASCVD risk reduction per 1 standard deviation (SD) of apoB than per 1SD reduction of LDL-C or per 1 SD reduction of non-HDL (39% vs 30% vs 32% respectively) [54]. Furthermore, a 2011 meta-analysis, performed by the same group, of 12 independent reports, including 233,455 patients and 22,950 events, reported that the relative risk reduction associated with a 1SD decrease in apoB was 5.7% greater than that of non-HDL-C and 12% greater than that of LDL-C (RRR 1.43, 1.34 and 1.25 for apoB, non-HDL-C and LDL-C, respectively). They also calculated the number of cardiovascular events that would be prevented among adult US residents by using apoB, non-HDL-C and LDL-C as risk markers in a NCEP Adult Treatment Plan-III-based prevention strategy. They found that using apoB as the primary ASCVD risk marker would prevent 800,000 more events over 10 years than using LDL-C [57].
More recently two other studies have further supported the superiority of apoB over other ASCVD risk biomarkers. In 2021, Johannesen et al analysed the data of 13,015 statin-treated patients from the Copenhagen general population study. They showed that elevated apoB and non-HDL-C were associated with increased risk of all-cause mortality and myocardial infarction (MI), but these associations were not found for elevated LDL-C [50]. Those with discordantly high apoB compared with LDL-C (apoB above median apoB and LDL-C below median LDL-C) had a HR of 1.21 for all-cause mortality and 1.49 for MI, while those with discordantly high apoB compared with non-HDL-C had a HR of 1.21 for all-cause mortality, and 0.93 for MI. Furthermore, when both apoB and non-HDL-C are discordantly high compared with LDL-C, the HR was 1.23 for all-cause mortality and 1.82 for MI. In 2021, Marston and colleagues published a prospective cohort analysis including 389,529 primary ASCVD prevention candidates from the UK Biobank, and 40,430 statin-treated patients from the Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER) trial and the Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) [9]. They examined the individual associations of baseline apoB, non-HDL-C, and TG with incident MI. In fully adjusted models, only apoB remained significantly associated with MI in the primary prevention cohort (adjusted HR 1.27 per 1 SD; 95% confidence interval [CI], 1.15-1.40; P < .001). In the secondary prevention cohort, apoB was again the only biomarker found to be independently associated with MI. It was also observed that there was no longer a significant association between the ratio of TG to LDL-C (a surrogate for the ratio of TRLs to LDL) and the risk of MI when the model was adjusted for apoB.
4. Clinical Utility of ApoB in Secondary Prevention
Secondary prevention of ASCVD generally entails increasing the intensity of statin treatment or adding a second lipid-lowering therapy to achieve a ≥50% reduction in LDL-C and an LDL-C threshold of ≤1.8 mmol/L. Most recent guidelines recommend considering adding either ezetimibe or a PCSK9i in cases where the patient fails to achieve the above LDL-C targets on the maximum dose, or maximum tolerated dose of statin therapy. Some have suggested that these medications should also be considered if the patient has achieved their LDL-C goal but not their apoB or non-HDL-C target [17,18,19]. Ezetimibe and PCSK9i both tend to reduce LDL-C more than they do apoB. Add-on ezetimibe typically achieves additional LDL-C reductions of about 20% and apoB reduction of about 17%, while an add-on PCSK9i achieves additional LDL-C reductions of 50-60% and apoB reductions of only 46-53% [58,59,60,61,62]. On PCSK9i therapy, it is now possible for patients to uniformly reach remarkably low LDL-C values, below 1.8 mmol/L, but the achieved apoB is not always correspondingly low. Thus, in high-risk individuals, measuring apoB and treating to apoB targets is emphasized to ensure optimal lipoprotein-associated risk reduction. In fact, it is now clear that discordance between LDL-C and apoB, and between non-HDL-C and apoB, exists across the range of values for these parameters, suggesting that apoB should be used more broadly [38].
In 2022, Hagström et al [37] analysed data from the ODYSSEY treatment trial, including 18,924 patients with a recent episode of acute coronary syndrome, who had not met treatment targets despite high-intensity or maximally tolerated statin therapy. The cohort was split into a treatment and a control arm, with the treatment group receiving a subcutaneous injection of the PCSK9i, alirocumab, 75 mg fortnightly, while controls received placebo and the baseline therapy. The investigators analysed the risk of MACE by baseline and achieved lipid parameters at 4 months. They found that while baseline apoB held independent prognostic value when the model was adjusted for the Friedewald LDL-C, it lost significance when adjusted for the Martin/Hopkins LDL-C. Continuous baseline apoB was a more sensitive marker of risk than non-HDL-C, which had an otherwise similar linear relationship. On the other hand, the benefit of 4 months’ treatment on alirocumab increased with decreasing apoB below 1.3 mmol/L after adjustment for LDL-C by both methods. Continuous achieved apoB had a significant linear relationship with risk, after adjustment for LDL-C and non-HDL-C, while the converse was not the case. Of note, the relationship between achieved non-HDL-C and risk was flat, with a rapidly widening confidence interval as non-HDL-C values increased such that it crossed the HR = 1 line for most values of non-HDL-C. When tertiles of achieved apoB were cross-tabulated with tertiles of achieved LDL-C, risk of MACE increased with increasing apoB for each tertile of LDL-C, but there was no relationship of MACE with achieved LDL-C.
Marston et al [9] performed a similar analysis on 40,430 statin-treated patients from the FOURIER and IMPROVE-IT studies, who were followed up for a median of 2.5 years. The study interventions were addition of the PCSK9i, evolocumab, or ezetimibe, respectively. Achieved apoB after add-on therapy was predictive of fatal MI after adjustment for non-HDL-C, HDL-C, TG and clinical factors. In contrast, non-HDL-C was no longer predictive after adjustment for apoB, and TG was not predictive after adjustment for clinical parameters, or both clinical and lipid parameters. The ratio of TG/LDL-C was also analysed in this cohort, to determine whether TRL or LDL have a greater association with MI. High ratios were achieved in this cohort due to LDL-C lowering, and the association line was flat up to a ratio of 2, meaning that neither lipoprotein type poses a higher risk of MI than the other. These findings were all consistent in sensitivity analyses using selected subgroups.
5. Assay Standardization
While many guidelines have now acknowledged that apoB is the most accurate lipid marker of ASCVD risk and response to therapy [17,18,19], and the European guideline has endorsed the assays as well-standardized and accurate and acknowledged that they are superior to the measurement or calculation of LDL-C and non-HDL-C [18], there are still those that question the reliability of apoB measurements [19]. To laboratorians, “accuracy” is a measure of how close a measurement or prediction is to the “truth”. In the case of analytical accuracy, this “truth” is determined by the “gold standard” or reference method, while in the case of diagnostic accuracy, the “truth” is the true diagnosis or prognosis. The accuracy of a lipid-associated risk prediction is a composite of the biological relationship of the actual plasma concentration of the lipid or lipoprotein with risk, and the ability of the assay to provide a true measurement of that concentration. The findings in the studies mentioned above represent this composite accuracy as they evaluate the relationships of the lipids with risk using measurements performed by the available assays with their current analytical performance. It is perplexing then that this evidence is discounted, particularly by clinicians, due to the supposed lack of standardization of apoB assays when it is these same assays that proved accurate across various manufacturers in these clinical trials.
ApoB may be routinely measured on automated clinical chemistry analysers using immunoturbidimetry or immunonephelometry. These same assay principles are used to accurately measure other proteins, such as C-reactive protein, immunoglobulins and transferrin in routine clinical chemistry laboratories. The World Health Organization (WHO) and International Federation of Clinical Chemistry (IFCC) have established a standardization program and a secondary reference material (SP3) for apoB, [63] and evidence from international proficiency testing programs suggests that apoB assays perform well [64,65]. The EAS and the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) Joint Consensus Initiative, in fact, reported that apoB assays have better analytical performance than do LDL-C and non-HDL-C [66]. While apoB is not yet standardized to a pure, higher order reference material, neither are other lipoprotein markers. Furthermore, because lipoproteins are heterogeneous, polydisperse particles, they cannot be isolated as a pure substance and will likely never be truly “standardized” according to the International Organization for Standardization standard 17511:2020 requirements. In contrast, the amino acid sequences recognized in apoB assays are well-defined. This has enabled the recent development of a primary reference method to accurately measure apoB by mass spectrometry [67], which should be put in place soon to further improve the standardization of apoB.
In addition to its better standardization, apoB is also largely unaffected by the fasting or non-fasting state. ApoB assays are also unaffected by high degrees of lipemia [66], and unit conversions are within the weight-based metric system (i.e. multiples of 10) and no further calculations are, therefore, required. In contrast, calculation of LDL-C is fraught with controversy. Most laboratories still use the Friedewald equation when TG ≤4.5 mmol/L and variably do not calculate it or use one of the alternative equations when TG >4.5 mmol/L. This is because the Friedewald equation assumes a fixed ratio of TG to VLDL-C and the error inherent in this assumption exceeds acceptability when TG >4.5 mmol/L. Owing to problems with lipoprotein specificity, direct LDL-C measurements do not necessarily improve the accuracy for LDL-C determinations, as shown by Miller et al [68]. While non-HDL-C calculation escapes the issue of the TG conversion factor, and thereby avoids this error in its calculation [69], it still includes HDL-C measurement, which is affected by elevated TG and other matrix effects that are common in dyslipidaemias [66]. Furthermore, the uncertainty of non-HDL-C determination is affected by the additive uncertainties in the TC and HDL-C measurement procedures, and it is also affected by unit conversion between the standard international and weight-based units.
6. Utility of ApoB Versus Non-HDL-C as ASCVD Risk Markers
The inclusion of cholesterol on all apoB-containing lipoproteins in non-HDL-C helps to partially correct for the underestimation of risk by LDL-C in hypertriglyceridemia. In addition, non-HDL-C may be calculated from the standard lipid panel at no additional cost. For these reasons, in the Canadian guidelines, routine calculation of non-HDL-C is recommended [17]. The EAS and the EFLM Joint Consensus Initiative also recommends non-HDL-C calculation for all patients [66]. Likewise, the US Multisociety guidelines allude to equivalence between non-HDL-C and apoB, and promote non-HDL-C calculation in place of apoB when LDL-C is inaccurate [19].
As shown in Figure 2, although non-HDL-C is less discordant with apoB than is LDL-C, it frequently results in a different risk assessment than apoB. In approximately one third of individuals in NHANES on a lipid-lowering medication, concentrations of non-HDL-C and apoB differ by more than ±10% on a population percentile basis. Although use of non-HDL-C in hypertriglyceridaemic patients correctly raises the risk assessment in most of these patients, it sometimes leads to overestimation of risk, particularly in those with the highest triglycerides (Figure 2: Sector A). In other cases of hypertriglyceridemia, risk is still underestimated by non-HDL-C (Figure 2: Sector B). Again, this relates to the complicated relationship between particle number, size and lipid composition, as well as analytical limitations, which often lead to a disconnect between these different parameters and the wide dispersion around the regression line between non-HDL-C and apoB.
Lipid test results from NHANES (N=1121) for individuals aged 18-75 years (mean age=62; 58% male) treated with lipid-lowering medication collected between 2005-2016 with LDL-C levels <100 mg/dL (mean LDL-C=76 mg/dL) were used to calculate the difference in percentile units between LDL-C and apoB (X-axis) and between nonHDL-C and apoB (Y-axis). Concordant LDL-C (Sectors B+E+H) and non-HDL-C results (Sectors D+E+F) were defined as being within ±10 percentile units of the apoB percentile value. Individual test results are color coded by the percentile of triglycerides. Percent of total population in each individual sector (next to sector designation) or combined sectors (column and row value headings) is indicated. LDL-C was calculated by the Friedewald equation. White background: concordant sectors; colored background: discordant sectors.
Numerous discordance studies, Mendelian randomization studies and prospective cohort studies already discussed have shown that non-HDL-C is not equivalent to apoB as a marker of risk. Whether non-HDL-C is adequate remains controversial, however. The main arguments in favour of its adequacy are its stronger correlation and reduced discordance with apoB compared with those of LDL-C - essentially, that it is “good enough”. Proponents of apoB raise the point that population-derived statistics, such as correlation coefficients, are not as meaningful on an individual level. The residual, or difference between observed and expected apoB after linear regression of apoB on non-HDL-C, provides additional risk information [49] and shows that for individual patients, non-HDL-C is not always adequate. In the large meta-analysis discussed above, Sniderman et al found that treating apoB targets could prevent 500,000 more cardiovascular events among US adults over 10 years than treating to non-HDL-C targets [57]. This, and the clinical trials already discussed, partially address the argument that there is a lack of evidence that treating to apoB or non-HDL-C targets improves outcomes [66]. While direct outcomes studies remain desirable, the evidence for LDL-C targets is also not robust [40].
The Canadian and European guidelines provide treatment target values for both non-HDL-C and apoB [17,18], whereas the US Multisociety guideline provides targets only for non-HDL-C [19]. In the Canadian guideline, the target values provided for both non-HDL-C and apoB are percentile equivalents of the recommended LDL-C targets [17]. The European guideline provides apoB treatment targets based on a study of 1154 diabetic patients treated with atorvastatin or placebo [18]. They performed linear regression of the achieved apoB on the achieved LDL-C to provide equivalent achieved apoB values to the target LDL-C values in the treatment arm of this cohort [70].
The US and European guidelines provide non-HDL-C targets that are a fixed 0.8 mmol/L greater than the LDL-C targets. This is based on an ideal TG value of 1.7 mmol/L and assumes a fixed ratio of TG to VLDL-C of 2.2, as per the Friedewald formula [44]. This is not ideal, as we know that this ratio is not fixed. Given that this is one of the major problems with LDL-C calculation, it is perplexing that this spurious ratio is still included in non-HDL-C target determination.
It has also been argued that apoB does not substantially improve the accuracy of risk prediction models that already include other risk factors. This argument no longer seems to hold once it is appreciated that the order in which markers are added to a model affects the incremental predictive value assigned to that marker [49,71,72] and several groups have shown impressive predictive value of apoB in risk models [49,73]. Aside from its role as a secondary risk marker, the European guideline states that apoB may be used as the primary marker for screening, diagnosis and management, where available. It also recommends apoB measurement in patients with hypertriglyceridemia, obesity, diabetes, the metabolic syndrome or very low LDL-C, and states that it may be preferred over non-HDL-C in these cases [18]. In Canada, apoB is an insured laboratory test in all provinces except Ontario, and while apoB is explicitly acknowledged as the better marker, it is left to clinicians to choose between apoB and non-HDL-C depending on their level of comfort with each measure, the availability of testing and concern regarding discordance between the markers [17].
In our view, non-HDL-C should be reported for all patients. Like apoB, it is superior to LDL-C because it is a more accurate proxy for atherogenic lipoprotein concentration. Also, it does not involve any additional cost and, provided the triglycerides are not high enough to affect the HDL-C assay, it is not affected by the non-fasting state [74]. ApoB, however, is superior to non-HDL-Cand it is cost-effective [75]. In this paradigm of precision medicine, it is likely that the value of the additional precision of apoB will ultimately result in its adoption as the primary risk marker for ASCVD.
5. Conclusions
The preponderance of evidence indicates that apoB is the superior biomarker for ASCVD prevention compared to other lipid and lipoprotein related measures. Its measurement is now adequately standardized, and it can be measured accurately and precisely by automated assays on clinical chemistry analysers [63,66]. Based on College of American Pathology proficiency surveys, however, apoB assays are not widely offered by clinical laboratories in the US and this may also be the case elsewhere. The poor availability reflects low demand for the assay, which may be attributed to several barriers, including the guideline recommendations, a lack of familiarity of clinicians with interpretation of apoB results, and with their value, and the cost of testing.
International guidelines are gradually promoting apoB measurement in specific contexts, and some have recommended initial assessment cut-offs and treatment targets (Table 1). With ongoing development of precision medicine approaches, and with more widespread acknowledgement of, and participation in the WHO/IFCC standardization program, further guidance on the use and interpretation of apoB may be expected. In the interim, clinical laboratories may illustrate the importance of apoB and assist clinicians in interpretation by converting apoB results to percentile equivalent LDL-C results [38]. This may also serve to improve familiarity with apoB values. It is imperative for laboratorians to continue to educate clinicians on the validity and standardization of the apoB assays that are routinely available.
Another challenge is the current cost of the assays. In fact, it was already shown that adding apoB to the lipid panel would not increase overall costs substantially due to its clinical effectiveness [75]. Given the much higher cost of drug therapy, particularly for the newer agents, and the even higher costs related to inadequate treatment of high-risk individuals, treating to apoB targets is the most reliable and cost-effective strategy in mitigating lipid-associated residual risk [9,37]. Most US insurance companies will reimburse apoB in high-risk individuals, and it is an insured test throughout most of Canada. With more demand from clinicians and patients, and with advocacy from clinical chemists, it may be possible to reduce the reimbursement rate for apoB, making it more financially accessible.
It is predicted that the next paradigm shift in ASCVD risk prediction in this era of precision medicine will involve individualized calculation of the potential for net benefit from treatment [76]. Although a step in the right direction, the pooled cohort equations applied in the US guidelines are known to regularly overestimate risk, and population-based risk scores often show poor specificity when applied on an individual basis [77,78,79]. Future individualized risk assessment will likely include current known risk markers and may include new, emerging risk markers, such as markers of HDL dysfunction and LDL oxidation, as well as genetic markers. Ideally, an individualized net treatment benefit prediction will also include an estimation of the risk of adverse events secondary to treatment [76]. Based on our analysis of the literature, the replacement of LDL-C with apoB is a step that should be taken now in managing lipid-lowering therapy and perhaps eventually for screening in primary prevention once the infrastructure is in place for its more widespread use.
Author Contributions
Conceptualization: AR, IJ and JC; Writing - Original draft preparation: JC, RZ and AW; Writing – Review & Editing: JC, IJ and AR; Supervision: AR and IJ.
Funding
Work by JC, AW and AR was funded by intramural research funds from National Heart, Lung and Blood Institute. RZ was funded by Fundación para la Salud y la Educación Salvador Zubirán and Asociación ALE.
Acknowledgments
We’d like to acknowledge Maureen Sampson for preparation of Figure 2.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jones, D.S.; Greene, J.A. The decline and rise of coronary heart disease: understanding public health catastrophism. American journal of public health 2013, 103, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Gofman, J.W.; Jones, H.B.; Lindgren, F.T.; Lyon, T.P.; Elliott, H.A.; Strisower, B. Blood lipids and human atherosclerosis. Circulation 1950, 2, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Olson, R.E. Prevention and control of chronic disease. I. Cardiovascular disease--with particular attention to atherosclerosis. American journal of public health and the nation's health 1959, 49, 1120–1128. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proceedings of the National Academy of Sciences of the United States of America 1974, 71, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Report of the National Cholesterol Education Program Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. The Expert Panel. Archives of internal medicine 1988, 148, 36–69. [CrossRef]
- Contois, J.H.; McConnell, J.P.; Sethi, A.A.; Csako, G.; Devaraj, S.; Hoefner, D.M.; Warnick, G.R.; Lipoproteins, A.; Vascular Diseases Division Working Group on Best, P. Apolipoprotein B and cardiovascular disease risk: position statement from the AACC Lipoproteins and Vascular Diseases Division Working Group on Best Practices. Clin Chem 2009, 55, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Cromwell, W.C.; Otvos, J.D.; Keyes, M.J.; Pencina, M.J.; Sullivan, L.; Vasan, R.S.; Wilson, P.W.; D'Agostino, R.B. LDL Particle Number and Risk of Future Cardiovascular Disease in the Framingham Offspring Study - Implications for LDL Management. J Clin Lipidol 2007, 1, 583–592. [Google Scholar] [CrossRef]
- Kuller, L.; Arnold, A.; Tracy, R.; Otvos, J.; Burke, G.; Psaty, B.; Siscovick, D.; Freedman, D.S.; Kronmal, R. Nuclear magnetic resonance spectroscopy of lipoproteins and risk of coronary heart disease in the cardiovascular health study. Arterioscler Thromb Vasc Biol 2002, 22, 1175–1180. [Google Scholar] [CrossRef]
- Marston, N.A.; Giugliano, R.P.; Melloni, G.E.M.; Park, J.G.; Morrill, V.; Blazing, M.A.; Ference, B.; Stein, E.; Stroes, E.S.; Braunwald, E.; et al. Association of Apolipoprotein B-Containing Lipoproteins and Risk of Myocardial Infarction in Individuals With and Without Atherosclerosis: Distinguishing Between Particle Concentration, Type, and Content. JAMA Cardiol 2022, 7, 250–256. [Google Scholar] [CrossRef]
- Otvos, J.D.; Collins, D.; Freedman, D.S.; Shalaurova, I.; Schaefer, E.J.; McNamara, J.R.; Bloomfield, H.E.; Robins, S.J. Low-density lipoprotein and high-density lipoprotein particle subclasses predict coronary events and are favorably changed by gemfibrozil therapy in the Veterans Affairs High-Density Lipoprotein Intervention Trial. Circulation 2006, 113, 1556–1563. [Google Scholar] [CrossRef]
- Lipoproteins, A.; Vascular Diseases Division Working Group on Best, P.; Cole, T.G.; Contois, J.H.; Csako, G.; McConnell, J.P.; Remaley, A.T.; Devaraj, S.; Hoefner, D.M.; Mallory, T.; et al. Association of apolipoprotein B and nuclear magnetic resonance spectroscopy-derived LDL particle number with outcomes in 25 clinical studies: assessment by the AACC Lipoprotein and Vascular Diseases Division Working Group on Best Practices. Clin Chem 2013, 59, 752–770. [Google Scholar] [CrossRef]
- Caixas, A.; Ordonez-Llanos, J.; de Leiva, A.; Payes, A.; Homs, R.; Perez, A. Optimization of glycemic control by insulin therapy decreases the proportion of small dense LDL particles in diabetic patients. Diabetes 1997, 46, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.M.; Perez, A.; Calvo, F.; Bonet, R.; Castellvi, A.; Ordonez, J. Apolipoprotein(B) identifies dyslipidemic phenotypes associated with cardiovascular risk in normocholesterolemic type 2 diabetic patients. Diabetes Care 1999, 22, 812–817. [Google Scholar] [CrossRef]
- Choi, C.U.; Seo, H.S.; Lee, E.M.; Shin, S.Y.; Choi, U.J.; Na, J.O.; Lim, H.E.; Kim, J.W.; Kim, E.J.; Rha, S.W.; et al. Statins do not decrease small, dense low-density lipoprotein. Texas Heart Institute journal 2010, 37, 421–428. [Google Scholar]
- Nigon, F.; Lesnik, P.; Rouis, M.; Chapman, M.J. Discrete subspecies of human low density lipoproteins are heterogeneous in their interaction with the cellular LDL receptor. Journal of lipid research 1991, 32, 1741–1753. [Google Scholar] [CrossRef] [PubMed]
- Sniderman, A.D. Differential response of cholesterol and particle measures of atherogenic lipoproteins to LDL-lowering therapy: implications for clinical practice. J Clin Lipidol 2008, 2, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.J.; Thanassoulis, G.; Anderson, T.J.; Barry, A.R.; Couture, P.; Dayan, N.; Francis, G.A.; Genest, J.; Gregoire, J.; Grover, S.A.; et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can J Cardiol 2021, 37, 1129–1150. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [CrossRef]
- Meeusen, J.W.; Ueda, M.; Nordestgaard, B.G.; Remaley, A.T. Lipids and lipoproteins. In ., Elsevier eBooks+, Edition. ed.; Rifai, N., Ed.; Elsevier - OHCE: 2022; pp. 1214–1229. In Tietz Textbook of Laboratory Medicine, Elsevier eBooks+, 7th ed.; Elsevier - OHCE, 2022; pp. 1214–1229. [Google Scholar]
- Phillips, M.L.; Pullinger, C.; Kroes, I.; Kroes, J.; Hardman, D.A.; Chen, G.; Curtiss, L.K.; Gutierrez, M.M.; Kane, J.P.; Schumaker, V.N. A single copy of apolipoprotein B-48 is present on the human chylomicron remnant. Journal of lipid research 1997, 38, 1170–1177. [Google Scholar] [CrossRef]
- Elovson, J.; Chatterton, J.E.; Bell, G.T.; Schumaker, V.N.; Reuben, M.A.; Puppione, D.L.; Reeve, J.R., Jr.; Young, N.L. Plasma very low density lipoproteins contain a single molecule of apolipoprotein B. Journal of lipid research 1988, 29, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Knott, T.J.; Pease, R.J.; Powell, L.M.; Wallis, S.C.; Rall, S.C., Jr.; Innerarity, T.L.; Blackhart, B.; Taylor, W.H.; Marcel, Y.; Milne, R.; et al. Complete protein sequence and identification of structural domains of human apolipoprotein B. Nature 1986, 323, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Lucero, D.; Wolska, A.; Aligabi, Z.; Turecamo, S.; Remaley, A.T. Lipoprotein Assessment in the twenty-first Century. Endocrinol Metab Clin North Am 2022, 51, 459–481. [Google Scholar] [CrossRef]
- Chen, S.H.; Habib, G.; Yang, C.Y.; Gu, Z.W.; Lee, B.R.; Weng, S.A.; Silberman, S.R.; Cai, S.J.; Deslypere, J.P.; Rosseneu, M.; et al. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science 1987, 238, 363–366. [Google Scholar] [CrossRef]
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. Journal of lipid research 2002, 43, 1363–1379. [Google Scholar] [CrossRef] [PubMed]
- Meyers, N.L.; Larsson, M.; Olivecrona, G.; Small, D.M. A Pressure-dependent Model for the Regulation of Lipoprotein Lipase by Apolipoprotein C-II. J Biol Chem 2015, 290, 18029–18044. [Google Scholar] [CrossRef]
- Wolska, A.; Dunbar, R.L.; Freeman, L.A.; Ueda, M.; Amar, M.J.; Sviridov, D.O.; Remaley, A.T. Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis 2017, 267, 49–60. [Google Scholar] [CrossRef]
- Nakajima, K.; Nakano, T.; Tokita, Y.; Nagamine, T.; Inazu, A.; Kobayashi, J.; Mabuchi, H.; Stanhope, K.L.; Havel, P.J.; Okazaki, M.; et al. Postprandial lipoprotein metabolism: VLDL vs chylomicrons. Clin Chim Acta 2011, 412, 1306–1318. [Google Scholar] [CrossRef]
- Hussain, M.M.; Kancha, R.K.; Zhou, Z.; Luchoomun, J.; Zu, H.; Bakillah, A. Chylomicron assembly and catabolism: role of apolipoproteins and receptors. Biochim Biophys Acta 1996, 1300, 151–170. [Google Scholar] [CrossRef]
- Wolska, A.; Remaley, A.T. Lipoproteins. In Handbook of Diagnostic Endocrinology, 3rd ed.; William Winter E, L.J.S., Brett Holmquist, Roger L. Bertholf, Ed.; Elsevier - Academic Press: Cambridge (MA), 2021; pp. 287–308. [Google Scholar]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017, 38, 2459–2472. [Google Scholar] [CrossRef]
- Segrest, J.P.; Jones, M.K.; De Loof, H.; Dashti, N. Structure of apolipoprotein B-100 in low density lipoproteins. Journal of lipid research 2001, 42, 1346–1367. [Google Scholar] [CrossRef] [PubMed]
- Sniderman, A.D.; Thanassoulis, G.; Glavinovic, T.; Navar, A.M.; Pencina, M.; Catapano, A.; Ference, B.A. Apolipoprotein B Particles and Cardiovascular Disease: A Narrative Review. JAMA Cardiol 2019, 4, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Sniderman, A.D.; Marcovina, S.M. Apolipoprotein A1 and B. Clin Lab Med 2006, 26, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Austin, M.A.; Breslow, J.L.; Hennekens, C.H.; Buring, J.E.; Willett, W.C.; Krauss, R.M. Low-density lipoprotein subclass patterns and risk of myocardial infarction. JAMA 1988, 260, 1917–1921. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, E.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Danchin, N.; Diaz, R.; Goodman, S.G.; Harrington, R.A.; Jukema, J.W.; et al. Apolipoprotein B, Residual Cardiovascular Risk After Acute Coronary Syndrome, and Effects of Alirocumab. Circulation 2022, 146, 657–672. [Google Scholar] [CrossRef]
- Cole, J.; Otvos, J.D.; Remaley, A.T. A Translational Tool to Facilitate Use of Apolipoprotein B for Clinical Decision-Making. Clin Chem 2023, 69, 41–47. [Google Scholar] [CrossRef]
- Michos, E.D.; McEvoy, J.W.; Blumenthal, R.S. Lipid Management for the Prevention of Atherosclerotic Cardiovascular Disease. N Engl J Med 2019, 381, 1557–1567. [Google Scholar] [CrossRef]
- Leibowitz, M.; Cohen-Stavi, C.; Basu, S.; Balicer, R.D. Targeting LDL Cholesterol: Beyond Absolute Goals Toward Personalized Risk. Current cardiology reports 2017, 19, 52. [Google Scholar] [CrossRef]
- Hoogeveen, R.C.; Ballantyne, C.M. Residual Cardiovascular Risk at Low LDL: Remnants, Lipoprotein(a), and Inflammation. Clin Chem 2021, 67, 143–153. [Google Scholar] [CrossRef]
- Wolska, A.; Remaley, A.T. Measuring LDL-cholesterol: what is the best way to do it? Current opinion in cardiology 2020, 35, 405–411. [Google Scholar] [CrossRef]
- Wadstrom, B.N.; Wulff, A.B.; Pedersen, K.M.; Nordestgaard, B.G. Remnant cholesterol in the era of intensive lipid-lowering therapies. Eur Heart J 2023. [CrossRef] [PubMed]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972, 18, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Mora, S.; Buring, J.E.; Ridker, P.M. Discordance of low-density lipoprotein (LDL) cholesterol with alternative LDL-related measures and future coronary events. Circulation 2014, 129, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, J.T.; Li, R.C.; Sniderman, A.; Chan, C.; Lloyd-Jones, D.M. Discordance Between Apolipoprotein B and LDL-Cholesterol in Young Adults Predicts Coronary Artery Calcification: The CARDIA Study. J Am Coll Cardiol 2016, 67, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Lawler, P.R.; Akinkuolie, A.O.; Ridker, P.M.; Sniderman, A.D.; Buring, J.E.; Glynn, R.J.; Chasman, D.I.; Mora, S. Discordance between Circulating Atherogenic Cholesterol Mass and Lipoprotein Particle Concentration in Relation to Future Coronary Events in Women. Clin Chem 2017, 63, 870–879. [Google Scholar] [CrossRef]
- Sniderman, A.D.; Islam, S.; Yusuf, S.; McQueen, M.J. Discordance analysis of apolipoprotein B and non-high density lipoprotein cholesterol as markers of cardiovascular risk in the INTERHEART study. Atherosclerosis 2012, 225, 444–449. [Google Scholar] [CrossRef]
- Pencina, M.J.; D'Agostino, R.B.; Zdrojewski, T.; Williams, K.; Thanassoulis, G.; Furberg, C.D.; Peterson, E.D.; Vasan, R.S.; Sniderman, A.D. Apolipoprotein B improves risk assessment of future coronary heart disease in the Framingham Heart Study beyond LDL-C and non-HDL-C. Eur J Prev Cardiol 2015, 22, 1321–1327. [Google Scholar] [CrossRef]
- Johannesen, C.D.L.; Mortensen, M.B.; Langsted, A.; Nordestgaard, B.G. Apolipoprotein B and Non-HDL Cholesterol Better Reflect Residual Risk Than LDL Cholesterol in Statin-Treated Patients. J Am Coll Cardiol 2021, 77, 1439–1450. [Google Scholar] [CrossRef]
- Cho, Y.; Lee, S.G.; Jee, S.H.; Kim, J.H. Hypertriglyceridemia is a major factor associated with elevated levels of small dense LDL cholesterol in patients with metabolic syndrome. Annals of laboratory medicine 2015, 35, 586–594. [Google Scholar] [CrossRef]
- Kang, H.S.; Gutin, B.; Barbeau, P.; Litaker, M.S.; Allison, J.; Le, N.A. Low-density lipoprotein particle size, central obesity, cardiovascular fitness, and insulin resistance syndrome markers in obese youths. International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity 2002, 26, 1030–1035. [Google Scholar] [CrossRef]
- Nikolic, D.; Katsiki, N.; Montalto, G.; Isenovic, E.R.; Mikhailidis, D.P.; Rizzo, M. Lipoprotein subfractions in metabolic syndrome and obesity: clinical significance and therapeutic approaches. Nutrients 2013, 5, 928–948. [Google Scholar] [CrossRef]
- Thanassoulis, G.; Williams, K.; Ye, K.; Brook, R.; Couture, P.; Lawler, P.R.; de Graaf, J.; Furberg, C.D.; Sniderman, A. Relations of change in plasma levels of LDL-C, non-HDL-C and apoB with risk reduction from statin therapy: a meta-analysis of randomized trials. Journal of the American Heart Association 2014, 3, e000759. [Google Scholar] [CrossRef] [PubMed]
- Emerging Risk Factors, C.; Danesh, J.; Erqou, S.; Walker, M.; Thompson, S.G.; Tipping, R.; Ford, C.; Pressel, S.; Walldius, G.; Jungner, I.; et al. The Emerging Risk Factors Collaboration: analysis of individual data on lipid, inflammatory and other markers in over 1.1 million participants in 104 prospective studies of cardiovascular diseases. European journal of epidemiology 2007, 22, 839–869. [Google Scholar] [CrossRef] [PubMed]
- Boekholdt, S.M.; Arsenault, B.J.; Mora, S.; Pedersen, T.R.; LaRosa, J.C.; Nestel, P.J.; Simes, R.J.; Durrington, P.; Hitman, G.A.; Welch, K.M.; et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. JAMA 2012, 307, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Sniderman, A.D.; Williams, K.; Contois, J.H.; Monroe, H.M.; McQueen, M.J.; de Graaf, J.; Furberg, C.D. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circulation. Cardiovascular quality and outcomes 2011, 4, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Soffer, G.; Pavlyha, M.; Ngai, C.; Thomas, T.; Holleran, S.; Ramakrishnan, R.; Karmally, W.; Nandakumar, R.; Fontanez, N.; Obunike, J.; et al. Effects of PCSK9 Inhibition With Alirocumab on Lipoprotein Metabolism in Healthy Humans. Circulation 2017, 135, 352–362. [Google Scholar] [CrossRef]
- Telford, D.E.; Sutherland, B.G.; Edwards, J.Y.; Andrews, J.D.; Barrett, P.H.; Huff, M.W. The molecular mechanisms underlying the reduction of LDL apoB-100 by ezetimibe plus simvastatin. Journal of lipid research 2007, 48, 699–708. [Google Scholar] [CrossRef]
- Toth, P.P.; Jones, S.R.; Monsalvo, M.L.; Elliott-Davey, M.; Lopez, J.A.G.; Banach, M. Effect of Evolocumab on Non-High-Density Lipoprotein Cholesterol, Apolipoprotein B, and Lipoprotein(a): A Pooled Analysis of Phase 2 and Phase 3 Studies. Journal of the American Heart Association 2020, 9, e014129. [Google Scholar] [CrossRef]
- Tremblay, A.J.; Lamarche, B.; Cohn, J.S.; Hogue, J.C.; Couture, P. Effect of ezetimibe on the in vivo kinetics of apoB-48 and apoB-100 in men with primary hypercholesterolemia. Arterioscler Thromb Vasc Biol 2006, 26, 1101–1106. [Google Scholar] [CrossRef]
- Waldmann, E.; Wu, L.; Busygina, K.; Altenhofer, J.; Henze, K.; Folwaczny, A.; Parhofer, K.G. Effect of PCSK9 inhibition with evolocumab on lipoprotein subfractions in familial dysbetalipoproteinemia (type III hyperlipidemia). PLoS One 2022, 17, e0265838. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Albers, J.J.; Kennedy, H.; Mei, J.V.; Henderson, L.O.; Hannon, W.H. International Federation of Clinical Chemistry standardization project for measurements of apolipoproteins A-I and B. IV. Comparability of apolipoprotein B values by use of International Reference Material. Clin Chem 1994, 40, 586–592. [Google Scholar] [CrossRef] [PubMed]
- College of American Pathologists. Surveys and Anatomic Pathology Education Programs. Chemistry/Therapeutic, Drug Monitoring. Participant Summary; CAP, 2022.
- Cobbaert, C.; Weykamp, C.; Baadenhuijsen, H.; Kuypers, A.; Lindemans, J.; Jansen, R. Selection, preparation, and characterization of commutable frozen human serum pools as potential secondary reference materials for lipid and apolipoprotein measurements: study within the framework of the Dutch project "Calibration 2000". Clin Chem 2002, 48, 1526–1538. [Google Scholar] [CrossRef]
- Langlois, M.R.; Chapman, M.J.; Cobbaert, C.; Mora, S.; Remaley, A.T.; Ros, E.; Watts, G.F.; Boren, J.; Baum, H.; Bruckert, E.; et al. Quantifying Atherogenic Lipoproteins: Current and Future Challenges in the Era of Personalized Medicine and Very Low Concentrations of LDL Cholesterol. A Consensus Statement from EAS and EFLM. Clin Chem 2018, 64, 1006–1033. [Google Scholar] [CrossRef] [PubMed]
- Cobbaert, C.M.; Althaus, H.; Begcevic Brkovic, I.; Ceglarek, U.; Coassin, S.; Delatour, V.; Deprez, L.; Dikaios, I.; Dittrich, J.; Hoofnagle, A.N.; et al. Towards an SI-Traceable Reference Measurement System for Seven Serum Apolipoproteins Using Bottom-Up Quantitative Proteomics: Conceptual Approach Enabled by Cross-Disciplinary/Cross-Sector Collaboration. Clin Chem 2021, 67, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.G.; Myers, G.L.; Sakurabayashi, I.; Bachmann, L.M.; Caudill, S.P.; Dziekonski, A.; Edwards, S.; Kimberly, M.M.; Korzun, W.J.; Leary, E.T.; et al. Seven direct methods for measuring HDL and LDL cholesterol compared with ultracentrifugation reference measurement procedures. Clin Chem 2010, 56, 977–986. [Google Scholar] [CrossRef]
- van Deventer, H.E.; Miller, W.G.; Myers, G.L.; Sakurabayashi, I.; Bachmann, L.M.; Caudill, S.P.; Dziekonski, A.; Edwards, S.; Kimberly, M.M.; Korzun, W.J.; et al. Non-HDL cholesterol shows improved accuracy for cardiovascular risk score classification compared to direct or calculated LDL cholesterol in a dyslipidemic population. Clin Chem 2011, 57, 490–501. [Google Scholar] [CrossRef]
- Charlton-Menys, V.; Betteridge, D.J.; Colhoun, H.; Fuller, J.; France, M.; Hitman, G.A.; Livingstone, S.J.; Neil, H.A.; Newman, C.B.; Szarek, M.; et al. Targets of statin therapy: LDL cholesterol, non-HDL cholesterol, and apolipoprotein B in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS). Clin Chem 2009, 55, 473–480. [Google Scholar] [CrossRef]
- Pencina, M.J.; Navar, A.M.; Wojdyla, D.; Sanchez, R.J.; Khan, I.; Elassal, J.; D'Agostino, R.B., Sr.; Peterson, E.D.; Sniderman, A.D. Quantifying Importance of Major Risk Factors for Coronary Heart Disease. Circulation 2019, 139, 1603–1611. [Google Scholar] [CrossRef]
- Sniderman, A.D.; Pencina, M.; Thanassoulis, G. Limitations in the conventional assessment of the incremental value of predictors of cardiovascular risk. Curr Opin Lipidol 2015, 26, 210–214. [Google Scholar] [CrossRef]
- Benn, M.; Nordestgaard, B.G.; Jensen, G.B.; Tybjaerg-Hansen, A. Improving prediction of ischemic cardiovascular disease in the general population using apolipoprotein B: the Copenhagen City Heart Study. Arterioscler Thromb Vasc Biol 2007, 27, 661–670. [Google Scholar] [CrossRef]
- Langlois, M.R.; Sniderman, A.D. Non-HDL Cholesterol or apoB: Which to Prefer as a Target for the Prevention of Atherosclerotic Cardiovascular Disease? Current cardiology reports 2020, 22, 67. [Google Scholar] [CrossRef] [PubMed]
- Kohli-Lynch, C.N.; Thanassoulis, G.; Moran, A.E.; Sniderman, A.D. The clinical utility of apoB versus LDL-C/non-HDL-C. Clin Chim Acta 2020, 508, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Ray, K. Moving Toward the Next Paradigm for Cardiovascular Prevention. Circulation 2016, 133, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.R.; Ridker, P.M. Calibration of the Pooled Cohort Equations for Atherosclerotic Cardiovascular Disease: An Update. Annals of internal medicine 2016, 165, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Kavousi, M.; Leening, M.J.; Nanchen, D.; Greenland, P.; Graham, I.M.; Steyerberg, E.W.; Ikram, M.A.; Stricker, B.H.; Hofman, A.; Franco, O.H. Comparison of application of the ACC/AHA guidelines, Adult Treatment Panel III guidelines, and European Society of Cardiology guidelines for cardiovascular disease prevention in a European cohort. JAMA 2014, 311, 1416–1423. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cook, N.R. Statins: new American guidelines for prevention of cardiovascular disease. Lancet 2013, 382, 1762–1765. [Google Scholar] [CrossRef]
Figure 1.
Triglyceride-Rich Lipoprotein Metabolism.
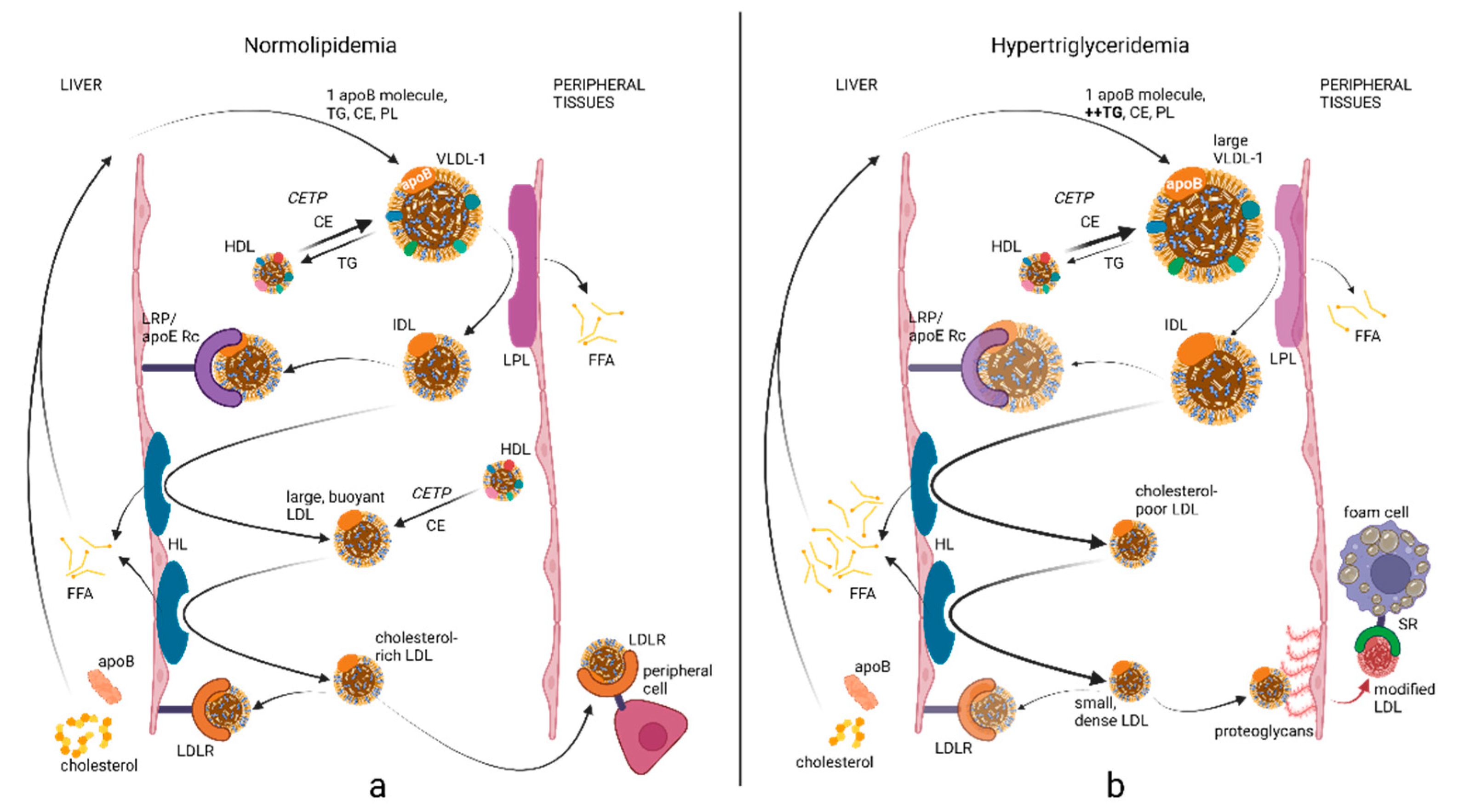
Figure 2.
Discordance of LDL-C and non-HDL-C with apoB.
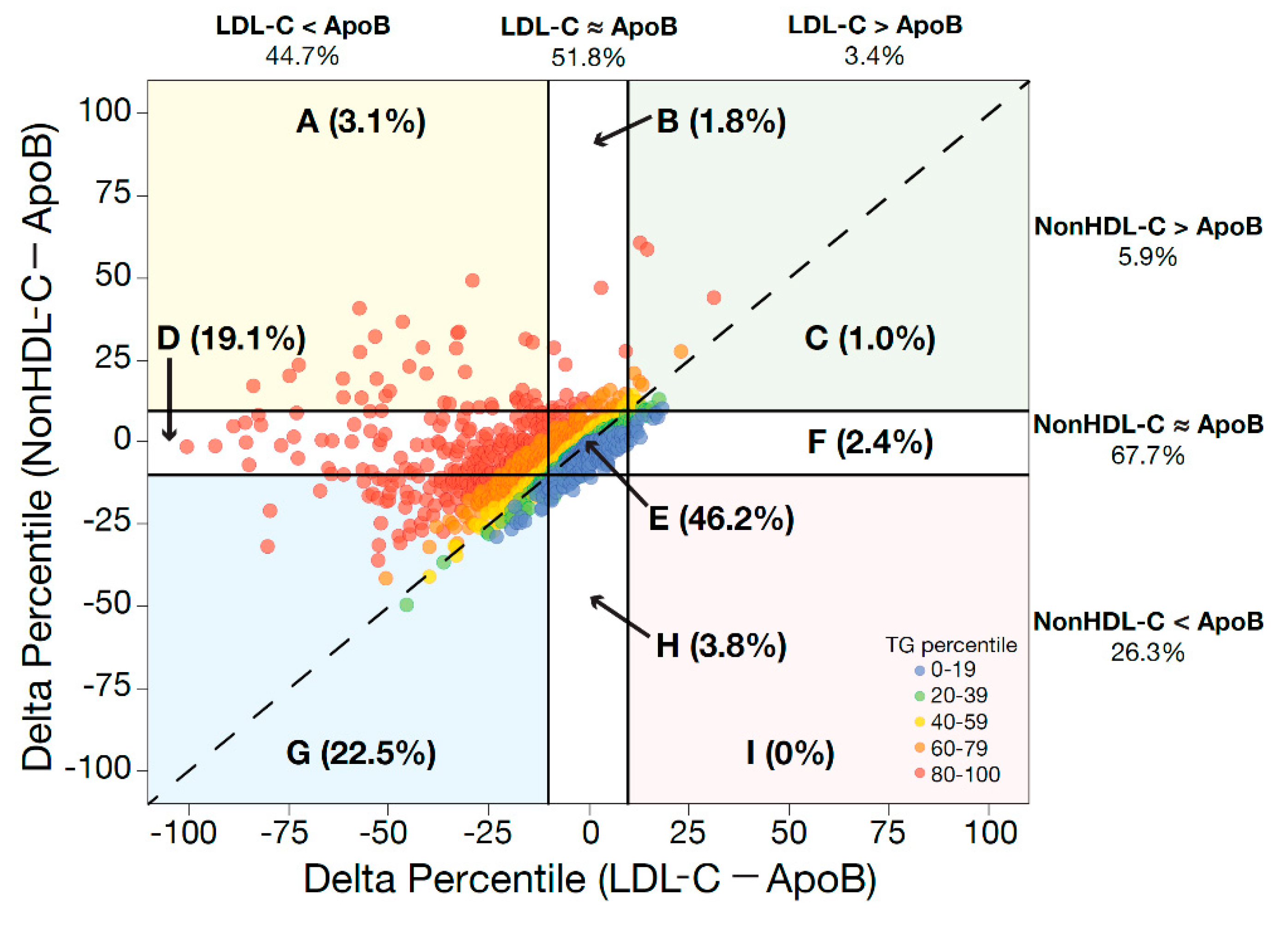

Table 1.
β-Lipoprotein Cut-Offs and Treatment Targets.
| β-Lipoprotein Cut-offs to Initiate Statins Defined in the 2021 Canadian Guideline (18) | |||
| Framingham Risk Score | LDL-C | ApoB | Non-HDL-C |
| <10% | ≥5.0 mmol/L | ≥1.45 g/L | ≥5.8 mmol/L |
| 5-9.9% with other CV risk factors | ≥3.5 mmol/L | ≥1.05 g/L | ≥4.2 mmol/L |
| 10-19.9% | ≥3.5 mmol/L | ≥1.05 g/L | ≥3.5 mmol/L |
| Statin Treatment Targets Recommended in the 2021 Canadian Guideline (18) | |||
| Statin indication | LDL-C | ApoB | Non-HDL-C |
| FH or genetic dyslipidaemia | <2.5 mmol/L | <0.85 g/L | <3.2 mmol/L |
| Intermediate or high risk, DM2, CKD | <2.0 mmol/L | <0.8 g/L | <2.6 mmol/L |
| ASCVD for ezetimibe | <1.8 mmol/L | <0.7 g/L | <2.4 mmol/L |
| ASCVD for PCSK9i | ≤2.2 mmol/L | ≤0.8 g/L | ≤2.9 mmol/L |
| Treatment Targets Recommended in the 2019 European Guideline (19) | |||
| Risk Group | LDL-C | ApoB | Non-HDL-C |
| Moderate | <2.6 mmol/L | <1.0 g/L | <3.4 mmol/L |
| High | <1.8 mmol/L | <0.8 g/L | <2.6 mmol/L |
| Very High | <1.4 mmol/L | <0.65 g/L | <2.2 mmol/L |
CV: cardiovascular, FH: familial hypercholesterolaemia, DM2: diabetes mellitus type II, CKD: chronic kidney disease, ASCVD: atherosclerotic cardiovascular disease, PCSK9i: proprotein convertase subtilisin/kexin type 9 inhibitor.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



