
Submitted:
03 August 2023
Posted:
07 August 2023
You are already at the latest version
Abstract
Peptide mapping is an important tool to confirm the correct sequence expressed for a protein and to evaluate protein post-translational modifications (PTMs) that may arise during production, processing, or storage of protein drugs. Our new orally administered drug (Ab-1), a single-domain antibody, is highly stable and resistant to proteolysis. Analysis by the commonly used tryptic map method did not generate sufficient sequence coverage. Alternative methods were needed to study the Ab-1 drug substance (75 mg/mL) and drug product (3 mg/mL). To meet analytical needs, we developed two new peptide map methods with lysyl endopeptidase (Lys-C) digestion. These newly developed protein digestion protocols do not require desalting/buffer-exchange steps, thereby reducing sample preparation time and improving method robustness. Additionally, the protein digestion was performed under neutral pH with methionine acting as a scavenger to minimize artifacts, such as deamidation and oxidation, which are induced during sample preparation. Further, the method for low concentration samples performs comparably to the method for high concentration samples. Both methods provide 100% sequence coverage for Ab-1, and therefore, enable comprehensive characterization for its product quality attributes (PQA) assessment. Both methods can be used to study other antibody modalities.
Keywords:
Biopharmaceuticals
; drug substances
; sample preparation
; mass spectrometry
1. Introduction
Biotherapeutic drugs, especially antibodies, dominate in pharmaceutical pipelines due to their suitability for modulating extracellular targets with high potency and selectivity [1,2,3]. Recent advancements in drug discovery allow us to witness the significant growth of next-generation antibodies in this regard [4,5,6]. These new antibody formats, which include bispecific (multi-specific) antibodies, nanobodies, antibody fragments, and modified antibodies, could help address unmet medical needs and provide treatments for cancer and inflammatory or immunological diseases [1,3]. Understanding the structural aspects of the new antibody formats is essential, because certain structural characteristics are strongly associated with the safety and efficacy of the antibody, thus affecting drug critical quality [7,8,9,10,11,12,13,14,15,16]. Despite the existence of a variety of analytical methods historically used to characterize biotherapeutics [17,18,19,20,21], next-generation antibodies need innovative analytical methods to characterize and monitor drug quality attributes, as new drug formats present new types of analytical challenges.
At the core of many analytical strategies, peptide map methods have been widely used for the structural characterization of antibody-based therapeutics. Few other assays can claim to match peptide mapping for sensitivity and specificity [2]. A typical peptide mapping workflow includes enzymatic digestion of a protein into peptides, peptide mixture separation and detection by liquid chromatography (LC) coupled with high-resolution accurate mass (HRAM) mass spectrometry (MS), and data analysis using cutting-edge software tools. The analysis can obtain specific and structurally resolved information on protein sequence, post-translational modifications (PTMs), and process impurities, all of which are important in molecule critical quality attribute (CQA) assessment, cell line selection, production process optimization, stability and release testing, and extended characterization [22]. Peptide mapping combined with new peak detection (NPD) allows for the simultaneous monitoring of multiple product quality attributes (PQAs) in a single analysis. This powerful approach is known as the multi-attribute method (MAM). NPD enables new impurities (as new peaks) to be detected when compared with a reference sample [23]. MAM has the potential to replace several conventional methods, thus increasing the efficiency of analytical testing throughout all stages of biopharmaceutical drug development processes [23,24,25].
One of the key components for MAM is a well-optimized and reproducible protein digestion. The digestion is performed with specific proteases. The most commonly used protease for conventional antibodies is trypsin, because it produces peptides in the size range that is most efficient for peptide identification via MS analysis [23,26]. Our platform MAM uses trypsin as a digestion enzyme [27]. However, trypsin is not suitable for the new pipeline molecule, Ab-1, which targets inflammatory bowel disease. Ab-1 is highly stable and tolerates extreme temperature and pH, which makes it a good candidate for oral administration via tablet or capsule. As Ab-1 is designed to be a proteolysis-resistant protein, trypsin does not digest it well. Moreover, based on the amino acid sequence of Ab-1, tryptic digestion generates some small peptides of only one or two amino acids near its complementarity-determining regions (CDRs) that will not be captured in the analysis. Alternatively, lysyl endopeptidase (Lys-C), which retains proteolytic activity under strong protein denaturing conditions, can be used to improve the digestion of proteolytic resistant proteins. Based on the in silico analysis of the Ab-1 amino acid sequence using Lys-C, complete sequence coverage is possible for this challenging molecule. A recently improved MAM with reduced Lys-C digestion was reported for the analysis of therapeutic monoclonal antibodies (mAbs) [28], in which the Lys-C digestion protocol eliminated the desalting step to increase the throughput. However, the digestion was done at pH 8.0, which is known to introduce sample preparation-related artifacts (e.g., increased deamidation). Other artifacts such as oxidation might be introduced as well. Moreover, the reported method is not suitable for low concentration protein drugs. Therefore, a robust digestion method is needed to characterize protein drugs at high and low concentrations with minimized method-introduced artifacts on PQA characterization.
In this work, we developed optimized Lys-C digestion protocols using the amino acid methionine (Met) as a scavenger and a neutral pH to minimize the artifacts induced during protein digestion. Additionally, we were able to establish a method for low concentration proteins with comparable method performance to that of high concentration protocols. Two new Lys-C digestion protocols were established to accommodate different concentrations of Ab-1, drug substance (DS; 75 mg/mL) and drug product (DP; 3 mg/mL). These Lys-C peptide map methods enable comprehensive characterization and monitor PQAs. They also serve as alternative or complementary methods to the platform MAM tryptic map method for many antibodies. Here, we also highlight the impact of the newly developed characterization methods and their applications in drug stability studies. Therefore, these new methods are valuable additions to our analytical toolbox for antibody characterization beyond traditional modalities.
2. Results and Discussion
2.1. Method Development: Lys-C Digestion Method 1
Lys-C digestion Method 1 was developed to be suitable for Ab-1 DS or any protein sample with a concentration ≥50 mg/mL. Our experiment design covered several considerations for the enzymatic digestion of the proteolysis-resistant protein Ab-1. (1) In this method, Ab-1 was denatured in 6 M guanidine HCl (GuHCl), reduced with dithiothreitol (DTT), and carbamidomethylated using iodoacetamide (IAM). (2) We chose DTT over tris(2-carboxyethyl)phosphine (TCEP) as the reducing agent because TCEP can reduce oxidized Met residues [29] on the molecule prior to digestion, resulting in less accurate quantitation of that attribute. (3) The most commonly used alkylation reagents are iodoacetic acid (IAA) and IAM. We used IAM because IAA is typically dissolved in 1 N NaOH, and that would increase the pH of the sample mixture and introduce a negative charge to peptides [30,31]. (4) The reduced and alkylated protein was digested with Lys-C enzyme after a simple dilution step to lower GuHCl to ≤2 M. GuHCl concentrations greater than 2 M affect enzyme activity. At a concentration ≤2 M GuHCl, Lys-C retains 90% of its activity while keeping the protein in a more denatured condition to help digestion of the protease resistant Ab-1, whereas trypsin only retains 50% of its activity and requires a desalting/buffer-exchange step to remove GuHCl prior to protein digestion [32,33]. Therefore, with a dilution step rather than a desalting/buffer-exchange, this Lys-C digestion protocol was simplified without the need for the additional desalting step.
To optimize the Lys-C digestion conditions, we focused on the following parameters: reagent concentration, buffer pH, incubation time and temperature during reduction, alkylation, and digestion. Table 1 provides a detailed list of the conditions tested. It is important that the antibody is sufficiently denatured, unfolded, and reduced prior to digestion, allowing Lys-C to easily access the molecule for a complete digestion. For the protein reduction to break its disulfide linkages and improve its digestion, we optimized the level of the reducing agent DTT to achieve complete reduction without an excess of DTT. We examined the amount of time required to fully reduce the protein, and we evaluated and selected a pH that would ensure complete reduction and minimize artificial deamidation. The optimal condition determined for each parameter is shown by a bold, underlined value in Table 1. The best reduction condition was achieved using 3.5 mM DTT and pH 7.5 at 37 °C for 1 h. Following reduction, alkylation of the sulfhydryl groups on the cysteine residues was required to prevent reformation of the disulfide bonds. IAM was used in 2.5-fold excess of DTT to ensure complete alkylation. However, if too much IAM was used, it would over-alkylate the protein, which would alkylate amino acid residues other than cysteine (e.g., lysine). We tested the alkylation conditions including IAM concentration and incubation time. The optimized condition for alkylation was 8.5 mM IAM for 15 min at 37 °C in the dark. The remaining (little) IAM after reaction with DTT was unstable under light and in the presence of excess water from a three-fold dilution immediately after the alkylation step. Therefore, there was no need to use additional DTT to quench the alkylation step.

Table 1.
Method 1 optimization of test conditions.
| Parameter | Test Conditiona | Note | |
|---|---|---|---|
| Reduction | DTT (mM) | 3.5, 5 | 6 M GuHCl for protein denaturation at 37 °C. Use Met (10–20 mM) as a scavenger throughout the procedure to reduce artificial oxidation |
| Buffer pH | 7, 7.5, 8 | ||
| Incubation time (min) | 30, 45, 60, 90 | ||
| Alkylation | IAM (mM) | 6.8, 8.5 | Alkylation at 37 °C in the dark |
| Incubation time (min) | 15, 30 | ||
| Dilution | Scheme 1. Dilute 3× before digestion and then dilute 3× before LC/MS injection Scheme 2. Dilute 9× before digestion |
Dilution to lower GuHCl level (≤2 M) in digestion and more dilution (<1 M) before LC/MS injection | |
| Digestion | Lys-C to protein ratio | 1:20, 1:10 | Digestion at pH 7 to minimize artificial deamidation Digestion temperature: 37 °C |
| Incubation time | 30 min, 1 h, 2 h | ||
a Bold, underlined value indicates the optimal test condition. DTT, dithiothreitol; GuHCl, guanidine HCl; IAM, iodoacetamide; LC/MS, liquid chromatography-mass spectrometry; Met, methionine.
As this protocol eliminated the desalting step, a simple dilution of the sample into the digestion buffer lowered the GuHCl concentration (≤2 M). Two dilution schemes were examined as shown in Table 1. Between them, Dilution Scheme 1 with a 3-fold dilution (before digestion) performed well, while the digestion efficiency was decreased using a 9-fold dilution (Dilution Scheme 2). After the dilution, the Lys-C digestion was carried out at pH 7 to minimize artificial deamidation occurring during protein digestion [34]. Lys-C to protein ratios (enzyme-weight: protein-weight) of 1:20 and 1:10 and digestion times were evaluated. The results revealed that efficient protein digestion was achieved after 2 h of Lys-C digestion at 37 °C. One important aspect when developing such a method was to choose the conditions that will minimize the degree of artificial modifications induced, balancing maximum digestion efficiency with minimum artificial modifications. Besides digestion at neutral pH for minimal artificial deamidation, free Met was added as a scavenger throughout the sample preparation to reduce artificial oxidation [35].
2.2. Method Optimization: Lys-C Digestion Method 2
The Ab-1 DP is formulated in a capsule form, i.e., an enterically coated solid form. After dissolving the capsule in liquid for analysis, the concentration is 3 mg/mL. With protein at such low concentration, any digestion using a dilution scheme is especially challenging; as the protein concentration in the digestion step is very low (0.03 mg/mL) if Method 1 was used. Our main focus was to find lower concentrations of the denaturing agent that were still effective for protein denaturing, so that a smaller dilution factor could be used. Therefore, lower concentrations of GuHCl (2–5 M) were tested, and the minimum concentration used to get complete reduction was 4.7 M. Another method modification was to dissolve DTT in Tris HCl buffer instead of water to avoid further dilution of the sample. Other conditions remained the same as those used in Method 1, including reduction and alkylation reagent concentrations, Lys-C enzyme-to-protein ratio, and incubation times. In parallel, Table 2 lists the reagent and the protein concentrations after each step for both methods. Using Method 2, the final protein concentration after the digestion was 0.2 mg/mL. Note that the Met levels were maintained in the range 10–20 mM to prevent protein oxidation during the sample preparation [35]. As the recommended injection amount for a protein sample is 2.5–5.0 µg for LC/MS analysis, the injection volume was modified accordingly. This Lys-C digestion protocol was suitable for use with Ab-1 DP lots or for samples with low concentrations (≥3 mg/mL).
Table 2.
Stepwise procedures and the concentration of the protein sample and reagents.
| Method 1 | Reagent | Concentration | Method 2 | Reagent | Concentration | |
|---|---|---|---|---|---|---|
| Reduction | 8 M GuHCl 85µL | GuHCl | 6.0 M | 8 M GuHCl 67µL | GuHCl | 4.7 M |
| 1 M Tris pH 7.5/0.2 M Met 6 µL | Tris HCl | 0.05 M | 1 M Tris HCl pH 7.5/0.2 M Met/65 mM DTT 6 µL | Tris HCl | 0.05 M | |
| Met | 11 mM | Met | 11 mM | |||
| 40 mM DTT 10 µL | DTT | 3.5 mM | DTT | 3.5 mM | ||
| 50 mg/mL Ab-1 12 µL | protein | 5.1 mg/mL | 3 mg/mL Ab-1 40 µL | protein | 1.0 mg/mL | |
| Incubate at 37 °C for 1 h | Incubate at 37 °C for 1 h | |||||
| Alkylation | 200 mM IAM 5 µL | IAM | 8.5 mM | 200 mM IAM 5 µL | IAM | 8.5 mM |
| Incubate at 37 °C for 15 min in dark | Incubate at 37 °C for 15 min in dark | |||||
| Digestion | Take 25 µLof the alkylated sample (125 µg) into a new tube | Take 68 µL of the reduced sample (68 µg) into a new tube | ||||
| Reduced sample 25 µL | protein | 1.5 mg/mL | Reduced sample 68 µL | protein | 0.4 mg/mL | |
| Add H2O 39 µL(GuHCl conc. <2 M) | GuHCl | 1.8 M | Add H2O 74 µL (GuHCl conc. <2M) | GuHCl | 2.0 M | |
| 1 M Tris pH 7/0.2 M Met 6 µL | Tris HCl | 0.1 M | 1 M Tris HCl pH7/0.2 M Met 12 µL | Tris HCl | 0.1 M | |
| Met | 18 mM | Met | 19 mM | |||
| 0.5 mg/mL Lys-C solution 13 µL | Lys-C: protein | 1:20 (w:w) | 0.5 mg/mL Lys-C solution 7 µL | Lys-C: protein | 1:20 (w:w) | |
| Incubate at 37°C for 2 h | Incubate at 37 °C for 2 h | |||||
| Dilution & Quench | Dilution (3x) | protein | 0.5 mg/mL | Pipette 79 µL digested sample, dilute 3× | protein | 0.2 mg/mL |
| Add H2O 140 µL (GuHCl conc. <1M) | GuHCl | 0.6 M | Add H2O 120 µL(GuHCl conc.<1M) | GuHCl | 0.7 M | |
| Add 20 µL0.2M Met | Met | 16 mM | Add20 µL 0.2M Met | Met | 18 mM | |
| Add 10% TFA 5 µL to quench digestion | TFA | 0.2% | Add 10%TFA 5 µL to quench digestion | TFA | 0.2% |
DTT, dithiothreitol; GuHCl, guanidine HCl; IAM, iodoacetamide; Met, methionine; TFA, trifluoroacetic acid.
2.3. Method Performance Evaluation
To evaluate the peptide map method performance, we focused on several aspects, such as protein reduction completion, alkylation efficiency, and digestion completion. Disulfide bonds are present in many proteins including Ab-1. Protein reduction and alkylation conditions play important roles for the complete reduction of disulfides. When optimizing the conditions for protein reduction, the reduction completion of disulfides was assessed and confirmed. Alkylation efficiency was also thoroughly evaluated. Efficiency decreased when alkylation of cysteine was not complete, i.e., <100% cysteine was alkylated. Over-alkylation was measured when the N-terminus or other amino acids besides cysteine (e.g., lysine) were alkylated. Therefore, we adjusted the alkylating agent and incubation time to balance under and over alkylation. The average alkylation efficiency was measured to be >95%. Optimizing reduction and alkylation conditions was also essential for improving protein digestion.
Protein sequence coverage is the key measurement when evaluating the performance of a peptide map method. In this study, the digestion conditions were optimized to achieve maximized protein sequence coverage. For Ab-1, Lys-C, as the enzyme choice, generated 100% sequence coverage as expected. However, the biggest challenge for this molecule in Lys-C digestion was the high level of missed cleavage peptides due to the enzymatic resistance of this molecule. The issue was the mis-cleaved peptide, L1+L2, which ranged from 9% to 15% in different sample preparations by different analysts or on different days. Mis-cleavage at such a high level leads to an inconsistency in method quantitation. Note that other mis-cleaved peptides (L3+L4 and L2+L3) were below 2%, which is less of a concern. Figure 1 shows the MS total ion current (TIC) chromatograms analyzed using the newly developed Lys-C peptide map method (Method 1) and illustrates the protein wild-type peptides L1 to L4 with some mis-cleaved peptide peaks. Note that the peptide mis-cleavages are common for peptide mapping. They are useful in some cases to improve sequence coverage. For example, a short peptide, usually three amino acids and less, is too hydrophilic to be retained in a reversed-phase (RP) high-performance liquid chromatography (HPLC) column. It often is washed out in the void volume with small molecule reagents used in sample preparation. With the peptide mis-cleavage event, the short peptide can be retained in a RP column and observed in LC/MS analysis when it attaches to another peptide. Therefore, the protein sequence coverage is improved. In contrast to mis-cleavages that cause incomplete digestion, over digesting the protein produces unspecific cuts, which is also undesirable. To minimize both mis-cleavages and unspecific cuts, we carefully optimized the digestion conditions and tested with higher amounts of enzyme and longer digestion times. In the end, with the optimized digestion conditions, we were able to bring down the level of mis-cleaved peptide L1+L2 to less than 5% from the 9–15% range.
Figure 1.
MS total ion current chromatograms of Ab-1 by Method 1 (L1-L4 =peptides by Lys-C).
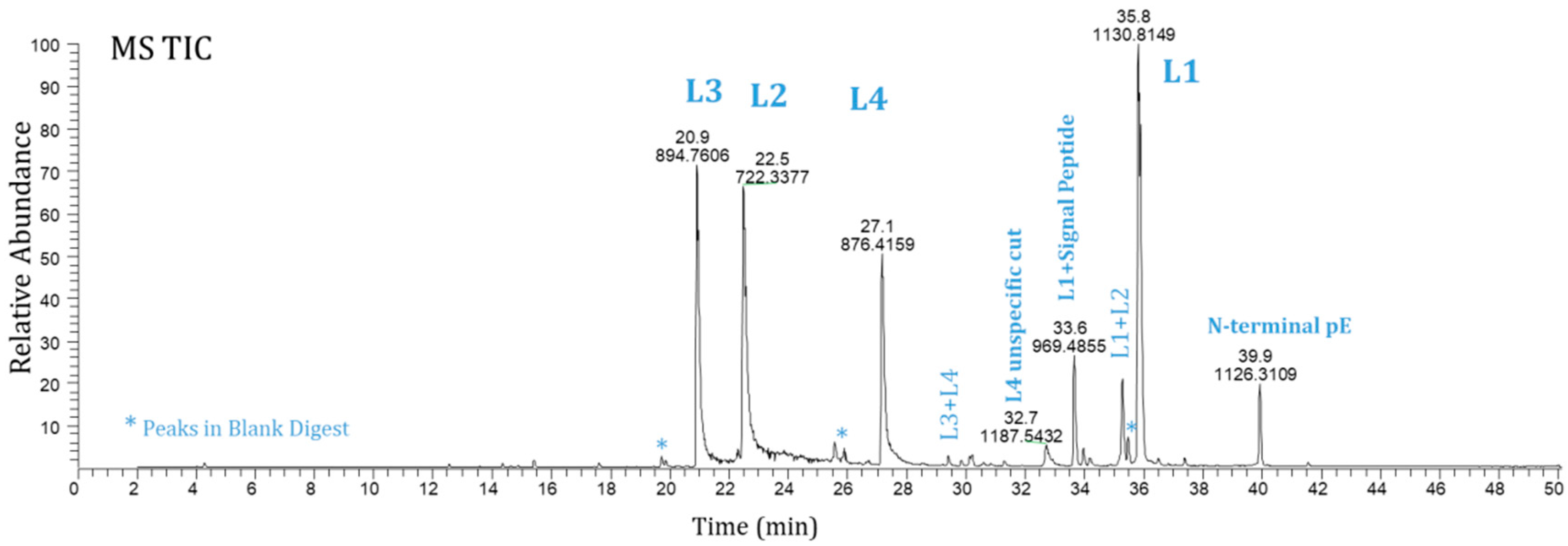
Figure 2 displays typical MS TIC chromatograms of Lys-C digested Ab-1 following the Method 1 protocol. Both methods provided comparable results, providing a choice for analysts to use. Moreover, using these methods, we tested seven other molecules, including mAbs, bispecific antibodies, an antibody drug conjugate, and a Fab antibody fragment. Figure 3 shows the TIC chromatograms of Ab-2 (a mAb) as an example. Therefore, these two newly developed Lys-C map methods can be applied to various antibody formats.
Figure 2.
MS total ion current chromatograms of Ab-1: Method 1 (bottom) and Method 2 (top). *Peaks in blank digest. (L1-L4 = peptides by Lys-C).
Figure 2.
MS total ion current chromatograms of Ab-1: Method 1 (bottom) and Method 2 (top). *Peaks in blank digest. (L1-L4 = peptides by Lys-C).
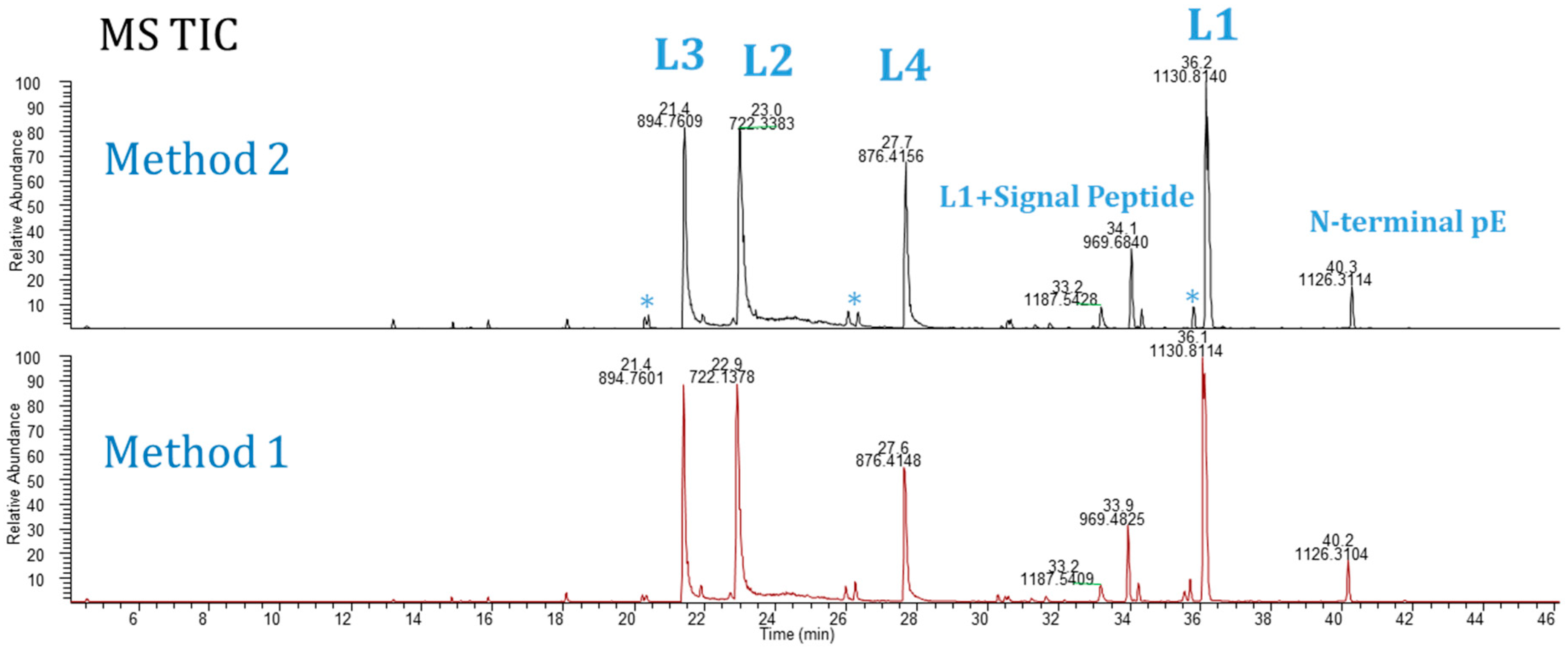
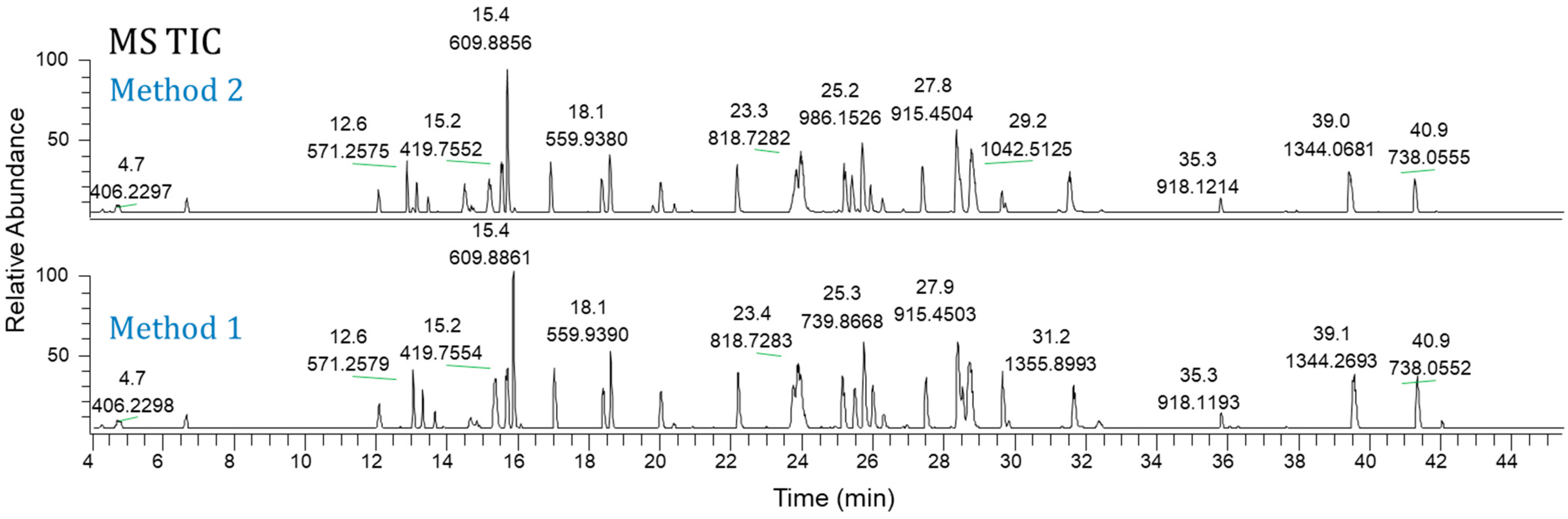
Figure 3.
MS total ion current chromatograms of Ab-2: Method 1 (bottom) and Method 2 (top).
2.4. Method Robustness
The optimized Lys-C digestion methods were evaluated and demonstrated to be robust. To assess the robustness, a half fractional factorial design of experiment (DoE) with five factors and two levels was performed (Table 3). Digest buffer pH, time, temperature, and reduction buffer pH were included because they are known to affect deamidation, pyroglutamate (pE) formation, isomerization, and succinimide formation. Injection volume changes would affect quantitation as sample load changes; therefore, it was also included in the DoE study. A total of 20 runs, including an additional 2 replicates and 2 center points, were completed for each method.
Table 3.
The DoE design with factors and levels.
| DoE Factor | Condition Specs | DoE Level (low/high) |
|---|---|---|
| Digest buffer (pH)a | 7.0 ± 0.1 | 6.9 / 7.1 |
| Digest time (min)a | 120 ± 10 | 110 / 130 |
| Digest temperature (°C)a | 37 ± 2 | 35 / 39 |
| Reduction buffer (pH)a | 7.5 ± 0.1 | 7.4 / 7.6 |
| Injection volume (µL)b | 5 for Method 1 17 for Method 2 |
4 / 6 15 / 19 |
- a This DoE factor can affect deamidation, pE formation, isomerization, and succinimide.
- b This DoE factor can affect quantitation as the load increases.
The DoE study demonstrated the robustness of Method 1 and Method 2. Table 4 lists the statistics for the DoE study with relative abundance maximum (Max), minimum (Min), difference (Max-Min), average, standard deviation (SD), and relative SD (%RSD). Based on these results, both methods are robust for Ab-1 PTM quantification, as the RSD was 5% or less.
Table 4.
DoE results: the statistics of relative abundance for Method 1 and Method 2.
| Statistics | Method 1 | Method 2 | ||
|---|---|---|---|---|
| Signal Peptide (%) | N-terminal pE (%) | Signal Peptide (%) | N-terminal pE (%) | |
| Max | 13.2 | 7.1 | 13.9 | 7.3 |
| Min | 11.7 | 6.3 | 11.9 | 6.1 |
| Difference (Max-Min) | 1.5 | 0.8 | 2.0 | 1.2 |
| Average | 12.5 | 6.7 | 12.8 | 6.7 |
| SD | 0.4 | 0.2 | 0.5 | 0.3 |
| % RSD | 3.3% | 2.8% | 4.2% | 5.1% |
Min, minimum; Max, maximum; pE, pyroglutamate; SD, standard deviation; %RSD, relative SD.
2.5. Suitability for Ab-1 Stability Study
The major PTMs of Ab-1 from the manufacturing process were N-terminal signal peptide and N-terminal pE. In the stability studies, Ab-1 samples were subjected to chemical and physical stresses including oxidation by 2,2-azobis (2-amidinopropane) dihydrochloride (AAPH), elevated temperatures, and low pH. AAPH is a free radical generator that produces alkoxyl and alkyl peroxyl radicals. The AAPH stress model is used as an indicator of oxidation susceptibility for antibodies. Such stress often induces protein oxidation of Met or tryptophan residues [8,9]. The stress under elevated temperatures and low pH may produce protein deamidation of asparagine [7,17], or pE formation at N-terminal glutamic acid [19,20], etc. Even if it is not known whether these PTMs would affect biological activity, monitoring PTMs occurs as part of routine quality testing because such quality control testing can ensure the safety and efficacy of the DP. Using the newly developed Lys-C maps, we can identify and quantify these modifications of Ab-1. Figure 4 illustrates the protein wild-type peptides L1-L4 with well-separated PTM-modified peptides.
Figure 4.
UV chromatograms of the protein digest of Ab-1 (L1-L4 = peptides by Lys-C; prepared using Method 1). M, methionine; W, tryptophan.
Figure 4.
UV chromatograms of the protein digest of Ab-1 (L1-L4 = peptides by Lys-C; prepared using Method 1). M, methionine; W, tryptophan.
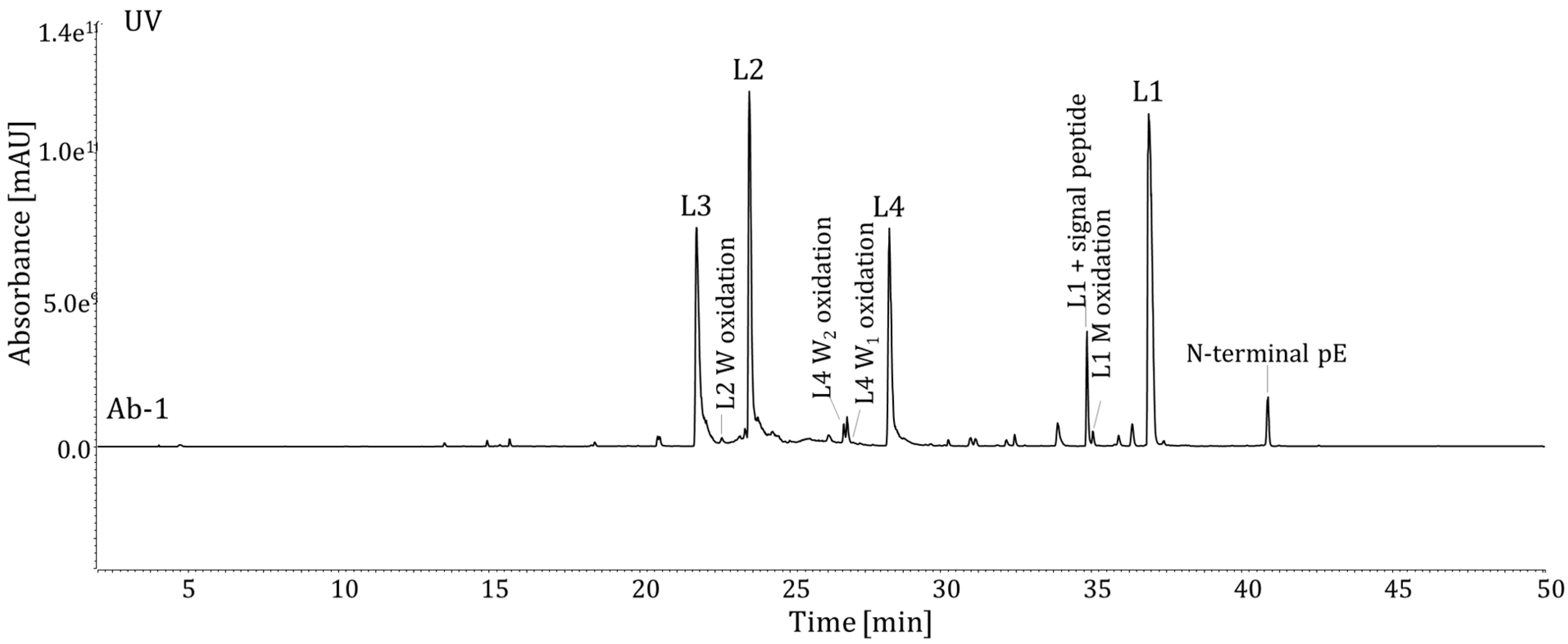
The Lys-C digestion method is stability indicating. Unstressed (control) and stressed samples were analyzed in parallel using the new method. The results of force-stressed Ab-1 DS samples using AAPH, high temperatures, and low pH are summarized in Table 5. The Ab-1 samples subjected to thermal stress conditions (storage at 40 °C and 50 °C) demonstrated an increase in N-terminal pE compared to the control sample. Deamidation was not detected at either of the two asparagine residues of Ab-1. It is worth noting, Ab-1 is particularly tolerant to high temperatures, and therefore, significant degradation upon thermal stress is not expected. The Ab-1 samples subjected to 10 mM AAPH demonstrated an increase in oxidation at two tryptophan residues, (W1, W2) in peptide L4 compared to the unstressed sample. No significant increase in oxidation was observed at other tryptophan or Met residues. The analysis of stressed samples demonstrated the Lys-C digestion method is stability indicating for attributes at critical sites for Ab-1. Relative levels of induced modifications were calculated as:
Table 5.
Attribute quantitation of the stressed Ab-1 DS (by Method 1).
| Attribute | Relative Abundance (%) | ||||||
|---|---|---|---|---|---|---|---|
| t=0 | 4 weeks at 40 °C | 2 weeks at 50 °C | pH Control | pH 3.25 | AAPH Control | 10 mM AAPH | |
| L1 + Signal peptide | 9.6 | 9.0 | 9.3 | 10.3 | 10.2 | 10.6 | 10.0 |
| N-terminal pE | 5.7 | 7.3 | 9.5 | 6.4 | 7.0 | 7.3 | 6.7 |
| L1 M oxidation | 2.3 | 2.3 | 2.2 | 2.2 | 2.1 | 2.2 | 2.1 |
| L1 W oxidation | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
| L2 W oxidation | <0.1 | ND | ND | 0.1 | 0.2 | 0.1 | 1.1 |
| L3 N1 deamidation | ND | ND | ND | ND | ND | ND | ND |
| L3 N2 deamidation | ND | ND | ND | ND | ND | ND | ND |
| L4 W1 oxidation | 0.3 | 0.3 | 0.4 | 0.3 | 0.2 | 0.3 | 3.7 |
| L4 W2 oxidation | 1.3 | 1.0 | 1.3 | 0.9 | 0.8 | 0.9 | 11.2 |
AAPH, 2,2’-azobis (2-amidinopropane) dihydrochloride; L1-L4 = peptides by Lys-C; ND, not detected; W, tryptophan.
3. Materials and Methods
3.1. Materials
This study used a camelid single variable domain on a heavy chain antibody (a fragment nanobody), Ab-1, which is a variable domain (VH) of a heavy-chain (HC) antibody. Ab-1 is derived from llama immunoglobulin G (IgG) and purified by the fed batch platform cell culture procedure without an affinity step. Ab-2 is a mAb derived from Chinese hamster ovary (CHO) cells. Both antibodies were made at the Roche/Genentech facilities. Pierce™ dithiothreitol (DTT), Pierce iodoacetamide (IAM), Pierce formic acid (FA), Pierce trifluoroacetic acid (TFA), and 8 M guanidine HCl (GuHCl) were obtained from Thermo Fisher Scientific (Sunnyvale, CA). Lysyl endopeptidase® MS grade (Lys-C) was purchased from Wako Chemicals (Richmond, VA). L-methionine (Met) and AAPH (2,2’-azobis (2-amidinopropane) dihydrochloride) were from Sigma-Aldrich (St. Louis, MO). All organic solvents were of analytical or HPLC grade.
3.2. Protein Digestion
3.2.1. Method 1: Lys-C Digestion for High Concentration Samples (≥50 mg/mL)
The optimized Lys-C digestion protocol for 50 mg/mL samples was as follows: in a sample tube, 50 mg/mL protein (12 µL) was mixed with 85 µL of 8 M GuHCl and 6 µL of 0.2 M Met in 1 M Tris HCl (pH 7.5). The reduction was started by adding 10 µL of 40 mM DTT. After 1 h incubation at 37 °C, alkylation was carried out by adding 5 µL of 200 mM IAM and incubating at 37 °C in the dark for 15 min. Then, an aliquot of 25 µL of the reduced/alkylated protein (125 µg) was removed to a new tube, in which the sample was diluted by adding 39 µL of water and 6 µL of 0.2 M Met in 1 M Tris HCl (pH 7.0). To digest the protein, 13 µL of 0.5 mg/mL Lys-C solution was added to the sample mixture (enzyme-to-substrate weight ratio of 1:20), and the sample was incubated at 37 °C for 2 h. After digestion, the sample was further diluted with 140 µL of water and 20 µL of 0.2 M Met, and the protein digestion was quenched with the addition of 5 µL of 10%TFA. The digested protein mixture was frozen until analysis. For LC/MS analysis, the injection volume was 5 µL (2.5 µg Ab-1).
3.2.2. Method 2: Lys-C Digestion for Low Concentration Samples (≥3 mg/mL)
In a sample tube, 3 mg/mL protein (40 µL) was mixed with 67 µL of 8 M GuHCl and 6 µL of 65 mM DTT, 0.2 M Met in 1 M Tris HCl (pH 7.5). Protein reduction was at 37 °C for a 1 h incubation. Then, the alkylation was carried out by adding 5 µL of 200 mM IAM and incubating at 37 °C in the dark for 15 min. A 68 µL aliquot of the reduced/alkylated protein (68 µg) was removed to a new tube, in which the sample was diluted by adding 74 µL of water and 12 µL of 0.2 M Met in 1 M Tris HCl (pH 7.0). To digest the protein, 7 µL of 0.5 mg/mL Lys-C solution was added to the sample mixture (enzyme-to-substrate weight ratio of 1:20), and the sample was incubated at 37 °C for 2 h. After digestion, 79 µL of the digested sample was pipetted into a new sample tube and diluted with 120 µL of water and 20 µL of 0.2 M Met. The protein digestion was quenched with the addition of 5 µL of 10% TFA. The digested protein mixture was frozen until analysis. For LC/MS analysis, the injection volume was 17 µL (2.5 µg Ab-1).
3.3. LC/MS Analysis
LC/MS analysis was performed using a Q Exactive™ Hybrid Quadrupole-Orbitrap™ mass spectrometer coupled with a Vanquish ultra high-performance liquid chromatography (UHPLC) system (Thermo Fisher Scientific, Sunnyvale, CA). A separation column, Acquity Premier Peptide CSH C18 Column 1.7 µm, 2.1 × 150 mm (Waters Corporation, Milford, MA), was used with column temperature of 77 °C. Mobile phases A and B consisted of 0.1% FA in water and 0.1% FA in acetonitrile (ACN), respectively. The flow rate was set to 0.2 mL/min. The pump gradient was as follows: an isocratic flow at 1% B for 8 min, a multi-step linear gradient from 1% B to 13% B for 5 min, 13% B to 35% B for 35 min, and 35% B to 95% B for 2 min. The column wash and equilibration was a 5-times step-wash with an isocratic flow at 95% B for 2 min and sharp gradient from 95% B to 1 % B in 2 min. The total run time was 86 min. The MS scan type was full MS with data dependent MS2 (dd-MS2) for the top eight highest intensity ions. The electrospray voltage was 3.5 kV in positive ion mode. Both capillary and aux gas heater temperatures were at 250 °C. The full mass scan range was 200–2000 (m/z) with full mass resolution at 35k and dd-MS2 resolution at 17.5k. The collision energy for data-dependent acquisition was high-energy collisional dissociation. The MS data collection started at 4 min and ended at 45 min for a total 86 min run time. The acquisition was controlled by the vendor’s Chromeleon™ software (Thermo Fisher Scientific, Sunnyvale, CA).
3.4. MS Data Analysis
The raw MS files were analyzed using Byos® software suite (Protein Metrics Inc., Cupertino, CA). Peptides were identified via searching against the Ab-1 sequence using the accurate mass of a full mass scan and assignments of product-ions in MS2 spectra. The cutoff MS2 score was 250 and the maximum precursor m/z error was ±10ppm. Carbamidomethylation (+57.0215 Da) was set as a fixed modification of cysteine residues. The modification list included oxidation (+15.9949 Da) of Met and tryptophan, deamidations (+0.9840 Da) of asparagine, pE (−18.0106 Da) of N-terminal glutamic acid, signal peptide at protein N-terminus (+323.1594), glycation (+162.0528) of lysine, carbamidomethylation (+57.0215 Da) of lysine and de-carbamidomethylation (-57.0215 Da) of cysteine. The modification percent per peptide in the software’s PTM default report was used to report the detected levels of PTMs.
4. Conclusion
To support characterization of a new pipeline molecule, Ab-1, we have developed two new Lys-C peptide map methods. Method 1 is suitable for relatively higher concentration protein samples, like Ab-1 DS (≥50mg/mL) and Method 2 for low concentration (≥3 mg/mL) samples such as Ab-1 DP. These methods were optimized using a neutral pH digestion buffer (pH 7) and a short digestion time (2 h) to minimize artificial deamidation and pE formation. Free Met was added throughout the sample preparation as a scavenger to reduce artificial oxidation. The protein digestion protocols without the desalting steps increase the sample preparation throughput and minimize process-induced modifications. Using these simple and reliable protein digestion procedures, we can analyze Ab-1 with 100% sequence coverage and good PTM peptide separations. Furthermore, we have demonstrated the robustness of the new methods via well-designed DoE studies. These methods were also successfully applied to the protein stability study, showing differences at the peptide level between stressed and unstressed material. As a well-optimized Lys-C digestion is one of the key components for MAM, these new methods lay the foundation for future applications to build Lys-C MAM methods. We have also applied these new methods to various antibody formats. Therefore, the methods are valuable additions in to our analytical toolbox for antibody characterization.
Author Contributions
Conceptualization, F.Y. and Y.D.L.; data curation, Y.D.L. and M.I.B.; writing—original draft preparation, Y.D.L.; writing—review and editing, all authors; visualization, Y.D.L.; supervision, F.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgements
We appreciate the help and support of this project from Lance Cadang, Galahad Deperalta, Tim Spirakes, Heather Lee, Luladey Ayalew, and Bingchuan Wei. We also thank Scott Chamberlain, John Briggs, and Aaron Wecklser for their critical reviews of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef]
- Robotham, A.C.; Kelly, F.J. LC-MS characterization of antibody-based therapeutics: recent highlights and future prospects, Approaches to the Purification, Analysis and Characterization of Antibody-Based Therapeutics, Publisher: Elsevier Limited, 2020; pp. 1–33.
- Grilo, A.L.; Mantalaris, A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019, 37, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. mAbs. 2020, 12, 1703531. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Caron, G. New therapeutic modalities in drug discovery and development: Insights & opportunities. ADMET DMPK 2021, 9, 227–230. [Google Scholar]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. mAbs 2022, 14, 2014296. [Google Scholar] [CrossRef] [PubMed]
- Vlasak, J.; Bussat, M.C.; Wang, S.; Wagner-Rousset, E.; Schaefer, M.; Klinguer-Hamour, C.; Kirchmeier, M.; Corvaïa, N.; Ionescu, R.; Beck, A. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal Biochem. 2009, 392, 145–154. [Google Scholar] [CrossRef]
- Bertolotti-Ciarlet, A.; Wang, W.; Lownes, R.; Pristatsky P; Fang Y; McKelvey T; Li Y; Li Y; Drummond J; Prueksaritanont T; Vlasak J. Impact of methionine oxidation on the binding of human IgG1 to Fc Rn and Fc gamma receptors. Mol Immunol. 2009, 46, 1878–1882.
- Pan H; Chen K; Chu L; Kinderman, F.;Apostol, I.; Huang, G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci. 2009, 18, 424–433. [CrossRef]
- Liu, Y.D.; Chen, X.; Enk, J.Z.; Plant, M.; Dillon, T.M.; Flynn, G.C. Human IgG2 antibody disulfide rearrangement in vivo. J Bio Chem 2008, 283, 29266–29272. [Google Scholar] [CrossRef]
- Wei, Z., Feng, J., Lin, H.Y., Mullapudi, S.; Bishop, E.; Tous, G.I.; Casas-Finet, J.; Hakki, F.; Strouse, R.; Schenerman, M.A. Identification of a single tryptophan residue as critical for binding activity in a humanized monoclonal antibody against respiratory syncytial virus. Anal Chem. 2007, 79, 2797–2805.
- Wei, B.; Berning, K.; Quan, C.; Zhang, Y.T. Glycation of antibodies: modification, methods and potential effects on biological functions. mAbs. 2017, 9, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.Q.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.D.; Flynn, G.C. The effect of Fc Glycan forms on human IgG2 antibody clearance in humans. Glycobiology 2009, 19, 240–249. [Google Scholar] [CrossRef]
- Liu, Y.D.; Flynn, G.C. Effect of high mannose glycan pairing on antibody clearance. Biologicals 2016, 44, 163–169. [Google Scholar] [CrossRef]
- Xu Y; Wang D; Mason B; Rossomando, T.; Li, N.; Liu, D.; Cheung, J.K.; Xu, W.; Raghava, S.; Katiyar, A.; Nowak, C.; Xiang, T.; Dong, D.D.; Sun, J.; Beck, A.; Liu, H. Structure, heterogeneity and developability assessment of therapeutic antibodies. mAbs. 2019, 11, 239–264.
- Liu, Y.D.; Enk, J.Z.; Flynn, G.C. Human antibody Fc deamidation in vivo. Biologicals 2009, 37, 313–322. [Google Scholar] [CrossRef]
- Goetze, A.M.; Liu, Y.D.; Arroll, T.; Chu, L.; Flynn, G.C. Rates and impact of human antibody glycation in vivo. Glycobiology 2012, 22, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.D.; Goetze, A.M.; Bass, R.B.; Flynn, G.C. N-terminal glutamate to pyroglutamate conversion in vivo. JBC 2011, 286, 11211–11217. [Google Scholar] [CrossRef]
- Liu, H.; Ponniah, G.; Zhang, H.-M.; Nowak, C.; Neill, A.; Gonzalez-Lopez, N.; Patel, R.; Cheng, G.; Kita, A.Z.; Andrien, B. In vitro and in vivo modifications of recombinant and human IgG antibodies. mAbs. 2014, 6, 1145–1154. [Google Scholar] [CrossRef]
- Liu YD; Cadang L.; Bol K; Pan X; Tschudi K; Jazayri M; Camperi J; Michels D; Stults J;Harris R.J; Yang F. Challenges and strategies for a thorough characterization of antibody acidic charge variants. Bioengineering 2022, 9, 641–655.
- Goetze,. A.M.; Schenauer, M.R.; Flynn, G.C. Assessing monoclonal antibody product quality attribute criticality through clinical studies. mAbs. 2010, 2, 500–507. [CrossRef] [PubMed]
- Rogstad, S.; Yan, H.; Wang, X.; Powers, D.; Brorson, K.; Damdinsuren, B.; Lee, S. . Multi-attribute method for quality control of therapeutic proteins. Anal Chem. 2019, 91, 14170–14177. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.E.; Dubois, H.; Hoffmann, M.; Eri, S.D.; Fromentin, Y.; Wiesner, J.; Pfenninger, A.; Clavier, S.; Pieper, A.; Duhau, L.; Roth, U. Automated mass spectrometry multi-attribute method analyses for process development and characterization of mAbs. J Chromatogr B Analyt Technol Biomed Life Sci. 2021, 1166, 122540. [Google Scholar] [CrossRef] [PubMed]
- Tajiri-Tsukada, M.; Hashii, N.; Ishii-Watabe, A. Establishment of a highly precise multi-attribute method for the characterization and quality control of therapeutic monoclonal antibodies. Bioengineered 2020, 11, 984–1000. [Google Scholar] [CrossRef]
- Ren, D. An improved trypsin digestion method minimizes digestion-induced modifications on proteins Anal. Biochem. 2009, 392, 12–21. [Google Scholar]
- Sadek, M.; Moore, B.N.; Yu, C.; Ruppe, N.; Abdun-Nabi, A.; Hao, Z.; Alvarez, M.; Dahotre, S.; Deperalta, G. A robust purity method for biotherapeutics using new peak detection in an LC–MS-based multi-attribute method. J. Am. Soc. Mass Spectrom. 2023, 34, 484–492. [Google Scholar] [CrossRef]
- Li, X.; Rawal, B.; Rivera, S.; Letarte, S.; Richardson, D.D. Improvements on sample preparation and peptide separation for reduced peptide mapping based multi-attribute method analysis of therapeutic monoclonal antibodies using lysyl endopeptidase digestion. J Chromatography A 2022, 1675, 463161. [Google Scholar] [CrossRef]
- Lundell, N.; Schreitmuller, T. Sample preparation for peptide mapping–a pharmaceutical quality-control perspective. Anal Biochem. 1999, 266, 31–47. [Google Scholar] [CrossRef]
- Suttapitugsakul, S.; Xiao, H.; Smeekens, J.; Wu, R. Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics. Mol Biosyst. 2017, 13, 2574–2582. [Google Scholar] [CrossRef]
- Hutterer, K.M.; Hong, R.W.; Lull, J.; Zhao, X.; Wang, T.; Pei, R.; Le, M.E.; Borisov, O.; Piper, R.; Liu, Y.D.; Petty, K.; Apostol, I.; Flynn, G.C. Monoclonal antibody disulfide reduction during manufacturing: Untangling process effects from product effects. MAbs 2013, 5, 608–613. [Google Scholar] [CrossRef]
- Jekel, P.A.; Weijer, W.J.; Beintema, J.J. Use of endoproteinase Lys-C from Lysobacter enzymogenes in protein sequence analysis. Anal. Biochem. 1983, 134, 347–354. [Google Scholar] [CrossRef]
- Poulsen, J.W.; Madsen, C.T.; Young, C.; Poulsen, F.M.; Nielsen, M.L. Using guanidine-hydrochloride for fast and efficient protein digestion and single-step affinity-purification mass spectrometry, J. Proteome Res. 2013, 12, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.D.; Chen, Y.; Tsui, G.; Wei, B.; Yang, F.; Yu, C.; Cornell, C. Predictive in vitro vitreous and serum models to assess thiol-related quality attributes in protein therapeutics, Anal. Chem. 2020, 92, 6869–6876. [Google Scholar]
- Yang, F.; Zhang, J.; Buettner, A.; Vosika, E.; Sadek, M.; Hao, Z.; Reusch, D.; Koenig, M.; Chan, W.; Bathke, A.; Pallat, H.; Lundin, V.; Kepert, J.F.; Bulau, P.; Deperalta, G.; Yu, C.; Beardsley, R.; Camilli, T.; Harris, R.; Stults, J. Mass spectrometry-based multi-attribute method in protein therapeutics product quality monitoring and quality control. mAbs 2023, 15, 2197668. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



