
Submitted:
04 August 2023
Posted:
07 August 2023
You are already at the latest version
Abstract
Organ transplantation remains the only treatment option for patients with end-stage organ dysfunction. However, there are numerous limitations that challenge its clinical application, including the shortage of organ donations, the quality of donated organs, injury during organ preservation and reperfusion, primary and chronic graft dysfunction, acute and chronic rejection, infection, and carcinogenesis in post-transplantation patients. Acute and chronic inflammation and cell death are two major underlying mechanisms for graft injury. Necroptosis is a type of programmed cell death involved in many diseases and has been studied in the setting of all major solid organ transplants, including the kidney, heart, liver, and lung. It is determined by the underlying donor organ conditions (e.g., age, alcohol consumption, fatty liver, hemorrhage shock, donation after circulatory death, etc.), preservation conditions and reperfusion, as well as allograft rejection. Specific molecular mechanisms of necroptosis have been uncovered in the organ transplantation setting, and potential targeting drugs have been identified. We hope this review article will promote more clinical research to determine the role of necroptosis and other types of programmed cell death in solid organ transplantation to alleviate the clinical burden of ischemia-reperfusion injury and graft rejection.
Keywords:
programmed cell death
; donor organ condition
; ischemia reperfusion injury
; allograft rejection
; inflammation
Introduction
Over the past several decades, organ transplantation has been performed of all the major organs and has become the most effective therapy for patients with end-stage organ failure. [1] However, the current practice still faces major challenges, including shortages in organ donation, the quality of donated organs, which is determined by the donor conditions (e.g., age, sex, smoking history, obesity and organ damage before donation), the types of donations (e.g., donation after brain death (DBD) vs. donation after circulatory death (DCD)), organ preservation conditions (e.g., temperature, time, static storage vs. machine perfusion), and reperfusion conditions (e.g., anesthesia, surgery, post-operative care). Primary graft dysfunction (PGD) is a major cause of early morbidity and mortality after organ transplantation. [2] It also contributes to acute and chronic rejection post-transplantation. Moreover, increased incidence and severity of infection (viral, bacterial, or fungal infection) and carcinogenesis are also challenges for transplant recipients. Among these pathological processes, acute and chronic inflammation and diverse types of cell death are two major underlying mechanisms for graft injury. Therapeutics targeting these processes may prevent and reduce organ injury and improve post-transplant outcomes.
Ischemia-reperfusion (IR) is an inevitable step in donor organ preservation and transplantation. IR injury is one of the primary causes of PGD. [3,4,5] Cellular damage resulting from IR is an important risk factor not only for PGD but also for acute and chronic rejection. [6] It consists of complex pathophysiological processes, including endothelial and epithelial cell dysfunction, acute inflammation, activation of programmed cell death (PCD), and triggers innate and adaptive immune responses. Recently, multiple different types of inflammation related PCD (e.g., necroptosis, pyroptosis, ferroptosis, autophagy associated cell death) have been reported in IR injury in lung transplantation. [7] Of which, necroptosis is particularly interesting, as it has been observed in other solid organ transplantation with clinical relevance.
Generally, necroptosis is defined as cell death mediated through a pathway that depends on the Receptor-Interacting Serine/Threonine-Protein Kinase 1/3 (RIPK1/3) complex (also called necrosome), which can be induced by a class of death receptors (DRs), including TNF receptor 1 (TNFR1), TNF-related apoptosis-inducing ligand-receptors (TRAIL-Rs), Fas and toll like receptors (TLRs). [8,9,10,11,12] The release of inflammatory cytokines, pathogen associated molecular patterns, and damage associated molecular patterns (DAMPs) in the donor and recipient graft can activate these DRs. Upon activation, the necrosome then recruits Mixed Lineage Kinase Domain-Like Protein (MLKL), which can be phosphorylated by RIPK3 [13] and translocated to lipid rafts in the plasma membrane, ultimately resulting in cell membrane rupture and cell death. [10,11,14] Necroptotic cells further release DAMPs and cytokines and propagate the inflammatory response to surrounding cells. [15] Necroptosis can also be initiated by a range of intrinsic factors, such as reactive oxygen species (ROS) and intracellular Ca2+ overload [16], which through p53 and cyclophilin D (CypD) exerts control over mitochondria permeability transition pore (mPTP) opening to trigger regulated necrosis (Figure 1). [17] Necroptosis is involved in many pathological conditions and diseases, [16,18] especially in IR injury [19].
In this review article, we summarized the scientific literatures on the prevalence of necroptosis in solid organ transplantation, focused on the regulatory mechanisms of necroptosis discovered from organ transplantation studies, introduced potential therapeutic targets for necroptosis, and proposed future directions on PCD related research in organ transplantation.
Materials and Methods
For this review, we conducted a literature search using PubMed and Web of Science databases. Our search included specific combinations of keywords such as "transplantation" or "transplant" with "necroptosis," "RIPK1/3," and "MLKL." We also included "ischemia-reperfusion injury" or “donor preservation” or “graft dysfunction” along with organ-specific terms like "liver," "kidney," "lung," and "heart." Studies focusing on necroptosis in other tissue or organ transplantations or those utilizing only ischemia-reperfusion injury models were excluded from this review.
Necroptosis in solid organ transplantation
Necroptosis has been studied in transplantation of all major solid organs, including the kidney, heart, liver, and lung. It can be induced in the donor organ, further enhanced by IR injury, and play an important role in allograft rejection.
Necroptosis in kidney transplantation
The first evidence that necroptosis is involved in organ transplantation was derived from kidney research. In 2013, a research group led by Dr. Zhu-Xu Zhang and Dr. Anthony Jevnikar at the University of Western Ontario, in collaboration with Dr. Linkermann (an expert in necroptosis research), demonstrated that Ripk3-/- mice are resistant to renal IR injury, with better renal function, less necrosis, and lower levels of HMGB-1 (high mobility group box 1, a chromatin protein that can be secreted by immune cells or released by dead cells) in kidney tissue. Ripk3-/- kidney allografts also had reduced histological injury scores, neutrophil infiltration, fibrosis, tubulitis, and vascular injury. Moreover, animals receiving Ripk3-/- kidney allografts achieved greater rejection-free survival. [20] As RIPK1/3 are the most important mediators of necroptosis, this important study built the foundation for the further investigations on necroptosis in organ transplantation.
Cold ischemia (CI) is a risk factor for acute kidney injury after kidney transplantation. Jain et al. used inbred mice to study the effects of CI on kidney transplants. After 1 h CI, the right kidney was transplanted to a recipient, 7 days later, the native left kidney was removed and the function of the right renal graft at postoperative day 8 was assessed. [21] After transplantation, acute tubular necrosis score was significantly higher in CI preserved than directly transplanted renal grafts. The expression of DRs including TNFR1 and TLR4 in the kidney were significantly increased in the CI preserved group. The expression of RIPK1, RIPK3 and phosphorylation of MLKL (pMLKL) were significantly higher in CI preserved kidneys after transplantation. [21] These results indicate that donor kidney preservation conditions may influence post-transplant renal function.
Hypothermic machine perfusion (HMP) is a method used to optimize the donor kidney quality during the preservation period. In rabbit kidneys, 25 minutes of warm ischemic exposure to simulate clinical DCD conditions were followed by either CI static preservation or HMP at 4-8°C for 4h. One day later, renal function was assessed and HMP significantly inhibited inflammation, apoptosis, and necroptosis in donor kidneys compared to those preserved statically. [22]
Necroptosis in heart transplantation
Zhang and Jevnikar’s group then studied necroptosis in heart transplantation extensively. To investigate the effects of necroptosis in donor hearts, B6 Ripk3-/- donor hearts were heterotopically transplanted into fully major histocompatibility complex (MHC) mismatched recipient BALB/c mice. Twelve days after transplantation, Ripk3-/- heart grafts showed significantly lower levels of endothelial damage, lymphocyte infiltration, necrosis, and HMGB1. Moreover, a brief immunosuppressive treatment with sirolimus markedly prolonged Ripk3-/- cardiac allograft survival. [23] They further studied the role of RIPK3 in chronic cardiac allograft rejection and demonstrated that RIPK3 deficiency protected donor cardiac grafts from CD4+ T cell-mediated chronic rejection and improved graft survival. Using a cell culture model, they also demonstrated that CD4+ T cells can induce RIPK3-dependent necroptosis in microvascular endothelial cells (MVEC) by releasing TNFa and inducing direct cell-cell contact cytotoxicity. Furthermore, Fas ligand and granzyme B may contribute to activated alloreactive CD4+ T cell-induced necroptosis in MVECs. [24]
This group further reported that the mPTP opening is an important hallmark of necroptosis in cardiac allografts. [25] Opening of mPTP is largely regulated by CypD, which has been shown to regulate both apoptotic and necrotic cell death. [26] Inhibition or deficiency of CypD protected MVECs from necroptosis in cell culture. Moreover, CypD-deficient cardiac grafts showed prolonged survival in mice. [25] To further elucidate the role of necroptosis in IR injury during cardiac transplant, a cold hypoxia and warm reoxygenation cell culture model was used. [27] In MVECs, after exposed to the simulated IR conditions, apoptosis-inducing factor (AIF), was translocated to the nucleus, which was prevented by the RIPK1 inhibitor, necrostatin-1 stable (Nec-1s), or by CypD-deficiency. CypD deficiency in donor cardiac grafts significantly mitigated graft IR injury and prolonged cardiac allograft survival. [27]
Additionally, IR induced cell death in transplantation can causes leakage of cellular RNA, which can interact with TLR3 on neighbouring cells and propagate inflammation and worsen graft injury. [28] The interaction between TLR3 and RNA leads to caspase-dependent apoptosis, RIPK-dependent necroptosis, and CypD-regulated mitochondrial damage in mouse MVECs. Moreover, TLR3 deficiency protects cardiac grafts from apoptotic and necroptotic cell death and tissue damage post-transplant in a Tlr3−/− heterotopic heart transplant model. [28]
Tuuminen et al. performed intra-abdominal heterotopic heart transplantation with fully MHC-mismatched rats, and they reported that 4h CI followed by 6h reperfusion increased mRNA and protein expression of RIPK1/3 in rat cardiac allografts. Pre-treatment of donor and recipients with a single dose of simvastatin (an HMG-CoA reductase inhibitor) 2h prior to allograft transplant reduced these changes and had a beneficial effect on IR injury. [29]
Necroptosis in liver transplantation
IR injury is also an important contributor to graft dysfunction in liver transplantation. [5] When C57BL6 mice were subjected to warm hepatic IR (90 min ischemia /240 min reperfusion), pre-treatment with RIPK1 inhibitor necrostatin-1 (Nec-1) before ischemic onset did not attenuate IR-induced leukocyte migration, perfusion failure, and hepatocellular injury. Western blot analysis showed baseline RIPK1 expression in livers from sham-operated mice, which was reduced in IR groups. They proposed that the activation of caspases seen in this experimental condition may negatively regulate necroptosis in the liver. [30]
Steatotic livers are prone to more severe IR injury, which has limited their usability for transplantation. In a fatty liver mouse model induced by a western diet, steatotic livers had increased levels of RIPK1, RIPK3, and MLKL. These mice had more severe liver injury and necrotic areas in the liver after IR than mice fed with a control diet. When Mlkl-/- mice were used, although the development of hepatic steatosis was not affected, these mice had decreased hepatic neutrophil infiltration and inflammation, and less IR injury, irrespective of diet. Moreover, Ripk3-/- or Ripk3 kinase-dead knock-in mice were protected against IR injury 24h after reperfusion, irrespective of diet. [31] The levels of liver MLKL and RIPK3 increased in alcohol-fed mice and corresponded with increased liver IR injury. [32]
Intriguingly, when young (8 weeks) and aged (100 weeks) mice were subjected to liver IR (90 min ischemia/6h reperfusion), there were significant increases in liver necroptosis in the aged group post IR injury. Nec-1 decreased hepatocyte necroptosis and liver IR injury in aged mice, without a significant reduction in younger mice. Furthermore, IR induced endoplasmic reticulum (ER) stress in the livers of both young and aged mice, especially so in aged mice. Administration of an ER stress antagonist, 4-phenylbutyrate, alleviated liver IR injury in both young and aged mice. ER stress inhibition reduced hepatocyte necroptosis mainly in aged but not in young mice. [33] These studies demonstrated that necroptosis is involved in the donor liver injury related to the pre-existing conditions (fatty liver, aging, alcohol consumption,etc.), and can be further activated by the IR conditions related to donor organ preservation.
Early allograft dysfunction (EAD) following liver transplantation is a major threat to the clinical outcome of recipients. [34] In a rat liver transplant model, donor livers were preserved statically at CI for 22h followed by transplantation. One day later, significantly higher pMLKL was observed in livers with IR injury. In human samples, the pMLKL score was significantly higher in grafts of patients who developed EAD after transplantation, compared to non-EAD grafts. pMLKL score at 1h after reperfusion and the ratio of pMLKL score between 1h of reperfusion and at the end of preservation are highly predictive for EAD. Liver grafts with a high pMLKL index had significantly higher serum levels of aminotransferases and lactate dehydrogenase 24 h after transplantation. Multivariate logistical regression analysis identified the pMLKL-index as a predictor of EAD development. [35]
Necroptosis in lung transplantation
Interestingly, the first report on transplant related necroptosis in the lung was from a study on so called kidney–lung cross talk. In a rat allogeneic transplantation model, IR injury in renal allografts caused pulmonary injury. RIPK1 and RIPK3 expression were significantly enhanced in the lung. Nec-1 given to recipients with ischemic renal grafts improved lung morphology with decreased hemorrhage and leukocyte infiltration. Acute immune rejection of renal allograft exacerbated the lung injury with enhanced RIPK1 expression. Treatment of animals with cyclosporine A significantly reduced lung injury, and when Nec-1 was combined with cyclosporine A, lung protection was further improved. [36] Cyclosporine A is not only an immunosuppressive drug, but also an effective inhibitor for mPTP opening related regulated necrosis. [37] It was further proposed that osteopontin, a multifunctional glycophosphoprotein that may mediate systemic inflammation, is involved in necroptosis and lung injury after transplantation of ischemic renal allografts. [38]
To determine the underlying mechanisms of IR injury in the lung transplant setting, Kim et al used a CI and warm reperfusion cell culture model that mimicked the ischemia-reperfusion process of transplantation. [39,40] Human lung epithelial cells were stored at 4°C in 50% oxygen to simulate donor lung preservation and cells were then returned to serum containing culture medium at body temperature (37°C) to simulate warm reperfusion. Reperfusion induced mPTP opening and regulated necrosis after prolonged CI, which is mediated by translocation of p53 into mitochondria to form a complex with CypD. Selective inhibition of PKCd by small interference RNA (siRNA) or dV1-1, a peptide PKCd inhibitor, reduced IR-induced inflammation, ER stress, and cell death. dV1-1 also reduced PKCd and p53 translocation to mitochondria. [39,41] dV1-1 and its nano formula also prevented lung IR injury in a left lung transplantation model and in a warm pulmonary IR injury model in rats. [39,41]
Kim et al. further found that the RIPK1 inhibitor Nec-1 reduced IR induced necroptosis in the cell culture model. [40] Necroptosis is usually triggered by the activation of DRs, [18] however, in this study blocking DRs did not affect IR-induced necroptosis. [40] Interestingly, N-acetyl-Leu-Leu-norleucinal (ALLN), a protease calpain inhibitor, reduced RIPK1/RIPK3 expression and pMLKL via signal transducer and activator of transcription 3 (STAT3) pathway. Blocking this calpain-STAT3-RIPK axis reduced ER stress and mitochondrial calcium dysregulation. In human lung transplants, mRNA levels of RIPK1, MLKL, and STAT3 were significantly increased at 2h of reperfusion. Moreover, the levels of pRIPK1, pMLKL and pSTAT3 are higher in human lung tissue samples from patients who developed PGD than those who did not. [40] Administration of Nec-1 to both donor lungs (preserved at 4ºC for 18h) and recipients significantly improved pulmonary gas exchange, reduced lung edema and necrosis in the grafts in rat lung transplant model. [42] Nec-1 significantly alleviated IR-induced lung injury, cytokine release, and necroptosis of epithelial cells in a left lung hilum clamping model in mice. [43] Similarly, necrosulfonamide, a MLKL inhibitor, attenuated IR injury in a rat left hilum clamping and reperfusion model. [44]
Wang et al. used a mouse left lung transplant model to determine the donor lung conditions that may promote the development of PGD. In a single-hit model, donor lungs from inbred C57BL/6 mice were CI preserved at 0°C for 1h, 72h, or 96h before engraftment. Multi-hit models were established by inducing 24h of hemorrhage shock and/or 3h of brain death before 24h of CI preservation. Extending CI to 96h led to increased necroptosis activation in lung grafts 24h post-transplantation. Animals in the multi-hit group showed increased lung injury, cellular infiltration, and activation of both necroptotic and apoptotic pathways. Nec-1 treatment significantly decreased necroptosis activation in both single- and multi-hit models of IR injury. [45]
The initial recruitment of neutrophils to the reperfused lung is a critical step in IR injury post-transplantation. Using a mouse left lung transplant model, Li et al. measured lipid peroxidation products and found that oxidized phosphatidylcholine species were rapidly increased after reperfusion, [46] which is a signal related to necroptosis. [47] Administration of Nec-1 to transplant recipients or using donor lungs from Ripk3-/- mice, significant improved graft function with less neutrophils extravasated and aggregated into large clusters, and better integrity of the subpleural vessels. Graft levels of oxidized phosphatidylcholine species were not elevated in RIPK3-deficient lungs. [46]
Mechanisms of necroptosis in organ transplantation
Organ transplantation may activate necroptosis in special manners. Understanding these features will help to develop strategies to prevent graft injury, dysfunction, and rejection.
DR-dependent and independent activation of necroptosis in organ transplantation
During organ transplantation, necroptosis can be activated via both extrinsic and intrinsic pathways. TNFa and other inflammatory cytokines have been used to induce necroptosis in murine renal tubular epithelial cells (TECs) [20] and murine MVECs [23] in culture. Additionally, proinflammatory cytokines up-regulated TNFR1 expression in murine MVECs. [23] Transplanted CI preserved kidney increased serum TNFa and expression of TNFR1 and TLR4 in the grafts during reperfusion. [21] Ischemic renal allograft transplant-induced remote lung injury in rats was associated with increased expression of TNFR1, TLR2 and TLR4 in the lung tissue, and acute immune rejection increased TNFa levels in serum and lung tissue. [36]
TNFR1 and TNFR2 double deficiency reduced graft arterial disease in murine cardiac allografts. [48] TLR3 can be activated by viral or endogenous RNA released from injured cells. Syngeneic cardiac transplants following 9h CI caused extracellular RNA release and activation of TLR3 in mice, and cardiac grafts from Tlr3-/- mice were protected from IR injury. [49] In IR injury, TLR9-dependent neutrophil extracellular trap formation is stimulated by mitochondrial DNA released from injured cells, [50] and may mediate neutrophil recruitment into ischemic tissue via a TLR9/MyD88/CXCL5 pathway. [51] The neutrophil rolling and adhesion to the vessel wall during reperfusion is mediated through TLR4/TRIF-dependent signaling in lung graft endothelial cells. [51,52] The roles of these TLRs in activation of necroptosis in IR injury and graft rejection should be further determined.
On the other hand, Kim et al. used a cell culture model and demonstrated that neutralization antibodies against TNF, Fas, and TRAIL-R, during both CI and warm reperfusion had no effect on cell death. Using TNFR1 siRNA to supress its expression also did not reduce IR-induced cell death. The expression of cell death receptor interaction proteins, such as XIAP, cIAP, and caspase 8 were not affected by Nec-1 treatment. [40] Therefore, the cold preservation and warm reperfusion conditions can activate necroptosis independent of DRs. Acute depletion of ATP during CI may dysregulate sodium channels, depolarize the plasma membrane, and increase intracellular Ca2+ levels, leading to calpain activation and mediating cell death during myocardial reperfusion. [53] In lung epithelial cells, activated calpain increased RIPK3 expression via STAT3 pathway. [40] A newly discovered highly selective RIPK1 inhibitor, 6E11, protected human aortic endothelial cells from cold hypoxia-warm reoxygenation induced cell death. [54] In MVECs, cold hypoxia and warm reperfusion enhanced TNFa and IFNg induced necroptosis. [27]
During organ transplantation, enriched inflammatory cytokines and DAMPs in donor grafts may interact with multiple DRs to activate necroptosis. On the other hand, the IR conditions imposed on the donor organs may activate intrinsic necroptotic pathways directly. Therefore, to effectively block necroptosis, therapeutics targeting both intrinsic and extrinsic mechanisms should be considered (Figure 1).
Relationship between necroptosis and apoptosis in organ transplantation
The IR process in organ transplantation may activate both apoptosis and necroptosis. Both apoptosis and necroptosis can be activated by DRs, of which caspase activity is crucial in switching between these two types of cell death. As mentioned above, caspase activation may negatively regulate necroptosis in a murine warm hepatic IR model. [30] In renal TECs, [20] or in MVECs, [23] TNFa induced necroptosis as caspase activity was inhibited. In mice, caspase 8 silencing with shRNA in donor kidneys decreased renal allograft survival with increased tissue necrosis. [20] However, inhibition of caspase 8 with shRNA attenuated IR injury induced by renal artery clamping at 32°C in mice. [58] Given the conflicting roles of caspase 8 in IRI, further investigations are needed.
In a murine model, tubular cell apoptosis and caspase 9 expression were induced by CI, while expression of both caspase 8 and necroptotic proteins (RIPK1, RIPK3, pMLKL, and TLR4) was significantly increased during reperfusion. [21,39] Similarly, apoptosis was observed after 18h of CI in human lung epithelial cells, which was switched to necrosis after reperfusion. [39] Therefore, apoptosis and necroptosis may be involved in IR injury at different stages.
Pretreatment of donors and recipients with a single dose of simvastatin 2h prior to allograft transplant reduced the mRNA expression and proteins of both apoptosis and necroptosis related molecules in cardiac allografts after CI (4h) followed by 6h reperfusion. [29] In a rat lung transplant model, apoptosis was observed after reperfusion from grafts preserved at cold temperature for 6h or 12h, but necrosis was observed in grafts preserved at 18h or 24h. [59] Blocking apoptosis with a pan-caspase inhibitor [60] or annexin V dimer were both effective in reducing apoptosis [61] and improving graft function in lung transplantation. Thus, blocking apoptosis may prevent its progression towards necroptosis. On the other hand, Nec-1 reduced CI-warm reperfusion induced necroptosis as well as apoptosis in a cell culture model. [43]
Taken together, these observations suggest that during organ transplantation under certain circumstance, apoptosis and necroptosis could co-exist, or be switched from one type to the other. The detailed mechanisms of these processes need to be further investigated. Recently, a new concept, “PANoptosis” has been introduced - as an inflammatory PCD pathway with key features of pyroptosis, apoptosis and/or necroptosis. It has been proposed that this is regulated by a so-called PANoptosome complex. [62] This and other types of PCD [7] should be further studied in the organ transplant setting.
Potential therapeutic targets and substances targeting necroptosis
Inhibition of necroptosis represents a therapeutic strategy for IR injury and allograft rejection. Several substances have been used as experimental tools and could be further developed as therapeutics (Table 1).
RIPK1 inhibitors
Necrostatins are a series of specific and potent small-molecule inhibitors of necroptosis. Necrostatin-1(Nec-1) has been used in various solid organ transplantation models to block RIPK1 activity. [20,23,24,40,42,43,45] The new Nec-1 analog, necrostatin-1 stable (Nec-1s), showed improved pharmacokinetic properties in vivo, [63] without any paradoxical toxicity at lower concentrations observed with Nec-1. [64] Nec-1s was very effective in reducing necroptosis and the release of HMGB1 in cardiac graft injury and rejection. [24,27]
GSK2982772 is an oral, selective, and specific inhibitor of RIPK1. [65] Its safety and tolerability have been tested in healthy human volunteers (Phase 1), [66] and in a phase 2a clinical study for the treatment of inflammatory diseases. [65,67] 6E11, a highly selective inhibitor of RIPK1, shows more potent protective effects against cold hypoxia/warm reoxygenation-induced necroptosis in human aortic endothelial cells than Nec-1 or Ned-1s. [54] Zharp1-211, another novel RIPK1 inhibitor, selectively inhibits RIPK1 kinase activity and reduces graft versus host disease in mice. [68]
RIPK3 inhibitors
MLKL inhibitor
Necrosulfonamide targets the pseudo kinase domain of MLKL, and it inhibited necroptosis in a rat pulmonary IR injury model. [44]
CypD inhibitor
Indirect inhibitors for necroptosis
dV1-1, a specific peptide PKCd inhibitor, ameliorated IR lung injury [39] and myocardial infarction. [72] N-acetyl-Leu-Leu-norleucinal (ALLN) is a strong, competitive inhibitor of the Ca2+-dependent neutral cysteine proteases calpain I and calpain II. It reduced IR-induced necroptosis in a cell culture model, [40] IR induced myocardial and cerebral injury, [73,74] and IR injury in rat lung transplants. [75]
Conclusion and future directions
Clinical relevance of necroptosis in organ transplantation
The roles of necroptosis in organ transplantation are complex (Figure 2). The incidence and severity of necroptosis is influenced by the donor conditions (e.g., age, fatty liver, hemorrhagic shock, brain death, circulatory death, etc.), and the systemic inflammatory responses, such as the crosstalk between the renal transplant and the lung. [36,38] The inflammatory responses in DBDs affect the quality of donor lung. [76] A proteomics study revealed that biomarkers in the donor plasma of DBDs predicts heart transplant outcome. [77] Transcriptomics studies showed that cell death pathways are more predominately activated in DCD donor lungs than that in DBD lungs. [78] Whether these pathways are related to necroptosis need to be determined.
The organ preservation conditions also influence the degree of necroptosis. CI time differentially affects donor organs depending on their condition. Transcriptomic studies have demonstrated that inflammatory responses and cell death are the two predominant mechanistic pathways activated in human lung grafts during the IR process. [76] Similar gene expression changes in other solid organ grafts may also be studied with focus on necroptosis.
Necroptosis is an important underlying mechanism for acute and chronic rejection after kidney [20] and heart [23,25,27,28] transplantation. Allografts from RIPK3, CypD, and TLR3 deficient donors exhibited prolonged graft survival. Similar studies should be conducted in other solid organ transplants.
Molecular mechanisms of necroptosis in organ transplantation
Several unique features of necroptosis in organ transplantation are discussed in this review article. Activation of DRs by inflammatory responses and activation of intrinsic pathways by IR conditions may have synergic effects in necroptosis. Crosstalk between the classical necroptotic pathways and mPTP opening related pathways may accelerate the necrosis in grafts. The therapeutic strategies should be developed accordingly. Different types of cell death are associated with IR-induced graft damage and dysfunction in the kidney, liver, lung, and heart. [79,80,81] The relationship among different types of cell death needs to be studied further.
Therapeutics and biomarkers of necroptosis in organ transplantation
Several potential drugs targeting necroptosis were described in this paper. While most of these drugs have been used experimentally to elucidate the importance of necroptosis and potential mechanisms in the organ transplant setting, their clinical applications need to be developed systematically with careful pharmacological, pharmacokinetic, and pharmacodynamic and toxicity studies in pre-clinical animal models and then in clinical trials. Necroptosis related biomarkers for clinical diagnosis and therapeutic guidance should of course be explored as well.
In conclusion, cell death plays a prominent role in the injury related to transplantation of all organs. More extensive research into mechanism of cell death and specifically necroptosis in organ transplantation is warranted. Examination of these pathway in clinical samples with clinical outcome data will reveal more opportunities for diagnosis and treatment in solid organ transplantation.
Author Contributions
YZ: KM and ML collected and reviewed references. YZ and ML drafted the manuscript. KM, TA, and SK edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This work is supported by research grants from University of Toronto’s Medicine by Design (MbDPEFR1-2021-01), and New Frontiers in Research Funding – Transformation grant (NFRFT-2020-00787). Dr. Mingyao Liu is James and Mary Davie Chair in Lung Injury, Repair and Regeneration at the University Health Network and University of Toronto.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO, Transplantation S, Organization Nacional de T. Third WHO Global Consultation on Organ Donation and Transplantation: Striving to achieve self-sufficiency, March 23-25, 2010, Madrid, Spain. Transplantation 2011;91 Suppl 11:S27-8. [CrossRef]
- Lee JC, Christie JD. Primary graft dysfunction. Proc Am Thorac Soc 2009;6(1):39-46. [CrossRef]
- Al-Adhami A, Avtaar Singh SS, De SD; et al. Primary Graft Dysfunction after Heart Transplantation - Unravelling the Enigma. Curr Probl Cardiol 2022;47(8):100941. [CrossRef]
- Gelman AE, Fisher AJ, Huang HJ; et al. Report of the ISHLT Working Group on Primary Lung Graft Dysfunction Part III: Mechanisms: A 2016 Consensus Group Statement of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2017;36(10):1114-1120. [CrossRef]
- Hirao H, Nakamura K, Kupiec-Weglinski JW. Liver ischaemia-reperfusion injury: A new understanding of the role of innate immunity. Nat Rev Gastroenterol Hepatol 2022;19(4):239-256. [CrossRef]
- Zhao H, Alam A, Soo AP, George AJT, Ma D. Ischemia-Reperfusion Injury Reduces Long Term Renal Graft Survival: Mechanism and Beyond. EBioMedicine 2018;28:31-42. [CrossRef]
- Capuzzimati M, Hough O, Liu M. Cell death and ischemia-reperfusion injury in lung transplantation. J Heart Lung Transplant 2022;41(8):1003-1013. [CrossRef]
- He S, Wang L, Miao L; et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009;137(6):1100-11. [CrossRef]
- Zhang DW, Shao J, Lin J; et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009;325(5938):332-6. [CrossRef]
- Zhao J, Jitkaew S, Cai Z; et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A 2012;109(14):5322-7. [CrossRef]
- Wang H, Sun L, Su L; et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 2014;54(1):133-146. [CrossRef]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014;15(2):135-47. [CrossRef]
- Sun L, Wang H, Wang Z; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012;148(1-2):213-27. [CrossRef]
- Cai Z, Jitkaew S, Zhao J; et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014;16(1):55-65. [CrossRef]
- Todd JL, Palmer SM. Danger signals in regulating the immune response to solid organ transplantation. J Clin Invest 2017;127(7):2464-2472. [CrossRef]
- Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature 2015;517(7534):311-20. [CrossRef]
- Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012;149(7):1536-48. [CrossRef]
- Linkermann A, Green DR. Necroptosis. N Engl J Med 2014;370(5):455-65. [CrossRef]
- Linkermann A, Brasen JH, Darding M; et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci U S A 2013;110(29):12024-9. [CrossRef]
- Lau A, Wang S, Jiang J; et al. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am J Transplant 2013;13(11):2805-18. [CrossRef]
- Jain S, Plenter R, Nydam T, Jani A. Injury Pathways That Lead to AKI in a Mouse Kidney Transplant Model. Transplantation 2020;104(9):1832-1841. [CrossRef]
- Yang Z, Zhong Z, Li M; et al. Hypothermic machine perfusion increases A20 expression which protects renal cells against ischemia/reperfusion injury by suppressing inflammation, apoptosis and necroptosis. Int J Mol Med 2016;38(1):161-71. [CrossRef]
- Pavlosky A, Lau A, Su Y; et al. RIPK3-mediated necroptosis regulates cardiac allograft rejection. Am J Transplant 2014;14(8):1778-90. [CrossRef]
- Kwok C, Pavlosky A, Lian D; et al. Necroptosis Is Involved in CD4+ T Cell-Mediated Microvascular Endothelial Cell Death and Chronic Cardiac Allograft Rejection. Transplantation 2017;101(9):2026-2037. [CrossRef]
- Gan I, Jiang J, Lian D; et al. Mitochondrial permeability regulates cardiac endothelial cell necroptosis and cardiac allograft rejection. Am J Transplant 2019;19(3):686-698. [CrossRef]
- Baines C, Kaiser, R., Purcell, N. et al. . Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005;434(7033):658-662. [CrossRef]
- Qamar A, Zhao J, Xu L; et al. Cyclophilin D Regulates the Nuclear Translocation of AIF, Cardiac Endothelial Cell Necroptosis and Murine Cardiac Transplant Injury. Int J Mol Sci 2021;22(20):11038. [CrossRef]
- Zhao J, Huang X, McLeod P; et al. Toll-like receptor 3 is an endogenous sensor of cell death and a potential target for induction of long-term cardiac transplant survival. Am J Transplant 2021;21(10):3268-3279. [CrossRef]
- Tuuminen R, Holmstrom E, Raissadati A; et al. Simvastatin pretreatment reduces caspase-9 and RIPK1 protein activity in rat cardiac allograft ischemia-reperfusion. Transpl Immunol 2016;37:40-45. [CrossRef]
- Rosentreter D, Funken D, Reifart J, Mende K, Rentsch M, Khandoga A. RIP1-Dependent Programmed Necrosis is Negatively Regulated by Caspases During Hepatic Ischemia-Reperfusion. Shock 2015;44(1):72-6. [CrossRef]
- Ni HM, Chao X, Kaseff J; et al. Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3)-Mixed Lineage Kinase Domain-Like Protein (MLKL)-Mediated Necroptosis Contributes to Ischemia-Reperfusion Injury of Steatotic Livers. Am J Pathol 2019;189(7):1363-1374. [CrossRef]
- Chen H, McKeen T, Chao X; et al. The role of MLKL in Hepatic Ischemia-Reperfusion Injury of Alcoholic Steatotic Livers. Int J Biol Sci 2022;18(3):1096-1106. [CrossRef]
- Zhong W, Wang X, Rao Z; et al. Aging aggravated liver ischemia and reperfusion injury by promoting hepatocyte necroptosis in an endoplasmic reticulum stress-dependent manner. Ann Transl Med 2020;8(14):869. [CrossRef]
- Olthoff KM, Kulik L, Samstein B; et al. Validation of a current definition of early allograft dysfunction in liver transplant recipients and analysis of risk factors. Liver Transpl 2010;16(8):943-9. [CrossRef]
- Shi S, Bonaccorsi-Riani E, Schurink I; et al. Liver Ischemia and Reperfusion Induce Periportal Expression of Necroptosis Executor pMLKL Which Is Associated With Early Allograft Dysfunction After Transplantation. Front Immunol 2022;13:890353. [CrossRef]
- Zhao H, Ning J, Lemaire A; et al. Necroptosis and parthanatos are involved in remote lung injury after receiving ischemic renal allografts in rats. Kidney Int 2015;87(4):738-48. [CrossRef]
- Piot C CP, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, André-Fouët X, Revel D, Kirkorian G, Monassier JP, Derumeaux G, Ovize M. Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction. N Engl J Med 2008;359(5):473-81. [CrossRef]
- Zhao H, Chen Q, Huang H; et al. Osteopontin mediates necroptosis in lung injury after transplantation of ischaemic renal allografts in rats. Br J Anaesth 2019;123(4):519-530. [CrossRef]
- Kim H, Zhao J, Zhang Q; et al. deltaV1-1 Reduces Pulmonary Ischemia Reperfusion-Induced Lung Injury by Inhibiting Necrosis and Mitochondrial Localization of PKCdelta and p53. Am J Transplant 2016;16(1):83-98. [CrossRef]
- Kim H, Zamel R, Bai XH; et al. Ischemia-reperfusion induces death receptor-independent necroptosis via calpain-STAT3 activation in a lung transplant setting. Am J Physiol Lung Cell Mol Physiol 2018;315(4):L595-L608. [CrossRef]
- Lee D, Zhao J, Yang H; et al. Effective delivery of a rationally designed intracellular peptide drug with gold nanoparticle-peptide hybrids. Nanoscale 2015;7(29):12356-60. [CrossRef]
- Kanou T, Ohsumi A, Kim H; et al. Inhibition of regulated necrosis attenuates receptor-interacting protein kinase 1-mediated ischemia-reperfusion injury after lung transplantation. J Heart Lung Transplant 2018;37(10):1261-1270. [CrossRef]
- Dong L, Liang F, Lou Z; et al. Necrostatin-1 Alleviates Lung Ischemia-Reperfusion Injury via Inhibiting Necroptosis and Apoptosis of Lung Epithelial Cells. Cells 2022;11(19):3139. [CrossRef]
- Ueda S, Chen-Yoshikawa TF, Tanaka S; et al. Protective effect of necrosulfonamide on rat pulmonary ischemia-reperfusion injury via inhibition of necroptosis. J Thorac Cardiovasc Surg 2022;163(2):e113-e122. [CrossRef]
- Wang X, O’Brien ME, Yu J; et al. Prolonged Cold Ischemia Induces Necroptotic Cell Death in Ischemia-Reperfusion Injury and Contributes to Primary Graft Dysfunction after Lung Transplantation. Am J Respir Cell Mol Biol 2019;61(2):244-256. [CrossRef]
- Li W, Terada Y, Tyurina YY; et al. Necroptosis triggers spatially restricted neutrophil-mediated vascular damage during lung ischemia reperfusion injury. Proc Natl Acad Sci U S A 2022;119(10):e2111537119. [CrossRef]
- Wiernicki B, Dubois H, Tyurina YY; et al. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis 2020;11(10):922. [CrossRef]
- Suzuki J CS, Batirel S, Kosuge H, Shimizu K, Isobe M, Libby P, Mitchell RN. Tumor necrosis factor receptor -1 and -2 double deficiency reduces graft arterial disease in murine cardiac allografts. Am J Transplant 2003;3(8):968-76. [CrossRef]
- Gollmann-Tepekoylu C, Graber M, Polzl L; et al. Toll-like receptor 3 mediates ischaemia/reperfusion injury after cardiac transplantation. Eur J Cardiothorac Surg 2020;57(5):826-835. [CrossRef]
- Mallavia B, Liu F, Lefrancais E; et al. Mitochondrial DNA Stimulates TLR9-Dependent Neutrophil Extracellular Trap Formation in Primary Graft Dysfunction. Am J Respir Cell Mol Biol 2020;62(3):364-372. [CrossRef]
- Li W, Hsiao HM, Higashikubo R; et al. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 2016;1(12). [CrossRef]
- Li W, Feng G, Gauthier JM; et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest 2019;129(6):2293-2304. [CrossRef]
- Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res 2012;94(2):168-80. [CrossRef]
- Delehouze C, Leverrier-Penna S, Le Cann F; et al. 6E11, a highly selective inhibitor of Receptor-Interacting Protein Kinase 1, protects cells against cold hypoxia-reoxygenation injury. Sci Rep 2017;7(1):12931. [CrossRef]
- Bopassa JC, Michel P, Gateau-Roesch O, Ovize M, Ferrera R. Low-pressure reperfusion alters mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 2005;288(6):H2750-5. [CrossRef]
- Mitochondrial permeability transition pore- an enigmatic gatekeeper. 2012. [CrossRef]
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta 2009;1787(11):1395-401. [CrossRef]
- Du C, Wang S, Diao H, Guan Q, Zhong R, Jevnikar AM. Increasing resistance of tubular epithelial cells to apoptosis by shRNA therapy ameliorates renal ischemia-reperfusion injury. Am J Transplant 2006;6(10):2256-67. [CrossRef]
- Fischer S MA, Liu M, Cardella JA, Slutsky AS, Suga M, Moreira JF, and Keshavjee S. Dynamic Changes in Apoptotic and Necrotic Cell Death Correlate with Severity of Ischemia-Reperfusion Injury in Lung Transplantation. Am J Respir Crit Care Med 2000;162(5):1932-1939. [CrossRef]
- Quadri SM, Segall L, de Perrot M; et al. Caspase inhibition improves ischemia-reperfusion injury after lung transplantation. Am J Transplant 2005;5(2):292-9. [CrossRef]
- Hashimoto K, Kim H, Oishi H; et al. Annexin V homodimer protects against ischemia reperfusion-induced acute lung injury in lung transplantation. J Thorac Cardiovasc Surg 2016;151(3):861-869. [CrossRef]
- Wang Y, Kanneganti TD. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J 2021;19:4641-4657. [CrossRef]
- Degterev A, Hitomi J, Germscheid M; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008;4(5):313-21. [CrossRef]
- Takahashi N, Duprez L, Grootjans S; et al. Necrostatin-1 analogues: Critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis 2012;3:e437. [CrossRef]
- Harris PA, Berger SB, Jeong JU; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J Med Chem 2017;60(4):1247-1261. [CrossRef]
- Weisel K, Scott NE, Tompson DJ; et al. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol Res Perspect 2017;5(6):e00365. [CrossRef]
- Weisel K, Berger S, Thorn K; et al. A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res Ther 2021;23(1):85. [CrossRef]
- Yu X MH, Li B; et al. . A Novel RIPK1 Inhibitor Reduces GVHD in Mice via a Non-immunosuppressive Mechanism that Restores Intestinal Homeostasis. Blood 2022;141(9). [CrossRef]
- Mandal P, Berger SB, Pillay S; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 2014;56(4):481-95. [CrossRef]
- Zhao H, Jaffer T, Eguchi S, Wang Z, Linkermann A, Ma D. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis 2015;6:e1975. [CrossRef]
- World Health Organization S, Marc C, Kouimtzi, Maria & Hill, Suzanne. WHO Model Formulary 2008. World Health Organization. 2009.
- Bates E, Bode C, Costa M; et al. Intracoronary KAI-9803 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction. Circulation 2008;117(7):886-96. [CrossRef]
- Hernando V, Inserte J, Sartorio CL, Parra VM, Poncelas-Nozal M, Garcia-Dorado D. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J Mol Cell Cardiol 2010;49(2):271-9. [CrossRef]
- Wang WY, Xie L, Zou XS; et al. Inhibition of extracellular signal-regulated kinase/calpain-2 pathway reduces neuroinflammation and necroptosis after cerebral ischemia-reperfusion injury in a rat model of cardiac arrest. Int Immunopharmacol 2021;93:107377. [CrossRef]
- Matsui Y, Kanou T, Matsui T; et al. Protective Effect of Calpain Inhibition During Cold Ischemia on Ischemia-reperfusion Injury After Lung Transplantation. Transplantation 2023;10.1097/TP.0000000000004515. [CrossRef]
- Wong A, Zamel R, Yeung J; et al. Potential therapeutic targets for lung repair during human ex vivo lung perfusion. Eur Respir J 2020;55(4):1902222. [CrossRef]
- Lukac J, Dhaygude K, Saraswat M; et al. Plasma proteome of brain-dead organ donors predicts heart transplant outcome. J Heart Lung Transplant 2022;41(3):311-324. [CrossRef]
- Baciu C, Sage A, Zamel R; et al. Transcriptomic investigation reveals donor-specific gene signatures in human lung transplants. Eur Respir J 2021;57(4):2000327. [CrossRef]
- Yamada N, Karasawa T, Wakiya T; et al. Iron overload as a risk factor for hepatic ischemia-reperfusion injury in liver transplantation: Potential role of ferroptosis. Am J Transplant 2020;20(6):1606-1618. [CrossRef]
- Zheng P, Kang J, Xing E, Zheng B, Wang X, Zhou H. Lung Inflation With Hydrogen During the Cold Ischemia Phase Alleviates Lung Ischemia-Reperfusion Injury by Inhibiting Pyroptosis in Rats. Front Physiol 2021;12:699344. [CrossRef]
- Lu J, Xu L, Zeng Z; et al. Normothermic ex vivo Heart Perfusion Combined With Melatonin Enhances Myocardial Protection in Rat Donation After Circulatory Death Hearts via Inhibiting NLRP3 Inflammasome-Mediated Pyroptosis. Front Cell Dev Biol 2021;9:733183. [CrossRef]
Figure 1.
Basic signaling pathways of necroptosis. Necroptosis can be triggered by the activation of death receptors, which induce the phosphorylation of receptor-interacting protein kinase 1 (RIPK1) and RIPK3 to form necrosome when the activity of caspase 8 is reduced. Necrosome then phosphorylate mixed lineage kinase domain-like protein (MLKL), causing it to translocate to the cell membrane and forms pore-like structures that disrupt the integrity of the plasma membrane. This leading to the release of cellular components and cell death. Aside from the activation of death receptors, necroptosis can also be triggered by reactive oxygen species (ROS). ROS can induce the translocation of p53 into the mitochondria, where it forms a complex with cyclophilin D, leading to the opening of the mitochondrial permeability transition pore (mPTP) and ultimately inducing regulated necrosis. Additionally, ROS can increase the accumulation of cytosolic Ca2+ and activate proteases such as calpain, which in turn activate necrosome formation.
Figure 1.
Basic signaling pathways of necroptosis. Necroptosis can be triggered by the activation of death receptors, which induce the phosphorylation of receptor-interacting protein kinase 1 (RIPK1) and RIPK3 to form necrosome when the activity of caspase 8 is reduced. Necrosome then phosphorylate mixed lineage kinase domain-like protein (MLKL), causing it to translocate to the cell membrane and forms pore-like structures that disrupt the integrity of the plasma membrane. This leading to the release of cellular components and cell death. Aside from the activation of death receptors, necroptosis can also be triggered by reactive oxygen species (ROS). ROS can induce the translocation of p53 into the mitochondria, where it forms a complex with cyclophilin D, leading to the opening of the mitochondrial permeability transition pore (mPTP) and ultimately inducing regulated necrosis. Additionally, ROS can increase the accumulation of cytosolic Ca2+ and activate proteases such as calpain, which in turn activate necrosome formation.
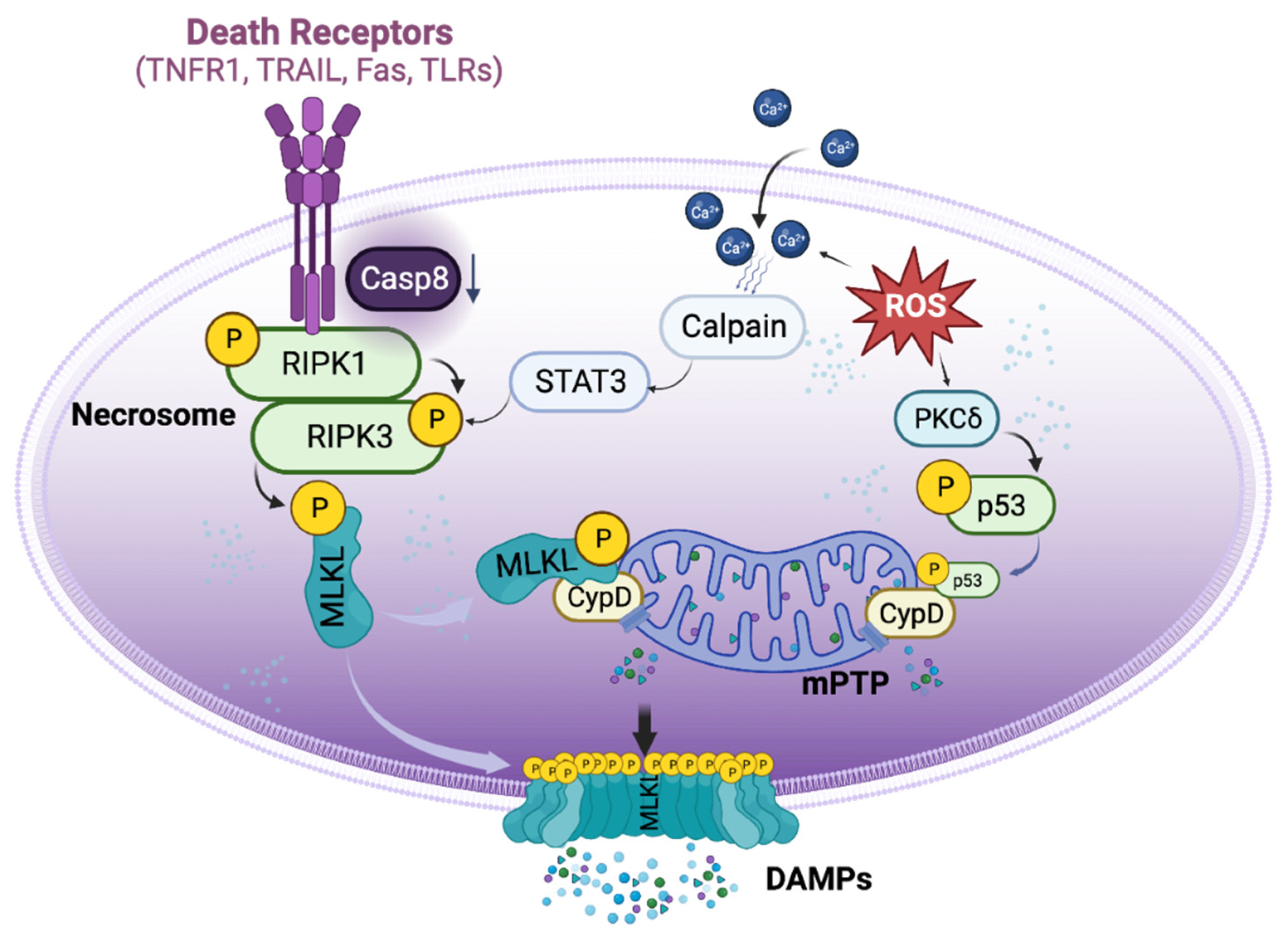
Figure 2.
The significance of necroptosis in organ transplantation. Necroptosis in the donor organ can be trigged by donor conditions and can further be affected by the conditions of organ preservation. Necroptosis contributes to ischemia-reperfusion injury, and to primary and chronic graft dysfunction and rejection.
Figure 2.
The significance of necroptosis in organ transplantation. Necroptosis in the donor organ can be trigged by donor conditions and can further be affected by the conditions of organ preservation. Necroptosis contributes to ischemia-reperfusion injury, and to primary and chronic graft dysfunction and rejection.
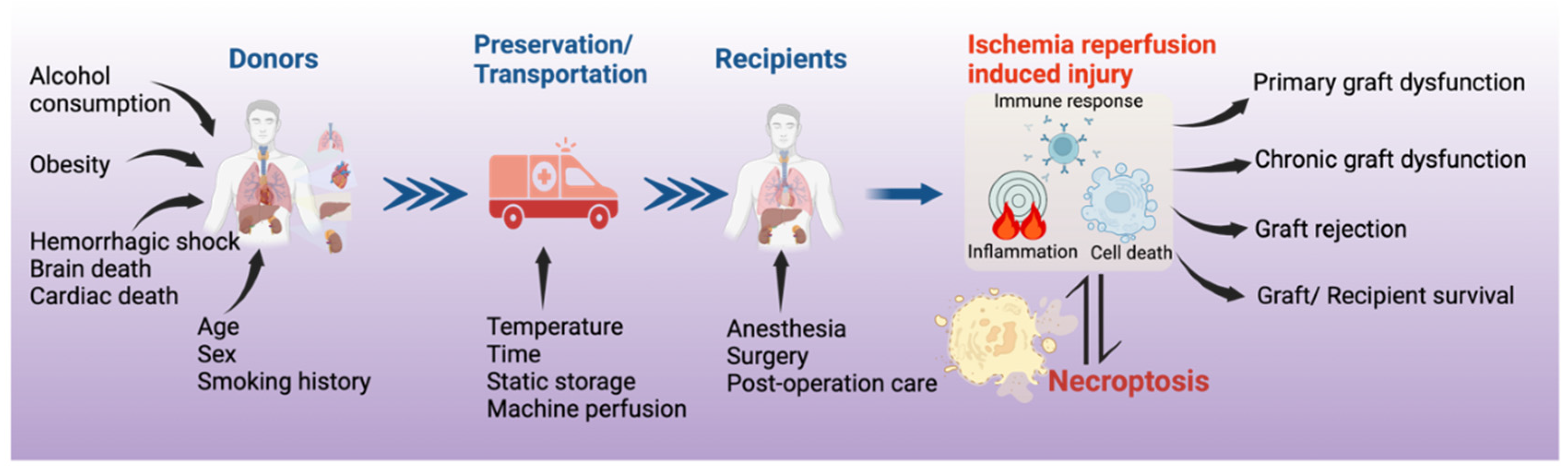

Table 1.
Substances that have inhibitory effects on necroptosis.
| Target | Substances | Research Setting | Models | References |
|---|---|---|---|---|
| RIPK1 | Necrostatin-1, Necrostatin-1 stable |
Renal/heart/lung transplant | Cell culture and animal models | 20, 23, 24, 40, 42, 43, 45 |
| RIPK1 | GSK2982772 | Inflammatory diseases | Phase 2a clinical trial | 65, 67 |
| RIPK1 | 6E11 | Hypoxia/reoxygenation | Cell culture | 54 |
| RIPK1 | Zharp1-211 | Graft versus host disease | Animal model | 68 |
| RIPK3 | GSK′840, GSK′843, GSK′872 | Colon Carcinoma | Cell culture | 69, 70 |
| MLKL | Necrosulfonamide | Lung ischemia/reperfusion | Animal model | 44 |
| cyclophilin D | Cyclosporin A | Heart transplant | Animal model | 27 |
| Protein Kinase C𝛿 | δV1-1 | Lung transplant | Cell culture and animal models | 39, 41 |
| Calpain | N-acetyl-Leu-Leu-norleucinal | Ischemia/reperfusion | Cell culture and animal model | 40, 73, 74, 75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



