
Submitted:
03 August 2023
Posted:
04 August 2023
You are already at the latest version
Abstract
Two-dimensional layered coordination polymers based on the hetero-substituted 3-chloro-6-cyano-2,5-dihydroxybenzoquinone ligand, hereafter ClCNAn2- anilate, and LnIII ions (Tb and Eu) are reported. Compounds 1, 2, formulated as Ln2(ClCNAn)3(DMSO)6 (LnIII = Tb, 1; Eu, 2) and their related intermediates 1’ and 2’, formulated as Ln2(ClCNAn)3(H2O)x.yH2O (x+y likely = 12, Ln = Tb, 1’; and Eu, 2’) were prepared by a conventional one-pot reaction (the latter) and recrystallized from DMSO solvent (the former). Polyhydrated intermediates 1’ and 2’ show very similar XRPD patterns, while, despite their common stoichiometry, 1 and 2 are not isostructural. 1 consists of a 2D coordination framework of 3,6 topology, where [Tb(DMSO)3]3+ moieties are bridged by three bis-chelating ClCNAn2- ligands, forming distorted hexagons. Ultrathin nanosheets of 1 were obtained by exfoliation, via liquid assisted sonication method and characterized by atomic force microscopy, confirming the 2D nature of 1. The crystal structure of 2, still showing the presence of 2D sheets with “hexagonal” mesh and a common (3,6) connectivity, is based onto flat, non-corrugated slabs. Indeed, at a larger scale, the different “rectangular tiles” show a clear roofing in 1, which is totally absent in 2. The magnetic behavior of 1 very likely indicates depopulation of the highest crystal-field levels, as expected for TbIII compounds.

Keywords:
Lanthanides
; 2D Coordination Polymers
; Nanosheets
; Anilates
1. Introduction
Lanthanide-based Coordination Polymers (Ln-CPs) and Metal-Organic Frameworks (Ln-MOFs) have attracted considerable interest in material science thanks to their peculiar supramolecular architectures (extending in one, two or three dimensions, 1D, 2D, 3D), their versatile optical properties, in the visible (EuIII, TbIII) and in the near-IR (NIR, for NdIII, ErIII, YbIII) regions, and also their peculiar magnetic (HoIII, DyIII and TbIII) properties. By virtue of these features, they find applications in several applied fields, ranging from telecommunication and data storage to drug delivery, sensing and catalysis [1,2,3,4]. Among these materials, two-dimensional coordination polymers (2D CPs), containing self-assembling metal-ligand-based sheets mutually interacting through weak(er) non-covalent type interactions as Van der Waals, dipolar and hydrogen-bonding, is of ever-growing interest in material chemistry, as their ultrathin nature favors unique optical, electronic, and magnetic properties [5,6,7], as well as chemical processing and rheology, through swelling or exfoliation [8,9].
Among the plethora of ligands which have been employed to fabricate multi-dimensional CPs, 3,6-disubstituted-2,5-dihydroxy-1,4-benzoquinone derivatives, commonly known as anilates, have been widely investigated, by virtue of their Janus-type ability of chelating different metal ions on two opposite sides of the central (and planar) benzoquinone core [10]. The introduction of Lewis-basic functional groups at the 3,6-positions has soon become a viable strategy to avoid the formation of polymeric ribbons, improving material’s dimensionality to sheets of the chess-board type [11]. The resulting 2D materials, thus, manifested interesting physical properties, as they were found to behave as organic ferroelectrics [12], magnetic conductors [13], or multifunctional MOFs [14].
In 2015, some of us reported on the synthesis and characterization of the first example of a heterosubstituted anilate ligand, 3-chloro-6-cyano-2,5-dihydroxybenzoquinone (in the form of its potassium salt, labeled as KHClCNAn)[15]. In 2018, by using this novel anilate Gomez-Garcia et al. obtained [Ln2(ClCNAn)3(solv)6]·CPs [LnIII = Ce, Pr, solv = N,N-dimethylformamide (DMF); Yb, Pr, solv = dimethyl sulfoxide (DMSO); Dy, solv = water (H2O)] [16], by strictly controlling the reaction conditions (solvent and LnIII size). Furthermore some of us highlighted the suitability of KHClCNAn to act as an efficient building block for functional (optical and magnetic) 2D CPs and as a sensitizer for NIR lanthanide emission (by the antenna effect). Specifically, by combining the doubly deprotonated ClCNAn2- ligand with ErIII, YbIII and NdIII ions, 2D [Ln2(ClCNAn)3(DMF)6]·nCH2Cl2 (Ln = Yb (n = 0), Nd, and Er (n = 2)) CPs, which manifested an efficient energy transfer from triplet states of ligand to the LnIII ions [17], have been prepared. Their 2D structure made it possible by the well-known top-down approach liquid-assisted exfoliation to ultrathin nanosheets [18,19]. Chart 1 illustrates the basic structural motif present in all these CPs.
In 2019, some of us investigated the structural polymorphism of chlorocyanoanilate-based DyIII CPs. Therein, it has been reported how, by changing the synthetic methods (layering technique, solvothermal or conventional one-pot reactions) and conditions (solvent, concentration, etc.), different types of (structurally and magnetically) characterized 2D extended networks could be selectively obtained. Later on, in 2020 YbIII-based nanosheets containing two mixed linkers (anilate/carboxylate) were obtained by exfoliation of bulk CPs via the solvent-assisted sonication method [5]. Ultrathin nanosheets were characterized by imaging (atomic force microscopy - AFM and high-resolution transmission electron microscopy - HRTEM) and diffraction (X-ray powder diffraction - XRPD) techniques, highlighting how the optical properties can be affected by the presence of different analytes.
As a further development and investigation of such materials, herein we report the synthesis, structural and magnetic characterization of two 2D CPs, formulated as Ln2(ClCNAn)3(DMSO)6 (LnIII = Tb, 1; Eu, 2), which were obtained by recrystallization in DMSO solvent of the related Ln2(ClCNAn)3(H2O)x.yH2O intermediates (x+y likely = 12, Ln = Tb, 1’; and Eu, 2’) synthesized by one-pot reaction in water.
2. Results
Lanthanide ClCNAn2- based polyhydrated compounds 1’ and 2’ are formed by self-assembly of LnIII and chlorocyananilate ions in aqueous solution (Figure 1). Precipitated as reddish polycrystalline powders, they showed very similar XRPD traces (shown in Figure 2a,b, together with their structureless Le Bail fit, obtained after successful indexing of isomorphous unit cells, see Methods and Table 1). Unfortunately, the quality of the diffraction data and the presence of unavoidable contaminant peaks, jointly to complexity of the material did not enable the determination of the crystal structure and molecular connectivity. This problem was not encountered in the structural determination of the 2D CPs 1 (from conventional single crystal X-ray diffraction data) and 2 (from PXRD data), later discussed. Notwithstanding, the cell volume of ca. 920 Å3 gives a clear indication, following Hofmann’s rules [20], of the material stoichiometry, which we propose to be Ln2(ClCNAn)3(H2O)x.yH2O, with x+y=12.
As illustrated in Figure 2, these hydrated species were further recrystallized in DMSO to obtain single crystals of 1, suitable for X-ray diffraction, and a monophasic polycrystalline material, 2, characterized by structural PXRD. Eventually, both these species were formulated as Ln2(ClCNAn)3(DMSO)6 (LnIII = Tb for 1, and Eu for 2).
Despite their common stoichiometry, compounds 1, Tb2(ClCNAn)3(DMSO)6, and 2, Eu2(ClCNAn)3(DMSO)6, are not isostructural and crystallize in the monoclinic P21/n and triclinic P-1 space groups, respectively. Given that the structure of compound 1 has been determined by conventional single crystal analysis (and not by the less accurate structural powder diffraction methods), the stereochemical description will be mostly focused on 1 (Tb-CP). In this structure, the asymmetric unit contains one TbIII ion, one and a half ClCNAn2- ligands and three DMSO molecules (see Figure 3a). In the ClCNAn2- ligands, chloro and cyano substituents are disordered in a 50:50 for the (crystallographically imposed) centrosymmetric anilate, and 58:42 for the fully independent ligand. Such a disorder, attributed to residues with similar steric requirements, is indeed commonly observed in ClCNAn2-based CPs [17,21,22].
In addition, also the DMSO solvent molecules (apart from the metal-bound oxygen atoms) were found to be severely disordered.
In 1, TbIII ion is ennea-coordinated by six oxygen atoms from three different (chelating) ClCNAn2- ligands and by three oxygen atoms from DMSO molecules (Figure 3a), forming a slightly distorted tri-capped trigonal prismatic geometry (Figure 4b). Tb−O bond lengths fall in the 2.332(4)-2.359(4) Å (O=S(CH3)2) and 2.432(4)-2.483(4) Å (anilate oxygen atoms) ranges, which then cluster into two well-defined classes, differing by ca 0.1 Å. The complex LnO9 cage (shown in Figure 4b) shows 36 O-Tb-O bond angles falling in the very wide 63.6(1)-146.6(1)° range, as expected for the very crowded coordination environment of the TbIII ion. Nevertheless, three distinct sets of angles can be envisaged: i) those near 135° (12×, attributable to 6 trans cap-Tb-prism and 6 cap-Tb-cap angles); ii) those below 90°, for 21 cis Ox-Tb-Oy angles (x, y = cap or prism), and iii) 3 intermediate ones (typically > 100°) for the remaining cis Ox-Tb-Oy angles. Two crystallographically distinct (but very similar) Tb···Tb distances are found at 8.807 and 8.825 Å, for terbium atoms bridged by μ2,η4-chlorocyananilates. These distances are similar to those observed in Dy-ClCNAn2- CP (8.167(1) Å) [7].
The whole CP structure contains a 2D coordination network (of 3,6 topology, Figure 3c) where three bis-chelating ClCNAn2- ligands bridge [Tb(DMSO)3]3+ moieties forming distorted hexagons. Indeed, the angles between TbIII ions, taken as the network connecting nodes, are 92.9, 100.5° and 162.9° (2× each, adding up to 712°, witnessing the substantially flat nature of such hexagons – ideal value = (n-2)×180°, that is, for n = 6, 720°). These values also offer the estimate of the large deviation of the structure of 2 (of the brick wall type, with 90 and 180° angles) from a regular (3,6) honeycomb structure, where all these angles are exactly 120°, towards degenerate hexagons, that is rectangles of 2:1 aspect ratios.
In the structure shown in Figure 3d, the corrugated 2D layers (drawn by omitting, for clarity, the disordered Cl/CN and DMSO atoms), are arranged parallel to the (10-1) plane and are highly corrugated, their nominal thickness being ca. 18 Å (the length of the a-c diagonal). As commonly seen in lanthanide-anilate based CPs, within these wavy 2D slabs the coordinated DMSO molecules stick out toward the concave potions of the neighboring layers (Figure 3d). A similar structure type was reported for Ln2(ClCNAn)3(DMSO)6 (Ln3+ = Dy [7] and Pr [16]).
In Figure 4a, one can appreciate the ca. 18 Å periodicity in the sequence of the layers (2×), which can be used to estimate the number of these slabs (ns) within an exfoliated 2D nanocrystal of thickness t (vide infra) as ns = t/9
Morphological characterization of the corresponding nanosheets of 1 CP was performed by AFM, on drop-casted suspensions, obtained by crystal sonication, confirming the 2D nature of 1 bulk size CP. Remarkably micrometer-sized nanosheets were obtained, with heights ranging from one to four layers, as clearly shown in Figure 4b.
The structure of compound 2, determined (in the absence of single crystal specimens of suitable size and quality) by unconventional powder diffraction methods, is here discussed only from a connectivity and topological point of view. Indeed, as several restraints were added to stabilize convergence to physically meaningful results, with an extensive usage of rigid bodies to describe the chlorocyananilate and the DMSO ligands, no substantial stereochemical (bond distances and angles) descriptors can be taken as being accurate enough. Nevertheless, the following discussion does not suffer of such inaccuracy, and includes fully reliable (packing) features. In the crystal structure of 2, the asymmetric unit contains one EuIII ion, three distinct half ClCNAn2- ligands and three DMSO molecules (see Figure 5a for a slightly larger fragment, excised from the whole structure). Being all chlorocyananilate ligands located onto three different inversion centers, they all have crystallographically imposed 50:50 disordered Cl/CN residues. Also in compound 2, the lanthanide ion is ennea-coordinated, with three chelating chlorocyananilates and three individual O-bound DMSO molecules.
Differently from the structure of 1, the overall crystal structure of Eu2(ClCNAn)3(DMSO)6, still showing the presence of 2D sheets with “hexagonal” mesh and a common (3,6) connectivity, is based onto flat, non-corrugated slabs (as per Figure 5b,c). The internal angles of these degenerate hexagons of 88.1, 109.5 and 156.9°(2×, adding up to 709°). All very similar to those found in compound 1 and presented above, the interionic Eu…Eu separations of 8.83, 8.84 and 8.89 Å alone do not provide any direct hint on the significantly different warping of the 2D slabs in 1 and 2. Indeed, the differences arise at a larger scale, where the different “rectangular tiles” show a clear roofing in 1 , which is totally absent in 2 (dihedral angles between tiles of 102.1° and 0°, respectively).
Magnetic measurements were carried out with a fine powder sample of 1 as a function of temperature (Figure 6). The χmT value at room temperature (25.1 cm3 K mol–1) corresponds to two magnetically independent TbIII cations (both exhibiting J = 6, g = 3/2 and theoretical χmT ≈ 11.81 cm3 K mol–1)[23]. As expected for TbIII, the χmT value decreases when the temperature is decreased, because of the depopulation of the highest crystal-field levels. The high anisotropy of the TbIII centers does not allow to detect any additional super-exchange interactions promoted by the bridging organic ligands. Dynamic (AC) magnetic susceptibility measurements were performed down to 2 K at different frequencies (Figure 7). No out-of-phase χ’’ signal was observed, not even when an additional DC magnetic field was applied, indicating no SMM (Single Molecular Magnet) behavior for this compound.
3. Conclusions
The ditopic capability of chlorocyanoanilate ligand to chelate metal ions on two opposite sides of the central (and planar) benzoquinone core has been successfully exploited in the construction of 2D lanthanide coordination polymers formulated as Ln2(ClCNAn)3(DMSO)6 (LnIII =Tb for 1 and Eu for 2), generated from corresponding hydrated intermediates. Remarkably, the polyhydrated intermediates are isostructural, while recrystallization from DMSO by slow evaporation affords 1 and 2, which are not isostructural despite their common stoichiometry and crystallize in the monoclinic P21/n and triclinic P-1 space groups, respectively. Single-crystal X-ray study of 1 shows that the TbIII ion is ennea-coordinated within a slightly distorted tri-capped trigonal prismatic geometry, where bis-chelating ClCNAn2- ligands bridge [Tb(DMSO)3]3+ moieties, providing 2D corrugated layers. The 2D character of the coordination network in 1 allowed its exfoliation into nanosheets imaged by AFM. The magnetic susceptibility measurements of 1 are in agreement with isolated TbIII centers without slow magnetic relaxation. The structure of the EuIII coordination polymer 2, as determined from PXRD measurements, is slighlty different compared to that of 1 since the slabs are not corrugated. These results nicely complete and complement the series of coordination polymers based on chlorocyananilate ligand and lanthanide ions. The variation of the anilate substituents and lanthanides co-ligands is envisaged in order to tailor the magnetic and optical properties of these 2D materials.
4. Materials and Methods
Materials Reagents were purchased from Zentek (TCI) and used without further purification. HPLC-grade solvents were purchased from Thermofisher Scientific Alfa-Aesar. KHClCNAn was synthesized as reported in literature[15].
Synthesis of Tb2(ClCNAn)3(DMSO)6. (12). An aqueous solution of Tb(NO3)3.5H2O (0.30 mmol; 103mg) was added dropwise to an aqueous red solution of KHClCN (0,15mmol; 36 mg) and NaOH (0,10 mmol; 7,2 mg), showing an immediate color change to purple. After stirring at 90 °C for ca. 4 h, a purple precipitate appeared (1’). The mixture was cooled down to room temperature and the powder was collected from the mother liquor by vacuum filtration. The solid was then washed several times with cold deionized water.
Analytical evidence (mention which) suggested a Tb2(ClCNAn)3(H2O)x chemical formula for this intermediate (1’). Finally, compound 1 was obtained by recrystallization from DMSO by slow evaporation, red crystals suitable for X-ray analysis being formed within one week.
Synthesis of Eu2(ClCNAn)3(DMSO)6. (2) This compound was synthesized with a similar synthetic approach, using Eu(NO3)3.5H2 O instead of Tb(NO3)3.5H2O. As for the above synthesis, the 2’ and 2 labels are associated to a red hydrated polycrystalline intermediate and the DMSO-containing recrystallized polycrystalline material, respectively.
Synthesis of Nanosheets of 1. Nanosheets were fabricated through the top-down sonication-assisted exfoliation method. Delamination was achieved by sonicating (Bandelin electronic equipment at 230 V) the dried powder of 1 CPs (1 mg) samples in isopropanol (1 mL) for 15 min at room temperature.
X-ray Single-Crystal Structure Determination. A single crystal of compound 1 was mounted on glass fiber loop using a viscous crystal-coating hydrocarbon oil and were immediately transferred to the diffractometer cradle equipped with a cold N2 stream. Data collection was performed at 150 K on an Agilent Supernova Diffractometer with monochromatized Cu Kα radiation (λ = 1.54184 Å). The structure was solved by direct methods with the SIR97 program [24] and refined against all F2 values with the SHELXL-97 program (G. M. Sheldrick, “SHELXS-97 and SHELXL-97, Program for Crystal Structure Solution and Refinement,” University of Gottingen, Gottingen, 1997) using the WinGX graphical user interface [25](. All H atoms were placed in calculated positions and refined iso-tropically with a riding model. Non-H atoms were refined anisotropically except for the disordered ones: C7, C8 and C12 within the cyano groups and the methyl residues of the DMSO molecules. Crystallographic data and refinement parameters for 1 are listed in Table 1. Full crystal data, in the standard Crystallographic Information File format, have been deposited at the Cambridge Crystallographic Data Centre (CCDC Code: 2284151).
X-ray Powder Diffraction-Crystal Structure Determination. Samples of 1’, 2’ and 2 were gently ground in an agate mortar and then deposited in the hollow of a silicon monocrystal zero-background-plate (supplied by Assing SpA, Monterotondo, Italy). XRPD measurements were performed using a Bruker AXS D8 Advance diffractometer in Bragg-Brentano θ:θ geometry, equipped with a Lynxeye position sensitive detector. DS: 0.5°; Generator setting: 40 kV, 40 mA; Ni-filtered Cu-Kα radiation, λ = 1.5418 Å, 2θ -range: 3-50°. XRPD data for structure solution and refinement of species 2 were collected the 3–105° 2θ range, sampling at 0.02°, with scan time lasting approximately 16 h.
Cell determination from X-ray diffraction data
Standard peak search methods followed by the accurate estimate of the low-angle peak position and the use of the singular value decomposition protocol [26] implemented in TOPAS-R (V.3.0, 2005, Bruker AXS, Germany) enabled the detection of triclinic unit cells with GOF(20) = 26.1 and 61.5, for 1’ and 2 respectively. The structureless Le Bail whole pattern profile fitting method was used to refine the lattice parameters of the isomorphous 1 and 2, species, evidencing the presence of unavoidable contaminants (perhaps differently hydrated species). Therefore, no structure solution attempt was found to be successful in retrieving a suitable model.
Ab-initio crystal structure solution from X-ray diffraction data
XRPD structure solution of the species 2 phases was performed in space group P-1 using the TOPAS-R software by Monte Carlo / Simulated Annealing technique using a single Eu3+ ion, rigid models for the ClCNAn2- and DMSO ligands described by the Z-matrix formalism with standard geometrical parameters. It was eventually found that all chlorocyananilate ligands lie on inversion centers with consequent 50:50 Cl/CN disorder. Due to the less-than-ideal quality of the XRPD data, no attempt in determining a (static or dynamic) disorder of the DMSO molecules was done.
The final refinements were eventually carried out by the Rietveld method[27]. maintaining the rigid bodies introduced at the structure solution stage and the crystallographically imposed symmetries. The background was modelled by a polynomial function of the Chebyshev type; peak profiles were described by the Fundamental Parameters Approach [28] and a common (refinable) isotropic thermal factor was attributed to all atoms. Fractional atomic coordinates and crystal structure details are supplied in the Supporting Information file. The final Rietveld plot is shown in Figure 8
Figure 7.
Final Rietveld refinement plot for species 2. Observed data in blue, calculated data in red. The difference plot (in grey) and tick markers for peak positions are shown at the bottom.
Figure 7.
Final Rietveld refinement plot for species 2. Observed data in blue, calculated data in red. The difference plot (in grey) and tick markers for peak positions are shown at the bottom.
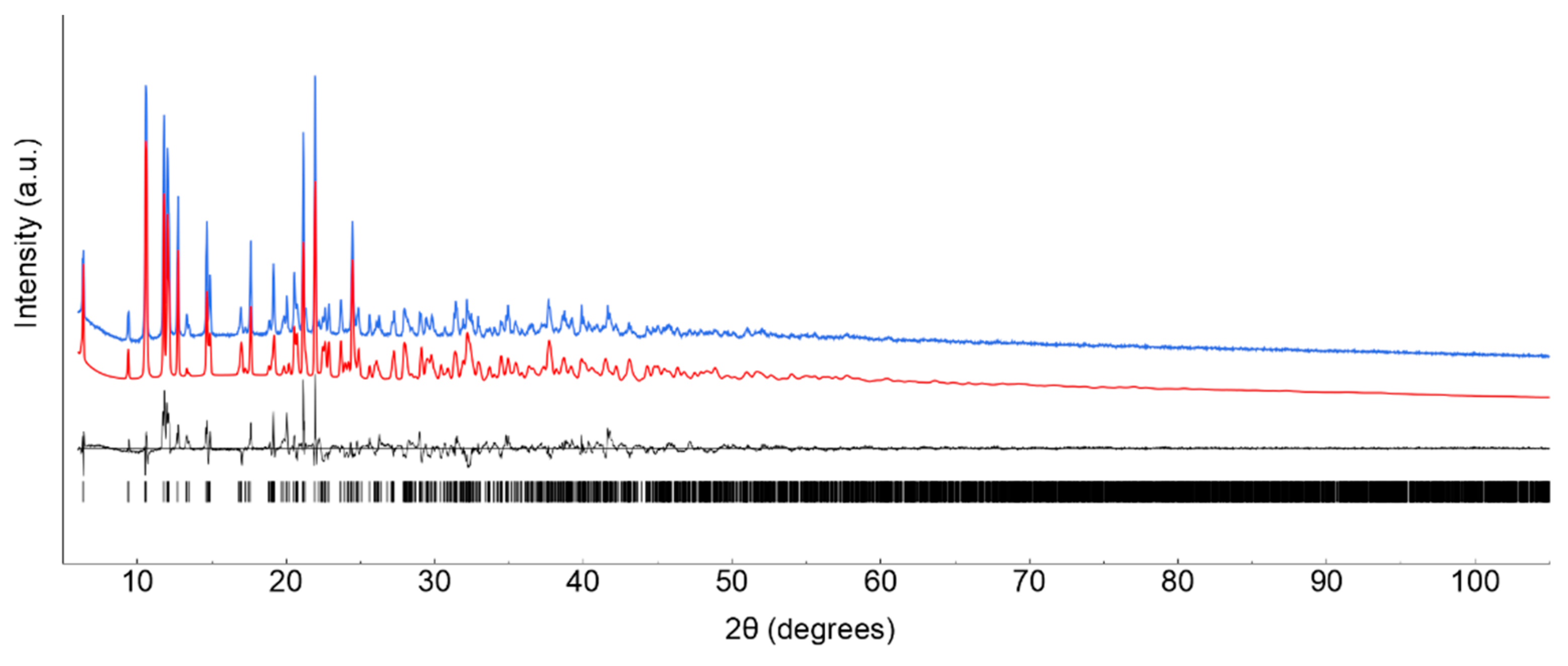

Table 1.
Crystallographic data and details of the refinement procedure for compounds 1 and 2.
| Compound | (1) | (2) |
| Formula | C33H36Cl3N3O18S6Tb2 | C33H36Cl3Eu2N3O18S6 |
| Few | 1379.20 | 1365.28 |
| Crystal color | Red | Red |
| Sample size (mm3) | 0.02*0.04*0.07 | 0.02*8.0*10.0 |
| Temperature (K) | 150(2) | 298(2) |
| Wavelength (Å) | 1.54184 | 1.54184 |
| Crystal system, Z | Monoclinic, 2 | Triclinic, 1 |
| Space group | P21/n | P-1 |
| a (Å) | 9.6868(2) | 9.704(1) |
| b (Å) | 16.3511(3) | 9.710(1) |
| c (Å) | 15.1558(3) | 14.146(1) |
| α (°) | 90 | 84.003(6) |
| β (°) | 93.683(2) | 97.145(6) |
| γ (°) | 90 | 78.526(5) |
| V (ų) | 2395.57(8) | 1284.6(4) |
| ρcalc (g.cm-³) | 1.912 | 1.764 |
| μ(CuKα) (mm-1) | 18.94 | 19.92 |
| θ range (°) | 3.981-73.587 | 2.5-52.5 |
| Data collected | 19515 | 5001 |
| Data unique | 4767 | - |
| Data observed | 4408 | - |
| Number of parameters / restraints | 388/65 | 58/9 |
| R(int) | 0.0486 | - |
| R1(F),a I > 2σ(I) | 0.0490 | (Rp) 0.0491 |
| wR2(F2),b all data | 0.1253 | (Rwp) 0.0755 |
| S(F2),c all data | 1.106 | 6.13 |
|
a R1(F) = ΣǁFo|-|Fcǁ/Σ|Fo|; b wR2(F2) = [Σw(Fo2-Fc2)2/ΣwFo4]1/2; c S(F2) = [Σw(Fo2-Fc2)2/(n+r-p)]1/2. | ||
Magnetic Measurements Magnetic measurements were carried out by using a Quantum Design MPMS-XL magnetometer. Magnetic susceptibility data were recorded in the 2 - 300 K temperature range in an external field of 1000 Oe. The diamagnetic corrections were calculated by using Pascal’s constants. (ref) Dynamic magnetic susceptibility data were obtained in the frequency range of 0.1-1000 Hz in an oscillating field of 3 Oe.
AFM characterization A NT-MDT Solver-Pro atomic-force microscopy (AFM) was used to study the topography and roughness of the nanosheets. AFM measurements were performed at 0.5-1 Hz scan speed in semicontact mode in air. Topographic image analysis and calculation of surface roughness were performed by using WSxM 5.0 Develop3.2 software. 10.1063/1.2432410.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
This research was funded in Italy by the Fondazione di Sardegna, Convenzione triennale tra la Fondazione di Sardegna e gli Atenei Sardi, Regione Sardegna, L.R. 7/2007 annualità 2020, through the SMAWRT project (CUP F75F21001260007).
Author Contribution
Conceptualization and project administration: MLM and MO. MO synthesized the CPs under the supervision of MLM. SC-XRD measurements have been performed and data analyzed by NA and AA. XRDP have been performed by FB and data analyzed by NM. JRGM performed the magnetic measurements and analyzed the data. DA and FQ performed AFM measurements. MLM, MO, NM and NA wrote the manuscript. All authors contributed to the critical revision of the manuscript. All authors have read and approved the final version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The SMAWRT project (CUP F75F21001260007) is acknowledged for M.O.'s post-doctoral grant. The work in France was supported by the CNRS and the University of Angers.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the writing of the manuscript; or in the decision to publish the results.
References
- James, S.L. Metal-Organic Frameworks. Chem. Soc. Rev. 2003, 32, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.C.J.; Kitagawa, S. Metal-Organic Frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–5418. [Google Scholar] [CrossRef] [PubMed]
- Oggianu, M.; Manna, F.; Sahadevan, S.A.; Avarvari, N.; Abhervé, A.; Mercuri, M.L. Metal-Organic Framework vs. Coordination Polymer—Influence of the Lanthanide on the Nature of the Heteroleptic Anilate/Terephtalate 3D Network. Crystals 2022, 12. [Google Scholar] [CrossRef]
- Oggianu, M.; Monni, N.; Mameli, V.; Cannas, C.; Sahadevan, S.A.; Mercuri, M.L. Designing Magnetic Nanomofs for Biomedicine: Current Trends and Applications. Magnetochemistry 2020, 6, 1–14. [Google Scholar] [CrossRef]
- Ashoka Sahadevan, S.; Monni, N.; Oggianu, M.; Abhervé, A.; Marongiu, D.; Saba, M.; Mura, A.; Bongiovanni, G.; Mameli, V.; Cannas, C.; et al. Heteroleptic NIR-Emitting YbIII/Anilate-Based Neutral Coordination Polymer Nanosheets for Solvent Sensing. ACS Appl. Nano Mater. 2020, 3, 94–104. [Google Scholar] [CrossRef]
- Ashoka Sahadevan, S.; Manna, F.; Abhervé, A.; Oggianu, M.; Monni, N.; Mameli, V.; Marongiu, D.; Quochi, F.; Gendron, F.; Le Guennic, B.; et al. Combined Experimental/Theoretical Study on the Luminescent Properties of Homoleptic/Heteroleptic Erbium(III) Anilate-Based 2D Coordination Polymers. Inorg. Chem. 2021, 60, 17765–17774. [Google Scholar] [CrossRef]
- Sahadevan, S.A.; Monni, N.; Abhervé, A.; Cosquer, G.; Oggianu, M.; Ennas, G.; Yamashita, M.; Avarvari, N.; Mercuri, M.L. Dysprosium Chlorocyanoanilate-Based 2D-Layered Coordination Polymers. Inorg. Chem. 2019, 58, 13988–13998. [Google Scholar] [CrossRef]
- Junggeburth, S.C.; Diehl, L.; Werner, S.; Duppel, V.; Sigle, W.; Lotsch, B. V. Ultrathin 2D Coordination Polymer Nanosheets by Surfactant-Mediated Synthesis. J. Am. Chem. Soc. 2013, 135, 6157–6164. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, J.; Fu, H.; Li, W.; Fan, X.; Xin, G.; Zheng, J.; Li, X. MOF Derived Catalysts for Electrochemical Oxygen Reduction. J. Mater. Chem. A 2014, 2, 14064–14070. [Google Scholar] [CrossRef]
- Kitagawa, S.; Kawata, S. Coordination Compounds of 1,4-Dihydroxybenzoquinone and Its Homologues. Structures and Properties. Coord. Chem. Rev. 2002, 224, 11–34. [Google Scholar] [CrossRef]
- Benmansour, S.; Gómez-Claramunt, P.; Vallés-García, C.; Mínguez Espallargas, G.; Gómez García, C.J. Key Role of the Cation in the Crystallization of Chiral Tris(Anilato)Metalate Magnetic Anions. Cryst. Growth Des. 2016, 16, 518–526. [Google Scholar] [CrossRef]
- Mencel, K.; Kinzhybalo, V.; Jakubas, R.; Zarȩba, J.K.; Szklarz, P.; Durlak, P.; Drozd, M.; Piecha-Bisiorek, A. 0D Bismuth(III)-Based Hybrid Ferroelectric: Tris(Acetamidinium) Hexabromobismuthate(III). Chem. Mater. 2021, 33, 8591–8601. [Google Scholar] [CrossRef]
- Souto, M.; Perepichka, D.F. Electrically Conductive Covalent Organic Frameworks: Bridging the Fields of Organic Metals and 2D Materials. J. Mater. Chem. C 2021, 9, 10668–10676. [Google Scholar] [CrossRef]
- Chakraborty, G.; Park, I.H.; Medishetty, R.; Vittal, J.J. Two-Dimensional Metal-Organic Framework Materials: Synthesis, Structures, Properties and Applications. Chem. Rev. 2021, 121, 3751–3891. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.; Artizzu, F.; Marchiò, L.; Loche, D.; Caneschi, A.; Serpe, A.; Deplano, P.; Avarvari, N.; Mercuri, M.L. Switching-on Luminescence in Anilate-Based Molecular Materials. Dalt. Trans. 2015, 44, 15786–15802. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Claramunt, P.; Benmansour, S.; Hernández-Paredes, A.; Cerezo-Navarrete, C.; Rodríguez-Fernández, C.; Canet-Ferrer, J.; Cantarero, A.; Gómez-García, C.J. Tuning the Structure and Properties of Lanthanoid Coordination Polymers with an Asymmetric Anilato Ligand. Magnetochemistry 2018, 4. [Google Scholar] [CrossRef]
- Ashoka Sahadevan, S.; Monni, N.; Abhervé, A.; Marongiu, D.; Sarritzu, V.; Sestu, N.; Saba, M.; Mura, A.; Bongiovanni, G.; Cannas, C.; et al. Nanosheets of Two-Dimensional Neutral Coordination Polymers Based on Near-Infrared-Emitting Lanthanides and a Chlorocyananilate Ligand. Chem. Mater. 2018, 30, 6575–6586. [Google Scholar] [CrossRef]
- Zhang, S.; Sunami, Y.; Hashimoto, H. Mini Review: Nanosheet Technology towards Biomedical Application. Nanomaterials 2017, 7, 1–7. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, C.; Chen, X.; Liu, J.; Yu, Q.; Liu, Y.; Liu, J. Drug Delivery System Based on Near-Infrared Light-Responsive Molybdenum Disulfide Nanosheets Controls the High-Efficiency Release of Dexamethasone to Inhibit Inflammation and Treat Osteoarthritis. ACS Appl. Mater. Interfaces 2019, 11, 11587–11601. [Google Scholar] [CrossRef]
- Hofmann, D.W.M. Fast Estimation of Crystal Densities. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 489–493. [Google Scholar] [CrossRef]
- Benmansour, S.; Gómez-García, C.J. Lanthanoid-Anilato Complexes and Lattices. Magnetochemistry 2020, 6, 1–44. [Google Scholar] [CrossRef]
- Benmansour, S.; Pérez-Herráez, I.; López-Martínez, G.; Gómez García, C.J. Solvent-Modulated Structures in Anilato-Based 2D Coordination Polymers. Polyhedron 2017, 135, 17–25. [Google Scholar] [CrossRef]
- Benelli, C.; Gatteschi, D. Magnetism of Lanthanides in Molecular Materials with Transition-Metal Ions and Organic Radicals. Chem. Rev. 2002, 102, 2369–2387. [Google Scholar] [CrossRef]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A New Tool for Crystal Structure Determination and Refinement. J. Appl. Crystallogr. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Coelho, A.A. Indexing of Powder Diffraction Patterns by Iterative Use of Singular Value Decomposition. J. Appl. Crystallogr. 2003, 36, 86–95. [Google Scholar] [CrossRef]
- Rietveld, H.M. A Profile Refinement Method for Nuclear and Magnetic Structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Cheary, R.W.; Coelho, A. Fundamental Parameters Approach to X-Ray Line-Profile Fitting. J. Appl. Crystallogr. 1992, 25, 109–121. [Google Scholar] [CrossRef]
Chart 1.
Potassium Chlorocyananilate (left) and Coordination Mode of the Ligand (right).
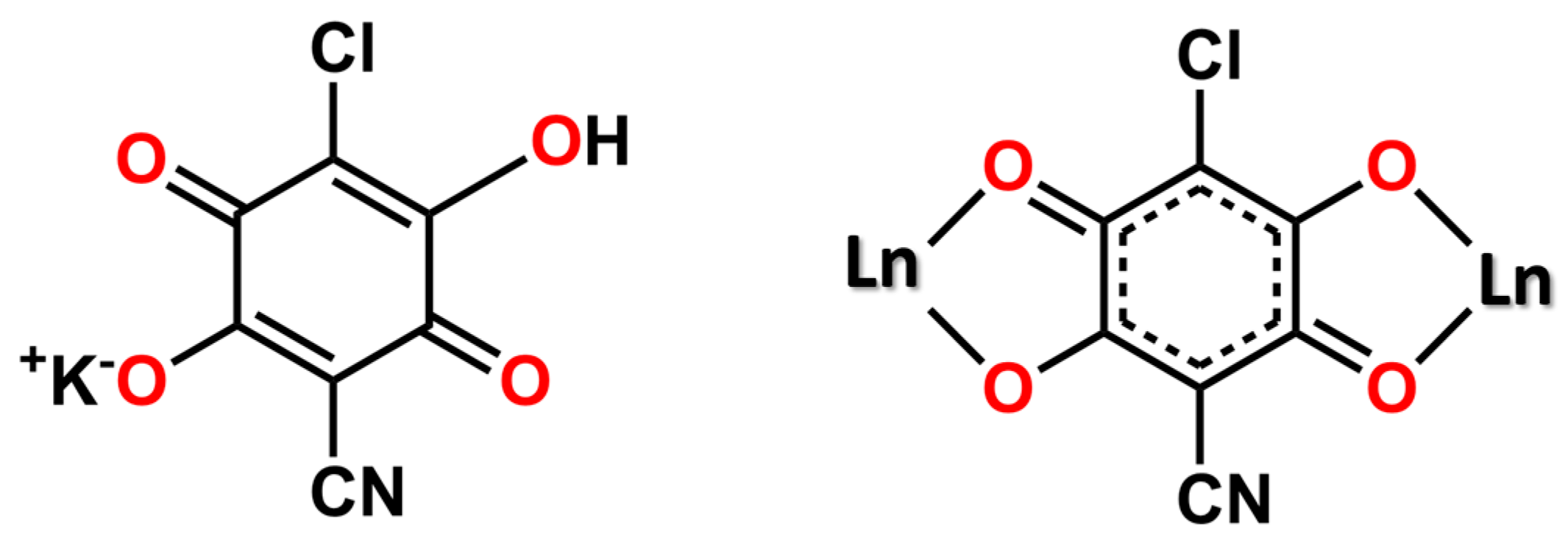
Figure 1.
Schematic representation of the synthesis of compounds 1 and 2, via the intermediacy of 1’ and 2’.
Figure 1.
Schematic representation of the synthesis of compounds 1 and 2, via the intermediacy of 1’ and 2’.
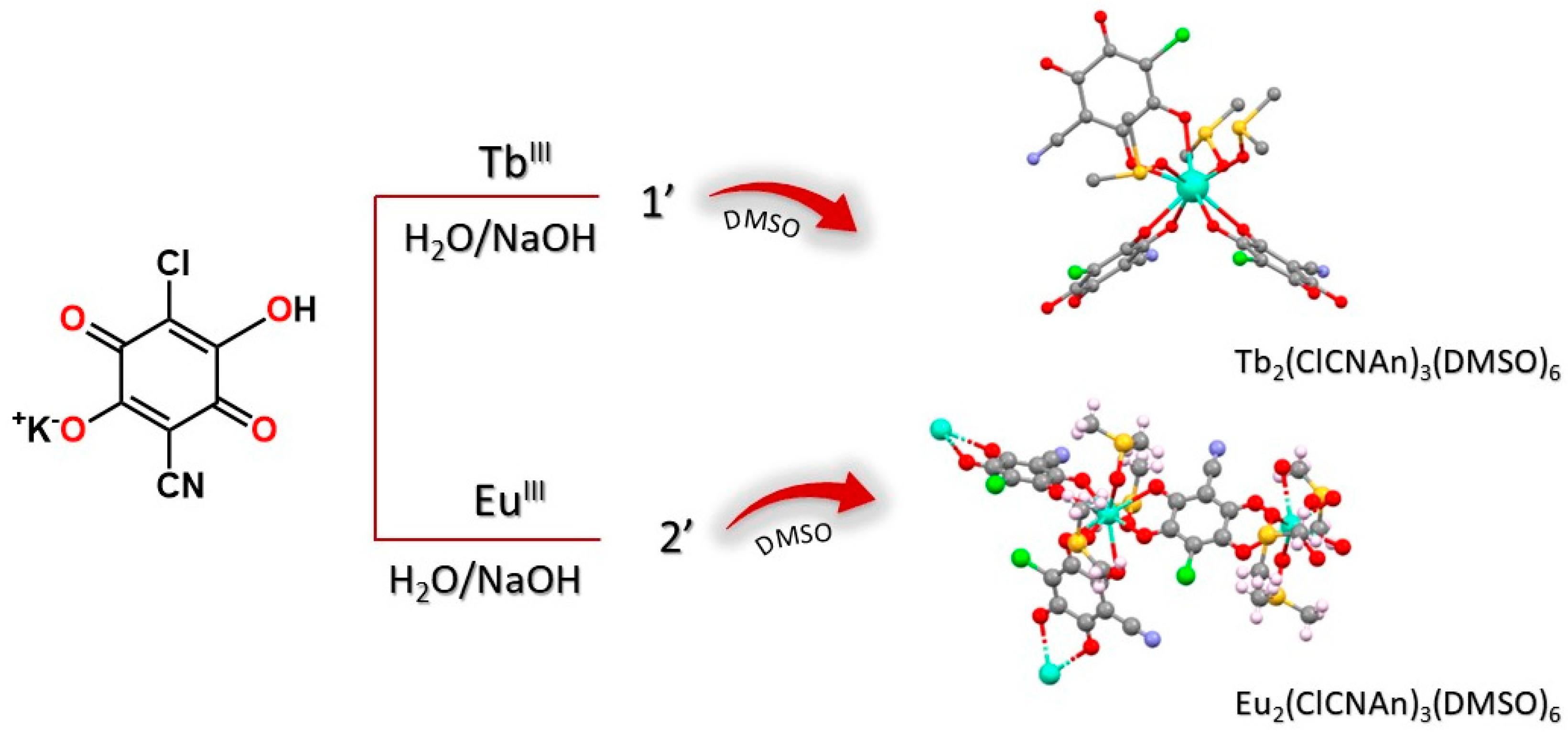
Figure 2.
XRPD raw data (blue line) and Le Bail fits (red line) for polycrystalline 1’ (a) and 2’ (b) species. Difference plot and tick markers are shown at the bottom.
Figure 2.
XRPD raw data (blue line) and Le Bail fits (red line) for polycrystalline 1’ (a) and 2’ (b) species. Difference plot and tick markers are shown at the bottom.
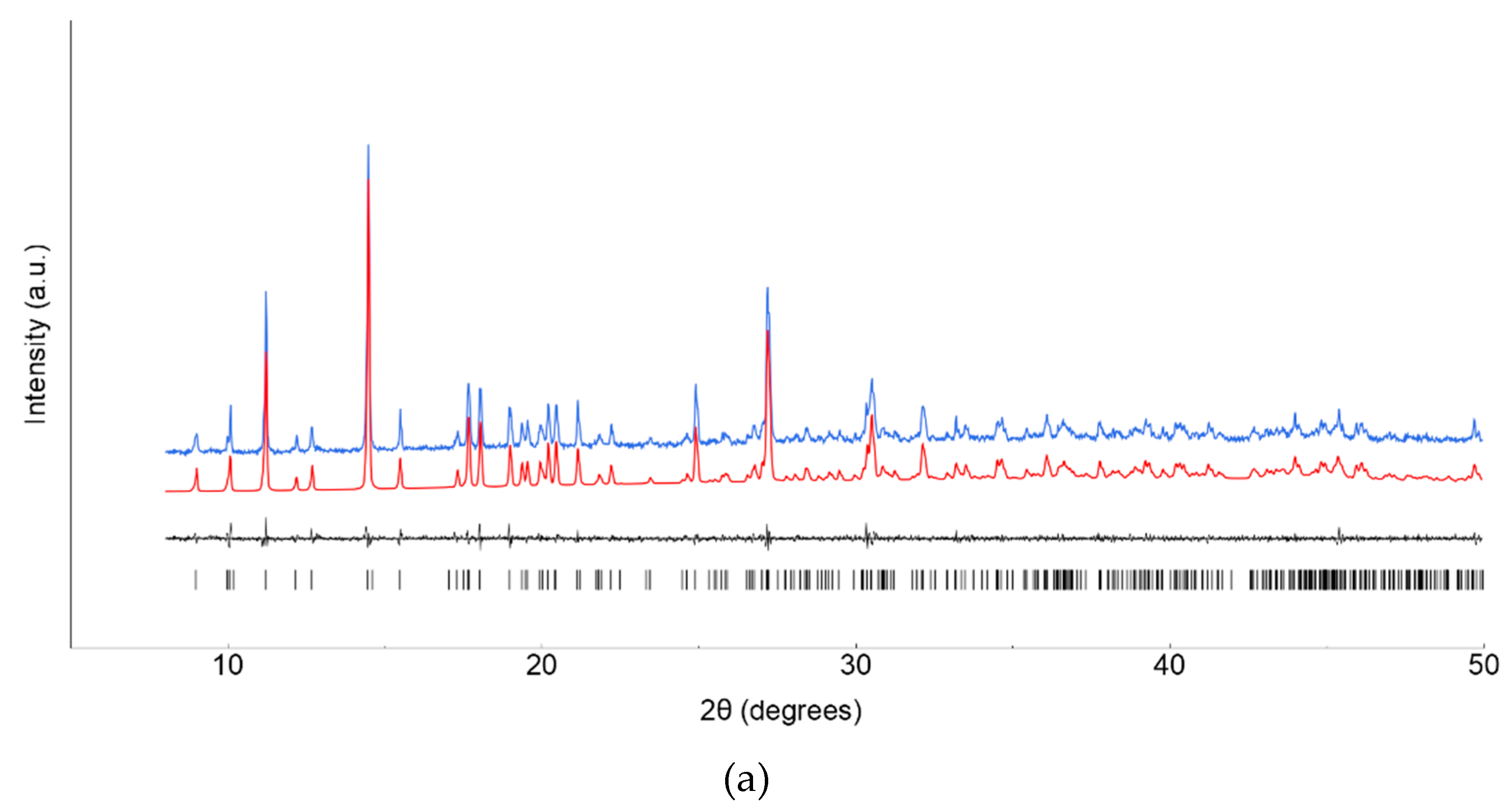
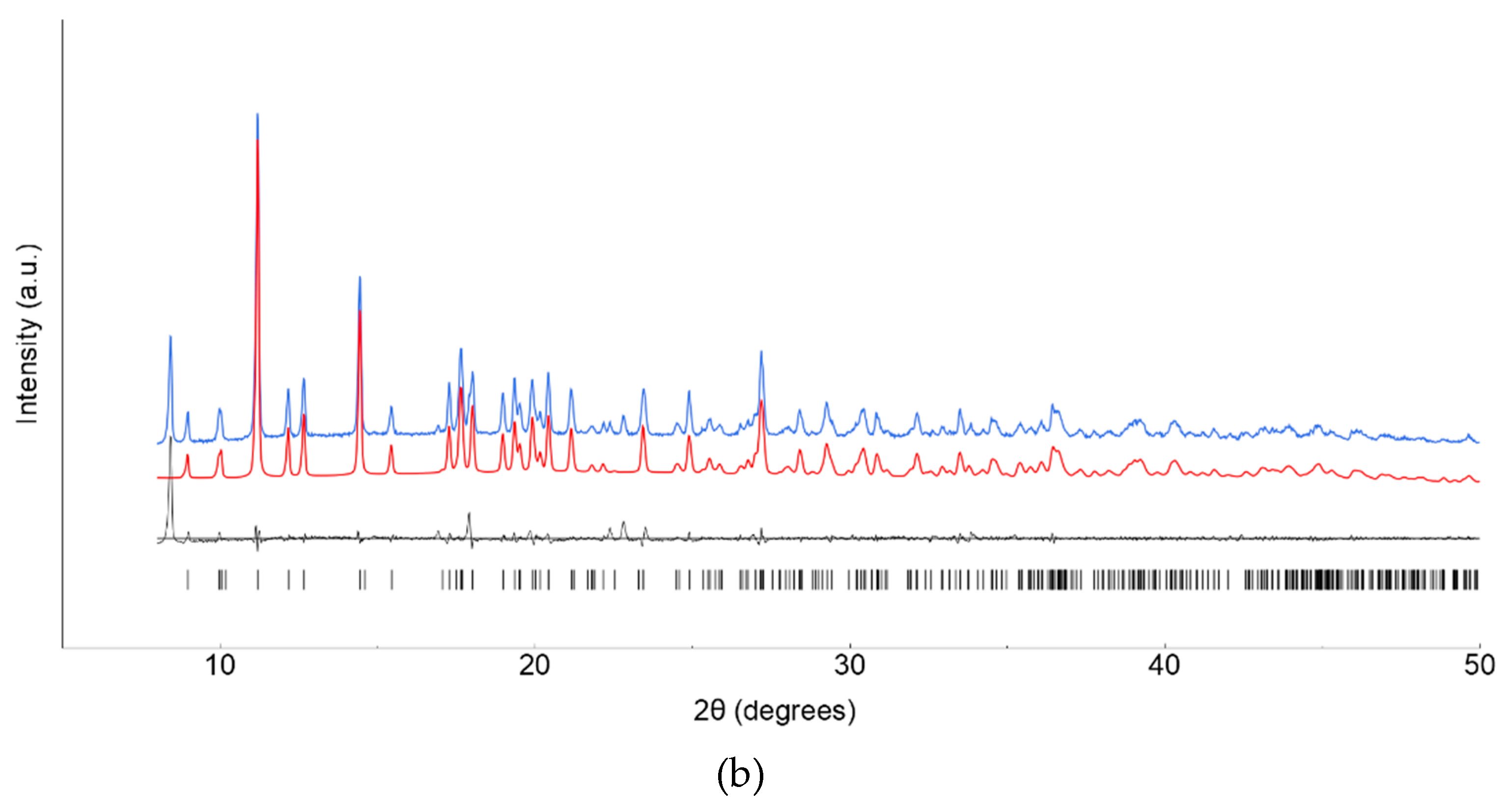
Figure 3.
a) Full coordination environment around TbIII; b) sketch of the distorted tricapped trigonal prismatic geometry around TbIII (hydrogen and disordered C,S ghosts removed for clarity); c) View of one pseudo-rectangular cavity in ac plane; d) View of three consecutive layers stacking parallel to the 3a-c vector. Colour code: Tb- dark green; O- red; C- grey; S-yellow; Cl- green; N- blue.
Figure 3.
a) Full coordination environment around TbIII; b) sketch of the distorted tricapped trigonal prismatic geometry around TbIII (hydrogen and disordered C,S ghosts removed for clarity); c) View of one pseudo-rectangular cavity in ac plane; d) View of three consecutive layers stacking parallel to the 3a-c vector. Colour code: Tb- dark green; O- red; C- grey; S-yellow; Cl- green; N- blue.
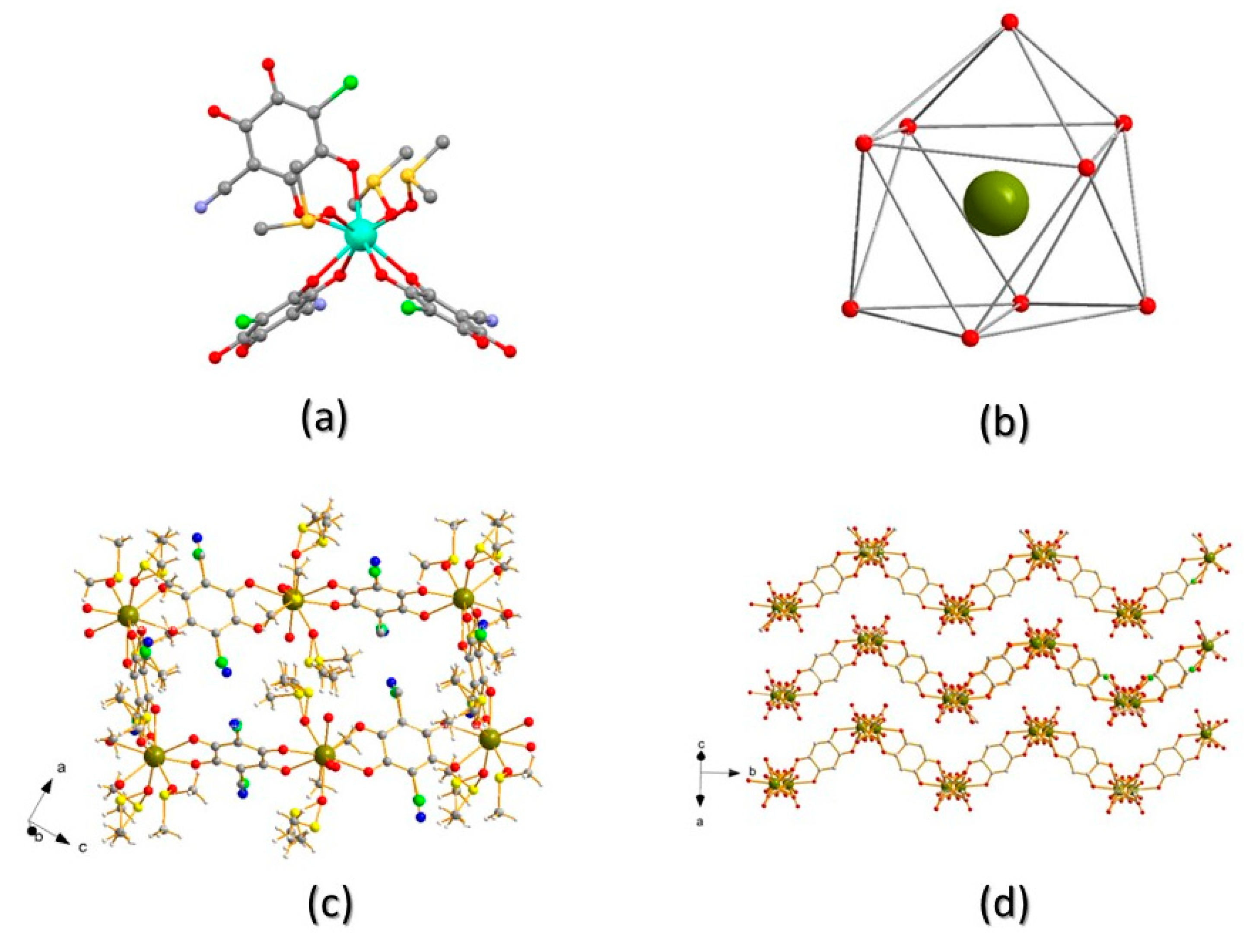
Figure 4.
a) The interdigitation of symmetry-related (and heavily corrugated) 2D layers of compound 1. The orientation of the crystal axes (not to scale) is also shown (bottom left). The double arrow addresses the length of the a-c vector (18 Å) corresponding to two corrugated layers; b) AFM characterization of drop-casted nanosheets of 1: topographic image and, in the inset, cross-sectional height profiles, showing that flat or terraced nanosheets are formed, with ca. 9 Å steps.
Figure 4.
a) The interdigitation of symmetry-related (and heavily corrugated) 2D layers of compound 1. The orientation of the crystal axes (not to scale) is also shown (bottom left). The double arrow addresses the length of the a-c vector (18 Å) corresponding to two corrugated layers; b) AFM characterization of drop-casted nanosheets of 1: topographic image and, in the inset, cross-sectional height profiles, showing that flat or terraced nanosheets are formed, with ca. 9 Å steps.
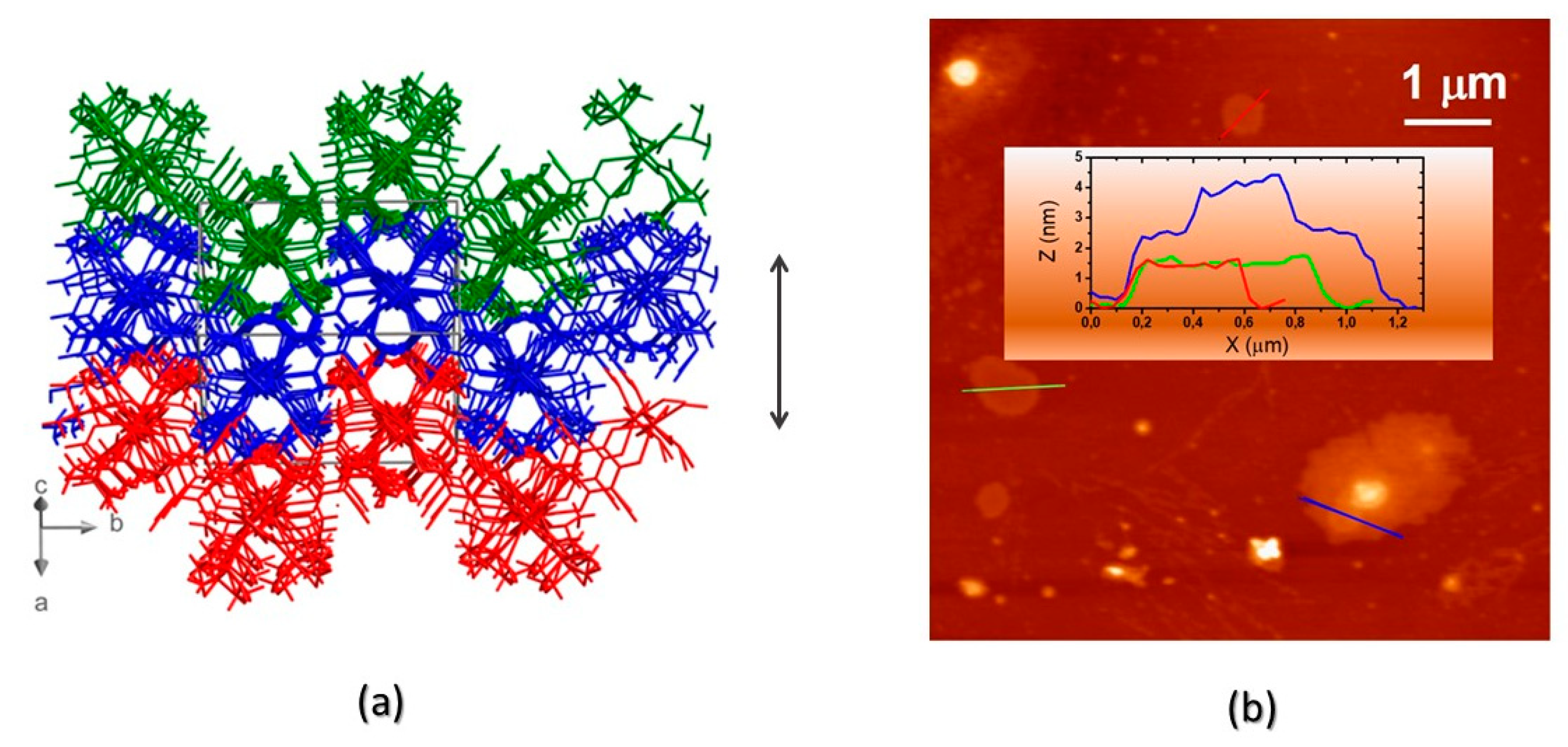
Figure 5.
Skectches of the main structural features of 1 CP. a) a fragment of the CP, showing the local environment of the EuIII ions; b) and c) the brick-wall connectivity of a flat slab in 2, viewed down the axis normal to the CP extension, and from the side, respectively; d) the overall crystal packing viewed down the a axis, showing that slabs, interacting only through weak van der Waals contacts mostly attibuted to the DMSO ligands, stack along b with a ca. 9.7 Å periodicity.
Figure 5.
Skectches of the main structural features of 1 CP. a) a fragment of the CP, showing the local environment of the EuIII ions; b) and c) the brick-wall connectivity of a flat slab in 2, viewed down the axis normal to the CP extension, and from the side, respectively; d) the overall crystal packing viewed down the a axis, showing that slabs, interacting only through weak van der Waals contacts mostly attibuted to the DMSO ligands, stack along b with a ca. 9.7 Å periodicity.
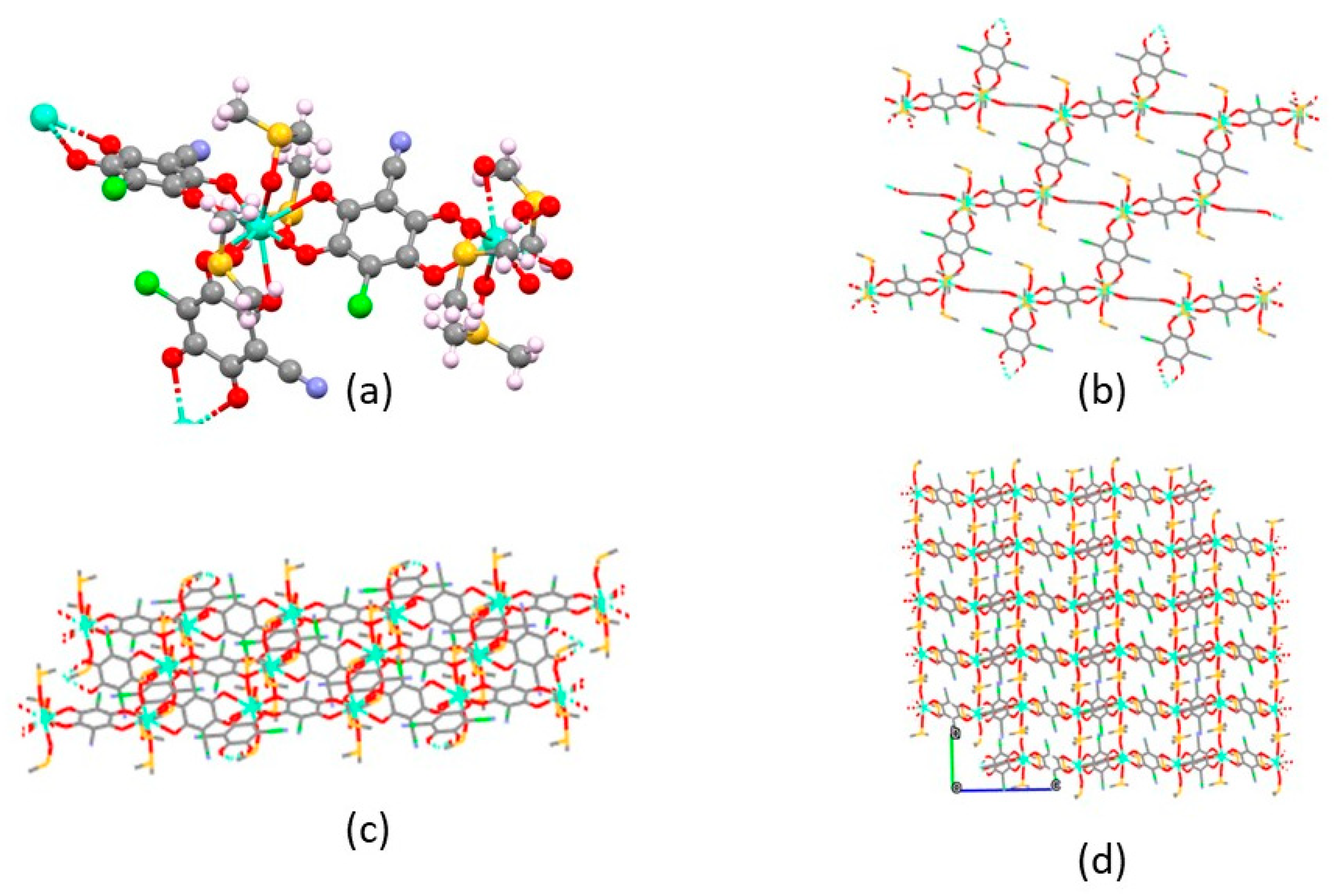
Figure 6.
a) Plot of the χmT product as a function of temperature for 1. b) In-phase (χ’) and out-of-phase (χ’’) dynamic susceptibility for 1 at different frequencies as a function of temperature.
Figure 6.
a) Plot of the χmT product as a function of temperature for 1. b) In-phase (χ’) and out-of-phase (χ’’) dynamic susceptibility for 1 at different frequencies as a function of temperature.
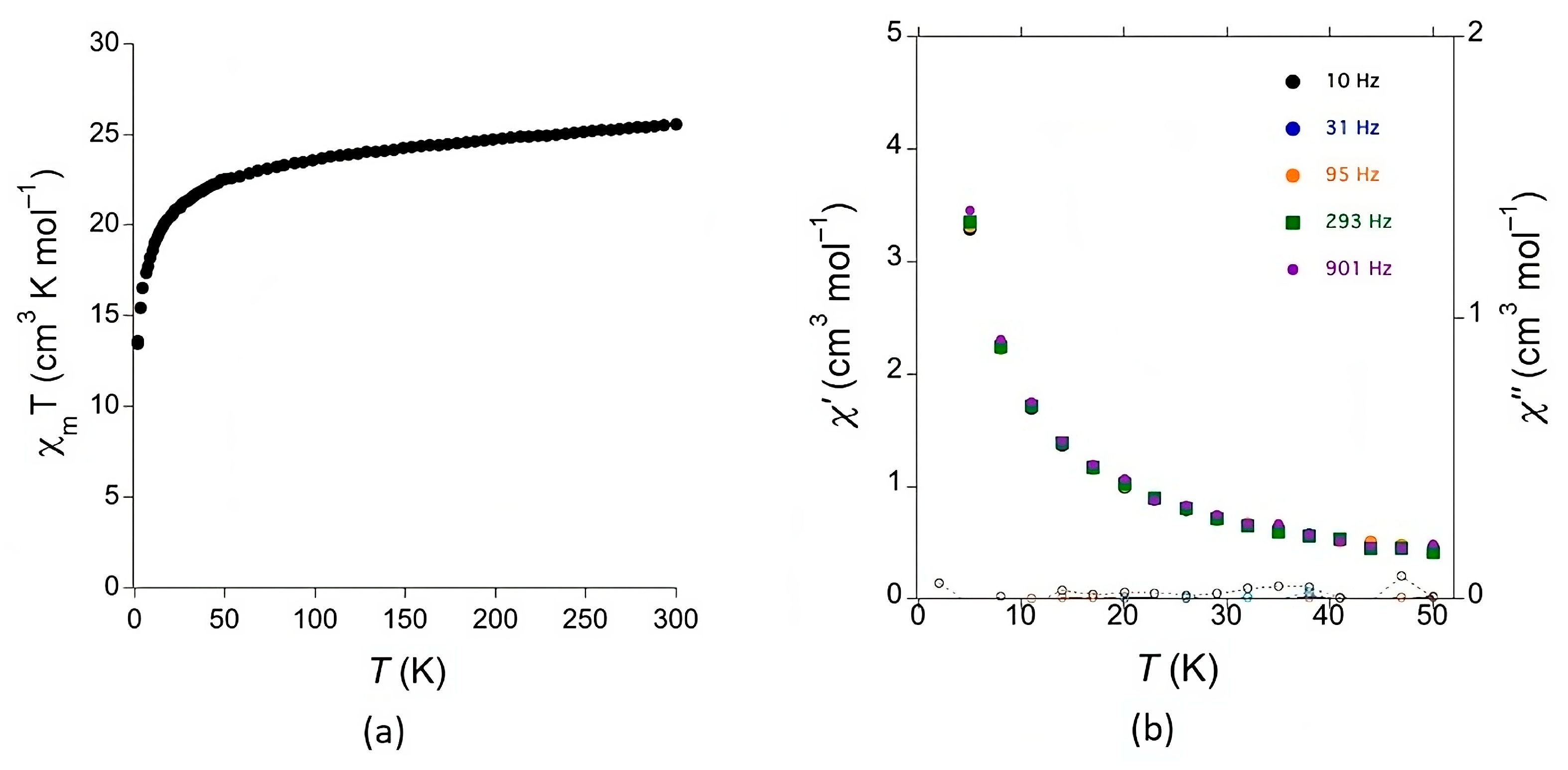
Table 1.
Crystal data for Compounds Ln2(ClCNAn)3(H2O)x. y(H2O) x+y= 12 (Ln = Tb, 1’; Eu, 2’).
| Species | Symmetry | a, Å | b, Å | c, Å | α, ° | β, ° | γ, ° | V, Å3 |
| 1’ | Triclinic | 10.12 | 10.68 | 10.36 | 73.3 | 88.0 | 60.0 | 920.7 |
| 2’ | Triclinic | 10.13 | 10.70 | 10.36 | 73.4 | 88.0 | 59.9 | 922.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



