
Submitted:
02 August 2023
Posted:
04 August 2023
You are already at the latest version
Abstract
Histoplasmosis is one of the most underdiagnosed and underreported endemic mycoses in the United States. Histoplasma capsulatum is the causative agent of this disease. To date, molecular epidemiologic studies detailing the phylogeographic structure of H. capsulatum in the United States have been limited. We conducted genomic sequencing using isolates from histoplasmosis cases reported in the United States. We identified North American Clade 2 (NAm2) as the most prevalent clade in the country. Despite high intra-clade diversity, isolates from Minnesota and Michigan cases predominately clustered by state. Future work incorporating environmental sampling and veterinary surveillance may further elucidate the molecular epidemiology of H. capsulatum in the United States and how genomic sequencing can be applied for surveillance and outbreak investigation of histoplasmosis.
Keywords:
Histoplasmosis
; Histoplasma capsulatum
; genome
; clades
; genomic
; epidemiology
1. Introduction
Histoplasma capsulatum is a thermally dimorphic fungus that can cause histoplasmosis when inhaled. It is non-contagious and affects humans and animals including dogs and cats [1]. The fungus predominately lives in soil, and infections are often linked with activities that disturb the environment, particularly where bird or bat droppings are present [2,3,4]. Clinical presentation ranges from mild self-resolving to moderate pneumonia-like symptoms to a severe, life-threatening, disseminated disease. Histoplasmosis can affect healthy individuals or those with compromised immune systems. In the case of disseminated histoplasmosis, the infection can affect several organs including the lungs, bone marrow, skin, brain, and gastrointestinal tract [5,6].
In the United States, H. capsulatum is endemic to central and eastern states around the Ohio River Valley and the Mississippi River Valley [7]. It is estimated that 60–90% of the population living in this area has been exposed to the fungus [7]. However, disease surveillance is limited, with histoplasmosis reportable to public health authorities in only 12 states [8]. Among reported cases in 2019 (>1,000), the high rate of hospitalization (54%) and death (5%) suggests that the actual number of cases is likely higher [9]. Furthermore, systematic environmental surveillance of H. capsulatum is not conducted. Therefore, due to under-detection of infections and limited surveillance, the true geographic distribution of H. capsulatum in the United States is poorly understood [10].
Based on morphology and pathogenic characteristics, genus Histoplasma was thought to consist of three distinct varieties: H. capsulatum, H. duboisii and H. farciminosum [11]. In 2003, Kasuga et al. utilized genealogical concordance–phylogenetic species concept (GC–PSC) to classify H. capsulatum into eight clades: North American clades 1 and 2 (NAm 1 and NAm 2), Latin American clades A and B (LAm A and LAm B), Eurasian, Netherlands, Australian, and African, as well as a distinct lineage (H81) comprised of Panamanian isolates [12]. LAm A and LAm B clades, which comprised isolates from Mexico, Suriname, Guatemala, Brazil, and Argentina [12], exhibited the highest genetic diversity. Furthermore, a few cases of LAm A, NAm 1, and NAm 2 clades co-occurred in the endemic areas of North America with different population dynamics [12,13]. More recently, Sepulveda et al. used genomic sequencing to classify H. capsulatum into five genetically distinct clades of which four could be considered as species: NAm 1 (also referred to as H. mississippiense species), NAm 2 (also referred to as H. ohiense species), LAm A (also referred to as H. suramericanum species), Panama lineage H81 (also referred to H. capsulatum sensu stricto species) and Africa [14]. In 2022, a new Indian lineage was reported [15]. However, it is important to note that these clades defined by genomic sequencing have not yet been accepted as valid species [16].
Whole-genome sequencing (WGS), compared with more traditional molecular typing methods, has proven to be a superior method for molecular surveillance and epidemiology of infectious diseases [17]. Specifically, it allows detection of genome-wide polymorphisms that can be highly correlated with epidemiologic data and spatio-temporal spread [18]. WGS also can help trace transmission, identify the source of an outbreak, and elucidate the evolution of a pathogen. In the case of H. capsulatum, WGS helped reclassify the five distinct major clades that were previously phenotypically identified as three clades, demonstrating its high resolution and ability to refine our understanding of pathogen diversity.
Here, we utilized WGS to better describe H. capsulatum in the United States. We present the phylogeographic structure of H. capsulatum within the United States by utilizing clinical isolates obtained from a previous enhanced surveillance study of histoplasmosis patients from eight U.S. states [8]
2. Materials and Methods
Culture and DNA extraction
H. capsulatum isolates were received at the Mycotic Diseases Branch laboratory at the U.S. Centers for Disease Control and Prevention (CDC) for routine fungal identification as part of ongoing fungal disease surveillance. Upon arrival, species identification was done by sequencing the ITS2 region of the rDNA and then isolates were stored in 20% glycerol at −70°C for further studies. Later, 48 isolates from this study were grown on brain heart infusion (BHI) agar at 25°C for ≤ 10 days. Genomic DNA was extracted using the Qiagen DNeasy Blood and Tissue kit (Qiagen, Gaithersburg, MD) according to the manufacturer’s instructions. All procedures were conducted in a Biosafety Level 3 laboratory.
Genomic sequencing
Genomic libraries were constructed and barcoded using the NEBNext Ultra DNA Library Prep kit (New England Biolabs, Ipswich, MA, USA) for Illumina sequencing following the manufacturer’s instructions. Libraries were sequenced on the Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) using the HiSeq Rapid SBS Kit v2 500 cycles. Raw sequence data were submitted to NCBI Sequence Read Archive (BioProject PRJNA868688).
Single-nucleotide polymorphism (SNP) and phylogenetic analysis
For the whole-genome SNP analysis, MycoSNP (v1.4) (https://github.com/CDCgov/mycosnp-nf), a reference-based SNP calling workflow was used [19]. The publicly available assembled genome (GenBank accession number: GCA_017607445.1) with 12 contigs and ~39 Mb length was used as the reference. Additionally, sequences from SRR8347492, SRR6243656, SRR6243645, SRR6243650, and SRR6243635 were included in the analysis. With MycoSNP, the genome was masked for repeats using nucmer command from mummer (v3.23) [20] and Bedtools (v2.30). The reference genome was indexed for downstream analysis [21,22]. Low-quality data trimming and filtering were performed using FaQCs (v 2.10) [23]. The trimmed reads were used for alignment using the BWA (0.7.17) MEM command [24]. Further, the aligned BAM files from each sample were pre-processed for variant calling using the genome analysis toolkit GATK (v 4.2.4.1) [25] with the haploid mode. GATK’s VariantFilteration tool was used to filter sites based on the filtering expression “QD < 2.0 || FS > 60.0 || MQ < 40.0”. A customized filtering criteria of minimum genotype quality <50, percentage alternate allele <0.80 and minimum depth of 10 was applied. For the phylogenetic analysis, variant sites were concatenated allowing a maximum of 10% of samples with ambiguous nucleotides for selecting a site. A pairwise distance matrix and a neighbor joining tree (NJ) were created using MEGA 7 [26], and the maximum likelihood (ML) tree was constructed using FastTreeMP (v 2.1.11) [27] using the GTR nucleotide substitution model and a bootstrap analysis based on 100 replicates. Multi-dimensional scaling (MDS) technique was used (R function cmdscale) [28] to visualize the clustering pattern of these samples. A patristic distance matrix was used to construct an MDS plot of the complete tree as well as specific clades.
3. Results
Among the 48 total isolates, 39 (81%) case patients were male, 25 (52%) were aged 20–65 years, 19 (40%) were immunosuppressed. (Table 1). Most (n=17, 34%) had a positive culture from bronchial specimens. Overall, cases resided in eight U.S. states (Indiana [N=2], Kentucky[N=4], Louisiana [N=1], Michigan[N=21], Minnesota[N=13], Nebraska[N=1], Pennsylvania[N=1], and Wisconsin[N=5]), most were from Michigan (44%), Minnesota (27%), and Wisconsin (10%)).

Table 1.
Associated case characteristics for Histoplasma capsulatum isolates collected from eight U.S. states.
Table 1.
Associated case characteristics for Histoplasma capsulatum isolates collected from eight U.S. states.
| Count | Percentage (%) | |
|---|---|---|
| Sex | ||
| Male | 39 | 81 |
| Female | 9 | 19 |
| Total | 48 | 100 |
| Age Group | ||
| <21 | 6 | 13 |
| >=21 & <65 | 25 | 52 |
| >=65 | 17 | 35 |
| Total | 48 | 100 |
| Immunosuppressed cases | ||
| Indiana | 1 | 5 |
| Kentucky | 3 | 16 |
| Michigan | 6 | 32 |
| Minnesota | 8 | 42 |
| Wisconsin | 1 | 5 |
| Total | 19 | 100 |
| Specimen Source | ||
| Sputum | 4 | 8 |
| Lymph node | 4 | 8 |
| Bronchial specimen | 17 | 35 |
| Lung tissue | 3 | 6 |
| Blood | 12 | 25 |
| Bone marrow | 3 | 6 |
| Others* | 5 | 10 |
| Total | 48 | 100 |
| States | ||
| Indiana | 2 | 4 |
| Kentucky | 4 | 8 |
| Louisiana | 1 | 2 |
| Michigan | 21 | 44 |
| Minnesota | 13 | 27 |
| Nebraska | 1 | 2 |
| Pennsylvania | 1 | 2 |
| Wisconsin | 5 | 10 |
| Total | 48 | 100 |
* Others includes Fluid Synovial, Wound, Tissues (arm, lungs) and Unknown sample source.
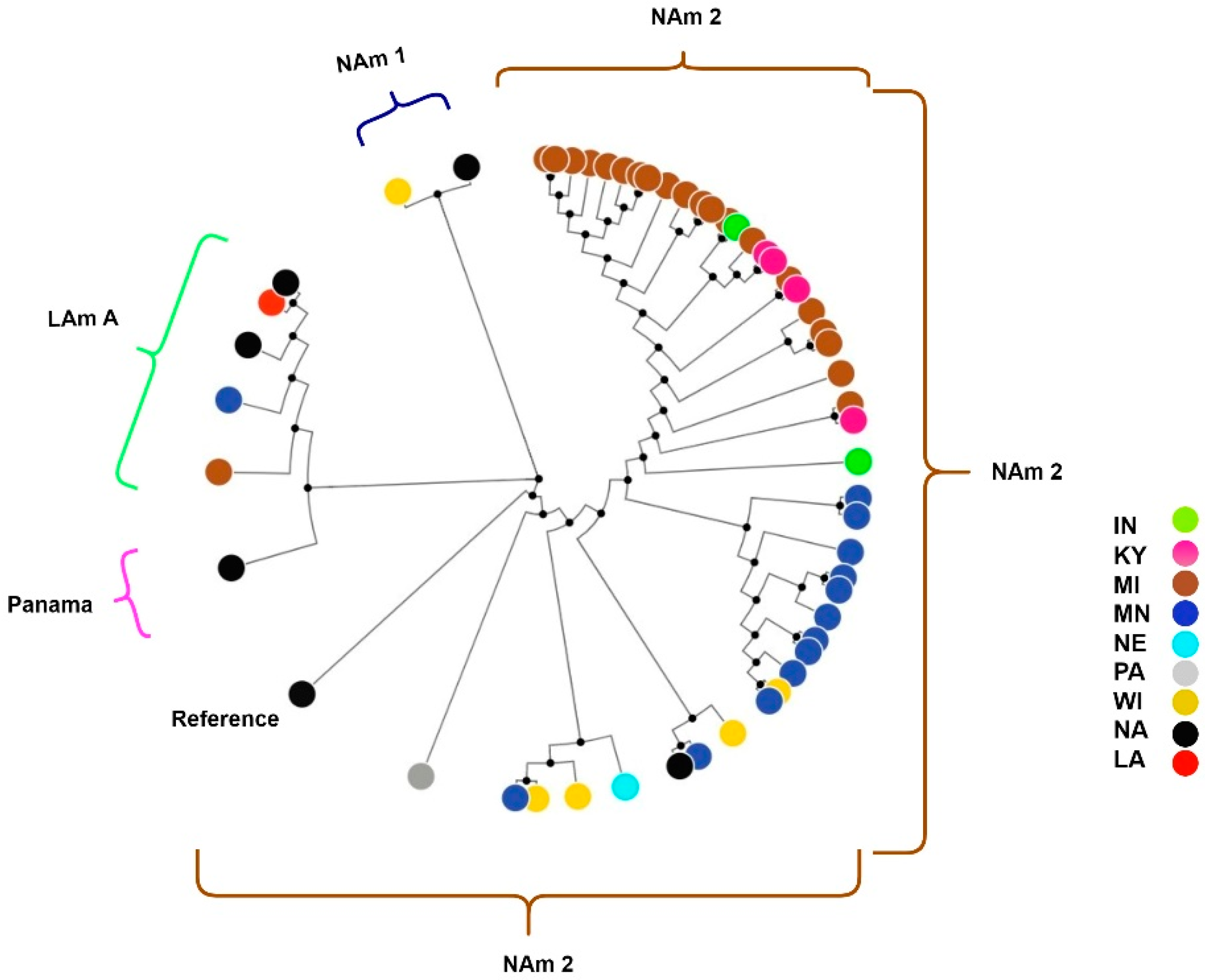
Genomic sequencing and SNP analysis identified 1,969,979 variant sites, which were used for constructing a NJ and ML phylogenetic tree. Phylogenetic analysis revealed that the isolates formed clades as previously described for H. capsulatum (Figure 1). Specifically, 44 (92%) samples clustered with the NAm 2 clade samples SRR6243656 and the reference genome (Figure 1). Three (6%) isolates clustered with the LAm A clade. One (2%) isolate clustered with the NAm 1 clade. Isolates from LAm A and NAm 2 clades were separated by ≤346,613 SNPs and isolates from the NAm1 and NAm 2 clades were separated by ≤600,854 SNPs.
Figure 1.
Phylogenetic analysis of H. capsulatum samples collected from eight U.S. states. The ML tree includes 54 isolates. Node color is based on the associated U.S. states where the patient resided.
Figure 1.
Phylogenetic analysis of H. capsulatum samples collected from eight U.S. states. The ML tree includes 54 isolates. Node color is based on the associated U.S. states where the patient resided.
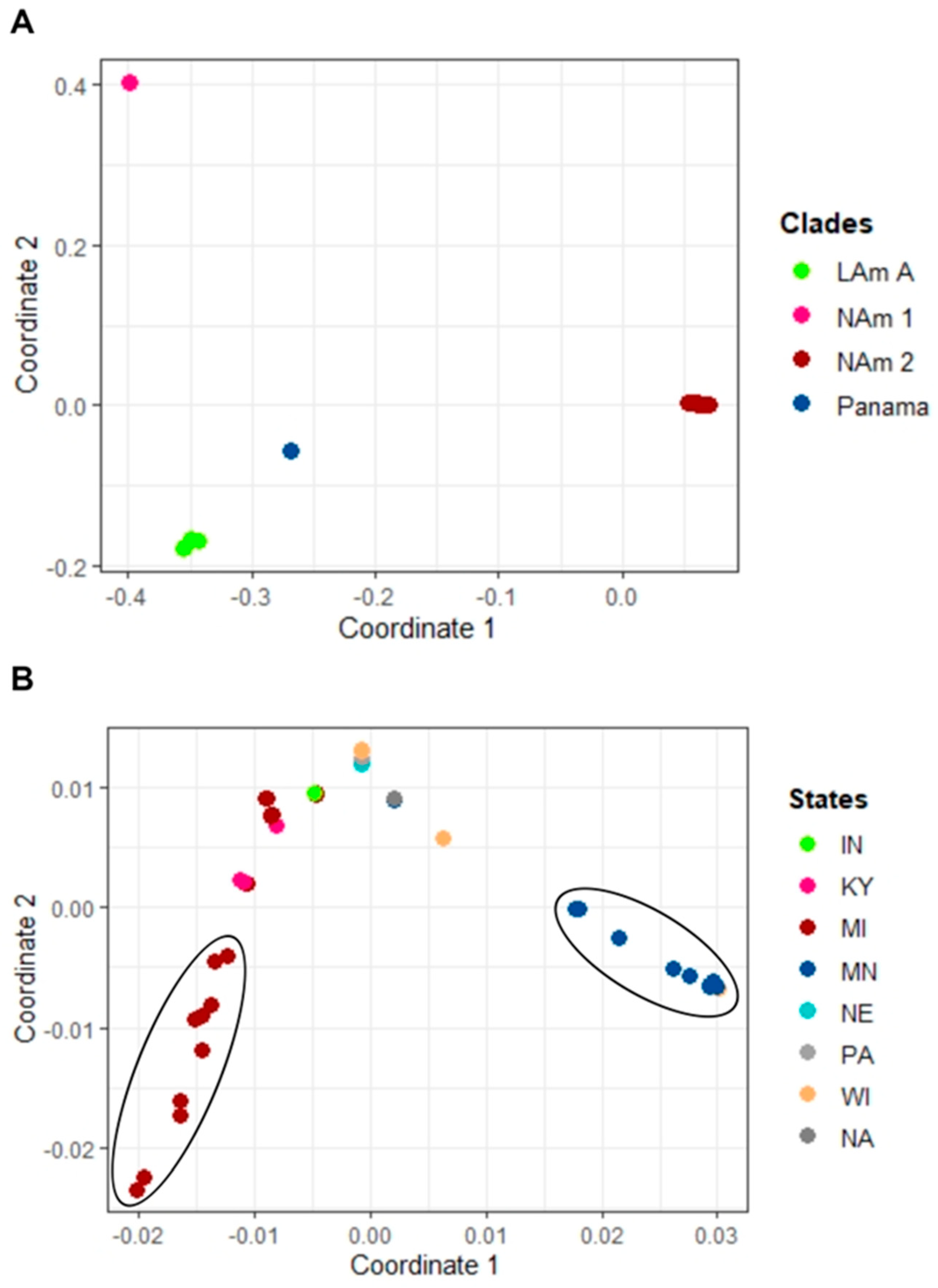
Additionally, MDS analysis showed similar findings to the phylogenetic tree whereby distinct clusters of NAm1, NAm2 and LAm A clades were observed (Figure 2A). The NAm 2 clade comprised two clusters whereby isolates primarily grouped by state. One cluster contained 11 isolates; 10 were from cases from Minnesota and one from a case from Wisconsin (Figure 2B). A second cluster contained 10 isolates from cases from Michigan. The remaining eleven cases from Michigan, four from Kentucky, two from Indiana, four from Wisconsin, two from Minnesota, one from Nebraska, and one from Pennsylvania clustered together in the third cluster. The reference sample was the most distant isolate within the NAm2 clade (not included in the MDS plot).
Figure 2.
Multi-dimensional (MDS) plot of Histoplasma isolates. A) MDS plot of the patristic distance with x axis being dimension 1 and y axis dimension 2. The plot which includes all isolates revealed four distinct clades. Most (92%) of the samples clustered with the NAm 2 clade, three with the LAm A clade, one with the NAm 1 clade, and none clustered with the Panama control sample. B) MDS plot of isolates from the NAm 2 clade. Two separate clusters of isolates belonging to MI and MN were observed.
Figure 2.
Multi-dimensional (MDS) plot of Histoplasma isolates. A) MDS plot of the patristic distance with x axis being dimension 1 and y axis dimension 2. The plot which includes all isolates revealed four distinct clades. Most (92%) of the samples clustered with the NAm 2 clade, three with the LAm A clade, one with the NAm 1 clade, and none clustered with the Panama control sample. B) MDS plot of isolates from the NAm 2 clade. Two separate clusters of isolates belonging to MI and MN were observed.
4. Discussion
The phylogeographic structure of H. capsulatum in the United States is poorly understood. Modeling studies have predicted potential shifts in the geographic distribution of H. capsulatum and other environmental fungal pathogens within the United States [29]. As previously observed, the expanding presence of fungal pathogens such as H. capsulatum in newer geographic areas could be attributed to changes in their ecological niche, alterations in the behavior of their natural reservoirs, and dispersers, [30]. Additionally, soil contamination via the guano of birds and bats is believed to play a crucial role in the dispersal of H. capsulatum [13]. To improve our understanding of lineages and clade specific genetic variations of H. capsulatum in the United States, we evaluated 48 histoplasmosis cases from eight U.S. states. By performing whole-genome analysis on associated isolates, we leveraged the power of WGS, which is recognized as a highly effective molecular epidemiologic tool that provides much greater epidemiologically relevant resolution than classical genotyping methods like MLST.
Our analysis revealed a single NAm 1 clade isolate from Wisconsin, which is not unexpected given that the NAm 1 clade has been previously reported in North America, both in the United States and Canada [12,31]. Likewise, we also found three isolates from Minnesota, Michigan, and Louisiana that grouped with the LAm A clade, which has also been previously reported in the United States [11].
Most isolates in this study belonged to the NAm 2 clade, a finding that is consistent with previous work [12]. Despite belonging to the same clade, isolates showed a high degree of SNP differences (maximum of 60,000 SNPs) highlighting high within-clade genetic diversity (Supplementary Figure 1). NAm 2 is amongst the oldest H. capsulatum clades which is distant from other clades and hypothesized to have emerged between 3.2–13 million years ago [12]. H. capsulatum is well known for its genetic complexity and the role played by geographical expansion in the creation of new lineages with notable phenotypic and virulence differences [12]; for example, the prevalence of extensive genetic variation of H. capsulatum in Latin America has been documented earlier [32]. A possible reason for large intra-clade diversity within the NAm 2 clade could be due to recombination and changes in selection pressures because of the expanding geographic boundaries, forcing the fungi to rapidly adapt to varying environmental changes [30,33].
Within NAm2, we identified two genetically distinct clusters, with isolates primarily grouping by state. Most cases from Minnesota were found in one cluster, while those from Michigan were in another cluster, with a few exceptions where isolates from Michigan clustered with those from other states. Moreover, one isolate from Wisconsin was found in the Minnesota cluster. This could be due to an exposure occurring in Minnesota when the patient resided in Wisconsin. It was not possible to determine whether cases were locally acquired or travel-related, such as from neighboring or visiting states. For future studies, interdisciplinary approaches that tap into environmental samples or samples from veterinary surveillance that have robust epidemiologic data may prove useful.
Regarding future applications of WGS for genomic surveillance and epidemiology of histoplasmosis, there may be a potential role for this technology as is performed for Coccidoides immitis. WGS has proven to be an effective method to identify locally acquired Valley fever due to the well-defined phylogeographic structure of C. immitis. Specifically, it is possible to identify cases of locally acquired Valley fever in Washington and delineate between exposures in Washington and California [34]. Histoplasmosis, like Valley fever, also has endemic and non-endemic areas. However, it is unknown whether H. capsulatum has a strong phylogeographic structure as described for C. immitis. Therefore, to understand whether WGS can be used to help determine locally acquired cases of histoplasmosis, a more robust characterization of the phylogeographic population structure is needed. Here, we show evidence that this may be possible for some endemic states such as Minnesota and Michigan, but further studies are needed that incorporate environmental sampling and comprehensive travel history to confirm this.
Overall, we employed WGS to investigate the prevalence of Histoplasma lineages in the United States. Our findings shed light on the phylogeographic structure of this significant pathogen and raise questions regarding the potential utility of WGS for genomic epidemiology of histoplasmosis.
Supplementary Materials
Figure S1: Neighbor-joining tree showing SNP differences.
Author Contributions
Conceptualization, Nancy Chow, Anastasia Litvintseva and Kaitlin Benedict; methodology, Lalitha Gade; Analysis and Manuscript writing, Ujwal Bagal
Funding
This research received no external funding.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
We would like to acknowledge Dr. Shawn Lockhart for providing valuable suggestions on clade nomenclature and Dr. Rory Welsh for providing help with the analysis. We thank the state and local health department personnel who participated in the enhanced surveillance study and submitted H. capsulatum isolates to CDC. We also thank CDC’s Office of Advanced Molecular Detection (OAMD).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brömel, Catharina, and Jane E. Sykes. "Histoplasmosis in Dogs and Cats." Clinical Techniques in Small Animal Practice 20, no. 4 (2005): 227-32. [CrossRef]
- Cano, MV, and Rana A Hajjeh. "The Epidemiology of Histoplasmosis: A Review." Paper presented at the Seminars in respiratory infections 2001.
- Deepe, G. S., Jr. "Outbreaks of Histoplasmosis: The Spores Set Sail." PLoS Pathog 14, no. 9 (2018): e1007213. [CrossRef]
- Gómez, Luisa F., Myrtha Arango, Juan G. McEwen, Oscar M. Gómez, Alejandra Zuluaga, Carlos A. Peláez, Jose M. Acevedo, María L. Taylor, and María del P. Jiménez. "Molecular Epidemiology of Colombian Histoplasma Capsulatum Isolates Obtained from Human and Chicken Manure Samples." Heliyon 5, no. 7 (2019): e02084. [CrossRef]
- Acir Rachid, Lucila Stange, Rezende, Suzelle Freitas de Moura, Paulo C. Loffy, Francisco Luiz Gomide M. Magalhães. "A Case Study of Disseminated Histoplasmosis Linked to Common Variable Immunodeficiency." Brazilian Journal of Infectious Diseases 7 (2003): 268-72. [CrossRef]
- Assi, Maha A., Mohamad S. Sandid, Larry M. Baddour, Glenn D. Roberts, and Randall C. Walker. "Systemic Histoplasmosis: A 15-Year Retrospective Institutional Review of 111 Patients." Medicine 86, no. 3 (2007): 162-69. [CrossRef]
- Manos, Nicholas E., Shirley H. Ferebee, and Winifred F. Kerschbaum. "Geographic Variation in the Prevalence of Histoplasmin Sensitivity." Diseases of the chest 29, no. 6 (1956): 649-68. [CrossRef]
- Benedict, Kaitlin, Stephanie McCracken, Kimberly Signs, Malia Ireland, Victoria Amburgey, Jose Antonio Serrano, Natalie Christophe, Suzanne Gibbons-Burgener, Sara Hallyburton, Kimberly A Warren, Alison Keyser Metobo, Racheal Odom, Matthew R Groenewold, and Brendan R Jackson. "Enhanced Surveillance for Histoplasmosis—9 States, 2018–2019." Open Forum Infectious Diseases 7, no. 9 (2020). [CrossRef]
- Dallas J. Smith; Samantha L. Williams; Endemic Mycoses State Partners Group; Kaitlin M. Benedict; Brendan R. Jackson; Mitsuru Toda. "Surveillance for Coccidioidomycosis, Histoplasmosis, and Blastomycosis — United States, 2019." MMWR Morb Mortal Wkly Rep 71, no. 7 (2022): 1-14. [CrossRef]
- Mazi, P. B., J. M. Sahrmann, M. A. Olsen, A. Coler-Reilly, A. M. Rauseo, M. Pullen, J. C. Zuniga-Moya, W. G. Powderly, and A. Spec. "The Geographic Distribution of Dimorphic Mycoses in the United States for the Modern Era." Clin Infect Dis 76, no. 7 (2023): 1295-301. [CrossRef]
- Kwon-Chung, K. J., and J. E. Bennett. "Medical Mycology." Revista do Instituto de Medicina Tropical de São Paulo 34 (1992).
- Kasuga, T., T. J. White, G. Koenig, J. McEwen, A. Restrepo, E. Castañeda, C. Da Silva Lacaz, E. M. Heins-Vaccari, R. S. De Freitas, R. M. Zancopé-Oliveira, Z. Qin, R. Negroni, D. A. Carter, Y. Mikami, M. Tamura, M. L. Taylor, G. F. Miller, N. Poonwan, and J. W. Taylor. "Phylogeography of the Fungal Pathogen Histoplasma Capsulatum." Mol Ecol 12, no. 12 (2003): 3383-401. [CrossRef]
- Teixeira, Marcus de M., José S. L. Patané, Maria L. Taylor, Beatriz L. Gómez, Raquel C. Theodoro, Sybren de Hoog, David M. Engelthaler, Rosely M. Zancopé-Oliveira, Maria S. S. Felipe, and Bridget M. Barker. "Worldwide Phylogenetic Distributions and Population Dynamics of the Genus Histoplasma." PLOS Neglected Tropical Diseases 10, no. 6 (2016): e0004732. [CrossRef]
- Sepúlveda, Victoria E., Roberto Márquez, David A. Turissini, William E. Goldman, and Daniel R. Matute. "Genome Sequences Reveal Cryptic Speciation in the Human Pathogen <I>Histoplasma Capsulatum</I>." mBio 8, no. 6 (2017): e01339-17. [CrossRef]
- Jofre, Gaston I., Ashutosh Singh, Heidi Mavengere, Gandhi Sundar, Emmanuel D’Agostino, Anuradha Chowdhary, and Daniel R. Matute. "An Indian Lineage of Histoplasma with Strong Signatures of Differentiation and Selection." Fungal Genetics and Biology 158 (2022): 103654. [CrossRef]
- Robert, V., D. Vu, A. B. Amor, N. van de Wiele, C. Brouwer, B. Jabas, S. Szoke, A. Dridi, M. Triki, S. Ben Daoud, O. Chouchen, L. Vaas, A. de Cock, J. A. Stalpers, D. Stalpers, G. J. Verkley, M. Groenewald, F. B. Dos Santos, G. Stegehuis, W. Li, L. Wu, R. Zhang, J. Ma, M. Zhou, S. P. Gorjón, L. Eurwilaichitr, S. Ingsriswang, K. Hansen, C. Schoch, B. Robbertse, L. Irinyi, W. Meyer, G. Cardinali, D. L. Hawksworth, J. W. Taylor, and P. W. Crous. "Mycobank Gearing up for New Horizons." IMA Fungus 4, no. 2 (2013): 371-9. [CrossRef]
- Köser, Claudio U., Matthew J. Ellington, Edward J. P. Cartwright, Stephen H. Gillespie, Nicholas M. Brown, Mark Farrington, Matthew T. G. Holden, Gordon Dougan, Stephen D. Bentley, Julian Parkhill, and Sharon J. Peacock. "Routine Use of Microbial Whole Genome Sequencing in Diagnostic and Public Health Microbiology." PLOS Pathogens 8, no. 8 (2012): e1002824. [CrossRef]
- Roetzer, Andreas, Roland Diel, Thomas A. Kohl, Christian Rückert, Ulrich Nübel, Jochen Blom, Thierry Wirth, Sebastian Jaenicke, Sieglinde Schuback, Sabine Rüsch-Gerdes, Philip Supply, Jörn Kalinowski, and Stefan Niemann. "Whole Genome Sequencing Versus Traditional Genotyping for Investigation of a Mycobacterium Tuberculosis Outbreak: A Longitudinal Molecular Epidemiological Study." PLOS Medicine 10, no. 2 (2013): e1001387. [CrossRef]
- Ujwal R. Bagal, John Phan, Rory M. Welsh, Elizabeth Misas, Darlene Wagner, Lalitha Gade, Anastasia P. Litvintseva, Christina A. Cuomo, Nancy A. Chow. "Mycosnp: A Portable Workflow for Performing Whole-Genome Sequencing Analysis of Candida Auris." In Candida Auris: Methods and Protocols, edited by Alexander Lorenz: Humana New York, NY, 2022. [CrossRef]
- Delcher, A. L., S. L. Salzberg, and A. M. Phillippy. "Using Mummer to Identify Similar Regions in Large Sequence Sets." Curr Protoc Bioinformatics Chapter 10 (2003): Unit 10.3. [CrossRef]
- Li, H., B. Handsaker, A. Wysoker, T. Fennell, J. Ruan, N. Homer, G. Marth, G. Abecasis, and R. Durbin. "The Sequence Alignment/Map Format and Samtools." Bioinformatics 25, no. 16 (2009): 2078-9. [CrossRef]
- Picard Toolkit: Repository. Http://Broadinstitute.Github.Io/Picard/.
- Lo, Chien-Chi, and Patrick S. G. Chain. "Rapid Evaluation and Quality Control of Next Generation Sequencing Data with Faqcs." BMC Bioinformatics 15, no. 1 (2014): 366. [CrossRef]
- Li, Heng. "Aligning Sequence Reads, Clone Sequences and Assembly Contigs with Bwa-Mem." arXiv (2013): 1303.3997.
- Cheng, A. Y., Y. Y. Teo, and R. T. Ong. "Assessing Single Nucleotide Variant Detection and Genotype Calling on Whole-Genome Sequenced Individuals." Bioinformatics 30, no. 12 (2014): 1707-13. [CrossRef]
- Kumar, S., G. Stecher, and K. Tamura. "Mega7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets." Mol Biol Evol 33, no. 7 (2016): 1870-4. [CrossRef]
- Price, Morgan N., Paramvir S. Dehal, and Adam P. Arkin. "Fasttree 2 – Approximately Maximum-Likelihood Trees for Large Alignments." PLOS ONE 5, no. 3 (2010): e9490. [CrossRef]
- R: A Language and Environment for Statistical Computing. Vienna, Austria.
- Hepler, S. A., K. A. Kaufeld, K. Benedict, M. Toda, B. R. Jackson, X. Liu, and D. Kline. "Integrating Public Health Surveillance and Environmental Data to Model Presence of Histoplasma in the United States." Epidemiology 33, no. 5 (2022): 654-9. [CrossRef]
- Taylor, Maria Lucia, María del Rocío Reyes-Montes, Daniel A. Estrada-Bárcenas, Rosely M. Zancopé-Oliveira, Gabriela Rodríguez-Arellanes, and José Antonio Ramírez. "Considerations About the Geographic Distribution of <I>Histoplasma</I> Species." Applied and Environmental Microbiology 88, no. 7 (2022): e02010-21. [CrossRef]
- Dingle, Tanis C., Matthew A. Croxen, Sumana Fathima, Sandy Shokoples, Ashlesha Sonpar, Lynora Saxinger, and Ilan S. Schwartz. "Histoplasmosis Acquired in Alberta, Canada: An Epidemiological and Genomic Study." The Lancet Microbe 2, no. 5 (2021): e191-e97. [CrossRef]
- Almeida-Silva, Fernando, Marcus de Melo Teixeira, Daniel R. Matute, Marcela de Faria Ferreira, Bridget M. Barker, Rodrigo Almeida-Paes, Allan J. Guimarães, and Rosely M. Zancopé-Oliveira. "Genomic Diversity Analysis Reveals a Strong Population Structure in Histoplasma Capsulatum Lama (Histoplasma Suramericanum)." Journal of Fungi 7, no. 10 (2021): 865. [CrossRef]
- Gusa, Asiya, and Sue Jinks-Robertson. "Mitotic Recombination and Adaptive Genomic Changes in Human Pathogenic Fungi." Genes 10, no. 11 (2019): 901. [CrossRef]
- Oltean, H. N., K. A. Etienne, C. C. Roe, L. Gade, O. Z. McCotter, D. M. Engelthaler, and A. P. Litvintseva. "Utility of Whole-Genome Sequencing to Ascertain Locally Acquired Cases of Coccidioidomycosis, Washington, USA." Emerg Infect Dis 25, no. 3 (2019): 501-06. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



