
Submitted:
04 August 2023
Posted:
07 August 2023
You are already at the latest version
Abstract
Neurodegenerative diseases encompass a broad spectrum of profoundly disabling disorders that impact millions of individuals globally. While their underlying causes and pathophysiol-ogy display considerable diversity and remain incompletely understood, a mounting body of evidence indicates that the disruption of blood-brain barrier (BBB) permeability, resulting in brain damage and neuroinflammation, constitutes a shared characteristic among them. Con-sequently, targeting the BBB has emerged as an innovative therapeutic strategy for address-ing neurological diseases. Within this review, we not only explore the neuroprotective, neu-rotrophic, and immunomodulatory benefits of mesenchymal stem cells (MSCs) in combating neurodegeneration but also delve into their recent role in modulating the BBB. We will delve into the cellular and molecular mechanisms through which MSC treatment impacts primary age-related neurological disorders like Alzheimer’s disease, Parkinson’s disease, and stroke as well as immune-mediated conditions such as multiple sclerosis. Our focus will center on how MSCs participate in the modulation of cell transporters, matrix remodeling, stabilization of cell-junction components, and restoration of BBB network integrity in these pathological con-texts.
Keywords:
blood-brain barrier
; mesenchymal stem cell
; neurodegenerative diseases
; Alzheimer’s disease
; neuroinflammation
; Parkinson’s disease
; multiple sclerosis
; stroke
1. Introduction
The evidence indicates that longevity is currently increasing worldwide and the occurrence of cumulative disabilities is the price to pay for living longer [1]. It is widely recognized that aging is a natural, progressive, and inevitable process that occurs in all organisms, although the functional and morphological changes affecting the body tissues and organs during its progression are highly variable [2]. Specifically, aging-associated changes in the central nervous system (CNS) are of crucial relevance, and the prevention and treatment of neurodegenerative diseases represent one of the greatest challenges for modern societies. However, therapeutic options to treat CNS-related diseases are very limited, mainly due to the fine-tuned status of the brain, the complexity of neurological diseases, and the lack of knowledge of their etiology and pathophysiology.
An additional challenge in this scenario comes from the singular presence in the CNS of the blood-brain barrier (BBB), a complex, dynamic, and structured network of cells and proteins responsible for protecting the brain and regulating the transport of substances and cells from the peripheral circulation to the CNS. Because keeping the integrity of BBB is critical for maintaining a constant environment in the CNS in healthy conditions, investigating how this structure changes during aging and specifically in pathological conditions, has lately received special attention worldwide. Although some researchers have demonstrated minor and variable BBB leakage in healthy aging without immune infiltration and neuropathological signs [3], evidence indicates that BBB integrity is compromised in most neurological diseases, including those associated with aging [4,5,6,7,8,9,10,11]. Therefore, BBB disruption emerges as a key innovative element to design new therapeutic approaches to treat neurodegeneration during aging and neurological disorders.
Recently, stem cells isolated from adult mesenchymal tissues (MSCs) have emerged as attractive candidates for the treatment of aging-associated neurological diseases [12,13]. While many studies have explored the involvement of the neuroprotective, neurotrophic, and immunomodulatory capacities of MSCs in their therapeutic actions in neuroinflammatory and neurodegenerative disorders, the potential and additional role played by MSCs in improving the sealing and modulation of the BBB has been scarcely addressed.
This review aims to examine the structure and functions of the BBB and how its impairment, along with changes in transporters, extracellular matrix, and cell-junctional components, influences the onset and progression of several neurodegenerative disorders. We will specially focus on four neurodegenerative disorders, namely Alzheimer´s disease (AD), Parkinson´s disease (PD), multiple sclerosis (MS), and stroke. These disorders exhibit different clinical and pathological signs, and although their etiologies remain mostly unknown, they all course with neuroinflammation and neurodegeneration, are significantly associated with aging, and display profound alterations in cerebral vasculature and microvessels components. Importantly, they all positively respond to MSC-based therapies. Our review will also delve into the molecular mechanisms involved in the MSCs-treatment of neurological pathologies, with special attention to their ability to restore the integrity of the BBB. Finally, we will discuss how to further improve MSCs therapies by specifically targeting BBB.
2. Structure and Role of the Blood-Brain Barrier
The BBB is a complex and dynamic microvascular structure composed of several types of cells, which maintains the homeostasis of the CNS by regulating the supply of molecules and filtering potentially harmful compounds from the bloodstream to brain tissues and back [14]. Its main scaffold is structured in a neurovascular unit (NVU) that is shaped by a tight layer of brain microvascular endothelial cells (BMVECs) surrounded by astrocyte end-feet and pericytes, all of them embedded in an extracellular matrix network and the basement membrane [15] (Figure 1).
The BMVECs are highly specialized endothelial cells that show unique structural and biological properties compared to peripheral endothelial cells [16], including low pinocytic activity, absence of fenestration, low levels of leukocyte adhesion molecules, high expression of intercellular junctions, mainly tight-junctions (TJs) and adherent-junctions (AJs) [17,18], and increased number of mitochondria to supply the energy that is required for the active transendothelial transport of molecules [19]. Additionally, BMVECs regulate BBB permeability by mainly controlling the intercellular and intracellular transport of cells and molecules via cellular junctions and specific membrane carriers, channels, and transporters [20]. Moreover, these cells present a very remarkable apicobasal polarity, based on a differential membrane composition (lipids, glycoproteins, receptors, and transporters) between the luminal and the abluminal sides [21]. For instance, enzymes like γ-glutamyl-transpeptidase [22] or alkaline phosphatases [23] are located in the luminal face of the endothelium, while Na+-K+ ATPase [24] and the Na+-dependent neutral amino acid transporter [25] are present at the basal membrane.
BMVECs are wrapped by pericytes which contribute to the regulation of endothelial cell proliferation, survival, migration, differentiation, vascular branching, and blood flow control [26]. Smooth muscle cells are also found around large vessels (arteries, arterioles, venules, and veins), providing strength and elasticity, and playing a significant role in basal tone maintenance, blood pressure and blood flow distribution [27,28]. Pericytes are localized along capillaries and imbedded within the BM. They regulate blood flow, modulate immune and phagocytic responses after brain injury, and promote angiogenesis in the adult CNS [29,30].
Astrocytes are the major glial cell that enfolds the endothelium of the BBB [31]. They connect to BMVECs through their end-feet, contributing to BBB consistency and determining its properties [30]. Astrocytic end-feet contain a set of proteins that interact with the vascular tube, such as dystroglycan-dystrophin complex or aquaporin 4 (AQP-4), among others. The latter is critical for regulating water homeostasis in the CNS, while the dystroglycan-dystrophin complex links the astrocytic skeleton to the BM [32]. Since astrocytes serve as the cellular linkage between the neuronal circuitry and the vascular system in the CNS, they release signals that regulate the blood flow in response to neuronal activity. For instance, they control the contraction and dilation of SMCs and pericytes [33]. Additionally, they contribute to the formation of endothelial cell TJs through vascular endothelial growth factor (VEGF)-mediated signals [34] and regulate tissue inhibitor metalloproteinases (TIMPs), which maintain the balance between deposition and degradation of the extracellular matrix components [35].
The basement membrane (BM) is a highly organized sheet composed prominently of extracellular matrix proteins (collagen IV, laminins, nidogen, and perlecan). It plays an important role in providing structural support, cell anchoring, and signaling transduction [36]. Two types of BM of the BBB have been characterized: the inner vascular BM, secreted by BMVECs and pericytes, which contains laminins α1 and α2; and the outer parenchymal BM, secreted by astrocytes, which contains laminins α4 and α5 [37,38]. Nevertheless, the BM is largely understudied, in comparison to the cellular components of BBB, probably due to its intrinsic complexity and the lack of research tools [39].
The exchange of molecules and cells across the BBB requires specific transporters, channels, and receptors. Two major mechanisms, named paracellular and transcellular transport have been identified. Table 1 describes some of the main transporters. Paracellular transport is the main pathway used for the exchange of small hydrophilic substances. On the other hand, the transcellular transport involves the movement of molecules through the cell membrane of BMVECs. It occurs via several mechanisms depending on the nature of the molecule, such as passive diffusion, facilitated diffusion, active transport, and receptor-mediated transport. Importantly, gases and small lipophilic molecules do not require transporters to cross the BBB and freely diffuse across the endothelium [40].
Both, paracellular and transcellular processes are precisely regulated by junctional structures, mainly TJs, with some contribution from gap junctions and AJs [41]. TJs are intricate structures located along the membranes of adjacent BMVECs, intermingled distributed with AJs, which provide stability and consistency to the BBB [42,43] (Figure 1). Among TJ proteins, claudin-1, -3, -5, -12, and occludin control the transportation of solutes and ions [44]. These proteins are associated with the cytoskeleton, primarily based on actin and vinculin, through scaffolding proteins, such as ZO-1, -2, and -3. Moreover, dystrophin acts as a scaffold protein that mobilizes actin and vinculin proteins [45]. AJs create inter-endothelial contact connections maintained by proteins such as VE-cadherin and platelet endothelial cell adhesion molecule-1 [46,47] (Figure 1), that contribute to the continuous crosstalk with TJs for paracellular transportation. Similarly to TJs, AJs are attached to the cytoskeleton, contributing to the regulation of cellular transportation of lymphocytes, monocytes, or neutrophils [48,49,50]. Pericyte-endothelial junctions also contain cadherins, with N-cadherins forming homophilic interactions between pericytes and BMVECs, thereby maintaining vascular integrity [51]. Furthermore, BMVECs interact with the BM stablishing AJs via α- and β-integrin receptors, which are transmembrane glycoproteins involved in the extracellular matrix connection to the endothelial cytoskeleton [44]. Conversely, gap junctions serve as intercellular channels facilitating cytoplasmic connections between neighboring cells, enabling selective communication of molecules primarily dependent on molecular size, driven by passive diffusion [52]. In the brain, endothelial cells express the gap junctions connexin 37 (Cx37), Cx40, and Cx43 [53], while astrocytes express Cx30 and Cx43 [54]. In addition to their channel function, certain connexins also play a regulatory role in the expression of other junctional molecules, such as Cx43 interacting with N-cadherin [55].
3. MSCs as a Therapeutic Option in CNS Diseases
Despite notable advancements in the management of symptoms of neurodegenerative diseases such as AD, PD, MS, and stroke, with treatments that enhance quality of life and increase lifespan, the available drugs only slow the progression of neuronal death. Given the multifactorial and complex nature of these diseases, the primary causal agent remains unclear, and it is imperative to develop multi-target therapies that address the different causes/consequences of these disorders such as neuroinflammation, neuronal cell death and dysfunction, and BBB disruption.
MSCs are emerging as one of the most promising cell therapies against different immune-mediated diseases due to their unique properties. MSCs are multipotent cells able to differentiate into mesodermal lineages (fibroblast, osteocyte, adipocyte, and chondrocyte) and in some cases, into endodermal or ectodermal (neuronal) fates [69]. The scarce expression of the major histocompatibility complex and other co-stimulatory molecules makes MSCs immune-privileged cells. This immune status allows MSCs to be used in an allogenic manner without requiring additional immunosuppression [70].
The International Society for Cellular Therapy has defined MSCs based on their expression of CD90, CD73, CD105, and CD44 while lacking the expression of CD45 and CD31 [71]. These markers help distinguishing MSCs from other cell types and are used to identify and isolate these cells for research and therapeutic purposes.
In adults, several tissues act as MSCs reservoirs [72]. The first type of MSCs to be described were bone marrow-derived mesenchymal stem cells (BM-MSCs) [73], making BM the primary source for MSCs isolation. However, the process of obtaining BM-MSCs involves a highly invasive and painful procedure that requires anesthesia, posing a risk of infection [74]. Alternatively, adipose tissue-derived MSCs (ASCs) can be isolated from biological material generated during liposuction or lipectomy after medical interventions. The natural abundance of MSCs in adipose tissue, which is approximately 500 times higher than in BM, accompanied by easier isolation, has led to an increased utilization of ASCs [75]. Additionally, a recent study has demonstrated that ASCs exhibit lower immunogenicity and transcriptomic heterogeneity compared to BM-MSCs [76]. Apart from adult tissues, MSCs can also be derived from birth-associated tissues, such as the umbilical cord Wharton's jelly (WJ). WJ-MSCs have emerged as an ideal source of MSCs for therapy due to several advantages: they can be harvested painlessly in abundance without causing donor site morbidity, are easy to isolate and culture, possess a high proliferative rate and retain their stemness properties in vitro [77].
In the context of neurodegeneration, there is growing interest and promise in therapies based on MSCs. As of July 2023, 249 clinical studies were found on www.clinicalstrials.gov [78] with the terms “Nervous System Diseases” and “Mesenchymal Stem Cell”, reflecting the potential of these cells in addressing neurodegenerative disorders. A summary of trials focused on AD, PD, MS and stroke identifying the tissue for MSC isolation, donor, route of administration, and pathological target are shown in Table 2.
The main mechanisms exerted for MSCs that contribute to their potential efficacy include:
- Neuroprotective effect: MSCs have demonstrated to have an important neuroprotective effect, as they secrete neurotrophic growth factors such as glial cell-derived neurotrophic factor, VEGF, brain-derived neurotrophic factor and nerve growth factor (NGF) [79], as well as anti-apoptotic factors like Bcl-2 [80]. These factors enable MSCs to promote nervous regeneration, inhibit neuronal apoptosis, and induce endogenous neurogenesis. For example, Oh et al. [81] demonstrated that intravenous injection of MSCs increased hippocampal neurogenesis and differentiation of neural progenitor cells into mature neurons in Aβ-treated mice (AD model) by augmenting the Wnt signaling pathway. Additionally, MSCs may inhibit stroke-associated apoptosis through the Bcl-2 pathway in neurons and astrocytes from rats [82]. Besides, MSCs can transfer healthy mitochondria to damaged cells, protecting neural stem cells from neurotoxic agents. MSCs may transfer this organelle in various ways, including gap junctions, cell fusion, microvesicles, and through tunnelling nanotube formation [83]. Mitochondria play a crucial role in maintaining metabolic homeostasis, and defects such as membrane leakage, electrolyte imbalances, activation of pro-apoptotic pathways, and mitophagy have been implicated in the pathogenesis of various CNS disorders [84]. It has been demonstrated that the ability of MSCs to transfer healthy mitochondria to damaged cells protects neural stem cells from neurotoxic agents [85], and has garnered significant attention in the field of cellular therapy for CNS disorders.
- Immunomodulatory role: MSCs can interact with the immune system and participating in both innate and adaptive immunity due to their significant immunoregulatory functions. This indicates that, depending on the environment in which MSCs are introduced, they can modulate the response. Thus, in an inflammatory environment, MSCs exhibit anti-inflammatory behavior. By expressing different molecules such as transforming growth factor β, indoleamine 2,3-dioxygenase, prostaglandin E2, nitric oxide, and interleukin-10 (IL-10), they can interact with immune cells either through direct cell-to-cell contact or via paracrine activity [86,87,88,89,90]. MSCs can also modulate the macrophage/microglia polarization upregulating the ratio of anti- versus pro-inflammatory responses [91], suppress Th1 and Th17 responses, enhance the maturation of DCs from monocytes, and enhance the Th2 response and the generation of Forkhead Box P3 positive Treg population. Moreover, some studies reported that the secretion of IL-6 by MSCs can inhibit astrocyte apoptosis, increase the neuroprotective population of astrocytes, and reduce neuron damage post-injury [92].
- Regulation of protein clearance: treatment with MSCs has been shown to induce the secretion of neprilysin in vitro and in vivo, improving the endogenous machinery for the degradation of Aβ-plaques and enhancing the clearance of these aggregates [93]. This is particularly relevant as abnormal protein aggregation is one of the major hallmarks of neurodegenerative diseases like PD and AD [94].
4. MSCs as Promising Modulators of the BBB in Neurodegenerative Diseases
As described before, while it remains unclear whether BBB disruption is a cause or a consequence of neuroinflammation, it is undoubtedly a crucial component of CNS pathologies. Unfortunately, the BBB is often viewed as a challenge that hinders the delivery of drugs to the CNS and reduces the efficacy of conventional treatment approaches for neurodegeneration, Therefore, the potential of pharmacological interventions targeting the BBB could represent a promising therapeutic strategy for the neuroinflammatory-mediated neurodegenerative diseases [12,13]. Besides the neuroprotective and immunomodulatory roles of MSCs in neurodegeneration, recent reports have pointed out a beneficial effect of MSCs on modulating the disrupted BBB.
In general, the delivery of MSCs to the CNS is highly diverse, although systemic administration, particularly intravenous infusion, is the preferred method (Table 2). When MSCs are infused intravenously, they transiently accumulate in the lungs for 1-3 hours, followed by a gradual movement to other tissues such as the liver, spleen, kidney, and bone marrow [95]. Interestingly, MSCs have shown the ability to reach brain vessels and adhere to them after 6 hours post-injection, according to Rüster et al. [96], thanks to specific interactions with endothelial cells through adhesion molecules such as P-selectin and VCAM-1/VLA-4. While some reports indicate that after a middle cerebral artery occlusion (MCAO) model of stroke, injected MSCs accumulate in the vessels of the infarcted region [97,98], other studies describe no MSCs were detected in the cerebral parenchyma after an intra-arterial injection in an Alzheimer’s disease mouse model [99]. Therefore, although MSCs are capable of rolling and executing a coordinated extravasation through activated endothelia in other tissues, allowing them to access sites of damage [96], it remains unclear whether this cellular therapy can cross the BBB and exert its function within the brain tissue.
Nevertheless, despite their uncertain ability to cross the BBB and penetrate to the brain, MSCs can directly contact with endothelial cells in the damaged area. This interaction likely enables MSCs to exert their paracrine functions to other cells of the NVU and the BBB from this location (Figure 1). In fact, the scientific community is currently exploring two interesting derivatives of MSCs: genetically modified MSCs and the use of MSC-derived extracellular vesicles (MSC-EVs). Genetically modified MSCs provide the opportunity to enhance the therapeutic effect of MSCs by improving their inherent functions or enabling them to synthesize drugs or active compounds. On the other hand, direct use of the secretome in the form of MSC-EVs improves the penetration through the BBB. In the following sections, we will provide examples of these strategies and the cellular and molecular beneficial effects exerted by MSCs in preclinical models of neurodegenerative diseases characterized by a severe disruption of the BBB (Figure 2, Figure 3 and Figure 4).
4.1. Alzheimer´s disease
Alzheimer´s disease (AD) is a progressive neurodegenerative disease characterized by a cerebrovascular and neuronal dysfunction, resulting in a gradual decrease in cognitive functions [100]. In 2019, it was estimated that 50 million people suffered from AD [101]. The principal pathological hallmarks are extracellular amyloid-β (Aβ) deposition and neuronal accretion of phosphorylated tau-forming neurofibrillary tangles [102]. Aβ is a proteolytic by-product derived from the amyloid precursor protein produced by several cleavages via β- and γ-secretases. There are two main types of AD: early onset AD, which is a rare form affecting <1% of AD cases in subjects <65 years-old and is caused by genetic mutations, and late onset AD, which is the most frequent form, and primarily affects to patients >65 years-old. While genetic factors may potentially contribute to its development, specific mutations that directly cause an increase of proteolytic cleavage in patients have not been observed [103].
4.1.1. Dysfunctional BBB in AD
BBB dysregulation plays a significant role in the pathogenesis of AD, affecting various components of the NVU (Figure 2).
For instance, the presence of Aβ disrupts the organization of TJs and AJs (i.e, occludin, claudin-5 and ZO-1) in BMVECs, leading to compromised barrier activity [104,105]. Additionally, the reduction of GLUT-1 in cerebral microvessels of AD patients contributes to vessel degeneration and further exacerbates the disease [106,107]. Conversely, patients with mild cognitive impairment, which is the precursor of AD, display increased BBB permeability that correlates with high levels of soluble platelet-derived growth factor receptor β in the cerebrospinal fluid, which is indicative of pericyte damage [108]. In fact, the decreased number of pericytes in AD patients may worsen the accumulation of Aβ both in brain parenchyma and blood vessels.
On the other hand, astrocytes in AD patients showed reduced expression of AQP4 in perivascular end-feet and increased levels of astrocytic activation markers [109]. In fact, the accumulation of Aβ in brain leads to pericyte degeneration and loss, dysregulated BM and astrocytic end-feet depolarization with loss of AQP4, which will decrease the Aβ clearance, fueling a pathogenic feedback loop. Thickening of BM [110] and increased collagen levels in these structures [111,112] are also common features in AD patients. MMP2 and MMP9 are significantly activated in the NVU, contributing to BM remodeling in AD [113,114].
Furthermore, transporters of the BBB, such as RAGE, LRP1, or P-gp, are key elements in the regulation of the Aβ clearance. In fact, analysis of microvessels and BMVECs in postmortem AD brains, showed high expression of RAGE, which mediates Aβ entry into the brain [115], and reduced expression of LRP1 and P-gp, involved in the clearance of cerebral Aβ [116,117,118]. The immune system is also compromised in AD and cells like monocytes, lymphocytes or neutrophils can cross the BBB in response to Aβ accumulation and the augmentation of vascular adhesion molecules, contributing to the pathogenesis of AD [119,120,121].
4.1.2. Therapeutic Opportunities for MSCs Targeting BBB in AD
Currently there are no effective treatments to cure or slow AD progression. However, emerging evidence suggest that MSCs therapy could be a promising approach. In general, MSCs transplantation has been found to decrease Aβ deposits and plaques, and tau-related cell death in vivo. The paracrine effects of MSCs stimulate neurogenesis, synaptogenesis, and neuronal differentiation, demonstrating neuroprotective functions. Beside this, their immunoregulatory properties, which modulate microglia/astrocytes activity state, can deactivate neuroinflammatory responses via several transcription factor signaling pathways [122].
The BBB represents a major challenge in treating AD, and different studies have focused on the action of MSCs on cerebral vasculature (Figure 2). For instance, Garcia et al. [123] demonstrated the ability of intracerebrally transplanted MSCs in a 2xTg-AD mouse model to promote neovascularization in the hippocampus. Specifically, they found that MSCs genetically modified to express VEGF, enhanced their therapeutic efficacy in promoting neovascularization. Focusing on transporters involved in AD, Son et al. [124] modified MSCs to express the secreted isoform of RAGE (sRAGE), which inhibits the interaction between RAGE and its ligands, thus preventing the adverse effects of this signaling pathway. To note, when activated by Aβ oligomers, RAGE can lead to cell stress, generation of ROS, and RAGE-mediated inflammation and neurodegeneration. Transplantation of sRAGE-MSCs into 5xFAD transgenic mice reduced the deposition of Aβ, cell death, and inflammation.
In a rat model of cerebral small vessel disease, a pathology characterized by Aβ deposition equivalent to AD, the intravenous infusion of MSCs restored the polarity/distribution of AQP4 to the end-feet of astrocytes, relieving cerebral edema and promoting the clearance of Aβ [125]. In another study, Tachibana et al. [126] implanted mouse MSC-derived pericytes into the brains of APP/PS1 mice and observed a reduction in Aβ levels in the hippocampus, an effect that was mediated by LRP1. Interestingly, a recent study has shown that, in a model of microfluidic BBB-like microvasculature, BM-MSCs emulate more efficiently the function of perivascular pericytes than induced pluripotent stem cell-derived pericytes, leading to greater restoration of TJs and the abluminal BMs [127]. In fact, there are several similarities between MSCs and pericytes. Pericytes express a similar pattern of immunological markers (CD44, CD90, CD73, CD105, and CD45), are self-renewable, and have the capacity to differentiate into nervous cells, mainly glial cells, in vivo [128]. Therefore, MSCs could potentially supply the loss of pericytes in AD. In summary, these results highlight the need for further research in this field, as understanding the role of MSCs in modulating the BBB in the context of AD is essential to develop effective therapies.
4.2. Parkinson´s disease
Parkinson´s disease (PD) is the second most common neurodegenerative disease after AD. This progressive disorder is characterized by the loss of dopamine neurons in the substantia nigra pars compacta (SNpc) and the accumulation of filamentous and oligomeric inclusion bodies (Lewy bodies) composed of misfolded α-synuclein proteins. These structures provoke cellular processes disruption and lead to neuronal degeneration mainly leading to motor dysfunction. Its etiology is still unknown and current treatments are mainly focused on symptoms [129].
4.2.1. Dysfunctional BBB in PD
Although decades ago, NVU was not recognized as an important element of the pathogenesis of PD, it is now well stablished that BBB disruption is associated with PD (Figure 3).
Nevertheless, further research is needed to completely understand the molecular mechanism of BBB disruption in this context [130]. Brain tissue from PD patients showed perivascular deposits of fibrinogen or fibrin, IgG and haemosiderin in specific regions, indicating BBB disruption [131,132,133]. In fact, degeneration of BMVECs and TJs, as well as disorganization of the components of the BM, have been reported in PD brain tissues [132]. In addition, patients with idiopathic PD show genetic mutations that affect NVU components. For example, mutations in leucine-rich repeat kinase 2 in BMVECs lead to increased monocyte attachment in PD patients [134]. Moreover, several PD patients have mutations in the MDR1 gene, which encodes P-gp in BMVECs, resulting in reduced pump function [135].
Angiogenesis is also affected in PD. Although PD patients show augmented vascular density in SNpc, in the proximity of neuronal damage, these new microvessels display impaired maturation processes and altered diameters [136]. While new microvascular architecture may allow the supply of nutrients and cellular debris, it also raises the risk of leakage and infiltration of toxins, drugs, and immune cells, potentially exacerbating the pathology [137,138]. Besides, proinflammatory cytokines secreted by activated immune and glial cells (such as TNF-α, IL-1β, and IL-6) [139] can decrease the expression of ZO-1 and occludin, leading to a disruptive state of TJs and subsequently contributing to BBB breakdown [140].
4.2.2. Therapeutic Opportunities for MSCs Targeting BBB in PD
Over the last decade, preclinical studies investigating the potential of MSCs in the context of PD have been performed (Figure 3). For instance, Chao et al. conducted an extensive study on the effect of MSCs using the preclinical model of PD induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [141]. They found that MSCs, intraperitoneally infused 24 hours after the last MPTP injection, migrated to the SNpc and efficiently rescued dopaminergic TH+ neurons. Moreover, the treatment with MSCs restored BBB integrity, as indicated by the retrieval of TJ-protein expression (claudin-1, claudin-5 and occludin).
Other studies have recently demonstrated that the treatment with MSC-derived exosomes regulates genes associated with the angiogenesis of human BMVECs in vitro, resulting in increased expression of Angpt1 and Flk1, as well as the secretion of the intercellular adhesion molecule 1 (ICAM1) protein. Injecting these exosomes into an MPTP-induced PD model resulted in their homing to the injured brain and a significant recovery from the disease. Additionally, there was an increase in the expression of ICAM1 and CD31 markers in the striatum and SNpc [142]. Conversely, in a LPS injection model into the SNpc, the treatment with MSCs increased the expression of P-gp in endothelial cells and restored BBB integrity [143]. The study suggested that MSCs decreased the proinflammatory activation of microglia and modulated the VEGF-A signalling through astrocytes, leading to an increase the astrocytic end-feet density. This process stabilized the expression of TJ-proteins. These results suggest the relevance of modulating the BBB in PD for developing effective therapies against this debilitating disease.
4.3. Multiple sclerosis
Multiple sclerosis (MS) is an inflammatory neurodegenerative autoimmune disease that affects the CNS. In MS, the immune system generates a complex response against myelin sheaths that wraps nerve axons, eventually leading to inflammation, demyelization, axonal degeneration and ultimately neuronal loss [144]. Immune cells cross the damaged BBB and release proinflammatory cytokines such as TNF-α, IFN-γ or IL-17 which directly attacks myelinating oligodendrocytes [145] or provoke a pro-inflammatory polarization of microglia and astrocytes that finally cause oligodendrocytes loss [146].
There are different types of MS depending on the evolution of the disease: relapsing-remitting MS (RRMS) and primary/secondary progressive MS (PPMS/SPMS). RRMS is characterized by alternating periods of symptoms enhancement (relapses) and partial or complete recovery of the neurologic function (remissions). Conversely, progressive MS (15% of MS patients) is marked by a gradual worsening of symptoms without periods of relapses or remissions. On the contrary, SPMS follows the initial relapsing-remitting course [144].
4.3.1. Dysfunctional BBB in MS
Although the etiology of the disease is not completely understood, a vast body of evidence suggests the importance of BBB disruption in the pathology of MS (Figure 4).
Inflammation affects to several components of the NVU and hampers the physiologic function of numerous transport mechanisms. BBB permeability may be an important early step that is correlated to the initiation of a CNS-specific immune response [147]. Pro-inflammatory mediators such as IL-1β [148], IL-6 [149], TNF-α [150], and chemokine (C-C motif) ligand 2 (CCL2) [151] reduces the expression of TJ and AJ proteins in different in vitro BBB models. Specifically, the expression of toll-like receptors, which play a significant role modulating MS, is significantly increased in BMVECs in response to ROS and TNF-α [152,153]. The expression of other transporters like GLUT-1 [154], LAT1 [155] or P-gp [156], is also affected by the exposure of BMVECs to inflammatory mediators. Moreover, pro-inflammatory molecules can cause pericyte detachment from BMVECs and undergo transformation into phagocytic or fibroblastic-like cells [147]. Regarding the autoimmune component of MS, activation of BMVECs with Th1 cytokines (IL-2, TNF-α, IFN-γ) modulates the BBB phenotype and stimulates the expression of endothelial cell adhesion molecules, such as ICAM-1 and VCAM-1 [157].
4.3.2. Therapeutic Opportunities for MSCs Targeting BBB in MS
The disruption of the BBB in MS allows immune cells to infiltrate into the CNS, contributing to the pathogenesis of MS, as previously mentioned. In vitro studies using a BBB model exposed to TNF-α have shown that treatment with embryonic MSCs modulates barrier permeability, increases the expression of TJ proteins, and decreases the expression of pro-inflammatory chemokines like CCL2 and CXCL12 [158] (Figure 4). In vivo studies have also demonstrated that co-administration of MSCs expressing IFN-β along with minocycline, tames the disruption of the Blood-Spinal Cord Barrier (the functional equivalent of the BBB in the spinal cord) [159]. Recent studies in the MS preclinical model (experimental autoimmune encephalomyelitis) have further shown that MSC transplantation reduced BBB disruption, as evidenced by reduced IgG leakage. Additionally, MSC transplantation led to the adequate expression of TJ-proteins occluding and ZO-1 in BMVECs and the restoration of AQP4 levels in astrocytes [160].
4.4. Stroke
Stroke is a complex and heterogeneous affection that is influenced by genetic predisposition, life habits, and chronic diseases. It is the second leading cause of death and third leading cause of disability worldwide. To note, 60% of the survived patients are affected by cognitive impairment, dementia, or depression. Stroke is classified into two types: hemorrhagic and ischemic, with the latter being the most prevalent (87% of the total). Ischemic stroke is characterized by a sudden cessation of oxygen and blood supply due to thrombus blocking blood flow in the brain vasculature [161].This initiates a rapid and complex cascade of pathophysiology events at genomic, molecular, and cellular levels that may evolve over hours to days and weeks after the onset, including energy failure, acidosis, loss of cell homeostasis, excitotoxicity, oxidative stress, activation of glial cells, inflammation, and disruption of the BBB with infiltration of leukocytes.[162,163]
4.4.1. Dysfunctional BBB in Brain Ischemia.
BBB plays a significant role in the pathophysiology of ischemic stroke, and its dysfunction varies depending on the severity, and duration of the ischemia. Predominantly, during human stroke, BBB presents a continuous opening pattern with biphasic peaks distributed along four stages [164] (Figure 5).
Hyperacute stage is the first phase that evolves within the first 6h after the ischemic onset. Along this phase, the first BBB opening is documented. Due to oxygen and glucose deprivation, Na+ and Ca+ accumulates inside the cells of the NVU, including astrocytes or BMVECs, leading to cytotoxic edema [165], glutamate excitotoxicity [166], oxidative damage associated with ROS generation, and mitochondrial dysfunction [164]. Moreover, MMPs (mainly MMP2) directly degrade TJ proteins and BM components, contributing to BBB leakage [167].
The next stage corresponds the acute phase, which occurs after the first 6h of the onset for a period of 72-96h. The second permeability peak is observed at this stage. Since this point, immune components start to participate more significantly to the stroke pathophysiology. Neutrophils are the predominant peripheral immune cells in the acute post-stroke period [168]. They contribute to the BBB disruption by producing excessive ROS [169], proteases (MMP9, proteinase-3, elastase) [170] and neutrophil gelatinase-associated lipocalin [171], generating neutrophil extracellular traps [172] and secreting inflammatory cytokines (IL-1β, IL-6, IL-8, TNF-α) and chemokines (CCL2, CCL3, and CCL5) [173]. Moreover, P-selectin glycoprotein ligand-1 and macrophage-1 antigen in neutrophils, and their respective receptors, P-selectin and ICAM-1, in BMVECs, are up-regulated shortly after ischemic stroke induced by IL-1β. Hence, paracellular BBB permeability is increased [174,175]. Natural killer (NK) cells are also present during this stage producing IFN-ϒ and ROS [176]. Monocytes and cerebral immune cells, microglia and astrocytes display a pro-inflammatory phenotype which exacerbate BBB breakdown through the secretion of proinflammatory factors (IL-1α, IL-1β, IL-6, TNF-α, IFN-γ, CCL2), MMP9, VEGF, ROS and activation of inducible nitric oxide synthase [162]. Finally, upregulation of MMP-9 is crucial in this phase (24-48h after onset) and it plays a dual role in the proteolytic degradation of the BM components of the BBB. Its capability to digest TJ proteins, enhances the disruption of the BBB [177]. Concomitantly, vascular remodeling is induced by VEGF, leading to the mobilization of progenitor BMVECs, which implies an immature and damaged BBB during this neovascularization process [178].
The subacute phase starts one week after the stoke onset, a timepoint from which BBB begins its recovery process. Monocytes and glial cells shift to anti-inflammatory phenotypes expressing cytokines (IL-10, IL-4, TNF-β) and neurotrophic factors that help prevent inflammation [179]. Moreover, during this phase, angiogenesis plays a vital role in restoring the blood flow and oxygen supply in ischemic tissues. Angiogenesis promotes the proliferation and migration of BMVECs and pericytes, enhances tube formation, branching, and anastomosis, all of which are modulated by the inflammatory microenvironment. VEGF, MMP9 and angiopoietin 2 (Ang-2), as well as its receptor Tie-2, are deeply involved in this process [164]. While these agents may temporarily contribute to BBB leakage, a higher degree of angiogenesis has been linked to increased survival in patients [180] and greater stability of the BBB [181].
Finally, the chronic post stroke phase starts approximately six weeks after the ischemic event, where the BBB is still disrupted but to a lesser extent compared to the previous phases. During this stage, NVU components are restored in order to seal the BBB, with an overexpression and new distribution of TJ proteins. Some of the factors that stabilize the BBB include Ang-1, which maintains BMVECs in a quiescent state and contributes to junction generation, and sphingosine-1 phosphate and activated protein C, which help balance junction proteins and cytoskeleton [182]. Immune cells also shift to anti-inflammatory phenotypes and develop mechanisms to diminish BBB breakdown. For example, anti-inflammatory astrocytes release pentraxin-3, which inhibits VEGF in the ischemic cerebral tissue and specifically supports BBB integrity [183]. At last, neuronal progenitor cells migrate to the ischemic tissue to favor neurogenesis and neuroplasticity and restore BBB components [184,185].
4.4.2. Therapeutic Opportunities for MSCs Targeting BBB in Brain Ischemia.
Several studies have demonstrated the therapeutic effect of MSCs in preclinical models of stroke (Figure 5). Thus, understanding their effect on the BBB has become a topic of great interest in research [186].
For example, MSCs have shown the ability to protect neurons against oxidative damage. A study conducted by Huang et al. [187] showed that MSCs express high levels of antioxidant enzymes from the peroxiredoxin (PRDX) family. In this report, MSCs were able to rescue BBB integrity in an in vitro model of oxidative damage with the bEnd.3 cell line, by reducing TJ degradation and excessive ROS generation. This effect was found to be partially mediated by the secretion of PRDX4. Interestingly, silencing PRDX4 in MSCs attenuated their protective effect on BBB integrity, both in vitro and in the in vivo MCAO ischemic model. Conversely, Cheng et al. [188] investigated the effect of the treatment of MSCs administered 15 minutes after MCAO and found a reduction in IgG leakage in the brain parenchyma, along with the reversal of the TJ-protein gap formation (ZO-1, occludin and claudin-5). This treatment also suppressed MMP-9 upregulation, reduced neuroinflammation, and decreased neutrophil infiltration with downregulation of ICAM-1 expression. In a related study, Liu et al. [189] demonstrated that co-culture of MSCs with HUVECs in an in vitro ischemic-reperfusion model, rescued injured endothelial cells via the generation of TNT-like structures, which was dependent on F-actin polymerization. They further investigated this effect in vivo in a model of MCAO in rats [190], in which the injection of MSCs 24 h after the ischemic induction led to a significant reduction in the infarct volume and higher microvessel densities in the peri-infarct areas. This was attributed to rescuing brain microvasculature through mitochondrial transfer with TNT formation in vivo. Moreover, Zacharek et al. [191] demonstrated that the administration of MSCs 24 h after MCAO in rats reduced BBB leakage and promoted angiogenesis and vascular stabilization in vivo and in vitro. This effect was achieved by increasing endogenous Ang-1/Tie2 and TJ proteins, and promoting the cross-talk between BMVECs and astrocytes.
Finally, different studies have demonstrated that the administration of MSC-derived EVs can lead to a reduction in the infarct volume, improve neurological recovery, and enhanced angiogenesis. These effects are particularly significant when MSCs are exposed to hypoxic conditions prior to EV isolation [192]. These results suggest that it is not necessary to use the whole MSC for the treatment of ischemic stroke [193,194], evidencing MSC-derived EVs as a promising alternative for stroke therapy. Overall, these studies suggest that MSC treatment has a significant impact on BBB integrity and a great potential to modulate microvasculature and reduce the pathologic processes associated to stroke.
5. Challenges and Future Directions
While preclinical studies have shown promising results regarding the therapeutic potential of MSCs in neurodegenerative and neuroinflammatory diseases, the clinical efficacy of using MSCs is still uncertain (Table 2). Recently, Kvistad et al. [195] conducted a meta-analysis to evaluate the safety and efficacy of MSC therapy in various neuroinflammatory diseases, including MS, ischemic stroke, and traumatic spinal cord injury. The study found that the treatment was generally safe and well-tolerated by patients. However, the efficacy results were inconclusive, with no significant improvements, likely due to a considerable placebo effect. This lack of efficacy may be attributed to several factors, including the retention of a significant proportion of the transplanted cells in the lungs rather than reaching the target brain tissue, as well as the absence of a robust mechanism for efficient arrival to the brain. Of note, the therapeutic role for MSCs seems to be not only based on their homing ability and/or to their capability to differentiate into diverse NVU cell types, but mainly dependent on their paracrine secretion of trophic factors, immunomodulatory molecules, microvesicles, microRNAs and mitochondrial transfer. Additionally, the specific source of MSCs, the dosage of transplanted cells, the timing and route of delivery, and the culture and isolation protocols [196] can also impact the therapeutic outcome. Interestingly, the clinical trials using ASCs against neurodegenerative disorders have been significantly increased in the last years (Table 2). These cells are easily available in adults, abundant, and readily to isolate and expand. Additionally, ASCs allow allogenic treatments characterized by many advantages from a therapeutical perspective as they can be used for generating a standardized allogenic cost-effective donor bank and avoid the autologous associated issues like donor-receiver diseases.
Besides, age, gender, genetic traits, and medical history of the donor are important factors that challenge the quality of the MSCs [197]. Therefore, the development of live imaging techniques to track and assess the bioavailability and biodistribution of these cells, the standardization of isolation and expansion protocols for MSCs, the incorporation of clear biomarkers that can accurately reflect the therapeutic effects of MSCs in patients, and the implementation of well-designed randomized controlled studies among others, will minimize variability and improve the reproducibility and reliability of MSCs-based therapies.
To address the challenges and improve the clinical outcomes of MSC therapy in neuroinflammatory pathologies, it is imperative gaining a clearer understanding of the underlying biology and mechanisms of action of MSCs as well as identifying new cellular and/or molecular targets involved in these diseases. This knowledge will serve as a foundation for optimizing the treatment approach and developing targeted strategies that can enhance therapeutic efficacy. On the other hand, understanding the role of the BBB in the pathophysiology of neurodegenerative and neuroinflammatory diseases is crucial for the development of new multi-target-based therapies. The ability of MSCs to restore the integrity and functionality of the disrupted BBB during aging-associated CNS diseases, together with their capacity to modulate the immune response and neurodegeneration makes them an attractive therapeutic option. By focusing on the BBB in the development of MSC-based therapies, we may be able to improve the clinical outcomes of patients suffering from these debilitating conditions.
Author Contributions
Conceptualization, E.G-R., J.C-G., and M.D.; writing—original draft preparation, EG-R., P.V-R. and A.C-M.; writing—review and editing, E.G-R, J.C-G., I.S-M., and M.D.; supervision, E.G-R and R.M.L.; project administration, E.G-R.; funding acquisition, E.G-R and R.M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Spanish Ministry of Science and Innovation (PID2020-119638RB-I00 to E.G-R., PhD fellowship FPU17/02616 to J.C-G., and PRE2021-100172 to P.V-R) and Next Generation EU (Investigo – E-14-2023-0079369) to A.C-M.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
AJs: Adherent-junctions
ASCs: Adipose tissue-derived MSCs
AD: Alzheimer´s disease
BM: Basement membrane
BBB: Blood-brain barrier
BM-MSCs: Bone marrow-derived mesenchymal stem cells
BMVECs: Brain microvascular endothelial cells
CNS: Central nervous system
CCL2: Chemokine (C-C motif) ligand 2
Aβ: Extracellular amyloid-β
ICAM1: Intercellular adhesion molecule 1
IL: Interleukin
MSC: Mesenchymal Stem Cells
MSC-EVs: MSC-derived extracellular vesicles
MCAO: middle cerebral artery occlusion
MS: Multiple sclerosis
NGF: Nerve growth factor
NVU: Neurovascular unit
PD: Parkinson´s disease
PRDX: Peroxiredoxin
PPMS/SPMS: Primary/secondary progressive MS
RRMS: Relapsing-remitting MS
sRAGE: Secreted isoform of RAGE
SNpc: Substantia nigra pars compacta
TJs: Tight junctions
TIMPs: Tissue inhibitor metalloproteinases
VEGF: Vascular endothelial growth factor
WJ: Wharton's jelly
References
- WHO reveals reading cause of death and disability worldwide: 2000-2019. Available online: https://www.who.int/news/item/09-12-2020-who-reveals-leading-causes-of-death-and-disability-worldwide-2000-2019 (accessed on 6 May 2023).
- Banks, W.A.; Reed, M.J.; Logsdon, A.F.; Rhea, E.M.; Erickson, M.A. Healthy aging and the blood-brain barrier. Nat Aging 2021, 1, 243–254. [Google Scholar] [CrossRef]
- Farrall, A.J.; Wardlaw, J.M. Blood-brain barrier: ageing and microvascular disease--systematic review and meta-analysis. Neurobiol Aging. 2009, 30, 337–352. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Elahy, M.; Jackaman, C.; Mamo, J.C.; Lam, V.; Dhaliwal, S.S.; Giles, C.; Nelson, D.; Takechi, R. Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun Ageing 2015, 12, 2. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Martinez, G.R.; Hu, A.; Choi, J.; Keep, R.F.; Andjelkovic, A.V. Decline in sirtuin-1 expression and activity plays a critical role in blood-brain barrier permeability in aging. Neurobiol Dis 2019, 126, 105–116. [Google Scholar] [CrossRef]
- Verheggen, I.C.M.; de Jong, J.J.A.; van Boxtel, M.P.J.; Postma, A.A.; Jansen, J.F.A.; Verhey, F.R.J.; Backes, W.H. Imaging the role of blood-brain barrier disruption in normal cognitive ageing. Geroscience 2020, 42, 1751–1764. [Google Scholar] [CrossRef]
- Verheggen, I.C.M.; de Jong, J.J.A.; van Boxtel, M.P.J.; et al. Increase in blood-brain barrier leakage in healthy, older adults. Geroscience 2020, 42, 1183–1193. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Morin, A.M.; Cipp, L.J.; Haspel, H.C. Glucose transport is reduced in the blood-brain barrier of aged rats. Brain Res 1991, 551, 145–149. [Google Scholar] [CrossRef]
- Banks, W.A.; Moinuddin, A.; Morley, J.E. Regional transport of TNF-alpha across the blood-brain barrier in young ICR and young and aged SAMP8 mice. Neurobiol Aging 2001, 22, 671–676. [Google Scholar] [CrossRef]
- Staff, N.P.; Jones, D.T.; Singer, W. Mesenchymal stromal cell therapies for neurodegenerative diseases. Mayo Clin. Proc. 2019, 94, 892–905. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, A.; Dabrowska, S.; Lukomska, B.; Janowski, M. Mesenchymal stem cells for neurological disorders. Adv Sci (Weinh) 2021, 8, 2002944. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb Perspect Biol 2015, 7, a020412. [Google Scholar] [CrossRef]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol 2006, 1, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Uwamori, H.; Ono, Y.; Yamashita, T.; Arai, K.; Sudo, R. Comparison of organ-specific endothelial cells in terms of microvascular formation and endothelial barrier functions. Microvasc Res 2019, 122, 60–70. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Greene, C.; Munnich, A.; et al. The CLDN5 gene at the blood-brain barrier in health and disease. Fluids Barriers CNS 2023, 20. [Google Scholar] [CrossRef]
- Wong, A. D; Ye, M.; Levy, A. F.; Rothstein, J. D.; Bergles, D. E.; Searson, P. C. The blood-brain barrier: an engineering perspective. Front Neuroeng 2013, 6, 7. [Google Scholar] [CrossRef]
- Oldendorf, W.H.; Cornford, M.E.; Brown, W.J. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol 1977, 1, 409–417. [Google Scholar] [CrossRef]
- Paolinelli, R.; Corada, M.; Orsenigo, F.; Dejana, E. The Molecular Basis of the blood brain barrier differentiation and maintenance. Is it still a mystery? Pharmacol Res 2011, 63, 165–171. [Google Scholar] [CrossRef]
- Artus, C.; Glacial, F.; Ganeshamoorthy, K.; et al. The Wnt/planar cell polarity signaling pathway contributes to the integrity of tight junctions in brain endothelial cells. J Cereb Blood Flow Metab 2014, 34, 433–440. [Google Scholar] [CrossRef]
- Ghandour, M.S.; Langley, O.K.; Varga, V. Immunohistological localization of γ glutamyltranspeptidase in cerebellum at light and electron microscope levels. Neurosci Lett 1980, 20, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Vorbrodt, A.W.; Lossinsky, A.S.; Wisniewski, H.M. Localization of alkaline phosphatase activity in endothelia of developing and mature mouse blood-brain barrier. Dev Neurosci 1986, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Betz, A.L. Sodium transport in capillaries isolated from rat brain. J Neurochem 1983, 41, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- van Tilborg, E.; van Kammen, C.M.; de Theije, C.G.M.; et al. A quantitative method for microstructural analysis of myelinated axons in the injured rodent brain. Sci Rep 2017, 7, 16492. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol Rev 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Badaut, J.; Bix, G.J. Vascular neural network phenotypic transformation after traumatic injury: potential role in long-term sequelae. Transl Stroke Res 2014, 5, 394–406. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 2018, 21, 1318–31. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef] [PubMed]
- Herndon, J.M.; Tome, M.E.; Davis, T.P. Primer on cerebrovascular diseases (Second Edition). In Primer on Cerebrovascular Diseases (Second Edition); Academic Press 2017, 2017; p. Chapter 9-Development and Maintenance of the Blood-Brain Barrier, pag. 53 ISBN 978-0-12-803059-2.
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Fallier-Becker, P.; Sperveslage, J.; Wolburg, H.; Noell, S. The Impact of agrin on the formation of orthogonal arrays of particles in cultured astrocytes from wild-type and agrin-null mice. Brain Res 2011, 1367, 2–12. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Guillamon, M.; Delgado, P.; Ortega, L.; et al. Neuronal TIMP-1 release accompanies astrocytic MMP-9 secretion and enhances astrocyte proliferation induced by beta-amyloid 25-35 fragment. J Neurosci Res 2009, 7, 2115–2125. [Google Scholar] [CrossRef]
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol 2008, 67, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ivars, F.; Anderson, P.; Hallmann, R.; Vestweber, D.; Nilsson, P.; Robenek, H.; Tryggvason, K.; Song, J.; Korpos, E.; et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med 2009, 15, 519–527. [Google Scholar] [CrossRef]
- Korpos, E.; Wu, C.; Song, J.; Hallmann, R.; Sorokin, L. Role of the extracellular matrix in lymphocyte migration. Cell Tissue Res 2010, 339, 47–57. [Google Scholar] [CrossRef]
- Xu, L.; Nirwane, A.; Yao, Y. Basement Membrane and blood–brain barrier. Stroke Vasc Neurol 2018, 4, 78–82. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug Transport across the blood-brain barrier. J Cereb Blood Flow Metab 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Barar, J.; Rafi, M.A.; Pourseif, M.M.; Omidi, Y. Blood-brain barrier transport machineries and targeted therapy of brain diseases. Bioimpacts 2016, 6, 225–248. [Google Scholar] [CrossRef]
- Schulze, C.; Firth, J.A. Immunohistochemical localization of adherens junction components in blood-brain barrier microvessels of the rat. J Cell Sci 1993, 104 Pt 3, 773–782. [Google Scholar] [CrossRef]
- Dejana, E.; Tournier-Lasserve, E.; Weinstein, B.M. The Control of Vascular Integrity by Endothelial Cell Junctions: Molecular Basis and Pathological Implications. Dev Cell 2009, 16, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Tietz, S.; Engelhardt, B. Brain Barriers: Crosstalk between complex tight junctions and adherens junctions. J Cell Biol 2015, 209, 493–506. [Google Scholar] [CrossRef]
- Ueda, H.; Baba, T.; Terada, N.; Kato, Y.; Fujii, Y.; Takayama, I.; Mei, X.; Ohno, S. Immunolocalization of dystrobrevin in the astrocytic endfeet and endothelial cells in the rat cerebellum. Neurosci Lett 2000, 283, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Vestweber, D. The Role of VE-Cadherin in vascular morphogenesis and permeability control. Prog Mol Biol Transl Sci 2013, 116, 119–144. [Google Scholar] [CrossRef]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.; et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef]
- Turowski, P.; Martinelli, R.; Crawford, R.; Wateridge, D.; Papageorgiou, A.-P.; Lampugnani, M.G.; Gamp, A.C.; Vestweber, D.; Adamson, P.; Dejana, E.; et al. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J Cell Sci 2008, 121, 10–1242. [Google Scholar] [CrossRef]
- Dickstein, D.L.; Biron, K.E.; Ujiie, M.; Pfeifer, C.G.; Jeffries, A.R.; Jefferies, W.A. Aβ peptide immunization restores blood-brain barrier integrity in Alzheimer disease. FASEB J 2006, 20, 426–433. [Google Scholar] [CrossRef]
- Wessel, F.; Winderlich, M.; Holm, M.; Frye, M.; Rivera-Galdos, R.; Vockel, M.; Linnepe, R.; Ipe, U.; Stadtmann, A.; Zarbock, A.; et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-Cadherin. Nat Immunol 2014, 15, 223–230. [Google Scholar] [CrossRef]
- Takeichi, M. Dynamic contacts: Rearranging adherens junctions to drive epithelial remodelling. Nat Rev Mol Cell Biol 2014, 15, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.J.; Weber, P.A.; Cao, F.; Chang, H.C.; Lampe, P.; Goldberg, G. The molecular basis of selective permeability of connexins is complex and includes both size and charge. Braz J Med Biol Res 2000, 33, 369–398. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Johnson, A.M.; Keep, R.F.; Andjelkovic, A.V. Junctional proteins of the blood-brain barrier: New insight into function and dysfunction. Tissue Barriers 2016, 4, e1154641. [Google Scholar] [CrossRef] [PubMed]
- Ezan, P.; André, P.; Cisternino, S.; Saubaméa, B.; Boulay, A.-C.; Doutremer, S.; Thomas, M.-A.; Quenech’du, N.; Giaume, C.; Cohen-Salmon, M. Deletion of astroglial connexins weakens the blood–brain barrier. J Cereb Blood Flow Metab 2012, 32, 1457. [Google Scholar] [CrossRef] [PubMed]
- Kotini, M.; Barriga, E.H.; Leslie, J.; et al. Gap junction protein Connexin-43 is a direct transcriptional regulator of N-cadherin in vivo. Nat Commun 2018, 9, 3846. [Google Scholar] [CrossRef]
- Devraj, K.; Klinger, M.E.; Myers, R.L.; Mokashi, A.; Hawkins, R.A.; Simpson, I.A. GLUT-1 Glucose transporters in the blood-brain barrier: Differential phosphorylation. J Neurosci Res 2011, 89, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Pines, G.; Danbolt, N.C.; Bjørås, M.; Zhang, Y.; Bendahan, A.; Eide, L.; Koepsell, H.; Storm-Mathisen, J.; Seeberg, E.; Kanner, B.I. Cloning and expression of a rat brain L-Glutamate transporter. Nature 1992, 360, 464–467. [Google Scholar] [CrossRef]
- Huttunen, J.; Peltokangas, S.; Gynther, M.; Natunen, T.; Hiltunen, M.; Auriola, S.; Ruponen, M.; Vellonen, K. S.; Huttunen, K. M. L-Type Amino Acid Transporter 1 (LAT1/Lat1)-Utilizing prodrugs can improve the delivery of drugs into neurons, astrocytes and microglia. Sci Rep 2019, 9, 12860. [Google Scholar] [CrossRef]
- Mann, G.E.; Yudilevich, D.L.; Sobrevia, L. Regulation of amino acid and glucose transporters in endothelial and smooth muscle cells. Physiol Rev 2003, 83, 183–252. [Google Scholar] [CrossRef]
- O’Kane, R.L.; Martı́nez-López, I.; DeJoseph, M.R.; Viña, J.R.; Hawkins, R.A. Na+-Dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain barrier: A mechanism for glutamate removal. J Biol Chem 1999, 274, 31891–31895. [Google Scholar] [CrossRef]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J Cereb Blood Flow Metab 2007, 27, 1766–1791. [Google Scholar] [CrossRef]
- Vijay, N.; Morris, M.E. Role of monocarboxylate transporters in drug delivery to the brain. Curr Pharm Des 2014, 20, 1487–1498. [Google Scholar] [CrossRef]
- Leclerc, M.; Bourassa, P.; Tremblay, C.; Caron, V.; Sugère, C.; Emond, V.; Bennett, D.A.; Calon, F. Cerebrovascular insulin receptors are defective in Alzheimer’s Disease. Brain 2023, 146, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Lillis, A.P.; Van Duyn, L.B.; Murphy-Ullrich, J.E.; Strickland, D.K. LDL Receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev 2008, 88, 887–918. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates Amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Giridharan, V.V.; Generoso, J.S.; Collodel, A.; Dominguini, D.; Faller, C.J.; Tardin, F.; Bhatti, G.S.; Petronilho, F.; Dal-Pizzol, F.; Barichello, T. Receptor for advanced glycation end products (RAGE) mediates cognitive impairment triggered by Pneumococcal Meningitis. Neurother 2021, 18, 640–653. [Google Scholar] [CrossRef]
- Ahmed Juvale, I.I.; Abdul Hamid, A.A.; Abd Halim, K.B.; Che Has, A.T. P-Glycoprotein: New insights into structure, physiological function, regulation and alterations in disease. Heliyon 2022, 8, e09777. [Google Scholar] [CrossRef]
- Wei, W.; Bodles-Brakhop, A.M.; Barger, S.W. A Role for P-Glycoprotein in clearance of Alzheimer Amyloid β-Peptide from the brain. Curr Alzheimer Res 2016, 13, 615–620. [Google Scholar] [CrossRef]
- Bourassa, P.; Alata, W.; Tremblay, C.; Paris-Robidas, S.; Calon, F. Transferrin receptor-mediated uptake at the blood-brain barrier is not impaired by Alzheimer’s Disease neuropathology. Mol Pharmaceutics 2019, 16. [Google Scholar] [CrossRef]
- Gimble, J.M.; Guilak, F.; Nuttall, M.E.; Sathishkumar, S.; Vidal, M.; Bunnell, B.A. In vitro differentiation potential of mesenchymal stem cells. Transfus Med Hemotherapy 2008, 35, 228–238. [Google Scholar] [CrossRef]
- Ryan, J.M.; Barry, F.P.; Murphy, J.M.; Mahon, B.P. Mesenchymal stem cells avoid allogeneic rejection. J Inflamm 2005, 2, 8. [Google Scholar] [CrossRef]
- Bourin, P.; Bunnell, B.A.; Casteilla, L.; Dominici, M.; Katz, A.J.; March, K.L.; Redl, H.; Rubin, J.P.; Yoshimura, K.; Gimble, J.M. Stromal cells from the adipose tissue-derived stromal vascular fraction and culture expanded adipose tissue-derived stromal/stem cells: A joint statement of the International Federation for Adipose Therapeutics and Science (IFATS) and the International Society for Cellular Therapy (ISCT). Cytotherapy 2013, 15, 641–648. [Google Scholar] [CrossRef]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.-W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Chailakhyan, R.K.; Gerasimov, U.V. Bone marrow osteogenic stem cells: In vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet 1987, 20, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Kasper, C.; Böhm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal 2011, 9, 12. [Google Scholar] [CrossRef]
- Fraser, J.K.; Wulur, I.; Alfonso, Z.; Hedrick, M.H. Fat Tissue: An underappreciated source of stem cells for biotechnology. Trends in Biotechnol 2006, 24, 150–154. [Google Scholar] [CrossRef]
- Liau, L.L.; Ruszymah, B.H.I.; Ng, M.H.; Law, J.X. Characteristics and clinical applications of Wharton’s Jelly-derived mesenchymal stromal cells. Curr Res. Transl Med 2020, 68, 5–16. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Available online: https://classic.clinicaltrials.gov (accessed on 26 July 2023).
- Cova, L.; Armentero, M.-T.; Zennaro, E.; Calzarossa, C.; Bossolasco, P.; Busca, G.; Lambertenghi Deliliers, G.; Polli, E.; Nappi, G.; Silani, V.; et al. Multiple neurogenic and neurorescue effects of human mesenchymal stem cell after transplantation in an experimental model of Parkinson’s Disease. Brain Res 2010, 1311, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, N.; Ong, L.-L.; Nesselmann, C.; Klopsch, C.; Ladilov, Y.; Furlani, D.; Piechaczek, C.; Moebius, J.M.; Lützow, K.; et al. Bcl-2 engineered MSCs inhibited apoptosis and improved heart function. Stem Cells 2007, 25, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Kim, H.N.; Park, H.-J.; Shin, J.Y.; Lee, P.H. Mesenchymal stem cells increase hippocampal neurogenesis and neuronal differentiation by enhancing the Wnt signaling pathway in an Alzheimer’s Disease model. Cell Transplant 2015, 24, 1097–1109. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, S.; Tuazon, J.P.; Lee, J.-Y.; Corey, S.; Kvederis, L.; Kingsbury, C.; Kaneko, Y.; Borlongan, C.V. Neuroprotective effects of human bone marrow mesenchymal stem cells against cerebral ischemia are mediated in part by an anti-apoptotic mechanism. Neural Regen Res 2019, 14, 597. [Google Scholar] [CrossRef]
- Paliwal, S.; Chaudhuri, R.; Agrawal, A.; Mohanty, S. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J Biomed Sci 2018, 25, 31. [Google Scholar] [CrossRef]
- Norat, P.; Soldozy, S.; Sokolowski, J. D.; Gorick, C. M.; Kumar, J. S.; Chae, Y.; Yağmurlu, K.; Prada, F.; Walker, M.; Levitt, M. R.; Price, R. J.; Tvrdik, P.; Kalani, M. Y. S. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. NPJ Regen 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Boukelmoune, N.; Chiu, G.S.; Kavelaars, A.; Heijnen, C.J. Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol Commun 2018, 6, 139. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, Q.; Wang, Z.; Tong, H.; Ma, L.; Zhang, Y.; Shan, F.; Meng, Y.; Yuan, Z. Comparative analysis of human mesenchymal stem cells from fetal-bone marrow, adipose tissue, and Warton’s Jelly as sources of cell immunomodulatory therapy. Hum Vaccines Immunother 2016, 12, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- François, M.; Romieu-Mourez, R.; Li, M.; Galipeau, J. Human MSC Suppression correlates with cytokine induction of indoleamine 2,3-Dioxygenase and bystander M2 macrophage differentiation. Mol Ther 2012, 20, 187–195. [Google Scholar] [CrossRef]
- Li, N.; Hua, J. Interactions between mesenchymal stem cells and the immune system. Cell. Mol. Life Sci. 2017, 74, 2345–2360. [Google Scholar] [CrossRef]
- Sato, K.; Ozaki, K.; Oh, I.; Meguro, A.; Hatanaka, K.; Nagai, T.; Muroi, K.; Ozawa, K. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood 2007, 109, 228–234. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, R.; Wang, Y.; Huang, W.; Hu, B.; Zhu, G.; Zhang, R.; Li, F.; Han, J.; Li, Y. Mesenchymal stem cells enhance microglia M2 polarization and attenuate neuroinflammation through TSG-6. Brain Res 2019, 1724, 146422. [Google Scholar] [CrossRef]
- Gu, Y.; He, M.; Zhou, X.; Liu, J.; Hou, N.; Bin, T.; Zhang, Y.; Li, T.; Chen, J. Endogenous IL-6 of mesenchymal stem cell improves behavioral outcome of hypoxic-ischemic brain damage neonatal rats by supressing apoptosis in astrocyte. Sci Rep 2016, 6, 18587. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Kim, D.H.; Kim, J.H.; Lee, D.; Jeon, H.B.; Kwon, S.-J.; Kim, S.M.; Yoo, Y.J.; Lee, E.H.; Choi, S.J.; et al. Soluble intracellular adhesion molecule-1 secreted by human umbilical cord blood-derived mesenchymal stem cell reduces Amyloid-β plaques. Cell Death Differ. 2012, 19, 680–691. [Google Scholar] [CrossRef]
- Sarkar, S. Protein aggregation in neurodegenerative disorders: A cause or consequence? Adv. Tech. Biol. Med. 2015, 02. [Google Scholar] [CrossRef]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first-pass effect. Stem Cells Dev 2009, 18, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Rüster, B.; Göttig, S.; Ludwig, R.J.; Bistrian, R.; Müller, S.; Seifried, E.; Gille, J.; Henschler, R. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood 2006, 108, 3938–3944. [Google Scholar] [CrossRef]
- Yilmaz, G.; Vital, S.; Yilmaz, C.E.; Stokes, K.Y.; Alexander, J.S.; Granger, D.N. Selectin-mediated recruitment of bone marrow stromal cells in the postischemic cerebral microvasculature. Stroke 2011, 42, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Mitkari, B.; Kerkelä, E.; Nystedt, J.; Korhonen, M.; Mikkonen, V.; Huhtala, T.; Jolkkonen, J. Intra-arterial infusion of human bone marrow-derived mesenchymal stem cells results in transient localization in the brain after cerebral ischemia in rats. Exp Neurol 2013, 239, 158–162. [Google Scholar] [CrossRef]
- Lee, N.K.; Yang, J.; Chang, E.H.; Park, S.E.; Lee, J.; Choi, S.J.; Oh, W.; Chang, J.W.; Na, D.L. Intra-arterially delivered mesenchymal stem cells are not detected in the brain parenchyma in an Alzheimer’s Disease mouse model. PLOS ONE 2016, 11, e0155912. [Google Scholar] [CrossRef]
- Albert, M.S. Changes in cognition. Neurobiol Aging 2011, 32 Suppl 1(0 1), S58–S63. [Google Scholar] [CrossRef]
- Whitaker, K.W.; LaFerla, F.M.; Steinbusch, H.W.M.; Lemere, C.A.; Bovenkamp, D.E. BrightFocus Alzheimer’s fast track 2019. Mol Neurodegener 2019, 14, 48. [Google Scholar] [CrossRef]
- Terry, R. D. Neuropathological changes in Alzheimer disease. Prog Brain Res 1994, 101, 383–390. [Google Scholar] [CrossRef]
- Bertram, L.; Tanzi, R.E. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci 2008, 9, 768–778. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.M.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J.M. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener Dis 2012, 10, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Marco, S.; Skaper, S.D. Amyloid Beta-peptide1-42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci Lett 2006, 401, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Chung, H.C.; Shah, G.N. GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol Aging 1997, 18, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Hooijmans, C.R.; Graven, C.; Dederen, P.J.; Tanila, H.; van Groen, T.; Kiliaan, A.J. Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res 2007, 1181, 93–103. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef]
- de Jong, G.I.; Jansen, A.S.; Horvath, E.; Gispen, W.H.; Luiten, P.G. Nimodipine effects on cerebral microvessels and sciatic nerve in aging rats. Neurobiol Aging 1992, 13, 73–81. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Pax, A.B. Increased collagen content of cerebral microvessels in Alzheimer’s disease. Brain Res 1995, 705, 349–352. [Google Scholar] [CrossRef]
- Christov, A.; Ottman, J.; Hamdheydari, L.; Grammas, P. Structural changes in Alzheimer’s disease brain microvessels. Curr Alzheimer Res 2008, 5, 392–395. [Google Scholar] [CrossRef]
- Jung, S.S.; Zhang, W.; Van Nostrand, W.E. Pathogenic a beta induces the expression and activation of matrix metalloproteinase-2 in human cerebrovascular smooth muscle cells. J Neurochem 2003, 85, 1208–1215. [Google Scholar] [CrossRef]
- Asahina, M.; Yoshiyama, Y.; Hattori, T. Expression of matrix metalloproteinase-9 and urinary-type plasminogen activator in Alzheimer’s disease brain. Clin Neuropathol 2001, 20, 60–63. [Google Scholar] [PubMed]
- Miller, M.C.; Tavares, R.; Johanson, C.E.; Hovanesian, V.; Donahue, J.E.; Gonzalez, L.; Silverberg, G.D.; Stopa, E.G. Hippocampal RAGE immunoreactivity in early and advanced alzheimer’s disease. Brain Res 2008, 1230, 273–280. [Google Scholar] [CrossRef] [PubMed]
- van Assema, D.M.E.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.I.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood-brain barrier p-glycoprotein function in alzheimer’s disease. Brain 2012, 135, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of alzheimer’s amyloid-Ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 2000, 106, 1489–1499. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/Amyloid beta-peptide interaction mediates differential brain efflux of abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J Cereb Blood Flow Metab 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Hohsfield, L.A.; Humpel, C. Migration of blood cells to β-amyloid plaques in Alzheimer’s disease. Exp Gerontol 2015, 65, 8–15. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, Z.; Zhang, J. Immunomodulatory role of mesenchymal stem cells in Alzheimer’s disease. Life Sci 2020, 246, 117405. [Google Scholar] [CrossRef]
- Garcia, K.; Ornellas, F.; Matsumoto, P.; Patti, C.; Mello, L.; Frussa-Filho, R.; Han, S.; Longo, B. Therapeutic effects of the transplantation of VEGF overexpressing bone marrow mesenchymal stem cells in the hippocampus of murine model of Alzheimer’s disease. Front Aging Neurosci 2014, 6, 30. [Google Scholar] [CrossRef]
- Son, M.; Oh, S.; Park, H.; Ahn, H.; Choi, J.; Kim, H.; Lee, H.S.; Lee, S.; Park, H.-J.; Kim, S.U.; et al. Protection against RAGE-mediated neuronal cell death by SRAGE-secreting human mesenchymal stem cells in 5xFAD transgenic mouse model. Brain Behav Immun 2017, 66, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. lu; Ouyang, F. bing; Hu, L. ting; Sun, P.; Yang, J.; Sun, Y. jing; Liao, M. shi; Lan, L. fang; Pei, Z.; Fan, Y. hua Mesenchymal stem cells improve cognitive impairment and reduce Aβ deposition via promoting AQP4 polarity and relieving neuroinflammation in rats with chronic hypertension-induced cerebral small-vessel disease. Front Aging Neurosci 2022, 14, 883503. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Yamazaki, Y.; Liu, C.-C.; Bu, G.; Kanekiyo, T. Pericyte implantation in the brain enhances cerebral blood flow and reduces amyloid-β pathology in amyloid model mice. Exp Neurol 2018, 300, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, S.; Lim, J.; Choi, H.; Kang, H.; Jeon, N.L.; Son, Y. Human bone marrow-derived mesenchymal stem cells play a role as a vascular pericyte in the reconstruction of human BBB on the angiogenesis microfluidic chip. Biomaterials 2021, 279, 121210. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.G.; Shinojima, N.; Hossain, A.; Gumin, J.; Yong, R.L.; Colman, H.; Marini, F.; Andreeff, M.; Lang, F.F. Isolation and perivascular localization of mesenchymal stem fells from mouse brain. Neurosurgery 2010, 67, 711. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat Rev Dis Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Al-Bachari, S.; Naish, J.H.; Parker, G.J.M.; Emsley, H.C.A.; Parkes, L.M. Blood–brain barrier leakage is increased in Parkinson’s disease. Front Physiol 2020, 11. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J Cereb Blood Flow Metab 2015, 35, 747–750. [Google Scholar] [CrossRef]
- Pienaar, I.S.; Lee, C.H.; Elson, J.L.; McGuinness, L.; Gentleman, S.M.; Kalaria, R.N.; Dexter, D.T. Deep-brain stimulation associates with improved microvascular integrity in the subthalamic nucleus in Parkinson’s disease. Neurobiol Dis 2015, 74, 392–405. [Google Scholar] [CrossRef]
- Loeffler, D.A.; Connor, J.R.; Juneau, P.L.; Snyder, B.S.; Kanaley, L.; DeMaggio, A.J.; Nguyen, H.; Brickman, C.M.; LeWitt, P.A. Transferrin and iron in normal, Alzheimer’s disease, and Parkinson’s disease brain regions. J Neurochem 1995, 65, 710–724. [Google Scholar] [CrossRef]
- Li, J.Q.; Tan, L.; Yu, J.T. The role of the LRRK2 gene in parkinsonism. Mol Neurodegener 2014, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Droździk, M.; Białecka, M.; Myśliwiec, K.; Honczarenko, K.; Stankiewicz, J.; Sych, Z. Polymorphism in the p-glycoprotein drug transporter MDR1 gene: A possible link between environmental and genetic factors in parkinson’s disease. Pharmacogenetics 2003, 13, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, B.A.; Bonnet, A.M.; Agid, Y.; Hirsch, E.C. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet 1999, 353, 981–982. [Google Scholar] [CrossRef] [PubMed]
- Barcia, C.; Emborg, M.E.; Hirsch, E.C.; Herrero, M.-T. Blood vessels and parkinsonism. Front Biosci 2004, 9, 277–282. [Google Scholar] [CrossRef]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: implications for drug therapy. Cell Transplant 2007, 16, 285–299. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J Neuroinflammation 2004, 1, 14. [Google Scholar] [CrossRef]
- Wong, D.; Dorovini-Zis, K.; Vincent, S.R. Cytokines, nitric oxide, and CGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol 2004, 190, 446–455. [Google Scholar] [CrossRef]
- Chao, Y.X.; He, B.P.; Tay, S.S.W. Mesenchymal stem cell transplantation attenuates blood brain barrier damage and neuroinflammation and protects dopaminergic neurons against MPTP toxicity in the substantia nigra in a model of Parkinson’s disease. J Neuroimmunol 2009, 216, 39–50. [Google Scholar] [CrossRef]
- Xue, C.; Li, X.; Ba, L.; Zhang, M.; Yang, Y.; Gao, Y.; Sun, Z.; Han, Q.; Zhao, R.C. MSC-derived exosomes can enhance the angiogenesis of human brain MECs and show therapeutic potential in a mouse model of Parkinson’s disease. Aging Dis 2021, 12, 1211–1222. [Google Scholar] [CrossRef]
- Park, H.J.; Shin, J.Y.; Kim, H.N.; Oh, S.H.; Song, S.K.; Lee, P.H. Mesenchymal stem cells stabilize the blood–brain barrier through regulation of astrocytes. Stem Cell Res Ther 2015, 6, 187. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N Engl J Med 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Pathology and disease mechanisms in different stages of multiple sclerosis. J Neurol Sci 2013, 333, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, T.G.; Romanescu, C.; Popescu, B.O. The blood–brain barrier—A key player in multiple sclerosis disease mechanisms. Biomolecules 2022, 12, 538. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, K.; Zhang, X.; Zhang, J.; Xu, B. ATP induces disruption of tight junction proteins via IL-1 beta-dependent MMP-9 activation of human blood-brain barrier in vitro. Neural Plast 2016, 2016, 8928530. [Google Scholar] [CrossRef]
- Desai, T.R.; Leeper, N.J.; Hynes, K.L.; Gewertz, B.L. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J Surg Res 2002, 104, 118–123. [Google Scholar] [CrossRef]
- Förster, C.; Burek, M.; Romero, I.A.; Weksler, B.; Couraud, P.O.; Drenckhahn, D. Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol 2008, 586, 1937–49. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Keep, R.F.; Wang, M.M.; Jankovic, I.; Andjelkovic, A.V. ; Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem 2009, 284, 19053–19066. [Google Scholar] [CrossRef]
- Nagyoszi, P.; Wilhelm, I.; Farkas, A.E.; Fazakas, C.; Dung, N.T.K.; Haskó, J.; Krizbai, I.A. Expression and regulation of Toll-like receptors in cerebral endothelial cells. Neurochem Int 2010, 57, 556–564. [Google Scholar] [CrossRef]
- Racke, M.K.; Drew, P.D. Toll-like receptors in multiple sclerosis. Curr Top Microbiol Immunol 2009, 336, 155–168. [Google Scholar] [CrossRef]
- Sheikh, M. H.; Henson, S. M.; Loiola, R. A.; Mercurio, S.; Colamatteo, A.; Maniscalco, G. T.; De Rosa, V.; McArthur, S.; Solito, E. Immuno-metabolic impact of the multiple sclerosis patients' sera on endothelial cells of the blood-brain barrier. J Neuroinflammation 2020, 17, 153. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, G.; Mohácsik, P.; Balkhi, M.Y.; Gereben, B.; Lechan, R.M. Endotoxin-induced inflammation down-regulates l-type amino acid transporter 1 (LAT1) expression at the blood–brain barrier of male rats and mice. Fluids Barriers CNS 2015, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Goralski, K.B. A Critical Overview of the Influence of inflammation and infection on p-glycoprotein expression and activity in the brain. Expert Opin Drug Metab Toxicol 2008, 4, 1245–1264. [Google Scholar] [CrossRef]
- Gelati, M.; Corsini, E.; Dufour, A.; Massa, G.; Giombini, S.; Solero, C.L.; Salmaggi, A. High-dose methylprednisolone reduces cytokine-induced adhesion molecules on human brain endothelium. Can. J Neurol. Sci. 2000, 27, 241–244. [Google Scholar] [CrossRef]
- Ge, S.; Jiang, X.; Paul, D.; Song, L.; Wang, X.; Pachter, J.S. Human ES-derived MSCs correct TNF-α-mediated alterations in a blood–brain barrier model. Fluids Barriers CNS 2019, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Heon Ryu, C.; Jun, J.A.; Kim, S.M.; Jeong, C.H.; Jeun, S.-S. Interferon β-secreting mesenchymal stem cells combined with minocycline attenuate experimental autoimmune encephalomyelitis. J Neuroimmunol 2014, 274, 20–27. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, Y.; Du, B.; Wang, Y.; Yang, G.-Y.; Bi, X. Mesenchymal stem cells attenuated blood-brain barrier disruption via downregulation of aquaporin-4 expression in EAE mice. Mol Neurobiol 2020, 57, 3891–3901. [Google Scholar] [CrossRef]
- Feigin, V.L.; Brainin, M.; Norrving, B.; Martins, S.; Sacco, R.L.; Hacke, W.; Fisher, M.; Pandian, J.; Lindsay, P. World Stroke Organization (WSO): global stroke fact sheet 2022. Int J Stroke 2022, 17, 18–29. [Google Scholar] [CrossRef]
- Peng, L.; Hu, G.; Yao, Q.; et al. Microglia autophagy in ischemic stroke: A double-edged sword. Front Immunol 2022, 13, 1013311. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Bernardo-Castro, S.; Sousa, J.A.; Brás, A.; Cecília, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of blood-brain barrier permeability throughout the different stages of ischemic stroke and its implication on hemorrhagic transformation and recovery. Front Neurol 2020, 11, 594672. [Google Scholar] [CrossRef]
- Magee, P.N.; Stoner, H.B.; Barnes, J.M. The experimental production in oedema in the central nervous system of the rat by triethyltin compounds. J Pathol 1957, 73, 107–124. [Google Scholar] [CrossRef]
- Liebeskind, D.S.; Jüttler, E.; Shapovalov, Y.; Yegin, A.; Landen, J.; Jauch, E.C. Cerebral edema associated with large hemispheric infarction. Stroke 2019, 50, 2619–2625. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rosenberg, G.A. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Price, C.J.; Menon, D.K.; Peters, A.M.; Ballinger, J.R.; Barber, R.W.; Balan, K.K.; Lynch, A.; Xuereb, J.H.; Fryer, T.; Guadagno, J.V.; Warburton, E.A. Cerebral neutrophil recruitment, histology, and outcome in acute ischemic stroke: an imaging-based study. Stroke 2004, 35, 1659–1664. [Google Scholar] [CrossRef]
- Krizbai, I.A.; Bauer, H.; Bresgen, N.; Eckl, P.M.; Farkas, A.; Szatmári, E.; Traweger, A.; Wejksza, K.; Bauer, H.-C. Effect of oxidative stress on the junctional proteins of cultured cerebral endothelial cells. Cell Mol Neurobiol 2005, 25, 129–139. [Google Scholar] [CrossRef]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front Cell Neurosci 2016, 10, 56. [Google Scholar] [CrossRef]
- Qiu, Y.M.; Zhang, C.L.; Chen, A.Q.; Wang, H.L.; Zhou, Y.F.; Li, Y.N.; Hu, B. Immune cells in the BBB disruption after acute ischemic stroke: targets for immune therapy? Front Immunol 2021, 12, 678744. [Google Scholar] [CrossRef]
- Kang, L.; Yu, H.; Yang, X.; Zhu, Y.; Bai, X.; Wang, R.; Cao, Y.; Xu, H.; Luo, H.; Lu, L.; Shi, M.J.; Tian, Y.; Fan, W.; Zhao, B.Q. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat Commun 2020, 11, 2488. [Google Scholar] [CrossRef]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab 2015, 35, 888–901. [Google Scholar] [CrossRef]
- Kataoka, H.; Kim, S.W.; Plesnila, N. Leukocyte-endothelium interactions during permanent focal cerebral ischemia in mice. J Cereb Blood Flow Metab 2004, 24, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.W.; Chang, W.N.; Shaw, C.F.; Jan, C.R.; Huang, C.R.; Chen, S.D.; Chuang, Y.C.; Lee, L.H.; Lu, C.H. The value of leukocyte adhesion molecules in patients after ischemic stroke. J Neurol 2009, 256, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Liu, Q.; Wu, W.; Yin, J.-X.; Bai, X.-F.; Shen, R.; Wang, Y.; Chen, J.; La Cava, A.; Poursine-Laurent, J.; et al. Ischemic neurons recruit natural killer cells that accelerate brain infarction. PNAS USA 2014, 111, 2704–2709. [Google Scholar] [CrossRef]
- Kurzepa, J.; Kurzepa, J.; Golab, P.; Czerska, S.; Bielewicz, J. The significance of matrix metalloproteinase (MMP)-2 and MMP-9 in the ischemic stroke. Int J Neurosci 2014, 124, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Stabile, E.; Kinnaird, T.; Shou, M.; Devaney, J.M.; Epstein, S.E.; Burnett, M.S. Temporal patterns of gene expression after acute hindlimb ischemia in mice: insights into the genomic program for collateral vessel development. J Am Coll Cardiol 2004, 43, 474–82. [Google Scholar] [CrossRef]
- Liu, R.; Pan, M.-X.; Tang, J.-C.; Zhang, Y.; Liao, H.-B.; Zhuang, Y.; Zhao, D.; Wan, Q. Role of neuroinflammation in ischemic stroke. Neurol Neuroimmunol Neuroinflamm 2017, 4, 158–166. [Google Scholar] [CrossRef]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Kumar, S.; Wang, J.M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 1994, 25, 1794–1798. [Google Scholar] [CrossRef]
- Navarro-Sobrino, M.; Rosell, A.; Hernández-Guillamon, M.; Penalba, A.; Boada, C.; Domingues-Montanari, S.; Ribó, M.; Alvarez-Sabín, J.; Montaner, J. A large screening of angiogenesis biomarkers and their association with neurological outcome after ischemic stroke. Atherosclerosis 2011, 216, 205–211. [Google Scholar] [CrossRef]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Shindo, A.; Maki, T.; Mandeville, E.T.; Liang, A.C.; Egawa, N.; Itoh, K.; Itoh, N.; Borlongan, M.; Holder, J.C.; Chuang, T.T.; et al. Astrocyte-derived pentraxin 3 supports blood-brain barrier integrity under acute phase of stroke. Stroke 2016, 47, 1094–1100. [Google Scholar] [CrossRef]
- Lin, R.; Cai, J.; Nathan, C.; Wei, X.; Schleidt, S.; Rosenwasser, R.; Iacovitti, L. Neurogenesis is enhanced by stroke in multiple new stem cell niches along the ventricular system at sites of high BBB permeability. Neurobiol Dis 2015, 74, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.L.; Chopp, M.; Roberts, C.; Liu, X.; Wei, M.; Nejad-Davarani, SP.; Wang, X.; Zhang, Z.G. Stroke increases neural stem cells and angiogenesis in the neurogenic niche of the adult mouse. PLoS One 2014, 9, 12e113972. [Google Scholar] [CrossRef]
- Do, P.T.; Wu, C.-C.; Chiang, Y.-H.; Hu, C.-J.; Chen, K.-Y. Mesenchymal stem/stromal cell therapy in blood–brain barrier preservation following ischemia: molecular mechanisms and prospects. Int J Mol Sci 2021, 22, 10045. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, J.; Cai, J.; Qiu, Y.; Zheng, H.; Lai, X.; Sui, X.; Wang, Y.; Lu, Q.; Zhang, Y.; et al. Targeted homing of CCR2-overexpressing mesenchymal stromal cells to ischemic brain enhances post-stroke recovery partially through PRDX4-mediated blood-brain barrier preservation. Theranostics 2018, 8, 5929–5944. [Google Scholar] [CrossRef]
- Cheng, Z.; Wang, L.; Qu, M.; Liang, H.; Li, W.; Li, Y.; Deng, L.; Zhang, Z.; Yang, G.-Y. Mesenchymal stem cells attenuate blood-brain barrier leakage after cerebral ischemia in mice. J Neuroinflammation 2018, 15, 135. [Google Scholar] [CrossRef]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia–reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res 2014, 92, 10–18. [Google Scholar] [CrossRef]
- Liu, K.; Guo, L.; Zhou, Z.; Pan, M.; Yan, C. Mesenchymal stem cells transfer mitochondria into cerebral microvasculature and promote recovery from ischemic stroke. Microvasc Res 2019, 123, 74–80. [Google Scholar] [CrossRef]
- Zacharek, A.; Chen, J.; Li, A.; Cui, X.; Li, Y.; Roberts, C.; Feng, Y.; Gao, Q.; Chopp, M. Angiopoietin1/TIE2 and VEGF/FLK1 induced by MSC treatment amplifies angiogenesis and vascular stabilization after stroke. J Cereb Blood Flow Metab 2007, 27, 1684–1691. [Google Scholar] [CrossRef]
- Gregorius, J.; Wang, C.; Stambouli, O.; Hussner, T.; Qi, Y.; Tertel, T.; Börger, V.; Mohamud Yusuf, A.; Hagemann, N.; Yin, D.; et al. Small extracellular vesicles obtained from hypoxic mesenchymal stromal cells have unique characteristics that promote cerebral angiogenesis, brain remodeling and neurological recovery after focal cerebral ischemia in mice. Basic Res Cardiol 2021, 116, 40. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Hu, X.; Li, L.; Fang, Y.; Yang, Y.; Gu, J.; Xu, J.; Chu, L. Exosomes derived from bone marrow mesenchymal stem cells promote angiogenesis in ischemic stroke mice via upregulation of miR-21-5p. Biomolecules 2022, 12, 883. [Google Scholar] [CrossRef]
- Xia, Y.; Ling, X.; Hu, G.; Zhu, Q.; Zhang, J.; Li, Q.; Zhao, B.; Wang, Y.; Deng, Z. Small extracellular vesicles secreted by human iPSC-derived MSC enhance angiogenesis through inhibiting STAT3-dependent autophagy in ischemic stroke. Stem Cell Res Ther 2020, 11, 313. [Google Scholar] [CrossRef] [PubMed]
- Kvistad, C.E.; Kråkenes, T.; Gjerde, C.; Mustafa, K.; Rekand, T.; Bø, L. Safety and clinical efficacy of mesenchymal stem cell treatment in traumatic spinal cord injury, multiple sclerosis and ischemic stroke – A systematic review and meta-analysis. Front Neurol 2022, 13, 891514. [Google Scholar] [CrossRef] [PubMed]
- Regmi, S.; Liu, D.D.; Shen, M.; Kevadiya, B.D.; Ganguly, A.; Primavera, R.; Chetty, S.; Yarani, R.; Thakor, A.S. Mesenchymal stromal cells for the treatment of Alzheimer’s disease: Strategies and Limitations. Front Mol Neurosci 2022, 15, 1011225. [Google Scholar] [CrossRef]
- Galipeau, J.; Sensébé, L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of the blood-brain barrier (BBB). It is located within the neurovascular unit (NVU) and constituted by endothelial cells interconnected by tight-junctions (TJ) and adherens-junctions (AJs), and neighboring cells, such as pericytes, astrocytes, neurons, and microglia. CNS: central nervous system; BMVECs: brain microvascular endothelial cells; ZO: zonula occludens; PECAM: Platelet endothelial cell adhesion molecule; VE-cadherin: vascular endothelial cadherin.
Figure 1.
Schematic representation of the blood-brain barrier (BBB). It is located within the neurovascular unit (NVU) and constituted by endothelial cells interconnected by tight-junctions (TJ) and adherens-junctions (AJs), and neighboring cells, such as pericytes, astrocytes, neurons, and microglia. CNS: central nervous system; BMVECs: brain microvascular endothelial cells; ZO: zonula occludens; PECAM: Platelet endothelial cell adhesion molecule; VE-cadherin: vascular endothelial cadherin.
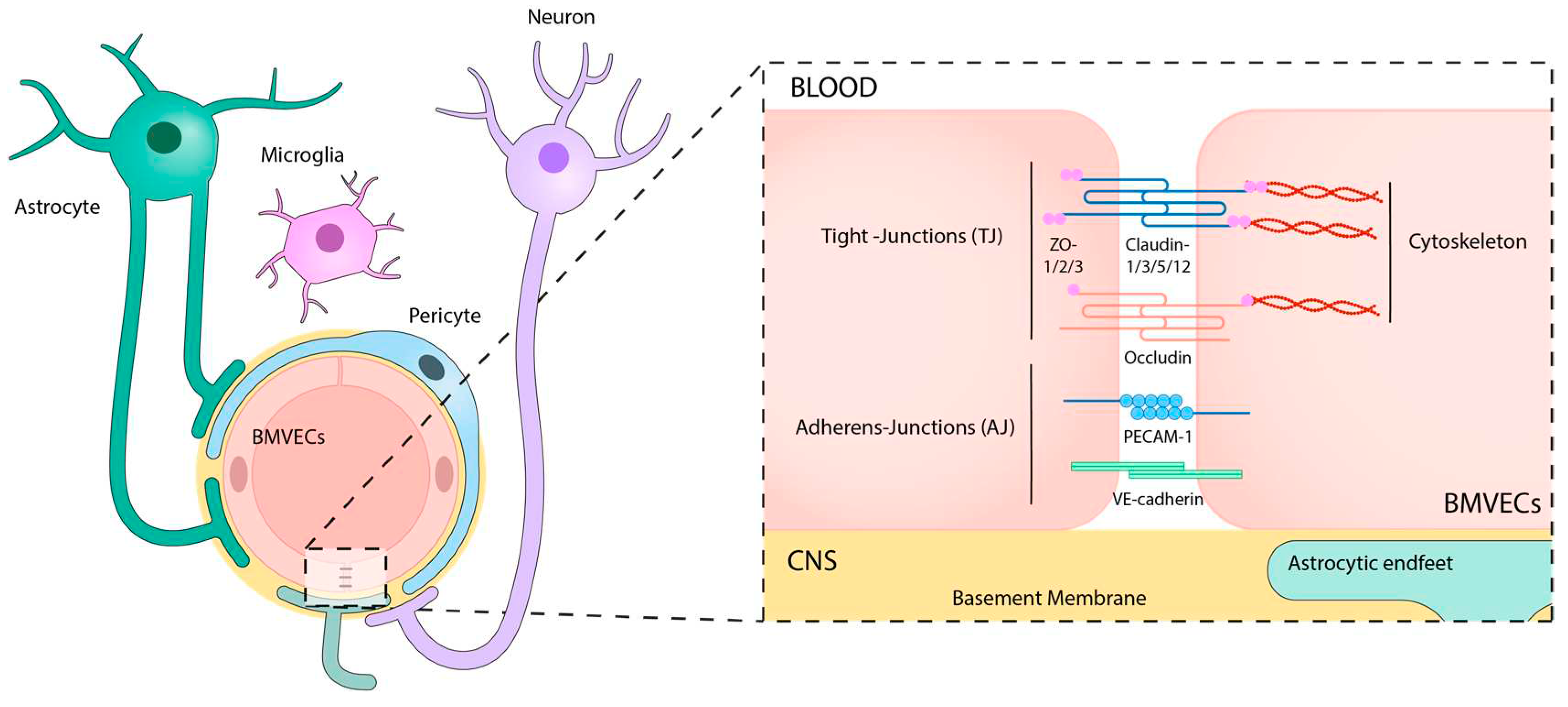
Figure 2.
Blood-brain barrier (BBB) disruption in Alzheimer’s disease (AD) pathogenesis. The breakdown of the BBB in AD is characterized by a cascade of events including loss of TJ integrity, disorganization of the basement membrane (BM) and extracellular matrix, pericyte degeneration and detachment, activation of glial cells, astrocyte depolarization, and alteration of BBB transporter expression (LRP1, P-gp and GLUT1 are reduced, whereas RAGE expression is increased). These modifications may lead directly or indirectly to the disturbed Amyloid β (Aβ) clearance in the neurovascular unit (NVU), contributing to further neuronal toxicity and AD pathogenesis. Treatment with MSCs restores BBB integrity by stabilizing TJ, BM and extracellular matrix (ECM) remodeling and reduces the neuroinflammation and Aβ accumulation. CNS: central nervous system; BMVECs: brain microvascular endothelial cells; BM: basement membrane; RAGE: receptor for advanced glycation endproducts; LRP1: Low density lipoprotein receptor-related protein 1; P-gp: permeability glycoprotein; GLUT1: glucose transporter 1; MMP: metalloproteinases; TJ: tight-junctions; AQP: aquaporin.
Figure 2.
Blood-brain barrier (BBB) disruption in Alzheimer’s disease (AD) pathogenesis. The breakdown of the BBB in AD is characterized by a cascade of events including loss of TJ integrity, disorganization of the basement membrane (BM) and extracellular matrix, pericyte degeneration and detachment, activation of glial cells, astrocyte depolarization, and alteration of BBB transporter expression (LRP1, P-gp and GLUT1 are reduced, whereas RAGE expression is increased). These modifications may lead directly or indirectly to the disturbed Amyloid β (Aβ) clearance in the neurovascular unit (NVU), contributing to further neuronal toxicity and AD pathogenesis. Treatment with MSCs restores BBB integrity by stabilizing TJ, BM and extracellular matrix (ECM) remodeling and reduces the neuroinflammation and Aβ accumulation. CNS: central nervous system; BMVECs: brain microvascular endothelial cells; BM: basement membrane; RAGE: receptor for advanced glycation endproducts; LRP1: Low density lipoprotein receptor-related protein 1; P-gp: permeability glycoprotein; GLUT1: glucose transporter 1; MMP: metalloproteinases; TJ: tight-junctions; AQP: aquaporin.
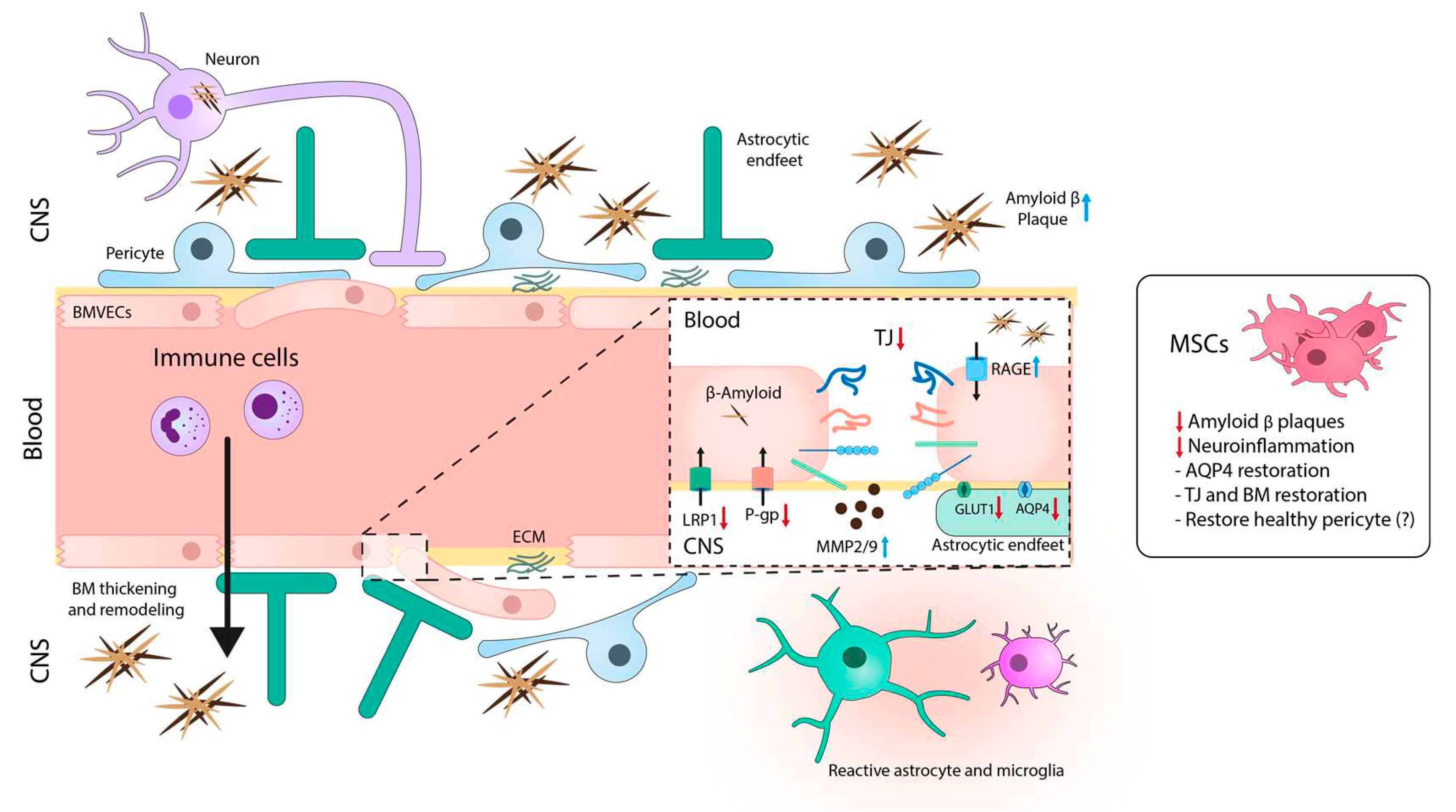
Figure 3.
Schematic image illustrating the blood-brain barrier dysfunction in PD. Alterations in BMVECs are characterized by decreased TJ and AJ proteins and disorganization of the basement membrane. In addition to the reduction of astrocytic end-feet and pericyte loss, these changes can lead to the BBB breakdown and subsequent accumulation of fibrinogen, thrombin, and hemosiderin, which together with α-synuclein can activate glial cells and injure dopaminergic neurons. Treatment with MSCs recovers the BBB integrity by stabilizing the TJ structure and decreases the production and accumulation of neuroinflammatory and neurotoxic mediators. CNS: central nervous system; BMVECs: brain microvascular endothelial cells; TJ: tight-junctions; P-gp: permeability glycoprotein; Ig: immunoglobulin.
Figure 3.
Schematic image illustrating the blood-brain barrier dysfunction in PD. Alterations in BMVECs are characterized by decreased TJ and AJ proteins and disorganization of the basement membrane. In addition to the reduction of astrocytic end-feet and pericyte loss, these changes can lead to the BBB breakdown and subsequent accumulation of fibrinogen, thrombin, and hemosiderin, which together with α-synuclein can activate glial cells and injure dopaminergic neurons. Treatment with MSCs recovers the BBB integrity by stabilizing the TJ structure and decreases the production and accumulation of neuroinflammatory and neurotoxic mediators. CNS: central nervous system; BMVECs: brain microvascular endothelial cells; TJ: tight-junctions; P-gp: permeability glycoprotein; Ig: immunoglobulin.
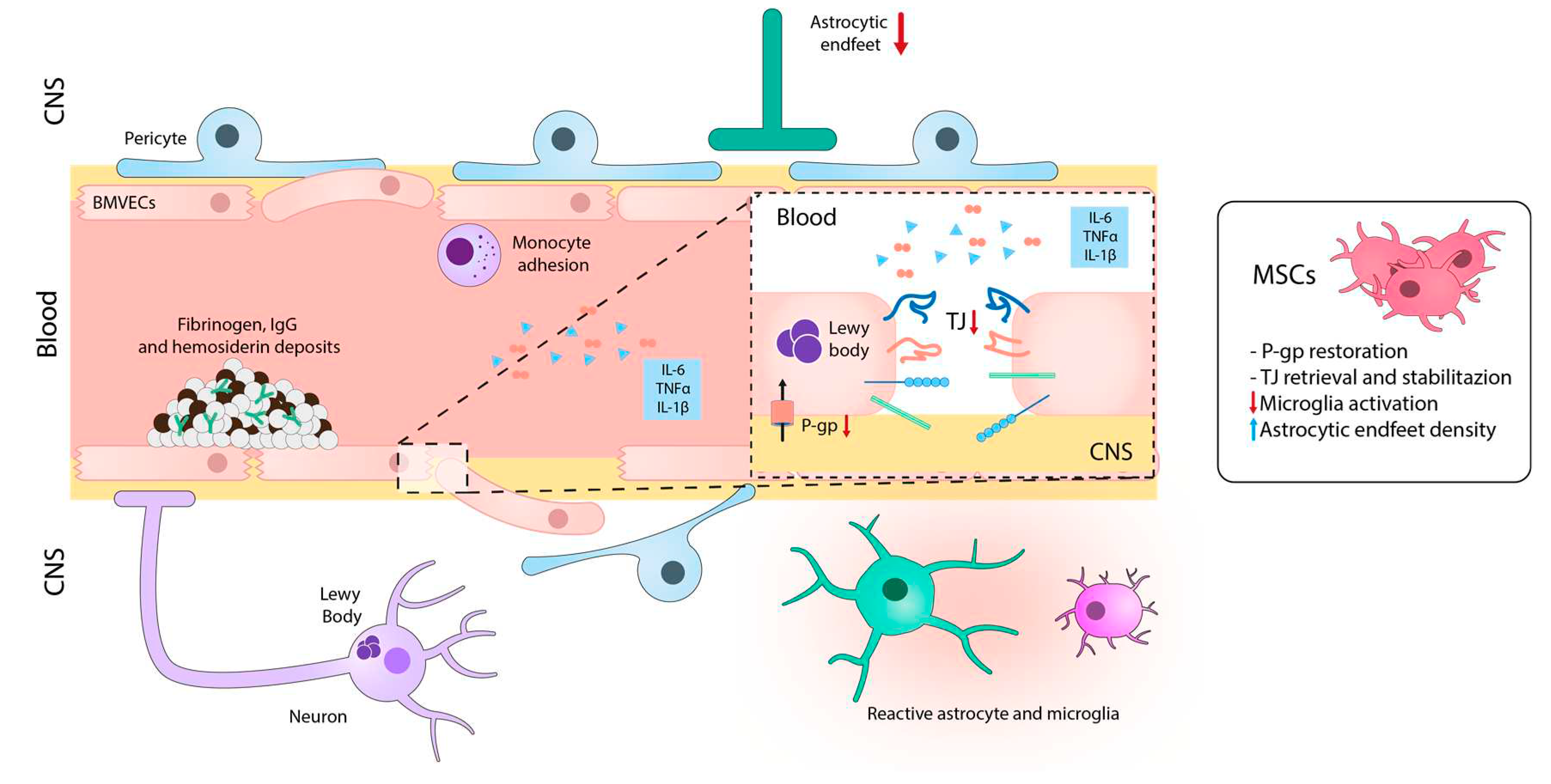
Figure 4.
Blood-brain barrier alterations in multiple sclerosis. Hallmark features of MS development include an early BBB breakdown accompanied by reduced TJ expression, endothelial degeneration, leukocyte infiltration and neuroimmune activation. Treatment with MSCs exerts immunomodulatory and neuroprotective roles in MS that in turn involve the stabilization of the BBB integrity. CNS: central nervous system; ECM: extracellular matrix; BMVECs: brain microvascular endothelial cells; ECAMs: endothelial cell adhesion molecules; ICAM: intercellular adhesion molecules; TJ: tight-junctions; TLR: Toll-like receptor; GLUT1: glucose transporter; Ig: immunoglobulin.
Figure 4.
Blood-brain barrier alterations in multiple sclerosis. Hallmark features of MS development include an early BBB breakdown accompanied by reduced TJ expression, endothelial degeneration, leukocyte infiltration and neuroimmune activation. Treatment with MSCs exerts immunomodulatory and neuroprotective roles in MS that in turn involve the stabilization of the BBB integrity. CNS: central nervous system; ECM: extracellular matrix; BMVECs: brain microvascular endothelial cells; ECAMs: endothelial cell adhesion molecules; ICAM: intercellular adhesion molecules; TJ: tight-junctions; TLR: Toll-like receptor; GLUT1: glucose transporter; Ig: immunoglobulin.
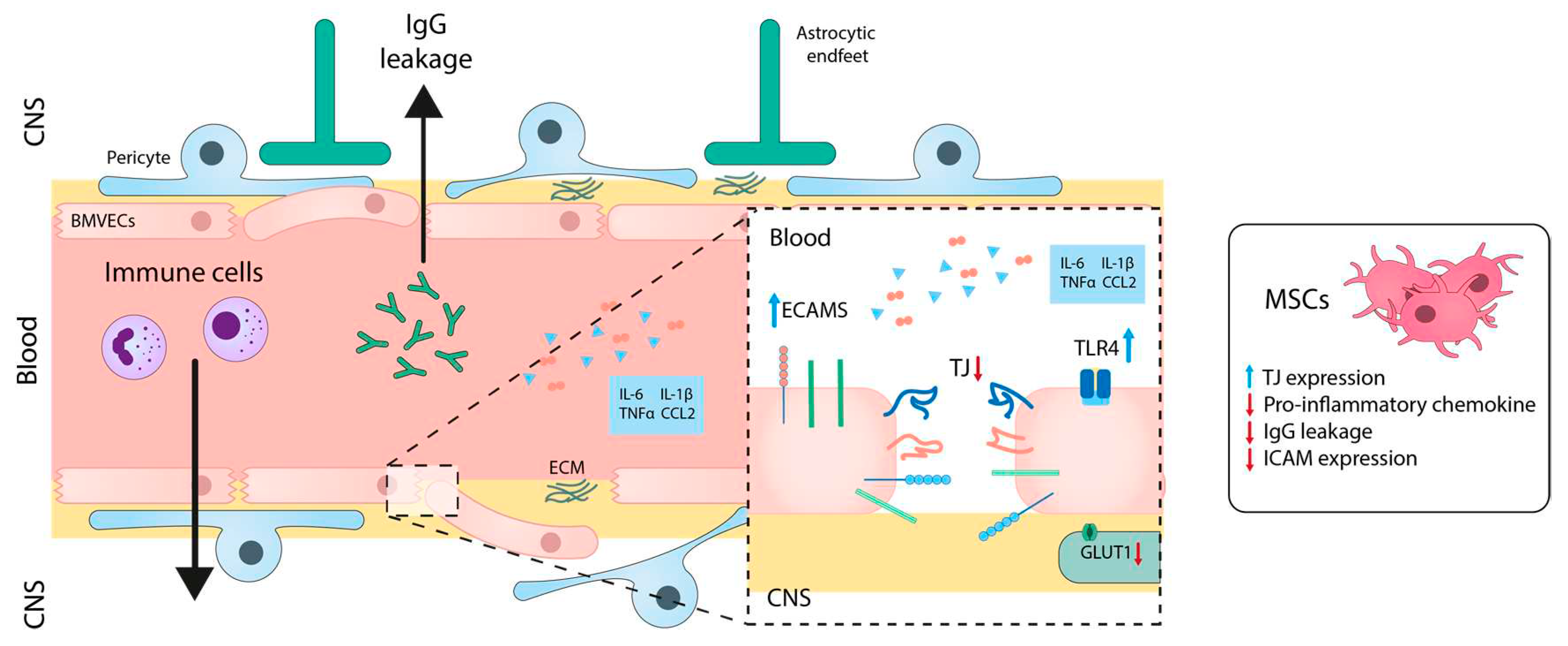
Figure 5.
Scheme illustrating pathophysiology of the blood-brain barrier permeability throughout stroke. Left panel, hyperacute and acute stages are characterized by endothelial cells destabilization and BBB disruption, edema, peripheral immune infiltration and glial immune activation. Right panel, subacute and chronic stages are involved in the recovery phase. This is characterized by vascular organization, restoration of BBB integrity, and by anti-inflammatory and reparative immune activities. Treatment with MSCs during the hyperacute and acute phases reduces the cytotoxic damage by the production of neurotrophic factors, the decrease of the pro-inflammatory mediators and the immune infiltration and the recovery of the BBB integrity. During the subacute and chronic stages, MSCs treatment may collaborate with the endogenous mechanisms of recovery by keeping immunomodulatory properties and by favoring the vascular stabilization. CNS: central nervous system; ECM: extracellular matrix; BMVECs: brain microvascular endothelial cells; ECAMs: endothelial cell adhesion molecules; NK: natural killer; MMP: metalloproteinases; VEGF: vascular endothelial growth factor; TJ: tight-junctions; Ang: angiopoietin; Tie2: tyrosine kinase receptor.
Figure 5.
Scheme illustrating pathophysiology of the blood-brain barrier permeability throughout stroke. Left panel, hyperacute and acute stages are characterized by endothelial cells destabilization and BBB disruption, edema, peripheral immune infiltration and glial immune activation. Right panel, subacute and chronic stages are involved in the recovery phase. This is characterized by vascular organization, restoration of BBB integrity, and by anti-inflammatory and reparative immune activities. Treatment with MSCs during the hyperacute and acute phases reduces the cytotoxic damage by the production of neurotrophic factors, the decrease of the pro-inflammatory mediators and the immune infiltration and the recovery of the BBB integrity. During the subacute and chronic stages, MSCs treatment may collaborate with the endogenous mechanisms of recovery by keeping immunomodulatory properties and by favoring the vascular stabilization. CNS: central nervous system; ECM: extracellular matrix; BMVECs: brain microvascular endothelial cells; ECAMs: endothelial cell adhesion molecules; NK: natural killer; MMP: metalloproteinases; VEGF: vascular endothelial growth factor; TJ: tight-junctions; Ang: angiopoietin; Tie2: tyrosine kinase receptor.
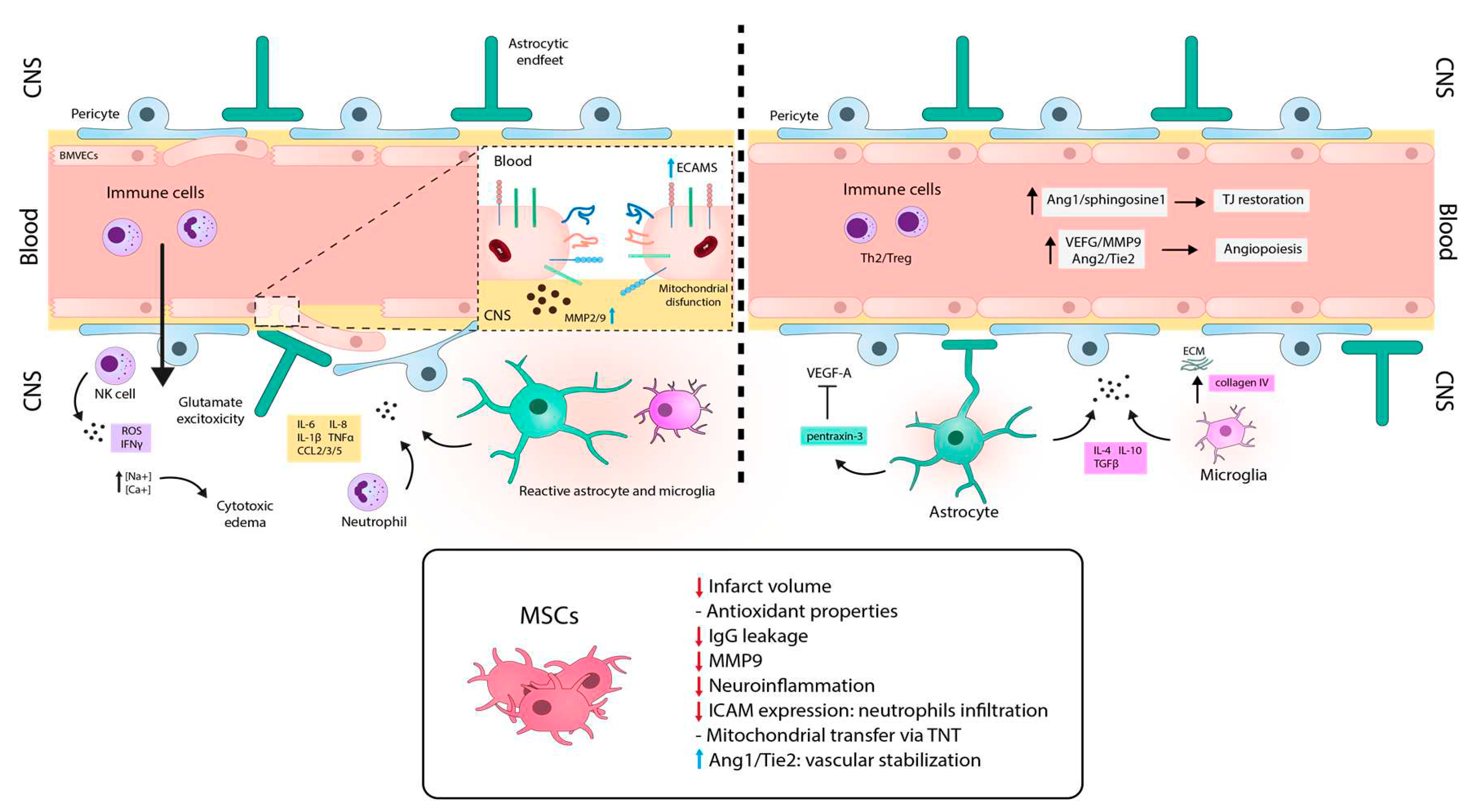

Table 1.
Proteins involved in transcellular transportation in BMVECs of BBB.
| Transporter | Cargo | Location | Description | Source |
|---|---|---|---|---|
| Glucose Transporter 1(GLUT-1) | Glucose | Abluminal and luminal side | Main glucose transporter of BMVECs. Also expressed in astrocytes but not in neurons. Na+ dependent transporters | [40] [56] |
| Large neutral amino acid transporter 1(LAT1) | Large neutral amino acids | Abluminal and luminal side | Abluminal side LAT1 transport is dependent of Na+ concentration. Bidirectional transport | [40] [57] |
| Cationic amino acid transporter 1 and 3 (CAT1/3) | Cationic amino acids | Abluminal and luminal side | CAT-1 is pH and Na+ independent but sensitive to changes in membrane potential | [58] |
| Na+-dependent transporters for glutamate exist on astrocytes 1 and 2 (EAAT1/2) | Glutamate | Abluminal side | Expressed in astrocytes. Possible protective mechanism against glutamate neurotoxicity | [59] |
| Monocarboxylate transporters (MCT1) | Monocarboxylic acids (lactate, pyruvate and acetoacetate and β-hydroxybutyrate) | Abluminal and luminal side | Intracerebral transport. Located in BMVECs and astrocytes. The transport mechanism is a H+ cotransporter or a monocarboxylate exchanger | [60] [61] |
| Insulin receptor (IR) | Insulin | Abluminal and luminal | Located in BMVECs. Insulin binding activates IR by phosphorylation of beta-chain region. Impaired phosphorylation response in AD | [62] |
| Low-density lipoprotein receptor–related protein 1 (LRP1) | APO2 and APO3 | Mainly in the abluminal side | Located in BMVECs. LRP1 binds to Aβ aggregates and mediates their clearance from brain to blood. LRP1 level diminished in AD patients leads to aggregates accumulation. | [26] [63] |
| Receptors for advanced glycation end-products (RAGE) | Advanced glycation end products (AGE), high mobility group box-1 (HMGB-1) protein | Mainly at the luminal side | Located in BMVECs, microglia and astrocytes. Upregulated in AD. It mediates the influx of Aβ into the brain | [64] [65] |
| P-glycoprotein, ATP-binding cassette 1(P-gp, ABCB1) | Xenobiotics and drugs | Expressed in the luminal side | P-gp is a unilateral efflux pump from blood to brain. It uses ATP in the active transport of substances. It is crucial in the ADMET properties of pharmaceutical drugs. In AD, P-gp is involved in accumulation of Aβ peptides in the CNS. | [66] [67] |
| Transferrin receptor protein (Tfr) | Transferrin (apo- and holo-transferrin) | Abluminal and luminal side | Primary iron transporting system. Highly enriched in BMVECs. Studied as a targeted transporter of therapeutics to the brain. | [68] |
Table 2.
Mesenchymal Stromal Cell-Studies selected from ClinicalTrials.gov for “Multiple Sclerosis”, “Ischemic Stroke”, “Alzheimer disease” and “Parkinson disease”.
Table 2.
Mesenchymal Stromal Cell-Studies selected from ClinicalTrials.gov for “Multiple Sclerosis”, “Ischemic Stroke”, “Alzheimer disease” and “Parkinson disease”.
| Components in the clinical trials | Categories | Studies (%) |
|---|---|---|
| MSC type | Bone Marrow | 27 (30) |
| Umbilical Cord | 24 (26.67) | |
| Adipose | 14 (15.56) | |
| Neural Progenitor derived | 4 (4.44) | |
| Embryonic | 1 (1.11) | |
| Exosomes | 1 (1.11) | |
| Not indicated | 20 (20.22) | |
| Disease | Multiple Sclerosis | 35 (38.89) |
| Ischemic Stroke | 25 (27.78) | |
| Alzheimer | 17 (18.89) | |
| Parkinson | 13 (14.44) | |
| Modality | Autologous | 41 (45.56) |
| Allogenic | 21 (23.33) | |
| Not indicated | 28 (31.11) | |
| Route | Intravenous | 48 (53.33) |
| Intrathecal | 8 (8.89) | |
| Intravenous/Intrathecal | 3 (3.33) | |
| Intraventricular | 1 (1.11) | |
| Intra-striatal | 1 (1.11) | |
| Intracerebral | 1 (1.11) | |
| Nasal | 1 (1.11) | |
| Not indicated | 27 (30) | |
| Target | Score | 76 (76.77) |
| Immune | 13 (13.13) | |
| Neurological | 10 (10.10) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



